Review article Molecular genetics of affective disorders Pierre Oswald * , Daniel Souery, Julien Mendlewicz Department of Psychiatry, Erasme Hospital, Free University of Brussels, 808 route de Lennik, B-1070, Brussels, Belgium Accepted 10 May 2004 Available online 23 July 2004 Abstract Evidence for familial aggregation in Affective Disorders (AD) has been provided in classical studies. Linkage and association genetic studies have been proposed to detect genetic factors implicated in AD. However, findings from molecular genetic studies remain inconclusive. Nevertheless, current research is focusing on the phenotypes, both sub- and endophenotypes. In addition, recent advances in technology, such as microarrays, provide new tools in psychiatric genetics. These different approaches offer a new optimism era in the search of genetic factors in AD. D 2004 Elsevier Inc. All rights reserved. Keywords: Affective Disorders; Anticipation; Association; Ethics; Linkage; Molecular genetics Contents 1. Introduction ........................................................... 865 2. The clinical evidence in favour of a genetic component for AD ................................. 866 3. Finding the genes: linkage and association methods ....................................... 866 4. Linkage studies with DNA markers in AD ........................................... 866 5. Association studies and candidate genes in AD ......................................... 867 5.1 Serotonergic and monoaminergic pathways ........................................ 867 5.2 GABAergic pathway ................................................... 868 5.3 Other candidate genes ................................................... 869 6. Anticipation and expanded trinucleotide repeat sequences .................................... 869 7 How to improve the genetic studies in affective disorders?.................................... 870 7.1 Search for phenotypes in affective disorders ....................................... 870 7.2 Improving genetic techniques and methods ........................................ 871 8 Ethical considerations ....................................................... 871 9 Conclusion ............................................................ 872 References ............................................................... 872 1. Introduction Despite significant advances in treatment strategies, Affective Disorders (AD) remain a problem not only in terms of quality of life but also of health economics. Research investigations have focused during the last decades on the aetiology of the disease. Historical obser- vations have consistently provided evidence for a genetic component in the vulnerability to AD (Mendlewicz, 1994; McGuffin et al., 1994). These evidences have demonstrat- ed that a single-gene dysfunction is not enough to explain the mode of inheritance, which is more complex and may include non-Mendelian patterns. We will review the current findings in the field of molecular genetic studies of Unipolar (UPAD) and Bipolar Affective Disorders (BPAD). In addition, we will discuss how to improve the current genetic studies in psychiatry. Finally, ethical aspects will be considered. 0278-5846/$ – see front matter D 2004 Elsevier Inc. All rights reserved. doi:10.1016/j.pnpbp.2004.05.028 Abbreviations: AD, Affective Disorder; BPAD, bipolar affective disorder; LD, linkage disequilibrium; RFLP, restriction fragment length polymorphism; UPAD, unipolar affective disorder. * Corresponding author. E-mail address: [email protected] (P. Oswald). www.elsevier.com/locate/pnpbp Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865 – 877

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

www.elsevier.com/locate/pnpbp
Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877
Review article
Molecular genetics of affective disorders
Pierre Oswald*, Daniel Souery, Julien Mendlewicz
Department of Psychiatry, Erasme Hospital, Free University of Brussels, 808 route de Lennik, B-1070, Brussels, Belgium
Accepted 10 May 2004
Available online 23 July 2004
Abstract
Evidence for familial aggregation in Affective Disorders (AD) has been provided in classical studies. Linkage and association genetic studies
have been proposed to detect genetic factors implicated in AD. However, findings from molecular genetic studies remain inconclusive.
Nevertheless, current research is focusing on the phenotypes, both sub- and endophenotypes. In addition, recent advances in technology, such as
microarrays, provide new tools in psychiatric genetics. These different approaches offer a new optimism era in the search of genetic factors in AD.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Affective Disorders; Anticipation; Association; Ethics; Linkage; Molecular genetics
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 865
2. The clinical evidence in favour of a genetic component for AD. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 866
3. Finding the genes: linkage and association methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 866
4. Linkage studies with DNA markers in AD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 866
5. Association studies and candidate genes in AD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 867
5.1 Serotonergic and monoaminergic pathways . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 867
5.2 GABAergic pathway . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 868
5.3 Other candidate genes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 869
6. Anticipation and expanded trinucleotide repeat sequences . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 869
7 How to improve the genetic studies in affective disorders?. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 870
7.1 Search for phenotypes in affective disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 870
7.2 Improving genetic techniques and methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 871
8 Ethical considerations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 871
9 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 872
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 872
1. Introduction
Despite significant advances in treatment strategies,
Affective Disorders (AD) remain a problem not only in
terms of quality of life but also of health economics.
Research investigations have focused during the last
0278-5846/$ – see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.pnpbp.2004.05.028
Abbreviations: AD, Affective Disorder; BPAD, bipolar affective
disorder; LD, linkage disequilibrium; RFLP, restriction fragment length
polymorphism; UPAD, unipolar affective disorder.
* Corresponding author.
E-mail address: [email protected] (P. Oswald).
decades on the aetiology of the disease. Historical obser-
vations have consistently provided evidence for a genetic
component in the vulnerability to AD (Mendlewicz, 1994;
McGuffin et al., 1994). These evidences have demonstrat-
ed that a single-gene dysfunction is not enough to explain
the mode of inheritance, which is more complex and may
include non-Mendelian patterns. We will review the
current findings in the field of molecular genetic studies
of Unipolar (UPAD) and Bipolar Affective Disorders
(BPAD). In addition, we will discuss how to improve
the current genetic studies in psychiatry. Finally, ethical
aspects will be considered.

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877866
2. The clinical evidence in favour of a genetic component
for AD
Evidence from classical studies demonstrates the presence
of a familial aggregation for AD (Sklar et al., 2002). Familial
aggregation means that a trait clusters among multiple
members of a family (Kamnasaran, 2003). Findings from
clinical chart data show that families with relatives with AD
have increased occurrence of the trait segregating among
them. However, familial aggregation studies are not sufficient
to indicate a genetic basis in AD, since the trait can also
possibly be related to environmental factors. Heritability
studies provide an interesting approach to estimate the
variance of the genetic component among affected families
(Kamnasaran, 2003). A genetic aetiology is proposed when
the variance is >30% for a trait in affected families. In recent
studies, the calculated variance is as high as 80% in BPAD
and around 50% in UPAD (Bennett et al., 2002; Maier et al.,
2003). Finally, twin and adoption studies have been imple-
mented to explore the heritability of AD by controlling
environmental factors. Evidence from twin studies has shown
that the concordance rate between monozygotic twin pairs
ranges from 50% for UPAD to 75% for BPAD, indicating that
these phenotypes are mainly, but not strictly, from genetic
origin. Finally, in adoption studies, AD have been described
to be higher in biological relatives of the adopted subject,
suffering fromAD than in adopted relatives (Mendlewicz and
Rainer, 1977; Bennett et al., 2002).
3. Finding the genes: linkage and association methods
The initial molecular genetic approach of AD involved
the parametric linkage studies of large families (Merikangas
et al., 1989; Risch and Merikangas, 1993; Souery et al.,
2001a). Linkage examines the cosegregation of a genetic
marker and disease in affected individuals within families,
that is, the non-random sharing of marker alleles between
affected members of each family. Two genetic loci are
linked if they are located closely together on a chromosome.
In linkage analysis, the frequency of meiotic recombinations
as an expression of the distance between marker locus and
the gene under investigation is used for gene mapping. The
classical method that has been successfully applied in
linkage studies is the LOD score (Schulze and McMahon,
2003). The LOD score is the log10 of the ratio LHa/LH10: the
likelihood (LHa) of the observed constellation of the disease
and marker data assuming linkage, compared to the likeli-
hood (LH10) of observing the same data assuming no
linkage. The traditional threshold of a LOD score >3.0
has been proposed to achieve a true significant linkage.
This threshold must not be considered as the golden
standard, since different methods have been studied, leading
to different values (Schulze and McMahon, 2003). More
recently, genome-wide linkage studies have been performed
on samples of families with multiply affected members
(Segurado et al., 2003; Maier et al., 2003). Marker systems
(restriction fragment length polymorphisms [RFLP] or
microsatellite marker systems) screen the whole genome
in search of candidate regions with predisposing genes,
which can be detected by subsequent sequencing and fine-
mapping or by focus on candidate genes located in these
intervals.
Considering the difficulties inherent in detecting genes
of small to modest effect using the linkage approach in
complex traits, the candidate gene association method
offers an alternative strategy of studying genetic factors
involved in complex diseases in which the mode of
inheritance is unknown. Association method compares
the allele frequencies between a control sample and sample
that suffers from the disease. Association between diseases
and markers may be found if the gene itself, or a locus in
linkage disequilibrium (LD) with the marker, is involved in
the pathophysiology of the disease (Hodge, 1994). Thus,
an association may imply a direct effect of the gene tested,
or the effect of another gene close to the marker examined.
This has important implication for replication in subse-
quent works. For example, as shown by Schulze and
McMahon (2003), if the associated marker is the causal
variant, the same allele should show association in other
populations. If the associated marker is in LD with the
causal variant, then different alleles may be implicated and
show association in other populations. However, the can-
didate gene approach remains a useful method to investi-
gate association between markers and disease. Two slightly
association different strategies are available: case-control
and family-based. Family-based approach seemed to be
more powerful, in reducing the risk of false-positive,
inherent to population stratification in case-control studies.
However, stratification problem seems to be less marked in
more homogenous populations such as those in Western
Europe (Pritchard and Rosenberg, 1999). Moreover, case-
control design permits to achieve a representative sample
of patients because family-based samples may show some
bias (Brunn and Ewald, 1999). As suggested by Craddock
et al. (2001), case-control and family-based association
samples have complementary roles in searching for genes
involved in AD.
4. Linkage studies with DNA markers in AD
In view of the plethora of linkage studies published, we
present here an update of replicated and/or representative
findings (previously reviewed in the works of Souery et al.,
2001a and Oswald et al., 2003a). Reviewing more than two
decades of linkage investigations in AD, it appears that
significant proportion of positive DNA findings involves
several chromosome regions (Turecki et al., 1996). Mend-
lewicz et al. (1987) first reported possible genetic linkage
between BPAD and coagulation Factor IX located at Xq27 in
11 pedigrees. Since then, several linkage studies with X

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877 867
markers revealed different results (Berrettini et al., 1990;
Bredbacka et al., 1993; Gejman et al., 1990; Lucotte et al.,
1992). Nevertheless, more recent studies showed that sug-
gestive results of X linkage and in particular the Xq26–28
region should be considered as a strong candidate region for
genetic studies in BPAD (De bruyn et al., 1994; Pekkarinen et
al., 1995; Stine et al., 1997). Another region of interest seems
to be the chromosome 18 where the pericentromeric region
was suggested to carry susceptibility genes (Berrettini et al.,
1994; Gershon et al., 1996; Stine et al., 1995; Kamnasaran,
2003). This result is of interest because genes coding for the a
unit of a GTP binding protein involved in neurotransmission,
a corticotrophin receptor gene, and RED-1 containing triplet
repeats have been mapped to this region. The John Hopkins
group also studied a set of 30 families supporting linkage at
18q21 (McMahon et al., 1997). Other groups also found
evidence for linkage at 18q12, 18q22 and 18q23 (De bruyn et
al., 1996; Ewald et al., 1997; Nothen et al., 1999). Concerning
18q23, findings from Freimer et al. (1996) were the strongest
at an estimated 80–82 Mb from the p-telomere. The chro-
mosome 11 has been thoroughly investigated in AD but
showed contradictory results (Egeland et al., 1987; Gurling
et al., 1995; Lim et al., 1993; Smyth et al., 1996; Byerley et
al., 1992; Holmes et al., 1991; Nanko et al., 1994; Souery and
Mendlewicz, 1995). Chromosomes 4, 6 and 10 were also
investigated with conflicting and/or unreplicated results
(Smeraldi et al., 1978; Stancer et al., 1988; Turner and King,
1981, 1983; Weitkamp et al., 1981; Blackwood et al., 1996;
Cichon et al., 2001). Darier’s disease (keratosis follicularis), a
rare autosomal dominant skin disorder associated with in-
creased prevalence of epilepsy and mental retardation, whose
gene was mapped on chromosome 12 (12q23–24.1), was
found to cosegregate with BPAD in one pedigree. This result
was replicated in several family studies (Aita et al., 1999;
Detera-Wadleigh et al., 1996, 1997; Straub et al., 1994).
