Original Contribution MULTIPLE SIGNALING EVENTS IN AMYLOID -INDUCED, OXIDATIVE STRESS-DEPENDENT NEURONAL APOPTOSIS ELENA TAMAGNO,* MAURIZIO PAROLA,* MICHELA GUGLIELMOTTO,* GIANNI SANTORO,* PAOLA BARDINI,* LAURA MARRA,* MASSIMO TABATON, † and OLIVIERO DANNI* *Department of Experimental Medicine and Oncology, General Pathology Section, University of Turin, Turin, Italy; and † Department of Neurological Sciences and Vision, University of Genoa, Genoa, Italy (Received 30 December 2002; Revised 17 March 2003; Accepted 10 April 2003) Abstract—Current evidence suggests that amyloid peptides (A) may play a major role in the pathogenesis of Alzheimer’s disease by eliciting oxidative stress and neuronal apoptosis. In this study we have used differentiated SK-N-BE neurons to investigate molecular mechanisms and regulatory pathways underlying apoptotic neuronal cell death elicited by A 1– 40 and A 1– 42 peptides as well as the relationships between apoptosis and oxidative stress. A peptides, used at concentrations able to induce oxidative stress, elicit a classic type of neuronal apoptosis involving mitochondrial regulatory proteins and pathways (i.e. affecting Bax and Bcl-2 protein levels as well as release of cytochrome c in the cytosol), poly-ADP rybose polymerase cleavage and activation of caspase 3. This pattern of neuronal apoptosis, that is significantly prevented by -tocopherol and N-acetylcysteine and completely abolished by specific inhibitors of stress-activated protein kinases (SAPK) such as JNKs and p38 MAPK , involved early elevation of p53 protein levels. Pretreatment of neurons with -pifithrin, a specific p53 inhibitor, resulted in a 50-60% prevention of A induced apoptosis. These results suggest that oxidative stress - mediated neuronal apoptosis induced by amyloid operates by eliciting a SAPK– dependent multiple regulation of pro-apoptotic mitochondrial pathways involving both p53 and bcl-2. © 2003 Elsevier Inc. Keywords—Amyloid peptides, Oxidative stress, p53, JNKs, p38 MAPK , Apoptosis, Free radicals INTRODUCTION Alzheimer’s disease (AD) is a neurodegenerative disor- der characterized by memory loss and cognitive impair- ment [1]. Although the ethiology of AD is not fully understood, an increasing body of evidence suggests the importance of amyloid (A) in the initiation/progres- sion of the disease. A, a 39 – 43 amino acid peptide, assembles into insoluble aggregates forming plaques [2]. Degeneration of neurons seems to be a fundamental process responsi- ble for clinical manifestations of many different neuro- logical disorders, including AD. It is generally accepted that A peptides may contribute to neuronal and synaptic loss during the course of the disease and “in vitro” data indicate that exposure of neuronal cells to A peptides is followed by apoptotic cell death [3,4]. The precise mechanism by which A induces neuro- nal apoptosis is still a matter of debate, but current literature suggests a central role for oxidative stress in AD pathogenesis. Reactive oxygen intermediates (ROI), such as superoxide anion (O 2 • ) and hydrogen peroxide (H 2 O 2 ), as well as 4-hydroxynonenal (HNE), a major aldehydic end product of lipid peroxidation, may mediate A neurotoxicity [5,6]. Aldehydic end products of poly- unsaturated fatty acids, such as 4-hydroxynonenal (HNE), colocalize with intraneuronal neurofibrillary tan- gles and contribute to the cytoskeletal derangement proper of the disease [7]. Protein oxidation, lipid peroxi- dation, and peroxynitrite formation have been unequiv- ocally reported in either human brain samples or in experimental models of AD [8]. Either ROI or HNE have been shown to trigger apoptosis in a large variety of cultured cells of different origin [9,10], including some cultured neuronal cells [11,12], and to stimulate stress- Address correspondence to: Dr. Elena Tamagno, Department of Experimental Medicine and Oncology, General Pathology Section, University of Turin, Corso Raffaello 30, 10125, Turin, Italy; Tel: 39 (11) 670-7763; Fax: 39 (11) 670-7753; E-Mail: elena.tamagno@ unito.it. Free Radical Biology & Medicine, Vol. 35, No. 1, pp. 45–58, 2003 Copyright © 2003 Elsevier Inc. Printed in the USA. All rights reserved 0891-5849/03/$–see front matter doi:10.1016/S0891-5849(03)00244-2 45

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Original Contribution
MULTIPLE SIGNALING EVENTS IN AMYLOID �-INDUCED, OXIDATIVESTRESS-DEPENDENT NEURONAL APOPTOSIS
ELENA TAMAGNO,* M AURIZIO PAROLA,* M ICHELA GUGLIELMOTTO,* GIANNI SANTORO,* PAOLA BARDINI,*LAURA MARRA,* M ASSIMO TABATON,† and OLIVIERO DANNI*
*Department of Experimental Medicine and Oncology, General Pathology Section, University of Turin, Turin, Italy; and†Department of Neurological Sciences and Vision, University of Genoa, Genoa, Italy
(Received 30 December 2002;Revised 17 March 2003;Accepted 10 April 2003)
Abstract—Current evidence suggests that amyloid� peptides (A�) may play a major role in the pathogenesis ofAlzheimer’s disease by eliciting oxidative stress and neuronal apoptosis. In this study we have used differentiatedSK-N-BE neurons to investigate molecular mechanisms and regulatory pathways underlying apoptotic neuronal celldeath elicited by A�1–40 and A�1–42 peptides as well as the relationships between apoptosis and oxidative stress. A�peptides, used at concentrations able to induce oxidative stress, elicit a classic type of neuronal apoptosis involvingmitochondrial regulatory proteins and pathways (i.e. affecting Bax and Bcl-2 protein levels as well as release ofcytochrome c in the cytosol), poly-ADP rybose polymerase cleavage and activation of caspase 3. This pattern ofneuronal apoptosis, that is significantly prevented by�-tocopherol and N-acetylcysteine and completely abolished byspecific inhibitors of stress-activated protein kinases (SAPK) such as JNKs and p38MAPK, involved early elevation ofp53 protein levels. Pretreatment of neurons with�-pifithrin, a specific p53 inhibitor, resulted in a 50-60% preventionof A� induced apoptosis. These results suggest that oxidative stress - mediated neuronal apoptosis induced by amyloid� operates by eliciting a SAPK–dependent multiple regulation of pro-apoptotic mitochondrial pathways involving bothp53 and bcl-2. © 2003 Elsevier Inc.
Keywords—Amyloid � peptides, Oxidative stress, p53, JNKs, p38MAPK, Apoptosis, Free radicals
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disor-der characterized by memory loss and cognitive impair-ment [1]. Although the ethiology of AD is not fullyunderstood, an increasing body of evidence suggests theimportance of� amyloid (A�) in the initiation/progres-sion of the disease.
A�, a 39–43 amino acid peptide, assembles intoinsoluble aggregates forming plaques [2]. Degenerationof neurons seems to be a fundamental process responsi-ble for clinical manifestations of many different neuro-logical disorders, including AD. It is generally acceptedthat A� peptides may contribute to neuronal and synapticloss during the course of the disease and “in vitro” data
indicate that exposure of neuronal cells to A� peptides isfollowed by apoptotic cell death [3,4].
