Leigh Disease Caused by the Mitochondrial DNA G14459A Mutation in Unrelated Families Denise M. Kirby, BSc(Hons),* Stephen G. Kahler, MD,² Mary-Louise Freckmann, FRACP,‡ Dinah Reddihough, FRACP,§ and David R. Thorburn, PhD* Leigh disease can be caused by defects of both nuclear and mitochondrially encoded genes. One mitochondrial DNA mutation, G14459A, has been associated with both respiratory chain complex I deficiency and Leber’s hered- itary optic neuropathy, with or without dystonia. Here, we report the occurrence of this mutation in 3 complex I–deficient patients from 2 separate pedigrees who pre- sented with Leigh disease, with no evidence or family his- tory of Leber’s hereditary optic neuropathy or dystonia. Kirby DM, Kahler SG, Freckmann M-L, Reddihough D, Thorburn DR. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann Neurol 2000;48:102–104 Leigh disease (LD), or subacute necrotizing encephalo- myelopathy, is a neurodegenerative disease caused by defects in mitochondrial energy generation, including pyruvate dehydrogenase and components of the respi- ratory chain. 1 It is characterized by progressive neuro- logical disease with motor and intellectual regression, with onset typically in infancy, and by characteristic lesions of the basal ganglia and brainstem. Elevated lac- tate levels in both blood and cerebrospinal fluid (CSF) are often present. Respiratory chain complex I defi- ciency is one of the most common causes of LD, affecting 12 of 35 patients in our study of LD in Aus- tralia. 1 We have now studied 57 patients with isolated complex I deficiency, 23 of whom presented with LD. A number of mutations of mitochondrial DNA (mtDNA) have been reported in LD, including some associated with complex I deficiency. Mutations of the mtDNA tRNA-leucine UUR gene as well as defects in the mitochondrial genes encoding complex I subunits can cause complex I deficiency. 2 Among the mtDNA point mutations in subunit genes affecting complex I activity are several that have been reported in Leber’s hereditary optic neuropathy (LHON), a condition characterized by the sudden onset of blindness, often in young adulthood. One of these point mutations, G14459A, in the gene encoding the ND6 subunit of complex I has been described in patients with LHON or dystonia, or a combination of both LHON and dys- tonia. 3–5 Here, we report on 3 complex I–deficient pa- tients from 2 unrelated families presenting with LD, in whom the G14459A mutation in the ND6 gene was found. Dystonia was not a prominent feature of their disease, and blindness was not present. Patients Patient 1 was investigated at 9 months of age because of delay in motor milestones and extreme hypotonia. He had no dysmorphic features. Blood and CSF lactate levels were moderately elevated at 3.4 and 3.6 mM, respectively (nor- mal , 2.5 mM). A computed tomography scan at this time showed bilateral low-density lesions of the basal ganglia con- sistent with LD. Electromyographic and nerve conduction studies were normal. At the age of 4 years and 5 months, he was having one to four seizures each day, and the clinical picture had changed from hypotonia to spasticity. At the age of 4 years and 10 months, he had an episode of constant head turn to the right which was attributed to a persistent asymmetrical tonic neck reflex but which, in retrospect, could be interpreted as dystonic posturing. His condition de- teriorated over the next few years, with increasing seizures, and he died of respiratory arrest at the age of 6 years and 11 months. Patient 2, the brother of Patient 1, presented at 3 months of age with roving eye movements, hypotonia, and neurolog- ical and developmental regression. At the age of 7 months, he was admitted to the hospital with an intercurrent infec- tion, and seizures were noted. Neurological opinion then was that his features were consistent with LD, although no neu- roimaging was performed. Blood and CSF lactates were 4.0 and 4.2 mM, respectively. He collapsed suddenly at the age of 10 months with profound metabolic acidosis and died. Patient 3, a girl, was the first child of nonconsanguineous parents. Developmental delay was apparent at the age of 6 months, with athetosis, abnormal extraocular movements, and hypotonia. Regression was apparent at the age of 8 months, when she presented for investigation. Blood and CSF lactates were 5.4 and 4.4 mM, respectively. Magnetic resonance imaging showed cerebral atrophy, involvement of basal ganglia, medial thalamic nuclei, anterior pons, and ce- rebral peduncles consistent with LD. She deteriorated rap- idly, with episodes of hypoventilation, and died at the age of 8 months. Results and Discussion Complex I deficiency was demonstrated in mitochon- dria from cultured fibroblasts of Patients 1 and 3 and From the *Murdoch Institute, ²Victorian Clinical Genetics Service, and §Department of Child Development and Rehabilitation, Royal Children’s Hospital, Melbourne, and ‡Department of Medical Ge- netics, Sydney Children’s Hospital, Sydney, Australia. Received Jul 23, 1999, and in revised form Jan 3, 2000. Accepted for publication Feb 29, 2000. Address correspondence to Dr Thorburn, Murdoch Institute, Royal Children’s Hospital, Flemington Road, Parkville, Victoria, 3052 Australia. BRIEF COMMUNICATIONS 102 Copyright © 2000 by the American Neurological Association

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Leigh Disease Caused by theMitochondrial DNAG14459A Mutation inUnrelated FamiliesDenise M. Kirby, BSc(Hons),* Stephen G. Kahler, MD,†Mary-Louise Freckmann, FRACP,‡Dinah Reddihough, FRACP,§and David R. Thorburn, PhD*
Leigh disease can be caused by defects of both nuclearand mitochondrially encoded genes. One mitochondrialDNA mutation, G14459A, has been associated with bothrespiratory chain complex I deficiency and Leber’s hered-itary optic neuropathy, with or without dystonia. Here,we report the occurrence of this mutation in 3 complexI–deficient patients from 2 separate pedigrees who pre-sented with Leigh disease, with no evidence or family his-tory of Leber’s hereditary optic neuropathy or dystonia.
Kirby DM, Kahler SG, Freckmann M-L,Reddihough D, Thorburn DR. Leigh diseasecaused by the mitochondrial DNA G14459A
mutation in unrelated families.Ann Neurol 2000;48:102–104
Leigh disease (LD), or subacute necrotizing encephalo-myelopathy, is a neurodegenerative disease caused bydefects in mitochondrial energy generation, includingpyruvate dehydrogenase and components of the respi-ratory chain.1 It is characterized by progressive neuro-logical disease with motor and intellectual regression,with onset typically in infancy, and by characteristiclesions of the basal ganglia and brainstem. Elevated lac-tate levels in both blood and cerebrospinal fluid (CSF)are often present. Respiratory chain complex I defi-ciency is one of the most common causes of LD,affecting 12 of 35 patients in our study of LD in Aus-tralia.1 We have now studied 57 patients with isolatedcomplex I deficiency, 23 of whom presented with LD.
A number of mutations of mitochondrial DNA(mtDNA) have been reported in LD, including someassociated with complex I deficiency. Mutations of the
mtDNA tRNA-leucine UUR gene as well as defects inthe mitochondrial genes encoding complex I subunitscan cause complex I deficiency.2 Among the mtDNApoint mutations in subunit genes affecting complex Iactivity are several that have been reported in Leber’shereditary optic neuropathy (LHON), a conditioncharacterized by the sudden onset of blindness, oftenin young adulthood. One of these point mutations,G14459A, in the gene encoding the ND6 subunit ofcomplex I has been described in patients with LHONor dystonia, or a combination of both LHON and dys-tonia.3–5 Here, we report on 3 complex I–deficient pa-tients from 2 unrelated families presenting with LD, inwhom the G14459A mutation in the ND6 gene wasfound. Dystonia was not a prominent feature of theirdisease, and blindness was not present.
PatientsPatient 1 was investigated at 9 months of age because ofdelay in motor milestones and extreme hypotonia. He hadno dysmorphic features. Blood and CSF lactate levels weremoderately elevated at 3.4 and 3.6 mM, respectively (nor-mal , 2.5 mM). A computed tomography scan at this timeshowed bilateral low-density lesions of the basal ganglia con-sistent with LD. Electromyographic and nerve conductionstudies were normal. At the age of 4 years and 5 months, hewas having one to four seizures each day, and the clinicalpicture had changed from hypotonia to spasticity. At the ageof 4 years and 10 months, he had an episode of constanthead turn to the right which was attributed to a persistentasymmetrical tonic neck reflex but which, in retrospect,could be interpreted as dystonic posturing. His condition de-teriorated over the next few years, with increasing seizures,and he died of respiratory arrest at the age of 6 years and 11months.
Patient 2, the brother of Patient 1, presented at 3 monthsof age with roving eye movements, hypotonia, and neurolog-ical and developmental regression. At the age of 7 months,he was admitted to the hospital with an intercurrent infec-tion, and seizures were noted. Neurological opinion then wasthat his features were consistent with LD, although no neu-roimaging was performed. Blood and CSF lactates were 4.0and 4.2 mM, respectively. He collapsed suddenly at the ageof 10 months with profound metabolic acidosis and died.
Patient 3, a girl, was the first child of nonconsanguineousparents. Developmental delay was apparent at the age of 6months, with athetosis, abnormal extraocular movements,and hypotonia. Regression was apparent at the age of 8months, when she presented for investigation. Blood andCSF lactates were 5.4 and 4.4 mM, respectively. Magneticresonance imaging showed cerebral atrophy, involvement ofbasal ganglia, medial thalamic nuclei, anterior pons, and ce-rebral peduncles consistent with LD. She deteriorated rap-idly, with episodes of hypoventilation, and died at the age of8 months.
Results and DiscussionComplex I deficiency was demonstrated in mitochon-dria from cultured fibroblasts of Patients 1 and 3 and
From the *Murdoch Institute, †Victorian Clinical Genetics Service,and §Department of Child Development and Rehabilitation, RoyalChildren’s Hospital, Melbourne, and ‡Department of Medical Ge-netics, Sydney Children’s Hospital, Sydney, Australia.
Received Jul 23, 1999, and in revised form Jan 3, 2000. Acceptedfor publication Feb 29, 2000.
Address correspondence to Dr Thorburn, Murdoch Institute, RoyalChildren’s Hospital, Flemington Road, Parkville, Victoria, 3052Australia.
BRIEF COMMUNICATIONS
102 Copyright © 2000 by the American Neurological Association

in skeletal muscle and liver homogenates from Patients2 and 3 (Table). The G14459A mutation was detectedby restriction fragment length polymorphism analysisas described2 and confirmed by sequencing part of theND6 gene. The mutation was present at a greater than95% mutant load in these tissues. The quantificationmethod cannot detect extremely small proportions(,1%) of one species of mtDNA, but blood from Pa-tient 3 seemed to be homoplasmic for mutantmtDNA, and blood from her mother and maternalgrandmother seemed to be homoplasmic for wild-typemtDNA. The mother of Patients 1 and 2 declinedmtDNA testing. Sequencing of nucleotides 1 to 391 ofthe hypervariable D-loop region of mtDNA from Pa-tients 1 and 3 showed that the two D-loop sequencesdiffered at three locations: nt 152 (T in Patient 1, C inPatient 3), nt 195 (C in Patient 1, T in Patient 3), andan insertion of two extra C’s in the string of seven C’sfrom nt 303 to nt 309 in Patient 1, indicating thatthese two families are not related.6
We have studied 23 complex I–deficient LD patientsand have found 3 patients from 2 unrelated pedigreeswith the G14459A mutation. These 3 patients had lac-tic acidosis, regression, and symptoms of brainstemdysfunction. Neuroimaging was performed in Patients1 and 3, and computed tomography or magnetic reso-nance imaging findings were consistent with LD. Dys-tonia was not present in Patients 2 and 3. There wasan episode in Patient 1 that could be interpreted asdystonic posturing, but this was not a prominent featureof his disease. There was no other history of blindness ordystonia in the patients or their families. Histochemicalexamination of skeletal muscle from Patients 1 and 3showed only nonspecific changes.
The G14459A mutant loads were greater than 95%in our 3 patients. There does not seem to be a simplecorrelation between mutant load and clinical features.In the initial family reported on,4 individuals with agreater than 99% load presented with only dystonia oronly LHON. One asymptomatic individual was re-
ported to have 99% mutant mtDNA in lymphoblasts;however, this could possibly be an artifact of transfor-mation and cell culture, where a clone with a high pro-portion of mutant mtDNA may have predominated. Inthe additional 2 families reported on by Shoffner andco-workers,5 some individuals were symptomatic witha 50% mutant load. These investigators propose thatthe G14459A mutation has a high degree of tissuespecificity, affecting optic nerve and basal ganglia. Theclinical features of our patients suggest that the muta-tion can have more systemic manifestations. It seemsthat the subunit affected by the G14459A mutation,ND6, is essential for the correct assembly of the mul-tisubunit complex I.7 At the high mutant loads foundin our patients, it is possible that complex I assembly iscompromised.
The G14459A mutation in the mtDNA gene encod-ing the ND6 subunit of complex I has not previouslybeen associated with LD. Transmitochondrial cybridstudies have shown that this mutation causes a defectof complex I activity.8 The G14459A mutation fulfillsaccepted criteria for pathogenicity, and there seems lit-tle doubt that it is causative of complex I–deficient LDin our 2 unrelated families. It is unclear whether thesevere phenotype in our 2 families is a result simply ofhigher mutant loads in certain brain regions or whethersome genetic or environmental cause has contributedto the more severe phenotype compared with that ofother reported G14459A families. The latter possibilityis suggested by the recent description of severe neuro-degenerative disease in a family with the commonLHON G11778A mutation,9 despite the fact thatmost people who are homoplasmic for this mutationare asymptomatic.
The 3 G14459A families already reported on haddifferent mtDNA haplotypes.5 Our 2 new families alsohave different mtDNA D-loop sequences, confirmingthat the mutation has occurred independently in mul-tiple mtDNA lineages. Furthermore, the mother andmaternal grandmother of Patient 3 were apparently ho-
Table. Residual Complex I Activities and Mutant Loads in Tissues from Patients with the G14459A Mutation
Family A Family B
Patient 1 Patient 2 Patient 3 MotherMaternal
Grandmother
Fibroblasts Muscle Liver Fibroblasts Muscle Liver Blood Blood Blood
Mutation (%) 97 97 97 99 95 98 .99 ,1 ,1Complex I (%)
(normal range)25 (50–145) 6 (36–167) 46 (65–137) 15 (50–145) 16 (36–167) 37 (65–137)
Complex I activity is expressed as a percentage of the control mean relative to citrate synthase.2 Mutant load is expressed as the percentage ofmutant genomes. Respiratory chain enzymology and mitochondrial DNA mutation analysis were performed as described elsewhere.1,2 Quan-tification of mutant loads was performed by labeling polymerase chain reaction (PCR) products with [a-33P]dCTP during the last PCR cycle,digesting the products with MaeIII, and analysis on a 5% nondenaturing polyacrylamide gel. The proportion of mutant load was quantifiedusing a Storm PhosphorImager with ImageQuant software (Molecular Dynamics, Sunnyvale, CA). Patients 1 and 2 are Patients 14a and 14bin the report by Kirby and co-workers2 and Patients 22a and 22b in the report by Rahman and colleagues.1
Brief Communication: Kirby et al: LD and mtDNA in G14459A Mutation 103