Finally, Blackwood et al. (2001) reported recently strong
evidence of linkage in a family with a (1;11)(q42;q14.3)
translocation. Genome-wide linkage analyses provide an
accurate tool to study regions of interest (see above). In
BPAD, early positive and promising results were contradicted
by further analyses. This fact is not surprising, since these
studies were performed on small samples sizes, insufficient to
replicate modest linkage signals (Suarez and Hampe, 1994).
Metaanalyses were thus performed on BPAD to increase the
power to detect modest linkage signals (Lewis et al., 2003;
Segurado et al., 2003; Maier et al., 2003). Bipolar loci with
evidence of linkage were found on the following arms: 9p,
10q, 14q and 18p–q (Maier et al., 2003).
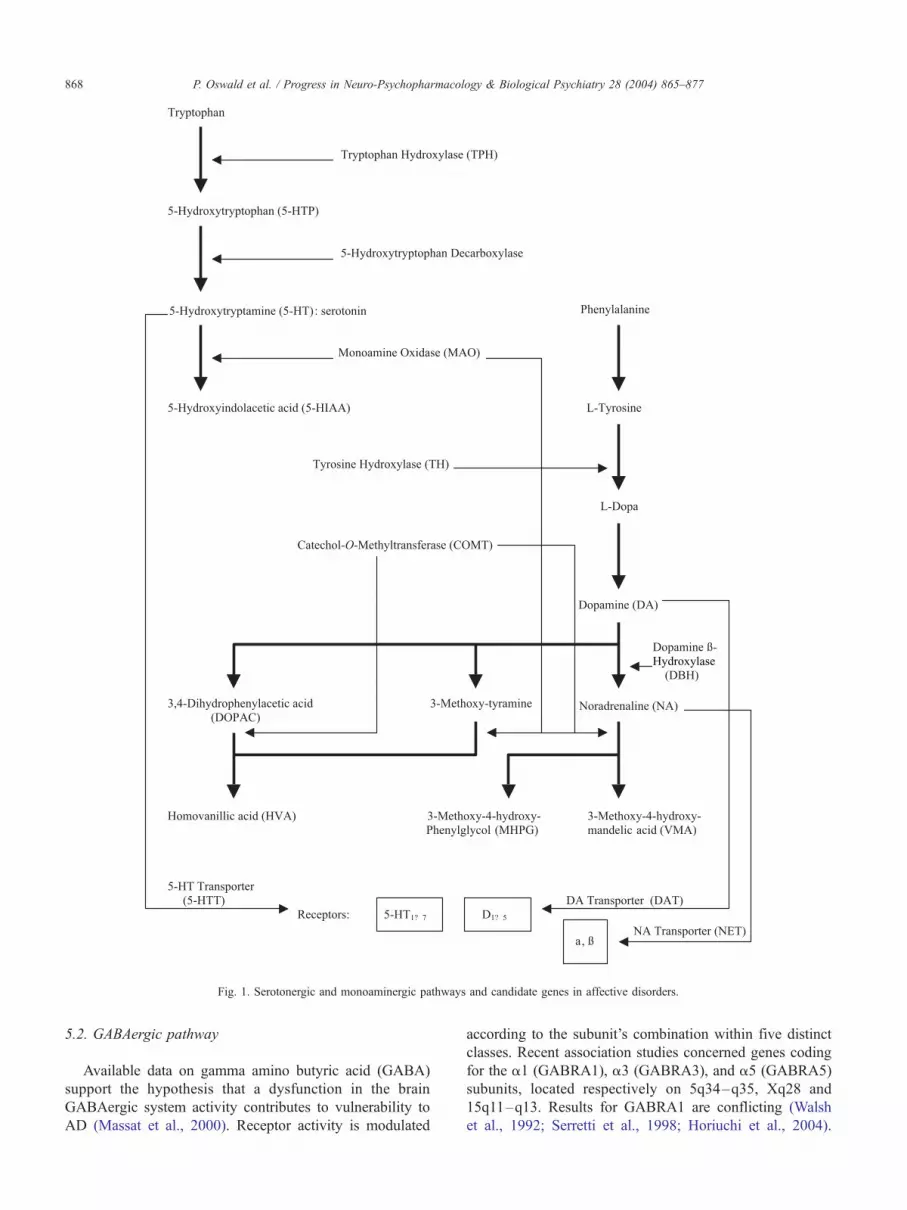
5. Association studies and candidate genes in AD
As mentioned above, association studies are best applied
if candidate genes are selected. Such candidate alleles are
chosen on the basis of the current understanding of the
biology of the disorder. Therefore, genes related to seroto-
nergic and monoaminergic pathways have been firstly
considered as the targets for association studies during the
last years (Fig. 1).
5.1. Serotonergic and monoaminergic pathways
Several polymorphisms in serotonergic and monoaminer-
gic related-genes have been studied (Potash and DePaulo,
2000). The tyrosine hydroxylase (TH) gene, located on
11p15.5, is a candidate gene extensively explored in associ-
ation studies in AD. Although some studies showed promis-
ing results (Perez et al., 1995; Oruc et al., 1997a), two meta-
analyses have failed to confirm the implication of TH gene
both in UPAD and BPAD (Furlong et al., 1999; Turecki et al.,
1997). The tryptophan hydroxylase gene (TPH), located on
11p15.3–p14, includes two identified polymorphisms
(A218C and A779C) (Bellivier et al., 1998; Nielsen et al.,
1997). Souery et al. (2001b), in a recent large multicenter
study, found no association between A218C polymorphism
and UPAD and BPAD. The monoamine oxidase A (MAOA)
gene, located on Xp11.23, has been studied in 16 and 7
association studies for BPAD and UPAD, respectively, with
conflicting results (Lim et al., 1995; Rubinsztein et al., 1996;
Kunugi et al., 1999; Ho et al., 2000; Lin et al., 2000; Preisig et
al., 2000; Syagailo et al., 2001). The catechol-O-methyltrans-
ferase (COMT) gene, located on 22q11.2, is implicated in
dopamine and noradrenaline degradation. A large number of
studies failed to show an implication of COMT gene in AD
(Biomed European Bipolar Collaborative Group, 1997;
Kunugi et al., 1997). Kirov et al. (1999) failed to show an
association between dopamine h-hydroxylase gene (DBH),
located on 9q34, and BPAD. The serotonin, dopamine and
noradrenaline transporter genes (5-HTT, DAT1 and NET),
located on 17q11.1–12, 5p15.3 and 16q12.2, respectively,
were also studied. A study concluded that 5-HTT has no
major role in the aetiology of BPAD (Craddock et al., 2001).
On the other hand, various studies support a relative influence
of 5-HTT in UPAD (Battersby et al., 1996; Collier et al.,
1996; Ogilvie et al., 1996). More recently, Mendlewicz et al.
(2004) showed in a sample of 539 UPAD, 572 BPAD and 821
controls a lack of association between 5-HTT and AD.
Studies on DAT1 and NET are largely negative (Craddock
et al., 2001). Serotonin receptors genes, principally 5-HT2A
(13q14–21), 5-HT2C (Xq24), 5-HT3A (11q23.1) and 5-HT7
(10q21–24) genes, were studied in several association stud-
ies but no definitive positive association was found (Oswald
et al., 2003b; Potash and DePaulo, 2000). D2 receptor gene
(DRD2), located on 11q22.2–22.3, was extensively studied
in BPAD and UPAD. In BPAD, most studies are negative
(Massat et al., 2002b). But a positive association was recently
found in a large sample of 716 subjects (Massat et al., 2002b).
D1, D3, D4 and D5 receptors genes (DRD1, DRD3, DRD4
and DRD5) located respectively on 5q35.1, 3q13.3, 11p15.5
and 4p15.3–16.1 have been shown not to be implicated in
AD in most studies (Asherson et al., 1998; Lim et al., 1994;
Savoye et al., 1998; Souery et al., 1996).
Hydroxylase
Fig. 1. Serotonergic and monoaminergic pathways and candidate genes in affective disorders.
P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877868
5.2. GABAergic pathway
Available data on gamma amino butyric acid (GABA)
support the hypothesis that a dysfunction in the brain
GABAergic system activity contributes to vulnerability to
AD (Massat et al., 2000). Receptor activity is modulated
according to the subunit’s combination within five distinct
classes. Recent association studies concerned genes coding
for the a1 (GABRA1), a3 (GABRA3), and a5 (GABRA5)
subunits, located respectively on 5q34–q35, Xq28 and
15q11–q13. Results for GABRA1 are conflicting (Walsh
et al., 1992; Serretti et al., 1998; Horiuchi et al., 2004).

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877 869
Massat et al. (2002a) showed a significant association
between the GABRA3 polymorphism and the occurrence
of BPAD, particularly in females. This result was not
replicated in UPAD (Massat et al., 2001). GABRA5 is
implicated in UPAD and BPAD in two association studies
but needs to be replicated (Papadimitriou et al., 1998; Oruc
et al., 1997a).
5.3. Other candidate genes
In addition to the classical pathways presented above, it is
now possible to investigate the genetics of enzymes and
receptors involved in novel and promising metabolic routes
such as neuroprotection or neurotrophy. Substance P (SP)
pathway has recently focused interest. In fact, SP antagonists
were found to be as effective as SSRIs in UPAD with limited
side effects (Kramer et al., 1998, 2004). The angiotensin
converting enzyme (ACE) gene is responsible of SP degra-
dation and is therefore a good candidate gene for association
studies in AD. An insertion/deletion polymorphism was
identified. Five association studies were conducted but only
one found an association with AD (Arinami et al., 1996;
Baghai et al., 2001; Furlong et al., 2000; Meira-Lima et al.,
2000; Pauls et al., 2000). Other genes of SP metabolism are
under investigation since their polymorphisms have been
identified (i.e. SP receptor (NK1R) gene, SP precursor
(TAC1) or peptidylglycine alpha-amidating monooxygenase
(PAM) gene). Neurotrophic factors have recently focused
interest since one of them, Brain-Derived Neurotrophic
Factor (BDNF) has been shown to be implicated in AD and
in response to mood stabilizers and antidepressants (Nibuya
et al., 1995; Duman, 1998; Fukumoto et al., 2001). Two
family-based association studies recently demonstrated an
association between BDNF and BPAD (Neves-Pereira et al.,
2002; Sklar et al., 2002). Also, these results were not
confirmed in two recent case-control association studies
(Nakata et al., 2003; Oswald et al., in press). More recently,
Tsai et al. (2003) found no association between BDNF and
UPAD. Most recent papers have provided consistent argu-
ments in favour of an influence of G72 gene in BPAD (Hattori
et al., 2003; Chen et al., 2004; Schumacher et al., 2004). This
gene, located in the 13q candidate region for schizophrenia,
suggests that these two psychiatric disorders may share some
of their etiologic background.
