Research Overview Metal Ion Chelation in Neurodegenerative Disorders Itzchak Angel, n Adina Bar, Tammy Horovitz, Galia Taler, Michael Krakovsky, Dalia Resnitsky, Gilad Rosenberg, Sarina Striem, Jonathan E. Friedman, and Alex Kozak D-Pharm Ltd., Kiryat Weizmann Science Park, Rehovot, Israel Strategy, Management and Health Policy Venture Capital Enabling Technology Preclinical Research Preclinical Development Toxicology, Formulation Drug Delivery, Pharmacokinetics Clinical Development Phases I-III Regulatory, Quality, Manufacturing Postmarketing Phase IV ABTRACT Disturbance in metallochemical reactions and metal-protein association are associated with chronic neurodegenerative conditions, such as Alzheimer’s and Parkinson’s disease, as well as with neurodegeneration triggered by acute cerebral ischaemia. Many neurological diseases have been linked directly or indirectly to perturbed homeostasis of Ca, Fe, Zn, or Cu ions. Consequently, acute or chronic neurodegenerative disorders represent excellent targets for exploiting metal ion chelator approaches. Drug Dev. Res. 56:300–309, 2002. c 2002 Wiley-Liss, Inc. Key words: chelation therapy; brain ischaemia; Alzheimer disease; Parkinson disease; BAPTA; calcium INTRODUCTION Metal ion chelators are being developed as a way of modulating metal dependent functions. BAPTA-AM, a cell permeant Ca 2þ chelator was highly neuropro- tective in cerebral ischemic models. Clioquinol, a Zn and Cu ion chelator, affected the stable markers Tau and GAP43 in patients with Alzheimer’s disease (AD) and clinical ratings after 3 weeks of treatment showed slight improvement. Desferrioxamine, a hydrophilic trivalent ion chelator, showed in a single blinded study significant reductions in the level of decline of daily living skills in patients with probable AD. DP-b99, a membrane activated bivalent ion chelator is in Phase II clinical trials for cerebral ischaemia and DP-109 is under preclinical development for chronic neurode- generation; both are lipid modified derivatives of the metal ion chelator, BAPTA. In vitro DP-b99 inhibits calpain activity in primary cortical neurons and attenuates matrix metalloproteinase 9 in primary cultured glial cells. It has demonstrated potent efficacy in several animal models of cerebral ischemia, with a large therapeutic window, and is effective for up to 8 h after ischemia. DP-109 was found to be effective in attenuating the progressive increase in apomorphine- induced rotations in an animal model for Parkinson’s disease. These novel approaches utilizing different metal ion chelators appear to offer a new promising mode for the treatment of diverse neurodegenerative disorders. METAL IONS IN MEDICINE Physiological Role The mechanisms by which organisms control metal ions and their role in cellular regulation has become an area of great scientific interest. Such metal ions include calcium, and the transition metals copper, zinc, manganese, and iron. These metals have been shown to be involved in cell-cell signaling, signal transduction, as well as, influencing transcription and translation via metal responsive regulators. There is an emerging view that cells are not buffer containers in which metals freely diffuse between thermodynamically controlled binding sites. Rather, a series of membrane metal transporters, metal DDR n Correspondence to: Itzchak Angel, D-Pharm Ltd, Kiryat Weizmann Science Park, P.O. Box 2313, Rehovot 76123, Israel. E-mail: [email protected] Published online in Wiley InterScience (www.interscience. wiley.com) DOI:10.1002/ddr.10083 DRUG DEVELOPMENT RESEARCH 56:300–309 (2002) c 2002 Wiley-Liss, Inc.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Research Overview
Metal Ion Chelation in Neurodegenerative DisordersItzchak Angel,n Adina Bar, Tammy Horovitz, Galia Taler, Michael Krakovsky, Dalia Resnitsky,
Gilad Rosenberg, Sarina Striem, Jonathan E. Friedman, and Alex KozakD-Pharm Ltd., Kiryat Weizmann Science Park, Rehovot, Israel
Strategy, Management and Health Policy
Venture Capital
Enabling
Technology
Preclinical
Research
Preclinical Development
Toxicology, Formulation
Drug Delivery,
Pharmacokinetics
Clinical Development
Phases I-III
Regulatory, Quality,
Manufacturing
Postmarketing
Phase IV
ABTRACT Disturbance in metallochemical reactions and metal-protein association are associatedwith chronic neurodegenerative conditions, such as Alzheimer’s and Parkinson’s disease, as well as withneurodegeneration triggered by acute cerebral ischaemia. Many neurological diseases have been linkeddirectly or indirectly to perturbed homeostasis of Ca, Fe, Zn, or Cu ions. Consequently, acute or chronicneurodegenerative disorders represent excellent targets for exploiting metal ion chelator approaches.Drug Dev. Res. 56:300–309, 2002. �c 2002 Wiley-Liss, Inc.
Key words: chelation therapy; brain ischaemia; Alzheimer disease; Parkinson disease; BAPTA; calcium
INTRODUCTION
Metal ion chelators are being developed as a wayof modulating metal dependent functions. BAPTA-AM,a cell permeant Ca2þ chelator was highly neuropro-tective in cerebral ischemic models. Clioquinol, a Znand Cu ion chelator, affected the stable markers Tauand GAP43 in patients with Alzheimer’s disease (AD)and clinical ratings after 3 weeks of treatment showedslight improvement. Desferrioxamine, a hydrophilictrivalent ion chelator, showed in a single blinded studysignificant reductions in the level of decline of dailyliving skills in patients with probable AD. DP-b99, amembrane activated bivalent ion chelator is in Phase IIclinical trials for cerebral ischaemia and DP-109 isunder preclinical development for chronic neurode-generation; both are lipid modified derivatives of themetal ion chelator, BAPTA. In vitro DP-b99 inhibitscalpain activity in primary cortical neurons andattenuates matrix metalloproteinase 9 in primarycultured glial cells. It has demonstrated potent efficacyin several animal models of cerebral ischemia, with alarge therapeutic window, and is effective for up to 8 hafter ischemia. DP-109 was found to be effective inattenuating the progressive increase in apomorphine-induced rotations in an animal model for Parkinson’s
disease. These novel approaches utilizing differentmetal ion chelators appear to offer a new promisingmode for the treatment of diverse neurodegenerativedisorders.
METAL IONS IN MEDICINE
Physiological Role
The mechanisms by which organisms controlmetal ions and their role in cellular regulation hasbecome an area of great scientific interest. Such metalions include calcium, and the transition metals copper,zinc, manganese, and iron. These metals have beenshown to be involved in cell-cell signaling, signaltransduction, as well as, influencing transcription andtranslation via metal responsive regulators.
