Fax +41 61 306 12 34 E-Mail [email protected] www.karger.com Original Paper Pharmacology 2006;76:19–33 DOI: 10.1159/000088929 Beta-Amyloid and Oxidative Stress Jointly Induce Neuronal Death, Amyloid Deposits, Gliosis, and Memory Impairment in the Rat Brain Laurent Lecanu a, b Janet Greeson c Vassilios Papadopoulos a, b a Departments of Biochemistry and Molecular Biology, b Samaritan Research Laboratories, Georgetown University Medical Center, Washington, D.C., and c Samaritan Pharmaceuticals, Las Vegas, Nev., USA Introduction Alzheimer’s disease (AD) is the most common form of dementia that occurs in the elderly, affecting about 10% of people older than 65 years and about 40% of people over 80 years of age. During the last decade, several hy- potheses have been set forth to explain the pathophysiol- ogy of AD. Currently, the main hypotheses concerning the origin of the disease are based on amyloidogenesis, disruption of calcium homeostasis, energetic failure, in- duction of oxidative stress, and most recently, hyperphos- phorylation of Tau protein [1–5]. Despite the large amount of data that has been generated upon testing of these hypotheses, the mechanisms underlying AD and the events responsible for its progression remain unclear. Transgenic mouse models, based on amyloid protein precursor (APP) mutations similar to those found in hu- man familial AD or on other proteins shown to be in- volved in AD, have led to the identification of factors that initiate and contribute to AD progression, and they have also led to the development of the above-mentioned hy- potheses [6, 7] . However, these transgenic models are closely related, physiologically, to cases of familial AD, which represent only about 5% of all AD cases. Thus, these models do not completely address the onset and progression of sporadic AD, which represents about 95% Key Words Alzheimer Amyloid deposit Oxidative stress Cerebrospinal fluid Protein Tau Neurodegenerative disease Abstract Infusion of Fe 2+ , A 42 , and buthionine-sulfoximine (FAB), but not A 42 alone or in combination with Fe 2+ , into the left cerebral ventricle of Long-Evans rats for 4 weeks in- duced memory impairment that was accompanied by increased hyperphosphorylated Tau protein levels in the CSF. FAB-infused animals displayed thioflavin-S-posi- tive amyloid deposits, hyperphosphorylated Tau pro- tein, neuronal loss, and gliosis. Animals treated with A 42 , Fe 2+ , or buthionine-sulfoximine alone or in combi- nation failed to show the histological modifications seen with FAB. This data suggests that A 42 is not sufficient to induce an Alzheimer’s disease-like symptomatology, and it supports a model whereby a decrease in the brain’s antioxidant defense system leads to the A 42 -indepen- dent oxidative stress necessary for the peptide to induce histopathological changes and memory loss. Copyright © 2006 S. Karger AG, Basel Received: June 28, 2005 Accepted after revision: August 15, 2005 Published online: October 13, 2005 Dr. V. Papadopoulos Department of Biochemistry, Georgetown University Medical Center 3900 Reservoir Road NW Washington, DC 20057 (USA) Tel. +1 202 687 8991, Fax +1 202 687 7855, E-Mail [email protected] © 2006 S. Karger AG, Basel 0031–7012/06/0761–0019$23.50/0 Accessible online at: www.karger.com/pha

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Fax +41 61 306 12 34E-Mail [email protected]
Original Paper
Pharmacology 2006;76:19–33 DOI: 10.1159/000088929
Beta-Amyloid and Oxidative Stress Jointly Induce Neuronal Death, Amyloid Deposits, Gliosis, and Memory Impairment in the Rat Brain
Laurent Lecanu
a,
b Janet Greeson
c Vassilios Papadopoulos
a,
b
a Departments of Biochemistry and Molecular Biology, b
Samaritan Research Laboratories, Georgetown University Medical Center, Washington, D.C., and c
Samaritan Pharmaceuticals, Las Vegas, Nev. , USA
Introduction
Alzheimer’s disease (AD) is the most common form of dementia that occurs in the elderly, affecting about 10% of people older than 65 years and about 40% of people over 80 years of age. During the last decade, several hy-potheses have been set forth to explain the pathophysiol-ogy of AD. Currently, the main hypotheses concerning the origin of the disease are based on amyloidogenesis, disruption of calcium homeostasis, energetic failure, in-duction of oxidative stress, and most recently, hyperphos-phorylation of Tau protein [1–5] . Despite the large amount of data that has been generated upon testing of these hypotheses, the mechanisms underlying AD and the events responsible for its progression remain unclear.
Transgenic mouse models, based on amyloid protein precursor (APP) mutations similar to those found in hu-man familial AD or on other proteins shown to be in-volved in AD, have led to the identifi cation of factors that initiate and contribute to AD progression, and they have also led to the development of the above-mentioned hy-potheses [6, 7] . However, these transgenic models are closely related, physiologically, to cases of familial AD, which represent only about 5% of all AD cases. Thus, these models do not completely address the onset and progression of sporadic AD, which represents about 95%
Key Words Alzheimer � Amyloid deposit � Oxidative stress � Cerebrospinal fl uid � Protein Tau � Neurodegenerative disease
Abstract Infusion of Fe 2+ , A � 42 , and buthionine-sulfoximine (FAB), but not A � 42 alone or in combination with Fe 2+ , into the left cerebral ventricle of Long-Evans rats for 4 weeks in-duced memory impairment that was accompanied by increased hyperphosphorylated Tau protein levels in the CSF. FAB-infused animals displayed thiofl avin-S-posi-tive amyloid deposits, hyperphosphorylated Tau pro-tein, neuronal loss, and gliosis. Animals treated with A � 42 , Fe 2+ , or buthionine-sulfoximine alone or in combi-nation failed to show the histological modifi cations seen with FAB. This data suggests that A � 42 is not suffi cient to induce an Alzheimer’s disease-like symptomatology, and it supports a model whereby a decrease in the brain’s antioxidant defense system leads to the A � 42 -indepen-dent oxidative stress necessary for the peptide to induce histopathological changes and memory loss.
Copyright © 2006 S. Karger AG, Basel
Received: June 28, 2005 Accepted after revision: August 15, 2005 Published online: October 13, 2005
Dr. V. PapadopoulosDepartment of Biochemistry, Georgetown University Medical Center3900 Reservoir Road NWWashington, DC 20057 (USA)Tel. +1 202 687 8991, Fax +1 202 687 7855, E-Mail [email protected]
© 2006 S. Karger AG, Basel0031–7012/06/0761–0019$23.50/0
Accessible online at:www.karger.com/pha

Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 20
of all cases and whose incidence is age-dependent and not obviously linked to genetic factors. Considering the events – amyloidosis, massive gliosis, mitochondrial im-pairment, vascularitis – that lead to neuronal death and loss of memory in AD patients, antioxidant defenses like-ly play a critical role in the onset of this disease [8, 9] .