6. Anticipation and expanded trinucleotide repeat
sequences
Anticipation implies that a disease occurs at a progres-
sively earlier age of onset and with increased severity in
successive generations. This phenomenon has been ob-
served in several neurological diseases including myotonic
dystrophy, fragile X syndrome and Huntington’s disease
(Paulson and Fischbeck, 1996; Trottier et al., 1995). Antic-
ipation has been found to correlate with a new class of
mutations, expanded trinucleotide repeat sequences. An
expanded repeat sequence is unstable and may increase in
size across generations, leading to an increased disease
severity of the disorder. Such unstable mutations could also
be an alternative explanation in addition to environmental
factors for discordance between monozygotic twins for AD
where the repeat amplification might be different during
mitosis in each of the two twins.
Evidence for anticipation has been observed in AD
(Engstrom et al., 1995; McInnis et al., 1993; Nylander et
al., 1994). Correlation between anticipation observed at the
phenotypic level with the number of dynamic mutations
may be the only way to confirm the implication of this
phenomenon in mood disorders. One study highlighted an
association between the number of CAG trinucleotide
repeats and severity of BPAD illness in Swedish and
Belgian patients (Lindblad et al., 1995). This study, repli-
cated subsequently in a different sample (O’Donovan et al.,
1995; Oruc et al., 1997b), showed for the first time in a
major psychiatric disorder that the length of CAG repeats
was significantly higher in BPAD compared to normal
controls. These molecular genetic findings may indicate a
genetic basis for anticipation in BPAD. However, no corre-
lation has been found between CAG/CTG repeats and
phenotypic measures of severity in several studies (Crad-
dock et al., 1997; Guy et al., 1999; Li et al., 1998; Vincent et
al., 1996; Zander et al., 1998). This hypothesis has recently
been tested in a sample of two-generation pairs with BPAD.
Globally, no significant differences were found in the mean
number of CAG repeats between parent and offspring
generations. A significant increase in CAG repeats between
parents and offspring was observed, however, when the
phenotype increased in severity, i.e. changed from major
depression, single episode or unipolar recurrent depression
to BPAD (Mendlewicz et al., 1997). A significant increase
in CAG repeats length between generations was also found
in female offspring with maternal inheritance, but not in
male offspring. This is the first evidence of genetic antici-
pation in BPAD families and has been followed by the
identification of loci within the genome containing triplet
repeats. CTG 18.1 on chromosome 18q21.1 and ERDA 1 on
chromosome 17q21.3 are two repeat loci recently identified
(Lindblad et al., 1998) which can be investigated in such
study. In this study, several hundreds of candidate loci
containing repeats were screened in a set of BPAD patients
and expanded alleles at ERDA1 and CTG18.1 loci were
found to be associated with BPAD phenotype. The authors
observed that in a Swedish sample, including both unrelated
and familial cases, 89% of expanded RED products corre-
late with expansions at these two loci and that expansion at
the CTG18.1 locus was associated to the phenotype. Using
the same method in a Belgian sample, Verheyen et al. (1999)
demonstrated that 86% of the RED expansions could be
accounted for by ERDA1 and CTG18.1 repeats (Verheyen
et al., 1999). Expanded alleles at ERDA1 were found to be
more frequent in bipolar patients. Eight CAG/CTG triplet

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877870
repeats located in the 18q21.33–q23, identified as a candi-
date region in bipolar families, have been investigated in
bipolar disorder by Goossens et al. (2000), but no expansion
has been found in the bipolar family and the case-control
sample.
7. How to improve the genetic studies in affective
disorders?
The plethora of results presented above may be viewed
as contradictory. As illustrated in this review, once a locus or
a polymorphism is claimed, one or more subsequent studies
cannot confirm it. Positive findings from association studies
are not always confirmed and more than one chromosomal
regions are proposed for the same phenotype. Different
reasons for the lack of unambiguously detected locus of
interest are evoked. First, the transmission patterns of AD
are complex. Environmental and genetic factors are in-
volved in the occurrence of such disorders. In addition, it
is anticipated that multiple deleterious genetic variants are
required in combination and that individual genes alone are
not sufficient (epistasis) (Souery et al., 2001a). Another
controversial issue is the phenotype definition. It is not
clearly established that the classical diagnostic categories
investigated, i.e. from the DSM-IV, are valid in the search
for genetic aetiology. Currently, available diagnostic
schemes and clinical symptoms have no proven biological
and/or genetic validity. As already raised by Leboyer et al.
(1998), the question is: do our modern definitions of clinical
syndromes (presently considered as phenotypes) accurately
reflect underlying genetic substrates (genotypes)? In a
recent review, Stoltenberg and Burmeister (2000) illustrate
that diagnostic categories may not accurately reflect the
underlying genetic condition with the velo-cardiofacial
syndrome (VCFS). This syndrome is usually caused by a
specific deletion on chromosome 22, and is associated with
comorbid psychiatric disorders, such as schizophrenia,
BPAD or UPAD. The authors conclude that at least some
genetic defects predispose to psychiatric disorders that do
not fall into a clearly defined DSM-IV category. Other
limitations are the locus heterogeneity which implies that
multiple disease genes may be acting with different genes
being implicated in different individuals, and the allelic
heterogeneity that indicates that multiple alleles at a single
disease locus may be implicated in the development of the
disorder and no one allele type alone is thus necessarily
related to the disease (Souery et al., 2001a). It is finally
important to note that the genetic transmission of AD may
involve mitochondrial genome, which would indicate a
maternal mode of transmission.
In order to improve the genetic studies, several authors
have proposed to develop innovative approaches based on
more accurate phenotypic definitions. On the other side,
there is also a considerable need for improving molecular
genetic techniques and methods, looking more carefully at
gene–gene and gene–environment interactions. We propose
here a short summary on promising methods.
7.1. Search for phenotypes in affective disorders
As suggested by Kidd and Matthysee (1978), molecular
genetic approaches should help in refining nosological
categories. However, initial genetic studies have focused
on broad phenotypic definitions, for example, affective
disorders or schizophrenia. Facing the heterogeneity of
results, it has been hypothesized that genetic factors could
explain some symptoms or clinical features of the syn-
dromes, such as severity of the disease, age at onset or
gender predominance. This method has been successfully
used in several somatic diseases. For example, the glyco-
gen-associated protein phosphatase 1 (PP-1) gene has
shown association in early diagnosed diabetes II, but not
with diabetes in general (Doney et al., 2003). In psychiatric
diseases, age at onset or disease severity has been found to
delineate more homogeneous subtypes (subphenotypes).
Among other, suicidal behavior has been considered as a
subphenotype genetically determined. Adoption and family
studies have confirmed the genetic implication in suicide
(Turecki, 2001). Linkowski et al. (1985) reported that 17%
of 713 depressed patients had a first- or second-degree
relative who had committed suicide. Nielsen et al. (1994,
1998) first reported an association between history of
suicide attempt and TPH gene. Bellivier et al. (1998)
examined TPH polymorphism in 152 patients with BPAD
and 94 normal controls. They found an association between
this polymorphism and suicidal behavior in BPAd patients.
Souery et al. (2001b) published data on a large sample of
927 patients with AD from several European populations in
a multicenter project. They observed that the CC genotype
in the A218C polymorphism was less frequent in the
subgroup of UPAD patients with a history of suicide attempt
compared to control subjects. In a recent meta-analysis,
Bellivier et al. (2004) found an overall association between
TPH and suicidal behavior. Bellivier et al. (1997) found no
association between an allele of the 5-HTT gene and suicide
attempts in BPAD patients. They later observed a significant
difference in 5-HTT allele distributions between patients
with AD who had made violent suicide attempts and
controls (Bellivier et al., 2000). Russ et al. (2000) found
no association in 5-HTT polymorphisms between a group of
suicidal patients and controls. In post-mortem studies, an
association was reported by Du et al. (1999). They found a
significantly higher frequency of the 5-HTT gene long allele
in depressed suicide victims compared with matched con-
trols. Bondy et al. (2000) showed a possible association of
the short allele of 5-HTT in patients with violent suicide.
Recently, Courtet et al. (2004) suggested that 5-HTT is
associated with further suicide attempts among patients who
have previously attempted suicide.
Early-onset, and more specifically pediatric-onset, psy-
chiatric disorders have been suggested to have their own

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877 871
pattern of genetic susceptibility factors. Family studies have
consistently found a higher rate of BPAD among the relatives
of early-onset BPAD patients than in relatives of later-onset
cases (Faraone et al., 2003). Here also, 5-HTT and MAOA
polymorphisms have been explored (Ospina-Duque et al.,
2000; Craddock et al., 2001). In pediatric-onset BPAD,
arguments have been put forward to support an anticipation
phenomenom (Vincent et al., 2000). On the other side, adult-
onset AD begins to focus interest (Kennedy et al., 2003).
Another strategy is to search for more specific neuro-
physiologic, neuroimaging, neurocognitive or neurochemi-
cal trait measures that might identify homogeneous groups
of patients. These ‘‘traits’’ are called ‘‘endophenotypes’’ and
are believed to represent the genetic liability of the disorder
among non-affected subjects (Leboyer et al., 1998). As
suggested by Gottesman and Gould (2003), criteria for
endophenotypes are (i) the trait is associated with the
disease in the population, (ii) it is heritable, (iii) it is not
state-dependent, (iv) it cosegregates with the illness within
families, and (v) it is found in nonaffected family members
at a higher rate than in the general population. Endopheno-
type studies have been highly developed in schizophrenia,
such as eye tracking, working memory, measures of neuro-
physiological response to various stimuli and evoked poten-
tials (Kennedy et al., 2003). Endophenotypes in AD are
more difficult to define. However, some studies have
focused on imaging as a marker of BPAD or UPAD. For
example, a recent work has studied the impact of 5-HTT on
the hippocampal volume in UPAD (Frodl et al., 2004).
Based on the well-studied link between the hippocampus
and mood, the authors have shown that the 5-HTT L-allele
is associated with decreased hippocampal volumes in UPAD
patients, as opposed to healthy controls. Future studies will
have to confirm the hypothesis that a genetic variant might
be related to an endophenotype, maybe transnosographical,
and not specifically to a clinical entity.
7.2. Improving genetic techniques and methods
Recent advances in technology have permitted to devel-
op genetic techniques that could screen several candidate
genes in a limited time. Microarrays constitute a promising
high-throughput method, allowing to screen the expression
of thousands of genes within specific tissues in a relatively
short period of time (Shoemaker and Linsley, 2002). Basi-
cally, microarray is able to monitor the collection of mRNA
in the cell (Bunney et al., 2003). Microarray technique is
based on the hypothesis that changes in mRNA expression,
directly linked to protein activity, can result in phenotypical
and morphological differences. The microarray technology
implies the availability of high quality tissue. For psychiat-
ric disorders, the acquisition, characterization and process-
ing of tissue are fundamental and are discussed elsewhere
(Bunney et al., 2003). Microarray, which has also been
applied to animal models, may be considered as the first step
to detect vulnerability genes, which also includes genome
wide-scans, linkage and association studies. Kakiuchi et al.
