Inhibitors of Aspartyl Proteinases Sherin S. Abdel-Meguid Department of Macromolecular Sciences, SrnithKline Beechum, 709 Swedeland Road, King of Prussia, Pennsylvania 19406 I. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11. Therapeutic Targets of Aspartyl Proteinase Inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . A. Himan Renin B. H :V Proteinase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111. Three-Dimensional Structure of Aspartyl Proteinases . . . . . . . . . . . . . . . . . . . . . . . . . . . A. Monomeric ............. . . . . . . . . . . . . . . . . ....................... . . . . . . . . . . . . . . . . . . . . . . . . ....................... ..................................................... onomeric and Dimeric Enzymes . . . . . . . . . . . . . . . . . . . . . . . IV. Featu 'es of Substrate Specificity that Influence Inhibitor Design ... A. Substrate Specificity of Human Renin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . B. Substrate Specificity of HIV-1 Proteinase A. Peptide Inhibitors B. Inhibitors Containing Statine or es ___.__ ............. C. Aininomethylene Isosteres (Red s) .............................. D. H jdroxyethylene Isosteres F. H jdroxyethylamine Isosteres ....................................... H. Glycol-Based . . . . . . . I. C!dic Inhibitors . . . . . . . . . . . . . . . . . . J. Sy mmetric Inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . K. Phosphorus-Containing ........................................... L. "in-Peptide-Based A. General Mode of Binding ....................... V. Design and Classification of Aspar . . . . . . . . . . . . ................................ . . . . . . . . . . . . . . ....................... E. Dihydroxyethylene Isosteres . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . G. Kttones and Difluoroketones . . . . . . . . . . . . . . . . _._ ............... ............................................. ............. ............. . . . . . . . . . . . . . . . . . . . . . ....................... VI. X-Ray -Diffraction Studies VII. Conformation of Inhibitors in the Active Site . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ............. B. B~ckbone Conformations ....................................... C. Si le-Chain Conformation D. Dfluoroketones and Implications for the Ca E. G.ycol-Based, Cyclic, Phosphorus-Containin F. Inhibitor-Induced Protein Conformation . . . ..................... G. Subsite Specificity . . . H. Comparison of Renin and HIV-1 Proteinase Inhibitors . . . . . . . . . . . . . . . . . . . . . . . VIII. Minirial Renin Inhibitor c . . . . . . . . . . . . . ............................... . . . . . . . . . . . . . . . . . . . . . . . . . . . . _._ ........... __... IX. Minirial HIV-1 Proteinase Inhibitor . . . . . . . . . , . . . . . . . . . . . . . . . . . . . . X. Aspaityl Proteinase Inhibitors as Potential Drugs .............. ........... Acknowledgments . . . . . . . . . ........................... ................................... .. ................ . .... 731 737 737 738 739 739 739 740 741 741 741 744 745 746 746 747 748 748 748 748 750 750 752 752 753 757 757 760 763 764 765 767 768 768 770 770 770 771 771 I. INTRODUCTION During the past two decades, attempts to combat two serious human health threats, hypertension and acquired immune deficiency syndrome (AIDS), Medicinal Iiesearch Reviews, Vol. 13, No. 6, 731-778 (1993) 0 1993 John Wiley & Sons, Inc. CCC 0198-6325/93/060731-48

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Inhibitors of Aspartyl Proteinases
Sherin S. Abdel-Meguid Department of Macromolecular Sciences, SrnithKline Beechum, 709 Swedeland Road, King of Prussia,
Pennsylvania 19406
I. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11. Therapeutic Targets of Aspartyl Proteinase Inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . .
A. Himan Renin B. H :V Proteinase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
111. Three-Dimensional Structure of Aspartyl Proteinases . . . . . . . . . . . . . . . . . . . . . . . . . . . A. Monomeric
. . . . . . . . . . . . .
. . . . . . . . . . . . . . . . .......................
. . . . . . . . . . . . . . . . . . . . . . . . ....................... . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . onomeric and Dimeric Enzymes . . . . . . . . . . . . . . . . . . . . . . .
IV. Featu 'es of Substrate Specificity that Influence Inhibitor Design . . . A. Substrate Specificity of Human Renin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . B. Substrate Specificity of HIV-1 Proteinase
A. Peptide Inhibitors B. Inhibitors Containing Statine or es _ _ _ . _ _ . . . . . . . . . . . . . C. Aininomethylene Isosteres (Red s) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . D. H jdroxyethylene Isosteres
F. H jdroxyethylamine Isosteres . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
H . Glycol-Based . . . . . . . I. C!dic Inhibitors . . . . . . . . . . . . . . . . . . J. Sy mmetric Inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
K. Phosphorus-Containing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . L. "in-Peptide-Based
A. General Mode of Binding
....................... V. Design and Classification of Aspar
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . ....................... E. Dihydroxyethylene Isosteres . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
G. Kttones and Difluoroketones . . . . . . . . . . . . . . . . _ . _ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . .
. . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . ....................... VI. X-Ray -Diffraction Studies
VII. Conformation of Inhibitors in the Active Site . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
B. B~ckbone Conformations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . C. Si le-Chain Conformation D. Dfluoroketones and Implications for the Ca E. G. ycol-Based, Cyclic, Phosphorus-Containin F. Inhibitor-Induced Protein Conformation . . . ..................... G. Subsite Specificity . . . H. Comparison of Renin and HIV-1 Proteinase Inhibitors . . . . . . . . . . . . . . . . . . . . . . .
VIII. Minirial Renin Inhibitor
c . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . _ . _ . . . . . . . . . . . _ _ . . . IX. Minirial HIV-1 Proteinase Inhibitor . . . . . . . . . , . . . . . . . . . . . . . . . . . . . . X. Aspaityl Proteinase Inhibitors as Potential Drugs . . . . . . . . . . . . . .
. . . . . . . . . . . Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
731 737 737 738 739 739 739 740 741 741 741 744 745 746 746 747 748 748 748 748 750 750 752 752 753 757 757 760 763 764 765 767 768 768 770 770 770 771 771
I. INTRODUCTION
During the past two decades, attempts to combat two serious human health threats, hypertension and acquired immune deficiency syndrome (AIDS),
Medicinal Iiesearch Reviews, Vol. 13, No. 6, 731-778 (1993) 0 1993 John Wiley & Sons, Inc. CCC 0198-6325/93/060731-48

732 ABDEL-MEGUID
have resulted in extensive research in both industry and academia to discover, design, and develop inhibitors of the class of enzymes known as aspartyl proteinases (also known as aspartic proteinases, aspartate proteinases, and acid proteinases, and frequently referred to as proteases instead of pro- teinases). Although the bulk of this research has historically focused on inhib- itors of remin, an aspartyl proteinase that plays a key physiologic role in regulating blood pressure and fluid balance, a growing number of recent studies have centered on inhibitors of the proteinase from the human immu- nodeficiency virus (HIV), the etiological agent of AIDS.'
Aspartyl proteinases represent one of the four main classes of proteolytic enzymes, the other three being serine proteinases, cysteine proteinases, and metalloproteinases. The aspartyl proteinase family includes many well-studied enzymes, such as pepsin (gastric and fungal), renin, retroviral proteinases, cathepsin D and E, and chymosin (formerly known as rennin). These en- zymes are characterized by an active-site cavity that contains two catalytic aspartic acids, inhibition by pepstatin2.3 (a naturally occurring hexapeptide from Streptornyces), and a conserved Asp-(Thr or Ser)-Gly amino acid sequence that occurs twice in the active molecule. Except for the retroviral aspartyl proteinases, which function as homodimers of approximately 100 amino acid residues in each monomer, all others are active as monomers of about 330 amino acid residues.
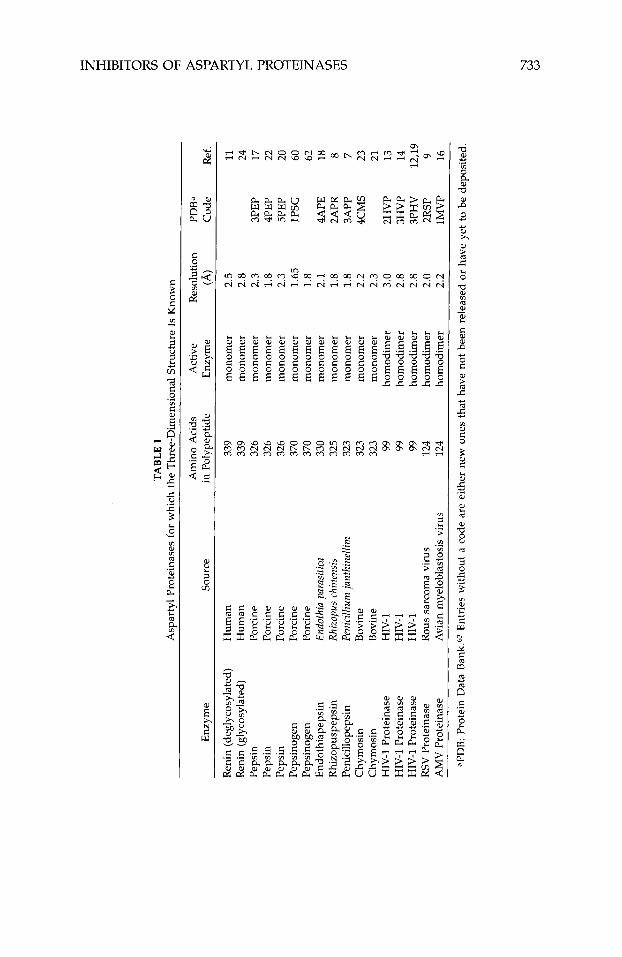
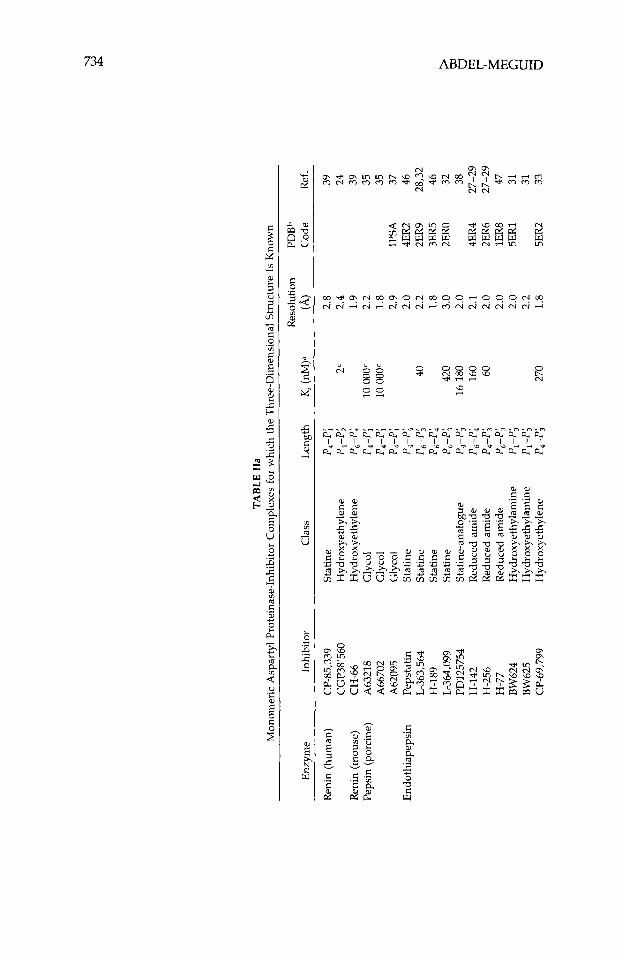
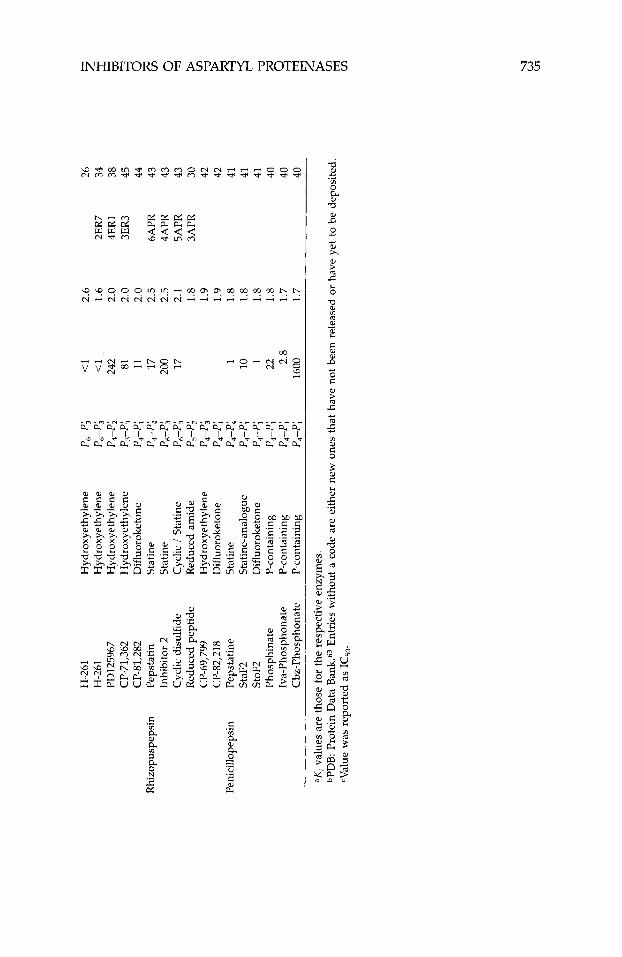
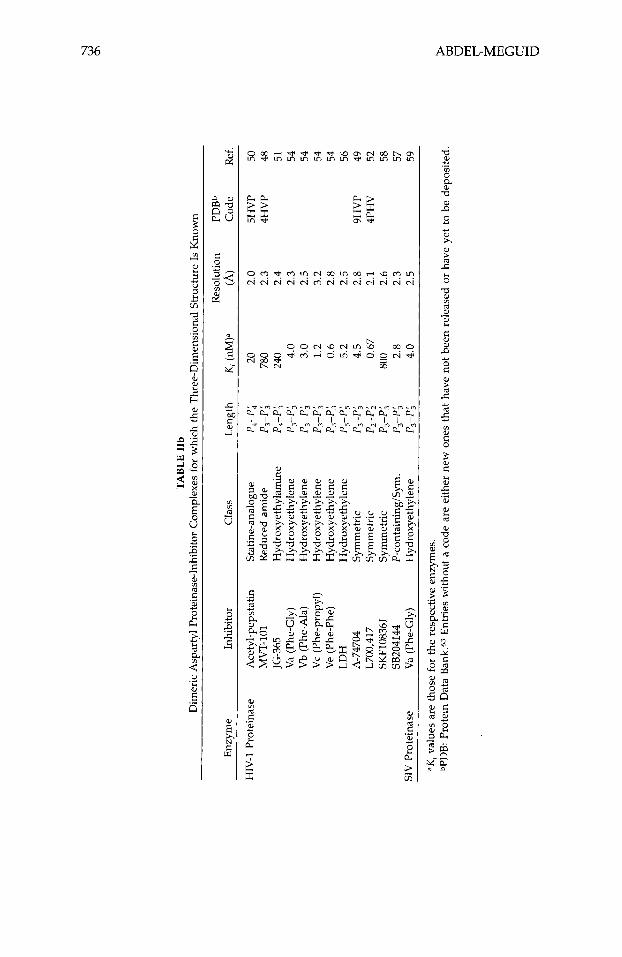
Although studies with antibodies4 first supplied evidence that interference with the action of renin had therapeutic potential, this review will not discuss this aspect of aspartyl proteinase inhibition, nor will it discuss inhibitors of HIV proteinase dimerization.5~6 Instead, it will focus entirely on inhibitors designed to bind in the active site of human renin and HIV type-1 proteinase (HIV-1 being the most common strain of this virus in the United States and Europe). Generally these inhibitors are peptide analogues designed to mimic a high-energy intermediate involved in peptidolysis. Various nonhydrolyz- able moieties, including the amino acid statine, secondary amines, and hy- droxyethylenes, have been incorporated in these peptides to replace the scissile peptide bond. Recently much of this work has been guided by the three- dimensional structures of a number of aspartyl proteinases.7-24 These struc- tures, determined using single-crystal x-ray-diffraction techniques, are listed in Table I. Numerous crystal structures of the inhibited form of these enzymes have also been rep0rted2~-~~ (Table 11), including the structure of pepsino- genm-G2 (Table I). The atomic coordinates of most of these structures can be obtained from the Protein Data Bank63 (PDB). Because of the wealth and availability of this information, highly potent inhibitors of HIV-1 proteinase
Sherin S. Abdel-Meguid received his Ph.D. degree in Chemistry with Professor Victor Day at the University of Nebraska and postdoctoral training in protein crystallography with Professor Michael Rossmann at Purdue University and Professor Thomas Steitz at Yale Univer- sity. After joining Monsanto Company in 1984, he soon became Senior Group Leader and Head of Biophysical Sciences. In 1990, he moved to SmithKline Beecham (SB) Pharmaceuticals, where he is an Associate Director in the Department of Macromolecular Sciences. He has been involved in a number of projects at Monsanto and S B including the design of renin and HIV-1 proteinase inhibitors.
U
TABLE
I z 9
Asp
arty
l Pr
otei
nase
s fo
r w
hich
the
Thr
ee-D
imen
sion
al S
truc
ture
Is K
now
n
Enz
yme
Sour
ce
Ren
in (
degl
ycos
ylat
ed)
Hum
an
Ren
in (
glyc
osyl
ated
) H
uman
Pe
psin
Po
rcin
e Pe
psin
Po
rcin
e Pe
psin
Po
rcin
e Pe
psin
ogen
Po
rcin
e Pe
psin
ogen
Po
rcin
e E
ndot
hiap
epsi
n En
doth
ia p
aras
itica
R
hizo
pusp
epsi
n Rh
izop
us c
hine
nsis
Peni
cillo
peps
in
Peni
cilliu
rn ja
nthi
nelli
m
Chy
mos
in
Bov
ine
Chy
mos
in
Bov
ine
HIV
-1 P
rote
inas
e H
IV-1
H
IV-1
Pro
tein
ase
HIV
-1
HIV
-1 P
rote
inas
e H
IV-1
RS
V P
rote
inas
e R
ous
sarc
oma
viru
s A
MV
Pro
tein
ase
Avi
an m
yelo
blas
tosi
s vi
rus
Am
ino
Aci
ds
in P
olyp
eptid
e
339
339
326
326
326
370
370
330
325
323
323
323 99
99
99
124
124
Act
ive
Enz
yme
mon
omer
m
onom
er
mon
omer
m
onom
er
mon
omer
m
onom
er
mon
omer
m
onom
er
mon
omer
m
onom
er
mon
omer
m
onom
er
hom
odim
er
hom
odim
er
hom
odim
er
hom
odim
er
hom
odim
er
% z R
esol
utio
n PD
Ba
(4
Cod
e R
ef.
2.5
2.8
2.3
1.8
2.3
1.65
1.
8 2.
1 1.
8 1.
8 2.
2 2.
3 3.
0 2.
8 2.
8 2.
0 2.
2
11
2 3
20
9 !3 %
24
3PE
P 17
4P
EP
22
5PE
P 1P
SG
60
62
4APE
18
3APP
7
4CM
S 23
21
2H
VP
13
3HV
P 14
3P
HV
12
,19
2RSP
9
lMV
P 16
2APR
8
m v,
aPD
B: P
rote
in D
ata
Ban
k.63
Ent
ries
with
out
a co
de a
re e
ither
new
one
s th
at h
ave
not
been
rel
ease
d or
hav
e ye
t to
be
depo
site
d.
TABL
E II
a M
onom
eric
Asp
arty
l Pro
tein
ase-
Inhi
bito
r C
ompl
exes
for
whi
ch t
he T
hree
-Dim
ensi
onal
Str
uctu
re I
S K
now
n
Res
olut
ion
PDB
b E
nzym
e In
hibi
tor
Cla
ss
Len
gth
Kz (n
M)a
(A
) C
ode
Ref
.
CG
P38'
560
Hyd
roxy
ethy
lene
P,
-P;
2c
2.4
24
Ren
in (
hum
an)
CP-
85,3
39
Stat
ine
P,-P
; 2.
8 39
Ren
in (
mou
se)
CH
-66
Hyd
roxy
ethy
lene
P,
-P;
1.9
39
Peps
in (
porc
ine)
A
6321
8 G
lyco
l P,
-P;
10 oo
oc
2.2
35
A66
702
Gly
col
P,-P
; 10
oooc
1.8
35
A62
095
Gly
col
P&
2.9
lPSA
37
E
ndot
hiap
epsi
n Pe
psta
tin
Stat
ine
P,-P
; 2.
0 4E
R2
46
L-36
3,56
4 St
atin
e P,
-P;
40
2.2
2ER
9 28
,32
H-1
89
Stat
ine
P,-P
; 1.
8 3E
R5
46
L-36
4,09
9 St
atin
e P,
-P;
420
3.0
2ER
0 32
I'D
1257
54
Stat
ine-
anal
ogue
P,
-Pj
16 1
80
2.0
38
H-1
42
Red
uced
am
ide
P,-P;
16
0 2.
1 4E
R4
27-2
9 H
-256
R
educ
ed a
mid
e P,
-P;
60
2.0
2ER
6 27
-29
H-7
7 R
educ
ed a
mid
e P,
-P;
2.0
lER
8 47
CP-
69,7
99
Hyd
roxy
e thy
lene
P,
-P;
270
1.8
5ER
2 33
BW
624
Hyd
roxy
ethy
lam
ine
P,-P
; 2.
0 5E
R1
31
BW
625
Hyd
roxy
ethy
lam
ine
Pl-P
; 2.
2 31
a
H-2
61
Hyd
roxy
ethy
lene
P,
-P;
<1
2.6
26
PD12
5967
H
ydro
xyet
hyle
ne
P,-P
; 24
2 2.
0 4E
R1
38
H-2
61
Hyd
roxy
ethy
lene
P,
-P;
1.6
2ER
7 34
CP-
71,3
62
Hyd
roxy
ethy
lene
P,
-P;
81
2.0
3ER
3 45
C
P-81
,282
D
iflu
orok
eton
e P,
-P;
11
2.0
44
Rhi
zopu
spep
sin
Peps
tatin
St
atin
e P,
-P;
17
2.5
6APR
43
In
hibi
tor
2 St
atin
e P,
-P;
200
2.5
4APR
43
C
yclic
dis
ulfi
de
Cyc
lic /
Stat
ine
P,-P
; 17
2.
1 5A
PR
43
Red
uced
pep
tide
R
educ
ed a
mid
e P,
-P;
1.8
3APR
30
C
P-69
,799
H
ydro
xyet
hyle
ne
P,-P;
1.
9 42
(3
82,2
18
Dif
luor
oket
one
P,-P
; 1.
9 42
Pe
nici
llope
psin
Pe
psta
tine
Stat
ine
P*-P
; 1
1.8
41
StaF
2 St
atin
e-an
alog
ue
P,-P
; 10
1.
8 41
St
oF2
Dif
luor
oket
one
P,-P
; 1
1.8
41
Phos
phin
ate
P-co
ntai
ning
P4
-P;
22
1.8
40
Iva-
Phos
phon
ate
P-co
ntai
ning
P,
-P;
2.8
1.7
40
Cbz
-Pho
spho
nate
P-
cont
aini
ng
P4-P
; 16
00
1.7
40
valu
es a
re th
ose
for
the
resp
ectiv
e en
zym
es.
bPD
B: P
rote
in D
ata
Ban
k.63
Ent
ries
with
out
a co
de a
re e
ither
new
one
s th
at h
ave
not
been
rel
ease
d or
hav
e ye
t to
be
depo
site
d.
=Val
ue w
as r
epor
ted
as IC
so.
> rn
v
w
cn
TABL
E II
b D
imer
ic A
spar
tyl
Prot
eina
se-I
nhib
itor
Com
plex
es f
or w
hich
the
Thr
ee-D
imen
sion
al S
truc
ture
Is
Kno
wn
Res
olut
ion
PDB
h E
nzym
e In
hibi
tor
Cla
ss
Len
gth
K, (n
M)a
(4
C
ode
Ref
.
HIV
-1 P
rote
inas
e A
cety
l-pe
psta
tin
Stat
ine-
anal
ogue
P,
-P;
20
2.0
5HV
P 50
M
VT-
101
Red
uced
am
ide
P,-P
; 78
0 2.
3 4H
VP
48
JG-3
65
Hyd
roxy
ethy
lam
ine
P,-P
; 24
0 2.
4 51
Va
(Ph
e-G
ly)
Hyd
roxy
ethy
lene
p,
-p;
4.0
2.3
54
Vb
(Phe
-Ala
) H
ydro
xyet
hyle
ne
P,-P
& 3.
0 2.
5 54
V
c (P
he-p
ropy
l)
Hyd
roxy
ethy
lene
P,
-P;
1.2
3.2
54
Ve (
Phe-
Phe)
H
ydro
xyet
hyle
ne
P,-P
; 0.
6 2.
8 54
A-7
4704
Sy
mm
etri
c P,
-P;
4.5
2.8
9HV
P 49
SB20
4144
P-
cont
aini
nglS
ym.
P,-P
; 2.
8 2.
3 57
SI
V P
rote
inas
e Va
(Ph
e-G
ly)
Hyd
roxy
ethy
lene
P,
-P;
4.0
2.5
59
LDH
H
ydro
xyet
hyle
ne
p5-p
; 5.
2 2.
5 56
L700
,417
Sy
mm
etri
c P,
-P;
0.67
2.
1 4P
HV
52
SK
F108
361
Sym
met
ric
P,-P
; 80
0 2.
6 58
=K, v
alue
s ar
e th
ose
for
the
resp
ectiv
e en
zym
es.
hPD
B: P
rote
in D
ata
Ban
k.63
Ent
ries
with
out
a co
de a
re e
ither
new
one
s th
at h
ave
not
been
rel
ease
d or
hav
e ye
t to
be
depo
site
d.

