Multiple Sequence Alignment & Phylogenetic Trees

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Multiple Sequence Alignment & Phylogenetic Trees

Multiple Sequence Alignment
Motivation:
• Indication of a common structure/function.
• A common evolutionary source (protein families, shared homologous regions).

High consensus colour: redLow consensus colour: blueNeutral colour: blackConsensus: the most common letter.
http://prodes.toulouse.inra.fr/multalin/multalin.html

Uses of Multiple Sequence Alignment
1. Determine consensus sequences• EMOTIF, Clustal, Pileup
2. Building gene families2. Blocks, Prints, Prodom, HSSP.
• Develop phylogenies
• clusters, evolutionary models.• PHYLIP, MACAU
• Model protein structures • Hidden Markov Models, PFAM• Profiles and templates, SCOP, FSSP• Neural Networks, PSI-PRED

EXAMPLE:LOON (bird): RED EYES, FEATHERS, 28 VERTEBRAEDOG: BROWN EYES, HAIR, 23 VERTEBRAECROC: GREEN EYES, SCALES, 28 VERTEBRAE
We would construct the matrix:
LOON (bird): 000DOG: 111CROC: 220
With DNA sequences each possible character has the same 4 possible states (A, C, G, T). Protein sequences have 20 possible states.
Multiple Alignment (Morphological Data):
http://research.amnh.org/~siddall/methods/align.html

Multiple Sequence Alignment - Definition
• A multiple alignment of sequences S1,S2,..,Sk is a series of sequences S1’, S2’, .., Sk’ with gaps such that:
–all Si’ sequences are of equal lengths.–Sj’ is an extension of Sj, obtained by insertion of gaps.
• Example: ACTCGT, CAGTG, ACATCG
AC__TCGT _CAGT_G_ ACA_TCG_

The Size Problem:
If we consider only short sequences and only two taxa, we can handle the comparison manually.
For example, 2 taxa matrix:
But if you were to do this for 75 taxa, you'd have touse 75 dimensional space !!!
In general, MSA methods are based on pairwise alignments between the sequences.
Taxa 2
Taxa 1

LOON: AACDOG: ACACROC: CCARAT: CAC
There is one difference (two states) in each of the columns, thus the column-score for the alignment is 3.
Determining Score:Most alignment algorithms determine the cost of an alignment column-wise. Example:
Usually we will align the sequences in pairs, and then align the pairs. Possible scoring schemes include:• Sum of pairs - sum of pairwise distances between all pairs of sequences.• Distance from consensus - the consensus is a string of the most common character in each column.

MSA Approaches• Progressive approach: Build MSA starting from most related sequences, and then progressively add less related sequences. ClustalW, Pileup.
• Iterative approach: Repeatedly realign subgroups of sequences. Objective: Improve the MSA score according to the scoring scheme, e.g., the sum of pairs score. Subgroups are based on phylogenetic tree or random selection. MultAlin, DiAlign.
Problem: Errors in the initial alignment are propagated to the MSA.

ClustalW Algorithm:• Compute pairwise alignment for all the pairs of sequences.
• Build a phylogenetic guide tree such that • similar sequences are neighbors in the tree• distant sequences are distant from each other in the tree.
• The sequences are progressively aligned according to the branching order in the guide tree.