The precise mechanism by which A� induces neuro-nal apoptosis is still a matter of debate, but currentliterature suggests a central role for oxidative stress inAD pathogenesis. Reactive oxygen intermediates (ROI),such as superoxide anion (O2
•�) and hydrogen peroxide(H2O2), as well as 4-hydroxynonenal (HNE), a majoraldehydic end product of lipid peroxidation, may mediateA� neurotoxicity [5,6]. Aldehydic end products of poly-unsaturated fatty acids, such as 4-hydroxynonenal(HNE), colocalize with intraneuronal neurofibrillary tan-gles and contribute to the cytoskeletal derangementproper of the disease [7]. Protein oxidation, lipid peroxi-dation, and peroxynitrite formation have been unequiv-ocally reported in either human brain samples or inexperimental models of AD [8]. Either ROI or HNE havebeen shown to trigger apoptosis in a large variety ofcultured cells of different origin [9,10], including somecultured neuronal cells [11,12], and to stimulate stress-
Address correspondence to: Dr. Elena Tamagno, Department ofExperimental Medicine and Oncology, General Pathology Section,University of Turin, Corso Raffaello 30, 10125, Turin, Italy; Tel:�39(11) 670-7763; Fax:�39 (11) 670-7753; E-Mail: [email protected].
Free Radical Biology & Medicine, Vol. 35, No. 1, pp. 45–58, 2003Copyright © 2003 Elsevier Inc.
Printed in the USA. All rights reserved0891-5849/03/$–see front matter
doi:10.1016/S0891-5849(03)00244-2
45

activated protein kinases [13–15] such as JNK andp38MAPK. The latter kinases are known to be activated inneuronal cells undergoing apoptosis after survival signalwithdrawal [16,17] or exposure to A� [18,19].
By investigating the mechanism(s) underlying oxida-tive stress-dependent, A�-induced apoptosis, we havepreviously reported that this event requires early andsimultaneous generation of hydrogen peroxide and HNEthat, in turn, are primarily responsible for JNK andp38MAPK activation as well as for apoptosis [20]. In thepresent study we show that A�-induced apoptosis notonly requires oxidative stress-mediated activation ofSAPKs, but operates through recruitment of classic ap-optotic mitochondrial regulatory proteins involving bothp53 and Bcl-2. Moreover, all the biological events in-duced by A� are mimicked by oxidative stress-relatedreactive intermediates, significantly prevented when cellsare pretreated with antioxidants and completely abol-ished by specific SAPKs inhibitors.
MATERIALS AND METHODS
Materials
All reagents used for cell culture and differentiationwere from Sigma Chemical Company (St. Louis, MO,USA). A�1–40 and A�1–42 were from Bachem (Buben-dorf, Switzerland). HNE, SB203580 inhibitor ofp38MAPK, and �-pifithrin were purchased from Calbio-chem (La Jolla, CA, USA). Polyclonal antibodies againstcaspase 3 and PARP were from Santa Cruz Biotechnol-ogy (La Jolla, CA, USA). Monoclonal antibody againstp53 was from Zymed Laboratories Inc. (San Francisco,CA, USA); polyclonal antibody against Bax and mono-clonal anti Bcl-2 were from Oncogene Research Prod-ucts (Boston, MA, USA); monoclonal antibody againstcytochrome c was from BD Pharmingen (San Diego,CA, USA). JNK inhibitor SP600125 was purchased fromBiomol Research Laboratories (Plymouth, PA, USA).
SK-N-BE differentiation
SK-N-BE neuroblastoma cell line used in this studyunderwent neuronal differentiation by means of chronictreatment with retinoic acid (RA) [21].
Cells were maintained in RPMI 1640 medium con-taining 2 mmol/l glutamine and supplemented with 100ml/l fetal bovine serum, 10 ml/l nonessential aminoacids, and 10 ml/l antibiotic mixture (penicillin-strepto-mycin-amphotericin), in a humidified atmosphere at37°C with 5% CO2.
For differentiation, 2 � 106 cells were plated in 75cm2 culture flasks (Costar) and exposed to 10 �mol/l RAfor 10 d. Growth medium was changed three times aweek.
Experimental protocol
In all experiments differentiated cells were left for24 h in serum-free RPMI medium and then treated withA�1–40 and A�1–42. A� fragments were dissolved inwater at 1 mg/ml directly before use or the solution wasaliquoted and stored frozen at �20°C. Refreezing wasavoided. Before use these peptides were maintained at37°C for 5 d, and at the beginning of each experimentwere diluted to the desired final concentration [22]. A�peptides used in this study were usually employed at 100nmol/l concentration; in one set of experiments, designedto follow apoptosis and cell death at the end of 1 week oftreatment, SK-N-BE cells were exposed to 25 as well as50 nmol/l concentrations of A�. In the experiments inwhich SK-N-BE were exposed only to HNE, H2O2, orboth, these reactive intermediates were used at concen-trations of 1 and 10 �mol/l and 10 �mol/l, respectively.Differentiated cells were exposed to A� peptides, HNE,and H2O2, as well as to HNE 1 �M/H2O2 10 �M mixturefor 4 h. To evaluate the morphological occurrence ofapoptosis, differentiated SK-N-BE cells were incubatedin the presence of the mentioned agents for 24 h.
When required, the antioxidants �-tocopherol succi-nate and N-acetylcysteine (final concentration 100�mol/l) as well as inhibitors of JNK and p38MAPK
(SP600125 20 �mol/l, SB203580 10 �mol/l, respective-ly), �-pifithrin (200 nmol/l) or the inhibitor of executioncaspases z-VAD.fmk (100 �mol/l) were added to culturemedium 1 h (antioxidants, �-pifithrin, and z-VAD.fmk)or 15 min (SAPK inhibitors) before exposure of cells tothe various experimental conditions (i.e., A� peptidesand H2O2/HNE mixture).
Oxidative stress determination
Thiobarbituric acid reactive substances (TBARS) for-mation was evaluated following the method described byEsterbauer et al. [23].
HNE was detected by means of an HPLC procedureessentially as previously described [24]. An aliquot ofculture media was added, in equal volume, to acetoni-trile-acetic acid (96:4,v:v). Samples were then centri-fuged at 250 � g for 20 min at 4°C and the supernatantwas directly injected to HPLC (Waters Associated,Milford, MA, USA) using a RP-18 column (Merck,Darmstadt, Germany). The mobile phase used was 42%acetonitrile:bidistilled water (v:v). Authentic 4-hy-droxynonenal (Calbiochem, La Jolla, CA, USA) wasused as a standard.
End products of lipid peroxidation were also evalu-ated in terms of fluorescent chromolipid adducts. Totallipids were extracted by the method of Folch et al. [25].Fluorescent intensity of samples was evaluated at 360/430 excitation/emission as described by Esterbauer et al.
46 E. TAMAGNO et al.

[26], using quinine sulphate (0.1 �g/ml in 0.05 H2SO4)as standard.
Oxidative stress was finally monitored, also measur-ing generation of hydrogen peroxide (H2O2), addinghorseradish peroxidase and acetylated ferrocytochrome cto cells. H2O2 release was evaluated as the increase ofacetylated ferrocytochrome c oxidation rate as describedby Zoccarato et al. [27].
