Aus der Klinik und Poliklinik für Neurologie Direktor: Herr Prof. Dr. Heinz Reichmann Mechanisms of Axonal Transport Defects in ALS Dissertationsschrift Zur Erlangung des akademischen Grades Doktor der Biomedizin Doctor rerum medicinalium (Dr. rer. medic.) vorgelegt bei der Medizinischen Fakultät Carl Gustav Carus der Technischen Universität Dresden von M.Sc. Anne Seifert geboren am 05.07.1992 in Dresden Dresden, Januar 2021

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Aus der Klinik und Poliklinik für Neurologie
Direktor: Herr Prof. Dr. Heinz Reichmann
Mechanisms of Axonal Transport Defects in ALS
Dissertationsschrift
Zur Erlangung des akademischen Grades
Doktor der Biomedizin
Doctor rerum medicinalium (Dr. rer. medic.)
vorgelegt bei
der Medizinischen Fakultät Carl Gustav Carus
der Technischen Universität Dresden
von
M.Sc. Anne Seifert
geboren am 05.07.1992 in Dresden
Dresden, Januar 2021

Für meine Mama.

i Anne Seifert
Gutachter:
1. Gutachter: Prof. Dr. Dr. Andreas Hermann, Sektion für Translationale Neurodegeneration
"Albrecht Kossel", Klinik und Poliklinik für Neurologie, Universitätsmedizin
Rostock, Gehlsheimer Straße 20, 18147 Rostock, Germany
2. Gutachter: Prof. Dr. Stefan Diez, Technische Universität Dresden, Center for Molecular and
Cellular Bioengineering (CMCB), B CUBE - Center for Molecular Bioengineering,
Tatzberg 41, 01307 Dresden, Germany
Weitere Mitglieder der Prüfungskommission:
Prof. Dr. Gerd Kempermann (Vorsitzender)
Prof. Dr. Björn Falkenburger
Prof. Dr. Mike O. Karl
Datum der Verteidigung: 23.04.2021
The work described in this thesis was performed at the Universitätsklinikum Carl Gustav Carus,
Fetscherstraße 74, 01307 Dresden and the B CUBE - Center for Molecular Bioengineering,
Technische Universität Dresden, Tatzberg 41, 01307 Dresden.

Mechanisms of Axonal Transport in ALS Table of Contents
Anne Seifert ii
Table of Contents List of Abbreviations ................................................................................................................... iv
1. Introduction ............................................................................................................................ 1
1.1 Amyotrophic lateral sclerosis (ALS) .................................................................................. 1
1.2 Axonal transport ............................................................................................................... 9
1.3 Modelling ALS and axonal transport in vitro .....................................................................18
1.4 Aim of this study ..............................................................................................................22
2. Materials and Methods ..........................................................................................................24
2.1 Materials ..........................................................................................................................24
2.2 Methods ..........................................................................................................................32
3. Results ..................................................................................................................................46
3.1 Modification of a kinesin-1-dependent microtubule gliding assay for the application of
whole cell lysates ..................................................................................................................46
3.2 Determination of assay sensitivity with the microtubule-associated protein tau ................53
3.3 Investigation of a direct interference of recombinant FUS-GFP with kinesin-1 motility and
microtubule gliding.................................................................................................................54
3.4 Kinesin-1-dependent microtubule gliding and assay sensitivity in the presence of whole
cell lysates expressing GFP-labelled FUS variants ................................................................58
3.5 Arrest in gliding of single microtubules caused by Tau-GFP compared to aging of the
assay .....................................................................................................................................60
3.6 Differential effects of 2N3R and 2N4R tau individually on kinesin-1-dependent microtubule
gliding velocity and microtubule binding .................................................................................64
3.7 Impact of increasing 4R:3R tau isoform ratios on kinesin-1-dependent microtubule gliding
and its microtubule binding ....................................................................................................68
3.8 Expression levels of 4R and 3R tau isoforms in whole cell lysates ..................................71
4. Discussion ............................................................................................................................73
4.1 The modified kinesin-1-dependent microtubule gliding assay detects nanomolar amounts
of recombinant human tau-GFP.............................................................................................74
4.2 Wildtype and ALS-associated FUS variants do not directly interfere with kinesin-1 motility
and do not bind to microtubules .............................................................................................76
4.3 An increase in 4R:3R tau isoform ratio might contribute to the axonal transport defects
observed in FUS-ALS ............................................................................................................79
4.4 FUS variants may indirectly affect microtubule-based axonal transport by acting on a
multitude of cellular processes ..............................................................................................88
4.5 Outlook ............................................................................................................................93
4.6 Conclusion .......................................................................................................................94

Mechanisms of Axonal Transport in ALS Table of Contents
Anne Seifert iii
References ...............................................................................................................................95
Supplementary Figures ........................................................................................................... 133
Supplementary Tables ............................................................................................................ 134
List of Figures ......................................................................................................................... 139
List of Tables .......................................................................................................................... 140
List of Supplementary Figures ................................................................................................. 141
List of Supplementary Tables .................................................................................................. 141
Summary ................................................................................................................................ 142
Zusammenfassung ................................................................................................................. 144
Acknowledgements ................................................................................................................. 147
Anlage 1.................................................................................................................................. 149
Anlage 2.................................................................................................................................. 150

Mechanisms of Axonal Transport in ALS List of Abbreviations
Anne Seifert iv
List of Abbreviations
AA ........................................................................................................................... Ascorbig acid ALS .................................................................................................. Amyotrophic lateral sclerosis APC .................................................................................................. Adenomatous polyposis coli APP ..................................................................................................... Amyloid precursor protein BCA ................................................................................................................. Bicinchoninic acid BDNF ........................................................................................ Brain-derived neurotrophic factor C9orf72 ............................................................................ Chromosome 9 open reading frame 72 CNS ........................................................................................................ Central nervous system CV ...................................................................................................................... Column volumes DAPT .................................. N-[N-(3,5-Difluorphenacetyl)-L-alanyl]-S-phenylglycin-tert-butylester DDB ...................................................................................................... Dynein dynactin BICD2N DDS .......................................................................................................... Dichlorodimethylsilane DHCs ........................................................................................................... Dynein heavy chains ER .......................................................................................................... Endoplasmatic reticulum FRET ............................................................................ Fluorescence resonance energy transfer FTD ....................................................................................................... Frontotemporal dementia FUS ................................................................................................................. Fused in sarcoma GMPCPP .................................................................. Guanosine 50-[a,b-methylene] triphosphate HDAC1 ...................................................................................................... Histone deacetylase 1 HDAC6 ...................................................................................................... Histone deacetylase 6 hESC ................................................................................................ Human embryonic stem cell HR .....................................................................................................Homologous recombination HRP ........................................................................................................ Horseradish peroxidase HSP60 ............................................................................................. heat shock protein of 60 kDa iPSCs ............................................................................................. Induced pluripotent stem cells JIP ............................................................................. c-Jun N-terminal kinase-interacting protein KHCs .......................................................................................................... Kinesin heavy chains KLCs .............................................................................................................. Kinesin light chains MAPs ........................................................................................... Microtubule-associated protein Miro .................................................................................................... Mitochondrial Rho GTPase N ................................................................................................................ Amino-terminal insert NDs .................................................................................................. Neurodegenerative diseases NHEJ ................................................................................................ Nonhomologous end joining NMR ................................................................................................ Nuclear magnetic resonance NPCs ..................................................................................................... Neuronal precursor cells PAR ....................................................................................... Poly adenosine diphosphate ribose PARP ................................................................. Poly adenosine diphosphate ribose polymerase PBS ....................................................................................................... Phosphate Buffer Saline PFS ............................................................................................................ Perfect Focus System PLO .................................................................................................................... Poly-L-Ornithine R ........................................................................................................ Microtubule-binding repeat RBPs .......................................................................................................... RNA-binding proteins rhBDNF ..................................................................................... Brain-derived neurotrophic factor rhGDNF ..................................................... Recombinant human glial derived neurotrophic factor RNP ................................................................................................................. Ribonucleoprotein ROS ...................................................................................................... Reactive oxygen species RT ................................................................................................................... Room temperature SAG ............................................................................................................. Smoothened agonist

Mechanisms of Axonal Transport in ALS List of Abbreviations
Anne Seifert v
SDS-PAGE ....................................... Sodium dodecyl sulfate-polyacrylamide gel electrophoresis SEC ............................................................................................ Size exclusion chromatography SEM .................................................................................................... standard error of the mean SFPQ ........................................................................... Splicing factor proline- and glutamine-rich SOD1 ...................................................................................................... Superoxide dismutase 1 TDP-43 ........................................................................................... TAR DNA-Binding Protein 43 TGFβ-3 ........................................................................................ Transforming growth factor β-3 TIRF ..................................................................................... Total internal reflection fluorescence TRAK ................................................................................................................ Trafficking kinase

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 1 / 157
1. Introduction
1.1 Amyotrophic lateral sclerosis (ALS)
Neurodegenerative diseases (NDs), such as Alzheimer’s, Parkinson’s, Huntington’s disease,
Frontotemporal dementia (FTD), and ALS, are among the most common causes for mortality
worldwide, with increasing tendency. NDs are highly age-dependent, occurring typically but not
exclusively in the elderly. It becomes progressively important to understand the underlying
pathomechanisms of these diseases, since the elderly population has increased over the last
decades (Heemels, 2016). To date, NDs are generally incurable and only limited treatment
options exist for most of them, partly because the underlying pathophysiology is very diverse.
Some disorders primarily cause cognitive impairment and/or memory loss as seen in FTD and
Alzheimer’s disease (Abeliovich and Gitler, 2016; Canter et al., 2016; Wyss-Coray, 2016), while
others mainly affect the motor system, causing movement, speech, and breathing deficits, as
seen in ALS (Taylor et al., 2016).
ALS is the most common ND specifically affecting cortical (upper) motor neurons in the
primary motor cortex, and spinal (lower) motor neurons in the brainstem and spinal cord. This
disease was first described by Jean-Martin Charcot and Alix Joffroy in 1869 (Charcot and
Joffroy, 1869). Various studies report an incidence for ALS ranging from 0.6 to 3.8 per 100 000
persons/year and a prevalence of between 4.1 and 8.4 per 100 000 persons, with regional
differences (reviewed by Longinetti and Fang, 2019). Men are at higher risk to develop ALS
than women, with reported male-to-female ratios between 1.2 and 1.5 (Manjaly et al., 2010). It is
a progressive disorder with a median age of onset between 51 and 66 years of age throughout
the world (Longinetti and Fang, 2019), in Germany around 61 years (Dorst et al., 2019), and
usually leads to death due to respiratory failure about 2-5 years after symptom onset (Naumann
et al., 2018). ALS commonly manifests as a spinal onset (e.g. weakness in the limbs, up to
82 % of cases), but patients may also show bulbar onset (e.g. difficulty in speaking or
swallowing), mixed spinal and bulbar onset, or other forms of onset, including thoracic onset
(D’Ovidio et al., 2019), dementia or respiratory symptoms (Longinetti et al., 2018; Leighton et
al., 2019). While all ALS patients suffer from motoric symptoms, only 30 % of patients also
develop symptoms of FTD (Lomen-Hoerth, 2011), which also include changes in mood and
behavior (Erkkinen et al., 2018), and those patients were found to face a shorter life expectancy
(Olney et al., 2005).
There is no cure for ALS to date and only two approved drugs, Riluzole and Edaravone.
For a long time, Riluzole, a presumed glutamate antagonist, has been the only authorized drug

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 2 / 157
for the treatment of ALS after its approval in 1995 (Wokke, 1996). It acts anti-excitotoxic, which
may be related to its ability to inhibit glutamate release, to inactivate voltage-gated sodium
channels, or to interfere with intracellular signal transmission following transmitter binding at
excitatory receptors (Jaiswal, 2019). Recently, Edaravone has been approved in some countries
as an alternative or additional treatment (Bhandari et al., 2018). Edaravone is thought to be a
scavenger for reactive oxygen species, and reportedly eliminates lipid peroxides and hydroxyl
radicals. In neurons, it presumably alleviates injuries caused by free oxygen radicals (Jaiswal,
2019). The exact cellular and molecular targets of Edaravone are however still unknown. Both
Riluzole and Edaravone are effective only in subpopulations of ALS patients and increase
survival time or slow down progression of the disease (Bensimon et al., 1994; Abe et al., 2017;
Jaiswal, 2019).
On the cellular level, ALS is a non-cell-autonomous disease and characterized by the
progressive atrophy of cortical and spinal motor neurons, a reduction in size of the remaining
neurons, hyperexcitability of the motor system (Ferraiuolo et al., 2011), as well as astro- and
microgliosis (Saberi et al., 2015). The latter implicates an inflammatory component in ALS,
which has been subject of a variety of studies in the past. ALS-associated mutations in
superoxide dismutase 1 (SOD1) expressed in astrocytes have been linked to enhanced
activation of microglia in a mouse model of ALS, which may play a role in disease progression
due to the enhanced production of nitric oxide and toxic cytokines (Yamanaka et al., 2008). In
contrast, reactive astrocytes and microglia were found to surround degenerating motor neurons
in ALS patients and mouse models (McGeer and McGeer, 2002; Boillee et al., 2006), where
they secreted proinflammatory cytokines (Saberi et al., 2015). This suggests that a
neuroinflammatory response may exert neuroprotective as well as neurodegenerative effects,
and provides additional evidence for an involvement of not only neurons, but also astrocytes,
microglia, and oligodendrocytes in ALS pathology (Scekic-Zahirovic et al., 2017).
On the molecular level, there is a general consensus that multiple pathogenic processes
contribute to the neuropathology of ALS, including abnormal RNA processing (Donnelly et al.,
2014), aberrant protein folding and the formation of stress granules (dense aggregates
containing proteins and RNA forming under stress conditions within the cell, Gutierrez-Beltran et
al., 2015; Parakh & Atkin, 2016), mitochondrial dysfunction (Smith et al., 2019) and impaired
axonal transport (Ferraiuolo et al., 2011; Ikenaka et al., 2012; Sama et al., 2014; De Vos and
Hafezparast, 2017a). The exact type and degree of neuropathology essentially depends on the
underlying cause for ALS.

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 3 / 157
About 90 % of cases are sporadic, where no specific genetic or environmental cause can
be identified. Common risk factors for sporadic ALS include older age (largely increasing above
age 50) and male sex. Other factors, such as physical fitness, repeated head injury,
occupational and environmental factors, and previous medical conditions (e.g. diabetes) are still
controversial, although they most likely strongly influence the development of the disease
(reviewed in Fang et al., 2015; van Rheenen et al., 2016). The remaining ~10 % of cases are
familial and are caused by specific mutations (Chen et al., 2013; reviewed in Renton et al.,
2014). The most common variations associated with ALS are within the genes of Chromosome
9 open reading frame 72 (C9orf72, ~30 %, DeJesus-Hernandez et al., 2011), SOD1 (~20 %,
first ALS gene to be identified in 1993 by Rosen et al.), TAR DNA-Binding Protein 43 (TDP-43,
5 %, Sreedharan et al., 2008), and fused in sarcoma (FUS, 5 %, (Kwiatkowski et al., 2009;
Vance et al., 2009; Naumann et al., 2019).
1.1.1 ALS-associated FUS
The FUS gene, also called translocated in liposarcoma, is located on chromosome 16 and
consists of 15 exons that encode a 526-amino-acid protein (Aman et al., 1996) and has first
been identified as a proto-oncogene causing liposarcoma by chromosomal translocation (Crozat
et al., 1993). It belongs to the FET protein family (the other members of that family being the
Ewing Sarcoma protein and TATA binding associated factor15, Svetoni et al., 2016), is
ubiquitously expressed in most tissues and mainly localizes to the nucleus (Andersson et al.,
2008; Sama et al., 2014). Its N-terminus is involved in transcriptional regulation and DNA
damage repair via a prion-like domain (rich in glutamine, glycine, serine, and tyrosine (QGSY)),
an additional glycine-rich region, and an RNA-recognition motif (Figure 1) (Sama et al., 2014).
The C-terminal region contains multiple arginine-glycine-glycine(RGG)-rich nucleic acid-binding
domains, a zinc-finger-binding domain, and a non-classical nuclear localization sequence
(Prasad et al., 1994) and attributes to e.g. RNA transport.

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 4 / 157
Figure 1: Functional domains of FUS.
The diversity of functional domains within FUS allows for its involvement in a multitude of cellular processes. Depicted are known domains of FUS and their annotated functions, as well as the most common mutations associated with familial ALS (adapted from Ticozzi et al., 2009; Sama et al., 2014; An et al., 2019). The mutation of interest in this study (P525L) is highlighted in red.
Due to this conserved, albeit complex structure, FUS interacts with DNA (Baechtold et al.,
1999) as well as RNA (Crozat et al., 1993; Zinszner et al., 1997) and its role in a variety of
cellular processes has long been proven. These include cell proliferation and DNA repair
(Bertrand et al., 1999), transcription regulation, RNA splicing (Yang et al., 1998) and transport
within the cell (Zinszner et al., 1997). FUS has been reported to bind RNA of more than 5500
genes in the central nervous system (CNS), primarily via a GUGGU-binding motif (Lagier-
Tourenne et al., 2012). By transporting mRNA to the distal axon or dendrites, e.g. those
encoding actin-related proteins, it may regulate local translation in response to external stimuli
(Fujii and Takumi, 2005; Fujii et al., 2005). Due to its diverse functions, it appears reasonable
that a series of pathological events are observed upon in its absence or malfunction.
1.1.2 Molecular pathology of ALS-FUS
Because FUS is involved in a variety of often interconnected processes within the cell, single
mutations in its genomic sequence can result in the occurrence of ALS (Figure 1), although it is
not yet clear whether the disease is caused by a loss-of-function or gain-of-function in FUS.
Mutations in FUS account for 35 % of cases manifesting before 40 years, typically presenting
classical ALS symptoms, and are responsible for the youngest case described (Conte et al.,
2012; Millecamps et al., 2012). These are autosomal-dominant mutations and primarily occur in
the C-terminal NLS, such as R521C or P525L, resulting in cytoplasmic mislocalization of FUS
due to reduced interaction with the nuclear import receptor Transportin-1 (Dormann et al., 2010)

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 5 / 157
and in impaired interaction of FUS with other RNA-binding proteins (RBPs) (Marrone et al.,
2019). Interestingly, there seems to be a direct relationship between the degree to which
nuclear import of FUS is diminished and the severity of the disease (defined by age of onset
and time till death) (Dormann et al., 2010; Kino et al., 2011; Niu et al., 2012).
FUS preferentially localizes to active chromatin (Immanuel et al., 1995), where it binds to
single-stranded DNA motifs in the promotor region of its target genes. Current evidence points
towards its function as a general transcriptional regulator, with only modest changes in mRNA
levels in its absence (Tan and Manley, 2010; Hoell et al., 2011; Lagier-Tourenne et al., 2012;
Rogelj et al., 2012; Tan et al., 2012; Li et al., 2013; Nakaya et al., 2013). FUS may also directly
bind to and recruit RNA polymerase II and III by preventing its phosphorylation at specific sites
(Immanuel et al., 1995; Bertolotti et al., 1996, 1998; Tan and Manley, 2010; Schwartz et al.,
2015). Additionally, FUS regulates transcription via interaction with specific transcription factors
(e.g. Runx2 or NF-κB, Du et al., 2011; Hallier et al., 1998; Kim et al., 2010; S. Sato et al., 2005;
Uranishi et al., 2001) and transcription initiation factor TFIID (Bertolotti et al., 1996; Hallier et al.,
1998; Uranishi et al., 2001; Li et al., 2010).
FUS modulates splicing of pre-mRNA in conjunction with other splicing regulators
machinery (e.g. SMN, U11, U11, U12, Sm proteins, SFPQ, and others) (Alliegro and Alliegro,
1996; Hackl and Lührmann, 1996; Zhou et al., 2002; Meissner et al., 2003; Yamazaki et al.,
2012; Gerbino et al., 2013; Tsuiji et al., 2013; Sun et al., 2015; Reber et al., 2016; Ishigaki and
Sobue, 2018) by binding to long introns within the prespliced RNA (Hoell et al., 2011; Ishigaki et
al., 2012; Lagier-Tourenne et al., 2012; Rogelj et al., 2012). As such, FUS is involved in the
splicing of actin pre-mRNA, as has been shown for example in mouse brain extracts (Fujii and
Takumi, 2005), but also of the microtubule-associated protein (MAP) tau, a neuron specific
cytoskeleton related protein (Ishigaki et al., 2012; Lagier-Tourenne et al., 2012; Orozco et al.,
2012; Rogelj et al., 2012). Altered splicing by mutant FUS variants hence might affect the
cytoskeletal architecture of neurons. FUS also regulates its own splicing (Lagier-Tourenne et al.,
2012; Nakaya et al., 2013), specifically by FUS-mediated skipping of exon 7 in FUS pre-mRNA,
which results in its nonsense-mediated decay (Ishigaki and Sobue, 2018). This process is
disrupted for mutant FUS variants due to their mislocalization to the cytoplasm. Impaired FUS
splicing may in turn exacerbate the pathogenic cytoplasmic accumulation of FUS (Zhou et al.,
2013). Further, FUS is involved in alternative splicing of more than 3200 exons, of which many
are part of mRNAs coding for proteins important in neuronal function or neurodegeneration
(Ishigaki et al., 2012).

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 6 / 157
There is evidence that the physical binding capability between FUS and its RNAs is not
altered between wildtype and mutant FUS variants, as 80 % of transcripts binding to mutant
(R521G and R521H) FUS can also bind to wildtype FUS (Hoell et al., 2011). This supports the
hypothesis of a gain-of- function phenotype for mislocalized cytoplasmic mutant FUS variants
with respect to RNA binding and processing. After splicing, FUS subsequently shuttles mRNA
between the nucleus and the cytoplasm (Fujii et al., 2005). FUS has been attributed to interact
with several actin- and microtubule-binding motor proteins, including Myosin-Va (Yoshimura et
al., 2006) as well as Myosin-VI (Takarada et al., 2009), and has been isolated as part of large
granules that associates with KIF5B (Kanai et al., 2004). This hints towards an involvement of
FUS in cellular transport of mRNAs to their dedicated locations for local translation. Indeed,
upon synaptic activation via the metabotropic glutamate receptor mGluR5, FUS translocates
into dendritic spines (by to date unknown mechanisms) and may facilitate the local translation of
actin-associated proteins (Fujii and Takumi, 2005). This is in line with the fact that loss of FUS
consequently results in abnormal spine morphology and attenuated spine density (Fujii et al.,
2005), since spine structure and stability is largely dependent on the actin cytoskeleton (Penzes
and Rafalovich, 2012; Basu and Lamprecht, 2018). Local translation is however not solely
regulated by the transport of mRNA, but also by sequestering mRNA together with its RBPs.
As a protein with primarily nuclear localization under physiological, non-stressed
conditions, FUS binds single- and double-stranded DNA (Baechtold et al., 1999; Liu et al.,
2013), thereby promoting DNA damage repair via homologous recombination (HR) and
nonhomologous end joining (NHEJ) (Mastrocola et al., 2013; Wang et al., 2013). For HR, the
pairing of homologous DNA is an essential step which has been proposed to be a crucial
function devoted especially to the C-terminal region of FUS (Akhmedov et al., 1995; Baechtold
et al., 1999; Liu et al., 2013). Phosphorylated FUS (Pezzano et al., 1996) is one of the first
proteins recruited to sites of DNA damage (Mastrocola et al., 2013; Wang et al., 2013), where it
interacts with histone deacetylase 1 (HDAC1) and poly-ADP ribose (Deng et al., 2014). A
subset of ALS-associated mutations impair FUS in its function in DNA damage response
(Rulten et al., 2014; Naumann et al., 2018). In the absence or in case of malfunction of FUS,
DNA double-strand repair by HR and NHEJ was decreased between 30-50 % (Mastrocola et al.,
2013; Wang et al., 2013). In primary mouse cortical neurons, the decrease in DNA repair
efficiency by NHEJ upon FUS depletion was even higher, namely 65 % (Wang et al., 2013).
Together, these results suggest a role of FUS in DNA damage repair in proliferating as well as
postmitotic cells. It has further been shown that FUS interacts with HDAC1 at sites of DNA
double-strand breaks (Miller et al., 2010; Dobbin et al., 2013; Thurn et al., 2013), while it is

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 7 / 157
recruited to single-strand breaks by poly adenosine diphosphate ribose polymerase (PARP),
which binds to these sites and subsequently polymerizes poly adenosine diphosphate ribose
(PAR) chains (Schreiber et al., 2006; De Vos et al., 2012). In a transgenic mouse model
expressing FUS-R521C next to its wildtype form, mutant FUS forms a complex with wildtype
FUS and alters its interaction with HDAC1 (Qiu et al., 2014), calling for a dominant negative
effect of this FUS mutation. This is in line with the finding that several mutant FUS variants
(R244C, R514S, H517Q, and R521C) showed deficiencies in HR--mediated repair relative to
wildtype FUS in U2OS cells, independent of their nuclear or cytoplasmic localization (Wang et
al., 2013). As DNA damage accumulates with increasing age (e.g. due to a lifetime exposure to
DNA-damaging factors and a decay of quality control mechanisms over decades, Gorbunova et
al., 2007), it seems logical that the pathological consequences, i.e. the phenotype of FUS -ALS,
becomes apparent only later in the life of patients. It also indicates why mainly neurons are
affected by the disease, since they lack the ability to replicate and self-renew and are therefore
more susceptible to accumulate DNA damage. This is in line with the finding of increased levels
of γH2AX in postmortem brain sections of patients harboring the FUS R521C or P525L mutation
(Wang et al., 2013). γH2AX is a marker for DNA damage, but also correlates with apoptosis
(Rogakou et al., 2000; Pasinelli and Brown, 2006).
The low complexity, prion-like domain of FUS, with its high content of glycine, accounts for
its tendency to aggregate (Han et al., 2012). Under physiological conditions, RBPs with such a
domain can accumulate together with mRNA, ribosome translation initiation factors, and other
proteins into membraneless compartments called stress granules (Dewey et al., 2012; Mateju et
al., 2017). Stress granules are stalled translational complexes evolving upon metabolic or
environmental stress involved in the triage of mRNAs, determining whether an mRNA is
translated, degraded, or suppressed in order to promote the expression of proteins essential to
reestablish homeostasis (Sama et al., 2014). Wildtype FUS can rapidly and reversibly shuttle to
the cytoplasm upon induction of cellular stress, where it associates with stress granules. Under
physiological conditions, it returns to the nucleus once stress is resolved (Dormann et al., 2010).
ALS-associated, consistently mislocalized FUS variants, however, have long been observed to
assemble within pathological stress granules upon protein overexpression, heat shock, and ER
or oxidative stress (Andersson et al., 2008; Bosco et al., 2010; Dormann et al., 2010; Gal et al.,
2011; Kino et al., 2011; Bentmann et al., 2012; Daigle et al., 2013). However, if mutated variants
of RBPs are incorporated into these structures, they undergo a pathological liquid-to-solid phase
transition, resulting in solidified SGs (Bowden and Dormann, 2016). Solidified SGs have lost
their dynamicity and show altered mechanical properties (Nötzel et al., 2018), and hence can no

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 8 / 157
longer fulfill their physiological roles. This results in impaired stress response and mRNA
transport, changes in local translation, and the formation of pathological aggregates, all of which
contributes to neuron dysfunction and the progression of neurodegeneration (Bowden and
Dormann, 2016). The formation of SGs and how severely it affects the cell therefore also largely
depends on the local concentration of a respective protein. For instance, cytoplasmic
accumulated mutant FUS colocalizes with these pathological aggregates (Dewey et al., 2012;
Wolozin, 2014). Hence, while FUS itself is not essential for the formation of SGs, the
cytoplasmic mislocalization of its mutant variants largely contributes to the transformation of
SGs.
Another class of cellular organelles that seems to be substantially influenced by mutant
FUS variants is mitochondria. Disruptions in various mitochondrial characteristics, such as
structure, dynamics, bioenergetics, calcium homeostasis, endoplasmatic reticulum (ER)-
mitochondrial contact, mitochondrial quality control, and cellular transport have been reported in
ALS patients and various model systems (reviewed in Smith et al., 2019). One of the first
observed changes in ALS patient motor neurons are structurally altered (i.e. swollen and
vacuolated) and aggregated mitochondria (Atsumi, 1981; Sasaki and Iwata, 2007).
Overexpression of FUS R521G/H or P525L variants results in mitochondrial shortening
(Tradewell et al., 2012; Deng et al., 2015; Sharma et al., 2016). The P525L mutation additionally
results in deformation and loss of mitochondrial cristae in mouse models (Deng et al., 2015;
Sharma et al., 2016). FUS seems to directly influence mitochondrial function through interaction
with the mitochondrial chaperone heat shock protein of 60 kDa (HSP60) (Deng et al., 2015) and
overexpression of FUS leads to reduced mitochondrial ATP production (Stoica et al., 2016) as
well as augmented levels of reactive oxygen species (ROS), causing oxidative stress (Deng et
al., 2015).
Non-functional and damaged mitochondria are subject to clearance by mitophagy under
physiological conditions and need to be replaced by functional mitochondria (Sterky et al., 2011;
Hamacher-Brady and Brady, 2016). This process requires a robust transport of mitochondria
throughout the cell. However, in motor neurons carrying the P525L mutation, generated from
ALS patient-derived induced pluripotent stem cells (iPSCs), virtual arrest of mitochondrial
movement in distal, but not proximal axons was observed, accompanied by a loss of membrane
potential in these mitochondria along with reduced mitochondrial length (Naumann et al., 2018).
In general, a defect in transport of mitochondrial and other organelles has reportedly been
observed, often as one of the earliest pathophysiological events in motor neurons affected by

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 9 / 157
ALS, which indicates that deficits in axonal transport might be a primary cause of motor neurons
loss (De Vos et al., 2008; De Vos and Hafezparast, 2017a).
1.2 Axonal transport
Intracellular transport of RNA, proteins, and organelles is essential to cell survival and correct
function, as well as neurotrophic and injury signaling. It becomes particularly important in
(motor) neurons, which have to facilitate long-range transport along dendrites and axons, latter
with lengths up to one meter (Grafstein and Forman, 1980; Fletcher and Theriot, 2004). In
addition to the large distance that needs to be covered by cellular transport mechanisms,
cellular homeostasis requires transport in two directions, namely anterograde (i.e. away from
the cell soma) and retrograde (i.e. towards the cell soma). Two main classes of axonal transport
can be distinguished based on the average speed of transport movement. Fast axonal transport
is characterized by a speed of ~50 - 400 mm/day or 0.6 - 5 µm/s. Slow axonal transport is
further subdivided into two parts, depending on the proteins transported and the speed, namely
slow component a and b, which cover a distance of 0.2 - 3 mm/day or 0.0002 - 0.03 µm/s and
2 - 10 mm/day or 0.02 - 0.1 µm/s, respectively (De Vos and Hafezparast, 2017a). Each of these
transport types delivers a distinct set of cargoes, and the differences in overall speed results
from prolonged pauses between movement phases in slow axonal transport (Black, 2016). Both
fast and slow transport is mediated by the same molecular motors moving along tracks of the
cytoskeletal network, defining motors and the cytoskeleton as the two major components of
axonal transport.
1.2.1 Microtubules as part of the neuronal cytoskeleton
The neuronal cytoskeleton consists of three major components, namely actin filaments,
neurofilaments, and microtubules. Actin filaments are composed of granular (G-) actin
monomers, which assemble to form a ~6 nm wide double-helix of filamentous (F-) actin (Boron
and Boulpaep, 2005) and are most prominent in the axonal growth cone and dendritic spines
(Schevzov et al., 2012). Neurofilaments are type IV intermediate filaments with an average
diameter of ~10 nm and are present in the perikarya and dendrites, but particularly in axons,
where they are vital for the radial growth of axons during development and the further
maintenance of axon caliber, as well as the transmission of electrical impulses along the axon
(Friede and Samorajski, 1970; Ohara et al., 1993; Eyer and Peterson, 1994; Zhu et al., 1997;
Yum et al., 2009; Yuan et al., 2012).
The major portion of axonal transport occurs along microtubules, which are rigid hollow
cylinders about 25 nm in diameter (Figure 2). They are dynamic filaments that frequently

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 10 / 157
assemble and disassemble within the cell. Microtubules polymerize from free tubulin, which
itself is a heterodimer of two closely related 55 kDa polypeptides, α- and β-tubulin. Microtubules
are formed by 13 linear chains of these heterodimers, so called protofilaments. Both tubulin
polypeptides bind GTP to regulate polymerization, but only GTP bound to β-tubulin is
hydrolyzed shortly after polymerization. GTP hydrolysis weakens the binding affinity of β-tubulin
to adjacent molecules, which favors depolymerization and enables dynamic behavior of
microtubules (Cooper, 2000a). This behavior is referred to as dynamic instability and describes
the alternating cycles of growth and shrinkage of microtubules at their individual protofilaments,
a process determined by the rate of GTP hydrolysis relative to the rate of tubulin dimer addition
(Mitchison and Kirschner, 1984). Since the sequential addition of tubulin heterodimers results in
so called head-to-tail arrays (Cooper, 2000b) with alternating α- and β-tubulin units in each
protofilament, microtubules are polar structures with a fast-growing plus end (protofilament
ending with a β-tubulin subunit) directed towards the cell cortex, and a slow-growing, more
stable minus end (protofilament ending with a α-tubulin subunit) directed towards the soma
(Maday et al., 2014). In axons, microtubules form a unipolar array with the plus end facing
towards the distal axon, while the microtubule organization in dendrites often consists of arrays
with mixed polarity (Baas et al., 1988; Kwan et al., 2008; Kleele et al., 2014). Hence, the correct
polarity and orientation of microtubules majorly contribute to the specific distribution of cargo
throughout the cell by a variety of motor proteins.
Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 11 / 157
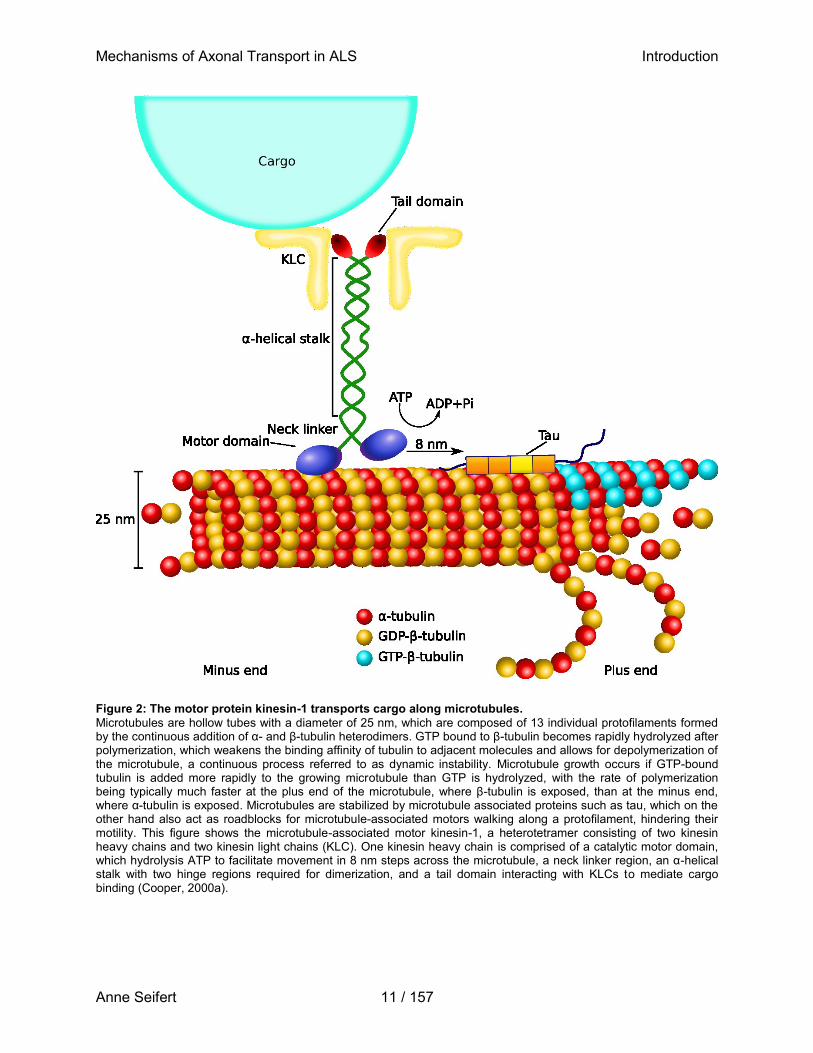
Figure 2: The motor protein kinesin-1 transports cargo along microtubules.
Microtubules are hollow tubes with a diameter of 25 nm, which are composed of 13 individual protofilaments formed by the continuous addition of α- and β-tubulin heterodimers. GTP bound to β-tubulin becomes rapidly hydrolyzed after polymerization, which weakens the binding affinity of tubulin to adjacent molecules and allows for depolymerization of the microtubule, a continuous process referred to as dynamic instability. Microtubule growth occurs if GTP-bound tubulin is added more rapidly to the growing microtubule than GTP is hydrolyzed, with the rate of polymerization being typically much faster at the plus end of the microtubule, where β-tubulin is exposed, than at the minus end, where α-tubulin is exposed. Microtubules are stabilized by microtubule associated proteins such as tau, which on the other hand also act as roadblocks for microtubule-associated motors walking along a protofilament, hindering their motility. This figure shows the microtubule-associated motor kinesin-1, a heterotetramer consisting of two kinesin heavy chains and two kinesin light chains (KLC). One kinesin heavy chain is comprised of a catalytic motor domain, which hydrolysis ATP to facilitate movement in 8 nm steps across the microtubule, a neck linker region, an α-helical stalk with two hinge regions required for dimerization, and a tail domain interacting with KLCs to mediate cargo binding (Cooper, 2000a).

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 12 / 157
1.2.3 Microtubule motor proteins
There are two major families of motor proteins that associate with microtubules, namely kinesins
and dyneins. The human kinesin superfamily consists of 45 members, of which 38 are
expressed in the brain (Miki et al., 2001), and can be subdivided into 15 subfamilies. Most
kinesins move towards the plus end of microtubules in a straight path along a single
protofilament (Maday et al., 2014), thereby mainly driving anterograde transport in neuronal
axons. Members of the kinesin-1 (also referred to as KIF5), kinesin-2 (i.e. KIF3A and B, KIF17),
and kinesin-3 (i.e. KIF1A, KIF1Bα, and KIF1Bβ) family collectively contribute to this anterograde
(Maday et al., 2014), with fast axonal transport mainly involving kinesin-1 and members of the
kinesin-3 family, while slow anterograde axonal transport appears to be mainly mediated by
kinesin-1 (Xia et al., 2003). Kinesin-1 is to date the best studied motor protein and was first
discovered in axons of the giant squid in 1985 (Vale et al., 1985). It is a heterotetramer
consisting of two kinesin heavy chains (KHCs), encoded by the three mammalian genes KIF5A,
B, and C, and in most cases two kinesin light chains (KLCs), which are substantial for cargo
binding and in part contribute to the autoinhibition of kinesin in the absence of cargo (Figure 2)
(Sun et al., 2011a). KHCs are comprised of the catalytic motor domain, which facilitates walking
along the microtubule by hydrolyzing ATP and conveys processivity and direction of movement
together with a neck linker region. The motor domain is linked to an α-helical stalk interrupted by
two hinge regions, which is required for dimerization. Further, a tail domain interacts with cargo
either on its own or through KLCs and facilitates autoinhibition of the motor domain (Williams et
al., 2014, reviewed in Nobutaka Hirokawa et al., 2010). Cargo binding often requires a variety of
adaptor proteins, such as c-Jun N-terminal kinase-interacting protein (JIP), trafficking kinase
(TRAK), or mitochondrial Rho GTPase (Miro) 1 and 2, which directly or indirectly (i.e. via KLCs)
link kinesin-1 to specific cargoes (Fu and Holzbaur, 2014). Kinesin-1 walks in a hand-over-hand
mechanism with a step size of ~8 nm (about the size of a tubulin dimer) for approximately 100
steps before it detaches, walks at a speed of ~0.5 - 1 µm/s and a stall force of 5 - 6 pN
(Svoboda and Block, 1994; Hackney, 1995; Hua et al., 1997; Coy et al., 1999; Yildiz et al.,
2004; Asbury, 2005) Cargoes transported by kinesin-1 include organelles such as mitochondria,
as well as vesicular and non-vesicular cargoes (i.e. lysosomes, signaling endosomes containing
e.g. brain-derived neurotrophic factor (BDNF), amyloid precursor protein vesicles, AMPA
vesicles, and mRNA/protein complexes (Hirokawa et al., 2010).
In contrast, members of the kinesin-2 family can assemble into both homodimeric and
heterodimeric motors (Scholey, 2013) and transport for example fodrin-positive plasma
membrane precursors (Takeda et al., 2000), N-cadherin and β-catenin (Teng et al., 2005),

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 13 / 157
choline acetyltransferase (Ray et al., 1999), and Rab7-positive late endo-lysosomes (Hendricks
et al., 2010; Castle et al., 2014). Members of the kinesin-3 family dimerize upon cargo binding to
form highly processive motors (Soppina et al., 2014) and drive the motility of dense core
vesicles and synaptic vesicle precursors (Hall and Hedgecock, 1991; Okada et al., 1995; Lo et
al., 2011).
While anterograde transport is driven by a pool of different kinesins, retrograde transport
is almost exclusively carried out by cytoplasmic dynein (Roberts et al., 2013). Cytoplasmic
dynein belongs to the family of AAA+ ATPases and can be subdivided into cytoplasmic dynein 1
and 2. Cytoplasmic dynein 1 (hereafter referred to as dynein) is the main retrograde molecular
motor in neurons. It is a large, ~1.5 kDa protein complex comprising two homodimeric dynein
heavy chains (DHCs) dimerizing by their N-terminal tail domain, and multiple dynein
intermediate chains, dynein light intermediate chains, and light chains (King, 2012). The latter
three are encoded by several genes giving rise to various isoforms, and therefore contribute to
cargo-specific recruitment (Kuta et al., 2010). Similar to kinesin-1, multiple adaptor proteins,
including the dynactin complex, Bicaudal D2, lissencephaly 1, or nuclear distribution protein,
regulate the correct function, specific cargo binding and localization of dynein (Kardon and Vale,
2009). Dynein is a fast motor with velocities ranging from 0.5 - 1 µm/s, but unlike kinesin, it
frequently takes back- and side-steps between microtubule protofilaments (Mallik et al., 2005;
Ross et al., 2006). The coordinated activity of multiple motor or the binding of activators such as
Bicaudal D2, however, converts dynein into a unidirectional, highly processive motor (Mallik et
al., 2005; McKenney et al., 2014; Schlager et al., 2014). With ~1 pN, the stall force of dynein is
much weaker than those of kinesins (e.g. 5-7 pN for processive kinesin-1 or 1.5-7 pN for weakly
processive kinesin-5 in vitro, Hesse et al., 2013; Schroeder et al., 2010).
Most cargoes are not transported by a single type of motor, but by an ensemble of various
oppositely directed kinesins and dyneins (Hendricks et al., 2010; Encalada et al., 2011), even if
they are processively transported in a single direction over long distances (Maday et al., 2012).
A single organelle may be transported by 1-2 kinesins and 6-12 dyneins (Hendricks et al., 2010;
Schroeder et al., 2012; Rai et al., 2013). Several models have been suggested as to how this
assembly of motors is organized (Gross, 2004; Welte, 2004; Gross et al., 2007). The simplest
model postulates an unregulated tug-of-war between opposing kinesins and dyneins, which
could be successfully modelled for late endo-lysosomes (Müller et al., 2008; Hendricks et al.,
2010). In this scenario, movement-of-directionality of cargoes might be determined by the
biophysical characteristics of motors themselves. Kinesin-2 for instance shows load
force-dependent attachment, indicating it may be less likely to win in a tug-of-war situation

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 14 / 157
(Schroeder et al., 2012). In contrast, other cargoes such as autophagosomes exhibit strong
unidirectional motility (Maday et al., 2012), suggesting a possible down-regulation of kinesin and
pointing towards two alternative models: i) the coordinated regulation of all motor types on a
single cargo, so that only one type of motor is active at a given time, or ii) a tight regulation of
only one type of motor, i.e. kinesin as it is the stronger motor, while the other, i.e. the weaker
dynein motor, might simply be overpowered if both motors are active simultaneously and only
becomes active when the stronger motor is downregulated. This is supported by evidence for a
regulatory role of scaffolding proteins to regulate opposing motors on the same cargo (Fu and
Holzbaur, 2014; Fu et al., 2014). In addition, the well-studied autoinhibitory mechanisms of
kinesin-1 provides further evidence for this model: in the absence of cargo, the kinesin-1 tail
domain binds to the motor domain and thereby blocks motor function (Kaan et al., 2011), which
is only relieved by binding of specific scaffolding proteins such as JIP1 and JIP3 (Blasius et al.,
2007; Sun et al., 2011a). Apart from kinesin-1, regulatory mechanisms for other kinesins or for
dynein have not been elucidated in such detail to date.
The exact regulatory mechanism for each cargo most likely results from combinations of
these models. Nevertheless, a few general commonalities can be found among the diverse
patterns of motility: i) during transport, motors remain stably associated with their cargo, even in
their inactive state; ii) cargo can be effectively transported over large distances (>1 µm) by a
small ensemble of (usually opposing) motors; iii) regulatory mechanisms to control motor activity
include specific recruitment by Rab-GTPases, scaffolding proteins, and upstream regulation by
kinases and phosphatases (Maday et al., 2014).
As motor proteins are often biased, if not limited, by their preference of walking towards
one or the other end of a microtubule, they are highly influenced by the assortment of MAPs and
other posttranslational modifications they encounter along their way. While MAPs often act as
physical barriers for motors walking on cytoskeletal tracks, they can also alter the stability of
these tracks and thereby influence motor motility.
1.2.2 Microtubule-associated protein tau
Because of their dynamic instability, microtubules frequently disassemble and assemble within
the cell. Disassembly is mediated either by microtubule severing proteins or by increasing the
rate of tubulin depolymerization at the microtubule ends. Continuous assembly, i.e. the growth
of microtubules, requires stabilization of joined tubulin heterodimers, which can be achieved
through posttranslational modifications of tubulin, such as detyrosination or acetylation, or
through tightly regulated interactions with MAPs. The best characterized MAPs are MAP-4 in

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 15 / 157
non-neuronal cells, and MAP-1, MAP-2 (which is absent in axons), and tau (which is absent in
dendrites, but abundant in axons) in neuronal cells (Weingarten et al., 1975; Witman et al.,
1976; Cooper, 2000a). The ability of MAPs to interact with microtubules highly depends on the
posttranslational modifications of MAPs, specifically phosphorylation in case of tau (Lindwall
and Cole, 1984; Mandelkow et al., 1995; Drewes et al., 1997; Stoothoff and Johnson, 2005;
Ramkumar et al., 2018). Hyperphosphorylation of tau is associated with a variety of diseases
(Ksiezak-Reding et al., 1992; Köpke et al., 1993), but also with fetal development (Matsuo et al.,
1994; Yu et al., 2009) or a state of hibernation (Arendt et al., 2003). Tau is to date the best
studied MAP, in part because of its implication in a group of neurodegenerative diseases called
tauopathies (e.g. AD) (Brion et al., 1986).
The tau gene is located on chromosome 17q21 and is comprised of 16 exons, of which
exon 2,3, and 10 are alternatively spliced to produce six different isoforms in the human brain
(Goedert et al., 1989a, 1989b). Exons 2 and 3 encode for two amino-terminal inserts (N), while
exon 10 encodes for one of a total four microtubule-binding repeats (R), followed by a short C-
terminal tail sequence (Schweers et al., 1994). An isoform hence contains 0N, 1N or 2N as well
as 3R or 4R and ranges from 352 to 441 amino acids (45-75 kDa) in length (Figure 3) (Bukar
Maina et al., 2016; Guo et al., 2017a; Pîrşcoveanu et al., 2017). The isoform 0N3R is the
predominant isoform expressed during fetal development, while all isoforms are expressed in
the adult brain (Goedert et al., 1989b; Hefti et al., 2018).The C-terminal microtubule-binding
repeats form the microtubule-binding domain of tau, with their in- or exclusion influencing the
tau microtubule binding affinity, while the N-terminal domain projects away from the microtubule
surface, possibly determining the space between microtubules and interacting with other
cytoskeletal components (Hirokawa et al., 1988; Chen et al., 1992; Lu and Kosik, 2001). Under
physiological conditions, tubulin is present in 10- to 20-fold molar excess of tau in mature adult
neurons, with a physiological concentration of tau ~2-4 µM (Butner and Kirschner, 1991;
Khatoon et al., 1992; Dixit et al., 2008; Avila, 2010) and a ratio of 3R to 4R isoforms of about 1:1
(Goedert et al., 1989a; Andreadis, 2005; Ishigaki et al., 2017; Pîrşcoveanu et al., 2017).
Interestingly, recent publications demonstrated an increase in 4R:3R ratio upon knockdown of
FUS and demonstrate that FUS, in combination with splicing factor proline- and glutamine-rich
(SFPQ), directly regulates the splicing of tau mRNA (Ishigaki et al., 2017), pointing towards an
involvement of tau isoform ratio changes in FUS-ALS pathology.