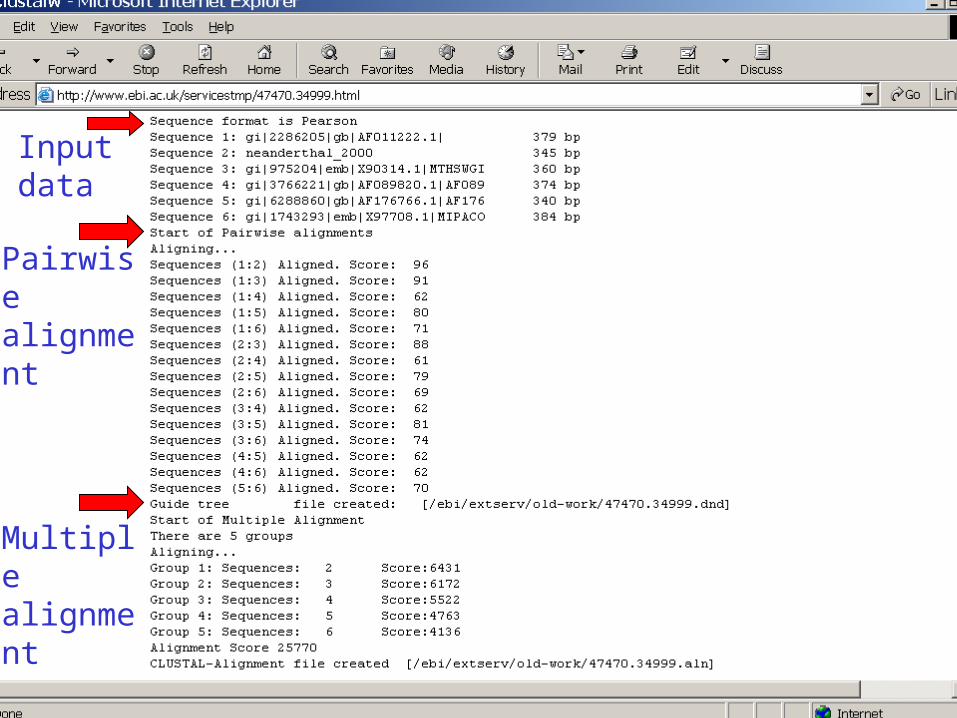
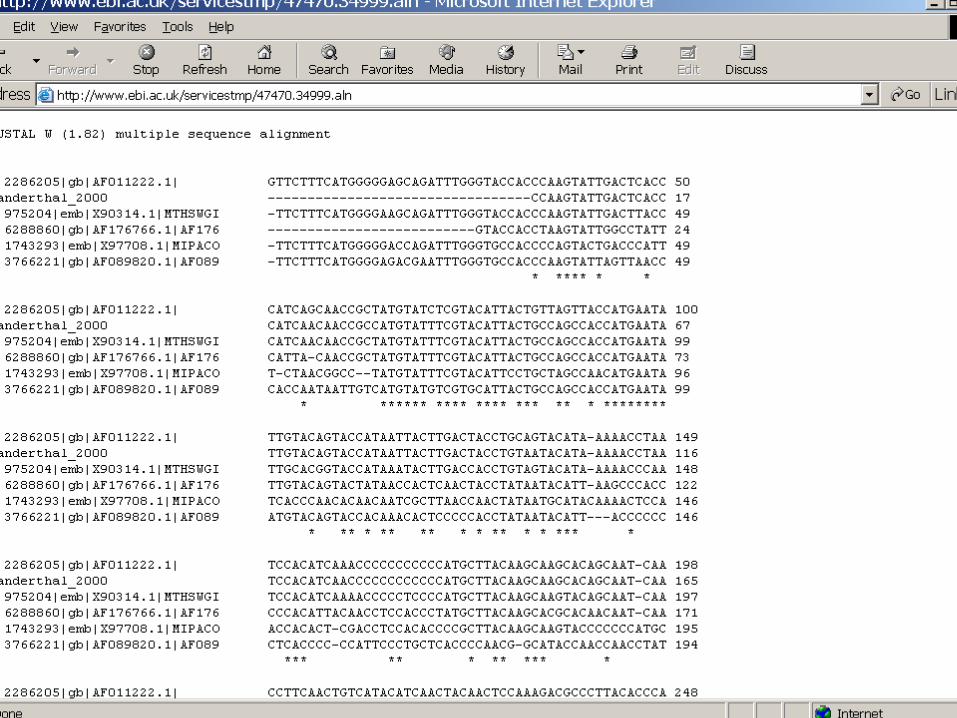
Input data
Pairwise alignment
Multiple alignment

PHYLOGENETIC RECONSTRUCTION
Goal: Given a set of species*, reconstruct the tree which best explains their evolutionary history.