Cell viability
The extent of necrosis was evaluated using lactatedehydrogenase (LDH) release in culture medium. Enzy-matic analysis of LDH activity released by necrotic cellsin culture medium was performed as previously de-scribed [20]. Values for control and treated cells wereexpressed as a percentage value of the total LDH activityreleased by untreated cells after exposure to TritonX-100.
Preparation of cell lysates and cytosolic extracts
Confluent differentiated cells were treated with theappropriate experimental conditions, quickly placed onice and washed with ice-cold PBS. Cell lysates andcytosolic extracts were obtained by the method of An-drew and Faller as previously described [28].
Preparation of mitochondria
Three 75 cm2 flasks for each condition at confluencewere harvested, treated with 0.1% Na-deoxycholate, andmaintained for 15 min to �80°C. The lysed cells werethen added to mannitol-sucrose buffer for a final strengthof 1� (210 mM mannitol, 70 mM sucrose, 5 mM EDTA,5 mM Tris, pH 7.5). The ice-cold suspension was thencentrifuged for 10 min at 800 � g to pellet nuclei. Thesupernatant was saved, the pelleted material was re-suspended in 1� mannitol-sucrose buffer, and centrifu-gation was repeated. This procedure was repeated three
Fig. 1. Induction of oxidative stress in differentiated SK-N-BE cells after treatment with A�1–40 100 nM and A�1–42 100 nM. Oxidativestress has been evaluated in terms of release of H2O2 and of HNE in the medium (panels A and B), of TBARS production (C) and offluorescent chromolipids formation (D). Data have been obtained at the end of 4 h of incubation at 37°C. Values are means � SD ofthree experiments performed in duplicate. � � significantly different from control (p � .02).
47A�, oxidative stress and apoptosis

times. The combined supernatants were then centrifugedto pellet any remaining nuclei, and the resulting super-natant was centrifuged at 10,000 � g to pellet mitochon-dria. Isolated mitochondria were re-suspended in a buffercontaining 20 mM HEPES, pH 7.6, 1 mM EDTA, 5 mMdithiothreitol, 300 mM KCl, and 5% glycerol. This prep-aration was briefly sonicated on ice and 5 �l of proteaseinhibitor mixture from Sigma (for mammalian extracts,100 mM AEBSF [4-(2-amynoethyl) benzenesulfonyl flu-oride], 4 mM bestatin, 1.4 mM E64, 2.2mM leupeptin,1.5 mM pepsatin and 80 �M aprotinin) were added permilliliter of buffer. The fractions were again centrifugedat 5000 � g to pellet any remaining cell debris andsupernatant proteins were used for Western blot analysis[29].
Western blot analysis
Total cell lysates, cytosolic and mitochondrial ex-tracts were subjected to SDS-PAGE on acrylamide gelsusing the mini-PROTEAN II electrophoresis cell (Bio-
Rad Laboratories, Segrate, Milano, Italy) according toLaemmli [30]. Proteins were transferred electrophoreti-cally to nitrocellulose membranes (Hybond-C extra; Am-ersham Life Science, Arlington Heights, IL, USA). Un-specific binding was blocked with 50 g/l nonfat dry milkin 50 mmol/l Tris-HCl, pH 7.4, containing 200 mmol/lNaCl and 0.5 ml/l Tween 20 (TBS-Tween). The blotswere incubated with the different primary antibodies,followed by incubation with peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulins in TBS-Tweencontaining 20 g/l nonfat dry milk. Immunoblots weredeveloped with ECL-plus reagents from Amersham ac-cording to manufacturer’s instructions. Comparativeanalysis of protein levels was performed by means ofcomputer-assisted densitometric analysis of proteinbands using an appropriate software program (Multi-analyst, Version 1.1, Bio-Rad Laboratories, Segrate Mi-lano); results were normalized to actin levels in thecorresponding blots and expressed as fold increase vs.respective control values; in crucial experiments con-
Fig. 2. Induction of caspase 3 (A) and caspase 3-like activity (B) obtained in lysates of differentiated SK-N-BE cells after 4 h oftreatment with A� peptides as well as HNE, H2O2, or HNE 1 �M/H2O2 10 �M (MIX). Induction was prevented by 1 h pretreatmentwith �-tocopherol (100 �M) and N-acetylcysteine (100 �M). Equal protein loading was confirmed by evaluating the levels of � actin.� � significantly different from control (p � .05); � � significantly different from control (p � .02); � � significantly different fromA� peptides or HNE 1 �M/H2O2 10 �M mixture (p � .02).
48 E. TAMAGNO et al.
cerning effects of �-pifithrin and SAPKs inhibitors, datawere also expressed as arbitrary units of optical densityand as means � SD of three independent experiments.
Caspase 3-like activity
Activity of caspase 3 was detected by using a color-imetric kit (Sigma Chemical Company). The colorimet-ric assay is based on the hydrolysis of the peptide sub-strate acetyl-Asp-Glu-Val-asp p-nitroanilide (Ac-DEVD-pNA) by caspase 3, resulting in the release ofp-nitroaniline (p-NA) moiety.
Morphological detection of apoptosis
The occurrence of apoptosis was evaluated by 4'-6diamidino-2-phenylindole (DAPI) staining. To identifyapoptotic nuclei with DAPI staining, cells were grown onglass coverslips, washed in PBS, fixed and permeabilizedwith 95% ethanol for 5 min, and then stained with DAPIsolution for 30 min at 37°C. After rinsing in PBS, cov-erslips were mounted on glass slides and observed byfluorescence microscopy (Leitz Dialux 20 Microscope)
[11]. All the experiments were repeated three times andthe number of stained cells was counted in 10 randomlyselected fields.
Statistical analysis of data
Where appropriate, statistical analysis was performedby means of Student’s t-test or ANOVA test followed bythe Bonferroni post test [31].
