MOLECULAR CARCINOGENESIS Functional Characterization of RAD52 as a Lung Cancer Susceptibility Gene in the 12p13.33 Locus Rachel Lieberman, 1 Donghai Xiong, 1 Michael James, 1 Younghun Han, 2 Christopher I. Amos, 2 Liang Wang, 3 and Ming You 1 * 1 Department of Pharmacology & Toxicology and Cancer Center, Medical College of Wisconsin, Milwaukee, Wisconsin 2 Department of Community and Family Medicine, Geisel School of Medicine, Dartmouth College, Lebanon, New Hampshire 3 Department of Pathology and Cancer Center, Medical College of Wisconsin, Milwaukee, Wisconsin Recent genome-wide association studies have identified variations in the recombination repair gene, RAD52, that are associated with increased lung cancer risk, and particularly with the development of lung squamous cell carcinomas (LUSC). As LUSC development is strongly associated with smoking, DNA repair is increased in the lung tissues of smokers, presumably because of ongoing DNA damage from exposure to tobacco smoke. A key player in the DNA damage response, RAD52 plays a role in DNA strand exchange and annealing during homologous recombination (HR) in mammalian cells. In this study, we discovered two cis-expression quantitative trait loci (eQTL) SNPs in the RAD52 gene that are associated with its expression and are also associated with LUSC risk. In addition, we report that amplification of the genomic region 12p13.33, which contains the RAD52 gene, is significantly associated with the development of LUSC in the TCGA database and that somatic overexpression of RAD52 was confirmed to be significant in LUSC tumors from our own patient cohort. Consistent with these genetic findings, we demonstrate that blockade of Rad52 slows cell growth and induces senescence in mouse bronchial epithelial cells. In contrast, overexpression of Rad52 leads to an increased rate of cell proliferation. We show that depletion of Rad52 in mouse lung tumor cells alters cell cycle distribution and increases DNA damage accumulation associated with increased tumor cell death. Our genetic and functional data implicate RAD52 as a significant determinant of risk in the development of LUSC. © 2015 Wiley Periodicals, Inc. Key words: eQTL; non-small cell lung carcinoma; lung cancer; DNA damage; cancer susceptibility gene; senescence; gene amplification INTRODUCTION Lung cancer currently ranks as the foremost cause of cancer deaths among men and women in the US with more than one-quarter of all cancer deaths due to this disease [1]. Squamous cell lung cancer (LUSC), which is particularly associated with smoking, com- prises about 25% to 30% of all lung cancers and begins in squamous cells which are flat cells that line the inside of the bronchial airways. Furthermore, while 80–90% of lung cancer patients have a history of smoking, only about 10% of heavy smokers develop lung cancer [2]. This suggests that although tobacco smoke undoubtedly increases one’s risk for lung cancer, genetic risk factors most likely predispose certain individuals to disease development as part of a complex interplay between carcinogens and genetic determinants. One would expect certain genes which influence exposure to, metabolism of, or response to carcinogens to play a role in lung cancer susceptibili- ty, especially in LUSC. Recent genome-wide association studies (GWAS) have identified a region on chromosome 12p13.33 as harboring a susceptibility locus for LUSC. This locus includes the recombination repair gene RAD52 which harbors a specific single nucleotide polymorphism (SNP)—rs6489769 that is associated with LUSC [3]. RAD52, originally described in yeast as playing a key role in recombination repair, is involved in strand exchange and annealing of strands during homolo- gous recombination and is predominantly recruited for DNA repair during S phase of the cell cycle [4,5]. Similarly, recent additions to the 1000 genomes project, including four genome-wide association studies of lung cancer in populations of European ancestry, identified large-effect genome-wide associ- ations for squamous cell lung cancer with the rare variant of BRCA2-K3326X [6]. While BRCA2 is the more recognized factor in human HR, studies per- formed in BRCA2-deficient mammalian cells impli- cate RAD52 as an independent and alternate No potential conflicts of interest were disclosed. Rachel Lieberman, Donghai Xiong, and Ming You contributed equally to this work. Grant sponsor: NIH; Grant number: U19CA148127 *Correspondence to: Medical College of Wisconsin, 8701 Water- town Plank Road, Cancer Center, Milwaukee, WI 53226. Received 9 December 2014; Revised 27 March 2015; Accepted 21 April 2015 DOI 10.1002/mc.22334 Published online in Wiley Online Library (wileyonlinelibrary.com). ß 2015 WILEY PERIODICALS, INC.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MOLECULAR CARCINOGENESIS
Functional Characterization of RAD52 as a Lung CancerSusceptibility Gene in the 12p13.33 LocusRachel Lieberman,1 Donghai Xiong,1Michael James,1 YounghunHan,2 Christopher I. Amos,2 LiangWang,3
and Ming You1*1Department of Pharmacology & Toxicology and Cancer Center, Medical College of Wisconsin, Milwaukee, Wisconsin2Department of Community and Family Medicine, Geisel School of Medicine, Dartmouth College, Lebanon, New Hampshire3Department of Pathology and Cancer Center, Medical College of Wisconsin, Milwaukee, Wisconsin
Recent genome-wide association studies have identified variations in the recombination repair gene, RAD52, that areassociated with increased lung cancer risk, and particularly with the development of lung squamous cell carcinomas(LUSC). As LUSC development is strongly associated with smoking, DNA repair is increased in the lung tissues of smokers,presumably because of ongoing DNA damage from exposure to tobacco smoke. A key player in the DNA damageresponse, RAD52 plays a role in DNA strand exchange and annealing during homologous recombination (HR) inmammalian cells. In this study, we discovered two cis-expression quantitative trait loci (eQTL) SNPs in the RAD52 gene thatare associated with its expression and are also associated with LUSC risk. In addition, we report that amplification of thegenomic region 12p13.33, which contains the RAD52 gene, is significantly associated with the development of LUSC inthe TCGA database and that somatic overexpression of RAD52 was confirmed to be significant in LUSC tumors from ourown patient cohort. Consistent with these genetic findings, we demonstrate that blockade of Rad52 slows cell growthand induces senescence inmouse bronchial epithelial cells. In contrast, overexpression of Rad52 leads to an increased rateof cell proliferation. We show that depletion of Rad52 in mouse lung tumor cells alters cell cycle distribution and increasesDNA damage accumulation associated with increased tumor cell death. Our genetic and functional data implicate RAD52as a significant determinant of risk in the development of LUSC. © 2015 Wiley Periodicals, Inc.
