DNA Repair 10 (2011) 556–566 Contents lists available at ScienceDirect DNA Repair j ourna l ho me pag e: www.elsevier.com/locate/dnarepair Minisatellite alterations in ZRT1 mutants occur via RAD52-dependent and RAD52-independent mechanisms in quiescent stationary phase yeast cells Maire K. Kelly 1 , Bonnie Alver 1 , David T. Kirkpatrick ∗ Department of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN 55455, United States a r t i c l e i n f o Article history: Received 4 May 2010 Received in revised form 21 February 2011 Accepted 4 March 2011 Available online 22 April 2011 Keywords: Minisatellites Recombination Genome stability Stationary phase a b s t r a c t Alterations in minisatellite DNA repeat tracts are associated with a variety of human diseases including Type 1 diabetes, progressive myoclonus epilepsy, and some types of cancer. However, in spite of their role in human health, the factors required for minisatellite alterations are not well understood. We pre- viously identified a stationary phase specific increase in minisatellite instability caused by mutations in the high affinity zinc transporter ZRT1, using a minisatellite inserted into the ADE2 locus in Saccharomyces cerevisiae. Here, we examined ZRT1-mediated minisatellite instability in yeast strains lacking key recom- bination genes to determine the mechanisms by which these alterations occur. Our analysis revealed that minisatellite alterations in a zrt1 mutant occur by a combination of RAD52-dependent and RAD52- independent mechanisms. In this study, plasmid-based experiments demonstrate that ZRT1-mediated minisatellite alterations occur independently of chromosomal context or adenine auxotrophy, and con- firmed the stationary phase timing of the events. To further examine the stationary phase specificity of ZRT1-mediated minisatellite alterations, we deleted ETR1 and POR1, genes that were previously shown to differentially affect the viability of quiescent or nonquiescent cells in stationary phase populations. These experiments revealed that minisatellite alterations in zrt1 mutants occur exclusively in quies- cent stationary phase cells. Finally, we show that loss of ZRT1 stimulates alterations in a derivative of the human HRAS1 minisatellite. We propose that the mechanism of ZRT1-mediated minisatellite instability during quiescence is relevant to human cells, and thus, human disease. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Minisatellite DNA repeat tracts are found throughout eukary- otic genomes [1,2]. They consist of repeat units ranging from 16 to 100 bp in length. The highly polymorphic nature of minisatellites makes them ideal for use as genetic markers, but they also have important biological functions. Minisatellites have been shown to act as enhancer elements for adjacent genes [2,3]. The human min- isatellite associated with the HRAS1 oncogene has been shown to bind the rel/NF-B transcription factor and can enhance expres- sion of a reporter gene [4,5]. Minisatellites also frequently act as fragile sites, possibly due to stalling of DNA polymerases at non-B DNA structures formed by the repeats (reviewed in [1,6]). It has also been suggested that minisatellites may have a role in chromosome pairing, specifically during male meiosis [2]. ∗ Corresponding author at: Department of Genetics, Cell Biology and Develop- ment, University of Minnesota, 6-160 Jackson Hall, 321 Church St. SE, Minneapolis, MN 55455, United States. Tel.: +1 612 624 9244; fax: +1 612 625 5754. E-mail addresses: [email protected], [email protected] (D.T. Kirkpatrick). 1 Co-first authors. In addition to their important biological functions, these repet- itive sequence elements also have a profound influence on human health. Rare altered alleles of human minisatellites have been corre- lated with increased risk of Type 1 diabetes, progressive myoclonus epilepsy, and various cancer subtypes [3,7–9]. Recently minisatel- lite alleles have also been associated with disorders as varied as asthma [10], ulcerative colitis [11], and attention-deficit hyperac- tivity disorder [12,13]. Little is known about how minisatellites alter to give rise to disease-associated minisatellite alleles. Human minisatellites have been shown to change in tract length and repeat composition dur- ing meiosis, while remaining relatively stable during mitotic cell cycles [1]. We previously demonstrated that these phenotypes are recapitulated in the budding yeast Saccharomyces cerevisiae [14,15]. Since the patterns of minisatellite alteration are similar in yeast and human cells, we used the more genetically tractable yeast to iden- tify factors that control minisatellite stability during meiosis. Our work demonstrated that meiotic minisatellite alterations require the meiosis-specific endonuclease Spo11p and the DNA loop repair protein Rad1p, while others have shown the recombination protein Rad50p is also critical [14,16]. Additional studies have demon- strated that mutation of the key replication genes RAD27, DNA2, POL3, or the PCNA complex can destabilize minisatellite tracts dur- 1568-7864/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.dnarep.2011.03.002

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MR
MD
a
ARRAA
KMRGS
1
o1miaibsfDbp
mM
(
1d
DNA Repair 10 (2011) 556– 566
Contents lists available at ScienceDirect
DNA Repair
j ourna l ho me pag e: www.elsev ier .com/ locate /dnarepai r
inisatellite alterations in ZRT1 mutants occur via RAD52-dependent andAD52-independent mechanisms in quiescent stationary phase yeast cells
aire K. Kelly1, Bonnie Alver1, David T. Kirkpatrick ∗
epartment of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN 55455, United States
r t i c l e i n f o
rticle history:eceived 4 May 2010eceived in revised form 21 February 2011ccepted 4 March 2011vailable online 22 April 2011
eywords:inisatellites
ecombinationenome stabilitytationary phase
a b s t r a c t
Alterations in minisatellite DNA repeat tracts are associated with a variety of human diseases includingType 1 diabetes, progressive myoclonus epilepsy, and some types of cancer. However, in spite of theirrole in human health, the factors required for minisatellite alterations are not well understood. We pre-viously identified a stationary phase specific increase in minisatellite instability caused by mutations inthe high affinity zinc transporter ZRT1, using a minisatellite inserted into the ADE2 locus in Saccharomycescerevisiae. Here, we examined ZRT1-mediated minisatellite instability in yeast strains lacking key recom-bination genes to determine the mechanisms by which these alterations occur. Our analysis revealedthat minisatellite alterations in a �zrt1 mutant occur by a combination of RAD52-dependent and RAD52-independent mechanisms. In this study, plasmid-based experiments demonstrate that ZRT1-mediatedminisatellite alterations occur independently of chromosomal context or adenine auxotrophy, and con-firmed the stationary phase timing of the events. To further examine the stationary phase specificity of
ZRT1-mediated minisatellite alterations, we deleted ETR1 and POR1, genes that were previously shownto differentially affect the viability of quiescent or nonquiescent cells in stationary phase populations.These experiments revealed that minisatellite alterations in �zrt1 mutants occur exclusively in quies-cent stationary phase cells. Finally, we show that loss of ZRT1 stimulates alterations in a derivative of thehuman HRAS1 minisatellite. We propose that the mechanism of ZRT1-mediated minisatellite instabilityduring quiescence is relevant to human cells, and thus, human disease.. Introduction
Minisatellite DNA repeat tracts are found throughout eukary-tic genomes [1,2]. They consist of repeat units ranging from 16 to00 bp in length. The highly polymorphic nature of minisatellitesakes them ideal for use as genetic markers, but they also have
mportant biological functions. Minisatellites have been shown toct as enhancer elements for adjacent genes [2,3]. The human min-satellite associated with the HRAS1 oncogene has been shown toind the rel/NF-�B transcription factor and can enhance expres-ion of a reporter gene [4,5]. Minisatellites also frequently act asragile sites, possibly due to stalling of DNA polymerases at non-B
NA structures formed by the repeats (reviewed in [1,6]). It has alsoeen suggested that minisatellites may have a role in chromosomeairing, specifically during male meiosis [2].∗ Corresponding author at: Department of Genetics, Cell Biology and Develop-ent, University of Minnesota, 6-160 Jackson Hall, 321 Church St. SE, Minneapolis,N 55455, United States. Tel.: +1 612 624 9244; fax: +1 612 625 5754.
E-mail addresses: [email protected], [email protected]. Kirkpatrick).
1 Co-first authors.
568-7864/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.dnarep.2011.03.002
© 2011 Elsevier B.V. All rights reserved.
In addition to their important biological functions, these repet-itive sequence elements also have a profound influence on humanhealth. Rare altered alleles of human minisatellites have been corre-lated with increased risk of Type 1 diabetes, progressive myoclonusepilepsy, and various cancer subtypes [3,7–9]. Recently minisatel-lite alleles have also been associated with disorders as varied asasthma [10], ulcerative colitis [11], and attention-deficit hyperac-tivity disorder [12,13].