RESULTS
Induction of oxidative stress by A� peptides
A� peptides have been reported to initiate multiplemembrane alterations including protein and lipid oxi-dation [5,32–34]. Oxidative stress induced by A� pep-tides resulted in an early and significant increase inHNE and H2O2; 170% and 400%, respectively (Fig. 1,A and B). Accordingly, exposure of differentiatedSK-N-BE cells to A� peptides was followed by asignificant increase in lipid peroxidation, as evaluated
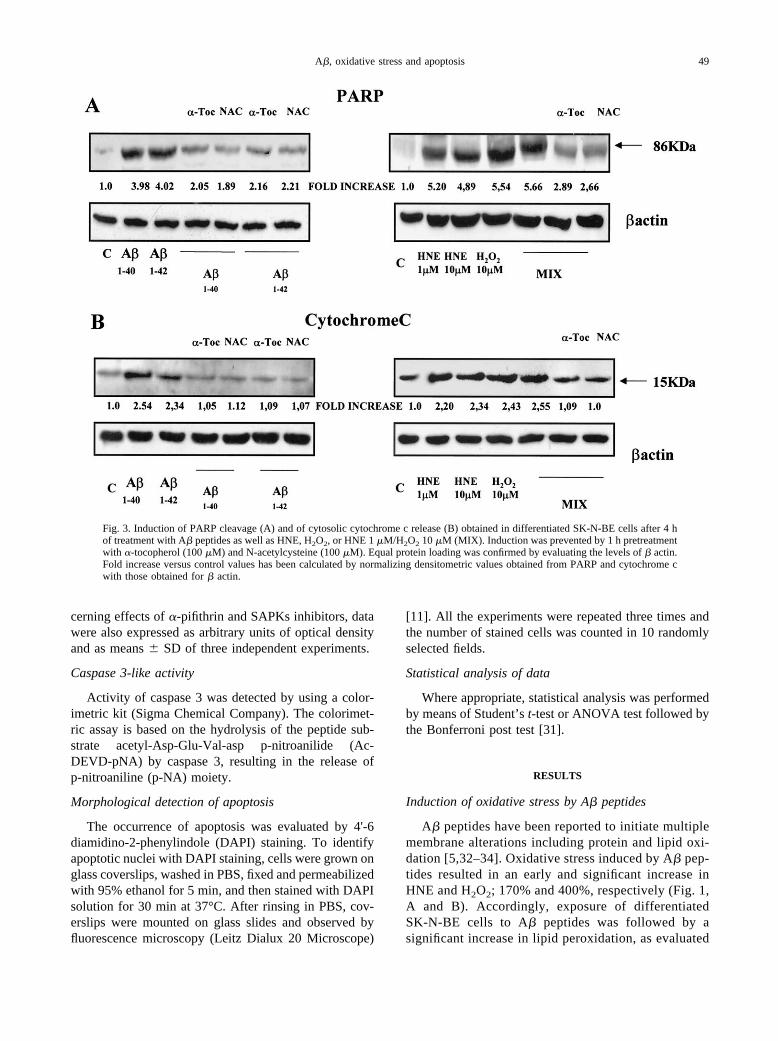
Fig. 3. Induction of PARP cleavage (A) and of cytosolic cytochrome c release (B) obtained in differentiated SK-N-BE cells after 4 hof treatment with A� peptides as well as HNE, H2O2, or HNE 1 �M/H2O2 10 �M (MIX). Induction was prevented by 1 h pretreatmentwith �-tocopherol (100 �M) and N-acetylcysteine (100 �M). Equal protein loading was confirmed by evaluating the levels of � actin.Fold increase versus control values has been calculated by normalizing densitometric values obtained from PARP and cytochrome cwith those obtained for � actin.
49A�, oxidative stress and apoptosis

by determining fluorescent chromolipid adducts(�150%) and TBARS production (� 400%) (Fig. 1, Cand D). All the parameters studied were significantlyincreased after 4 h of incubation with A� peptides ascompared to control cells.
Induction of caspase 3 and PARP cleavage by A�peptides and oxidative stress-related intermediates
Caspase 3 activation was evaluated either by quanti-fying both 32 kDa proenzyme and 20 kDa active isoformin SK-N-BE cell lysates by Western blot analysis or byevaluating spectrophotometrically caspase 3-related en-zymatic activity. A significant increase (90–100%) in 20kDa active isoform levels was detected 4 h after treat-ment with either A� peptides or HNE and H2O2 (alone orin combination), suggesting proteolytic activation ofcaspase-3 in our experimental conditions. This featurewas associated with the concomitant decrease in the levelof pro-caspase-3 (Fig. 2A). Ac-DEVD-pNa cleavage ac-tivity (i.e., a measure of caspase 3-like activity) was
again significantly increased (400–600%) after treat-ment with A� peptides or exposure to the combination ofHNE and H2O2. Furthermore, cleavage of PARP (anendogenous substrate of caspase 3) to the 86 kDa C-terminal fragment resulted similarly increased (�400–500%) after treatment with A� peptides as well as pro-oxidant conditions (Fig. 3). The induction of classicapoptotic cell death was confirmed by data obtainedmonitoring cytochrome c release in the cytosol (Fig. 3B).Results obtained in cells treated with A� peptides or withHNE and/or H2O2 are in agreement with those alreadyobtained for the other parameters of apoptosis, indicatingthat all the experimental conditions used were effectivein increasing cytochrome c release (approximately250%).
Pretreatment of differentiated SK-N-BE cells with�-tocopherol and N-acetylcysteine significantly pre-vented activation of caspase 3, release of cytochrome c,and cleavage of PARP (Fig. 2, A and B; Fig. 3, A and B),suggesting a direct relationship between A� peptides-
Fig. 4. Increase of intracellular p53 levels (A) as evaluated by Western blot analysis obtained in differentiated SK-N-BE cells after 4 hof treatment with A� peptides as well as HNE, H2O2, or HNE 1 �M/H2O2 10 �M (MIX). The increase was prevented by 1 hpretreatment with �-tocopherol and N-acetylcysteine. Pretreatment of cells with specific inhibitors of JNKs (SP600125) and p38MAPK
(SB203580) or with �-pifithrin completely prevented the increase of p53 protein levels (panels B and C). Equal protein loading wasconfirmed by evaluating the levels of � actin. Fold increase versus control values has been calculated by normalizing densitometricvalues obtained from p53 with those obtained for � actin.
50 E. TAMAGNO et al.

mediated activation of classic caspase-dependent execut-ing pathway and oxidative stress.
Role of p 53 in the induction of pro-apoptoticpathways by A� peptides and oxidative stress
Exposure of SK-N-BE differentiated cells to A� pep-tides, HNE, and/or H2O2 resulted in an early and signif-icant increase (approx. �400%) in p53 protein levels, asevaluated by Western blot analysis (Fig. 4A). Pretreat-ment with antioxidants was again able to significantlyprevent this induction.
To evaluate the relative role of p53 and of SAPKs inthe apoptotic pathway induced by A� peptides, cellswere pretreated with a mixture of two specific inhibitorsfor JNKs and p38MAPK (SB203580�SP600125) or witha specific inhibitor for p53 (�-pifithrin).
Specific inhibitors for JNKs and p38MAPK were usedsimultaneously because we found in a previous study[20] that only this procedure was followed by an almostcomplete prevention of apoptosis induced by A� pep-tides or by oxidative stress intermediates. Treatment of
cells with the single inhibitors resulted in a significantbut incomplete (approximately 50%) prevention of apo-ptosis, suggesting equal causative involvement of bothpathways [20].
As shown in Fig. 4B, inhibition of the two kinasesresulted in the complete prevention of p53 induction.Moreover, the pretreatment of cells with �-pifithrin alsois able to inhibit p53 induction (Fig. 4C).
Exposure of cells to A� peptides and to HNE andH2O2, alone or in association, resulted in a significantincrease (�300–400%) of the protein levels of the pro-apoptotic effector Bax (Fig. 5); moreover, antioxidantswere able to significantly prevent this induction (Fig.5A). As expected, the induction of Bax was completelyprevented by either SAPKs inhibitors or by �-pifithrin,suggesting that Bax was under the control of SAPKs thatoperated through activation of p53. Data reported in Fig.6 indicate that the same experimental conditions thatwere able to elicit an increase of Bax levels also resultedin a significant decrease of Bcl-2 (�50%) (Fig. 6A).Interestingly, Bcl-2 levels were strongly affected by
Fig. 5. Increase of Bax levels (A) by Western blot analysis obtained in differentiated SK-N-BE cells after 4 h of treatment with A�peptides as well as HNE, H2O2, or HNE 1 �M/H2O2 10 �M (MIX). The increase was prevented by 1 h pretreatment with �-tocopheroland N-acetylcysteine. Pretreatment of cells with specific inhibitors of JNKs (SP600125) and p38MAPK (SB203580) or with �-pifithrin,completely prevented the increase of proapoptotic Bax protein levels (panels B and C). Equal protein loading was confirmed byevaluating the levels of � actin. Fold increase versus control values has been calculated by normalizing densitometric values obtainedfrom Bax with those obtained for � actin.