All organisms undergo a slow process of transformation through the ages - Evolution. The process of speciation (creating new species) is described by phylogenetic trees.
Trees are acyclic connected graphs.
Example: Primate phylogenetic tree
The common ancestorof human and chimp
chimpanzeehumangorillaorangutangibbonsiamang
EVOLUTION and PHYLOGENY
The common ancestorof all six primates

Nodes: External nodes (tips of tree) represent extant (existing) species. Internal nodes represent ancestral species (usually extinct). Branches: Length correspond to number of mutations. Longer branch means more mutations, usually implying longer evolutionary time. Typical time scale is mya (millions years ago).
chimpanzeehumangorillaorangutangibbonsiamang
External nodes
Internal nodesBranch
Tree Features:

Phylogenetic Reconstruction Goal: Given a set of taxa (a group of related biological species), build a tree which best represents the course of evolution for this set over time.
Trees: Rooted or unrooted. Most reconstruction methods produce unrooted trees. To root a tree we need “external information’’ (e.g. outgroup).
human
chimpanzee
Unrooted
chimpanzeehuman
gorillaorangutan
Rooted
orangutan
gorilla

Classical phylogenetic analysis: Darwin (origin of species, November 24, 1859) and his contemporaries based their work on morphological and physiological properties (e.g. cold/warm blood, existence of scales, number of teeth, existence of wings, etc., etc.)
Modern biological methods arebased on molecular features: homologous sequences (e.g., globins) in different species;use DNA or protein sequences.
Trees are Based on What?

Homologous genes have a common ancestor. However geneduplications and losses events obscure evolutionary events.
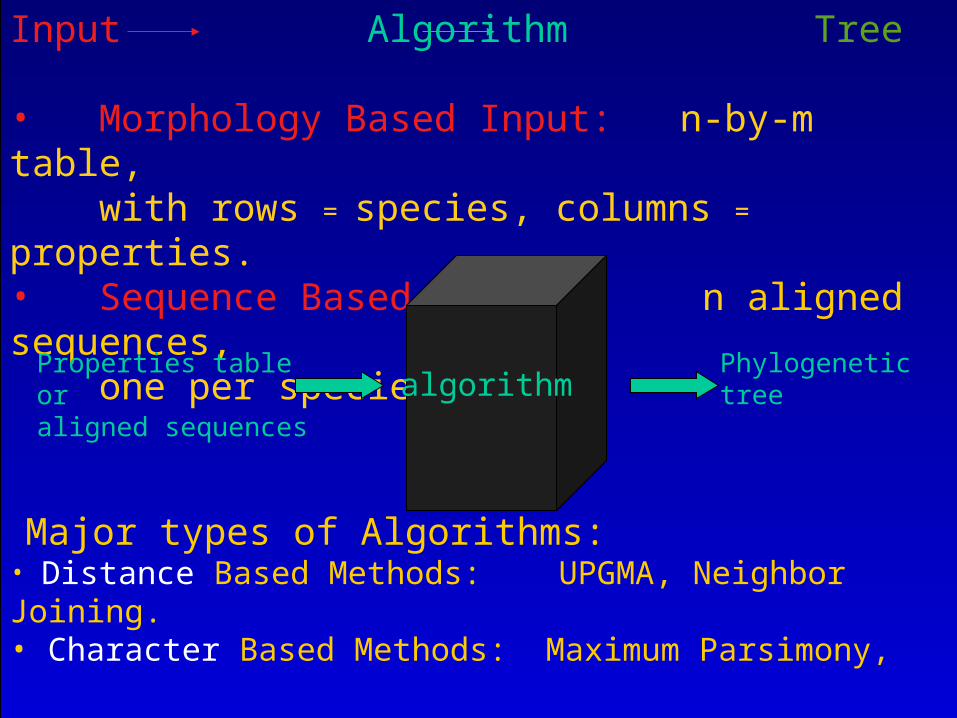
Input Algorithm Tree
• Morphology Based Input: n-by-m table, with rows = species, columns = properties.• Sequence Based Input: n aligned sequences, one per species.
algorithmPhylogenetic tree
Properties tableoraligned sequences
Major types of Algorithms:• Distance Based Methods: UPGMA, Neighbor Joining.• Character Based Methods: Maximum Parsimony, Maximum Likelihood.