moplasmic for the normal sequence at nt 14459. Al-though this does not exclude the possibility of highermutant loads in other tissues, it suggests that the mu-tation may have arisen de novo in Patient 3.
In our group of 23 complex I–deficient LD patients,the G14459A mutation is present in the 3 patients wedescribe here, representing 2 pedigrees in a total of 21.We have screened all patients referred to us in whomdystonia was a feature for investigation of possible mi-tochondrial disease as well as all patients found to havecomplex I deficiency, a total of 93 patients. No furtherpatients have been found to harbor the mutation.Shoffner and co-workers5 screened 19 patients withLD and found no patients with the mutation; hence,they concluded that this was not a common cause ofLD. Our results demonstrate that this mutation shouldbe tested for in LD patients with proven complex Ideficiency. Furthermore, because dystonia has been re-ported as a major feature of LD,10 it should be testedfor in patients presenting with dystonia or other move-ment abnormalities that may reflect basal ganglia dam-age and LD. Several other mtDNA point mutationshave been implicated in LD both in subunit genes11–14
and in tRNA genes.15–17 Although maternally inher-ited mtDNA mutations are probably not a major causeof complex I–deficient LD, it is important to identifythose cases in which they are causative. There are con-siderable implications for genetic counselling and pre-natal diagnosis.
This study was supported in part by an institute grant from theNational Health and Medical Research Council of Australia.
References1. Rahman S, Blok RB, Dahl HHM, et al. Leigh syndrome: clin-
ical features and biochemical and DNA abnormalities. AnnNeurol 1996;39:343–351
2. Kirby DM, Crawford M, Cleary MA, et al. Respiratory chaincomplex I deficiency: an underdiagnosed energy generation dis-order. Neurology 1999;52:1255–1264
3. Novotny EJ Jr, Singh G, Wallace DC, et al. Leber’s disease anddystonia: a mitochondrial disease. Neurology 1986;36:1053–1060
4. Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mu-tation at nucleotide pair 14459 of the NADH dehydrogenasesubunit 6 gene associated with maternally inherited Leber he-reditary optic neuropathy and dystonia. Proc Natl Acad SciUSA 1994;91:6206–6210
5. Shoffner JM, Brown MD, Stugard C, et al. Leber’s hereditaryoptic neuropathy plus dystonia is caused by a mitochondrialDNA point mutation. Ann Neurol 1995;38:163–169
6. Stoneking M, Hedgecock D, Higuchi RG, et al. Populationvariation of human mtDNA control region sequences detectedby enzymatic amplification and sequence-specific oligonucleo-tide probes. Am J Hum Genet 1991;48:370–382
7. Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mi-tochondrial NADH dehydrogenase is essential for the assemblyof the membrane arm and the respiratory function of the en-zyme. EMBO J 1998;17:4848–4858
8. Jun AS, Trounce IA, Brown MD, et al. Use of transmitochon-drial cybrids to assign a complex I defect to the mitochondrialDNA-encoded NADH dehydrogenase subunit 6 gene mutationat nucleotide pair 14459 that causes Leber hereditary optic neu-ropathy and dystonia. Mol Cell Biol 1996;16:771–777
9. Simon DK, Pulst SM, Sutton JP, et al. Familial multisystemdegeneration with parkinsonism associated with the 11778 mi-tochondrial DNA mutation. Neurology 1999;53:1787–1793
10. Lera G, Bhatia K, Marsden CD. Dystonia as the major mani-festation of Leigh’s syndrome. Mov Disord 1994;9:642–649
11. Tatuch Y, Christodoulou J, Feigenbaum A, et al. HeteroplasmicmtDNA mutation (T–-G) at 8993 can cause Leigh diseasewhen the percentage of abnormal mtDNA is high. Am J HumGenet 1992;50:852–858
12. De Vries DD, van Engelen BGM, Gabreels FJM, et al. A sec-ond missence mutation in the mitochondrial ATPase 6 gene inLeigh’s syndrome. Ann Neurol 1993;34:410–412
13. Campos Y, Martin MA, Rubio JC, et al. Leigh syndrome asso-ciated with the T9176C mutation in the ATPase 6 gene ofmitochondrial DNA. Neurology 1997;49:595–597
14. De Meirleir L, Seneca S, Lissens W, et al. Bilateral striatal ne-crosis with a novel point mutation in the mitochondrial ATPase6 gene. Pediatr Neurol 1995;13:242–246
15. Chalmers RM, Lamont PJ, Nelson I, et al. A mitochondrialDNA tRNA(Val) point mutation associated with adult-onsetLeigh syndrome. Neurology 1997;49:589–592
16. Berkovic SF, Shoubridge EA, Andermann F, et al. Clinicalspectrum of mitochondrial DNA mutation at base pair 8344.Lancet 1991;338:457
17. Santorelli FM, Tanji K, Sano M, et al. Maternally inheritedencephalopathy associated with a single-base insertion in themitochondrial tRNATrp gene. Ann Neurol 1997;42:256–260
104 Annals of Neurology Vol 48 No 1 July 2000

[18F]FluorodeoxyglucosePositron EmissionTomography in theDiagnosis of Cancer inPatients with ParaneoplasticNeurological Syndrome andAnti-Hu AntibodiesJ. C. Antoine, MD,* L. Cinotti, MD,† C. Tilikete,‡F. Bouhour, MD,§ J. P. Camdessanche, MD,*C. Confavreux, MD,§ A. Vighetto, MD,‡V. Renault-Mannel, MD,‡ D. Michel, MD,*and J. Honnorat, MDi
The diagnosis of cancer is often difficult in patients withparaneoplastic neurological syndrome and anti-Hu anti-bodies. Fluorodeoxyglucose 18 positron emission tomog-raphy scanning is a highly sensitive and specific methodto detect lung tumors. We investigated 15 patients withparaneoplastic neurological syndrome and anti-Hu anti-bodies. Radiological methods led to the diagnosis of can-cer in 12 patients, and test results were negative in 3.Whole-body [18F]fluorodeoxyglucose positron emissiontomography showed abnormal uptake in the mediasti-num in these 3 patients in accordance with the expectedlocation of the malignancy.
Antoine JC, Cinotti L, Tilikete C, Bouhour F,Camdessanche JP, Confavreux C, Vighetto A,
Renault-Mannel V, Michel D, Honnorat J.[18F]Fluorodeoxyglucose positron emissiontomography in the diagnosis of cancer inpatients with paraneoplastic neurological
syndrome and anti-Hu antibodies.Ann Neurol 2000;48:105–108
In patients with anti-Hu antibodies and paraneoplasticneurological syndrome (PNS), the neurological disor-der usually precedes the diagnosis of cancer by monthsor even years.1,2 This diagnosis is often difficult withconventional radiological investigations. In 75 to 85%of cases, the tumor is a small cell lung carcinoma
(SCLC); however, in 50 to 60% of cases, the initialradiological workup is negative, and in 5 to 10% ofpatients, the tumor is not detected despite repetitiveinvestigations.1,2 An early diagnosis of the tumor maybe important because any delay could have deleteriouseffects on the evolution of both the cancer and theneurological syndrome. We report a study showing theusefulness of [18F]fluorodeoxyglucose (FDG) positronemission tomography (PET) imaging in the diagnosisof tumors in patients with PNS and anti-Hu antibod-ies when the cancer was not detected after an initialradiological workup.
Cases and MethodsFrom January 1994 to May 1999, we investigated 15 con-secutive patients (13 men and 2 women aged 55–80 years)with PNS and anti-Hu antibodies. The neurological syn-dromes included sensory neuropathy, paraneoplastic enceph-alomyelitis, the association of paraneoplastic encephalomyeli-tis and sensory neuropathy, and limbic encephalitis. Anti-Huantibodies were detected by immunohistochemistry and con-firmed by Western blot analysis using the recombinant HuDprotein kindly furnished by Dr Josep Dalmau (Sloan Ketter-ing Cancer Center, New York, NY).3 Once anti-Hu antibod-ies were discovered in the serum of the patients, the diagno-sis of PNS was made, and patients were investigated forSCLC by chest radiography, chest computed tomography(CT) scanning, and bronchofibroscopy. If the test resultswere negative or inconclusive, another tumor was searchedfor, particularly prostate cancer by means of abdominal andpelvic CT scanning and echography. In 12 of 15 patients,the diagnosis of cancer was obtained at first investigation andconfirmed by biopsy within 2 months of the detection ofanti-Hu antibodies. In 11 cases, the tumor was SCLC, and 1patient had small cell prostate carcinoma. In 3 patients, in-vestigation results were negative or inconclusive. In these pa-tients, FDG-PET body scanning was then performed using210 mBq of FDG and a Siemens exact HR1 tomograph.Scanning involved chin to pelvis regions, starting from thepelvis 45 minutes after injection. Transverse, coronal, andsagittal planes were reconstructed.
ResultsIn the 3 patients undergoing FDG-PET scanning, ab-normal uptake of FDG was demonstrated in the me-diastinum. In 2 of these patients, renewed investiga-tions led to the diagnosis of the cancer within 2additional months. Investigation results were still neg-ative in the third patient after 18 months. The detaileddata on these patients are now discussed.
Case 1A 57-year-old man, who was a heavy cigarette smoker,was referred for a 6-month history of anxiety and epi-lepsy as well as recent memory loss consistent withlimbic encephalitis. General and neurological examina-tion findings were normal. Electroencephalographyrecorded epileptic discharges in temporal lobes. Brain
From the *Service de Neurologie, Hopital de Bellevue and EA3063, Faculte de Medecine Jacques Lisfranc, Saint-Etienne, and†Centre d’Exploration et de Recherche Medicale par Emission dePositons and ‡Service de Neurologie C, §Service de Neurologie A,and iService de Neurologie B, Hopital Neurologique, Lyon, France.
Received Oct 28, 1999, and in revised form Feb 29, 2000. Acceptedfor publication Feb 29, 2000.
Address correspondence to Dr Antoine, Service de Neurologie, Ho-pital de Bellevue, 42 055, Saint-Etienne, Cedex O2, France.
Copyright © 2000 by the American Neurological Association 105
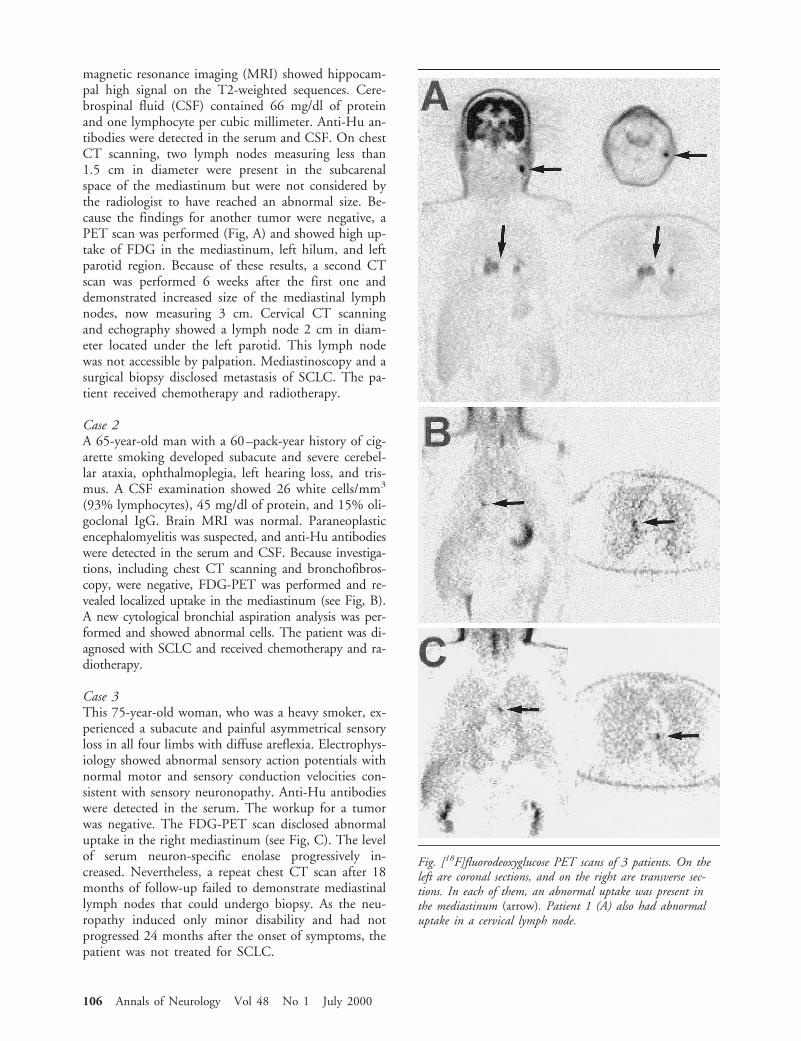
magnetic resonance imaging (MRI) showed hippocam-pal high signal on the T2-weighted sequences. Cere-brospinal fluid (CSF) contained 66 mg/dl of proteinand one lymphocyte per cubic millimeter. Anti-Hu an-tibodies were detected in the serum and CSF. On chestCT scanning, two lymph nodes measuring less than1.5 cm in diameter were present in the subcarenalspace of the mediastinum but were not considered bythe radiologist to have reached an abnormal size. Be-cause the findings for another tumor were negative, aPET scan was performed (Fig, A) and showed high up-take of FDG in the mediastinum, left hilum, and leftparotid region. Because of these results, a second CTscan was performed 6 weeks after the first one anddemonstrated increased size of the mediastinal lymphnodes, now measuring 3 cm. Cervical CT scanningand echography showed a lymph node 2 cm in diam-eter located under the left parotid. This lymph nodewas not accessible by palpation. Mediastinoscopy and asurgical biopsy disclosed metastasis of SCLC. The pa-tient received chemotherapy and radiotherapy.
Case 2A 65-year-old man with a 60–pack-year history of cig-arette smoking developed subacute and severe cerebel-lar ataxia, ophthalmoplegia, left hearing loss, and tris-mus. A CSF examination showed 26 white cells/mm3
(93% lymphocytes), 45 mg/dl of protein, and 15% oli-goclonal IgG. Brain MRI was normal. Paraneoplasticencephalomyelitis was suspected, and anti-Hu antibodieswere detected in the serum and CSF. Because investiga-tions, including chest CT scanning and bronchofibros-copy, were negative, FDG-PET was performed and re-vealed localized uptake in the mediastinum (see Fig, B).A new cytological bronchial aspiration analysis was per-formed and showed abnormal cells. The patient was di-agnosed with SCLC and received chemotherapy and ra-diotherapy.
Case 3This 75-year-old woman, who was a heavy smoker, ex-perienced a subacute and painful asymmetrical sensoryloss in all four limbs with diffuse areflexia. Electrophys-iology showed abnormal sensory action potentials withnormal motor and sensory conduction velocities con-sistent with sensory neuronopathy. Anti-Hu antibodieswere detected in the serum. The workup for a tumorwas negative. The FDG-PET scan disclosed abnormaluptake in the right mediastinum (see Fig, C). The levelof serum neuron-specific enolase progressively in-creased. Nevertheless, a repeat chest CT scan after 18months of follow-up failed to demonstrate mediastinallymph nodes that could undergo biopsy. As the neu-ropathy induced only minor disability and had notprogressed 24 months after the onset of symptoms, thepatient was not treated for SCLC.
Fig. [18F]fluorodeoxyglucose PET scans of 3 patients. On theleft are coronal sections, and on the right are transverse sec-tions. In each of them, an abnormal uptake was present inthe mediastinum (arrow). Patient 1 (A) also had abnormaluptake in a cervical lymph node.
106 Annals of Neurology Vol 48 No 1 July 2000