51A�, oxidative stress and apoptosis

SAPKs inhibitors but were essentially p53-independent:treatment of cells with SB203580/SP600125 mixtureprevented the decrease of Bcl-2 induced by A� peptidesor by HNE/H2O2 (Fig. 6B), whereas �-pifithrin wasineffective.
Similarly, pretreatment with SB203580/SP600125mixture was able to offer a complete prevention in termsof cyt.c release, caspase 3 activation, and PARP cleavageby A� peptides and oxidative stress intermediates (Fig.7, panels A, B, and C). However, �-pifithrin pretreatmentresulted only in a significant but incomplete preventionof the mentioned parameters. As documented by densi-tometric analyses, a residual increase in cytochrome crelease, caspase 3 activation, and PARP cleavage wasdetected in the presence of �-pifithrin. The antiapoptoticaction of SAPKs inhibitors and �-pifithrin was con-firmed morphologically by means of DAPI staining (Fig.8) and once again �-pifithrin resulted in an approximate50–60% prevention of A� peptides and oxidative stressintermediates mixture-induced apoptosis.
A� peptides as well as HNE and/or H2O2 do notinduce a necrotic type of cell death
In order to assess whether A� peptides as well asHNE and/or H2O2 may elicit necrotic cell death in SK-N-BE cells, the release of lactate dehydrogenase hasbeen evaluated at 24 (Fig. 9, panel A) and 48 h (Fig. 9,panel B) time points in the presence as well as in theabsence of the inhibitor of execution caspases z-VAD-.fmk (i.e., to inhibit specifically caspase-mediated apo-ptosis). Our data indicate that no significant change inLDH release was found, suggesting that A�- as well asHNE- and/or H2O2-induced apoptosis represents the ma-jor phenomenon in our experimental conditions.
Chronic exposure of SK-N-BE cells to lower doses ofA� peptides is again followed by induction of p53 andactivation of caspase 3
Because AD is a chronic disease, we exposed SK-N-BE cells to lower concentrations of A� peptides for
Fig. 6. Decrease of Bcl-2 levels (A) by Western blot analysis obtained in differentiated SK-N-BE cells after 4 h of treatment with A�peptides as well as HNE, H2O2, or HNE 1 �M/H2O2 10 �M (MIX). The decrease was prevented by 1 h pretreatment with �-tocopheroland N-acetylcysteine. Pretreatment of cells with specific inhibitors of JNKs (SP600125) and p38MAPK (SB203580) completelyprevented the decrease of antiapoptotic Bcl-2 protein levels (B). Pretreatment with � pifithrin was ineffective (C). Equal protein loadingwas confirmed by evaluating the levels of � actin. Fold increase versus control values has been calculated by normalizing densitometricvalues obtained from Bcl-2 with those obtained for � actin.
52 E. TAMAGNO et al.

as long as 1 week to evaluate in a preliminary waywhether the features previously described may still berecognized. In order to do so we evaluated, as relevantparameters, active form of caspase 3, p53 levels, andLDH release. In these “chronic” experimental condi-tions we found once again that A� peptides were stillable to induce both activation of caspase 3 (monitoredas a significant increase in the 20 KDa active form)
and recruitment of p53 (Fig. 10, panels A and B)without eliciting any significant increase in LDH re-lease (Fig. 10).
DISCUSSION
Although recent evidence suggests the importance ofA� in AD-related neuronal death, the signaling mecha-
Fig. 7. Prevention of cytosolic cytochrome c release (A), caspase 3-like activity (B) and PARP cleavage (C) observed by pretreatingdifferentiated SK-N-BE cells with SAPK inhibitors and �-pifithrin, before exposure to A� peptides or HNE 1 �M/H2O2 10 �Mmixture. Cell lysates were obtained after 4 h of the latter treatment. Equal protein loading was confirmed by evaluating the levels of� actin. Optical density values of cytochrome c and PARP, normalized against � actin, are means � SD of three independentexperiments. � � significantly different from control (p � .05); � � significantly different from control (p � .02); � � significantlydifferent from A� peptides or HNE 1 �M/H2O2 10 �M mixture (p � .02).
53A�, oxidative stress and apoptosis

nisms controlling this event are still unclear and a matterof debate.
The present study indicates that A� peptides caninduce classic apoptosis (i.e., caspase 3-dependent)throughout oxidative stress-mediated activation ofSAPKs that, in turn, can affect proapoptotic mitochon-drial regulatory pathways by involving independentlyboth p53- and Bcl-2-dependent processes. Moreover, inour experimental conditions, apoptotic cell death is themajor feature observed in SK-N-BE cells exposed to A�peptides as well as to HNE and H2O2; necrotic cell deathdoes not significantly occur even if the cells are exposedchronically to A� peptides.
All the major events induced by A� peptides, includ-ing induction of Bax, decrease of Bcl-2, recruitment ofp53, cytosolic release of cytochrome c as well as ofPARP cleavage, can be mimicked by the simultaneousadministration of low concentrations of HNE and H2O2
that are known to be generated early after exposure ofSK-N-BE to A� peptides [20]. Moreover, since all theparameters affected by A� peptides are significantlyprevented by pretreatment with �-tocopherol and NAC, adirect involvement of oxidative stress is stronglysupported.
Previous data and present findings indicate that stress-activated protein kinases JNKs and p38MAPK are primar-
Fig. 8. Prevention of morphological signs of apoptosis induced by A�1–40 and HNE 1 �M/H2O2 10 �M mixture. Nuclear SK-N-BEmorphology has been evaluated in terms of DAPI staining 24 h after treatments with A�1–40 and HNE 1 �M/H2O2 10 �M mixture.Inhibitors of JNKs (SP600125) and p38MAPK (SB203580) and �-pifithrin were added 15 min and 1 h, respectively, before treatmentwith A�1–40 or mixture. Apoptotic nuclei are indicated by arrows (A). In panel B apoptosis is expressed as the number of apoptoticcells on the coverslips 24 h after treatment. Data shown are the means � SD of three independent experiments. � � significantlydifferent from control (p � .05); � � significantly different from control (p � .02); � � significantly different from A� peptides orHNE 1 �M/H2O2 10 �M mixture (p � .02).
54 E. TAMAGNO et al.

ily involved in A�-induced neuronal apotosis. Althoughno clear consensus has been achieved in the literatureconcerning the role of JNKs and p38MAPK in determiningneuronal apoptosis, Shoji et al. [19] have recently shownthat intracellular A� accumulation can indeed triggerJNK activation leading to neuronal cell death. Moreover,evidence for a sequential activation of ERK, JNK/SAPK,and p38MAPK in brain tissue from subjects with AD hasbeen provided [35]. In this connection, it has been clearlyestablished that in the brain of AD patients bothp38MAPK and JNKs are activated [36,37]. Moreover, inanother study Zhu et al. [38] have shown evidence for theactivation in AD patient’s brain of MKK6, an upstreamkinase activator of p38MAPK. More recently, Wei et al.[39] reported that JNK was rapidly activated by A�treatment and that this activation appeared to be criticalfor A�-induced neuronal death. However, these authorsreported no activation of p38MAPK by A� in their modelof cultured SH-SY5Y cells.