Little is known about how minisatellites alter to give rise todisease-associated minisatellite alleles. Human minisatellites havebeen shown to change in tract length and repeat composition dur-ing meiosis, while remaining relatively stable during mitotic cellcycles [1]. We previously demonstrated that these phenotypes arerecapitulated in the budding yeast Saccharomyces cerevisiae [14,15].Since the patterns of minisatellite alteration are similar in yeast andhuman cells, we used the more genetically tractable yeast to iden-tify factors that control minisatellite stability during meiosis. Ourwork demonstrated that meiotic minisatellite alterations requirethe meiosis-specific endonuclease Spo11p and the DNA loop repair
protein Rad1p, while others have shown the recombination proteinRad50p is also critical [14,16]. Additional studies have demon-strated that mutation of the key replication genes RAD27, DNA2,POL3, or the PCNA complex can destabilize minisatellite tracts dur-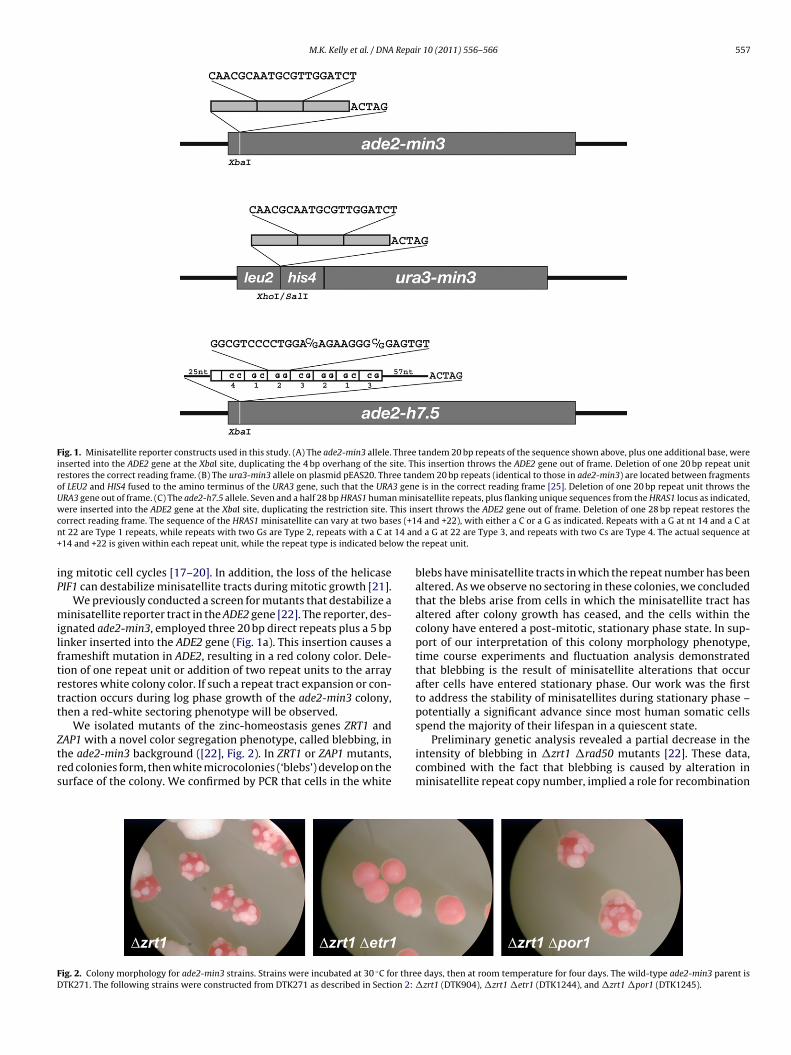
M.K. Kelly et al. / DNA Repair 10 (2011) 556– 566 557
Fig. 1. Minisatellite reporter constructs used in this study. (A) The ade2-min3 allele. Three tandem 20 bp repeats of the sequence shown above, plus one additional base, wereinserted into the ADE2 gene at the XbaI site, duplicating the 4 bp overhang of the site. This insertion throws the ADE2 gene out of frame. Deletion of one 20 bp repeat unitrestores the correct reading frame. (B) The ura3-min3 allele on plasmid pEAS20. Three tandem 20 bp repeats (identical to those in ade2-min3) are located between fragmentsof LEU2 and HIS4 fused to the amino terminus of the URA3 gene, such that the URA3 gene is in the correct reading frame [25]. Deletion of one 20 bp repeat unit throws theURA3 gene out of frame. (C) The ade2-h7.5 allele. Seven and a half 28 bp HRAS1 human minisatellite repeats, plus flanking unique sequences from the HRAS1 locus as indicated,were inserted into the ADE2 gene at the XbaI site, duplicating the restriction site. This insert throws the ADE2 gene out of frame. Deletion of one 28 bp repeat restores thec es (+1n 14 an+ ow the
iP
milftrtt
Ztrs
FD
orrect reading frame. The sequence of the HRAS1 minisatellite can vary at two bast 22 are Type 1 repeats, while repeats with two Gs are Type 2, repeats with a C at14 and +22 is given within each repeat unit, while the repeat type is indicated bel
ng mitotic cell cycles [17–20]. In addition, the loss of the helicaseIF1 can destabilize minisatellite tracts during mitotic growth [21].
We previously conducted a screen for mutants that destabilize ainisatellite reporter tract in the ADE2 gene [22]. The reporter, des-
gnated ade2-min3, employed three 20 bp direct repeats plus a 5 bpinker inserted into the ADE2 gene (Fig. 1a). This insertion causes arameshift mutation in ADE2, resulting in a red colony color. Dele-ion of one repeat unit or addition of two repeat units to the arrayestores white colony color. If such a repeat tract expansion or con-raction occurs during log phase growth of the ade2-min3 colony,hen a red-white sectoring phenotype will be observed.
We isolated mutants of the zinc-homeostasis genes ZRT1 andAP1 with a novel color segregation phenotype, called blebbing, in
he ade2-min3 background ([22], Fig. 2). In ZRT1 or ZAP1 mutants,ed colonies form, then white microcolonies (‘blebs’) develop on theurface of the colony. We confirmed by PCR that cells in the whiteig. 2. Colony morphology for ade2-min3 strains. Strains were incubated at 30 ◦C for threTK271. The following strains were constructed from DTK271 as described in Section 2:
4 and +22), with either a C or a G as indicated. Repeats with a G at nt 14 and a C atd a G at 22 are Type 3, and repeats with two Cs are Type 4. The actual sequence at
repeat unit.
blebs have minisatellite tracts in which the repeat number has beenaltered. As we observe no sectoring in these colonies, we concludedthat the blebs arise from cells in which the minisatellite tract hasaltered after colony growth has ceased, and the cells within thecolony have entered a post-mitotic, stationary phase state. In sup-port of our interpretation of this colony morphology phenotype,time course experiments and fluctuation analysis demonstratedthat blebbing is the result of minisatellite alterations that occurafter cells have entered stationary phase. Our work was the firstto address the stability of minisatellites during stationary phase –potentially a significant advance since most human somatic cellsspend the majority of their lifespan in a quiescent state.
Preliminary genetic analysis revealed a partial decrease in the
intensity of blebbing in �zrt1 �rad50 mutants [22]. These data,combined with the fact that blebbing is caused by alteration inminisatellite repeat copy number, implied a role for recombinatione days, then at room temperature for four days. The wild-type ade2-min3 parent is�zrt1 (DTK904), �zrt1 �etr1 (DTK1244), and �zrt1 �por1 (DTK1245).

5 Repai
imrRto
pmammaapduoaot
2
2
wfd
2tlawAwsmros(AGGTAGG
fwSfwt
DTflDfSs
58 M.K. Kelly et al. / DNA
n the minisatellite alterations of ZRT1 mutants. In S. cerevisiae, theajority of mitotic recombination requires RAD52 [23], but some
are recombination events are RAD52-independent – these requireAD50 or RAD51. It was clear that the mechanism responsible forhese events would be of significant interest given the limited datan DNA repair in quiescent cells.
In this study, we examine the requirements for stationaryhase minisatellite tract alterations in the �zrt1 zinc homeostasisutant. We show that stationary phase minisatellite alter-
tions occur by both RAD52-dependent and RAD52-independentechanisms. In addition, we demonstrate that ZRT1-dependentinisatellite alterations likely have broad genomic relevance,
s they occur independently of chromosomal context, adenineuxotrophy, or marker assay gene. We confirm the stationaryhase timing of minisatellite alterations in the ZRT1 mutant andemonstrate that these events are limited to the quiescent pop-lation of stationary phase cells. Finally, we establish that lossf ZRT1 can destabilize a tract derived from the human HRAS1-ssociated minisatellite. Our findings indicate that the mechanismf ZRT1-dependent minisatellite alteration is likely to be relevanto minisatellites found in a broad range of biological contexts.
. Materials and methods
.1. Media, plasmids and strains
Media was made as previously described [24]. YPD + G418 mediaas composed of standard YPD solid media + 200 mg/l of G418 sul-
ate (geneticin). Sporulation and dissection protocols were used asescribed [14].
Plasmid pKK055 was used to integrate the ade2-hras7.5 allele’s8 bp minisatellite repeats into the ADE2 gene (Fig. 1). To constructhis plasmid, a PCR product containing the HRAS1 minisatel-ite repeats from DTK759a [15] and XbaI-cleavable ends wasmplified using primers 35138391 and 42180832. This cassetteas digested with XbaI and ligated into the XbaI site of theDE2 fragment contained in the plasmid pEAS8 [25]. Insertionas verified by sequencing. The insert includes 28 bp of unique
equence from the human genome on the 5′ end, one 14 bp HRAS1inisatellite repeat fragment, seven 28 bp HRAS1 minisatellite
epeats, and 57 bp of unique sequence from the human genomen the 3′ end. With the 6 bp duplication of the XbaI restrictionite, the insert is 301 bp in length. The sequence of the insert5′ to 3′ and excluding XbaI sites) is: 5′ GAAGCTGGTCACTCGG-GGCTGCTGGGGAGAAGGGGGAGTGTGGCATTCCCTGGACAGAAGG-CAAGTGTGGCGTCCCCTGGAGAGAAGGGCGAGTGTGGCGTCCCCT-GAGAGAAGGGGGAGTGTGGCGTCCCCTGGACAGAAGGGGGAGTG-GGCGTCCCCTGGAGAGAAGGGGGAGTGTGGCGTCCCCTGGAGAGA-GGGCGAGTGTGGCGTCCCCTGGACAGAAGGGGGAGTGTCCCTGCA-CCCTGGCCAGCCAGCGGCATGGCCTACTAGCTCTCCCCCAACCTCA-GG 3′.
S. cerevisiae strains used in this study (Table 1) were derivedrom EAS28 (MATa his7-2 trp1-289 ura3-52) from [25]. Those strainshose construction is not mentioned here are described in [22].
train DTK1188, containing the ade2-h7.5 allele, was made by trans-orming DTK260 [22] with BglII digested pKK055. Ura− derivativesere then selected on 5-fluoroorotic acid (5-FOA) plates. Integra-
ion of the ade2-h7.5 allele was verified by sequencing.The following strains were constructed by amplifying genomic
NA as template from the Yeast Deletion Consortium strains.he PCR products are composed of the G418 resistance geneanked by 3′ and 5′ homology to the target sequence. DTK944 and
TK945, bearing deletions of RAD1, were constructed by ampli-ying a �rad1::KAN cassette using oligos 6934207 and 6934208.trains DTK1074 and DTK1076, bearing deletions of RAD51, andtrains DTK1309 and DTK1316, bearing deletions of DNL4, were
r 10 (2011) 556– 566
constructed in a similar manner using oligos 26876476 and26876477 for �rad51::KAN and oligos 39724850 and 39724851 for�dnl4::KAN. Strains DTK1244 and DTK1360, bearing a deletion ofETR1, were constructed by amplifying an �etr1::KAN cassette usingoligos 37616572 and 37616573. DTK1245 and DTK1361, bearing adeletion of POR1, were constructed using �por1::KAN cassette DNAwith oligos 37616574 and 37616575. Strain DTK1666, bearing adeletion of DNL4, was constructed using genomic DNA from strainDTK1316 using oligos 39724850 and 39724851 for �dnl4::KAN.DTK1690, bearing a deletion of RAD52, was constructed usinggenomic DNA amplified from DTK1253 using oligos 34756508 and34756507 for �rad52::URA3. Strain DTK1200, ade2-h7.5 with adeletion of ZRT1, was also constructed using a PCR construct gen-erated using oligos 12966370 and 12966371 for �zrt1::KAN withgenomic DNA from strain DTK878 as a template. Parental strainswere transformed with PCR product, grown for 4 h in liquid YPDand then plated on YPD + G418 or SD-ura solid media to select forintegration events. Transformants were checked by PCR.