INHIBITORS OF ASPARTYL PROTEINASES 737
have been rapidly developed. The synthesis of numerous potent inhibitors of the enzyme was reported@-67 shortly after the discovery that the proteinases from retroviruses were aspartyl proteinases.68-72
Determination of the three-dimensional structures of inhibitors bound to the various aspartyl proteinases played an important role not only in the development of potent inhibitors but also in understanding the mechanism of action of these enzymes. Based on the crystal structure of rhizopuspepsin bound to a reduced amide inhibitor, Suguna et aI. proposed a mechanism of action for these enzymes.30 Their proposal is consistent with a “general acid- general base” mechanism of the two aspartyl residues, with a central water molecule bound between the carboxyl groups of these two residues as the nucleophile. This mechanism has recently been supported by a series of crys- tallographic structure determinations of fungal aspartyl proteinases with in- hibitors containing a difluoroketone moiety.4*,*2,44
Aspartyl proteinases and their inhibitors have been extensively reviewed in the past ten years. A number of general reviews have summarized work in this area,73-79 while numerous others focused exclusively on renin80-84 and its inhibitors.85-94 Studies on HIV-1 proteinase and its inhibitors have also been reviewed.95-102 This article will focus on studies, particularly those of a struc- tural nature, that have led to further advances in the design of renin and HIV-1 proteinase inhibitors.
11. THERAPEUTIC TARGETS OF ASPARTYL PROTEINASE INHIBITORS
Extensive research efforts have focused on the development of inhibitors of human renin and HIV-1 proteinase as potential agents to combat hyperten- sion and AIDS, respectively. Each of these two enzymes is believed to be a unique target for therapeutic intervention. This uniqueness, coupled with the seriousness and prevalence of these two diseases, assures continued interest in the discovery and development of aspartyl proteinase inhibitors as poten- tial drugs.
A. Human Renin
Human renin is the first enzyme in the renin-angiotensin system, a pro- teolytic cascade responsible for the formation of the potent vasoconstrictor octapeptide angiotensin I1 (Fig. 1). It is a glycoprotein of 339 amino acid residues that has two glysylation sites and is active as a monomer.103-107 It is synthesized in the juxtaglomerular cells of the kidney as an inactive precur- sor, prorenin.108 A renin-processing enzyme converts prorenin to the active form by cleaving the first 45 amino acid residues.109
The success of angiotensin converting enzyme (ACE) inhibitors as anti- hypertensive drugs has fueled the race to develop novel, potent inhibitors of renin. Unlike ACE, which works on a host of substrates such as enkephalins, neurotensin, bradykinin, and substance P, as well as angiotensin I,11o renin is believed to have no biological substrates other than angiotensinogen. This feature makes renin a unique drug target, since its inhibition should affect only the renin-angiotensin system. Thus it is postulated that drugs that are renin inhibitors will lack the side effects associated with ACE inhibitors, most

738 ABDEL-MEGUID
P17
HlUllm Angiotensinogen
A s p - Arg - Val - Tyr - Ile - His - Pro - Phe - His ~ Leu *Val - Ile - His -Protein
4 Renin
Angiotensin 1 Asp - Arg - Val - Tyr - Ile - His - Pro - Phe * His - Leu
4 Angiotensin-Converting Enzyme (ACE)
Angiotensin I1 Asp*Arg - Val - Tyr - Ile - His - Pro - Phe
4 Aminopeptidase
Angiotensin 111 Arg - Val - Tyr - Ile - His - Pro - Phe
Figure 1. Renin-angiotensin system showing cleavage sites of human angiotensinogen2z2; aster- isk (*) denotes position of the scissile bonds.
I P24 P7 P6 PR RT I H IN
of which are presumed to be unrelated to the inhibition of the renin- angiotensin system. These side effects include coughing,l'l eurticaria,ll2,1*3 and angioneurotic edema.112
B. HIV Proteinase
It is estimated that as many as 20 million individuals world-wide may become infected with HIV by the year 2000.114 This growing health threat has led to extensive research to identify treatment strategies to combat this virus. One strategy is to inhibit the enzymatic action of the virus-encoded proteinase that is essential for viral replication.115 HIV proteins are translated as polypro- *
I I pd I I in I Pr I rt I
I I Frameshin /
/ ./MA CA NC
1 2 3 4 5 6 7 8
P I 7 * P24 P24 * X
x * P7 P7 P6
* PR PR * RT
RT51 * RNase H RT IN
P, P, P, P, P,' P,' P,' P,' SER GLN ASN TYR *PRO ILE VAL GLN ALA *ARG VAL LEU * ALA *GLU* ALA MET ALA *THR ILE *MET*MET *GLN*ARG* GLY PRO GLY ASN PHE LEU *GLN SER ARG SER PHE ASN PHE *PRO *GLN ILE THR THR LEU ASN PHE PRO ILE SER PRO ALA *GLU THR *PHE* TYR -VAL* ASP GLY ARG LYS * ILE 9 LEU * PHE LEU ASP GLY
Figure 2. Organization of HIV-1 proteins in the gag and pol polyproteins, and the cleavage sites for HIV-I proteinase (from reference 99). Amino acid sequences flanking each of the 8 proteolytic cleavage sites are listed, with the asterisk (*) indicating position of the scissile bond. Substrate residues are designated P,-Pi according to the notation of Schechter and Berger.IZ9

INHIBITORS OF ASPARTYL PROTEINASES 739
teins that must be cleaved during the maturation of the virions.71 The pro- teinase processes the gug and gug-pol polyproteins (Fig. 2) into mature struc- tural proteins and the enzymes required for viral replication: proteinase, re- verse transcriptase, and integrase. The necessity of these enzymes for the proper assembly and maturation of fully infectious virions makes them im- portant targets in the design of antiviral agents for AIDS, particularly the proteinase that plays a pivotal role in this process.
HIV proteinase is a 99-amino acid residue protein that is active as a homo- dimer.72 It was classified as a member of the aspartyl proteinase family based on its active-site sequence similarity68~~~ and structural analogy70 to the well- characterized, monomeric enzymes renin and pepsin. Interestingly, Tang ef al. predicted, in 1978, that such an aspartyl proteinase homodimer might exist based on analysis of molecular symmetry within the structures of the mono- meric pepsin enzymes.116
111. THREE-DIMENSIONAL STRUCTURE OF ASPARTYL PROTEINASES
Aspartyl proteinases are divided into two subclasses, the monomeric and the dimeric. Pepsin (gastric and fungal), renin, cathapsin D and E, and chymosin are monomeric enzymes, while the retroviral proteinases are di- meric.
A. Monomeric
The three-dimensional structures of all monomeric aspartyl proteinases predominantly consist of p strands that fold in two structurally similar do- mains related by an approximate twofold axis116 [Fig. 3(a)]. Within each of the domains a less precise twofold axis has been reported.117 The active site is at the junction of the two domains, each of which contributes one of the two catalytic aspartic acids at the center of the substrate binding cleft. The overall fold of all the monomeric enzymes is strikingly similar. For example, super- position of the a-carbon atoms of human renin on those of penicillopepsin, rhizopuspepsin, endothiapepsin, and porcine pepsinogen gives root-mean- square (rms) deviations of 1.6, 1.4, 2.0, and 1.3 A, respectively.11 When only the active-site residues are compared, the similarities are even greater, with rms deviation less than 0.5 A. Less similar, however, are the folds of the amino- and carboxy-terminal domains within the same enzyme.
B. Dimeric
The three-dimensional structures of the various dimeric retroviral enzymes are predominantly composed of p strands showing exact twofold symmetry [Fig. 3(b)J; their active site is formed at the interface of the dimer and consists of two aspartyl residues, one contributed by each subunit.12-16 These proteins undergo considerable conformational change upon complexation with inhibi- tors, particularly in the two "flaps" (flexible P-hairpin structures) which move by as much as 7 A to tightly embrace the ligands.48
Similarity of three-dimensional structures among the retroviral proteinases is high. Superpositon of the a-carbon atoms of HIV-1 and Rous sarcoma virus (RSV) proteinases gives 1.5 A for 86 common a-carbon atoms of one mono- mer.14 The RSV proteinase is 25 amino acid residues longer than HIV enzyme.

740 ABDEL-MEGUID
(b)
Figure 3. The structures of (a) rhizopuspepsin (PDB code 2APR) and (b) HIV-1 proteinase (PDB code 3HVP); only a-carbon atoms are shown. Each structure is shown with its twofold axis (quasi-twofold axis in rhizopuspepsin) pointing up and down in the plane of the paper.
As common with all analogous three-dimensional structures, the additional residues of RSV are found in surface loops.
C. Comparison of the Monomeric and Dimeric Enzymes
Inspection of Fig. 3 indicates considerable similarity between the three- dimensional structures of the monomeric and dimeric aspartyl proteinases. Superposition of 57 pairs of a-carbon atoms of HIV-1 proteinase (more than one-half of all such atoms in a monomer) and rhizopuspepsin gives a rms diviation of 1.4 A.14 More significant is the strong structural analogy of the amino acid residues consituting the active sites in both classes. Comparison of 88 atom pairs of active-site residues of HIV-1 proteinase with those of rhizo- puspepsin yielded a rms difference of 0.59 A.14
Although the overall fold is conserved in the two classes of aspartyl pro- teinases, there are striking differences. The active forms of retroviral enzymes are considerably smaller than the monomeric ones. While HIV-1, RSV, and avian myeloblastosis virus (AMV) dimers are made up of 198, 248, and 248 amino acid residues each, respectively, all monomeric aspartyl proteinases contain more than 300 residues in their polypeptide chain. The additional residues are found in surface loops between p strands of the monomeric proteinases. Another difference is that dimeric enzymes have two flaps, one on each monomer, while the monomeric enzymes have a single long flap on the amino-terminal domain. Although these structural differences may reflect potential differences in their mechanism of acti0n,~9,96 it is unclear how that would be, given the great similarity in active-site geometry of the two classes.

INHIBITORS OF ASPARTYL PROTEINASES 741
IV. FEATURES OF SUBSTRATE SPECIFICITY THAT INFLUENCE INHIBITOR DESIGN
Traditionally, the discovery of drugs has focused on screening for com- pounds in nature and in synthetic chemical libraries. The success of this approach relies on the sensitivity and specificity of the assays used and the ability to configure them for high throughput. Although screening continues to be a very powerful tool in drug discovery, knowledge-based or "rational" approaches are gaining rapid acceptance, either to complement screening or for use independently in the design of novel therapeutic molecules. One rational approach used in the design of enzyme inhibitors is based on knowl- edge of substrate(s) sequence and the substrate-specificity of the target en- zyme. Another, known as structure-based, requires knowledge of the target- enzyme three-dimensional structure preferably in the liganded state.118.119 These two approaches have been used in the design of both renin and HIV-1 proteinase inhibitors.
Earlier efforts to design inhibitors of aspartyl proteinases emanated from studies to identify the essential structural requirements for substrates. The pioneering work by Skeggs and CO-workers120-124 aimed at understanding renin substrate specificity, was critical to the rational design of inhibitors. Similarly, attempts to identify HIV-1 proteinase inhibitors initially focused on understanding the substrate requirements of the en~yme.~25-1*8 However, early recognition that HIV proteinase is an aspartyl proteinase considerably facilitated the task of designing potent inhibitors because of the large body of knowledge then available on renin inhibition. Also, the early availability of the three-dimensional structure of HIV-1 proteinasel2-14 and its inhibitor complex48 has greatly accelerated the process.
The notation first described by Schechter and Berger will be used in the succeeding sections of this article to describe the binding of ligands to the aspartyl proteinases.129 In this notation, residues of a ligand are designated P1,P2, . . . ,P, and Pi, Pi, . . . , PA from the scissile bond towards the amino- and carboxy-terminal, respectively, while the corresponding enzyme binding sites (subsites) are designated Sl,S2, . . . ,S, and S;,S;, . . . ,S;.
A. Substrate Specificity of Human Renin
A potential therapeutic advantage of renin inhibitors compared to those of ACE is their unique substrate specificity. Human renin cleaves at Leu-Val while the enzymes from other mammalian species cleave at Leu-Leu. Al- though human renin can cleave human as well as other mammalian angioten- sinogens, only the human enzyme can efficiently cleave the human substrate. Using synthetic peptides, Skeggs et al. defined an octapeptide A-1 (Table 111) as the minimum substrate that is efficiently hydrolyzed by renin.I24 Upon removal of the carboxy-terminal tyrosine, the resulting peptide was no longer a substrate. Thus the octapeptide A-1 became the template on which further inhibitor design was undertaken.
B. Substrate Specificity of HIV-1 Proteinase
Unlike renin with only one substrate (Fig. l), HIV proteinase cleaves the gag and gag-pol polyproteins at numerous sites spanning a heterogeneous amino

TABL
E 11
1 Pe
ptid
e, S
tatin
e, A
min
omet
hyl
Isos
tere
(R
educ
ed-B
ond)
, and
Hyd
roxy
ethy
lene
Iso
ster
e In
hibi
tors
of
Ren
iwb
p,
P6
Ps
p4
p3
P
2 PI
1
P;
Pi
Pi
PA
IC,O
(n
M)
Ref
.
Hum
an s
ubst
rate
.I
. -
Ile
- H
is
- Pr
o -
Phe
- H
is
- Le
u V
al -
Ile
- H
is
- ...
22
2 Po
rcin
e su
bstr
ate
... -
Ile
-
His
-
Pro
- Ph
e -
His
-
Leu
- L
eu
- V
al -
Tyr
- ...
22
3 A
-1
His
-
Pro
- Ph
e -
His
-
Leu
- L
eu
- V
al -
Tyr
200
000
124
A-2
Le
u -
Leu
- V
al -
Tyr
- O
Me
143
A-3
(R
IP)
Pro
- H
is
- Pr
o -
Phe
- H
is
- Ph
e -
Phe
- V
al -
Tyr
- Ly
s 2
000
144
A-4
(H-1
12)
His
-
Pro
- Ph
e -
His
-
Leu
Val
- Il
e -
His
33
0 16
1 B
-1 (
Peps
tatin
) Iv
a -
Val
- V
al -
Stat
ine
- A
la
- St
atin
e 22
000
14
7 8-
2 (H
-176
) H
is
- Pr
o -
Phe
- H
is
- St
atin
e -
Val
- Il
e -
His
17
16
1 C
-1 (
H-7
6)
His
-
Pro
- Ph
e -
His
-
Leu
R L
eu
- V
al -
Tyr
1000
16
1 C
-2 (
H-1
13)
His
-
Pro
- Ph
e -
His
-
Leu
R V
al -
Ile
- H
is
190
161
C-3
(H
-142
) Pr
o -
His
-
Pro
- Ph
e -
His
-
Leu
R V
al -
Ile
- H
is
- Ly
s 10
16
1 D
-1 (
H-1
94)
His
-
Pro
- Ph
e -
His
-
Leu
OH
V
al -
Ile
- H
is
3 16
1 D
-2 (H
-261
) B
oc
- H
is
- Pr
o -
Phe
- H
is
- Le
u O
H
Val
- Il
e -
His
0.
7 16
2
=Arr
ow in
dica
tes
posi
tion
of sc
issi
le b
ond
or s
urro
gate
. bI
va, i
sova
lery
l; B
oc, t
-but
oxyc
arbo
nyl;
R, a
min
omet
hyle
ne is
oste
re (
redu
ced
amid
e); O
H, h
ydro
xyet
hyle
ne is
oste
re.

TABL
E IV
C
lass
ific
atio
ns of
Sub
stra
tes
of R
etro
vira
l Pro
tein
ases
Bas
ed o
n th
e Ty
pe o
f A
min
o A
cids
Fla
nkin
g th
e Sc
issi
le B
onda
p4
p3
p2
PI
1 p;
p;
0;
Cla
ss 1
b
Gly
lpol
ar
Asn
T
yrIP
he
Pro
hydr
opho
bic
Cla
ss 2
b A
rg
pola
r hy
drop
hobi
c Ph
e L
eu
Cla
ss 3
b hy
drop
hobi
c hy
drop
hobi
c hy
drop
hobi
c G
lulG
ln
Typ
e 1c
sm
alld
un
char
gede
al
ipha
tic'
Tyr
lPhe
lLeu
Pr
o al
ipha
tic
alip
hatic
al
ipha
tic8
alip
hatic
h T
ype
2c
smal
ld
unch
arge
de
alip
hatic
' L
eu
Ala
ILeu
lVal
aArr
ow in
dica
tes
posi
tion
of s
ciss
ile b
ond.
bR
efer
ence
125
. =R
efer
ence
142.
dS
mal
l am
ino
acid
s in
bot
h ty
pes
favo
ring
Pro
, Se
r, T
hr, G
ly, a
nd A
la.
eGen
eral
ly u
ncha
rged
; G
ln s
een
in s
ome
type
1, w
hile
Arg
lLys
see
n in
som
e ty
pe 2
. 'A
lipha
tic f
or b
oth
type
s; A
sn o
ccas
iona
lly se
en a
nd V
al la
rgel
y ex
clud
ed f
rom
type
1
gIle
is e
xclu
ded.
hA
la is
fav
ored
.