Oxidative stress has been observed in several trans-genic AD mouse model strains [10] and brain tissues from transgenic mice expressing the Alzheimer’s linked PS1 mutated protein exhibit decreased antioxidant enzyme activity [11] . However, the relationship between oxida-tive stress and amyloid deposition and their respective roles in A � 42 -induced pathology in the transgenic model are incompletely understood. Moreover, the origin of A � 42 -dependent and -independent oxidative stress has yet to be fully established. Here we report results of ex-periments examining the effect of chronic intracerebro-ventricular (i.c.v.) delivery of A � 42 alone, or A � 42 in com-bination with the pro-oxidant, Fe 2+ , or the glutathione synthesis inhibitor, buthionine sulfoximine (BSO), on brain histopathology and memory loss.
Material and Methods
All experimental protocols were approved by the Georgetown University Animal Care and Use Committee. Experiments were performed according to the code of practice for animal experimen-tation of the Animal Welfare Act and the Public Health Service Policy on Laboratory Animal Care.
Animals Male Long-Evans rats weighing 300–325 g (3–4 months old)
were housed following a natural day-night cycle with food and wa-ter ad libitum.
Morris Water Maze Protocol Prior to surgery, rats were trained in a standard Morris spatial
navigation task in a black water tank (200 cm diameter). The water was rendered opaque by water-miscible, nontoxic white paint (Crayola). Starting from four separate, randomly assigned ‘start positions’, rats were trained to fi nd an invisible platform (20 cm diameter) placed at a fi xed position. Water temperature was ap-proximately 24 ° C. Trials continued until the animal found the plat-form or until 120 s had elapsed. If the animal did not fi nd the plat-form within 120 s, then it was placed on the platform for 20 s and removed from the water tank. Rats were trained for 4 consecutive days, receiving 4 trials per day at 30-min intervals. Surgery was peformed on the fi fth day. Rats were re-tested for memory reten-tion after being subjected to 4 weeks of intracerebroventricular per-fusion. The mean platform retrieval times at the fourth day of train-ing and at postinfusion were calculated for each group and used for statistical analysis.
Surgical Procedure Rats were anesthetized with equithesin (3 ml/kg i.p.) and placed
on a stereotaxic frame. Using an electrode micromanipulator, the outlet of an osmotic micropump (Durect Corp., Cupertino, Calif., USA) was implanted into the left cerebral ventricle according to the following coordinates: D = 3.4 mm, L = 1.4 mm, and AP = 0.92 mm caudal to bregma [12] . The osmotic pump tanks were im-planted into a subcutaneous pocket in the midscapular area of the animals’ back. During recovery, rats were placed on a heating blan-ket and their body temperature was maintained at 37 ° C.
Four weeks after surgery, the rats were sacrifi ced by intracar-diac perfusion – fi rst with a washing solution (NaCl 8 g/l, dextrose 4 g/l, sucrose 8 g/l, calcium chloride 0.23 g/l, sodium cacodylate anhydrous 0.25 g/l, in deionized water) and then with fi xative cac-odylate buffer (sucrose 40 g/l, paraformaldehyde 40 g/l, sodium cacodylate anhydrous 10.72 g/l, in deionized water). Brains were removed and stored in fi xative cacodylate buffer until further pro-cessing.
Perfusion Rats were randomly assigned to one of four separate groups
(n = 6–8 animals per group): group I (control), artifi cial cerebrospi-nal fl uid (CSF); group II, 15 � mol/l A � 42 , pH = 4.8 8 0.1; group III, A � 42 + 1 mmol/l ferrous sulfate (FeSO 4 ), pH = 4.4 8 0.1; group IV, A � 42 + FeSO 4 + 12 mmol/l buthionine sulfoximine (BSO),pH = 5.1 8 0.1; group V, BSO, pH = 6.2 8 0.1; group VI, FeSO 4 , pH = 4.7 8 0.1; group VII, BSO + FeSO 4 , pH = 5.7 8 0.1 (values are mean 8 SEM from three independent experiments, determined in triplicates). The different solutions were made in distilled water. The perfusion rate of the pump was 2.5 � l/h. At the end of the 4-week perfusion period, each respective group received 25.2 nmol of A � 42 , 1.68 � mol of FeSO 4 and 20.16 � mol of BSO. Each group was composed of 6–8 animals.
Immunoblot Analysis of A � Polymerization in A � 42 , A � 42 + Fe 2+ , and A � 42 + Fe 2+ + BSO Solutions A � 42 , A � 42 + Fe 2+ , and A � 42 + Fe 2+ + BSO solutions were incu-
bated in pump tanks for 14 days at 37 ° C. Samples were then sepa-rated on 4–20% Tris-glycine gels (Invitrogen, Carlsbad, Calif., USA) under native conditions for 2 h at 125 V and electrotrans-ferred to nitrocellulose membranes (Hybond™ ECL™, Amersham Pharmacia Biotech, Piscataway, N.J., USA) for 30 min at 130 Å. Non-specifi c adsorption of the antibodies was blocked by incubat-ing the nitrocellulose in 5% milk. The blots were probed for A � species using a polyclonal antibody that recognizes a 30 amino acid peptide of the A � protein (Zymed Laboratories, San Francisco, Calif., USA). Membranes were incubated in primary antibody for 1 h at room temperature at a dilution of 1: 2,000. Membranes were then incubated in the secondary antibody at a dilution of 1: 1,000 for 1.5 h at room temperature. The blots were visualized using the ECL™ Western Blotting Analysis System (Amersham Bio-sciences).
Immunohistochemistry and Other Histochemical Staining Immediately after excision, brains were embedded in a gelatin
block and sectioned into 40- � m-thick slices. The brain sections were processed with the following primary antibodies: anti-P-Tau protein recognizing phosphorylated Ser202 (BioSource Interna-tional, Camarillo, Calif., USA) or the AT-8 clone (Innogenetics, Belgium), polyclonal anti-neurofi brillary tangles (Chemicon Inter-

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 21
national, Temecula, Calif., USA), anti-glial fi brillary acidic protein (GFAP; Santa Cruz Biotechnology, Santa Cruz, Calif., USA) for activated astrocytes, anti-A � 42 – which specifi cally recognizes ami-no acids 33–42 in the C-terminus of the peptide (Signet Laborato-ries, Dedham, Mass., USA), anti-CD11b (clone OX42) (Novus Bi-ologicals) for activated microglia, and polyclonal anti-8-hydroxy-deoxyguanosine (Chemicon International) to assess oxidative DNA damage.
Consecutive sections were also stained with cresyl violet, De Olmos amino cupric silver stain, and Campbell-Switzer silver stain. Sections were stained with 0.5% cresyl violet (pH = 3.7) for 10 min before being mounted using aqueous mounting medium. Cresyl violet selectively stains Nissl bodies that are lost when neurons die, leading to a decrease or a lack of staining. De Olmos amino cupric silver stain was employed to detect degenerating neuronal peri-karya, dendrites, axons, and synaptic terminals and was performed according to the method of Switzer [13] . Campbell-Switzer silver stain was employed to visualize neuritic plaques and neurofi brillary tangles (NFTs) and was carried, without modifi cation, according to the original method devised by Campbell et al. [14] . Finally, thiofl avin S staining was performed in order to detect amyloid plaques.