There is an emerging view that cells are notbuffer containers in which metals freely diffusebetween thermodynamically controlled binding sites.Rather, a series of membrane metal transporters, metal
DDR
nCorrespondence to: Itzchak Angel, D-Pharm Ltd, KiryatWeizmann Science Park, P.O. Box 2313, Rehovot 76123, Israel.E-mail: [email protected]
Published online in Wiley InterScience (www.interscience.wiley.com) DOI:10.1002/ddr.10083
DRUG DEVELOPMENT RESEARCH 56:300–309 (2002)
�c 2002 Wiley-Liss, Inc.

chaperones, and assembly complexes have recentlybeen identified that regulate the uptake and delivery ofmetal ions to specific sites [Outten and O’Halloran,2001]. Metal ion dependent functions appears to beemerging as a new area of cell biology and is a potentialsite for therapeutic interventions. Normal metalmetabolism maintains free metal ion concentrationsat a very low level and delivers metals very selectivelyto their sites of action, while maintaining tight controlover their reactivity. The macromolecular players andvesicular compartments involved in metal ion home-ostasis and metal trafficking are only just beingdiscovered.
The brain is an example of a specialized organthat concentrates metal ions such as Fe, Zn, and Cu inconcentrations in the order of 0.1–0.5 mM [Lovellet al., 1998]. In healthy tissue, efficient homeostaticmechanisms and buffers are in place, compartmenta-lizing and regulating metal ion release [Bush, 2000].Thus, despite reasonably high total concentrations, theconcentration of free metal is very low. In the case ofZn2þ, for example, the balance between the cellularredox state, the concentration of other biologicalchelating agents, and the energy status of the cell,has been shown to determine Zn2þ distribution inthe cell [Jacob et al., 1998].
Physiological function relies, to a large extent, onthe universal intracellular messenger calcium and cellshave evolved a versatile calcium signaling toolkit[Berridge et al., 2000]. The calcium concentration incells is controlled by reversible binding to specificclasses of proteins that act as Ca2þ sensors to decode itsinformation before passing it on to targets. Thedecoding operation is based on specific conformationalchanges in the sensor proteins. Other proteins intrinsicto membranes simply control Ca2þ concentrationwithout processing its message, by transporting itacross membrane boundaries. They are located in theplasma membrane and in the membranes of theendoplasmic reticulum (ER), the mitochondria, andmost likely calcium containing organelles [Korkotianand Segal, 1997] and the nuclear envelope, which playdistinctive roles in the cellular homeostasis of Ca2þ
[Carafoli et al., 2001]. This versatile system is exploitedto control processes as diverse as fertilization, prolif-eration, development, learning and memory, contrac-tion, and secretion. These normal processes must,however, be accomplished within the context of calciumbeing highly toxic, since excess calcium can result incell death through both necrosis and apoptosis.
In neuronal systems, the endoplasmic reticulum(ER) localized to dendrites, axons, and their terminals,provides for local control of Ca2þ signals that effectchanges in the structure and function of neuronal
circuits [Mattson et al., 2000]. Following activation ofmost excitatory synapses, calcium is released fromintracellular stores of central neurons. There isincreasing evidence for the presence of local calciumsignals caused by calcium-induced calcium release,coupling store signaling to activity-dependent synapticplasticity [Rose and Konnerth, 2001]. Calcium plays animportant role in the regulation of neuronal geneexpression; moreover, calcium influx has been shown toinduce transcription of particular genes, which isregulated by the route of calcium entry into the cell[West et al., 2001]. Altered calcium homeostasis is alsoimplicated in the normal aging process [Griffith et al.,2000]. In addition, zinc-mediated neuron signaling hasbeen identified as a process analogous to otherneurotransmitter mechanisms. Zinc is stored in pre-synaptic vesicles, released in brief pulses into thesynaptic cleft, and acts on recognition sites in the post-synaptic membrane. Zinc-containing nerves are anato-mically widely distributed in the brain [Fredericksonet al., 2000], with the highest concentrations in thehippocampus.
Metals ions are important cofactors for manytransporters, transcription factors, and enzymes. Forexample, the metal-dependent matrix metalloprotei-nases (MMPs) are dependent on calcium and zinc,calpain cysteine proteases are calcium dependent, andthe Cu/Zn-superoxide dismutases are copper and zincdependent. MMPs are a family of calcium requiringand zinc containing proteinases that include gelati-nases, collagenases, thermolysin, stromalysin, andmembrane-bound proteases; they are involved inextracellular matrix degradation. MMPs are normallypresent in the brain in latent forms that requireactivation, which occurs at the cell surface bymembrane-type metalloproteinases (MT-MMPs) andother proteases [Bryce, 1998]. Their targets includeother proteinases, proteinase inhibitors, clottingfactors, chemotactic molecules, latent growthfactors, growth factor-binding proteins, cell surfacereceptors, cell-cell adhesion molecules, and virtually allstructural extracellular matrix proteins [Sternlicht andWerb, 2001]. More than 20 structurally homologousMMPs are known that differ in substrate specificity.Two calcium ions and a structural zinc site contributeto the stability of the enzyme structure, and an activesite zinc is essential for catalytic activity [Bode et al.,1999].
Calpains are a family of non-lysosomal neutralcysteine proteases [White et al., 2000] that form agrowing family of structurally related intracellularmultidomain cysteine proteinases containing a pa-pain-related catalytic domain, whose activity dependson calcium. The calpains are believed to play important
CHELATION IN NEURODEGENERATION 301

roles in cytoskeletal remodeling processes, cell differ-entiation, apoptosis, and signal transduction, but arealso implicated in muscular dystrophy, cardiac andcerebral ischemia, platelet aggregation, restenosis,neurodegenerative diseases, rheumatoid arthritis, andcataract formation. Current evidence points to acooperative interaction of several sites, which, uponcalcium binding, trigger the reformation of a papain-similar catalytic domain [Reverter et al., 2001].
Copper, zinc superoxide dismutase (CuZnSOD),is an important antioxidant enzyme that requires twoessential metals for catalytic function. It converts superoxide free radicals to the less active peroxide, whichthen can be further converted by other antioxidantenzymes into water. The balance of the Cu/Zn ratio isimportant in maintaining proper functioning of SODand any imbalance can be damaging.
Metal ions in Pathology
Any disturbance in the sophisticated metal ionhomeostasis of the body can lead to severe pathology.Examples of such pathologies include the chronicneurodegenerative conditions, for example, Alzhei-mer’s and Parkinson’s disease, Amyotrophic LateralSclerosis, as well as, neurodegeneration triggered byacute ischaemia (stroke).