744 ABDEL-MEGUID
acid sequence (Fig. 2). Although it appears that there is little or no consensus for amino acids at most of the substrates’ subsites, the predominance of Phe- Pro or Tyr-Pro at the P,-P; in retroviral proteinases was noted, since hydro- lysis at the N-terminal of proline is not common for endopeptidases.101
Henderson et al. have grouped substrates of HIV-1 and other closely related retroviral proteinases into three classes based on the amino acid sequence flanking the scissile bond125 (Table IV). Class 1 is characterized by Phe-Pro or Tyr-Pro at P,-P;, Asn at P,, and a preference for hydrophobic residues at Pi. Class-2 substrates contain an Arg at P4 and the sequence Phe-Leu at Pi-P;. Class-3 substrates show a preference for hydrophobic residues at P2, PI, and Pi and for Glu or Gln at P;. Interestingly, almost all the nonviral protein substrates of HIV-1 proteinase identified to date661130-134 belong to class 3. When amino acid sequences of substrates from 10 diverse retroviral pro- teinases were compared,lQ the classes were narrowed to two (Table IV), of which the first is similar to that of Henderson et in that it contains Pro at Pi and a preference for Phe or Tyr at PI (type 1, Table IV). The second class, however, has either Leu, Ala, or Val at Pi, while PI favors Leu over Phe or Tyr (type 2, Table IV). Furthermore, a statistical analysis of amino acid sequences of viral and nonviral substrates of HIV-1 proteinase suggested that the highest stringency for particular amino acid residues is at the P,, PI, and P2’ sites.135
The lack of a consensus sequence beyond the Pl-P; has led to the sugges- tion that the primary sequence at other subsites within the substrate is of limited importance, and that it is features of secondary and tertiary structure of the polyprotein substrates that contribute significantly to their recognition by HIV-1 proteinase.101 This assertion is contradicted by studies showing that synthetic octapeptides corresponding to amino acid sequences found in non- viral protein substrates of HIV-1 proteinase are also efficiently hydrolyzed, while some containing a central Tyr-Pro sequence are not hydrolyzed.66 Also, the ability of HIV-1 proteinase to cleave on either side of a phenylalanine residue (Fig. 2; Table IV), depending on the flanking sequence,l25 supports the assumption that it is amino acid sequence, not conformation, that is critical to substrate recognition and cleavage.
Studies with synthetic peptides128,135-142 have helped define the minimum peptide that can function as a specific and efficient substrate to be a heptapep- tide spanning the region from P, to PA. Thus much of the data on substrate specificity indicates that a template for inhibitor design would optimally be a heptapeptide spanning the P, to P i sites, in which the P, and Pi are hydro- phobic residues, P, favors a hydrophobic, particularly p branched, residue, or Asn, P; is a hydrophobic or anionic group, P3 and PA are large residues of variable polarity, and P4 is a small, hydrophilic residue.101
V. DESIGN AND CLASSIFICATION OF ASPARTYL PROTEINASE INHIBITORS
As indicated above, inhibitors of renin were designed using the minimum substrate of Skeggs et aI. as the template.124 Initially that involved the replace- ment of the Pl-P; dipeptide with a number of nonhydrolyzable moieties. Thus one classification of aspartyl proteinase inhibitors is based on the nature of this P,-P,’ pseudodipeptide. Inhibitors can be further subclassified as linear, cyclic,

INHIBITORS OF ASPARTYL PROTEINASES 745
H 0 P,'
Peptide Tetrahedral Intermediate
El H I \N&$J( ,?+
H OH P,' '"W' H I OH 0 I
P,'
Statlne Aminomethylene Hydroxyethylene (X I H) lsostere Dihydroxyethylene (X = OH)
lsosteres
Hydroxyethylamine Ketone (X = H) Glycol 1sostere Difluoroketone (X = F)
Figure 4. Stereochemical relationships of peptide, tetrahedral intermediate, and their isosteric replacements.
or symmetric, with the latter applying to inhibitors of retroviral proteinases. The stereochemical relationships of the different classes are shown in Fig. 4.
Because a number of recent reviews describe in detail the inhibitors of aspartyl proteinases,90-102 I will describe the inhibitors in each class only briefly, focusing on the specific compounds that were critical for advancement of the field.
A. Peptide Inhibitors
Historically, the design of inhibitors of aspartyl proteinases focused on the inhibition of renin. In 1968, Skeggs et al. demonstrated that an amino-terminal octapeptide (A-1; Table 111) of horse angiotensinogen was a weak competitive inhibitor of renin,124 while Kokubu et a l . defined the minimum inhibitory peptide for the enzyme143 (A-2; Table 111). Armed with this knowledge, Burton et al. identified a decapeptide inhibitor of renin (A-3; Table 111) in which the Leu-Leu dipeptide at the scissile bond of A-1 was replaced by Phe-Phe; an amino-terminal proline and carboxy-terminal lysine were added, the latter to increase solubility.144 This peptide, named renin-inhibitory peptide (RIP), was found to exhibit considerable selectivity for the renin of primates, and was effective in lowering blood pressure in animal models14 and in man.145 Al- though these peptides were only micromolar inhibitors of renin and exhibited poor pharmacokinetics, they inspired much of the later work to develop more metabolically stable and potent inhibitors.
It addition to designing peptide inhibitors of renin based on its substrate sequence, attempts were made to identify inhibitors derived from the "pro" segment of the renin precursor.146,147 Although inhibition was observed. their potencies were not high enough to warrant further studies.
Little was done to design peptide inhibitors of HIV-1 proteinase, since it was recognized from work with renin that peptide inhibitors have poor phar- macokinetics.

746 ABDEL-MEGUID
B. Inhibitors Containing Statine or Its Analogues
Pepstatin (B-1; Table 111) is a nonspecific inhibitor of aspartyl proteinases. With the notable exception of renin, it is an extremely potent inhibitor of most members of this family.148~149 Pepstatine, isolated from culture filtrates of various Streptomyces, was found to contain the unusual amino acid statine3 [ (3S,4S)-4-amino-3-hydroxy-6-methylheptanoic acid]. It was proposed that pepstatin is a transition-state analogue inhibitor in which statine is a dipep- tide isostere replacing the P,-P; portion of the substrate,150,151 and that the 3s-hydroxyl group is a mimic of a hydroxyl group in the tetrahedral inter- mediate.152-154 Later, Rich proposed that pepstatin serves as a ”collected- substrate” inhibitor (that is, a collection of both the peptide and water substrates) rather than a transition-state analogue, and that the statine 3S- hydroxyl group displaces the enzyme-bound water molecule that is necessary for catalysis.75.155
Numerous potent and highly specific inhibitors of human renin that contain statine or its analogues have been identified. This work was recently detailed in an excellent review by Greenleego and will not be reviewed in this article.
Attempts to design potent inhibitors of HIV-1 proteinase that contain statine were not encouraging. This class of molecules only poorly inhibit the enzyme, with inhibition constants mostly in the micromolar range.64,128,138,156,157 The design of inhbitors containing alkylated statines (hydroxymethylene isosteres), however, has been more successful, probably due to the presence of Pi side chains.
C. Aminomethylene Isosteres (Reduced Amides)
One of the first design concepts introduced to enhance the potency of peptide inhibitors of renin was the formation of secondary amines (Fig. 4) in which the carbonyl group of the scissile peptide bond (--CO-NH-) is re- duced to give the methylene-containing group (-CH2-NH-).158-161 These nonhydrolyzable peptides were designed to mimic the transition state of the enzyme reaction.162 The high affinity of this class of inhibitors (also known as reduced peptide, reduced-bond, and reduced peptide-bond isostere) appar- ently stems from the tetrahedral nature of the carbon atom that replaces the carbonyl, and from the electrostatic interaction between the basic amine and the active site aspartates.
Although an aminomethylene isostere analogue of Kokubu’s inhibitory peptide A-2 was not a potent one, it was effective in lowering blood pressure when tested in ~ i v 0 . l ~ ~ A dramatic increase in binding affinity was observed when the aminomethylene isostere was introduced into Skeggs’ octapeptide A-1, giving the analogue C-1 (Table 111), bearing the code H-76. The latter inhibitor and its analogues were based on the equine substrate sequence. Thus, when the human substrate sequence became known, it was possible to synthesize inhibitors such as C-2 and C-3 (code named H-113 and H-142; Table 111), which showed a marked preference for human renin.164,165
Attempts to design potent aminomethylene isostere inhibitors of HIV-1 proteinase were disappointing, with most of the resulting inhibitors having Ki values in the micromolar range.64.128

INHIBITORS OF ASPARTYL PROTEINASES 747
D. Hydroxyethylene Isosteres
A variation on the transition-state mimics containing reduced amide inhibi- tors was the introduction by Szelke and col-workers of the hydroxyethylene isosteres (Fig. 4), in which the scissile bond (-CCF-NH-) of the substrate is replaced with a (-CHOH--CH,-) moiety.160 As was proposed for statine and its analogues, the hydroxyl group in this class of inhibitors also serves as a mimic of the hydrated carbonyl (gem-diol) of the tetrahedral intermediate (Fig. 4) involved in peptidolysis. Unlike statine, however, the hydroxy- ethylene moiety preserves the atomic spacing between P, and Pi and is an isosteric replacement for that dipeptide (Fig. 4). This fact, combined with the absence of a Pi side chain in statine, may be responsible for the lower affinity seen when the hydroxyethylene moiety of a particular inhibitor is replaced by a statine (Table 111).
The hydroxyethylene class of inhibitors has received considerable atten- tion. Numerous members of this class have been reported to be potent inhibi- tors of both renin (D-1, D-2; Table 111) and HIV-1 proteinase. In a series of renin inhibitors differing only in the nature of the scissile-bond surrogate, Szelke compared inhibitors containing a peptide bond (A-4), statine (B-Z), reduced amide (C-Z), and hydroxyethylene to find that the latter is the most potent in the series161 (Table 111).
In a systematic study with HIV-1 proteinase inhibitors of this class Dreyer et aZ.54 showed a dramatic increase in potency as the length of the inhibitors and the size of their side chains at Pi increased (Table V). At one extreme the inhibitor spanning P,-P; and containing a Gly residue at P i has a K j of 6500 nM, at the other extreme the inhibitor spanning P,-P; and containing a Phe residue at Pi has a Ki of 0.4 nM (Table V).
TABLE V Inhibition of HIV-1 Proteinase by Hydroxyethylene Isosteresa
Inhibitorb Ki (nM)
X Y R = H R = Me R = n-Pr R = i-Bu R = Bn
I Boc Val-NH, 6500 260 50 23 1.4 I1 Cbz-Ala Val-NH, 2500 50 20 3.9 0.6 111 Cbz-Ala Val-Val-OMe 118 1.6 0.6 0.9 0.6 IV Cbz-Ala-Ala Val-Val-OMe 44 0.6 0.4 0.8 0.4
aReference 54. ~Boc, t-butoxycarbonyl; Cbz, carbobenzyloxy; Me, methyl; n-Pr, n-propyl; i-Bu, isobutyl; Bn,
benzyl.

ABDEL-MEGUID
E. Dihydroxyethylene Isosteres
Given the success of the hydroxyethylene isostere class of inhibitors that contain only one hydroxyl, it was hoped that the introduction of a second hydroxyl (--CHOH--CHOH-) would considerably enhance the potency of aspartyl proteinase inhibitors. However, inhibitors of renin166 and HIV-1 pro- teinase167.168 containing the dihydroxyethylene isostere (Fig. 4) do not appear to show the expected enhancement in potency.
F. Hydroxyethylamine Isosteres
This class of inhibitors combines elements from the aminomethylene and hydroxyethelene isostere classes to give potent inhibitors of both renin169-174 and HIV-1 proteinase.67,175,176 The scissile-bond surrogate in this case is (-CHOH-CH,-NH-), containing one more atom along the backbone be- tween P, and Pi than reduced amides, hydroxyethylene isosteres, or a sub- strate.
G . Ketones and Difluoroketones
Interest in the ketone class of inhibitors, in which the peptide bond (-CO-NH-) is replaced by (-CO-CH,-), has focused on their ability to serve, upon hydration, as realistic transition-state mimics of the tetrahe- dral proteolysis intermediate [-C(OH),-NH-1. It has been shown177J78 that the form of these inhibitors that binds to porcine pepsin is the hydrate [-C(OH),-CH,-1. The weaker binding of the keto-containing inhibitors to procine pepsin and renin was attributed to the unfavorable equilibrium for hydration of the ketone moiety.155 This was remediedlso-183 by the synthesis of inhibitors containing the difluoroketone (-CO-CF,-) moiety, which fa- vors the formation of [-C(OH),-CF,-].
Although this class of inhibitors offers little advantage over hydroxy- ethylene analogues, it has played an important role in support of the mecha- nism of action of aspartyl p r o t e i n a s e ~ ~ ~ - ~ ~ ~ ~ ~ first proposed by Suguna et al. based on crystallographic studies with rhizopuspepsin.30
Difluoroketone inhibitors of HIV-1 proteinase having Ki values ranging from 1 to 100 nM have been reported.64.184
H. Glycol-Based
For renin inhibitors to effectively compete with ACE inhibitors as drugs, they must be orally bioavailable. One approach to achieve that goal is to produce enzymatically resistant lipophilic inhibitors.185 Another approach, based on the belief that substances of low molecular weight (<600) have a higher proba- bility of gastrointestinal absorption, is to minimize the molecular weight by eliminating most of the carboxy-terminal portion of these inhibitors.lg6 In an attempt to design such inhibitors of low molecular weight, Hanson and co- workers186-188 and Matsueda et aZ.189 independently introduced the metaboli- cally stable glycol as a carboxy-terminal moiety. Although inhibitors ending in an unsubstituted glycol (H-la) were not exceedingly potent, the addition of substituents that can fit in the S; pocket (H-1 and H-2) improved their potency toward renin (Table VI). The renin inhibitory potency was highest when the

TABL
E V
I In
hibi
tion
of H
uman
Ren
in b
y G
lyco
l-B
ased
Inhi
bito
ra
Inhi
bito
rb
X R
2 R,
H-1
BO
C i-B
u be
nzyl
H
-2
BOC
i-Bu
cycl
ohex
ylm
ethy
l H
-3
mor
phol
ino-
CO
i-B
u cy
cloh
exyl
met
hyl
H-4
m
orph
olin
o-C
O
CH
,-im
idaz
ole
cycl
ohex
ylm
ethy
l H
-5
Me-
pipe
razi
ne-S
O,
thia
zole
cy
cloh
exyl
me th
y1
a b
C
d e
R;
= H
R;
= M
e R;
= E
t R;
= n
-Pr
R; =
i-B
u
8700
17
00
200
250
720
190
210
90
60 5 1 1
aRef
eren
ces a
re 1
87, 1
87, 1
88, a
nd 1
90 fo
r H
-1, H
-2, H
-3, a
nd H
-4, r
espe
ctiv
ely.
~
Bo
c, t-bu
toxy
carb
onyl
; Cbz
, car
bobe
nzyl
oxy;
Me,
met
hyl;
Et,
ethy
l; n-
Pr, n
-pro
pyl;
i-Bu,
iso
buty
l.