A direct link between oxidative stress and SAPKs hasbeen shown demonstrating that JNK and p38MAPK areactivated by HNE and H2O2 in various cell types[13,40,41]. In a previous study we reported that thesimultaneous addition of exogenous HNE and H2O2,used at the same low concentrations detected within thefirst 3 h after A� exposure, can elicit activation of JNKand p38MAPK as well as apoptosis [20].
The use of specific inhibitors of JNKs and p38MAPK
(SP600125 and SB203580) or of the specific inhibitor ofp53 �-pifithrin, that selectively blocks the ability of p53to activate target genes in neurons [42], pointed out thatactivation of SAPKs is crucial since inhibition of JNKsand p38MAPK completely prevented apoptosis induced byA� peptides and mediated by oxidative stress interme-diates. However, inhibition of p53 function by �-pifithrinstill resulted in a significant (approximately 50–60%),but not complete, prevention of apoptotic neuronal celldeath.
p53 is known to play a crucial role in the regulation ofapoptosis in many cell types by stimulating expressionand/or mitochondrial translocation of the death effectorprotein Bax [43]. Recently, involvement of p53 in neu-ronal death occurring in Alzheimer’s disease [44], Par-kinson’s disease [45], and traumatic brain injury [46] hasbeen detected. Cell culture studies have establishedstrong relationships between p53 expression and neuro-nal death induced by DNA damaging agents and gluta-mate [47]. A critical role for p53 in neuronal apoptosisresulting from ischemic and excitotoxic insults has beenconfirmed by studies performed in p53-deficient mice[48,49]. However, results by Giovanni et al. [50] de-scribed that apoptosis by A� in cultured embryonic cor-tical neurons obtained from p53 knockout mice wasmainly p53-independent. In our experimental model theinvolvement of p53 in A�-induced apoptotic cell deathseems critical since it accounts for approximately 50–60% of total cell number, as suggested by data obtainedwith the specific inhibitor �-pifithrin.
Accumulating evidence suggests that the regulation ofp53’s transcriptional activities depends on the rate of itsphosphorylation [51,52]. Stability of p53, a key factorable to determine its ability to mediate multiple activi-ties, is known to be tightly regulated by JNK in a phos-phorylation-dependent manner [53]. A phosphoacceptorsite of p53 for JNK phosphorylation, tyrosine 81, seemsto be crucial for p53 activities [54]. Moreover, phosphor-ylation by p38MAPK on residues 33 and 46 is required forUV-induced p53-mediated apoptosis [55], and inhibitionof p38MAPK has been found to attenuate transcriptionalactivities of p53 and p53-dependent apoptosis inducedby chemotherapeutic agents [56].
Results obtained with SAPKs inhibitors and�-pifithrin suggest that in SK-N-BE cells exposed to
Fig. 9. Cell death induced by 24 h (A) or 48 h (B) treatment with A�peptides as well as HNE, H2O2, or HNE 1 �M/H2O2 10 �M (MIX) asevaluated in terms of LDH release. Pretreatment with z-VAD.fmk didnot modify the parameter.
55A�, oxidative stress and apoptosis

A�, p53 involvement is downstream to SAPKs acti-vation; in this connection, activation of SAPKs, par-ticularly of JNK isoforms, may act as proapoptoticfactor also by phosphorylating and inactivating theantiapoptotic protein Bcl-2 [57]. The involvement ofthis pathway may explain the 40 –50% residual, p53-independent apoptosis. Data presented in this study,including negative effects of pretreatment with�-pifithrin on bcl-2 levels and complete prevention ofA�-induced apoptosis by SAPK inhibitors stronglysupport this view. Other authors have also demon-strated that UV-induced classic apoptosis in mouse
fibroblasts, mediated by cytochrome c release, can becompletely abrogated in the absence of JNK signaling[58]. The scheme in Fig. 11 summarizes results ob-tained in this study suggesting that oxidative stress-mediated neuronal apoptosis induced by amyloid �peptides operates by eliciting a SAPK-dependent mul-tiple regulation of proapoptotic mitochondrial path-ways involving both p53 and bcl-2. Finally, since theinvolvement of p53 has been reported in neurodegen-erative diseases, including AD, our findings seem toconfirm that chemical inhibitors of p53 activation maybe efficient in hampering neurodegeneration [42].
Fig. 10. Induction of caspase 3 (A) and of p53 protein levels (B) obtained in lysates of differentiated SK-N-BE cells after a chronic(1 week) treatment with low concentrations of A� peptides (25–50 nM). Equal protein loading was confirmed by evaluating � actinlevels. Densitometric analysis of caspase 3 and p53 levels, normalized against � actin, are means � SD of three independentexperiments. Cell death has been evaluated in terms of LDH release (C). � � significantly different from control (p � .05); � �significantly different from control (p � .02).
56 E. TAMAGNO et al.

Acknowledgements — This study was supported by Ministerodell’Universita e della Ricerca Scientifica e Tecnologica (MURST,Rome), National Project: Alteration of signaling systems induced byglyco-oxidative stress: potential substrates of Alzheimer’s diseasepathogenesis.
REFERENCES
[1] Rosemblum, W. I. The presence, origin and significance of Abetapeptide in the cell bodies of neurons. J. Neuropathol. Exp. Neurol.58:575–581; 1999.
[2] Selkoe, D. J. Alzheimer’s disease: genes, proteins, and therapy.Physiol. Rev. 81:741–766; 2001.
[3] Shimohama, S. Apoptosis in Alzheimer’s disease-an update. Ap-optosis 5:9–16; 2000.
[4] Roth, K. A. Caspases, apoptosis, and Alzheimer’s disease: cau-sation, correlation, and confusion. J. Neuropathol. Exp. Neurol.60:829–838; 2001.
[5] Mark, R. J.; Lovell, M. A.; Markesbery, W. R.; Uchida, K.;Mattson, M. P. A role for 4-hydroxynonenal, an aldehydic prod-uct of lipid peroxidation, in disruption of ion homeostasis andneuronal cell death induced by amyloid �-peptide. J. Neurochem.68:255–264; 1997.
[6] Camandola, S.; Poli, G.; Mattson, M. P. The lipid peroxidationproduct 4-hydroxy-2,3-nonenal increases AP-1-binding activitythrough caspase activation in neurons. J. Neurochem. 74:159–168; 2000.
[7] Takeda, A.; Smith, M. A.; Avila, J.; Nunomura, A.; Siedlak, S. L.;Zhu, X.; Perry, G.; Sayre, L. M. In Alzheimer’s disease, hemeoxygenase is coincident with Alz50, an epitope of � induced by
4-hydroxy-2-nonenal modification. J. Neurochem. 75:1234–1241; 2000.
[8] Pratico, D. Alzheimer’s disease and oxygen radicals: new in-sights. Biochem. Pharmacol. 63:563–567; 2002.