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 16 / 157
Figure 3: Functional domains of tau.
Six different isoforms of tau are expressed in the adult human brain. Since there is only one gene coding for tau, isoforms are created by alternative splicing of exons 2 and 3, coding for one of two N-terminal repeats (N1 or N2), and exon 10, coding for the second of four microtubule binding repeats (R2). The projection domain, facing away from the microtubule surface, is linked to the microtubule binding domain through a proline-rich region. Depicted are known domains of tau and their annotated functions (adapted from Ambadipudi et al., 2017; Bukar Maina et al., 2016; T. Guo et al., 2017; Polverino de Laureto et al., 2019; Tepper et al., 2014).
The constant association and disassociation of tau with microtubules, along with the
different microtubule binding affinities of each tau isoform, provides microtubule stabilization as
well as flexibility as needed for correct cytoskeletal organization, especially in the distal part of
the axon (Black et al., 1996; Kempf et al., 1996). Tau localizes to labile, unstable domains of
microtubules (Black et al., 1996), where it increases its stability, thereby promoting assembly
while preventing depolymerization (Brandt and Lee, 1993; Panda et al., 1995). On the other
hand, MAPs bound to microtubules may act as roadblocks for microtubule-associated motor
proteins walking along individual protofilaments. Especially kinesin-1 has been shown to detach
at patches of microtubule-bound tau, while dynein tends to reverse direction upon encountering
lattice bound tau (Dixit et al., 2008). In addition, kinesin-1 has been proposed to compete with
tau for the same binding site on microtubules (Stamer et al., 2002; Dubey et al., 2008).
The sensitive interplay between motors, cargoes, microtubules and their posttranslational
modifications as described above can be disturbed, leading to perturbed axonal transport
perturbed as observed in for example ALS.

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 17 / 157
1.2.4 Axonal transport in ALS
First evidence for defects in axonal transport as part of ALS pathology came from post mortem
studies that reported abnormal accumulations of cytoskeletal filaments and organelles, such as
mitochondria and lysosomes (Hirano et al., 1984b, 1984a; Rouleau et al., 1996). Since then,
numerous studies have been carried out on endosome and mitochondrial trafficking or the
distribution of mRNA-containing granules (reviewed in (De Vos and Hafezparast, 2017a),
highlighting the importance of axonal transport defects in ALS pathology as a pathological event
preceding symptoms of ALS in a mouse model (Bilsland et al., 2010). However, most studies
focused on axonal transport defects associated with mutant SOD1 variants. Only little is known
about the extent and underlying pathological mechanisms of axonal transport defects caused by
abnormal FUS variants. The FUS-P525L variant causes severe impairment in axonal transport
of mitochondria in a Drosophila melanogaster model of ALS and in spinal motor neurons
differentiated from ALS patient-derived iPSCs (Baldwin et al., 2016; Naumann et al., 2018).
FUS has been proven to directly bind and regulate expression of mRNAs for several
motor proteins such as KIF5C and KIF1B (Guo et al., 2020), which are part of the ensemble that
regulates mitochondrial movement and distribution in neurons (Schwarz, 2013; Campbell et al.,
2014). In addition, stress granules undergoing a pathological liquid-to-solid phase transition
caused by FUS-NLS mutant variants, as described above (see 1.1.2), sequester RNA and
proteins in the cytoplasm, including those of the axonal transport machinery (Aulas and Vande
Velde, 2015). A more indirect influence of FUS on axonal transport has been proposed due to
the fact that expression levels of histone deacetylase 6 (HDAC6) are decreased when FUS is
silenced in mammalian cells, leading to cytoskeletal changes due to aberrant acetylation and
hence ultra-stabilization of microtubules (Hubbert et al., 2002; Kim et al., 2010). Conversely,
Guo et. al. could show that axonal transport is restored in FUS-ALS patient-specific
iPSC-derived motor neurons upon HDAC6 inhibition (Guo et al., 2017b), suggesting that the
role of tubulin acetylation in causing axonal transport defects is more complex and needs further
investigation. Changes in the underlying cytoskeletal tracks likely influence the motility of motors
walking on them. Apart from impaired transport itself, improper functioning of mitochondria has
also been suggested to contribute to ALS pathophysiology. Several FUS variants with a
mutation in their NLS disrupt mitochondrial ATP production and calcium homeostasis. The latter
is likely caused by dysfunctional communication between the ER and mitochondria due to a
reduction of their contact sites (Stoica et al., 2016). A disruption in mitochondrial ATP production
might indicate that impaired transport is due to a local depletion of ATP, the essential energy
source for motor motility.

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 18 / 157
Our limited understanding of the pathomechanisms involved in axonal transport defects in
FUS-ALS highlight the importance for further studies addressing this matter. A number of model
systems have been established that mimic FUS-ALS pathology and/or axonal transport in
general.
1.3 Modelling ALS and axonal transport in vitro
Age-related NDs are human-specific, as non-human primates or rodents do not readily develop
comparable neuropathological or clinical phenotypes (Mullane and Williams, 2019). Despite this
fact, especially mouse and rat models have long been the gold standard to study NDs in general
(Petrov et al., 2017; Van Damme et al., 2017), and although the results obtained from these
studies have been fundamental in uncovering disease pathogenesis, potential drugs identified in
animal trials have failed in clinical tests (Matus et al., 2014).
1.3.1 FUS-ALS specific phenotypes in iPSC-derived motor neurons
A more physiological model system for NDs developed with the availability of iPSCs. iPSCs are
generated by transfecting e.g. the so called Yamanaka transcription factors (Oct3/4, Sox2, Klf4,
and c-Myc) into human adult somatic cells, which induce the reprogramming and conversion of
these somatic cells to pluripotent stem cells (Takahashi and Yamanaka, 2006; Takahashi et al.,
2007). This technique allows for the generation of patient-specific iPSCs from e.g. human
fibroblasts, which are easily obtained by a skin biopsy. After successful regeneration, iPSCs can
be differentiated into virtually every cell type (Yu et al., 2007; Aasen et al., 2008), including
neuronal subtypes (Emdad et al., 2012; Japtok et al., 2015; Maury et al., 2015; Douvaras et al.,
2016), and have as such been used to model ALS (Chen et al., 2014; Kiskinis et al., 2014;
Wainger et al., 2014; Japtok et al., 2015). One of the most important advantages of this model
system is that neurons, or every other cell type, derived from patient-specific iPSCs exhibit
endogenous pathomechanisms. Such might be masked in conventional transgenic models due
to unphysiological overexpression of proteins, species-specific differences in protein expression
profiles, or the lack of availability of cell types of interest (e.g. human postmitotic neurons). The
emergence of improved gene-engineering techniques, such as CRISPR/Cas9, further increased
the capability of iPSC models to study intrinsic and endogenous cellular mechanisms, as it
allows for the reversion of single mutations to their wildtype state or the introduction of a specific
mutation into the wildtype genome (Deveau et al., 2010; Deltcheva et al., 2011; Jinek et al.,
2012; Hsu et al., 2013; Ran et al., 2013). Pairs of so called isogenic cell lines can thereby be
created from a single donor line, which contain either the wildtype or the mutated form of a
specific gene in an otherwise identical genetic background. Hence, differences observed

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 19 / 157
between those two lines can quite precisely be attributed to the individual and direct influence of
a genetic mutation.
Although motor neurons derived directly from iPSCs have been used in the past for drug
screening purposes (Egawa et al., 2012), the technique required further improvement as the
cultivation of iPSCs requires expensive growth factors, frequent feeding and splitting at narrow
ratios and needs significant time for differentiation, while it often results in inefficient
differentiation. Therefore, protocols were developed that prime iPSCs towards certain cell
lineages, e.g. neuronal cell types, using only a limited set of small molecules, i.e. growth factors
or inhibitors influencing WNT and sonic hedgehog signaling (Reinhardt et al., 2013). Using such
protocols, neuronal precursor cells (NPCs) can be derived from iPSCs, which are much easier
to propagate and can be efficiently and repeatedly differentiated into e.g. spinal motor neurons
using a different set of small molecules (Reinhardt et al., 2013; Naumann et al., 2018). Spinal
motor neurons differentiated from iPSC-derived NPCs of two isogenic cell lines can hence be
utilized to specifically and reproducibly investigate the direct influence of a single ALS-
associated mutation on cellular (patho-) mechanisms in an endogenous environment.
1.3.2 Reconstitution of axonal transport in vitro
In cell culture models, it is difficult to study the contribution of a single protein to pathological
changes in its endogenous environment. One particular protein is often involved in various
cellular pathways and in its absence or malfunctioning, it can in some cases be substituted by
other proteins. The involvement of this protein in a particular cellular mechanism might hence be
masked in cell culture models. To address this limitation, minimal systems have emerged to
study the direct interaction of a few much defined components. One of these systems is the so
called stepping assay, where single fluorescently labelled motors move across
surface-immobilized microtubules. First experimental setups involved binding of motors to
micron-sized beads, which can be tracked with nanometer precision (Howard, 2001), and
subsequent video imaging (Sheetz and Spudich, 1983; Yanagida et al., 1984; Spudich et al.,
1985) or optical trap experiments to measure forces generated by motors (Svoboda et al., 1993;
Mehta et al., 1999; Rief et al., 2000; Schnitzer et al., 2000). With advances in microscopy
techniques, e.g. TIRF microscopy, the use of smaller cyanine-based fluorophores, such as Cy3
(Funatsu et al., 1995; Vale et al., 1996), and GFP (Pierce et al., 1997) coupled to individual
kinesin molecules became possible. To date, these single molecule experiments have not only
been used to study the stepping characteristics of motors, but also (de-) polymerizing activities
and diffusion of motors and various MAPs (Yildiz et al., 2003; Helenius et al., 2006; Varga et al.,

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 20 / 157
2006, 2009; Bieling et al., 2007; Brouhard et al., 2008; Fink et al., 2009; Bugiel and Schäffer,
2018; Mitra et al., 2018).
Another method to reconstitute cellular transport in vitro has been developed with gliding
motility assays, where fluorescently labelled microtubules or actin filaments are propelled by
underlying surface-immobilized molecular motors, using ATP as an energy source. Motors are
typically immobilized on surfaces either via antibodies or by adsorption (Figure 4), while the
number of motors can vary and even be reduced to the point where a microtubule is propelled
by a single motor (Howard et al., 1989). The setup can be easily observed and analyzed using
conventional widefield fluorescence microscopy (Nitzsche et al., 2010). In the past, actin
filaments as narrow as 6 nm could observed by dark-field (Nagashima and Asakura, 1980) or
epifluorescence microscopy (Yanagida et al., 1984).
Figure 4: Principle of a microtubule gliding motility assay.
Fluorescently labeled microtubules are propelled by truncated kinesin-1 motors that are immobilized on a glass surface. As a first step in assays preparation, the assay’s glass surface is blocked with casein to prevent unspecific binding (Nitzsche et al., 2010).
By tracking the distance a microtubule filament is propelled by multiple underlying motors within
a defined time, the integrated motility of the underlying motors can be determined (Ruhnow et
al., 2011). The best understood and hence most widely used motor in these assays is kinesin-1,
but other motors can and have also been employed (Braun et al., 2011, 2017; Herold et al.,
2012; Monzon et al., 2019). Microtubules in vivo are characterized by high dynamic instability,
but often need to be highly stabilized for the use in microtubule gliding assays. This can be
achieved by growing them in the presence of the slowly hydrolysable GTP analogue guanylyl-
(α, β)-methylene-diphosphonate (GMP-CPP) or by adding microtubule stabilizing drugs such as

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 21 / 157
taxol after polymerization (Schiff et al., 1979; Korten et al., 2010), a drug which has also been
used for in vivo applications.
Taxol, or paclitaxel, was first isolated from Taxus brevifolia (pacific yew) in 1971 (Fischer
and Ganellin, 2006) and has since become one of the most widely used chemotherapeutics to
treat a variety of common solid tumors (Hagiwara and Sunada, 2004; Speyer et al., 2017). It
induces mitotic arrest and consequently apoptosis in rapidly dividing cancer cells by binding
along the length of microtubules, thereby stabilizing them, promoting microtubule assembly, and
suppressing microtubule dynamics (Schiff et al., 1979; Kumar, 1981; Howard and Timasheff,
1988; Wang et al., 2016; Zhu et al., 2016) by strengthening the lateral protofilament interactions
between tubulin dimers (Downing and Nogales, 1998; Nogales et al., 1999). There is also
evidence that taxol decreases the number of protofilaments and increases filament flexibility
(Dye et al., 1993; Felgner et al., 1996; Díaz et al., 1998). What is crucial for the elimination of
malignant tumor cells is, however, detrimental for other, healthy cells within the body that are
also susceptible to taxol, e.g. in neurons, where it leads to microtubule hyper-stabilization and
subsequent axonal degeneration (Shichinohe et al., 2015; Tasnim et al., 2016; Gornstein and
Schwarz, 2017). Although taxol is known to cause an array of sometimes severe side-effects
(e.g. sensory and painful neuropathy, Kuroi and Shimozuma, 2004; Mielke et al., 2006;
Scripture et al., 2006; Reyes-Gibby et al., 2009; Li et al., 2015; Gornstein and Schwarz, 2017), it
has recently been suggested to exploit its microtubule-stabilizing characteristics for the
treatment of diseases other than cancer (reviewed in Baas and Ahmad, 2013),
neurodegenerative diseases (Zhang et al., 2005; Michaelis et al., 2006; Ballatore et al., 2012),
as well as nerve injury (Hellal et al., 2011; Sengottuvel et al., 2011), despite the fact that it has a
poor blood-brain-barrier permeability (Brunden et al., 2011). The microtubule-stabilizing feature
of taxol has been exploited for decades to stabilize microtubules assembled from purified tubulin
dimers in vitro (Nitzsche et al., 2010)
Microtubule gliding assays have been used in a wide range of studies for varying
purposes, including understanding the active self-assembly of the transport machinery (Lam et
al., 2016), or different nanotechnological applications (Bachand et al., 2014; Hess and Saper,
2018), such as high-throughput compound screening for modulators of motor activity (Korten et
al., 2018) or establishing molecular detection devices for e.g. diagnostic purposes (Korten et al.,
2010). A microtubule gliding motility assay thereby provides insights into the biophysical
properties of molecular motors, but, when additional proteins are introduced, also makes it
possible to investigate potential direct interactions of these proteins with microtubules and/or
motors (Howard et al., 1989; Böhm et al., 1997; Korten and Diez, 2008; Scharrel et al., 2014).

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 22 / 157
While many studies previously used gliding assays to investigating the role of MAPs like tau on
microtubule-dependent motor motility (Peck et al., 2011; Yu et al., 2014), a potential interaction
of FUS with microtubules or motors has not been evaluated in this setup to date. Microtubule
gliding assays therefore provide a suitable tool to model the translocation of motors along
microtubules, and hence to model microtubule-dependent axonal transport. In contrast to in vivo
model systems, the direct and sole interaction between proteins and microtubules or motors can
be investigated. This allows for studying the influence of either a single protein of interest (i.e.
recombinant protein) or complex solutions comprising a mixture of proteins (i.e. cell lysates) on
microtubule-dependent axonal transport. Thereby, using microtubule gliding assays,
disturbances in the axonal transport machinery can be investigated which might otherwise be
masked in in vivo models of axonal transport (e.g. cell culture models).
1.4 Aim of this study
Although axonal transport defects have long been described as a pathological hallmark of FUS-
ALS, little is known about the cause and exact underlying mechanisms. FUS protein colocalizes
with kinesin-1 mRNA and was isolated as part of an RNA-transporting granule associating with
kinesin-1 protein (Kanai et al., 2004; Yasuda et al., 2017), but whether FUS directly interacts
with kinesin-1 or microtubules remains unknown to date. This thesis hence aims at developing a
novel and robust assay mimicking axonal transport in vitro in a complex environment such as
neuronal whole cell lysates. Using this assay, two hypotheses on the pathomechanisms that
might contribute to axonal transport defects will be tested.
I) FUS directly interacts with kinesin-1 or microtubules in order to transport e.g. RNA, and its
mislocalized, mutant form directly alters this interaction and impairs kinesin-1 motility on
microtubules. This would indicate that disturbances in axonal transport are a direct
consequence of cytoplasmic mislocalization of FUS.
II) An increased ratio of 4R:3R tau isoforms, as previously observed in FUS-ALS, is sufficient to
impair kinesin-1 motility on microtubules, indicating an indirect disturbance of axonal transport
as a consequence of the nuclear loss-of-function of FUS.
To address these hypotheses, axonal transport will be modelled utilizing a
kinesin-1-dependent microtubule gliding assay. The assay will be modified to be compatible with
and provide robust results in the presence of physiological buffers and complex solutions such
as whole cell lysates. First, in order to test the direct interference of wildtype FUS-GFP or the
FUS-P525L-GFP variant with kinesin-1 and/or microtubules, recombinant FUS-GFP variants, as

Mechanisms of Axonal Transport in ALS Introduction
Anne Seifert 23 / 157
well as cell lysates generated of spinal motor neurons differentiated from isogenic iPSC lines
expressing the same FUS variants, will be generated and administered to the modified gliding
assay. Second, to test the influence of increasing 4R:3R tau isoform ratios on the kinesin-1
motility on microtubules, recombinant human 2N4R tau-mScarlet and 2N3R tau-GFP isoforms
will be purified from insect cells and administered individually or combined at different ratios to
the modified kinesin-1-dependent microtubule gliding assay.
In both scenarios, the relative microtubule gliding velocity in the presence or absence of
proteins is assessed, which is determined as a measure for kinesin-1 motility. In addition, the
binding of recombinant proteins is either qualitatively assed by widefield-epifluorescence
microscopy (in the case of recombinant FUS variants) or quantitatively by total internal reflection
fluorescence (TIRF) microscopy (in the case of recombinant tau variants).
This study will hence investigate the direct or indirect interference of ALS-associated FUS
on kinesin-1-dependent transport. It will thereby provide important contributions to
understanding the pathomechanisms leading to axonal transport defects in FUS-ALS.

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 24 / 157
2. Materials and Methods
2.1 Materials
2.1.1 Technical equipment
Table 1: Technical equipment
Instrument Company
70 mL Polycarbonate Bottle Assembly Beckman Coulter GmbH, Krefeld, Germany
ÄKTA pure protein purification system General Electric Company, Boston, MA, USA
Analytical Balance - CP225D-0CE Analytical Balance - CP225D-0CE
Axio Observer Z1 microscope with a 1. 63x oil
immersion objective
Carl Zeiss, Jena, Germany
Azure c600 Gel Imaging System Azure Biosystems Inc., Dublin, CA, USA
Balance - SBA 52 Scaltec Instruments GmbH, Heiligenstadt,
Germany
BAT-10 Multipurpose Thermometer Physitemp Instruments, LLC, Clifton, NJ, USA
Biosafety cabinet HERAsafe® HS Heraeus Holding GmbH, Hanau, Germany
Cell culture Microscope – Axiovert 35 Carl Zeiss, Jena, Germany
Centrifuge – 5403 Eppendorf, Hamburg, Germany
Centrifuge - MiniSpinPlus Eppendorf, Hamburg, Germany
Centrifuge Heraeus® Biofuge® Pico Thermo Fisher Scientific, Massachusetts, USA
Centrifuge Heraeus® Biofuge® Primo Thermo Fisher Scientific, Massachusetts, USA
Centrifuge Heraeus™ Biofuge™ Stratos™ Thermo Fisher Scientific, Massachusetts, USA
CORIO CP-200F Thermostat JULABO GmbH, Seelbach, Germany
Elektro Automatik EA-PS 3016-10B Power
Supply
Elektro-Automatik GmbH & Co. KG, Viersen,
Germany
EmulsiFlex-C5 High Pressure Homogenizer AVESTIN Inc., Ottawa, ON, Canada
Fluorescence Microscope - Observer.Z1 Carl Zeiss, Jena, Germany
Fluorescence Microscope – Ti-E Nikon GmbH, Düsseldorf, Germany
Freezer -80°C HERAfreeze® Thermo Fisher Scientific, Massachusetts, USA
Hemocytometer - Neubauer improved Paul Marienfeld GmbH Co. KG, Lauda-
Königshofen, Germany
Hybridization oven - OV3 Biometra GmbH, Göttingen, Germany
iBlot® Gel Transfer Device Thermo Fisher Scientific, Massachusetts, USA

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 25 / 157
Imager - LAS3000 Fujifilm, Tokyo, Japan
Incubator - Heracell 150 Thermo Fisher Scientific, Massachusetts, USA
iXon EM+ DU-897 BV back-illuminated
EMCCD camera
Andor Technology Ltd., Belfast, UK
iXon Ultra back-illuminated EMCCD camera Andor Technology Ltd., Belfast, UK
Liquid Nitrogen Tank K Series Worthington Industries, Columbus, Ohio, USA
Microcentrifuge Heraeus™ Fresco™ 17 Thermo Fisher Scientific, Massachusetts, USA
Microscope CKX41 Olympus Deutschland GmbH, Hamburg,
Germany
MINIPULS 3 Peristaltic Pump Gilson Incorporated, Middleton, WI, USA
Multi-channel micropipette Transferpette®-8
electronic
Brand GmbH & Co. KG, Wertheim, GER
neoVAQ Maxi Vacuum Pump neoLab Migge GmbH, Heidelberg, Germany
Nikon Eclipse Ti microscope with a 1.49
PlanApo 100x oil immersion objective
Nikon, Tokio, Japan
OptimaTM LE-80K Ultracentrifuge
with Type45 Ti Rotor
Beckman Coulter GmbH, Krefeld, Germany
pH meter - inoLab ph 720 Wtw GmbH, Weilheim, Germany
Sunrise™ Absorbance Reader Tecan Trading AG, Switzerland
Thermomixer – Thermomixer 5436 Eppendorf, Hamburg, Germany
Thermomixer – Thermomixer Comfort Eppendorf, Hamburg, Germany
Water bath Julambo SW22 JULAMBO Labortechnik GmbH, Seelbach,
Germany
Water purification system - GenePure Thermo Fisher Scientific, Massachusetts, USA
XCell SureLock Mini-Cell Electrophoresis
System
Thermo Fisher Scientific, Massachusetts, USA
2.1.2 General equipment
Table 2: General equipment
Equipment Company
15 mL and 50 mL Falcons BD, New Jersey, USA
96-Well White Clear Flat Bottom Plates Corning Inc., Tewksbury MA, USA
Amicon® Ultra-4 centrifugal filters Ultracel® 30K Merck Millipore Ltd., Tullagreen, Ireland

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 26 / 157
Axygen® Filter Tips 10-200 µL Corning Inc., Tewksbury MA, USA
BD Luer-Lok 50 mL syringe Becton Dickinson S.A., Madrid, Spain
BD MicrolanceTM Canula 27 G 3/4 0.4 x 19 mm Becton Dickinson GmbH, Heidelberg,
Germany
Biosphere® Filter Tips 1000µl Sarstedt AG & Co, Nürnberg, Germany
Bio-Spin® Columns Bio-Rad Laboratories Inc., Hercules, CA, USA
Cell scraper TPP, Trasadingen, Switzerland
Costar® 6-well cell culture plates Corning Inc., Tewksbury MA, USA
Cryotubes Greiner Bio-One, Frickenhausen, Germany
Disposable Serological Pipettes Corning Inc., Tewksbury MA, USA
Eppendorf Tubes, 1.5mL, 2mL Eppendorf AG, Hamburg, Germany
iBlot® Nitrocellulose transfer stack Thermo Fisher Scientific, Massachusetts, USA
Mikrozid® Liquid Schülke&Mayr, Norderstedt, Germany
Millex HV low binding PVDV membrane filters
0.45 μm
Merck Millipore, Billerica, USA
Nalgene® Mr. Frosty Freezing Sigma Aldrich, St. Louis, USA
Omnifix® Syringes, 2-3 mL B. Braun Medical Inc., Bethlehem, PA, USA
Parafilm® Carl Roth GmbH + Co. KG, Karlsruhe,
Germany
ROTI®Nanoquant Protein Assays Carl Roth GmbH + Co. KG, Karlsruhe,
Germany
SuperdexTM 200 Increase 10/300 GL GE Healthcare Bio-Sciences AB, Uppsala,
Sweden
Surgical mask Paul Hartmann AG, Heidenheim, Germany
T25 Cell Culture Flasks Thermo Fisher Scientific, Massachusetts, USA
2.1.3 Cell culture ingredients, chemicals and reagents
Table 3: Cell culture ingredients, chemicals and reagents
Chemical Company
Accutase Life Technologies Corporation, Carlsbad, USA
Activin A Biomol GmbH, Hamburg, Germany
Amersham ECL Prime Western Blotting
Detection Reagent
GE Healthcare Life Sciences, Marlborough,
MA, USA

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 27 / 157
Ascorbic Acid Sigma-Aldrich, St. Louis, USA
B-27 Supplement minus Vitamin A Life Technologies Corporation, Carlsbad, USA
Benzonase(R) Nuclease Sigma-Aldrich, St. Louis, USA
BSA Fraction V Thermo Fisher Scientific, Massachusetts, USA
CHIR99021 Cayman Chemical, Michigan, USA
Chromatography paper grade 3mm GE Healthcare UK Limited, Buckinghamshire,
UK
cOmplete EDTA -free Protease Inhibitor
Cocktail Tablets
Roche Holding AG, Basel, Switzerland
DAPT Cayman chemical company, Ann Arbor, USA
dbcAMP Sigma-Aldrich, St. Louis, USA
Dichlorodimethylsilane Sigma-Aldrich, St. Louis, USA
DMEM/F-12 Life Technologies Corporation, Carlsbad, USA
DMSO Sigma-Aldrich, St. Louis, USA
DPBS without Ca2+/Mg2+ Life Technologies Corporation, Carlsbad, USA
Ethanol VWR International GmbH, Dresden, Germany
Glycerol MP biomedicals, Santa Ana, USA
Glycine Carl Roth GmbH & Co. KG, Karlsruhe,
Germany
HaltTM Phosphatase inhibitor Cocktail (100x) Thermo Fisher Scientific, Massachusetts, USA
HaltTM Protease Inhibitor Cocktail (100x) Thermo Fisher Scientific, Massachusetts, USA
HisTrap™ HP Columns GE Healthcare Bio-Sciences AB, Uppsala,
Sweden
Hydrofluoric acid ACS reagent, 48 % Sigma-Aldrich, St. Louis, USA
Imidazole Sigma-Aldrich, St. Louis, USA
Isopropanol VWR International GmbH, Darmstadt,
Germany
Kel-F Hub needle, 30 gauge, 2 inches, point
style 3
Hamilton Bonaduz AG, Bonaduz, Switzerland
KnockoutTM DMEM Thermo Fisher Scientific, Massachusetts, USA
Laminin from EHS sarcoma (mouse) Sigma-Aldrich, St. Louis, USA
Laminin from EHS sarcoma (mouse) Roche Holding AG, Basel, Switzerland
Luer-LokTM syringe Becton, Dickinson and Company, Franklin
Lakes, NJ, USA

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 28 / 157
Magnesium chloride hexahydrate Merck Millipore, Billerica, USA
MatrigelTM Basement Membrane Corning Inc., Tewksbury MA, USA
MenzelTM glass coverslips, 18 x 18 mm,
22 x 22 mm
Gerhard Menzel Glasbearbeitungswerk GmbH
& Co. KG, Braunschweig, Germany
Mikrozid Schülke&Mayr, Norderstedt, Germany
Monoclonal Anti-β-Tubulin I antibody,
clone SAP.4G5
Sigma-Aldrich, St. Louis, USA
N2-Supplement Life Technologies Corporation, Carlsbad, USA
NeurobasalTM Medium Life Technologies Corporation, Carlsbad, USA
Ni-NTA agarose beads Qiagen, Hilden, Germany
Non-Silicone Thermal Interface Compound Electrolube, Leicestershire, UK
Novex™ MagicMark™ XP Western Protein
Standard
Thermo Fisher Scientific, Massachusetts, USA
Novex™ NuPAGE™ LDS-Probenpuffer (4X) Thermo Fisher Scientific, Massachusetts, USA
Novex™ SeeBlue™ Plus2 Prestained
Proteinmarker
Thermo Fisher Scientific, Massachusetts, USA
Novex™ Sharp Pre-stained Protein Standard Thermo Fisher Scientific, Massachusetts, USA
NuPAGE™ MOPS SDS Running Buffer Thermo Fisher Scientific, Massachusetts, USA
NuPAGE™ Novex™ 4-12 % Bis-Tris-
Proteingel
Thermo Fisher Scientific, Massachusetts, USA
Parafilm Bemis Company Inc., Neenah, WI, USA
Penicillin-Streptomycin Life Technologies Corporation, Carlsbad, USA
Phosphatase Inhibitor (100x) Thermo Fisher Scientific, Massachusetts, USA
Phosphate Buffer Saline (PBS) Thermo Fisher Scientific, Massachusetts, USA
Pierce™ BCA Protein Assay Kit Thermo Fisher Scientific, Massachusetts, USA
Pluronic F127 Sigma-Aldrich, St. Louis, USA
Poly-L-Ornithine Sigma-Aldrich, St. Louis, USA
Potassium chloride Merck Millipore, Billerica, USA
PreScission (HRV 3C + GST) Protein Expression Facility, MPI-CBG,
Dresden Germany
Purmorphamine Cayman Chemical, Michigan, USA
Retinoic Acid Sigma-Aldrich, St. Louis, USA
rhBDNF Promega GmbH, Mannheim, Germany
rhGDNF Sigma-Aldrich, St. Louis, USA

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 29 / 157
SAG Cayman chemical company, Ann Arbor, USA
SDS Sigma-Aldrich, St. Louis, USA
Sf9-ESF Expression Systems, LLC, Davis, CA, USA
SimpleBlue Safe Stain Invitrogen, Carlsbad, CA, USA
SuperSignal™ West Femto Maximum
Sensitivity Substrate
Thermo Fisher Scientific, Massachusetts, USA
TGFβ-3 Peprotech, London, UK
Tris-HCL Carl Roth GmbH & Co. KG, Karlsruhe,
Germany
Tween 20 Serva Elektrophoresis GmbH, Heidelberg,
Germany
β-Mercaptoethanol Sigma-Aldrich, St. Louis, USA
2.1.4 Media and buffers
Table 4: Media and buffers, all amounts given in percent refer to volume/volume
Medium Composition
N2B27 medium 48.75 % DMEM/F12, 48.75 % NeurobasalTM Medium, 1 %
Penicillin/Streptomycin, 1 % B-27 Supplement without
Vitamin A, 0.5 % N2-Supplement
Normal culture (expansion)
medium
N2B27 medium with 3 µM Chiron99021, 150 µM AA,
0.5 µM SAG
Patterning medium N2B27 medium with 1 ng/mL rhBDNF, 0.2 mM AA, 1 µM
retinoic acid, 1 ng/mL rhGDNF, 0.5 µM SAG
Maturation medium N2B27 medium with 0.1 mM cAMP, 2 ng/mL rhBDNF,
0.2 mM AA, 1 ng/mL TGFβ-3, 2 ng/mL rhGDNF, 5 ng/mL
Activin A (on day 1 of maturation), 5 µM DAPT (on day 4
of maturation)
Lysis buffer for whole cell lysates PBS with 1 x protease inhibitor, 10 % glycerol, 10 mM β-
glycerophosphate
FUS-GFP storage buffer 50 mM Tris-HCL, 500 mM KCL, 1 mM DTT, 5 % glycerol
in H2O
BRB80 80 mM PIPES, 1 mM EGTA, 1 mM MgCl2
Standard motility buffer PBS with 10 µM taxol, 10 % glycerol, 10 mM β-
glycerophosphate, 0.3 % methylcellulose, 1 mM ATP,
Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 30 / 157
20 mM D-glucose, 20 μg/mL glucose oxidase, 10 μg/mL
catalase, 10 mM DTT (and 0.2 mg/mL casein in absence
of recombinant protein, BSA, or cell lysate)
VALAP Vaseline, lanolin, and paraffin in a 1:1:1 ratio
Transfer buffer for western blot 200 mM glycine, 25 mM Tris-HCL, 6.7 % methanol (in
cathode buffer) or 0.1 % SDS (in anode buffer) in H2O, pH
8.3
PBST PBS with 0.05 % Tween20
Stripping buffer 3 g glycine, 0.1 % SDS, 1 % Tween20, in H2O, pH 2.2
HisTrap storage buffer 2x 50 mM HEPES, 300 mM KCL, 0.2 % Tween20 (v/v) in
H2O
Equilibration buffer
HisTrap storage buffer with 30 mM imidazole, 10 mM β-
mercaptoethanol in H2O
Lysis buffer for the purification of
tau-GFP from Sf9 cells
50 mL equilibration buffer with one protease inhibitor
tablet
High salt buffer Equilibration buffer with 200 mM KCL
Elution buffer Equilibration buffer with 200 mM imidazole
2.1.5 Antibodies
Table 5: Primary antibodies
Antibody Host Dilution Company Reference Number
FUS Mouse 1:1000 Sigma-Aldrich, St. Louis, USA AMAB90549-100UL 3R Tau Mouse 1:2000 Merck KGaA, Darmstadt,
Germany 05-803
4R Tau Mouse 1:2000 Merck KGaA, Darmstadt, Germany
05-804
β-actin Rabbit 1:8000 Cell Signaling Technology, Inc, Beverly, MA, USA
#4967
Table 6: Secondary antibodies
Antibody Host & Reactive Species
Dilution Company Reference Number
HRP anti Mouse Donkey 1:6000 DIANOVA GmbH, Hamburg, Germany
711-035-150
HRP anti Rabbit Donkey 1:6000 DIANOVA GmbH, Hamburg, Germany
711-035-152
Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 31 / 157
2.1.6 Software
Table 7: Software
Product Type Company
FIJI (ImageJ) 1.52p Image analysis Open source (Schindelin et al., 2012)
Matlab R2017a Data evaluation and graphics The MathWorks, Inc., Natick, MA,USA
Inkscape 0.91 Figure preparation Open source (www.inkscape.org)
GraphPad Prism 7 Statistical analysis GraphPad Software Inc., La Jolla, USA
Microsoft Office 2010 Data analysis and writing Microsoft Corporation, Redmond,
Washington, USA
MetaMorph 7.8.4.0 Image analysis Molecular Devices, Sunnyvale, CA,
USA
Image StudioTM Lite,
5.2.5
Western blot image analysis LI-COR Biosciences, Lincoln, Nebraska
USA
OriginLab 2019b Statistical analysis OriginLab Corporation, Northampton,
MA, USA
2.1.7 Patient-derived isogenic iPS cell lines
Isogenic cell lines used in this study were generated and characterized previously by members
of the group of Prof. Andreas Hermann. The specifications for both lines are summarized in
Table 8. This study was approved by the local ethics committee (EK45022009, EK393122012).
Table 8: Characteristics for isogenic cell lines used in this study
Name Genotype Gender Year of
birth Age at biopsy Reference
Wildtype
FUS-EGFP
FUS-WT-
EGFP/WT F 1952 58 (Naumann et al., 2018)
FUS-P525L-
EGFP
FUS-P525L-
EGFP/WT F 1952 58 (Naumann et al., 2018)

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 32 / 157
2.2 Methods
2.2.1 Coating of cell culture plates and flasks
NPCs were maintained and primed for differentiation in T25 flasks. Flasks were coated with
Matrigel diluted 1:100 in KnockOut™ DMEM and incubated at room temperature (RT) for
30 min. For maturation, cells were cultured on 6-well plates coated first with 15 % Poly-L-
Ornithine (PLO) in PBS, then with 0.005 mg/ml laminin in PBS at 37 °C overnight, respectively.
2.2.2 NPC thawing, culture, and cryoconservation
NPCs were thawed by suspending them in pre-warmed N2B27 medium, centrifuged with
230 x g for 5 min and subsequent seeded into Matrigel-coated culture flasks in expansion
medium. Cells were maintained at standard cell culture conditions (37 °C in a humidified
atmosphere containing 5 % CO2) in N2B27 medium supplemented with 3 µM Chiron99021,
150 µM ascorbic acid (AA), and 0.5 µM smoothened agonist (SAG). Medium was changed three
times a week, while cells were split at 85 % confluency, on average once per week. For splitting,
cells were detached from the surface via incubation with accutase at 37 °C for 5 min. Detached
cells were taken up in pre-warmed N2B27 medium and either reseeded at an appropriate
density or cryopreserved at -80°C. For this, cells were resuspended in N2B27 medium
containing 10 % DMSO as a cryoprotectant. Cells were frozen over night at a cooling rate of
~1 °C per hour, and subsequently transferred to liquid nitrogen (-180 °C) for long-term storage.
2.2.3 Differentiation and maturation of NPCs towards spinal motor neurons
The generation of spinal motor neurons out of NPCs requires the addition of different small
molecules to the culture medium at specified time points (Figure 5). After splitting, NPCs were
reseeded in N2B27 medium supplemented with 1 ng/mL recombinant human brain-derived
neurotrophic factor (rhBDNF), 0.2 mM AA, 1 µM retinoic acid, 1 ng/mL recombinant human
glial-derived neurotrophic factor (rhGDNF), and 0.5 µM SAG in order to prime their
differentiation into spinal motor neurons. Medium was exchanged every two days until day six
post induction, where the maturation phase was started by the addition of N2B27 medium
supplemented with 0.1 mM cAMP, 2 ng/mL rhBDNF, 0.2 mM AA, 1 ng/mL Transforming growth
factor β-3 (TGFβ-3), and 2 ng/mL rhGDNF, which marked day 0 of maturation. On this day only,
5 ng/mL Activin A was additionally supplemented to the maturation medium. Primed NPCs were
split one more time between days 2-4 of maturation and reseeded in maturation medium into a
6-well plate coated with PLO and laminin, at a concentration of 1 x 106 cells/well (determined
using a Neubauer counting chamber). On day 4 of maturation only, 5 µM N-[N-(3,5-

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 33 / 157
Difluorphenacetyl)-L-alanyl]-S-phenylglycin-tert-butylester (DAPT) was added to the maturation
medium. DAPT is a γ-secretase inhibitor that inactivates Notch signaling and thereby results in
fewer cells in culture with progenitor morphology while increasing cells with a neuronal
morphology (Nelson et al., 2007). Maturing spinal motor neurons were kept in maturation
medium without any additional supplements, with medium changes twice a week, until they
were lysed on day 21 of maturation.
Figure 5: Timeline for the differentiation and maturation of NPCs towards spinal motor neurons
Different sets of small molecules are added to N2B27 medium during the differentiation and maturation of NPCs towards spinal motor neurons.
2.2.4 Generation of cell lysates – isolation techniques
At day 21 of maturation, spinal motor neurons were gently washed with PBS once, scrapped off
the dish surface and resuspended in fresh PBS. Cells were then lysed by following methods:
A | Chemical lysis using the Active Motif® Nuclear Extraction Kit according to the
manufacturer’s instructions for the generation of whole cell lysates. For the exchange of buffers
after chemical lysis, PD Minitrap™ G-10 desalting columns or Vivaspin® 500 centrifugal
concentrators were used according to the manufacturer’s instructions. Protein in PD Minitrap
columns was eluted in PBS. Centrifugation speed and time for Vivaspin columns was 13.000 x g
and 30 min, respectively.
B | Lysis by grinding in liquid nitrogen: Cells were pelleted at 14.000 x g for 5 min at 4°C
and the cell pellet was resuspended in 1 ml fresh PBS supplemented with 1 x protease inhibitor
cocktail (containing 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, aprotinin, bestatin,
E-64, leupeptin and pepstatin A), dropwise frozen in liquid nitrogen and ground to a fine powder
using a pre-chilled mortar.

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 34 / 157
C | Mechanical lysis with glass beads: Cells were pelleted at 14.000 x g for 5 min and the
cell pellet resuspended in 1 ml fresh PBS supplemented with 1 x protease inhibitor. Sterile glass
beads were added to each Eppendorf tube to an approximate volume of 400 µL, and the tube
was vortexed for 5 min. Glass beads were allowed to sediment on ice for 5 min and the
supernatant was aspirated.
D | Mechanical lysis using a dounce homogenizer: Cells were pelleted at 14.000 x g for
5 min and the cell pellet was resuspended in 1 ml fresh PBS supplemented with 1 x protease
inhibitor. Resuspended cell were sheared in a dounce homogenizer on ice using ten strokes.
E | Mechanical lysis using a syringe: Cells were pelleted at 14.000 x g for 5 min. The cell
pellet was resuspended in 1 ml fresh PBS supplemented with 1 x protease inhibitor and cells
were sheared by pulling them ten times through a thin needle (0.4 x 19 mm; 27G ¾) using a
2 ml syringe while on ice.
Lysates prepared by methods B through E were subsequently centrifuged at 4 °C and
70.000 x g for 5 min. The supernatant was then transferred to a new tube, supplemented with
1 x protease inhibitor, 10 mM β-glycerophosphate, and 10 % glycerol, and snap-frozen in liquid
nitrogen for long term storage at -80 °C.
2.2.5 Determining protein concentration
Protein concentrations were determined using the Pierce™ bicinchoninic acid (BCA) protein
assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly,
standards were prepared with BSA diluted in lysis buffer to concentrations ranging from
25 µg / ml to 1 mg / m and measured in duplicates in a white 96-well transparent flat bottom
plate. Lysates were measured in triplicates. All probes were mixed with the kit’s working reagent
within the plate and subsequently incubated at 37 °C for 30 min. After incubation, the plate was
allowed to cool down to RT, the absorbance of samples at 562 nm was measured with a
TECAN plate reader and corrected for the values of blank measurements. A standard curve was
created in Microsoft Excel 2010, and the sample concentration was calculated from a linear
regression line. The BCA assay is a highly sensitive, two-step reaction. In the first step, cupric
ions (Cu2+) are reduced to cuprous ions (Cu+) by proteins under alkaline conditions, a reaction
referred to as the Biuret reaction (Gornall et al., 1949), while two BCA molecules are chelated in
the second step by one cuprous ion, resulting in a water-soluble, stable BCA/copper complex
(Smith et al., 1985). This complex absorbs light at 562 nm, which causes a color change of the
solution from blue to intense purple, measurable using a spectrophotometer. The measured

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 35 / 157
absorbance is directly proportional to the amount of protein in solution. The assay allows for the
measurement of protein over a wide range of concentrations (between 0.5 ng/µL and 1.5 µg/µL)
and is compatible with many detergents and other substances. However, exogenous reducing
agents, such as DTT or mercaptoethanol, or copper chelating agents in particular greatly
interfere with the reaction and hence may influence the results. Similarly, the presence of
certain amino acids (i.e. cysteine, tyrosine, and tryptophan, Wiechelman et al., 1988) influences
the accuracy of the results. In addition, the assay requires comparably long incubation times of
30 min up to two hours and incubation at 37°C, as the reaction is temperature sensitive (Lowry
et al., 1951; Smith et al., 1985; Krohn, 2002; Brady and Macnaughtan, 2015).
For reasons of comparison, the protein concentration of lysates produced by chemical
lysis was additionally determined using the ROTI®Nanoquant Bradford assay in a microtiter
plate according to the manufacturer’s instructions. In brief, standards were prepared with BSA
diluted in PBS ranging from a concentration of 1 to 100 µg / ml and again prepared in duplicates
in a white 96-well transparent flat-bottom plate. Lysates were diluted 1:20 and measured in
triplicates. All probes were then mixed with working reagent diluted 1:5 in H2O and incubated at
RT for 5 min, after which absorbance was measured three times at 450 nm and 590 nm, each
and respectively, in a TECAN plate reader. For each well, the measured value at 590 nm was
divided by the value measured at 490 nm, and the average of three subsequent measurements
was taken to calculate the final concentration. A standard curve was created in this way,
through which the sample concentration could be estimated using a linear regression line. The
Bradford assay is comparably sensitive, but more straightforward as it deploys the ability of the
dye Coomassie blue to directly bind to proteins and subsequently change its color from red
(absorbance at 465 nm) to blue (absorbance at 595 nm). The absorbance can be measured at
both wavelengths, and the change in color is directly related to the amount of protein present in
the sample. Since this method does not require a series of reactions to indicate protein
concentration, the incubation time is much faster (5 min) compared to BCA, although the range
of protein amount that can be detected is limited (between 1 and 20 µg). The reagent is
however compatible with a large variety of buffers and solvents, as well as reducing and
chelating agents, which makes it a suitable alternative to BCA if such agents cannot be
removed from solution. The downside is that the Coomassie reagent precipitates in the
presence of surfactants and is highly acidic, indicating that proteins with poor acid solubility
cannot be measured with this dye. Similar to BCA, it is also very sensitive to certain amino acids
(i.e. aromatic acids such as arginine, histidine or lysine, Compton and Jones, 1985; de Moreno
et al., 1986; Fountoulakis et al., 1992). In addition, it adsorbs to glassware or quartz cuvettes,

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 36 / 157
which require thorough cleansing after detection in order to be reusable (Bradford, 1976; Zor
and Selinger, 1996; Krohn, 2002; Georgiou et al., 2008; Brady and Macnaughtan, 2015).
2.2.6 Acquisition of kinesin-1, tubulin, and recombinant human FUS-GFP variants
Porcine tubulin was purified from porcine brain (obtained from Vorwerk Podemus, Dresden,
Germany) using previously established protocols (Castoldi and Popov, 2003) and fluorescently
labeled using mixed isomers of 5-(and 6-) carboxytetramethylrhodamine, succinimidyl ester
(5(6)-TAMRA,SE, Invitrogen, C1171), a member of the rhodamine family of dyes (Gessner and
Mayer, 2000). Drosophila melanogaster kinesin-1 heavy chains were expressed in full length in
insect cells and purified as previously described (Korten et al., 2016). Recombinant human
FUS-GFP variants were kindly provided by Dr. Jie Wang, Max Planck Institute of Molecular Cell
Biology and Genetics, Dresden. The first batch of recombinant human 2N4R tau-GFP protein
used for the determination of assay sensitivity was kindly provided by Dr. Marcus Braun,
Institute of Biotechnology CAS, Prague.
2.2.7 Expression and purification of recombinant human tau variants
The construct for recombinant human 2N3R tau-GFP was kindly provided by Prof. Eckhard
Mandelkow, Deutsches Zentrum für Neurodegenerative Erkrankungen e.V., Bonn and is
described in Gustke et al., 1994 and Seitz et al., 2002. The construct for recombinant human
2N4R tau-GFP was kindly provided by Dr. Amayra Hernandez Vega, Max Planck Institute of
Molecular Cell Biology and Genetics, Dresden, and is characterized in Hernández-Vega et al.,
2017. In order to express both constructs in insect cells, the gene of interest was amplified by
PCR and flanked with the restriction sites NotI and AscI using the respective
5’ (AATAATAACATGCGGCCGCAATGGCTGAGCCCCGCC) and
3’ (AATAATAACATGGCGCGCCCAAACCCTGCTTGGCCAG) primers. The resulting product
was inserted into the vector pOCC61, containing a C-terminal monoGFP tag and a
3C-cleavable 6xHis-tag for affinity purification. Baculovirus for the expression in insect cells was
generated from these constructs by Régis Lemaitre at the Protein Expression Purification and
Characterization (PEPC) facility at the Max Planck Institute of Molecular Cell Biology and
Genetics, Dresden (Lemaitre et al., 2019). Insect cells (Sf9-ESF) were infected with the
respective virus with a ratio of 1:100 (v/v) and harvested 72 hours post infection by
centrifugation at 350 x g for 10 min.

