Mutations in microphthalmia, the mouse homolog of the human deafness gene MITF, affect neuroepithelial and neural crest-derived melanocytes differently Atsuo Nakayama 1 , Minh-Thanh T. Nguyen, Catherine C. Chen, Karin Opdecamp 2 , Colin A. Hodgkinson 3 , Heinz Arnheiter* Laboratory of Developmental Neurogenetics, National Institutes of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA Received 24 September 1997; accepted 11 November 1997 Abstract The mouse microphthalmia (Mitf) gene encodes a basic-helix-loop-helix-zipper transcription factor whose mutations are associated with abnormalities in neuroepithelial and neural crest-derived melanocytes. In wild type embryos, Mitf expression in neuropithelium and neural crest precedes that of the melanoblast marker Dct, is then co-expressed with Dct, and gradually fades away except in cells in hair follicles. In embryos with severe Mitf mutations, neural crest-derived Mitf-expressing cells are rare, lack Dct expression, and soon become undetectable. In contrast, the neuroepithelial-derived Mitf-expressing cells of the retinal pigment layer are retained, express Dct, but not the melanogenic enzyme genes tyrosinase and Tyrp1, and remain unpigmented. The results show that melanocyte development critically depends on functional Mitf and that Mitf mutations affect the neural crest and the neuroepithelium in different ways. 1998 Elsevier Science Ireland Ltd. Keywords: Retinal pigment epithelium; Stria vascularis; Basic-helix-loop-helix-zipper protein; DOPAchrome tautomerase; Kit 1. Introduction Mutations that are associated with pigment cell abnorm- alities occur in many species, including mouse and man. In the mouse, more than 80 different loci are known to influ- ence the development or function of pigment cells (Green, 1989). Approximately one-quarter of these loci have been analyzed at the molecular level and a plethora of transcrip- tion factors, signaling systems, motor proteins and pigment enzymes have been identified that are required for normal melanogenesis (Spritz and Hearing, 1994; Mouse Genome Database, 1997). Mutant alleles at these loci may not only affect pigment cells but also other cells, either because of shared expression of the mutant genes or because abnormal pigment cells affect the physiology of other cells. Conse- quently, disturbances of pigmentation are often part of syn- dromes that may include abnormalities in hematopoiesis, gametogenesis or sensory organs such as eyes or ears (Spritz and Hearing, 1994; Online Mendelian Inheritance in Man, 1997). Thus, the analysis of these mutations is important far beyond the goal of understanding pigment cell develop- ment. The melanocytes that make up the retinal pigment epithe- lium (RPE), and those that are found in part of the iris, Harderian gland, choroid, inner ear and skin have distinct developmental origins. The RPE cells are derived from the proximal parts of the budding diencephalic neural epithe- lium while the other pigment cells are derived from the neural crest. As a consequence, mutations in some genes, such as those encoding the tyrosine kinase receptor Kit or its ligand Mgf, may affect neural crest-derived melanocytes but spare neuroepithelial melanocytes (Geissler et al., 1988; Mechanisms of Development 70 (1998) 155–166 0925-4773/98/$19.00 1998 Elsevier Science Ireland Ltd. All rights reserved PII S0925-4773(97)00188-3 * Corresponding author. LDN, NINDS, NIH, Building 36/Room 5D04, 36 Convent Drive MSC 4160, Bethesda, MD 20892-4160, USA. Tel.: +1 301 4961645; fax: +1 301 4960899; e-mail: [email protected] 1 Present address: Department of Pathology, Nagoya University School of Medicine, Nagoya 466, Japan. 2 Present address: CNRS EP 560/Institut Pasteur de Lille, Diffe ´renciation Cellulaire et Mole ´culaire 1, rue Calmette, B.P. 245, Lille Cedex, France. 3 Present address: Howard Hughes Medical Institute, University of Michigan, 1150 West Medical Center Drive, Ann Arbor, MI 48109- 0650, USA.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Mutations inmicrophthalmia,the mouse homolog of the human deafness geneMITF, affect neuroepithelial and neural crest-derived melanocytes differently
Atsuo Nakayama1, Minh-Thanh T. Nguyen, Catherine C. Chen, Karin Opdecamp2,Colin A. Hodgkinson3, Heinz Arnheiter*
Laboratory of Developmental Neurogenetics, National Institutes of Neurological Disorders and Stroke,National Institutes of Health, Bethesda, MD 20892, USA
Received 24 September 1997; accepted 11 November 1997
Abstract
The mousemicrophthalmia (Mitf)gene encodes a basic-helix-loop-helix-zipper transcription factor whose mutations are associated withabnormalities in neuroepithelial and neural crest-derived melanocytes. In wild type embryos,Mitf expression in neuropithelium and neuralcrest precedes that of the melanoblast markerDct, is then co-expressed withDct, and gradually fades away except in cells in hair follicles.In embryos with severeMitf mutations, neural crest-derivedMitf-expressing cells are rare, lackDct expression, and soon becomeundetectable. In contrast, the neuroepithelial-derivedMitf-expressing cells of the retinal pigment layer are retained, expressDct, but notthe melanogenic enzyme genes tyrosinase andTyrp1,and remain unpigmented. The results show that melanocyte development criticallydepends on functionalMitf and thatMitf mutations affect the neural crest and the neuroepithelium in different ways. 1998 ElsevierScience Ireland Ltd.
Keywords:Retinal pigment epithelium; Stria vascularis; Basic-helix-loop-helix-zipper protein; DOPAchrome tautomerase; Kit
1. Introduction
Mutations that are associated with pigment cell abnorm-alities occur in many species, including mouse and man. Inthe mouse, more than 80 different loci are known to influ-ence the development or function of pigment cells (Green,1989). Approximately one-quarter of these loci have beenanalyzed at the molecular level and a plethora of transcrip-tion factors, signaling systems, motor proteins and pigmentenzymes have been identified that are required for normalmelanogenesis (Spritz and Hearing, 1994; Mouse Genome
Database, 1997). Mutant alleles at these loci may not onlyaffect pigment cells but also other cells, either because ofshared expression of the mutant genes or because abnormalpigment cells affect the physiology of other cells. Conse-quently, disturbances of pigmentation are often part of syn-dromes that may include abnormalities in hematopoiesis,gametogenesis or sensory organs such as eyes or ears (Spritzand Hearing, 1994; Online Mendelian Inheritance in Man,1997). Thus, the analysis of these mutations is important farbeyond the goal of understanding pigment cell develop-ment.
The melanocytes that make up the retinal pigment epithe-lium (RPE), and those that are found in part of the iris,Harderian gland, choroid, inner ear and skin have distinctdevelopmental origins. The RPE cells are derived from theproximal parts of the budding diencephalic neural epithe-lium while the other pigment cells are derived from theneural crest. As a consequence, mutations in some genes,such as those encoding the tyrosine kinase receptorKit or itsligandMgf, may affect neural crest-derived melanocytes butspare neuroepithelial melanocytes (Geissler et al., 1988;
Mechanisms of Development 70 (1998) 155–166
0925-4773/98/$19.00 1998 Elsevier Science Ireland Ltd. All rights reservedPII S0925-4773(97)00188-3
* Corresponding author. LDN, NINDS, NIH, Building 36/Room 5D04,36 Convent Drive MSC 4160, Bethesda, MD 20892-4160, USA. Tel.: +1301 4961645; fax: +1 301 4960899; e-mail: [email protected]
1 Present address: Department of Pathology, Nagoya University Schoolof Medicine, Nagoya 466, Japan.
2 Present address: CNRS EP 560/Institut Pasteur de Lille, Diffe´renciationCellulaire et Moleculaire 1, rue Calmette, B.P. 245, Lille Cedex, France.
3 Present address: Howard Hughes Medical Institute, University ofMichigan, 1150 West Medical Center Drive, Ann Arbor, MI 48109-0650, USA.

Chabot et al., 1988; Copeland et al., 1990; Zsebo et al.,1990). Evidently, Kit signaling is only crucial for the devel-opment of neural crest-derived melanocytes. However,mutations in other genes, such as that encoding tyrosinase,which is involved in melanin synthesis, may affect melano-cytes of either origin (Spritz and Hearing, 1994). Mutationsin still others, such as that encoding the transcription factorMitf, also affect both neuroepithelial and neural crest-derived melanocytes but, as will be shown here, by patho-genetic mechanisms that differ between these two celltypes.