750 ABDEL-MEGUID
substituent was an isobutyl and when the stereochemistry of the two hy- droxyls was (3R,4S).94,189 The replacement of Boc-L-phenylalanine of H-2e with an O-(N-morpholinocarbonyl)-3-~-phenyllactic acid (H-3e; Table VI) gave rise to highly potent human renin inhibitors that are orally efficacious in reducing plasma renin activity in salt-depleted marmosets.188 Further substi- tutions at P4 and P,, as exemplified by H-5e (Table VI), enhanced oral bio- availability. This inhibitor was reported to have 8, 24, 32, and 53% oral bio- availability in the monkey, rat, ferret, and dog, respectively.190 This and similar inhibitors show considerable promise for therapeutic use.
Although inhibitors of this class can be viewed as belonging to the dihy- droxyethylene isostere class, they are considered here separately, because they were conceived as a carboxy-terminal modification intended to reduce the size and molecular weight of inhibitors, not as an incremental improve- ment to the hydroxyethylenes needed to maximize interaction with the en- zyme. Removal of the second hydroxyl from glycol-containing inhibitors of renin significantly decreased their potency as renin inhibitors.94
Considering both the C,-symmetric nature of the active site of HIV pro- teinase and the need to have inhibitors that extend beyond Pi to obtain rea- sonable potency, it is not surprising that little has been done to develop HZV-1 proteinase inhibitors of this class.
I. Cyclic Inhibitors
Although cyclic inhibitors can potentially belong to any of the above classes, they are considered here separately due to their unique design fea- tures. Because of their greater rigidity, it was hoped that they would be more potent and metabolically stable than their acyclic analogues.
A number of cyclic renin inhibitors with moderate potency have been re- ported. They vary in the number of atoms within the ring, the largest being 20 atoms representing cyclization through the side chains of P, and P6,191’192 and the smallest being 14 atoms representing cyclization between P, side chain and P, amide nitrogen.193-195 Smaller P1-P, rings containing 10 and 12 atoms were found to be much less active.
Based on the poor potency of cyclic renin inhibitors relative to their acyclic counterparts, the design of cyclic inhibitors of HIV-1 proteinase was not en- thusiastically pursued. However, given the small size of this enzyme and its symmetric nature, it is possible to design cyclic inhibitors in which the cycliza- tion is between P, and P, and/or Pi and Pj. Crystal structures of complexes of HIV-1 proteinase and inhibitors show54 close proximity of the side chains at the €‘,/Pi and P,lP$, enabling the design of cyclic compounds.
The identification of a central water molecule that bridges the enzyme and the inhibitor (Fig. 5) in the crystal structures of all HIV-1 proteinase-inhibitor complexes (Table IIb), presents exciting possibilities for the formation of cyclic inhibitors that incorporates this water. Although no such inhibitors have been reported, it is possible to generate models of such molecules that contain 6 to 10 atoms in the ring.196
J. Symmetric Inhibitors
Despite the presence of a quasi-two-fold axis that relates the amino- and carboxy-terminal halves of renin and other monomeric aspartyl protein-
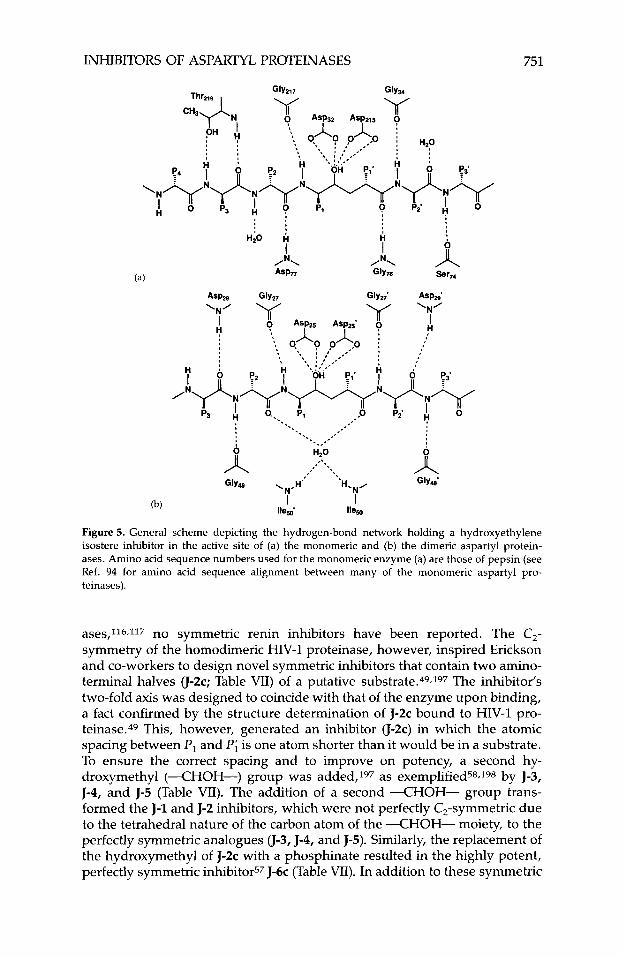
INHIBITORS OF ASPARTYL PROTEINASES 751
Figure 5. General scheme depicting the hydrogen-bond network holding a hydroxyethylene isostere inhibitor in the active site of (a) the monomeric and (b) the dimeric aspartyl protein- ases. Amino acid sequence numbers used for the monomeric enzyme (a) are those of pepsin (see Ref. 94 for amino acid sequence alignment between many of the monomeric aspartyl pro- teinases).
ases,116,117 no symmetric renin inhibitors have been reported. The C,- symmetry of the homodimeric HIV-1 proteinase, however, inspired Erickson and co-workers to design novel symmetric inhibitors that contain two amino- terminal halves u - 2 ~ ; Table VII) of a putative substrate.49.197 The inhibitor's two-fold axis was designed to coincide with that of the enzyme upon binding, a fact confirmed by the structure determination of J-2c bound to HIV-1 pro- teinase.49 This, however, generated an inhibitor (J-2c) in which the atomic spacing between P , and Pi is one atom shorter than it would be in a substrate. To ensure the correct spacing and to improve on potency, a second hy- droxymethyl (--CHOH-) group was added,197 as exemplified58.198 by J-3, J-4, and J-5 (Table VII). The addition of a second --CHOH- group trans- formed the J-1 and J-2 inhibitors, which were not perfectly C,-symmetric due to the tetrahedral nature of the carbon atom of the X H O H - moiety, to the perfectly symmetric analogues 0-3, J-4, and J-5). Similarly, the replacement of the hydroxymethyl of J-2c with a phosphinate resulted in the highly potent, perfectly symmetric inhibitor57 J-6c (Table VII). In addition to these symmetric
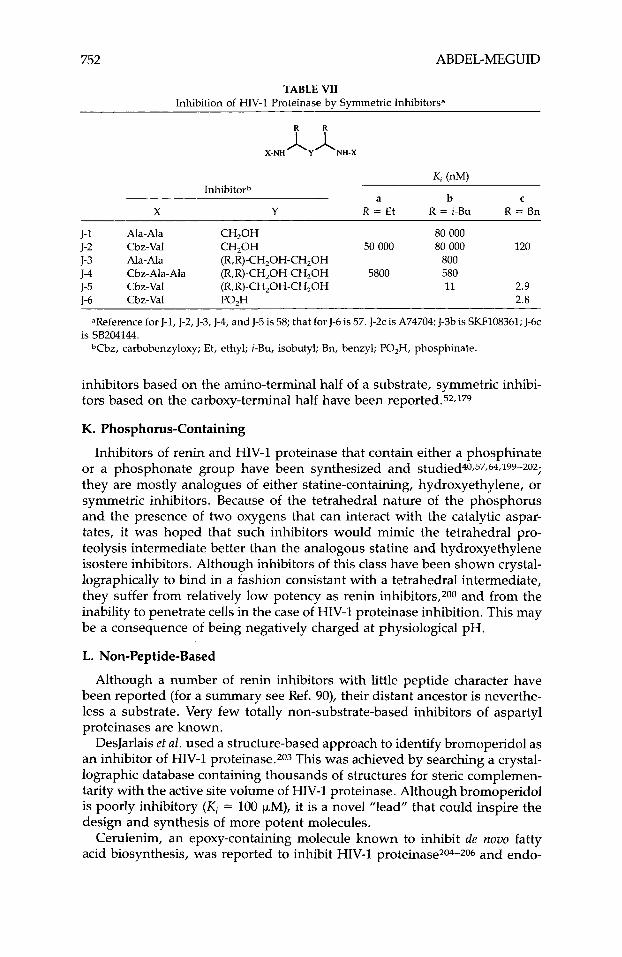
752 ABDEL-MEGUID
TABLE VII Inhibition of HIV-I Proteinase by Symmetric Inhibitors”
K , (nM) Inhibitorh
a b C
X Y R = Et R = i-Bu R = Bn
J- 1 Ala- Ala CH,OH 80 000
J-3 Ala- Ala (R,R)-CH,OH-CH,OH 800 J-4 Cbz-Ala-Ala (R,R)-CH,OH-CH,OH 5800 580 J-5 Cbz-Val (R,R)-CH,OH-CH,OH 11 2.9 1-6 Cbz-Val PO,H 2.8
J-2 Cbz-Val CH,OH 50 000 80 000 120
~~~~~~~~~ ~~
aReference for J-I, J-2, J-3, J-4, and J-5 is 58; that for J-6 is 57. J-2c is A74704; J-3b is SKF108361; J-6c
T b z , carbobenzyloxy; Et, ethyl; i-Bu, isobutyl; Bn, benzyl; PO,H, phosphinate. is SB204144.
inhibitors based on the amino-terminal half of a substrate, symmetric inhibi- tors based on the carboxy-terminal half have been reported.52.179
K. Phosphorus-Containing
Inhibitors of renin and HIV-1 proteinase that contain either a phosphinate or a phosphonate group have been synthesized and studied40,57,64,199-207-;
they are mostly analogues of either statine-containing, hydroxyethylene, or symmetric inhibitors. Because of the tetrahedral nature of the phosphorus and the presence of two oxygens that can interact with the catalytic aspar- tates, it was hoped that such inhibitors would mimic the tetrahedral pro- teolysis intermediate better than the analogous statine and hydroxyethylene isostere inhibitors. Although inhibitors of this class have been shown crystal- lographically to bind in a fashion consistant with a tetrahedral intermediate, they suffer from relatively low potency as renin inhibitors,7-00 and from the inability to penetrate cells in the case of HIV-1 proteinase inhibition. This may be a consequence of being negatively charged at physiological pH.
L. Non-Peptide-Based
Although a number of renin inhibitors with little peptide character have been reported (for a summary see Ref. 90), their distant ancestor is neverthe- less a substrate. Very few totally non-substrate-based inhibitors of aspartyl proteinases are known.
DesJarlais et al. used a structure-based approach to identify bromoperidol as an inhibitor of HIV-1 proteinase.203 This was achieved by searching a crystal- lographic database containing thousands of structures for steric complemen- tarity with the active site volume of HIV-1 proteinase. Although bromoperidol is poorly inhibitory (Ki = 100 FM), it is a novel ”lead” that could inspire the design and synthesis of more potent molecules.
Cerulenim, an epoxy-containing molecule known to inhibit de novo fatty acid biosynthesis, was reported to inhibit HIV-1 proteinase204-7-06 and endo-

INHIBITORS OF ASPARTYL PROTEINASES 753
thiapepsin.206 It is, however, highly cytotoxic.204 Another epoxy-containing compound, 1,2-epoxy-3-(p-nitrophenoxy) propane (EPNP), was shown to be an inhibitor of monomeric207-209 and dimeric” aspartyl proteinases. This mol- ecule is proposed to inactivate these enzymes through the formation of a covalent attachment to the catalytic carboxylates.101,208-210
VI. X-RAY-DIFFRACTION STUDIES OF ENZYME/INHIBITOR COMPLEXES
The crystal structures of a large number of aspartyl proteinases complexed with inhibitors have been reported (Table 11). Many more have been deter- mined at industrial institutions, but have yet to be published. Table VIII shows the primary structure of the inhibitors listed in Table 11. Many of the published three-dimensional structures have been determined at, or better than, 2.0-A resolution. Numerous tightly bonded water molecules were identified in these structures and included as part of the final atomic model. These water molecules bind either directly to the protein or inhibitor, or indirectly, as part of a water cluster. Atomic coordinates for all these structures, except for the most recent, are available from the PDB.63 The inhibitors in these studies represent different classes, viz., they are statine, reduced amide, hydroxy- ethylamine, hydroxyethylene, difluoroketone, glycol-based, cyclic, phosphorus- containing, and symmetric.
Endothiapepsin/inhibitor complexes have been the most extensively stud- ied and reported by a single laboratory. Blundell’s laboratory has reported 16 crystal structures of these complexes, most of which have been determined to 2.0-A resolution or better (Table IIa). Crystals of these complexes belong to two different crystalline forms; one is isomorphous with crystals of native (unliganded) endothiapepsin, while the other is not. The complex with H-261 crystallizes in two different crystalline forms, one of which is isomorphous to native endothiapepsin. The crystals in all of these studies were grown from solutions of preformed enzymehhibitor complexes (co-crystallization).
The crystal structures of six rhizopuspepsin/inhibitor complexes have been determined (Table IIA), most of which have been reported recently by Davies’ laboratory. Nearly all the crystals used in these studies were of the native, unliganded enzyme in which the inhibitors were soaked. The structure of rhizopuspepsin complexed to CP-69,799, however, was determined twice; once from crystals soaked in the inhibitor and the other from protein crystal- lized in the presence of excess inhibitor.42 Both structures were found to be essentially identical.
Recently, five crystal structures of penicillopepsin/inhibitor complexes were reported from James’ laboratory (Table IIa). The crystals used in these experi- ments, as well as the previously reported crystal structure containing pep- statin,211 were isomorphous to crystals of the native, uncomplexed enzyme, and all were grown as co-crystals.
The co-crystallization of four remain inhibitors bound to procine pepsin has been published37 (Table IIa). Crystals for all four were obtained under condi- tions similar to those used to grow crystals of the native enzyme; none how- ever, were isomorphous to the native (Table 11). Details of three of these structures were reported.35.37
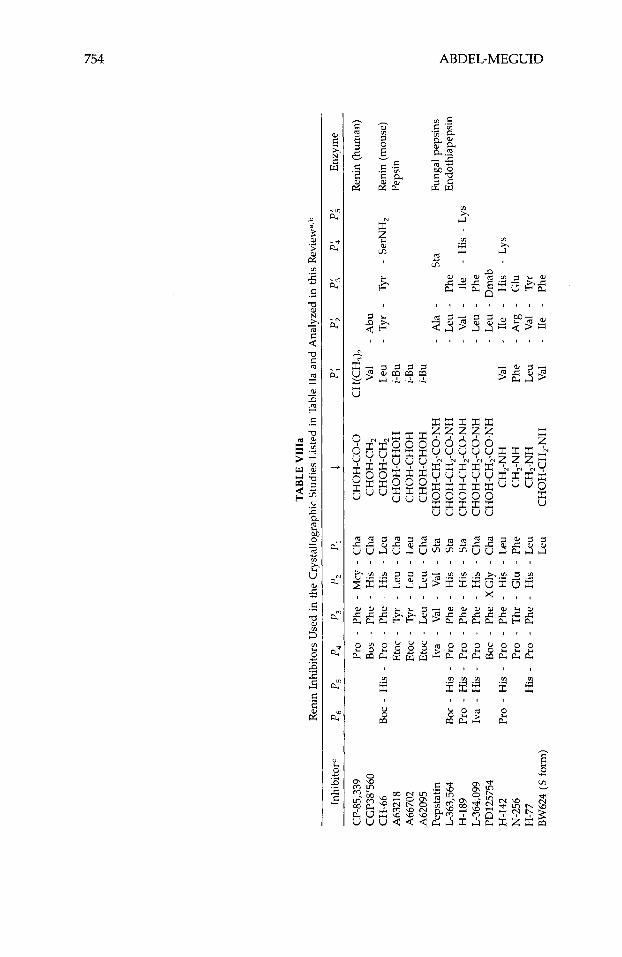
TABL
E V
IIIa
R
enin
Inh
ibito
rs U
sed
in t
he C
ryst
allo
grap
hic
Stud
ies
Lis
ted
in T
able
IIa
and
Ana
lyze
d in
thi
s
Inhi
bito
rc
P6
p5
p4
p,
p,
p,
4 p;
P;
P
j P;
P;
E
nzym
e
(3'45
,339
Pro
- Ph
e -
Mcy
- C
ha
CH
OH
-CO
-0
CH
(CH
,),
CG
P38'
560
CH
-66
A63
218
A66
702
A62
095
Peps
tatin
L-
363,
564
H-1
89
L-36
4,09
9 I'D
1257
54
H-1
42
N-2
56
H-7
7 B
W62
4 (5
form
)
Bos
-
Phe
- H
is
- C
ha
Boc
- H
is -
Pr
o -
Phe
- H
is
- L
eu
Eto
c -
Tyr
- Le
u -
Cha
E
toc
- Ty
r -
Leu
- L
eu
Eto
c -
Leu
- Le
u -
Cha
Iv
a -
Val
- V
al -
Sta
Boc
- H
is -
Pr
o -
Phe
- H
is
- St
a Pr
o -
His
-
Pro
- Ph
e -
His
-
Sta
Iva
- H
is -
Pr
o -
Phe
- H
is
- C
ha
Boc
-
Phe
XG
ly
- C
ha
Pro
- His
- Pr
o -
Phe
- H
is
- L
eu
Pro
- T
hr
- G
lu -
Phe
H
is -
Pr
o -
Phe
- H
is
- L
eu
Leu
C H
0 H
- C H
2 C
HO
H-C
HZ
CH
OH
-CH
OH
C
HO
H-C
HO
H
CH
OH
-CH
OH
C
HO
H-C
HZ
-CO
-NH
C
HO
H-C
HI-
CO
-NH
C
HO
H-C
HZ
-CO
-NH
C
HO
H-C
H,-C
O-N
H
CH
OH
-CH
,-CO
-NH
C
HZ-
NH
C
HZ-
NH
C
HI-
NH
C
HO
H-C
H,-N
H
Val
Leu
i-B
u i-B
u i-B
u
Val
Phe
Leu
V
al
- A
bu
- Ty
r -
Tyr
- Se
rNH
,
- A
la
- St
a -
Leu
-
Phe
- V
al -
Ile
- H
is -
Lys
-
Leu
-
Phe
- L
eu
- D
mab
-
Ile
- H
is
- Ly
s -
Arg
-
Glu
-
Val
- Ty
r -
Ile
- Ph
e
Ren
in (
hum
an)
Ren
in (
mou
se)
Peps
in
Fung
al p
epsi
ns
End
othi
apep
sin

BW
625
(R fo
rm)
CP-
69,7
99
H-2
61
PD12
5967
C
P-71
,362
C
P-81
,282
In
hibi
tor
2 C
yclic
dis
ulfi
de
Red
uced
pep
tide
StaF
, St
oF,
Phos
phin
ate
Iva-
phos
phon
ate
Cbz
-uho
snho
nate
CP-
82,2
18
Leu
Bo
c -
Phe
- H
is
- C
ha
Boc
- H
is -
Pr
o -
Phe
- H
is
- Le
u N
ap -
Nap
-
His
-
Cha
Bo
c -
Phe
- H
is
- C
ha
Mor
- P
he -
Nle
-
Cha
Iv
a -
His
-
Pro
- Ph
e -
His
-
Sta
Ibu
- H
is -
Pr
o -
Phe
- C
ys -
St
a H
is -
Pr
o -
Phe
- H
is
- Ph
e Pi
p -
Phe
- N
le
- C
ha
Iva
- V
al -
Val
- St
a Iv
a -
Val
- V
al -
Sta
Iva
- V
al -
Val
- S
ta
Iva
- V
al -
Val
- Le
u C
bz -
A
la
- A
la
- L
eu
CH
OH
-CH
2-N
H
CH
OH
-CH
,-N( i
-Bu)
C
HO
H-C
H,
CH
OH
-CH
Z C
HO
H-C
H,
CH
( OH
),-C
F,-C
O-N
H
CH
OH
-CH
2-C
O-N
H
CH
OH
-CH
2-C
O-N
H
CH
OH
-CH
Z-C
O-N
H
CH
(OH
),-C
F,-C
O-N
H
CH
OH
-CF2
-CO
-NH
C
H(O
H)z
-CF,
-CO
-NH
P0
2H-C
H2-
CO
-0
POZ
H-0
PO
qH-0
Val
- Ile
-
Phe
- Ly
s -
Phe
Val
- Il
e -
His
L
eu
- Il
e Le
u -
Lys
Me
- L
eu
- Ph
e -
Leu
-
Phe
Phe
- V
al -
Tyr
Me
Me
Me
Et
Phe
- O
Me
Phe
- O
Me
rea
Rhi
zopu
spep
sin
Peni
cillo
peps
in
0
L! 2 >
I1
=Ref
eren
ces a
re li
sted
in T
able
IIa
. bA
rrow
indi
cate
s po
sitio
n of
scis
sile
-bon
d su
rrog
ate.
C
Abu
, am
inob
utyl
; Boc
, t-b
utox
ycar
bony
l; B
os,
t-bu
tyls
ulfi
nyl;
Cbz
, car
bobe
nzyl
oxy;
Cha
, cy
cloh
exyl
alan
ine;
Dm
ab,
(dim
ethy
1am
ino)
benz
ene;
Et,
v,
M
v,
ethy
l; Et
oc,
etho
xyca
rbon
yl;
Hai
, 2-
hydr
oxy-
1-am
inoi
ndan
e; i
-Bu,
iso
buty
l; Ib
u, i
sobu
tary
l; Iv
a, i
sova
lery
l; M
cy,
met
hylc
ycte
inyl
; Me,
met
hyl;
Mor
, m
orph
olin
ocar
bony
l; N
ap,
1-na
phth
ylm
ethy
l; N
le,
L-n
orle
ucin
e; P
ip, p
iper
azin
e; S
ta, s
tatin
e; T
ea, t
hioe
thyl
amid
e; X
, hyd
roxy
ethy
lene
link
age.
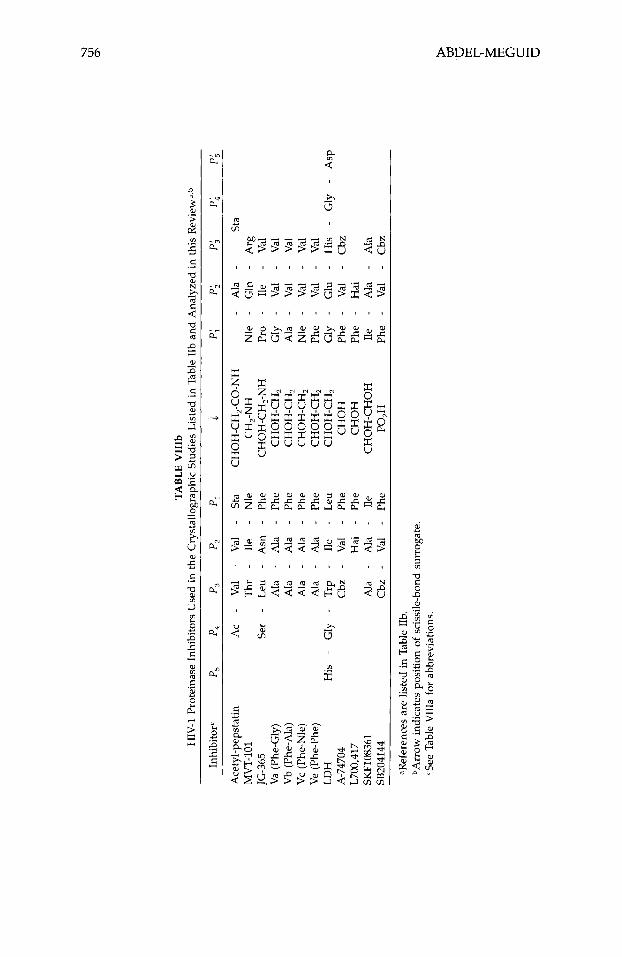
TABL
E V
IIIb
H
IV-1
Pro
tein
ase
Inhi
bito
rs U
sed
in t
he C
ryst
allo
grap
hic
Stud
ies
Lis
ted
in T
able
IIb
and
Ana
lyze
d in
thi
s R
evie
wa,
b
Inhi
bito
rc
p5
p4
p3
p2
PI
4 p;
P;
P
j 0
; P;
Ace
tyl-
peps
tatin
A
c -
Val
- V
al -
Sta
CH
OH
-CH
,-CO
-NH
-
Ala
-
Sta
MV
T-10
1 T
hr
- Il
e -
Nle
C
HZ-
NH
N
le
- G
ln
- A
rg
JG-3
65
Ser
- L
eu
- A
sn
- Ph
e C
HO
H-C
H,-N
H
Pro
- Ile
-
Val
Va (
Phe-
Gly
) A
la
- A
la
- Ph
e C
HO
H-C
Hz
Gly
-
Val
- V
al V
b (P
he-A
la)
Ala
-
Ala
-
Phe
CH
OH
-CH
Z A
la
- V
al -
Val
Vc
(Phe
-Nle
) A
la
- A
la
- Ph
e C
HO
H-C
H,
Nle
-
Val
- V
al Ve
(Ph
e-Ph
e)
Ala
-
Ala
-
Phe
CH
OH
-CH
, Ph
e -
Val
- V
al LD
H
His
-
Gly
-
Trp
-
Ile
- Le
u C
HO
H-C
HI
Gly
-
Glu
-
His
-
Gly
-
Asp
A
-747
04
Cbz
-
Val
- Ph
e C
HO
H
Phe
- V
al -
Cbz
L7
00,4
17
Hai
-
Phe
CH
OH
Ph
e -
Hai
SK
F108
361
Ala
-
Ala
-
Ile
CH
OH
-CH
OH
Il
e -
Ala
-
Ala
SB
2041
44
Cbz
-
Val
- Ph
e PO
,H
Phe
- V
al -
Cbz
aRef
eren
ces a
re li
sted
in T
able
IIb
. bA
rrow
indi
cate
s po
sitio
n of
scis
sile
-bon
d su
rrog
ate.
4
ee
Tab
le V
IIIa
for
abbr
evia
tions
.

INHIBITORS OF ASPARTYL PROTEINASES 757
Three structures of renidinhibitor complexes have been recently reported by two different laboratories24.39 (Table IIa). Two of these were structures of glycosylated, recombinant human renin and the third was of mouse subman- dibular renin. Crystals of human renin complexed to CP-85,339 were iso- morphous to crystals of the native enzyme.212 All crystals used in these stud- ies were grown as co-crystals.
Twelve crystal structures of inhibitors bound to HIV-1 proteinase have been reported (Table IIb). As indicated above, HIV-1 proteinase undergoes consid- erable conformational change upon binding of inhibitors, thus all crystals used in these studies were non-isomorphous to the native enzyme, and were produced through co-crystallization.
VII. CONFORMATION OF INHIBITORS IN THE ACTIVE SITE
The wealth of information obtained from the crystal structures of aspartyl proteinase/inhibitor complexes has provided considerable detail of both the mode of binding of the inhibitors and the subsite specificity of the enzymes. It also enables one to devise general and specific rules to guide in the design of additional inhibitors with improved properties. Here, I will summarize much of the information gained from these crystal structures, concentrating on the general rules gained from these studies regarding inhibitor conformations.