[9] Kalinich, J. F.; Ramakrishnan, R.; McClain, D. E.; Ramakrishnan,N. 4-hydroxynonenal, an end product of lipid peroxidation, in-duces apoptosis in human leukemic T- and B-cell lines. FreeRadic. Res. 33:349–358; 2000.
[10] Han, H.; Wang, H.; Long, H.; Nattel, S.; Wang, Z. Oxidativepreconditioning and apoptosis in L-cells: roles of protein kinase Band mitogen-activated protein kinases. J. Biol. Chem. 276:26357–26364; 2001.
[11] Kaltschmidt, B.; Uherek, M.; Wellmann, H.; Volk, B.;Kaltschmidt, C. Inhibition of NF-kB potentiates amyloid �-me-diated neuronal apoptosis. Proc. Natl. Acad. Sci. USA 96:9409–9414; 1999.
[12] Zhou, L.-J.; Zhu, X.-Z. Reactive oxygen species-induced apopto-sis in PC12 cells and protective effect of bilobalide. J. Pharmacol.Exp. Ther. 293:982–988; 2000.
[13] Parola, M.; Robino, G.; Marra, F.; Pinzani, M.; Bellomo, G.;Leonarduzzi, G.; Chiarugi, P.; Camandola, S.; Poli, G.; Waeg, G.;Gentilini, P.; Dianzani, M. U. HNE interacts directly with JNKisoforms in human hepatic stellate cells. J. Clin. Invest. 102:1942–1950; 1998.
[14] Soh, Y.; Jeong, K.-S.; Lee, I. J.; Bae, M.-A.; Kim, Y.-C.; Song, B.J. Selective activation of the c-Jun N-terminal protein kinasepathway during 4-hydroxynonenal-induced apoptosis of PC12cells. Mol. Pharmacol. 58:535–541; 2000.
[15] Kurata, S. Selective activation of p38 MAPK cascade and mitoticarrest caused by low level oxidative stress. J. Biol. Chem. 275:23413–23416; 2000.
[16] Kummer, J. L.; Rao, R. K.; Heidenreich, K. A. Apoptosis inducedby withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J. Biol. Chem. 272:20490–20494; 1997.
[17] Watson, A.; Eilers, A.; Lallemand, D.; Kyriakis, J.; Rubin, L. L.;Ham, J. Phosphorylation of c-Jun is necessary for apoptosisinduced by survival signal withdrawal in cerebellar granule neu-rons. J. Neurosci. 18:751–762; 1998.
[18] McDonald, D. R.; Bamberger, M. E.; Combs, C. K. M.; Landreth,G. E. � amyloid fibrils activate parallel mitogen-activated proteinkinase pathways in microglia and THP1 monocytes. J. Neurosci.18:4451–4460; 1998.
[19] Shoji, M.; Iwakami, N.; Takeuchi, S; Waragai, M.; Suzuki, M.;Kanazawa, I.; Lippa, C. F.; Ono, S.; Okazawa, H. JNK activationis associated with intracellular beta-amyloid accumulation. BrainRes. Mol. Brain Res. 8:221–233; 2000.
[20] Tamagno, E.; Robino, G.; Obbili, A.; Bardini, P.; Aragno, M.;Parola, M.; Danni, O. H2O2 and 4-hydroxynonenal mediate amy-loid �-induced neuronal apoptosis by activating JNKs andp38MAPK. Exp. Neurol. 180:144–155; 2003.
[21] Condorelli, F.; Sortino, M. A.; Stella, A. M.; Canonico, P. L.Relative contribution of different receptor subtypes in the re-sponse of neuroblastoma cells to tumor necrosis factor-alpha.J. Neurochem. 75:1172–1179; 2000.
[22] Kaltschmidt, B.; Uherek, J. K.; Volk, B.; Bauerle, P. A.;Kaltschmidt, C. Transcription factor NF-kB is activated in pri-mary neurons by amyloid � peptides and in neurons surroundingearly plaques from patients with Alzheimer disease. Proc. Natl.Acad. Sci. USA 94:2642–2647; 1997.
[23] Esterbauer, H.; Cheeseman, K. H.; Dianzani, M. U.; Poli, G.;Slater, T. F. Separation and characterization of the aldehydicproducts of lipid peroxidation stimulated by ADP/Fe�� in ratliver microsomes. Biochem. J. 208:129–140; 1982.
[24] Esterbauer, H.; Schaur, R. J.; Zollner, H. Chemistry and biochem-istry of 4-hydroxynonenal, malondialdehyde and related alde-hydes. Free Radic. Biol. Med. 11:81–128; 1991.
[25] Folch, J.; Lees, M.; Sloane-Stanley, G. H. A simple method forthe isolation and purification of total lipids from animal tissues.J. Biol. Chem. 226:497–509; 1957.
[26] Esterbauer, H.; Koller, E.; Slee, R. G.; Koster, J. F. Possibleinvolvement of the lipid peroxidation product 4-hydroxynonenal
Fig. 11. Proposed pro-apoptotic sequence elicited by A� peptidesthroughout the generation of oxidative stress-related intermediates,related activation of SAPK, and recruitment of p53 protein.
57A�, oxidative stress and apoptosis

in the formation of fluorescent chromolipids. Biochem. J. 239:405–409; 1986.
[27] Zoccarato, F.; Valente, M.; Alexandre, A. Identification of anNADPH plus iron dependent, Ca2� activated peroxide productionin synaptosomes. Biochim. Biophys. Acta 1176:208–214; 1993.
[28] Andrew, N. C.; Faller, D. V. A rapid micropreparation techniquefor extraction of DNA-binding proteins from limiting numbers ofmammalian cells. Nucleic Acids Res. 19:2499; 1991.
[29] Dobson, A. W.; Xu, Y.; Kelley, M. R.; LeDoux, S. P.; Wilson, G.L. Enhanced mitochondrial DNA repair and cellular survival afteroxidative stress by targeting the human 8-oxoguanine glycosylaserepair enzyme to mitochondria. J. Biol. Chem. 275:37518–37523;2000.
[30] Laemmli, U. K. Cleavage of structural proteins during the assem-bly of the head of bacteriophage T4. Nature 227:680–685; 1970.
[31] Mattwes, D. E.; Farewell, V. T. Using and understanding medicalstatistics, 2nd rev. ed. Basel: Karger; 1988.
[32] Smith, M. A.; Hirai, K.; Hsiao, K.; Pappolla, M. A.; Harris, P. L.;Siedlak, M.; Tabaton, M.; Perry, G. Amyloid-beta deposition inAlzheimer transgenic mice is associated with oxidative stress.J. Neurochem. 70:2212–2215; 1998.
[33] Markesbery, W. R.; Carney, J. H. Oxidative alterations in Alz-heimer’s disease. Brain Pathol. 9:133–146; 1999.
[34] Christen, Y. Oxidative stress and Alzheimer’s disease. Am. J.Clin. Nutr. 71:621S–629S; 2000.
[35] Zhu, X.; Castellani, R. J.; Takeda, A.; Nunomura, A.; Atwood,C. S.; Perry, G.; Smith, M. A. Differential activation of neuronalJNK/SAPK and p38 in Alzheimer disease: the “ two hit” hypoth-esis. Mech. Ageing Dev. 123:39–46; 2001.