TheMitf gene, originally cloned from a transgenic inser-tional mutation at themicrophthalmialocus (Hodgkinson etal., 1993; Hughes et al., 1993), encodes a basic-helix-loop-helix-zipper protein that forms homodimers and heterodi-mers with related proteins, interacts with other transcriptionregulators and specifically binds E-box motifs present in thepromoter elements of pigment cell-specific genes (Hodgkin-son et al., 1993; Hughes et al., 1993; Hemesath et al., 1994;Yavuzer et al., 1995; Sato et al., 1997). In vitro co-transfec-tion assays with Mitf expression and appropriate reporterplasmids suggest that Mitf serves as a positive transcriptionfactor for tyrosinase (Tyr) and tyrosinase-related protein-1(Tyrp1) (Bentley et al., 1994; Yasumoto et al., 1994, 1997).The human DOPAchrome tautomerase (Dct or Tyrp2 ortyrosinase-related protein-2) promoter also contains E-boxmotifs but, based on co-transfection experiments, Mitf doesnot transactivate the Dct promoter efficiently (Yokoyama etal., 1994; Yasumoto et al., 1997).
Mutations in Mitf have occurred in several species. Inhumans, they are found in families with Waardenburg syn-drome IIa whose key features are congenital deafness andpigment disturbances in skin and eye (Tassabehji et al.,1994).Mitf is also the gene mutated inmib (microphthal-mie-blanc) rats (Moutier et al., 1989; Opdecamp et al.,unpublished data) andWh (anophthalmic white) hamsters(Knapp and Polivanov, 1958; Asher, 1968; Hodgkinson etal., unpublished data). In the mouse, there are over 20 dif-ferentMitf mutations, many of them leading to amino acidsubstitutions in critical molecular domains (Steingrı´mssonet al., 1994; Mouse Genome Database, 1997). Common toall of these mutations is a deficiency in skin or coat mela-nocytes that in the mouse may range in severity from minorreductions in coat tyrosinase activity but with normal eyes(as with the alleleMitfmi-sp) to total lack of coat and eyepigmentation, small, colobomatous eyes, deafness and addi-tional disturbances such as osteopetrosis (as inMitfmi)(Moore, 1995). In fact, in the mouse alone, the series ofindependent alleles atMitf is the largest collection ofmutations in any member of this class of transcriptionfactor.
Here, we have analyzed the developmental profile ofMitfexpression in the neural crest and neuroepithelium of wildtype mouse embryos and embryos homozygous for severalMitf mutations. The analyses revealed crucial differences inthe way the two types of melanocyte are affected byMitf.
2. Results and discussion
2.1. Mitf expression in wild type neural crest
2.1.1. Rostro-caudal sequence of Mitf expressionRecent experiments in cultured cells derived from trunk
neural crest indicated that the Mitf transcription factor isrequired early on in development, before the generation ofmelanoblasts (Opdecamp et al., 1997). We now examinedwhether the in vivoMitf expression pattern was consistentwith such an early role in the trunk and other areas of thecrest. Wild type embryos and embryos homozygous forseveral differentMitf mutations were harvested at differentstages of development and serial cryostat sections were pre-pared. The sections were then processed for in situ hybridi-zation or immunolabeling, using probes and antibodiesspecific for Mitf and a number of additional markers ofthe melanocyte lineage.
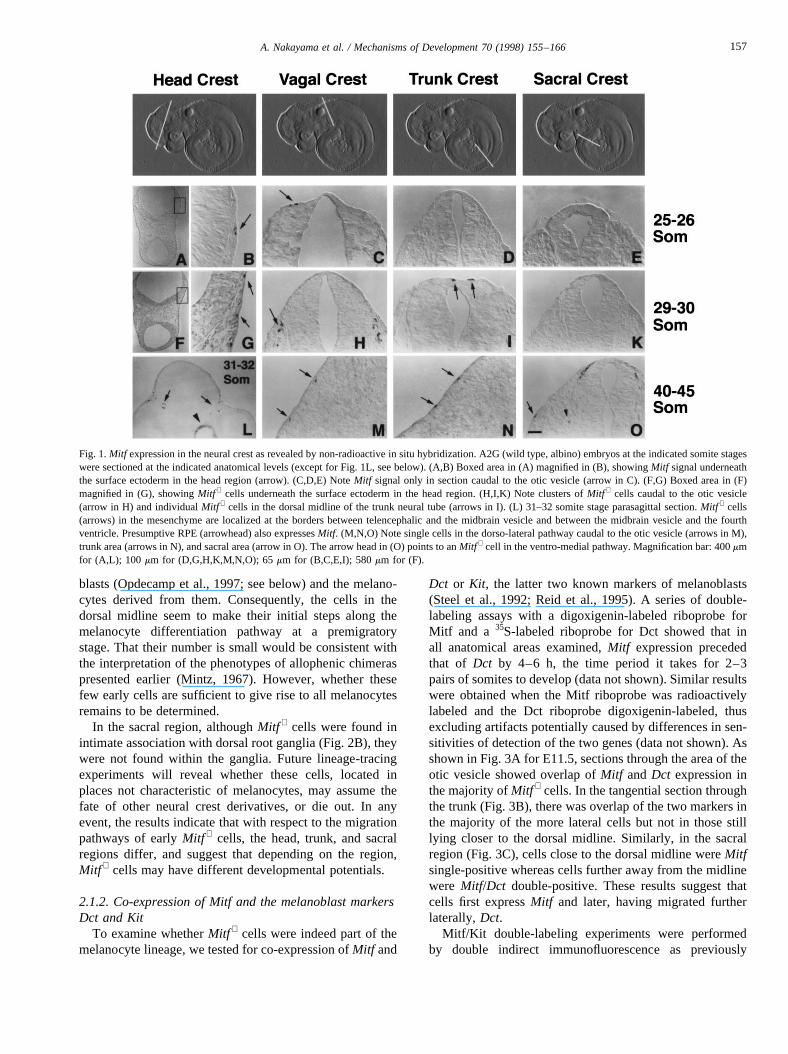
Fig. 1 documents the early appearance of wild typeMitf +
cells as identified by a digoxigenin-labeled Mitf riboprobein four anatomical areas. The firstMitf + cells were seen atthe 25–26 somite stage in the narrow space between thesurface ectoderm and the neuroepithelium of the brain(marked head crest) and in the region immediately caudalto the otic vesicle (marked vagal crest) (Fig. 1A–C, arrow inB,C). In this region, they were soon seen in clusters close tothe neural tube (arrow, Fig. 1H for the 29–30 somite stage).Sagittal sections of the head region showed cells in themesenchyme at the forebrain/midbrain and midbrain/hind-brain boundaries (arrows, Fig. 1L for the 31–32 somitestage). In the trunk region,Mitf + cells were not seen untilthe 27–28 somite stage. Soon thereafter, some rareMitf +
cells appeared close to the dorsal midline of the neural tube(arrows, Fig. 1I for the 29–30 somite stage). Some of thesecells directly overlay the roof plate of the neural tube asshown in the high power view of Fig. 2A. In the sacralarea, the firstMitf + cells were observed at the 33–34 somitestage and were more numerous at the 40–45 somite stage.Interestingly, in this region,Mitf + cells were found in loca-tions characteristic of the dorsolateral neural crest migrationpathway (arrow in Fig. 10) as well as the ventro-medialpathway (arrowhead in Fig. 10) where they came to lieclose to the dorsal root ganglia (dotted line in Fig. 2B). Atthese later stages, formerly clustered cells in the more ros-tral areas were now dispersed into individual cells andlocated more laterally from the dorsal midline (Fig. 1M,N,arrows).
The above analysis revealed thatMitf is expressed earlyin development, first in the head and last in the tail, and atlocations where previous work had identified neural crest-derived cells. With the embryos’ increasing age, the numberof Mitf + cells initially increased but then decreased suchthat at birth only hair bulb melanocytes were still positivefor Mitf (see below).
Mitf + cells likely are precursors to melanocytes sincemice with severeMitf mutations lack authentic melano-
156 A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
blasts (Opdecamp et al., 1997; see below) and the melano-cytes derived from them. Consequently, the cells in thedorsal midline seem to make their initial steps along themelanocyte differentiation pathway at a premigratorystage. That their number is small would be consistent withthe interpretation of the phenotypes of allophenic chimeraspresented earlier (Mintz, 1967). However, whether thesefew early cells are sufficient to give rise to all melanocytesremains to be determined.
In the sacral region, althoughMitf + cells were found inintimate association with dorsal root ganglia (Fig. 2B), theywere not found within the ganglia. Future lineage-tracingexperiments will reveal whether these cells, located inplaces not characteristic of melanocytes, may assume thefate of other neural crest derivatives, or die out. In anyevent, the results indicate that with respect to the migrationpathways of earlyMitf + cells, the head, trunk, and sacralregions differ, and suggest that depending on the region,Mitf + cells may have different developmental potentials.