Key words: eQTL; non-small cell lung carcinoma; lung cancer; DNA damage; cancer susceptibility gene; senescence;gene amplification
INTRODUCTION
Lung cancer currently ranks as the foremost causeof cancer deaths among men and women in the USwithmore thanone-quarter of all cancer deaths due tothis disease [1]. Squamous cell lung cancer (LUSC),which is particularly associated with smoking, com-prises about 25% to 30%of all lung cancers and beginsin squamous cells which are flat cells that line theinside of the bronchial airways. Furthermore, while80–90% of lung cancer patients have a history ofsmoking, only about 10% of heavy smokers developlung cancer [2]. This suggests that although tobaccosmoke undoubtedly increases one’s risk for lungcancer, genetic risk factors most likely predisposecertain individuals to disease development as part of acomplex interplay between carcinogens and geneticdeterminants. One would expect certain genes whichinfluence exposure to, metabolism of, or response tocarcinogens to play a role in lung cancer susceptibili-ty, especially in LUSC.Recent genome-wide association studies (GWAS)
have identified a region on chromosome 12p13.33 asharboring a susceptibility locus for LUSC. This locusincludes the recombination repair gene RAD52whichharbors a specific single nucleotide polymorphism(SNP)—rs6489769 that is associated with LUSC [3].
RAD52, originally described in yeast as playing a keyrole in recombination repair, is involved in strandexchange and annealing of strands during homolo-gous recombination and is predominantly recruitedfor DNA repair during S phase of the cell cycle [4,5].
Similarly, recent additions to the 1000 genomesproject, including four genome-wide associationstudies of lung cancer in populations of Europeanancestry, identified large-effect genome-wide associ-ations for squamous cell lung cancer with the rarevariant of BRCA2-K3326X [6]. While BRCA2 is themore recognized factor in human HR, studies per-formed in BRCA2-deficient mammalian cells impli-cate RAD52 as an independent and alternate
No potential conflicts of interest were disclosed.Rachel Lieberman, Donghai Xiong, and Ming You contributed
equally to this work.Grant sponsor: NIH; Grant number: U19CA148127*Correspondence to: Medical College of Wisconsin, 8701 Water-
town Plank Road, Cancer Center, Milwaukee, WI 53226.Received 9 December 2014; Revised 27 March 2015; Accepted 21
April 2015DOI 10.1002/mc.22334Published online in Wiley Online Library
(wileyonlinelibrary.com).
� 2015 WILEY PERIODICALS, INC.

participant in HR [7]. Upon RAD52 depletion, boththe frequency of double-strand break-inducedHR andionizing radiation-induced RAD51 foci decreasedsignificantly. Likewise, RAD52-RAD51 foci wereshown to form equally in the presence or absence ofBRCA2 [8]. As well, inactivation of RAD52 in BRCA2competent mouse ES cells did result in a modestreduction in HR [9].
In humans, RAD52 is a key player in the regulationof HR-related genomic instability that may lead to anincreased LUSC risk. Furthermore, when combinedwith depletion of BRCA2 or PALB2, depletion ofhuman RAD52 is synthetically lethal [10]. Thus, inhuman cells deficient in the PALB2 or BRCA2 genes,RAD52 depletion may decrease cell survival byreducing rates of homologous recombination andby increasing damage-induced chromosomal abnor-malities [3]. However, the exact role of RAD52 inhuman lung cancer remains elusive andmore effort isneeded to fill this gap in the knowledge base.
In this study, we sought to investigate the role ofRAD52 in lung cancer susceptibility. As mentionedpreviously, squamous cell lung carcinoma is particu-larly associated with smoking. Thus, it fits thatvariation in this DNA repair gene would impactone’s risk for lung cancer, suggestive of a potentiallydecreased ability to repair carcinogen-induced dam-age. Surprisingly, our copy number variation (CNV)analysis of LUSC tumors revealed a copy number gainin the region of RAD52, resulting in a statisticallysignificant level of amplification for RAD52 in lungsquamous cell carcinoma patients. We also identifiedtwo correlated cis-eQTL SNPs that exhibit cis-regula-tion of RAD52 expression and significant associationwith LUSC. Elevated RAD52 protein expression wasalso significant in LUSC tumors compared to that inmatched normal lung tissues.
In linewith the statistical evidence portraying a rolefor RAD52 in developing lung cancer, we functionallydemonstrate that upon Rad52 depletion, the rate ofcell proliferation was attenuated and senescence wasinduced in non-tumorigenic bronchial epithelialmouse cells. We also show that loss of RAD52 slowscell proliferation while overexpression of RAD52enhances proliferation in mouse lung cancer cells.Additionally, in mouse tumor cell lines we demon-strate that depletion of RAD52 alters cell cycledistribution by decreasing the fraction of cells inG0/G1 and significantly increasing the fraction ofcells in G2/M. Finally, we demonstrate that loss ofRad52 expression increases DNA damage accumula-tion in mouse normal bronchial and tumor cells.
MATERIALS AND METHODS
Study Subjects
The cis-eQTL analysis of RAD52was conducted on acohort of 70 normal lung tissue samples provided bythe Mayo Clinic [11]. The genetic association study
conducted between RAD52 SNPs and lung cancerphenotypes was performed by analyzing a patientpopulation of European ancestry (n¼28 998) used ina recent large-scale GWAS study for lung cancer [6].Gene expression analyses for RAD52 used the RNA-seq data for a cohort of tumor-normal paired RNAsamples from 45 non-small cell lung cancer patients(36 lung adenocarcinoma (LUAD) and nine lungsquamous cell carcinoma (LUSC) recruited at theMedical College of Wisconsin.
eQTL and Expression Analysis of RAD52
The genotyping of the RAD52 SNPs for cis-eQTLanalysis was part of a previous GWAS project usingIllumina HumanHap 370 and 610k BeadChips [11](Illumina, San Diego, CA). CEPH DNA samples (afamily trio) were included in each 96-well plate tomonitor genotyping, and the concordance betweenthe replicates was 99.5%. The RAD52 gene expressiondata in 70 normal lung tissues were part of the resultsof the genome microarray analysis done with theIllumina Human WG DASL array (Illumina) asdetailed previously [11]. The genetic associationanalysis between RAD52 and lung cancer was partof the recent large-scale lung cancer GWAS researchdetailed previously [6]. RNA sequencing for thetumor-normal paired RNA samples from 45 NSCLCpatients followed the protocol from our previouspublication [12]. Briefly, for data analysis, we usedTopHat to conduct alignment of reads to the humanreference genome (hg19). We used Cufflinks tocompute expression levels of transcripts. Fragmentsper kilo base of exon per million fragments mapped(FPKM) values were summarized to the gene level bytaking the sum of Cufflinks FPKMs estimated for thealternative transcripts of each Ensemble gene [12].Wespecifically extracted expression data for RAD52 andconducted a pairwise comparison of the expression ofthis gene for the 45 lung tumor-normal pairs.