DiscussionThe diagnosis of cancer is frequently difficult in pa-tients with PNS and anti-Hu antibodies. SCLC devel-ops in most, and the tumor is often limited to the me-diastinal lymph nodes.1,2 Chest CT scanning or MRIis commonly used for the diagnosis of the tumor, butin 50 to 60% of cases, the findings are negative at theinitial workup,2 probably because the size of the met-astatic lymph nodes does not allow detection by thesemethods.4 When SCLC is not apparent, other tumorsmust be suspected such as prostate cancer, neuroblas-toma, or sarcoma,1,2 and when careful investigationsare negative, it is recommended to repeat them every 6months.5 It is probably important that the cancer bediagnosed as soon as possible. Indeed, in patients withanti-Hu antibodies, the tumor frequently has limitedextension1 and responds better to chemotherapy thanin patients without anti-Hu antibodies.6 Furthermore,early treatment of the cancer probably affords a betterchance of stabilizing the PNS, which is frequently themain cause of death and usually does not respond toimmunosuppressive treatment.7,8
In contrast to CT scanning, which relies on anatom-ical and morphological criteria, FDG-PET scanningdepends mainly on the metabolic characteristics of thestudied tissue. Because glycolysis is increased in malig-nancies, FDG-PET scanning seems to be a useful toolfor detecting tumors. A direct relation between tumorgrowth and FDG uptake has been demonstrated inlung cancer.9 Recent studies show that FDG-PET im-aging is accurate in predicting mediastinal involvementin patients with lung cancer. In most studies, themethod has at least a 90% sensitivity and specificity todemonstrate the malignant nature of a radiologicallydetected chest abnormality.9–12 Nevertheless, false-positive results can occur with infectious or inflamma-tory lesions.
In our 15 patients, the neurological disorders consis-tent with PNS and the presence of anti-Hu antibodiesmade the diagnosis of cancer highly probable. It wasconfirmed in 12 of our patients by the usual radiolog-ical methods, but it could not be established in 3 pa-tients. In these 3 patients, FDG-PET scanning showedabnormal uptake in the mediastinum that suggestedmetastatic lymph nodes of SCLC. These 3 cases illus-trate several aspects of the use of FDG-PET imaging inpatients with PNS. In Patient 1, the mediastinal lymphnodes did not have a clearly abnormal size on the CTscan. The FDG-PET scan prompted us to renew ra-diological investigations long before the recommendedtime, leading to the diagnosis of SCLC. In the 2 otherpatients, chest CT scanning failed to disclose enlargedlymph nodes. Because of the topography of FDG up-take and the absence of a radiological target, thoracos-copy and surgery were dismissed. In Patient 2, how-ever, the combination of a single cytological positive
aspiration with a positive FDG-PET study was consid-ered sufficient to justify treating the patient for SCLC.In the last patient, pathological or cytological evidenceof SCLC is still pending 24 months after the onset ofsymptoms. Because of the specificity of both anti-Huantibodies and FDG-PET imaging, this case raises thequestion as to whether similar patients should be con-sidered as having SCLC, despite the absence of patho-logical evidence, with the possibility of treatment withchemotherapy and radiotherapy if the neurological dis-order progresses and becomes disabling.
We did not observe patients with anti-Hu antibodiesand negative PET studies. False-negative results are ex-tremely rare with PET investigations and may corre-spond to tumors with slow growth or a diameter lessthan 1 cm.13 In this situation, it is recommended thatthe PET study be repeated every 6 months. Indeed,one should be extremely cautious when diagnosing apatient with anti-Hu antibodies as not having under-lying tumor because some patients with anti-Hu anti-bodies develop an open cancer up to 8 years after theonset of the neurological disorder.2
In conclusion, our study illustrates the use of whole-body FDG-PET scanning in the diagnosis of cancer inpatients with PNS and anti-Hu antibodies who havenegative test results after an initial workup using radio-logical methods.
This work was supported by La Ligue Contre le Cancer de le Loire,La Ligue Contre le Cancer du Rhone, and l’Association pour laRecherche Contre le Cancer.
References1. Dalmau J, Graus F, Rosenblum MK, et al. Anti-Hu associated
paraneoplastic encephalomyelitis/sensory neuronopathy. A clin-ical study of 71 patients. Medicine 1991;71:59–72
2. Luchinetti CF, Kimmel DW, Lennon V. Paraneoplastic andoncologic profiles of patients seropositive for type 1 antineuro-nal nuclear antibodies. Neurology 1998;50:652–657
3. Moll JWB, Antoine JC, Brashear HR, et al. Guidelines on thedetection of paraneoplastic anti-neuronal antibodies. Neurology1995;45:1937–1941
4. Chartran-Lefebvre C, Howarth HN, Grenier P, et al. Associa-tion of small cell lung cancer and the anti-Hu paraneoplasticsyndrome: radiological and CT findings. AJR Am J Roentgenol1998;170:1513–1517
5. Molinuevo JL, Graus F, Serrano C, et al. Utility of anti-Huantibodies in the diagnosis of paraneoplastic sensory neuropa-thy. Ann Neurol 1998;44:976–980
6. Graus F, Dalmau J, Rene R, et al. Anti-Hu antibodies in pa-tients with small cell lung cancer: association with complete re-sponse to therapy and improved survival. J Clin Oncol 1997;15:2866–2872
7. Mason WP, Graus F, Lang B, et al. Small cell lung cancer,paraneoplastic cerebellar degeneration, and the Lambert-Eatonmyasthenic syndrome. Brain 1997;120:1279–1300
8. Keime-Guibert F, Graus F, Broet P, et al. Clinical outcome ofpatients with anti-Hu-associated encephalomyelitis after treat-ment of the tumor. Neurology 1999;53:1719–1723
Brief Communication: Antoine et al: FDG-PET in the Diagnosis of Cancer 107

9. Duhaylongsod FG, Lowe VJ, Patz EF Jr, et al. Lung tumorgrowth correlates with glucose metabolism measured byfluoride-18 fluorodeoxyglucose positron emission tomography.Ann Thorac Surg 1995;60:1348–1352
10. Sazon DA, Santiago SM, Soo Hoo GW, et al. Fluorodeoxy-glucose-positron emission tomography in the detection andstaging of lung cancer. Am J Respir Crit Care Med 1996;153:417–421
11. Chin R Jr, Ward R, Keyes JW, et al. Mediastinal staging ofnon-small-cell lung cancer with positron emission tomography.Am J Respir Crit Care Med 1995;152:2090–2106
12. Valk PE, Pounds TR, Hopkins DM, et al. Staging non–smallcell lung cancer by whole-body positron emission tomographicimaging. Ann Thorac Surg 1995;60:1573–1581
13. Knight SB, Delbeke D, Stewart JR, et al. Evaluation of pulmonary lesions with FDG-PET: comparison of findings in patientswith and without a history of prior malignancy. Chest 1996;109:982–988
Linkage of AutosomalRecessive Hereditary SpasticParaplegia with MentalImpairment and ThinCorpus Callosum toChromosome 15q13–15Yoko Shibasaki,* Hajime Tanaka,* Kiyoshi Iwabuchi,†Sari Kawasaki,* Hiroshi Kondo,* Kazutoshi Uekawa,‡Masayuki Ueda,§ Tatsushi Kamiya,§ Yasuo Katayama,§Akinori Nakamura,i Hiroshi Takashima,i
Masanori Nakagawa,i Masayuki Masuda,¶ Hiroya Utsumi,¶Takuya Nakamuro,# Kazuo Tada,** Kazuhiro Kurohara,††Ken Inoue,‡‡ Fumihiko Koike,§§ Tetsuo Sakai,§§Shoji Tsuji,* and Hisashi Kobayashi*
To date, three loci for autosomal recessive hereditary spas-tic paraplegia (ARHSP) linked to chromosomes 8p12-q13,
16qter, and 15q13–15 have been characterized. We haveclinically characterized 13 Japanese ARHSP families andperformed genetic linkage analyses. All 13 families wereclassified as having the “complicated” form, which man-ifests with mental impairment and thin corpus callosum.Linkage to the 8p12-q13 and 16qter loci was excluded,although 10 of the 13 families showed marker data con-sistent with linkage to the 15q13–15 locus. The multi-point LOD score of the 10 families linked to chromo-some 15 was above 9.00 in the 3-centimorgan segmentflanked by D15S994 and D15S659, with a maximummultipoint LOD score of 9.68 at a position 1.2 centimor-gans telomeric from D15S994 to D15S659. We haveshown that ARHSP with thin corpus callosum, a subtypeof recessive spastic paraplegia, maps to chromosome15q13–15.
Shibasaki Y, Tanaka H, Iwabuchi K, Kawasaki S,Kondo H, Uekawa K, Ueda M, Kamiya T,
Katayama Y, Nakamura A, Takashima H,Nakagawa M, Masuda M, Utsumi H,
Nakamuro T, Tada K, Kurohara K, Inoue K,Koike F, Sakai T, Tsuji S, Kobayashi H. Linkage
of autosomal recessive hereditary spastic paraplegiawith mental impairment and thin corpus
callosum to chromosome 15q13–15.Ann Neurol 2000;48:108–112
Familial or hereditary spastic paraplegia (HSP) is aclinically and genetically heterogeneous group of neu-rodegenerative disorders of the motor system character-ized by slowly progressive spasticity and weakness ofthe lower extremities. Clinically, in addition to the“pure” form of HSP, which solely affects the legs,“complicated” forms of HSP also exist. These latterforms may show additional symptoms such as mentalretardation and deterioration, ataxia, epilepsy, optic at-rophy, retinal changes, ichthyosis, and peripheral neu-ropathy.1,2
To date, three loci for autosomal recessive HSP(ARHSP), 8p12-q13 (SPG5A: MIM 270800), 16q24.3(SPG7: MIM 602783), and 15q13–15 (SPG11: MIM604360), have been reported.3–5 The first described lo-cus for ARHSP showed a pure HSP phenotype withno other associated symptoms and was linked to chro-mosome 8p12-q13 between PLAT and D8S279, a32.2-centimorgan (cM) region, in 4 Tunisian families.4
The second locus was identified on chromosome16q24.3 in a large consanguineous Italian family.3 Thegene at this 16q24 locus, paraplegin, was then identi-fied by Casari and co-workers,6 and several mutationsof this nuclear-encoded mitochondrial metalloprotease(paraplegin) gene were shown to be responsible for
From the *Department of Neurology, Brain Research Institute, Ni-igata University, Niigata, †Department of Neurology and Psychia-try, Kanagawa Rehabilitation Center, Kanagawa, ‡National Kuma-moto Minami Hospital, Kumamoto, §Division of Neurology,Second Department of Internal Medicine, Nippon Medical School,and ¶Third Department of Internal Medicine, Tokyo Medical Uni-versity, Tokyo, iThird Department of Internal Medicine, Ka-goshima University, Kagoshima, #Department of Neurology, NaraMedical University, Nara, **National Sanatorium Hyogo ChuoHospital, Hyogo, ††Department of Internal Medicine, Saga Medi-cal School, Saga, and §§Department of Neurology, National Chi-kugo Hospital, Fukuoka, Japan; and ‡‡Department of Molecularand Human Genetics, Baylor College of Medicine, Houston, TX.
Received Dec 8, 1999, and in revised form Feb 29, 2000. Acceptedfor publication Mar 22, 2000.
Address correspondence to Dr Kobayashi, Department of Neurol-ogy, Brain Research Institute, Niigata University, 1 Asahimachi-dori, Niigata, Niigata 951-8585, Japan.
108 Copyright © 2000 by the American Neurological Association

pure and complicated forms of HSP. The third locusfor ARHSP was recently identified as mapping to chro-mosome 15q13–15 in 7 spastic paraplegia familiesfrom North America and Europe whose membersshowed both pure and complicated forms of the dis-ease.5 In 2 of the 7 families, magnetic resonance imag-ing (MRI) revealed thin corpus callosum (TCC); how-ever, the other 5 families did not show TCC.
Iwabuchi and colleagues7 have reported on a seriesof Japanese patients with a clinical subtype of compli-cated ARHSP with TCC and mental retardation.7
There have been reports on approximately 30 Japanesepatients with this unique complicated form of AR-HSP.8–11 Overall, these patients showed slowly pro-gressive spastic paraparesis with mental retardation anddeterioration, thalamic degeneration, and TCC.7–10
In this study, we have analyzed 13 Japanese familieswith ARHSP with TCC accumulated by linkage anal-ysis from a series of candidate regions. We found evi-dence that 10 of the 13 families mapped to the chro-mosome 15q13–15 locus. Our data suggest that thisrecessive locus may be the most common HSP subtypein Japan.
Subjects and MethodsFamiliesWe examined 42 individuals from 13 Japanese families withARHSP with mental impairment and TCC: 24 family mem-
bers were affected, and the rest were not affected. Informedconsent was obtained from all studied family members. Allfamilies were newly ascertained to have ARHSP, with theexception of Families 11, 14, 27, and 310, whose clinicalfeatures have been reported elsewhere.8–11 Each of the familymembers studied was clinically examined by two or moreneurologists from a total of 10 different institutions.
The diagnostic criteria for ARHSP with mental impair-ment and TCC included the following: (1) inheritance con-sistent with an autosomal recessive trait, (2) slowly progres-sive spastic paraparesis and mental impairment, (3) thinningof the corpus callosum as revealed by brain computed to-mography or MRI, and (4) exclusion of other disorders byMRI of the spine and brain as well as other laboratorytests.5,8
GenotypingBlood samples were obtained after receiving informed con-sent from the family members. Genomic DNA was extractedfrom whole blood using standard protocols. Polymorphicmicrosatellite markers were chosen based on information ob-tained from the Genome Database and previously publishedstudies.3–5 Polymerase chain reaction amplification was per-formed using the 59-fluorescently labeled oligonucleotideprimers for DNA polymorphic markers on chromosomes8p12-q13, 16qter, and 15q13–15. We further investigatedadditional markers on chromosome 15q13–21 mapping te-lomeric to the candidate locus. The DNA markers weregenotyped as previously described.12 Analysis of the polymer-ase chain reaction products was performed using an auto-
Fig 1. Pedigrees of the 13 investigated autosomal recessive hereditary spastic paraplegia with thin corpus callosum families.
Brief Communication: Shibasaki et al: Linkage of ARHSP to Chromosome 15q13–15 109