To construct the RAD52 null mutant DTK1191, the�rad52::URA3 strain DNY101 [26] was crossed to DTK904,sporulated, and dissected as previously described [14]. A sporebearing the ade2-min3 allele, the �zrt1::LEU2 allele, and the�rad52::URA3 allele was isolated by color and survival on SD-leu,SD-ura, and YPD + G418 sulfate media. This isolate was backcrossedtwice to DTK904 and dissected as above to generate DTK1191. TheRAD59 null strain DTK1229 was constructed by crossing DTK1195,a haploid spore isolate from sporulation and dissection of the�rad59::KAN Yeast Deletion Consortium strain, to DTK904 andsporulating and dissecting. A spore bearing the ade2-min3 allele,the �zrt1::LEU2 allele, and the �rad59::KAN allele was isolatedby red color, survival on SD-leu, and G418 sulfate resistance.This isolate was backcrossed twice to DTK904 and dissected togenerate DTK1229. DTK1230 was constructed in a similar fashion,by crossing DTK1196, a haploid spore isolate of the �rad59::KANYeast Deletion Consortium strain, to DTK271 and sporulating anddissecting as above. A spore bearing ade2-min3 and �rad59::KANwas isolated by color and G418 sulfate resistance, then backcrossedtwice to DTK271. The following strains were constructed by cross-ing, sporulating, and dissecting: DTK1253 (DTK1191 × DTK284),DTK1254 (DTK1057 × DTK1076), DTK1268 (DTK1253 × DTK1057),DTK1269 (DTK1191 × DTK1076), DTK1289 (DTK1268 × DTK284),DTK1290 (DTK1269 × DTK284), DTK1299 (DTK1254 × DTK284),DTK1320 (DTK1191 × DTK1299), DTK1323 (DTK1191 × DTK1299),DTK1400 (DTK1188 × DTK1056), DTK1401 (DTK1188 × DTK1074),DTK1403 (DTK1200 × DTK1191), DTK1406 (DTK1200 × DTK1056),DTK1407 (DTK1400 × DTK1191), DTK1426 (DTK1400 × DTK1191),DTK1427 (DTK1426 × DTK1188), DTK1428 (DTK1401 × DTK1200),DTK1438 (DTK1401 × DTK1407), DTK1677 (DTK1316 × 1229) andDTK1678 (DTK1316 × DTK1229).
2.2. PCR primers
The following primers were used in this study:
Primer 6934207 (Rad1F): TCTGTTTGCCTTTATTTTGCPrimer 6934208 (Rad1R): GAAGATGAATTGCGGATGPrimer 12966370 (Zrt1F): TACGCACGGCATTAGCTCPrimer 12966371 (Zrt1R): ACTCGTAGATGGCACGGTCPrimer 26876476 (Rad51F): GTGTAGCGACAAAGAGCAGACGTAPrimer 26876477 (Rad51R): GCAGTAGGGTTGCGAGGTATATGAPrimer 34756508 (Rad52F): AAGAGTCTGCTCTTCCCGTTAGPrimer 34756507 (Rad52R): ACGACACATGGAGGAAAGAA
Primer 37616572 (Etr1R): TTGAAGGGTCGACGTCCCCTTTTAPrimer 37616573 (Etr1F): TGTACCCAGGGGTGGTTTCCATPrimer 37616574 (Por1R): TTCTCACTGCCAAGCAACCAPrimer 37616575 (Por1F): CCAATCAAACACCGCCATTTCG
M.K. Kelly et al. / DNA Repair 10 (2011) 556– 566 559
Table 1Yeast strains.
Strain Relevant genotype Construction details
EAS28 Wild-type MATa his7-2 trp1-289 ura3-52 [25]DNY101 rad52::URA3 [23]DTK260 leu2::HisG EAS28 with pNKY85 [22]DTK264 ade2-min3 DTK260 with pDTK123 [22]DTK271 ade2-min3, MAT� DTK264 mating-type switch [22]DTK284 ade2-min3, arg8::HisG DTK264 with pDS27 [22]DTK878 ade2-min3, zrt1::KAN DTK271 with zrt1::KAN [22]DTK904 ade2-min3, zrt1::LEU2 DTK284 with zrt1::LEU2 [22]DTK944 ade2-min3, rad1::KAN DTK271 with rad1::KANa
DTK945 ade2-min3, zrt1::LEU2, rad1::KAN DTK904 with rad1::KANa
DTK1056 ade2-min3, rad50::KAN DTK271 with rad50::KAN [22]DTK1057 ade2-min3, zrt1::LEU2, rad50::KAN DTK904 with rad50::KAN [22]DTK1074 ade2-min3, rad51::KAN DTK271 with rad51::KANa
DTK1076 ade2-min3, zrt1::LEU2, rad51::KAN DTK904 with rad51::KANa
DTK1188 ade2-h7.5 DTK260 with pKK055, FOAR isolateDTK1191 ade2-min3, zrt1::LEU2, rad52::URA3 DNY101 × DTK904, isolated sporeDTK1195 rad59::KAN Spore isolated from Yeast Deletion Consortium strain dissectionDTK1196 rad59::KAN Spore isolated from Yeast Deletion Consortium strain dissectionDTK1200 ade2-h7.5, zrt1::KAN DTK1188 with zrt1::KANa
DTK1229 ade2-min3, zrt1::LEU2, rad59::KAN DTK904 × DTK1195, isolated sporeDTK1230 ade2-min3, rad59::KAN DTK271 × DTK1196, isolated sporeDTK1244 ade2-min3, zrt1::LEU2, �etr1::KAN DTK904 with etr1::KANa
DTK1245 ade2-min3, zrt1::LEU2, �por1::KAN DTK904 with por1::KANa
DTK1253 ade2-min3, rad52::URA3 DTK1191 × DTK284, isolated sporeDTK1254 ade2-min3, zrt1::LEU2, rad50::KAN, rad51::KAN DTK1057 × DTK1076, isolated sporeDTK1268 ade2-min3, zrt1::LEU2, rad50::KAN, rad52::URA3 DTK1253 × DTK1057, isolated sporeDTK1269 ade2-min3, zrt1::LEU2, rad51::KAN, rad52::URA3 DTK1191 × DTK1076, isolated sporeDTK1289 ade2-min3, rad50::KAN, rad52::URA3 DTK1268 × DTK284, isolated sporeDTK1290 ade2-min3, rad51::KAN, rad52::URA3 DTK1269 × DTK284, isolated sporeDTK1299 ade2-min3, rad50::KAN, rad51::KAN DTK1254 × DTK284, isolated sporeDTK1309 ade2-min3, zrt1::LEU2, dnl4::KAN DTK904 with dnl4::KANa
DTK1316 ade2-min3, dnl4::KAN DTK271 with dnl4::KANa
DTK1320 ade2-min3, rad50::KAN, rad51::KAN, rad52::URA3 DTK1191 × DTK1299, isolated sporeDTK1323 ade2-min3, zrt1::LEU2, rad50::KAN, rad51::KAN, rad52::URA3 DTK1191 × DTK1299, isolated sporeDTK1360 ade2-min3, �etr1::KAN DTK271 with etr1::KANa
DTK1361 ade2-min3, �por1::KAN DTK271 with por1::KANa
DTK1400 ade2-h7.5, �rad50::KAN DTK1188 × DTK1056, isolated sporeDTK1401 ade2-h7.5, �rad51::KAN DTK1188 × DTK1074, isolated sporeDTK1403 ade2-h7.5, �zrt1::LEU2, �rad52::URA3 DTK1200 × DTK1191, isolated sporeDTK1406 ade2-h7.5, �zrt1::KAN, �rad50::KAN DTK1200 × DTK1056, isolated sporeDTK1407 ade2-h7.5, �zrt1::LEU2, �rad50::KAN, �rad52::URA3 DTK1400 × DTK1191, isolated sporeDTK1426 ade2-h7.5, �rad50::KAN, �rad52::URA3 DTK1400 × DTK1191, isolated sporeDTK1427 ade2-h7.5, �rad52::URA3 DTK1426 × DTK1188, isolated sporeDTK1428 ade2-h7.5, �zrt1::KAN, �rad51::KAN DTK1401 × DTK1200, isolated sporeDTK1438 ade2-h7.5, �zrt1::LEU2, �rad50::KAN, �rad51::KAN, �rad52::URA3 DTK1401 × DTK1407, isolated sporeDTK1612 ade2-min3 with pEAS20 (ura3-min3) DTK271 transformed with pEAS20DTK1613 ade2-min3, zrt1::LEU2 with pEAS20 (ura3-min3) DTK904 transformed with pEAS20DTK1666 ade2-min3, zrt1::LEU2, rad52::URA3, dnl4::KAN DTK1191 with dnl4::KANa
DTK1677 ade2-min3, zrt1::LEU2, rad59::KAN, dnl4::KAN DTK1316 × DTK1229, isolated sporeDTK1678 ade2-min3, dnl4::KAN, rad59::KAN DTK1316 × DTK1229, isolated spore
2
fmatifpu
DTK1690 ade2-min3, dnl4::KAN, rad52::URA3
a Indicates that the strain was made using a PCR-generated construct.
Primer 39724850 (Dnl4F): GAGTTAAGATCGTTTTCGATCCCTPrimer 39724851 (Dnl4R): GCGCATCTTCCACTCTTATTGPrimer 35138391 (Hras1 minisatellite F): AAATCTAGACCCTGAG-GTTGGGGGAGAGCPrimer 42180832 (Hras1 minisatellite R): AAATCTAGAGAAGCTG-GTCACTCGGAGGCTG
.3. Fluctuation analysis
Fluctuation analysis was performed as described [27], with theollowing modifications: plasmid pEAS20 [25], containing the ura3-in3 construct (Fig. 1), was transformed into DTK271 and DTK904
nd plated on SD-ura to select for plasmid uptake. Ten Ade+ anden Ade− transformants for each strain were picked and used to
noculate liquid cultures. SD-trp media, rather than YPD, was usedor growing liquid cultures in order to maintain selection for thelasmid, without selecting for a particular conformation of thera3-min3 repeats. SD-trp liquid cultures of strains bearing pEAS20DTK1316 with rad52::URA3a
were grown at 30 ◦C. Cultures were diluted and plated on YPD andSD-trp 5-FOA plates at 0, 4, 8, and 24 h after inoculation. The OD600of the cultures was measured at each timepoint to monitor culturegrowth. Three independent trials were conducted for each strainand the average frequency of minisatellite alteration and stan-dard error for each strain was calculated. In contrast to previousapplications of this technique, we did not extend our calculationsto determine rate of minisatellite alteration over generations; asdescribed in Section 3, we determined that minisatellite alterationsoccur specifically in quiescent cells in stationary phase, thus makingit impossible to calculate a rate measurement.