Statistical Analysis
The linkage disequilibrium analysis for the RAD52regions was conducted using Haploview software(http://www.broadinstitute.org/scientific-communi-ty/science/programs/medical-and-population-genet-ics/haploview/haploview) and the data from theCaucasian population of the 1000 genomes project.For the association testing between RAD52 SNPs andlung cancer risk, we calculated odds ratios (ORs) and95% confidence intervals (CIs) under a fixed-effectmodel for each SNP and the associated per-allele Pvalues. To explore variability in associations accord-ing to tumor histology, we derived ORs for all lungcancer, LUAD and LUSC in the same way as in ourprevious study [6]. For the eQTL analysis, a linearregression model adjusted for age and sex was used toassess the correlation between genotypes (indepen-dent variable, coded as 0, 1, or 2) and transcriptexpression levels of RAD52 (eQTL, dependent
2 LIEBERMAN ET AL.
Molecular Carcinogenesis

variable) in 70 samples of normal lung tissue. Theanalysis was done with SAS version 9.0. Confoundingfactors thatmay be associated with lung cancer risk orgene expression were adjusted in the same way as inour previous publications (9,10). Therefore, combin-ing data from different cohorts did not introduceconfounding information. In terms of RNA-seq-baseddifferential gene expression analysis of RAD52, apaired Student’s t-test was done to compare RAD52expression between tumor samples and samples ofadjacent normal tissue. We analyzed 36 LUAD (datanot shown) and 9 LUSC tumor samples withmatchednormal tissue. The Wilcoxon signed rank test for theRAD52 gene showed differential expression using thetumor-normal pairs. We also downloaded somaticcopy number alteration data in the 12p13.33 regioncontaining RAD52 and somatic gene expression dataof RAD52 generated by TCGA (The Cancer GenomeAtlas, http://cghub.ucsc.edu/) to check the somaticcopy number change and its association with somaticRAD52 expression changes in LUSC samples.
Cell Culture
Cell lines used include: the immortalized, non-tumorigenic mouse lung epithelial cell line, E10, aswell as themouse lung tumor cell lines obtained fromspontaneous lung tumors in the A/J mouse (Spon2and Spon6), MC7 cells established from a methylenechloride-induced nonmetastatic lung tumor fromB6C3F1 and the metastatic nicotine-derived nitrosa-mine ketone (NNK)-induced A/J mouse lung tumor(CL25M cell line). All cell lines were cultured aspreviously described [13].
Lentivirus Construction and Transduction for Short HairpinRNA (shRNA) Expression
The pLKO.1 lentiviral system (GE Healthcare, PA)was used to generate replication-deficient lentiviralparticles for stable transductionof shRNA. Thematureanti-sense strand of the shRNA sequences are asfollows, Rad52-01: 50- TAACTGCACCTTTACAAATGC-30, Rad52-03: 50- TAGTCCTCTAAAGAGTTCAGC -30,Rad52-04: 50- ATTCATCCGCTGTATACTGGC -30,shSCRAM:50- CCTAAGGTTAAGTCGCCCTCGCTC-GAGCGAGGGCGACTTAACCTTAGG -30. Lentivirusstockswere prepared in 293T cells, infected into targetcells with 8mg/ml polybrene and selected with 2mg/ml puromycin. Puromycin selected lineswere used forexperimental analysis from 4–25d post infection.
Overexpression of RAD52
The plasmids encoding empty vector pOPUR andpOPUR/Rad52 were a generous gift from Dr. Jac A.Nickoloff and were constructed according to hisprotocol [14]. Briefly, mammalian expression vectorpOPUR was constructed to carry the RSV LTRpromoter and triplicated lac operators from pOPI3-CAT (Stratagene, La Jolla, CA) and the puromycinresistance gene from pPUR (Clontech, Palo Alto, CA).
Human Rad52 cDNA was inserted downstream of theRSV LTR promoter in order to create a Rad52 over-expression plasmid that would retain its stability andnot degrade post-transcription due to mRNA degra-dation signals in the 30-untranslated region [14].
Immunoblot Analysis of Cellular Lysates
Protein from E10, CL25M, Spon6, and Spon2 cellsexpressing either shRNA targeting RAD52, constructsfor overexpression of RAD52, scrambled vectorcontrol shRNAor empty pOPURvectorwere extractedat 72–96h post transfection for transient transfec-tions and in line with each experiment for stablestransfections. Cells were lysedwith 100ml of 1� RIPAlysis buffer containing proteinase inhibitor cocktails(Fisher Scientific, Pittsburg, PA), sonicated for 10 s,and spun at 13000 rpm for 30min at 48C. Purifiedprotein concentration was then calculated by theBradford method, and boiled for 5min. Normalizedlysate was resolved by 4–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (In-vitrogen, Carlsbad, CA) and transferred to PVDFmembranes. Commercial antibodies used included:Rad52 (Cell Signaling, Danvers, MA), Rad52 (SantaCruz, Dallas, TX), Anti-phospho-Histone H2A.X(Ser139) (EMD Millipore, Billerica, MA), p21 (F-5)(Santa Cruz), and b-actin (Santa Cruz). Immunoblotswere developed using Clarity Western ECL Substrate(BioRad, Hercules, CA) and quantitated on a Chem-iDoc XRSþ Imaging System (BioRad).
Cellular Senescence by SA-b-Gal Staining
Cellular senescence was assayed as described previ-ously [15]. Briefly, equalnumbersofE10 Immortalized,nontumorigenic mouse lung epithelial cells stablyinfected with either scrambled pLKO.1 vector orshRNA constructs targeting the Rad52 gene(shRad52-03,04) were plated in 6-well sterile tissueculture plates 48 hours prior to analysis and at aconfluence of approximately 30%. Senescence wasmeasured according to manufacturer’s protocol (CellBiolabs, San Diego, CA). Briefly, the cells were washedwith PBS. After washing, 1� Fixing Solution (25%glutaraldehyde) was added to each well and the cellswere incubated at room temperature for 5min.Fixation solution was removed, the cells were washedin PBS and cell stainingworking solutionwas preparedfresh and added to each well. The cells were thenincubated overnight at 378C, protected from light. Thedevelopment of blue color was detected visually undera light microscope. Data correspond to the percentageof SA-b-gal-positive cells counted for each cell popula-tion. Numbers on the graphs indicate a percentage ofSA-b-gal-positive cells andwere derived from counting200 cells in triplicate plates.