A. General Mode of Binding
All inhibitors bind in an extended conformation in the active site cavity formed between the amino- and carboxy-terminal domains of the monomeric enzymes, and between the two monomers of the dimeric retroviral pro- teinases. They are held by a set of hydrophobic and hydrogen-bonding inter- actions, most of which are conserved within each of the two aspartyl pro- teinase classes, the monomeric and the dimeric. Inhibitor residues P3-P, form a short antiparallel p sheet with residues 217-219 of the monomeric and 27-29 of the dimeric enzymes, respectively. Almost every heteroatom of an inhibitor backbone is hydrogen-bonded to an atom of the protein, either directly or indirectly through a water molecule (Fig. 5). A water molecule, centrally lo- cated on the enzyme’s twofold axis near the flaps, is found in all liganded structures of HIV-1 proteinase [Fig. 5(b)]. It bridges the ligands and the pro- tein by forming hydrogen bonds with the carbonyl oxygens at P, and Pi on one side, and with the amide hydrogens of Ile-50 and Ile-50’ of the flaps on the other side. Table IX lists these hydrogen-bond distances for monomeric and dimeric aspartyl proteinases, for which atomic coordinates are available from the PDB.63 Residue numbers for the monomeric enzymes are those of pepsin (see Ref. 94, for amino acid sequence alignment); the pepsin number- ing system will be used for monomeric aspartyl proteinases throughout this review. Correlations of hydrogen-bond strength with potency is not obvious. For example CP-69,799 is more than 270-fold weaker in binding to endo- thiapepsin than H-261, however, their hydrogen-bond distances are not sig- nificantly different (Table IXa). This confirms the view that binding affinity is a consequence of both hydrophilic and hydrophobic interactions.
Some heteroatoms of the side chains of inhibitors also participate in hydro- gen bond interactions, thus contributing toward higher binding affinity and
TABL
E IX
a Po
tent
ial H
ydro
gen
Bon
ds B
etw
een
Inhi
bito
rs a
nd T
heir
Res
pect
ive M
onom
eric
Enz
ymes
a,b,
c
P,
P3
P2
P,
P,
P,
P,
P,
PI
PI
P;
P;
P;
Pj
I I
I I
I I
I I
I I
I I
I i
NH
0
NH
0
0
NH
O
H
OH
O
H
OH
0
NH
0
N
H
OG
1 N
H
0
NH
N
H
0
OD
2 O
D1
OD
1 O
D2
N
0
0
0
Thr
T
hr
H,O
G
ly
Asp
G
ly
Asp
A
sp
Asp
A
sp
Gly
G
ly
H,O
Se
r In
hibi
tor
219
219
76
77
217
32
32
215
215
76
34
74
Peps
tatin
L-
363,
564
H-1
89
L364
,099
H
-142
H
-256
H
-77
BW
624
CP-
69,7
99
H-2
61
I'D12
5967
C
P-71
,362
Peps
tatin
In
hibi
tor
2 C
yclic
dis
ulfi
de
Red
uced
pep
tide
3.10
2.
92
2.71
2.
80
2.75
3.
04
2.91
3.
34
2.39
3.
24
2.82
2.
94
2.91
3.
34
2.70
3.
03
2.71
3.
07
3.03
2.
69
2.86
3.08
3.
02
2.59
3.
24
2.79
3.
09
2.92
2.
97
3.26
3.
69
3.27
3.
15
3.61
3.
44
2.69
3.
04
2.70
3.
41
2.70
3.
55
3.20
2.
70
3.47
3.12
3.
43
3.84
3.
17
End
othi
apep
sin
3.31
3.
31
3.08
2.
89
3.26
3.
15
3.24
3.
29
3.21
3.
02
3.75
3.
14
2.71
3.
43
2.89
3.
70
3.00
3.
28
3.11
3.
19
3.25
3.
20
3.26
3.
13
3.21
3.
35
3.26
3.
21
3.51
2.
82
3.60
Rhi
zopu
spep
sin
3.07
3.
20
3.40
2.
77
3.39
3.
22
3.07
3.
32
3.01
3.
59
2.90
2.52
2.
66
2.68
2.
75
2.72
2.
70
2.67
2.
66
2.83
2.79
2.
56
2.55
2.65
2.
75
2.69
2.
65
3.08
2.
67
2.68
2.
74
2.70
2.49
2.
58
3.22
3.03
2.
95
2.95
2.
84
3.04
3.
08
3.09
2.
87
2.96
2.93
2.
90
3.02
2.87
3.
09
2.92
3.
37
2.80
2.
94
2.75
3.
49
3.24
2.
88
3.09
2.
90
2.97
2.
90
2.96
3.
31
3.09
3.
09
2.83
3.
09
3.13
2.
72
2.79
2.
99
2.67
3.
09
2.75
3.
53
2.72
3.
19
2.83
2.
97
Mea
n 2.
79
3.06
2.
70
3.38
3.
24
3.17
3.
21
2.67
2.
74
2.97
2.
90
3.10
aRef
eren
ces a
re li
sted
in T
able
IIa
. bA
min
o ac
id r
esid
ue n
umbe
rs a
re th
ose
of po
rcin
e pe
psin
(se
e R
ef. 9
4 fo
r se
quen
ce a
lignm
ent b
etw
een
peps
in a
nd o
ther
apa
rtyl
pr
otei
nase
s). R
esid
ues 3
2, 3
4, 7
4, 7
6, 7
7, 2
15, 2
17, a
nd 2
19 o
f po
rcin
e pe
psin
are
equ
ivel
ant t
o re
sidu
es 3
5, 3
7, 7
7, 7
9,80
, 21
8,22
0, a
nd
222
of b
oth
endo
thia
peps
in a
nd r
hizo
pusp
epsi
n.
=Dat
a for
H-2
61 a
re d
eriv
ed f
rom
the
cry
stal
str
uctu
re h
avin
g PD
B c
ode
2ER
7 (s
ee T
able
Ha)
.
3.34
2.94
3.
13
2.79
2.
91
2.79
2.
99
2.92
3.
15
2.94
2.67
3.
29
2.59
2.96
2.
96
u
wl
m
TA
BL
E IX
b Po
tent
ial H
ydro
gen
Bon
ds B
etw
een
Inhi
bito
rs a
nd H
IV-1
Pro
tein
asea
p3
p2
p2
PI
PI
PI
PI
p,
p;
P;
0;
Pj
I I
I I
I I
I I
I I
I I
0
NH
0
NH
O
H
OH
O
H
OH
0
NH
0
N
H
0
NH
0
0
OD
2 O
D1
OD
1 O
D2
0
NH
0
Asp
G
ly
H2
0
Gly
A
sp
Asp
A
sp
Asp
H
20
G
ly
Asp
G
ly
Inhi
bito
r 29
48
27
25
25
25
' 25
' 27
' 29
' 48
0
Ace
tyl-
peps
tatin
(A
)b
Ace
tyl-
peps
tatin
(B
)b
MV
T-10
1 A
-747
04 (
1)'
A-7
4704
(2)'
L700
,417
(A
l)b,
d L7
00,4
17 (A
2)b,
d L7
00,4
17 (
Bl)
b,d
L700
,417
(B2)
b,d
Mea
n
~ ~
2.81
2.
89
2.75
2.
90
2.72
1.
99
3.88
3.
27
2.37
3.
54
3.06
3.
11
3.26
2.
88
2.88
3.
03
2.55
2.
46
3.61
3.
55
2.34
3.
12
3.06
2.
74
3.09
2.
59
3.04
2.
81
2.48
2.
89
2.97
3.
00
3.05
2.
73
2.64
3.
43
2.84
2.
87
3.22
3.
58
2.80
3.
09
2.70
3.
72
2.96
2.
61
2.75
3.
13
2.59
3.
22
3.11
2.
49
3.21
3.
09
3.01
2.
76
2.66
3.
08
2.59
3.
27
3.07
2.
50
3.22
3.
04
3.00
2.
84
2.80
3.
18
2.82
2.
54
3.22
3.
32
2.48
3.
21
3.06
2.
95
__
__
_
.Ref
eren
ces
are
liste
d in
Tab
le II
b.
bAce
tyl-
peps
tatin
and
L70
0,41
7 ar
e fo
und
in tw
o or
ient
atio
ns,
desi
gnat
ed (
A) a
nd (8
). T
he tw
o or
ient
atio
ns a
re re
late
d by
the
dim
er
cA-7
4704
is a
sym
met
ric
inhi
bito
r th
at c
onta
ins
two
amin
o-te
rmin
al h
alve
s de
sign
ated
(1)
and
(2).
dL70
0,41
7 is
a s
ymm
etri
c in
hibi
tor
that
con
tain
s tw
o ca
rbox
y-te
rmin
al h
alve
s de
sign
ated
(1)
and
(2).
twof
old
axis
.

760 ABDEL-MEGUID
specificity. Inhibitors with a histidine side chain at P2 appear to have optimum potency and selectivity for human renin94; substitutions of other basic amino acids for histidine at the P2 position caused significant loss of potency.213 From molecular-modeling studies using a model of human renin, it was pro- posed213 that this enhanced selectivity is due to a hydrogen bond between one of the imidazole nitrogens and the amide hydrogen of Tyr-220. An alter- native explanation was put forth by Green et uZ.214 in which they attributed the enhancement to a hydrogen-bond interaction with a carboxylate oxygen of the catalytic Asp-215. Two of the structures of renin bound to inhibitors con- tain a histidine at P,, viz., the human renin/CGP 38,560 complex24 and the mouse renin/CH-66 complex.39 In both of these structures one of the im- idizole nitrogens hydrogen bond to the side chain hydroxyl of Ser-222.
Novel interactions that contribute to greater binding affinity were also ob- served. For example, the P, and PA carbobenzyloxy (Cbz) groups of the HIV-1 protease inhibitor SB204144 (Table VIIIb) participate in a novel interaction in which a terminal nitrogen of Arg-8 points toward the center of its benzene ring, being 2.15 A above the center of the ring.57
The water molecules observed in the active site of the native, unliganded structures are displaced upon binding, including the catalytic water molecule located between the two active-site carboxylates.8 Thus, in all liganded struc- tures, the central residues of each inhibitor (P,-P;) are shielded from bulk solvent.
B. Backbone Conformations
Superpositon of the structures of 12 endothiapepsin/inhibitor complexes reveals a striking similarity between the inhibitor’s backbone conformation from P, to Pi [Fig. 6(a)]. These superpositions were achieved by aligning only the backbone atoms of the proteins. Although it is not surprising that inhibi- tors within the same class have very similar backbone conformations [Fig. 6(b)], it is surprising to see that inhibitors containing statine as a scissile-bond surrogate superpose well with those containing reduced amides and hy- droxyethylenes, considering that statine is one atom shorter along the back- bone. Comparison of the conformations of H-189 (statine class) and H-261 (hydroxyethylene class) bound to endothiapepsin [Fig. 6(c)] shows remark- able superposition of the backbone atoms from P4 to P,, except for an approx- imately 0.5-A shift. This difference becomes larger around Pi, but realigns again at Pi. In these two inhibitors the side chains of P4, P3, P2, P,, and P; are practically superposable [Fig. 6(c)].
Similar conclusions were drawn from the overlay of the structures of rhizo- puspepsin/inhibitor complexes.42 Furthermore, when the backbone confor- mations of pepstatin bound to endothiapepsin and rhizopuspepsin, respec- tively, were compared, they were found to be remarkably similar [Fig. 6(d)].
In general, the backbone conformation of inhibitors bound to the mono- meric enzymes remains essentially the same for all inhibitors on the amino- terminal side of the scissile bond between P3 and P,, and on both sides of it for inhibitors of a given ~ lass .7~ Backbone conformations beyond P, and P; toward the amino- and carboxy-terminals show much less similarity, even for inhibitors of the same class. This is due to the solvent accessibility of these
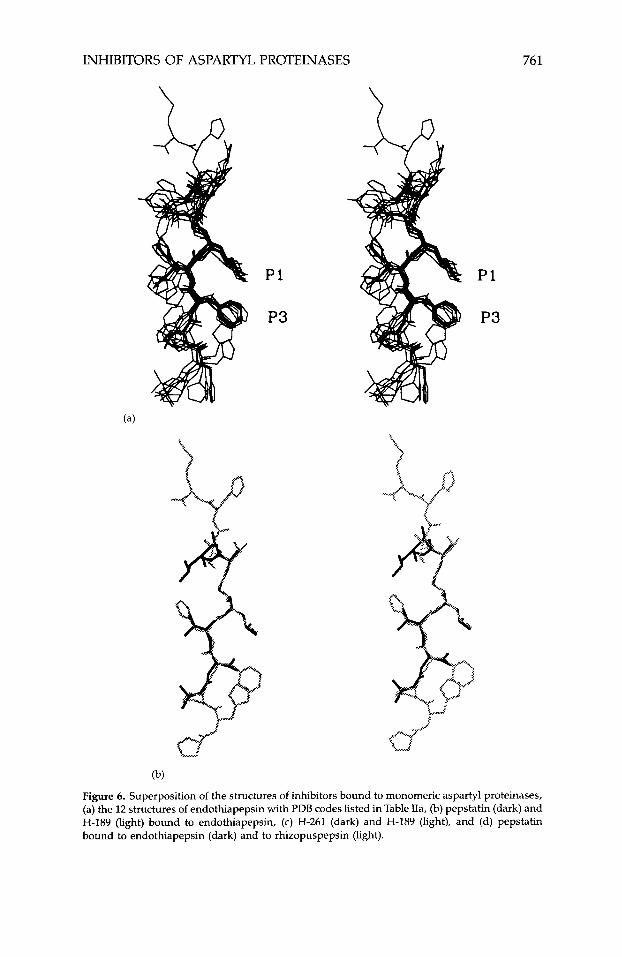
INHIBITORS OF ASPARTYL PROTEINASES 761
P1
P3
(b)
Figure 6. Superposition of the structures of inhibitors bound to monomeric aspartyl proteinases, (a) the 12 structures of endothiapepsin with PDB codes listed in Table IIa, (b) pepstatin (dark) and H-189 (light) bound to endothiapepsin, (c) H-261 (dark) and H-189 (light), and (d) pepstatin bound to endothiapepsin (dark) and to rhizopuspepsin (light).
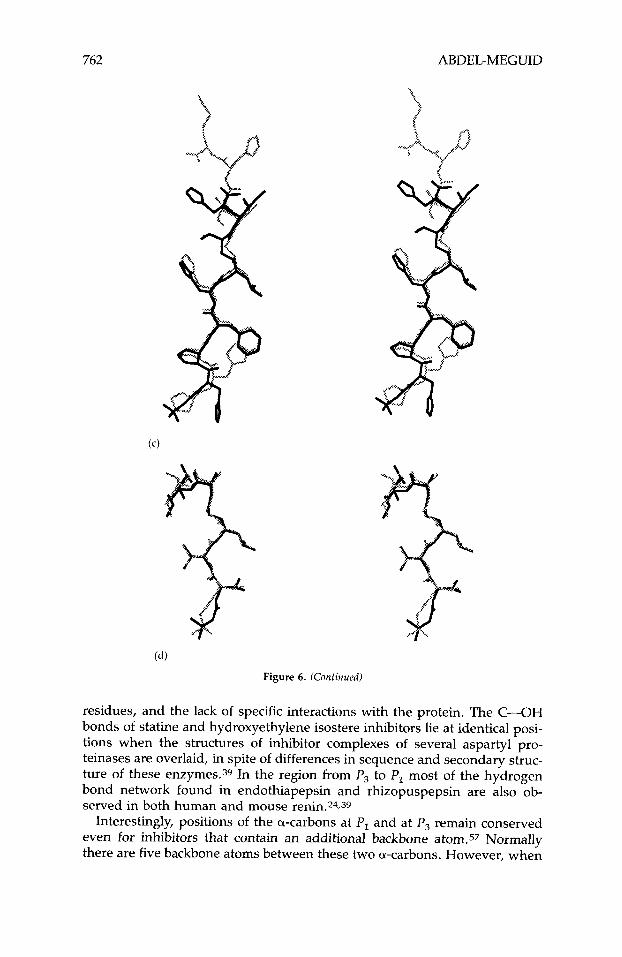
762 ABDEL-MEGUID
Figure 6. (Continued)
residues, and the lack of specific interactions with the protein. The C-OH bonds of statine and hydroxyethylene isostere inhibitors lie at identical posi- tions when the structures of inhibitor complexes of several aspartyl pro- teinases are overlaid, in spite of differences in sequence and secondary struc- ture of these enzymes.39 In the region from P3 to PI most of the hydrogen bond network found in endothiapepsin and rhizopuspepsin are also ob- served in both human and mouse renin.24.39
Interestingly, positions of the a-carbons at PI and at P3 remain conserved even for inhibitors that contain an additional backbone atoms7 Normally there are five backbone atoms between these two a-carbons. However, when

INHIBITORS OF ASPARTYL PROTEINASES 763
an additional sixth atom was introduced, the positions of the a-carbon and side chain atoms at P , and P3 remained the same. The conformation of the backbone atoms between those two a-carbons bulged toward the P, side- chain positions to accommodate the additional atom.196
Dreyer and co-workers have reported the crystal structures of four HIV-1 protease-inhibitor complexes.54 The overlay of these structures shows similar inhibitor backbone conformations for all four structures, and a good consewa- tion of the hydrogen-bonding network between the backbone atoms of these inhibitors and atoms of the proteins from P, to P i (Table IXb). A similar overlay between the structures of MVT-101 and JG-365 show excellent agree- ment in backbone conformations.51
C. Side-Chain Conformations
Most side chains of the HIV-1 proteinase inhibitors listed in Table VIIIb are small, hence very few general conclusions can be drawn regarding their con- formation. The only exception is at PI where most side chains are Phe. In the four structures reported by Dreyer et al . , the P , Phe side chains have essen- tially the same conformation.54
Similarly, the conformation of the side-chains at P , is highly conserved for all monomeric enzymes. When all the available endothiapepsin structures (Table IIa) were overlaid, the side chains at P, were found, remarkably, to occupy the same three-dimensional space, even when they were as different as cyclohexylmethyl and isobutyl [Fig. 6(a)]. The side chain at P,, however, assumes multiple conformations, even within the same structure, as was observed33 for the complex of endothiapepsin with CP-69,799. Histidine side chains at P, can adopt conformations separated by about 120" on x2.2,S The S, pocket is large and shallow, extending to the S; pocket; as a result, the orienta- tion of histidines at P, is dictated by the size of the side chain at Pi, and the absence of specific interactions with the protein. There are no direct, specific interactions between any of the P, histidines and the protein, except for renin where the side chain of the histidine at P, forms a hydrogen bond with the Ser-222 hydroxyl. Of the inhibitors listed in Table VIIIa, 12 have a histidine residue at P,. Of those, six point toward Pi (CGP38'560, L-363,564, H-189, L-364,099, H-261, and Inhibitor 2), another five point toward P4 (H-142, H-77, PD125967, CP-71,362 and Reduced Peptide) and (369,799 assumes both con- formations. Of the six pointing toward Pi and partially occupying the s;, only CGP38'560 and H-261 have P; side chains (Val), while the other four are statine inhibitors with no Pi side chain. The absence of a P; side chain pro- vides room for the histidine to partially occupy the S; pocket. On the other hand, the five inhibitors with histidines pointing away from Pi, toward P4, have mostly Leu or Phe as their side chain at Pi, thus a totally filled S; pocket. In this case the histidine must orient away from Pi to avoid unfavorable van der Waals contacts. The orientation of the histidine side chain at P, in the structure of mouse renin/CH-66 complex was not reported.39 However, the authors' reported that it forms a hydrogen-bond interaction with Ser-222, leading one to assume that it is pointing toward Pi as in the structure of the human renin/CGP38560 complex. The CH-66 inhibitor has a leucine at Pi; thus one may speculate that in general the availability of the specific interac-

764 ABDEL-MEGUID
tion with Ser-222 will direct histidines at P, to adopt the conformation point- ing toward Pi in renin complexes even when a larger side chain than that of valine is present at Pi. This should be facilitated by the presence of a larger S; pocket in renin, relative to other monomeric aspartyl proteinases,39 allowing for reorientation of the Pi side chain to relieve any unfavorable van der Waals contacts between the P, and Pi side chains.
Most of the inhibitors of the monomeric enzymes listed in Table VIIIa have a Phe at P,. These Phe side chains assume one of two orientations, related by approximately 55" rotation along x2.33 The energetically less favorable orienta- tion of the two is found in inhibitors having bulky Phe, cyclohexyl, or naphthyl side chains, at P, and at P, (L-364,099, CP-69,799, PD125967, CP-71,362, CP-81,282, Reduced Peptide, and CP-82,218; Table VIIIa). In these inhibitors the ring of P, assumes an orientation that is essentially parallel to that of PI in order to minimize van der Waals contacts. The P3 side chain assumes an energetically more favorable orientation in those inhibitors hav- ing a smaller P, side chain (L-363,564, H-189, H-142, H-77, H261, Inhibitor 2, and Cyclic disulfide; Table VIIIa). The only exception is the structure of PD125754, which has a cyclohexylmethyl at PI and a Phe at P,; however, its Phe assumes the energetically more favorable orientation. Cooper et al. have suggested that this is a result of having a hydroxyethylene moiety, rather than a peptide bond, between P, and P2.38 The absence of the planar restraints imposed by the peptide bond would allow for subtle changes in the confor- mation of the backbone resulting in relief of the unfavorable interactions between the two bulky side chains at PI and P,.
Many of the inhibitors of the monomeric aspartyl proteinases, listed in Table VIIIa, have a statine or its analogue as a scissile bond surrogate and thus have no Pi side chain. Of those which have one, it appears that their confor- mations are not necessarily similar; however, it is difficult to generalize since many of these are short, Val side chains.
As with the backbone of the monomeric enzymes, side-chain conforma- tions of the inhibitor beyond P, and P i toward the amino- and carboxy- terminals, respectively, show much less similarity.
D. Difluoroketones and Implications for the Catalytic Mechanism