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 37 / 157
Purification of recombinant human tau-GFP variants
Harvested cell pellets were resuspended in 20 mL lysis buffer (50 mL equilibration buffer with
one protease inhibitor tablet) and the suspension lysed by passing them three times through an
Emulsiflux French press on ice. All further steps were performed at 4°C or on ice. The lysate
was collected in cold Beckmann centrifuge bottles, supplemented with benzonase (at least
1:6000), and cleared of cell debris by centrifugation at 14000 x g at 4°C for 45 min. The cleared
lysate was then passed through a 0.45 µm filter in order to completely remove cell debris before
protein purification via nickel-sepharose affinity chromatography. His-trap columns were
equilibrated with 10 column volumes (CV) of equilibration buffer before the protein was loaded
onto the column by passing the supernatant through the column at a flow rate of about
0.75 ml/min with the help of a peristaltic pump. The column was washed once with 10 CV high
salt buffer or equilibration buffer (50 mM HEPES, 300 mM KCL, 0.2 % Tween20 (v/v),
10 mM β-mercaptoethanol in H2O) without imidazole. The protein was then eluted by flowing
elution buffer through the column and collecting 1 ml fractions. Eluted fractions were pooled and
supplemented with PreScission 3C-6xHis protease (1 mg/ml) for GST(His)-tag removal, shaking
at 4°C overnight. On the next day, the sample was concentrated and the elution buffer
exchanged for equilibration buffer by centrifugation through 30K AmiconUltra filters at 7000 rpm
for 10 min three times. In order to remove the cleaved His-tag from the cleaved protein fraction,
the concentrated sample was incubated with 1 mL Ni-NTA agarose beads shaking at 4°C for
one hour. This solution was then passed through a 10 mL gravity flow filter column which
retained the beads and the flow through was concentrated again by centrifugation as described
above. 50 µL samples were collected after each step to determine purification efficiency in a
subsequent sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel (Figure S1). The protein of
interest was further purified by size exclusion chromatography (SEC) with equilibration buffer
through a superdex200 column on an ÄKTA pure liquid chromatography system at 4°C.
Absorbance was measured at 480 nM for eGFP construct and 569 nM for the mScarlet
construct and the fractions under the peak (0.5 mL per fraction) were collected. These pooled
fractions were again concentrated by centrifugation as described above, and the concentrated
protein was flash frozen in liquid nitrogen and stored at -80°C in 5 µL aliquots.
Protein quantification by SDS-PAGE gel electrophoresis and Coomassie staining
Purified proteins and samples taken throughout the purification procedure were analyzed by
SDS-PAGE for purification efficiency and concentration measurement. SDS loading buffer,
supplemented with 0.1 M DTT, was mixed 1:4 with the respective samples to be analyzed and

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 38 / 157
boiled at 95°C for 5 min. 10 µL of sample was loaded onto a 4-12 % Bis-Tris gel, together with
an appropriate protein ladder. The gel was assembled into an SDS gel electrophoresis running
chamber filled with 1 x MOPS buffer and run at 120 V, 85W, and 60 mA until all protein had
passed to the end of the gel (at least one hour). Gels were then stained with Coomassie
SimplyBlue SafeStain for at least 45 min up to overnight. Afterwards, gels were destained in
ddH2O until bands were clearly visible.
Protein quantification on SDS-PAGE gels
Purified 6xHis-eGFP protein at a known concentration was run as a standard on SDS-PAGE
gels, together with different dilutions of the protein of interest and stained with Coomassie blue
as described above. Gels were scanned with an Azure c600 Gel Imaging System and the
integrated density of protein bands was quantified using ImageJ. A rectangular area was
selected enclosing the first lane. The same rectangular shape was the applied onto all lanes on
the gel. The intensity profiles of all lanes were then plotted using the ‘analyze gel’ function of
ImageJ and the integrated intensities were recorded by selecting the area under each peak. A
calibration curve was then obtained from the linear fit to the integrated intensity for different
concentrations of the 6xHis-eGFP protein. Protein sample concentration were then calculated
for each dilution and averaged to get the undiluted protein concentration.
2.2.7 Kinesin-1-dependent microtubule gliding assay
Flow chamber and imaging assembly
Kinesin-1-dependent microtubule gliding assays were adapted from previously described
protocols (Nitzsche et al., 2010). Assays were performed in manually assembled flow chambers
(Figure 6), which were constructed by cutting out seven stripes of parafilm of about 25 x 1 mm2
size and sandwiching them in between 18 x 18 mm2 (top) and 22 x 22 mm2 (bottom) glass
coverslips, forming parallel channels. In addition, two 25 x 3 mm2 parafilm stripes were cut and
placed orthogonal to the channels on the sides of the top coverslip, just above the entry and exit
sites of channels, to prevent immersion oil from the objective from flowing into the channels
during later imaging. Overhanging parafilm was cut off using a razor blade. The sandwich was
heated until the parafilm got soft and six channels were sealed leak-proof by gently applying
pressure onto the coverslips. Molecular motor motility is very sensitive to changes in
temperature (Böhm et al., 2000; Hong et al., 2016). In order to minimize temperature
fluctuations, and therefore improve reproducibility of motility measurements, an objective heater
was used at the microscope and flow chambers were mounted onto a custom-build Peltier
element (Figure 6), and both devices were set to 30 °C. The Peltier element makes use of the

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 39 / 157
Peltier effect, which describes the transfer of heat from one junction to another if a voltage is
applied across joined conductors, creating an electrical current, with consumption of electrical
energy. The effect was discovered in 1834 by Jean Charles Athanase Peltier (Peltier, 1834) and
allows for heating as well as cooling, depending on the direction of the current (Taylor and
Solbrekken, 2008). A Peltier element therefore has two major advantages: i) it does not
overheat, but can actively cool down its surface by changing the electrical polarity when
connected to a conventional power supply, and ii) reaches desired temperatures on a very fast
time scale (e.g. a temperature change of 8 °C is achieved in about 25 s) (Ionov et al., 2006;
Nitzsche et al., 2010). The temperature of the herein used Peltier element was constantly
monitored using a thermometer with analogue readout capability and the current was adjusted if
needed to maintain a steady temperature of 30 °C within flow chambers with an accuracy of
0.1 °C. The Peltier element and the flow chambers were coupled by applying, electrolube, a
silicon-free heat transfer compound, in a thin layer on the surface of the Peltier element. The
flow chamber was stuck with its bottom coverslip onto the lube and firmly attached by gentle
pressure on the flow chamber. Because of this setup, flow channels were images upside down
through the 18 x 18 mm2 top glass slip. To prevent light reflecting from the surface of the Peltier
element, bottom coverslips of all flow chambers were darkened with a black permanent marker
before fixation to the Peltier element.

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 40 / 157
Figure 6: Assembly, mounting, and imaging of flow channels.
Flow chambers are assembled by stacking two coverslips of different sizes on top of each other, with parallel stripes of parafilm in between to form individual channels. Two wider stripes of parafilm are mounted orthogonal to flow channels on the edges of the top coverslip to prevent immersion oil from flowing into the channels during imaging. By heating this assembly, the parafilm melts and tightly seals channels. The backside of the bottom coverslip was painted with a black permanent marker to prevent reflection of light after the assembly was mounted on a custom-built Peltier element, using electrolube as a temperature conducting adhesive. The Peltier element was equipped with a temperature sensor at its surface to constantly monitor the temperature. All solutions used in the experiments are pipetted onto one side of the flow chamber and aspirated at the other end of the channel using chromatography filter paper. Flow chambers in this setup were installed at the microscope upside-down, enabling imaging through the top coverslip.
Microtubule assembly and kinesin-1-dependent gliding motility assay
Microtubules were polymerized at 37 °C for 15 min from 32 μM rhodamine-labeled tubulin in
BRB80 supplemented with 4 mM MgCl2, 1 mM Mg-GTP, and 5 % DMSO in a total volume of
6.25 μL. Polymerized microtubules were immediately stabilized with taxol by adding 10 µM taxol
in BRB80 to a final volume of 100 μL, and further kept at RT in the dark. Directly before use,
microtubules were diluted 1:10 in standard motility buffer and carefully sheared twice through a
Kel-F Hub needle (gauge 30, point style 3) using a 1 mL Luer-LokTM syringe.
For the assay itself, a series of solutions was flushed through each flow channel by
carefully pipetting 20 μL of each solution on one end of the channel while sucking the previous
solution out of the channel at the other end using chromatography paper. All solutions were
applied at RT. In a first step, flow channels were perfused with a casein-containing solution

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 41 / 157
(0.5 mg/mL) in PBS and left to adsorb for 5 min in order to block unspecific protein binding sites
on the glass surface. Next, a solution containing Drosophila full-length kinesin-1 heavy chains
(4 μg/mL), casein (0.2 mg/mL), 1 mM ATP, and 10 mM DTT in PBS was flushed through the
channels and again left to adsorb for 5 min in order for kinesin-1 to attach to the glass surface.
Following this, diluted and sheared microtubules in standard motility buffer were perfused into
the channels and allowed to attach to motors for 5 min. Flow channels were then mounted onto
a Nikon Eclipse Ti microscope equipped with a Perfect Focus System (PFS) and a 1.49
PlanApo 100 x oil immersion objective heated to 30 °C, the same temperature as the Peltier
element. During imaging, the Peltier element was continuously monitored and adjusted to
ensure a constant temperature of 30 °C with a precision of 0.1 °C. Images were acquired using
widefield-epifluorescence and excited by LED lamp and respective filter sets for excitation at
550 nm for rhodamine-labeled microtubules, 470 nm for eGFP-labeled proteins, or 640 nm for
mScarlet- and mCherry-labeled proteins, respectively. Time lapse images were recorded at ten
different positions within a channel, at a rate of one frame per second and 10 frames at each
position. Images were recorded with an exposure time of 100 ms using an iXon Ultra back-
illuminated EMCCD camera in conjunction with Nikon NIS-Elements imaging software.
After imaging rhodamine-labeled microtubules alone, channels were perfused depending
on the experiment with either recombinant human tau isoforms, recombinant human wildtype
FUS-GFP or FUS-P525L-GFP, standard motility buffer, BSA, or the respective cell lysate. Each
of these solutions was supplemented with 0.3 % methylcellulose, 20 mM D-glucose, 20 μg/mL
glucose oxidase, 10 μg/mL catalase, 10 mM DTT, and 1 mM ATP to ensure optimal conditions
for efficient kinesin-1 motility. Each channel was then sealed off with VALAP (Vaseline, lanolin,
and paraffin in a 1:1:1 ratio) to inhibit evaporation. Flow chambers were allowed to equilibrate
for 5 min before running the imaging workflow as described above. In channels where tau or
FUS-GFP variants were administered, single frames were captured via excitation with 470 nm
or 640 nm light and an exposure time of 100 ms at every position immediately before acquiring
time laps movies of rhodamine-labelled microtubules. For the first batch of 2N4R tau-GFP,
single frames of the first imaged positions were captured via excitation with 470 nm light and an
exposure time of 100 ms immediately before acquiring time laps movies of rhodamine-labelled
microtubules, while single frames of the remaining nine imaging positions were captured via
excitation with 470 nm light after time lapse acquisition. This workflow takes into consideration
that analyzing the colocalization of moving microtubules with potentially bound proteins was
only possible if overlaid images were captured virtually simultaneously in both channels. In case
of FUS-GFP variants, the buffer control experiments were performed with standard motility

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 42 / 157
buffer to which FUS-protein storage buffer was added in equal dilutions as required for the
respective protein concentrations tested. When flow chambers were imaged over the course of
three hours, channels were sealed with VALAP to prevent evaporation.
2.2.8 Detection of protein binding to microtubules
Microtubules were polymerized at 37 °C for 30 min from 40 μM Atto647-labeled tubulin in
BRB80 supplemented with 4 mM MgCl2, 1 mM Mg-GTP, and 5 % DMSO in a total volume of
6.25 μL. Polymerized microtubules were immediately stabilized with taxol by adding 10 µM taxol
in BRB80 to a final volume of 100 μL, and further kept at RT in the dark. Microtubules were
cleared from free tubulin by centrifugation in a table top centrifuge at 13.3 rpm for 30 min at RT,
the supernatant was removed and the pellet resuspended in 400 μL BRB80 supplemented with
10 μM Taxol.
For the assay itself, flow chambers were assembled as described above, except that the
coverslips used for this assay were silanized with dichlorodimethylsilane (DDS) in order to
increase the hydrophobicity of the surface.
The first solution containing α-tubulin antibodies, 1:200 diluted in BRB80, had to be
sucked through each channel with the help of a vacuum pump due to the strong hydrophobicity
of the glass surface, and was left to adsorb for 15 min. Next, a solution containing the 1 % of the
non-ionic detergent F127 was flushed through the channels and again left to adsorb for at least
one hour in order to reduce non-specific protein binding to the glass surface. Flow chambers
containing F127 were then either used directly or kept in a sealed humidified container at 4°C
for up to two days. Channels were washed once with BRB80 directly before use, and then
perfused with 20 μL undiluted Atto-647-labeled microtubules for 1 min. Unbound microtubules
were then washed out with standard motility buffer without ATP, before channels were perfused
with recombinant 2N3R tau-GFP or 2N4R tau-mScarlet or standard motility buffer without ATP.
Each channel was then sealed off with VALAP to inhibit evaporation. Flow channels were then
mounted onto an Axio Observer Z1microscope equipped with a Zeiss TIRF slider and a 1.46
PlanApochromat 63 x oil immersion objective combined with a 1.6x optovar and heated to
30 °C. Images were acquired by TIRF using a 532 nm laser line for Atto647-labeled
microtubules, 488 nm for eGFP-labeled proteins, or 642 nm for mScarlet- labeled tau isoforms,
respectively, with an exposure time of 100 ms. Single images at each wavelength were
recorded at ten different positions within one channel using an iXon Ultra back-illuminated
EMCCD camera in conjunction with MetaMorph MicroManager imaging software.

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 43 / 157
2.2.9 Data analysis and statistics
Kinesin-1-dependent microtubule gliding velocities were derived from time lapse movies using
an automated MATLAB script based on a microtubule tip-tracking algorithm developed in-house
(Ruhnow et al., 2011). In detail, the position of a microtubule tip was localized in each frame
with sub-pixel resolution and compared to its position in the subsequent frame one second later,
thereby determining the frame-to-frame velocities for this microtubule between two frames. All
microtubule frame-to-frame velocities determined in each of the ten time lapse movies for one
experimental condition are combined in a MATLAB cell format for further analysis. The median
microtubule gliding velocity was determined from this data set, as well as fraction of gliding
microtubules. The median gliding velocity of microtubules in one channel after perfusion with
recombinant protein, BSA, cell lysate, or standard motility buffer, respectively, was then
normalized to the median gliding velocity of microtubules in the same channel in standard
motility buffer before perfusion. This way of analysis yields the relative speed for microtubule
gliding in a certain experimental condition, a more robust parameter for comparison among
experiments than the absolute microtubule gliding velocity. The fraction of non-gliding
microtubules describes the number of microtubules gliding with a frame-to-frame velocity less
than 20 % of the gliding velocity in standard motility buffer in that channel.
The binding affinity of 2N3R tau-GFP and 2N4R tau-mScarlet to microtubules was
determined by measuring the integrated intensity in the respective image channel along at least
20 microtubules in each of the ten positions imaged using the line tool of ImageJ, and the
fluorescence intensity along each microtubule was background corrected locally. Therefore, at
least 200 microtubules were measured per condition and experiment to obtain the average
fluorescence intensity for every tested concentration of tau isoforms. Intensities for each tau
isoform were normalized in every experiment to the respective highest concentration tested in
order to determine the fraction of bound tau protein to microtubules. The fraction of bound tau
was plotted against the concentration of tau protein in solution and fit with the Hill model using
OriginLab 2019.
Significance was determined using either one-way or two-way ANOVA and Tukey’s
multiple comparison post-hoc test (see supplementary tables). Kymographs were created using
the Kymograph plugin for ImageJ created by J. Rietdorf (FMI Basel) and A. Seitz (EMBL
Heidelberg), while overlay images were generated using MetaMorph.

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 44 / 157
2.2.10 Western blot analysis
SDS-PAGE gel electrophoresis and blotting
Probes for Western blot analysis were mixed with NuPage loading buffer in a 3:4 ratio, heated
to 95 °C for 5 min to denature all proteins and put on ice immediately after. 12 µL of this mixture
were loaded on a 4-12 % Bis-Tris SDS-PAGE gel, as well as at least one lane with 5 µL of a 1:5
mixture of two markers, one visible by eye (SeeBlue) and one visible during detection
(MagicMarker). The gel was assembled into an electrophoresis running chamber filled with
1 x MOPS buffer and electrophoresis run for at least one hour with the following parameters:
140 V, 85W, and 60 mA for one and 140 mA for two gels simultaneously. Following
electrophoresis, gels were kept in transfer buffer supplemented with 0.1 % SDS for 8 min to
increase protein release from the gel during blotting. In contrast, the PVDF membrane used to
blot on and the thick filter paper within the iBlot transfer stack were soaked in transfer buffer
containing 6.7 % methanol prior to blotting in order to activate the membrane and increase
retention of proteins on the membrane. iBlot transfer stacks were assembled according to the
manufacturer’s instructions in the iBlot transfer system and blotted for 7 min with 19 V. After
blotting, membranes were transferred into 50 mL falcons and blocked for one hour at RT with
PBST containing 5 % milk powder. Membranes were then incubated with primary antibodies
over night at 4 °C and washed twice with PBST. After incubation with horseradish peroxidase
(HRP)-coupled secondary antibodies for one hour at RT, membranes were washed again with
PBST five times before detection.
As a means to accurately determine whether the expression level of a protein of interest
differs between two samples, the detected intensity for bands for this protein of interest should
be normalized to the intensity of a band for a housekeeping protein within the same sample. In
order to not mask any bands of the protein of interest, membranes were incubated with
antibodies to detect the protein of interest, as described above, imaged, and subsequently
stripped. Membranes were stripped at RT by incubating them in fresh stripping buffer twice for
10 min each, two subsequent washing steps with PBS for 10 min each, and two final washing
steps with PBST for 5 min each. After this procedure membranes were again blocked and
incubated with respective primary and secondary antibodies for the detection of the
housekeeping protein GAPDH.
Detection, imaging and quantification of protein bands
Chemiluminescent detection was achieved by applying either Amersham ECL Prime or
SuperSignalTM West Femto substrate according to the manufacturer’s instructions, depending

Mechanisms of Axonal Transport in ALS Materials and Methods
Anne Seifert 45 / 157
on the strength of the signal as the latter is more sensitive. Imaging was immediately performed
after substrate application on an LAS3000 imager with continuous exposure and image
acquisition every 10 sec. The integrated intensity of protein bands was determined using Image
StudioTM Lite as follows: a rectangular area was created around the largest band and all other
bands subsequently selected with the same rectangular area. The background was computed
as the median pixel intensity within a three pixel area bordering the top and bottom of each
rectangular area around the corresponding band. The background value was hence calculated
for every band individually and locally and subtracted from the total intensity in the respective
rectangular area. To determine the concentration of FUS variants and tau isoforms in whole cell
lysates, recombinant FUS-WT-GFP, 2N3R tau-GFP or 2N4R tau-mScarlet of known
concentrations were loaded on SDS-PAGE gels in combination with whole cell lysates. A
standard curve was created in Microsoft Excel 2010, and the protein concentration in whole cell
lysates was calculated from a linear regression line.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 46 / 157
3. Results
3.1 Modification of a kinesin-1-dependent microtubule gliding assay for the
application of whole cell lysates
Defects in axonal transport have long been recognized as part of the pathology in FUS-ALS, but
only little is known about the causes and mechanisms involved in its development (De Vos and
Hafezparast, 2017a). This thesis aimed at investigating possible direct engagement of FUS
variants with the transport machinery, as well as the importance of an altered tau isoform ratio
caused by FUS variants. For this purpose, a previously established kinesin-1-dependent
microtubule gliding assay was modified to run reliably in the presence whole cell lysates, so that
the kinesin-1 motility can be monitored in a minimal system. In this assay, stabilized and
fluorescently labelled microtubules are propelled by a layer of underlying, surface bound
kinesin-1 motor proteins (Figure 7A). The collective motility of kinesins, however, depends on a
variety of external factors such as temperature, ionic strength, the presence of microtubule or
motor interacting proteins, or whole cell lysates. To study the impact of FUS variants or different
ratios of tau on this machinery, recombinant FUS-GFP and tau-GFP protein or whole cell
lysates of isogenic iPSC-derived spinal motor neurons with either wildtype FUS-GFP or
FUS-P525L-GFP are added to the assay (Figure 7B).
Figure 7: Reconstituting axonal transport in vitro.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 47 / 157
A) ALS-patient-derived and isogenic control iPSC lines were differentiated towards spinal motor neurons. On day 21 of maturation, cells were mechanically lysed and centrifuged to obtain the soluble fraction of cell lysates, further referred to as whole cell lysates. B) Depending on the experiment, whole cell lysates or recombinant human protein (FUS-GFP or tau-GFP) were applied to a kinesin-1-dependent microtubule gliding assay in manually assembled flow chambers (see materials and methods for details).
To achieve efficient cell lysis and a high protein yield in the lysates, different lysis methods
have been tested (Table 9). Chemical lysis using the Active Motif Nuclear Extract Kit and a
variety of mechanical lysis protocols were assessed for their efficiency in terms of protein yield.
PBS was used in herein assessed mechanical lysis protocols as a preliminary lysis buffer, since
microtubule gliding velocity was as efficient in the presence of this physiological buffer as in the
presence of the most commonly used BRB80 buffer (Figure 8B) (Nitzsche et al., 2010). In
addition, two methods for determining the total protein concentration were evaluated for their
accuracy using lysates produced by chemical lysis, one based on the detection of
BCA/copper-complexes and one modified Bradford assay based on the direct binding of
Coomassie blue to proteins (Table 9B). In general, higher quantities of total protein were
detected using the BCA detection kit than with the Bradford detection. Importantly, the average
relative error for measurements performed with the Bradford assay was much larger
(0.078 ± 0.035) than for those performed with the BCA assay (0.013 ± 0.005), and hence total
protein levels in all mechanically prepared lysates were only quantified using the latter.
Previously established and widely used methods for mechanical lysis, such as glass bead
shearing, dounce homogenization, and liquid nitrogen grinding (Mackall et al., 1979; Harlow and
Lane, 2006; Walker, 2009), resulted in low protein yields as determined by BCA or did not
results in sufficient cell lysis, since the detected amount of the housekeeping protein β-actin was
very low as determined by western blot (Table 9A). A high yield, together with sufficient
detection of the housekeeping protein and hence cell lysis, was accomplished by shearing the
cells via passage through a syringe needle with a diameter of 400 µm or by dounce
homogenization. Although dounce homogenization yielded similar or even higher protein
concentrations, a lot of lysate stuck to the walls of the glass tube, so that overall lower volumes
of lysate were collected. This was not the case for mechanical lysis using a syringe and needle,
since all lysate could be dispensed with the plunger.
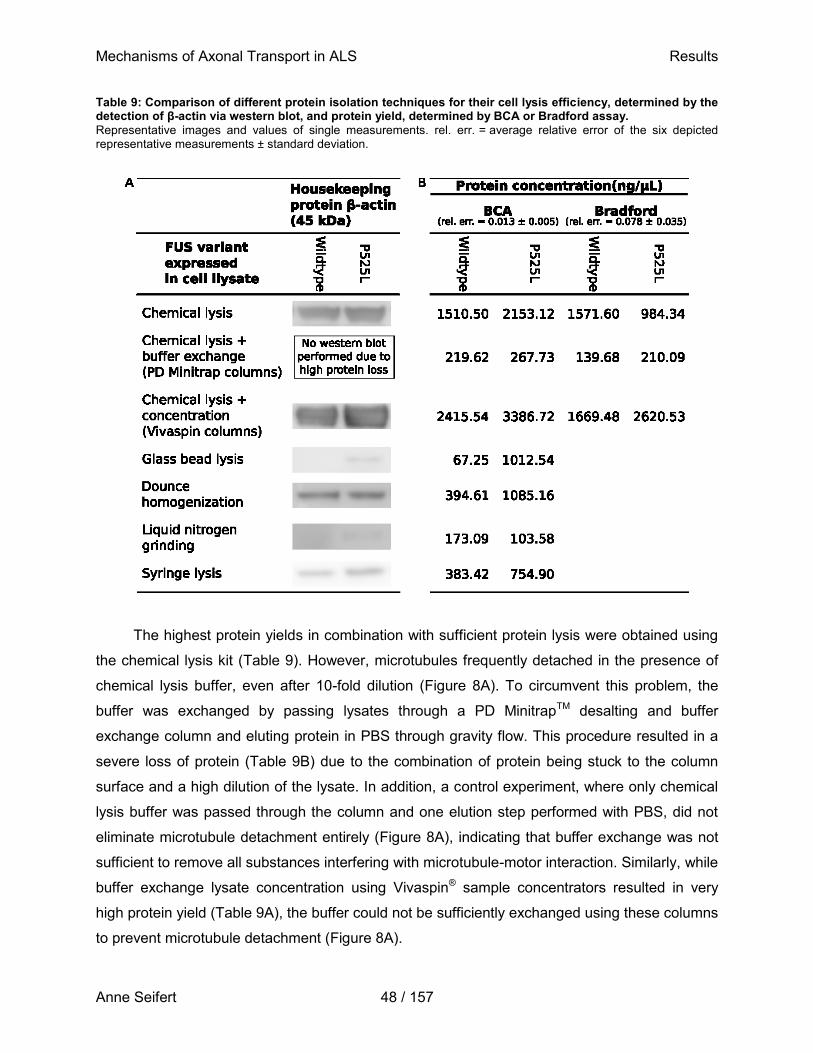
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 48 / 157
Table 9: Comparison of different protein isolation techniques for their cell lysis efficiency, determined by the detection of β-actin via western blot, and protein yield, determined by BCA or Bradford assay.
Representative images and values of single measurements. rel. err. = average relative error of the six depicted representative measurements ± standard deviation.
The highest protein yields in combination with sufficient protein lysis were obtained using
the chemical lysis kit (Table 9). However, microtubules frequently detached in the presence of
chemical lysis buffer, even after 10-fold dilution (Figure 8A). To circumvent this problem, the
buffer was exchanged by passing lysates through a PD MinitrapTM desalting and buffer
exchange column and eluting protein in PBS through gravity flow. This procedure resulted in a
severe loss of protein (Table 9B) due to the combination of protein being stuck to the column
surface and a high dilution of the lysate. In addition, a control experiment, where only chemical
lysis buffer was passed through the column and one elution step performed with PBS, did not
eliminate microtubule detachment entirely (Figure 8A), indicating that buffer exchange was not
sufficient to remove all substances interfering with microtubule-motor interaction. Similarly, while
buffer exchange lysate concentration using Vivaspin® sample concentrators resulted in very
high protein yield (Table 9A), the buffer could not be sufficiently exchanged using these columns
to prevent microtubule detachment (Figure 8A).

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 49 / 157
Therefore, whole cell lysates used in the herein presented study were prepared by
shearing cells in PBS via passage through a syringe needle, as this presented the most efficient
mechanical lysis method. Lysates were supplemented with a commercially available protease
inhibitor cocktail to prevent protein degradation. Using a commercially available phosphatase
inhibitor cocktail, however, immediately led to diminished microtubule gliding (Figure 8C).
Therefore, β-glycerophosphate was added to the lysate to steer phosphatase activity towards
the excess substrate. In addition, 10 % glycerol was added as a cryoprotectant. Glycerol by
itself did not alter microtubule gliding characteristics, but the addition of
10 mM β-glycerophosphate caused microtubules to detach from surface-bound motors (Figure
8D). To prevent this, 0.3 % methylcellulose was added to the assay to allow cross-comparison
as well to experiments involving recombinant proteins. The addition of methylcellulose did not
affect microtubule gliding, but effectively eliminated microtubule detachment (Figure 8D).
Methylcellulose due to its high viscosity acts as a cushion to keep microtubules close to the
imaging surface. Since microtubules sporadically detach from the surface-bound motors,
methylcellulose facilitates fast microtubule reattachment to motors (Inoue et al., 2015; Saito et
al., 2017). Cells were hence lysed in the presence of PBS and lysates supplemented with a
commercially available protease inhibitor, 10 % glycerol, 10 mM β-glycerophosphate, and
0.3 % methylcellulose (further referred to as lysis buffer). Similarly, standard motility buffer used
in subsequent microtubule gliding assays was prepared by addition of 10 µM taxol, 1 mM ATP,
20 mM D-glucose, 20 μg/mL glucose oxidase, 10 μg/mL catalase, 10 mM DTT, and
0.2 mg/mL casein to the lysis buffer.
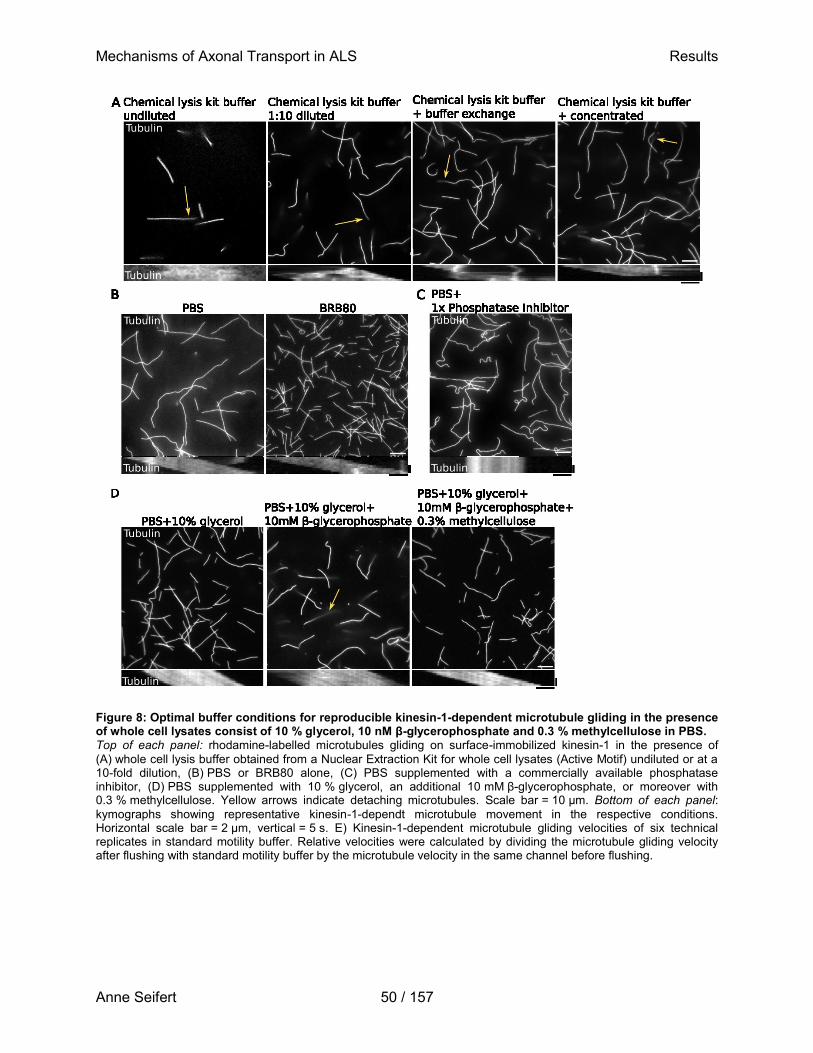
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 50 / 157
Figure 8: Optimal buffer conditions for reproducible kinesin-1-dependent microtubule gliding in the presence of whole cell lysates consist of 10 % glycerol, 10 nM β-glycerophosphate and 0.3 % methylcellulose in PBS. Top of each panel: rhodamine-labelled microtubules gliding on surface-immobilized kinesin-1 in the presence of
(A) whole cell lysis buffer obtained from a Nuclear Extraction Kit for whole cell lysates (Active Motif) undiluted or at a 10-fold dilution, (B) PBS or BRB80 alone, (C) PBS supplemented with a commercially available phosphatase inhibitor, (D) PBS supplemented with 10 % glycerol, an additional 10 mM β-glycerophosphate, or moreover with 0.3 % methylcellulose. Yellow arrows indicate detaching microtubules. Scale bar = 10 µm. Bottom of each panel:
kymographs showing representative kinesin-1-dependt microtubule movement in the respective conditions. Horizontal scale bar = 2 µm, vertical = 5 s. E) Kinesin-1-dependent microtubule gliding velocities of six technical replicates in standard motility buffer. Relative velocities were calculated by dividing the microtubule gliding velocity after flushing with standard motility buffer by the microtubule velocity in the same channel before flushing.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 51 / 157
Having optimized the buffer conditions, microtubule gliding assays were set up using
manually assembled flow chambers with six individual flow channels separated and tightly
sealed by stripes of parafilm in between two differently sized glass coverslips (Figure 6). Flow
channels were first perfused with a casein-containing solution to prevent unspecific protein
adsorption. This was followed by perfusion with a solution containing full-length constitutively-
active Drosophila melanogaster kinesin-1 motors. Both solutions were allowed to settle within
the channels at RT for 5 min each, before rhodamine-labelled microtubules in standard motility
buffer were flushed through the channels. Because the microtubule gliding velocity is highly
sensitive to temperature changes (Böhm et al., 2000), flow chambers were mounted onto a
custom-built Peltier element to minimize temperature fluctuations during an experiment and
between single technical replicates. Flow chambers were imaged using
widefield-epifluorescence microscopy with 10 s time lapse movies (Figure 9A, B) with an
exposure time of 100 ms and at an acquisition rate of one frame per second. From these time
lapses, frame-to-frame velocities of gliding microtubules were calculated using a previously
developed automated filament tracking algorithm (FIESTA, Ruhnow et al., 2011). The median
microtubule gliding velocity, as well as the 25th and 75th percentile (Figure 9C), were calculated
for microtubules in a given condition (i.e. presence of standard motility buffer, recombinant
protein, or whole cell lysates). Normalizing the median velocity to the initial median velocity (i.e.
before addition of recombinant FUS-GFP variants, recombinant tau isoforms, BSA, or standard
motility buffer) of microtubule in the same channel allows for cross-comparison of different
conditions and takes account of small experiment-to-experiment differences in e.g. the motor
density or motor activity. This normalization procedure further reduces the error arising from
possible fluctuations in microtubule number between experiments or technical replicates. With
this multitude of optimization steps, a robust assay to model axonal transport in vitro could be
established that generated highly reproducible measurements (Figure 9D).
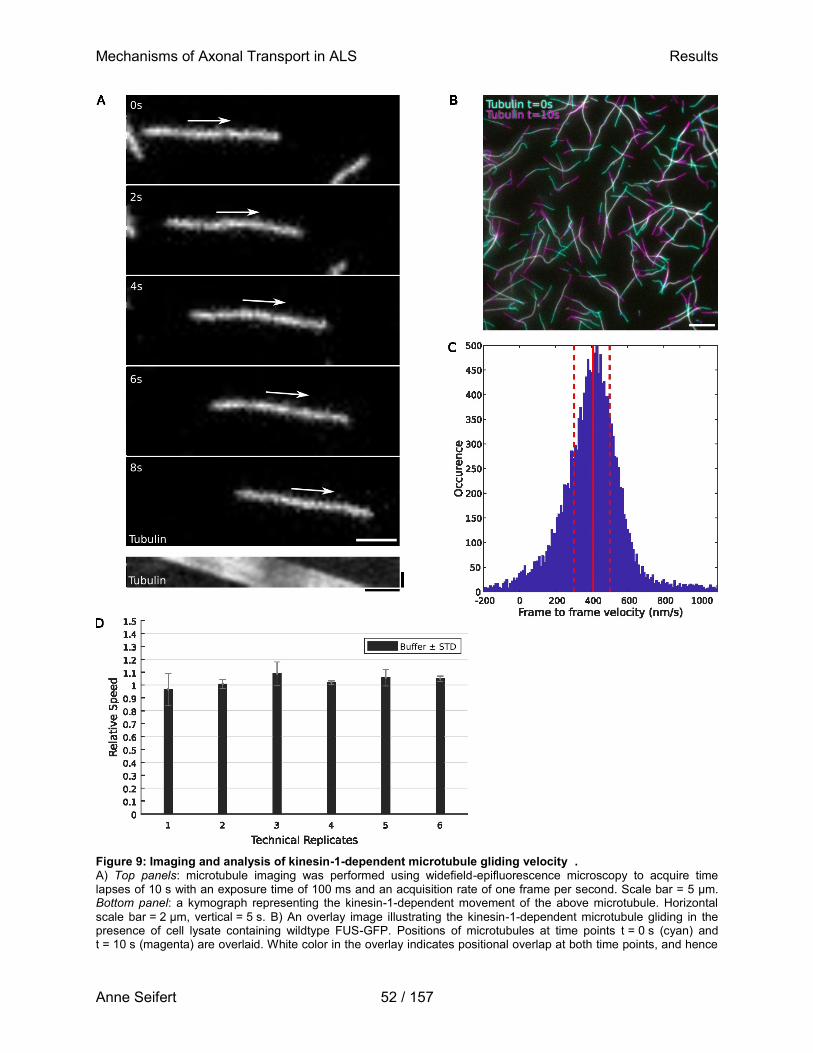
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 52 / 157
Figure 9: Imaging and analysis of kinesin-1-dependent microtubule gliding velocity . A) Top panels: microtubule imaging was performed using widefield-epifluorescence microscopy to acquire time lapses of 10 s with an exposure time of 100 ms and an acquisition rate of one frame per second. Scale bar = 5 µm. Bottom panel: a kymograph representing the kinesin-1-dependent movement of the above microtubule. Horizontal
scale bar = 2 µm, vertical = 5 s. B) An overlay image illustrating the kinesin-1-dependent microtubule gliding in the presence of cell lysate containing wildtype FUS-GFP. Positions of microtubules at time points t = 0 s (cyan) and t = 10 s (magenta) are overlaid. White color in the overlay indicates positional overlap at both time points, and hence

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 53 / 157
no microtubule movement. Scale bar = 10 µm. C) A previously developed automated MATLAB script was used to calculate frame-to-frame velocities from 10 s time lapse movies. Frame-to-frame velocities of all time lapse movies in a given condition (e.g. cell lysate containing wildtype FUS-GFP) of one technical replicate were combined in a single histogram. The red solid line marks the median, while dashed red lines mark the 25
th and 75
th percentile, respectively.
D) Flow channels containing gliding rhodamine-labelled microtubules in standard motility buffer were flushed with buffer to rule out that the process of flushing solutions through a channel changes microtubule gliding velocity. Relative speed of gliding microtubules was calculated by dividing the microtubule gliding velocity after flushing with standard motility buffer by the velocity of microtubules in the same flow channel before flushing. Six technical replicates are shown.
3.2 Determination of assay sensitivity with the microtubule-associated
protein tau
In order to estimate the sensitivity of this modified kinesin-1-dependent microtubule gliding
motility assay for proteins interfering with motor motility on microtubules, human recombinant
full length (2N4R) tau-GFP was added to the assay at varying concentrations (Figure 10A). Tau
is a MAP known to inhibit kinesin-1 stepping on microtubules by a simple road-block mechanism
(Tarhan et al., 2013), thereby significantly reducing kinesin-1 run-length and velocity (Hoeprich
et al., 2014). To see whether tau shows the same behavior in our modified microtubule gliding
assay, recombinant 2N4R tau-GFP was added at final concentrations of 2 to 500 nM in
standard motility buffer, and the gliding velocity as well as microtubule binding were assessed
by widefield-epifluorescence microscopy. No significant binding of 2N4R tau-GFP to
microtubules was evident at low tau concentrations, while binding was observed at a
concentration of 50 nM and above (Figure 10B). The strong background signal of unbound tau-
GFP, however, prevented quantification of the exact amount of microtubule-bound tau, and
therefore binding was assessed only qualitatively.
In line with the observed microtubule-binding behavior of tau, kinesin-1-dependent
microtubule gliding velocity was not altered below a tau-GFP concentration of 24 nM, but
microtubules considerably slowed down in the presence of 59 nM 2N4R tau-GFP and
completely ceased movement at concentrations above 118 nM (Figure 10C). Hence, the
modified kinesin-1-dependent microtubule gliding assay is sensitive enough to detect the
presence of 60 nM 2N4R tau-GFP, which is equivalent to 2.65 µg/ml.
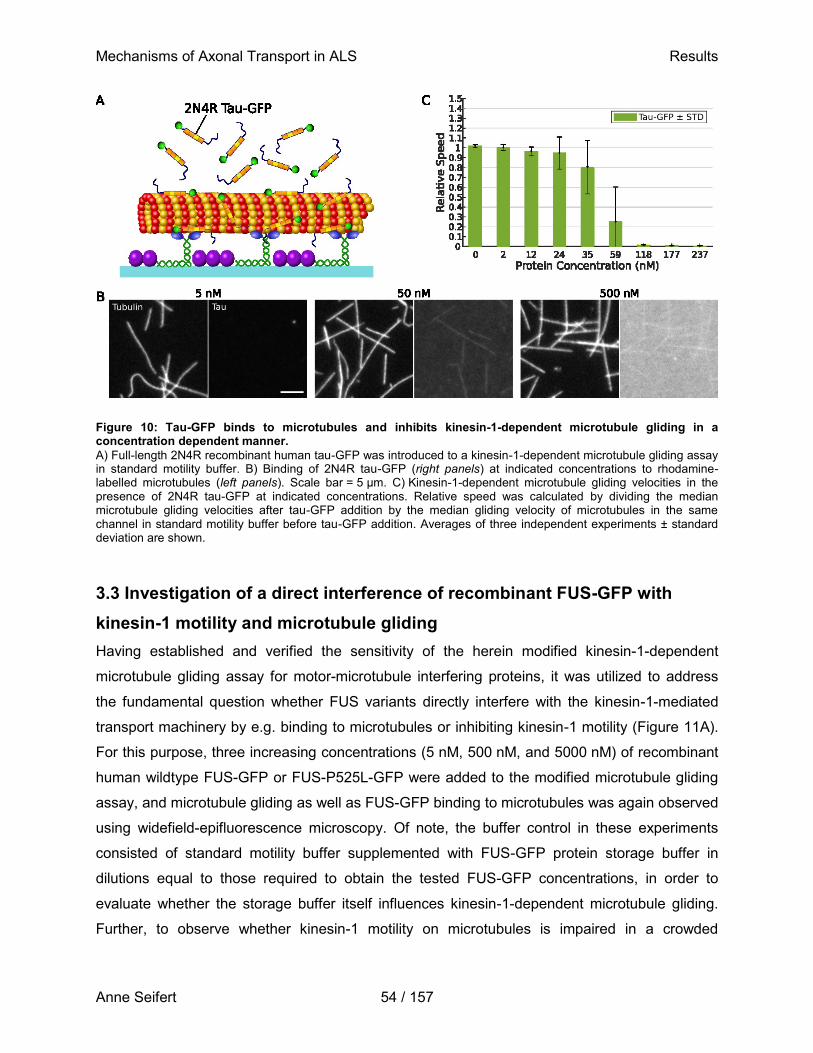
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 54 / 157
Figure 10: Tau-GFP binds to microtubules and inhibits kinesin-1-dependent microtubule gliding in a concentration dependent manner.
A) Full-length 2N4R recombinant human tau-GFP was introduced to a kinesin-1-dependent microtubule gliding assay in standard motility buffer. B) Binding of 2N4R tau-GFP (right panels) at indicated concentrations to rhodamine-labelled microtubules (left panels). Scale bar = 5 µm. C) Kinesin-1-dependent microtubule gliding velocities in the presence of 2N4R tau-GFP at indicated concentrations. Relative speed was calculated by dividing the median microtubule gliding velocities after tau-GFP addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before tau-GFP addition. Averages of three independent experiments ± standard deviation are shown.
3.3 Investigation of a direct interference of recombinant FUS-GFP with
kinesin-1 motility and microtubule gliding
Having established and verified the sensitivity of the herein modified kinesin-1-dependent
microtubule gliding assay for motor-microtubule interfering proteins, it was utilized to address
the fundamental question whether FUS variants directly interfere with the kinesin-1-mediated
transport machinery by e.g. binding to microtubules or inhibiting kinesin-1 motility (Figure 11A).
For this purpose, three increasing concentrations (5 nM, 500 nM, and 5000 nM) of recombinant
human wildtype FUS-GFP or FUS-P525L-GFP were added to the modified microtubule gliding
assay, and microtubule gliding as well as FUS-GFP binding to microtubules was again observed
using widefield-epifluorescence microscopy. Of note, the buffer control in these experiments
consisted of standard motility buffer supplemented with FUS-GFP protein storage buffer in
dilutions equal to those required to obtain the tested FUS-GFP concentrations, in order to
evaluate whether the storage buffer itself influences kinesin-1-dependent microtubule gliding.
Further, to observe whether kinesin-1 motility on microtubules is impaired in a crowded
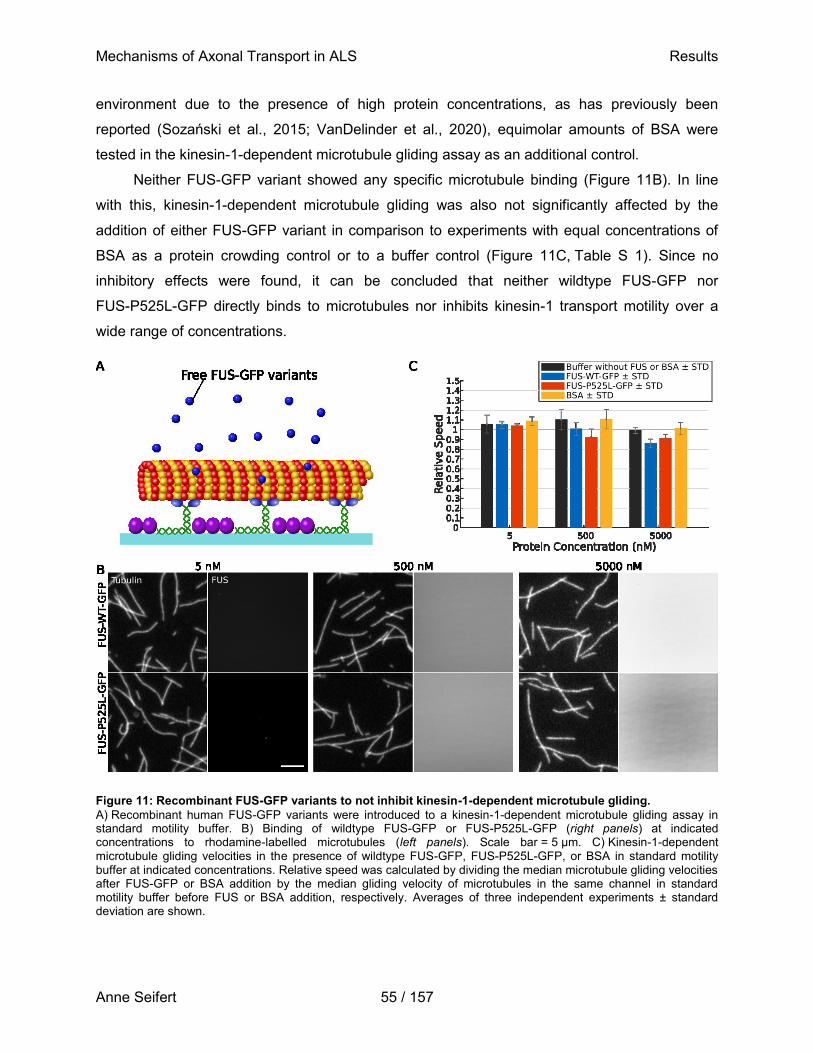
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 55 / 157
environment due to the presence of high protein concentrations, as has previously been
reported (Sozański et al., 2015; VanDelinder et al., 2020), equimolar amounts of BSA were
tested in the kinesin-1-dependent microtubule gliding assay as an additional control.
Neither FUS-GFP variant showed any specific microtubule binding (Figure 11B). In line
with this, kinesin-1-dependent microtubule gliding was also not significantly affected by the
addition of either FUS-GFP variant in comparison to experiments with equal concentrations of
BSA as a protein crowding control or to a buffer control (Figure 11C, Table S 1). Since no
inhibitory effects were found, it can be concluded that neither wildtype FUS-GFP nor
FUS-P525L-GFP directly binds to microtubules nor inhibits kinesin-1 transport motility over a
wide range of concentrations.
Figure 11: Recombinant FUS-GFP variants to not inhibit kinesin-1-dependent microtubule gliding.
A) Recombinant human FUS-GFP variants were introduced to a kinesin-1-dependent microtubule gliding assay in standard motility buffer. B) Binding of wildtype FUS-GFP or FUS-P525L-GFP (right panels) at indicated concentrations to rhodamine-labelled microtubules (left panels). Scale bar = 5 µm. C) Kinesin-1-dependent microtubule gliding velocities in the presence of wildtype FUS-GFP, FUS-P525L-GFP, or BSA in standard motility buffer at indicated concentrations. Relative speed was calculated by dividing the median microtubule gliding velocities after FUS-GFP or BSA addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before FUS or BSA addition, respectively. Averages of three independent experiments ± standard deviation are shown.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 56 / 157
This study focused on the interference of free-in-solution, non-aggregated FUS variants
with kinesin-1 motility on microtubules. In vivo, cytoplasmic mislocalized mutant FUS variants
form aggregates (Patel et al., 2015) over the course of hours, developing from liquid
compartments in the cytoplasm that undergo a liquid-to-solid phase transition (Mateju et al.,
2017; Nötzel et al., 2018). These aggregation events can be reproduced to some extent in vitro,
where they occur at a time scale of seconds in buffer with high ionic strength (Marrone et al.,
2019). Buffers used in the present study were of low to moderate ionic strength, and hence
FUS-GFP aggregate formation was, as expected, not observed directly after the addition of
FUS-GFP variants to the kinesin-1-dependent microtubule gliding assay (Figure 11B). However,
it cannot be excluded that – similar to the in vivo situation – FUS-GFP variants aggregate over
time and may further only in this state exhibit direct inhibitory effects on kinesin-1 motility.
Therefore, kinesin-1-dependent microtubule gliding assays were repeated as above, but the
microtubule binding and gliding velocity in the presence of wildtype FUS-GFP or
FUS-P525L-GFP was now observed over the course of three hours. Although the microtubule
gliding velocities in the presence of either FUS-GFP variant declined linearly over time, this
effect can be attributed to assay ageing since all control experiments (i.e. presence of standard
motility buffer or BSA at equivalent concentrations) showed the same age-dependent decline in
microtubule gliding velocity (Figure 12A, Table S 1). This is again in line with the finding that
neither FUS-GFP variant bound to microtubules over the course of three hours, and neither
could aggregate formation be observed in the presence of those FUS-GFP variants during the
same time course (Figure 12B). From both experiments can hence be concluded that neither
wildtype FUS-GFP nor the FUS-P525L-GFP variant in its non-aggregated form interferes with
kinesin-1 motility on or binds to microtubules, even over the course of hours.
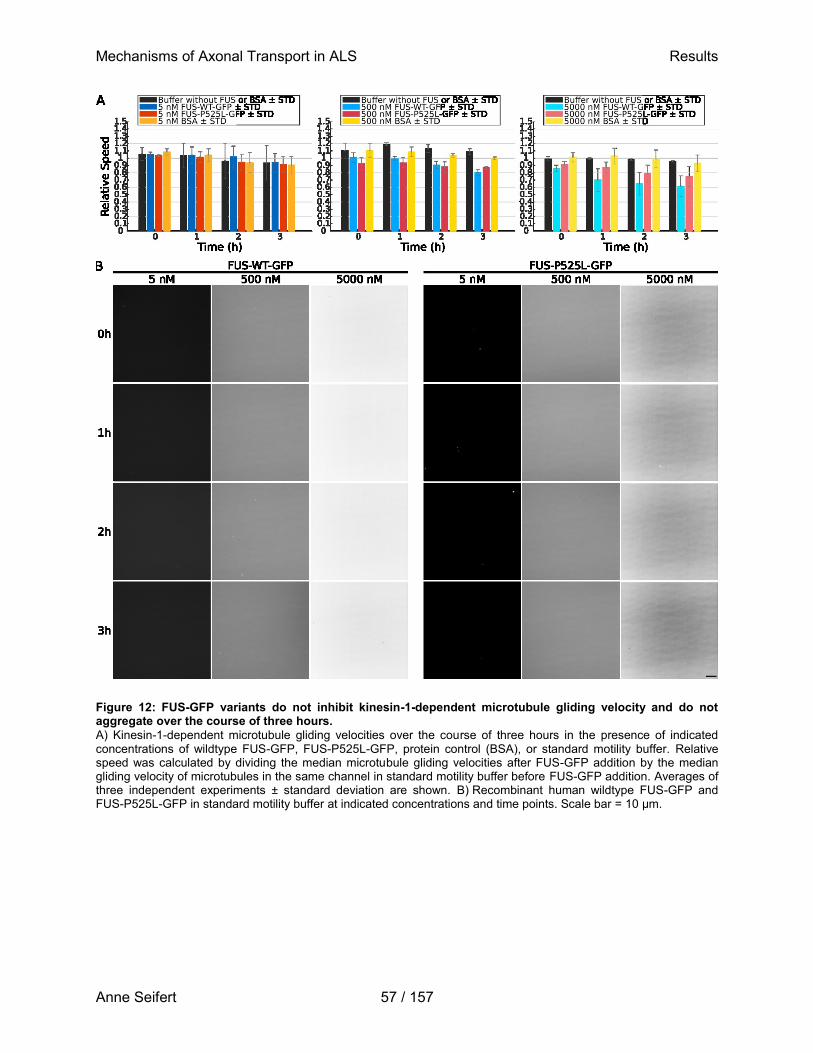
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 57 / 157
Figure 12: FUS-GFP variants do not inhibit kinesin-1-dependent microtubule gliding velocity and do not aggregate over the course of three hours.
A) Kinesin-1-dependent microtubule gliding velocities over the course of three hours in the presence of indicated concentrations of wildtype FUS-GFP, FUS-P525L-GFP, protein control (BSA), or standard motility buffer. Relative speed was calculated by dividing the median microtubule gliding velocities after FUS-GFP addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before FUS-GFP addition. Averages of three independent experiments ± standard deviation are shown. B) Recombinant human wildtype FUS-GFP and FUS-P525L-GFP in standard motility buffer at indicated concentrations and time points. Scale bar = 10 µm.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 58 / 157
3.4 Kinesin-1-dependent microtubule gliding and assay sensitivity in the
presence of whole cell lysates expressing GFP-labelled FUS variants
FUS-GFP variants did not directly interfere with kinesin-1-dependent microtubule gliding (e.g. by
binding to microtubules or motors). However, kinesin-1 often requires a multitude of adaptor
proteins in order to efficiently bind its cargo (Hirokawa et al., 2009; Fu and Holzbaur, 2014).
Using whole cell lysates from spinal motor neurons expressing GFP-labelled FUS variants in the
modified kinesin-1-dependent microtubule gliding assay, we aimed to investigate whether FUS
requires endogenous adaptors or whether there are indirect effects (e.g. changes in expression
levels of other motor or microtubule effector proteins (Fujioka et al., 2013; Akiyama et al., 2019)
or sequestering of MAPs etc. in aggregates (Yasuda et al., 2017) that may impact the
kinesin-1-dependent transport on microtubules. Therefore, previously generated and
characterized ALS-patient-derived and CRISPR/Cas9-engineered isogenic iPSCs (Naumann et
al., 2018; Pal et al., 2018) were differentiated via NPCs to form spinal motor neurons expressing
either wildtype FUS-GFP or FUS-P525L-GFP. Whole cell lysates were obtained after 21 days of
neuron maturation by mechanical lysis through a thin needle. Lysates were cleared by
centrifugation in order to obtain only soluble parts within the lysate, especially free floating,
cytoplasmic FUS. For application of whole cell lysates in the kinesin-1-dependent microtubule
gliding assay (Figure 13A), the total protein concentration in the lysates was determined using
the BCA assay, and lysates were diluted in cell lysis buffer to a final concentration of
50, 80, 110, and 140 ng/µL. Due to low yields after protein isolation, higher concentrations could
not be tested. Imaging and analysis was performed as described above, with respective
concentrations of BSA in standard motility buffer again serving as a protein crowding control.
However, neither lysates from wildtype FUS-GFP nor FUS-P5525L-GFP expressing cells had
any significant effect on microtubule gliding compared to controls (Figure 13B, Table S 3).
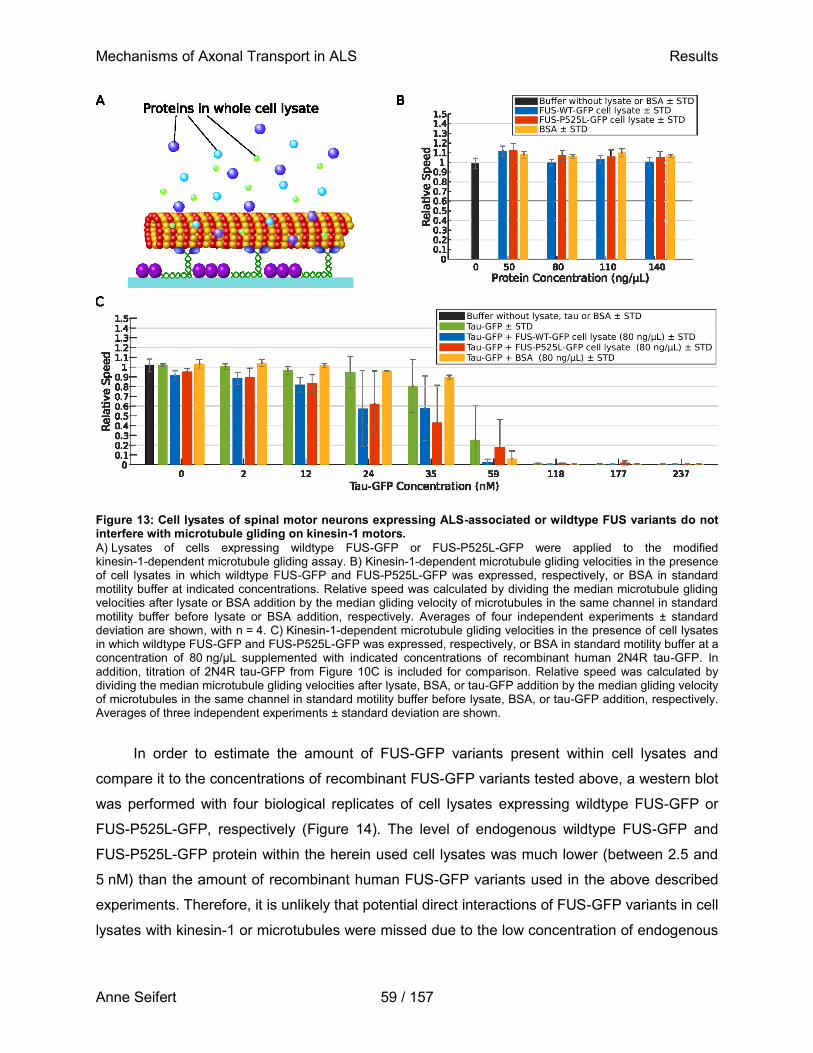
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 59 / 157
Figure 13: Cell lysates of spinal motor neurons expressing ALS-associated or wildtype FUS variants do not interfere with microtubule gliding on kinesin-1 motors.
A) Lysates of cells expressing wildtype FUS-GFP or FUS-P525L-GFP were applied to the modified kinesin-1-dependent microtubule gliding assay. B) Kinesin-1-dependent microtubule gliding velocities in the presence of cell lysates in which wildtype FUS-GFP and FUS-P525L-GFP was expressed, respectively, or BSA in standard motility buffer at indicated concentrations. Relative speed was calculated by dividing the median microtubule gliding velocities after lysate or BSA addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before lysate or BSA addition, respectively. Averages of four independent experiments ± standard deviation are shown, with n = 4. C) Kinesin-1-dependent microtubule gliding velocities in the presence of cell lysates in which wildtype FUS-GFP and FUS-P525L-GFP was expressed, respectively, or BSA in standard motility buffer at a concentration of 80 ng/µL supplemented with indicated concentrations of recombinant human 2N4R tau-GFP. In addition, titration of 2N4R tau-GFP from Figure 10C is included for comparison. Relative speed was calculated by dividing the median microtubule gliding velocities after lysate, BSA, or tau-GFP addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before lysate, BSA, or tau-GFP addition, respectively. Averages of three independent experiments ± standard deviation are shown.
In order to estimate the amount of FUS-GFP variants present within cell lysates and
compare it to the concentrations of recombinant FUS-GFP variants tested above, a western blot
was performed with four biological replicates of cell lysates expressing wildtype FUS-GFP or
FUS-P525L-GFP, respectively (Figure 14). The level of endogenous wildtype FUS-GFP and
FUS-P525L-GFP protein within the herein used cell lysates was much lower (between 2.5 and
5 nM) than the amount of recombinant human FUS-GFP variants used in the above described
experiments. Therefore, it is unlikely that potential direct interactions of FUS-GFP variants in cell
lysates with kinesin-1 or microtubules were missed due to the low concentration of endogenous