2.4. Bleb quantitation
To quantify the amount of blebbing in our strains we countedthe number of blebs on the surface of each colony after 7 days (30 ◦Cfor 3 days and then room temperature for 4 days). For each strainwe surveyed at least 100 colonies over 3 independent experiments,

5 Repai
cdwtcdfcwttocoiti
3
3o
bicrfc
Zcmmdl[a
TB
60 M.K. Kelly et al. / DNA
alculating the mean number of blebs per colony and the 95% confi-ence interval for the mean. The size of the 95% confidence intervalsas examined to determine the repeatability of the assay. In order
o ensure accurate comparisons, we measured the diameter of eacholony and calculated the mean colony diameter and the 95% confi-ence interval for the mean for each strain. Mean colony diametersor all strains in this analysis ranged from 1.24 to 1.29 mm and 95%onfidence intervals for all strains overlapped, indicating that thereas not a significant difference in colony size between strains. The
imepoint at which we counted blebs was chosen because at thatime blebs are readily detected but still small enough to precludeverlap between blebs, which could potentially result in an under-ount. We note that the extremely small 95% confidence intervalsbserved in our measurements demonstrate that our bleb countsn this assay are highly reproducible and are therefore likely to be arue indication of the alteration frequency of the minisatellite tractn the strain.
. Results
.1. Minisatellite alterations in stationary phase �zrt1 mutantsccur in quiescent cells
The ade2-min3 allele allows us to monitor minisatellite sta-ility in S. cerevisiae (Fig. 1a, [22]). Three minisatellite repeats
nserted into the ADE2 gene disrupt the reading frame; cellsarrying this allele are Ade− and form red colonies. Loss of aepeat unit from the minisatellite tract restores the correct readingrame; cells carrying these altered alleles are Ade+ and form whiteolonies.
Using the ade2-min3 reporter, we previously demonstrated thatRT1 mutants exhibit a novel color segregation phenotype that wealled blebbing (Fig. 2). The blebbing phenotype consists of whiteicrocolonies growing on the surface of the red colony. Colonyorphology examination, PCR analysis, and timing experiments
etermined that blebbing in an ade2-min3 strain results from theoss of one minisatellite repeat specifically during stationary phase22]. Previous studies have shown that stationary phase culturesre composed of quiescent cells that have exited the cell cycle and
able 2lebbing frequency of ade2-min3 and ade2-h7.5 strains.
Straina Genotype
ade2-min3 strainsDTK271/DTK904 ParentalDTK1360/DTK1244 �etr1
DTK1361/DTK1245 �por1
DTK1056/DTK1057 �rad50
DTK1074/DTK1076 �rad51
DTK1253/DTK1191 �rad52
DTK1289/DTK1268 �rad50 �rad52
DTK1290/DTK1269 �rad51 �rad52
DTK1299/DTK1254 �rad50 �rad51
DTK1320/DTK1323 �rad50 �rad51 �rad52
DTK1230/DTK1229 �rad59
DTK944/DTK945 �rad1
DTK1316/DTK1309 �dnl4
DTK1678/DTK1677 �dnl4�rad59
DTK1690/DTK1666 �dnl4�rad52
ade2-h7.5 strainsDTK1188/DTK1200 Parental
DTK1400/DTK1406 �rad50
DTK1401/DTK1428 �rad51
DTK1427/DTK1403 �rad52
DTK1426/DTK1407 �rad50 �rad52
NA/DTK1438 �rad50 �rad51 �rad52
a WT strain number/�zrt1 strain number.
r 10 (2011) 556– 566
nonquiescent cells which may still be undergoing some cell division[28]. These studies also demonstrated that loss of ETR1 specifi-cally reduces the capacity of the quiescent population of stationaryphase cells to reenter the cell cycle when plated on rich media.The loss of POR1 differentially affects the reproductive capacityof nonquiescent cells in a similar manner. ETR1 encodes a 2-enoyl thioester reductase, which is involved in fatty acid synthesis[29], while POR1 is a mitochondrial porin, necessary for mainte-nance of mitochondrial osmotic stability and aerobic respiration[30].
These distinct genetic requirements for cell cycle reentry in qui-escent and nonquiescent stationary phase cells provide a means todistinguish which cell population is responsible for ZRT1-mediatedminisatellite alteration. If the reproductive capacity of the cell frac-tion in which the minisatellite alterations are occurring is reduced,then the cells will be unable to return to growth after minisatellitetract changes restore adenine biosynthesis, preventing blebs fromforming. In order to determine whether minisatellite alterationsin a �zrt1 mutant occur in quiescent or nonquiescent stationaryphase cells, we deleted ETR1 or POR1 in a �zrt1 strain background.
The parental �zrt1 strain has 20.6 blebs/colony (Table 2). Theextremely small 95% confidence interval for this strain (20.6 ± 0.8),as well as all other strains examined, demonstrated that our blebcounts for this assay are highly repeatable. Loss of ETR1 in a �zrt1strain reduced blebbing to 0.09 blebs/colony (Table 2, Fig. 2). Thisindicates that most, if not all, minisatellite alterations in a �zrt1mutant occur in quiescent cells. In contrast, loss of POR1 had littleeffect on blebbing, with 17.9 blebs/colony in a �zrt1 �por1 strain,indicating that minisatellite alterations in a �zrt1 mutant likely donot occur in nonquiescent cells.
We also examined the effect of ETR1 or POR1 deletion onminisatellite alteration in wild-type ZRT1 cells. A wild-typeade2-min3 strain has 3.7 blebs/colony. Loss of ETR1 completelysuppressed bleb formation in this strain background, reduc-ing it to 0.06 blebs/colony (Table 2). Loss of POR1 reduced
blebbing to 1.8 blebs/colony. Therefore, while it is likely that back-ground minisatellite alterations occur primarily in quiescent cells,some events may be occurring in nonquiescent stationary phasecells.Mean blebs/colony ± 95% confidence interval
WT �zrt1
3.7 ± 0.4 20.6 ± 0.80.06 ± 0.05 0.09 ± 0.05
1.8 ± 0.06 17.9 ± 0.70.9 ± 0.2 9.0 ± 0.52.1 ± 0.3 10.0 ± 0.71.4 ± 0.2 9.4 ± 0.60.6 ± 0.2 3.9 ± 0.31.7 ± 0.2 9.8 ± 0.61.2 ± 0.3 9.1 ± 0.50.3 ± 0.1 0.5 ± 0.12.0 ± 0.3 16.0 ± 0.64.5 ± 0.5 17.2 ± 0.61.8 ± 0.3 18.4 ± 0.93.8 ± 0.4 18.9 ± 0.50.9 ± 0.2 2.4 ± 0.3
0.7 ± 0.2 4.8 ± 0.40.3 ± 0.08 1.1 ± 0.20.4 ± 0.1 1.8 ± 0.20.2 ± 0.09 2.5 ± 0.30.1 ± 0.06 0.9 ± 0.2
NA 0.3 ± 0.09

M.K. Kelly et al. / DNA Repair 10 (2011) 556– 566 561
Table 3Frequency of 5-FOA resistance (events/cells) ± standard error.
Time after inoculation (h) WT strain �zrt1 strain Fold difference WT vs. �zrt1
0 1.26 × 10−4 ± 1.19 × 10−4 1.25 × 10−2 ± 2.96 × 10−3 99×4 3.16 × 10−4 ± 2.39 × 10−4 2.71 × 10−2 ± 1.57 × 10−2 86×8 4.04 × 10−4 ± 2.25 × 10−4 4.05 × 10−2 ± 2.60 × 10−2 100×
−4 −5 −1 −1
3c
egtimiTiflc(
dflctopiffacmfdwsqfaeotp8pfcoDtimscA2Za
24 4.06 × 10 ± 6.38 × 10
.2. ZRT1-dependent minisatellite instability is independent ofhromosomal context and adenine auxotrophy
Minisatellite stability can be affected by cis-acting sequencelements [1]. In addition, loss of ADE2 function can be highly muta-enic [31,32]. To determine if local DNA context or the use ofhe ADE2 gene as the marker in our minisatellite stability assaynfluences the stability of the minisatellite tract, we used another
inisatellite assay system in which the min3 minisatellite tract isncorporated into the URA3 gene on a plasmid, pEAS20 [25]. ThisRP1 CEN plasmid contains three tandem repeats, identical to thosen the ade2-min3 allele, in-frame with the URA3 coding region,orming the ura3-min3 allele (Fig. 1b). Alterations in the minisatel-ite repeats will throw the URA3 gene out of frame, making cellsarrying the altered plasmid Ura− and resistant to 5-fluoorotic acid5-FOA).