(2003) recently applied this process to find that XBP1 gene
(on chromosome 22), implicated in endoplasmic reticulum
stress response signalling, can be considered as a vulnera-
bility factor in BPAD. DNA Microarray was first used in
lymphoblastoid cells from discordant monozygotic twins
with respect to BPAD. XBP1, among others, was identified
in this first step. The next step was to identify a single
nucleotide polymorphism (SNP) within the XBP1 upstream
region. This SNP was studied in a case-control association
study, involving 197 BPAD patients and 451 controls, and
was found to be associated to BPAD. This kind of strategy,
combining microarray and association studies, will have to
be generalized in the future. Microarray can be considered
as one of the functional genomics approaches, gathering a
set of technologies and strategies directed at the problem of
determining the function of genes, and understanding how
the genome works together to generate whole patterns of
biological function (Shilling and Kelsoe, 2002).
Finally, there is considerable need to take into account
environmental factors in genetic studies , such as life stress
and others, since it is accepted that AD are multifactorial
diseases, involving both genetic and environmental factors.
The gene–environment interaction was studied by Caspi et
al. (2003) in a prospective-longitudinal study of a represen-
tative birth cohort, involving subjects assessed nine times
between ages 3 to 26 years. Stressful life events were
systematically screened. In addition, DNA was extracted
for 5-HTT genotyping. A moderated regression framework
was used to test the association between depression and (i)
5-HTT genotype, (ii) stressful life events and (iii) their
interaction. The authors found that the 5-HTT polymor-
phism moderated the influence of stressful life events on
depression. This study provides evidence for a gene/envi-
ronment interaction and implies that environmental factors
might be evaluated in future genetic studies.
8. Ethical considerations
Ethical questions arise from genetic research on complex
diseases, such as AD, as well from clinical management of a
complex disorder, involving both genetic and environmental
components. The approaches and ethical rules on genetic
studies vary between countries, as is the case for the informed
consent (Shore, 1993). The Department of Health andHuman
Services Office for Protection from Research Risks (OPRR)
released a few years ago an evaluation on human subjects
issues in a study at the University of California at Los
Angeles (US) involving outpatients with schizophrenia. Sev-
eral faults with the informed consent documents were found.
In particular, some investigators estimated that people who
have mental disorders should be considered incapable of
providing valid informed consent. This opinion is not cur-
rently accepted in many countries. In a NIMH consensus, it
has been established that consent documents, and the process

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877872
by which informed consent is obtained, are state-of-the-art
(Shore, 1996). Parker (2002) recently summarized the diffi-
culties in the enrolment of patients with BPAD. The study of
bipolar disorders presents particular challenges because of
the uncertainty and stigma that surrounds the disorders and
because some of the relevant subjects may have diminished
capacity to consent to participation. During a severe manic or
a depressive episode, the subject will not be able to correctly
judge the cost and benefits of the study. Therefore, inves-
tigators must wait to approach the patient when his symptoms
are controlled by medication. Furthermore, genetic studies do
not offer direct benefit to the patient. This point must be taken
into account and explained to the patient. The disclosure of
familial genetic information has been widely discussed. The
clinician has often to decide when or whether an ethical duty
to inform at-risk family members about an increased genetic
risk overrides the duty to maintain patient confidentiality
(Lehmann et al., 2000). Recently, the American Society of
Human Genetics argued that confidentiality can be breached
in situations in which ‘‘serious and foreseeable harm’’ is
highly likely to occur to the at-risk relative, assuming that the
relative is identifiable and the disease is preventable, treatable
or can be detected in its early stages (American Society of
Human Genetics, 1998). In BPAD, some individuals who
have never considered themselves as affected may be found
to fit the diagnostic criteria. From our experience, it appears
that the clinicians must be aware that the disclosure of such an
information has psychosocial implications for the subjects
and his family. More generally, the clinicians must take time
to explain the nature of the genetic risk, the virtual risk within
the family and the psychosocial consequences.
9. Conclusion
The amazing developments in molecular genetics during
the last decade made possible the search for susceptibility
genes in AD. Nevertheless, results so far remain preliminary
in both linkage and association studies. The growing eluci-
dation of biochemical pathways implicated in AD will
provide new candidate genes to be tested. Advances in the
definition of disease phenotypes and the identification of
endophenotypes will certainly be helpful in the future.
Finally, the application of modern technology to global
studies, involving both clinical and environmental factors,
is predictive of an optimistic era of discovery of suscepti-
bility genes of AD.
References
Aita, V.M., Liu, J., Knowles, J.A., Terwilliger, J.D., Baltazar, R., Grunn, A.,
Loth, J.E., Kanyas, K., Lerer, B., Endicott, J., Wang, Z., Penchaszadeh,
G., Gilliam, T.C., Baron, M., 1999. A comprehensive linkage analysis
of chromosome 21q22 supports prior evidence for a putative bipolar
affective disorder locus. Am. J. Hum. Genet. 64, 210–217.
American Society of Human Genetics, 1998. Professional disclosure of
familial genetic information. Am. J. Hum. Genet. 62, 474–483.
Arinami, T., Li, L., Mitsushio, H., Itokawa, M., Hamaguchi, H., Toru, M.,
1996. An Insertion–Deletion polymorphism in the angiotensin convert-
ing enzyme gene is associated with both brain substance P contents and
affective disorders. Biol. Psychiatry 40, 1122–1127.
Asherson, P., Mant, R., Williams, N., Cardno, A., Jones, L., Murphy, K.,
Collier, D.A., Nanko, S., Craddock, N., Morris, S., Muir, W., Black-
wood, B., McGuffin, P., Owen, M.J., 1998. A study of chromosome 4p
markers and dopamine D5 receptor gene in schizophrenia and bipolar
disorder. Mol. Psychiatry 3, 310–320.
Baghai, T.C., Schule, C., Zwanzger, P., Minov, C., Schwarz, M.J., de
Jonge, S., Rupprecht, R., Bondy, B., 2001. Possible influence of the
insertion/deletion polymorphism in the angiotensin I-converting en-
zyme gene on therapeutic outcome in affective disorders. Mol. Psy-
chiatry 6, 258–259.
Battersby, S., Ogilvie, A.D., Smith, C.A., Blackwood, D.H., Muir, W.J.,
Quinn, J.P., Fink, G., Goodwin, G.M., Harmar, A.J., 1996. Structure of
a variable number tandem repeat of the serotonin transporter gene and
association with affective disorder. Psychiatr. Genet. 6, 177–181.
Bellivier, F., Laplanche, J.L., Leboyer, M., Feingold, J., Bottos, C., Alli-
laire, J.F., Launay, J.M., 1997. Serotonin transporter gene associated
and manic depressive illness: an association study. Biol. Psychiatry
41, 750–752.
Bellivier, F., Leboyer, M., Courtet, P., Buresi, C., Beaufils, B., Samolyk, D.,
Allilaire, J.F., Feingold, J., Mallet, J., Malafosse, A., 1998. Association
between the tryptophan hydroxylase gene and manic-depressive illness.
Arch. Gen. Psychiatry 55, 33–37.
Bellivier, F., Szoke, A., Henry, C., Lacoste, J., Bottos, C., Nosten-Bertrand,
M., Hardy, P., Rouillon, F., Launay, J.M., Laplanche, J.L., Leboyer, M.,
2000. Possible association between serotonin transporter gene polymor-
phism and violent suicidal behavior in mood disorders. Biol. Psychiatry
48, 319–322.
Bellivier, F., Chaste, P., Malafosse, A., 2004. Association between the TPH
gene A218C polymorphism and suicidal behavior: a meta-analysis. Am.
J. Med. Genet. 124B, 87–91.
Bennett, P., Segurado, R., Jones, I., Bort, S., McCandless, F., Lambert, D.,
Heron, J., Comerford, C., Middle, F., Corvin, A., Pelios, G., Kirov, G.,
Larsen, B., Mulcahy, T., Williams, N., O’Connell, R., O’Mahony, E.,
Payne, A., Owen, M., Holmans, P., Craddock, N., Gill, M., 2002. The
Wellcome trust UK-Irish bipolar affective disorder sibling-pair genome
screen: first stage report. Mol. Psychiatry 7, 189–200.
Berrettini, W.H., Goldin, L.R., Gelernter, J., Gejman, P.V., Gershon, E.S.,
Detera-Wadleigh, S., 1990. X chromosome markers and manic-depres-
sive illness: rejection of linkage to Xq28 in nine bipolar pedigrees.
Arch. Gen. Psychiatry 47, 366–373.
Berrettini, W.H., Ferraro, T.N., Goldin, L.R., Weeks, D.E., Detera-
Wadleigh, S., Nurnberger Jr., J.I., Gershon, E.S. 1994. Chromosome
18 DNA markers and manic-depressive illness: evidence for a suscep-
tibility gene. Proc. Natl. Acad. Sci. U. S. A. 91, 5918–5921.
Biomed European Bipolar Collaborative Group, 1997. No association be-
tween bipolar disorder and alleles at a functional polymorphism in the
COMT gene. Br. J. Psychiatry 170, 526–528.
Blackwood, D.H., He, L., Morris, S.W., McLean, A., Whitton, C., Thom-
son, M., Walker, M.T., Woodburn, K., Sharp, C.M., Wright, A.F.,
Shibasaki, Y., St Clair, D.M., Porteous, D.J., Muir, W.J., 1996. A
locus for bipolar affective disorder on chromosome 4p. Nat. Genet.
12, 427–430.
Blackwood, D.H., Fordyce, A., Walker, M.T., 2001. Schizophrenia and
affective disorders-cosegregation with a translocation at chromosome
1q42 that directly disrupts brain-expressed genes: clinical and P300
findings in a family. Am. J. Hum. Genet. 69, 428–433.
Bondy, B., Erfurth, A., de Jonge, S., Kruger, M., Meyer, H., 2000. Possible
association of the short allele of the serotonin transporter promoter gene
polymorphism (5-HTTLPR) with violent suicide. Mol. Psychiatry 5,
193–195.
Bredbacka, P.E., Pekkarinen, P., Peltonen, L., 1993. Bipolar disorder in an

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877 873
extended pedigree with a segregation pattern compatible with X-linked
transmission: exclusion of the previously reported linkage to F9. Psy-
chiatr. Genet. 3, 79–87.
Brunn, T.G., Ewald, H., 1999. Selection bias of susceptibility genes possi-
ble when using parent–offspring trios in genetic association studies.
Mol. Psychiatry 4 (5), 415–416 (Sep.).
Bunney, W.E., Bunney, B.G., Vawter, M.P., Tomita, H., Li, J., Evans, S.J.,
Choudary, P.V., Myers, R.M., Jones, E.G., Watson, S.J., Akil, H., 2003.
Microarray technology: a review of new strategies to discover candidate
vulnerability genes in psychiatric disorders. Am. J. Psychiatry 160,
657–666.
Byerley, W., Plaetke, R., Hoff, M., Jensen, S., Holik, J., Reimherr, F.,
Mellon, C., Wender, P., O’Connell, P., Leppert, M., 1992. Tyrosine
hydroxylase gene not linked to manic-depression in seven of eight
pedigrees. Hum. Hered. 42, 259–263.
