Methodology challenges in studying human gut microbiota – comparison of illumina and Ion Torrent NGS platforms Hana Čipčić Paljetak 1 , Anja Barešić 2 , Mihaela Perić 1 , Marina Panek 1 , Mario Matijašić 1 , Darija Vranešić Bender 3 , Ana Kunović 3 , Željko Krznarić 3 , Donatella Verbanac 1 1 University of Zagreb School of Medicine, Zagreb, Croatia; 2 Imperial College London, London, UK; 3 University Hospital Centre Zagreb, Croatia [email protected] INTRODUCTION The human gut contains dense and diverse microbial communities which have profound impact on human health. To understand microbiota complexity, reliable technological solutions for determination of bacterial community structure are needed. The quality of final output is influenced by the methodologies along the pipeline – sample collection, storage, DNA extraction and sequencing approach [1]. As number of microbiota studies increases continually, it is important to ensure comparability and meaningful data interpretation both within and between studies. Therefore, efforts aiming at comparing different workflows are useful for much needed methodology standardisation. REFERENCES [1] Salipante et al. Appl. Environ. Microbiol. 2014, 80 (24), 7583-7591. [2] Caporaso et al. Nat. Methods 2010, 7 (5), 335-336. [3] DeSantis et al. Applied and Environmental Microbiology 2006, 72 (7), 5069-5072. [4] Perić et al. Evaluation of human faecal microbiota content by 16S rRNA analysis using different collection, storage and DNA extraction methods – OMNIgene.GUT case study. 3rd World Congress on Targeting Microbiota, Paris, France, Oct 21-23, 2015. This work has been fully supported by the Croatian Science Foundation (project number 5467) AIM The objective of this study was to compare results obtained by 16S rRNA gene sequencing of human faeces samples on two NGS platforms – illumina MiSeq and Ion Torrent PGM. MATERIALS & METHODS Samples from 4 healthy donors were collected fresh and in OMNIgene.GUT device, then stored for 14 days at -20°C and at room temperature, respectively. Three commercially available DNA extraction kits (MO BIO Power faecal DNA isolation kit, QIAamp Fast DNA Stool Mini Kit and MP Biomedicals Fast DNA spin kit for feces) were employed. In total, six samples per donor were prepared. After extraction DNA samples integrity was tested on 1% agarose gel electrophoresis while purity and concentration were verified using Qubit fluorometer. Faecal bacterial communities were profiled by 16S rRNA amplicon sequencing using two NGS platforms MiSeq (illumina MiSeq, regions V3-V4) and IT (Ion Torrent PGM, regions V2-V9). Manufacturer recommended reagents and protocols were applied: for MiSeq Nextera XT Index Kit, MiSeq Reagent Kit v3 and pair end sequencing protocol and for IT Ion Hi-Q Sequencing kit and 4 316 chip. Sequence analyses were performed using QIIME protocol [2]: OTU assignment procedure for demultiplexed and trimmed reads was performed against the Greengenes reference database version 13_8 (May 2013) [3] by the uclust algorithm. RESULTS In our previous work, we have shown that DNA yield and quality varied between DNA extraction kits (MP Biomedicals>QIAGEN>MO BIO) [4]. Here we compare MiSeq and Ion Torrent NGS platforms at the level of overall discrimination at different taxonomic levels, as well as in terms of correlation with other experimental parameters. Donor-specific patterns of bacterial diversity were maintained irrespective of the collection, storage or DNA extraction method used (principal component analysis (PCA), Figure 1). Platform-based differentiation could be observed (PC2 vs PC3). Kit- based trends are indicated as well and the main differentiation seems to occur between MP Biomedicals and MO BIO kits. Variability due to collection and storage methodology was not observed in PCA (not shown). The number of families detected in the entire sample pool, as presented on Venn diagram, was 118 and 130 for MiSeq and Ion Torrent, respectively, while the number of overlapping families was 86 (Figure 2A). When families detected only once per platform were removed, the number of overlapping families was 74 (MiSeq detected 109 and IT 92 families) (Figure 2B). The total number of families per sample detected on both platforms is shown in Figure 2C. To check if distinct relative abundance patterns due to sequencing on different platforms can be observed, relative abundance of the overlapping families within the entire sample