Metallochemical reactions now appear to be acommon denominator underlying several prevalentdiseases; namely, Alzheimer’s disease (AD), amytrophiclateral sclerosis (ALS), prion disease, cataracts, mito-chondrial disorders, and Parkinson’s disease (PD)[Bush et al., 1994]. In these disorders, an abnormalreaction between a protein and a redox-active metal ionpromotes the formation of Reactive Oxygen Species(ROS) or radicalization. Several major neurologicaldiseases have been linked either directly or indirectlyto the role of Fe, Cu, Zn, and Ca in the generation ofROS. Furthermore, metal-catalyzed protein oxidationleads to protein damage and denaturation. b-amyloid(Ab), which accumulates in the brain in AD, was shownto reduce Cu2þ and Fe3þ and then produce H2O2
(peroxide) using oxygen as a substrate. Oxidation islikely to mediate neurodegeneration in AD, hence, Ab-mediated hydrogen peroxide production could be thecause of this problem [Huang et al., 1999] . Recentevidence implicates Cu2þ and Fe3þ in the overproduc-tion of free radicals as a possible causal factor in thedeath of nigral cells associated with PD. Iron depositsselectively involving neuromelanin in substantia nigraneurons of PD patients have been found [Kienzl et al.,1995].
Metal-protein association leading to proteinaggregation is of particular relevance to neurodegen-erative disease [Bush, 2000]. For example, in AD one
of the major characteristics is the deposition of b-amyloid (Ab) within the neocortex. It has beenobserved that Cu and Zn are elevated in the neocortexand particularly concentrated in amyloid plaques[Cherny et al., 2001]. Experiments using fluorescencemethods have determined there to be an abundance ofzinc in Ab amyloid plaques in human AD brain [Suh etal., 2000]. Furthermore, Ab possesses selective high-and low-affinity Cu2þ and Zn2þ binding sites [Atwoodet al., 2000; Bush et al.,1994] , which play an importantrole in Ab precipitation.
PD is characterized by a-synuclein aggregatesthat form Lewy bodies [Duda et al., 2000] . The abilityof iron to induce aggregation and toxicity of a-synucleinhave reinforced the critical role of iron in thepathogenesis of nigrostriatal injury and PD [Berget al., 2001]. Interestingly, it was shown that theA53T alpha-synuclein mutation, involved in familialPD, increases iron-dependent aggregation and toxicity[Ostrerova-Golts et al., 2000].
MMPs have been implicated in many neurologi-cal diseases that involve the neuroinflammatory re-sponse, and have a major role in blood brain barrierbreakdown following ischemia. Shortly after ischaemiabegins, a secondary inflammatory response follows,which is fueled by the release of cytokines, chemo-kines, and immediately early genes. These mediatorsactivate astrocytes and microglia, which release extra-cellular matrix degrading proteases. Neurological dis-ease, such as brain tumors, ischemia, and AD lead to anincrease in the matrix-degrading proteases. In thesecondary injury that accompanies the neuroinflamma-tory response of cerebrovascular diseases, activatedMMPs are believed to participate and have beenlocalized to the site of the insult [Lukes et al., 1999].Ab was a potent stimulator of MMP-9 and MMP-2activity, suggesting a role for MMPs in the develop-ment or progression of neuritic plaques, suggesting thatMMPs might be increased in Alzheimer’s afflictedbrain tissue. It is evident that MMPs have significanteffects on the brain micro-environment and the issue ofMMPs as causative factors in disease is an active area ofinvestigation.
Observations of proteolytic degradation of calpainsubstrates were utilized to indirectly infer pathologiccalpain activation in ischemic neuronal injury [Seubertet al., 1989; Lee et al.,1991], AD [Saito et al., 1993],and apoptosis [Squier et al., 1994]. A recent studyindicated that in AD, abundant amyloid plaques in thebrain can cause calpain activation and conversion ofp35 to p25 [Lee et al., 2000], suggesting onemechanism by which calcium can activate atau-phosphorylating kinase. Activated m-calpain andm-calpain are both abnormally upregulated in the
302 ANGEL ET AL.

brains of patients with AD. The calpain-mediated p35cleavage pathway may serve as a target for pharmaco-logical intervention.
Prion disease is thought to involve disturbance tobrain copper homeostasis [Brown, 2001]. One aspect ofprion disease is the conversion of functional prionprotein into an aggregated amyloid. The prion proteinis a cell surface glyco-protein expressed by neurons,shown to cooperatively bind copper at four identicalbinding sites [Viles et al., 1999] . Prion protein has arole in normal brain copper metabolism; its expressionalters copper uptake into cells and enhances copperincorporation into superoxide dismutase (SOD).
Another disorder associated with neurotoxicaggregates is familial amyotrophic lateral sclerosis(FALS). In FALS, a mutation in Cu/Zn SOD(SOD 1) causes this antioxidant enzyme to aggregatein motor neurons and glia, which is associated with celldeath [Bruijn et al., 1998].
Disruption of neuronal Ca2þ homeostasis isknown to occur in many different disorders, includingstroke and AD, and emerging data from experimentalmodels indicate a central role for perturbed ER-mediated Ca2þ regulation in the neurodegenerativeprocess [Mattson et al., 2000; Frederickson et al., 2000;Paschen and Doutheil, 1999]. In addition, it appearsthat mitochondria have a key role in the neurotoxicityresulting from rapid NMDA-receptor-dependent in-tracellular Ca2þ accumulation. Mitochondria serve ashigh capacity Ca2þ buffers, and when overloaded withCa2þ, generation of ROS and irrecoverable loss offunction can result [Weiss and Sensi, 2000].
Brain injury following stroke develops from acomplex series of events that evolve in time and space.Substantial attention has focused on the possibility thatexcessive Ca2þ influx through several channel- andtransporter-mediated routes, leading to intracellularCa2þ overload, is a key factor underlying ischaemicneuronal necrosis. More recently, it was found thatother alterations in cellular ionic homeostasis alsocontribute to neuronal death, in particular, whereprogrammed cell death is induced [Lee et al., 1999].Calcium homeostasis is intimately related to thefunctions of the plasma membrane and its associatedproteins, as well as to different cellular compartmentsor organelles. It has been suggested that functional‘‘unit’’ composed of plasma membrane and ER calciumchannels as well as mitochondria, are recruitedsimultaneously during physiological cell activation[Montero et al., 2000]. With respect to Ca2þ dependentcell injury, both mitochondria and the endoplasmicreticulum (ER) play key roles. In the case of calciumoverload in mitochondria, ATP generation is shut downand calcium pumps fail, resulting in massive calcium
output from mitochondria, reduced calcium intake intothe ER, and calcium extrusion across the plasmamembrane. Under these circumstances, cytochrome cis released from mitochondria into the cytoplasm,triggering apoptosis. It appears that programmed celldeath and excitotoxicity are triggered in parallel in theischaemic brain accounting for the observed mixedmorphological features and biochemical markers.Survival of neuronal cells appears critically dependentupon an optimal intracellular ‘‘calcium set-point’’consistent with the hypothesis that excitotoxic Ca2þ
overload may predominate close to the ischaemic core,whereas further from the core Ca2þ starvation andapoptosis may predominate at later time intervals.