Apparent Doubling Time
E10 cells were infected with either Scr. pLKO.1 orshRad52-03. Cells underwent puromycin selection for
AMPLIFICATION OF RAD52 EXPRESSION IN LUSC TUMORS 3
Molecular Carcinogenesis

48h until all cells in the mock infected plate weredead. Puromycin selected populations were thenseeded at equal densities, while remaining in mediawith puromycin, and incubated under normal growthconditions for 48–72h. Cell number and viabilitywere determined by the Countess Automated CellCounter (Invitrogen) and apparent doubling timewascalculated. 2x¼n final/n start for the number ofdoublings (x) and (h/x) for the apparent doublingtime, where h is the varying periods of time.
Cell Proliferation Curves and Plating Efficiency
Cells were harvested with trypsin, counted on theCountess automated cell counter (Invitrogen) andplated at a density of 3000 cells per well in multiplereplicates on a 96-well tissue culture plate. Cellnumbers were measured starting 4 d after infectionand 2d after completing selection in puromycin,although cells remained on puromycin throughoutthe length of the experiment to ensure measurementof the correct population. Cell confluence wasmeasured every two hours on an Incucyte live cellimager (Essen Biosciences, Ann Arbor, MI), throughphotomicrographs. Cell culture confluence wasmeasured using Incucyte software (EssenBiosciences).
Analysis of Cell Cycle Distribution by Propidium IodideStaining
Mouse tumor cell lines were cultured in RPMI with10% serum over a period of 14d. Flow cytometry wasemployed to analyze cell cycle distribution ontriplicate samples of cells using ethanol fixing andpropidium iodide staining with RNAse to assessrelative DNA content. Sub-confluent cultures of cellswere harvested by trypsinization. Post harvest cellswere centrifuged in 15ml conical tubes at 1000 rpmfor 5min, re-suspended in cold PBS, then centrifugedagain in the 15ml conical at 500�g for 20min. Next,the supernatant was discarded and 1ml cold PBS wasadded to wash the cells. The cells were thencentrifuged again at 500�g for 20min, the superna-tant was discarded and the cells were thoroughlysuspended in 500ml cold PBS, avoiding formation ofcell clumps. Next, using the Cell Countess, cells weremeasured to a concentration of 2�106 cells/ml inPBS. For fixation, 5ml of ice cold 95%/absoluteethanol was added to the cells in PBS while gentlyvortexing. Cells were then held on ice for 2h tocomplete fixation. For staining, cells were spun downat 1500 rpm for 5min to pellet. The ethanol wasaspirated and the cells were washed twice with PBS,leaving a small amount to re-suspend the cells in. Onemilliliter of PI staining solution (900ml PBS, 50ml PIstock solution, 50ml RNase stock solution) was thenadded to the cell pellet. Finally, the cells wereincubated in the dark for 3h at 48C to ensure theRNase had digested all the RNA. Flow cytometry wasperformed on the Accuri C6 cytometer (BD
Biosciences, San Jose, CA). Data were compiled andanalyzed using CFlow Plus and FlowJo software.
Comet Assay
A Comet Assay kit (Trevigen, Helgermen CT) wasused to quantify and analyze DNA damage inindividual E10 and Spon2 cells. The image that isobtained resembles a “comet” with a distinct headand tail. The head is composed of intact DNA, whilethe tail consists of damaged DNA. Cells are scoredbased on their Olive Tail Moment (OTM). The OTM isdefined as the product of the tail length and thefraction of total DNA in the tail. One group ofshSCRAM cells in each cell line was treated withhypochlorous acid (HOCL) at 48C in suspension toserve as a positive control for DNA damageaccumulation.
RESULTS
eQTL Containing RAD52 is Associated With Lung CancerDevelopment
We found two SNPs located in RAD52 that areassociated with RAD52 gene expression in both lungtumor tissue aswell as with non-small cell lung cancerphenotypes, especially LUSC. The first SNP,rs6413436, is significantly associated with LUSC inour GWAS cohort (P¼1.0�10�3 after multipletesting adjustment, odds ratio (OR)¼1.06 for overalllung cancer (95%CIs: 1.024–1.10), n¼28–998; P¼2.7�10�3, OR¼1.09 for LUSC (95%CIs: 1.03–1.15),n¼19,437). Although this SNP is not genotyped inour eQTL cohort of 70 lung tissues, it is in stronglinkage disequilibrium (LD) (D0 ¼1 and r2¼0.82) witha nearby SNP, rs10744729, which we have genotypedin our eQTL cohort and found to be significantlyassociated with RAD52 gene expression (P¼0.01,n¼70). Figure 1A shows the linkage disequilibriumplot of the tagged SNPs for the RAD52 region based ondata from the Caucasian population of the 1000genomes project. SNPs rs6413436 and rs10744729 arelocated in RAD52 intron 11 and intron 9, and thedistance between them is relatively small, at about1.9 kb. These are the two tagged SNPmarkers in Block1 of RAD52 (marker #4 and #6 in Figure 1A red box).The SNP rs10744729 also showed a significantassociation with LUSC in our GWAS study (P¼0.02after multiple testing adjustment). And the linkagedisequilibriumbetween rs10744729 and rs6413436 inour dataset is close to complete LD (r2¼0.82). There iscomplete LD between these two SNPs as revealed byHapMap data for whites (r2¼1, Figure 1A). It is thusreasonable to conclude that rs10744729 is a proxySNP for rs6413436. The significant association of bothrs10744729 and rs6413436 with LUSC could beexplained by the strong LD between these twoSNPs. This plus the fact that rs10744729 wassignificantly associated with RAD52 expression inlung tissues, suggests that these two SNPs were eQTL
4 LIEBERMAN ET AL.
Molecular Carcinogenesis

SNPs that associate with LUSC risk by influencingRAD52 gene expression.Figure 1B demonstrates that the risk allele “A” of
rs10744729 (in LDwith the lung cancer risk allele “C”of rs6413436) is associated with increased expressionof RAD52 in a dose-dependent manner (homozygousreference alleles “CC” < heterozygous “AC” <homozygous mutants “AA”). ANOVA analysisshowed that this SNP explains 9.2% of the expressionvariance of RAD52. Thus, the eQTL for rs10744729and its associated SNP, rs6413436, accounts for asmall proportion of the variance in RAD52 expressionin lung tissues, an observation consistent with therelativelymodest genetic risk observedwith these twovariants for LUSC (OR¼1.09). By contrast, nosignificant association with lung adenocarcinoma(LUAD) was found for these two SNPs (P¼0.24,n¼19589), suggesting that these two eQTL SNPswerenot related to the LUAD etiology.