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 60 / 157
protein, as recombinant FUS-GFP variants were supplied at a wide range of much higher
concentrations (5-5000 nM) and did not interfere with kinesin-1-dependent microtubule gliding.
Figure 14: Western blot of FUS-GFP variants expressed in whole cell lysates of spinal motor neurons.
Representative western blot of recombinant wildtype FUS-GFP at indicated concentrations and four biological replicates (n) of whole cell lysates obtained from spinal motor neurons expressing either wildtype FUS-GFP or FUS-P525L-GFP.
Due to the protein-rich environment in cell lysates, however, the sensitivity of the
kinesin-1-dependent microtubule gliding assay might be reduced. Hence, to test assay
sensitivity, recombinant human 2N4R tau-GFP was titrated at low amounts (2 – 237 nM) into the
lysates (which were diluted to a total protein concentration of 80 ng/µL). Assay performance was
re-evaluated to see if a slowdown of microtubule gliding velocity could still be detected below a
concentration of 118 nM tau-GFP in the presence of whole cell lysates. In contrast to
experiments with tau-GFP alone, kinesin-1-dependent relative microtubule gliding velocity
started to decrease already in the presence of 12 nM 2N4R tau-GFP in lysates, but also
completely ceased at a concentration of 118 nM substituted tau-GFP (Figure 13C), similar to
what was observed in the absence of cell lysates. This behavior was also independent of the
FUS variant expressed within the lysate. This implies that, even in a protein-rich environment
such as in the presence of whole cell lysates, the assay is still sufficiently sensitive to inhibitors
of kinesin-1 motility. Further, it can be concluded that neither lysates carrying wildtype FUS-GFP
nor those containing ALS-associated FUS-P525L-GFP negatively affect kinesin-1 motility.
3.5 Arrest in gliding of single microtubules caused by Tau-GFP compared
to aging of the assay
Since aging of the assay over the course of three hours and the presence of 2N4R tau-GFP
both reduced the relative microtubule gliding velocity to a similar extent (Figure 10 and Figure
12), the mode of action by which this slowdown occurred was further investigated. In the

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 61 / 157
presence of standard motility buffer, microtubules of one technical replicate glide with
frame-to-frame velocities around 600 nm/s, while microtubule gliding was entirely arrested in the
presence of 118 nM 2N4R tau-GFP (Figure 15A). When 24 nM 2N4R tau-GFP are
supplemented to the assay, microtubules display two heterogeneously moving populations, one
non-gliding fraction indicated by a peak in frame-to-frame velocities around zero, and one
fraction of microtubules that are gliding with frame-to-frame velocities broadly distributed
between 200 and 800 nm/s, resulting in a median frame-to-frame velocity of about 250 nm/s
(Figure 15A). A similar median frame-to-frame velocity of about 300 nm/s could be observed in
the presence of 5000 nM recombinant FUS-P525L-GFP two hours after protein administration,
although the peak of frame-to-frame velocities around zero was much smaller. Rather, most
microtubule frame-to-frame velocities lay between 100 and less than 600 nm/s (Figure 15A).
This indicates that microtubules collectively glide at a reduced frame-to-frame velocity when the
assay is aging, possibly due to subsiding motility of the underlying kinesin-1 motors, while the
presence of small amounts of tau-GFP causes an arrest in gliding of individual microtubules
while others are still propelled at about similar speed as in standard motility buffer. In order to
investigate this hypothesis further, the fraction of non-gliding microtubules in the presence of
2N4R tau-GFP, FUS-GFP variants, cell lysate or BSA, respectively, was calculated as the
number of microtubules gliding at a velocity less than 20 % of the median microtubule velocity in
standard motility buffer, and normalized to the total number of microtubules in one time-lapse
movie (Figure 15B-F). In the presence of standard motility buffer, the fraction of non-gliding
microtubules consistently appears around 0.1. Corresponding to a decrease in relative
microtubule gliding velocity upon 2N4R tau-GFP addition (Figure 10C), the fraction of
non-gliding microtubules begins to increase in the presence of 24 nM 2N4R tau-GFP and
continues to grow with increasing tau-GFP concentration until virtually all microtubules have
stopped gliding above 118 nM tau-GFP (Figure 15B). In contrast, the fraction of non-gliding
microtubules remained around 0.1 in the presence of either FUS-GFP variant at a concentration
of 5, 500, or 5000 nM, or equal concentrations of BSA (Figure 15C). Even over the course of
three hours, the fraction of non-gliding microtubules under these conditions only slightly
increased to a maximum of 0.20 ± 0.04 (i.e. for 5000 nM wildtype FUS-GFP after three hours,
Figure 15D). Similarly, the presence of cell lysates where either wildtype FUS-GFP or
FUS-P525L-GFP was expressed resulted in a low fraction of non-gliding microtubules around
0.1, independent of the total protein concentration (Figure 15E). When varying concentrations of
2N4R tau-GFP were added to either cell lysates, however, the fraction of non-gliding
microtubules started to increase already at a concentration of 12 nM supplemented tau-GFP,
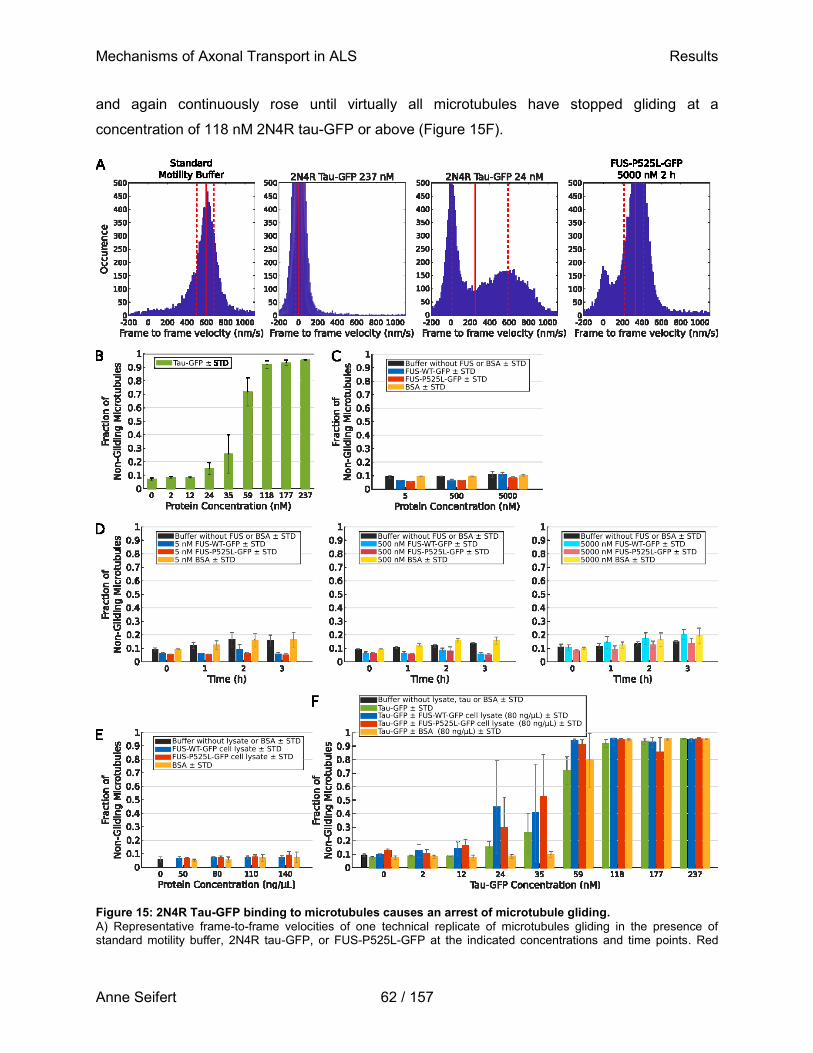
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 62 / 157
and again continuously rose until virtually all microtubules have stopped gliding at a
concentration of 118 nM 2N4R tau-GFP or above (Figure 15F).
Figure 15: 2N4R Tau-GFP binding to microtubules causes an arrest of microtubule gliding.
A) Representative frame-to-frame velocities of one technical replicate of microtubules gliding in the presence of standard motility buffer, 2N4R tau-GFP, or FUS-P525L-GFP at the indicated concentrations and time points. Red

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 63 / 157
solid lines mark the median, while dashed red lines mark the 25th and 75
th percentile, respectively. B-F) The fraction
of non-gliding microtubules describes the number of microtubules in one technical replicate gliding with a frame-to-frame velocity less than 20 % of the gliding velocity in standard motility buffer, normalized to the total number of microtubules in one time lapse movie, before the addition of B) 2N4R tau-GFP at the indicated concentrations, C) recombinant human wildtype FUS-GFP, FUS-P525L-GFP, or BSA, at the indicated concentrations, D) standard motility buffer, recombinant human wildtype FUS-GFP, FUS-P525L-GFP, or BSA at the indicated concentrations over the course of three hours, E) cell lysates expressing wildtype FUS-GFP or the FUS-P525L-GFP variant, or BSA, at the indicated concentrations, and F) cell lysates expressing wildtype FUS-GFP or the FUS-P525L-GFP variant, or BSA at a concentration of 80 ng/µL supplemented with 2N4R tau-GFP at the indicated concentrations.
Of note, while the relative microtubule gliding velocity in the presence of recombinant
wildtype FUS-GFP decreases over the course of three hours down to 0.65 ± 0.14 (Figure 12A),
the fraction of non-gliding microtubules in this condition only increases up to 0.20 ± 0.04 within
the same time frame (Figure 15D). In contrast, the relative microtubule gliding velocity in the
presence of 35 nM and 59 nM 2N4R tau-GFP decreases from 0.80 ± 0.27 to 0.25 ± 0.35,
respectively (Figure 10C), accompanied by a drastic increase in the fraction of non-gliding
microtubules from 0.26 ± 0.14 to 0.72 ± 0.10, accordingly (Figure 15B). Interestingly, the relative
microtubule gliding velocity tends to decrease stronger when cell lysates supplemented with
2N4R tau-GFP are administered to gliding microtubules compared to equal concentrations of
tau-GFP alone (Figure 13C), while the fraction of non-gliding microtubules correspondingly also
tends to increase at lower concentrations of tau-GFP supplemented to cell lysates (Figure 15F).
Together, these findings suggest that 2N4R tau-GFP causes an arrest in the gliding of individual
microtubules, while others seem to be propelled with almost equal velocities as in standard
motility buffer. Although tau-decoration on microtubules often corresponded to arrest of
microtubule gliding, some tau-decorated microtubules were still propelled by underlying
kinesin-1 motors in the presence of low amounts of tau-GFP, e.g. 35 nM (Figure 16), indicating
that microtubule gliding is only reduced when a threshold amount of tau binds to microtubules
and further declines the more tau attaches to the filament.
Figure 16: 2N4R Tau-GFP binding to microtubules at low concentrations leads to an arrest in gliding of individual microtubules, while others are still motile.
A) Binding of2N4R tau-GFP at a concentration of 35 nM to rhodamine-labelled microtubule. Scale bar = 5 µm. B) Top panels: rhodamine-labelled microtubules gliding (1) or non-gliding (2) in the presence of 35 nM 2N4R tau-GFP. Scale bar = 5 µm. Bottom panels: kymographs representing the kinesin-1-dependent movement of the above gliding (1) or non-gliding (2) microtubule. Horizontal scale bar = 2 µm, vertical = 5 s.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 64 / 157
3.6 Differential effects of 2N3R and 2N4R tau individually on
kinesin-1-dependent microtubule gliding velocity and microtubule binding
While this work shows that FUS variants have no direct effect on kinesin-1-dependent transport,
they might indirectly disturb axonal transport by changing the equilibrium of tau isoforms in the
axon due to nuclear loss-of-function of FUS-P525L (Fujioka et al., 2013; Ishigaki et al., 2017).
Under physiological conditions, FUS directly binds to tau pre-mRNA in the nucleus and,
together with the splicing factor SFPQ, regulates its alternative splicing by skipping exon 10,
resulting in 3R tau isoforms (Orozco et al., 2012; Ishigaki et al., 2017; Ishigaki and Sobue,
2018). This is in line with the observation that upon FUS mislocalization to the cytoplasm, exon
10 is more frequently incorporated into the mature tau-mRNA, which results in an increased
translation of 4R tau isoforms. The increase in 4R:3R isoform ratio has previously been
indicated to cause a variety of pathological changes within the cell, such as slightly shorter
axons and highly enlarged growth cones with less bundled and further spread microtubules
(Orozco et al., 2012). This toxic effect on neurite outgrowth could be rescued by silencing 4R
tau (Ishigaki et al., 2017). This indicates how detrimental a shift in 4R:3R tau isoform ratio can
be for the physiological function of the cell. However, it is not known to date if and to what
extend the shift in 4R:3R tau isoform ratio, caused by the cytoplasmic mislocalization of FUS-
P525L, contributes to the axonal transport defects observed in ALS pathology.
This study therefore aimed at investigating the direct effect of 2N4R and 2N3R tau
isoforms, alone or in combination, on the kinesin-1-dependent microtubule gliding. For this
purpose, recombinant human 2N3R tau-GFP and 2N4R tau-mScarlet labelled isoforms were
expressed in and purified from insect cells (see 2.2.7). Each tau variant was at first added
individually to the modified kinesin-1-dependent microtubule gliding assay at increasing
concentrations (Figure 17A and B), and microtubule gliding again observed using widefield-
epifluorescence microscopy. BSA was again used as a protein crowding control at the highest
concentration of tau variants tested (5.25 µM). While the relative microtubule gliding velocity
goes down with increasing concentrations of either isoform (Figure 17C) compared to the
microtubule gliding in standard motility buffer or BSA, the 4R tau-mScarlet isoform causes a
slowdown of microtubule gliding already at much lower concentrations (starting at 0.05 µM tau)
compared to the 3R tau-GFP isoform. At 0.8 µM 4R tau-mScarlet, the relative microtubule
gliding speed has decreased to 0.31 ± 0.08, while the 3R tau-GFP isoforms causes a similar
drop of relative microtubule gliding speed only at a more than 5-fold higher concentration
(5.25 µM).
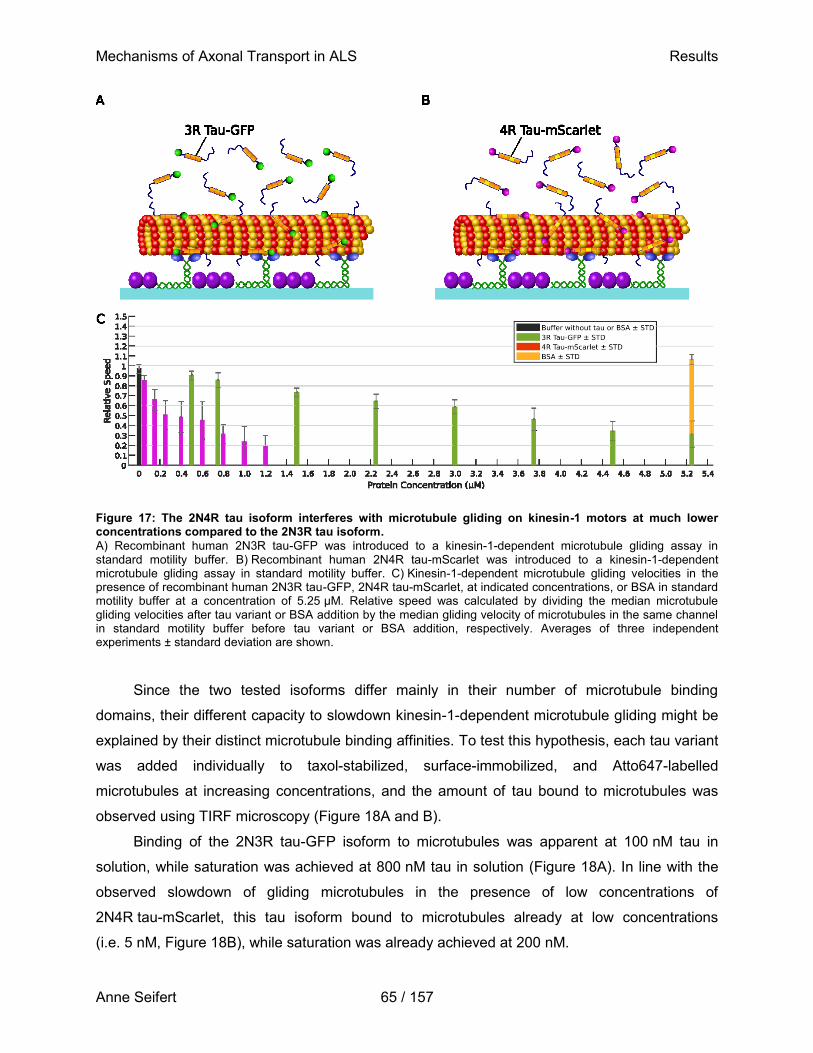
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 65 / 157
Figure 17: The 2N4R tau isoform interferes with microtubule gliding on kinesin-1 motors at much lower concentrations compared to the 2N3R tau isoform.
A) Recombinant human 2N3R tau-GFP was introduced to a kinesin-1-dependent microtubule gliding assay in standard motility buffer. B) Recombinant human 2N4R tau-mScarlet was introduced to a kinesin-1-dependent microtubule gliding assay in standard motility buffer. C) Kinesin-1-dependent microtubule gliding velocities in the presence of recombinant human 2N3R tau-GFP, 2N4R tau-mScarlet, at indicated concentrations, or BSA in standard motility buffer at a concentration of 5.25 µM. Relative speed was calculated by dividing the median microtubule gliding velocities after tau variant or BSA addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before tau variant or BSA addition, respectively. Averages of three independent experiments ± standard deviation are shown.
Since the two tested isoforms differ mainly in their number of microtubule binding
domains, their different capacity to slowdown kinesin-1-dependent microtubule gliding might be
explained by their distinct microtubule binding affinities. To test this hypothesis, each tau variant
was added individually to taxol-stabilized, surface-immobilized, and Atto647-labelled
microtubules at increasing concentrations, and the amount of tau bound to microtubules was
observed using TIRF microscopy (Figure 18A and B).
Binding of the 2N3R tau-GFP isoform to microtubules was apparent at 100 nM tau in
solution, while saturation was achieved at 800 nM tau in solution (Figure 18A). In line with the
observed slowdown of gliding microtubules in the presence of low concentrations of
2N4R tau-mScarlet, this tau isoform bound to microtubules already at low concentrations
(i.e. 5 nM, Figure 18B), while saturation was already achieved at 200 nM.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 66 / 157
The fraction of tau protein that was bound to microtubules when a specific concentration
of tau was present in solution, relative to the amount of the respective tau isoform bound at the
highest concentration tested, was plotted and the data fitted using the Hill equation (Hill, 1910)
in order to describe the binding kinetics of the respective tau isoform (Gesztelyi et al., 2012)
(Figure 18C and D). The respective value for the highest concentration tested for each tau
isoform (800 nM for 3R tau-GFP and 200 nM for 4R tau-mScarlet, respectively) was excluded
from the fit due to the fact that it was used for normalization and yielded a standard error of the
mean (SEM) of zero after averaging the experiments. Interestingly, the Hill equation determined
a Hill coefficient (n) larger than 1 for both binding curves for 3R tau-GFP and 4R tau-mScarlet,
namely 2.38 ± 0.67 and 1.83 ± 0.29, respectively. This indicates a cooperative binding
mechanism for both tau isoforms (Weiss, 1997). The dissociation constant Kd (or k in Figure
18C and D), corresponding to the concentration at which half of the tau molecules in solution
are bound to microtubules, for 3R tau-GFP (Kd = 137.12 ± 29.7 nM) was determined to be
approximately 20-fold higher compared to 4R tau-mScarlet (Kd = 6.79 ± 0.41 nM), indicating that
the binding affinity of 4R tau-mScarlet to microtubules is approximately 20-fold higher than that
of 3R tau-GFP.
Interestingly, although the microtubule was heavily decorated with 2N3R tau-GFP
molecules, the relative microtubule gliding speed at 750 nM 2N3R tau-GFP was only reduced to
0.85 ± 0.07 (Figure 17C). Similarly, microtubule saturation with 2N4R tau-mScarlet at 250 nM
corresponded to a decrease in relative kinesin-1-dependent microtubule gliding speed only
down to 0.48 ± 0.13 (Figure 17C). For both recombinant tau isoforms, the relative microtubule
gliding velocity decreased further at higher concentrations than those needed to saturate
microtubules with tau. We hence suggest that tau molecules do not just form a monolayer on
microtubules, but can cluster on the surface and impair kinesin-1 motility stronger than single
tau molecules.
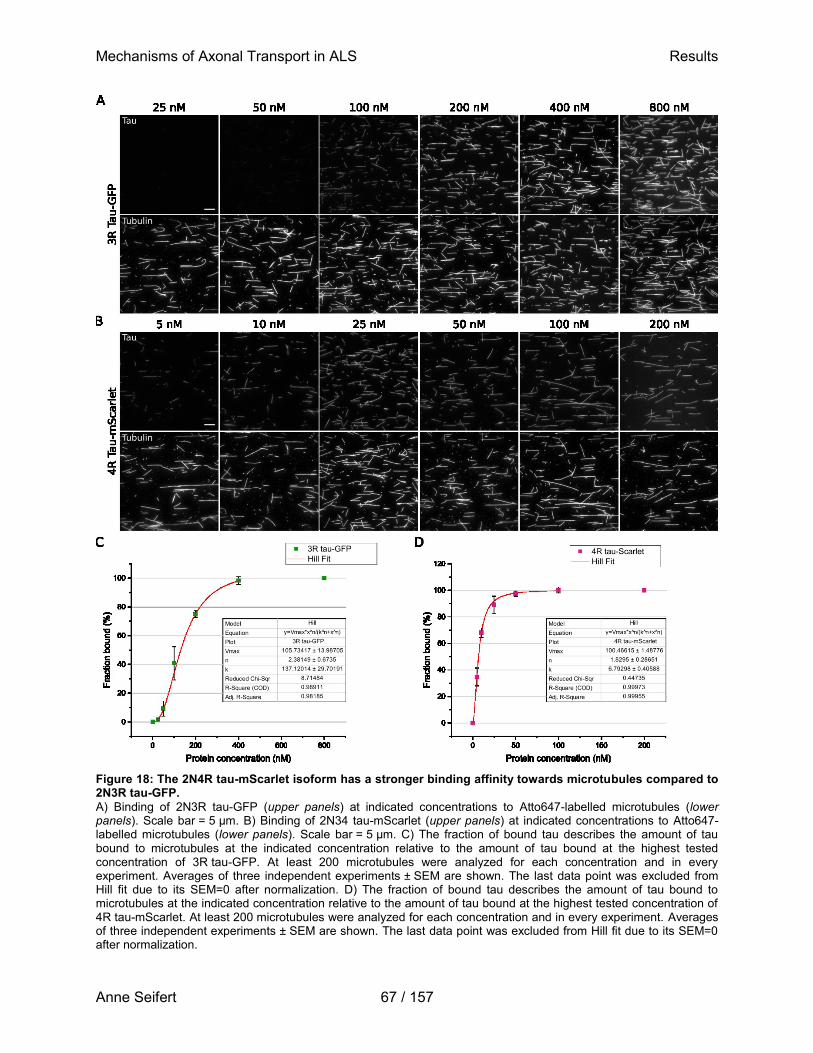
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 67 / 157
Figure 18: The 2N4R tau-mScarlet isoform has a stronger binding affinity towards microtubules compared to 2N3R tau-GFP.
A) Binding of 2N3R tau-GFP (upper panels) at indicated concentrations to Atto647-labelled microtubules (lower panels). Scale bar = 5 µm. B) Binding of 2N34 tau-mScarlet (upper panels) at indicated concentrations to Atto647-labelled microtubules (lower panels). Scale bar = 5 µm. C) The fraction of bound tau describes the amount of tau bound to microtubules at the indicated concentration relative to the amount of tau bound at the highest tested concentration of 3R tau-GFP. At least 200 microtubules were analyzed for each concentration and in every experiment. Averages of three independent experiments ± SEM are shown. The last data point was excluded from Hill fit due to its SEM=0 after normalization. D) The fraction of bound tau describes the amount of tau bound to microtubules at the indicated concentration relative to the amount of tau bound at the highest tested concentration of 4R tau-mScarlet. At least 200 microtubules were analyzed for each concentration and in every experiment. Averages of three independent experiments ± SEM are shown. The last data point was excluded from Hill fit due to its SEM=0 after normalization.
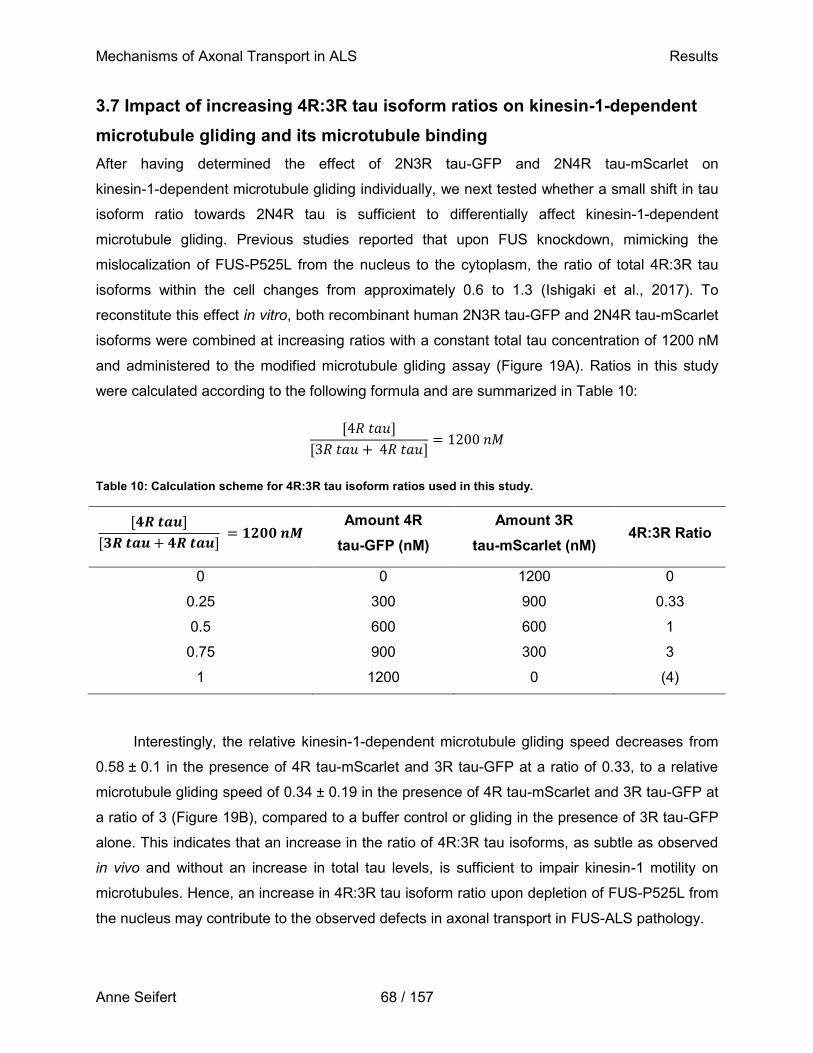
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 68 / 157
3.7 Impact of increasing 4R:3R tau isoform ratios on kinesin-1-dependent
microtubule gliding and its microtubule binding
After having determined the effect of 2N3R tau-GFP and 2N4R tau-mScarlet on
kinesin-1-dependent microtubule gliding individually, we next tested whether a small shift in tau
isoform ratio towards 2N4R tau is sufficient to differentially affect kinesin-1-dependent
microtubule gliding. Previous studies reported that upon FUS knockdown, mimicking the
mislocalization of FUS-P525L from the nucleus to the cytoplasm, the ratio of total 4R:3R tau
isoforms within the cell changes from approximately 0.6 to 1.3 (Ishigaki et al., 2017). To
reconstitute this effect in vitro, both recombinant human 2N3R tau-GFP and 2N4R tau-mScarlet
isoforms were combined at increasing ratios with a constant total tau concentration of 1200 nM
and administered to the modified microtubule gliding assay (Figure 19A). Ratios in this study
were calculated according to the following formula and are summarized in Table 10:
[4𝑅 𝑡𝑎𝑢]
[3𝑅 𝑡𝑎𝑢 + 4𝑅 𝑡𝑎𝑢]= 1200 𝑛𝑀
Table 10: Calculation scheme for 4R:3R tau isoform ratios used in this study.
[𝟒𝑹 𝒕𝒂𝒖]
[𝟑𝑹 𝒕𝒂𝒖 + 𝟒𝑹 𝒕𝒂𝒖] = 𝟏𝟐𝟎𝟎 𝒏𝑴
Amount 4R
tau-GFP (nM)
Amount 3R
tau-mScarlet (nM) 4R:3R Ratio
0 0 1200 0
0.25 300 900 0.33
0.5 600 600 1
0.75 900 300 3
1 1200 0 (4)
Interestingly, the relative kinesin-1-dependent microtubule gliding speed decreases from
0.58 ± 0.1 in the presence of 4R tau-mScarlet and 3R tau-GFP at a ratio of 0.33, to a relative
microtubule gliding speed of 0.34 ± 0.19 in the presence of 4R tau-mScarlet and 3R tau-GFP at
a ratio of 3 (Figure 19B), compared to a buffer control or gliding in the presence of 3R tau-GFP
alone. This indicates that an increase in the ratio of 4R:3R tau isoforms, as subtle as observed
in vivo and without an increase in total tau levels, is sufficient to impair kinesin-1 motility on
microtubules. Hence, an increase in 4R:3R tau isoform ratio upon depletion of FUS-P525L from
the nucleus may contribute to the observed defects in axonal transport in FUS-ALS pathology.
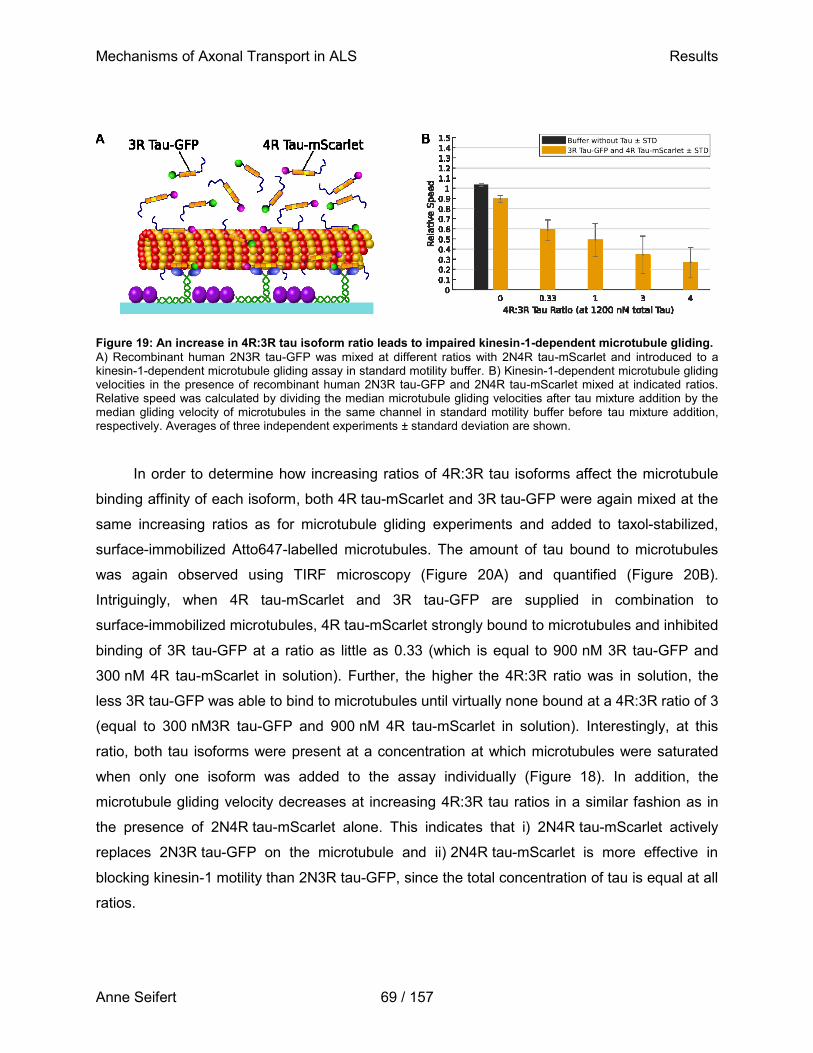
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 69 / 157
Figure 19: An increase in 4R:3R tau isoform ratio leads to impaired kinesin-1-dependent microtubule gliding.
A) Recombinant human 2N3R tau-GFP was mixed at different ratios with 2N4R tau-mScarlet and introduced to a kinesin-1-dependent microtubule gliding assay in standard motility buffer. B) Kinesin-1-dependent microtubule gliding velocities in the presence of recombinant human 2N3R tau-GFP and 2N4R tau-mScarlet mixed at indicated ratios. Relative speed was calculated by dividing the median microtubule gliding velocities after tau mixture addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before tau mixture addition, respectively. Averages of three independent experiments ± standard deviation are shown.
In order to determine how increasing ratios of 4R:3R tau isoforms affect the microtubule
binding affinity of each isoform, both 4R tau-mScarlet and 3R tau-GFP were again mixed at the
same increasing ratios as for microtubule gliding experiments and added to taxol-stabilized,
surface-immobilized Atto647-labelled microtubules. The amount of tau bound to microtubules
was again observed using TIRF microscopy (Figure 20A) and quantified (Figure 20B).
Intriguingly, when 4R tau-mScarlet and 3R tau-GFP are supplied in combination to
surface-immobilized microtubules, 4R tau-mScarlet strongly bound to microtubules and inhibited
binding of 3R tau-GFP at a ratio as little as 0.33 (which is equal to 900 nM 3R tau-GFP and
300 nM 4R tau-mScarlet in solution). Further, the higher the 4R:3R ratio was in solution, the
less 3R tau-GFP was able to bind to microtubules until virtually none bound at a 4R:3R ratio of 3
(equal to 300 nM3R tau-GFP and 900 nM 4R tau-mScarlet in solution). Interestingly, at this
ratio, both tau isoforms were present at a concentration at which microtubules were saturated
when only one isoform was added to the assay individually (Figure 18). In addition, the
microtubule gliding velocity decreases at increasing 4R:3R tau ratios in a similar fashion as in
the presence of 2N4R tau-mScarlet alone. This indicates that i) 2N4R tau-mScarlet actively
replaces 2N3R tau-GFP on the microtubule and ii) 2N4R tau-mScarlet is more effective in
blocking kinesin-1 motility than 2N3R tau-GFP, since the total concentration of tau is equal at all
ratios.
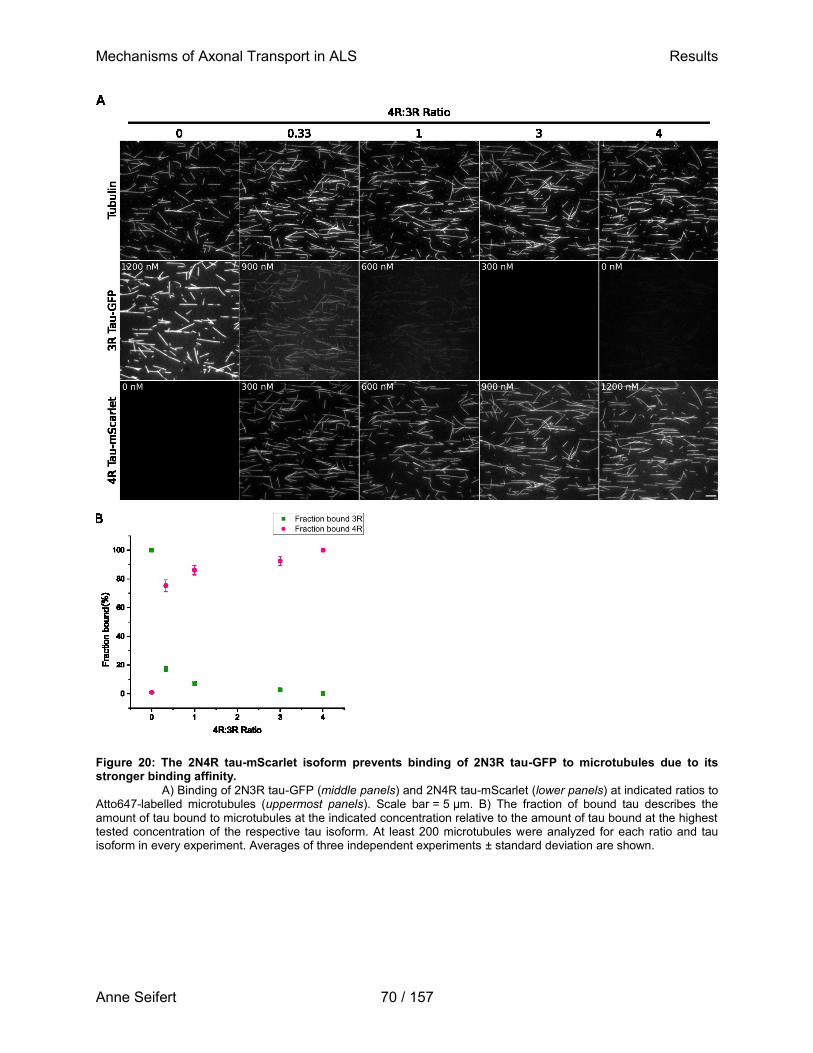
Mechanisms of Axonal Transport in ALS Results
Anne Seifert 70 / 157
Figure 20: The 2N4R tau-mScarlet isoform prevents binding of 2N3R tau-GFP to microtubules due to its stronger binding affinity.
A) Binding of 2N3R tau-GFP (middle panels) and 2N4R tau-mScarlet (lower panels) at indicated ratios to Atto647-labelled microtubules (uppermost panels). Scale bar = 5 µm. B) The fraction of bound tau describes the amount of tau bound to microtubules at the indicated concentration relative to the amount of tau bound at the highest tested concentration of the respective tau isoform. At least 200 microtubules were analyzed for each ratio and tau isoform in every experiment. Averages of three independent experiments ± standard deviation are shown.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 71 / 157
3.8 Expression levels of 4R and 3R tau isoforms in whole cell lysates
The so far presented experimental data strongly points towards the fact that an increase in
4R:3R tau isoform ratio within the cell contributes to the observed defects in axonal transport.
This is likely due to a higher decoration of microtubules with 4R tau isoforms and their larger
roadblock effect compared to 3R tau isoforms. An increased 4R:3R ratio likely results from the
cytoplasmic mislocalization of the FUS-P525L mutant as observed in the used cell lines
(Dormann et al., 2010; Ishigaki et al., 2017). This would indicate that the 4R:3R tau isoform ratio
should be increased in whole cell lysates expressing the FUS-P525L-GFP variant compared to
lysates from cells expressing the wildtype FUS-GFP variant. However, when both lysates were
added to the herein used kinesin-1-dependent microtubule gliding assay, they did not
differentially influence the relative microtubule gliding speed (Figure 13). Therefore, western
blots were performed to determine the level of each tau isoform within the herein used whole
cell lysates (Figure 21). In order to detect 3R and 4R tau isoforms individually, primary
pan-specific antibodies detecting either 0N-, 1N-, and 2N3R tau isoforms or 0N-, 1N-, and 2N4R
tau isoforms were used in these blots. Recombinant human 2N3R tau-GFP and
2N4R tau-mScarlet was used as a positive control, respectively. Interestingly, none of the 4R
tau isoforms could be detected in four biological replicates of whole cell lysates expressing the
wildtype FUS-GFP or FUS-P525L-GFP variants, respectively (Figure 21B), as well as the 1N3R
and 2N3R tau isoforms (Figure 21A).The 0N3R tau isoform, however, was consistently present
in all biological replicates with a concentration between 10 and 30 nM. This indicates that the
expression level of tau isoforms in the herein used iPSC-derived spinal motor neurons has not
(yet) been influenced by the FUS-P525L-GFP variant, at least not within the detection limit of
this western blot technique. However, the presence of small amounts of 0N3R tau within lysates
might contribute to the non-significant, but consistently lower relative microtubule gliding speeds
observed in the kinesin-1-dependent microtubule gliding assay in the presence of whole cell
lysates supplemented with recombinant human 2N4R tau-GFP, compared to the presence of
2N4R tau-GFP alone (Figure 13C). Alternatively, unknown factors present in whole cell lysates
may interfere with kinesin-1-dependent microtubule gliding, which could be determined by e.g.
pulldown experiments with microtubules and motors in the presence of whole cell lysates and
subsequent mass spectrometry.