Evidence indicates that Ca2þ may not be the onlydivalent cation whose toxic influx contributes toischaemic brain-cell death. The pathophysiological roleof zinc has been gradually elucidated [Choi and Koh,1998], revealing that, like calcium, disrupted zinchomeostasis can be neurotoxic and lead to neuronaldeath. It has been suggested that upon excessivesynaptic Zn2þ release, its accumulation in post-synapticneurons contributes to the selective neuronal loss thatis associated with acute conditions such as stroke[Weiss et al., 2000].
Chelator approach
The earliest reported research of the use ofchelation therapy was in 1946, using Ethylene DiamineTetracetic Acid (EDTA) for the removal of plaque-producing calcium deposits [Trowbridge and Walker,1991]. Metal ion chelators have been suggested aspotential therapies for diseases involving metal ionimbalance. The obvious use for this approach has beenin diseases involving iron overload, such as occurs insickle cell anemia and thalassemia. Such treatment isreadily available and includes desferrioxamine Btherapy. Unfortunately, this drug is slow acting, rapidlyexcreted, and not orally active. Metal complexation isalso the basis for chelation therapy to rectify abnormalmetal accumulations or toxic metal exposures (e.g.,lead, cadmium and mercury poisoning). Improvedchelator designs are needed to enhance selectivity,affinity, stability, renal clearance, and oral activity, whilemaintaining low toxicity and low cost.
Neurodegeneration represents an excellent targetfor exploiting the metal chelator approach to thera-peutics. In contrast to the direct chelation approach inmetal ion overload disorders, in neurodegeneration thegoal seems to be a better and subtle modulation ofmetal ion homeostasis, aimed at restoring ionicbalance. The rationale behind this approach involvestwo aspects related to metal induced neuronal damage.
CHELATION IN NEURODEGENERATION 303

Blocking Metal Dependent Aggregation
The chelator approach to blocking Ab aggregationwas recently substantiated in vivo in transgenic micetreated with a metal ion chelator [Cherny et al., 2001].A 49% decrease in brain Ab deposition was detectedfollowing 9 weeks of oral treatment with a Cu/Znchelator (Clioquinol). Moreover, in an earlier clinicaltrial conducted on Alzheimer’s patients, it was con-cluded that sustained administration of desferrioxa-mine, a chelator with affinity for Fe, Cu, Zn Al, mayslow the clinical progression of the dementia associatedwith AD [McLachlan, 1991].
Reducing Free Radical Damage
The toxic influx of zinc, known to initiate ROSrelated damage, may be a key mechanism underlyingselective neuronal death after stroke. In rats, it wasshown that after transient forebrain ischemia, chelatablezinc accumulated specifically in degenerating neurons.This accumulation was found to precede neurodegenera-tion, and was prevented by the intraventricular injectionof a zinc chelating agent (Ca-EDTA) [Koh et al., 1996].Other chelators, such as a-Lipoic acid, have been shown,when orally administered to rats, to improve mitochon-drial function, decrease oxidative damage, and increasemetabolic rate [Hagen et al., 1999].
BAPTA-AM
BAPTA-AM (1,2-bis-(2-amino-phenoxy)ethane-N,N,N0,N0-tetraacetic acid acetoxymethyl ester) is acell-permeable form of the calcium chelator BAPTAdesigned in the early 1980s [Tsien, 1981]. When usedto treat rats prior to the induction of focal corticalischemia, BAPTA-AM was highly neuroprotective, asgauged by significant reductions in cortical infarctvolumes and neuronal sparing. It has been shown toattenuate excitotoxic and hypoxic/ischaemic neurode-generation in vitro and in vivo [Tymianski et al., 1993].BAPTA-AM’s neuroprotective mechanism may involveattenuating excitatory neurotransmission without af-fecting Ca2þ signaling in the submicromolar [Ca2þ] irange [Spigelman et al., 1996] . BAPTA-AM has beensuggested for the treatment of ischemic CNS injury. Sofar, this agent has proven useful as a research tool buthas not been promoted to clinical use.
Clioquinol
Clioquinol (5-chloro-7-iodo-8-hydroxyquinoline)is a lipophilic chelator that freely crosses the bloodbrain barrier. Clioquinol selectively binds Zn2þ andCu2þ with greater affinity than for Ca2þ and Mg2þ.
Clioquinol was used extensively in the past as an anti-
infective, especially for diarrhea. In a recent blindedstudy, AD transgenic mice (APP2576) were treatedorally for 9 weeks with clioquinol. This resulted in a49% decrease in b-amyloid deposition. Moreover, thegeneral health and body weight parameters weresignificantly more stable in the treated animals [Chernyet al., 2001]. The first results from an open clinicalstudy of clioquinol in patients with AD are nowavailable [Regland et al., 2001]. This was a single-center study of the efficacy and safety of clioquinol in20 patients randomized to receive a daily dose of either20 or 80 mg. Treatment was for 21 days, blinded to thedosages but included no controls. Two stable markersfor AD, Tau protein and growth associated protein(GAP43), were significantly increased at day 7 andreturned to baseline values at day 21. The authorsspeculate that the initial increase could be due totemporary cytotoxicity to the brain and/or an increasedrelease into the CSF from stores, possibly from senileplaques. Clinical rating showed slight improvementafter 3 weeks of treatment. Improvements according toAlzheimer’s Disease Assessment Scale-Cognitive Sub-scale (ADAS-Cog) totaled 2.7 points (Po0.07) betweentreatment days 0 and 21. Of the ADAS-Cog subsets,naming, instructions, and comprehension scalesshowed significant improvement (Po0.05). Clioquinolis currently under development by Lundbeck (Den-mark), who licensed the compound from P.N. Ger-olymatos S.A (Greece). In parallel, Prana Biotechnolgy(Australia) are developing their lead compound PBT-1(Iodochlorhydroxyquin), a metal ion chelator that issimilar to clioquinol, following the encouraging resultsobtained in mice with clioquinol [Spigelman et al.,1996]. PBT-1 has just completed its first clinical trial inpatients with AD, and interim findings reveal that b-amyloid was significantly reduced in the blood ofpatients randomized to the treatment group, anddisease progression, as measured by the ADAS-COGassessment scale, was delayed in PBT-1 treatedpatients.