2.1.2. Co-expression of Mitf and the melanoblast markersDct and Kit
To examine whetherMitf + cells were indeed part of themelanocyte lineage, we tested for co-expression ofMitf and
Dct or Kit, the latter two known markers of melanoblasts(Steel et al., 1992; Reid et al., 1995). A series of double-labeling assays with a digoxigenin-labeled riboprobe forMitf and a 35S-labeled riboprobe for Dct showed that inall anatomical areas examined,Mitf expression precededthat of Dct by 4–6 h, the time period it takes for 2–3pairs of somites to develop (data not shown). Similar resultswere obtained when the Mitf riboprobe was radioactivelylabeled and the Dct riboprobe digoxigenin-labeled, thusexcluding artifacts potentially caused by differences in sen-sitivities of detection of the two genes (data not shown). Asshown in Fig. 3A for E11.5, sections through the area of theotic vesicle showed overlap ofMitf andDct expression inthe majority ofMitf + cells. In the tangential section throughthe trunk (Fig. 3B), there was overlap of the two markers inthe majority of the more lateral cells but not in those stilllying closer to the dorsal midline. Similarly, in the sacralregion (Fig. 3C), cells close to the dorsal midline wereMitfsingle-positive whereas cells further away from the midlinewere Mitf/Dct double-positive. These results suggest thatcells first expressMitf and later, having migrated furtherlaterally,Dct.
Mitf/Kit double-labeling experiments were performedby double indirect immunofluorescence as previously
Fig. 1.Mitf expression in the neural crest as revealed by non-radioactive in situ hybridization. A2G (wild type, albino) embryos at the indicated somite stageswere sectioned at the indicated anatomical levels (except for Fig. 1L, see below). (A,B) Boxed area in (A) magnified in (B), showingMitf signal underneaththe surface ectoderm in the head region (arrow). (C,D,E) NoteMitf signal only in section caudal to the otic vesicle (arrow in C). (F,G) Boxed area in (F)magnified in (G), showingMitf + cells underneath the surface ectoderm in the head region. (H,I,K) Note clusters ofMitf + cells caudal to the otic vesicle(arrow in H) and individualMitf + cells in the dorsal midline of the trunk neural tube (arrows in I). (L) 31–32 somite stage parasagittal section.Mitf + cells(arrows) in the mesenchyme are localized at the borders between telencephalic and the midbrain vesicle and between the midbrain vesicle and the fourthventricle. Presumptive RPE (arrowhead) also expressesMitf. (M,N,O) Note single cells in the dorso-lateral pathway caudal to the otic vesicle (arrows in M),trunk area (arrows in N), and sacral area (arrow in O). The arrow head in (O) points to anMitf + cell in the ventro-medial pathway. Magnification bar: 400mmfor (A,L); 100 mm for (D,G,H,K,M,N,O); 65mm for (B,C,E,I); 580mm for (F).
157A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166

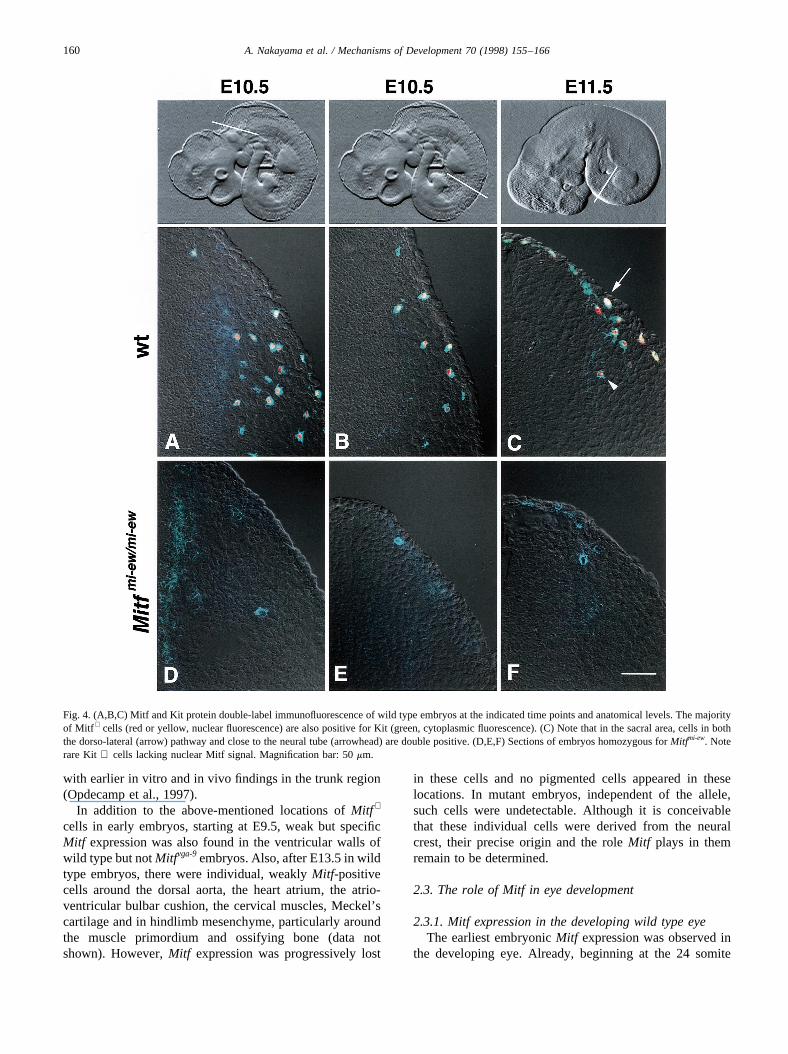
described (Opdecamp et al., 1997). As shown in Fig. 4A,B,in a region caudal to the otic vesicle as well as in the trunk ofan E10.5 embryo, there were many Mitf+ cells (nuclear redfluorescence) that were also Kit+ (cytoplasmic green fluor-escence). UnlikeDct expression which was delayed relativeto that of Mitf, Kit expression was seen as soon as Mitfbecame detectable, i.e. from E10.5, consistent with previousKit expression studies (Keshet et al., 1991; Matsui et al.,1990). As shown in Fig. 4C, the sacral area contained dou-ble-positive cells in locations corresponding to the dorso-lateral migration pathway (arrows) but also close to theneural tube (arrowhead) whereMitf + cells have beenobserved by in situ hybridization (Fig. 2B). Thus, it appearsthat in the neural crest, the majorityof Mitf + cells developinto melanoblasts – expressing at least two additional mel-anoblast markers. However there were also some cells thatwere single-labeled for any of the markers (see for instanceKit single-labeled cells in Fig. 4A). Whether these cellssimply reflect dynamic changes in the sequence of geneexpression or whether they belong to separate lineagesawaits further analyses.