Mechanisms of Axonal Transport in ALS Results
Anne Seifert 72 / 157
Figure 21: Western blot of tau variants expressed in whole cell lysates of spinal motor neurons.
Representative western blot of A) recombinant human 2N3R tau-GFP and b) recombinant human 2N4R tau-mScarlet
at indicated concentrations and four biological replicates (n) of whole cell lysates (at a concentration of 80 ng/µL)
obtained from spinal motor neurons expressing either wildtype FUS-GFP or FUS-P525L-GFP. The blot in A) was incubated with primary antibodies against 0N-, 1N-, and 2N3R tau isoforms, while the blot in B) was incubated with primary antibodies against 0N-, 1N-, and 2N4R tau isoforms.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 73 / 157
4. Discussion
The pathology of ALS has been subject to many previous studies, and yet there is no clear
consensus about the exact mechanisms leading to this devastating disease. Since mutations in
SOD1 were the first discovered in ALS patients, most research results to date are based on
models with this mutation (Rosen et al., 1993; Huai and Zhang, 2019), but intensive studies
have also been performed on the role of mutant FUS variants (Groen et al., 2010; Shang and
Huang, 2016; Naumann et al., 2019). Although defects in axonal transport have long been
observed in FUS-ALS (De Vos et al., 2008; Ikenaka et al., 2012; De Vos and Hafezparast,
2017b), little is known about underlying pathomechanisms that lead to these detrimental
defects, and whether they are causative or occur as a consequence of other pathological
events. The work done for this thesis therefore aimed at investigating the fundamental
hypothesis of a direct influence of NLS-mutant, cytoplasmic mislocalized FUS on the axonal
transport machinery involving kinesin-1. For this purpose, a kinesin-1-dependent microtubule
gliding assay was modified to be compatible with and robustly deliver reproducible results in the
presence of protein-rich solutions such as whole cell lysates. To evaluate the direct influence of
FUS variants on kinesin-1 motility, recombinant human GFP-labelled FUS variants, as well as
cell lysates from iPSC-derived spinal motor neurons expressing the same GFP-labelled FUS
variants, were analyzed in this assay. In addition, a more indirect effect of mutant FUS variants
on axonal transport was hypothesized and investigated: FUS is known to regulate splicing of
mRNA coding for the microtubule-binding protein tau, which suggests that the ratio of 4R:3R tau
is increased in cells carrying a mutation in FUS leading to its cytoplasmic mislocalization
(Ishigaki et al., 2017). Therefore, the modified kinesin-1-dependent microtubule gliding assay
was utilized to study whether and which 3R:4R tau ratio is sufficient to alter kinesin-1 motility on
microtubules.
This study therefore presents the first to address the direct role of FUS variants on
kinesin-1 motility in a bottom-up in vitro reconstruction of axonal transport. Likewise, using the
same reconstruction approach, it provides further evidence for the relevance of tau isoform
variations in disease progression. The results presented herein therefore largely contribute to
our understanding of the underlying pathomechanisms in FUS-ALS.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 74 / 157
4.1 The modified kinesin-1-dependent microtubule gliding assay detects
nanomolar amounts of recombinant human tau-GFP
The buffer typically used in standard kineins-1-dependent microtubule gliding assays is BRB80
(with PIPES as a buffering agent), which we aimed to substitute with PBS for optimal cell lysis at
physiological pH (7.4). To prevent water from crystallizing and hence damage proteins during
snap freezing, glycerol was added to every cell lysate, as well as a commercially available
protease inhibitor cocktail. Since the phosphorylation state of proteins, in this case especially of
tau, is important for their activity, phosphatases within lysates needed to be inhibited as well.
However, a commercially available phosphatase inhibitor cocktail introduced to the microtubule
gliding assay led to an immediate arrest of microtubule gliding (Figure 8C). According to the
manufacturer, the tested phosphatase inhibitor cocktail was a mixture of the broad-spectrum
inhibitors sodium fluoride, sodium pyrophosphate, sodium orthovanadate and
β-glycerophosphate. Sodium fluoride and sodium pyrophosphate are general inhibitors for
phosphoseryl and phosphothreonyl phosphatases and commonly used in the purification of
protein kinases (Gordon, 1991; Hardie, 1993; Mercan and Bennett, 2010). The serine-threonine
phosphatase inhibitor β-glycerophosphate acts as a bait substrate (Belfield and Goldberg, 1968;
Chung et al., 1992; Lecanda et al., 1997), thereby protecting a protein’s phosphorylation status
by attracting phosphatases away from endogenous proteins. Sodium orthovanadate is a general
inhibitor for phosphotyrosyl phosphatases, alkaline phosphatases and a variety of ATPases
(Gordon, 1991; Wang et al., 1994; Yun et al., 2001). It has been repeatedly reported to inhibit
dynein (D’Souza et al., 2006; Kharkwal et al., 2014) and kinesin motility at a concentration as
low as 100 mM (Scholey, 1993; Dentler et al., 1995). Orthovanadate hence is very likely to
mediate the observed complete arrest of kinesin-1 motility. In consequence, an excess of
β-glycerophosphate alone was used to inhibit dephosphorylation of proteins within whole cell
lysates by serving as an alternative substrate. These high concentrations of
β-glycerophosphate, however, interfered with microtubule-motor binding and led to detachment
of microtubules (Figure 8D). Most likely, negatively charged phosphate groups of
β-glycerophosphate (Kolawole et al., 2019) disturb the electrostatic interaction between the
lattice-exposed acidic C-termini of tubulin subunits (E-hook) and the positively charged K-loop of
kinesin-1 (Okada and Hirokawa, 2000). These E-hook/K-loop interactions are crucial to stabilize
the weak binding state of kinesin motor domains (Okada and Hirokawa, 2000), resulting in more
frequent detachment of microtubules from surface-bound kinesin-1 in the presence of
β-glycerophosphate. To prevent this, methylcellulose was added to all solutions in the gliding
assay, which increases the viscosity of the assay solution. This prevents detached microtubules

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 75 / 157
from escaping into solution, keeps them closer to the imaging surface and facilitates their
efficient recapture by surface-bound motors (Wiesner et al., 2003; Saito et al., 2017; Farhadi et
al., 2018).
Implementation of these adjustments in the used kinesin-1-dependent microtubule gliding
assay produced highly reproducible results (Figure 9D). In line with previous studies,
kinesin-1-dependent microtubule gliding was completely inhibited at concentrations of
recombinant human 2N4R tau-GFP of around 118 nM and higher (Figure 10C) (Hagiwara et al.,
1994; Peck et al., 2011; Parimalam et al., 2016). The same sensitivity still applies to the used
kinesin-1-dependent microtubule gliding assay in the presence of whole cell lysates, underlining
the robustness and suitability of the modified assay to study the influence of proteins in such a
protein-rich solution on kinesin-1 motility.
Comparison of chemical and mechanical protein isolation techniques
Various protein isolation techniques were utilized in this study to determine the method yielding
the most sufficient cell lysis in combination with a high concentration of total protein in the final
lysate, while being compatible with the herein used kinesin-1-dependent microtubule gliding
assay. Buffers used for chemical cell lysis yielded the highest amounts of total protein and
sufficiently lysed cells so that the housekeeping protein β-actin could be detected (Table 9).
However, those buffers interfered with microtubule gliding on kinesin-1 motors, leading to
microtubule detachment even in the presence of very highly diluted buffer or after buffer
exchange (Figure 8). Hence, different mechanical lysis methods, utilizing PBS as a buffer that is
not interfering with kinesin-1-dependent microtubule gliding (Figure 8), were tested for their lysis
efficiency and protein yield.
In this study, the housekeeping protein β-actin could not be detected in motor neuron
lysates produced by glass bead shearing (Table 9), suggesting that lysis was incomplete or that
a major portion of protein got lost during lysate processing. Evidence for the latter comes from
the fact that beads are generally provided to the cell suspension and mixed at high speeds to
break open the cell membrane by shear force. This process generates heat, which may cause
degradation of proteins to some degree (Shehadul Islam et al., 2017) and cause the observed
loss of protein. This is in line with the finding that protein denaturation and aggregation has been
observed in yeast cell lysates produced by glass bead shearing (Papanayotou et al., 2010).
Likewise, β-actin could not be detected in lysates prepared by grinding of motor neurons in the
presence of liquid nitrogen (Table 9). Although grinding in liquid nitrogen has been successfully
used in previous studies, e.g. for the isolation of protein from Chlorella vulgaris (Zheng et al.,

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 76 / 157
2011), the procedure presented several disadvantages in this study. For instance, slight thawing
of the lysate could not be avoided during collection of the finely ground powder from the mortar,
which might have caused some protein degradation. Also, a full recovery of the lysates from the
mortar was not possible. A similar problem arose when preparing cell lysates using a dounce
homogenizer. Consistent with previously reported efficient use of dounce homogenization
(Osborne and Neuhoff, 1972; Gozes et al., 1977; Krishnaswami et al., 2016), dounce
homogenization of single motor neurons yielded comparably high amounts of protein as well as
well detectable levels of the housekeeping protein β-actin in this study (Table 9). However, a
small fraction of the lysate always stuck to the walls of the homogenization tube, resulting in a
reduced volume of recovered lysate. Therefore, mechanical lysis of motor neurons was chosen
as a suitable method for whole cell lysate production in this study, by passing the cell
suspension several times through a thin needle with the help of a syringe. This method resulted
in similar protein yields and detection of β-actin in western blot as compared to dounce
homogenization (Table 9), but nearly full lysate volume recovery since the lysate could be
pressed out of the syringe and needle using the plunger.
4.2 Wildtype and ALS-associated FUS variants do not directly interfere with
kinesin-1 motility and do not bind to microtubules
Defects in the axonal transport of a variety of organelles and nucleic acids have long been
observed in ALS (Fujii and Takumi, 2005; Bilsland et al., 2010; Ikenaka et al., 2012; De Vos and
Hafezparast, 2017b; Naumann et al., 2018; Pal et al., 2018; Smith et al., 2019), the underlying
mechanism for impaired transport is, however, still enigmatic. In this study, a modified
kinesin-1-dependent microtubule gliding assay was used to investigate a direct influence of
ALS-associated FUS variants alone or in combination with their endogenous interaction
partners present in whole cell lysates on the kinesin-1-driven transport machinery. Results
presented herein provide clear evidence that neither recombinant human wildtype FUS-GFP nor
FUS-P525L-GFP by itself directly binds to microtubules or inhibits kinesin-1 motility over a wide
range of concentrations. Although relative microtubule gliding velocity in the presence of FUS
variants slightly reduced over the course of three hours (Figure 12A), this phenomenon can be
likely attributed to aging of the assay components. Aging occurs as a result of changes in pH,
depletion of ATP, or oxidative damage due to depletion of oxygen scavenging components
glucose oxidase, catalase or DTT (Rasnik et al., 2006; Aitken et al., 2008; Landry et al., 2009;
Swoboda et al., 2012). Interestingly, although the median gliding velocity was comparable in the

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 77 / 157
presence of recombinant FUS variants over the course of three hours and in the presence of
low amounts (15-25 nM) of 2N4R tau-GFP (Figure 12A and Figure 10C), aging of the assay
resulted in a uniform decrease in the gliding velocity of all microtubules gliding in the field of
view, while the introduction of tau-GFP caused only individual microtubules to slow down or fully
stop (Figure 15B and D). The fraction of non-gliding microtubules sharply increased after a
threshold concentration of 15 nM tau-GFP until virtually all microtubules have stopped gliding at
a concentration of 50 nM 2N4R tau-GFP or above. A very similar distribution of gliding and
non-gliding microtubules has been observed in a system employing active and inactive
kinesin-1 motors at different ratios. When 30% of kinesin-1 motors immobilized on a glass
surface were inactive, microtubules displayed a bistable movement with one portion of
microtubules gliding at a peak velocity around 500 nm/s and another portion with peak velocity
at 0 nm/s. Slightly higher or lower amounts of inactive motors shifted the distribution of
microtubule gliding velocities towards the slower or the faster peak, respectively (Scharrel et al.,
2014). Whether a microtubule is propelled by the underlying motors therefore depends on the
number of inactive motors that attach to the filament, where a stop of motion is achieved when
inactive motors dominate. Similarly, whether a microtubule is propelled by underlying kinesin-1
motors in the presence of 2N4R tau-GFP depends on the amount of tau molecules creating
roadblocks on the microtubule, thereby impairing kinesin-1 motility. Since neither FUS variant
tested in the present work caused a bistable kinesin-1-dependent gliding of microtubules, this
suggests that wildtype FUS and FUS-P525L do not impair kinesin-1 motility on microtubules by
direct interaction with either microtubules or kinesin-1 motors.
FUS by itself hence does not interfere with the kinesin-1-driven transport machinery.
Further, whole cell lysates of iPSC-derived spinal motor neurons expressing the same FUS
variants did not interfere with kinesin-1-driven microtubule gliding at the tested concentrations.
This indicates that FUS variants did not form complexes with other proteins in whole cell lysates
that would interfere with kinesin-1 motility. Taken together, these results strongly suggest that
neither the wildtype form nor the mislocalized cytoplasmic FUS-P525L variant directly or by
interaction with other proteins interfere with kinesin-1-driven (anterograde) axonal transport in
neurons.
Yet, there are some limitations to the herein used assay setup which have to be taken into
consideration. Although there were no aggregates of recombinant or endogenous FUS variants
apparent in the presented experiments, even over the course of three hours, ALS-associated
FUS aggregation has repeatedly been observed in vivo and in vitro (Shelkovnikova et al., 2014;
Murakami et al., 2015; Patel et al., 2015; Marrone et al., 2019) and may very well be involved in

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 78 / 157
hindering axonal transport in neurons of ALS-patients, e.g. by sterical hindrance (Sau et al.,
2011) or sequestering of proteins (Aulas and Vande Velde, 2015; Yasuda et al., 2017). In case
aggregates composed of cytoplasmic mislocalized FUS-P525L-GFP were initially present in
iPSC-derived spinal motor neurons, they were most likely lost during cell lysate clearance by
centrifugation (>100K g in pellet, Shelkovnikova et al., 2014) and hence no longer present in
whole cell lysates. A possible impact of endogenous cytoplasmic FUS aggregates on axonal
transport is difficult to study in a minimal system such as microtubule gliding assays, since
compartmentalization of the neuron is lost during lysates generation. Furthermore, a loss of
compartmentalization after cell lysis also abolishes the possibility to study pathological events
with local impact. For instance, axonal transport of mitochondria is significantly impaired and
their membrane potential broken down in iPSC-derived spinal motor neurons expressing
FUS-P525L-GFP (Naumann et al., 2018), indicating a potential local depletion of ATP and
hence impaired kinesin-1 motility. Local ATP deficiencies, however, are lost during lysis, and an
excess of ATP is additionally supplied to cell lysates used in kinesin-1-dependent microtubule
gliding assays to ensure efficient kinesin-1-motility. Another challenge for studying the impact of
cytosolic inclusions in microtubule gliding assays presents the generation of FUS-containing
aggregates. In vitro assembled FUS aggregates do not form uniformly in size and do not fully
resemble all components present in endogenous pathological FUS aggregates (Marrone et al.,
2019). Endogenous FUS granules could be enriched by centrifugation in fractions from cell
lysates (Shelkovnikova et al., 2014) in the past, however, this required the introduction of the
detergent Triton X to lysates, which has been shown to influence kinesin-1-dependent
microtubule gliding velocity and causes detachment of actin filaments from myosin motors
(Kellermayer, 1997; Kuramitsu, 1999). Endogenous cytoplasmic FUS aggregates are also likely
to sequester proteins and RNA interacting with cytoplasmic FUS under physiological conditions,
which results in severe reduction in their concentration or virtual exclusion from herein used
whole cell lysates during clearance by centrifugation or because the neuronal cytoplasm
becomes highly diluted after lysis. Lack of such factors might hence contribute to the absence of
a potential interference of FUS with kinesin-1-dependent microtubule gliding in this study.
Finally, some interactions of proteins in cell lysates with kinesin-1 might have been missed due
to the inaccessibility of motors in a gliding assay.
Gliding assays present a useful tool to study the effect that proteins have on the transport
machinery, in particularly if they interact with microtubules or the motor domain or motor
proteins, and imaging of large polymeric filaments is not technically challenging. Yet, the actual
imaging of single motors walking across cytoskeletal filaments in stepping assays presents a

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 79 / 157
more direct way of measuring motor characteristics and how these may change in the presence
of proteins engaging with the motor. Depending on the motor construct that is used (i.e.
full-length kinesin-1) in a stepping assay, the motor itself becomes much more accessible for
interaction with proteins in solution, compared to its surface-bound state in a gliding assay.
Proteins present within the herein used whole cell lysates may have been prevented from
interacting with kinesin-1 due to the inaccessibility of the surface-bound motor. Hence, applying
whole cell lysates of isogenic iPSC-derived spinal motor neurons carrying different FUS variants
in a stepping assay may yield different results than those presented here and provide further
insights into how mutations in FUS impact the kinesin-1-driven transport machinery directly.
In addition, there is evidence that FUS associates with ATP-dependent actin-binding
motor Myo5A (Yoshimura et al., 2006) and Myo6 (Takarada et al., 2009), and has been isolated
as part of a large granule associating with KIF5B (Kanai et al., 2004). Although the herein
presented results demonstrate that FUS by itself does not interfere with kinesin-1 (KIF5B)
motility, it remains to be elucidated whether this holds true for actin-associated myosin motors
as well. Similarly to the kinesin-1-dependent microtubule gliding assay used in this study, actin
filaments can glide over myosin-coated surfaces (Rock et al., 2000; Vilfan, 2009; Hariadi et al.,
2016), and single myosin molecules can be tracked while stepping on immobilized actin
filaments (Ricca and Rock, 2010; Bao et al., 2013). By utilizing such actin and myosin based
gliding or stepping assays in future experiments, the direct interaction of FUS variants with
these components of the transport machinery may be investigated.
Taken together, the present data provide strong evidence that mislocalized, free
cytoplasmic FUS-P525L itself does not directly interfere with kinesin-1-driven axonal transport
on microtubules. Apart from the fact that ALS-associated FUS aggregates sequester proteins
involved in RNA transport (Kamelgarn et al., 2016) as well as mRNA and protein of kinesin-1
itself (Yasuda et al., 2017), there is also little evidence in the literature to date about a direct
involvement of mislocalized cytoplasmic ALS-associated FUS in microtubule-based axonal
transport.
4.3 An increase in 4R:3R tau isoform ratio might contribute to the axonal
transport defects observed in FUS-ALS
Since the herein presented results show that FUS is not directly interfering with the microtubule
based kinesin-1 mediated transport machinery, this study also investigated possible effects of
FUS-dependent changes in the expression of tau isoforms on axonal transport. Defects in

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 80 / 157
axonal transport have been observed early in the progression of many neurodegenerative
diseases, and might partly be caused by hyperphosphorylated mutant tau variants or by altered
expression levels of tau isoforms (Ebneth et al., 1998; Stamer et al., 2002; De Vos et al., 2008;
Goldstein, 2012; Qian and Liu, 2014; Correia et al., 2016; Fernández-Nogales and Lucas,
2020). Overexpression of single wildtype tau isoforms (0N3R, 2N3R, 0N4R, and 2N4R) in H4
human neuroglioma cells have previously been associated with defects in axonal transport of
mitochondria (Stoothoff et al., 2009). Previous studies could show that the FUS, together with
the transcription factor SFPQ, is directly involved in the splicing of tau pre mRNA, and a
knockdown of FUS results in in a higher expression of the 4R tau isoform in mice (Ishigaki et al.,
2017; Bourefis et al., 2020). Furthermore, pathological behavioral phenotypes caused by FUS
knockdown in these mice could be weakened by silencing of 4R tau with small hairpin RNAs
(Ishigaki and Sobue, 2018), indicating that an increased 4R:3R ratio within the cell may indeed
contribute to pathology. In line with this, changes in the 4R:3R in favor of 4R have been
observed in diverse tauopathies (D’Souza and Schellenberg, 2005; Goedert and Jakes, 2005;
Sergeant et al., 2005). However, the role of changing ratios of wildtype 4R and 3R tau isoforms,
without increased total tau levels, in contributing to axonal transport defects is not well
investigated to date.
We hence hypothesized that cytoplasmic mislocalization of FUS and the resulting loss of
function in splicing of tau might as well increase the 4R:3R isoform ratio of tau in vivo, which in
turn might affect kinesin-1-dependdent transport in axons. In order to test this hypothesis, the
microtubule binding affinity of purified recombinant human 2N3R tau-GFP and
2N4R tau-mScarlet and their effect on kinesin-1-dependent microtubule gliding was analyzed in
the modified microtubule gliding assay.
Although the largest tau isoforms utilized in this study are the least expressed within the
human brain (~9%, Hong, 1998), the ratio between respective 3R and 4R isoforms with varying
N-termini (e.g. 0N3R and 0N4R) is similar and hence results obtained with 2N3R and
2N4R tau isoforms are expected to be transferable to all other tau isoforms (He et al., 2020).
While both tau variants used in this study impaired microtubule gliding in the modified
kinesin-1-dependent microtubule gliding assay, a 5-fold lower concentration of
2N4R tau-mScarlet than of 2N3R tau-GFP variant was sufficient to slow relative microtubule
gliding speed down to 0.19 ± 0.09 and 0.31 ± 0.13, respectively (Figure 17C). In addition, an
approximately 20-fold higher microtubule binding affinity was measured for 2N4R tau-mScarlet
(Kd = 6.79 ± 0.41 nM) compared to 2N3R tau-GFP (Kd = 137.12 ± 29.7 nM) (Figure 18C, D). In
both cases the binding data was best fitted with a Hill-model fit with a Hill coefficient of

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 81 / 157
2.38 ± 0.67 for 3R tau GFP and 1.83 ± 0.29 for 4R tau mScarlet (Figure 18C). Both coefficients
are larger than 1, suggesting a cooperative binding mechanism for 2N3R and 2N4R tau
isoforms (Weiss, 1997). This means that the binding of tau molecules to microtubules may not
be independent, but that binding of one tau molecule increases the binding-probability of
another tau molecule. While the exact mechanism by which tau binds soluble tubulin or
microtubules is still elusive, Fung et al. have proposed a model for its cooperative binding to
tubulin dimers (Fung et al., 2020), and other recent studies also suggest a cooperative binding
mechanism of 2N4R tau within non diffusive, but dynamic tau condensates on microtubules
(Siahaan et al., 2019; Tan et al., 2019).
Previous studies often used co-sedimentation assays to determine the binding affinity of
3R and 4R tau isoforms to microtubules, and results varied greatly from up to 6 µM down to less
than 100 nM (Butner and Kirschner, 1991; Goode and Feinstein, 1994; Hong, 1998; Ackmann et
al., 2000; Goode et al., 2000). However, bound tau was in those cases assessed by SDS-PAGE
gel analysis, which is not suitable to reliably detect tau concentrations below 1 µM (Gustke et
al., 1994b). And yet, studies performing a much more sensitive fluorescence resonance energy
transfer (FRET) assay or nuclear magnetic resonance (NMR) also reported largely varying
dissociation constants for comparable tau isoforms, ranging from 2.5 µM down to 14 nM
(Makrides et al., 2004; Sillen et al., 2007; Elbaum-Garfinkle et al., 2014; Maïo et al., 2014). Also,
in contrast to these studies, microtubule bundles were excluded from the analysis in this study.
The quantification of dissociation constants for tau hence largely depends on the experimental
procedures and can be compared only to a very limited extent.
However, while the exact Kd-values may vary, these studies consistently reported a
stronger microtubule binding affinity for 4R compared to 3R tau variants, which is consistent
with the herein presented results. This suggests that when the ratio of 4R:3R tau isoforms
changes, the microtubule decoration with tau isoforms and hence the motility of kinesin-1
walking across these microtubules may change as well. To test this hypothesis, we analyzed
the relative microtubule-binding behavior of mixed recombinant human 4R tau-mScarlet to
3R tau-GFP and their isoform-ratio dependent effect on kinesin-1-dependent microtubule
gliding. Increasing 4R:3R tau isoform ratios led to decreased relative microtubule gliding
velocities (Figure 19), suggesting that a shift towards higher 4R tau isoform levels within a
neuron negatively affects axonal transport along microtubules. Interestingly, the fact that
2N4R tau-mScarlet in solution seems to prevent 3R tau-GFP from binding to microtubules, even
at low 4R:3R ratios (Figure 20), suggests that transport defects are caused by a higher
decoration of axonal microtubules with 4R tau isoforms due to its higher roadblock efficiency.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 82 / 157
Together, these results suggest that an increased 4R:3R tau isoform ratio within neurons
expressing the FUS-P525L variant likely contributes to, or might even be sufficient to cause the
axonal transport defects observed in FUS-ALS pathology.
Due to the higher net positive charge of the microtubule binding domain, 4R tau also
shields the negative surface charge of microtubules more efficiently when compared to 3R tau.
This results in a reduced kinesin-1 landing rate and decreased kinein-1 velocities (Parimalam et
al., 2016). In line with these findings in vitro, 0N4R tau largely co-localized with microtubules of
NIH 3T3 cells when co-transfected with differently labeled 0N3R and 0N4R tau isoforms at a 1:1
ratio, while 0N3R tau diffusely distributed to the cytosol (Lu and Kosik, 2001). Studying the
effect of 4R:3R tau isoform imbalances in mouse models is difficult, as 4R tau is the
predominantly expressed isoform in the adult rodent brain while 3R tau is hardly expressed
(Lacovich et al., 2017; Tuerde et al., 2018). Therefore, Lacovich et al. created a trans-splicing
model in neurons differentiated from human embryonic stem cells (hESCs), which allowed for
selective in- and exclusion of exon 10 leading to an increase in relative endogenous levels of 3R
or 4R tau isoforms, respectively. Using this model system, they could show that an increased
level of 3R tau isoforms relative to 4R tau favored anterograde transport of the amyloid
precursor protein (APP), while an increased 4R:3R tau isoform ratio promoted
(dynein-dependent) retrograde transport by decreasing (kinesin-dependent) anterograde
transport velocities (Lacovich et al., 2017). Although these neurons differentiated from hESCs
predominantly express 3R tau isoforms, this data suggests that a shift in 4R:3R tau isoform ratio
is sufficient to interfere with the transport of APP in vivo. Taken together, previous studies in line
with the data presented herein provide clear evidence that a slight shift in 4R:3R tau isoform
ratio, e.g. caused by the nuclear loss of function of FUS-P525L, impairs kinesin-1-dependent
transport on microtubules.
Interference of other tau isoforms with kinesin-1 motility
Although the effect of tau on motor motility has been studied in detail, studies often investigated
only one tau isoform or variant, preventing any direct cross-comparison of tau isoforms and their
ability to interfere with kinesin-1 motility on microtubules. In addition, taxol-dependent
microtubule stabilization or the decoration of microtubules with tau differentially affects kinesin-1
motility. For instance, microtubules assembled in the presence of 0N3R tau glide moderately,
but significantly faster (0.54 ± 0.01 µm/s) compared to microtubules assembled in the presence
of 0N4R tau (0.48 ± 0.01 µm/s) (Yu et al., 2014). Similarly, another study reported a significantly
faster gliding of microtubules assembled in the presence of 0N3R tau (0.52 µm/s) than in the

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 83 / 157
presence of 0N4R tau (0.43 µm/s) (Peck et al., 2011). When microtubules are, however,
assembled in the presence of taxol and subsequently decorated with these tau isoforms, the
gliding velocity is slower in the presence of 0N3R tau (0.29 ± 0.01 µm/s) compared to 0N4R tau
(0.32 ± 0.01 µm/s) at tau concentrations of 100-1000 nM (Yu et al., 2014).
The properties of molecular motors are also often studied in so called stepping assays,
where either single motor molecules or cargo-attached motors are allowed to bind to and
translocate along surface-immobilized microtubules, while they are visualized by TIRF
microscopy. These assays are more suitable to investigate the characteristics of single motors
when encountering proteins interfering with motor motility on microtubules compared to
microtubule gliding assays, where motors act in ensemble to propel microtubules across the
surface. In single molecule stepping assays, kinesin-1 moves at similar velocities in the
presence of 0N3R tau (0.36 ± 0.02 µm/s) or 0N4R tau (0.33 ± 0.02 µm/s), but shows a
decreased run length at these conditions (Vershinin et al., 2007; Dixit et al., 2008; Yu et al.,
2014). Conversely, concentrations of 2N4R tau up to 1.5 µM did not decrease the run length of
recombinant rat kinesin-1, but drastically reduced its landing on microtubules, leading to the
hypothesis that the crucial step in the motility of this kinesin-1 is its attachment to microtubules,
where it translocates undisturbed by tau roadblocks once it is bound (Seitz et al., 2002). A
similar effect of 2N4R tau has been observed in CHO cells. The velocities of a variety of cellular
organelles and vesicles were unaffected upon microinjection of recombinant 2N4R tau into
these cells, but attachment and detachment frequencies were altered causing impaired
transport of these organelles and vesicles throughout the cell (Trinczek et al., 1999).
Interestingly, low concentrations of 2N4R tau (0.25-20 nM) lead to the reversible formation of
cohesive islands or condensates on taxol-stabilized (but not GMP-CPP-stabilized) microtubules
(Siahaan et al., 2019; Tan et al., 2019). These condensates seem to have a kind of regulatory
effect on motor translocation across microtubules, as they are constitute impermeable barriers
for kinesin-1 motors but not for processive dynein-dynactin-BICD2N (DDB) complexes, while
they are disassembled by kinesin-8. Tau islands further protect microtubules from severing
enzymes such as katanin (Siahaan et al., 2019) or spastin (Tan et al., 2019). In vitro studies
suggest that alternative splicing of tau does not seem to affect tau condensate formation on
microtubules, since 2N3R and 0N3R tau readily incorporated into preformed 2N4R tau
condensates (Tan et al., 2019). Further, it has been hypothesized that impaired motor motility
on microtubules upon higher 4R levels in solution is not caused by an overall higher decoration
of microtubules with tau molecules, but due to the replacement of tau molecules with three

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 84 / 157
microtubule binding repeats by those with four repeats, as it has been observed in vivo (Lu and
Kosik, 2001).
Interestingly, the presence of increasing concentrations (1-100 nM) of the longest tau
isoform (2N4R) caused a more subtle decrease the run length of unloaded, GFP-labeled kinesin
in stepping assays, compared to the presence of the shortest isoform (0N3R) (Vershinin et al.,
2007; Dixit et al., 2008), indicating that the projection domain of tau may actually counteract the
roadblock effect caused by tau binding to microtubules and aid in kinesin translocation. In line
with this, Tarhan et al. have investigated all six human tau isoforms for their capacity to act as
roadblocks for bead-attached kinesin-1 motors. Increasing concentrations of 2N4R tau
(0.14 - 140 nM) resulted in a significant decrease of bead velocity and hence kinesin-1
translocation speed. While all 4R tau isoforms had a higher microtubule binding affinity and
decreased kinesin-1 velocity along microtubules stronger compared to their respective 3R tau
N-terminal counterparts (e.g. 0N3R compared to 0N4R), the kinesin-1 velocity increased for tau
isoforms with more N-terminal domains (0N<1N<2N). This suggests that the negatively
charged projection domain of tau aids in recruiting and keeping positively charged kinesin-1
motor domains to the microtubule lattice (Tarhan et al., 2013), and in combination with a
previously proposed sidestepping mechanism allowing kinesin to bypass roadblocks on
microtubules (Dixit et al., 2008; Yildiz et al., 2008; Dreblow et al., 2010). Intriguingly, an
additional N-terminal domain (i.e. in 1N3R tau) was reported to cause a considerably larger
slowdown in microtubule gliding velocity compared to 0N3R tau (on taxol-stabilized
microtubules decorated with 20, 200, or 400 nM of the respective recombinant tau isoform)
(Schmidt et al., 2012).
In light of these sometimes opposing results in previous studies, it would be interesting to
investigate the interference of the different tau isoforms, alone or in combination with each
other, utilized in this present study with individual kinesin-1 motors in the reversed assay
geometry of stepping assays. In addition, the fact that the projection domain of tau may
influence the stepping behavior of kinesin-1 on microtubules suggests that stepping assay in the
presence of different ratios of all six 3R and 4R isoforms would add valuable knowledge as to
how a shift in 4R:3R tau isoform ratio could impair kinesin-1 motility on microtubules within the
cell.
Batch-to-batch differences in purified recombinant 2N4R tau variants
Initial experiments of this study were performed with recombinant human 2N4R tau-GFP, kindly
provided by Dr. Marcus Braun from the Institute of Biotechnology CAS in Prague, while later

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 85 / 157
experiments were performed with 2N4R tau-mScarlet, purified by myself. Both batches exhibit
significantly different inhibitory potential in the kinesin-1-dependent microtubule gliding assay.
While kinesin-1-dependent microtubule gliding completely ceased in the presence of 118 nM of
the 2N4R tau-GFP variant (Figure 10), about 10-fold more protein (1200 nM) of the
2N4R tau-mScarlet variant was needed to reduce the relative microtubule gliding velocity to
0.18 ± 0.09 (Figure 17). In order to determine to what degree different batches of tau differ in
their inhibitory potential and to exclude variations due to the different fluorophores, two more
2N4R tau batches, i.e. 2N4R tau-mCherry and 2N4R tau-GFP, were purified and tested in the
modified kinesin-1-dependent microtubule gliding assay (Figure S2). Significantly larger
amounts of 2N4R tau-mScarlet (1200 nM), -mCherry (1500 nM), or –GFP (1500 nM, second
batch) were required to reduce microtubule gliding velocity down to 0.18 ± 0.09, 0.15 ± 0.01, or
0.44 ± 0.01, respectively. However, the qualitative effect on kinesin-1 motility was the same for
all tested tau isoform variants, since the addition of increasing amounts of tau consistently
decreased kinesin-1-dependent microtubule gliding velocity. Together with above discussed
previous studies, where the concentration of tau used ranges from nM to mM, it indicates that
the inhibitory potential of tau varies between different purification batches (Trinczek et al., 1999;
Seitz et al., 2002; Vershinin et al., 2007; Dixit et al., 2008; Peck et al., 2011; Schmidt et al.,
2012; Tarhan et al., 2013; Yu et al., 2014; Siahaan et al., 2019; Tan et al., 2019).
The absolute amount of a protein needed to reproduce an effect or degree of activity often
differs between individual batches of purified recombinant protein. During the purification of the
protein of interest, co-purification of endogenous proteins (in this case from insect cells)
commonly occurs, while the composition and amount of these co-purified proteins differs from
batch to batch (Structural Genomics Consortium et al., 2008). Such proteins could include
MAPs or motors, which are unlabeled and hence invisible for widefield-epifluorescence, but
would interfere with kinesin-1 motility in a microtubule gliding assay. Further, intrinsically
disordered proteins like tau (KrishnaKumar and Gupta, 2017) tend to form aggregates, a
process often aggravated by long term storage of proteins (Chakraborty et al., 2018).
Aggregated tau likely impairs kinesin-1 motility on microtubules differently compared to
non-aggregated, soluble tau. In addition, the activity of tau strongly depends on its
phosphorylation state. The microtubule binding affinity of tau is decreased upon its
hyperphosphorylation (Ballatore et al., 2012). When purified from insect cell, tau is heavily
phosphorylated (Tepper et al., 2014), although the exact degree of phosphorylation is not
consistent between individual purifications. Hence, different phosphorylation states between the

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 86 / 157
earlier purified 2N4R tau-GFP and the later purified 2N4R tau-GFP or -mScarlet, which likely
influence its ability to hinder kinesin-1 motility on microtubules, cannot be excluded.
The up- and downsides of using Taxol in combination with tau
The modified kinesin-1-dependent microtubule gliding assay presented in this study used in
vitro polymerized microtubules that were stabilized by the addition of taxol. Previous studies
showed that taxol can migrate through 2-nm pores within the microtubule wall, where it binds to
the luminal face of β-tubulin (Meurer-Grob et al., 2001; Li et al., 2002; Kar et al., 2003; Ross and
Fygenson, 2003) and counteracts the effect of GTP hydrolysis (i.e. tubulin destabilization) on
the outward facing side of this β-tubulin molecule (Amos and Löwe, 1999; Prota et al., 2013).
Interestingly, recent reports suggest that tau molecules occupy a similar, if not the same binding
site in the microtubule lumen as taxol (Kar et al., 2003; Ross et al., 2004; Yu et al., 2014), with
the N-terminus of tau projecting outside the microtubule wall (Amos, 2004a, 2004b), a process
which primarily occurs if microtubules are assembled in the presence of tau and is thought to be
irreversible (Makrides et al., 2004). This indicates that, because microtubules in the herein used
assay were stabilized with taxol before tau addition, some binding sites for tau on the
microtubules were likely already occupied by taxol molecules, which would result in less tau
molecules interacting with the microtubule lattice. However, the incorporation of tau into the
microtubule lumen is expected to be especially important for the stabilization of dynamic
microtubules, and only to a lesser extent for the interaction with other MAPs such as motors. In
addition, while tau was found to induce cooperative binding of Taxol to microtubules, the inverse
was not observed as binding of tau to microtubules was independent of the presence or
absence of Taxol during microtubule assembly (Ross et al., 2004). Tau has also been found to
reversibly bind to the outer surface of microtubules (Makrides et al., 2004; Martinho et al.,
2018), along as well as across filaments (Santarella et al., 2004; Duan et al., 2017), a process
more likely to influence microtubule-dependent transport. Hence, the stabilization of
microtubules with taxol before tau addition, as performed in the herein presented experiments,
is considered to have only minor impact on the herein measured microtubule gliding velocities.
Tau isoform expression profile in iPSC-derived neuronal cultures
Intriguingly, whole cell lysates obtained from patient-specific iPSC-derived spinal motor neurons
expressing either the wildtype FUS-GFP or the FUS-P525L-GFP variant did not differentially
interfere with kinesin-1-dependent microtubule gliding (Figure 13B). Due to the cytoplasmic
mislocalization of FUS-P525L-GFP, the 4R:3R tau isoform ratio is expected to be increased in
lysates from cells expressing this variant. Hence we would expect the relative microtubule

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 87 / 157
gliding velocity to decrease in lysates from neurons expressing FUS-P525L-GFP compared to
lysates from cells expressing wildtype FUS-GFP. Therefore, the level of total 3R and 4R tau
isoforms in the used whole cell lysates was determined by western blot using 3R or 4R isoform
specific antibodies, respectively (Figure 21).
However, only the shortest, 0N3R isoform could be detected in cell lysates irrespective of
the expressed FUS variant, indicating that all other isoforms are not expressed above the
detection limit of this technique (low pg range, Wiltfang et al., 1991). This is in line with other
studies on iPSC-derived neurons (Iovino et al., 2015; Verheyen et al., 2015, 2018; Biswas et al.,
2016; Imamura et al., 2016; Silva et al., 2016; Hallmann et al., 2017; Sato et al., 2018) and
reflects the relative immaturity of these reprogrammed neurons, since 0N3R tau is the only
isoform expressed in the fetal human brain (Kosik et al., 1989; Goedert and Jakes, 1990). While
other tau isoforms can be detected on the RNA level in iPSC-derived neurons (Ehrlich et al.,
2015), detection of the 0N4R, 1N3R, and 1N4R isoforms on the protein level is only possible
when neurons are allowed to mature for up to 365 days (Iovino et al., 2015; Sposito et al.,
2015). In fact, recent evidence suggests that iPSC-derived neurons display transcriptional
profiles similar to those found in human fetal brains rather than adult brains (Handel et al., 2016;
Sobol et al., 2019; Griesi-Oliveira et al., 2020).
Taken together, we have to assume that the whole cell lysates used in this study, obtained
from iPSC-derived spinal motor neurons at day 21 of maturation, did not contain endogenous
tau isoforms other than the shortest, fetal 0N3R isoform due to the relative immaturity of
differentiated neurons. Although, previous studies reported defects in axonal transport in
mitochondria in the iPSC-derived spinal motor neurons as early as day 21 of maturation
(Naumann et al., 2018). This implies that the axonal transport defects observed in iPSC-derived
spinal motor neurons are not caused by a shift in tau isoform splicing. However, further
evidence on the transcriptional and protein level would be needed to consolidate this
hypothesis.
In order to obtain more mature neurons, the time that neurons are allowed to differentiate
in culture could be increased, while the course of tau isoform expression levels within these
neurons after defined time points should be evaluated. Other studies have demonstrated the
culture of iPSC-derived spinal motor neurons up to almost 130 days (Devlin et al., 2015), when
the fetal 0N3R tau isoform is however still predominantly expressed (Iovino et al., 2015; Sposito
et al., 2015). Unfortunately, with increasing culture time, iPSC-derived spinal motor neurons
tend to form clusters over time and detach from the herein used PLO-laminin coated surface.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 88 / 157
Another recently evolved method to obtain disease- or patient-specific neurons, and to
circumvent the rejuvenation or reset in maturation state during the production of iPSCs, is direct
programming of neuronal subtypes from human somatic cells. A number of somatic cell types,
including mouse and human fibroblasts, but also astrocyte and human retina-derived fibroblasts,
can be transformed into a variety of neuronal subtypes using different combinations of
overexpressed transcription factors, small molecules or the application of microRNAs (reviewed
in Liou et al., 2020).
In addition, cells cultured in a 3D culture system have recently proven to relate to the
characteristics, including transcriptional and expression profiles, of their in vivo counterparts
more closely compared to cells grown in conventional 2D culture systems (Kapałczyńska et al.,
2016; Duval et al., 2017). Especially the reconstruction of retinal development and regeneration
has been extensively studied using organoids created in a 3D culture of human iPSCs (Eiraku
et al., 2011; Zhong et al., 2014; Hallam et al., 2018; Shrestha et al., 2019). However, 3D culture
systems have also been used to model AD and have enhanced the maturation of iPSC-derived
cortical neurons. Interestingly, neurons grown in this 3D culture exhibit an increased RNA
expression level of 4R tau isoforms already after 49 days in culture (Choi et al., 2014). A
systemic analysis of tau isoform protein expression levels in a 3D culture system revealed an
expression of 0N3R, 0N4R, 1N3R and 1N4R tau isoforms after 15 weeks in culture (105 days),
while 2N3R and 2N4R tau isoforms were expressed after 25 weeks in culture (175 days). As in
the 2D culture system though, the 0N3R tau isoform remained the predominantly expressed
isoform throughout all time points (Miguel et al., 2019).
Although the generation of mature spinal motor neurons can hence be improved through
the above discussed culture conditions, the tau isoform expression profile within these human
iPSC-derived neuronal subtypes presents the major limitation for studying the interference of an
increased 4R:3R tau isoform ratio on kinesin-1-dependent axonal transport. However, the fact
that defects in axonal transport have been observed in iPSC-derived neurons at early stages of
maturation indicates that those defects must be due to another, non-tau related pathogenesis.
4.4 FUS variants may indirectly affect microtubule-based axonal transport
by acting on a multitude of cellular processes
As FUS affects multiple cellular pathways, cytoplasmic mislocalized ALS-associated FUS may
also indirectly interfere with microtubule-based axonal transport. Due to its diverse roles in DNA
damage repair (Bertrand et al., 1999), RNA metabolism and transcriptional regulation (Yang et

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 89 / 157
al., 1998), and mRNA transport within the cell (Zinszner et al., 1997), mutant FUS variants can
trigger many, not mutually exclusive, pathological events within the cell, which in turn may
indirectly affect axonal transport.
Nuclear loss of function - DNA damage repair and RNA splicing
Wildtype FUS localizes to sites of laser-induced DNA damage prior to the recruitment of other
key proteins involved in DNA-repair, whereas recruitment of FUS-P525L to sites of
laser-induced DNA damage is impaired in iPSC-derived spinal motor neurons, subsequently
leading to defects in mitochondrial transport (Naumann et al., 2018). Interestingly, the inhibition
of PARG, which degrades the PAR chains attracting FUS to DNA damage (Lin et al., 1997),
rescued not only FUS recruitment to laser-induced DNA damage sites, but also mitochondrial
transport defects, suggesting that impaired DNA damage repair causes defects in axonal
transport (Naumann et al., 2018). In addition, it was shown that DNA damage enhances
cytoplasmic mislocalization of FUS-P525L and induces FUS aggregation as well as distal
axonal trafficking defects (especially of mitochondria) and subsequent distal axonopathy (dying
back of axons) (Higelin et al., 2016; Naumann et al., 2018). This supports the notion that defects
in DNA-repair mechanisms due to cytoplasmic mislocalization of FUS inevitably result in the
long-term accumulation of DNA damage, which coincides with the rather late mean age of onset
in ALS. As neurons lack the ability to self-renew or replicate, they are particularly susceptible
DNA damage accumulating over time which consequently likely causes disruptions in other
cellular pathways, such as axonal transport (Guo et al., 2020).
Next to role in DNA damage repair, FUS is also involved in the expression and splicing of
a variety of motor proteins (Colombrita et al., 2012), such as KIF5B. Accordingly, cytoplasmic
mislocalized mutant FUS variants (i.e. FUS-R521G and FUS-R521H) can no longer partake in
proper splicing of these motors (Hoell et al., 2011). Additionally, FUS binds to mRNAs coding for
β-actin and the actin-stabilizing protein Nd1-L (Fujii and Takumi, 2005). Altered splicing caused
by mutant FUS variants hence likely directly affects cytoskeletal composition, which might affect
axonal transport.
Endosomal trafficking and axonal transport
Clear evidence for how FUS is involved in both, short- and long-distance cellular transport is to
date still sparse. Only few studies have focused on the direct influence of FUS variants on the
axonal transport machinery. One study found that several FUS mutant variants (G230C, R495X,
and R521G) disrupt bidirectional fast axonal transport via activation of the mitogen-activated

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 90 / 157
protein kinase p38 when expressed in squid axoplasm and in mouse primary motor neurons
(Sama 2017). p38 phosphorylates kinesin-1 and thereby inhibits its translocation along
microtubules, possibly by reducing the landing rate on microtubules (Morfini et al., 2013).
Apart from long-distance transport along the dendritic arbor or axon, short-distance
transport of vesicles, e.g. from the ER to the Golgi apparatus, was also found to be impaired in
FUS-ALS. In particular, Rab-1-dependent transport of coat protein complex II-coated vesicles
from the ER to the Golgi was found to be disrupted upon expression of several FUS-NLS
mutant variants (but not wildtype FUS) (Soo et al., 2015). The same study also showed
inhibition of Golgi-associated vesicular trafficking. Defects in axonal transport of ER-vesicles
was also observed in spinal motor neurons that were differentiated from ALS-patient-derived
iPSCs carrying the FUS-R521H or FUS-P525L variants (Guo et al., 2017b). Interestingly, mice
expressing a frame-shift mutant FUS variant did not show major deficits in endosomal trafficking
(Sleigh et al., 2020), indicating that impaired transport mechanisms in FUS-ALS might not affect
endosomal trafficking in this model.
Mitochondrial damage and trafficking defects
Mutant FUS variants have extensively been linked to the impaired transport of larger organelles,
such as mitochondria. Defective mitochondrial transport may result from the fact that FUS
regulated the expression of mRNAs coding for several motor proteins, such as KIF5C and
KIF1B (Schwarz, 2013; Campbell et al., 2014; Guo et al., 2020). An increase in stationary
mitochondria was observed upon expression of human FUS-P525L, but also wildtype FUS, in
mouse cortical neurons. The same was observed for the fly homologues cabezaP398L and
Cabeza in Drosophila motor neurons (Baldwin et al., 2016; Chen et al., 2016a), suggesting that
overexpression and hence increased cytoplasmic abundance of either FUS variant might
already impair mitochondrial axonal transport. Similarly, the number of motile mitochondria was
significantly decreased in ALS-patient-specific iPSC-derived spinal motor neurons expressing
FUS-R521H or FUS-P525L when compared to isogenic controls (Guo et al., 2017b).
Interestingly , mitochondrial transport defects could be rescued by HDAC6 inhibition, leading to
increased α-tubulin acetylation and hence stabilizing microtubule protofilaments (Janke and
Montagnac, 2017). In addition, an arrest of mitochondria (and lysosome) motility was observed
in the distal axon of ALS-patient specific iPSC-derived spinal motor neurons expressing the
FUS-P525L variant, accompanied by a breakdown in membrane potential and reduced
mitochondrial length (Naumann et al., 2018). ATP production ceases upon the breakdown of
mitochondrial membrane potential, leading to a local depletion of energy around the damaged