Desferrioxamine
Desferrioxamine is a hydrophilic chelator withaffinity for Cu2þ, Zn2þ, Fe3þ, and Al3þ and does notpass across the blood brain barrier [Cherny et al.,2001]. In 1991, a 2-year, single-blind study toinvestigate whether the progression of dementia couldbe slowed by the trivalent ion chelator, desferrioxa-mine, was performed on 48 patients with probable AD,randomly assigned to receive desferrioxamine (125 mgintramuscularly twice daily, 5 days per week, for 24months), oral placebo (lecithin), or no treatment[McLachlan, 1991]. Parenteral administration ofdesferrioaxamine led to significant reduction in the
304 ANGEL ET AL.

rate of decline of daily living skills. The mean rate ofdecline was twice as rapid for the no-treatment group.Currently, this drug is not being pursued clinicallyfor AD.
SELECTIVE METAL CHELATION IN THE VICINITY OFCELL MEMBRANES
Until recently, Ca2þ neurotoxicity was thought todepend on the Ca2þ load. However, current under-standing of mechanisms of cell damage now supportthe notion that Ca2þ neurotoxicity is rather a functionof the Ca2þ influx pathways and intracellular Ca2þ
compartmentalization rather than total Ca2þ load[Sattler et al., 1998]. In addition to Ca2þ, othertransition ions, such as Zn, Cu, and Fe, play a pivotalrole in neurotoxicity [Bush, 2000]. Calcium-dependentexcitotoxicty involves, for example, Zn-dependentmembrane associated matrix metalloproteinases thatare involved in degeneration, or metal-dependentproteases that cause the release of the inflammatoryagent TNFa, in addition to ion exchangers (e.g.,Na/Ca) [Chidekel et al., 1997; Hershfinkel et al.,2001]. Moreover distinct, membrane receptor-specific,neurotoxic signaling pathways have been proposed thattransduce Ca2þ dependent excitotoxicity [Sattler andTymianski, 2000]. Membranes are the site for theexchange of ions and metabolites in and out of cells.This provides an excellent pharmacological target toinfluence critical biological processes, based on ionhomeostasis. In addition, metal ion chelation in thevicinity of membranes avoids disrupting the delicatebalance of cytoplasmic metal ions by unrestrictedchelation.
NEW FAMILY Of MEMBRANE ACTIVE CHELATORS
A new family of membrane active chelators hasbeen synthesized, which involves the lipid modificationof the known calcium Ca2þ chelator, BAPTA. Thiscreates a new structure that alters the chelator’sphysicochemical properties, thus, restricting its chela-tion capability to lipophilic environments such as cellmembranes, as well as altering its affinity for particularmetals. Restricting drug action to specific cellularmembranes provides several important benefits. Thefocused activity within membranes positions the drugat critical junctions of physiological activity, allowing forincreased potency. Furthermore, due to the lack ofpotentially harmful activity in body fluids, such asplasma or within the aqueous intracellular environ-ment, safer drugs may be developed. This novelapproach is illustrated schematically in Fig. 1, withthe lead compound, DP-b99.
DP-b99
The need to control levels of free intracellularcalcium in a wide range of physiological and biochem-ical systems has led to the widespread use of differentcalcium chelators [Donkor, 2000; Tsien, 1980]. BAPTA,one of the high-affinity calcium chelators has a numberof advantages over other chelators; high selectivity forcalcium over magnesium ions, low pH sensitivity, and afast calcium binding rate. The inherent problem withthe BAPTA salt is that it is highly hydrophilic andcannot pass through membranes [Tsien, 1980].
A lipophilic BAPTA diester, DP-b99 (1,2-Bis(2-aminophenoxy)ethane- N,N,N0N0-tetraacetic acid,N,N0-di(octyloxyethyl ester),N,N0-disodium salt) iscurrently under Phase II clinical development and
Fig. 1. The metal ion interacts with the lipid modified chelator mainly in the lipophilic-membranousenvironment. M represents a metal ion and R the attached lipid moiety (2-octyloxy-ethyl).
CHELATION IN NEURODEGENERATION 305
has great potential in biological systems in treatingpatients with cerebral ischemia. DP-b99 has addedlipid moieties to the BAPTA back-bone, therebyreducing its affinity to calcium in the water environ-ment, such as plasma or extracellular fluids, and alsochelates Cu, Zn, and Fe ions. Partitioning logD datasuggests that DP-b99 distributes more effectively to theorganic octanol vs. the water phase compared to theBAPTA molecule, irrespective of whether sodium orcalcium ions are bound. Potentiometric and spectro-photometric titration [Tsien, 1980] used to determinethe dissociation constant (KD) show that BAPTA CaH2
is a better chelator of calcium than DP-b99 in water,whereas in organic solvents, DP-b99 is a more effectivecalcium ion chelator than in water.
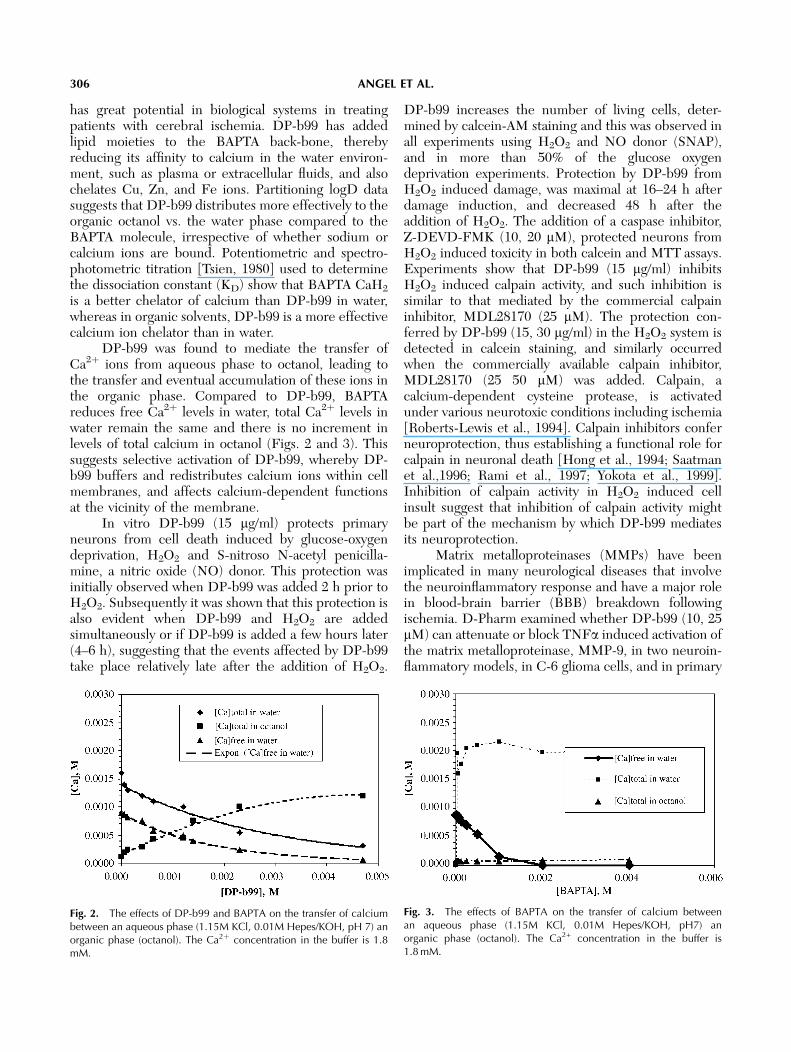
DP-b99 was found to mediate the transfer ofCa2þ ions from aqueous phase to octanol, leading tothe transfer and eventual accumulation of these ions inthe organic phase. Compared to DP-b99, BAPTAreduces free Ca2þ levels in water, total Ca2þ levels inwater remain the same and there is no increment inlevels of total calcium in octanol (Figs. 2 and 3). Thissuggests selective activation of DP-b99, whereby DP-b99 buffers and redistributes calcium ions within cellmembranes, and affects calcium-dependent functionsat the vicinity of the membrane.