ELISA Cerebrospinal fl uid (125 � l) was withdrawn from the magna
cisternae the day of the surgery, and at the end of the 4-week infu-sion period, immediately following the last water maze test. Levels of hyperphosphorylated Tau protein were then measured using the INNOTEST™ ELISA kit (Innogenetics, Belgium) according to manufacturer’s instructions. Samples were run in triplicates.
Statistical Analysis Group means were compared using a one-way analysis of vari-
ance (ANOVA). Multiple comparisons testing was performed using the Dunnett test ( � = 0.05). p ̂ 0.05 was accepted as signifi cant.
Results
Effect of A � 42 , A � 42 + Fe2 + , and Fe2 + + A � 42 + BSO Infusion on Memory Retention To assess memory impairment, mean platform re-
trieval times obtained during the fourth day of training were compared to those obtained after the 4-weeks infu-sion period for each treatment group. At the end of the fourth day of training, the mean platform retrieval times were similar among all groups. Postinfusion platform re-trieval times for the groups that have been infused with A � 42 , A � 42 + Fe 2+ or Fe 2+ + A � 42 + BSO (FAB) were com-pared to the post-infusion times obtained from the con-trol group (CSF infusion). As shown in fi gure 1 , animals that received FAB exhibited a statistically signifi cant in-crease in platform retrieval time compared to control an-imals (p ! 0.001, n = 8). Although intra-group comparison revealed an increase in the post-infusion retrieval time
compared to the pre-infusion time for groups perfused with A � 42 alone or in combination with Fe 2+ , no differ-ence was observed when these groups’ post-infusion re-trieval times were compared to the control groups’ times (p 1 0.05).
Effect of A � 42 , A � 42 + Fe2 + , and Fe2 + + A � 42 + BSO Infusion on Amyloid Deposits No silver staining was detected in hippocampus of
control rats, or rats perfused with A � 42 , BSO, Fe 2+ , or A � 42 + Fe 2+ ( fi g. 2 a–d, f). Silver staining also allowed for the detection of activated glial cells, and a few activated astrocytes were detected in rats infused with BSO + Fe 2+ ( fi g. 2 e, arrow). In contrast, a pronounced dense silver staining was observed in FAB-treated rats ( fi g. 2 g). High-er power views revealed that FAB-treated rats displayed amyloid deposits in the CA1 and CA2 subregions of the hippocampus as well as in the dentate gyrus, which ex-hibited extensive gliosis ( fi g. 3 c). Amyloid deposits could
Fig. 1. Effect of A � 42 , A � 42 + Fe2 + , and Fe2 + + A � 42 + BSO infusion on Morris water maze scores. Rats were trained for 4 consecutive days, with 4 trials per day at 30 min intervals. Surgery was pe-formed on the fi fth day. Rats were re-tested for memory retention after being subjected to 4 weeks of intracerebroventricular perfu-sion. Each group is composed of 6–8 animals. Rats were divided into four groups: group I, control (artifi cial cerebrospinal fl uid; CSF); group II, 15 � mol/l A � 42 , pH = 4.8 8 0.1; group III, A � 42 + 1 mmol/l ferrous sulfate (FeSO 4 ), pH = 4.4 8 0.1; group IV, A � 42
+ FeSO 4 + 12 mmol/l buthionine sulfoximine (BSO), pH = 5.1 8 0.1 (pH values are means 8 SEM from three independent experi-ments determined in triplicates). Results are expressed as mean 8 SD. *** p ! 0.001 compared to control group. Open bars = Re-trieval time measured at the fourth day of training, before the sur-gery; closed bars = retrieval time measured after 4 weeks perfu-sion.

Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 22
also be seen in blood vessels in the CA2 ( fi g. 3 b, arrow). These deposits were further identifi ed in the FAB rats CA1 as being amyloid plaques where the thiofl avin-S staining was extremely intense ( fi g. 3 e) whereas no stain-ing was observed in the control rats ( fi g. 3 d).
Outside of the hippocampus, amyloid deposits con-taining remnants of dead neurons were observed in the cingulate cortex of FAB brains ( fi g. 4 b and insert). They were also present in blood vessels situated in the temporal cortex ( fi g. 4 c). Neurons containing strong silver staining
were also found in the temporal cortex of FAB-treated rats ( fi g. 8 f), whereas no positive staining was observed in the control rat cortex ( fi g. 8 e). At low magnifi cation the fl ame-shaped silver staining observed in the neuron body suggests the presence of NFTs.
Effect of A � 42 , A � 42 + Fe2 + , and Fe2 + + A � 42 + BSO Infusion on A � 42 Immunostaining No positive immunoreactivity for A � 42 was detected
in control rats, in rats infused with A � 42 , or rats infused
b
f
amu 0001
gmu 0001
c d
e
1000 µm 1000 µm 1000 µm
1000 µm 1000 µm 1000 µm
Fig. 2. Effect of A � 42 , Fe2 + , BSO infusion on Campbell-Switzer staining of amyloid deposits in the hippocampus. Representative, low magnifi cation images of the left hippocampus of the animals infused with CSF ( a ); A � 42 ( b ); BSO ( c ); Fe 2+ ( d ) ; BSO + Fe 2+ ( e ); A � 42 + Fe 2+ ( f ); A � 42 + Fe2 + + BSO (FAB) ( g ). Silver-based Campbell-Switzer impregnation allows the visualization of plaques and tangles. When rats were infused with A � 42 , BSO, Fe 2+ , or A � 42 + Fe 2+ no silver impregnation was observed and the images obtained were identical to the control. In con-trast, note the pronounced dense silver staining observed in FAB-treated rats. When a combination of BSO and Fe 2+ was infused, only a few activated astrocytes were detectable ( e , black arrow).

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 23
with A � 42 + Fe 2+ ( fi g. 5 ). In contrast to control sections, sections from FAB-infused rats displayed a strong A � 42 immunoreactivity in the CA1, CA2, and CA3 sub-regions of the hippocampus ( fi g. 5 e, f – high power views). This immunostaining in the CA1 and CA3 of the FAB, but not control rats was confi rmed by immunofl uorescence.
Effect of A � 42 , A � 42 + Fe2 + , and Fe2 + + A � 42 + BSO Infusion on Oxidative DNA Damage No 8-hydroxydeoxyguanosine was detected in control
rats, rats perfused with A � 42 either alone or in combina-tion with Fe 2+ ( fi g. 6 a–c). In contrast, FAB rats displayed a strong 8-hydroxydeoxyguanosine immunoreactivity re-vealing oxidative DNA damage ( fi g. 6 d).
Effect of A � 42 , A � 42 + Fe2 + , and Fe2 + + A � 42 + BSO Infusion on CSF P-Tau Levels For each animal, CSF was withdrawn from the magna
cisterna on the day of the surgery (day 0) and following
the infusion period (day 28). Hyperphosphorylated Tau (P-Tau) could only be detected in the CSF of rats that re-ceived infusion of FAB, but not in control animals or in animals infused with A � 42 or with A � 42 + Fe 2+ (429 8 175 pg/ml, p ! 0.001, n = 6–8; mean 8 SEM).