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 91 / 157
mitochondrion (Stoica et al., 2016). A recent study has linked ALS-associated FUS-P525L to
aberrant mitophagy, since the expression of members of the mitochondrial quality control PINK1
and Parkin, as well as ubiquitination of Miro1, is increased in cells expressing this FUS variant
(Park et al., 2006; Sarraf et al., 2013; Chen et al., 2016a, 2016b). This indicates a higher degree
of mitophagy in the presence of FUS-P525L, which also leads to a reduced ATP production.
ATP depletion has been reported to lead to microtubule depolymerization (Park et al., 2013) and
may also locally impair axonal transport due to the lack of the motor’s energy source. Further,
ATP has recently been identified to inhibit fibrillization of pathological FUS aggregates by
specifically binding to its RRM domain (Kang et al., 2019). This implicates that mitochondrial
damage might not only affect axonal transport through ATP depletion, but also by worsening
pathogenic effects of aggregated FUS.
RNP and stress granules
mRNAs assemble with RBPs into ribonucleoprotein (RNP) granules, which are transported
through the axon by active bi-directional transport (Knowles et al., 1996; Tiruchinapalli et al.,
2003; Leung et al., 2006; Nalavadi et al., 2012; Alami et al., 2014; Medioni et al., 2014; Gopal et
al., 2017; Wong et al., 2017; De Graeve and Besse, 2018; Turner-Bridger et al., 2018;
Vijayakumar et al., 2019). Several studies have previously demonstrated that the transport of
RNPs containing FUS depends on the interaction with other RNA transport-related proteins,
such as TDP-43 and SMN (Groen et al., 2013), IMP1 (Kamelgarn et al., 2016), Sam 68 (Belly et
al., 2005), or the adenomatous polyposis coli (APC) tumor-suppressor protein, which targets
β2B-tubulin mRNA to the plus end of detyrosinated microtubules where APC promotes tubulin
synthesis (Mili et al., 2008). Perturbing this interaction leads to disrupted dynamic microtubules
within axonal growth cones (Yasuda et al., 2013; Preitner et al., 2014), and suggests that
structural changes in the cytoskeleton might be a potential cause for axonal transport defects.
Other studies have proposed that RNP granules hitchhike on membrane-bound organelles
when those are transported throughout the cell (Jansen et al., 2014; Salogiannis and Reck-
Peterson, 2017). For instance, recent work has demonstrated a role for kinesin- and
dynein-dependent endosome trafficking in mRNA transport (Gould and Lippincott-Schwartz,
2009; Baumann et al., 2012; Pohlmann et al., 2015). The lysosome has been proposed as
another cellular organelle acting as a platform for RNP granule transport, with coupling possibly
mediated by Annexin A11 (Markmiller et al., 2018; Liao et al., 2019). It will be interesting to
investigate whether mutant FUS variants show perturbed interaction with membrane-bound
organelles and thereby hinder the transport of RNP granules.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 92 / 157
After liquid-to-solid phase transition (Murakami et al., 2015; Patel et al., 2015; Bowden
and Dormann, 2016), solidified granules have lost their dynamic properties and can hence no
longer perform their physiological functions (Bowden and Dormann, 2016). The incorporation of
ALS-associated FUS variants into RNP or stress granules and their subsequent pathogenic
phase transition indicates that FUS may impact axonal transport in several distinct, but not
mutually exclusive ways. These include the sequestration of mRNAs and proteins directly
involved in the axonal transport machinery or inhibition of translation of proteins important for
cell survival, such as antiapoptotic factors under prolonged stress conditions (Nevins et al.,
2003). Additionally, when stress granules increase in size (Baron et al., 2013) to form
pathological aggregates, e.g. through the continuous incorporation of RBPs and mRNA, they
might sterically impair cellular processes, i.e. cytoskeletal assembly or axonal transport. A
similar process has been described due to the swelling of mitochondria, which impaired
mitochondrial and lysosomal transport in rat primary neurons (Kaasik et al., 2007).
While it is known that the cytoplasmic localization of FUS on its own is not sufficient to
trigger FUS aggregation or inclusion into RNP and stress granules (Bosco et al., 2010;
Dormann et al., 2010; Ito et al., 2011; Bentmann et al., 2012), there is no clear consensus yet
as to which domain is mainly responsible for the association of FUS with stress granules. Some
evidence points towards its low-complexity, prion-like domain (Sun et al., 2011b; Han et al.,
2012), while others suggest the methylation of arginine residues within the RGG domain RGG
domain(s) (Bentmann et al., 2012; Baron et al., 2013) to be essential for incorporation
(Rappsilber et al., 2003; Dormann et al., 2012). However, its capacity to bind mRNA is clearly
essential for FUS incorporation into granules, as mutations in FUS abolishing mRNA-binding
greatly reduce aggregate-related toxicity (Andersson et al., 2008; Elden et al., 2010; Sun et al.,
2011b; Bentmann et al., 2012; Daigle et al., 2013). This led to the hypothesis that FUS might
undergo conformational changes upon binding to mRNA (Wang et al., 2015; Hamad et al.,
2020), after which it might be capable to interact with an extended set of proteins including
motor proteins or tubulin. It would hence be interesting to see whether microtubule gliding
characteristics would change or whether FUS aggregation occurs in our kinesin-1-dependent
microtubule gliding assay in the presence of sufficient amounts of mRNA transcripts that are
known to interact with FUS.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 93 / 157
4.5 Outlook
The present results strongly indicate that FUS does not directly interfere with kinesin-1 or
microtubules. However, it is possible that structurally changed FUS protein or its aggregate
form, e.g. induced by its interaction with mRNAs, may alter its potential to interact with motors or
cytoskeletal filaments. It will therefore be interesting to test whether FUS interferes with
kinesin-1-dependent transport in the presence of mRNA. Further, may not only interfere with
microtubule-associated motors, but also with actin-associated motors, such as myosin. For both
motor types, possible interactions with FUS might be masked in a gliding assay due to lacking
accessibility of motors. Hence, both scenarios should additionally be investigated in stepping
assays, where motors are more accessible.
Axonal transport in this study was reconstituted by modifying a kinesin-1-dependent
microtubule gliding assay to be compatible with protein rich solutions such as whole cell lysates.
Cell lysates of iPSC-derived spinal motor neurons expressing wildtype FUS-GFP or
FUS-P525L-GFP did not differentially interfere with kinesin-1-dependent microtubule gliding.
However, kinesin-1 motility was more strongly impaired in the presence of cell lysates
containing endogenous 0N3R tau and supplemented with 2N4R tau-GFP compared to the
presence of 2N4R tau-GFP alone, indicating that different tau isoform levels in cell lysates can
potentially be detected using the modified kinesin-1-dependent microtubule gliding assay.
However, further evidence would be needed to confirm this hypothesis. For instance, lysates of
primary cells could be tested in this assay, e.g. using a mouse model of tauopathies (such as
FTD or progressive supranuclear palsy, where differences in tau isoform composition are a
major hallmark of the disease (Hu et al., 2007).
The precise impact of increasing 2N4R:2N3R tau isoform ratios on individual motors as
well as the contribution of the N-terminal projection domain of tau on kinesin-1 motility has to be
investigated in further stepping assays using different ratios of shorter tau isoforms (i.e. 0N, 1N).
Of particular interest in future experiments should be the interplay of these different isoforms
present at different ratios with microtubules in a gliding or stepping assay, rather than the
influence of each isoform alone.

