Review Immune system targeting by biodegradable nanoparticles for cancer vaccines ☆ Joana M. Silva a, b , Mafalda Videira a , Rogério Gaspar a , Véronique Préat b , Helena F. Florindo a, ⁎ a iMed.UL, Research Institute for Medicines and Pharmaceutical Sciences, Faculty of Pharmacy, University of Lisbon, 1649-003 Lisbon, Portugal b Université Catholique de Louvain, Louvain Drug Research Institute, Pharmaceutics and Drug Delivery, Avenue Mounier, B1 73.12, 1200 Brussels, Belgium abstract article info Article history: Received 10 November 2012 Accepted 14 March 2013 Available online 21 March 2013 Keywords: Cancer vaccines Immunotherapy Nanoparticles Active targeting Adjuvants The concept of therapeutic cancer vaccines is based on the activation of the immune system against tumor cells after the presentation of tumor antigens. Nanoparticles (NPs) have shown great potential as delivery systems for cancer vaccines as they potentiate the co-delivery of tumor-associated antigens and adjuvants to dendritic cells (DCs), insuring effective activation of the immune system against tumor cells. In this review, the immunolog- ical mechanisms behind cancer vaccines, including the role of DCs in the stimulation of T lymphocytes and the use of Toll-like receptor (TLR) ligands as adjuvants will be discussed. An overview of each of the three essential com- ponents of a therapeutic cancer vaccine – antigen, adjuvant and delivery system – will be provided with special emphasis on the potential of particulate delivery systems for cancer vaccines, in particular those made of biode- gradable aliphatic polyesters, such as poly(lactic-co-glycolic acid) (PLGA) and poly-ε-caprolactone (PCL). Some of the factors that can influence NP uptake by DCs, including size, surface charge, surface functionalization and route of administration, will also be considered. © 2013 Elsevier B.V. All rights reserved. 1. Introduction It is well known that the immune system is able to mount responses against tumors and that this effect can be enhanced using a number of strategies [1]. The cancer immunotherapy approach uses the specificity of the immune system to provide a more efficacious and better tolerated therapy than more conventional treatments, such as chemotherapy and radiotherapy. Current approaches to cancer immunotherapy can be categorized as those that attempt to enhance the host's own immune response to a tumor, such as the development of prophylactic and therapeutic cancer vaccines, cytokine therapy, administration of immune activating antibodies and radioimmunotherapy, and those that use adoptive cell therapies from a non-tumor-bearing syngeneic or, more often, allogeneic donor [2]. Landmark advances have promoted interest in cancer vaccine development following the identification of defined T cell tumor antigens at the molecular level and the demonstration in mice and humans that T cells of well-defined specificity can mediate the complete elimination of large solid tumors. Evidence supports the general concept of immune surveillance in experimental mice models and in cancer patients [3]. Cancer vaccine development is focusing on different forms of cancer, but melanoma has been one of the main targets of research and, thus, most of the immunogenic antigens have been identified in melanoma [4]. In fact, there are strong rational arguments to expect good outcomes for therapeutic melanoma vaccines, including documented spontaneous remissions, lymphocytic infiltration of tumors, expression of developmental and melanoma-specific antigens on tumor tissue, and responses to biological agents, such as interleukin (IL)-2 and interferon alpha (IFN-α) [5–8]. Several vaccination strategies have been tested in vivo, including whole tumor cells, cell lysates, and proteins or specific peptide fragments, leading to the generation of tumor-specific T cell responses [9]. Nevertheless, the therapeutic benefit of these strategies remains unclear and further well-designed studies are needed to clarify the potential of each approach. In the last decades, convergence of many scientific disciplines explor- ing nanotechnology and nanomedicine has successfully driven the first generation of nanomedicines to the clinic [10]. A great number of prom- ising nano-sized tools for biomedical applications have been emerging. Among them, biodegradable nanoparticles (NPs) are gaining increased attention for their ability to serve as specialized biocompatible delivery systems of a multitude of molecules, including proteins, peptides, nucleic