Effect of Fe 2+ + A � 42 + BSO Infusion on P-Tau and NFT Immunoreactivity Using two distinct antibodies against P-Tau [Ser202
and Ser199/202 (AT-8)], no P-Tau immunoreactivity was detected in control animals ( fi g. 7 ). In contrast, in FAB rats immunoreactive P-Tau was detected in neurons and in the parenchyma of the CA1 and CA2-CA3 using the anti-Ser202 antibody or the AT-8 antibody ( fi g. 7 c, d). Higher magnifi cation pictures clearly show the presence of P-Tau inside the neuronal body and also in the axons and dendrites. To confi rm the presence of microtubule-associated histological modifi cations in FAB-infused rat brains, we employed a polyclonal antibody raised against
a
b
c
50 µm
50.0 µm50.0 µm
50 µm50 µm
ed
Fig. 3. High magnifi cation of the Campbell-Switzer staining of amyloid deposits in hip-pocampus. Higher magnifi cation images of fi gure 2 g demonstrate the histological mod-ifi cations that occurred in the rat hippocam-pus after 4 weeks of the FAB solution perfu-sion. FAB-perfused rats displayed amyloid deposits (red arrows) in the CA2 ( b ), in the CA1 ( a ), and in the dentate gyrus ( c ). Vas-cular amyloid deposits (black arrows) were also seen in CA2 ( b ). Thiofl avin-S staining revealed that the amyloid deposits seen in the CA1 have the characteristics of amyloid plaques formation. d Control; e FAB.

Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 24
ba
c
50 µm 50 µm
50 µm
Fig. 4. Campbell-Switzer staining of amy-loid deposits in the cortex of a control and FAB-infused rat. High magnifi cation im-ages of the cortex of control and FAB-in-fused rats demonstrate the histological modifi cations that occurred after 4 weeks of the FAB solution perfusion. FAB-treated rats displayed amyloid deposits in the cin-gulate cortex ( b ), as compared to control rat ( a ). Vascular amyloid deposits (black ar-rows) were also seen in the temporal cortex of FAB rats ( c ).
a
c
b
d
fe
e
f
1000 µm
1000 µm 1000 µm
1000 µm
Fig. 5. Effect of A � 42 , A � 42 + Fe2 + , and Fe2 +
+ A � 42 + BSO infusion on A � 42 immuno-staining in the hippocampus. An antibody raised against the amino acids 33 to 42 of A � 42 and a secondary antibody labeled with horseradish peroxidase was employed to vi-sualize A � 42 in the hippocampus of control ( a ); A � 42 -infused ( b ); A � 42 + Fe 2+ -infused ( c ), and FAB-infused animals ( d ). No posi-tive immunoreactivity for A � 42 was detect-ed in control rats, in rats perfused with A � 42 or with A � 42 + Fe 2+ . FAB-infused rats dis-played a strong A � 42 immunoreactivity in the CA1, CA2, and CA3 sub-regions of the hippocampus. e High power view of fi g. 5a insert; f high power view of fi g. 5d insert.

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 25
200 mm 200 mm
200 mm200 mm
a b
c d
Fig. 6. Detection of the FAB-induced oxidative DNA damage in hippocampus. Oxidative DNA damage was detected using an antibody raised against 8-hydroxydeoxyguanosine and a secondary antibody conjugated to Cy-2. No 8-hydroxydeoxy-guanosine adducts were detected in control rats ( a ), in rats perfused by A � 42 ( b ), or in combination with Fe 2+ ( c ). In contrast, FAB rats displayed a strong 8-hydroxydeoxyguanosine immunoreactivity ( d ) sug-gesting that A � 42 , infused alone is not suffi cient to induce an oxidative stress in a healthy brain.
a
c d
FAB
Co
ntr
ol
b
CA1 CA2/CA3
50 µm
50 µm
200 µm
200 µm
Fig. 7. Effect of Fe2 + + A � 42 + BSO infusion on P-Tau immunoreactivity in the hippocampus. Immunofl uorescent staining for P-Tau was performed using antibodies raised against the hyperphosphorylated Ser 202 and raised against the hyperphosphorylated Ser 199/202 (clone AT-8). Images of the CA1 obtained with this antibody are shown in a and c . No P-Tau was detected in control sections. In contrast, there was a marked increase in P-Tau immu-noreactivity in FAB-infused animals. These results were confi rmed using the AT-8 clone as shown in the CA2-CA3 ( b , d ). The bodies and dendritic formations of the neurons which contain P-Tau can be clearly seen in the high power view of the fi eld ( d ).
Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 26
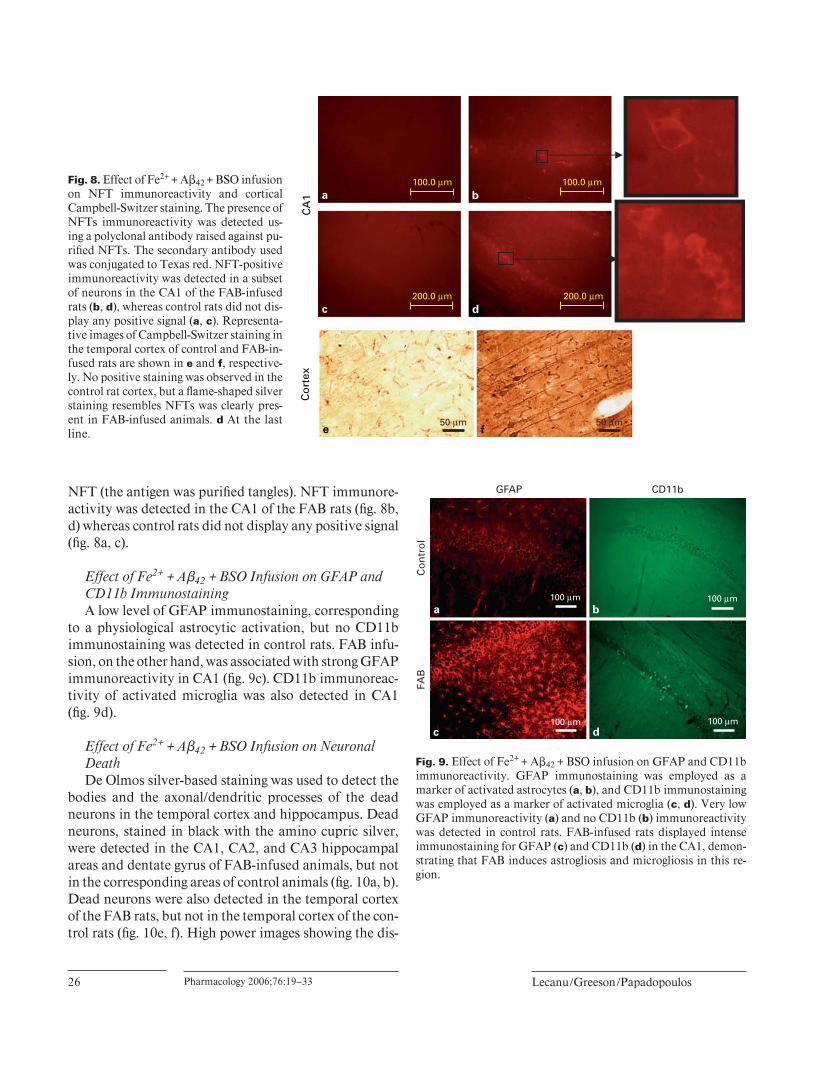
NFT (the antigen was purifi ed tangles). NFT immunore-activity was detected in the CA1 of the FAB rats ( fi g. 8 b, d) whereas control rats did not display any positive signal ( fi g. 8 a, c).