Somatic Copy Number Analysis in the RAD52 GeneRegion Using TCGA Data
Using GISTIC software, somatic copy numberanalysis in 358 TCGA lung squamous cell carcinomapatients showed that the genomic region of 12p13.33containing RAD52 reached a statistically significantlevel for focal amplification (q-value¼2.56�10�3,much less than the genome wide cutoff q¼0.25,Figure 2). Thirty-eight of 358 lung SCC tumors (11%)
showed copy number gains in the RAD52 region(segments were reported as amplified if the corre-sponding estimated copy number ratio was greaterthan 1.25, i.e., log2 ratio > 0.32). These 38 identifiedTCGA LUSC tumors and their copy number amplifi-cation at the RAD52 gene region are listed inSupplemental Table S1. Gene expression levels ofRAD52 are also positively correlated with somaticcopy number gains in the RAD52 region in the TCGALUSC samples (Supplementary Figure S1), suggestingthat the somatic copy number gains in the LUSCtumors led to the corresponding increase in RAD52expression.
Other Significant RAD52 SNPs Influencing LUSC
We identified a number of RAD52 SNPs whichshowed more significant P values (P values<10�6
(Supplemental Table S2) for the associations withsquamous cell lung cancer phenotype. However,these SNPs were not associated with RAD52 geneexpression in our samples, suggesting that othermechanisms may underlie their associations withLUSC. The RAD52 SNPs we presented here are theones associatedwith bothRAD52 gene expression andLUSC risk. As well, these are cis-eQTL SNPs that mayinfluence LUSC phenotype by affecting the expres-sion level of RAD52. We replicated the significantassociation between RAD52 SNP rs6489769 and therisk of squamous cell carcinoma P¼9.4�10�7,
Figure 1. (A) Linkage disequilibrium (LD) analysis of tagging SNPsbased on the data of the Caucasian population used in 1000 genomeproject. The two cis-eQTL SNPs for RAD52 expression in lung tissuewere rs6413436 and rs10744729, which were in strong linkagedisequilibrium (D0 ¼ 1 and r2¼ 0.82 in our own sample and r2¼ 1 inHapMap Caucasian sample). SNPs rs6413436 and rs10744729 are
located in RAD52 intron 11 and intron 9, and the distance betweenthem is about 1.9 kb. They are the two tagging SNP markers in Block 1of RAD52 (marker #4 and #6 in red box). Therefore, rs10744729 is a“proxy” SNP for rs6413436. (B) The cis-eQTL SNP rs10744729 wassignificantly associated with the RAD52 gene expression (P¼ 0.01,n¼ 70).
AMPLIFICATION OF RAD52 EXPRESSION IN LUSC TUMORS 5
Molecular Carcinogenesis

(Supplemental Table S2) which was originally re-ported by Shi et al. [3]; however, this SNP does notshow significant association with our RAD52 geneexpression data, suggesting it may not be a cis-eQTLSNP in RAD52. In fact, Shi et al. [3] did not find thatthe SNP rs6489769 was associated with RAD52 geneexpression. Current evidence suggests that rs6489769may influence one’s risk of developing LUSC viamechanisms other than RAD52 expression.
Increased RAD52 Gene Expression in LUSC Samples FromNSCLC Cohort
Based on our independent non-small cell lungcancer (NSCLC) patient dataset, we found that RAD52was significantly overexpressed in the tumors of LUSCpatients (P¼0.05, n¼9, Figure 3). Specifically, therewas an average of a 50% increase in RAD52 expressionin LUSC tumors compared to that in normal tissuesamples (Figure 3). However, there was no significantdifference in RAD52 expression between the tumorand normal lung tissues of LUAD patients (P¼0.29,n¼36). This data suggests that RAD52 overexpressioncould contribute to LUSC etiology but not to LUAD
Figure 2. Somatic copy number analysis using the GISTIC program on 358 TCGA lung squamous cell carcinomapatients showed that the genomic region of 12p13.33 containing RAD52 reached a statistically significant level forfocal amplification (q-value¼ 2.56� 10�3, much less than the genome-wide cutoff q¼ 0.25).
Figure 3. Overexpression of RAD52 in lung squamous cell carcinomatumors observed in our independent NSCLC patient dataset. RAD52was significantly overexpressed in the tumors of LUSC patients(P¼ 0.05, n¼ 9). There was an average of a 50% increase in RAD52gene expression in LUSC tumors compared to normal tissues.
6 LIEBERMAN ET AL.
Molecular Carcinogenesis

carcinogenesis, which is consistent with our eQTLstudy showing that the identified eQTL SNPs inRAD52 are significantly associated with LUSC but notLUAD.
Loss of Rad52 Induces Senescence in Mouse ImmortalizedLung Epithelial Cells
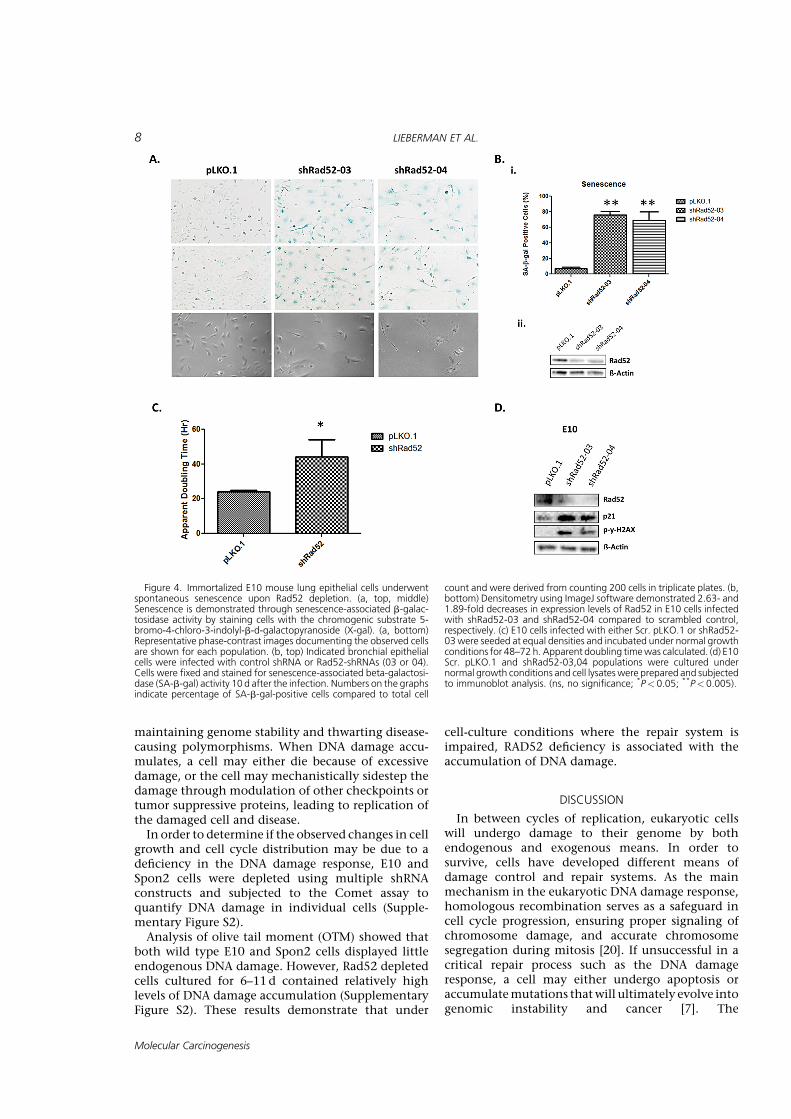
Although Rad52 acts in a parallel fashion to theknown tumor suppressor BRCA2, depletion of Rad52has actually been shown to slow cell proliferation inMCF7 breast cancer cells [8]. This suggests thatalthough the inherent function of Rad52 is to repairdamaged DNA, in a pro-tumorigenic setting, it mayactually function to enhance cell proliferation or toprotect tumor cells from apoptosis. Given Rad52’srecent identification as a susceptibility candidateassociated with squamous cell lung carcinoma, wehypothesized that depletion of Rad52 might be afactor in the survival and proliferation of lung andlung tumor cells. To determine the effect of Rad52depletion in mouse bronchial cells, we treatedimmortalized, non-tumorigenic mouse bronchialepithelial cells with either shRNA targeting RAD52or a scrambled vector. RAD52 was depleted at theprotein level from the E10 cells by 2.63- and 1.89-fold using shRNA constructs 03 and 04, respectively.The cells were cultured for 7–10 d post-infectionbefore they stopped dividing, became morphologi-cally round and flattened, and stained positive forsenescence associated b-galactosidase activity, indi-cating that loss of RAD52 resulted in senescence(Figure 4).Phosphorylation of g-H2AX is an early sign of DNA