pool detected by each NGS platform was compared, in order to exclude variances due to other experimental parameters. Different levels of abundance were observed depending on the platform, especially among the less abundant families (Figure 3). However, comparison at the phylum level reveals the ten most abundant phyla, covering 99,9% of the assigned OTUs, occurred in similar ratios on both platforms, with the exception of Fusobacteria and Verrucomicrobia (Figure 4). These phyla were represented in higher abundance in the samples sequenced on MiSeq. CONCLUSION The results reveal differentiation between NGS platforms as indicated by PCA analysis. However, donor specific microbiota composition is maintained irrespective of the platform, DNA extraction kit, collection and storage methodology. Although comparable numbers of families are identified across platforms, the families identified are not fully overlapping. The relative abundance ratio for the most abundant phyla was comparable across platforms, with the exception of Fusobacteria and Verrucomicrobia. Figure 2. A) Venn diagram of number of families detected using MiSeq and Ion Torrent NGS platforms. B) Number of families detected when those detected only once per platform are removed. C) Number of families detected on each platform per sample, coloured by donor. C Figure 3: Relative abundance ratio of overlapping families detected on both NGS platforms. 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Lachnospiraceae Ruminococcaceae Bacteroidaceae Clostridiaceae Streptococcaceae Prevotellaceae Porphyromonadaceae Coriobacteriaceae Enterobacteriaceae Rikenellaceae Veillonellaceae [Barnesiellaceae] Erysipelotrichaceae Pasteurellaceae Alcaligenaceae Bifidobacteriaceae Christensenellaceae [Odoribacteraceae] [Mogibacteriaceae] Peptostreptococcaceae Desulfovibrionaceae Enterococcaceae Lactobacillaceae Staphylococcaceae Actinomycetaceae Fusobacteriaceae Verrucomicrobiaceae Micrococcaceae Turicibacteraceae [Paraprevotellaceae] Victivallaceae Carnobacteriaceae Dehalobacteriaceae Oxalobacteraceae Pseudomonadaceae [Tissierellaceae] Corynebacteriaceae Leuconostocaceae Gemellaceae Moraxellaceae Xanthomonadaceae Peptococcaceae Caulobacteraceae Eubacteriaceae Sphingomonadaceae Campylobacteraceae At425EubF1 Planococcaceae Comamonadaceae Rhodobacteraceae S24-7 Burkholderiaceae Neisseriaceae Bradyrhizobiaceae Microbacteriaceae Rhodospirillaceae Vibrionaceae Aerococcaceae Rhodocyclaceae Synergistaceae Sphingobacteriaceae Paenibacillaceae [Chromatiaceae] Aeromonadaceae Leptotrichiaceae Listeriaceae Nitrosomonadaceae Mycobacteriaceae Syntrophobacteraceae Halomonadaceae Planctomycetaceae Rs-045 Cardiobacteriaceae Streptomycetaceae MiSeq Ion Torrent Figure 4: Relative abundance ratio of overlapping phyla detected on both NGS platforms. 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Firmicutes Bacteroidetes Proteobacteria Actinobacteria Cyanobacteria Tenericutes Fusobacteria Verrucomicrobia Lentisphaerae Acidobacteria MiSeq Ion Torrent Figure 1: Principle component analysis (PCA) of families detected. MiSeq samples are designated by dots and Ion Torrent by triangles. A) Coloured by donor B) Coloured by DNA extraction kit. First four components cover 99,88 % of total variance. A B

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Methodology challenges in studying human gut microbiota – comparison of illumina and Ion Torrent NGS platforms
Hana Čipčić Paljetak1, Anja Barešić 2, Mihaela Perić 1, Marina Panek 1, Mario Matijašić 1, Darija Vranešić Bender 3, Ana Kunović 3, Željko Krznarić 3, Donatella Verbanac1
1 University of Zagreb School of Medicine, Zagreb, Croatia; 2 Imperial College London, London, UK; 3 University Hospital Centre Zagreb, Croatia [email protected]
INTRODUCTION
The human gut contains dense and diverse microbial communities which have profound impact on human health. To understand microbiota complexity, reliable technological solutions for determination of bacterial community structure are needed. The quality of final output is influenced by the methodologies along the pipeline – sample collection, storage, DNA extraction and sequencing approach [1]. As number of microbiota studies increases continually, it is important to ensure comparability and meaningful data interpretation both within and between studies. Therefore, efforts aiming at comparing different workflows are useful for much needed methodology standardisation.