An interesting feature of the difluoroketone inhibitors is their apparent ability to serve as mimics of the tetrahedral intermediate involved in peptide- bond hydrolysis. In this class of inhibitors the carbonyl is hydrated in solu- tions, binding to the enzyme as a geminal diol (Fig. 7). The structures of difluoroketones bound to three different aspartyl proteinases have been re- ported (Tables IIa and VIIIa); these are endothiapepsin,M rhizopuspepsin,42 and penicillopepsin.41 Except for the tetrahedral hydrate (gem-diol) portion, these inhibitors interact with the protein in a fashion similar to that found in other monomeric enzyme complexes. One of the geminal diol two hydroxyls mimics the attacking water molecule. It is almost equidistant from the two internal oxygen atoms of the catalytic aspartic acids (Fig. 7)' making one hydrogen bond with the internal oxygen atom of Asp-32 and another bond with the external oxygen atom of Asp-215. The other hydroxyl makes only one hydrogen bond to the external oxygen atom of Asp-32 (Fig. 7). This mode

INHIBITORS OF ASPARTYL PROTEINASES 765
Figure 7. Schematic representation of the hydrogen-bonding network between the gem-diol por- tion of a difluoroketone inhibitor and a monomeric aspartyl proteinase, based on the structure of CP-81,282 bound to endothiapepsin.&
of binding supports a catalytic mechanism for aspartyl proteinases in which Asp-32 polarizes the carbonyl oxygen atom of the substrate and donates a proton. Simultaneously, the central water molecule, polarized into a nucleo- philic state by the partial transfer of a proton to Asp-215, attacks the carbonyl carbon atom, resulting in a transfer of the proton to that aspartic acid. Proto- nation of the nitrogen atom in the tetrahedral intermediate then results in proteolysis, with the proton coming from A ~ p - 2 1 5 . ~ ~ It is important to note that proton positions were not determined from these crystallographic stud- ies, but rather, were inferred from hydrogen bond distances and geometry of nonhydrogen atoms.
E. Glycol-Based, Cyclic, Phosphorus-Containing, and Symmetric
Three crystal structures of porcine pepsin complexed to glycol-based inhibi- tors have been reported,35,37 as well as a molecular modeling study in which the glycol-based renin inhibitor SC-46944 was docked in the active site of endothiapepsin.215 Focusing on the P,-P; region, the glycol portion adopts a conformation resembling an inverse y turn, with the P , and Pi side chains antiperiplanar to each other and the two glycol hydroxyl groups in an anti arrangement (Fig. 8). The (3X) hydroxyl interacts with the catalytic aspartates, while the (4s) hydroxyl points in the opposite direction binding to Gly-76 of the flap, and in so doing, mimics the Pi carbonyl of a substrate (Fig. 8). Similar results were obtained from crystal structures of a number of glycol-based inhibitors bound to endothiapepsin. 196
The only crystal structure reported for an aspartyl proteinase complexed to a cyclic ligand is that of rhizopuspepsin with the cyclic disulfide inhibitor (Tables IIa and VIIIa). The backbone and side-chain conformations in this inhibitor are very similar to those observed in the less restrained ligands, forming similar hydrogen bonds and van der Waals contacts in the segment from P4 to Pi.43
Similar to the difluoroketones, the phosphorus-containing inhibitors can be viewed as mimics of the tetrahedral intermediate of peptide bond hydrolysis. Four structures of such inhibitors bound to aspartyl proteinases have been determined; three to penicillopepsin,40 and the fourth to HIV-1 p r ~ t e i n a s e . ~ ~ All three inhibitors bound to penicillopepsin have virtually identical confor-

766 ABDEL-MEGUID
PZ - flap region
Figure 8. Model of the structure of the glycol-based inhibitor SC-46944 bound to endothiapepsin showing hydrogen-bonding interactions (from reference 215).
mations in the active site; they bind in a manner that closely approximates that expected for the transition state.40 Comparison of the mode of binding of these inhibitors with that of similar difluoroketones41 shows the same confor- mation in both, despite an approximate 0.6-A shift toward the N-terminal in the phosphonate inhibitors.40 Similarly, the bound conformation of phosphi- nate inhibitor (SB204144) of HIV-1 p r ~ t e i n a s e ~ ~ is very similar to that of its hydroxymethylene analogue49 A74704. An interesting feature of the phosphorus- containing inhibitors is the very short (2.2-2.6-A) oxygen-oxygen contacts formed between the phosphinic or phosphonic acid moieties and the catalytic carboxylates. These short distances and the stereochemical environment of the two interacting oxygen atoms were interpreted in terms of hydrogen bonds that have symmetric, single-well, potential-energy curves, with the proton located midway between the two oxygen atoms.40,57 Such short dis- tances between oxygen atoms, observed in small molecule structures, are indicative of strong hydrogen bonds.216
Four structures of symmetric inhibitors bound to HIV-1 proteinase have been reported (Tables IIb and VIIIb); the phosphinate inhibitor57 described above is one. Of these, SB204144, and SKF108361 are perfectly symmetric inhibitors, the latter being a dihydroxyethylene.58.198 Although the P, to P; region of SB204144 binds symmetrically to the active site, that of SKF108361, surprisingly, does not. Dreyer et al. reported that one of the hydroxyls of the SKF108361 diol interacts with the two catalytic aspartates, while the other interacts with only 0ne.58 The central hydroxyl is situated near the twofold axis of the complex, thus the midpoint of the inhibitor is offset by nearly 0.8 A from that axis.58
The two other inhibitors, A-74704 and L700,417, are not perfectly sym- metric as a result of the presence of only one hydroxyl at the central carbon. However, their P,-P; regions show essentially symmetric binding to the ac- tive site, in a conformation similar to other, asymmetric inhibitors. The novel indanol ring of L700,417 projects into the SJS; pocket providing good hydro- phobic and hydrophilic interactions with the protein.52

INHIBITORS OF ASPARTYL PROTEINASES 767
F. Inhibitor-Induced Protein Conformation
Initial comparison of the structures of endothiapepsinlinhibitor complexes with that of the uncomplexed enzyme18 revealed no major conformational changes in the enzyme upon binding of the inhibitor, except for small move- ments of the flap.*8 However, recently, Sali et al. have analyzed 16 of these structures more extensively. They reported the existence of two forms of the enzyme differing by a rigid-body rotation and a translation of a domain com- prising residues 190-302 relative to the rest of the structure.217 The first form is nativelike; it was observed in the structures of endothiapepsin containing L-363,564, H-142, H-256, BW624, BW625, H-261 (one crystal form), and CP-81,282. The second one differs from the native by approximately 4" rota- tion and 0.3-A translation; it was observed in the structures containing pepstatin, H-189, L-364,099, H-77, CP-69,799, H-261 (a second crystal form), PD125967 and CP-71,362. Surprisingly, there was no correlation between the class of the inhibitor and the form of the enzyme, except that most of the structures containing statine inhibitors belonged to the latter.
One is tempted to believe that these two forms are a consequence of crystal packing, since most of the nativelike structures are determined from crystals that are isomorphous with those of native endothiapepsin. However, crystals of the BW625 complex are nonisomorphous to those of the native, yet its structure has a nativelike conformation. This, plus the fact that the protein of the H-261 complex was observed in both forms, suggests that endothiapepsin may exist in solution in an equilibrium between the two forms. This view is supported by the structure determination of unliganded, glycosylated human renin,*4 in which the structure of renin was found to adopt two distinct conformations within the same crystal lattice. Similar to endothiapepsin, the two conformations of human renin are related by approximately 4" rigid-body rotation and less than 1 A translation of part of the C-terminal domain relative to the N-terminal domain. The necessity for conformational change upon substrate binding has been proposed as an important step in the catalysis by aspartic proteinases.218,219
No similar conformational changes have been reported in the structures of complexes with rhizopuspepsin or penicillopepsin, except for a significant movement of the flap so that hydrogen bonds and van der Waals contacts can form between the protein and the inhibitor. This movement is accompanied by a reduction in the thermal motion ( B value) of all three aspartyl pro- teinases, endothiapepsin,217 rhizopuspepsin,43 and penicillopepsin.41 Fur- thermore, inhibitor binding results in a general reduction of thermal motion of most atoms of the protein.41
The most dramatic ligand-induced conformational change is observed with HIV-1 protease. As indicated above, the two flaps move by as much as 7 to tightly embrace the ligand.48-50 Inspection of these structures indicates that three sets of hydrogen bonds play a critical role in stabilizing ligand binding and in inducing the closure of the flaps (residues 42-58 of each monomer). In the first set, hydrogen bonds are between the peptide backbone amide- hydrogen of Gly-51 in one monomer and the carbonyl-oxygen of Gly-51 in the other monomer. In the second set, the peptide backbond amide-hydrogens of Ile-50 in both monomers hydrogen bond to a bridging water molecule that also participates in hydrogen bonds with the ligand. In the third, the back-

768 ABDEL-MEGUID
bone amide-hydrogen and the carbonyl-oxygen of Gly-48 in each of the two monomers hydrogen-bond directly to the ligand. Movement of the flaps was analyzed by Harte et 61.220; they proposed a model in which closing of the flaps is mediated by a “cantilever” (residues 59-75) and a ”fulcrum” (residues 9-21). According to this model, the movement of the flap regions upon ligand binding is accompanied by compensatory movement in the cantilever region. Mutations in the cantilever region result in nonfunctional protease and nonin- fectious virions,127 indicating that this region is important for protease struc- ture and function.
G . Subsite Specificity
Because portions of inhibitors beyond P,-P; of the monomeric enzymes and P2-P; of the dimeric enzymes are considerably solvent exposed, thus they make few van der Waals contacts with atoms of the protein. Each residue of an inhibitor’s central portion, however, fits in a pocket (subsite) defined by a number of protein atoms (Table X). These protein atoms are usually identified as those that are within 4.1 A of atoms of the inhibitor. The protein atoms identified using such a definition vary, depending on the size and conforma- tion of the side chains of the inhibitor. Smaller side chains, for example, have fewer atoms within 4.1 A than larger ones. For a particular aspartyl pro- teinase, however, the positions of these protein atoms in various structures of enzyme-inhibitor complexes appear to be invariant, indicating that they de- fine a reasonably constant volume in which the inhibitor’s central portion must fit in. Thus intuitively one can suggest that the degree to which an inhibitor fills this invariant volume should correlate with its potency. Given the number of high-resolution structures of aspartyl proteinase-inhibitor com- plexes available, it should be possible to determine whether such correlations are valid. The major difficulty, however, with such an analysis is that the binding constants available for many of the inhibitors were obtained in vari- ous laboratories, under different assay conditions; thus they are not neces- sarily on the same scale.
Although many of the residues that contribute atoms to the various S, to S; pockets are conserved among the monomeric enzymes, a few are not. As a consequence the volume available for a particular inhibitor will differ from one enzyme to another, contributing to differences in the conformation and binding affinity of an inhibitor when complexed to these various enzymes.
H. Comparison of Renin and HIV-1 Proteinase Inhibitors
Dreyer and co-workers reported58 a molecular-modeling study in which they docked the structure of the inhibitor A-74704, derived from the structure of its complex with HIV-1 proteinase,@ into the active site of pepsin, derived from the crystal structure of the renin inhibitor A62095 bound to pepsin.37 They found it to fit well without alterations; most of the hydrogen bonds observed in the crystal structure were also conserved in the model. Several adjustments improved the fit; these mostly centered around the PJP; Cbz groups of A-74704.
No experimentally based, direct comparison of an inhibitor conformation in both HIV-1 proteinase and any pepsin enzyme is available. However,
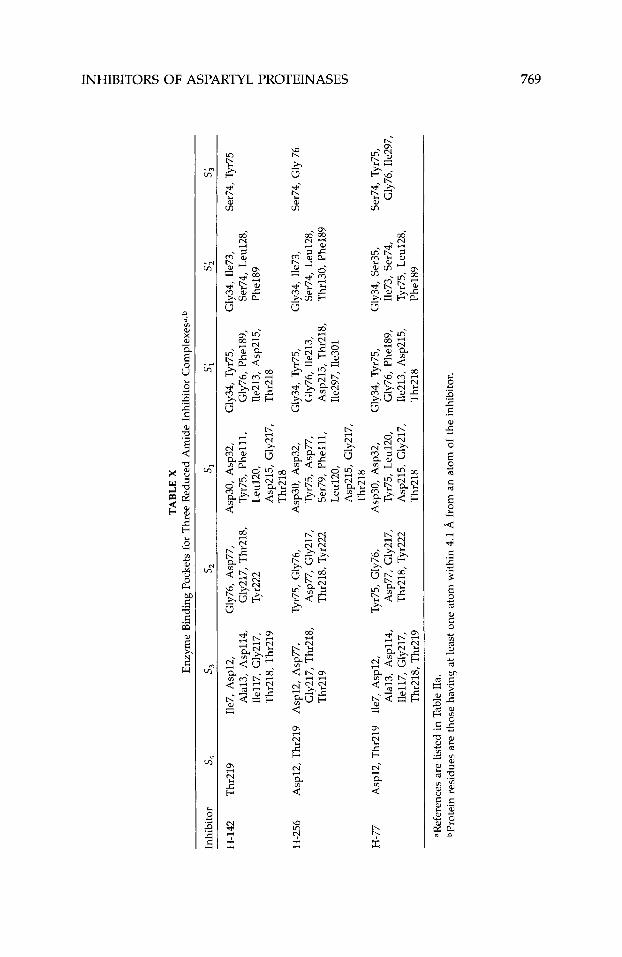
2 z E 8 z 8 % 2
H-2
56
Asp
l2, T
hr21
9 A
spl2
, A
sp77
, Ty
r75,
Gly
76,
Asp
30,
Asp
32,
Gly
34, T
yr75
, G
ly34
, lle
73,
Ser7
4, G
ly 7
6 3
Gly
217,
Thr
218,
A
sp77
, G
ly21
7,
Tyr7
5, A
sp77
, G
ly76
, Ile
213,
Se
r74,
Leu
128,
2 %
Leu
l20,
Ile
297,
Ile
301
m
Asp
215,
Gly
217,
v,
TABL
E X
E
nzym
e B
indi
ng P
ocke
ts f
or T
hree
Red
uced
Am
ide
Inhi
bito
r C
ompl
exes
a,b
z 8
Inhi
bito
r s.l
s3
s2
s,
s;
s; s;
H-1
42
Thr2
19
Ile7,
Asp
l2,
Gly
76, A
sp77
, A
sp30
, A
sp32
, G
ly34
, Ty
r75,
G
ly34
, Ile
73,
Ser7
4, T
yr75
r
Ala
13,
Asp
ll4,
G
ly21
7, T
hr21
8,
Tyr7
5, P
hell
l,
Gly
76,
Phe1
89,
Ser7
4, L
eu12
8,
Ile11
7, G
ly21
7,
Tyr2
22
Leu
l20,
Ile
213,
Asp
215,
Ph
e189
Th
r218
, Thr
219
Asp
215,
Gly
217,
Th
r218
Th
r218
Thr2
19
Thr2
18, T
yr22
2 Se
r79,
Phe
lll,
A
sp21
5, T
hr21
8,
Thr
l30,
Phe
189
Thr2
18
H-7
7 A
spl2
, Th
r219
Ile
7, A
spl2
, Ty
r75,
Gly
76,
Asp
30,
Asp
32,
Gly
34,
Tyr7
5,
Gly
34,
Ser3
5,
Ser7
4, T
yr75
, A
la13
, A
spll
4,
Asp
77,
Gly
217,
Ty
r75,
Leu
l20,
G
ly76
, Ph
e189
, Ile
73,
Ser7
4,
Gly
76, I
le29
7,
Ile11
7, G
ly21
7,
Thr2
18, T
yr22
2 A
sp21
5, G
ly21
7,
Ile21
3, A
sp21
5,
Tyr7
5, L
eu12
8,
Thr2
18, T
hr21
9 Th
r218
Th
r218
Ph
e189
aRef
eren
ces
are
liste
d in
Tab
le II
a.
bPro
tein
res
idue
s ar
e th
ose
havi
ng a
t lea
st o
ne a
tom
with
in 4
.1 A
from
an
atom
of
the
inhi
bito
r.

770 ABDEL-MEGUID
molecular-modeling studies have shown that, in general, the conformation of an inhibitor is similar in both enzyme classes, particularly between P, and Pi.
VIII. MINIMAL RENIN INHIBITOR
It is difficult to determine the number of renin inhibitors synthesized to date. However, it is probably safe to say that they are in the tens of thou- sands. Inspection of the binding constants and chemical structures of many of these inhibitors leads one to suggest that the smallest inhibitor able to bind to renin with high affinity is equivalent to a tetrapeptide spanning from P, to Pi. Ideally, this inhibitor would have Phe, His, and cyclohexylmethyl side chains at P,, P,, and P,, respectively. It would have a glycol group as the scissile- bond surrogate, and would begin with a methyl and terminate with an iso- butyl group. The former group is intended to reduce the peptide character of the inhibitor while the latter group should fit in the S; pocket, thus substitut- ing for the valine side chain of the sub~trate.35,37,~96,215 Hanson221 has synthe- sized this inhibitor and found it to be a 10-nM (IC50) inhibitor of human renin. Removal of either the methyl group from the amino-terminal end or the isobutyl group from the carboxy-terminal end resulted in more than tenfold reduction in potency. Replacement of the cyclohexylmethyl with a phe- nylalanine side chain, also results in lower potency.187
Although this inhibitor has a molecular weight less than 600 and relatively high potency, it does not have the degree of oral bioavailability hoped for by reducing the size.221 The addition of groups that fit in the S4 pocket was necessary to confer oral bioavilability. 787,190 The choice of the amino-terminal end for adding substituents is dictated by the relative tolerance of the P4 position for differing substituents resulting from its considerable solvent ac- cessibility.