damage induced by replication stalling [16]. The roleof g-H2AX foci in theDNA damage signaling pathwayinduced by double-strand breaks is to act as a dockingarea for the recruitment of repair factors and to bringthe broken DNA ends closer so that DNA repair isaccomplished [17]. Beard et al. found that uponinduction ofDNAdamage, g-H2AX-proficient cells, aswell as H2AX-complemented H2AX(�/�), cells reactedby increasing p21 levels and arresting the cell cycle. Asour results also suggest, (Figure 4D), H2AX is requiredfor p21-induced cell cycle arrest after replicationstalling. In addition, g-H2AX has been shown to beessential for concentrating repair proteins and main-taining the integrity of the DNA damage responsefoci [18]. Finally, the significance ofH2AX in cell cyclearrest was previously demonstrated by Fernandez-Capetillo et al. [19]. This serves to confirm theobserved senescence in our E10 bronchial epithelialcells and to suggest a further role for the DNA damageresponse in the aforementioned growth arrest discov-ered upon Rad52 depletion.
Expression of RAD52 Affects Growth in Mouse LungEpithelial and Tumor Cells
Utilizing distinct viral transfections, equal numbersof viable Spon2, Spon6, CL25M, and E10 wild-type
and RAD52-depleted cells were seeded in the Incucytelive cell imager, and the growth ratewas observedovera period of 3–5d. Upon depletion of RAD52 in mouselung epithelial cells, we observed the rate of cellproliferation begin to decrease around 96h postinfection and 48h post completion of puromycinselection, prior to the appearance of a senescentphenotype. Cellular proliferation in lung tumor celllines (Spon2, Spon6, and CL25M) also decreased withdepletion of RAD52 (Figure 4A). Admittedly, werecognize that percent confluence is not a puremeasure of cell number; however, doubling timeassays in E10 cells (Figure 4) along with MTSmeasurements of cell proliferation confirm that cellmorphology did not affect proliferation rates. Ineffect, any error caused by differences in cellmorphology would likely function to enlarge cellsdepleted of RAD52, enhancing their rate ofconfluence.
In comparing the apparent doubling time of E10cells, we determined that depletion of RAD52 led to asignificant increase in apparent doubling time(*P<0.05), increasing 1.83-fold from those infectedwith a scrambled control shRNA (Figure 4C). More-over, although E10 cells overexpressing RAD52 didnot show a growth difference, all tumor cell linesoverexpressing RAD52 showed a significant increasein growth rate around 3d post infection.
Depletion of RAD52 Alters Cell-Cycle Distribution in LungTumor Cells and Induces Accumulation of Cells in G2/M
To further investigate whether changes in cell-cycle regulation correlate with the observedchanges in cell growth, we compared cell-cycleprofiles of different mouse lung tumor cell linesthrough flow cytometry with propidium iodide (PI)staining. The E10 mouse bronchial epithelial cellline became senescent post-RAD52 knockdown,which prevented the acquisition of enough cellsfor flow cytometry. Results from these studiesdemonstrate accumulation of tumor cell popula-tions in the G2/M fraction and a decrease in cellpopulations in the G0/G1 fraction consistent withdeficient DNA repair-induced senescence (Figure 6).These data strongly support a role for RAD52 in G2/M progression and/or passage through the G2/Mcheckpoint in mouse lung tumor cells. This G2delay is likely to be the major determinant of theslower growth rate seen for the RAD52-depletedmouse cells (Figure 5). A less severe profile was seenin the Spon2 cell line (Figure 6A). This indicatesthat other factors besides Rad52 expression may berate-limiting in the passage of Spon2 cells beyondthe G2 phase.
Loss of RAD52 Induces Spontaneous DNA Damage inImmortalized Bronchial Epithelial and Lung Tumor Cells
Highly efficient and accurate DNA repair through-out the replication cycle of a cell is critical in
AMPLIFICATION OF RAD52 EXPRESSION IN LUSC TUMORS 7
Molecular Carcinogenesis
maintaining genome stability and thwarting disease-causing polymorphisms. When DNA damage accu-mulates, a cell may either die because of excessivedamage, or the cell may mechanistically sidestep thedamage through modulation of other checkpoints ortumor suppressive proteins, leading to replication ofthe damaged cell and disease.
In order to determine if the observed changes in cellgrowth and cell cycle distribution may be due to adeficiency in the DNA damage response, E10 andSpon2 cells were depleted using multiple shRNAconstructs and subjected to the Comet assay toquantify DNA damage in individual cells (Supple-mentary Figure S2).
Analysis of olive tail moment (OTM) showed thatboth wild type E10 and Spon2 cells displayed littleendogenous DNA damage. However, Rad52 depletedcells cultured for 6–11d contained relatively highlevels of DNA damage accumulation (SupplementaryFigure S2). These results demonstrate that under
cell-culture conditions where the repair system isimpaired, RAD52 deficiency is associated with theaccumulation of DNA damage.