[36] Hensley, K.; Floyd, R. A.; Zheng, N. Y.; Nael, R.; Robinson,K. A.; Nguyen, X.; Pye, Q. N.; Stewart, C. A.; Geddes, J.;Markesbery, W. R.; Patel, E.; Johnson, G. V.; Bing G. p38 kinaseis activated in the Alzheimer’s disease brain. J. Neurochem.72:2053–2058; 1999.
[37] Zhu, X.; Raina, A. K.; Rottkamp, C. A.; Aliev, G.; Perry, G.;Boux, H.; Smith, M. A. Activation and redistribution of c-junN-terminal kinase/stress activated protein kinase in degeneratingneurons in Alzheimer’s disease. J. Neurochem. 76:435–441;2001.
[38] Zhu, X.; Rottkamp, C. A.; Hartzler, A.; Sun, Z.; Takeda, A.;Boux, H.; Shimohama, S.; Perry, G.; Smith, M. A. Activation ofMKK6, an upstream activator of p38, in Alzheimer’s disease.J. Neurochem. 79:311–318; 2001.
[39] Wei, W.; Wang, X.; Kusiak, W. Signalling events in amyloid�-peptide-induced neuronal death and insulin-like growth factor Iprotection. J. Biol. Chem. 277:17649–17656; 2002.
[40] Lo, Y. Y. C.; Wong, J. M. S.; Cruz, T. F. Reactive oxygen speciesmediate cytokine activation of c-jun NH2-terminal kinases.J. Biol. Chem. 271:15703–15707; 1996.
[41] Uchida, K.; Shiraishi, M.; Naito, Y.; Torii, Y.; Nakamura, Y.;Osawa, T. Activation of stress signaling pathways by the endproduct of lipid peroxidation. 4-hydroxy-2-nonenal is a potentialinducer of intracellular peroxide production. J. Biol. Chem. 274:2234–2242; 1999.
[42] Culmsee, C.; Zhu, X.; Yu, Q.-S.; Chan, S. L.; Camandola, S.;Guo, Z.; Greig, N. H.; Mattson, M. P. A synthetic inhibitor of p53protects neurons against death induced by ischemic and excito-toxic insults, and amyloid �-peptide. J. Neurochem. 77:220–228;2001.
[43] Sheikh, M. S.; Fornace, A. J. Jr. Role of p53 family members inapoptosis. J. Cell. Physiol. 182:171–181; 2000.
[44] Zhang, Y.; McLaughlin, R.; Goodyer, C.; LeBlanc, A. Selectivecytotoxicity of intracellular amyloid beta peptide 1–42 throughp53 and Bax in cultured primary human neurons. J. Cell Biol.156:519–529; 2002.
[45] Blum, D.; Wu, Y.; Nissou, M. F.; Arnaud, S.; Benabid, A.; Verna,J. M. P53 and Bax in 6-hydroxydopamine-induced apoptosis inPC12 cells. Brain Res. 751:139–142; 1997.
[46] Napieralski, J. A.; Raghupathi, R.; McIntosh, T. K. The tumor-suppressor gene, p53, is induced in injured brain regions follow-ing experimental traumatic brain injury. Brain Res. Mol. BrainRes. 71:78–86; 1999.
[47] Inamura, N.; Araki, T.; Enokido, Y.; Nishio, C.; Aizawa, S.;Hatanaka, H. Role of p53 in DNA strand break-induced apoptosisin organotypic slice culture from the mouse cerebellum. J. Neu-rosci. Res. 60:450–457; 2000.
[48] Maeda, K.; Hata, R.; Gillardon, F.; Hossmann, K. A. Aggravationof brain injury after transient focal ischemia in p53-deficient mice.Brain Res. Mol. Brain Res. 88:54–61; 2001.
[49] Cregan, S. P.; Fortin, A.; MacLaurin, J. G.; Callaghan, S. M.;Cecconi, F.; Yu, S. W.; Dawson, T. M.; Dawson, V. L.; Park,D. S.; Kroemer, G.; Slack, R. S. Apoptosis-inducing factor isinvolved in the regulation of caspase-independent neuronal celldeath. J. Cell Biol. 158:507–517; 2000.
[50] Giovanni, A.; Keramaris, E.; Morris, E. J.; Hou, S. T.; O’Hare,M.; Dyson, N.; Robertson, G. S.; Slack, R. S.; Park, D. S. E2F1mediates death of B-amyloid-treated cortical neurons in a mannerindependent of p53 and dependent on Bax and Caspase 3. J. Biol.Chem. 275:11553–11560; 2000.
[51] Giaccia, A.; Kastan, M. B. The complexity of p53 modulation:emerging patterns from divergent signals. Genes Dev. 12:2973–2983; 1998.
[52] Ashcroft, M.; Taya, Y.; Vousden, K. H. Stress signals utilizemultiple pathways to stabilize p53. Mol. Cell. Biol. 20:3224–3233; 2000.
[53] Buschmann, T.; Adler, V.; Matusevich, E.; Fuchs, S. Y.; Ronai, Z.P53 phosphorylation and association with murine double minute2, c-jun NH2-terminal kinase, p13ARF, and p300/CBP during thecell cycle and after exposure to ultraviolet irradiation. CancerRes. 60:896–900; 2000.
[54] Buschmann, T.; Popatova, O.; Bar-Shira, A.; Ivanov, V. N.;Fuchs, S. Y.; Henderson, S.; Fried, V. A.; Minamoto, T.; Alarcon-Vargas, D.; Pincus, M. R.; Gaarde, W. A.; Holbrook, N. J.;Shiloh, Y.; Ronai, Z. Jun NH2-terminal kinase phosphorylation ofp53 on Thr-81 is important for p53 stabilization and transcrip-tional activities in response to stress. Mol. Cell. Biol. 21:2743–2754; 2001.
[55] Bulavin, D. V.; Saito, S.; Hollander, M. C.; Sakaguchi, K.;Anderson, C. W.; Appella, E.; Fornace, A. J. Phosphorylation ofhuman p53 by p38 kinase coordinates N-terminal phosphorylationand apoptosis in response to UV radiation. EMBO J. 18:6845–6854; 1999.
[56] Sanchez-Prieto, R.; Rojas, J. M.; Taya, Y.; Gutkind, J. S. A rolefor the p38 mitogen-activated protein kinase pathway in thetranscriptional activation of p53 on genotoxic stress by chemo-therapeutic agents. Cancer Res. 60:2464–2472; 2000.
[57] Yamamoto, K.; Ichijo, H.; Korsmeyer, S. J. Bcl-2 is phosphory-lated and inactivated by an ASK/Jun N-terminal protein kinasepathway normally activated at G(2)/M. Mol. Cell. Biol. 19:8469–8478; 1999.
[58] Tournier, C.; Hess, P.; Yang, D. D.; Xu, J.; Turner, T. K.;Nimnual, A.; Bar-Sagi, D.; Jones, S. N.; Flavell, R. A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cyto-chrome c-mediated death pathway. Science 288:870–874; 2000.
ABBREVIATIONS
HNE—4-hydroxynonenalJNK—c-Jun NH2- terminal kinasep38MAPK—p38-mitogen activated protein kinasePARP—poly-ADP rybose polymeraseSAPK—stress-activated protein kinase
58 E. TAMAGNO et al.
Related Documents