Although substituting other moieties for the two peptide bonds between PI and P, is desirable to reduce the peptide bond character of renin inhibitors, it has recently been shown that this is not necessary to obtain inhibitors with moderate-to-high oral bioavailability. 190 Given the importance of the hydrogen- bonding network generated between the heteroatoms of these two amide groups and the enzyme (Table IXa; Fig. 5), it is easy to see that their modifica- tion could result in loss of potency. Alternatively, the reduction of the peptide character of these inhibitors has been achieved by introducing nonpeptidic side chains, such as cyclohexylmethyl and thiazole at PI and P,, respec- tively. 190
IX. MINIMAL HIV-1 PROTEINASE INHIBITOR
The diversity in number and class diversity among published HIV-1 pro- teinase inhibitors is considerably less than for renin. Of the aspartyl pro- teinase classes listed in Table 11, molecules containing a hydroxyethylene moiety have perhaps been most extensively studied as HIV-1 proteinase in- hibitors. One can propose a minimal hydroxyethylene isostere inhibitor based on the available data, that has bulky side chains such as Phe at PI and Pi, and Val at P, and P;. The peptide bonds in the inhibitor appear to be important for tight interaction with the enzyme. Dreyer et a1.54 proposed that the minimum hydrogen-bonding interactions required for tight-binding inhibition starts

INHIBITORS OF ASPARTYL PROTEINASES 771
with the carbonyl carbon of P, and ends at the amide nitrogen of Pi, with each of the backbone heteroatoms involved in a hydrogen bond with the enzyme. This proposal was deduced from a study of the effects of changes within a series of hydroxyethylene isosteres on their potency (Table V). The minimal inhibitor I, where R = Bn (Table V), is a 1.4-nM (Ki) inhibitor of the purified en~yme.5~ Additional interactions that confer still greater potency can be ob- tained by extending the minimal inhibitor beyond P, and P;.
X. ASPARTYL PROTEINASE INHIBITORS AS POTENTIAL DRUGS
For a renin inhibitor to be a commercially successful drug a number of requirements must be achieved. The inhibitor must be (1) able to lower renin blood pressure in hypertensive patients, (2) orally bioavailable to compete in the market place with ACE inhibitors, (3) advantageous relative to other anti- hypertensive agents, (4) potent, (5) specific for renin versus pepsin, (6) non- toxic, and (7) metabolically stable. Progress toward achieving most of these requirements has been substantial. The greatest stumbling block remains the lack of high oral bioavailability caused by either low gastrointestinal absorp- tion or high hepatic extraction, or both. A recent report of an inhibitor having high oral bioavailabilityl90 is reassuring.
As with renin inhibitors, an HIV proteinase inhibitor must meet a number of requirements to be a commercially successful drug. It must (1) inhibit viral replication in vivo, (2) efficiently penetrate cells, (3) be potent, (4) be synergis- tic with, or advantageous, relative to other antiviral agents, (5) be metaboli- cally stable, and (6) be specific. Although oral bioavailability is not absolutely necessary for the treatment of AIDS, it is highly desirable.
The ability of many of the HIV-1 proteinase inhibitors to exert potent anti- viral activity in vitro is reassuring. Not only can these inhibitors block the action of the enzyme in virions shed from chronically infected cells, but they also can penetrate and inhibit the enzyme within cells. It is also reassuring that many of these inhibitors show low cytotoxicity and high selectivity to- ward HIV-1 protease relative to other aspartyl proteinases, especially renin and pepsin. As with renin inhibitors, these peptide-based molecules show generally low oral bioavailability, biliary clearance, and short serum half-life. It is encouraging to see that systematic structural modifications can lead to molecules with significantly improved properties, as in the case of the devel- opment of orally bioavailable renin inhibitors, and at least one is undergoing clinical trials.190 Unlike the development of renin inhibitors, however, the possible emergence of clinical resistance to HIV proteinase inhibitors must be addressed.
I am grateful to Dr. Susan Dendinger, Dr. Renee DesJarlais, Dr. Geoffrey Dreyer, Dr. John Erickson, Dr. David Green, and Or. Gunnar Hanson for reading the manuscript and offering insightful suggestions. I am also grateful to Dr. Christine Debouck and Dr. Gunnar Hanson for Figs. 2 and 7, respectively.
REFERENCES
1. R. C. Gallo and L. Montagnier, Sci. Am. 259, 40, (1988). 2. H. Umezawa, T. Aoyagi, H. Morishima, M. Matsuzaki, and M. Hamada, 1. Antibiot. (Tokyo)
23, 295 (1970).

772 ABDEL-MEGUID
3. H. Horishima, T. Takita, T. Aoyagi, T. Takeuchi, and H. Umezawa, J. Antibiot. (Tokyo) 23,263
4. G. E. Wakerlin, Circulation 17, 653 (1958). 5. Z.-Y. Zhang, R. A. Poorman, L. L. Maggiora, R. L. Heinrikson, and F. J. Kezdy, J. Biol. Chem.
6. H. J. Schramm, H. Nakashima, W. Schramm, H. Wakayama, and N. Yamamoto, Biochem.
7. M. N. James and A. R. Sielecki, J. Mol. Biol. 163, 299 (1983). 8. K. Suguna, R. R. Bott, E. A. Padlan, E. Subramanian, S. Sheriff, G. H. Cohen, and
9. M. Jaskolski, M. Miller, J. K. Rao, J. Leis, and A. Wlodawer, Biochemistry 29, 5889 (1990). 10. I. T. Weber, M. Miller, M. Jaskolski, J. Leis, A. M. Skalka, and A. Wlodawer, Science 243,928
11. A. R. Sielecki, K. Hayakawa, M. Fujinaga, M. E. Murphy, M. Fraser, A. K. Muir,
12. R. Lapatto, T. Blundell, A. Hemmings, J. Overington, A. Wilderspin, S. Wood, J. R. Merson,
13. M. A. Navia, P. M. Fitzgerald, B. M. McKeever, C. T. Leu, J. C. Heimbach, W. K. Herber,
14. A. Wlodawer, M. Miller, M. Jaskolski, B. K. Sathyanarayana, E. Baldwin, 1. T. Weber,
15. M. Miller, M. Jaskolski, J. K. Rao, J. Leis, and A. Wlodawer, Nature 337, 576 (1989). 16. S. I. Foundling, J. Cooper, F. E. Watson, B. Korant, J. J. Wendoloski, P. C. Weber,
A. C. Treharne, M. C. Schadt, M. Jaskolski, M. Miller, A. Wlodawer, P. Strop, V. Kostka, Sedlacek and D. H. Ohlendorf, in Viral Proteinuses as Targets for Chemotherapy, H.-G. Krausslich, S. Oroszlan, E. Wimmer, Eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989, p. 181.
(1970).
266, 15591 (1991).
Biophys. Res. Commun. 179, 847 (1991).
D. R. Davies, 1. Mot. Bid . 196, 877 (1987).
(1989).
C. T. Carilli, J. A. Lewicki, J. D. Baxter, and M. N. James, Science 243, 1346 (1989).
P. J. Whittle, D. E. Danley, K. F. Geoghegan, et al., Nature 342, 299 (1989).
I. S. Sigal, P. L. Darke, and J. P. Springer, Nature 337, 615 (1989).
L. M. Selk, L. Clawson, J. Schneider, and S. B. Kent, Science 245, 616 (1989).
17. C. Abad-Zapatero, T. J. Rydel, and J. Erickson, Proteins 8, 62 (1990). 18. T. L. Blundell, J. A. Jenkins, B. T. Sewell, L. H. Pearl, J. 8 . Cooper, I. J. Tickle, 8 . Veerapan-
19. T. L. Blundell, R. Lapatto, A. F. Wilderspin, A. M. Hemmings, P. M. Hobart, D. E. Danley,
20. J. B. Cooper, G. Khan, G. Taylor, I . J. Tickle, and T. L. Blundell, J. Mol. Bid . 214, 199 (1990). 21. G. L. Gilliland, E. L. Winborne, J. Nachman, and A. Wlodawer, Proteins 8, 82 (1990). 22. A. R. Sielecki, A. A. Fedorov, A. Boodhoo, N. S. Andreeva, and M. N. James, J. Mol. Bid.
23. M. Newman, M. Safro, C. Frazao, G. Khan, A. Zdanov, I. J. Tickle, T. L. Blundell, and
24. J. Rahuel, J. P. Priestle, and M. G. Grutter, J. Struct. Bid. 107, 227 (1991). 25. M. N. G. James, A. Sielecki, F. Salituro, D. H. Rich, and T. Hofmann, Proc. Natl. Acad. Sci.
U S A 79, 6137 (1982). 26. T. L. Blundell, J. Cooper, S. I. Foundling, D. M. Jones, B. Atrash, and M. Szelke, Biochemistry
26, 5585 (1987). 27. J. Cooper, S. Foundling, A. Hemmings, T. Blundell, D. M. Jones, A. Hallett, and M. Szelke,
Eur. I. Biochem. 169, 215 (1987). 28. S. I. Foundling, J. Cooper, F. E. Watson, A. Cleasby, L. H. Pearl, 8 . L. Sibanda, A. Hem-
mings, s. P. Wood, T. L. Blundell, M. J. Valler, et al., Nature 327, 349 (1987). 29. S. I. Foundling, J. Cooper, F. E. Watson, L. H. Pearl, A. Hemmings, S. P. Wood, T. Blundell,
A. Hallett, D. M. Jones, J. Sueiras, et al., Cardiovasc. Pharmacol. 7, (1987). 30. K. Suguna, E. A. Padlan, C. W. Smith, W. D. Carlson, and D. R. Davies, Proc. Natl. Acad.
Sci. U S A 84, 7009 (1987). 31. J. B. Cooper, S. I. Foundling, T. L. Blundell, R. J. Arrowsmith, C. J. Hams, and N. J. Champ-
ness, in Topics in Medicinul Chemistry, P. R. Leeming, Ed., Royal Soaety of Chemistry Speaal Publication, London, 1988, p. 308.
32. J. 8 . Cooper, S. I. Foundling, T. L. Blundell, J. Boger, R. A. Jupp, and J. Kay, Biochemistry 28, 8596 (1989).
33. A. Sali, B. Veerapandian, J. B. Cooper, S. I. Foundling, D. J. Hoover, and T. L. Blundell, Embo. J. 8, 2179 (1989).
34. B. Veerapandian, J. 8 . Cooper, A. Sali, and T. L. Blundell, J. Mol. Biol. 216, 1017 (1990).
dian, and S. P. Wood, J. Mol. Bid. 211, 919 (1990).
and P. J. Whittle, Trends Biochem. Sci. 15, 425 (1990).
214, 143 (1990).
N. Andreeva, I . Mol. Biol. 221, 1295 (1991).

INHIBITORS OF ASPARTYL PROTEINASES 773
35. C. Abad-Zapatero, T. J. Rydel, D. J. Neidhart, J. Luly, and J. W. Erickson, Adv. Exp. Med. Biol.
36. K. D. Parris, D. J. Hoover, and D. R. Davies, Adv. Exp. Med. Biol. 306, 217 (1991). 37. L. Chen, J. W. Erickson, T. J. Rydel, C. H. Park, D. Neidhart, J. Luly, and C . Abad-Zapatero,
38. J. Cooper, W. Quail, C. Frazao, S. I. Foundling, T. L. Blundell, C. Humblet, E. A. Lunney,
39. V. Dhanaraj, C. G. Dealwis, C. Frazao, M. Badasso, B. L. Sibanda, I. J. Tickle, J. B. Cooper,
40. M. E. Fraser, N. C. Strynadka, P. A. Bartlett, J. E. Hanson, and M. N. James, Biochemistry 31,
41. M. N. James, A. R. Sielecki, K. Hayakawa, and M. H. Gelb, Biochemistry 31, 3872 (1992). 42. K. D. Parris, D. J. Hoover, D. B. Damon, and D. R. Davies, Biochemistry 31, 8125 (1992). 43. K. Suguna, E. A. Padlan, R. Bott, J. Boger, K. D. Parris, and D. R. Davies, Proteins 13, 195
(1992). 44. B. Veerapandian, J. B. Cooper, A. Sali, T. L. Blundell, R. L. Rosati, B. W. Dominy,
D. 8. Damon, and D. J. Hoover, Protein Sci. 1, 322 (1992). 45. S. Al-Karadaghi, J. B. Cooper, 8 . Veerapandian, D. Hoover, and T. L. Blundell, atomic
coordinates deposited in the Protein Data Bank (see Ref. 63), code 3ER3. 46. D. Bailey, 8 . Veerapandian, J. Cooper, M. Szelke, and T. L. Blundell, atomic coordinates
deposited in the Protein Data Bank (see Ref. 63), code 3ER5. 47. A. M. Hemmings, 8. Veerapandian, M. Szelke, J. B. Cooper, and T. L. Blundell, atomic
coordinates deposited in the Protein Data Bank (see Ref. 63), code 1ER8. 48. M. Miller, J. Schneider, 8. K. Sathyanarayana, M. V. Toth, G. R. Marshall, L. Clawson,
L. Selk, S. B. Kent, and A. Wlodawer, Science 246, 1149 (1989). 49. J. Erickson, D. J. Neidhart, J. VanDrie, D. J. Kempf, X. C. Wang, D. W. Norbeck, J. J. Plattner,
J. W. Rittenhouse, M. Turon, N. Wideburg, et ul., Science 249, 527 (1990). 50. P. M. Fitzgerald, 8 . M. McKeever, J. F. VanMiddlesworth, J. P. Springer, J. C. Heimbach,
C. T. Leu, W. K. Herber, R. A. Dixon, and P. L. Darke, J. Biol. Chem. 265, 14209 (1990). 51. A. L. Swain, M. M. Miller, J. Green, D. H. k c h , J. Schneider, S. B. Kent, and A. Wlodawer,
PYOC. Nutl. Acud. Sci. U S A 87, 8805 (1990). 52. R. Bone, J. P. Vacca, P. S. Anderson, and M. K. Holloway, J. A m . Chem. Soc. 113,9382 (1991). 53. M. Jaskolski, A. G. Tomasselli, T. K. Sawyer, D. G. Staples, R. L. Heinrikson, J. Schneider,
S. B. Kent, and A. Wlodawer, Biochemistry 30, 1600 (1991). 54. G. B. Dreyer, D. M. Lambert, T. D. Meek, T. J. Carr, T. A. Tomaszek, Jr., A. V. Fernandez,
H. Bartus, E. Cacciavillani, A. M. Hassell, M. Minnich, et al. , Biochemistry 31, 6646 (1992). 55. K. H. M. Murthy, E. L. Winborne, M. Minnich, J. S. Culp, and C. Debouck, J. Biol. Chem.
267, 22770 (1992). 56. T. A. Tomaszek, Jr., M. L. Moore, J. E. Strickler, R. L. Sanchez, J. S. Dixon, B. W. Metcalf,
A. Hassell, G. 8 . Dreyer, I. Brooks, C. Debouck, and T. D. Meek, Biochemistry 31, 10153 (1992).
57. S. S. Abdel-Meguid, B. Zhao, K. H. M. Murthy, E. Winborne, H.-K. Choi, R. L. DesJarlais, M. D. Minnich, J. S. Culp, C. Debouck, T. A. Tomaszek, Jr., T. D. Meek, and G. B. Dreyer, Biochemistry 32, 7972 (1993).
58. G. B. Dreyer, J. C. Boehm, B. Chenera, R. L. DesJarlais, A. M. Hassell, T. D. Meek, T. A. Tomaszek, Jr., and M. Lewis, Biochemistry 32, 937 (1993).
59. B. Zhao and S. S. Abdel-Meguid (unpublished). 60. J. Hartsuck and J. Remington, Abstr. 18th Linderstrom-Lung Conf. (1988). 61. M. N. G. James and A. R. Sielecki,Nuture 319, 33 (1986). 62. A. R. Sielecki, M. Fujinaga, R. J. Read, and M. N. James, J. Mol. Bio/. 219, 671 (1991). 63. F. C. Bernstein, T. F. Koetzle, G. J. Williams, E. E. J. Meyer, M. D. Brice, J. R. Rodgers,
0. Kennard, T. Shimanouchi, and M. Tasumi, J. Mol. Biol. 112, 535 (1977). 64. G. B. Dreyer, B. W. Metcalf, T. A. Tomaszek, Jr., T. J. Carr, A. C. Chandler, 111, L. Hyland,
S. A. Fakhoury, V. W. Magaard, M. L. Moore, J. E. Strickler, et ul., Proc. Nutl. Acud. Sci. USA 86, 9752 (1989).
65. T. J. McQuade, A. G. Tomasselli, L. Liu, V. Karacostas, B. Moss, T. K. Sawyer, R. L. Heinrikson, and W. G. Tarpley, Science 247, 454 (1990).
66. A. G. Tomasselli, J. 0. Hui, T. K. Sawyer, D. J. Staples, D. J. FitzGerald, V. K. Chaudhary, I. Pastan, and R. L. Heinrikson, J. Biol. Ckem. 265, 408 (1990).
306, 9 (1991).
Acta Crystullogr. Sec. B 48, 476 (1992).
W. T. Lowther, and B. M. Dunn, Biochemistry 31, 8142 (1992).
H. P. Driessen, M. Newman, C. Aguilar, et ul., Nature, 357, 466 (1992).
5201 (1992).

774 ABDEL-MEGUID
67. N. A. Roberts, J. A. Martin, D. Kinchington, A. V. Broadhurst, J. C. Craig, I. B. Duncan,
68. H. Toh, M. Ono, K. Saigo, and T. Miyata, Nature 315, 691 (1985). 69. M. D. Power, P. A. Marx, M. L. Bryant, M. B. Gardner, P. J. Barr, and P. A. Luciw, Science
70. L. H. Pearl and W. R. Taylor, Nature 329, 351 (1987). 71. N. E. Kohl, E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Heimbach, R. A. Dixon,
E. M. Scolnick, and I. S. Sigal, Proc. Nutl. Acad. Sci. U S A 85, 4686 (1988). 72. T. D. Meek, B. D. Dayton, B. W. Metcalf, G. 8. Dreyer, J. E. Strickler, J. G. Gorniak,
M. Rosenberg, M. L. Moore, V. W. Magaard, and C. Debouck, Proc. Nutl. Acud. Sci. U S A 86, 1841 (1989).
S. A. Galpin, B. K. Handa, J. Kay, A. Krohn, et al., Science 248, 358 (1990).
231, 1567 (1986).
73. J. S. Fruton, Adv. Enzymol. Relut. Areas Mol. Biol. 44, 1 (1976). 74. V. Kostka, in Aspartic Proteinases and Their Inhibitors, V. Kostka, Ed., de Gruyter, Berlin, 1985. 75. D. H. Rich, J. Med. Chem. 28, 263 (1985). 76. J. S. Fruton, in Hydrolytic Enzymes, A. Neuberger, and K. Brocklehurst, Eds., Elsevier,
77. L. Polgar, FEBS Lett. 219, 1 (1987). 78. M. N. G. James and A. R. Sielecki, in Biofogical Mucromolecules uitd Assemblies, F. A. Jurnak,
79. D. R. Davies, Annu. Rev. Biophys. Biophys. Chem. 19, 189 (1990). 80. T. Inagami and Y. Suketa, Methods Enzymol. 46, 235 (1977). 81. R. Day and E. Haber, in Handbook of Hypertension, A. Zanchetti, and R. C. Tarazi, Eds.,
82. T. Inagami, in Kidney Hormones, J. W. Fisher, Ed., Acadamic, London, 1986, Vol. 3, p. 217. 83. H. R. Brunner, B. Waeber, and J. Nussberger, Kidney Internut. 34, S80 (1988). 84. T. Inagami, J. Hypertens Suppl. 7, S3 (1989). 85. M. J. Antonaccio and D. W. Cushman, Fed. Proc. 40, 2275 (1981). 86. M. J. Antonaccio and 1. J. Wright, Prog. Drug Res. 31, 161 (1987). 87. W. J. Greenlee, Pharm. Res. 4, 364 (1987). 88. L. M. Wood, J. L. Stanton, and K. G. Hofbauer, J. Enzym. Inhib. 1, 169 (1987). 89. H. D. Kleinert, A m . J. Hypertens. 2, 800 (1989). 90. W. J. Greenlee, Med. Res. Rev. 10, 173 (1990). 91. H. D. Kleinert, W. R. Baker, and H. H. Stein, Adv. Pharmacol. 22, 207 (1991). 92. T. D. Ocain and M. Abou-Gharbia, Drugs Future 16, 37 (1991). 93. P. Corvol, D. Chauveau, X. Jeunemaitre, and J. Menard, Hypertension. 16, 1 (1990). 94. C. Hutchins and J. Greer, Crit. Rev. Biochem. Mol. Biol. 26, 77 (1991). 95. C. Debouck and B. W. Metcalf, Drug Develop. Res. 21, 1 (1990). 96. P. M. D. Fitzgerald and J. P. Springer, Annu. Rev. Biophys. Biophys. Chem. 20, 299 (1991). 97. J. R. Huff, J. Med. Chem. 34, 2305 (1991). 98. D. W. Norbeck and D. J. Kempf, Ann. Rep. Med. Chem. 26, 141 (1991). 99. C. Debouck, AIDS Res. Hum. Retroviruses 8, 153 (1992).
Amsterdam, 1987, p 1.
and A. McPherson, Eds., Wiley, New York, 1987, p. 413.
North-Holland, Amsterdam, 1986, Vol. 8, p. 315.
100. J. A. Martin, Antiviral Res. 17, 265 (1992). 101. T. D. Meek, J. Enz. Inhib. 6, 65 (1992). 102. A. Wlodawer and J. W. Erickson, Annu. Rev. Biochem. 62, 543 (1993). 103. J. J. Panthier, S. Foote, 8. Chambraud, A. D. Strosberg, P. Corvol, and F. Rougeon, Nature