Caspi, A., Sugden, K., Moffitt, T.E., Taylor, A., Craig, I.W., Harrington, H.,
McClay, J., Mill, J., Martin, J., Braithwaite, A., Poulton, R., 2003.
Influence of life stress on depression: moderation by a polymorphism
in the 5-HTT gene. Science 301, 386–389.
Chen, Y.S., Akula, N., Detera-Wadleigh, S.D., Schulze, T.G., Thomas, J.,
Potash, J.B., DePaulo, J.R., McInnis, M.G., Cox, N.J., McMahon, F.J.,
2004. Findings in an independent sample support an association be-
tween bipolar affective disorder and the G72/G30 locus on chromosome
13q33. Mol. Psychiatry 9, 87–92.
Cichon, S., Schmidt-Wolf, G., Schumacher, J., Muller, D.J., Hurter, M.,
Schulze, T.G., Albus, M., Borrmann-Hassenbach, M., Franzek, E.,
Lanczik, M., Fritze, J., Kreiner, R., Weigelt, B., Minges, J., Lichter-
mann, D., Lerer, B., Kanyas, K., Strauch, K., Windemuth, C., Baur,
M.P., Wienker, T.F., Maier, W., Rietschel, M., Propping, P., Nothen, M.,
2001. A possible susceptibility locus for bipolar affective disorder in
chromosomal region 10q25–q26. Mol. Psychiatry 6, 342–349.
Collier, D.A., Stober, G., Li, T., Heils, A., Catalano, M., Di Bella, D.,
Arranz, M.J., Murray, R.M., Vallada, H.P., Bengel, D., Muller, C.R.,
Roberts, G.W., Smeraldi, E., Kirov, G., Sham, P., Lesch, K.P., 1996. A
novel functional polymorphism within the promoter of the serotonin
transporter gene: possible role in susceptibility to affective disorders.
Mol. Psychiatry 1, 453–460.
Courtet, P., Picot, M.C., Bellivier, F., Torres, S., Jollant, F., Michelon, C.,
Castelnau, D., Astruc, B., Buresi, C., Malafosse, A., 2004. Serotonin
transporter gene may be involved in short-term risk of subsequent sui-
cide attempts. Biol. Psychiatry 55, 46–51.
Craddock, N., McKeon, P., Moorhead, S., Guy, C., Harrison, D., Mynett-
Johnson, L., Claffey, E., Feldman, E., McGuffin, P., Owen, M.J., O’Do-
novan, M.C., 1997. Expanded CAG/CTG repeats in bipolar disorder: no
correlation with phenotypic measures of illness severity. Biol. Psychi-
atry 42, 876–881.
Craddock, N., Dave, S., Greening, J., 2001. Association studies of bipolar
disorder. Bipolar Disord. 3, 284–298.
De bruyn, A., Raeymaekers, P., Mendelbaum, K., Sandkuijl, L.A., Raes, G.,
Delvenne, V., Hirsch, D., Staner, L., Mendlewicz, J., Van Broeckhoven,
C., 1994. Linkage analysis of bipolar illness with X-chromosome DNA
markers: a susceptibility gene in Xq27–28 cannot be excluded. Am. J.
Med. Genet. 54, 411–419.
De bruyn, A., Souery, D., Mendelbaum, K., Mendlewicz, J., Van Broeck-
hoven, C., 1996. Linkage analysis of families with bipolar illness and
chromosome 18 markers. Biol. Psychiatry 39, 689–696.
Detera-Wadleigh, S.D., Badner, J.A., Goldin, L.R., Berrettini, W.H., Sand-
ers, A.R., Rollins, D.Y., Turner, G., Moses, T., Haerian, H., Muniec, D.,
Nurnberger Jr., J.I., Gershon, E.S., 1996. Affected sib-pair analyses
reveal support of prior evidence for a susceptibility locus for bipolar
disorder on 21q. Am. J. Hum. Genet. 58, 1279–1285.
Detera-Wadleigh, S.D., Badner, J.A., Yoshikawa, T., Sanders, A.R., Goldin,
L.R., Turner, G., Rollins, D.Y., Moses, T., Guroff, J.J., Kazuba, D.,
Maxwell, M.E., Edenberg, H.J., Foroud, T., Lahiri, D., Nurnberger
Jr., J.I., Stine, O.C., McMahon, F., Meyers, D.A., MacKinnon, D.,
Simpson, S., McInnis, M., DePaulo, J.R., Rice, J., Goate, A., Gershon,
E.S., 1997. Initial genome screen for bipolar disorder in the NIMH
genetics initiative pedigrees: chromosomes 4, 7, 9, 18, 19, 20 and
21q. Am. J. Med. Genet. 74, 254–262.
Doney, A.S., Fischer, B., Cecil, J.E., Cohen, P.T., Boyle, D.I., Leese, G.,
Morris, A.D., Palmer, C.N., 2003. Male preponderance in early diag-
nosed type 2 diabetes is associated with the ARE insertion/deletion
polymorphism in the PPP1R3A locus. BMC Genet. 28, 11.
Du, L., Faludi, G., Palkovits, M., Demeter, E., Bakish, D., Lapierre, Y.D.,
Sotonyi, P., Hrdina, P.D., 1999. Frequency of long allele in serotonin
transporter gene is increased in depressed suicide victims. Biol. Psychi-
atry 46, 196–201.
Duman, R.S., 1998. Novel therapeutic approaches beyond the serotonin
receptor. Biol. Psychiatry 44, 324–335.
Egeland, J.A., Gerhard, D.S., Pauls, D.L., Sussex, J.N., Kidd, K.K., Allen,
C.R., Hostetter, A.M., Housman, D.E., 1987. Bipolar affective disorders
linked to markers on chromosome 11. Nature 325, 783–787.
Engstrom, C., Thornlund, A.S., Johansson, E.L., Langstrom, M., Chotai, J.,
Adolfsson, R., Nylander, P.O., 1995. Anticipation in unipolar affective
disorder. J. Affect. Disord. 35, 31–40.
Ewald, H., Mors, O., Koed, K., Eiberg, H., Kruse, T.A., 1997. Susceptibil-
ity loci for bipolar affective disorder on chromosome 18? A review and
a study of Danish families. Psychiatr. Genet. 7, 1–12.
Faraone, S.V., Glatt, S.J., Tsuang, M.T., 2003. The genetics of pediatric-
onset bipolar disorder. Biol. Psychiatry 53, 970–977.
Freimer, N.B., Reus, V.I., Escamilla, M.A., McInnes, L.A., Spesny, M.,
Leon, P., Service, S.K., Smith, L.B., Silva, S., Rojas, E., Gallegos,
A., Meza, L., Fournier, E., Baharloo, S., Blankenship, K., Tyler, D.J.,
Batki, S., Vinogradov, S., Weissenbach, J., Barondes, S.H., Sandkuijl,
L.A., 1996. Genetic mapping using haplotype, association and linkage
methods suggests a locus for severe bipolar disorder (BPI) at 18q22–
q23. Nat. Genet. 2, 436–441.
Frodl, T., Meisenzahl, E.M., Zill, P., Baghai, T., Rujescu, D., Leinsinger,
G., Bottlender, R., Schule, C., Zwanzger, P., Engel, R.R., Rupprecht, R.,
Bondy, B., Reiser, M., Moller, H.J., 2004. Reduced hippocampal vol-
umes associated with the long variant of the serotonin transporter poly-
morphism in major depression. Arch. Gen. Psychiatry 61, 177–183.
Fukumoto, T., Morinobu, S., Okamoto, Y., Kagaya, A., Yamawaki, S.,
2001. Chronic lithium treatment increases the expression of brain-de-
rived neurotrophic factor in the rat brain. Psychopharmacology 158,
100–106.
Furlong, R.A., Rubinsztein, J.S., Ho, L., Walsh, C., Coleman, T.A., Muir,
W.J., Paykel, E.S., Blackwood, D.H., Rubinsztein, D.C., 1999. Analysis
and meta-analysis of two polymorphisms within the tyrosine hydroxy-
lase gene in bipolar and unipolar affective disorders. Am. J. Med.
Genet. 88, 88–94.
Furlong, R.A., Keramatipour, M., Ho, L.W., Rubinsztein, J.S., Michael, A.,
Walsh, C., Paykel, E.S., Rubinsztein, D.C., 2000. No association of an
insertion/deletion polymorphism in the angiotensin I converting enzyme
gene with bipolar or unipolar affective disorders. Am. J. Med. Genet.
96, 733–735.
Gejman, P.V., Detera-Wadleigh, S., Martinez, M.M., Berrettini, W.H.,
Goldin, L.R., Gelernter, J., Hsieh, W.T., Gershon, E.S., 1990. Manic
depressive illness not linked to factor IX in a independent series of
pedigrees. Genomics 8, 648–655.
Gershon, E.S., Badner, J.A., Detera-Wadleigh, S.D., Ferraro, T.N., Ber-
rettini, W.H., 1996. Maternal inheritance and chromosome 18 allele
sharing in unilineal bipolar illness pedigrees. Am. J. Med. Genet. 67,
202–207.
Goossens, D., Villafuerte, S., Tissir, F., Van Gestel, S., Claes, S., Souery,
D., Massat, I., Van den Bossche, D., Van Zand, K., Mendlewicz, J., Van
Broeckhoven, C., Del-Favero, J., 2000. No evidence for the involve-
ment of CAG/CTG repeats from within 18q21.33–q23 in bipolar dis-
order. Eur. J. Hum. Genet. 8, 385–388.
Gottesman, I.I., Gould, T.D., 2003. The endophenotype concept in psychia-
try: etymology and strategic intentions. Am. J. Psychiatry 160, 636–645.
Gurling, H., Smyth, C., Kalsi, G., Moloney, E., Rifkin, L., O’Neill, J.,
Murphy, P., Curtis, D., Petursson, H., Brynjolfsson, J., 1995. Linkage
findings in bipolar disorders. Nat. Genet. 10, 8–9.

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877874
Guy, C.A., Bowen, T., Jones, I., McCandless, F., Owen, M.J., Craddock,
N., O’Donovan, M.C., 1999. CTG18.1 and ERDA-1 CAG/CTG repeat
size in bipolar disorder. Neurobiol. Dis. 6, 302–307.
Hattori, E., Liu, C., Badner, J.A., Bonner, T.I., Christian, S.L., Maheshwari,
M., Detera-Wadleigh, S.D., Gibbs, R.A., Gershon, E.S., 2003. Poly-
morphisms at the G72/G30 gene locus, on 13q33, are associated with
bipolar disorder in two independent pedigree series. Am. J. Hum. Genet.
72, 1131–1140.
Ho, L.W., Furlong, R.A., Rubinsztein, J.S., Walsh, C., Paykel, E.S.,
Rubinsztein, D.C., 2000. Genetic associations with clinical character-
istics in bipolar affective disorder and recurrent unipolar depressive
disorder. Am. J. Med. Genet. 96, 36–42.
Hodge, S.E., 1994. What association analysis can and cannot tell us about
the genetics of complex disease. Am. J. Med. Genet. 54, 318–323.
Holmes, D., Brynjolfsson, J., Brett, P., Curtis, D., Petursson, H., Sherring-
ton, R., Gurling, H., 1991. No evidence for a susceptibility locus pre-
disposing to manic depression in the region of the Dopamine (D2)
receptor gene. Br. J. Psychiatry 158, 635–641.