We employed a modified fluctuation analysis assay [27] toetermine the stability of the minisatellite tract in pEAS20. In ouructuation assay, we calculated the frequency of 5-FOA resistantolony formation (and thus minisatellite alteration) rather thanhe rate over generations, as the events being measured do notccur until after cells have ceased dividing and entered stationaryhase. Cells bearing pEAS20 were grown in SD-trp liquid media
n order to maintain selection for the plasmid without selectionor a particular minisatellite conformation. We determined therequency of minisatellite alteration in cells at 0, 4, 8, and 24 hfter inoculation, while monitoring the growth of the cells in theultures to determine entry into stationary phase. An increase ininisatellite alteration, as indicated by a dramatic increase in the
requency of 5-FOA-resistant colonies in the �zrt1 strain, was notetected until the 24 h timepoint (Table 3), which correspondedith the onset of stationary phase (data not shown). Entry into
tationary phase and the increase in minisatellite alteration fre-uency in ura3-min3 �zrt1 cells was consistent with prior datarom ade2-min3 �zrt1 cells [22], although stationary phase onset,s determined by OD600 observation of the liquid cultures, wasarlier in the ura3-min3 experiments, presumably due to the usef synthetic drop-out media (required to maintain selection forhe ura3-min3 pEAS20 plasmid) rather than rich media. Wild-typeEAS20 cells maintained a level of minisatellite alteration at least6-fold lower than the corresponding �zrt1 cells at every timeoint, demonstrating that loss of ZRT1 significantly increases therequency of minisatellite alterations independent of chromosomalontext (Table 3). Control experiments indicated that the low levelf white colonies present at early timepoints in both the wild-typeTK271 and �zrt1 DTK904 strains were likely derived from cells
hat entered stationary phase in the colony that was chosen for thenitial liquid media inoculation, as their frequency could be dra-
atically reduced by using colonies that were grown for a muchhorter period of time prior to use. Finally, there was no signifi-ant difference in the frequency of minisatellite alteration between
de+ and Ade− �zrt1 transformants (2.87 × 10−1 events/cell vs..47 × 10−1 events/cell at 24 h after inoculation), indicating thatRT1-dependent minisatellite instability is also independent ofdenine auxotrophy.2.47 × 10 ± 1.50 × 10 608×
3.3. Multiple recombination pathways facilitate minisatellitealterations in �zrt1 mutants
Blebbing in the �zrt1 mutant is partially suppressed by lossof RAD50 [22], which encodes a recombination protein [33]. In S.cerevisiae, mitotic recombination can occur by the canonical RAD52-dependent pathway, or one of two RAD52-independent pathwaysrequiring either RAD50 or RAD51 [23]. To determine which recom-bination pathways are required for minisatellite alteration in the�zrt1 mutant, we constructed ade2-min3 �zrt1 strains contain-ing deletions for key recombination genes. We then quantified theaverage number of blebs per colony for each of our strains (Table 2).
We examined �zrt1 strains bearing deletions of RAD50, RAD51,and RAD52, along with double and triple mutant combinationsof these genes. The parental �zrt1 strain has 20.6 blebs/colony(Table 2). Minisatellite alterations in the �zrt1 �rad50 doublemutant are reduced over 50% to 9.0 blebs/colony. Similarly, min-isatellite alterations in the �zrt1 �rad51 and �zrt1 �rad52 doublemutants are reduced to 10.0 blebs/colony and 9.4 blebs/colony,respectively. Therefore, minisatellite alterations in a �zrt1 strainare partially dependent on RAD51 and RAD52, as well as RAD50.
To distinguish between the relative contributions of the RAD52-dependent and the RAD52-independent pathways, we examinedmutants with combinations of recombination gene deletionsin the �zrt1 background. The extent of blebbing in a �zrt1�rad50 �rad52 triple mutant was reduced to 3.9 blebs/colony(Table 2), lower than any of the double mutants, suggesting thatZRT1-dependent minisatellite alterations can occur by a RAD52-dependent mechanism or a RAD52-independent mechanism thatrequires RAD50. In contrast, a �zrt1 �rad51 �rad52 strain wasphenotypically identical to the �zrt1 �rad51 and �zrt1 �rad52parents (9.8 blebs/colony vs. 10.0 and 9.4 blebs/colony). Thisresult indicates that the RAD52-independent pathway that requiresRAD51 does not significantly contribute to minisatellite alter-ations in a �zrt1 mutant. A �zrt1 �rad50 �rad51 strain was alsophenotypically identical to the corresponding parent strains (9.1blebs/colony vs. 9.0 and 10.0 blebs/colony). These data indicatethat RAD51 does not contribute to ZRT1-dependent minisatellitealterations as long as wild-type RAD50 or RAD52 is present.
In order to explain the residual minisatellite alterations in the�zrt1 �rad50 �rad52 triple mutant, we examined a �zrt1 �rad50�rad51 �rad52 quadruple mutant. Blebbing in this strain wasreduced to 0.5 blebs per colony (Table 2). Thus, any remainingevents in a �zrt1 �rad50 �rad52 mutant are dependent on aRAD52-independent mechanism that requires RAD51, although thispathway does not contribute to minisatellite alterations in the pres-ence of wild-type RAD50 or RAD52.
A significant loss in cell viability could be one possible expla-nation for the observed decrease in blebbing in the recombinationmutants, rather than loss of recombination activity. To address thispossibility we examined cell viability, by comparing cell numbervia hemocytometer counts with viable colony forming units, in
colonies that were beginning to exhibit blebbing. We observed nosignificant difference in cell viability in wild type (DTK271), �zrt1(DTK904), �rad50 �rad51 �rad52 (DTK1320), and �zrt1 �rad50
5 Repair 10 (2011) 556– 566
�2omd
bstR1imtm
wrdgbsimwTaoa
iwpw1ttrj
3d
u(ofmhaplir
tattahai
m
Fig. 3. Analysis of tract alterations in the ade2-h7.5 minisatellite allele in �zrt1strains. The ade2-h7.5 allele was sequenced in 27 independent white bleb isolatesfrom DTK1200 (ade2-h7.5 �zrt1). The parental repeat structure, using the repeattype designations from Fig. 1, is given for reference. Added or modified repeats areindicated in grey, while the location of deleted repeats is depicted as a break in therepeat tract. Added repeats are indicated to the right of the repeats they duplicate
62 M.K. Kelly et al. / DNA
rad51 �rad52 (DTK1323) strains – all exhibited viability between5% and 32%. These data also indicate that it is unlikely that the lossf ZRT1 is causing a mutator phenotype, as significantly increasedutation in these haploid strains would likely lead to increased cell
eath.Single-strand annealing (SSA) is a RAD52-dependent recom-
ination pathway that can result in repeat deletions like thoseeen in ZRT1-dependent minisatellite alteration [34]. SSA is par-ially dependent on RAD59 and RAD1 [35,36]. When RAD59 orAD1 is deleted in a �zrt1 background, blebbing is reduced to6.0 blebs/colony and 17.2 blebs/colony, respectively. While this
s a smaller reduction than is seen in the �zrt1 �rad52 doubleutant (9.4 blebs/colony, Table 2), it indicates that at least some of
he RAD52-dependent minisatellite alterations in the �zrt1 mutantay occur by SSA.As RAD50 is also involved in non-homologous end joining, we
anted to determine if non-homologous end-joining (NHEJ) has aole in ZRT1-dependent minisatellite alterations. We introduced aeletion of DNL4, a critical NHEJ ligase [37], into the �zrt1 back-round. The �zrt1 �dnl4 double mutant had an average of 18.4lebs/colony, which was not substantially different from the �zrt1ingle mutant (Table 2). A �zrt1 �dnl4 �rad59 triple mutant exhib-ted a similar blebbing frequency. However, a �zrt1 �dnl4 �rad52
utant had a significantly reduced frequency (2.4 blebs/colony),hich is well below the �zrt1 �rad52 mutant (9.4 blebs/colony).
hus, while NHEJ does not play a major role in the minisatellitelterations occurring in �zrt1 strains, nor does it affect eventsccurring by SSA, it does influence some of the events remainingfter the RAD52-dependent pathway has been eliminated.
We also determined if recombination plays a role in thenfrequent minisatellite alterations occurring in a strain with a
ild-type ZRT1 gene. The parental ade2-min3 strain has 3.7 blebser colony (Table 2). Loss of RAD50, RAD51, RAD52 or DNL4 in aild-type background reduced minisatellite alterations to 0.9, 2.1,
.4, and 1.8 blebs/colony, respectively. These results indicate thathe small number of minisatellite tract alterations seen in the wild-ype ade2-min3 strain involve DNA double-strand break repair, butequire both homologous recombination and non-homologous endoining (NHEJ).
.4. A tract derived from the human HRAS1 minisatellite isestabilized in �zrt1 mutants
Both the ade2-min3 and the ura3-min3 minisatellite constructstilize three identical tandem repeats with roughly 50% GC contentFig. 1a and b). However, human minisatellites are often composedf variable repeats that are enriched for CG content [2], and there-ore may be altered by different mechanisms than our reporter
inisatellite constructs. To determine whether the stability of auman minisatellite is affected by loss of ZRT1, we constructed thede2-h7.5 allele, which is composed of one partial and seven com-lete 28 bp repeats of the human HRAS1 minisatellite plus HRAS1
ocus flanking DNA inserted into the ADE2 gene (Fig. 1c). This min-satellite has a CG content of 68%, and variable nucleotides in eachepeat at the 14th and 22nd positions.
The insertion of the HRAS1 minisatellite-derived DNA throwshe ADE2 gene out of frame, making the cells carrying this allele rednd Ade−. Alterations in the minisatellite tract can restore ADE2 tohe correct reading frame, making cells in which alterations haveaken place white and Ade+. Thus, colony color segregation servess a reporter for minisatellite alteration in strains with the ade2-7.5 allele, as with the ade2-min3 allele. The ade2-h7.5 minisatellite
llele is very stable, with a blebbing frequency of 0.7 blebs/colonyn a WT strain background (Table 2).To examine the effect of ZRT1 on the stability of the ade2-h7.5inisatellite repeats, we deleted ZRT1 in the ade2-h7.5 background.
but, as it is not possible to distinguish which repeats are original and which havebeen added, the designation is done in this manner only for consistency.
The ade2-h7.5 �zrt1 strain displayed a blebbing phenotype similarto the ade2-min3 �zrt1 strain, but the frequency of minisatellitealterations in the ade2-h7.5 �zrt1 strain (4.8 blebs/colony) waslower than the ade2-min3 �zrt1 strain (20.6 blebs/colony) (Table 2).However, the 7-fold increase seen in the �zrt1 ade2-h7.5 strain rel-ative to the wild-type parental ade2-h7.5 strain is comparable tothe 6-fold increase seen in ade2-min3 strains (Table 2). Thus, theZRT1-dependent pathway of minisatellite alteration in yeast mayalso be relevant to minisatellite stability in human cells. The lowerfrequency of minisatellite alterations in the ade2-h7.5 �zrt1 strainmay be due to the fact that the HRAS1 minisatellite repeats arevariable at two nucleotides in their sequence (Fig. 1c), which couldinfluence homologous recombination events occurring betweenrepeats. Prior work in our lab demonstrated that single nucleotideheterozygosities in HRAS1 minisatellite repeats act to stabilize theDNA tracts [15].