mated fluorescent capillary DNA sequencer (ABI 310;Perkin-Elmer Applied Biosystems, Foster City, CA).
Linkage AnalysisPair-wise and multipoint LOD scores were calculated usingthe MLINK program of the FASTLINK 4.1P package and theGENEHUNTER 2.0B r2 package, respectively.13–15 The dis-ease was assumed to be an autosomal recessive trait with adisease gene frequency of 0.001. Penetrance was assumed to becomplete by the age of 20 years. The genetic intermarker dis-tances used in the analysis were sex-averaged distances basedon published maps (Marshfield Medical Research Foundation:http://www.marshmed.org/genetics/). Allele frequencies formarkers were determined using a minimum of 37 unrelatedJapanese individuals. Multipoint linkage analysis using GENE-HUNTER was performed based on the following map forchromosome 15q13–21: D15S971-(1.1 cM)-D15S118-(3.4cM)-D15S1012-(3.8 cM)-D15S146-(0.5 cM)-D15S994-(3.2cM)-D15S659-(2.2 cM)-D15S123-(5.6 cM)-D15S117.
ResultsClinical StudyAffected individuals from all 13 families were classifiedas having the complicated form of HSP. Consanguin-eous marriage was present in 8 of the 13 families (Fig1). The overall clinical picture of the families studied isshown in the Table. The mean age at onset was 14years but ranged from 6 to 30 years. The disease wasslowly progressive. Disease duration ranged from 4 togreater than 42 years. Disease duration, as expected,influenced the clinical features. In the clinical coursesof all patients, the primary symptom was spastic para-
paresis, and the secondary symptom was mental dete-rioration. The frequency of associated signs such as de-creased vibration sense or urinary disturbance increasedwith the duration of the disease.
Linkage AnalysisWe first genotyped the families by using linked mark-ers from chromosome 8p12-q13 and 16q loci. Bothloci showed a negative LOD score in all 13 families,with the exception that Families 8 and 26 showed anonsignificant positive LOD score (data not shown).We then performed two-point linkage analysis for the15q SPG11 locus. We detected evidence for linkagebetween ARHSP TCC and D15S118 on chromosome15q, with a maximum LOD score of 5.24 at a recom-bination fraction of U 5 0.07 under the hypothesis ofgenetic homogeneity. We then saturated this region bygenotyping an additional seven markers that map tothe 15q13–21 region and confirmed linkage to this re-gion. A maximum LOD score of 6.16 was obtained formarker D15S994 at U 5 0.05.
Multipoint linkage analyses were performed in all 13families by means of the GENEHUNTER program.Multipoint LOD score analysis of each individual ped-igree suggested that the disease gene in 3 of the 13families studied (Families 27, 150, and 292) did notmap to chromosome 15q13–15 because these 3 fami-lies showed a LOD score less than 22.0 over all ormost of the 15q13–15 region. No recombination wasdetected with D15S118 (LOD score 5 5.81),D15S1012 (LOD score 5 2.18), D15S146 (LODscore 5 3.27), D15S994 (LOD score 5 6.52),D15S659 (LOD score 5 6.65), or D15S123 (LODscore 5 5.12) in 8 families (Families 11, 12, 14, 17,26, 136, 300, and 320) (data not shown). It was notpossible to assign the other 2 families (Families 8 and310) with confidence to chromosome 15q13–15 or toexclude them for the locus.
The heterogeneity hypothesis was therefore tested byuse of the HOMOG program. The HOMOG pro-gram identified no evidence in support of genetic het-erogeneity for the tightly linked markers D15S994(a 5 1, x2 5 0, p 5 0.5) and D15S118 (a 5 0.87,x2 5 1.59, p 5 0.1).
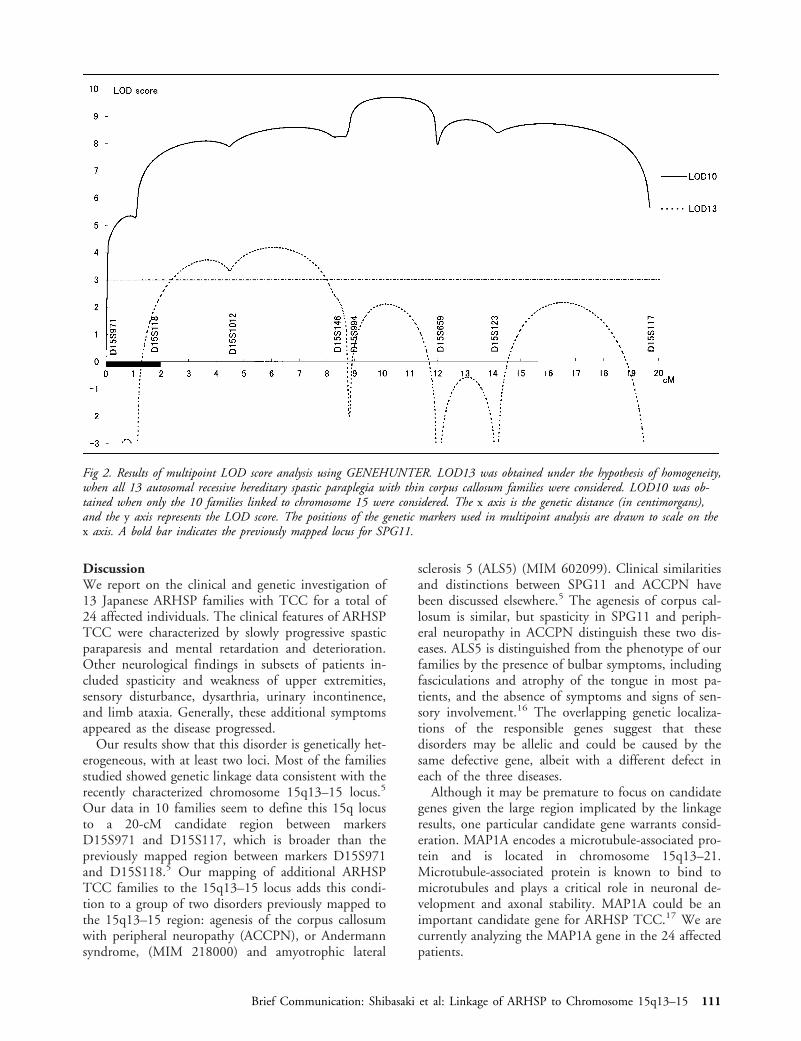
In conclusion, multipoint analysis for all 13 familiesprovided a multipoint LOD score above 3.00 for a6-cM region flanked by D15S118 and D15S146 underthe hypothesis of genetic homogeneity (Fig 2). Multi-point analysis restricted to the set of 10 families inwhich the disease gene is associated with chromosome15 showed ARHSP TCC linkage to the region be-tween D15S994 and D15S659, with multipoint LODscores (LOD10) greater than 9.00 (see Fig 2). Themaximum multipoint LOD score was 9.68 at a posi-tion 1.2 cM telomeric from D15S994 to D15S659.
Table. Autosomal Recessive Hereditary Spastic Paraplegia withThin Corpus Callosum: Summary of Clinical Features
Number of families 13Number of patients 24Consanguineous marriage 62%Mean age at onset of gait distur-
bance14 yr (range, 6–30 yr)
Mean disease duration 20 yr (range, 4–42 yr)Mental impairment 100%Thinning of corpus callosum 100%Spasticity in the upper limbs 62%b
Spasticity in the lower limbs 100%Hyperreflexia in the upper limbs 77%b
Hyperreflexia in the lower limbs 100%Extensor plantar reflexes 92%Weakness in the upper limbs 77%b
Weakness in the lower limbs 85%Muscle atrophy 77%b
Pes cavus 54%Superficial sensory disturbancea 33%b
Impaired vibration sensea 62%b
Urinary disturbance 38%b
Dysarthria 77%b
Nystagmus 15%
aImpossible to evaluate in 2 of 24 patients because of their mentallyretarded state.bThe symptoms appeared as the disease progressed.
110 Annals of Neurology Vol 48 No 1 July 2000
DiscussionWe report on the clinical and genetic investigation of13 Japanese ARHSP families with TCC for a total of24 affected individuals. The clinical features of ARHSPTCC were characterized by slowly progressive spasticparaparesis and mental retardation and deterioration.Other neurological findings in subsets of patients in-cluded spasticity and weakness of upper extremities,sensory disturbance, dysarthria, urinary incontinence,and limb ataxia. Generally, these additional symptomsappeared as the disease progressed.
Our results show that this disorder is genetically het-erogeneous, with at least two loci. Most of the familiesstudied showed genetic linkage data consistent with therecently characterized chromosome 15q13–15 locus.5
Our data in 10 families seem to define this 15q locusto a 20-cM candidate region between markersD15S971 and D15S117, which is broader than thepreviously mapped region between markers D15S971and D15S118.5 Our mapping of additional ARHSPTCC families to the 15q13–15 locus adds this condi-tion to a group of two disorders previously mapped tothe 15q13–15 region: agenesis of the corpus callosumwith peripheral neuropathy (ACCPN), or Andermannsyndrome, (MIM 218000) and amyotrophic lateral
sclerosis 5 (ALS5) (MIM 602099). Clinical similaritiesand distinctions between SPG11 and ACCPN havebeen discussed elsewhere.5 The agenesis of corpus cal-losum is similar, but spasticity in SPG11 and periph-eral neuropathy in ACCPN distinguish these two dis-eases. ALS5 is distinguished from the phenotype of ourfamilies by the presence of bulbar symptoms, includingfasciculations and atrophy of the tongue in most pa-tients, and the absence of symptoms and signs of sen-sory involvement.16 The overlapping genetic localiza-tions of the responsible genes suggest that thesedisorders may be allelic and could be caused by thesame defective gene, albeit with a different defect ineach of the three diseases.
Although it may be premature to focus on candidategenes given the large region implicated by the linkageresults, one particular candidate gene warrants consid-eration. MAP1A encodes a microtubule-associated pro-tein and is located in chromosome 15q13–21.Microtubule-associated protein is known to bind tomicrotubules and plays a critical role in neuronal de-velopment and axonal stability. MAP1A could be animportant candidate gene for ARHSP TCC.17 We arecurrently analyzing the MAP1A gene in the 24 affectedpatients.
Fig 2. Results of multipoint LOD score analysis using GENEHUNTER. LOD13 was obtained under the hypothesis of homogeneity,when all 13 autosomal recessive hereditary spastic paraplegia with thin corpus callosum families were considered. LOD10 was ob-tained when only the 10 families linked to chromosome 15 were considered. The x axis is the genetic distance (in centimorgans),and the y axis represents the LOD score. The positions of the genetic markers used in multipoint analysis are drawn to scale on thex axis. A bold bar indicates the previously mapped locus for SPG11.
Brief Communication: Shibasaki et al: Linkage of ARHSP to Chromosome 15q13–15 111

This study shows that disease inheritance was notassociated with the chromosome 15q13–15 locus(LOD , 22.0) in 3 of 13 families, indicating thatARHSP TCC is probably a genetically heterogeneousdisease. To date, however, formal tests for genetic het-erogeneity are not conclusive with the relatively linkedfamily resources. Study of additional families is re-quired to evaluate the existence of genetic heterogene-ity in ARHSP TCC.
The initial part of this study was supported by a grant from the Min-istry of Health and Welfare of Japan (H10-010) (Dr Kobayashi).
We thank the patients and their family members for their partici-pation in the study. We are grateful to Yasuyuki Aoyagi, MiyukiTsuchiya, and Kyoko Suzuki for excellent technical assistance. Wealso thank Francisco Martinez-Murillo and Prof Eric P. Hoffmanfrom the Research Center for Genetic Medicine, Children’s Na-tional Medical Center, Washington, DC, for helpful comments onthis manuscript.
References1. Bruyn RPM, Scheltens P. Hereditary spastic paraparesis
(Strumpell-Lorrain). In: Vinken PJ, Bruyn GW, Klawans HL,eds. Handbook of clinical neurology, vol 59. Amsterdam:Elsevier Science, 1991:301–318
2. Sutherland JM. Familial spastic paraplegia. In: Vinken PJ,Bruyn GW, eds. Handbook of clinical neurology, vol 22.Amsterdam: North Holland Publishing, 1975:421–431
3. De Michele G, De Fusco M, Cavalcanti F, et al. A new locusfor autosomal recessive hereditary spastic paraplegia maps tochromosome 16q24.3. Am J Hum Genet 1998;63:135–139
4. Hentati A, Pericak-Vance MA, Hung W-Y, et al. Linkage ofthe “pure” autosomal recessive familial spastic paraplegia tochromosome 8 markers and evidence of genetic locus heteroge-neity. Hum Mol Genet 1994;3:1263–1267
5. Martinez Murillo F, Kobayashi H, Pegoraro E, et al. Geneticlocalization of a new locus for recessive familial spastic parapa-resis to 15q13–15. Neurology 1999;53:50–56
6. Casari G, De Fusco M, Ciarmatori S, et al. Spastic paraplegiaand OXPHOS impairment caused by mutations in paraplegin,a nuclear-encoded mitochondrial metalloprotease. Cell 1998;93:973–983
7. Iwabuchi K, Yagishita S, Amano N, Kosaka K. A new type ofcomplicated form of hereditary spastic paraplegia showing men-tal deterioration, quadriplegia with muscular atrophy, sensorydisturbance, extrapyramidal disorders and epilepsy. RinshoShinkeigaku 1991;31:945–952
8. Iwabuchi K, Kubota Y, Hanihara T, Nagatomo H. Three pa-tients of complicated form of autosomal recessive hereditaryspastic paraplegia associated with hypoplasia of the corpus cal-losum. No To Shinkei 1994;46:941–947
9. Ueda M, Katayama Y, Kamiya T, et al. Hereditary spastic para-plegia with a thin corpus callosum and thalamic involvement inJapan. Neurology 1998;51:1751–1754
10. Nakamura A, Izumi K, Umehara F, et al. Familial spastic para-plegia with mental impairment and thin corpus callosum.J Neurol Sci 1995;131:35–42
11. Morishita S, Konagaya M, Konagaya Y, Takayanagi T. MRIstudy of three siblings of suspicious Sjogren-Larsson syndrome.Rinsho Shinkeigaku 1990;30:1118–1122
12. Kobayashi H, Matise TC, Marks HG, et al. Towards fully au-tomated genotyping: use of an X linked recessive spastic para-plegia family to test alternative analysis methods. Hum Genet1995;95:483–490
13. Cottingham RW Jr, Idury RM, Schaffer AA. Faster sequentialgenetic linkage computations. Am J Hum Genet 1993;53:252–263
14. Schaffer AA, Gupta SK, Shriram K, Cottingham RW Jr. Avoid-ing recomputation in linkage analysis. Hum Hered 1994;44:225–237
15. Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametricand nonparametric linkage analysis: a unified multipoint ap-proach. Am J Hum Genet 1996;58:1347–1363
16. Hentati A, Ouahchi K, Pericak-Vance MA, et al. Linkage of acommoner form of recessive amyotrophic lateral sclerosis tochromosome 15q15–q22 markers. Neurogenetics 1998;2:55–60
17. Rainier S, Jones SM, Esposito C, et al. Analysis of microtubule-associated protein 1a gene in hereditary spastic paraplegia. Neu-rology 1998;51:1509–1510
112 Annals of Neurology Vol 48 No 1 July 2000