Horiuchi, Y., Nakayama, J., Ishiguro, H., Ohtsuki, T., Detera-Wadleigh, S.D.,
Toyota, T., Yamada, K., Nankai, M., Shibuya, H., Yoshikawa, T., Ari-
nami, T., 2004. Possible association between a haplotype of the GABA-A
receptor alpha 1 subunit gene (GABRA1) and mood disorders. Biol.
Psychiatry 55, 40–45.
Kakiuchi, C., Iwamoto, K., Ishiwata, M., Bundo, M., Kasahara, T., Kusumi,
I., Tsujita, T., Okazaki, Y., Nanko, S., Kunugi, H., Sasaki, T., Kato, T.,
2003. Impaired feedback regulation of XBP1 as a genetic risk factor for
bipolar disorder. Nat. Genet. 35, 171–175.
Kamnasaran, D., 2003. Genetic analysis of psychiatric disorders associated
with human chromosome 18. Clin. Invest. Med. 26, 285–302.
Kennedy, J.L., Farrer, L.A., Andreasen, N.C., Mayeux, R., St George-Hys-
lop, P., 2003. The genetics of adult-onset neuropsychiatric disease:
complexities and conundra? Science 302, 822–826.
Kidd, K.K., Matthysee, S., 1978. Research designs for the study of gene–
environment interactions in psychiatric disorders. Report of a Founda-
tions Fund for Research in Psychiatry Panel. Arch. Gen. Psychiatry 35,
925–932.
Kirov, G., Jones, I., McCandless, F., 1999. Family-based association studies
of bipolar disorder with candidate genes in dopamine neurotransmis-
sion: DBH, DAT1, COMT, DRD2, DRD3 and DRD5. Mol. Psychiatry
4, 558–565.
Kramer, M.S., Cutler, N., Feighner, J., Shrivastava, R., Carman, J., Sramek,
J.J., Reines, S.A., Liu, G., Snavely, D., Wyatt-Knowles, E., Hale, J.J.,
Mills, S.G., MacCoss, M., Swain, C.J., Harrison, T., Hill, R.G., Hefti,
F., Scolnick, E.M., Cascieri, M.A., Chicchi, G.G., Sadowski, S., Wil-
liams, A.R., Hewson, L., Smith, D., Rupniak, N.M., 1998. Distinct
mechanism for antidepressant activity by blockade of central substance
P receptors. Science 281, 1640–1645.
Kramer, M.S., Winokur, A., Kelsey, J., Preskorn, S.H., Rothschild, A.J.,
Snavely, D., Ghosh, K., Ball, W.A., Reines, S.A., Munjack, D., Apter,
J.T., Cunningham, L., Kling, M., Bari, M., Getson, A., Lee, Y., 2004.
Demonstration of the efficacy and safety of a novel substance P (NK1)
receptor antagonist in major depression. Neuropsychopharmacology 29,
385–392.
Kunugi, H., Vallada, H.P., Hoda, F., Kirov, G., Gill, M., Aitchison, K.J.,
Ball, D., Arranz, M.J., Murray, R.M., Collier, D.A., 1997. No evidence
for an association of affective disorders with high- or low-activity allele
of catechol-o-methyltransferase gene. Biol. Psychiatry 42, 282–285.
Kunugi, H., Ishida, S., Kato, T., Tatsumi, M., Sakai, T., Hattori, M., Hirose,
T., Nanko, S., 1999. A functional polymorphism in the promoter region
of monoamine oxidase-A gene and mood disorders. Mol. Psychiatry 4,
393–395.
Leboyer, M., Bellivier, F., Nosten-Bertrand, M., Jouvent, R., Pauls, D.,
Mallet, J., 1998. Psychiatric genetics: search for phenotypes. Trends
Neurosci. 21, 102–105.
Lehmann, L.S., Weeks, J.C., Klar, N., Biener, L., Garber, J.E., 2000. Dis-
closure of Familial genetic information: perceptions of the duty to in-
form. Am. J. Med. 109, 705–711.
Lewis, C.M., Levinson, D.F., Wise, L.H., DeLisi, L.E., Straub, R.E., Hov-
atta, I., Williams, N.M., Schwab, S.G., Pulver, A.E., Faraone, S.V.,
Brzustowicz, L.M., Kaufmann, C.A., Garver, D.L., Gurling, H.M.,
Lindholm, E., Coon, H., Moises, H.W., Byerley, W., Shaw, S.H.,
Mesen, A., Sherrington, R., O’Neill, F.A., Walsh, D., Kendler, K.S.,
Ekelund, J., Paunio, T., Lonnqvist, J., Peltonen, L., O’Donovan, M.C.,
Owen, M.J., Wildenauer, D.B., Maier, W., Nestadt, G., Blouin, J.L.,
Antonarakis, S.E., Mowry, B.J., Silverman, J.M., Crowe, R.R., Clo-
ninger, C.R., Tsuang, M.T., Malaspina, D., Harkavy-Friedman, J.M.,
Svrakic, D.M., Bassett, A.S., Holcomb, J., Kalsi, G., McQuillin, A.,
Brynjolfson, J., Sigmundsson, T., Petursson, H., Jazin, E., Zoega, T.,
Helgason, T., 2003. Genome scan meta-analysis of schizophrenia and
bipolar disorder, part II: Schizophrenia. Am. J. Hum. Genet. 73, 34–48.
Li, T., Vallada, H.P., Liu, X., Xie, T., Tang, X., Zhao, J., O’Donovan, M.C.,
Murray, R.M., Sham, P.C., Collier, D.A., 1998. Analysis of CAG/CTG
repeat size in Chinese subjects with schizophrenia and bipolar affective
disorder using the repeat expansion detection method. Biol. Psychiatry
44, 1160–1165.
Lim, L.C., Gurling, H., Curtis, D., Brynjolfsson, J., Petursson, H., Gill, M.,
1993. Linkage between tyrosine hydroxylase gene and affective disor-
der cannot be excluded in two of six pedigrees. Am. J. Med. Genet. 48,
223–228.
Lim, L.C., Nothen, M.M., Korner, J., Rietschel, M., Castle, D., Hunt, N.,
Propping, P., Murray, R., Gill, M., 1994. No evidence of association
between dopamine D4 receptor variants and bipolar affective disorder.
Am. J. Med. Genet. 54, 259–263.
Lim, L.C., Powell, J., Sham, P., Castle, D., Hunt, N., Murray, R., Gill, M.,
1995. Evidence for a genetic association between alleles of monoamine
oxidase A gene and bipolar affective disorder. Am. J. Med. Genet. 60,
325–331.
Lin, S., Jiang, S., Wu, X., Qian, Y., Wang, D., Tang, G., Gu, N., 2000.
Association analysis between mood disorder and monoamine oxidase
gene. Am. J. Med. Genet. 96, 12–14.
Lindblad, K., Nylander, P.O., De bruyn, A., Souery, D., Zander, C., Eng-
strom, C., Holmgren, G., Hudson, T., Chotai, J., Mendlewicz, J., 1995.
Expansion of trinucleotide CAG repeats detected in Bipolar Affective
Disorder by the RED-(rapid expansion detection) method. Neurobiol.
Dis. 2, 55–62.
Lindblad, K., Nylander, P.O., Zander, C., Yuan, Q.P., Stahle, L., Engstrom,
C., Balciuniene, J., Pettersson, U., Breschel, T., McInnis, M., Ross,
C.A., Adolfsson, R., Schalling, M., 1998. Two commonly expanded
CAG/CTG repeat loci: involvement in affective disorders? Mol. Psy-
chiatry 3, 405–410.
Linkowski, P., De Maertelaer, V., Mendlewicz, J., 1985. Suicidal behavior
in major depressive illness. Acta Psychiatr. Scand. 72, 233–238.
Lucotte, G., Landoulsi, A., Berriche, S., David, F., Babron, M.C., 1992.
Manic depressive illness is linked to factor IX in a French pedigree.
Ann. Genet. 35, 93–95.
Maier, W., Zobel, A., Rietschel, M., 2003. Genetics of schizophrenia and
affective disorders. Pharmacopsychiatry 36 (Suppl 3), 195–202.
Massat, I., Souery, D., Papadimitriou, G.N., Mendlewicz, J., 2000. The
GABAergic hypothesis of mood disorders. In: Soares, J.C., Gershon,
S. (Eds.), Bipolar Disorders. Marcel Dekker, New York, pp. 143–165.
Massat, I., Souery, D., Del-Favero, J., Oruc, L., Jakovljevic, M., Folne-
govic, V., Adolfsson, R., Kaneva, R., Papadimitriou, G., Dikeos, D.,
Jazin, E., Milanova, V., Van Broeckhoven, C., Mendlewicz, J., 2001.
Lack of association between GABRA3 and unipolar affective disorder:
a multicentre study. Int. J. Neuropsychopharmacol. 4, 273–278.
Massat, I., Souery, D., Del-Favero, J., Oruc, L., Noethen, M.M., Black-
wood, D., Thomson, M., Muir, W., Papadimitriou, G.N., Dikeos, D.G.,
Kaneva, R., Serretti, A., Lilli, R., Smeraldi, E., Jakovljevic, M., Folne-
govic, V., Rietschel, M., Milanova, V., Valente, F., Van Broeckhoven,
C., Mendlewicz, J., 2002a. Excess of allele1 for alpha3 subunit GABA
receptor gene (GABRA3) in bipolar patients: a multicentric association
study. Mol. Psychiatry 7, 201–207.
Massat, I., Souery, D., Del-Favero, J., Van Gestel, S., Serretti, A., Mac-
ciardi, F., Smeraldi, E., Kaneva, R., Adolfsson, R., Nylander, P.O.,

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877 875
Blackwood, D., Muir, W., Papadimitriou, G.N., Dikeos, D., Oruc, L.,
Segman, R.H., Ivezic, S., Aschauer, H., Ackenheil, M., Fuchshuber, S.,
Dam, H., Jakovljevic, M., Peltonen, L., Hilger, C., Hentges, F., Staner,
L., Milanova, V., Jazin, E., Lerer, B., Van Broeckhoven, C., Mendle-
wicz, J., 2002b. Positive association of dopamine D2 receptor polymor-
phism with bipolar affective disorder in a European multicenter
association study of affective disorders. Am. J. Med. Genet. 114,
177–185.
McGuffin, P., Asherson, P., Owen, M., Farmer, A., 1994. The strength of
the genetic effect. Is there room for an environmental influence in the
aetiology of schizophrenia? Br. J. Psychiatry 164, 593–599.
McInnis, M.G., McMahon, F.J., Chase, G.A., Simpson, S.G., Ross, C.A.,
DePaulo Jr., J.R., 1993. Anticipation in bipolar affective disorder. Am.
J. Hum. Genet. 53, 385–390.
McMahon, F.J., Hopkins, P.J., Xu, J., McInnis, M.G., Shaw, S., Cardon, L.,
Simpson, S.G., MacKinnon, D.F., Stine, O.C., Sherrington, R., Meyers,
D.A., DePaulo, J.R., 1997. Linkage of bipolar disorder to chromosome
18markers in a new pedigree series. Am. J. Hum.Genet. 61, 1397–1404.
Meira-Lima, I.V., Pereira, A.C., Mota, G.F., Krieger, J.E., Vallada, H.,
2000. Angiotensinogen and angiotensin converting enzyme gene poly-
morphisms and the risk of bipolar affective disorder in humans. Neuro-
sci. Lett. 293, 103–106.