Effect of Fe2 + + A � 42 + BSO Infusion on GFAP and CD11b Immunostaining A low level of GFAP immunostaining, corresponding
to a physiological astrocytic activation, but no CD11b immunostaining was detected in control rats. FAB infu-sion, on the other hand, was associated with strong GFAP immunoreactivity in CA1 ( fi g. 9 c). CD11b immunoreac-tivity of activated microglia was also detected in CA1 ( fi g. 9 d).
Effect of Fe 2+ + A � 42 + BSO Infusion on Neuronal Death De Olmos silver-based staining was used to detect the
bodies and the axonal/dendritic processes of the dead neurons in the temporal cortex and hippocampus. Dead neurons, stained in black with the amino cupric silver, were detected in the CA1, CA2, and CA3 hippocampal areas and dentate gyrus of FAB-infused animals, but not in the corresponding areas of control animals ( fi g. 10 a, b). Dead neurons were also detected in the temporal cortex of the FAB rats, but not in the temporal cortex of the con-trol rats ( fi g. 10 e, f). High power images showing the dis-
a
cC
A1
fe
b
dC
ort
ex
50 µm 50 µm
100.0 µm100.0 µm
200.0 µm200.0 µm
Fig. 8. Effect of Fe2 + + A � 42 + BSO infusion on NFT immunoreactivity and cortical Campbell-Switzer staining. The presence of NFTs immunoreactivity was detected us-ing a polyclonal antibody raised against pu-rifi ed NFTs. The secondary antibody used was conjugated to Texas red. NFT-positive immunoreactivity was detected in a subset of neurons in the CA1 of the FAB-infused rats ( b, d ), whereas control rats did not dis-play any positive signal ( a, c ). Representa-tive images of Campbell-Switzer staining in the temporal cortex of control and FAB-in-fused rats are shown in e and f , respective-ly. No positive staining was observed in the control rat cortex, but a fl ame-shaped silver staining resembles NFTs was clearly pres-ent in FAB-infused animals. d At the last line.
Co
ntr
ol
FAB
b
d
a
c
GFAP CD11b
100 µm
100 µm 100 µm
100 µm
Fig. 9. Effect of Fe2 + + A � 42 + BSO infusion on GFAP and CD11b immunoreactivity. GFAP immunostaining was employed as a marker of activated astrocytes ( a, b ), and CD11b immunostaining was employed as a marker of activated microglia ( c, d ). Very low GFAP immunoreactivity ( a ) and no CD11b ( b ) immunoreactivity was detected in control rats. FAB-infused rats displayed intense immunostaining for GFAP ( c ) and CD11b ( d ) in the CA1, demon-strating that FAB induces astrogliosis and microgliosis in this re-gion.

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 27
Co
ntr
ol
FAB
50.0 mm 50.0 mm
50.0 mm 50.0 mm
500 µm
500 µm 500 µm 500 µm
500 µm 500 µm 100 µm
100 µm
Fig. 10. Effect of Fe2 + + A � 42 + BSO infusion on De Olmos amino cupric silver and cresyl violet staining in cortex and hippocampus. Images of De Olmos cupric silver staining in the hippocampus are shown in a and b , and staining in the cortex is shown in e and f . Dead neurons stain in black with the amino cupric silver method and are detected in the CA1, CA2, CA3, and dentate gyrus ( b ) areas of FAB-infused rats, but not in control rats ( a ). At higher magnifi -cation view of the fi eld indicated in b and f, the silver impregnation is clearly shown in neuronal bodies (red arrow) and in the dendrit-ic processes of dead neurons of the CA2-CA3 region; whereas, no staining is visible in the control group. This neuronal loss was con-
fi rmed by the cresyl violet staining ( c , d hippocampus; g , h cortex). Decreased staining density was observed in CA1, CA3, and the crest of the dentate gyrus of FAB-infused rats compared to their control counterparts. Signifi cant neuronal loss also occurred in the temporal cortex of FAB rats, as shown by the two different staining methods ( f vs. e and g vs. h ). High magnifi cation views of the tem-poral cortex (inserts) of the FAB-infused rats revealed a strong sil-ver impregnation of the neuronal dendritic processes and of the large pyramidal neurons bodies (red arrow); whereas, no neuronal loss was observed in the corresponding regions of control rats.

Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 28
integrated neurons in the hippocampus and temporal cor-tex of the FAB rats are shown from inserts of fi gures 10 b and 10f. As shown in fi gure 11 , rats that had been infused with BSO, Fe 2+ , or a combination of both did not display any neuronal death, either in the cortex or in the hippo-campus. The loss of neurons in the FAB group was con-fi rmed with cresyl violet staining. As shown in fi gures 10 d and 10c, as compared to the staining density in control rats, there is a decreased staining density in the CA1, CA3, and the crest of dentate gyrus in FAB-infused rats. The cresyl violet staining also revealed a loss in large py-ramidal neurons from the temporal cortex of FAB-in-fused rats ( fi g. 10 g, h).
a b c
d e f
Co
rtex
Hip
po
cam
pu
s
1000 µm 1000 µm 1000 µm
1000 µm 1000 µm 1000 µm
Fe2+ BSO Fe2+/BSO
Fig. 11. Effect of Fe 2+ , BSO, or a mixture of both on De Olmos amino cupric silver staining in the cortex and the hippocampus. Low magnifi cation images of the silver-based De Olmos staining in the cortex ( a–c ) and in the hip-pocampus ( d–f ) of rats that received for 4 weeks by i.c.v. BSO ( a , d ), Fe 2+ ( b , e ), or both ( c , f ). These pictures have to be compared to the control and to the FAB group displayed on fi gure 12. These pictures showed that Fe 2+ and BSO alone or combined, infused at their respective concentrations and rates, did not induce any neuronal death in the cortex or in the hippocampus of Long-Evans rats.
OSB
20 kD
10 kD
Aβ42 Aβ42 Aβ42
Fe2+ Fe2+
Fig. 12. Immunoblot analysis of Aß polymerization in the infusion mixtures. Immunoblot analysis was performed on the content of the pump at 2 weeks did not reveal any difference on the polymer-ization state of A � 42 when the peptide was infused alone or in as-sociation with Fe 2+ or Fe 2+ /BSO. The predominant A � species among all mixtures was approximately 9 kDa in weight and repre-sented the dimerized species.