Multifocal or GeneralizedTonic Dystonia of ComplexRegional Pain Syndrome:A Distinct Clinical EntityAssociated with HLA-DR13J. J. van Hilten, MD, PhD,* W. J. T. van de Beek, MSc,*and B. O. Roep, MD, PhD†
We report on 26 patients with a distinct phenotype ofcomplex regional pain syndrome that progressed towarda multifocal or generalized tonic dystonia. The dystoniainitiated distally, involved mainly flexor muscles, and wasassociated with sensory and autonomic symptoms. Dry-ness of the eyes or mouth and bladder and bowel distur-bances were frequently reported. There was no increasein the familial prevalence of autoimmune-mediated dis-eases. Compared with controls, a significant elevation ofHLA-DR13 was found in the patients. Thus, HLA-DR13may be a factor indicating susceptibility to this distinctphenotype of complex regional pain syndrome.
van Hilten JJ, van de Beek WJT, Roep BO.Multifocal or generalized tonic dystonia ofcomplex regional pain syndrome: a distinctclinical entity associated with HLA-DR13.
Ann Neurol 2000;48:113–116
Complex regional pain syndrome (CRPS) is a syn-drome characterized by sensory, autonomic, and motorfeatures that may occur spontaneously, after trauma, orin the setting of neurological or rheumatic disease.1 Al-though the underlying pathophysiology still remainsobscure, the clinical hallmarks, including pain, edema,discolored skin, and loss of function, have led someauthors to hypothesize that CRPS is the result of anexaggerated inflammatory response to tissue injury.2,3
Associations of CRPS with the human leukocyte anti-gen (HLA) class II loci DR15 and DQ1 have beenreported.4,5
In this study, we evaluated the clinical aspects of thedisease, the familial prevalence of autoimmune-mediated diseases, and the association with the DQ
and DR alleles in 26 CRPS patients whose conditionprogressed toward a multifocal or generalized dystonia.
Patients and MethodsTwenty-six consecutive caucasian patients (24 women, 2men) were recruited at the outpatient clinic during the pe-riod 1998 through 1999. Thirteen patients were referredfrom other hospitals. All patients met the criteria of CRPS(Table 1).6 The diagnosis of CRPS was made on the presen-tation of symptoms in the extremity that was first affected.All patients had to show a progressive course, and at leasttwo extremities had to be affected with tonic dystonia.
All patients were evaluated with regard to their clinicalfeatures, disease course, and (family) history of autoimmune-mediated diseases. In the affected limbs, particular attentionwas given to the perception of light touch with cotton wool,pinprick sensation using the sharp side of a wooden stick,and vibratory sense using a tuning fork. To evaluate distur-bances of temperature perception in the affected limbs, allpatients were questioned about their daily experiences withrespect to temperature sensation when using water (ie, whentaking a bath or shower).
A history of autoimmune-mediated disease was verified bychecking the records of the family physician. The diagnosticcriteria for the identified autoimmune diseases were as fol-lows: rheumatoid arthritis was based on the American Rheu-matism Association criteria,7 thyroid disorders were based onclinical data and measurements of thyroid antibodies, andpsoriasis was based on clinical data.
HLA TypingHLA-DRB1, HLA-DQA1, and HLA-DQB1 polymorphismwas tested by polymerase chain reaction using sequence-specific oligonucleotides as probes on locus-specific amplifiedDNA according to the recommended protocol of the Elev-enth International Histocompatibility Workshop.8 A panelof 2,355 healthy blood donors was used for comparison ofphenotypes with those in the CRPS patient group.9,10 TheHLA phenotype frequencies were compared using Fisher’s x2
test for 2 3 2 contingency tables. Probability values werecorrected for the number of comparisons.
ResultsClinical CharacteristicsThe mean age at onset of disease was 35 years (range,16–58 years), and the mean disease duration was 11years (range, 2–37 years). In 10 patients, the onset ofCRPS was preceded by an injury or operation. Seven-teen patients had an abrupt onset that was followed bygradual or intermittent progression. Nine patients hadgradual progression from onset without remission. Atthe time of evaluation, CRPS affected three or four ex-tremities in 7 and 19 patients, respectively.
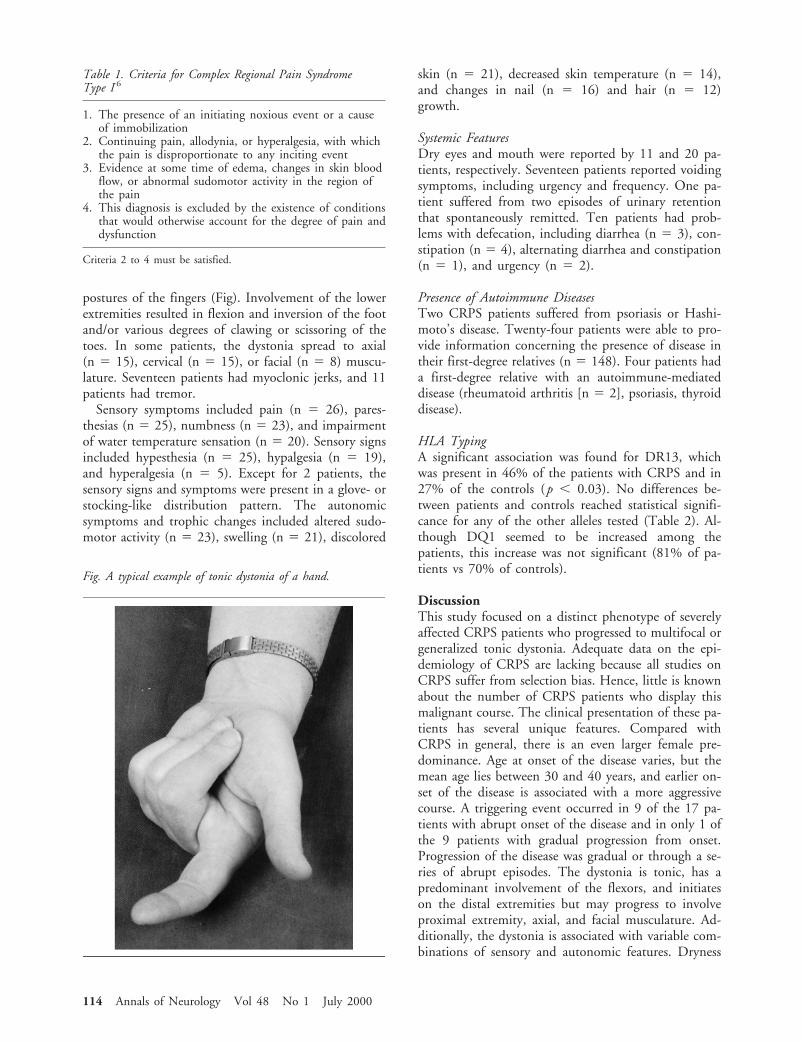
The dystonia was tonic in all patients. In 12 pa-tients, dystonia occurred together with sensory and au-tonomic symptoms at onset, although in 14 patients,dystonia developed at a later phase of the disease. In allbut 1 patient, dystonia of the hands resulted in flexor
From the *Departments of Neurology and †Immunohematologyand Blood Bank, Leiden University Medical Center, Leiden, TheNetherlands.
Received Dec 14, 1999, and in revised form Feb 21, 2000. Ac-cepted for publication Mar 27, 2000.
Address correspondence to Dr van Hilten, Department of Neurol-ogy, Leiden University Medical Center, PO Box 9600, 2300 RCLeiden, The Netherlands.
Copyright © 2000 by the American Neurological Association 113
postures of the fingers (Fig). Involvement of the lowerextremities resulted in flexion and inversion of the footand/or various degrees of clawing or scissoring of thetoes. In some patients, the dystonia spread to axial(n 5 15), cervical (n 5 15), or facial (n 5 8) muscu-lature. Seventeen patients had myoclonic jerks, and 11patients had tremor.
Sensory symptoms included pain (n 5 26), pares-thesias (n 5 25), numbness (n 5 23), and impairmentof water temperature sensation (n 5 20). Sensory signsincluded hypesthesia (n 5 25), hypalgesia (n 5 19),and hyperalgesia (n 5 5). Except for 2 patients, thesensory signs and symptoms were present in a glove- orstocking-like distribution pattern. The autonomicsymptoms and trophic changes included altered sudo-motor activity (n 5 23), swelling (n 5 21), discolored
skin (n 5 21), decreased skin temperature (n 5 14),and changes in nail (n 5 16) and hair (n 5 12)growth.
Systemic FeaturesDry eyes and mouth were reported by 11 and 20 pa-tients, respectively. Seventeen patients reported voidingsymptoms, including urgency and frequency. One pa-tient suffered from two episodes of urinary retentionthat spontaneously remitted. Ten patients had prob-lems with defecation, including diarrhea (n 5 3), con-stipation (n 5 4), alternating diarrhea and constipation(n 5 1), and urgency (n 5 2).
Presence of Autoimmune DiseasesTwo CRPS patients suffered from psoriasis or Hashi-moto’s disease. Twenty-four patients were able to pro-vide information concerning the presence of disease intheir first-degree relatives (n 5 148). Four patients hada first-degree relative with an autoimmune-mediateddisease (rheumatoid arthritis [n 5 2], psoriasis, thyroiddisease).
HLA TypingA significant association was found for DR13, whichwas present in 46% of the patients with CRPS and in27% of the controls (p , 0.03). No differences be-tween patients and controls reached statistical signifi-cance for any of the other alleles tested (Table 2). Al-though DQ1 seemed to be increased among thepatients, this increase was not significant (81% of pa-tients vs 70% of controls).
DiscussionThis study focused on a distinct phenotype of severelyaffected CRPS patients who progressed to multifocal orgeneralized tonic dystonia. Adequate data on the epi-demiology of CRPS are lacking because all studies onCRPS suffer from selection bias. Hence, little is knownabout the number of CRPS patients who display thismalignant course. The clinical presentation of these pa-tients has several unique features. Compared withCRPS in general, there is an even larger female pre-dominance. Age at onset of the disease varies, but themean age lies between 30 and 40 years, and earlier on-set of the disease is associated with a more aggressivecourse. A triggering event occurred in 9 of the 17 pa-tients with abrupt onset of the disease and in only 1 ofthe 9 patients with gradual progression from onset.Progression of the disease was gradual or through a se-ries of abrupt episodes. The dystonia is tonic, has apredominant involvement of the flexors, and initiateson the distal extremities but may progress to involveproximal extremity, axial, and facial musculature. Ad-ditionally, the dystonia is associated with variable com-binations of sensory and autonomic features. Dryness
Fig. A typical example of tonic dystonia of a hand.
Table 1. Criteria for Complex Regional Pain SyndromeType I 6
1. The presence of an initiating noxious event or a causeof immobilization
2. Continuing pain, allodynia, or hyperalgesia, with whichthe pain is disproportionate to any inciting event
3. Evidence at some time of edema, changes in skin bloodflow, or abnormal sudomotor activity in the region ofthe pain
4. This diagnosis is excluded by the existence of conditionsthat would otherwise account for the degree of pain anddysfunction
Criteria 2 to 4 must be satisfied.
114 Annals of Neurology Vol 48 No 1 July 2000

of the mouth and eyes was frequently reported. Manyof our patients suffered from urological symptoms,which is in agreement with the findings of a previousstudy.11 Ten patients suffered from unexplained boweldisturbances.
Because chronic widespread pain, stiffness, paresthe-sias, and fatigue are also commonly associated with fi-bromyalgia, chronic fatigue syndrome, and Lyme dis-ease, one may question how the clinical features of ourpatients compare with those of patients with these syn-dromes. Lyme disease has other well-defined dermato-logical, neurological, cardiac, and rheumatological signsand symptoms and can be confirmed by serologicaltesting.12 Diagnostic criteria sets for fibromyalgia andchronic fatigue syndrome show overlap but place em-phasis on the most prominent symptom, that is, spe-cific tender points and extreme fatigue, respective-ly.13,14 The motor features as well as the distributionpatterns of the combinations of motor, sensory, andautonomic features are unique for our sample of pa-tients and, to the best of our knowledge, have not beendescribed in fibromyalgia or chronic fatigue syndrome.
The uniqueness of this clinical presentation of CRPSis underscored by a significant increase in HLA-DR13among these patients. As the major histocompatibilitycomplex class II subregion contains genes for class IImolecules in addition to other functional and nonfunc-tional genes, the clinical significance of the association
between DR13 and CRPS remains to be elucidated.15
Our study could not confirm the previously reportedincrease of DQ1 in CRPS.5 A cohort bias or flawedanalysis of data was most likely responsible for this pre-vious finding, however, because the controls in thestudy by Kemler and co-workers5 were largely homozy-gous for HLA-DQ (95%), and DQ1 was underrepre-sented (42%) compared with that detected in our con-trols (70%).
Although it has been suggested that CRPS is the re-sult of an exaggerated inflammatory response to tissueinjury, studies searching for evidence in favor of im-mune system involvement have yielded variable results.Calder and colleagues16 reported an increased numberof Langerhans cells in skin biopsies of 5 patients withCRPS. Langerhans cells express significant levels of classII antigens and serve a dedicated role in immune re-sponse induction.17 Ribbers and co-workers18 found noabnormalities in lymphocyte populations and activatedT cells on flow cytometry in 13 patients with CRPS.
The female predominance among our CRPS patientsis in accordance with the findings in other studies onCRPS and is also observed in autoimmune disorders.Additionally, the trajectories of the disease course ofour patients show some similarities with those estab-lished for multiple sclerosis, in which the immune sys-tem plays an important role.19 If one assumes that anincreased prevalence of autoimmune disease among
Table 2. Clinical and Major Histocompatibility Complex Class II Characteristics of 26 Complex Regional Pain Syndrome Patientswith Tonic Dystonia
Patient No. Sex Age at Onset (yr)AutonomicSymptoms
SensorySymptoms
MotorSymptoms HLA-DRB1 HLA-DQA1 HLA-DQB1
1 F 18 C P, Nu, It D, W 11 13 0103/0501 0301/06032 F 20 T, E, C P, Pa, It D, M, W 15 04 0102/0301 0302/06023 F 37 T, C P, Pa, Nu, It D 15 13 0102/01 0602/06044 F 50 T, H, N, E, S, C P, Pa, Nu, It D, M, W 03 04 0302/0303 0201/03015 F 28 T, E, S, C P, Pa, Nu D, Tr, M, W 13 13 0102/0501 0301/06046 F 36 T, E, C P, Pa, Nu, It D, M, W 01 15 0101/0102 0501/06027 M 50 E, S P, Pa, Nu D, W 01 07 0101/0201 0202/05018 F 41 T, H, N, E, S, C P, Pa, Nu, It D, W 15 13 0102/01 0602/06049 F 33 T, E, S, C P, It D, W 04 07 0201/0301 0202/0302
10 F 32 T, S, C P, Pa, Nu, It D, M, W 01 03 0101/0501 0201/050111 M 46 H, N, E, S P, Pa, Nu, It D, Tr, M 13 14 0102/0103 0503/060312 F 38 T, N, E, S, C P, Nu, It D, W 04 13 0102/0301 0302/060413 F 16 T, H, N, S, C P, Pa, Nu, It D, M, W 04 13 0103/0301 0302/060314 F 20 T, H, E, S, C P, Pa, Nu D, Tr, W 01 11 0101/0501 0301/050115 F 21 N, E, S P, Pa, Nu, It D, Tr, M, W 04 13 0102/03 0301/060416 F 38 T, E, C P, Nu D, W 13 08 0103/0401 0402/060317 F 20 T, H, E, C P, Pa, Nu, It D, Tr, M, W 04 04 0301/0301 0302/030218 F 50 T, H, N, E, S, C P, Pa, Nu D, Tr, M, W 03 03 0501/0501 0201/020119 F 44 T, N, E, S P, Pa, Nu D, W 01 04 0101/0301 0302/050120 F 46 T, E, S P, It D, W 01 13 ND 05 /0621 F 46 T, S, C P, Pa D, W 03 04 03 /0501 0201/030222 F 25 T, N, E, S, C P, Pa, Nu, It D, Tr, M, W 01 13 0101/0103 0501/060323 F 38 T, S P, Pa, It D, W 15 13 0102/0103 0602/060324 F 46 T, N, E, S, C P, Pa, Nu, It D, Tr, M, W 01 14 0101/0104 0501/050325 F 58 T, S P, Pa, Nu D, M, W 01 03 0101/0501 0201/050126 F 19 T, N, E, S, C P, Pa, Nu, It D, Tr, W 15 08 0102/0401 0402/0602
F 5 female; M 5 male; T 5 temperature changes; H 5 hair growth changes; N 5 nail growth changes; E 5 edema; S 5 sudomotor changes;C 5 color changes; P 5 pain; Pa 5 paresthesias; Nu 5 numbness; It 5 impaired temperature sensation; D 5 dystonia; Tr 5 tremor; M 5myoclonic jerks; W 5 weakness; ND 5 not done.
Brief Communication: van Hilten et al: Tonic Dystonia of CRPS 115