2.1.3. Expression in the otic vesicleIt has been noted earlier thatMitf mutant mice may be
deaf due to lack of neural crest-derived melanocytes in thestria vascularis of the cochlea (Tachibana et al., 1992;
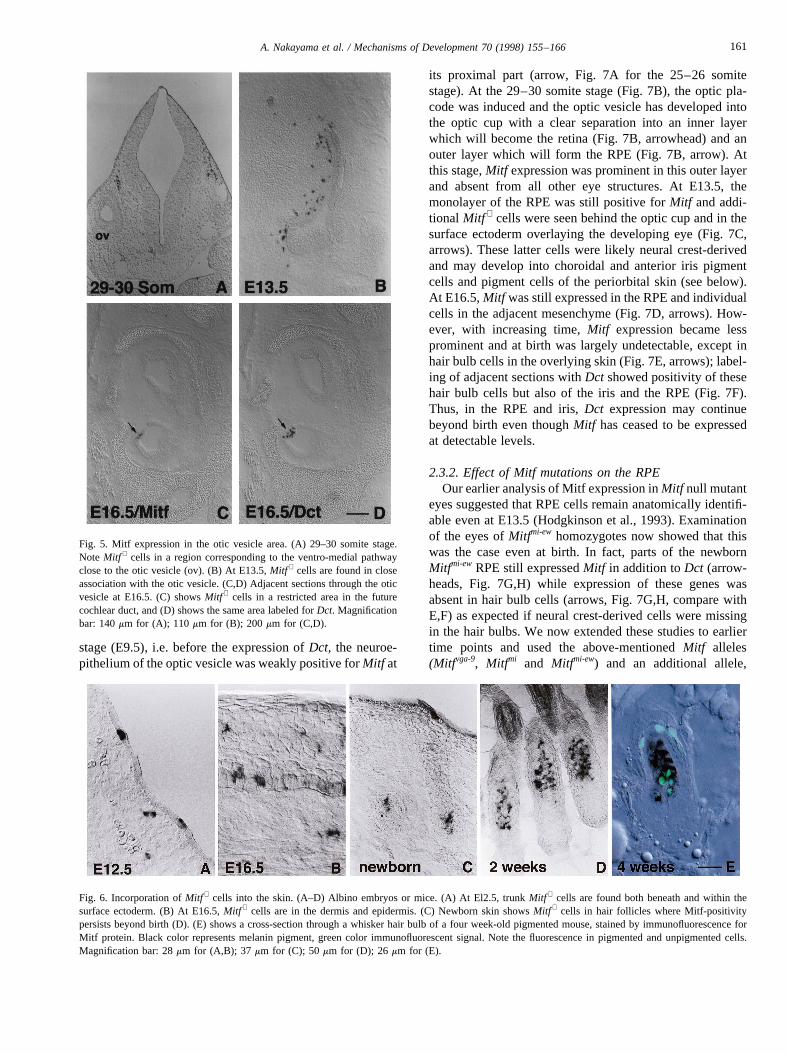
Motohashi et al., 1994). Lack of these melanocytes maylead to absence of endocochlear potential, secondary degen-eration of hair cells and hearing impairment (Steel andBarkway, 1989; Cable et al., 1994). Fig. 5A shows wildtypeMitf + cells at the 29–30 somite stage located betweenthe otic vesicle and the neuroepithelium of the hindbrain.During the following days, these cells increased in numberand became intimately associated with the otic vesicle (Fig.5B, see also Fig. 3A). Three days later, they were concen-trated in a small area that represented the presumptive striavascularis (Fig. 5C); in a serial section, this area was alsopositive for Dct (Fig. 5D), consistent with earlier results(Steel et al., 1992). Later, theMitf signal in the stria vascu-laris was less intense and at birth was undetectable while theDct signal remained intact (data not shown). These observa-tions suggest that in the area of the otic vesicle,Mitf + cellsmigrate on the ventro-medial pathway toward the otic vesi-cle where they give rise to pigmented cells in the inner ear,including the intermediate cells of the stria vascularis. Inaddition, they are also found in the dorso-lateral migrationpathway where they give rise to skin melanocytes.
2.1.4. Expression in the skinAs shown in Fig. 1,Mitf + cells on the lateral pathway
were seen beneath the surface ectoderm until the 40–45somite stage. At E12.5, such cells became incorporated inthe epithelial layer as shown for the forelimb bud area inFig. 6A. Four days later,Mitf + cells were found in thedermis and epidermis (Fig. 6B). We cannot exclude, how-ever, that some of these cells represented mast cells, whichare known to expressMitf (Hodgkinson et al., 1993) andpopulate the embryonic skin (Isozaki et al., 1994). In new-born skin,Mitf expression was restricted to hair follicles(Fig. 6C) where it persisted postnatally (Fig. 6D). In fact,immunofluorescent assays showed that a subset of the pig-mented cells in hair follicles stayed positive for Mitf (Fig.6E), in contrast to pigmented cells in other locations. This isconsistent with the observation that mice homozygous fortheMitfvit allele show an age-dependent progressive loss ofcoat pigmentation (Lerner et al., 1986; Boissy et al., 1991)but apparently no postnatal changes in the RPE (Boissy etal., 1987; Nir et al., 1995) whereMitf is not expressedpostnatally (see below).
2.2. Mitf expression in mutant neural crest
Mitf expression in the neural crest was examined inembryos homozygous for three differentMitf alleles:Mitfvga-9, a transgenic insertional allele that accumulates lit-tle if any Mitf mRNA (Hodgkinson et al., 1993),Mitfmi, anallele that encodes a protein with a deletion of a criticalarginine in its DNA-binding domain (Steingrı´msson et al.,1994), andMitfmi-ew, which encodes a protein in which 25residues, including most of the basic DNA-binding domain,are replaced by a single valine (Steingrı´msson et al., 1994).Previous examination of cultured trunk neural crest cells of
Fig. 2. (A) High power view of Fig. 1I. SingleMitf + cell in the dorsalmidline of the trunk neural tube. The positive cell overlays the neural tubejust underneath the surface ectoderm. (B) Sacral crest at E12.5.Mitf + cellsare found on the dorso-lateral neural crest migration pathway (left-handside of the figure) and on the ventro-medial pathway close to a dorsal rootganglion (d). Magnification bar: 8mm for (A); 50 mm for (B).
158 A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
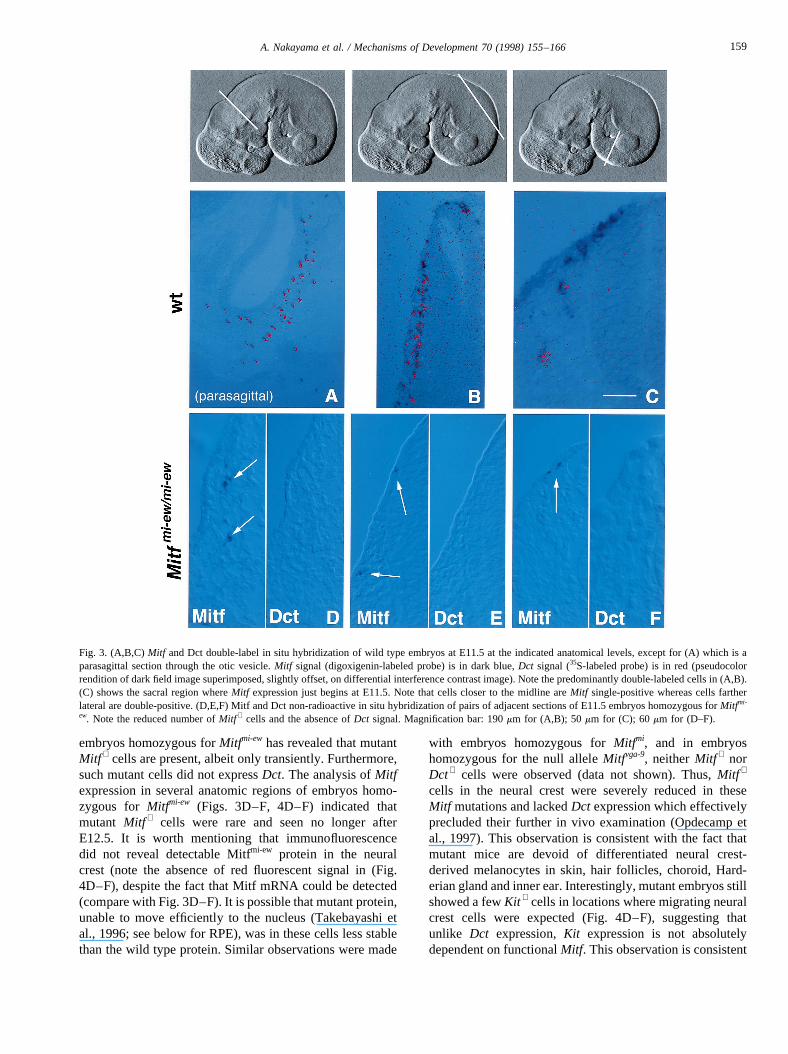
embryos homozygous forMitfmi-ew has revealed that mutantMitf + cells are present, albeit only transiently. Furthermore,such mutant cells did not expressDct. The analysis ofMitfexpression in several anatomic regions of embryos homo-zygous for Mitfmi-ew (Figs. 3D–F, 4D–F) indicated thatmutant Mitf + cells were rare and seen no longer afterE12.5. It is worth mentioning that immunofluorescencedid not reveal detectable Mitfmi-ew protein in the neuralcrest (note the absence of red fluorescent signal in (Fig.4D–F), despite the fact that Mitf mRNA could be detected(compare with Fig. 3D–F). It is possible that mutant protein,unable to move efficiently to the nucleus (Takebayashi etal., 1996; see below for RPE), was in these cells less stablethan the wild type protein. Similar observations were made
with embryos homozygous forMitfmi, and in embryoshomozygous for the null alleleMitfvga-9, neitherMitf + norDct+ cells were observed (data not shown). Thus,Mitf +
cells in the neural crest were severely reduced in theseMitf mutations and lackedDct expression which effectivelyprecluded their further in vivo examination (Opdecamp etal., 1997). This observation is consistent with the fact thatmutant mice are devoid of differentiated neural crest-derived melanocytes in skin, hair follicles, choroid, Hard-erian gland and inner ear. Interestingly, mutant embryos stillshowed a fewKit + cells in locations where migrating neuralcrest cells were expected (Fig. 4D–F), suggesting thatunlike Dct expression,Kit expression is not absolutelydependent on functionalMitf. This observation is consistent
Fig. 3. (A,B,C)Mitf and Dct double-label in situ hybridization of wild type embryos at E11.5 at the indicated anatomical levels, except for (A) which is aparasagittal section through the otic vesicle.Mitf signal (digoxigenin-labeled probe) is in dark blue,Dct signal (35S-labeled probe) is in red (pseudocolorrendition of dark field image superimposed, slightly offset, on differential interference contrast image). Note the predominantly double-labeledcells in (A,B).(C) shows the sacral region whereMitf expression just begins at E11.5. Note that cells closer to the midline areMitf single-positive whereas cells fartherlateral are double-positive. (D,E,F) Mitf and Dct non-radioactive in situ hybridization of pairs of adjacent sections of E11.5 embryos homozygous for Mitfmi-
ew. Note the reduced number ofMitf + cells and the absence ofDct signal. Magnification bar: 190mm for (A,B); 50 mm for (C); 60mm for (D–F).