Mendlewicz, J., 1994. The search for a manic depressive gene: from clas-
sical tomolecular genetics. Prog. Brain Res. 100, 255–259.
Mendlewicz, J., Rainer, J.D., 1977. Adoption study supporting genetic
transmission in manic-depressive illness. Nature 268, 327–329.
Mendlewicz, J., Simon, P., Sevy, S., Charon, F., Brocas, H., Legros, S.,
Vassart, G., 1987. Polymorphic DNA marker on chromosome X and
manic-depression. Lancet 1, 1230–1232.
Mendlewicz, J., Lindbald, K., Souery, D., Mahieu, B., Nylander, P.O., De
Bruyn, A., Zander, C., Engstrom, C., Adolfsson, R., Van Broeck-
hoven, C., Schalling, M., Lipp, O., 1997. Expanded trinucleotide
CAG repeats in families with bipolar affective disorder. Biol. Psychi-
atry 42, 1115–1122.
Mendlewicz, J., Massat, I., Souery, D., Del-Favero, J., Oruc, L., Nothen,
M.M., Blackwood, D., Muir, W., Battersby, S., Lerer, B., Segman, R.H.,
Kaneva, R., Serretti, A., Lilli, R., Lorenzi, C., Jakovljevic, M., Ivezic,
S., Rietschel, M., Milanova, V., Van Broeckhoven, C., 2004. Serotonin
transporter 5HTTLPR polymorphism and affective disorders: no evi-
dence of association in a large European multicenter study. Eur. J. Hum.
Genet. 12, 377–382.
Merikangas, K.R., Spence, M.A., Kupfer, D.J., 1989. Linkage studies of
bipolar disorder: methodologic and analytic issues. Arch. Gen. Psychi-
atry 46, 1137–1141.
Nakata, K., Ujike, H., Sakai, A., Uchida, N., Nomura, A., Imamura, T.,
Katsu, T., Tanaka, Y., Hamamura, T., Kuroda, S., 2003. Association
study of the brain-derived neurotrophic factor (BDNF) gene with bipo-
lar disorder. Neurosci. Lett. 337, 17–20.
Nanko, S., Fukuda, R., Hattori, M., Sasaki, T., Dai, X.Y., Kanba, S., Kato,
T., Kazamatsuri, H., 1994. Linkage studies between affective disorder
and dopamine D2, D3, and D4 receptor gene loci in four Japanese
pedigrees. Psychiatry Res. 52, 149–157.
Neves-Pereira, M., Mundo, E., Muglia, P., King, N., Macciardi, F., Ken-
nedy, J.L., 2002. The brain-derived neurotrophic factor gene confers
susceptibility to bipolar disorder: evidence from a family-based associ-
ation study. Am. J. Hum. Genet. 71, 651–655.
Nibuya, M., Morinobu, S., Duman, R.S., 1995. Regulation of BDNF and
trkB mRNA in rat brain by chronic electroconvulsive seizure and anti-
depressant drug treatments. J. Neurosci. 15, 7539–7547.
Nielsen, D.A., Goldman, D., Virkkunen, M., Tokola, R., Rawlings, R.,
Linnoila, M., 1994. Suicidality and 5-Hydroxyindoleacetic acid concen-
tration association with a tryptophan hydroxylase polymorphism. Arch.
Gen. Psychiatry 51, 34–38.
Nielsen, D.A., Jenkins, G.L., Stefanisko, K.M., Jefferson, K.K., Goldman,
D., 1997. Sequence, splice site and population frequency distribution
analyses of the polymorphic human tryptophan hydroxylase intron 7.
Brain Res. Mol. Brain Res. 45, 145–148.
Nielsen, D.A., Virkkunen, M., Lappalainen, J., Eggert, M., Brown, G.L.,
Long, J.C., Goldman, D., Linnoila, M., 1998. A tryptophan hydroxylase
gene marker for suicidality and alcoholism. Arch. Gen. Psychiatry 55,
593–602.
Nothen, M.M., Cichon, S., Rohleder, H., Hemmer, S., Franzek, E., Fritze,
J., Albus, M., Borrmann-Hassenbach, M., Kreiner, R., Weigelt, B.,
Minges, J., Lichtermann, D., Maier, W., Craddock, N., Fimmers, R.,
Holler, T., Baur, M.P., Rietschel, M., Propping, P., 1999. Evaluation of
linkage of bipolar affective disorder to chromosome 18 in a sample of
57 German families. Mol. Psychiatry 4, 76–84.
Nylander, P.O., Engstrom, C., Chotai, J., Wahlstrom, J., Adolfsson, R.,
1994. Anticipation in Swedish families with bipolar affective disorder.
Am. J. Med. Genet. 9, 686–689.
O’Donovan, M.C., Guy, C., Craddock, N., Murphy, K.C., Cardno, A.G.,
Jones, L.A., Owen, M.J., McGuffin, P., 1995. Expanded CAG repeats in
schizophrenia and bipolar disorder. Nat. Genet. 10, 380–381.
Ogilvie, A.D., Battersby, S., Bubb, V.J., Fink, G., Harmar, A.J., Good-
wim, G.M., Smith, C.A., 1996. Polymorphism in serotonin transporter
gene associated with susceptibility to major depression. Lancet 347,
731–733.
Oruc, L., Verheyen, G.R., Furac, I., Ivezic, S., Jakovljevic, M., Raey-
maekers, P., Van Broeckhoven, C., 1997a. Positive association between
the GABRA5 gene and unipolar recurrent depression. Neuropsychobi-
ology 36, 62–64.
Oruc, L., Lindblad, K., Verheyen, G.R., Ahlberg, S., Jakovljevic, M.,
Ivezic, S., Raeymaekers, P., Van Broeckhoven, C., Schalling, M.,
1997b. CAG expansions in bipolar and unipolar disorders. Am. J.
Hum. Genet. 60, 730–732.
Ospina-Duque, J., Duque, C., Carvajal-Carmona, L., Ortiz-Barrientos, D.,
Soto, I., Pineda, N., Cuartas, M., Calle, J., Lopez, C., Ochoa, L., Garcia,
J., Gomez, J., Agudelo, A., Lozano, M., Montoya, G., Ospina, A.,
Lopez, M., Gallo, A., Miranda, A., Serna, L., Montoya, P., Palacio,
C., Bedoya, G., McCarthy, M., Reus, V., Freimer, N., Ruiz-Linares,
A., 2000. An association study of bipolar mood disorder (type I) with
the 5-HTTLPR serotonin transporter polymorphism in a human popu-
lation isolate from Colombia. Neurosci. Lett. 292, 199–202.
Oswald, P., Souery, D., Mendlewicz, J., 2003a. Molecular genetics of af-
fective disorders. Int. J. Neuropsychopharmacol. 6, 155–169.
Oswald, P., Souery, D., Massat, I., Del-Favero, J., Linotte, S., Papadimi-
triou, G., Dikeos, D., Kaneva, R., Milanova, V., Oruc, L., Ivezic, S.,
Serretti, A., Lilli, R., Van Broeckhoven, C., Mendlewicz, J., 2003b.
Lack of association between the 5HT2A receptor polymorphism
(T102C) and unipolar affective disorder in a multicentric European
study. Eur. Neuropsychopharmacol. 13, 365–368.
Oswald, P., Del-Favero, J., Massat, I., Souery, D., Claes, S., Van Broeck-
hoven, C., Mendlewicz, J., 2004. Non replication of the Brain-Derived
Neurotrophic Factor (BDNF) association in bipolar affective disorder: a
Belgian patient-control study. Am. J. Med. Genet. (in press).
Papadimitriou, G.N., Dikeos, D.G., Karadima, G., Avramopoulos, D., Das-
kalopoulou, E.G., Vassilopoulos, D., Stefanis, C.N., 1998. Association
between the GABA(A) receptor alpha5 subunit gene locus (GABRA5)
and bipolar affective disorder. Am. J. Med. Genet. 81, 73–80.
Parker, L.S., 2002. Ethical issues in bipolar disorders pedigree research:
privacy concerns, informed consent and grounds for waiver. Bipolar
Disord. 4, 1–16.
Pauls, J., Bandelow, B., Ruther, E., Kornhuber, J., 2000. Polymorphism of
the gene of angiotensin converting enzyme: lack of association with
mood disorder. J. Neural Transm. 107, 1361–1366.
Paulson, H.L., Fischbeck, K.H., 1996. Trinucleotide repeats in neurogenetic
disorders. Annu. Rev. Neurosci. 19, 79–107.
Pekkarinen, P., Terwilliger, J., Bredbacka, P.E., Lonnqvist, J., Peltonen,
L., 1995. Evidence of a predisposing locus to bipolar disorder on
Xq24–q27.1 in an extended Finnish pedigree. Genome Res. 5,
105–115.
Perez, dC., Santos, J., Torres, P., 1995. Aweak association between TH and
DRD2 genes and bipolar affective disorder in a Spanish sample. Am. J.
Med. Genet. 32, 131–134.

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877876
Potash, J.B., DePaulo Jr., J.R., 2000. Searching high and low: a review of
the genetics of bipolar disorder. Bipolar Disord. 2, 8–26.
Preisig, M., Bellivier, F., Fenton, B.T., Baud, P., Berney, A., Courtet, P.,
Hardy, P., Golaz, J., Leboyer, M., Mallet, J., Matthey, M.L., Mouthon,
D., Neidhart, E., Nosten-Bertrand, M., Stadelmann-Dubuis, E., Gui-
mon, J., Ferrero, F., Buresi, C., Malafosse, A., 2000. Association be-
tween bipolar disorder and monoamine oxidase A gene polymorphisms:
results of a multicenter study. Am. J. Psychiatry 157, 948–955.
Pritchard, J.K., Rosenberg, N.A., 1999. Use of unlinked genetic markers to
detect population stratification in association studies. Am. J. Hum.
Genet. 65, 220–228.
Risch, N., Merikangas, K.R., 1993. Linkage studies of psychiatric disor-
ders. Eur. Arch. Psychiatry Clin. Neurosci. 243, 143–149.
Rubinsztein, D.C., Leggo, J., Goodburn, S., Walsh, C., Jain, S., Paykel,
E.S., 1996. Genetic association between monoamine oxidase A micro-
satellite and RFLP alleles and bipolar affective disorder: analysis and
meta-analysis. Hum. Mol. Genet. 5, 779–782.
Russ, M.J., Lachman, H.M., Kashdan, T., Saito, T., Bajmakovic-Kacila, S.,
2000. Analysis of catechol-o-methyltransferase and 5-Hydroxytrypta-
mine transporter polymorphisms in patients at risk for suicide. Psychi-
atry Res. 93, 73–78.
Savoye, C., Laurent, C., Amadeo, S., Gheysen, F., Leboyer, M., Lejeune, J.,
Zarifian, E., Mallet, J., 1998. No association between dopamine D1,
D2, D3 receptor genes and manic- depressive illness. Biol. Psychiatry
44, 644–647.
Schulze, T.G., McMahon, F.J., 2003. Genetic linkage and association stud-
ies in bipolar affective disorder: a time for optimism. Am. J. Med.
Genet. 123C (1), 36–47.