family members of patients supports the contention ofa predisposing genetic factor with an immune compo-nent, our findings fail to suppport this assumption.Although we did not use control pedigrees, the lownumber of affected family members (2.7%) makes itunlikely that our approach would have missed a true in-crease in autoimmune disease among family members.
An unexpectedly high percentage of the patients suf-fered from dryness of the eyes and mouth. Sicca symp-toms can result from Sjogren syndrome, deficiency ofthe lacrimal glands, or trigeminal sensory abnormali-ties. The cause of the sicca complaints in our patientsrequires further investigation.
In conclusion, we have identified a distinct pheno-type of CRPS with progressive tonic dystonia that isassociated with HLA-DR13. The discordance of thelatter finding with those of previous studies illustratesthat the clinical phenotype defined in this report re-flects a specific entity of CRPS.
We thank Prof R. R. P. de Vries and Dr G. Th. M. Schreuder fortheir useful opinions and advice.
References1. Blumberg H, Griesser HJ, Hornyak M. Neurologische aspekte
der klinik, pathophysiologie und therapie der sympathischen re-flexdystrophie (kausalgie, morbus sudeck). Nervenarzt 1991;62:205–211
2. Oyen WJG, Arntz IE, Claessens AMJ, et al. Reflex sympatheticdystrophy of the hand: an excessive inflammatory response?Pain 1993;55:151–157
3. Van der Laan L, Goris RJA. Reflex sympathetic dystrophy. Anexaggerated regional inflammatory response? Hand Clin 1997;13:373–385
4. Mailis A, Wade J. Profile of Caucasian women with possiblegenetic predisposition to reflex sympathetic dystrophy: a pilotstudy. Clin J Pain 1994;10:210–217
5. Kemler MA, Van de Vusse AC, Van den Berg-Loonen EM, etal. HLA-DQ1 associated with reflex sympathetic dystrophy.Neurology 1999;53:1350–1351
6. Merskey H, Bogduk N. Classification of chronic pain: descrip-tions of chronic pain syndromes and definitions of pain terms.2nd ed. Seattle: IASP Press, 1994
7. Arnett FC, Edworthy SM, Bloch DA, et al. The AmericanRheumatism Association 1987 revised criteria for the classifica-tion of rheumatoid arthritis. Arthritis Rheum 1988;31:315–324
8. Kimura A, Sasazuki T. Eleventh International Histocompatibil-ity Workshop reference protocol for the HLA DNA typingtechnique. In: Tsuji K, Aizawa M, Sasazuki T, eds. HLA 1991:Proceedings of the eleventh international histocompatibilityworkshop and conference. Oxford, UK: Oxford UniversityPress, 1992:379–419
9. Izaks GJ, Van Houwelingen HC, Schreuder GMT, LigthartGJ. The association between human leucocyte antigens (HLA)and mortality in community residents aged 85 and older. J AmGeriatr Soc 1997;45:56–60
10. Lagaay AM, D’Amro J, Ligthart GJ, et al. Longevity and he-redity in humans. Association with the human leucocyte anti-gen phenotype. Ann NY Acad Sci 1991;621:78–89
11. Chancellor MB, Shenot PJ, Rivas DA, et al. Urological symp-tomatology in patients with reflex sympathetic dystrophy.
J Urol 1996;155:634–63712. Paulson E, ed. Consensus conference on Lyme disease. CMAJ
1991;144:1627–163213. Wolfe F. The fibromyalgia syndrome: a consensus report on
fibromyalgia and disability. J Rheumatol 1996;23:534–53914. Bell DS. Chronic fatigue syndrome. Recent advances in diag-
nosis and treatment. Postgrad Med 1992;91:245–25215. Beck S, Abdula S, Alderton RP, et al. Evolutionary dynamics of
non-coding sequences within the class II region of the humanMHC. J Mol Biol 1996;255:1–13
16. Calder JS, Holten I, McAllister RMR. Evidence for immunesystem involvement in reflex sympathetic dystrophy. J HandSurg [Br] 1998;23:147–150
17. Kaufman JF, Auffray C, Korman AJ, et al. The class II mole-cules of the human and murine major histocompatibility com-plex. Cell 1984;36:1–13
18. Ribbers GM, Oosterhuis WP, Limbeek van J, Metz de M. Re-flex sympathetic dystrophy: is the immune system involved?Arch Phys Med Rehabil 1998;79:1549–1552
19. Lublin FD, Reingold SC. Defining the clinical course of mul-tiple sclerosis: results of an international survey. Neurology1996;46:907–911
116 Annals of Neurology Vol 48 No 1 July 2000

Dual Epileptic Foci in aSingle Patient ExpressDistinct Temporal PatternsDependent on Limbic versusNonlimbic Brain LocationMark Quigg, MD,*† and Martin Straume, PhD†‡
How timing information is transferred from the supra-chiasmatic nucleus to other regions of the brain to me-diate activity, either physiological or pathological, islargely unclear. A patient with medically refractory epi-lepsy and a well-documented, long-term seizure diaryprovided a unique means to demonstrate how suscepti-bility to chronobiological modulation varies with brainregion. Evaluation for epilepsy surgery disclosed two in-dependent epileptic foci, one limbic and the other non-limbic. Seizures from both foci occurred periodically witha dominant period of 24 hours but were out of phasewith each other. Temporal lobe seizures occurred maxi-mally in the light portion of the daily light-dark cycle,and parietal lobe seizures occurred nocturnally and outof phase with limbic seizures. These data suggest thatneuronal excitation and inhibition, depending on the an-atomical system involved in epilepsy, may be differentlyaffected by circadian modulation.
Quigg M, Straume M. Dual epileptic foci in asingle patient express distinct temporal patterns
dependent on limbic versus nonlimbic brainlocation. Ann Neurol 2000;48:117–120
Although molecular mechanisms underlying mamma-lian circadian machinery continue to be elucidated,how timing information is transduced to the myriadcircadian rhythms remains largely unclear. Even lessclear is how specific diseases are inhibited or facilitatedby the clock or its subordinate rhythms. One exampleis the periodic occurrence of epileptic seizures.1–4 Theoccurrence of partial seizures can be influenced bysleep-wake state,5 and their distribution throughout theday influenced by the location of foci6 or pathophysi-ology.1 Although these observations were suggested by
studies of groups of patients with epilepsy, a clear il-lustration of the differing susceptibility of brain regionsto circadian modulation has never been demonstratedin a single individual.
Patient and MethodsClinical DataA 57-year-old right-handed woman presented with a 12-yearhistory of two types of seizures that we identified as emanat-ing from separate brain foci. The first consisted of complexpartial seizures with confusion, orofacial automatisms, anddystonic posturing of the left hand; the second consisted ofabrupt episodes of pain of the left hand and arm. In evalu-ation for epilepsy surgery, scalp ictal electroencephalography(EEG) documented right temporal onset of complex partialseizures. Seizures of ictal pain were poorly seen because ofartifact. Magnetic resonance imaging disclosed dual pathol-ogy of right hippocampal atrophy and a subtle right parietalcortical malformation. Because scalp EEG was insufficientfor surgery, intracranial monitoring was performed for 17days using subdural strip electrodes and bilateral, occipitallyinserted intrahippocampal depth electrodes. Intracranial EEGdisclosed two seizure foci, one right temporal correspondingto complex partial seizures and one right parietal correspond-ing to ictal pain (Figs 1 and 2).
MethodsBefore epilepsy surgery evaluation, the patient maintained along-term seizure diary containing date, hour, and type ofseizure from 1991 to 1997. We confined analyses to com-plete years (January 1, 1992 to December 31, 1996). Weconverted entries to serial hours containing presence or ab-sence of at least one seizure. Only 5 hours contained morethan one seizure (“seizure clusters”), resulting in the exclu-sion of seven seizures from further analysis. To test for sta-tionarity, we fitted plots of cumulative seizure number byhour using simple regression.2–4 To determine whether sei-zures occurred in an exponential, Poisson random manner,plots of interseizure intervals i against the log10 of intervalsgreater than i were fitted with simple regression.2–4 Althoughintervals may be random, periodicities of occurrence may stillbe present. To quantify the degree of irregularity within datausing a model-independent measure, we analyzed data usingapproximate entropy (ApEn).7,8 Spectral analysis using fastFourier transform-nonlinear least squares (FFT-NLLS) objec-tively determined rhythmic components that were significantat a 95% confidence limit.9 To summarize 24-hour histo-grams of seizure occurrence, we used cosinor-nonlinear leastsquares analysis.1 To test whether resulting daily distribu-tions of seizures were not uniform, we calculated the x2 ofobserved values compared with the expected mean of a uni-form distribution.
ResultsStochastic probability models2–4 determined that sei-zures occurred without discontinuities and at stableand linear rates. Log transform plots of interseizure in-tervals2–4 revealed mixed evidence of Poisson randomoccurrence. By ApEn, both temporal and parietal sei-
From the Departments of *Neurology, Comprehensive EpilepsyProgram, and ‡Internal Medicine, Division of Endocrinology andMetabolism, and †NSF Center for Biological Timing, University ofVirginia, Charlottesville, VA.
Received Feb 9, 2000, and in revised form March 27. Accepted forpublication March 27, 2000.
Address correspondence to Dr Quigg, Department of Neurology,Comprehensive Epilepsy Program, Box 394, Health Sciences Cen-ter, University of Virginia, Charlottesville, VA 22908.
Copyright © 2000 by the American Neurological Association 117
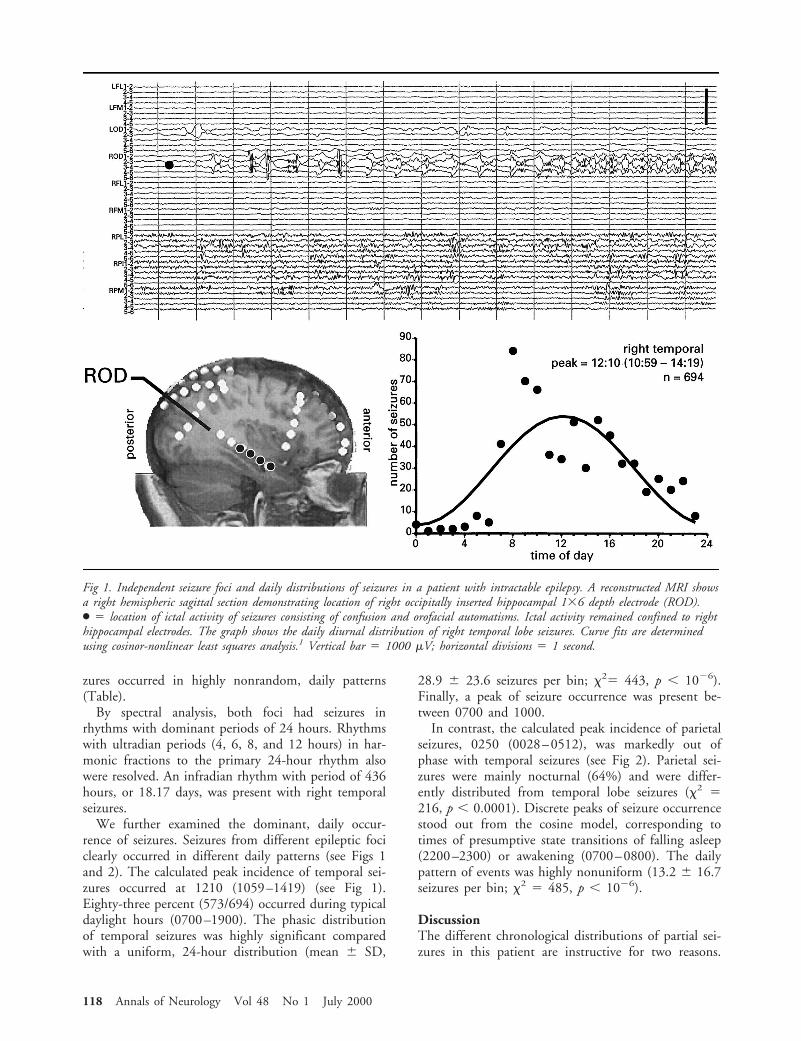
zures occurred in highly nonrandom, daily patterns(Table).
By spectral analysis, both foci had seizures inrhythms with dominant periods of 24 hours. Rhythmswith ultradian periods (4, 6, 8, and 12 hours) in har-monic fractions to the primary 24-hour rhythm alsowere resolved. An infradian rhythm with period of 436hours, or 18.17 days, was present with right temporalseizures.
We further examined the dominant, daily occur-rence of seizures. Seizures from different epileptic fociclearly occurred in different daily patterns (see Figs 1and 2). The calculated peak incidence of temporal sei-zures occurred at 1210 (1059–1419) (see Fig 1).Eighty-three percent (573/694) occurred during typicaldaylight hours (0700–1900). The phasic distributionof temporal seizures was highly significant comparedwith a uniform, 24-hour distribution (mean 6 SD,
28.9 6 23.6 seizures per bin; x25 443, p , 1026).Finally, a peak of seizure occurrence was present be-tween 0700 and 1000.
In contrast, the calculated peak incidence of parietalseizures, 0250 (0028–0512), was markedly out ofphase with temporal seizures (see Fig 2). Parietal sei-zures were mainly nocturnal (64%) and were differ-ently distributed from temporal lobe seizures (x2 5216, p , 0.0001). Discrete peaks of seizure occurrencestood out from the cosine model, corresponding totimes of presumptive state transitions of falling asleep(2200–2300) or awakening (0700–0800). The dailypattern of events was highly nonuniform (13.2 6 16.7seizures per bin; x2 5 485, p , 1026).
DiscussionThe different chronological distributions of partial sei-zures in this patient are instructive for two reasons.
Fig 1. Independent seizure foci and daily distributions of seizures in a patient with intractable epilepsy. A reconstructed MRI showsa right hemispheric sagittal section demonstrating location of right occipitally inserted hippocampal 136 depth electrode (ROD).● 5 location of ictal activity of seizures consisting of confusion and orofacial automatisms. Ictal activity remained confined to righthippocampal electrodes. The graph shows the daily diurnal distribution of right temporal lobe seizures. Curve fits are determinedusing cosinor-nonlinear least squares analysis.1 Vertical bar 5 1000 mV; horizontal divisions 5 1 second.
118 Annals of Neurology Vol 48 No 1 July 2000
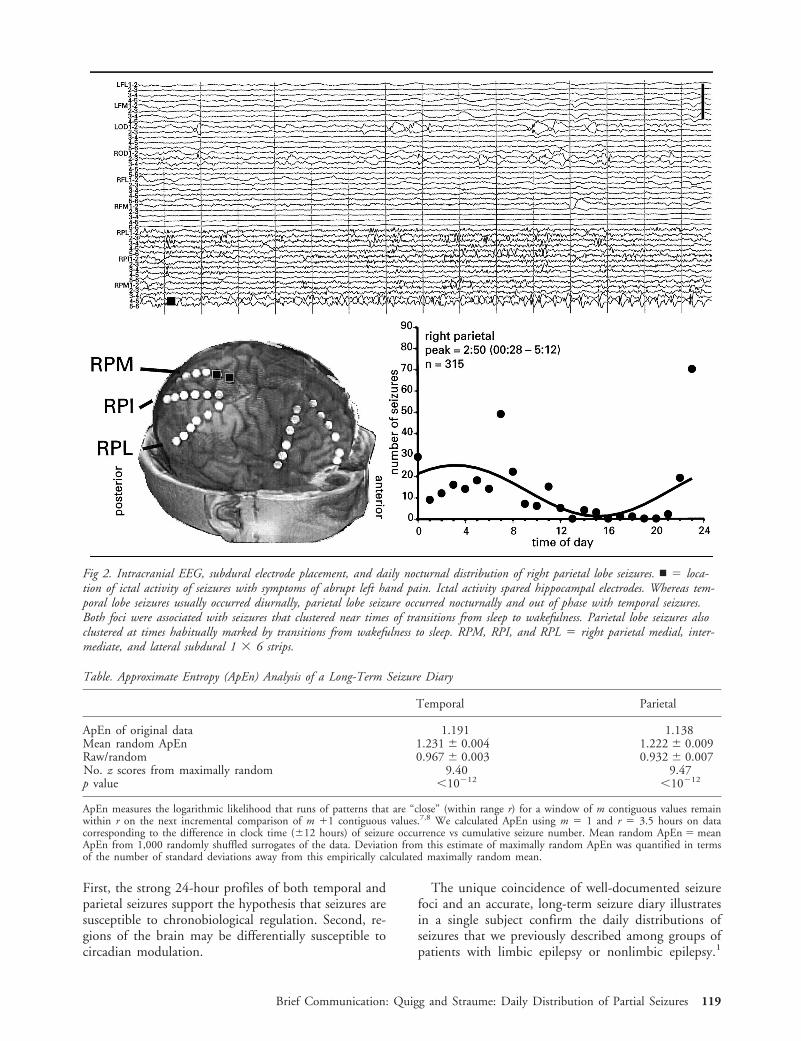
First, the strong 24-hour profiles of both temporal andparietal seizures support the hypothesis that seizures aresusceptible to chronobiological regulation. Second, re-gions of the brain may be differentially susceptible tocircadian modulation.
The unique coincidence of well-documented seizurefoci and an accurate, long-term seizure diary illustratesin a single subject confirm the daily distributions ofseizures that we previously described among groups ofpatients with limbic epilepsy or nonlimbic epilepsy.1
Table. Approximate Entropy (ApEn) Analysis of a Long-Term Seizure Diary
Temporal Parietal
ApEn of original data 1.191 1.138Mean random ApEn 1.231 6 0.004 1.222 6 0.009Raw/random 0.967 6 0.003 0.932 6 0.007No. z scores from maximally random 9.40 9.47p value ,10212 ,10212
ApEn measures the logarithmic likelihood that runs of patterns that are “close” (within range r) for a window of m contiguous values remainwithin r on the next incremental comparison of m 11 contiguous values.7,8 We calculated ApEn using m 5 1 and r 5 3.5 hours on datacorresponding to the difference in clock time (612 hours) of seizure occurrence vs cumulative seizure number. Mean random ApEn 5 meanApEn from 1,000 randomly shuffled surrogates of the data. Deviation from this estimate of maximally random ApEn was quantified in termsof the number of standard deviations away from this empirically calculated maximally random mean.
Fig 2. Intracranial EEG, subdural electrode placement, and daily nocturnal distribution of right parietal lobe seizures. n 5 loca-tion of ictal activity of seizures with symptoms of abrupt left hand pain. Ictal activity spared hippocampal electrodes. Whereas tem-poral lobe seizures usually occurred diurnally, parietal lobe seizure occurred nocturnally and out of phase with temporal seizures.Both foci were associated with seizures that clustered near times of transitions from sleep to wakefulness. Parietal lobe seizures alsoclustered at times habitually marked by transitions from wakefulness to sleep. RPM, RPI, and RPL 5 right parietal medial, inter-mediate, and lateral subdural 1 3 6 strips.
Brief Communication: Quigg and Straume: Daily Distribution of Partial Seizures 119