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 29
Immunoblot Analysis of A � Polymerization in the Infusion Mixtures Immunoblot analysis of the content of the pump at
after weeks did not reveal any difference in the polymer-ization state of A � 42 when the peptide was infused alone or in association with Fe 2+ or Fe 2+ /BSO ( fi g. 12 ). The main band seen had an apparent molecular weight of9 kDa, corresponding to an A � 42 dimer. More slowly mi-grating oligomers were also observed (molecular sizes up to 20 kDa).
Discussion
Young adult male Long-Evans rats were directly in-fused, in the left cerebral ventricle, with a solution con-taining either A � 42 alone or in combination with pro-oxidants. Long-Evans rats were selected for these studies because of their high susceptibility to neurodegenerative diseases. Indeed, the Long-Evans strain carries a muta-tion of the Cblb gene that has been demonstrated to ren-der the encoded protein inactive, and Cblb -defi cient mouse strains are highly sensitive to experimental en-cephalomyelitis after immunization with myelin basic
Fig. 13. Schematic representation of the proposed mechanism underlying plaque and tangle formation in the presence of A � 42 . The data presented in this manuscript support a unifying ‘amyloidoxidative’ hypothesis to ex-plain the onset and progression of AD. a Healthy patient neuron. b Increase in A � 42 concentration in the brain of AD patients. The peptide displays oxidative properties that do not have biochemical or pathological conse-quences, probably because of a functional antioxidant system. c The occurrence of oxidative stress, either because of disabled antioxidant defenses or because radical species production creates a favorable environment for A � 42 aggregation, plaque formation, and formation of NFTs, leading to neuronal death ( d ).

Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 30
protein [15, 16] . That the rodent protein is 96% homolo-gous with the human protein, makes fi ndings from this rat strain even more pertinent to human neurodegenera-tive diseases [17] .
Here we demonstrate that administration of A � 42 alone was not suffi cient to induce memory impairment or the appearance of P-Tau protein in the CSF, a well-characterized marker of AD in humans [18, 19] . How-ever, administration of A � 42 with ferrous and buthio-nine-sulfoximine (FAB) resulted in memory loss and the appearance of P-Tau in CSF. Although these data are consistent with the amnesic effect of i.c.v. injection of A � previously described by others, they differ in that A � 42 , singly applied, was ineffective – as opposed to results ob-tained with infusion with A � 40 or with the A � 25–35 frag-ment [20–23] . This discrepancy might be due to a differ-ence in the rat strain used and/or the infused peptide employed. That is, A � 42 , A � 40 , and A � 25–35 may differ with respect to their exact neuropathological roles. Taken together, these data suggest that A � and amyloidogenesis may not represent the sole (or most important) factors at the origin of the neuropathological changes that occur in AD.
Several cellular and molecular events participate in the onset and the development of AD. Thus, inducing histo-logical modifi cations in a rat resembling those described in AD patients’ brain would require the use of a variety of pharmacological tools able to trigger the range of events reported to occur in AD. This multi-targeted pharmaco-logical approach is similar to the multigene targeting ap-proach used to generate transgenic AD models. In these transgenic mouse models, multiple familial AD-related genes are targeted with the goal of triggering the different pathways responsible for the progression of the disease [24, 25] . In the present study, ferrous ion (Fe 2+ ) was add-ed to the infusion solution because iron is a constituent of neuritic plaques and because it is a pro-oxidant [26] . Infusion of a solution containing both A � 42 and Fe 2+ did not signifi cantly affect the memory, nor did it induce his-tological alteration suggesting that the pathological mod-ifi cations observed in FAB-infused rats were not the re-sult of a Fe 2+ -induced neurotoxicity. The lack of effective-ness of the A � 42 /Fe 2+ solution was overcome with the addition of BSO, an inhibitor of glutathione synthesis [27] . Infusion of Fe 2+ + A � 42 + BSO (FAB) solution into the lateral ventricles signifi cantly decreased the animals’ ability to locate the submerged platform in the water maze task, indicating that the FAB infusion had inter-fered with spatial learning. We therefore postulate that the oxidative properties of the amyloid peptide, singly or
in combination with Fe 2+ , are not suffi cient to create a microenvironment, or oxidative stress, favorable to the development of the events leading to memory loss in Long-Evans rats [28, 29] . These results contrast with those showing that a two-week perfusion with A � 42 in-duced memory impairment that could be improved by antioxidants [30] . Importantly, in these studies A � 42 did not induce neuronal loss and the memory impairment was reversible, unlike AD [31] . The idea that encumber-ing antioxidant defenses renders the brain susceptible to A � 42 toxicity is supported by the positive 8-hydroxyde-oxyguanosine immunostaining in FAB-infused rats. These data support previously published data showing that oxidative stress induced by a aluminum-enriched diet increases amyloidosis in brains of Tg2576 mice and that an acute i.c.v. injection of Al 3+ prior to a chronic in-fusion of A � 40 is necessary to induce a cognitive impair-ment in rats [32, 33] . Moreover, the antioxidant vitamin E has been recently reported to reduce A � 42 levels and amyloid deposition in a transgenic model of AD [34] . Therefore, impairing the brain’s antioxidant defenses ap-pears to be required for the A � 42 -induced damage. In this regard, it is interesting that oxidative stress was recently proposed to play a crucial role in the age-associated cog-nitive impairment that occurs in klotho gene mutant mice, a genetic model of aging [35] .
FAB-induced memory impairment was accompanied by a signifi cant increase in CSF P-Tau protein concentra-tions, a feature observed in AD patients [18, 19] . This fi nding was confi rmed with immunohistochemical data showing that FAB-infused rats displayed intense P-Tau immunoreactivity in the hippocampal neurons. The use of an antibody, specifi cally raised against NFTs, positive-ly labeled neurons in the CA1 suggesting the presence of microtubule alterations in these cells. In addition, the Campbell-Switzer staining of the pyramidal neurons in the temporal cortex revealed a fl ame-shaped pattern of staining in the neuronal body, suggesting that these cells also contained NFTs. Our data is consistent with results obtained by Sigurdsson et al. [36] who induced Tau pro-tein hyperphosphorylation in rats by injecting A � 25–35 into the amygdala and showed that A � 42 can induce NFT formation. FAB rats displayed amyloid deposits mainly in the CA1 fi eld of the hippocampus, a region known to be highly sensitive to A � 42 toxicity. It is of interest that no silver staining was found in brains of rats perfused with A � 42 in association with Fe 2+ , thus ruling out any contribution of Fe 2+ -induced toxicity in our results. In addition, the rats infused with BSO, Fe 2+ , or a mixture of both did not display any positive Campbell-Switzer label-

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 31
ing, demonstrating the specifi city of the reaction induced by FAB. Thiofl avin-S staining revealed that the amyloid deposits we observed correspond to amyloid plaques. Amyloid deposits were also observed in the CA2 and CA3 fi elds of the hippocampus and in the cingulate and tem-poral cortices. The presence of A � 42 in these brain areas was also confi rmed by immunohistochemistry. Of note, the morphology and the distribution of the amyloid de-posits we observed here were different than what has been reported in human AD brains. It is possible that the neu-ropathological changes described here may take on the appearance of those associated with AD brains over lon-ger periods of time. The deposits we described in the cin-gulate cortex of the rat resemble something intermediate to a diffuse and a core neuritic plaque – as has been de-scribed in human brain by Thal et al. [37] – and more ‘maturation’ might lead to a fully AD-featured deposit. In addition, the anatomical distribution of these patho-logical changes is undoubtedly affected by the access of the infused amyloid to different regions. Nevertheless, these results suggest that A � 42 is necessary, but not suf-fi cient, to induce histopathological modifi cations in the rat brain. Moreover, the presence of 8-hydroxydeoxy-guanosine in brains of FAB-infused rats indicate that the generation of oxidative stress that depletes the antioxi-dant defense system in a manner independent of A � 42 was a prerequisite for induction of the observed patho-logical features. Our results are supported those obtained by Pratico et al. [38] , who showed that lipid peroxidation occurred prior to the onset of amyloid deposition in the brains of Tg2576 transgenic mice overexpressing mutant human amyloid precursor protein. These authors also demonstrated that aluminum modulates brain amyloido-sis by increasing oxidative stress in Tg2576 mice [32] .