The Methods:
Distance- A tree that recursively combines two nodes of the smallest distance.
Parsimony – A tree with a total minimum number of character changes between nodes.
Maximum likelihood - Finds the most probable tree under a mutation model. The method of choice nowadays.

Distance Based MethodsIterative process, n-1 stages.Each stage consists of two steps:• Step 1: Determine the closest pair of species v,
u. “Merge’’ together these two
“neighbors” to a new species w.
• Step 2: Update the distance matrix. Determine the distances from the new species w to the n-2 other.
There are many distance based methods. Most popular are UPGMA and Bio-NJ.
Different choices of the closest pair, and the ways to resolve ties.

UPGMA –Unweighted Pair Group Method with Arithmetic mean
Algorithm - 2 stages:1. Build a simple distance matrix: Distance
between a pair of species may be the number of sites in which they differ.
2. Construct a tree by iteratively clustering species with small distances (“neighbors”).
ABCD
B6
C57
D10127

EXAMPLE for UPGMA
• Find the pair with the closets distance: AC.• Calculate distance between A and C:
2.5----A|
----C 2.5• Merge A and C to AC and update distance matrix.
Dist(AC,x) = [dist(A,x) + dist(C,x)]/2.ABCD
B6
C57
D10127
ACBD
B6.5
D8.512

EXAMPLE for UPGMA• Next pair: AC,B.
2.5 0.75 ----A------- |
| ----C | 2.5 | ------------B 3.25
ACB
D10.25
ACBD
B6.5
D8.512
• Next pair: ACB.D2.5
0.75 ----A------- |
1.875| ----C ------| 2.5 | | | ------------B | 3.25 | ------------------D 5.125

UPGMA Properties
• Builds a rooted tree.
• The output tree is ultrametric: the distance between the root and any leaf is the same.
• This leads to a similar molecular clock assumption, which is too good to be true.
• The tree is additive: the distance between any two nodes equals the sum of the lengths of the branches connecting them.

Neighbor Joining
• Builds an additive tree which does not assume an equal molecular clock.
• The tree is unrooted.
• Algorithm is similar: merge the pair of nodes whose distance is smallest.
• Merge nodes A and B such that M(A,B) is smallest:r(A) = [xd (A,x)]/(N-2). M(A,B) = d (A,B)-[r(A)+r(B)].
d (A,AB) = 0.5[ d(A,B)+r(A)-r(B)] d (B,AB) = d (A,B) – d (A,AB).

Neighbor Joining
• Set N to contain all leavesIteration: Choose i,j such that M(i,j) is minimal Create new node k, and set
remove i,j from N, and add kTerminate:
when |N| =2, connect two remaining nodes
)),(),(),((),(
),(),(),(
)),((),(
jidmjdmid21
mkd
kidjidkjd
rrjid21
kid ji
ij
k
m