We examined the nature of the tract alterations that led to blebformation in the �zrt1 ade2-h7.5 strain. Whole-cell PCR was usedto examine the ade2-h7.5 allele in 104 independent white blebs.While almost all alleles (99/104 – 95%) were contractions of therepetitive tract, we did observe tract expansions in 5% (5/104) ofthe blebs examined. All expansion alleles gained two repeat units.Of the 99 tracts exhibiting loss, 91 had lost a single repeat while 8lost four repeats.
We sequenced the tracts in 27 of the 104 ade2-h7.5 alleles exam-ined by PCR. As shown in Fig. 3, both expansions sequenced addedrepeat units to the middle of the tract. The majority of single repeatlosses occurred at the fourth or fifth repeat. The first allele with afour-repeat contraction lost the second through fifth repeats, whilethe second four-repeat contraction allele lost DNA from the middleof the first repeat (between the variable 14th and 22nd nucleotides)
to the middle of the fifth repeat (between the variable nucleotides)to generate a hybrid Type 3 repeat.Finally, we asked if the ZRT1-dependent alterations in the ade2-h7.5 minisatellite, like alterations in the ade2-min3 minisatellite,

Repai
oaahorb�bbaTmRm
4
sZstacfqsRtii
cDioionscct�itsot
tetooppacesoaa
M.K. Kelly et al. / DNA
ccur via RAD52-dependent and -independent pathways. As withde2-min3 strains, we deleted key recombination factors in thede2-h7.5 parent and ade2-h7.5 �zrt1 mutant strains. The ade2-7.5 �zrt1 mutant displays an average of 4.8 blebs/colony. Lossf RAD50, RAD51, or RAD52 in the ade2-h7.5 �zrt1 backgroundeduced blebbing to 1.1 blebs/colony, 1.8 blebs/colony, and 2.5lebs/colony, respectively (Table 2). An ade2-h7.5 �zrt1 �rad50rad52 strain showed a further reduction in blebbing – to 0.9
lebs/colony. Finally, loss of RAD50, RAD51, and RAD52 reducedlebbing in the ade2-h7.5 �zrt1 background to 0.3 blebs/colony,
level not significantly higher than the ZRT1 wild-type strain.his indicates that HRAS1 minisatellite alterations in the �zrt1utant likely occur via a combination of RAD52-dependent and
AD52-independent mechanisms, as is the case in for ade2-min3inisatellite alterations.
. Discussion
In this work we examined minisatellite alterations duringtationary phase in S. cerevisiae. First, we demonstrated thatRT1-dependent stationary phase minisatellite alterations occurpecifically in quiescent cells, and not in nonquiescent cells withinhe stationary phase population. We showed that minisatellitelterations in �zrt1 mutants are not dependent on chromosomalontext or adenine auxotrophy. We determined that recombinationactors are required for alterations in minisatellite tracts duringuiescence. The majority of minisatellite alterations in a �zrt1train occurs by RAD52-dependent homologous recombination or aAD52-independent pathway that requires RAD50. Finally, we findhat mutation of ZRT1 can destabilize a human minisatellite, imply-ng that zinc homeostasis may play a role in minisatellite stabilityn human cells.
Stationary phase cultures of S. cerevisiae consist of a quies-ent cell fraction, in which cells are uniformly arrested and bulkNA synthesis does not occur, and a nonquiescent cell fraction,
n which some budded cells are present and DNA replication mayccur [28]. Therefore, we considered that ZRT1-dependent min-satellite alterations during stationary phase might occur as a resultf polymerase slippage during whole-genome DNA synthesis inonquiescent cells. However, we found that loss of ETR1, whichpecifically reduces the ability of quiescent cells to reenter theell cycle, completely eliminated blebbing in a �zrt1 mutant. Inontrast, loss of POR1, which specifically reduces the reproduc-ive capacity of nonquiescent cells, has little effect on blebbing in a
zrt1 mutant. Therefore, our data argue that ZRT1-dependent min-satellite alterations occur as a result of events in quiescent cellshat do not require bulk DNA synthesis. Recent work [38] demon-trates limited DNA synthesis at specific locations in the genomef stationary phase yeast cells; the events that lead to minisatelliteract alterations may utilize this type of limited repair synthesis.
Using the chromosomal ade2-min3 allele (Fig. 1), we determinedhat loss of ZRT1 triggers an increase in minisatellite alterationsxclusively in quiescent cells. We utilized the ura3-min3 alleleo show that minisatellite alterations in a �zrt1 mutant strainccur independently of chromosomal context and adenine aux-trophy. In stationary phase �zrt1 cells, we see an increase inlasmid-borne ura3-min3 minisatellite alterations, eliminating theossibility that the �zrt1 minisatellite instability phenotype is anrtifact of cis-acting sequences surrounding ADE2 and implying thathromosomal context is not likely to have an influence on thesevents. Alterations occurred with equal frequency in Ade+ or Ade−
trains, indicating that the alterations are not due to the disruptionf the adenine biosynthetic pathway. Taken together, these resultsrgue that loss of ZRT1 could potentially destabilize minisatellitest many loci and in many genetic backgrounds.
r 10 (2011) 556– 566 563
Our data demonstrate that most minisatellite alterations in a�zrt1 mutant require homologous recombination. Loss of RAD50,RAD51, or RAD52 in a �zrt1 mutant strain reduces the minisatel-lite alterations by approximately 50% compared to the parental�zrt1 single mutant (Table 2). Similarly, loss of both RAD50 andRAD51 or both RAD51 and RAD52 in a �zrt1 mutant also reduceminisatellite alterations by ∼50%. However, loss of both RAD50and RAD52 in a �zrt1 strain reduces minisatellite alterations by∼81%, indicating that both RAD52-dependent recombination anda RAD50-dependent, RAD52-independent mechanism are requiredfor minisatellite alterations in the �zrt1 mutant. Finally, loss ofRAD50, RAD51, and RAD52 in a �zrt1 mutant reduces minisatellitealterations to a level not significantly different from the parental�rad50 �rad51 �rad52 strain, indicating RAD51 plays a role inZRT1-dependent minisatellite alterations in the absence of RAD50and RAD52. Finally, deletion of the non-homologous end-joining(NHEJ) ligase DNL4 in a �zrt1 mutant strain does not reduce thefrequency of minisatellite alterations, but a significant reductionis seen when RAD52 is also deleted, indicating that NHEJ can playa role in stability maintenance if the homologous recombinationpathway has been compromised. These results are consistent witha model in which two homologous recombination pathways, oneRAD52-dependent and the other RAD52-independent and requir-ing RAD50, are required for most alterations occurring in the �zrt1mutant cells, with other pathways becoming active when homolo-gous recombination has been compromised.
While recombinational mechanisms have previously beenimplicated in minisatellite instability during yeast mitosis [1,22],minisatellite alterations in �zrt1 mutants occur during station-ary phase, a stage at which most yeast cells are quiescent [39].Recombination modulation previously has been linked to genomealterations during stationary phase in prokaryotic Escherichia colicells (see, for example [40–42]); our data extend these findings toeukaryotic yeast cells.
Stationary phase in our �zrt1 strain consists of a relatively uni-form G1 arrest; visual inspection of the ade2-min3 �zrt1 strainshows that by the time the culture enters stationary phase, whichoccurs at ∼48 h after inoculation [22], approximately 98% of thecells are unbudded (G1) (data not shown). G1 cells have only onecopy of each chromosome while G2 cells, which have undergoneDNA replication, have two sister chromatids. Thus, minisatellitealterations in a �zrt1 mutant are not likely to be a result of recombi-nation between sister chromatids. This is significant because all thestrains we have used to examine minisatellite stability in this studyare haploid and therefore cannot undergo recombination betweenhomologous chromosomes.
What is the molecular mechanism underlying stationary phaseminisatellite tract alteration? Since we have shown that ZRT1-dependent minisatellite alterations occur in quiescent, G1-arrestedstationary phase haploid cells, the mechanism for these eventsmust not rely on bulk DNA synthesis or involve exchange betweensister chromatids or homologous chromosomes. Therefore, wepropose that minisatellite alterations in �zrt1 mutants during sta-tionary phase occur by two mechanisms: single strand annealing(SSA) and intramolecular repair events. Simple misalignment ofrepeats during SSA could easily result in minisatellite repeat dele-tion, and the role of SSA in repeat array contraction has been wellestablished [34]. An intramolecular repair event could be initiatedby a single-strand gap (Fig. 4). During the limited DNA synthesisneeded to repair the gap, polymerase slippage and misalignmentcould result in repeat units forming a single-stranded loop. If theloop forms on the template DNA strand and is removed, a dele-
tion of repeat units will result. If the loop forms on the newlysynthesized DNA strand and is repaired by nicking the templateDNA strand opposite the loop, an expansion of the repeat tractwill result (Fig. 4). The frequency of this type of event is depen-
564 M.K. Kelly et al. / DNA Repair 10 (2011) 556– 566
Fig. 4. A model for intramolecular minisatellite alterations. One strand of a minisatellite DNA tract (with repeats shown as a series of vertical lines) is damaged, generating asingle-stranded gap. Repair synthesis initiates (red arrow), but the polymerase dissociates during replication. In (A) the polymerase re-associates further along the templatestrand, forming a single-stranded loop in the template. This loop is cleaved and the resulting nick is repaired, generating a minisatellite tract that has lost repeat units. In(B) the polymerase re-associates with repeat units that have already been replicated, forming a loop on the nascent strand. The template strand subsequently is nicked, andt t withl
domtciilFs
�sotsewaRRifabuRRmctwfaatT
he single-stranded loop undergoes repair synthesis, generating a minisatellite tracegend, the reader is referred to the web version of this article.)
ent on the frequency of polymerase slippage and the formationf single-stranded nicks or gaps. It is quite possible that the poly-erase slippage frequency is significantly increased at repetitive
racts in stationary phase cells. Nucleotide reserves in quiescentells are likely to be small relative to the nucleotide pools availablen actively dividing cells, leading to increased polymerase paus-ng (especially at repetitive DNA tracts) thereby increasing theikelihood of polymerase dissociation and re-association events.inally, defects in zinc transport have been shown to elevate single-tranded DNA damage in mammalian cells [43,44].