In vitro DP-b99 (15 mg/ml) protects primaryneurons from cell death induced by glucose-oxygendeprivation, H2O2 and S-nitroso N-acetyl penicilla-mine, a nitric oxide (NO) donor. This protection wasinitially observed when DP-b99 was added 2 h prior toH2O2. Subsequently it was shown that this protection isalso evident when DP-b99 and H2O2 are addedsimultaneously or if DP-b99 is added a few hours later(4–6 h), suggesting that the events affected by DP-b99take place relatively late after the addition of H2O2.
DP-b99 increases the number of living cells, deter-mined by calcein-AM staining and this was observed inall experiments using H2O2 and NO donor (SNAP),and in more than 50% of the glucose oxygendeprivation experiments. Protection by DP-b99 fromH2O2 induced damage, was maximal at 16–24 h afterdamage induction, and decreased 48 h after theaddition of H2O2. The addition of a caspase inhibitor,Z-DEVD-FMK (10, 20 mM), protected neurons fromH2O2 induced toxicity in both calcein and MTT assays.Experiments show that DP-b99 (15 mg/ml) inhibitsH2O2 induced calpain activity, and such inhibition issimilar to that mediated by the commercial calpaininhibitor, MDL28170 (25 mM). The protection con-ferred by DP-b99 (15, 30 mg/ml) in the H2O2 system isdetected in calcein staining, and similarly occurredwhen the commercially available calpain inhibitor,MDL28170 (25 50 mM) was added. Calpain, acalcium-dependent cysteine protease, is activatedunder various neurotoxic conditions including ischemia[Roberts-Lewis et al., 1994]. Calpain inhibitors conferneuroprotection, thus establishing a functional role forcalpain in neuronal death [Hong et al., 1994; Saatmanet al.,1996; Rami et al., 1997; Yokota et al., 1999].Inhibition of calpain activity in H2O2 induced cellinsult suggest that inhibition of calpain activity mightbe part of the mechanism by which DP-b99 mediatesits neuroprotection.
Matrix metalloproteinases (MMPs) have beenimplicated in many neurological diseases that involvethe neuroinflammatory response and have a major rolein blood-brain barrier (BBB) breakdown followingischemia. D-Pharm examined whether DP-b99 (10, 25mM) can attenuate or block TNFa induced activation ofthe matrix metalloproteinase, MMP-9, in two neuroin-flammatory models, in C-6 glioma cells, and in primary
Fig. 2. The effects of DP-b99 and BAPTA on the transfer of calciumbetween an aqueous phase (1.15M KCl, 0.01M Hepes/KOH, pH 7) anorganic phase (octanol). The Ca2þ concentration in the buffer is 1.8mM.
Fig. 3. The effects of BAPTA on the transfer of calcium betweenan aqueous phase (1.15M KCl, 0.01M Hepes/KOH, pH7) anorganic phase (octanol). The Ca2+ concentration in the buffer is1.8 mM.
306 ANGEL ET AL.

rat cortical glial cells. Preliminary findings show thatDP-b99 blocks basal activation of MMP-9 as well asreducing TNFa-induced expression of MMP-9 inprimary cultured glial cells.
In vivo studies with DP-b99 demonstrate efficacyin several animal models of cerebral ischaemia[Tolmasov et al., 1999; Krakovsky et al., 2000, 2001]and indicate the therapeutic potential of this com-pound. Survival was improved by 10–30% in Sprague-Dawley rats subject to permanent focal cerebralischaemia and treated with DP-b99 (5 or 10 mg/kg) 1h after occlusion. A significant reduction was observedin infarct volume in Sprague-Dawley rats administeredDP-b99 (1, 5, 10, 30, or 100 mg/kg); this attenuation wasalso observed when animals were treated 6 h (5 mg/kg)following the onset of ischemia. Findings from atransient model of ischaemia show a significantreduction of cortical and total infarct volumes inSprague-Dawley rats treated (i.v.) immediately afterreperfusion, and in animals treated i.p. 3 h followingreperfusion (i.e., 2 and 5 h following the onset ofischaemia). In a model of transient forebrain ischaemiain Mongolian gerbils, a single dose of 5 mg/kg DP-b99significantly increased the number of survivingneurons in the hippocampus. Serum neuron-specificenolase (NSE) is a sensitive marker of neuronaldamage and its levels following permanent or transientMCAO usually increase significantly [Barone et al.,1993]. Serum neuron specific enolase (NSE) levelswere reduced significantly in Sprague-Dawley ratsand Mongolian gerbils administered DP-b99 afterthe start of occlusion. The primary endpoint forall potentially neuroprotective drugs is functionalimprovement, and neuromuscular function is one ofthe important outcomes of neuroprotection afterstroke and cerebral ischaemia. Neurological scorestwo days following ischemia were significantlyimproved in Wistar rats given a single dose ofDP-b99 (5 mg/kg).
In Phase 1 studies using single and repeatedescalating doses, DP-b99 were found to be safe andwell-tolerated in young or elderly volunteers in thedose range of 0.003–1.0 mg/kg/day, when administeredfor up to 4 consecutive days. Adverse events were mildor moderate and most of these were localized eventsattributed to the injection site (phlebitis). The drugseems to be better tolerated in elderly than in youngsubjects, despite a greater systemic exposure. In bothstudy parts, there was no evidence of any drug or dose-related changes detected in the 12-lead ECG results,vital signs parameters, or the results of laboratoryevaluations including clinical chemistry, hematology(blood count and coagulation) and urinalysis. A PhaseII exploratory study of 2-day repeated (i.v) administra-
tion of DP-b99 in stroke patients with corticalinvolvement is now in progress in Europe.