Mechanisms of Axonal Transport in ALS Discussion
Anne Seifert 94 / 157
4.6 Conclusion
Defects in axonal transport are one of the major pathological events observed early in FUS-ALS
and other neurodegenerative diseases. The exact cause and mechanism of these defects in
neurons expressing the mutant FUS-P525L variant is largely unknown to date. This study
provides evidence that neither wildtype FUS nor the mutant FUS-P525L variant directly
interferes with kinesin-1-dependent axonal transport. Further, the presented results suggest that
neither FUS variant impedes kinesin-1 motility by interacting with adaptor proteins present in
whole cell lysates of ALS patient-specific iPSC-derived spinal motor neurons.
We hence hypothesized that mislocalization of FUS-P525L to the cytoplasm corresponds
to a loss of FUS-dependent splicing activity of tau mRNA. This leads to an increased ratio of
4R:3R tau isoforms within neurons expressing this FUS mutant, affecting the kinesin-1 motility
on microtubules. In line with recent literature, the presented results show that an increased
4R:3R tau isoform ratio is indeed sufficient to impede microtubule-dependent kinesin-1 motility.
Due to the 20-fold higher microtubule binding affinity of 2N4R tau compared to 2N3R tau, the
4R tau isoform efficiently displaces the 3R tau isoform from the microtubule lattice at saturating
tau concentrations. Since 4R tau affects kinesin-1 motility more strongly than 3R tau, an
increase in the 4R:3R tau isoform ratio results in a 4R dose dependent reduction of the
observed gliding velocity.
Finally, defects in axonal transport are just one of multiple pathological processes
occurring in neurons expressing ALS-associated FUS-P525L or other mutant FUS variants.
These are not mutually exclusive, but rather interdependent and strongly connected. Hence,
one malfunctioning cellular process might affect other essential processes, thereby processively
worsening ALS pathogenesis. Whether defects in axonal transport are one of the first events
caused by mutant FUS variant expression, leading to harmful disturbances in other cellular
pathways, or whether it arises as a consequence of prior pathogenic developments, remains to
be investigated in future studies. In summary, we conclude that the axonal transport defects
seen in FUS-associated ALS arise indirectly as a consequence of FUS-P525L involvement in
one or several of the above discussed cellular mechanisms such as DNA damage repair, RNA
metabolism (especially splicing of tau), endosomal trafficking, mitochondrial homeostasis, and
stress granule dynamics.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 95 / 157
References Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilić J, Pekarik V, Tiscornia G, Edel M, Boué S, et al. 2008. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26:1276–1284.
Abe K, Aoki M, Tsuji S, Itoyama Y, Sobue G, Togo M, Hamada C, Tanaka M, Akimoto M, Nakamura K, Takahashi F, Kondo K, et al. 2017. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. The Lancet Neurology 16:505–512.
Abeliovich A, Gitler AD. 2016. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539:207–216.
Ackmann M, Wiech H, Mandelkow E. 2000. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J Biol Chem 275:30335–30343.
Aitken CE, Marshall RA, Puglisi JD. 2008. An Oxygen Scavenging System for Improvement of Dye Stability in Single-Molecule Fluorescence Experiments. Biophysical Journal 94:1826–1835.
Akhmedov AT, Bertrand P, Corteggiani E, Lopez BS. 1995. Characterization of two nuclear mammalian homologous DNA-pairing activities that do not require associated exonuclease activity. Proc Natl Acad Sci USA 92:1729–1733.
Akiyama T, Suzuki N, Ishikawa M, Fujimori K, Sone T, Kawada J, Funayama R, Fujishima F, Mitsuzawa S, Ikeda K, Ono H, Shijo T, et al. 2019. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine 45:362–378.
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, et al. 2014. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81:536–543.
Alliegro MC, Alliegro MA. 1996. A Nuclear Protein Regulated during the Transition from Active to Quiescent Phenotype in Cultured Endothelial Cells. Developmental Biology 174:288–297.
Aman P, Panagopoulos I, Lassen C, Fioretos T, Mencinger M, Toresson H, Höglund M, Forster A, Rabbitts TH, Ron D, Mandahl N, Mitelman F. 1996. Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 37:1–8.
Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M. 2017. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun 8:275.
Amos LA. 2004a. Microtubule structure and its stabilisation. Org Biomol Chem 2:2153.
Amos LA. 2004b. Bending at Microtubule Interfaces. Chemistry & Biology 11:745–747.
Amos LA, Löwe J. 1999. How Taxol stabilises microtubule structure. Chem Biol 6:R65-69.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 96 / 157
Andersson MK, Ståhlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O, Aman P. 2008. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 9:37.
Andreadis A. 2005. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1739:91–103.
Arendt T, Stieler J, Strijkstra AM, Hut RA, Rüdiger J, Van der Zee EA, Harkany T, Holzer M, Härtig W. 2003. Reversible Paired Helical Filament-Like Phosphorylation of Tau Is an Adaptive Process Associated with Neuronal Plasticity in Hibernating Animals. J Neurosci 23:6972–6981.
Asbury CL. 2005. Kinesin: world’s tiniest biped. Curr Opin Cell Biol 17:89–97.
Atsumi T. 1981. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol 55:193–198.
Aulas A, Vande Velde C. 2015. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Frontiers in Cellular Neuroscience 9:423.
Avila J. 2010. Intracellular and extracellular tau. Front Neurosci 4:.
Baas PW, Ahmad FJ. 2013. Beyond taxol: microtubule-based treatment of disease and injury of the nervous system. Brain 136:2937–2951.
Baas PW, Deitch JS, Black MM, Banker GA. 1988. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci USA 85:8335–8339.
Bachand GD, Bouxsein NF, VanDelinder V, Bachand M. 2014. Biomolecular motors in nanoscale materials, devices, and systems. Wiley Interdiscip Rev Nanomed Nanobiotechnol 6:163–177.
Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS, Akhmedov AT. 1999. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem 274:34337–34342.
Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ. 2016. Axonal transport defects are a common phenotype in Drosophila models of ALS. Human Molecular Genetics 25:2378–2392.
Ballatore C, Brunden KR, Huryn DM, Trojanowski JQ, Lee VM-Y, Smith AB. 2012. Microtubule stabilizing agents as potential treatment for Alzheimer’s disease and related neurodegenerative tauopathies. J Med Chem 55:8979–8996.
Bao J, Huck D, Gunther LK, Sellers JR, Sakamoto T. 2013. Actin Structure-Dependent Stepping of Myosin 5a and 10 during Processive Movement. PLoS ONE 8:e74936.
Baron DM, Kaushansky LJ, Ward CL, Sama RRK, Chian R-J, Boggio KJ, Quaresma AJC, Nickerson JA, Bosco DA. 2013. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol Neurodegener 8:30.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 97 / 157
Basu S, Lamprecht R. 2018. The Role of Actin Cytoskeleton in Dendritic Spines in the Maintenance of Long-Term Memory. Front Mol Neurosci 11:143.
Baumann S, Pohlmann T, Jungbluth M, Brachmann A, Feldbrügge M. 2012. Kinesin-3 and dynein mediate microtubule-dependent co-transport of mRNPs and endosomes. J Cell Sci 125:2740–2752.
Belfield A, Goldberg DM. 1968. Inhibition of the nucleotidase effect of alkaline phosphatase by beta-glycerophosphate. Nature 219:73–75.
Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y. 2005. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett 379:152–157.
Bensimon G, Lacomblez L, Meininger V. 1994. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N Engl J Med 330:585–591.
Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. 2012. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem 287:23079–23094.
Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L. 1996. hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. The EMBO Journal 15:5022–5031.
Bertolotti A, Melot T, Acker J, Vigneron M, Delattre O, Tora L. 1998. EWS, but not EWS-FLI-1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol 18:1489–1497.
Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS. 1999. Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 18:4515–4521.
Bhandari R, Kuhad A, Kuhad A. 2018. Edaravone: a new hope for deadly amyotrophic lateral sclerosis. Drugs Today 54:349.
Bieling P, Laan L, Schek H, Munteanu EL, Sandblad L, Dogterom M, Brunner D, Surrey T. 2007. Reconstitution of a microtubule plus-end tracking system in vitro. Nature 450:1100–1105.
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. 2010. Deficits in axonal transport precede ALS symptoms in vivo. Proceedings of the National Academy of Sciences of the United States of America 107:20523–20528.
Biswas MHU, Almeida S, Lopez-Gonzalez R, Mao W, Zhang Z, Karydas A, Geschwind MD, Biernat J, Mandelkow E-M, Futai K, Miller BL, Gao F-B. 2016. MMP-9 and MMP-2 Contribute to Neuronal Cell Death in iPSC Models of Frontotemporal Dementia with MAPT Mutations. Stem Cell Reports 7:316–324.
Black MM. 2016. Axonal transport: The orderly motion of axonal structures. Methods Cell Biol 131:1–19.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 98 / 157
Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I. 1996. Tau Is Enriched on Dynamic Microtubules in the Distal Region of Growing Axons. J Neurosci 16:3601–3619.
Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ. 2007. Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol 176:11–17.
Böhm KJ, Steinmetzer P, Daniel A, Baum M, Vater W, Unger E. 1997. Kinesin-driven microtubule motility in the presence of alkaline-earth metal ions: indication for a calcium ion-dependent motility. Cell Motil Cytoskeleton 37:226–231.
Böhm KJ, Stracke R, Baum M, Zieren M, Unger E. 2000. Effect of temperature on kinesin-driven microtubule gliding and kinesin ATPase activity. FEBS letters 466:59–62.
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. 2006. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 312:1389–1392.
Boron WF, Boulpaep EL. 2005. Medical Physiology: A Cellular and Molecular Approach. Philadelphia, Pa: Elsevier Saunders. 1319 p.
Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Sapp P, McKenna-Yasek D, Brown RH, Hayward LJ. 2010. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19:4160–4175.
Bourefis A-R, Campanari M-L, Buee-Scherrer V, Kabashi E. 2020. Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS. Neurobiology of Disease 142:104935.
Bowden HA, Dormann D. 2016. Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem 138:112–133.
Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72:248–254.
Brady PN, Macnaughtan MA. 2015. Evaluation of colorimetric assays for analyzing reductively methylated proteins: Biases and mechanistic insights. Anal Biochem 491:43–51.
Brandt R, Lee G. 1993. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J Biol Chem 268:3414–3419.
Braun M, Lansky Z, Fink G, Ruhnow F, Diez S, Janson ME. 2011. Adaptive braking by Ase1 prevents overlapping microtubules from sliding completely apart. Nat Cell Biol 13:1259–1264.
Braun M, Lansky Z, Szuba A, Schwarz FW, Mitra A, Gao M, Lüdecke A, Wolde PR ten, Diez S. 2017. Changes in microtubule overlap length regulate kinesin-14-driven microtubule sliding. Nat Chem Biol 13:1245–1252.
Brion J-P, Flament-Durand J, Dustin P. 1986. Alzheimers’s disease and tau proteins. The Lancet 328:1098.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 99 / 157
Brouhard GJ, Stear JH, Noetzel TL, Al-Bassam J, Kinoshita K, Harrison SC, Howard J, Hyman AA. 2008. XMAP215 is a processive microtubule polymerase. Cell 132:79–88.
Brunden KR, Yao Y, Potuzak JS, Ferrer NI, Ballatore C, James MJ, Hogan A-ML, Trojanowski JQ, Smith AB, Lee VM-Y. 2011. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol Res 63:341–351.
Bugiel M, Schäffer E. 2018. Three-Dimensional Optical Tweezers Tracking Resolves Random Sideward Steps of the Kinesin-8 Kip3. Biophysical Journal 115:1993–2002.
Bukar Maina M, Al-Hilaly YK, Serpell LC. 2016. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 6:9.
Butner KA, Kirschner MW. 1991. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol 115:717–730.
Campbell PD, Shen K, Sapio MR, Glenn TD, Talbot WS, Marlow FL. 2014. Unique Function of Kinesin Kif5A in Localization of Mitochondria in Axons. Journal of Neuroscience 34:14717–14732.
Canter RG, Penney J, Tsai L-H. 2016. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539:187–196.
Castle MJ, Perlson E, Holzbaur EL, Wolfe JH. 2014. Long-distance axonal transport of AAV9 is driven by dynein and kinesin-2 and is trafficked in a highly motile Rab7-positive compartment. Mol Ther 22:554–566.
Castoldi M, Popov AV. 2003. Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expression and Purification 32:83–88.
Chakraborty M, Tarasovetc EV, Grishchuk EL. 2018. In vitro reconstitution of lateral to end-on conversion of kinetochore–microtubule attachments. Methods in Cell Biology, Elsevier, p 307–327.
Charcot J-M, Joffroy A. 1869. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere. Arch Physiol Neurol Pathol 2:744–760.
Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn LW, Huang C-L, Errigo A, Yin Y, Lu J, Ayala M, et al. 2014. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14:796–809.
Chen J, Kanai Y, Cowan NJ, Hirokawa N. 1992. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360:674–677.
Chen M, Li Y, Yang M, Chen X, Chen Y, Yang F, Lu S, Yao S, Zhou T, Liu J, Zhu L, Du S, et al. 2016a. A new method for quantifying mitochondrial axonal transport. Protein Cell 7:804–819.
Chen S, Sayana P, Zhang X, Le W. 2013. Genetics of amyotrophic lateral sclerosis: An update. Molecular neurodegeneration 8:28.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 100 / 157
Chen Y, Deng J, Wang P, Yang M, Chen X, Zhu L, Liu J, Lu B, Shen Y, Fushimi K, Xu Q, Wu JY. 2016b. PINK1 and Parkin are genetic modifiers for FUS-induced neurodegeneration. Hum Mol Genet 25:5059–5068.
Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, et al. 2014. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515:274–278.
Chung C-H, Golub EE, Forbes E, Tokuoka T, Shapiro IM. 1992. Mechanism of action of ?-glycerophosphate on bone cell mineralization. Calcif Tissue Int 51:305–311.
Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. 2012. TDP-43 and FUS RNA-binding Proteins Bind Distinct Sets of Cytoplasmic Messenger RNAs and Differently Regulate Their Post-transcriptional Fate in Motoneuron-like Cells. J Biol Chem 287:15635–15647.
Compton SJ, Jones CG. 1985. Mechanism of dye response and interference in the Bradford protein assay. Analytical Biochemistry 151:369–374.
Conte A, Lattante S, Zollino M, Marangi G, Luigetti M, Del Grande A, Servidei S, Trombetta F, Sabatelli M. 2012. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscular disorders : NMD 22:73–75.
Cooper GM. 2000a. The Cell: A Molecular Approach. Washington, DC: ASM Press [u.a.]. 689 p.
Cooper GM. 2000b. Microtubules. The Cell: A Molecular Approach, 2. ede. Washington, DC: ASM Press [u.a.],.
Correia SC, Perry G, Moreira PI. 2016. Mitochondrial traffic jams in Alzheimer’s disease - pinpointing the roadblocks. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1862:1909–1917.
Coy DL, Wagenbach M, Howard J. 1999. Kinesin takes one 8-nm step for each ATP that it hydrolyzes. J Biol Chem 274:3667–3671.
Crozat A, Aman P, Mandahl N, Ron D. 1993. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363:640–644.
Daigle JG, Lanson NA, Smith RB, Casci I, Maltare A, Monaghan J, Nichols CD, Kryndushkin D, Shewmaker F, Pandey UB. 2013. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Human Molecular Genetics 22:1193–1205.
De Graeve F, Besse F. 2018. Neuronal RNP granules: from physiological to pathological assemblies. Biol Chem 399:623–635.
De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ. 2008. Role of Axonal Transport in Neurodegenerative Diseases. Annu Rev Neurosci 31:151–173.
De Vos KJ, Hafezparast M. 2017a. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol Dis 105:283–299.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 101 / 157
De Vos KJ, Hafezparast M. 2017b. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiology of disease 105:283–299.
De Vos M, Schreiber V, Dantzer F. 2012. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochemical Pharmacology 84:137–146.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, et al. 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256.
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607.
Deng J, Yang M, Chen Y, Chen X, Liu J, Sun S, Cheng H, Li Y, Bigio EH, Mesulam M, Xu Q, Du S, et al. 2015. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet 11:e1005357.
Deng Q, Holler CJ, Taylor G, Hudson KF, Watkins W, Gearing M, Ito D, Murray ME, Dickson DW, Seyfried NT, Kukar T. 2014. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J Neurosci 34:7802–7813.
Dentler W, Witman G, American Society for Cell Biology. 1995. Cilia and Flagella. San Diego: Academic Press.
Deveau H, Garneau JE, Moineau S. 2010. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol 64:475–493.
Devlin A-C, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S, Miles GB. 2015. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun 6:5999.
Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. 2012. TDP-43 Aggregation In Neurodegeneration: Are Stress Granules The Key? Brain Research 1462:16–25.
Díaz JF, Valpuesta JM, Chacón P, Diakun G, Andreu JM. 1998. Changes in microtubule protofilament number induced by Taxol binding to an easily accessible site. Internal microtubule dynamics. J Biol Chem 273:33803–33810.
Dixit R, Ross JL, Goldman YE, Holzbaur ELF. 2008. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science (New York, NY) 319:1086–1089.
Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao P-C, Qiu Y, Zhao Y, Tsai L-H. 2013. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci 16:1008–1015.
Donnelly CJ, Grima JC, Sattler R. 2014. Aberrant RNA homeostasis in amyotrophic lateral sclerosis: potential for new therapeutic targets? Neurodegener Dis Manag 4:417–437.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 102 / 157
Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IRA, Neumann M, Haass C. 2012. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J 31:4258–4275.
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IRA, Capell A, Schmid B, Neumann M, Haass C. 2010. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857.
Dorst J, Chen L, Rosenbohm A, Dreyhaupt J, Hübers A, Schuster J, Weishaupt JH, Kassubek J, Gess B, Meyer T, Weyen U, Hermann A, et al. 2019. Prognostic factors in ALS: a comparison between Germany and China. J Neurol 266:1516–1525.
Douvaras P, Rusielewicz T, Kim KH, Haines JD, Casaccia P, Fossati V. 2016. Epigenetic Modulation of Human Induced Pluripotent Stem Cell Differentiation to Oligodendrocytes. Int J Mol Sci 17:.
D’Ovidio F, Rooney JPK, Visser AE, Manera U, Beghi E, Logroscino G, Vermeulen RCH, Veldink JH, Berg LH van den, Hardiman O, Chiò A, Euro-MOTOR consortium. 2019. Association between alcohol exposure and the risk of amyotrophic lateral sclerosis in the Euro-MOTOR study. J Neurol Neurosurg Psychiatry 90:11–19.
Downing KH, Nogales E. 1998. Tubulin structure: insights into microtubule properties and functions. Curr Opin Struct Biol 8:785–791.
Dreblow K, Kalchishkova N, Böhm KJ. 2010. Kinesin passing permanent blockages along its protofilament track. Biochemical and Biophysical Research Communications 395:490–495.
Drewes G, Ebneth A, Preuss U, Mandelkow E-M, Mandelkow E. 1997. MARK, a Novel Family of Protein Kinases That Phosphorylate Microtubule-Associated Proteins and Trigger Microtubule Disruption. Cell 89:297–308.
D’Souza I, Schellenberg GD. 2005. Regulation of tau isoform expression and dementia. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1739:104–115.
D’Souza VM, Bareford LM, Ray A, Swaan PW. 2006. Cytoskeletal scaffolds regulate riboflavin endocytosis and recycling in placental trophoblasts. J Nutr Biochem 17:821–829.
Du K, Arai S, Kawamura T, Matsushita A, Kurokawa R. 2011. TLS and PRMT1 synergistically coactivate transcription at the survivin promoter through TLS arginine methylation. Biochem Biophys Res Commun 404:991–996.
Duan AR, Jonasson EM, Alberico EO, Li C, Scripture JP, Miller RA, Alber MS, Goodson HV. 2017. Interactions between Tau and Different Conformations of Tubulin: Implications for Tau Function and Mechanism. J Mol Biol 429:1424–1438.
Dubey M, Chaudhury P, Kabiru H, Shea TB. 2008. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil Cytoskeleton 65:89–99.
Duval K, Grover H, Han L-H, Mou Y, Pegoraro AF, Fredberg J, Chen Z. 2017. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 32:266–277.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 103 / 157
Dye RB, Fink SP, Williams RC. 1993. Taxol-induced flexibility of microtubules and its reversal by MAP-2 and Tau. J Biol Chem 268:6847–6850.
Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E-M, Mandelkow E. 1998. Overexpression of Tau Protein Inhibits Kinesin-dependent Trafficking of Vesicles, Mitochondria, and Endoplasmic Reticulum: Implications for Alzheimer’s Disease. The Journal of Cell Biology 143:777–794.
Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, Aoi T, Watanabe A, et al. 2012. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med 4:145ra104.
Ehrlich M, Hallmann A-L, Reinhardt P, Araúzo-Bravo MJ, Korr S, Röpke A, Psathaki OE, Ehling P, Meuth SG, Oblak AL, Murrell JR, Ghetti B, et al. 2015. Distinct Neurodegenerative Changes in an Induced Pluripotent Stem Cell Model of Frontotemporal Dementia Linked to Mutant TAU Protein. Stem Cell Reports 5:83–96.
Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y. 2011. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472:51–56.
Elbaum-Garfinkle S, Cobb G, Compton JT, Li X-H, Rhoades E. 2014. Tau mutants bind tubulin heterodimers with enhanced affinity. Proceedings of the National Academy of Sciences 111:6311–6316.
Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, et al. 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075.
Emdad L, D’Souza SL, Kothari HP, Qadeer ZA, Germano IM. 2012. Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev 21:404–410.
Encalada SE, Szpankowski L, Xia C, Goldstein LSB. 2011. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell 144:551–565.
Erkkinen MG, Kim M-O, Geschwind MD. 2018. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb Perspect Biol 10:a033118.
Eyer J, Peterson A. 1994. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-beta-galactosidase fusion protein. Neuron 12:389–405.
Fang F, Ingre C, Roos P, Kamel F, Piehl F. 2015. Risk factors for amyotrophic lateral sclerosis. CLEP 181.
Farhadi L, Fermino Do Rosario C, Debold EP, Baskaran A, Ross JL. 2018. Active Self-Organization of Actin-Microtubule Composite Self-Propelled Rods. Front Phys 6:75.
Felgner H, Frank R, Schliwa M. 1996. Flexural rigidity of microtubules measured with the use of optical tweezers. J Cell Sci 109:509.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 104 / 157
Fernández-Nogales M, Lucas JJ. 2020. Altered Levels and Isoforms of Tau and Nuclear Membrane Invaginations in Huntington’s Disease. Front Cell Neurosci 13:574.
Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. 2011. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7:616–630.
Fink G, Hajdo L, Skowronek KJ, Reuther C, Kasprzak AA, Diez S. 2009. The mitotic kinesin-14 Ncd drives directional microtubule-microtubule sliding. Nat Cell Biol 11:717–723.
Fischer J, Ganellin CR. 2006. Analogue-Based Drug Discovery. Weinheim: Wiley-VCH.
Fletcher DA, Theriot JA. 2004. An introduction to cell motility for the physical scientist. Physical biology 1:T1-10.
Fountoulakis M, Juranville J-F, Manneberg M. 1992. Comparison of the Coomassie brilliant blue, bicinchoninic acid and Lowry quantitation assays, using non-glycosylated and glycosylated proteins. Journal of Biochemical and Biophysical Methods 24:265–274.
Friede RL, Samorajski T. 1970. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec 167:379–387.
Fu M, Holzbaur ELF. 2014. Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends in Cell Biology 24:564–574.
Fu M-M, Nirschl JJ, Holzbaur ELF. 2014. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell 29:577–590.
Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. 2005. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15:587–593.
Fujii R, Takumi T. 2005. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118:5755–5765.
Fujioka Y, Ishigaki S, Masuda A, Iguchi Y, Udagawa T, Watanabe H, Katsuno M, Ohno K, Sobue G. 2013. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci Rep 3:2388.
Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature 374:555–559.
Fung HYJ, McKibben KM, Ramirez J, Gupta K, Rhoades E. 2020. Structural Characterization of Tau in Fuzzy Tau:Tubulin Complexes. Structure 28:378-384.e4.
Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. 2011. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging 32:2323.e27–40.
Georgiou CD, Grintzalis K, Zervoudakis G, Papapostolou I. 2008. Mechanism of Coomassie brilliant blue G-250 binding to proteins: a hydrophobic assay for nanogram quantities of proteins. Anal Bioanal Chem 391:391–403.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 105 / 157
Gerbino V, Carrì MT, Cozzolino M, Achsel T. 2013. Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol Dis 55:120–128.
Gessner T, Mayer U. 2000. Triarylmethane and Diarylmethane Dyes. In: Wiley-VCH Verlag GmbH & Co. KGaA, editor. Ullmann’s Encyclopedia of Industrial Chemistry, Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, p a27_179.
Gesztelyi R, Zsuga J, Kemeny-Beke A, Varga B, Juhasz B, Tosaki A. 2012. The Hill equation and the origin of quantitative pharmacology. Arch Hist Exact Sci 66:427–438.
Goedert M, Jakes R. 1990. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9:4225–4230.
Goedert M, Jakes R. 2005. Mutations causing neurodegenerative tauopathies. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1739:240–250.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. 1989a. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3:519–526.
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. 1989b. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. The EMBO journal 8:393–399.
Goldstein LSB. 2012. Axonal transport and neurodegenerative disease: Can we see the elephant? Progress in Neurobiology 99:186–190.
Goode B, Feinstein S. 1994. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. Journal of Cell Biology 124:769–782.
Goode BL, Chau M, Denis PE, Feinstein SC. 2000. Structural and Functional Differences between 3-Repeat and 4-Repeat Tau Isoforms: IMPLICATIONS FOR NORMAL TAU FUNCTION AND THE ONSET OF NEURODEGENERATIVE DISEASE. J Biol Chem 275:38182–38189.
Gopal PP, Nirschl JJ, Klinman E, Holzbaur ELF. 2017. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc Natl Acad Sci USA 114:E2466–E2475.
Gorbunova V, Seluanov A, Mao Z, Hine C. 2007. Changes in DNA repair during aging. Nucleic Acids Res 35:7466–7474.
Gordon JA. 1991. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in enzymology 201:477–482.
Gornall AG, Bardawill CJ, David MM. 1949. Determination of serum proteins by means of the biuret reaction. J Biol Chem 177:751–766.
Gornstein EL, Schwarz TL. 2017. Neurotoxic mechanisms of paclitaxel are local to the distal axon and independent of transport defects. Exp Neurol 288:153–166.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 106 / 157
Gould GW, Lippincott-Schwartz J. 2009. New roles for endosomes: from vesicular carriers to multi-purpose platforms. Nat Rev Mol Cell Biol 10:287–292.
Gozes I, Walker MD, Kaye AM, Littauer UZ. 1977. Synthesis of tubulin and actin by neuronal and glial nuclear preparations from developing rat brain. J Biol Chem 1819–1825.
Grafstein B, Forman DS. 1980. Intracellular transport in neurons. Physiological reviews 60:1167–1283.
Griesi-Oliveira K, Fogo MS, Pinto BGG, Alves AY, Suzuki AM, Morales AG, Ezquina S, Sosa OJ, Sutton GJ, Sunaga-Franze DY, Bueno AP, Seabra G, et al. 2020. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol Psychiatry.
Groen EJN, Es MA van, Vught PWJ van, Spliet WGM, Engelen-Lee J van, Visser M de, Wokke JHJ, Schelhaas HJ, Ophoff RA, Fumoto K, Pasterkamp RJ, Dooijes D, et al. 2010. FUS Mutations in Familial Amyotrophic Lateral Sclerosis in the Netherlands. Arch Neurol 67:.
Groen EJN, Fumoto K, Blokhuis AM, Engelen-Lee J, Zhou Y, Heuvel DMA van den, Koppers M, Diggelen F van, Heest J van, Demmers JAA, Kirby J, Shaw PJ, et al. 2013. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet 22:3690–3704.
Gross SP. 2004. Hither and yon: a review of bi-directional microtubule-based transport. Phys Biol 1:R1-11.
Gross SP, Vershinin M, Shubeita GT. 2007. Cargo transport: two motors are sometimes better than one. Curr Biol 17:R478-486.
Guo T, Noble W, Hanger DP. 2017a. Roles of tau protein in health and disease. Acta Neuropathol 133:665–704.
Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R, Ordovás L, Patel A, Welters M, Vanwelden T, Geens N, Tricot T, et al. 2017b. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nature Communications 8:861.
Guo W, Stoklund Dittlau K, Van Den Bosch L. 2020. Axonal transport defects and neurodegeneration: Molecular mechanisms and therapeutic implications. Seminars in Cell & Developmental Biology 99:133–150.
Gustke N, Trinczek B, Biernat J, Mandelkow E-M, Mandelkow E. 1994a. Domains of tau Protein and Interactions with Microtubules. Biochemistry 33:9511–9522.
Gustke N, Trinczek B, Biernat J, Mandelkow E-M, Mandelkow E. 1994b. Domains of tau Protein and Interactions with Microtubules. Biochemistry 33:9511–9522.
Gutierrez-Beltran E, Moschou PN, Smertenko AP, Bozhkov PV. 2015. Tudor Staphylococcal Nuclease Links Formation of Stress Granules and Processing Bodies with mRNA Catabolism in Arabidopsis. Plant Cell 27:926–943.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 107 / 157
Hackl W, Lührmann R. 1996. Molecular cloning and subcellular localisation of the snRNP-associated protein 69KD, a structural homologue of the proto-oncoproteins TLS and EWS with RNA and DNA-binding properties. J Mol Biol 264:843–851.
Hackney DD. 1995. Highly processive microtubule-stimulated ATP hydrolysis by dimeric kinesin head domains. Nature 377:448–450.
Hagiwara H, Sunada Y. 2004. Mechanism of taxane neurotoxicity. Breast Cancer 11:82–85.
Hagiwara H, Yorifuji H, Sato-Yoshitake R, Hirokawa N. 1994. Competition between motor molecules (kinesin and cytoplasmic dynein) and fibrous microtubule-associated proteins in binding to microtubules. J Biol Chem 269:3581–3589.
Hall DH, Hedgecock EM. 1991. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell 65:837–847.
Hallam D, Hilgen G, Dorgau B, Zhu L, Yu M, Bojic S, Hewitt P, Schmitt M, Uteng M, Kustermann S, Steel D, Nicholds M, et al. 2018. Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency: Generating Light Responsive Retinal Organoids from iPSC. Stem Cells 36:1535–1551.
Hallier M, Lerga A, Barnache S, Tavitian A, Moreau-Gachelin F. 1998. The Transcription Factor Spi-1/PU.1 Interacts with the Potential Splicing Factor TLS. J Biol Chem 273:4838–4842.
Hallmann A-L, Araúzo-Bravo MJ, Mavrommatis L, Ehrlich M, Röpke A, Brockhaus J, Missler M, Sterneckert J, Schöler HR, Kuhlmann T, Zaehres H, Hargus G. 2017. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci Rep 7:42991.
Hamacher-Brady A, Brady NR. 2016. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 73:775–795.
Hamad N, Mashima T, Yamaoki Y, Kondo K, Yoneda R, Oyoshi T, Kurokawa R, Nagata T, Katahira M. 2020. RNA sequence and length contribute to RNA-induced conformational change of TLS/FUS. Sci Rep 10:2629.
Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL. 2012. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149:768–779.
Handel AE, Chintawar S, Lalic T, Whiteley E, Vowles J, Giustacchini A, Argoud K, Sopp P, Nakanishi M, Bowden R, Cowley S, Newey S, et al. 2016. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum Mol Genet 25:989–1000.
Hardie DGeditor. 1993. Protein Phosphorylation: A Practical Approach. Oxford ; New York: Oxford University Press. 300 p.
Hariadi RF, Appukutty AJ, Sivaramakrishnan S. 2016. Engineering Circular Gliding of Actin Filaments Along Myosin-Patterned DNA Nanotube Rings To Study Long-Term Actin-Myosin Behaviors. ACS Nano 10:8281–8288.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 108 / 157
Harlow E, Lane D. 2006. Immunoprecipitation: Lysing yeast cells using glass beads. CSH protocols 2006:.
He Z, McBride JD, Xu H, Changolkar L, Kim S, Zhang B, Narasimhan S, Gibbons GS, Guo JL, Kozak M, Schellenberg GD, Trojanowski JQ, et al. 2020. Transmission of tauopathy strains is independent of their isoform composition. Nat Commun 11:7.
Heemels M-T. 2016. Neurodegenerative diseases. Nature 539:179.
Hefti MM, Farrell K, Kim S, Bowles KR, Fowkes ME, Raj T, Crary JF. 2018. High-resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS ONE 13:e0195771.
Helenius J, Brouhard G, Kalaidzidis Y, Diez S, Howard J. 2006. The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature 441:115–119.
Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F. 2011. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331:928–931.
Hendricks AG, Perlson E, Ross JL, Schroeder HW, Tokito M, Holzbaur ELF. 2010. Motor Coordination Via Tug-Of-War Mechanism Drives Bidirectional Vesicle Transport. Current biology : CB 20:697–702.
Hernández-Vega A, Braun M, Scharrel L, Jahnel M, Wegmann S, Hyman BT, Alberti S, Diez S, Hyman AA. 2017. Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Rep 20:2304–2312.
Herold C, Leduc C, Stock R, Diez S, Schwille P. 2012. Long-Range Transport of Giant Vesicles along Microtubule Networks. ChemPhysChem 13:1001–1006.
Hess H, Saper G. 2018. Engineering with Biomolecular Motors. Acc Chem Res 51:3015–3022.
Hesse WR, Steiner M, Wohlever ML, Kamm RD, Hwang W, Lang MJ. 2013. Modular Aspects of Kinesin Force Generation Machinery. Biophysical Journal 104:1969–1978.
Higelin J, Demestre M, Putz S, Delling JP, Jacob C, Lutz A-K, Bausinger J, Huber A-K, Klingenstein M, Barbi G, Speit G, Huebers A, et al. 2016. FUS Mislocalization and Vulnerability to DNA Damage in ALS Patients Derived hiPSCs and Aging Motoneurons. Front Cell Neurosci 10:290.
Hill AV. 1910. The possible effects of the aggregation of the molecules of hæmoglobin on its dissociation curves. The Journal of Physiology 40:iv–vii.
Hirano A, Donnenfeld H, Sasaki S, Nakano I. 1984a. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 43:461–470.
Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G. 1984b. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 43:471–480.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 109 / 157
Hirokawa N, Niwa S, Tanaka Y. 2010. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 68:610–638.
Hirokawa N, Noda Y, Tanaka Y, Niwa S. 2009. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10:682–696.
Hirokawa N, Shiomura Y, Okabe S. 1988. Tau proteins: the molecular structure and mode of binding on microtubules. J Cell Biol 107:1449–1459.
Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. 2011. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18:1428–1431.
Hoeprich GJ, Thompson AR, McVicker DP, Hancock WO, Berger CL. 2014. Kinesin’s neck-linker determines its ability to navigate obstacles on the microtubule surface. Biophysical journal 106:1691–1700.
Hong M. 1998. Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary FTDP-17. Science 282:1914–1917.
Hong W, Takshak A, Osunbayo O, Kunwar A, Vershinin M. 2016. The Effect of Temperature on Microtubule-Based Transport by Cytoplasmic Dynein and Kinesin-1 Motors. Biophysical journal 111:1287–1294.
Howard J. 2001. Mechanics of Motor Proteins and the Cytoskeleton. Sunderland, Mass: Sinauer Associates, Publishers. 367 p.
Howard J, Hudspeth AJ, Vale RD. 1989. Movement of microtubules by single kinesin molecules. Nature 342:154–158.
Howard WD, Timasheff SN. 1988. Linkages between the effects of taxol, colchicine, and GTP on tubulin polymerization. J Biol Chem 263:1342–1346.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832.
Hu WT, Parisi JE, Knopman DS, Boeve BF, Dickson DW, Ahlskog JE, Petersen RC, Josephs KA. 2007. Clinical Features and Survival of 3R and 4R Tauopathies Presenting as Behavioral Variant Frontotemporal Dementia: Alzheimer Disease & Associated Disorders 21:S39–S43.
Hua W, Young EC, Fleming ML, Gelles J. 1997. Coupling of kinesin steps to ATP hydrolysis. Nature 388:390–393.
Huai J, Zhang Z. 2019. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front Neurol 10:527.
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang X-F, Yao T-P. 2002. HDAC6 is a microtubule-associated deacetylase. Nature 417:455–458.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 110 / 157
Ikenaka K, Katsuno M, Kawai K, Ishigaki S, Tanaka F, Sobue G. 2012. Disruption of axonal transport in motor neuron diseases. Int J Mol Sci 13:1225–1238.
Imamura K, Sahara N, Kanaan NM, Tsukita K, Kondo T, Kutoku Y, Ohsawa Y, Sunada Y, Kawakami K, Hotta A, Yawata S, Watanabe D, et al. 2016. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci Rep 6:34904.
Immanuel D, Zinszner H, Ron D. 1995. Association of SARFH (sarcoma-associated RNA-binding fly homolog) with regions of chromatin transcribed by RNA polymerase II. Mol Cell Biol 15:4562–4571.
Inoue D, Mahmot B, Kabir AMR, Farhana TI, Tokuraku K, Sada K, Konagaya A, Kakugo A. 2015. Depletion force induced collective motion of microtubules driven by kinesin. Nanoscale 7:18054–18061.
Ionov L, Stamm M, Diez S. 2006. Reversible Switching of Microtubule Motility Using Thermoresponsive Polymer Surfaces. Nano Lett 6:1982–1987.
Iovino M, Agathou S, González-Rueda A, Del Castillo Velasco-Herrera M, Borroni B, Alberici A, Lynch T, O’Dowd S, Geti I, Gaffney D, Vallier L, Paulsen O, et al. 2015. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138:3345–3359.
Ishigaki S, Fujioka Y, Okada Y, Riku Y, Udagawa T, Honda D, Yokoi S, Endo K, Ikenaka K, Takagi S, Iguchi Y, Sahara N, et al. 2017. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Reports 18:1118–1131.
Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K. 2012. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2:529.
Ishigaki S, Sobue G. 2018. Importance of Functional Loss of FUS in FTLD/ALS. Front Mol Biosci 5:44.
Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. 2011. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69:152–162.
Jaiswal MK. 2019. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev 39:733–748.
Janke C, Montagnac G. 2017. Causes and Consequences of Microtubule Acetylation. Current Biology 27:R1287–R1292.
Jansen R-P, Niessing D, Baumann S, Feldbrügge M. 2014. mRNA transport meets membrane traffic. Trends Genet 30:408–417.
Japtok J, Lojewski X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J, Putz S, Demestre M, Boeckers TM, Ludolph AC, Liebau S, Storch A, et al. 2015. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiology of disease 82:420–429.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 111 / 157
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821.
Kaan HYK, Hackney DD, Kozielski F. 2011. The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science 333:883–885.
Kaasik A, Safiulina D, Choubey V, Kuum M, Zharkovsky A, Veksler V. 2007. Mitochondrial swelling impairs the transport of organelles in cerebellar granule neurons. The Journal of biological chemistry 282:32821–32826.
Kamelgarn M, Chen J, Kuang L, Arenas A, Zhai J, Zhu H, Gal J. 2016. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1862:2004–2014.
Kanai Y, Dohmae N, Hirokawa N. 2004. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 43:513–525.
Kang J, Lim L, Song J. 2019. ATP binds and inhibits the neurodegeneration-associated fibrillization of the FUS RRM domain. Commun Biol 2:223.
Kapałczyńska M, Kolenda T, Przybyła W, Zajączkowska M, Teresiak A, Filas V, Ibbs M, Bliźniak R, Łuczewski Ł, Lamperska K. 2016. 2D and 3D cell cultures – a comparison of different types of cancer cell cultures. aoms.
Kar S, Fan J, Smith MJ, Goedert M, Amos LA. 2003. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J 22:70–77.
Kardon JR, Vale RD. 2009. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol 10:854–865.
Kellermayer MSZ. 1997. Delayed dissociation of in vitro moving actin filaments from heavy meromyosin induced by low concentrations of Triton X-100. Biophysical Chemistry 67:199–210.
Kempf M, Clement A, Faissner A, Lee G, Brandt R. 1996. Tau Binds to the Distal Axon Early in Development of Polarity in a Microtubule- and Microfilament-Dependent Manner. J Neurosci 16:5583–5592.
Kharkwal H, Smith CG, Wilson DW. 2014. Blocking ESCRT-Mediated Envelopment Inhibits Microtubule-Dependent Trafficking of Alphaherpesviruses In Vitro. Journal of Virology 88:14467–14478.
Khatoon S, Grundke-Iqbal I, Iqbal K. 1992. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem 59:750–753.
Kim SH, Shanware NP, Bowler MJ, Tibbetts RS. 2010. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. The Journal of biological chemistry 285:34097–34105.
King SMeditor. 2012. Dyneins: Structure, Biology and Disease. Amsterdam ; Boston: Academic Press. 639 p.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 112 / 157
Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N. 2011. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res 39:2781–2798.
Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, Mellin C, Merkle FT, et al. 2014. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14:781–795.
Kleele T, Marinković P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F, Bishop D, Herms J, et al. 2014. An assay to image neuronal microtubule dynamics in mice. Nat Commun 5:4827.
Knowles RB, Sabry JH, Martone ME, Deerinck TJ, Ellisman MH, Bassell GJ, Kosik KS. 1996. Translocation of RNA granules in living neurons. J Neurosci 16:7812–7820.
Kolawole OM, Lau WM, Khutoryanskiy VV. 2019. Chitosan/β-glycerophosphate in situ gelling mucoadhesive systems for intravesical delivery of mitomycin-C. International Journal of Pharmaceutics: X 1:100007.
Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. 1993. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268:24374–24384.
Korten T, Chaudhuri S, Tavkin E, Braun M, Diez S. 2016. Kinesin-1 Expressed in Insect Cells Improves Microtubule in Vitro Gliding Performance, Long-Term Stability and Guiding Efficiency in Nanostructures. IEEE transactions on nanobioscience 15:62–69.
Korten T, Diez S. 2008. Setting up roadblocks for kinesin-1: mechanism for the selective speed control of cargo carrying microtubules. Lab Chip 8:1441–1447.
Korten T, Månsson A, Diez S. 2010. Towards the application of cytoskeletal motor proteins in molecular detection and diagnostic devices. Current Opinion in Biotechnology 21:477–488.
Korten T, Tavkin E, Scharrel L, Kushwaha VS, Diez S. 2018. An automated in vitro motility assay for high-throughput studies of molecular motors. Lab Chip 18:3196–3206.
Kosik KS, Orecchio LD, Bakalis S, Neve RL. 1989. Developmentally regulated expression of specific tau sequences. Neuron 2:1389–1397.
KrishnaKumar VG, Gupta S. 2017. Simplified method to obtain enhanced expression of tau protein from E. coli and one-step purification by direct boiling. Preparative Biochemistry & Biotechnology 47:530–538.
Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, Linker SB, Pham S, Erwin JA, Miller JA, Hodge R, McCarthy JK, et al. 2016. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc 11:499–524.
Krohn RI. 2002. The colorimetric detection and quantitation of total protein. Curr Protoc Cell Biol Appendix 3:Appendix 3H.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 113 / 157
Ksiezak-Reding H, Liu W-K, Yen S-H. 1992. Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Research 597:209–219.
Kumar N. 1981. Taxol-induced polymerization of purified tubulin. Mechanism of action. J Biol Chem 256:10435–10441.
Kuramitsu T. 1999. Enhancement of kinesin-driven microtubule gliding by bile acids. Hepatology Research 15:124–135.
Kuroi K, Shimozuma K. 2004. Neurotoxicity of taxanes: Symptoms and quality of life assessment. Breast Cancer 11:92–99.
Kuta A, Deng W, Morsi El-Kadi A, Banks GT, Hafezparast M, Pfister KK, Fisher EMC. 2010. Mouse cytoplasmic dynein intermediate chains: identification of new isoforms, alternative splicing and tissue distribution of transcripts. PLoS ONE 5:e11682.
Kwan AC, Dombeck DA, Webb WW. 2008. Polarized microtubule arrays in apical dendrites and axons. Proc Natl Acad Sci USA 105:11370–11375.
Kwiatkowski TJ, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, et al. 2009. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 323:1205–1208.
Lacovich V, Espindola SL, Alloatti M, Pozo Devoto V, Cromberg LE, Čarná ME, Forte G, Gallo J-M, Bruno L, Stokin GB, Avale ME, Falzone TL. 2017. Tau Isoforms Imbalance Impairs the Axonal Transport of the Amyloid Precursor Protein in Human Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 37:58–69.
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling S-C, Liang TY, Mazur C, Wancewicz E, Kim AS, et al. 2012. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15:1488–1497.
Lam AT, VanDelinder V, Kabir AMR, Hess H, Bachand GD, Kakugo A. 2016. Cytoskeletal motor-driven active self-assembly in in vitro systems. Soft Matter 12:988–997.
Landry MP, McCall PM, Qi Z, Chemla YR. 2009. Characterization of Photoactivated Singlet Oxygen Damage in Single-Molecule Optical Trap Experiments. Biophysical Journal 97:2128–2136.
Lecanda F, Avioli LV, Cheng SL. 1997. Regulation of bone matrix protein expression and induction of differentiation of human osteoblasts and human bone marrow stromal cells by bone morphogenetic protein-2. J Cell Biochem 67:386–396.
Leighton DJ, Newton J, Stephenson LJ, Colville S, Davenport R, Gorrie G, Morrison I, Swingler R, Chandran S, Pal S, CARE-MND Consortium. 2019. Changing epidemiology of motor neurone disease in Scotland. J Neurol 266:817–825.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 114 / 157
Lemaitre RP, Bogdanova A, Borgonovo B, Woodruff JB, Drechsel DN. 2019. FlexiBAC: a versatile, open-source baculovirus vector system for protein expression, secretion, and proteolytic processing. BMC Biotechnol 19:20.
Leung K-M, Horck FPG van, Lin AC, Allison R, Standart N, Holt CE. 2006. Asymmetrical beta-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat Neurosci 9:1247–1256.
Li H, DeRosier DJ, Nicholson WV, Nogales E, Downing KH. 2002. Microtubule Structure at 8 Å Resolution. Structure 10:1317–1328.
Li X, Decker M, Westendorf JJ. 2010. TEThered to Runx: Novel binding partners for runx factors. Blood Cells, Molecules, and Diseases 45:82–85.
Li Y, Adamek P, Zhang H, Tatsui CE, Rhines LD, Mrozkova P, Li Q, Kosturakis AK, Cassidy RM, Harrison DS, Cata JP, Sapire K, et al. 2015. The Cancer Chemotherapeutic Paclitaxel Increases Human and Rodent Sensory Neuron Responses to TRPV1 by Activation of TLR4. J Neurosci 35:13487.
Li YR, King OD, Shorter J, Gitler AD. 2013. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201:361–372.
Liao Y-C, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, Patel R, Qamar S, Nixon-Abell J, Shen Y, Meadows W, Vendruscolo M, et al. 2019. RNA Granules Hitchhike on Lysosomes for Long-Distance Transport, Using Annexin A11 as a Molecular Tether. Cell 179:147-164.e20.
Lin W, Amé JC, Aboul-Ela N, Jacobson EL, Jacobson MK. 1997. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J Biol Chem 272:11895–11901.
Lindwall G, Cole RD. 1984. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259:5301–5305.
Liou RH-C, Edwards TL, Martin KR, Wong RC-B. 2020. Neuronal Reprogramming for Tissue Repair and Neuroregeneration. IJMS 21:4273.
Liu X, Niu C, Ren J, Zhang J, Xie X, Zhu H, Feng W, Gong W. 2013. The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochim Biophys Acta 1832:375–385.
Lo KY, Kuzmin A, Unger SM, Petersen JD, Silverman MA. 2011. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci Lett 491:168–173.
Lomen-Hoerth C. 2011. Clinical Phenomenology and Neuroimaging Correlates in ALS-FTD. J Mol Neurosci 45:656–662.
Longinetti E, Fang F. 2019. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol 32:771–776.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 115 / 157
Longinetti E, Regodón Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi L-O, Kierkegaard M, Andersen PM, Hillert J, Fang F, Ingre C. 2018. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener 19:528–537.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275.
Lu M, Kosik KS. 2001. Competition for Microtubule-binding with Dual Expression of Tau Missense and Splice Isoforms. MBoC 12:171–184.
Mackall J, Meredith M, Lane MD. 1979. A mild procedure for the rapid release of cytoplasmic enzymes from cultured animal cells. Analytical biochemistry 95:270–274.
Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur ELF. 2014. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 84:292–309.
Maday S, Wallace KE, Holzbaur ELF. 2012. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196:407–417.
Maïo I, Barbier P, Allegro D, Brault C, Peyrot V. 2014. Quantitative Analysis of Tau-Microtubule Interaction Using FRET. IJMS 15:14697–14714.
Makrides V, Massie MR, Feinstein SC, Lew J. 2004. Evidence for two distinct binding sites for tau on microtubules. Proc Natl Acad Sci USA 101:6746–6751.
Mallik R, Petrov D, Lex SA, King SJ, Gross SP. 2005. Building complexity: an in vitro study of cytoplasmic dynein with in vivo implications. Curr Biol 15:2075–2085.
Mandelkow E-M, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. 1995. Tau domains, phosphorylation, and interactions with microtubules. Neurobiology of Aging 16:355–362.
Manjaly ZR, Scott KM, Abhinav K, Wijesekera L, Ganesalingam J, Goldstein LH, Janssen A, Dougherty A, Willey E, Stanton BR, Turner MR, Ampong M-A, et al. 2010. The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotroph Lateral Scler 11:439–442.
Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo E-C, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, et al. 2018. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172:590-604.e13.
Marrone L, Drexler HCA, Wang J, Tripathi P, Distler T, Heisterkamp P, Anderson EN, Kour S, Moraiti A, Maharana S, Bhatnagar R, Belgard TG, et al. 2019. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathologica 138:67–84.
Martinho M, Allegro D, Huvent I, Chabaud C, Etienne E, Kovacic H, Guigliarelli B, Peyrot V, Landrieu I, Belle V, Barbier P. 2018. Two Tau binding sites on tubulin revealed by thiol-disulfide exchanges. Sci Rep 8:13846.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 116 / 157
Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. 2013. The RNA-binding Protein Fused in Sarcoma (FUS) Functions Downstream of Poly(ADP-ribose) Polymerase (PARP) in Response to DNA Damage. J Biol Chem 288:24731–24741.
Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, Lee HO, Carra S, Hyman AA, Alberti S. 2017. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J 36:1669–1687.
Matsuo ES, Shin R-W, Billingsley ML, Van deVoorde A, O’Connor M, Trojanowski JQ, Lee VMY. 1994. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 13:989–1002.
Matus S, Medinas DB, Hetz C. 2014. Common Ground: Stem Cell Approaches Find Shared Pathways Underlying ALS. Cell Stem Cell 14:697–699.
Maury Y, Côme J, Piskorowski RA, Salah-Mohellibi N, Chevaleyre V, Peschanski M, Martinat C, Nedelec S. 2015. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol 33:89–96.
McGeer PL, McGeer EG. 2002. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 26:459–470.
McKenney RJ, Huynh W, Tanenbaum ME, Bhabha G, Vale RD. 2014. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 345:337–341.
Medioni C, Ramialison M, Ephrussi A, Besse F. 2014. Imp promotes axonal remodeling by regulating profilin mRNA during brain development. Curr Biol 24:793–800.
Mehta AD, Rief M, Spudich JA, Smith DA, Simmons RM. 1999. Single-molecule biomechanics with optical methods. Science 283:1689–1695.
Meissner M, Lopato S, Gotzmann J, Sauermann G, Barta A. 2003. Proto-oncoprotein TLS/FUS is associated to the nuclear matrix and complexed with splicing factors PTB, SRm160, and SR proteins. Exp Cell Res 283:184–195.
Mercan F, Bennett AM. 2010. Analysis of protein tyrosine phosphatases and substrates. Curr Protoc Mol Biol Chapter 18:Unit 18.16.
Meurer-Grob P, Kasparian J, Wade RH. 2001. Microtubule structure at improved resolution. Biochemistry 40:8000–8008.
Michaelis ML, Georg G, Telikepalli H, McIntosh M, Rajewski RA. 2006. Ongoing in vivo studies with cytoskeletal drugs in tau transgenic mice. Curr Alzheimer Res 3:215–219.
Mielke S, Sparreboom A, Mross K. 2006. Peripheral neuropathy: a persisting challenge in paclitaxel-based regimes. Eur J Cancer 42:24–30.
Miguel L, Rovelet-Lecrux A, Feyeux M, Frebourg T, Nassoy P, Campion D, Lecourtois M. 2019. Detection of all adult Tau isoforms in a 3D culture model of iPSC-derived neurons. Stem Cell Research 40:101541.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 117 / 157
Miki H, Setou M, Kaneshiro K, Hirokawa N. 2001. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA 98:7004–7011.
Mili S, Moissoglu K, Macara IG. 2008. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature 453:115–119.
Millecamps S, Boillée S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, Moigneu C, Vandenberghe N, Danel-Brunaud V, Corcia P, Pradat P-F, Le Forestier N, et al. 2012. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet 49:258–263.
Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. 2010. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 17:1144–1151.
Mitchison T, Kirschner M. 1984. Dynamic instability of microtubule growth. Nature 312:237–242.
Mitra A, Ruhnow F, Girardo S, Diez S. 2018. Directionally biased sidestepping of Kip3/kinesin-8 is regulated by ATP waiting time and motor–microtubule interaction strength. Proc Natl Acad Sci USA 115:E7950–E7959.
Monzon GA, Scharrel L, Santen L, Diez S. 2019. Activation of mammalian cytoplasmic dynein in multimotor motility assays. J Cell Sci 132:jcs220079.
Moreno MR de, Smith JF, Smith RV. 1986. Mechanism Studies of Coomassie Blue and Silver Staining of Proteins. Journal of Pharmaceutical Sciences 75:907–911.
Morfini GA, Bosco DA, Brown H, Gatto R, Kaminska A, Song Y, Molla L, Baker L, Marangoni MN, Berth S, Tavassoli E, Bagnato C, et al. 2013. Inhibition of Fast Axonal Transport by Pathogenic SOD1 Involves Activation of p38 MAP Kinase. PLoS ONE 8:e65235.
Mullane K, Williams M. 2019. Preclinical Models of Alzheimer’s Disease: Relevance and Translational Validity. Current Protocols in Pharmacology 84:e57.
Müller MJI, Klumpp S, Lipowsky R. 2008. Tug-of-war as a cooperative mechanism for bidirectional cargo transport by molecular motors. Proc Natl Acad Sci USA 105:4609–4614.
Murakami T, Qamar S, Lin JQ, Schierle GSK, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FTS, Michel CH, Kronenberg-Versteeg D, Li Y, et al. 2015. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 88:678–690.
Nagashima H, Asakura S. 1980. Dark-field light microscopic study of the flexibility of F-actin complexes. Journal of Molecular Biology 136:169–182.
Nakaya T, Alexiou P, Maragkakis M, Chang A, Mourelatos Z. 2013. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA 19:498–509.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 118 / 157
Nalavadi VC, Griffin LE, Picard-Fraser P, Swanson AM, Takumi T, Bassell GJ. 2012. Regulation of zipcode binding protein 1 transport dynamics in axons by myosin Va. J Neurosci 32:15133–15141.
Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, Naujock M, Gunther R, Jin M, Stanslowsky N, Reinhardt P, Sterneckert J, et al. 2018. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nature Communications 9:335.
Naumann M, Peikert K, Günther R, Kooi AJ van der, Aronica E, Hübers A, Danel V, Corcia P, Pan-Montojo F, Cirak S, Haliloglu G, Ludolph AC, et al. 2019. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann Clin Transl Neurol 6:2384–2394.
Nelson BR, Hartman BH, Georgi SA, Lan MS, Reh TA. 2007. Transient inactivation of Notch signaling synchronizes differentiation of neural progenitor cells. Dev Biol 304:479–498.
Nevins TA, Harder ZM, Korneluk RG, Holcík M. 2003. Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J Biol Chem 278:3572–3579.
Nitzsche B, Bormuth V, Bräuer C, Howard J, Ionov L, Kerssemakers J, Korten T, Leduc C, Ruhnow F, Diez S. 2010. Studying Kinesin Motors by Optical 3D-Nanometry in Gliding Motility Assays. In: Wilson L, Correia JJ, editors. Microtubules, in Vitro, s.l.: Elsevier textbooks, p 247–271.
Niu C, Zhang J, Gao F, Yang L, Jia M, Zhu H, Gong W. 2012. FUS-NLS/Transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS ONE 7:e47056.
Nogales E, Whittaker M, Milligan RA, Downing KH. 1999. High-resolution model of the microtubule. Cell 96:79–88.
Nötzel M, Rosso G, Möllmert S, Seifert A, Schlüßler R, Kim K, Hermann A, Guck J. 2018. Axonal Transport, Phase-Separated Compartments, and Neuron Mechanics - A New Approach to Investigate Neurodegenerative Diseases. Frontiers in Cellular Neuroscience 12:358.
Ohara O, Gahara Y, Miyake T, Teraoka H, Kitamura T. 1993. Neurofilament deficiency in quail caused by nonsense mutation in neurofilament-L gene. J Cell Biol 121:387–395.
Okada Y, Hirokawa N. 2000. Mechanism of the single-headed processivity: Diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proceedings of the National Academy of Sciences of the United States of America 97:640–645.
Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. 1995. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81:769–780.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 119 / 157
Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, Kohn MA, Lomen-Hoerth C. 2005. The effects of executive and behavioral dysfunction on the course of ALS. Neurology 65:1774–1777.
Orozco D, Tahirovic S, Rentzsch K, Schwenk BM, Haass C, Edbauer D. 2012. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep 13:759–764.
Osborne NN, Neuhoff V. 1972. Use of dansyl chloride and microchromatography to detect and study the metabolism of suspected transmitter substances and amino acids in single neurons. Biochemical Journal 128:82P-83P.
Pal A, Glaß H, Naumann M, Kreiter N, Japtok J, Sczech R, Hermann A. 2018. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci Data 5:180241.
Panda D, Goode BL, Feinstein SC, Wilson L. 1995. Kinetic Stabilization of Microtubule Dynamics at Steady State by Tau and Microtubule-Binding Domains of Tau. Biochemistry 34:11117–11127.
Papanayotou I, Sun B, Roth AF, Davis NG. 2010. Protein aggregation induced during glass bead lysis of yeast. Yeast 27:801–816.
Parakh S, Atkin JD. 2016. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res 1648:633–649.
Parimalam SS, C. Tarhan M, L. Karsten S, Hiroyuki Fujita, Hirofumi Shintaku, Hidetoshi Kotera, Ryuji Yokokawa. 2016. On-chip microtubule gliding assay for parallel measurement of tau protein species. Lab on a Chip 16:1691–1697.
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim J-M, Chung J. 2006. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161.
Park JY, Jang SY, Shin YK, Koh H, Suh DJ, Shinji T, Araki T, Park HT. 2013. Mitochondrial swelling and microtubule depolymerization are associated with energy depletion in axon degeneration. Neuroscience 238:258–269.
Pasinelli P, Brown RH. 2006. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723.
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, et al. 2015. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162:1066–1077.
Peck A, Sargin ME, LaPointe NE, Rose K, Manjunath BS, Feinstein SC, Wilson L. 2011. Tau isoform–specific modulation of kinesin–driven microtubule gliding rates and trajectories as determined with tau–stabilized microtubules. Cytoskeleton 68:44–55.
Peltier JCA. 1834. Nouvelles expériences sur la caloricité des courants électrique. Annales de Chimie et de Physique, p 371–386.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 120 / 157
Penzes P, Rafalovich I. 2012. Regulation of the Actin Cytoskeleton in Dendritic Spines. In: Kreutz MR, Sala C, editors. Synaptic Plasticity, Vienna: Springer Vienna, p 81–95.
Petrov D, Mansfield C, Moussy A, Hermine O. 2017. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front Aging Neurosci 9:68.
Pezzano M, Philp D, Stephenson S, Li Y, Reid V, Maitta R, Guyden JC. 1996. Positive selection by thymic nurse cells requires IL-1 beta and is associated with an increased Bcl-2 expression. Cell Immunol 169:174–184.
Pierce DW, Hom-Booher N, Vale RD. 1997. Imaging individual green fluorescent proteins. Nature 388:338.
Pîrşcoveanu DFV, Pirici I, Tudorică V, Bălşeanu TA, Albu VC, Bondari S, Bumbea AM, Pîrşcoveanu M. 2017. Tau protein in neurodegenerative diseases - a review. Rom J Morphol Embryol 58:1141–1150.
Pohlmann T, Baumann S, Haag C, Albrecht M, Feldbrügge M. 2015. A FYVE zinc finger domain protein specifically links mRNA transport to endosome trafficking. Elife 4:.
Polverino de Laureto P, Palazzi L, Acquasaliente L. 2019. Polyphenols as Potential Therapeutic Drugs in Neurodegeneration. Neuroprotection - New Approaches and Prospects [Working Title], IntechOpen,.
Prasad DD, Ouchida M, Lee L, Rao VN, Reddy ES. 1994. TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t(16;21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene 9:3717–3729.
Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young-Pearse TL, Flanagan JG. 2014. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158:368–382.
Prota AE, Bargsten K, Zurwerra D, Field JJ, Díaz JF, Altmann K-H, Steinmetz MO. 2013. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 339:587–590.
Qian W, Liu F. 2014. Regulation of alternative splicing of tau exon 10. Neurosci Bull 30:367–377.
Qiu H, Lee S, Shang Y, Wang W-Y, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, Tang AA, Oldham MC, et al. 2014. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124:981–999.
Rai AK, Rai A, Ramaiya AJ, Jha R, Mallik R. 2013. Molecular adaptations allow dynein to generate large collective forces inside cells. Cell 152:172–182.
Ramkumar A, Jong BY, Ori-McKenney KM. 2018. ReMAPping the microtubule landscape: How phosphorylation dictates the activities of microtubule-associated proteins: Phosphorylation of Microtubule-Associated Proteins. Dev Dyn 247:138–155.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 121 / 157
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308.
Rappsilber J, Friesen WJ, Paushkin S, Dreyfuss G, Mann M. 2003. Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal Chem 75:3107–3114.
Rasnik I, McKinney SA, Ha T. 2006. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat Methods 3:891–893.
Ray K, Perez SE, Yang Z, Xu J, Ritchings BW, Steller H, Goldstein LS. 1999. Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J Cell Biol 147:507–518.
Reber S, Stettler J, Filosa G, Colombo M, Jutzi D, Lenzken SC, Schweingruber C, Bruggmann R, Bachi A, Barabino SM, Mühlemann O, Ruepp M-D. 2016. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J 35:1504–1521.
Reinhardt P, Glatza M, Hemmer K, Tsytsyura Y, Thiel CS, Höing S, Moritz S, Parga JA, Wagner L, Bruder JM, Wu G, Schmid B, et al. 2013. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PloS one 8:e59252.
Renton AE, Chiò A, Traynor BJ. 2014. State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience 17:17–23.
Reyes-Gibby CC, Morrow PK, Buzdar A, Shete S. 2009. Chemotherapy-induced peripheral neuropathy as a predictor of neuropathic pain in breast cancer patients previously treated with paclitaxel. J Pain 10:1146–1150.
Rheenen W van, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, Spek RAA van der, Võsa U, Jong S de, Robinson MR, Yang J, Fogh I, et al. 2016. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48:1043–1048.
Ricca BL, Rock RS. 2010. The stepping pattern of myosin X is adapted for processive motility on bundled actin. Biophys J 99:1818–1826.
Rief M, Rock RS, Mehta AD, Mooseker MS, Cheney RE, Spudich JA. 2000. Myosin-V stepping kinetics: a molecular model for processivity. Proc Natl Acad Sci USA 97:9482–9486.
Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA. 2013. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol 14:713–726.
Rock RS, Rief M, Mehta AD, Spudich JA. 2000. In vitro assays of processive myosin motors. Methods 22:373–381.
Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 275:9390–9395.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 122 / 157
Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, Tollervey J, Fujii R, et al. 2012. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2:603.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62.
Ross JL, Fygenson DK. 2003. Mobility of taxol in microtubule bundles. Biophys J 84:3959–3967.
Ross JL, Santangelo CD, Makrides V, Fygenson DK. 2004. Tau induces cooperative Taxol binding to microtubules. Proc Natl Acad Sci U S A 101:12910.
Ross JL, Wallace K, Shuman H, Goldman YE, Holzbaur ELF. 2006. Processive bidirectional motion of dynein-dynactin complexes in vitro. Nat Cell Biol 8:562–570.
Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien JP, Figlewicz D. 1996. SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol 39:128–131.
Ruhnow F, Zwicker D, Diez S. 2011. Tracking single particles and elongated filaments with nanometer precision. Biophysical journal 100:2820–2828.
Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DAQ, Gómez-Herreros F, Hafezparast M, Caldecott KW. 2014. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res 42:307–314.
Saberi S, Stauffer JE, Schulte DJ, Ravits J. 2015. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin 33:855–876.
Saito A, Farhana TI, Kabir AMdR, Inoue D, Konagaya A, Sada K, Kakugo A. 2017. Understanding the emergence of collective motion of microtubules driven by kinesins: Role of concentration of microtubules and depletion force. RSC Adv 7:13191–13197.
Salogiannis J, Reck-Peterson SL. 2017. Hitchhiking: A Non-Canonical Mode of Microtubule-Based Transport. Trends Cell Biol 27:141–150.
Sama RRK, Ward CL, Bosco DA. 2014. Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro 6:.
Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow E-M, Mandelkow E, Hoenger A. 2004. Surface-decoration of microtubules by human tau. J Mol Biol 339:539–553.
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. 2013. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376.
Sasaki S, Iwata M. 2007. Mitochondrial Alterations in the Spinal Cord of Patients With Sporadic Amyotrophic Lateral Sclerosis: Journal of Neuropathology and Experimental Neurology 66:10–16.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 123 / 157
Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, Kanaan NM, Yarasheski KE, et al. 2018. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 98:861–864.
Sato S, Idogawa M, Honda K, Fujii G, Kawashima H, Takekuma K, Hoshika A, Hirohashi S, Yamada T. 2005. Beta-catenin interacts with the FUS proto-oncogene product and regulates pre-mRNA splicing. Gastroenterology 129:1225–1236.
Sau D, Rusmini P, Crippa V, Onesto E, Bolzoni E, Ratti A, Poletti A. 2011. Dysregulation of axonal transport and motorneuron diseases. Biology of the cell 103:87–107.
Scekic-Zahirovic J, Oussini HE, Mersmann S, Drenner K, Wagner M, Sun Y, Allmeroth K, Dieterlé S, Sinniger J, Dirrig-Grosch S, René F, Dormann D, et al. 2017. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol 133:887–906.
Scharrel L, Ma R, Schneider R, Jülicher F, Diez S. 2014. Multimotor transport in a system of active and inactive kinesin-1 motors. Biophys J 107:365–372.
Schevzov G, Curthoys NM, Gunning PW, Fath T. 2012. Functional Diversity of Actin Cytoskeleton in Neurons and its Regulation by Tropomyosin. International Review of Cell and Molecular Biology, Elsevier, p 33–94.
Schiff PB, Fant J, Horwitz SB. 1979. Promotion of microtubule assembly in vitro by taxol. Nature 277:665–667.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682.
Schlager MA, Hoang HT, Urnavicius L, Bullock SL, Carter AP. 2014. In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J 33:1855–1868.
Schmidt C, Kim B, Grabner H, Ries J, Kulomaa M, Vogel V. 2012. Tuning the “Roadblock” Effect in Kinesin-Based Transport. Nano Lett 12:3466–3471.
Schnitzer MJ, Visscher K, Block SM. 2000. Force production by single kinesin motors. Nat Cell Biol 2:718–723.
Scholey JM. 1993. Motility Assays for Motor Proteins. San Diego: Academic Press.
Scholey JM. 2013. Kinesin-2: a family of heterotrimeric and homodimeric motors with diverse intracellular transport functions. Annu Rev Cell Dev Biol 29:443–469.
Schreiber V, Dantzer F, Ame J-C, Murcia G de. 2006. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7:517–528.
Schroeder HW, Hendricks AG, Ikeda K, Shuman H, Rodionov V, Ikebe M, Goldman YE, Holzbaur ELF. 2012. Force-dependent detachment of kinesin-2 biases track switching at cytoskeletal filament intersections. Biophys J 103:48–58.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 124 / 157
Schroeder HW, Mitchell C, Shuman H, Holzbaur ELF, Goldman YE. 2010. Motor number controls cargo switching at actin-microtubule intersections in vitro. Curr Biol 20:687–696.
Schwartz JC, Cech TR, Parker RR. 2015. Biochemical Properties and Biological Functions of FET Proteins. Annu Rev Biochem 84:355–379.
Schwarz TL. 2013. Mitochondrial Trafficking in Neurons. Cold Spring Harbor Perspectives in Biology 5:a011304–a011304.
Schweers O, Schönbrunn-Hanebeck E, Marx A, Mandelkow E. 1994. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem 269:24290–24297.
Scripture CD, Figg WD, Sparreboom A. 2006. Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives. Curr Neuropharmacol 4:165–172.
Seitz A, Kojima H, Oiwa K, Mandelkow E-M, Song Y-H, Mandelkow E. 2002. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J 21:4896–4905.
Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D. 2011. Taxol facilitates axon regeneration in the mature CNS. J Neurosci 31:2688–2699.
Sergeant N, Delacourte A, Buée L. 2005. Tau protein as a differential biomarker of tauopathies. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1739:179–197.
Shang Y, Huang EJ. 2016. Mechanisms of FUS Mutations in Familial Amyotrophic Lateral Sclerosis. Brain Research 1647:65–78.
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA. 2016. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7:10465.
Sheetz MP, Spudich JA. 1983. Movement of myosin-coated fluorescent beads on actin cables in vitro. Nature 303:31–35.
Shehadul Islam M, Aryasomayajula A, Selvaganapathy P. 2017. A Review on Macroscale and Microscale Cell Lysis Methods. Micromachines 8:83.
Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, Buchman VL. 2014. Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Human Molecular Genetics 23:5211–5226.
Shichinohe H, Ishihara T, Takahashi K, Tanaka Y, Miyamoto M, Yamauchi T, Saito H, Takemoto H, Houkin K, Kuroda S. 2015. Bone Marrow Stromal Cells Rescue Ischemic Brain by Trophic Effects and Phenotypic Change Toward Neural Cells. Neurorehabil Neural Repair 29:80–89.
Shrestha R, Wen Y-T, Ding D-C, Tsai R-K. 2019. Aberrant hiPSCs-Derived from Human Keratinocytes Differentiates into 3D Retinal Organoids that Acquire Mature Photoreceptors. Cells 8:36.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 125 / 157
Siahaan V, Krattenmacher J, Hyman AA, Diez S, Hernández-Vega A, Lansky Z, Braun M. 2019. Kinetically distinct phases of tau on microtubules regulate kinesin motors and severing enzymes. Nat Cell Biol 21:1086–1092.
Sillen A, Barbier P, Landrieu I, Lefebvre S, Wieruszeski J-M, Leroy A, Peyrot V, Lippens G. 2007. NMR Investigation of the Interaction between the Neuronal Protein Tau and the Microtubules †. Biochemistry 46:3055–3064.
Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, Zhang Z, Huang Y, Temple S, Coppola G, Geschwind DH, Karydas A, et al. 2016. Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Reports 7:325–340.
Sleigh JN, Tosolini AP, Gordon D, Devoy A, Fratta P, Fisher EMC, Talbot K, Schiavo G. 2020. Mice Carrying ALS Mutant TDP-43, but Not Mutant FUS, Display In Vivo Defects in Axonal Transport of Signaling Endosomes. Cell Reports 30:3655-3662.e2.
Smith EF, Shaw PJ, De Vos KJ. 2019. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 710:132933.
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. 1985. Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85.
Sobol M, Klar J, Laan L, Shahsavani M, Schuster J, Annerén G, Konzer A, Mi J, Bergquist J, Nordlund J, Hoeber J, Huss M, et al. 2019. Transcriptome and Proteome Profiling of Neural Induced Pluripotent Stem Cells from Individuals with Down Syndrome Disclose Dynamic Dysregulations of Key Pathways and Cellular Functions. Mol Neurobiol 56:7113–7127.
Soo KY, Sultana J, King A, Atkinson R, Warraich ST, Sundaramoorthy V, Blair I, Farg MA, Atkin JD. 2015. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discovery 1:15030.
Soppina V, Norris SR, Dizaji AS, Kortus M, Veatch S, Peckham M, Verhey KJ. 2014. Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proc Natl Acad Sci USA 111:5562–5567.
Sozański K, Ruhnow F, Wiśniewska A, Tabaka M, Diez S, Hołyst R. 2015. Small Crowders Slow Down Kinesin-1 Stepping by Hindering Motor Domain Diffusion. Phys Rev Lett 115:218102.
Speyer CL, Bukhsh MA, Jafry WS, Sexton RE, Bandyopadhyay S, Gorski DH. 2017. Riluzole synergizes with paclitaxel to inhibit cell growth and induce apoptosis in triple-negative breast cancer. Breast Cancer Res Treat 166:407–419.
Sposito T, Preza E, Mahoney CJ, Setó-Salvia N, Ryan NS, Morris HR, Arber C, Devine MJ, Houlden H, Warner TT, Bushell TJ, Zagnoni M, et al. 2015. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum Mol Genet 24:5260–5269.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 126 / 157
Spudich JA, Kron SJ, Sheetz MP. 1985. Movement of myosin-coated beads on oriented filaments reconstituted from purified actin. Nature 315:584–586.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, Belleroche J de, et al. 2008. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow E-M. 2002. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. Journal of Cell Biology 156:1051–1063.
Sterky FH, Lee S, Wibom R, Olson L, Larsson N-G. 2011. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proceedings of the National Academy of Sciences 108:12937–12942.
Stoica R, Paillusson S, Gomez‐Suaga P, Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay‐Barrena G, De Vos KJ, Shaw CE, Hanger DP, Noble W, et al. 2016. ALS/FTD‐associated FUS activates GSK‐3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep 17:1326–1342.
Stoothoff W, Jones PB, Spires-Jones TL, Joyner D, Chhabra E, Bercury K, Fan Z, Xie H, Bacskai B, Edd J, Irimia D, Hyman BT. 2009. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. Journal of Neurochemistry 111:417–427.
Stoothoff WH, Johnson GVW. 2005. Tau phosphorylation: physiological and pathological consequences. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1739:280–297.
Structural Genomics Consortium, Architecture et Fonction des Macromolécules Biologiques, Berkeley Structural Genomics Center, China Structural Genomics Consortium, Integrated Center for Structure and Function Innovation, Israel Structural Proteomics Center, Joint Center for Structural Genomics, Midwest Center for Structural Genomics, New York Structural GenomiX Research Center for Structural Genomics, Northeast Structural Genomics Consortium, Oxford Protein Production Facility, Protein Sample Production Facility, Max Delbrück Center for Molecular Medicine, et al. 2008. Protein production and purification. Nat Methods 5:135–146.
Sun F, Zhu C, Dixit R, Cavalli V. 2011a. Sunday Driver/JIP3 binds kinesin heavy chain directly and enhances its motility. EMBO J 30:3416–3429.
Sun S, Ling S-C, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, Huang EJ, Eggan K, et al. 2015. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6:6171.
Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD. 2011b. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 9:e1000614.
Svetoni F, Frisone P, Paronetto MP. 2016. Role of FET proteins in neurodegenerative disorders. RNA Biol 13:1089–1102.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 127 / 157
Svoboda K, Block SM. 1994. Force and velocity measured for single kinesin molecules. Cell 77:773–784.
Svoboda K, Schmidt CF, Schnapp BJ, Block SM. 1993. Direct observation of kinesin stepping by optical trapping interferometry. Nature 365:721–727.
Swoboda M, Henig J, Cheng H-M, Brugger D, Haltrich D, Plumeré N, Schlierf M. 2012. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano 6:6364–6369.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872.
Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676.
Takarada T, Tamaki K, Takumi T, Ogura M, Ito Y, Nakamichi N, Yoneda Y. 2009. A protein-protein interaction of stress-responsive myosin VI endowed to inhibit neural progenitor self-replication with RNA binding protein, TLS, in murine hippocampus. J Neurochem 110:1457–1468.
Takeda S, Yamazaki H, Seog DH, Kanai Y, Terada S, Hirokawa N. 2000. Kinesin superfamily protein 3 (KIF3) motor transports fodrin-associating vesicles important for neurite building. J Cell Biol 148:1255–1265.
Tan, Manley JL. 2010. TLS Inhibits RNA Polymerase III Transcription. Molecular and Cellular Biology 30:186–196.
Tan R, Lam AJ, Tan T, Han J, Nowakowski DW, Vershinin M, Simó S, Ori-McKenney KM, McKenney RJ. 2019. Microtubules gate tau condensation to spatially regulate microtubule functions. Nat Cell Biol 21:1078–1085.
Tan, Riley TR, Coady T, Bussemaker HJ, Manley JL. 2012. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proceedings of the National Academy of Sciences 109:6030–6035.
Tarhan MC, Orazov Y, Yokokawa R, Karsten SL, Fujita H. 2013. Biosensing MAPs as “roadblocks”: Kinesin-based functional analysis of tau protein isoforms and mutants using suspended microtubules (sMTs). Lab on a Chip 13:3217–3224.
Tasnim A, Rammelkamp Z, Slusher AB, Wozniak K, Slusher BS, Farah MH. 2016. Paclitaxel causes degeneration of both central and peripheral axon branches of dorsal root ganglia in mice. BMC Neurosci 17:47.
Taylor JP, Brown RH, Cleveland DW. 2016. Decoding ALS: from genes to mechanism. Nature 539:197–206.
Taylor RA, Solbrekken GL. 2008. Comprehensive system-level optimization of thermoelectric devices for electronic cooling applications. IEEE Trans Comp Packag Technol 31:23–31.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 128 / 157
Teng J, Rai T, Tanaka Y, Takei Y, Nakata T, Hirasawa M, Kulkarni AB, Hirokawa N. 2005. The KIF3 motor transports N-cadherin and organizes the developing neuroepithelium. Nat Cell Biol 7:474–482.
Tepper K, Biernat J, Kumar S, Wegmann S, Timm T, Hübschmann S, Redecke L, Mandelkow E-M, Müller DJ, Mandelkow E. 2014. Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem 289:34389–34407.
Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. 2013. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther 12:2078–2087.
Tiruchinapalli DM, Oleynikov Y, Kelic S, Shenoy SM, Hartley A, Stanton PK, Singer RH, Bassell GJ. 2003. Activity-dependent trafficking and dynamic localization of zipcode binding protein 1 and beta-actin mRNA in dendrites and spines of hippocampal neurons. J Neurosci 23:3251–3261.
Tradewell ML, Yu Z, Tibshirani M, Boulanger M-C, Durham HD, Richard S. 2012. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Human Molecular Genetics 21:136–149.
Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. 1999. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci 112 ( Pt 14):2355–2367.
Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, Sobue G, Yamanaka K. 2013. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 5:221–234.
Tuerde D, Kimura T, Miyasaka T, Furusawa K, Shimozawa A, Hasegawa M, Ando K, Hisanaga S. 2018. Isoform-independent and -dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J Biol Chem 293:1781–1793.
Turner-Bridger B, Jakobs M, Muresan L, Wong HH-W, Franze K, Harris WA, Holt CE. 2018. Single-molecule analysis of endogenous β-actin mRNA trafficking reveals a mechanism for compartmentalized mRNA localization in axons. Proc Natl Acad Sci USA 115:E9697–E9706.
Uranishi H, Tetsuka T, Yamashita M, Asamitsu K, Shimizu M, Itoh M, Okamoto T. 2001. Involvement of the Pro-oncoprotein TLS (Translocated in Liposarcoma) in Nuclear Factor-κB p65-mediated Transcription as a Coactivator. J Biol Chem 276:13395–13401.
Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida T. 1996. Direct observation of single kinesin molecules moving along microtubules. Nature 380:451–453.
Vale RD, Reese TS, Sheetz MP. 1985. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42:39–50.
Van Damme P, Robberecht W, Van Den Bosch L. 2017. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech 10:537–549.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 129 / 157
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, et al. 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211.
VanDelinder V, Sickafoose I, Imam ZI, Ko R, Bachand GD. 2020. The effects of osmolytes on in vitro kinesin-microtubule motility assays. RSC Adv 10:42810–42815.
Varga V, Helenius J, Tanaka K, Hyman AA, Tanaka TU, Howard J. 2006. Yeast kinesin-8 depolymerizes microtubules in a length-dependent manner. Nat Cell Biol 8:957–962.
Varga V, Leduc C, Bormuth V, Diez S, Howard J. 2009. Kinesin-8 motors act cooperatively to mediate length-dependent microtubule depolymerization. Cell 138:1174–1183.
Verheyen A, Diels A, Dijkmans J, Oyelami T, Meneghello G, Mertens L, Versweyveld S, Borgers M, Buist A, Peeters P, Cik M. 2015. Using Human iPSC-Derived Neurons to Model TAU Aggregation. PLoS ONE 10:e0146127.
Verheyen A, Diels A, Reumers J, Van Hoorde K, Van den Wyngaert I, Outryve d’Ydewalle C van, De Bondt A, Kuijlaars J, De Muynck L, De Hoogt R, Bretteville A, Jaensch S, et al. 2018. Genetically Engineered iPSC-Derived FTDP-17 MAPT Neurons Display Mutation-Specific Neurodegenerative and Neurodevelopmental Phenotypes. Stem Cell Reports 11:363–379.
Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP. 2007. Multiple-motor based transport and its regulation by Tau. Proceedings of the National Academy of Sciences 104:87–92.
Vijayakumar J, Perrois C, Heim M, Bousset L, Alberti S, Besse F. 2019. The prion-like domain of Drosophila Imp promotes axonal transport of RNP granules in vivo. Nat Commun 10:2593.
Vilfan A. 2009. Twirling motion of actin filaments in gliding assays with nonprocessive Myosin motors. Biophys J 97:1130–1137.
Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SSW, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, et al. 2014. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7:1–11.
Walker JMeditor. 2009. The Protein Protocols Handbook. New York, NY: Humana Press.
Wang, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IRA, Huang EJ, Tsai L-H. 2013. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16:1383–1391.
Wang R-X, Chen S, Jin X, Shao Z-M. 2016. Value of Ki-67 expression in triple-negative breast cancer before and after neoadjuvant chemotherapy with weekly paclitaxel plus carboplatin. Sci Rep 6:30091.
Wang W, Himes RH, Dentler WL. 1994. The binding of a ciliary microtubule plus-end binding protein complex to microtubules is regulated by ciliary protein kinase and phosphatase activities. The Journal of biological chemistry 269:21460–21466.
Wang X, Schwartz JC, Cech TR. 2015. Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res 43:7535–7543.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 130 / 157
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. 1975. A protein factor essential for microtubule assembly. Proceedings of the National Academy of Sciences 72:1858–1862.
Weiss JN. 1997. The Hill equation revisited: uses and misuses. FASEB j 11:835–841.
Welte MA. 2004. Bidirectional transport along microtubules. Curr Biol 14:R525-537.
Wiechelman KJ, Braun RD, Fitzpatrick JD. 1988. Investigation of the bicinchoninic acid protein assay: Identification of the groups responsible for color formation. Analytical Biochemistry 175:231–237.
Wiesner S, Helfer E, Didry D, Ducouret G, Lafuma F, Carlier M-F, Pantaloni D. 2003. A biomimetic motility assay provides insight into the mechanism of actin-based motility. J Cell Biol 160:387–398.
Williams LS, Ganguly S, Loiseau P, Ng BF, Palacios IM. 2014. The auto-inhibitory domain and ATP-independent microtubule-binding region of Kinesin heavy chain are major functional domains for transport in the Drosophila germline. Development 141:176–186.
Wiltfang J, Arold N, Neuhoff V. 1991. A new multiphasic buffer system for sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100 000-1000, and their detection with picomolar sensitivity. Electrophoresis 12:352–366.
Witman GB, Cleveland DW, Weingarten MD, Kirschner MW. 1976. Tubulin requires tau for growth onto microtubule initiating sites. Proceedings of the National Academy of Sciences 73:4070–4074.
Wokke J. 1996. Riluzole. Lancet 348:795–799.
Wolozin B. 2014. Physiological protein aggregation run amuck: Stress granules and the genesis of neurodegenerative disease. Discovery medicine 17:47–52.
Wong HH-W, Lin JQ, Ströhl F, Roque CG, Cioni J-M, Cagnetta R, Turner-Bridger B, Laine RF, Harris WA, Kaminski CF, Holt CE. 2017. RNA Docking and Local Translation Regulate Site-Specific Axon Remodeling In Vivo. Neuron 95:852-868.e8.
Wyss-Coray T. 2016. Ageing, neurodegeneration and brain rejuvenation. Nature 539:180–186.
Xia C-H, Roberts EA, Her L-S, Liu X, Williams DS, Cleveland DW, Goldstein LSB. 2003. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. Journal of Cell Biology 161:55–66.
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. 2008. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253.
Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, Sharma A, Chandran S, et al. 2012. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2:799–806.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 131 / 157
Yanagida T, Nakase M, Nishiyama K, Oosawa F. 1984. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature 307:58–60.
Yang L, Embree LJ, Tsai S, Hickstein DD. 1998. Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273:27761–27764.
Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, Mili S. 2017. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. The Journal of Cell Biology 216:1015–1034.
Yasuda K, Zhang H, Loiselle D, Haystead T, Macara IG, Mili S. 2013. The RNA-binding protein Fus directs translation of localized mRNAs in APC-RNP granules. J Cell Biol 203:737–746.
Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science 300:2061–2065.
Yildiz A, Tomishige M, Gennerich A, Vale RD. 2008. Intramolecular Strain Coordinates Kinesin Stepping Behavior along Microtubules. Cell 134:1030–1041.
Yildiz A, Tomishige M, Vale RD, Selvin PR. 2004. Kinesin walks hand-over-hand. Science 303:676–678.
Yoshimura A, Fujii R, Watanabe Y, Okabe S, Fukui K, Takumi T. 2006. Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr Biol 16:2345–2351.
Yu D, LaPointe NE, Guzman E, Pessino V, Wilson L, Feinstein SC, Valentine MT. 2014. Tau proteins harboring neurodegeneration-linked mutations impair kinesin translocation in vitro. Journal of Alzheimer’s disease : JAD 39:301–314.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920.
Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, Iqbal K, Grundke-Iqbal I, Gong C-X. 2009. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. Journal of Neurochemistry 108:1480–1494.
Yuan A, Rao MV, Veeranna null, Nixon RA. 2012. Neurofilaments at a glance. J Cell Sci 125:3257–3263.
Yum SW, Zhang J, Mo K, Li J, Scherer SS. 2009. A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy. Ann Neurol 66:759–770.
Yun M, Zhang X, Park CG, Park HW, Endow SA. 2001. A structural pathway for activation of the kinesin motor ATPase. The EMBO journal 20:2611–2618.
Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM-Y, et al. 2005. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA 102:227–231.