159A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
with earlier in vitro and in vivo findings in the trunk region(Opdecamp et al., 1997).
In addition to the above-mentioned locations ofMitf +
cells in early embryos, starting at E9.5, weak but specificMitf expression was also found in the ventricular walls ofwild type but notMitfvga-9embryos. Also, after E13.5 in wildtype embryos, there were individual, weaklyMitf-positivecells around the dorsal aorta, the heart atrium, the atrio-ventricular bulbar cushion, the cervical muscles, Meckel’scartilage and in hindlimb mesenchyme, particularly aroundthe muscle primordium and ossifying bone (data notshown). However,Mitf expression was progressively lost
in these cells and no pigmented cells appeared in theselocations. In mutant embryos, independent of the allele,such cells were undetectable. Although it is conceivablethat these individual cells were derived from the neuralcrest, their precise origin and the roleMitf plays in themremain to be determined.
2.3. The role of Mitf in eye development
2.3.1. Mitf expression in the developing wild type eyeThe earliest embryonicMitf expression was observed in
the developing eye. Already, beginning at the 24 somite
Fig. 4. (A,B,C) Mitf and Kit protein double-label immunofluorescence of wild type embryos at the indicated time points and anatomical levels. The majorityof Mitf + cells (red or yellow, nuclear fluorescence) are also positive for Kit (green, cytoplasmic fluorescence). (C) Note that in the sacral area, cells in boththe dorso-lateral (arrow) pathway and close to the neural tube (arrowhead) are double positive. (D,E,F) Sections of embryos homozygous forMitfmi-ew. Noterare Kit + cells lacking nuclear Mitf signal. Magnification bar: 50mm.
160 A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
stage (E9.5), i.e. before the expression ofDct, the neuroe-pithelium of the optic vesicle was weakly positive forMitf at
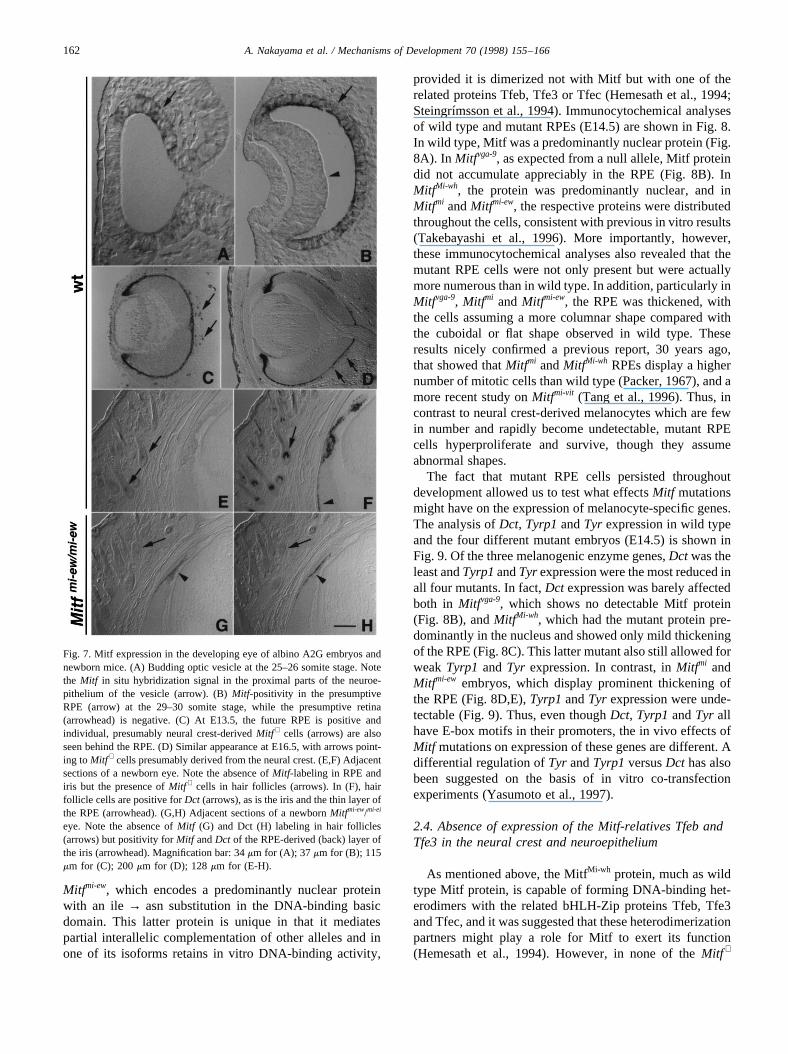
its proximal part (arrow, Fig. 7A for the 25–26 somitestage). At the 29–30 somite stage (Fig. 7B), the optic pla-code was induced and the optic vesicle has developed intothe optic cup with a clear separation into an inner layerwhich will become the retina (Fig. 7B, arrowhead) and anouter layer which will form the RPE (Fig. 7B, arrow). Atthis stage,Mitf expression was prominent in this outer layerand absent from all other eye structures. At E13.5, themonolayer of the RPE was still positive forMitf and addi-tional Mitf + cells were seen behind the optic cup and in thesurface ectoderm overlaying the developing eye (Fig. 7C,arrows). These latter cells were likely neural crest-derivedand may develop into choroidal and anterior iris pigmentcells and pigment cells of the periorbital skin (see below).At E16.5,Mitf was still expressed in the RPE and individualcells in the adjacent mesenchyme (Fig. 7D, arrows). How-ever, with increasing time,Mitf expression became lessprominent and at birth was largely undetectable, except inhair bulb cells in the overlying skin (Fig. 7E, arrows); label-ing of adjacent sections withDct showed positivity of thesehair bulb cells but also of the iris and the RPE (Fig. 7F).Thus, in the RPE and iris,Dct expression may continuebeyond birth even thoughMitf has ceased to be expressedat detectable levels.
2.3.2. Effect of Mitf mutations on the RPEOur earlier analysis of Mitf expression inMitf null mutant
eyes suggested that RPE cells remain anatomically identifi-able even at E13.5 (Hodgkinson et al., 1993). Examinationof the eyes ofMitfmi-ew homozygotes now showed that thiswas the case even at birth. In fact, parts of the newbornMitfmi-ew RPE still expressedMitf in addition toDct (arrow-heads, Fig. 7G,H) while expression of these genes wasabsent in hair bulb cells (arrows, Fig. 7G,H, compare withE,F) as expected if neural crest-derived cells were missingin the hair bulbs. We now extended these studies to earliertime points and used the above-mentionedMitf alleles(Mitfvga-9, Mitfmi and Mitfmi-ew) and an additional allele,
Fig. 5. Mitf expression in the otic vesicle area. (A) 29–30 somite stage.Note Mitf + cells in a region corresponding to the ventro-medial pathwayclose to the otic vesicle (ov). (B) At E13.5,Mitf + cells are found in closeassociation with the otic vesicle. (C,D) Adjacent sections through the oticvesicle at E16.5. (C) showsMitf + cells in a restricted area in the futurecochlear duct, and (D) shows the same area labeled forDct. Magnificationbar: 140mm for (A); 110 mm for (B); 200mm for (C,D).
Fig. 6. Incorporation ofMitf + cells into the skin. (A–D) Albino embryos or mice. (A) At El2.5, trunkMitf + cells are found both beneath and within thesurface ectoderm. (B) At E16.5,Mitf + cells are in the dermis and epidermis. (C) Newborn skin showsMitf + cells in hair follicles where Mitf-positivitypersists beyond birth (D). (E) shows a cross-section through a whisker hair bulb of a four week-old pigmented mouse, stained by immunofluorescence forMitf protein. Black color represents melanin pigment, green color immunofluorescent signal. Note the fluorescence in pigmented and unpigmented cells.Magnification bar: 28mm for (A,B); 37 mm for (C); 50mm for (D); 26 mm for (E).