DP-109
DP-109 is another lipid derivative of the metalion chelator, BAPTA, from the new family of mem-brane active chelators and is under preclinical devel-opment by D-Pharm for the treatment of chronicneurodegeneration such as Parkinson’s and Alzheimer’sDiseases. Whereas DP-b99 was designed for IVadministration, in the case of DP-109, it was designedfor oral administration and for an increased penetrationand residence time in the brain. The properties of thiscompound extended to being non-mutagenic, stable inserum, with a favorable pharmacokinetic profile andacceptable bioavailability.
DP-109, due to its lipophilic nature, can prefer-entially locate to the membrane and modulate levels ofthese transition metal ions in the neighbouring aqueousenvironment, thus restoring normal homeostasis.
DP-109 was found to be effective in attenuatingthe progressive increase in apomorphine-inducedrotations following a unilateral 6-OHDA lesion, ananimal model for PD [Ungersstedt, 1973; Emmi 1996;Kreiss et al., 1997]. Whereas 10 mg/kg was not effective,doses of 100 mg/kg (p.o.) and 500 mg/kg (p.o) DP-109were significantly effective at 15 days post-lesion.Additional studies are currently ongoing for the precli-nical development of DP-109 in neurodegenerativedisorders.
Conclusions
Several approaches aimed at modifying metal ionfunctions by chelation are currently under develop-ment for both acute and chronic neurodegenerativedisorders. They offer a multi-functional, novel ap-proach to these disorders and are promising modes oftreatment for these devastating conditions.
REFERENCES
Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP,Tanzi RE, Bush AI. 2000. Characterization of copper interactionswith alzheimer amyloid beta peptides: identification of anattomolar-affinity copper binding site on amyloid beta1-42.J Neurochem 75:1219–1233.
Barone FC, Clark RK, Price WJ, White RF, Feuerstein GZ, StorerBL, Ohlstein EH. 1993. Neuron-specific enolase increases incerebral and sytemic circulation following focal ischemia. BrainRes 623:77–82.
Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P,Becker G. 2001. Brain iron pathways and their relevance toParkinson’s disease. J Neurochem 79:225–236.
Berridge MJ, Lipp P, Bootman MD. 2000. The versatility anduniversality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21.
CHELATION IN NEURODEGENERATION 307

Bode W, Fernandez-Catalan C, Tschesche H, Grams F, Nagase H,Maskos K. 1999. Structural properties of matrix metalloprotei-nases. Cell Mol Life Sci 55:639–652.
Brown DR. 2001. Copper and prion disease. Brain Res Bull 55:165–173.
Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD,Ohama E, Reaume AG, Scott RW, Cleveland DW. 1998.Aggregation and motor neuron toxicity of an ALS-linked SOD1mutant independent from wild-type SOD1. Science 281:1851–1854.
Bryce SM, Rosenberg G. 1998. Matrix Metalloproteinases inCerebrovascular Disease. J Cereb Blood Flow Metab 18:1163–1172.
Bush AI. 2000. Metals and neuroscience. Curr Opin Chem Biol4:184–191.
Bush AI, Pettingell WH, Multhaup G, Paradis M, Vonsattel JP,Gusella JF, Beyreuther K, Masters CL, Tanzi RE. 1994. Rapidinduction of Alzheimer A beta amyloid formation by zinc. Science265:1464–1467.
Carafoli E, Santella L, Branca D, Brini M. 2001. Generation,control, and processing of cellular calcium signals. Crit RevBiochem Mol Biol 36:107–260.
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLeanCA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X,Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, TanziRE, Masters CL, Bush AI. 2001. Treatment with a copper-zincchelator markedly and rapidly inhibits beta-amyloid accumulationin Alzheimer’s disease transgenic mice. Neuron 30:665–676.
Chidekel AS, Friedman JE, Haddad GG. 1997. Anoxia-inducedneuronal injury: role of Na+ entry and Na+-dependent transport.Exp Neurol 146:403–413.
Choi DW, Koh JY. 1998. Zinc and brain injury. Annu Rev Neurosci21:347–375.
Donkor IO. 2000. A survey of calpain inhibitors. Curr Med Chem7:1171–1188.
Duda JE, Lee VM, Trojanowski JQ. 2000. Neuropathology ofsynuclein aggregates. J Neurosci Res 61:121–127.
Emmi A, Rajabi H, Stewart J. 1996. Behavioral and neurochemicalrecovery from partial 6-hydroxydopamine lesions of the substantianigra is blocked by daily treatment with glutamate receptorantagonists MK-801 and CPP. J Neurosci 16.
Frederickson CJ, Suh SW, Silva,D, Thompson RB. 2000. Impor-tance of zinc in the central nervous system: the zinc-containingneuron. J Nutr 130:1471S–1483S.
Griffith WH, Jasek MC, Bain SH, Murchison D. 2000. Modificationof ion channels and calcium homeostasis of basal forebrainneurons during aging. Behav Brain Res 115:219–233.
Hagen TM, Ingersoll RT, Lykkesfeldt J, Liu J, Wehr CM, Vinarsky V,Bartholomew JC, Ames AB. 1999. (R)-alpha-lipoic acid-supple-mented old rats have improved mitochondrial function, decreasedoxidative damage, and increased metabolic rate. Faseb J 13:411–418.
Hershfinkel M, Moran A, Grossman N, Sekler I. 2001. A zinc-sensing receptor triggers the release of intracellular Ca2+and regulates ion transport. Proc Natl Acad Sci USA 98:11749–11754.
Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF, Lee KS. 1994.Neuroprotection with a calpain inhibitor in a model of focalcerebral ischemia. Stroke 25:663–669.
Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE,Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE,Bush AI. 1999. The A beta peptide of Alzheimer’s disease directlyproduces hydrogen peroxide through metal ion reduction.Biochemistry 38:7609–7616.
Jacob C, Maret W, Vallee BL. 1998. Control of zinc transfer betweenthionein, metallothionein, and zinc proteins. Proc Natl Acad SciUSA 95:3489–3494.
Kienzl E, Puchinger L, Jellinger K, Linert W, Stachelberger H,Jameson RF. 1995. The role of transition metals in thepathogenesis of Parkinson’s disease. J Neurol Sci 134Suppl.:69–78.
Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. 1996. Therole of zinc in selective neuronal death after transient globalcerebral ischemia. Science 272:1013–1016.
Korkotian E, Segal M. 1997. Calcium-containing organelles displayunique reactivity to chemical stimulation in cultured hippocampalneurons. J Neurosci 17:1670–1682.
Krakovsky M, Beit-Yannai E, Angel I, ShemeshT, Kozak A. 2000.In: Pharmacology of cerebral ischemia. DP-b99 improvesthe neurological and histological outcome following occlusionof the middle cerebral artery in rats. Abstract. Marburg,Germany.