DISCUSSION
In between cycles of replication, eukaryotic cellswill undergo damage to their genome by bothendogenous and exogenous means. In order tosurvive, cells have developed different means ofdamage control and repair systems. As the mainmechanism in the eukaryotic DNA damage response,homologous recombination serves as a safeguard incell cycle progression, ensuring proper signaling ofchromosome damage, and accurate chromosomesegregation during mitosis [20]. If unsuccessful in acritical repair process such as the DNA damageresponse, a cell may either undergo apoptosis oraccumulatemutations thatwill ultimately evolve intogenomic instability and cancer [7]. The
Figure 4. Immortalized E10 mouse lung epithelial cells underwentspontaneous senescence upon Rad52 depletion. (a, top, middle)Senescence is demonstrated through senescence-associated b-galac-tosidase activity by staining cells with the chromogenic substrate 5-bromo-4-chloro-3-indolyl-b-d-galactopyranoside (X-gal). (a, bottom)Representative phase-contrast images documenting the observed cellsare shown for each population. (b, top) Indicated bronchial epithelialcells were infected with control shRNA or Rad52-shRNAs (03 or 04).Cells were fixed and stained for senescence-associated beta-galactosi-dase (SA-b-gal) activity 10 d after the infection. Numbers on the graphsindicate percentage of SA-b-gal-positive cells compared to total cell
count and were derived from counting 200 cells in triplicate plates. (b,bottom) Densitometry using ImageJ software demonstrated 2.63- and1.89-fold decreases in expression levels of Rad52 in E10 cells infectedwith shRad52-03 and shRad52-04 compared to scrambled control,respectively. (c) E10 cells infected with either Scr. pLKO.1 or shRad52-03were seeded at equal densities and incubated under normal growthconditions for 48–72 h. Apparent doubling timewas calculated. (d) E10Scr. pLKO.1 and shRad52-03,04 populations were cultured undernormal growth conditions and cell lysateswere prepared and subjectedto immunoblot analysis. (ns, no significance; *P< 0.05; **P< 0.005).
8 LIEBERMAN ET AL.
Molecular Carcinogenesis

complementary evidence determined by our geneticand functional studies suggests a potential role for theDNA repair gene, RAD52, in lung tumorigenesis. Ourresearch into the role of RAD52 in susceptibility tolung carcinogenesis emerged from prior findingsimplicating variation in RAD52 as a factor in LUSCdevelopment [3]. Here, we have identified twocandidate regions in RAD52 where variations in thegene are associated with increased copy number aswell as increased RAD52 expression in lung tumortissue compared to normal.A study performed by the Barlow group demon-
strated that by reducing homologous recombinationinATM-deficientmice through genetic deletionof theRad52 gene, not only did the development of T-celllymphomas decrease, butmice depleted of Rad52 alsolived longer [21]. This report suggests that excessivehomologous recombination, termed hyperrecombi-nation, by a member of the normal DNA damageresponse may contribute to tumorigenesis. Thishyperrecombinogenic phenotype is known to betoxic to the cell, leading to chromosomal instabilityand disease through an accumulation of lethalrecombination variants [21,22]. In A-T, diseasephenotypes are believed to arise from a loss of ATMkinase activity as well as disrupted signaling toproteins such as p53, chk2, and DNA-repair proteins,
such as Rad52, resulting in cell cycle checkpointdefects and impaired DNA repair [21]. Barlow’s grouptested the hypothesis that excessive recombinationmight be responsible for the overlapping phenotypesin A-T and Blooms in vivo by crossing Atm�/� micewith mice deficient in the HR protein Rad52 [21]. Hergroup clearly demonstrated that by reducing homol-ogous recombination through genetic deletion of theRad52 gene, they were able to significantly decreaseand delay the incidence of thymic tumors in Atm�/�
mice: more than half of the Atm�/� mice developedtumors by 4months of age compared to less than 20%of the Atm�/� Rad52�/� mice [21].
Therefore, when considering current GWAS studies,our eQTL analysis, and recent experimental studies, itis reasonable to suggest that although chromosomalinstability is not rescued in the absence of Rad52,genetic deletion of Rad52 may result in a reduction ofabnormally high levels of intrachromosomal recombi-nation and that this reduction by Rad52 reducestumorigenesis by protecting genome integrity. Giventhe recent genomic data linking Rad52 to lung canceralong with our data demonstrating Rad52 amplifica-tion inLUSCcases, alongwith theknownroleofRad52in recombination, it is rational to suggest that over-expressionof Rad52would aid in the development of alung cancer phenotype.
Figure 5. Cellular proliferation of lung tumor and bronchialepithelial cells with altered RAD52 expression. (a) Cellular proliferationin lung tumor cells (Spon2, Spon6, andCL25M) and bronchial epithelialcells (E10) transfected with shRNA against Rad52 compared toscrambled control. (b) Cellular proliferation in lung tumor cells(Spon2, Spon6, and CL25M) and bronchial epithelial cells (E10)
transfected with the pOPUR Rad52 overexpression plasmid comparedto vector. (c, d) Transfected populations from all cell lines were culturedunder normal growth conditions for 7–10 d, Cell lysates were preparedand subjected to immunoblot analysis. (ns, no significance; *P< 0.05;**P< 0.005).
AMPLIFICATION OF RAD52 EXPRESSION IN LUSC TUMORS 9
Molecular Carcinogenesis

As expected from the Barlow study, we found thatnot only did lung tumor tissue samples express higherlevels of RAD52 compared to normal lung tissue, butdepleting RAD52 in functional in vitro studies led tophenotypes associated with decreased tumorigenesis.As our functional studies show, overexpression ofRad52 leads to an increased rate of cell growth, aprominent tumorigenic phenotype, further support-ing a role for Rad52 in the development of lungcancer.
As mentioned above, in this study we sought todetermine the effect of loss and overexpression of
RAD52 on such phenotypes associated with lungtumorigenesis including induction of senescence,rates of cell proliferation, altered cell cycle distribu-tion and accumulation of DNA damage. Currentliterature implicates a connection between a highfrequency of structural variation in the genome withphenotypic diversity and predisposition to dis-ease [23]. Hence, it would be reasonable to believethat DNA repair genes such as RAD52 may modulateoncogenic phenotypes related to DNA damage andgenomic variation. We observed that E10 immortal-ized mouse lung epithelial cells went into a state of
Figure 6. Depletion of Rad52 alters the cell-cycle distribution of lungtumor cells inducing a decrease in cells in G0/G1 and an accumulationof cells in G2/M. (a) Cell cycle progression was assessed by propidiumiodide staining as a measure of DNA content. Flow cytometry wasperformed on CL25M, Spon2, and MC7 Rad52-depleted andscrambled vector control cells. Fixed cells were acquired using theAccuri C6 Cytometer. (b) Cells were then analyzed using FlowJo
software and theWatson–Pragmatic cell cycle method and percentagevalues for G0/G1 (2 NDNA content), S (intermediate DNA content), andG2/M (4N DNA content) cells. (c) Confirmation of knockdown ofRad52 in mouse tumor cell lines using the shRad52-03 construct andscrambled control shRNA by Western blot. (ns, no significance;*P< 0.05; **P< 0.005).