REFERENCES
[1] Salipante et al. Appl. Environ. Microbiol. 2014, 80 (24), 7583-7591. [2] Caporaso et al. Nat. Methods 2010, 7 (5), 335-336. [3] DeSantis et al. Applied and Environmental Microbiology 2006, 72 (7), 5069-5072. [4] Perić et al. Evaluation of human faecal microbiota content by 16S rRNA analysis using different collection, storage and DNA extraction methods – OMNIgene.GUT case study. 3rd World Congress on Targeting Microbiota, Paris, France, Oct 21-23, 2015.
This work has been fully supported by the Croatian Science Foundation (project number 5467)
AIM
The objective of this study was to compare results obtained by 16S rRNA gene sequencing of human faeces samples on two NGS platforms – illumina MiSeq and Ion Torrent PGM.
MATERIALS & METHODS
Samples from 4 healthy donors were collected fresh and in OMNIgene.GUT device, then stored for 14 days at -20°C and at room temperature, respectively. Three commercially available DNA extraction kits (MO BIO Power faecal DNA isolation kit, QIAamp Fast DNA Stool Mini Kit and MP Biomedicals Fast DNA spin kit for feces) were employed. In total, six samples per donor were prepared. After extraction DNA samples integrity was tested on 1% agarose gel electrophoresis while purity and concentration were verified using Qubit fluorometer.
Faecal bacterial communities were profiled by 16S rRNA amplicon sequencing using two NGS platforms MiSeq (illumina MiSeq, regions V3-V4) and IT (Ion Torrent PGM, regions V2-V9). Manufacturer recommended reagents and protocols were applied: for MiSeq Nextera XT Index Kit, MiSeq Reagent Kit v3 and pair end sequencing protocol and for IT Ion Hi-Q Sequencing kit and 4 316 chip.
Sequence analyses were performed using QIIME protocol [2]: OTU assignment procedure for demultiplexed and trimmed reads was performed against the Greengenes reference database version 13_8 (May 2013) [3] by the uclust algorithm.
RESULTS
In our previous work, we have shown that DNA yield and quality varied between DNA extraction kits (MP Biomedicals>QIAGEN>MO BIO) [4].
Here we compare MiSeq and Ion Torrent NGS platforms at the level of overall discrimination at different taxonomic levels, as well as in terms of correlation with other experimental parameters.
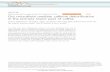
Donor-specific patterns of bacterial diversity were maintained irrespective of the collection, storage or DNA extraction method used (principal component analysis (PCA), Figure 1). Platform-based differentiation could be observed (PC2 vs PC3). Kit-based trends are indicated as well and the main differentiation seems to occur between MP Biomedicals and MO BIO kits. Variability due to collection and storage methodology was not observed in PCA (not shown).
The number of families detected in the entire sample pool, as presented on Venn diagram, was 118 and 130 for MiSeq and Ion Torrent, respectively, while the number of overlapping families was 86 (Figure 2A). When families detected only once per platform were removed, the number of overlapping families was 74 (MiSeq detected 109 and IT 92 families) (Figure 2B). The total number of families per sample detected on both platforms is shown in Figure 2C.
To check if distinct relative abundance patterns due to sequencing on different platforms can be observed, relative abundance of the overlapping families within the entire sample pool detected by each NGS platform was compared, in order to exclude variances due to other experimental parameters. Different levels of abundance were observed depending on the platform, especially among the less abundant families (Figure 3). However, comparison at the phylum level reveals the ten most abundant phyla, covering 99,9% of the assigned OTUs, occurred in similar ratios on both platforms, with the exception of Fusobacteria and Verrucomicrobia (Figure 4). These phyla were represented in higher abundance in the samples sequenced on MiSeq.
CONCLUSION
The results reveal differentiation between NGS platforms as indicated by PCA analysis. However, donor specific microbiota composition is maintained irrespective of the platform, DNA extraction kit, collection and storage methodology. Although comparable numbers of families are identified across platforms, the families identified are not fully overlapping. The relative abundance ratio for the most abundant phyla was comparable across platforms, with the exception of Fusobacteria and Verrucomicrobia.