A considerable body of evidence indicates that the physiopathology of AD has a vascular component simi-lar to that already described for vascular dementia [39–42] . This vascular component co-exists with NFTs in the AD brain and might be related to the severity of AD; it could, therefore, be of importance in the prognosis and diagnosis of the disease [39, 40] . FAB-infused rats dis-played blood vessel-associated amyloid deposits in the CA3 and in the temporal cortex suggesting a vascular amyloidosis in the brain of our animals. The massive astrogliosis and microgliosis found in the CA1, CA3, and the dentate gyrus areas of the hippocampus, mainly in the vicinity of neuritic plaques, indicates that an im-portant infl ammatory process occurs in the FAB-infused rat brain. These histopathological modifi cations were associated with a signifi cant degree of neuronal death.
Interestingly, the cupric silver-based black staining, which labels degenerating neurons, perfectly comple-mented the decrease in cresyl violet staining (Nissl stain-ing), which normally labels the undamaged neurons by staining the Nissl bodies. The majority of neuronal death occurred in the CA1 fi eld of the hippocampus, although dead neuronal bodies were also observed in the CA3 fi eld and in the cingulate cortex. These results are com-parable to the loss of synaptophysin previously reported in aged female rats infused with amyloid peptide [43] and confi rmed the highest vulnerability of the hippo-campal neurons to oxidative stress [44] . Interestingly, the infusion of BSO or Fe 2+ alone, or in combination, did not induce any neuronal death indicating that, under our experimental conditions, these two chemicals are not neurotoxic. These data suggest that in the FAB mod-el neuronal death is triggered by the presence of A � 42 in the FAB solution.
Immunoblot analysis of the A � 42 confi rmed the forma-tion of the amyloid-derived diffusible ligands (ADDLs) in the three solutions containing the amyloid peptide. Al-though neurotoxic [45, 46] , ADDLs did not induce neu-ronal death in the rat brains infused with A � 42 alone, or in combination only with Fe 2+ , suggesting that the oxida-tive stress generated by the FAB solution made the rat brain vulnerable to the neurotoxic effects of the amyloid oligomers.
In summary, the results provided herein show that a continuous 4-week intracerebroventricular infusion of rats with a solution containing the amyloidogenic peptide A � 42 , the pro-oxidative cation Fe 2+ and the glutathione synthesis inhibitor BSO has the capacity to create the memory loss, the increase in CSF P-Tau protein levels and the histopathological profi les – including neuronal loss – seen in AD. In revealing that a signifi cant level of oxidative stress, occurring independently of A � 42 , is man-datory for the amyloid peptide to exert its detrimental effects, our data suggest that oxidative processes develop in AD patient brains before A � 42 begins to aggregate. On this basis, we believe that the FAB-infused rats constitute an interesting alternative for examining the molecular mechanisms underlying the onset and progression of AD. A number of issues concerning this model require further study. As examples, the changes that might occur in cho-linergic neurotransmission and in other neurotransmitter systems, the behavioral and histological changes that might occur after the 4-week infusion period, the revers-ibility of the phenomena upon arrest of the treatment, and the sensitivity of aged animals to the treatment, will be addressed in future studies. Moreover, considering the

Lecanu /Greeson /Papadopoulos
Pharmacology 2006;76:19–33 32
References
1 Hardy JD, Higgins GA: Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992;
256: 184–185. 2 Kachaturian ZS: Hypothesis on the regulation
of cytosol calcium concentration and the aging brain. Neurobiol Aging 1987; 8: 345–346.
3 Beal MF: Does impairment of energy metabo-lism result in excitotoxic neuronal death in neurodegenerative disease? Ann Neurol 1992;
31: 119–130. 4 Volicer L, Crino PB: Involvement of free radi-
cals in dementia of the Alzheimer type: a hy-pothesis. Neurobiol Aging 1990; 11: 567–571.
5 Mudher A, Lovestone S: Alzheimer’s disease – do Tauists and Baptists fi nally shake hands? TiNS 2002; 25: 22–26.
6 Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M: Neuronal loss in APP transgenic mice. Nature 1998; 395:
755–756. 7 Callahan MJ, Lipinski WJ, Bian F, Durham
RA, Pack A, Walker LC: Augmented senile plaques load in aged female � -amyloid precur-sor protein-transgenic mice. Am J Pathol 2001;
158: 1173–1177. 8 Christen Y: Oxidative stress and Alzheimer
disease. Am J Clin Nutr 2000; 71: 621S–626S. 9 Smith MA, Hirai K, Hsiao K, Pappolla MA,
Harris PLR, Siedlack SL, Tabaton M, Perry G: Amyloid- � deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neu-rochem 1998; 70: 2212–2215.
10 Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G: Oxidative stress in Alz-heimer’s disease. Biochim Biophys Acta 2000;
1502: 139–144. 11 Leutner S, Czech C, Schindowski K, Touchet
N, Eckert A, Muller, WE: Reduced antioxidant enzyme activity in brains of mice transgenic for human presenilin-1 with single or multiple mutations. Neurosci Lett 2000; 292: 87–90.
12 Paxinos G, Watson C: The Rat Brain in Ste-reotaxic Coordinates, ed 4. New York, Aca-demic Press, 1998.
13 Switzer RC III: Application of silver degenera-tion stains for neurotoxicity testing. Toxicol Pathol 2000; 28: 70–83.
14 Campbell SK, Switzer III RC, Martin TL: Alz-heimer’s plaques and tangles: a controlled and enhanced silver staining method. US Patent Offi ce ‘Histological Analysis Method’ 1993; patent #5,192,688.