Neighbor Joining Example
• Compute r for every node, N=4. r(A)=0.5*(6+5+10); r(B)=0.5*(6+7+12);
r(C) = 0.5*(5+7+7); r(D) = 0.5*(10+12+7);
• Compute M for every pair of nodes. M(A,B) = dist(A,B)-[r(A)+r(B)]=6-
(10.5+12.25).• In this example C and D are merged first.ABCD
B6
C57
D10127
AB
CD
24
16
2
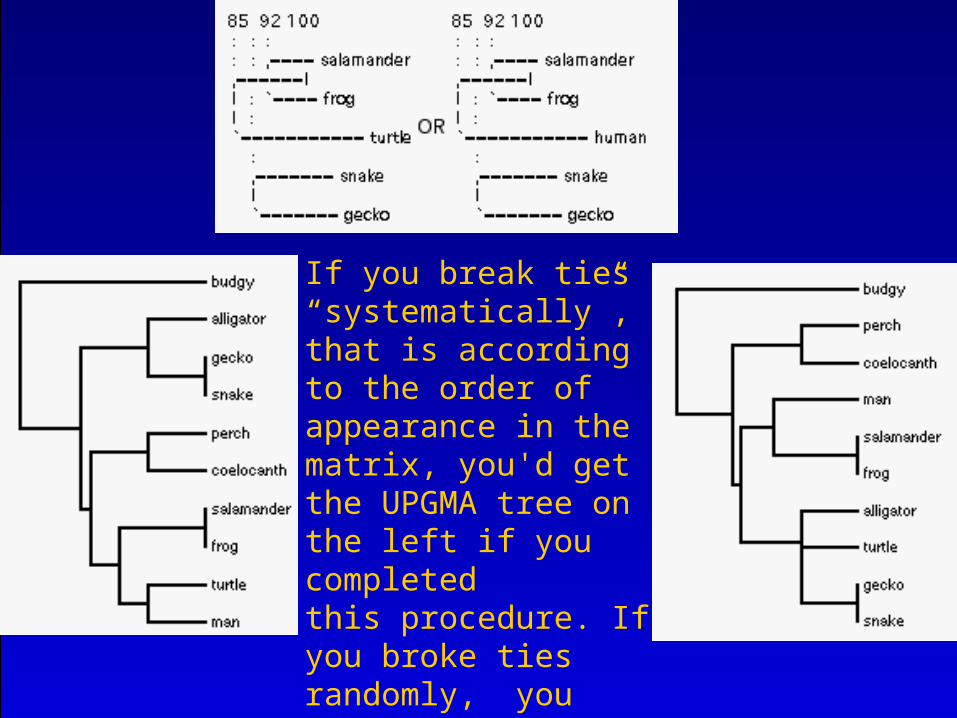
If you break ties “systematically”,that is according to the order of appearance in the matrix, you'd get the UPGMA tree on the left if you completedthis procedure. If you broke ties randomly, you might get the treeon the right here.

Maximum Parsimony
• We are looking for an “evolutionary explanation” for existing species that will minimize the number of mutations.
• Evolutionary explanation - a tree and series in internal nodes. The internal nodes stand for steps required to generate the observed variation in the sequences.
• This problem is NP-hard. However, for a given tree it is easy to find an assignment for the internal nodes that minimizes the number of mutations.

Calculating the minimal number of steps
The intersection of C, T and C is (of course) C
The intersection set of A, C and C is C
We add a length of 1 Length=2
An intersection of A andA, it is A, thus we applyA to the node. Length =0
We add a length of 1 Length=1

Maximum Parsimony Problems
• It is possible for small datasets to evaluate all possible tree topologies.
• Done by adding taxa to the growing tree in all possible locations. Specifically, where the number of taxa t = 4, there are 3 un-rooted trees.
• The number of possible trees rapidly increases with increasing t. Number of trees: (2t - 5)!/[2t-3(t - 3)!]
• When t = 10, the number is more than two million.
• Maximum parsimony is not always real.

Maximum Likelihood• Uses probability calculations to find a tree that best accounts for the variation in a set of sequences. • In each tree the number of sequence changes is considered.
• Allows for variation in mutation rates, and can incorporate evolutionary models such as Jukes-Cantor. • Like Maximum parsimony - analysis is performed on each column in a series, and all possible trees are considered. Computational intensive!

Comparison
• When the sequences are very similar all methods will produce a tree close to the real tree.
• When sequences are less related, neighbor joining and maximum likelihood are usually better than maximum parsimony.
Related Documents