Our data on the types of minisatellite alterations observed inzrt1 mutants are consistent with these models. We previously
howed that minisatellite alterations in �zrt1 mutants consistf deletions of repeat units from the ade2-min3 tract [22]. Inhis study we observed both repeat deletions and tract expan-ion in the ade2-h7.5 minisatellite (Fig. 3). Intramolecular repairvents and SSA can both lead to deletions in direct repeat tractshen repeats are misaligned [34]. We find that minisatellite alter-
tions in a �zrt1 mutant during stationary phase occur by bothAD52-dependent and RAD52-independent mechanisms. SSA is aAD52-dependent process for shorter repeat arrays [45,46]. RAD52-
ndependent mechanisms of SSA have been demonstrated onlyor large CUP1 and rDNA repeat tracts [47]. Since our ade2-min3nd ade2-h7.5 reporters are significantly smaller than those tractsoth in terms of repeat unit length and number of repeats, it isnlikely that RAD52-independent SSA contributes to our events.AD59 and RAD1 are involved in SSA [36,48,49]. Deletion of eitherAD1 or RAD59 in a �zrt1 strain leads to a ∼20% reduction ininisatellite alterations (Table 2, data not shown), clearly impli-
ating SSA in these events. Since some SSA has been demonstratedo occur even without RAD59 [49], it is difficult to estimatehat proportion of ZRT1-dependent minisatellite alterations result
rom SSA. It may be that all RAD52-dependent minisatellite alter-
tions in the �zrt1 mutant occur by SSA. However, as we havergued above, RAD52-independent SSA is not likely to contributeo minisatellite alterations in the ade2-min3 minisatellite tract.herefore, all RAD52-independent minisatellite alterations in theadditional repeat units. (For interpretation of the references to color in this figure
ZRT1 mutant may occur via some form of intramolecular repair.Consistent with this, we find that most RAD52-independent min-isatellite alterations require RAD50; prior studies demonstrateda requirement for RAD50 in RAD52-independent intramolecu-lar recombination [34]. Rad50p possesses zinc hooks that areemployed in linking DNA ends [50,51], which could play a role informing the single stranded DNA loops in our model of intramolec-ular minisatellite repair. Previous work has demonstrated thatsingle stranded DNA loops over 16 nt can be repaired by loopremoval, but the genetic requirements for this process remain elu-sive, likely because redundant pathways facilitate it [52]. Therefore,it is difficult to provide evidence for this aspect of our model.Finally, our DNL4 data show that NHEJ can be used as a repairmechanism when homologous recombination has been compro-mised.
Importantly, we demonstrate here that a human HRAS1 min-isatellite tract alters in quiescent cells, and that loss of ZRT1 furtherdestabilizes the minisatellite (Table 2). The yeast ZRT1 protein is amember of the ZIP (Zrt-, Irt-like Protein) family of zinc transporters,which are found throughout bacteria and eukaryotes, includinghumans [53]. Mammals have three orthologs of the yeast ZRT1gene: ZIP1 (also known as ZIRTL), ZIP2 and ZIP3 [54–59]. The ZIP1protein in humans (hZip1) is likely to be the major zinc transporterfor most of the cells in the body, as it is expressed in most cell types[59]. The human Zip2 protein is expressed in prostate and uter-ine epithelial cells. Mutations in ZIP family zinc transporters leadto zinc deficiency and associated health problems in humans [60].For example, mutation of Zip2 or Zip3 has been linked to prostatecancers [54].
Our data provide a mechanism to link zinc deficiencies withhuman disease generation. DNA strand breaks, triggered by zincdeficiency, could be the initiating lesions for minisatellite alter-ations in post-mitotic cells. In agreement with this model, zinc
deficiency has been linked to increased DNA strand breakage inmammals [43,44]. Variations in the HRAS1 minisatellite tract areknown to alter HRAS1 transcription [4,61,62], modifying the activityof this important oncogene and influencing the onset of particular
Repai
cms
zPrbhtomi
5
tiloadrimc
C
A
tNm
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
M.K. Kelly et al. / DNA
ancers. These alteration events may be elevated in quiescent, post-itotic cells, as our data demonstrate that they are particularly
ensitive to loss of zinc transporters.Links that support this hypothesis have been detected between
inc homeostasis and minisatellite stability in human disease.rostate cancer has been linked to both zinc deficiency andare HRAS1 minisatellite alleles [8,43]. Type 1 diabetes has longeen associated with rare alleles of the IDDM1 minisatellite, andas recently been correlated with SNPs in the ZIP family zincransporter SLC30A8 [3,63]. Thus, the proposal that zinc home-stasis may directly affect minisatellite stability and influenceinisatellite-correlated disease in humans clearly merits further
nvestigation.
. Conclusions
We previously discovered that a reporter minisatellite DNAract is destabilized in stationary phase yeast cells with defectsn zinc homeostasis [22]. Here we demonstrate that minisatel-ites are destabilized specifically in quiescent cells. Tract alterationsccur regardless of minisatellite context, and can also take place in
human minisatellite inserted into the yeast genome. We haveetermined that both RAD52-dependent and RAD52-independentecombination pathways are necessary to alter minisatellite tractsn quiescent �zrt1 cells. Our findings indicate that analogous zinc-
ediated minisatellite rearrangements may be occurring in humanells, leading to minisatellite-associated pathologies such as cancer.
onflict of interest statement
The authors declare that there are no conflicts of interest.
cknowledgements
We thank Laura Brosnan for technical assistance in constructinghe ade2-h7.5 allele. This work was sponsored by a grant from theational Institutes of Health (5RO1-GM072598) and ARRA supple-ent 3R01-GM072598-05S1.
eferences
[1] G.F. Richard, A. Kerrest, B. Dujon, Comparative genomics and molecular dynam-ics of DNA repeats in eukaryotes, Microbiol. Mol. Biol. Rev. 72 (2008) 686–727.
[2] G. Vergnaud, F. Denoeud, Minisatellites: mutability and genome architecture,Genome Res. 10 (2000) 899–907.
[3] G.C. Kennedy, M.S. German, W.J. Rutter, The minisatellite in the diabetes sus-ceptibility locus IDDM2 regulates insulin transcription, Nat. Genet. 9 (1995)293–298.
[4] M. Green, T.G. Krontiris, Allelic variation of reporter gene activation by theHRAS1 minisatellite, Genomics 17 (1993) 429–434.
[5] W.L. Trepicchio, T.G. Krontiris, Members of the rel/NF-kappa B family of tran-scriptional regulatory proteins bind the HRAS1 minisatellite DNA sequence,Nucl. Acids Res. 20 (1992) 2427–2434.
[6] G.R. Sutherland, E. Baker, R.I. Richards, Fragile sites still breaking, Trends Genet.14 (1998) 501–506.
[7] Y.H. Jeong, M.C. Kim, E.K. Ahn, S.Y. Seol, E.J. Do, H.J. Choi, I.S. Chu, W.J. Kim, Y.Sunwoo, S.H. Leem, Rare exonic minisatellite alleles in MUC2 influence suscep-tibility to gastric carcinoma, PLoS ONE 2 (2007) e1163.
[8] T.G. Krontiris, Minisatellites and human disease, Science 269 (1995)1682–1683.
[9] K. Virtaneva, E. D’Amato, J. Miao, M. Koskiniemi, R. Norio, G. Avanzini, S.Franceschetti, R. Michelucci, C.A. Tassinari, S. Omer, L.A. Pennacchio, R.M.Myers, J.L. Dieguez-Lucena, R. Krahe, A. de la Chapelle, A.E. Lehesjoki, Unsta-ble minisatellite expansion causing recessively inherited myoclonus epilepsy,EPM1, Nat. Genet. 15 (1997) 393–396.
10] H.J. Kirkbride, J.G. Bolscher, K. Nazmi, L.E. Vinall, M.W. Nash, F.M. Moss, D.M.Mitchell, D.M. Swallow, Genetic polymorphism of MUC7: allele frequencies and
association with asthma, Eur. J. Hum. Genet. 9 (2001) 347–354.11] K. Kyo, M. Parkes, Y. Takei, H. Nishimori, P. Vyas, J. Satsangi, J. Simmons, H.Nagawa, S. Baba, D. Jewell, T. Muto, G.M. Lathrop, Y. Nakamura, Association ofulcerative colitis with rare VNTR alleles of the human intestinal mucin gene,MUC3, Hum. Mol. Genet. 8 (1999) 307–311.
[
[
r 10 (2011) 556– 566 565
12] S.V. Faraone, A.E. Doyle, E. Mick, J. Biederman, Meta-analysis of the associationbetween the 7-repeat allele of the dopamine D(4) receptor gene and attentiondeficit hyperactivity disorder, Am. J. Psychiatry 158 (2001) 1052–1057.
13] B. Yang, R.C. Chan, J. Jing, T. Li, P. Sham, R.Y. Chen, A meta-analysis of associationstudies between the 10-repeat allele of a VNTR polymorphism in the 3′-UTR ofdopamine transporter gene and attention deficit hyperactivity disorder, Am. J.Med. Genet. B: Neuropsychiatr. Genet. 144B (2007) 541–550.
14] P.A. Jauert, S.N. Edmiston, K. Conway, D.T. Kirkpatrick, RAD1 controls the mei-otic expansion of the human HRAS1 minisatellite in Saccharomyces cerevisiae,Mol. Cell. Biol. 22 (2002) 953–964.
15] P.A. Jauert, D.T. Kirkpatrick, Length and sequence heterozygosity differentiallyaffect HRAS1 minisatellite stability during meiosis in yeast, Genetics 170 (2005)601–612.
16] H. Debrauwere, J. Buard, J. Tessier, D. Aubert, G. Vergnaud, A. Nicolas, Meioticinstability of human minisatellite CEB1 in yeast requires DNA double-strandbreaks, Nat. Genet. 23 (1999) 367–371.
17] R.J. Kokoska, L. Stefanovic, A.B. Buermeyer, R.M. Liskay, T.D. Petes, A mutation ofthe yeast gene encoding PCNA destabilizes both microsatellite and minisatelliteDNA sequences, Genetics 151 (1999) 511–519.
18] R.J. Kokoska, L. Stefanovic, H.T. Tran, M.A. Resnick, D.A. Gordenin, T.D. Petes,Destabilization of yeast micro- and minisatellite DNA sequences by mutationsaffecting a nuclease involved in Okazaki fragment processing (rad27) and DNApolymerase delta (pol3-t), Mol. Cell. Biol. 18 (1998) 2779–2788.
19] J. Lopes, H. Debrauwere, J. Buard, A. Nicolas, Instability of the human minisatel-lite CEB1 in rad27Delta and dna2-1 replication-deficient yeast cells, EMBO J. 21(2002) 3201–3211.