Figure 2. A) Venn diagram of number of families detected using MiSeq and Ion Torrent NGS platforms. B) Number of families detected when those detected only once per platform are removed. C) Number of families detected on each platform per sample, coloured by donor.
C
Figure 3: Relative abundance ratio of overlapping families detected on both NGS platforms.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Lach
no
spir
acea
e
Ru
min
oco
ccac
eae
Bac
tero
idac
eae
Clo
stri
dia
ceae
Stre
pto
cocc
acea
e
Pre
vote
llace
ae
Po
rph
yro
mo
nad
acea
e
Co
rio
bac
teri
acea
e
Ente
rob
acte
riac
eae
Rik
enel
lace
ae
Vei
llon
ella
ceae
[Bar
nes
iella
ceae
]
Erys
ipe
lotr
ich
acea
e
Pas
teu
rella
ceae
Alc
alig
enac
eae
Bif
ido
bac
teri
acea
e
Ch
rist
ense
nel
lace
ae
[Od
ori
bac
tera
ceae
]
[Mo
gib
acte
riac
eae]
Pe
pto
stre
pto
cocc
ace
ae
Des
ulf
ovi
bri
on
ace
ae
Ente
roco
ccac
eae
Lact
ob
acill
acea
e
Stap
hyl
oco
ccac
eae
Act
ino
myc
etac
eae
Fuso
bac
teri
acea
e
Ver
ruco
mic
rob
iace
ae
Mic
roco
ccac
eae
Turi
cib
acte
race
ae
[Par
apre
vote
llace
ae]
Vic
tiva
llace
ae
Car
no
bac
teri
acea
e
Deh
alo
bac
teri
acea
e
Oxa
lob
acte
race
ae
Pse
ud
om
on
adac
eae
[Tis
sier
ella
ceae
]
Co
ryn
ebac
teri
acea
e
Leu
con
ost
oca
ceae
Gem
ella
ceae
Mo
raxe
llace
ae
Xan
tho
mo
nad
acea
e
Pe
pto
cocc
acea
e
Cau
lob
acte
race
ae
Eub
acte
riac
eae
Sph
ingo
mo
nad
acea
e
Cam
pyl
ob
acte
race
ae
At4
25E
ub
F1
Pla
no
cocc
acea
e
Co
mam
on
adac
eae
Rh
od
ob
acte
race
ae
S24
-7
Bu
rkh
old
eri
acea
e
Nei
sser
iace
ae
Bra
dyr
hiz
ob
iace
ae
Mic
rob
acte
riac
eae
Rh
od
osp
irill
acea
e
Vib
rio
nac
eae
Aer
oco
ccac
eae
Rh
od
ocy
clac
eae
Syn
ergi
stac
eae
Sph
ingo
bac
teri
acea
e
Pae
nib
acill
acea
e
[Ch
rom
atia
ceae
]
Aer
om
on
adac
eae
Lep
totr
ich
iace
ae
List
eria
ceae
Nit
roso
mo
nad
acea
e
Myc
ob
acte
riac
eae
Syn
tro
ph
ob
acte
race
ae
Hal
om
on
adac
eae
Pla
nct
om
ycet
acea
e
Rs-
045
Car
dio
bac
teri
acea
e
Stre
pto
myc
etac
eae
MiSeq
Ion Torrent
Figure 4: Relative abundance ratio of overlapping phyla detected on both NGS platforms.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Firm
icu
tes
Bac
tero
idet
es
Pro
teo
bac
teri
a
Act
ino
bac
teri
a
Cya
no
bac
teri
a
Ten
eric
ute
s
Fuso
bac
teri
a
Ver
ruco
mic
rob
ia
Len
tisp
hae
rae
Aci
do
bac
teri
a
MiSeq
Ion Torrent
Figure 1: Principle component analysis (PCA) of families detected. MiSeq samples are designated by dots and Ion Torrent by triangles. A) Coloured by donor B) Coloured by DNA extraction kit. First four components cover 99,88 % of total variance.
A
B
Related Documents