104. T. Imai, H. Miyazaki, S. Hirose, H. Hori, T. Hayashi, R. Kageyama, H. Ohkubo, S. Na-
105. J. A. Hardman, Y. J. Hort, D. F. Catanzaro, J. T. Tellam, J. D. Baxter, 8. J. Morns, and
106. H. Miyazaki, A. Fukamizu, S. Hirose, T. Hayashi, H. Hori, H. Ohkubo, S. Nakanishi, and
107. P. M. Hobart, M. Fogliano, B. A. OConnor, I. M. Schaefer, and J. M. Chirgwin, Proc. Nut / .
108. F. X. Gallen, M. T. Corvol, C. Devaux, M. C. Gubler, F. Mounier, J. P. Camilleri,
109. R. T. Pratt, A. J. Onelette, and V. J. Dzau, Proc. Nutl. Acad. Sci. U S A 80, 6809 (1983). 110. E. G. Erdos, Hypertension 16, 363 (1990). 111. D. M. Coulter and I. R. Edwards, Br. Med. 1. 294, 1521 (1987).
298, 90 (1982).
kanishi, and K. Murakami, Proc. Natl. Acad. Sci. U S A 80, 7405 (1983).
J. Shine, D N A 3, 457 (1984).
K. Murakami, Proc. Natl. Acud. Sci. U S A 81, 5999 (1984).
Acad. Sci. U S A 81, 5026 (1984).
A. M. Houot, J. Menard, and P. Corvol, J. Clin. Invest. 73, 1144 (1984).

INHIBITORS OF ASPARTYL PROTEINASES 775
112. S. M. Wood, R. D. Mann, and M. D. Rawlins, Br. Med. 1. 294, 91 (1987). 113. J. K. Wilkin, J. J. Hammond, and W. M. Kirkendall, Arch. Dermatol. 116, 902 (1980). 114. G. W. Shannon, G. F. Pyle, and R. L. Bashshur, in The Geography of Aids: Origins and Course of
115. H. Mitsuya, R. Yarchoan, S. Kageyama, and S. Broder, FASEB J. 5, 2369 (1991). 116. J. Tang, M. N. G. James, I. N. Hsu, J. A. Jenkins, and T. L. Blundell, Nature 271, 618 (1978). 117. T. L. Blundell, B. T. Sewell, and A. D. McLachlan, Biochim. Biophys. Acta 580, 24 (1979). 118. K. Appelt, R. J. Bacquet, C. A. Bartlett, C. L. J. Booth, S. T. Freer, M. A. M. Fuhry,
M. R. Gehring, S. M. Herrmann, E. F. Howland, C. H. Janson, T. T. Jones, C.-C. Kan, V. Kathardekar, K. K. Lewis, G. P. Marzoni, D. A. Matthews, C. Mohr, E. W. Moomaw, C. A. Morse, S. J. Oatley, R. C. Ogden, M. R. Reddy, S. H. Reich, W. S. Schoettlin, W. W. Smith, M. D. Varney, J. E. Villafranca, R. W. Ward, S. Webber, S. E. Webber, K. M. Welsh, and J. White, J. Med. Chem. 34, 1925 (1991).
an Epidemic, Guilford, New York, 1991.
119. J. W. Erickson and S. W. Fesik, Annu. Rep. Med. Chem. 27, 271 (1992). 120. K. E. Lentz, L. T. Skeggs, K. R. Woods, J. R. Kahn, and N. P. Shumway, J. Exp. Med. 104,183
121. L. T. Skeggs, K. E. Lentz, J. R. Kahn, N. P. Shumway, andK. R. Woods, J. Exp. Med. 104,193
122. L. T. Skeggs, J. R. Kahn, K. E. Lentz, and N. P. Shumway, J. Exp. Med. 106, 439 (1957). 123. L. T. Skeggs, K. E. Lentz, J. R. Kahn, and N. P. Shumway, J. Exp. Med. 108, 283 (1958). 124. L. T. Skeggs, K. E. Lentz, J. R. Kahn, and H. Hochstrasser, J. Exp. Med. 128, 13 (1968). 125. L. E. Henderson, R. E. Benveniste, R. Sowder, T. D. Copeland, A. M. Schultz, and S.
126. S. F. J. Le Grice, R. Ette, J. Mills, and J. Mous, J. Bid. Chem. 264, 14902 (1989). 127. D. D. Loeb, C. A. Hutchison, 111, M. H. Edgell, W. G. Farmerie, and R. Swanstrom, J. Vzrol.
63, 111 (1989). 128. M. L. Moore, W. M. Bryan, S. A. Fakhoury, V. W. Magaard, W. F. Huffman, B. D. Dayton, T.
D. Meek, L. Hyland, G. 8 . Dreyer, B. W. Metcalf, et al., Biochem. Biophys. Res. Commun. 159, 420 (1989).
(1956).
(1956).
Oroszlan, J. Virol. 62, 2587 (1988).
129. I. Schechter and A. Berger, Biochem. Biophys. Res. Commun. 27, 157 (1967). 130. R. L. Shoeman, B. Honer, T. J. Stoller, C. Kesselmeier, M. C . Miedel, P. Traub, and
131. R. L. Shoeman, C. Kesselmier, E. Mothes, B. Honer, and P. Traub, F E B S Lett. 278,199 (1991). 132. R. 0. Hui, A. G. Tomasselli, N. H. A. Zurcher, and R. L. Heinrikson, J. Bid. Chem. 265,
21 386 (1990). 133. A. G. Tomasselli, J. 0. Hui, T. K. Sawyer, S. Thaisrivongs, J. B. Hester, and R. L. Heinrik-
son, Adv. Exp. Med. Bid. 306, 469 (1991). 134. A. G. Tomasselli, J. 0. Hui, L. Adams, J. Chosay, D. Lowery, B. Greenberg, A. Yem,
M. R. Deibel, N. H. Zurcher, and R. L. Heinrikson, 1. Bid. Chem. 266, 14 548 (1991). 135. R. A. Poorman, A. G. Tomasselli, R. L. Heinrikson, and F. J. Kezdy, J. Bid. Chem. 266,14 554
(1991). 136. P. L. Darke, R. F. Nutt, S. F. Brady, V. M. Garsky, T. M. Ciccarone, C.-T. Leu, P. K. Lumma,
R. M. Freidinger, D. F. Veber, and I . S. Sigal, Biochem. Biophys. Res. Commun. 156,297 (1988). 137. P. L. Darke, C. T. Leu, L. J. Davis, J. C. Heimbach, R. E. Diehl, W. S. Hill, R. A. Dixon, and
I. S. Sigal, J. Bid. Chem. 264, 2307 (1989). 138. S. Billich, M.-T. Knoop, J. Hansen, P. Strop, J. Sedlacek, R. Mertz, and K. Moelling, J. Biol.
Chem. 263, 17 905 (1988). 139. L. H. Phylip, A. D. Richards, J. Kay, J. Konvalinka, P. Strop, I. Blaha, J. Velek, V. Kostka,
A. J. Ritchie, A. V. Broadhurst, W. G. Farmerie, P. E. Scarborough, and 8 . Dunn, Biochern. Biophys. Res. Commun. 171, 439 (1990).
140. A. D. Richards, R. Roberts, B. M. Dunn, M. C. Graves, and J. Kay, F E B S Lett. 247,113 (1990). 141. J. Konvalinka, P. Strop, J. Velek, V. Cerna, V. Kostka, L. H. Phylip, A. D. Richards,
142. S. C. Pettit, J. Simsic, D. D. Loeb, L. Everitt, C. A. Hutchison 111, and R. Swanstrom, J. Bid.
143. T. Kokubu, E. Ueda, S. Fujimoto, K. Hiwada, and A. Kato, Nature 217, 456 (1968). 144. J. Burton, R. J. Cody, J. A. Herd, and E. Haber, Proc. Natl. Acad. Sci. USA 77, 5476 (1980). 145. R. M. Zusman, J. Burton, D. Christensen, J. Nussberger, A. Dodds, and E. Haber, Trans.
M. C. Graves, Proc. Natl. Acad. Sci USA 87, 6336 (1990).
B. M. Dunn, and J. Kay, FEBS Lett. 268, 35 (1990).
Chem. 266, 14 539 (1991).
Am. Assoc. Physicians 96, 365 (1983).

776 ABDEL-MEGUID
146. G. Evin, J. Devin, B. Castro, J. Menard, and P. Corvol, Proc. Natl. Acad. Sci. U S A 81, 48
147. F. Cumin, G. Evin, J. A. Fehrentz, R. Seyer, B. Castro, J. Menard, and P. Corvol, 1. Biol.
148. F. Gross, J. Lazar, and H. Orth, Science 175, 656 (1972). 149. R. P. Miller, C. P. Poper, C. W. Wilson, and E. DeVito, Biochem. Pharmacol. 21, 2941 (1972). 150. J. C. Powers, A. D. Harley, and D. V. Myers, Adv. Exp. Med. Bid . 95, 141 (1977). 151. J. Boger, N. S. Lohr, E. H. Ulm, M. Poe, E. H. Blaine, G. M. Fanelli, T. Y. Lin, L. S. Payne,
T. W. Schorn, 8 . I. LaMont, T. C. Vassil, I. I. Stabilito, D. F. Veber, D. H. Rich, and A. S. Bopari, Nature 303, 81 (1983).
152. J. J. Marciniszyn, J. A. Hartsuck, and J. Tang, J. Bid. Chem. 251, 7088 (1976). 153. G. R. Marshall, Fed. Proc. 35, 2494 (1976). 154. J. J. Marciniszyn, J. A. Hartsuck, and J. Tang, Adv. Exp. Med. Biol. 95, 199 (1977). 155. D. H. Rich, M. S. Bernatowicz, N. S. Agarwal, M. Kawai, F. G. Salituro, and P. G. Schmidt,
Biochemistry 24, 3165 (1985). 156. A. G. Tomasselli, M. K. Olson, J. 0. Hui, D. J. Stapples, T. K. Saweyr, R. L. Heinrikson, and
C. M. Tomich, Biochemistry 29, 264 (1990). 157. K. Y. Hui, J. V. Manetta, T. Gygi, J. B. Bowdon, K. A. Keith, W. M. Shannon, and
M.-H. T. Lai, FASEB J. 5, 2606 (1991). 158. M. Szelke, D. Hudson, R. Sharpe, I. MacIntyre, G. Fink, and A. M. C . Pickering, in
Molecular Endocrinology, I. MacIntyre and M. Szelke, Eds., North-Holland, Amsterdam, 1977, p. 57.
159. M. Szelke, B. Leckie, A. Hallett, D. M. Jones, J. Sueiras, B. Atrash, and A. F. Lever, Nature 299, 555 (1982).
160. M. Szelke, D. M. Jones, B. Atrash, A. Hallett, and B. J. Leckie, in Peptides, Structure and Function, Proceedings of the 8th American Peptide Symposium, V. J. Hruby and D. H. Rich, Eds., Pierce Chemical Co., Rockford, lL, 1983, p. 579.
161. M. Szelke, in Aspartic Proteinases and Their Inhibtors, V. Kostka, Ed., de Gruyter, New York, 1985, p. 421.
162. M. Szelke, M. Tree, 8 . J. Leckie, D. M. Jones, B. Atrash, S. Beattie, B. Donovan, A. Hallett, M. Hughes, A. F. Lever, et al., 1. Hypertens. 3, 13 (1985).
163. M. J. Parry, A. B. Russell, M. Szelke, in Chemistry and Biology of Peptides, J. Meienhofer, Ed., Ann Arbor Science, Ann Arbor, MI, 1972, p. 541.
164. B. Leckie, M. Szelke, A. Hallett, M. Hughes, A. F. Lever, G. McIntyre, J. J. Morton, and M. Tree, Clin. Exp. Hypertens. A 5, 1221 (1983).
165. B. J. Leckie, M. Szelke, B. Atrash, S. R. Beattie, A. Hallett, D. M. Jones, G. D. McIntyre, J. Suerias, and D. J. Webb, Biochem. Soc. Trans. 13, 1029 (1985).
166. S. Thaisrivongs, D. T. Pals, L. T. Kroll, S. R. Turner and F. S. Han, 1. Med. Chem. 30, 976 (1987).
167. P. Ashorn, T. J. McQuade, S. Thaisrivongs, A. G. Tomasselli, W. G. Tarpley and B. Moss, Proc. Natl. Acad. Sci. U S A 87, 7472 (1990).
168. S. Thaisrivongs, A. G. Tomasselli, J. B. Moon, J. Hui, T. J. McQuade, S. R. Turner, J. W. Strohbach, W. J. Howe, W. G. Tarpley and R. L. Heinrikson, 1. Med. Chem. 34, 2344 (1991).
169. D. E. Ryono, C. A. Free, R. Neubeck, S. C. Samaniego, J. D. Godfrey, and E. W. Petrillo, Jr., in Peptides, Structure and Function. Proceedings of the Ninth American Peptide Symposium, C. M. Deber, V. J. Hruby, and K. D. Kopple, Eds., Pierce Chemical Co, Rockford, IL, 1985, p. 739.
170. J. G. Dann, D. K. Stammers, C. J. Harris, R. J. Arrowsmith, D. E. Davies, G. W. Hardyand J. A. Morton, Biochem. Biophys. Res. Commun. 134, 71 (1986).
171. J. R. Luly, N. Yi, J. Soderquist, H. Stein, J. Cohen, T. J. Perunand J. J. Plattner, 1. Med. Chem. 30, 1609 (1987).
172. S. Natarajan, C. A. Free, E. F. Sabo, L. Lin, E. R. Spitzmiller, S. G. Samaniego, S. A. Smith, and L. M. Zanoni, in Peptides, Chemistry and Biology. Proceedings of the Tenth American Peptide Symposium, G. R. Marshall, Ed., ESCOM, Leiden, 1988, p. 131.
173. J. B. Cooper, S. I. Foundling, T. Blundell, R. J. Arrowsmith, C. J. Harris, and J. N. Chapness, in Topics in Medicinal Chemistry, Fourth SCI-RSC Medicinal Chemistry Symposium, P. R. Leening, Ed., Special Publication No. 65, The Royal Society of Chemistry, London, 1988, p. 309.
(1984).
Chem. 260, 9154 (1985).

INHIBITORS OF ASPARTYL PROTEINASES 777
174. G. Breipohl, R. Geiger, S. Henke, H.-W. Kleeman, J. Knolle, D. Ruppert, B. A. Scholkens, H. Urbach, A. Wagner, and H. Wegmann, in Topics in Medicinal Chemistq, Fourth SCI-RSC Medicinal Chemistry Symposium, P. R. Leening, Ed., Special Publication No. 65, The Royal Society of Chemistry, London, 1988, p. 101.
175. D. H. Rich, J. Green, M. V. Toth, G. R. Marshall, and S. B. Kent, J. Med. Chem. 33, 1285 (1990).
176. D. H. Rich, C.-Q. Sun, V. J. V. N. Prasad, A. Pathiasseril, M. V. Toh, G. R. Marshall, M. Clare, R. A. Mueller, and K. Houseman, J. Med. Chem. 34, 1222 (1991).
177. D. H. Rich, M. S. Bernatowicz, and P. G. Schmidt, J. Am. Chem. SOC. 104,3535 (1982). 178. M. W. Holladay, F. G. Salituro, P. G. Schmidt, and D. H. Rich, Biochem. SOC. Trans. 13, 1046
179. R. E. Babine, N. Zhang, A. R. Jurgens, S. R. S-Chow, P. R. Desai, J. C. James, and M. F.
180. M. H. Gelb, J. P. Svaren, and R. H. Abeles, Biochemistry 24, 1813 (1985). 181. S. Thaisrivongs, D. T. Pals, W. M. Kati, S. R. Turner, and L. M. Thomasco, J. Med. Chem. 28,
182. S . Thaisrivongs, D. T. Pals, W. M. Kati, S . R. Turner, L. M. Thomasco, and W. Watt, J. Med.
183. K. Fearon, A. Spaltenstein, P. B. Hopkins, and M. H. Gelb, 1. Med. Chem. 30, 1617 (1987). 184. H. L. Sham, D. A. Betebenner, N. E. Wideburg, A. C. Saldivar, W. E. Kohlbrenner,
S. Vasavanonda, D. J. Kempf, D. W. Norbeck, C. Zhao, J. J. Clement, J. E. Erickson, and J. J. Plattner, Biochem. Biophys. Res. Commun. 175, 914 (1991).
185. D. T. Pals, D. Thaisrivongs, J. A. Lawson, W. M. Kati, S. R. Turner, G. L. DeGraaf, D. W. Harris, and G. A. Johnson, Hypertension 8, 1105 (1986).
186. G. J. Hanson, J. S. Baran, T. Lindberg, G. M. Walsh, S. E. Papaioannou, M. Babler, S. E. Bittner, P. C. Yang, and C. M. Dal, Biochem. Biophys. Res. Commun. 132, 155 (1985).
187. G. J. Hanson, J. S . Baran, H. S . Lowrie, S . J. Sarussi, P. C. Yang, M. Babler, S . E. Bittner, S. E. Papaioannou, and G. M. Walsh, Biochem. Biophys. Res. Commun. 146, 959 (1987).
188. G. J. Hanson, J. S. Baran, H. S. Lowrie, M. A. Russell, S. J. Sarussi, K. Williams, M. Babler, S. E. Bittner, S. E. Papaioannou, P. C. Yang, et al., Biochem. Biophys. Res. Commun. 160,l (1989).
189. R. Matsueda, Y. Yabe, H. Kogen, S. Higashida, H. Koike, Y. Iijima, T. Kokubu, K. Hiwada, E. Murakami, and Y. Imamura, Chem. Lett. 1041 (1985).
190. H. D. Kleinert, S . H. Rosenberg, W. R. Baker, H. H. Stein, V. Klinghofer, J. Barlow, K. Spina, J. Polakowski, P. Kovar, J. Cohen, and J. Denissen, Science 257, 1940 (1992).
191. C. R. Nakaie, M. C. F. Oliveira, L. Juliano, and A. C. Paiva, Biochem. 1. 205, 43 (1982). 192. C. R. Nakaie, J. L. Pesquero, M. C. F. Oliveira, L. Juliano, and A. C. Paiva, in Peptides,
Structure and Function. Proceedings of the Ninth American Peptide Symposium, C. M. Deber, V. J. Hruby, and K. D. Kopple Eds., Pierce Chemical Co, Rockford, IL, 1985, p. 755.
193. H. L. Sham, G. Bolis, H. H. Stein, S. W. Fesik, P. A. Marcotte, J. J. Plattner, C. A. Rempel, and J. Greer, J. Med. Chem. 31, 284 (1988).
194. A. E. Weber, T. A. Halgren, J. J. Doyle, R. J. Lynch, P. K. Siegl, W. H. Parsons, W. J. Greenlee, and A. A. Patchett, J. Med. Chem. 34,2692 (1991).
195. A. E. Weber, M. G. Steiner, L. Yang, D. S. Dhanoa, J. R. Tata, T. A. Halgren, P. K. S. Siegl, W. H. Parsons, W. J. Greenlee, and A. A. Patchett, in Peptides, Chemistry and Biology. Proceed- ings of the Twelfth American Peptide Symposium, J. A. Smith, and J. E. Rivier, Eds., ESCOM, Leiden, 1991, p. 794.
(1985).
Semmelhack, Bioorg. Med. Chem. Lett. 2, 541 (1992).
1553 (1985).
Chem. 29, 2080 (1986).
196. S. S. Abdel-Meguid (unpublished). 197. D. J. Kempf, D. W. Norbeck, L. M. Codacovi, X. C. Wang, W. E. Kohlbrenner,
N. E. Wideburg, D. A. Paul, M. F. Knigge, S. Vasavanonda, A. Craig-Kennard, A. Saldivar, W. Rosenbrook, Jr., J. J. Clement, J. J. Plattner, and J. Erickson, J. Med. Chem. 33,2687 (1990).
198. 8 . Chenera, J. C. Boehem, and G. B. Dreyer, Bioorg. Med. Chem. Lett. 1, 219 (1991). 199. P. A. Bartlett and W. B. Kezer, J. Am. Chem. SOC. 106, 4282 (1984). 200. M. C. Allen, W. Fuhrer, 8. Tuck, R. Wade, and J. M. Wood, J. Med. Chem. 32, 1652
201. P. A. Bartlett, J. E. Hanson, and P. P. Giannousis, J. Org. Chem. 55, 6268 (1990). 202. D. Grobelny, E. M. Wondrak, R. E. Galardy, and S. Oroszlan, Biochem. Biophys. Res. Com-
203. R. L. DesJarlais, G. L. Seibel, I. D. Kuntz, P. S. Furth, J. C. Alvarez, P. R. Ortiz de Mon-
(1989).
mun. 169, 1111 (1990).

778 ABDEL-MEGUID
tellano, D. L. Decamp, L. M. Babe, and C. S. Craik, Proc. Natl. Acad. Sci. USA 87, 6644 (1990).
204. R. Pal, R. C. Gallo, and M. G. Sarngadharan, Proc. Natl. Acad. Sci. USA 85, 9283 (1988). 205. J. J. Blumenstein, T. D. Copeland, S. Oroszlan, and C. J. Michejda, Biochem. Biophys. Res.
206. K. Moelling. T. Schulze, M. T. Knoop, J. Kay, R. Jupp, G. Nicolaou, and L. H. Pearl, FEBS
207. J. Tang, J. Biol. Chem. 246, 4510 (1971). 208. J. A. Hartsuck and J. Tang, J . Bid. Chem. 247, 2575 (1972). 209. K. C. S. Chen and J. Tang, J . Biol. Chem. 247, 2566 (1972). 210. M. N. James, I. N. Hsu, and L. T. Delbaere, Nature 267, 808 (1977). 211. M. N. G. James, A. R. Sielecki, and J. Moult, in Peptides, Structure and Function, Proceedings of
the 8th American Peptide Symposium, V. J. Hruby and D. H. Rich, Eds., Pierce Chemical Co, Rockford, IL, 1983, p. 521.
212. L. W. Lim, R. A. Stegeman, N. K. Leimgruber, J. K. Gierse, and S. 5. Abdel-Meguid, J. Mol. Biol. 210, 239 (1989).
213. K. Y. Hui, W. D. Carlson, M. S. Bernatowicz, and E. Haber, J. Med Chem. 30, 1287 (1987). 214. D. W. Green, S. Aykent, J. K. Gierse, and M. E. Zupec, Biochemistry 29, 3126 (1990). 215. G. J. Hanson, J. S. Baran, M. Clare, K. Williams, M. Babler, S. E. Bittner, M. A. Russell, S. E.
Papaioannou, P.-C. Yang, and G. M. Walsh, in Peptides, Chemistry and Biology. Proceedings of the Eleventh American Peptide Symposium, J. E. Rivier and G. R. Marshall, Eds., ESCOM, Leiden, 1990, p. 396.
Commun. 163, 980 (1989).
Lett. 261, 373 (1990).
216. G. A. Jeffrey and H. Maluszynska, Int. J. Biol. Macromol. 4, 173 (1982). 217. A. Sali, B. Veerapandian, J. B. Cooper, D. S. Moss, T. Hofmann, and T. L. Blundell, Proteins
218. J. S. Fruton, Mol. Cell. Biol. 32, 105 (1980). 219. T. Hofmann, 8. Allen, M. Bendinger, M. Blum, A. Cuningham, Biochemistry 27,1140 (1988). 220. W. E. J. Harte, S. Swaminathan, M. M. Mansuri, J. C. Martin, I. E. Rosenberg, and
221. G. J. Hanson (personal communication). 222. D. A. Tewksbury, R. A. Dart, and J. Travis, Biochem. Biophys. Res. Commun. 99, 1311 (1981).
12, 158 (1992).
D. L. Beveridge, Proc. Natl. Acad. Sci. USA 87, 8864 (1990).
Related Documents