Mechanisms of Axonal Transport in ALS References
Anne Seifert 132 / 157
Zheng H, Yin J, Gao Z, Huang H, Ji X, Dou C. 2011. Disruption of Chlorella vulgaris Cells for the Release of Biodiesel-Producing Lipids: A Comparison of Grinding, Ultrasonication, Bead Milling, Enzymatic Lysis, and Microwaves. Appl Biochem Biotechnol 164:1215–1224.
Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao L-H, Peters A, Park TS, Zambidis ET, Meyer JS, Gamm DM, Yau K-W, et al. 2014. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 5:4047.
Zhou Y, Liu S, Liu G, Öztürk A, Hicks GG. 2013. ALS-Associated FUS Mutations Result in Compromised FUS Alternative Splicing and Autoregulation. PLoS Genet 9:e1003895.
Zhou Z, Licklider LJ, Gygi SP, Reed R. 2002. Comprehensive proteomic analysis of the human spliceosome. Nature 419:182–185.
Zhu M, Li W, Lu Y, Dong X, Chen Y, Lin B, Xie X, Guo J, Li M. 2016. Alpha fetoprotein antagonizes apoptosis induced by paclitaxel in hepatoma cells in vitro. Sci Rep 6:26472.
Zhu Q, Couillard-Després S, Julien JP. 1997. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol 148:299–316.
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. 1997. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110 ( Pt 15):1741–1750.
Zor T, Selinger Z. 1996. Linearization of the Bradford Protein Assay Increases Its Sensitivity: Theoretical and Experimental Studies. Analytical Biochemistry 236:302–308.
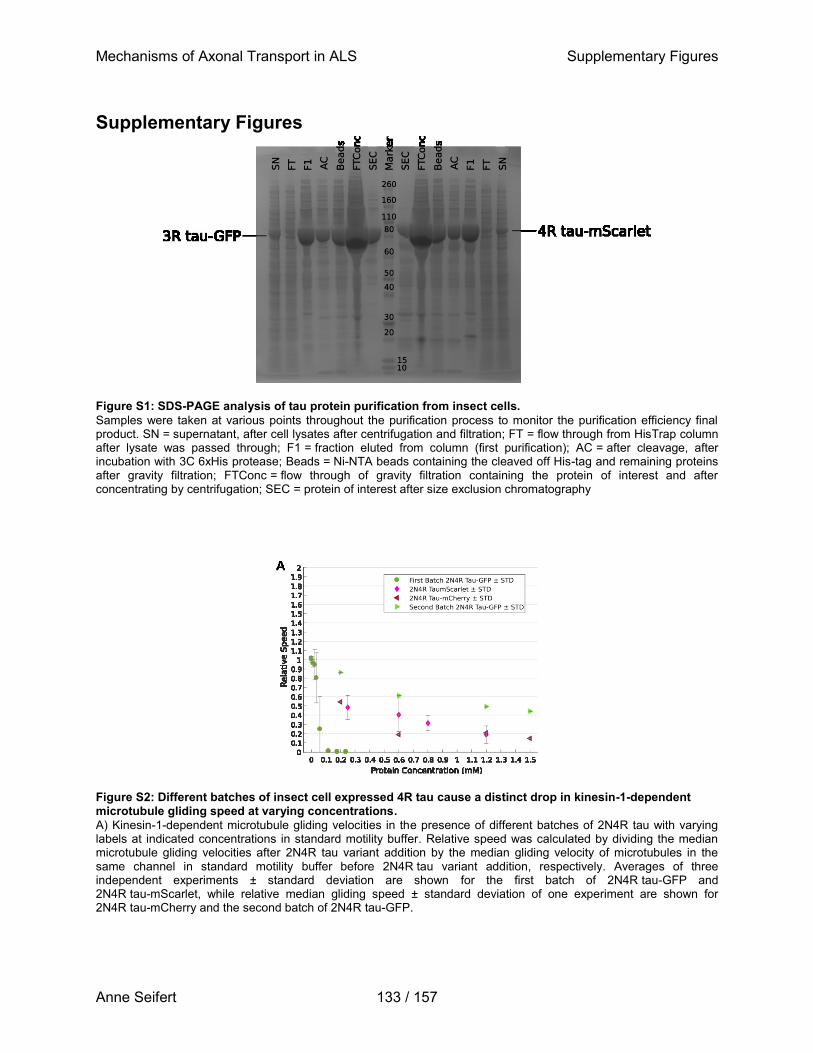
Mechanisms of Axonal Transport in ALS Supplementary Figures
Anne Seifert 133 / 157
Supplementary Figures
Figure S1: SDS-PAGE analysis of tau protein purification from insect cells.
Samples were taken at various points throughout the purification process to monitor the purification efficiency final product. SN = supernatant, after cell lysates after centrifugation and filtration; FT = flow through from HisTrap column after lysate was passed through; F1 = fraction eluted from column (first purification); AC = after cleavage, after incubation with 3C 6xHis protease; Beads = Ni-NTA beads containing the cleaved off His-tag and remaining proteins after gravity filtration; FTConc = flow through of gravity filtration containing the protein of interest and after concentrating by centrifugation; SEC = protein of interest after size exclusion chromatography
Figure S2: Different batches of insect cell expressed 4R tau cause a distinct drop in kinesin-1-dependent microtubule gliding speed at varying concentrations.
A) Kinesin-1-dependent microtubule gliding velocities in the presence of different batches of 2N4R tau with varying labels at indicated concentrations in standard motility buffer. Relative speed was calculated by dividing the median microtubule gliding velocities after 2N4R tau variant addition by the median gliding velocity of microtubules in the same channel in standard motility buffer before 2N4R tau variant addition, respectively. Averages of three independent experiments ± standard deviation are shown for the first batch of 2N4R tau-GFP and 2N4R tau-mScarlet, while relative median gliding speed ± standard deviation of one experiment are shown for 2N4R tau-mCherry and the second batch of 2N4R tau-GFP.
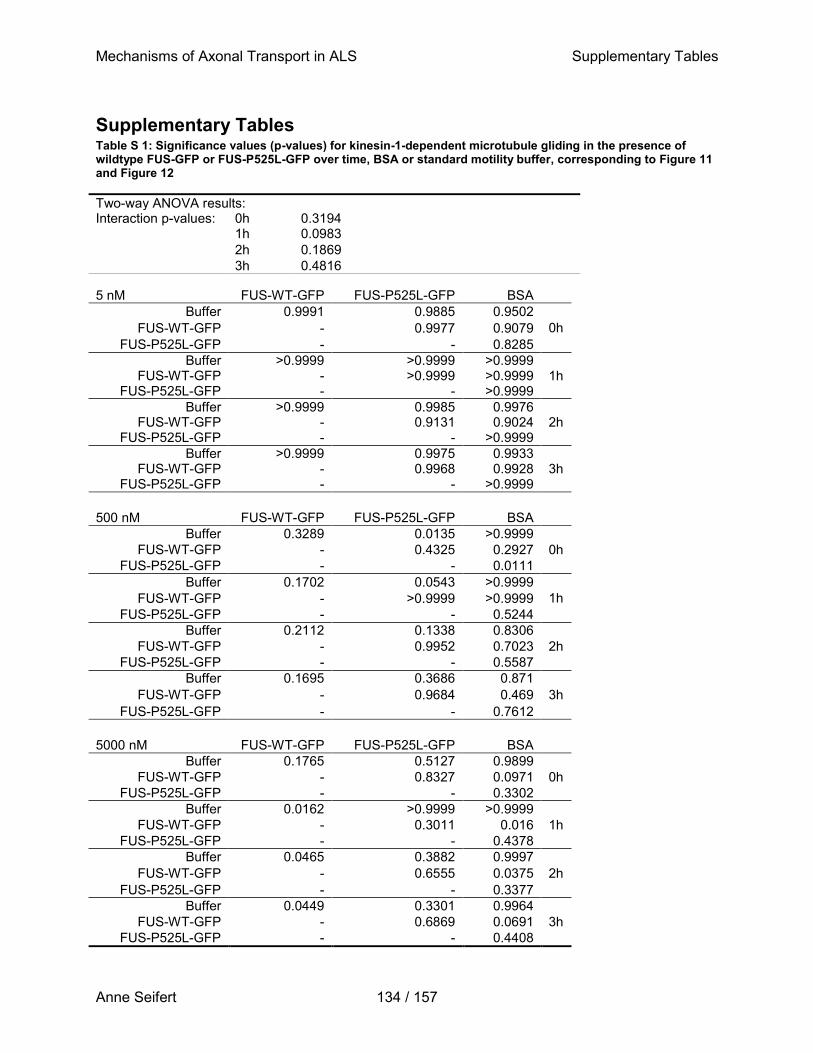
Mechanisms of Axonal Transport in ALS Supplementary Tables
Anne Seifert 134 / 157
Supplementary Tables Table S 1: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of wildtype FUS-GFP or FUS-P525L-GFP over time, BSA or standard motility buffer, corresponding to Figure 11 and Figure 12
Two-way ANOVA results: Interaction p-values: 0h 0.3194
1h 0.0983
2h 0.1869
3h 0.4816
5 nM FUS-WT-GFP FUS-P525L-GFP BSA
Buffer 0.9991 0.9885 0.9502
0h FUS-WT-GFP - 0.9977 0.9079
FUS-P525L-GFP - - 0.8285
Buffer >0.9999 >0.9999 >0.9999 1h FUS-WT-GFP - >0.9999 >0.9999
FUS-P525L-GFP - - >0.9999
Buffer >0.9999 0.9985 0.9976 2h FUS-WT-GFP - 0.9131 0.9024
FUS-P525L-GFP - - >0.9999
Buffer >0.9999 0.9975 0.9933 3h FUS-WT-GFP - 0.9968 0.9928
FUS-P525L-GFP - - >0.9999
500 nM FUS-WT-GFP FUS-P525L-GFP BSA
Buffer 0.3289 0.0135 >0.9999
0h FUS-WT-GFP - 0.4325 0.2927
FUS-P525L-GFP - - 0.0111
Buffer 0.1702 0.0543 >0.9999
1h FUS-WT-GFP - >0.9999 >0.9999
FUS-P525L-GFP - - 0.5244
Buffer 0.2112 0.1338 0.8306
2h FUS-WT-GFP - 0.9952 0.7023
FUS-P525L-GFP - - 0.5587
Buffer 0.1695 0.3686 0.871
3h FUS-WT-GFP - 0.9684 0.469
FUS-P525L-GFP - - 0.7612
5000 nM FUS-WT-GFP FUS-P525L-GFP BSA
Buffer 0.1765 0.5127 0.9899
0h FUS-WT-GFP - 0.8327 0.0971
FUS-P525L-GFP - - 0.3302
Buffer 0.0162 >0.9999 >0.9999
1h FUS-WT-GFP - 0.3011 0.016
FUS-P525L-GFP - - 0.4378
Buffer 0.0465 0.3882 0.9997
2h FUS-WT-GFP - 0.6555 0.0375
FUS-P525L-GFP - - 0.3377
Buffer 0.0449 0.3301 0.9964
3h FUS-WT-GFP - 0.6869 0.0691
FUS-P525L-GFP - - 0.4408
Mechanisms of Axonal Transport in ALS Supplementary Tables
Anne Seifert 135 / 157
Table S 2: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of 2N4R tau-GFP, corresponding to Figure 10
Two-way ANOVA results: Interaction p-value: >0.0001
1 nM 5 nM 10 nM 15 nM 25 nM 50 nM 75 nM 100 nM
0 nM >0.9999 >0.9999 >0.9999 0.8786 0.016 0.0005 0.0004 0.0004 1 nM - >0.9999 >0.9999 0.909 0.0189 0.0005 0.0005 0.0005 5 nM - - >0.9999 0.9537 0.0258 0.0007 0.0007 0.0007
10 nM - - - 0.9828 0.0366 0.001 0.001 0.001 15 nM - - - 0.9828 0.2066 0.0067 0.0063 0.0062 25 nM - - - - - 0.6482 0.6255 0.6216 50 nM - - - - - - >0.9999 >0.9999 75 nM - - - - - - - >0.9999
Table S 3: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of whole cell lysates from spinal motor neurons expressing wildtype FUS-GFP or FUS-P525L-GFP, BSA or standard motility buffer, corresponding to Figure 13B
Two-way ANOVA results: Interaction p-value: 0.0696
FUS-WT-GFP FUS-P525L-GFP BSA
Buffer 0.0691 0.0544 0.3116
50 ng/µL FUS-WT-GFP - 0.9992 0.8558
FUS-P525L-GFP - - 0.7956 Buffer 0.9973 0.3495 0.48
80 ng/µL FUS-WT-GFP - 0.2831 0.4451 FUS-P525L-GFP - - 0.998
Buffer 0.827 0.5112 0.1648 110 ng/µL FUS-WT-GFP - 0.9117 0.4083
FUS-P525L-GFP - - 0.7726 Buffer 0.9889 0.5873 0.5245
140 ng/µL FUS-WT-GFP - 0.6499 0.5884
FUS-P525L-GFP - - 0.9891
Table S 4: Significance values (p-values) for kinesin-1-depenent microtubule gliding in the presence of whole cell lysates from spinal motor neurons expressing wildtype FUS-GFP or FUS-P525L-GFP, BSA or standard motility buffer supplemented with 2N4R tau-GFP at indicated concentrations, corresponding to Figure 13C
Two-way ANOVA results: Interaction p-value: 0.6536
Tau-GFP FUS-WT-GFP FUS-P525L-GFP BSA
Buffer >0.9999 0.9876 0.9974 >0.9999
0 nM Tau-GFP - 0.9544 0.9895 >0.9999
FUS-WT-GFP - - 0.9993 0.9279
FUS-P525L-GFP - - - 0.9786
Buffer >0.9999 0.967 0.9748 >0.9999
1 nM Tau-GFP - 0.9223 0.9416 0.9991 FUS-WT-GFP - - >0.9999 0.8187
FUS-P525L-GFP - - - 0.8505
Mechanisms of Axonal Transport in ALS Supplementary Tables
Anne Seifert 136 / 157
Tau-GFP FUS-WT-GFP FUS-P525L-GFP BSA
Buffer 0.9997 0.8977 0.9003 >0.9999
5 nM Tau-GFP - 0.858 0.8625 0.9989
FUS-WT-GFP - - >0.9999 0.719
FUS-P525L-GFP - - - 0.725
Buffer 0.997 0.2203 0.3247 0.9987
10 nM Tau-GFP - 0.0966 0.1882 >0.9999
FUS-WT-GFP - - 0.9978 0.0775
FUS-P525L-GFP - - - 0.1555
Buffer 0.7365 0.1383 0.0462 0.977
15 nM Tau-GFP - 0.4992 0.1674 0.8932
FUS-WT-GFP - - 0.9636 0.0992
FUS-P525L-GFP - - - 0.0182
Buffer 0.0002 <0.0001 0.0029 0.0002
25 nM Tau-GFP - 0.9991 0.7958 >0.9999
FUS-WT-GFP - - 0.6478 0.9995
FUS-P525L-GFP - - - 0.776
Buffer <0.0001 <0.0001 <0.0001 <0.0001
50 nM Tau-GFP - >0.9999 >0.9999 >0.9999
FUS-WT-GFP - - >0.9999 >0.9999 FUS-P525L-GFP - - - >0.9999
Buffer <0.0001 <0.0001 <0.0001 <0.0001
75 nM Tau-GFP - >0.9999 >0.9999 >0.9999
FUS-WT-GFP - - >0.9999 >0.9999
FUS-P525L-GFP - - - >0.9999
Buffer <0.0001 <0.0001 <0.0001 <0.0001
100 nM Tau-GFP - >0.9999 >0.9999 >0.9999
FUS-WT-GFP - - >0.9999 >0.9999
FUS-P525L-GFP - - - >0.9999
Table S 5: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of 2N3R tau-GFP and 2N4R tau-mScarlet, corresponding to Figure 17
Two-way ANOVA results: Interaction p-value: 0.9739 Concentration p-value: <0,0001 Tau variant p-value: 0.0009
3R-GFP 750 nM 1500 nM 2250 nM 3000 nM 3750 nM 4500 nM 5250 nM
500 nM 0.9999 0.7801 0.3063 0.1222 0.0082 0.0004 0.0002 750 nM - 0.9491 0.5552 0.2729 0.0245 0.0014 0.0007
1500 nM - - 0.9922 0.8952 0.265 0.0268 0.0139 2250 nM - - - 0.9996 0.7307 0.158 0.0922 3000 nM - - - 0.9449 0.3739 0.2446 3750 nM - - - - - 0.9571 0.8772 4500 nM - - - - - - >0,9999 5250 nM - - - - - - -
4R-mScarlet 150 nM 250 nM 400 nM 600 nM 800 nM 1000 nM 1200 nM
50 nM 0.6572 0.0675 0.044 0.0212 0.0008 0.0001 <0,0001
150 nM - 0.8682 0.7776 0.5955 0.0718 0.0128 0.0042 250 nM - - >0,9999 0.9997 0.6744 0.2605 0.1172
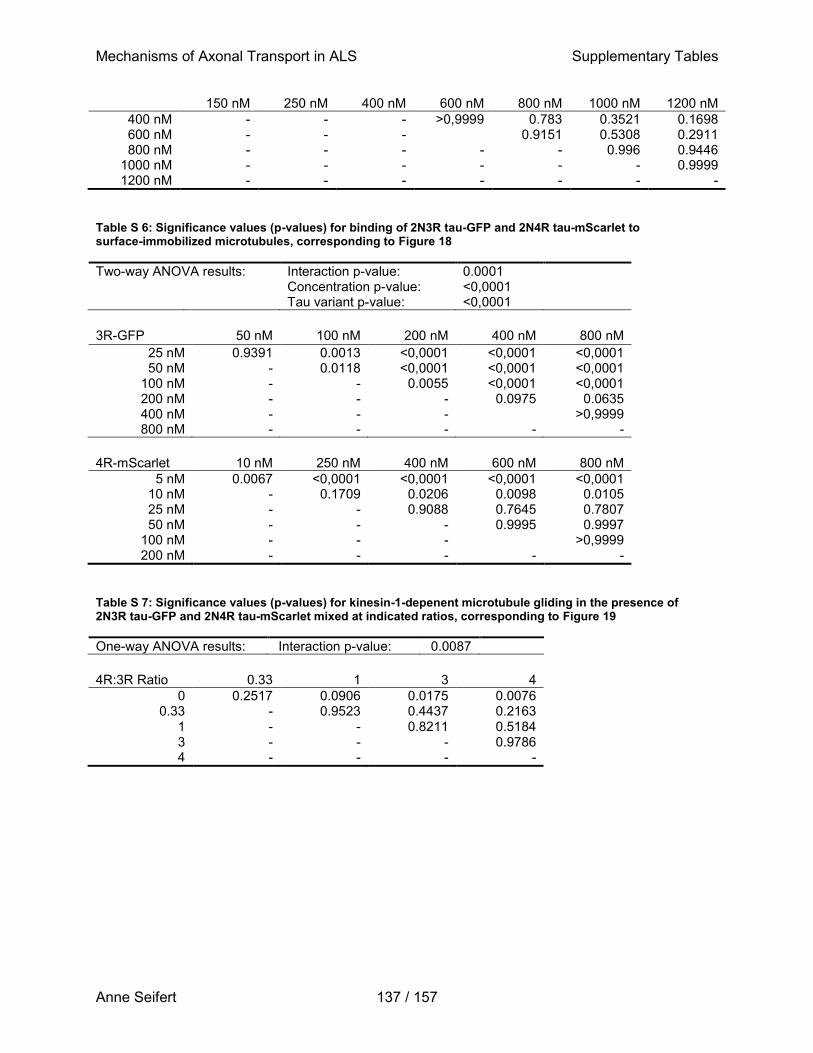
Mechanisms of Axonal Transport in ALS Supplementary Tables
Anne Seifert 137 / 157
150 nM 250 nM 400 nM 600 nM 800 nM 1000 nM 1200 nM
400 nM - - - >0,9999 0.783 0.3521 0.1698 600 nM - - -
0.9151 0.5308 0.2911
800 nM - - - - - 0.996 0.9446 1000 nM - - - - - - 0.9999 1200 nM - - - - - - -
Table S 6: Significance values (p-values) for binding of 2N3R tau-GFP and 2N4R tau-mScarlet to surface-immobilized microtubules, corresponding to Figure 18
Two-way ANOVA results: Interaction p-value: 0.0001 Concentration p-value: <0,0001 Tau variant p-value: <0,0001
3R-GFP 50 nM 100 nM 200 nM 400 nM 800 nM
25 nM 0.9391 0.0013 <0,0001 <0,0001 <0,0001 50 nM - 0.0118 <0,0001 <0,0001 <0,0001
100 nM - - 0.0055 <0,0001 <0,0001 200 nM - - - 0.0975 0.0635 400 nM - - - >0,9999 800 nM - - - - -
4R-mScarlet 10 nM 250 nM 400 nM 600 nM 800 nM
5 nM 0.0067 <0,0001 <0,0001 <0,0001 <0,0001 10 nM - 0.1709 0.0206 0.0098 0.0105 25 nM - - 0.9088 0.7645 0.7807 50 nM - - - 0.9995 0.9997
100 nM - - - >0,9999 200 nM - - - - -
Table S 7: Significance values (p-values) for kinesin-1-depenent microtubule gliding in the presence of 2N3R tau-GFP and 2N4R tau-mScarlet mixed at indicated ratios, corresponding to Figure 19
One-way ANOVA results: Interaction p-value: 0.0087
4R:3R Ratio 0.33 1 3 4
0 0.2517 0.0906 0.0175 0.0076 0.33 - 0.9523 0.4437 0.2163
1 - - 0.8211 0.5184 3 - - - 0.9786 4 - - - -
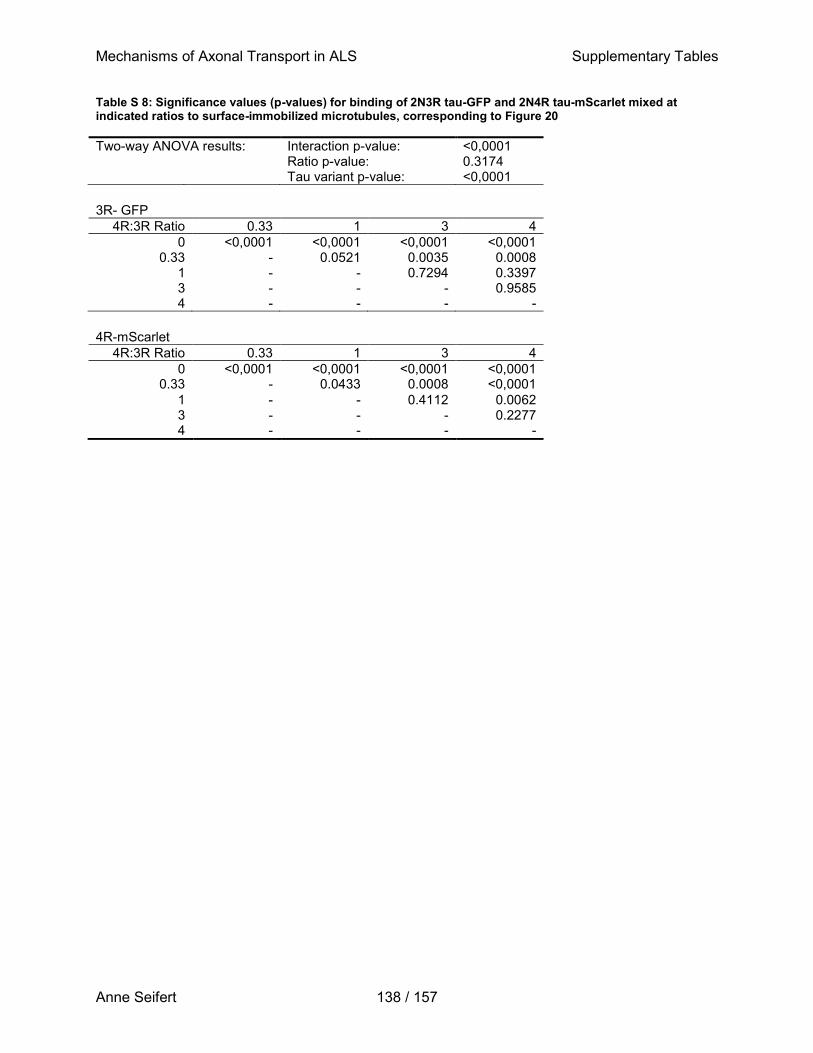
Mechanisms of Axonal Transport in ALS Supplementary Tables
Anne Seifert 138 / 157
Table S 8: Significance values (p-values) for binding of 2N3R tau-GFP and 2N4R tau-mScarlet mixed at indicated ratios to surface-immobilized microtubules, corresponding to Figure 20
Two-way ANOVA results: Interaction p-value: <0,0001 Ratio p-value: 0.3174 Tau variant p-value: <0,0001
3R- GFP
4R:3R Ratio 0.33 1 3 4
0 <0,0001 <0,0001 <0,0001 <0,0001 0.33 - 0.0521 0.0035 0.0008
1 - - 0.7294 0.3397 3 - - - 0.9585 4 - - - -
4R-mScarlet
4R:3R Ratio 0.33 1 3 4
0 <0,0001 <0,0001 <0,0001 <0,0001 0.33 - 0.0433 0.0008 <0,0001
1 - - 0.4112 0.0062 3 - - - 0.2277 4 - - - -

Mechanisms of Axonal Transport in ALS List of Figures
Anne Seifert 139 / 157
List of Figures Figure 1: Functional domains of FUS. ........................................................................................ 4 Figure 2: The motor protein kinesin-1 transports cargo along microtubules. ..............................11 Figure 3: Functional domains of tau. .........................................................................................16 Figure 4: Principle of a microtubule gliding motility assay. .........................................................20 Figure 5: Timeline for the differentiation and maturation of NPCs towards spinal motor neurons .................................................................................................................................................33 Figure 6: Assembly, mounting, and imaging of flow channels. ..................................................40 Figure 7: Reconstituting axonal transport in vitro. ......................................................................46 Figure 8: Optimal buffer conditions for reproducible kinesin-1-dependent microtubule gliding in the presence of whole cell lysates consist of 10 % glycerol, 10 nM β-glycerophosphate and 0.3 % methylcellulose in PBS. ...................................................................................................50 Figure 9: Imaging and analysis of kinesin-1-dependent microtubule gliding velocity . .............52 Figure 10: Tau-GFP binds to microtubules and inhibits kinesin-1-dependent microtubule gliding in a concentration dependent manner. ......................................................................................54 Figure 11: Recombinant FUS-GFP variants to not inhibit kinesin-1-dependent microtubule gliding. ......................................................................................................................................55 Figure 12: FUS-GFP variants do not inhibit kinesin-1-dependent microtubule gliding velocity and do not aggregate over the course of three hours. ......................................................................57 Figure 13: Cell lysates of spinal motor neurons expressing ALS-associated or wildtype FUS variants do not interfere with microtubule gliding on kinesin-1 motors. ......................................59 Figure 14: Western blot of FUS-GFP variants expressed in whole cell lysates of spinal motor neurons. ....................................................................................................................................60 Figure 15: 2N4R Tau-GFP binding to microtubules causes an arrest of microtubule gliding......62 Figure 16: 2N4R Tau-GFP binding to microtubules at low concentrations leads to an arrest in gliding of individual microtubules, while others are still motile. ..................................................63 Figure 17: The 2N4R tau isoform interferes with microtubule gliding on kinesin-1 motors at much lower concentrations compared to the 2N3R tau isoform. .........................................................65 Figure 18: The 2N4R tau-mScarlet isoform has a stronger binding affinity towards microtubules compared to 2N3R tau-GFP. .....................................................................................................67 Figure 19: An increase in 4R:3R tau isoform ratio leads to impaired kinesin-1-dependent microtubule gliding. ...................................................................................................................69 Figure 20: The 2N4R tau-mScarlet isoform prevents binding of 2N3R tau-GFP to microtubules due to its stronger binding affinity. .............................................................................................70 Figure 21: Western blot of tau variants expressed in whole cell lysates of spinal motor neurons. .................................................................................................................................................72

Mechanisms of Axonal Transport in ALS List of Tables
Anne Seifert 140 / 157
List of Tables Table 1: Technical equipment ...................................................................................................24 Table 2: General equipment ......................................................................................................25 Table 3: Cell culture ingredients, chemicals and reagents .........................................................26 Table 4: Media and buffers, all amounts given in percent refer to volume/volume .....................29 Table 5: Primary antibodies.......................................................................................................30 Table 6: Secondary antibodies ..................................................................................................30 Table 7: Software ......................................................................................................................31 Table 8: Characteristics for isogenic cell lines used in this study ...............................................31 Table 9: Comparison of different protein isolation techniques for their cell lysis efficiency, determined by the detection of β-actin via western blot, and protein yield, determined by BCA or Bradford assay. .........................................................................................................................48 Table 10: Calculation scheme for 4R:3R tau isoform ratios used in this study. ..........................68

Mechanisms of Axonal Transport in ALS List of Supplementary Figures and Tables
Anne Seifert 141 / 157
List of Supplementary Figures Figure S1: SDS-PAGE analysis of tau protein purification from insect cells. ........................... 133 Figure S2: Different batches of insect cell expressed 4R tau cause a distinct drop in kinesin-1-dependent microtubule gliding speed at varying concentrations. ............................. 133
List of Supplementary Tables Table S 1: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of wildtype FUS-GFP or FUS-P525L-GFP over time, BSA or standard motility buffer, corresponding to Figure 11 and Figure 12 ............................................................................... 134 Table S 2: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of 2N4R tau-GFP, corresponding to Figure 10 ......................................................... 135 Table S 3: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of whole cell lysates from spinal motor neurons expressing wildtype FUS-GFP or FUS-P525L-GFP, BSA or standard motility buffer, corresponding to Figure 13B .................... 135 Table S 4: Significance values (p-values) for kinesin-1-depenent microtubule gliding in the presence of whole cell lysates from spinal motor neurons expressing wildtype FUS-GFP or FUS-P525L-GFP, BSA or standard motility buffer supplemented with 2N4R tau-GFP at indicated concentrations, corresponding to Figure 13C ........................................................... 135 Table S 5: Significance values (p-values) for kinesin-1-dependent microtubule gliding in the presence of 2N3R tau-GFP and 2N4R tau-mScarlet, corresponding to Figure 17 ................... 136 Table S 6: Significance values (p-values) for binding of 2N3R tau-GFP and 2N4R tau-mScarlet to surface-immobilized microtubules, corresponding to Figure 18 ........................................... 137 Table S 7: Significance values (p-values) for kinesin-1-depenent microtubule gliding in the presence of 2N3R tau-GFP and 2N4R tau-mScarlet mixed at indicated ratios, corresponding to Figure 19 ................................................................................................................................. 137 Table S 8: Significance values (p-values) for binding of 2N3R tau-GFP and 2N4R tau-mScarlet mixed at indicated ratios to surface-immobilized microtubules, corresponding to Figure 20 .... 138

Mechanisms of Axonal Transport in ALS Summary
Anne Seifert 142 / 157
Summary Background
Neurodegenerative diseases have become one of the most common causes of death worldwide
over the last couple of decades, with increasing tendency. Amyotrophic lateral sclerosis (ALS) is
the most common neurodegenerative disease affecting specifically spinal (lower) and cortical
(upper) motor neurons in the spinal cord and brainstem, respectively. It is usually a late onset
disorder (average age of onset in Germany is 61 years) and leads to death within 2-5 years
after symptoms onset due to respiratory failure. To date, there is no cure for ALS and only two
drugs have been approved for its treatment, which prolong the lifespan for up to six months or
slow down disease progression in a subpopulation of patients. About 90 % of ALS cases are
sporadic, while about 10 % are familial and hence caused by mutations in specific genes,
among them fused in sarcoma (FUS), a DNA- and RNA-binding protein. Mutations in FUS
account for roughly 5 % of familial cases and occur predominantly in its nuclear localization
sequence (NLS), such as the FUS-P525L mutation. Neurons expressing this variant display a
strong cytoplasmic mislocalization of FUS and hence a loss of its nuclear function. Among other
pathological events, defects in axonal transport along microtubules have been observed early in
disease progression in several models of FUS-ALS, indicating its role as a major hallmark of the
disease. However, the mechanism of how transport is impaired within these neurons remains
unknown to date.
Hypothesis
This study aimed at investigating two possible mechanisms how the FUS-P525L mutant variant
affects microtubule-based axonal transport. First, it was analyzed whether FUS directly interacts
with microtubules or motors and if the mislocalized, mutant variant alters this interaction.
Secondly, cytoplasmic mislocalized FUS-P525L can no longer fulfil its regular role in the splicing
of pre-mRNAs, among them the mRNA coding for the microtubule-associated protein tau. This
reportedly leads to an increased ratio of translated tau isoforms containing four microtubule
binding repeats (4R) to those containing three repeats (3R). 4R tau isoforms are known to have
a stronger binding affinity towards microtubules and may hence impair transport more severely
by acting as a roadblock for motor proteins. Towards this end, this study investigated whether
an increase in 4R:3R tau isoform ratio is sufficient to impair microtubule based transport.
Methods
Axonal transport was reconstituted in vitro using a kinesin-1-dependent microtubule gliding
assays, in which microtubules are propelled by surface-immobilized kinesin-1 motors. The
assay was modified and optimized to operate sensitively and robust in the presence of complex
solutions such as whole cell lysates and the microtubule gliding velocity analyzed as a measure
for motility of the underlying motors. To determine the direct interaction of FUS variants with

Mechanisms of Axonal Transport in ALS Summary
Anne Seifert 143 / 157
kinesin-1 or microtubules, recombinant human wildtype FUS-GFP and FUS-P525L-GFP was
added to the assay. In addition, ALS patient-specific induced pluripotent stem cells (iPSCs)
expressing the same FUS variants were differentiated towards spinal motor neurons and their
cell lysates applied to this assay in order to determine whether FUS variants need endogenous
adaptors or interaction partners to interfere with kinesin-1 motility on microtubules. Further, to
investigate the interference of tau isoforms with kinesin-1 motility, recombinant human 2N3R
tau-GFP and 2N4R tau-mScarlet was purified from insect cells and added to the modified
kinesin-1-dependent microtubule gliding assay, either individually or combined at different
ratios. In addition, the binding of these tau variants to microtubules was assessed.
Results
The kinesin-1-dependent microtubule gliding assays was modified to operate sensitively and
robustly in the presence of β-glycerophosphate (to inhibit endogenous phosphatases in whole
cell lysates), and methylcellulose (to prevent microtubule detachment from kinesin-1 motors due
to presence of β-glycerophosphate). Under these conditions, neither recombinant human
FUS-GFP nor endogenous FUS-GFP variants in lysates of spinal motor neurons bound to
microtubules or interfered with kinesin-1 motility. In contrast, both tau isoforms used in the
present study bound to microtubules and impaired kinesin-1 motility, while 2N4R tau-mScarlet
was a much more potent inhibitor of microtubule gliding and displayed a 20-fold stronger binding
affinity to microtubules compared to 2N3R tau-GFP. Interestingly, increasing ratios of 4R:3R tau
isoforms impaired kinesin-1-dependent microtubule gliding. In addition, the presence of 2N4R
tau-mScarlet strongly prevented 2N3R tau-GFP from binding to microtubules.
Conclusion
This study provides evidence that neither wildtype FUS nor the FUS-P525L variant directly
interfere with axonal transport by interacting with kinesin-1 motors or microtubules. Further, the
present data suggests that neither FUS variant impedes kinesin-1 motility on microtubules by
interacting with endogenous adaptor proteins present in cell lysates of iPSC-derived spinal
motor neurons. Therefore, it is proposed that axonal transport defects are not directly caused by
interaction of cytoplasmic mislocalized FUS with the motors or microtubules, but rather arise as
a consequence of other pathological events triggered by mutant FUS variants. In particular, this
study demonstrates that an increased ratio of 4R:3R tau isoforms is sufficient to impair kinesin-1
motility on microtubules due to increased decoration of microtubules with 4R tau isoforms,
preventing 3R tau isoforms from binding to microtubules. This strongly suggests that an
increased ratio of 4R:3R tau isoforms, since FUS no longer regulates splicing of tau pre-mRNA
upon its cytoplasmic mislocalization, may be sufficient to cause or contribute to the axonal
transport defects observed early in FUS-ALS pathology.

Mechanisms of Axonal Transport in ALS Zusammenfassung
Anne Seifert 144 / 157
Zusammenfassung Hintergrund
Neurodegenerative Erkrankungen sind in den letzten Jahrzehnten mit zunehmender Tendenz
zu einer der häufigsten Todesursachen weltweit geworden. Amyotrophe Lateralsklerose (ALS)
ist die häufigste neurodegenerative Erkrankung, die spezifisch spinale (untere) und kortikale
(obere) Motoneuronen im Rückenmark bzw. im Hirnstamm betrifft. Es handelt sich in der Regel
um eine spät einsetzende Krankheit (das mittlere Erkrankungsalter in Deutschland beträgt 61
Jahre) und führt innerhalb von 2-5 Jahren nach Auftreten der Symptome zum Tod aufgrund von
Atemversagen. Bisher gibt es keine Heilung für ALS und es wurden nur zwei Medikamente für
die Behandlung zugelassen, die die Lebensdauer um bis zu sechs Monate verlängern oder das
Fortschreiten der Krankheit bei einer Subpopulation von Patienten verlangsamen. Ungefähr
90% der ALS-Fälle sind sporadisch, während ungefähr 10% familiär sind und daher durch
Mutationen in bestimmten Genen verursacht werden, darunter fused in sarcoma (FUS), einem
DNA- und RNA-bindenden Protein. Mutationen in FUS machen etwa 5% der familiären Fälle
aus und treten überwiegend in der Kernlokalisierungssequenz (NLS) auf, wie beispielsweise die
FUS-P525L Mutation. Neuronen, die diese Mutante exprimieren, zeigen eine starke
zytoplasmatische Fehllokalisierung von FUS und damit einen Verlust seiner Funktionen im
Zellkern. Neben anderen pathologischen Ereignissen wurden in mehreren FUS-ALS
Modellsystemen Defekte im Mikrotubuli-basierenden axonalen Transport früh im
Krankheitsverlauf beobachtet, was auf seine Rolle als eines der Hauptmerkmale dieser
Krankheit hindeutet. Der Mechanismus, wie der Transport innerhalb dieser Neuronen
beeinträchtigt wird, ist jedoch bis heute unbekannt.
Hypothese
Ziel dieser Studie ist es, zwei mögliche Mechanismen zu untersuchen, wie das mutierte
FUS-P525L Protein den axonalen Transport entlang von Mikrotubuli beeinflusst. Zunächst
wurde analysiert, ob FUS direkt mit Mikrotubuli oder Motorproteinen interagiert und ob
zytoplasmatische fehllokalisierte FUS-P525L Protein diese Interaktion verändert. Ferner kann
zytoplasmatische fehllokalisiertes FUS-P525L seine reguläre Rolle beim Spleißen von
Prä-mRNAs nicht mehr erfüllen, darunter die mRNA, die für das mit Mikrotubuli-assoziierte
Protein Tau kodiert. Dies führt zu einem erhöhten Verhältnis von translatierten Tau-Isoformen,
die vier Mikrotubuli-Bindestellen (4R) enthalten, zu solchen mit drei Bindestellen (3R). Es ist
bekannt, dass 4R-Tau-Isoformen eine stärkere Bindungsaffinität zu Mikrotubuli im Vergleich zu
3R-Tau-Isoformen aufweisen und daher den Transport stärker beeinträchtigen können, indem
sie als Hindernis für Motorproteine agieren. In dieser Studie wurde daher untersucht, ob eine
Erhöhung des Verhältnisses von 4R:3R-Tau-Isoform ausreicht, um den Mikrotubuli-basierenden
Transport zu beeinträchtigen.

Mechanisms of Axonal Transport in ALS Zusammenfassung
Anne Seifert 145 / 157
Methoden
Der axonale Transport wurde in vitro unter Verwendung eines Kinesin-1-gestuerten Mikrotubuli
Motilitätsassay rekonstruiert, bei welchem Mikrotubuli von darunterliegenden
oberflächenimmobilisierte Kinesin-1 Motorproteinen transportiert werden, also über die
Oberfläche gleiten. Der Assay wurde modifiziert und optimiert, um in Gegenwart komplexer
Lösungen wie Ganzzelllysaten sensitiv und robust zu funktionieren, und die
Gleitgeschwindigkeit der Mikrotubuli wurde als Maß für die Motilität der darunterliegenden
Motoren analysiert. Um die direkte Wechselwirkung von FUS-Varianten mit Kinesin-1
Motorproteinen oder Mikrotubuli zu bestimmen, wurde dem Assay rekombinantes menschliches
Wildtyp-FUS-GFP und FUS-P525L-GFP hinzugegeben. Zusätzlich wurden ALS-
patientenspezifische, induzierte pluripotente Stammzellen (iPSCs), welche dieselben FUS-
Varianten exprimieren, zu spinalen Motoneuronen differenziert und ihre Zelllysate in diesem
Assay angewendet, um zu bestimmen, ob FUS-Varianten endogene Adapter oder
Interaktionspartner für die Interaction mit Kinesin-1 oder Mikrotubuli benötigen. Um den Einfluss
von Tau-Isoformen auf die Kinesin-1 Motilität zu untersuchen, wurde rekombinantes
menschliches 2N3R Tau-GFP und 2N4R Tau-mScarlet aus Insektenzellen aufgereinigt und
dem modifizierten Kinesin-1-gestuerten Mikrotubuli Motilitätsassay entweder einzeln oder in
unterschiedlichen Verhältnissen kombiniert hinzugegeben. Zusätzlich wurde die Bindung dieser
Tau-Varianten an Mikrotubuli analysiert.
Ergebnisse
Die Kinesin-1-gesteuerte Mikrotubuli Motilitätsassay wurden so modifiziert, dass er in
Gegenwart von β-Glycerophosphat (zur Hemmung endogener Phosphatasen in
Ganzzelllysaten) und Methylcellulose (zur Verhinderung der Ablösung von Mikrotubuli von
Kinesin-1 Motoren aufgrund von β-Glycerophosphat) empfindlich und robust funktioniert. Unter
diesen Bedingungen zeigten weder rekombinantes menschliches FUS-GFP noch endogene
FUS-GFP-Varianten in Lysaten von spinalen Motoneuronen eine Wechselwirkung mit
Mikrotubuli und beeinträchtigten auch nicht die Kinesin-1 Motilität. Im Gegensatz dazu banden
beide in der vorliegenden Studie verwendeten Tau-Isoformen an Mikrotubuli und
beeinträchtigten die Kinesin-1-Motilität, wobei 2N4R Tau-mScarlet das Gleitens von Mikrotubuli
viel stärkerer beeinträchtigte und eine 20-fach stärkere Bindungsaffinität zu Mikrotubuli im
Vergleich zu 2N3R Tau-GFP zeigte. Ferner beeinträchtigten steigende Verhältnisse von 4R:3R
Tau-Isoformen über Kinesin-1 gleitende Mikrotubuli, während die Präsenz von 2N4R
Tau-mScarlet die Bindung von 2N3R Tau-GFP an Mikrotubuli stark verminderte.
Schlussfolgerung
Diese Studie liefert Hinweise darauf, dass weder Wildtyp-FUS noch die FUS P525L-Variante
den axonalen Transport direkt beeinflussen, da sie nicht mit Kinesin-1 Motorproteinen oder
Mikrotubuli interagieren. Ferner legen die vorliegenden Daten nahe, dass keine der FUS-

Mechanisms of Axonal Transport in ALS Zusammenfassung
Anne Seifert 146 / 157
Varianten die Kinesin-1 Motilität auf Mikrotubuli durch Wechselwirkung mit endogenen
Adapterproteinen behindert, die in Zelllysaten von iPSC-differenzierte spinalen Motoneuronen
vorhanden sind. Dies legt nahe, dass axonale Transportdefekte nicht durch direkte
Wechselwirkung von zytoplasmatisch fehllokalisiertem FUS Protein mit Motorproteinen oder
Mikrotubuli verursacht werden, sondern als Folge anderer pathologischer Ereignisse auftreten,
die durch mutierte FUS-Varianten entstehen. Insbesondere zeigt diese Studie, dass ein
erhöhtes Verhältnis von 4R:3R Tau-Isoformen ausreicht, um die Kinesin-1 Motilität auf
Mikrotubuli zu behindern. Dies geschieht vermutlich aufgrund der erhöhten Bindung von 4R
Tau-Isoformen an Mikrotubuli, weil dadurch die Bindung von 3R Tau-Isoformen an Mikrotubuli
verhindert wird. Dies deutet stark darauf hin, dass ein erhöhtes Verhältnis von 4R:3R
Tau-Isoformen, verursacht durch die fehlende regulatorische Beteiligung von FUS am Spleißen
von Tau-Prä-mRNA aufgrund der zytoplasmatischen Fehllokalisation von FUS, wahrscheinlich
zu den axonalen Transportdefekten beiträgt, die früh in der FUS-ALS-Pathologie beobachtet
wurden.

Mechanisms of Axonal Transport in ALS Acknowledgements
Anne Seifert 147 / 157
Acknowledgements I would like to thank Prof. Dr. Dr. Andreas Hermann and Prof. Dr. Stefan Diez for the
opportunity to pursue my doctoral studies in their labs. I wish to thank them for their continuous
support, constructive criticism, scientific input and motivation during the last years. Further, I
would like to thank Prof. Kempermann, Prof. Falkenburger, and Prof. Karl for their participation
in my thesis committee.
This work was supported in part by the NOMIS foundation, German Federal Ministry of
Education and Research BMBF OptiZeD 03Z22E511, by a seed grant of the Center For
Regenerative Therapies Dresden (CRTD), the Helmholtz Virtual Institute “RNA dysmetabolism
in ALS and FTD (VH-VI-510)”, an unrestricted grant by a family of a deceased ALS patient, and
the Stiftung zur Förderung der Hochschulmedizin in Dresden, and I am very grateful for the
support of all these funding sources.
Additionally, I would like to thank Dr. Jie Wang from the Max Planck Institute of Molecular
Cell Biology and Genetics, Dresden, for providing recombinant human FUS-GFP variant protein,
and Dr. Marcus Braun from the Institute of Biotechnology CAS, Prague, for the first batch of
recombinant human 2N4R tau-GFP protein. In line with this, I would like to thank Prof. Eckhardt
Mandelkow at the German Center for Neurodegenerative Diseases e.V. Bonn for the provision
of a DNA construct coding for recombinant human 2N3R tau-GFP and Dr. Amayra Hernandez
Vega for the construct coding for 2N4R tau-GFP. I am extremely grateful to Dr. Jens Ehrig from
the Molecular Imaging and Manipulation facility at B CUBE, who always took the time to
optimize my imaging workflows in the best way possible.
Further, I would like to voice my heartfelt appreciation to everyone who supported me in
one way or another during the course of my thesis. My deepest gratitude goes to Dr. Hauke
Drechsler, who significantly contributed to the success of this project through his scientific input,
but also through his continuous motivation (both verbally and with chocolate) and cure of panic
attacks. On the same note, I’m very grateful to Corina Bräuer for her unconditional support in
the lab, but also for always having an open ear and looking after me during tough times.
Together with Nora Hartmann, we always had a fun time in the lab, especially (but not
exclusively) on Fridays, and I will miss our almost philosophic discussions.
A huge thank you goes to Dr. Rahul Grover for his help in generating recombinant human
2N4R tau-mScarlet and 2N3R tau-GFP constructs and for his help and supervision during the
purification process. Likewise, my sincere gratitude goes to all Diez lab members who
supported me and this project during the last four years for the fruitful discussions and fun times

Mechanisms of Axonal Transport in ALS Acknowledgements
Anne Seifert 148 / 157
in and outside the lab: Dr. Till Korten, Foram Joshi, Dr. Veikko Geyer, Dr. Cordula Reuther,
Ashwin D’ Souza, Laura Meißner. I would also like to point out the amazing Barbara Lindemann
and thank her for her intensive administrational support and help with every not science-related
question.
I also would like to thank former and current members of the Hermann lab for their
continuous support in the lab and our fun lab nights at various restaurants and Christmas
markets: Barbara Szewczyk, Marie Anskat, Dr. Hannes Glaß, Yu Niu, Marcel Naumann,
Dr. Arun Pal, and Dr. Anne-Karen Meyer. In particular, I would like to thank Anett, Sylvia,
Andrea, Katja and Conny for their help with all organizational matters in the lab and their
sympathy. Special appreciation goes to Dr. Julia Japtok for her scientific, but especially her
emotional support throughout the last four years, even after she finished her doctoral thesis.
Schließlich möchte ich mich bei meiner Familie für ihre unendliche Unterstützung während
meiner Doktorzeit bedanken. Besonders danke ich dabei meiner Astaoma, die mich jederzeit
bei sich willkommen geheißen hat, mit und ohne Wäsche, mit und ohne Nala.
Außerdem danke ich meinem Freund Tino dafür, dass er mir in stressigen Zeiten viel
abgenommen und mich unterstützt hat, wann immer er konnte.
Vor allem aber danke ich meiner Mama für ihre bedingungslose Unterstützung während
meiner gesamten Ausbildung. Du hattest immer ein offenes Ohr, hast immer eine Lösung
gefunden oder wusstest Rat, und du hast mir immer Mut zum Weitermachen gegeben. Du hast
mir die Kraft gegeben bis hierhin und noch viel weiter zu kommen, und dafür danke ich dir von
ganzem Herzen.

Mechanisms of Axonal Transport in ALS Anlage 1
Anne Seifert 149 / 157
Anlage 1 Erklärungen zur Eröffnung des Promotionsverfahrens,
Technische Universität Dresden
Medizinische Fakultät Carl Gustav Carus
Promotionsordnung vom 24. Juli 2011
1. Hiermit versichere ich, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und
ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe; die aus fremden
Quellen direkt oder indirekt übernommenen Gedanken sind als solche kenntlich gemacht.
2. Bei der Auswahl und Auswertung des Materials sowie bei der Herstellung des Manuskripts
habe ich Unterstützungsleistungen von folgenden Personen erhalten:
Prof. Dr. Dr. Andreas Hermann
Prof. Dr. Stefan Diez
Dr. Hauke Drechsler
3. Weitere Personen waren an der geistigen Herstellung der vorliegenden Arbeit nicht beteiligt.
Insbesondere habe ich nicht die Hilfe eines kommerziellen Promotionsberaters in Anspruch
genommen. Dritte haben von mir weder unmittelbar noch mittelbar geldwerte Leistungen für
Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen.
4. Die Arbeit wurde bisher weder im Inland noch im Ausland in gleicher oder ähnlicher Form
einer anderen Prüfungsbehörde vorgelegt.
5. Die Inhalte dieser Dissertation wurden in folgender Form veröffentlicht: Noch nicht zutreffend.
6. Ich bestätige, dass es keine zurückliegenden erfolglosen Promotionsverfahren gab.
7. Ich bestätige, dass ich die Promotionsordnung der Medizinischen Fakultät der Technischen
Universität Dresden anerkenne.
8. Ich habe die Zitierrichtlinien für Dissertationen an der Medizinischen Fakultät der
Technischen Universität Dresden zur Kenntnis genommen und befolgt.
9. Ich bin mit den "Richtlinien zur Sicherung guter wissenschaftlicher Praxis, zur Vermeidung
wissenschaftlichen Fehlverhaltens und für den Umgang mit Verstößen" der Technischen
Universität Dresden einverstanden.
Ort, Datum Unterschrift des Doktoranden

Mechanisms of Axonal Transport in ALS Anlage 2
Anne Seifert 150 / 157
Anlage 2 Erklärung über die Einhaltung gesetzlicher Bestimmungen.
Hiermit bestätige ich die Einhaltung der folgenden aktuellen gesetzlichen Vorgaben im Rahmen
meiner Dissertation
Untersuchungen mit Personenbezug oder Sachverhalten, die das Medizinproduktegesetz
betreffen
Aktenzeichen der zuständigen Ethikkommission: EK45022009; EK393122012
Entfällt
Az. 55-8811.71/170
mungen der Medizinischen Fakultät und des
Universitätsklinikums Carl Gustav Carus.
Ort, Datum Unterschrift des Doktoranden
Related Documents