20] S. Maleki, H. Cederberg, U. Rannug, The human minisatellites MS1, MS32,MS205 and CEB1 integrated into the yeast genome exhibit different degreesof mitotic instability but are all stabilised by RAD27, Curr. Genet. 41 (2002)333–341.
21] C. Ribeyre, J. Lopes, J.B. Boule, A. Piazza, A. Guedin, V.A. Zakian, J.L. Mergny,A. Nicolas, The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo, PLoS Genet. 5 (2009) e1000475.
22] M.K. Kelly, P.A. Jauert, L.E. Jensen, C.L. Chan, C.S. Truong, D.T. Kirkpatrick, Zincregulates the stability of repetitive minisatellite DNA tracts during stationaryphase, Genetics 177 (2007) 2469–2479.
23] E. Coic, T. Feldman, A.S. Landman, J.E. Haber, Mechanisms of Rad52-independent spontaneous and UV-induced mitotic recombination in Saccha-romyces cerevisiae, Genetics 179 (2008) 199–211.
24] C. Guthrie, G.R. Fink, Guide to Yeast Genetics and Molecular Biology, AcademicPress, San Diego, 1991.
25] E.A. Sia, M. Dominska, L. Stefanovic, T.D. Petes, Isolation and characterizationof point mutations in mismatch repair genes that destabilize microsatellites inyeast, Mol. Cell. Biol. 21 (2001) 8157–8167.
26] D.K. Nag, T.D. Petes, Physical detection of heteroduplexes during meioticrecombination in the yeast Saccharomyces cerevisiae, Mol. Cell. Biol. 13 (1993)2324–2331.
27] R.M. Spell, S. Jinks-Robertson, Determination of mitotic recombination ratesby fluctuation analysis in Saccharomyces cerevisiae, Methods Mol. Biol. 262(2004) 3–12.
28] A.D. Aragon, A.L. Rodriguez, O. Meirelles, S. Roy, G.S. Davidson, P.H. Tapia, C.Allen, R. Joe, D. Benn, M. Werner-Washburne, Characterization of differentiatedquiescent and nonquiescent cells in yeast stationary-phase cultures, Mol. Biol.Cell. 19 (2008) 1271–1280.
29] J.M. Torkko, K.T. Koivuranta, I.J. Miinalainen, A.I. Yagi, W. Schmitz, A.J. Kas-taniotis, T.T. Airenne, A. Gurvitz, K.J. Hiltunen, Candida tropicalis Etr1p andSaccharomyces cerevisiae Ybr026p (Mrf1′p) 2-enoyl thioester reductasesessential for mitochondrial respiratory competence, Mol. Cell. Biol. 21 (2001)6243–6253.
30] N.S. Sanchez, D.A. Pearce, T.S. Cardillo, S. Uribe, F. Sherman, Requirements ofCyc2p and the porin, Por1p, for ionic stability and mitochondrial integrity inSaccharomyces cerevisiae, Arch. Biochem. Biophys. 392 (2001) 326–332.
31] A Achilli, N. Matmati, E. Casalone, G. Morpurgo, A. Lucaccioni, Y.I. Pavlov, N.Babudri, The exceptionally high rate of spontaneous mutations in the poly-merase delta proofreading exonuclease-deficient Saccharomyces cerevisiaestrain starved for adenine, BMC Genet. 5 (2004) 34.
32] A.L. Todeschini, A. Morillon, M. Springer, P. Lesage, Severe adenine starvationactivates Ty1 transcription and retrotransposition in Saccharomyces cerevisiae,Mol. Cell. Biol. 25 (2005) 7459–7472.
33] J. San Filippo, P. Sung, H. Klein, Mechanism of eukaryotic homologous recom-bination, Annu. Rev. Biochem. 77 (2008) 229–257.
34] H.L. Klein, Genetic control of intrachromosomal recombination, Bioessays 17(1995) 147–159.
35] A.P. Davis, L.S. Symington, The yeast recombinational repair protein Rad59interacts with Rad52 and stimulates single-strand annealing, Genetics 159(2001) 515–525.
36] E.L. Ivanov, J.E. Haber, RAD1 and RAD10, but not other excision repair genes,are required for double-strand break-induced recombination in Saccharomycescerevisiae, Mol. Cell. Biol. 15 (1995) 2245–2251.
37] T.E Wilson, U. Grawunder, M.R. Lieber, Yeast DNA ligase IV mediates non-homologous DNA end joining, Nature 388 (1997) 495–498.
38] A. de Morgan, L. Brodsky, Y. Ronin, E. Nevo, A. Korol, Y. Kashi, Genome-wideanalysis of DNA turnover and gene expression in stationary phase Saccha-romyces cerevisiae, Microbiology 156 (2010) 1758–1771.
39] C. Allen, S. Buttner, A.D. Aragon, J.A. Thomas, O. Meirelles, J.E. Jaetao, D. Benn,S.W. Ruby, M. Veenhuis, F. Madeo, M. Werner-Washburne, Isolation of quies-

5 Repai
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
66 M.K. Kelly et al. / DNA
cent and nonquiescent cells from yeast stationary-phase cultures, J. Cell Biol.174 (2006) 89–100.
40] H.J. Bull, M.J. Lombardo, S.M. Rosenberg, Stationary-phase mutation in thebacterial chromosome: recombination protein and DNA polymerase IV depen-dence, Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 8334–8341.
41] R.G. Ponder, N.C. Fonville, S.M. Rosenberg, A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation, Mol.Cell 19 (2005) 791–804.
42] J. Torkelson, R.S. Harris, M.-J. Lombardo, J. Nagendran, C. Thulin, S.M. Rosenberg,Genome-wide hypermutation in a subpopulation of stationary-phase cellsunderlies recombination-dependent adaptive mutation, EMBO J. 16 (1997)3303–3311.
43] E. Ho, Zinc deficiency, DNA damage and cancer risk, J. Nutr. Biochem. 15 (2004)572–578.
44] E. Ho, B.N. Ames, Low intracellular zinc induces oxidative DNA damage, disruptsp53, NFkappa B, and AP1 DNA binding, and affects DNA repair in a rat gliomacell line, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 16770–16775.
45] J. Fishman-Lobell, J.E. Haber, Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1,Science 258 (1992) 480–484.
46] N. Sugawara, J.E. Haber, Characterization of double-strand break-inducedrecombination: homology requirements and single-stranded DNA formation,Mol. Cell. Biol. 12 (1992) 563–575.
47] B.A. Ozenberger, G.S. Roeder, A unique pathway of double-strand breakrepair operates in tandemly repeated genes, Mol. Cell. Biol. 11 (1991) 1222–1231.
48] N. Sugawara, G. Ira, J.E. Haber, DNA length dependence of the single-strandannealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair, Mol. Cell. Biol. 20 (2000) 5300–5309.
49] L.S. Symington, Role of RAD52 epistasis group genes in homologous recom-bination and double-strand break repair, Microbiol. Mol. Biol. Rev. 66 (2002)
630–670.50] K.P. Hopfner, L. Craig, G. Moncalian, R.A. Zinkel, T. Usui, B.A. Owen, A. Karcher, B.Henderson, J.L. Bodmer, C.T. McMurray, J.P. Carney, J.H. Petrini, J.A. Tainer, TheRad50 zinc-hook is a structure joining Mre11 complexes in DNA recombinationand repair, Nature 418 (2002) 562–566.
[
r 10 (2011) 556– 566
51] J.J. Wiltzius, M. Hohl, J.C. Fleming, J.H. Petrini, The Rad50 hook domain is a crit-ical determinant of Mre11 complex functions, Nat. Struct. Mol. Biol. 12 (2005)403–407.
52] D.T. Kirkpatrick, T.D. Petes, Repair of DNA loops involves DNA mismatch andnucleotide excision repair proteins, Nature 387 (1997) 929–931.
53] D.J. Eide, Zinc transporters and the cellular trafficking of zinc, Biochim. Biophys.Acta 1763 (2006) 711–722.
54] M.M. Desouki, J. Geradts, B. Milon, R.B. Franklin, L.C. Costello, hZip2 and hZip3zinc transporters are down regulated in human prostate adenocarcinomatousglands, Mol. Cancer 6 (2007) 37.
55] J. Dufner-Beattie, S.J. Langmade, F. Wang, D. Eide, G.K. Andrews, Structure, func-tion, and regulation of a subfamily of mouse zinc transporter genes, J. Biol.Chem. 278 (2003) 50142–50150.
56] R.B Franklin, J. Ma, J. Zou, Z. Guan, B.I. Kukoyi, P. Feng, L.C. Costello, Human ZIP1is a major zinc uptake transporter for the accumulation of zinc in prostate cells,J. Inorg. Biochem. 96 (2003) 435–442.
57] L.A. Gaither, D.J. Eide, Functional expression of the human hZIP2 zinc trans-porter, J. Biol. Chem. 275 (2000) 5560–5564.
58] L.A. Gaither, D.J. Eide, The human ZIP1 transporter mediates zinc uptake inhuman K562 erythroleukemia cells, J. Biol. Chem. 276 (2001) 22258–22264.
59] M. Lioumi, C.A. Ferguson, P.T. Sharpe, T. Freeman, I. Marenholz, D. Mischke, C.Heizmann, J. Ragoussis, Isolation and characterization of human and mouseZIRTL, a member of the IRT1 family of transporters, mapping within the epi-dermal differentiation complex, Genomics 62 (1999) 272–280.
60] C Murgia, C.J. Lang, A.Q. Truong-Tran, D. Grosser, L. Jayaram, R.E. Ruffin, G. Per-ozzi, P.D. Zalewski, Zinc and its specific transporters as potential targets inairway disease, Curr. Drug Targets 7 (2006) 607–627.
61] J.B. Cohen, M.V. Walter, A.D. Levinson, A repetitive sequence element 3′ of thehuman c-Ha-ras1 gene has enhancer activity, J. Cell. Physiol. Suppl. 5 (1987)75–81.
62] D.A. Spandidos, L. Holmes, Transcriptional enhancer activity in the variable
tandem repeat DNA sequence downstream of the human Ha-ras1 gene, FEBSLett. 218 (1987) 41–46.63] H. Gohlke, U. Ferrari, K. Koczwara, E. Bonifacio, T. Illig, A.G. Ziegler, SLC30A8(ZnT8) polymorphism is associated with young age at type 1 diabetes onset,Rev. Diabetes Stud. 5 (2008) 25–27.
Related Documents