161A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
Mitfmi-ew, which encodes a predominantly nuclear proteinwith an ile → asn substitution in the DNA-binding basicdomain. This latter protein is unique in that it mediatespartial interallelic complementation of other alleles and inone of its isoforms retains in vitro DNA-binding activity,
provided it is dimerized not with Mitf but with one of therelated proteins Tfeb, Tfe3 or Tfec (Hemesath et al., 1994;Steingrımsson et al., 1994). Immunocytochemical analysesof wild type and mutant RPEs (E14.5) are shown in Fig. 8.In wild type, Mitf was a predominantly nuclear protein (Fig.8A). In Mitfvga-9, as expected from a null allele, Mitf proteindid not accumulate appreciably in the RPE (Fig. 8B). InMitfMi-wh, the protein was predominantly nuclear, and inMitfmi andMitfmi-ew, the respective proteins were distributedthroughout the cells, consistent with previous in vitro results(Takebayashi et al., 1996). More importantly, however,these immunocytochemical analyses also revealed that themutant RPE cells were not only present but were actuallymore numerous than in wild type. In addition, particularly inMitfvga-9, Mitfmi andMitfmi-ew, the RPE was thickened, withthe cells assuming a more columnar shape compared withthe cuboidal or flat shape observed in wild type. Theseresults nicely confirmed a previous report, 30 years ago,that showed thatMitfmi andMitfMi-wh RPEs display a highernumber of mitotic cells than wild type (Packer, 1967), and amore recent study onMitfmi-vit (Tang et al., 1996). Thus, incontrast to neural crest-derived melanocytes which are fewin number and rapidly become undetectable, mutant RPEcells hyperproliferate and survive, though they assumeabnormal shapes.
The fact that mutant RPE cells persisted throughoutdevelopment allowed us to test what effectsMitf mutationsmight have on the expression of melanocyte-specific genes.The analysis ofDct, Tyrp1andTyr expression in wild typeand the four different mutant embryos (E14.5) is shown inFig. 9. Of the three melanogenic enzyme genes,Dct was theleast andTyrp1andTyr expression were the most reduced inall four mutants. In fact,Dct expression was barely affectedboth in Mitfvga-9, which shows no detectable Mitf protein(Fig. 8B), andMitfMi-wh, which had the mutant protein pre-dominantly in the nucleus and showed only mild thickeningof the RPE (Fig. 8C). This latter mutant also still allowed forweak Tyrp1 and Tyr expression. In contrast, inMitfmi andMitfmi-ew embryos, which display prominent thickening ofthe RPE (Fig. 8D,E),Tyrp1andTyr expression were unde-tectable (Fig. 9). Thus, even thoughDct, Tyrp1andTyr allhave E-box motifs in their promoters, the in vivo effects ofMitf mutations on expression of these genes are different. Adifferential regulation ofTyr andTyrp1versusDct has alsobeen suggested on the basis of in vitro co-transfectionexperiments (Yasumoto et al., 1997).
2.4. Absence of expression of the Mitf-relatives Tfeb andTfe3 in the neural crest and neuroepithelium
As mentioned above, the MitfMi-wh protein, much as wildtype Mitf protein, is capable of forming DNA-binding het-erodimers with the related bHLH-Zip proteins Tfeb, Tfe3and Tfec, and it was suggested that these heterodimerizationpartners might play a role for Mitf to exert its function(Hemesath et al., 1994). However, in none of theMitf +
Fig. 7. Mitf expression in the developing eye of albino A2G embryos andnewborn mice. (A) Budding optic vesicle at the 25–26 somite stage. Notethe Mitf in situ hybridization signal in the proximal parts of the neuroe-pithelium of the vesicle (arrow). (B)Mitf-positivity in the presumptiveRPE (arrow) at the 29–30 somite stage, while the presumptive retina(arrowhead) is negative. (C) At E13.5, the future RPE is positive andindividual, presumably neural crest-derivedMitf + cells (arrows) are alsoseen behind the RPE. (D) Similar appearance at E16.5, with arrows point-ing toMitf + cells presumably derived from the neural crest. (E,F) Adjacentsections of a newborn eye. Note the absence ofMitf-labeling in RPE andiris but the presence ofMitf + cells in hair follicles (arrows). In (F), hairfollicle cells are positive forDct (arrows), as is the iris and the thin layer ofthe RPE (arrowhead). (G,H) Adjacent sections of a newbornMitfmi-ew/mi-ei
eye. Note the absence ofMitf (G) and Dct (H) labeling in hair follicles(arrows) but positivity forMitf andDct of the RPE-derived (back) layer ofthe iris (arrowhead). Magnification bar: 34mm for (A); 37mm for (B); 115mm for (C); 200mm for (D); 128mm for (E-H).
162 A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
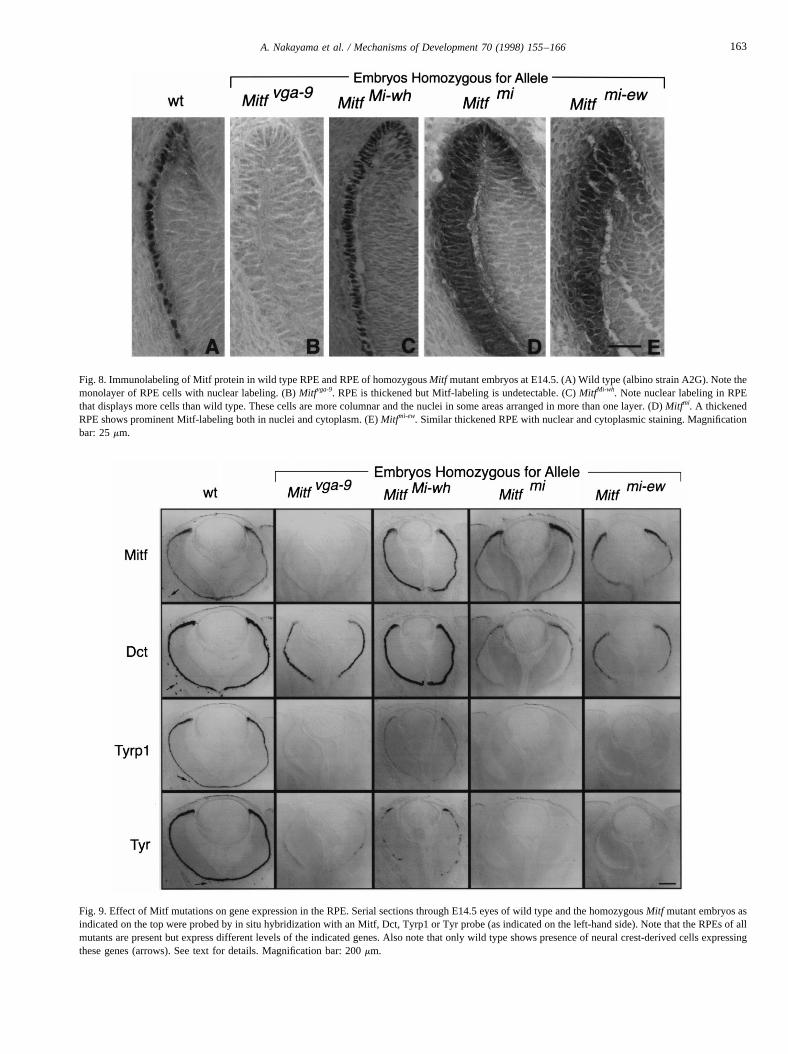
Fig. 8. Immunolabeling of Mitf protein in wild type RPE and RPE of homozygousMitf mutant embryos at E14.5. (A) Wild type (albino strain A2G). Note themonolayer of RPE cells with nuclear labeling. (B)Mitfvga-9. RPE is thickened but Mitf-labeling is undetectable. (C)MitfMi-wh. Note nuclear labeling in RPEthat displays more cells than wild type. These cells are more columnar and the nuclei in some areas arranged in more than one layer. (D)Mitfmi. A thickenedRPE shows prominent Mitf-labeling both in nuclei and cytoplasm. (E)Mitfmi-ew. Similar thickened RPE with nuclear and cytoplasmic staining. Magnificationbar: 25mm.