Schumacher, J., Jamra, R.A., Freudenberg, J., Becker, T., Ohlraun, S., Otte,
A.C., Tullius, M., Kovalenko, S., Bogaert, A.V., Maier, W., Rietschel,
P., Propping, P., Nothen, M.M., Cichon, S., 2004. Examination of G72
and D-amino-acid oxidase as genetic risk factors for schizophrenia and
bipolar affective disorder. Mol. Psychiatry 9, 203–207.
Segurado, R., Detera-Wadleigh, S.D., Levinson, D.F., Lewis, C.M., Gill, M.,
Nurnberger Jr., J.I., Craddock, N., DePaulo, J.R., Baron, M., Gershon,
E.S., Ekholm, J., Cichon, S., Turecki, G., Claes, S., Kelsoe, J.R., Scho-
field, P.R., Badenhop, R.F., Morissette, J., Coon, H., Blackwood, D.,
McInnes, L.A., Foroud, T., Edenberg, H.J., Reich, T., Rice, J.P., Goate,
A., McInnis, M.G., McMahon, F.J., Badner, J.A., Goldin, L.R., Bennett,
P., Willour, V.L., Zandi, P.P., Liu, J., Gilliam, C., Juo, S.H., Berrettini,
W.H., Yoshikawa, T., Peltonen, L., Lonnqvist, J., Nothen, M.M., Schu-
macher, J., Windemuth, C., Rietschel, M., Propping, P., Maier, W., Alda,
P., Grof, P., Rouleau, G.A., Del-Favero, J., Van Broeckhoven, C., Mend-
lewicz, J., Adolfsson, R., Spence, M.A., Luebbert, H., Adams, L.J.,
Donald, J.A., Mitchell, P.B., Barden, N., Shink, E., Byerley, W., Muir,
P.M., Visscher, P.M., Macgregor, S., Gurling, H., Kalsi, G., McQuillin,
M.A., Escamilla, M.A., Reus, V.I., Leon, P., Freimer, N.B., Ewald, H.,
Kruse, T.A., Mors, O., Radhakrishna, U., Blouin, J.L., Antonarakis,
N., Akarsu, N., 2003. Genome scan meta-analysis of schizophrenia
and bipolar disorder: Part III. Bipolar disorder. Am. J. Hum. Genet.
73, 49–62.
Serretti, A., Macciardi, F., Cusin, C., Lattuada, E., Lilli, R., Di Bella, D.,
Catalano, M., Smeraldi, E., 1998. GABAA alpha-A subunit gene not
associated with depressive symptomatology in mood disorders. Psy-
chiatr. Genet. 8, 251–254.
Shilling, P.D., Kelsoe, J.R., 2002. Functional genomics approaches to un-
derstanding brain disorders. Pharmacogenomics 3, 31–45.
Shoemaker, D.D., Linsley, P.S., 2002. Recent developments in DNA micro-
arrays. Curr. Opin. Microbiol. 5, 334–337.
Shore, D., 1993. Legal and ethical issues in psychiatric genetic research.
Am. J. Med. Genet. 48, 17–21.
Shore, D., 1996. Ethical principles and informed consent: an NIMH per-
spective. Psychopharmacol. Bull. 32, 7–10.
Sklar, P., Gabriel, S.B., McInnis, M.G., Bennett, P., Lim, Y.M., Tsan, G.,
Schaffner, S., Kirov, G., Jones, I., Owen, M., Craddock, N., DePaulo,
J.R., Lander, E.S., 2002. Family-based association study of 76 candi-
date genes in bipolar disorder: BDNF is a potential risk locus. Brain-
derived neutrophic factor. Mol. Psychiatry 7, 579–593.
Smeraldi, E., Negri, F., Melica, A.M., Scorza-Smeraldi, R., 1978. HLA
system and affective disorders: a sibship genetic study. Tissue Antigens
12, 270–274.
Smyth, C., Kalsi, G., Brynjolfsson, J., O’Neill, J., Curtis, D., Rifkin, L.,
Moloney, E., Murphy, P., Sherrington, R., Petursson, H., Gurling, H.,
1996. Further tests for linkage of bipolar affective disorder to the tyro-
sine hydroxylase gene locus on chromosome 11p15 in a new series of
multiplex British affective disorder pedigrees. Am. J. Psychiatry 153,
271–274.
Souery, D., Papadimitriou, G.N., Mendlewicz, J., 1995. New genetic
approaches in Affective Disorders. In: Papadimitriou, G.N., Mendle-
wicz, J. (Eds.), Genetics of Mental Disorders Part II: Clinical Issues-
Bailliere’s Clinical Psychiatry, International Practice and Research, vol.
2 No. 1. Bailliere Tindall, London, pp. 73–85.
Souery, D., Lipp, O., Mahieu, B., Mendelbaum, K., De Martelaer, V., Van
Broeckhoven, C., Mendlewicz, J., 1996. Association study of bipolar
disorder with candidate genes involved in catecholamine neurotransmis-
sion: DRD2, DRD3, DAT1, and TH genes. Am. J. Med. Genet. 67,
551–555.
Souery, D., Rivelli, S.K., Mendlewicz, J., 2001a. Molecular genetic and
family studies in affective disorders: state of the art. J. Affect. Disord.
62, 45–55.
Souery, D., Van Gestel, S., Massat, I., Blairy, S., Adolfsson, R., Blackwood,
D., Del-Favero, J., Dikeos, D., Jakovljevic, M., Kaneva, R., Lattuada,
E., Lerer, B., Lilli, R., Milanova, V., Muir, W., Nothen, M., Oruc, L.,
Papadimitriou, G., Propping, P., Schulze, T., Serretti, A., Shapira, B.,
Smeraldi, E., Stefanis, C., Thomson, M., Van Broeckhoven, C., Mend-
lewicz, J., 2001b. Tryptophan hydroxylase polymorphism and suicidal-
ity in unipolar and bipolar affective disorders: a multicenter association
study. Biol. Psychiatry 49, 405–409.
Stancer, H.C., Weitkamp, L.R., Persad, E., Flood, C., Jorna, T., Guttormsen,
S.A., Yagnow, R.L., 1988. Confirmation of the relationship of HLA
(chromosome 6) genes to depression and manic depression: II. The
Ontario follow-up and analysis of 117 kindreds. Ann. Hum. Genet.
52, 279–298.
Stine, O.C., Xu, J., Koskela, R., McMahon, F.J., Gschwend, M., Friddle,
C., Clark, C.D., McInnis, M.G., Simpson, S.G., Breschel, T.S., 1995.
Evidence for linkage of bipolar disorder to chromosome 18 with a
parent-of-origin effect. Am. J. Hum. Genet. 57, 1384–1394.
Stine, O.C., McMahon, F.J., Chen, L., Xu, J., Meyers, D.A., MacKinnon,
D.F., Simpson, S., McInnis, M.G., Rice, J.P., Goate, A., Reich, T.,
Edenberg, H.J., Foroud, T., Nurnberger Jr., J.I., Detera-Wadleigh,
S.D., Goldin, L.R., Guroff, J., Gershon, E.S., Blehar, M.C., DePaulo,
J.R. 1997. Initial genome screen for bipolar disorder in the NIMH
genetics initiative pedigrees: chromosomes 2, 11, 13, 14 and X. Am.
J. Med. Genet. 74, 263–269.
Stoltenberg, S.F., Burmeister, M., 2000. Recent progress in psychi-
atric genetics—some hope but no hype. Hum. Mol. Genet. 9,
927–935.
Straub, R.E., Lehner, T., Luo, Y., Loth, J.E., Shao, W., Sharpe, L., Alex-
ander, J.R., Das, K., Simon, R., Fieve, R.R., 1994. A possible vulner-
ability locus for bipolar affective disorder on chromosome 21q22.3.
Nat. Genet. 8, 291–296.
Suarez, B.K., Hampe, C.L., 1994. Linkage and association. Am. J. Hum.
Genet. 54, 554–559.
Syagailo, Y.V., Stober, G., Grassle, M., Reimer, E., Knapp, M., Jungkunz,
G., Okladnova, O., Meyer, J., Lesch, K.P., 2001. Association analysis of
the functional monoamine oxidase A gene promoter polymorphism in
psychiatric disorders. Am. J. Med. Genet. 105, 168–171.
Trottier, Y., Lutz, Y., Stevanin, G., Imbert, G., Devys, D., Cancel, G.,
Saudou, F., Weber, C., David, G., Tora, L., 1995. Polyglutamine ex-
pansion as a pathological epitope in Huntington’s disease and four
dominant cerebellar ataxias. Nature 378, 403–406.
Tsai, S.J., Cheng, C.Y., Yu, Y.W., Chen, T.J., Hong, C.J., 2003. Association

P. Oswald et al. / Progress in Neuro-Psychopharmacology & Biological Psychiatry 28 (2004) 865–877 877
study of a brain-derived neurotrophic-factor genetic polymorphism and
major depressive disorders, symptomatology, and antidepressant re-
sponse. Am. J. Med. Genet. 123, 19–22.
Turecki, G., 2001. Suicidal behavior: is there a genetic predisposition?
Bipolar Disord. 3, 335–349.
Turecki, G., Rouleau, G.A., Mari, J.J., Morgan, K., 1996. A systematic
evaluation of linkage studies in bipolar disorder. Acta Psychiatr. Scand.
93, 317–326.
Turecki, G., Rouleau, G.A., Mari, J., Joober, R., Morgan, K., 1997. Lack of
association between bipolar disorder and tyrosine hydroxylase: a meta-
analysis. Am. J. Med. Genet. 74, 348–352.
Turner, W.J., King, S., 1981. Two genetically distinct forms of bipolar
affective disorder? Biol. Psychiatry 16, 417–439.
Turner, W.J., King, S., 1983. BPD2. An autosomal dominant form of bi-
polar affective disorder. Biol. Psychiatry 18, 63–88.
Verheyen, G.R., Del-Favero, J., Mendlewicz, J., Lindblad, K., Van Zand,
K., Aalbregtse, M., Schalling, M., Souery, D., Van Broeckhoven, C.,
1999. Molecular interpretation of expanded RED products in bipolar
disorder by CAG/CTG repeats located at chromosomes 17q and 18q.
Neurobiol. Dis. 6, 424–432.
Vincent, J.B., Klempan, T., Parikh, S.S., Sasaki, T., Meltzer, H.Y., Sirugo,
G., Cola, P., Petronis, A., Kennedy, J.L., 1996. Frequency analysis of
large CAG/CTG trinucleotide repeats in schizophrenia and bipolar af-
fective disorder. Mol. Psychiatry 1, 141–148.
Vincent, J.B., Paterson, A.D., Strong, E., Petronis, A., Kennedy, J.L., 2000.
The unstable trinucleotide repeat story of major psychosis. Am. J. Med.
Genet. 97, 77–97.
Walsh, C., Hicks, A., Sham, P., 1992. GABA receptor subunits genes as
candidate genes for bipolar affective disorder: an association analysis.
Psychiatr. Genet. 2, 239–247.
Weitkamp, L.R., Stancer, H.C., Persad, E., Flood, C., Guttormsen, S., 1981.
Depressive disorders and HLA: a gene on chromosome 6 that can affect
behavior. N. Engl. J. Med. 305, 1301–1306.
Zander, C., Schurhoff, F., Laurent, C., 1998. CAG repeat sequences in
bipolar affective disorder: no evidence for association in a French pop-
ulation. Am. J. Med. Genet. 81, 338–341.
Related Documents