Krakovsky M, Polyak M, Angel I, Kozak A. 2001. A novel membranetargeted compound active against global and focal ischemia. In:Gjedde SH, Kundsen GM, Paulson OB, editors. PhysiologicalImaging of the Brain with PET. New York: Academic Press.p 347–352
Kreiss DS, Mastropietro CW, Rawji SS, Walters J. 1997. Theresponse of subthalamic nucleus neurons to dopamine receptorstimulation in a rodent model of Parkinson’s disease. J Neurosci17.
Lee JM, Zipfel GJ, Choi DW. 1999. The changing landscape ofischaemic brain injury mechanisms. Nature 399:A7–14.
Lee KS, Frank S, Vanderklish P, Arai A, Lynch G. 1991. Inhibition ofproteolysis protects hippocampal neurons from ischemia. ProcNatl Acad Sci USA 88:7233–7237.
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. 2000.Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature405:360–364.
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, MarkesberyWR. 1998. Copper, iron and zinc in Alzheimer’s disease senileplaques. J Neurol Sci 158:47–52.
Lukes A, Mun-Bryce S, Lukes M, Rosenberg GA. 1999. Extra-cellular matrix degradation by metalloproteinases and centralnervous system diseases. Mol Neurobiol 19:267–284.
Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN,Geiger JD. 2000. Calcium signaling in the ER: its role in neuronalplasticity and neurodegenerative disorders. Trends Neurosci23:222–229.
McLachlan JC. 1991. Transcutaneous electrical nerve stimulation.Lancet 337:742.
Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A,Garcia AG, Garcia-Sancho J, Alvarez J. 2000. Chromaffin-cellstimulation triggers fast millimolar mitochondrial Ca2+ transientsthat modulate secretion. Nat Cell Biol 2:57–61.
Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, WolozinB. 2000. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 20:6048–6054.
308 ANGEL ET AL.

Outten CE, O’Halloran TV. 2001. Femtomolar sensitivity ofmetalloregulatory proteins controlling zinc homeostasis.Science29:2488–2492.
Paschen W, Doutheil J. 1999. Disturbances of the functioning ofendoplasmic reticulum: a key mechanism underlying neuronalcell injury? J Cereb Blood Flow Metab 19:1–18.
Rami A, Ferger D, Krieglstein J. 1997. Blockade of calpainproteolytic activity rescues neurons from glutamate excitotoxicity.Neurosci Res 27:93–97.
Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M,Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG. 2001.Treatment of Alzheimer’s disease with clioquinol. Dement GeriatrCogn Disord 12:408–414.
Reverter D, Sorimachi H, Bode W. 2001. The structure of calcium-free human m-calpain: implications for calcium activation andfunction. Trends Cardiovasc Med 11:222–229.
Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R.1994. Immunolocalization of calpain I-mediated spectrin degra-dation to vulnerable neurons in the ischemic gerbil brain. JNeurosci 14:3934–3944.
Rose CR, Konnerth A. 2001. Stores not just for storage. intracellularcalcium release and synaptic plasticity. Neuron 31:519–522.
Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, PerriBR, McIntosh TK. 1996. Calpain inhibitor AK295 attenuatesmotor and cognitive deficits following experimental brain injury inthe rat. Proc Natl Acad Sci USA 93:3428–3433.
Saito K, Elce JS, Hamos JE, Nixon RA. 1993. Widespread activationof calcium-activated neutral proteinase (calpain) in the brain inAlzheimer disease: a potential molecular basis for neuronaldegeneration. Proc Natl Acad Sci USA 90:2628–2632.
Sattler R, Charlton MP, Hafner M, Tymianski M. 1998. Distinctinflux pathways, not calcium load, determine neuronal vulner-ability to calcium neurotoxicity. J Neurochem 71:2349–2364.
Sattler R, Tymianski M. 2000. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med 78:3–13.
Seubert P, Lee K, Lynch G. 1989. Ischemia triggers NMDAreceptor-linked cytoskeletal proteolysis in hippocampus. BrainRes 492:366–370.
Spigelman I, Tymianski M, Wallace CM, Carlen PL, Velumian AA.1996. Modulation of hippocampal synaptic transmission by lowconcentrations of cell-permeant Ca2+ chelators: effects of Ca2+affinity, chelator structure and binding kinetics. Neuroscience75:559–572.
Squier MK, Miller AC, Malkinson AM, Cohen JJ. 1994. Calpainactivation in apoptosis. J Cell Physiol 159:229–237.
Sternlicht MD, Werb Z. 2001. How matrix metalloproteinasesregulate cell behavior. Annu Rev Cell Dev Biol 17:463–516.
Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G,Frederickson C. Histochemically-reactive zinc in amyloid pla-ques, angiopathy, and degenerating neurons of Alzheimer’sdiseased brains. J 2000. Brain Res 852:274–278.
Tolmasov M, Krakovsky M, Angel I, Kozak A. 1999. In E.A.C.P.TDP-b99: A novel membrane-targeted compound with a strongneuroprotective action in cerebral ischemia. (Abstract. Eilat,Israel.
Trowbridge JP, Walker M. 1991. The healing powers of chelationtherapy. New Way of Life, Inc., CT.
Tsien RY. 1980. New calcium indicators and buffers with highselectivity against magnesium and protons: design, synthesis,and properties of prototype structures. Biochemistry 19:2396–2404.
Tsien RY. 1981. A non-disruptive technique for loading calciumbuffers and indicators into cells. Nature 290:527–528.
Tymianski M, Wallace MC, Spigelman I, Uno M, Carlen PL, TatorCH, Charlton MP. 1993. Cell-permeant Ca2+ chelators reduceearly excitotoxic and ischemic neuronal injury in vitro and in vivo.Neuron 11:221–235.
Ungersstedt U, Avemo A, Avemo E, Ljungber T, Rouge C. 1973.Animal models of Parkinsonism. Adv Biochem Psycopharmacol 9.
Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, DysonHJ. 1999. Copper binding to the prion protein: structuralimplications of four identical cooperative binding sites. Proc NatlAcad Sci USA 96:2042–2047.
Weiss JH, Sensi SL. 2000. Ca2+ permeable AMPA or kainatechannels and selective neurodegeneration. Trends Neurosci23:365–371.
Weiss JH, Sensi SL, Koh JY. 2000. Zn(2+): a novel ionic mediatorof neural injury in brain disease. Trends Pharmacol Sci 21:395–401.
West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM,Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. 2001. Calciumregulation of neuronal gene expression. Proc Natl Acad Sci USA98:11024–11031.
White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW,Grossman LI, Rafols JA, Krause GS. 2000. Brain ischemia andreperfusion: molecular mechanisms of neuronal injury. J NeurolSci 179:1–33.
Yokota M, Tani E, Tsubuki S, Yamaura I, Nakagaki I, Hori S, SaidoTC. 1999. Calpain inhibitor entrapped in liposome rescuesischemic neuronal damage. Brain Res 819:8–14.
CHELATION IN NEURODEGENERATION 309
Related Documents