Fig. 9. Effect of Mitf mutations on gene expression in the RPE. Serial sections through E14.5 eyes of wild type and the homozygousMitf mutant embryos asindicated on the top were probed by in situ hybridization with an Mitf, Dct, Tyrp1 or Tyr probe (as indicated on the left-hand side). Note that the RPEs ofallmutants are present but express different levels of the indicated genes. Also note that only wild type shows presence of neural crest-derived cells expressingthese genes (arrows). See text for details. Magnification bar: 200mm.
163A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166

cells did we observe any appreciable expression of Tfeb orTfe3 at stages when theMitf mutant phenotype becomesmanifest (data not shown). Thus, it is unlikely that theserelated proteins are relevant forMitf to function duringdevelopment and we predict that knock-out mice in whichthese related genes are deleted will not show a neural crestor eye phenotype similar toMitf mutant mice.
2.5. Summary and conclusion
The results from these studies are summarized in Fig. 10.The RPE represents the first place ofMitf expression, fol-lowed by expression in the neural crest in a rostro-caudalsequence. The periods during which wild type neural crestcells and their derivatives are positive forMitf, and the typesof their positive derivatives, are shown in the lower half ofFig. 10. Our results show thatMitf mutations severely affect
Mitf + cells in the neural crest. These cells are rare and lackDct expression. In contrast, the initial steps of RPE devel-opment are not affected but RPE cells show abnormal pro-liferation, shape, andTyrp1 and Tyr expression but theylargely retainDct expression. This clearly establishes thatmelanocytes of different origin, though dependent on thesameMitf transcription factor gene for their development,respond differently to mutations in this gene and suggeststhatMitf plays different roles in these two types of pigmentcell.
3. Experimental procedures
3.1. Mice and harvest of embryos
Mice of the strains C57BL/6 (pigmented) and A2G
Fig. 10. Summary of the expression pattern of wild typeMitf. The top row of pictures shows a schematic representation of the expression pattern in eye andneural crest at the indicated developmental stages as obtained by non-radioactive in situ hybridization. The lower part of the figure lists theMitf-positivestages for the neuroepithelium and the different parts of the neural crest and their derivatives. As mentioned in the text, there areMitf + cells in additionalanatomical areas, including in the heart, whose origin has not yet been determined. NC, neural crest.
164 A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166

(albino) were used asMitf wild type controls. For in situhybridizations, A2G embryos were used predominantlysince we observed that the presence of melanin in pigmen-ted mouse embryos may result in non-specific labeling.Embryos homozygous forMitf mutant alleles were obtainedeither by homozygous× homozygous mating or by hetero-zygous× heterozygous mating and identification of homo-zygous embryos by absence of eye pigmentation. Thealleles used were:Mitfvga-9 (background: mixed C57BL/6/C3H) (Tachibana et al., 1992; Hodgkinson et al., 1993);Mitfmi-ew (background: C57BL/6) (Hertwig, 1942);MitfMi-
wh (background: C57BL/6) (Grobman and Charles, 1947);and Mitfmi-ew (background: Naw) (Miner, 1968). The noonon which a vaginal plug was found upon mating was desig-nated embryonic day 0.5 (E0.5). At the indicated timepoints, embryos were harvested and placed into minimalessential medium, containing 5% fetal bovine serum beforefurther processing. For accurate staging of embryos youngerthan E11.5, the number of somites were counted. All animalprocedures were approved by the institutional review board.
3.2. Riboprobes
Riboprobes were made by in vitro transcription of appro-priate plasmids with either T3 or T7 RNA polymerase andincorporation of digoxigenin-labeled UTP or35S a-labeledUTP. The template for the Mitf probe, MC1, was describedpreviously (Hodgkinson et al., 1993), as was that for themouseDct probe (Steel et al., 1992; Opdecamp et al.,1997). For the mouse Tfe3 probe, a 1.1 kb fragment corre-sponding to the 3′-UTR of the Tfe3 cDNA (Roman et al.,1991) kindly provided by K. Calame was subcloned in pBS.Mouse TfeB cDNA clones were obtained by screening algt10 library derived from adult mouse heart (Clonetech)using human cDNA as a probe. Positive clones were sub-cloned into pBluescript KS- and further characterized. Oneclone containing a 1.9 kb insert was sequenced and identi-fied as mouse TfeB by sequence comparison with the humanTfeB sequence. A fragment coding for the carboxyl 184amino acids (excluding the conserved bHLH-Zip region)and 373 bases of the 3′-UTR was further subcloned andused to generate antisense riboprobes. For all probes, corre-sponding sense probes were generated and used as controls.
3.3. In situ hybridization
Non-radioactive in situ hybridization was performedessentially as described previously (Hodgkinson et al.;1993; Opdecamp et al., 1997). For double-labeling in situhybridization, a mixture of digoxigenin-labeled and35S-labeled probes was applied. Digoxigenin-labeling was firstvisualized in the standard manner, followed by coating ofthe glass slides with Ilford K-5 D emulsion (PolysciencesInc.). After appropriate times of exposure, the hybridizationsignals were visualized by standard development and fixa-tion procedures. The slides were viewed and photographed
using a Polyvar microscope set for differential interferencecontrast (DIC) mode to view the digoxigenin-labeled cellsand for dark field to view the grains. Both images were thenscanned and the dark field image was converted into pseu-docolors and overlayed over the DIC image.
3.4. Immunofluorescence and immunocytochemistry
Double indirect immunofluorescence of cryostat sectionsusing a rabbit anti-mouse Mitf serum was performed asdescribed (Opdecamp et al., 1997). For immunocytochem-istry, cryostat sections were postfixed in 4.0% formaldehydefor 10 min, treated with 10% normal goat serum and thenexposed to a 1:2000 dilution of the rabbit anti-Mitf anti-body. The presence of the antibody was revealed by theavidin-biotin complex method (Vectastain).
Acknowledgements
We thank Drs. Ian Jackson and David Fisher for plasmids,Dr. Vincent Hearing for Dct antibodies, Dr. Nitin Gogate forinitial help with in situ hybridization and Drs. Eirı´kurSteingrımsson, William Pavan, Monique Dubois-Dalcqand Lynn Hudson for valuable comments on the manuscript.During the course of this work, C.C. was a HHMI-NIHResearch Scholar.
References
Asher, J.H., Jr. 1968. A partial biochemical and morphological descriptionof the action of the gene Wh causing anophthalmia in the Syrian ham-ster,Mesocricetus auratus, 1968. MA Thesis, California State Collegeat Long Beach, USA, p. 360.
Bentley, N.J., Eisen, T., Goding, C.R., 1994. Melanocyte-specific expres-sion of the human tyrosinase promoter: activation by the microphthal-mia gene product and role of the initiator. Mol. Cell. Biol. 14, 7996–8006.
Boissy, R.E., Moellmann, G.E., Lenzer, A.B., 1987. Morphology of mel-anocytes in hair bulbs and eyes of vitiligo mice. Am. J. Pathol. 127,380–388.
Boissy, R.E., Beato, K.E., Nordlund, J.J., 1991. Dilated rough endoplasmicreticulum and premature death in melanocytes cultured from the vitiligomouse. Am. J. Pathol. 138, 1511–1525.
Cable, J., Huszar, D., Jaenisch, R., Steel, K.P., 1994. Effects of mutationsat the W locus (c-kit) on inner ear pigmentation and function in themouse. Pigment Cell Res. 7, 17–32.
Chabot, B., Stephenson, D.A., Chapman, V.M., Besmer, B., Bernstein, A.,1988. The proto-oncogenec-kit encoding a transmembrane tyrosinekinase receptor maps to the mouse W locus. Nature 335, 88–89.
Copeland, N., Gilbert, D.J., Cho, B.C., Donovan, P.J., Jenkins, N.A.,Cosman, D., Anderson, D., Lyman, S.D., Williams, D.E., 1990. Mastcell growth factor maps near the steel locus on mouse chromosome 10and is deleted in a number of steel alleles. Cell 63, 175–183.
Geissler, E.N., Ryan, M.A., Housman, D.E., 1988. The dominant-whitespotting (W) locus of the mouse encodes thec-kit proto-oncogene. Cell55, 185–192.
Green, M.C., 1989. Catalog of mutant genes and polymorphic loci. In:Lyon, M.F., Searle, A.G. (Eds.), Genetic Variants and Strains of theLaboratory Mouse. Oxford University Press, New York, pp. 12–403.
165A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166

Grobman, A.B., Charles, D.R., 1947. Mutant white mice. A new dominantautosomal mutant affecting coat color in Mus musculus. J. Hered. 38,381–384.
Hemesath, T.J., Steingrı´msson, E., McGill, G., Hansen, M.J., Vaught, J.,Hodgkinson, C.A., Arnheiter, H., Copeland, N.G., Jenkins, N.A., Fisher,D.E., 1994.microphthalmia, a critical factor in melanocyte develop-ment, defines a discrete transcription factor family. Genes Dev. 8,2770–2780.