The present case is also consistent with studies of thedistribution of partial seizures that demonstrated vary-ing distributions of seizures between states dependingon location of seizure foci.5,6 Analogous to these find-ings are in vivo recordings of circadian electrical activ-ity that show that multiple unit activity within supra-chiasmatic nuclei and other regions occur out ofphase.10 Seizures involving the limbic system may beespecially sensitive to circadian modulation as mediatedby the hypothalamus, since the limbic system and hy-pothalamus share anatomical and functional intercon-nections.11,12 Extratemporal, cortically based seizuresthat spare the limbic system may be more susceptibleto mediators of cortical excitation, such as the rhythmof the sleep-wake cycle.13 Furthermore, the sleep-wakecycle may contribute to both limbic and nonlimbic sei-zure patterns, with limbic seizures facilitated by wake-fulness (or resistant to effects of sleep) and extralimbicseizures promoted by sleep (or inhibited by the wakingstate). Transition states are particularly seizure provok-ing for a variety of epileptic syndromes13 and may be astrong factor independent of syndrome.
A circadian rhythm, in the strict sense, is any self-sustained activity that oscillates with a period of about24 hours.14 Examples of circadian rhythms include ac-tivity of the suprachiasmatic nuclei, cortisol and mela-tonin secretion, and the daily oscillation of body tem-perature. Seizure diary data, however, are not sufficientto determine the source of the circadian modulation ofseizure occurrence. It is reasonable to postulate that thestrong, daily distribution of partial seizures in this pa-tient is circadian and susceptible to circadian modula-tion, but we cannot exclude the possibility that exoge-nous factors confound the different 24-hour patterns ofseizures. Although underreporting of seizures duringsleep is a valid concern, the patient accurately con-firmed the times of seizure occurrence during inpatientmonitoring and was aided in documentation by herhusband. Previous studies of seizure diaries did not dis-criminate patients by location of foci, were limited inresolution (1 day) and in duration (usually ,1year),2–4 and thereby could not resolve the daily, peri-odic patterns determined in this study with hourly res-olution and 5-year duration. Finally, ApEn comple-ments spectral analysis and can be more sensitive inpattern detection than stochastic models in quantifyingrandomness.
In addition to 24-hour rhythms, temporal and pari-etal seizures had significant ultradian components,which were likely harmonic artifacts of spectral analy-sis. An infradian period of about 18 days did not cor-respond to obvious cycles such as the 7-day week orlunar, menstrual, or other clear hormonal cyclic influ-
ences. In one study, seizure cycles of approximately 20days were reported in patients with complex partial sei-zures.4 In summary, the remarkable coincidence of areliable seizure diary, intractable epilepsy, and well-documented, independent seizure foci from a single in-dividual illustrate that circadian seizure expression mayvary with limbic or nonlimbic locations of the epilep-togenic zones. Mechanisms that allow biological clocksto transduce their timing signals may vary with the re-gion of brain involved, and this transduction has rele-vance in the dysfunction of neuronal excitability thatcharacterizes epilepsy.
Dr Quigg receives support from grants from NIH NS K0802021.Data analysis was supported by the NSF Center for BiologicalTiming.
We thank Hunter Downs, PhD of the University of Virginia Neu-rovisualization Laboratory for the initial three-dimensional magneticresonance images, and Sue Moenter, PhD, for editorial suggestions.
References1. Quigg M, Straume M, Menaker M, Bertram E. Temporal dis-
tribution of partial seizures: comparison of an animal modelwith human partial epilepsy. Ann Neurol 1998;43:748–755
2. Taubøll E, Lundervold A. Temporal distribution of seizures inepilepsy. Epilepsy Res 1991;8:153–165
3. Milton J, Gotman J, Remillard G, Andermann F. Timing ofseizure recurrence in adult epileptic patients: a statistical analy-sis. Epilepsia 1987;28:471–478
4. Binnie C, Aarts J, Houtkooper M, et al. Temporal characteris-tics of seizures and epileptiform discharges. ElectroencephalogrClin Neurophysiol 1984;58:498–505
5. Bazil CW, Walczak TS. Effects of sleep and sleep stage on ep-ileptic and nonepileptic seizures. Epilepsia 1997;38:56–62
6. Janz D. Epilepsy and the sleeping-waking cycle. In: Magnus O,Lorentz De Haas A, eds. The epilepsies, vol 15. Amsterdam:North Holland, 1974:457–490
7. Pincus S, Singer B. Randomness and degrees of irregularity.Proc Natl Acad Sci USA 1996;93:2083–2088
8. Pincus S. New test sizes up randomness. Science 1997;276:5329. Quigg M, Clayburn H, Straume M, et al. Hypothalamic neu-
ronal loss and altered circadian rhythm of temperature in a ratmodel of mesial temporal lobe epilepsy. Epilepsia 1999;40:1688–1696
10. Yamazaki S, Kerbeshian M, Hocker C, et al. Rhythmic prop-erties of the hamster suprachiasmatic nucleus in vivo. J Neuro-sci 1998;15:10709–10723
11. Buijs R. The anatomical basis for the expression of circadianrhythms: the efferent projections of the suprachiasmatic nu-cleus. Progr Brain Res 1996;111:229–240
12. Peng Z, Grassi-Zucconi G, Bentivoglio M. Fos-related proteinexpression in the midline paraventricular nucleus of the rathypothalamus: basal oscillation and relationship with limbic ef-ferents. Exp Brain Res 1995;104:21–29
13. Shouse M, Martins da Silva A, Sammaritano M. Circadianrhythms, sleep and epilepsy. J Clin Neurophysiol 1996;13:32–50
14. Schwartz W. Understanding circadian clocks: from c-fos to fly-balls. Ann Neurol 1997;41:289–297
120 Annals of Neurology Vol 48 No 1 July 2000

Pipecolic Acid Elevation inPlasma and CerebrospinalFluid of Two Patients withPyridoxine-DependentEpilepsyBarbara Plecko, MD,*† Sylvia Stockler-Ipsiroglu, MD,†and Eduard Paschke, PhD,* Wolfgang Erwa, MD,*Eduard A. Struys, MSc,‡ and Cornelis Jakobs, PhD‡
Diagnosis of pyridoxine-dependent epilepsy is based onthe clinical response to high-dosage application of pyri-doxine. Here, we report on 2 patients with pyridoxine-dependent epilepsy with significant elevation of pipecolicacid concentrations in plasma and cerebrospinal fluid(CSF) and further increase of pipecolic acid in CSF dur-ing a 72-hour pyridoxine withdrawal in 1 of them. Pa-tients with non–pyridoxine-dependent epilepsy had nor-mal pipecolic acid concentrations in plasma andsignificantly lower concentrations in CSF. High plasmaand CSF pipecolic acid concentrations might provide adiagnostic marker in pyridoxine-dependent epilepsy.
Plecko B, Stockler-Ipsiroglu S, Paschke E,Erwa W, Struys EA, Jakobs C. Pipecolic acid
elevation in plasma and cerebrospinal fluid of twopatients with pyridoxine-dependent epilepsy.
Ann Neurol 2000;48:121–125
Pyridoxine-dependent epilepsy is a rare (1 in 100,000)autosomal recessive disorder (McKusick 266100)1 withunclear biochemical background. Inborn errors in pyr-idoxine metabolism have widely been excluded.2,3 Apostulated defect at the binding site of glutamate-decarboxylase (GAD) to pyridoxalphosphate (PLP)2
was supported by single reports on high glutamate butlow GABA levels in the cortex4 and cerebrospinal fluid(CSF)5,6 of affected patients, but so far no mutationsin the coding genes of the two isoenzymes GAD 65and GAD 67 have been identified.7,8 Pipecolic acid to-gether with piperideine compounds is an intermediary
product in central nervous system (CNS)-specific lysinedegradation.9–11 A pyridoxine-dependent enzymaticstep is involved in pipecolic acid degradation at thelevel of a-aminoadipic acid conversion (Fig 1). Pipe-colic acid accumulates with various compounds indisorders of peroxisomal biogenesis (eg, Zellweger syn-drome). Disorders with isolated pipecolic acid accumu-lation have not been specified so far.12 We report on 2patients with pyridoxine-dependent epilepsy and isolatedelevation of pipecolic acid in plasma and CSF.
Patients and MethodsCase ReportsPatient 1 was born after a first uncomplicated 37-week preg-nancy. Apgar scores were 9/10/10, umbilical artery pH 7.38.Generalized tonic-clonic seizures started on the second day oflife. The child was severely hypotonic with poor sucking andpoor visual contact. Cranial ultrasound revealed unilateralgrade II intraventricular hemorrhage and moderately dilatedventricles. Electroencephalography (EEG) showed focalspike-wave discharges over the right frontotemporal regionwith normal background activity. Urinary and plasma pipe-colic acid concentrations were repeatedly elevated on bothqualitative amino acid screening in urine (by thin-layer chro-matography) and on quantitative determination in plasma(Table). Disorders of peroxisomal biogenesis were excludedby normal levels of very-long-chain fatty acids (VLCFAs),phytanic acid and bile acids in serum, and normal catalaselatency and de novo biosynthesis of plasmalogens in fibro-blasts. A possible isolated defect of pipecolic acid oxidase wasexcluded by normal enzyme activity in liver tissue. He con-tinued to have short, secondary generalized seizures 3 to 10times a day. At the age of 3.5 months, seizures stopped com-pletely after the first single intravenous administration of 300mg of pyridoxine, recurred after 3 weeks, and again re-sponded promptly to 300 mg of intravenous pyridoxine. Un-der continuous oral supplementation of pyridoxine (200 mg/day), the boy remained seizure-free for the next 6 years.When he started attending a school for handicapped chil-dren, intake of pyridoxine became irregular, and after a weekof complete withdrawal he was referred with generalized sta-tus epilepticus, which had even necessitated anesthesia withthiopentone. Seizures were promptly interrupted by a singleintravenous dose of 300 mg of pyridoxine. Under regularsubstitution of pyridoxine, the patient again remainedseizure-free and had complete neurological recovery from sta-tus epilepticus.
Patient 2 was the product of a first pregnancy complicatedby gestational toxicosis. He was delivered by cesarean sectionat term. Apgar scores were 8/9/10; birth weight was 3,260 g.Focal seizures (myoclonic jerks of both arms) started withinhours, and his EEG showed paroxysms of polyspike-wave ac-tivity and intermittent burst suppression patterns. Cranialimaging was normal. Routine laboratory examinations, CSFanalysis, organic acids in urine, amino acids in serum, andVLCFAs were normal, but pipecolic acid in plasma wasmarkedly elevated (see Table). Seizures ceased immediatelyafter a single intravenous administration of 100 mg pyridox-ine on the third day of life. Apathy with recurrent apnea
From the *Department of Pediatrics, University Hospital Graz,Graz, †Department of Pediatrics, University Hospital Vienna, Vi-enna, Austria; and ‡Department of Clinical Chemistry, Free Uni-versity Hospital Amsterdam, The Netherlands.
Received Feb 1, 2000, and in revised form Mar 27. Accepted forpublication Mar 28, 2000.
Address correspondence to Dr Stockler-Ipsiroglu, Department ofPediatrics, University Hospital Vienna, Wahringergurtel 18-20A-1090 Vienna, Austria.
Copyright © 2000 by the American Neurological Association 121

necessitated assisted ventilation for the following 3 days. Atthe age of 3 weeks, focal seizures recurred and promptly re-sponded to oral treatment with pyridoxine, 10 mg/kg/day.When last seen at the age of 5 months, Patient 2 was stillseizure-free on pyridoxine therapy but was mildly hypotonicwith moderate developmental delay.
MethodsIn Patient 1, a controlled 72-hour pyridoxine withdrawal wasperformed at the age of 7.5 years. Venous blood samples fordetermination of pipecolic acid and pyridoxalphosphate weretaken 24, 48, and 72 hours after withdrawal. Lumbar punc-ture for determination of pipecolic acid, amino acids, GABA,and vitamin B6 vitamers in CSF was performed 24 hours and72 hours after withdrawal. Piperidine and a-aminoadipicacid levels in CSF were determined before and 72 hours afterwithdrawal. Plasma and CSF samples taken before and dur-ing pyridoxine supplementation and during the uncontrolled
pyridoxine withdrawal and stored at 280°C were availablefor retrospective investigations. In Patient 2, plasma sampleswere taken before and during pyridoxine supplementation. ACSF sample was taken at the age of 5 months while thepatient was receiving pyridoxine. Because of the patient’syoung age, pyridoxine withdrawal has not been performedyet. Pipecolic acid was also determined in plasma of 26 pa-tients with various forms of non–pyridoxine-dependent epi-lepsy. CSF samples were additionally available from 9 ofthem. Pipecolic acid in plasma and CSF was determined asdescribed.13,14
D- and L-separation of pipecolic acid wasachieved according to Struys and Jakobs.15 For determina-tion of piperidines, CSF was derivatized with ethylchlorofor-mate, extracted with ethylacetate, and analyzed by a gaschromatography method using d11-piperidine as an internalstandard. a-Aminoadipic acid in CSF was detected asFMOC derivative by RP-HPLC. Pyridoxalphosphate inplasma was determined by a commercially available HPLC
Fig 1. Conversion of lysine to acetoacetyl-CoAis predominant via saccharopine in liver andvia pipecolic acid in the brain. Beyond theconversion of the two pathways degradation ofa-aminoadipic acid is catalyzed by apyridoxine-dependent transaminase.
122 Annals of Neurology Vol 48 No 1 July 2000