15 Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T: New inbred strain of Long-Evans Tokushima lean rats with IDDM without lymphopenia. Diabetes 1991; 40:
1375–1381. 16 Chiang YJ, Kole HK, Brown K, Naramura M,
Fukuhara S, Hu RJ, Jang IK, Gutkind JS, She-vach E, Gu H: Cbl-b regulates the CD28 de-pendence of T-cell activation. Nature 2000;
403: 216–220. 17 Yokoi N, Komeda K, Wang HY, Yano H,
Kitada K, Saitoh Y, Seino Y, Yasuda K, Seri-kawa T, Seino S: Cblb is a major susceptibility gene for rat type 1 diabetes mellitus. Nat Gen-et 2002; 31: 391–394.
18 Green AJE: Cerebrospinal fl uid brain-derived proteins in the diagnosis of Alzheimer’s disease and Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol 2002; 28: 427–440.
19 Mulder C, Schoonenboom SNM, Wahlund LO, Scheltens P, van Kamp GJ, Veerhuis R, Hack CE, Blomberg M, Schutgens RBH, Eikelenboom P: CSF markers related to patho-genetic mechanisms in Alzheimer’s disease. J Neural Transm 2002; 109: 1491–1498.
20 Nag S, Tang F, Yee BK: Chronic intracerebro-ventricular exposure to � -amyloid (1–40) im-pairs object recognition but does not affect spontaneous locomotor activity or sensorimo-tor gating in the rat. Exp Brain Res 2001; 136:
93–100. 21 Nag S, Yee BK, Tang F: Reduction in soma-
tostatin and substance P levels and choline acetyltransferase activity in the cortex and hip-pocampus of the rat after chronic intracerebro-ventricular infusion of � -amyloid (1–40). Brain Res Bull 1999; 50: 251–262.
22 Nitta A, Itoh A, Hasegawa T, Nabeshima T: Beta-amyloid protein-induced Alzheimer’s disease animal model. Neurosci Lett 1994; 170:
63–66. 23 Olariu A, Tran MH, Yamada K, Mizuno M,
Hefco V, Nabeshima T: Memory defi cits and increased emotionality induced by � -amy-loid(25–35) are correlated with the reduced acetylcholine release and altered phorbol dibu-tyrate binding in the hippocampus. J Neural Transm 2001; 108: 1065–1079.
24 Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Celacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L: Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated A � 42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 2004; 165: 1289–1300.
25 Higgins GA, Jacobsen H: Transgenic mouse models of Alzheimer’s disease: phenotype and application. Behav Pharmacol 2003; 14: 419–438.
26 Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR: Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 1998; 158: 47–52
27 Griffi th OW, Meister A: Potent and specifi c inhibition of glutathione synthesis by buthio-nine sulfoximine (S-n-butyl homocysteine sulf-oximine). J Biol Chem 1979; 254: 7558–7560.
28 Butterfi eld DA, Kanski J: Review: methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid � -peptide 1–42. Peptides 2002; 23: 1299–1309.
29 Goodman Y, Bruce AJ, Cheng B, Mattson MP: Estrogens attenuate and corticosterone exacer-bates excitotoxicity, oxidative injury, and am-yloid beta-peptide toxicity in hippocampal neurons. J Neurochem 1996; 66: 1836–1844.
30 Yamada K, Tanaka T, Han D, Senzaki K, Ka-meyama T, Nabeshima T: Protective effects of idebenone and alpha-tocopherol on beta-amy-loid-(1–42)-induced learning and memory def-icits in rats: implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur J Neurosci 1999; 11: 83–90.
31 Nitta A, Fukuta T, Hasegawa T, Nabeshima T: Continuous infusion of beta-amyloid protein into he rat cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn J Pharmacol 1997; 73: 51–57.
32 Pratico D, Uryu K, Sung S, Tang S, Trojanows-ki JQ, Lee VMY: Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J 2002; 16: 1138–1140.
33 Hashimoto M, Tanabe Y, Fujii Y, Kikuta T, Shibata H, Shido O: Chronic administration of docosahexaenoic acid ameliorates the impair-ment of spatial cognition learning ability in amyloid � -infused rats. J Nutr 2004; 135: 549–555.
34 Sung S, Yao Y, Uryu K, Yang H, Lee VM, Tro-janowski JQ, Pratico D: Early vitamin E sup-plementation in young but not aged mice re-duces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J 2004; 18: 323–325.
35 Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima YI, Nabeshima T: Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxida-tive stress. FASEB J 2003; 17: 50–52.
rationale used to develop the FAB solution, the data pre-sented herein are in favor of a unifying ‘amyloidoxida-tive’ hypothesis to explain the onset and progression of AD.
Acknowledgements
We are grateful to Dr. Luis Dettin for his helpful assistance and advice in immunohistochemistry. This work has been supported by a contract from Samaritan Pharmaceuticals.

Amyloid Peptide and Oxidative Stress Pharmacology 2006;76:19–33 33
36 Sigurdsson EM, Lee JM, Dong XW, Hejna MJ, Lorens SA: Bilateral injections of amyloid- � 25–35 into amygdala of young Fischer rats: be-havioral, neurochemical, and time dependent histopathological effects. Neurobiol Aging 1997; 18: 591–608.
37 Thal DR, Rub U, Schultz C, Sassin I, Ghe-brenedhin E, Del Tredici K, Braak E, Braak H: Sequence of A � -protein deposition in the hu-man medial temporal lobe. J Neuropathol Exp Neurol 2000; 59: 733–748.
38 Pratico D, Uryu K, Leight S, Trojanowski JQ, Lee VMY: Increased lipid peroxidation pre-cedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci 2001; 21: 4183–4187.
39 De La Torre JC: Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:
196–215. 40 Matsubara E, Shoji M, Abe K, Frangione B,
Ghiso J: Vascular amyloidosis in neurodegen-erative conditions. Drug News Perspect 2002;
15: 439–444. 41 Miyakawa T: Vascular pathology in Alzheim-
er’s disease. Ann NY Acad Sci 2002; 977: 303–305.
42 Weller RO, Nicoll JA, Cerebral amyloid angi-opathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res 2003; 26:
611–616. 43 Frautschy SA, Hu W, Kim P, Miller SA, Chu
T, Harris-White ME, Cole GM: Phenolic anti-infl ammatory antioxidant reversal of A � -in-duced cognitive defi cit and neuropathology. Neurobiol Aging 2001; 22: 993–1105.
44 Savory J, Rao JK, Huang Y, Letada PR, Her-man MM: Age-related hippocampal changes in Bcl-2:Bax ratio, oxidative stress, redox-active iron and apoptosis associated with aluminium-induced neurodegeration: increased suscepti-bility with aging. Neurotoxicology 1999; 20:
805–817. 45 Lambert MP, Barlow AK, Chromy BA, Ed-
wards C, Freed R, Liosatos M, et al: Diffusible, nonfi brillar ligands derived from A � 1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998; 95: 6448–6453.
46 Lecanu L, Yao W, Teper GL, Yao ZX, Greeson J and Papadopoulos V: Identifi cation of natu-rally occurring spirostenols preventing � -amy-loid-induced neurotoxicity. Steroids 2004; 69:
1–16.
Related Documents