10 LIEBERMAN ET AL.
Molecular Carcinogenesis

senescence 7–10d post infection with shRNA con-structs targeting Rad52. E10 cells express high levelsof the known oncogene MDM2 and are ARF/p53-inactivated [13]. Thus, it makes sense that depletionof Rad52 in E10 cells lead to upregulation of p21 andphospho-g H2AX expression, similar to observationsmade by the MacLaren group upon knockdown of c-Jun [24]. This group found a potential role for c-Jun inthe DNA damage response, linking its depletion topremature senescence, an accumulation of cells inG2/Mphase of the cell cycle, and a persistence of DNAdamage [24].Furthermore, inmouse lung tumor cells, knockdown
of Rad52 curbed proliferation and led to an accumula-tionof cells in theG2/Mphaseof thecell cycle (Figures 5and 6). Alteration of cell-cycle regulation by RAD52 hasnotbeendemonstratedpreviously. TheobservedG2/M-accumulation upon RAD52 depletion may be due tovariation in expression of other proteins or increasedsensitivity to culture conditions.Little is known of the effects of variation in RAD52
in regards to susceptibility to lung cancer and tumordevelopment. However, genetic variations that alterhomologous recombination, coupled with a carcino-genic environment, may plausibly contribute totumorigenesis. Further understanding of the rolethat RAD52 plays in the development of NSCLC willultimately yield clinically significant results throughdetermination of prediction markers and prognosticfactors in regard to LUSC therapeutics.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CACancer J Clin 2014;64:9–29.
2. Halmos B, Boiselle PM, Karp DD. Lung cancer. Prim CareUpdate Ob Gyns 2003;10:87–94.
3. Shi J, Chatterjee N, Rotunno M, et al. Inherited variation atchromosome 12p13.33, including RAD52, influences the risk ofsquamous cell lung carcinoma. Cancer Discov 2012;2:131–139.
4. Lisby M, Antunez de Mayolo A, Mortensen UH, Rothstein R.Cell cycle-regulated centers of DNA double-strand breakrepair. Cell Cycle 2003;2:479–483.
5. LisbyM, Rothstein R, Mortensen UH. Rad52 forms DNA repairand recombination centers during S phase. Proc Natl Acad SciUSA 2001;98:8276–8282.
6. WangY,McKay JD, Rafnar T, et al. Rare variants of large effectin BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet2014;46:736–741.
7. Lok BH, Powell SN. Molecular pathways: Understanding therole of Rad52 in homologous recombination for therapeuticadvancement. Clin Cancer Res 2012;18:6400–6406.
8. Feng Z, Scott SP, Bussen W, et al. Rad52 inactivation issynthetically lethal with BRCA2 deficiency. Proc Natl Acad SciUSA 2011;108:686–691.
9. Rijkers T, Van Den Ouweland J, Morolli B, et al. Targetedinactivation of mouse RAD52 reduces homologous recombi-nation but not resistance to ionizing radiation. Mol Cell Biol1998;18:6423–6429.
10. Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation issynthetically lethal with deficiencies in BRCA1 and PALB2 inaddition to BRCA2 through RAD51-mediated homologousrecombination. Oncogene 2013;32:3552–3558.
11. Li Y, Sheu CC, Ye Y, et al. Genetic variants and risk of lungcancer in never smokers: A genome-wide association study.Lancet Oncol 2010;11:321–330.
12. Govindan R, Ding L, Griffith M, et al. Genomic landscape ofnon-small cell lung cancer in smokers and never-smokers. Cell2012;150:1121–1134.
13. McDoniels-Silvers AL, Herzog CR, Tyson FL, Malkinson AM,You M. Inactivation of both Rb and p53 pathways in mouselung epithelial cell lines. Exp Lung Res 2001;27:297–318.
14. Kim PM, Allen C, Wagener BM, Shen Z, Nickoloff JA.Overexpression of human RAD51 and RAD52 reducesdouble-strand break-induced homologous recombination inmammalian cells. Nucleic Acids Res 2001;29:4352–4360.
15. Zhuang D, Mannava S, Grachtchouk V, et al. C-MYCoverexpression is required for continuous suppression ofoncogene-induced senescence in melanoma cells. Oncogene2008;27:6623–6634.
16. Fragkos M, Jurvansuu J, Beard P. H2AX is required for cellcycle arrest via the p53/p21 pathway. Mol Cell Biol2009;29:2828–2840.
17. Franco S, Gostissa M, Zha S, et al. H2AX prevents DNA breaksfrom progressing to chromosome breaks and translocations.Mol Cell 2006;21:201–214.
18. Celeste A, Fernandez-Capetillo O, Kruhlak MJ, et al. HistoneH2AX phosphorylation is dispensable for the initial recogni-tion of DNA breaks. Nat Cell Biol 2003;5:675–679.
19. Fernandez-Capetillo O, Chen HT, Celeste A, et al. DNAdamage-induced G2-M checkpoint activation by histoneH2AX and 53BP1. Nat Cell Biol 2002;4:993–997.
20. Liu J, HeyerWD.Who's who in human recombination: BRCA2and RAD52. Proc Natl Acad Sci USA 2011;108:441–442.
21. Treuner K, Helton R, Barlow C. Loss of Rad52 partially rescuestumorigenesis and T-cell maturation in Atm-deficient mice.Oncogene 2004;23:4655–4661.
22. Heyer WD, Ehmsen KT, Liu J. Regulation of homologousrecombination in eukaryotes. AnnuRevGenet 2010;44:113–139.
23. Feuk L, Carson AR, Scherer SW. Structural variation in thehuman genome. Nat Rev Genet 2006;7:85–97.
24. MacLaren A, Black EJ, Clark W, Gillespie DA. C-Jun-deficientcells undergo premature senescence as a result of spontane-ous DNA damage accumulation. Mol Cell Biol 2004;24:9006–9018.
SUPPORTING INFORMATION
Additional supporting information may be found inthe online version of this article at the publisher’sweb-site.
AMPLIFICATION OF RAD52 EXPRESSION IN LUSC TUMORS 11
Molecular Carcinogenesis
Related Documents