Hertwig, P., 1942. Neue Mutationen und Kopplungsgruppen bei derHausmaus. Z. Indukt. Abstammungs-Vererbungsl. 80, 220–246.
Hodgkinson, C.A., Moore, K.J., Nakayama, A., Steingrı´msson, E.,Copeland, N.G., Jenkins, N.A., Arnheiter, H., 1993. Mutations at themouse microphthalmia locus are associated with defects in a geneencoding a novel basic-helix-loop-helix-zipper protein. Cell 74, 395–404.
Hughes, M.J., Lingrel, J.B., Krakowsky, J.M., Anderson, K.P., 1993. Ahelix-loop-helix transcription factor-like gene is located at themi locus.J. Biol. Chem. 268, 20687–20690.
Isozaki, K., Tsujimura, T., Nomura, S., Morii, E., Koshimizu, U.,Nishimune, Y., Kitamura, Y., 1994. Cell type-specific deficiency of c-kit gene expression in mutant mice of mi/mi genotype. Am. J. Pathol.145, 827–836.
Keshet, E., Lyman, S.D., Williams, D.E., Anderson, D.M., Jenkins, N.A.,Copeland, N.G., Parada, L.F., 1991. Embryonic RNA expression pat-terns of the c-kit receptor and its cognate ligand suggest multiple func-tional roles in mouse development. EMBO J. 10, 2425–2435.
Knapp, B.H., Polivanov, S., 1958. Anophthalmic albino: a new mutation inthe Syrian hamster. Am. Naturalist 92, 317–318.
Lerner, A.B., Shiohara, T., Boissy, R.E., Jacobson, K.A., Lamoreux, L.M.,Moellmann, G.E., 1986. A mouse model for vitiligo. J. Invest.Dermatol. 87, 299–304.
Matsui, Y., Zsebo, K.M., Hogan, B.L., 1990. Embryonic expression of ahaematopoietic growth factor encoded by the S1 locus and the ligand forc-kit. Nature 347, 667–669.
Miner, G., 1968. Mouse News Lett. 38, 25.Mintz, B., 1967. Gene control of mammalian pigmentary differentiation, I.
Clonal origin of melanocytes. Proc. Natl. Acad. Sci. USA 58, 344–351.Moore, K.J., 1995. Insight into the microphthalmia gene. Trends Genet.
11, 442–448.Motohashi, H., Hozawa, K., Oshima, T., Takeuchi, T., Takasaka, T., 1994.
Dysgenesis of melanocytes and cochlear dysfunction in mutant micro-phthalmia (mi) mice. Hear. Res. 80, 10–20.
Mouse Genome Database (MGD), 1997. Mouse Genome Informatics,Jackson Laboratory, Bar Harbor, Maine, World Wide Web (URL:http:/ /www.informatics jax.org/).
Moutier, R., Ostrowski, K., Lamendin, H., 1989. Microphthalmia: a newrecessive mutation in the Norway rat. J. Hered. 80, 76–78.
Nir, I., Ransom, N., Smith, S.B., 1995. Ultrastructural features of retinaldystrophy in mutant vitiligo mice. Exp. Eye Res. 61, 363–377.
Online Mendelian Inheritance in Man, OMIM (TM), 1997. Center forMedical Genetics, Johns Hopkins University, Baltimore, MD andNational Center for Biotechnology Information, National Library ofMedicine, Bethesda, MD. World Wide Web URL http:/ /www.ncbi.nlm.nih.gov/Omim/.
Opdecamp, K., Nakayama, A., Nguyen, M.-T.T., Hodgkinson, C.A.,Pavan, W.J., Arnheiter, H., 1997. Melanocyte development in vivoand in neural crest cell cultures: crucial dependence on the Mitfbasic-helix-loop-helix-zipper transcription factor. Development 124,2377–2386.
Packer, S.O., 1967. The eye and skeletal effects of two mutant alleles atthe microphthalmia locus of Mus musculus. J. Exp. Zool. 165, 21–46.
Reid, K., Nishikawa, S.-I., Bartlett, P.F., Murphy, M., 1995. Steel factordirects melanocyte development in vitro through selective regulation ofthe number of c-kit+ progenitors. Dev. Biol. 169, 568–579.
Roman, C., Cohn, L., Calame, K., 1991. A dominant negative form oftranscription activator mTFE3 created by differential splicing. Science254, 94–97.
Sato, S., Roberts, K., Gambino, G., Cook, A., Kouzarides, T., Goding,C.R., 1997. CBP/p300 as a co-factor for the microphthalmia transcrip-tion factor. Oncogene 14, 3083–3092.
Spritz, R.A., Hearing, V.J. Jr., 1994. Genetic disorders of pigmentation.Adv. Hum. Genet. 22, 1–45.
Steel, K.P., Barkway, C., 1989. Another role for melanocytes: their impor-tance for normal stria vascularis development in the mammalian innerear. Development 107, 453–463.
Steel, K.P., Davidson, D.R., Jackson, I.J., 1992. TRP-2/DT a new earlymelanoblast marker, shows that steel growth factor (c-kit ligand) is asurvival factor. Development 115, 1111–1119.
Steingrı´msson, E., Moore, K.J., Lynn Lamoreux, M., Ferre´-D’Amare,A.R., Burley, S.K., Sanders Zimring, D.C., Skow, L.C., Hodgkinson,C.A., Arnheiter, H., Copeland, N.G., Jenkins, N.A., 1994. Molecularbasis of mousemicrophthalmia(mi) mutations helps explain their devel-opmental and phenotypic consequences. Nature Genet. 8, 256–263.
Tachibana, M., Hara, Y., Vyas, D., Hodgkinson, C., Fex, J., Grundfast, K.,Arnheiter, H., 1992. Cochlear disorder associated with melanocyteanomaly in mice with a transgenic insertional mutation. Mol. Cell.Neurosci. 3, 433–445.
Takebayashi, K., Chida, K., Tsukamoto, I., Morii, E., Munakata, H.,Arnheiter, H., Kuroki, T., Kitamura, Y., Nomura, S., 1996. The reces-sive phenotype displayed by a dominant negative microphthalmia-asso-ciated transcription factor mutant is a result of impaired nuclearlocalization potential. Mol. Cell. Biol. 16, 1203–1211.
Tang, M., Ruiz, M., Kosaras, B., Sidman, R.L., 1996. Increased cell gen-esis in retinal pigment epithelium of perinataI vitiligo mutant mice.Invest. Ophthalmol. Vis. Sci. 37, 1116–1124.
Tassabehji, M., Newton, V.E., Read, A.P., 1994. MITF gene mutations inpatients with Type 2 Waardenburg Syndrome. Nature Genet. 8, 251–255.
Yasumoto, K.-I., Yokoyama, K., Shibata, K., Tomita, Y., Shibahara, S.,1994. Microphthalmia-associated transcription factor as a regulator formelanocyte-specific transcription of the human tyrosinase gene. Mol.Cell. Biol. 14, 8058–8070.
Yasumoto, K.-I., Yokoyama, K., Takahashi, K., Tomita, Y., Shibahara, S.,1997. Functional analysis of microphthalmia-associated transcriptionfactor in pigment cell-specific transcription of the human tyrosinasefamily genes. J. Biol. Chem. 272, 503–509.
Yavuzer, U., Keenan, E., Lowings, P., Vachtenheim, J., Currie, G.,Goding, C.R., 1995. The microphthalmia gene product interacts withthe retinoblastoma protein in vitro and is a target for deregulation ofmelanocyte-specific transcription. Oncogene 10, 123–134.
Yokoyama, K., Yasumoto, K.-I., Suzuki, H., Shibahara, S., 1994. Cloningof the human DOPAchrome tautomerase/tyrosinase-related protein 2gene and identification of two regulatory regions required for its pig-ment cell-specific expression. J. Biol. Chem. 43, 27080–27087.
Zsebo, K., Williams, D.A., Geissler, E.N., Broudy, V.C., Martin, F.H.,Atkins, H.L., Hsu, R.-Y., Birkett, N.C., Okino, K.H., Murdock, D.C.,Jacobsen, F.W., Langley, K.E., Smith, K.A., Takeishi, T., Cattanach, B.,Galli, S.J., Suggs, S.V., 1990. Stem cell factor is encoded at theSl locusof the mouse and is the ligand for thec-kit tyrosine kinase receptor. Cell63, 213–224.
166 A. Nakayama et al. / Mechanisms of Development 70 (1998) 155–166
Related Documents