method (Chromsystems, Munich, Germany). Vitamin B6 vi-tamers in CSF were determined as described elsewhere.16
ResultsPipecolic AcidElevated concentrations of pipecolic acid in plasma andCSF of our 2 patients with pyridoxine-dependent epi-lepsy in comparison with normal values and to concen-trations in patients with various forms of non–pyridoxine-dependent epilepsy are shown in the Table.The pipecolic acid was exclusively in the L-form.
a-Aminoadipic Acid and Piperidine in CSFPipecolic acid elevation in patients with pyridoxine-dependent epilepsy might be caused by deficiency ofpyridoxal-dependent a-aminoadipic acid transaminase,which might be indicated by high concentrations ofa-aminoadipic acid and of D1piperideine-6 carboxylicacid. In both patients, a-aminoadipic acid concentra-tions (0.22 and 0.20 mmol/L) were comparable to val-ues obtained from one age-matched control (0.31mmol/L) and below the value obtained from a CSFpool (1.18 mmol/L) while on pyridoxine, and 72 hoursafter pyridoxine withdrawal (0.24 mmol/L) in Patient1. In the same samples, piperidine concentrations (0.03
and 0.05 mmol/L) were within the normal range(0.03–0.18 mmol/L, n 5 5) while on pyridoxine and72 hours after withdrawal (0.05 mmol/L) in Patient 1.
PyridoxalphosphateConcentrations of pyridoxalphosphate in the plasma ofPatient 1 were above or within normal range (Fig 2)during periods on and off pyridoxine supplementation.Pyridoxalphosphate in CSF was high (90 nmol/L; nor-mal,5 26 6 14) while on pyridoxine and within thenormal range after the 72-hour withdrawal (31 nmol/L).Values for pyridoxal and pyridoxic acid in CSF wereinconclusive because of missing data on normal values.Inverse correlation of pyridoxalphosphate versus pipe-colic acid in plasma of Patient 1 is shown in Figure 2.
DiscussionPipecolic acid elevation seems to be a specific findingin pyridoxine-dependent epilepsy as plasma concentra-tions of pipecolic acid were continuously elevated inboth patients with pyridoxine-dependent epilepsy, whilethey were normal in patients with non–pyridoxine-dependent seizures. The specificity of these findings isfurther supported by the fact that the degree of pipe-colic acid elevation is inversely correlated with pyridox-
Table. Pipecolic Acid Concentrations in Plasma and Cerebrospinal Fluid of Patients 1 and 2 with Pyridoxine-Dependent Epilepsyand of Age-Matched Patients with Non–Pyridoxine-Dependent Epilepsy
Normal Range Patient 1 Patient 2 Non–vitamin B6-Dependent Patients
Plasma1st wk 1–3.2 mmol/L13 49 mmol/La 3.2 mmol/L (n 5 1b)1–6 mo 0.6–4.1 mmol/L13 17.6 mmol/La 14.3 mmol/L 2.1 mmol/L (n 5 1b)6–12 mo 0.8–5.3 mmol/L13 9.87 mmol/L 10.1 mmol/L 2.52 mmol/L (n 5 1b)1–5 yr 0.4–4.9 mmol/L13 5.7 mmol/L Mean 6 SD, 1.18 mmol/L 6 0.27
(n 5 7/1b)Range, 0.65–1.5 mmol/L
5–8 yr 0.7–2.6 mmol/L13 6 mmol/L Mean 6 SD, 1.19 mmol/L 6 0.72,16.4 mmol/La (n 5 6/1b)5.8 mmol/L Range, 0.73–2.6 mmol/L5.3 mmol/L4.07 mmol/L
.8 yr 0.7–2.6 mmol/L13 4.0 mmol/L Mean 6 SD, 1.44 mmol/L 6 0.47,(n 5 10/2b)
Range, 1.0–2.6 mmol/LCSF
0–1 yr 3.38 mmol/L 0.195 mmol/L (n 5 1b,c) 0.140 mmol/L(n 5 1)
1–5 yr 0.009–0.12 mmol/L14 1.93 mmol/L 0.033–0.046 mmol/L (n 5 3)5–8 yr 0.57 mmol/L 0.039–0.066 mmol/L (n 5 3/1b)
0.67 mmol/La (24 hr)d
0.88 mmol/La (72 hr)d
.8 yr 0.051 mmol/L (n 5 1)
Note age-dependency of normal values for pipecolic acid concentrations in plasma.aValues off pyridoxine therapy.bValues of patients with seizure activity within the past 24 hours.cBlood contaminated.dHours of pyridoxine withdrawal in Patient 1.
Brief Communication: Plecko et al: Pipecolic Acid Elevation in Plasma 123
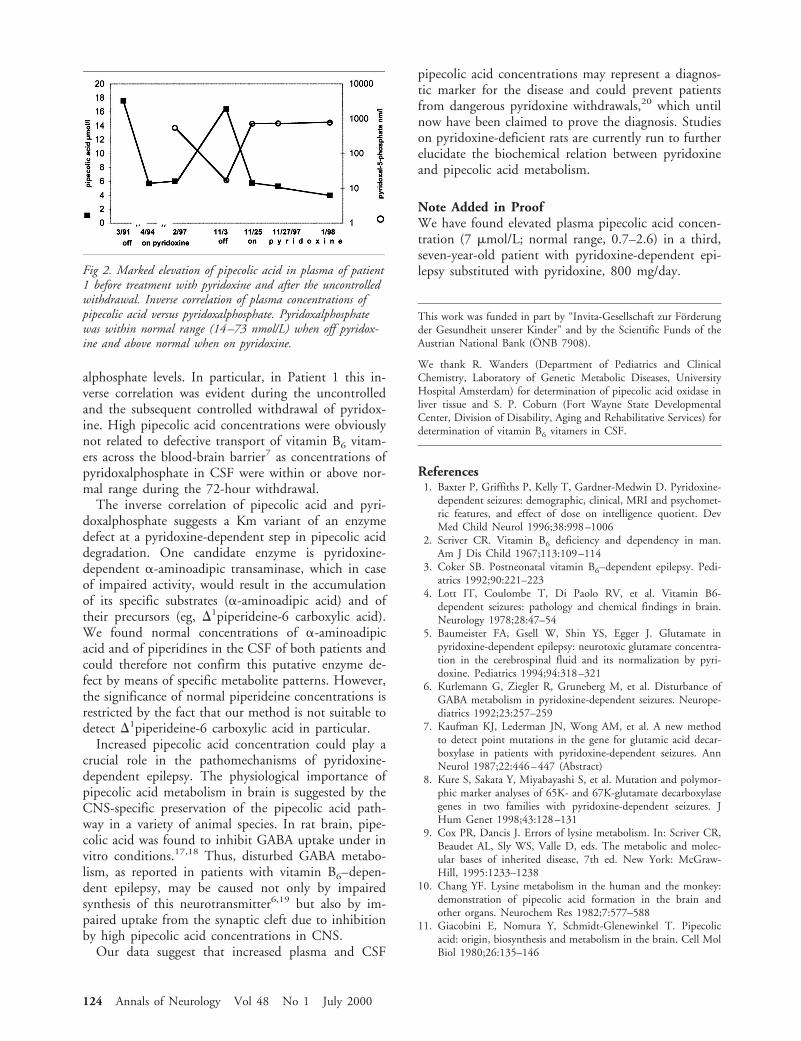
alphosphate levels. In particular, in Patient 1 this in-verse correlation was evident during the uncontrolledand the subsequent controlled withdrawal of pyridox-ine. High pipecolic acid concentrations were obviouslynot related to defective transport of vitamin B6 vitam-ers across the blood-brain barrier7 as concentrations ofpyridoxalphosphate in CSF were within or above nor-mal range during the 72-hour withdrawal.
The inverse correlation of pipecolic acid and pyri-doxalphosphate suggests a Km variant of an enzymedefect at a pyridoxine-dependent step in pipecolic aciddegradation. One candidate enzyme is pyridoxine-dependent a-aminoadipic transaminase, which in caseof impaired activity, would result in the accumulationof its specific substrates (a-aminoadipic acid) and oftheir precursors (eg, D1piperideine-6 carboxylic acid).We found normal concentrations of a-aminoadipicacid and of piperidines in the CSF of both patients andcould therefore not confirm this putative enzyme de-fect by means of specific metabolite patterns. However,the significance of normal piperideine concentrations isrestricted by the fact that our method is not suitable todetect D1piperideine-6 carboxylic acid in particular.
Increased pipecolic acid concentration could play acrucial role in the pathomechanisms of pyridoxine-dependent epilepsy. The physiological importance ofpipecolic acid metabolism in brain is suggested by theCNS-specific preservation of the pipecolic acid path-way in a variety of animal species. In rat brain, pipe-colic acid was found to inhibit GABA uptake under invitro conditions.17,18 Thus, disturbed GABA metabo-lism, as reported in patients with vitamin B6–depen-dent epilepsy, may be caused not only by impairedsynthesis of this neurotransmitter6,19 but also by im-paired uptake from the synaptic cleft due to inhibitionby high pipecolic acid concentrations in CNS.
Our data suggest that increased plasma and CSF
pipecolic acid concentrations may represent a diagnos-tic marker for the disease and could prevent patientsfrom dangerous pyridoxine withdrawals,20 which untilnow have been claimed to prove the diagnosis. Studieson pyridoxine-deficient rats are currently run to furtherelucidate the biochemical relation between pyridoxineand pipecolic acid metabolism.
Note Added in ProofWe have found elevated plasma pipecolic acid concen-tration (7 mmol/L; normal range, 0.7–2.6) in a third,seven-year-old patient with pyridoxine-dependent epi-lepsy substituted with pyridoxine, 800 mg/day.
This work was funded in part by “Invita-Gesellschaft zur Forderungder Gesundheit unserer Kinder” and by the Scientific Funds of theAustrian National Bank (ONB 7908).
We thank R. Wanders (Department of Pediatrics and ClinicalChemistry, Laboratory of Genetic Metabolic Diseases, UniversityHospital Amsterdam) for determination of pipecolic acid oxidase inliver tissue and S. P. Coburn (Fort Wayne State DevelopmentalCenter, Division of Disability, Aging and Rehabilitative Services) fordetermination of vitamin B6 vitamers in CSF.
References1. Baxter P, Griffiths P, Kelly T, Gardner-Medwin D. Pyridoxine-
dependent seizures: demographic, clinical, MRI and psychomet-ric features, and effect of dose on intelligence quotient. DevMed Child Neurol 1996;38:998–1006
2. Scriver CR. Vitamin B6 deficiency and dependency in man.Am J Dis Child 1967;113:109–114
3. Coker SB. Postneonatal vitamin B6–dependent epilepsy. Pedi-atrics 1992;90:221–223
4. Lott IT, Coulombe T, Di Paolo RV, et al. Vitamin B6-dependent seizures: pathology and chemical findings in brain.Neurology 1978;28:47–54
5. Baumeister FA, Gsell W, Shin YS, Egger J. Glutamate inpyridoxine-dependent epilepsy: neurotoxic glutamate concentra-tion in the cerebrospinal fluid and its normalization by pyri-doxine. Pediatrics 1994;94:318–321
6. Kurlemann G, Ziegler R, Gruneberg M, et al. Disturbance ofGABA metabolism in pyridoxine-dependent seizures. Neurope-diatrics 1992;23:257–259
7. Kaufman KJ, Lederman JN, Wong AM, et al. A new methodto detect point mutations in the gene for glutamic acid decar-boxylase in patients with pyridoxine-dependent seizures. AnnNeurol 1987;22:446–447 (Abstract)
8. Kure S, Sakata Y, Miyabayashi S, et al. Mutation and polymor-phic marker analyses of 65K- and 67K-glutamate decarboxylasegenes in two families with pyridoxine-dependent seizures. JHum Genet 1998;43:128–131
9. Cox PR, Dancis J. Errors of lysine metabolism. In: Scriver CR,Beaudet AL, Sly WS, Valle D, eds. The metabolic and molec-ular bases of inherited disease, 7th ed. New York: McGraw-Hill, 1995:1233–1238
10. Chang YF. Lysine metabolism in the human and the monkey:demonstration of pipecolic acid formation in the brain andother organs. Neurochem Res 1982;7:577–588
11. Giacobini E, Nomura Y, Schmidt-Glenewinkel T. Pipecolicacid: origin, biosynthesis and metabolism in the brain. Cell MolBiol 1980;26:135–146
Fig 2. Marked elevation of pipecolic acid in plasma of patient1 before treatment with pyridoxine and after the uncontrolledwithdrawal. Inverse correlation of plasma concentrations ofpipecolic acid versus pyridoxalphosphate. Pyridoxalphosphatewas within normal range (14–73 nmol/L) when off pyridox-ine and above normal when on pyridoxine.
124 Annals of Neurology Vol 48 No 1 July 2000

12. Fournier B, Smeitink JAM, Dorland L, et al. Peroxisomaldisorders: a review. J Inherit Metab Dis 1994;17:470–486
13. Kelley RI. Quantification of pipecolic acid in plasma and urineby isotope-dilution gas chromatography/mass spectrometry. In:Hommes FA, ed. Techniques in diagnostic human biochemicalgenetics. New York: Wiley-Liss, 1991:205–218
14. Kok RM, Kaster L, de Long APJM, et al. Stable isotope dilu-tion analysis of pipecolic acid in cerebrospinal fluid, plasma,urine and amniotic fluid using electron capture negative ionmass fragmentography. Clin Chim Acta 1987;168:143–152
15. Struys EA, Jakobs C. Enantiomeric analysis of D- and L- pipe-colic acid in plasma using a chiral capillary gas chromatographycolumn and mass fragmentography. J Inherit Metab Dis 1999;22:677–678
16. Mahuren JD, Coburn S. B6 vitamers: cation exchange HPLC. JNutr Biochem 1990;1:659–663
17. Nomura Y, Okuma Y, Segawa T. Influence of piperidine andpipecolic acid on the uptake of monoamines, GABA and gly-cine into P2 fractions of the rat brain and the spinal cord.J Pharm Dyn 1978;1:251–255
18. Gutierrez MC, Delgado-Coello BA. Influence of pipecolic acidon the release and uptake of [3H] GABA from brain slices ofmouse cerebral cortex. Neurochem Res 1989;14:405–408
19. Gospe SM, Olin KL, Keen CL. Reduced GABA synthesis inpyridoxine-dependent seizures. Lancet 1994;343:1133–1134
20. Bass NE, Wyllie E, Cohen B, Joseph SA. Pyridoxine-dependentepilepsy: the need for repeated pyridoxine trials and the risk ofsevere electrocerebral suppression with intravenous pyridoxineinfusion. J Child Neurol 1996;11:422–444
Brief Communication: Plecko et al: Pipecolic Acid Elevation in Plasma 125
Related Documents