acids and oligonucleotides that constitute the main vaccine components. There are a considerable number of advantages in using biodegradable NPs as vaccine delivery systems that will be presented throughout the manuscript. One aspect that stands out is their unique capacity of targeting the immune system and co-deliver antigen and adjuvant simultaneously to the same cell, enabling the generation of an effective immune response [11]. Despite all the challenges in the development of cancer vaccines and the many mechanisms by which tumors can evade or subvert im- mune responses, immunotherapy is still an attractive strategy for Journal of Controlled Release 168 (2013) 179–199 ☆ Conflict of interest: The authors declare no conflicts of interest. ⁎ Corresponding author at: Nanomedicines & Drug Delivery Systems Group, iMed.UL, Faculty of Pharmacy, University of Lisbon, Av. Prof. Gama Pinto, Edf. E, 1649-003 Lisboa, Portugal. Tel.: +351 217946400x14245. E-mail address: hfl[email protected] (H.F. Florindo). 0168-3659/$ – see front matter © 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.jconrel.2013.03.010 Contents lists available at SciVerse ScienceDirect Journal of Controlled Release journal homepage: www.elsevier.com/locate/jconrel

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Journal of Controlled Release 168 (2013) 179–199
Contents lists available at SciVerse ScienceDirect
Journal of Controlled Release
j ourna l homepage: www.e lsev ie r .com/ locate / jconre l
Review
Immune system targeting by biodegradable nanoparticles forcancer vaccines☆
Joana M. Silva a,b, Mafalda Videira a, Rogério Gaspar a, Véronique Préat b, Helena F. Florindo a,⁎a iMed.UL, Research Institute for Medicines and Pharmaceutical Sciences, Faculty of Pharmacy, University of Lisbon, 1649-003 Lisbon, Portugalb Université Catholique de Louvain, Louvain Drug Research Institute, Pharmaceutics and Drug Delivery, Avenue Mounier, B1 73.12, 1200 Brussels, Belgium
☆ Conflict of interest: The authors declare no conflicts⁎ Corresponding author at: Nanomedicines & Drug De
Faculty of Pharmacy, University of Lisbon, Av. Prof. GamaPortugal. Tel.: +351 217946400x14245.
E-mail address: [email protected] (H.F. Florindo).
0168-3659/$ – see front matter © 2013 Elsevier B.V. Allhttp://dx.doi.org/10.1016/j.jconrel.2013.03.010
a b s t r a c t
a r t i c l e i n f oArticle history:Received 10 November 2012Accepted 14 March 2013Available online 21 March 2013
Keywords:Cancer vaccinesImmunotherapyNanoparticlesActive targetingAdjuvants
The concept of therapeutic cancer vaccines is based on the activation of the immune system against tumor cellsafter the presentation of tumor antigens. Nanoparticles (NPs) have shown great potential as delivery systemsfor cancer vaccines as they potentiate the co-delivery of tumor-associated antigens and adjuvants to dendriticcells (DCs), insuring effective activation of the immune system against tumor cells. In this review, the immunolog-ical mechanisms behind cancer vaccines, including the role of DCs in the stimulation of T lymphocytes and the useof Toll-like receptor (TLR) ligands as adjuvants will be discussed. An overview of each of the three essential com-ponents of a therapeutic cancer vaccine – antigen, adjuvant and delivery system – will be provided with specialemphasis on the potential of particulate delivery systems for cancer vaccines, in particular those made of biode-gradable aliphatic polyesters, such as poly(lactic-co-glycolic acid) (PLGA) and poly-ε-caprolactone (PCL). Someof the factors that can influence NP uptake by DCs, including size, surface charge, surface functionalization androute of administration, will also be considered.
© 2013 Elsevier B.V. All rights reserved.
1. Introduction
It is well known that the immune system is able tomount responsesagainst tumors and that this effect can be enhanced using a number ofstrategies [1]. The cancer immunotherapy approach uses the specificityof the immune system to provide amore efficacious and better toleratedtherapy than more conventional treatments, such as chemotherapy andradiotherapy. Current approaches to cancer immunotherapy can becategorized as those that attempt to enhance the host's own immuneresponse to a tumor, such as the development of prophylactic andtherapeutic cancer vaccines, cytokine therapy, administration of immuneactivating antibodies and radioimmunotherapy, and those that useadoptive cell therapies from a non-tumor-bearing syngeneic or, moreoften, allogeneic donor [2]. Landmark advances have promoted interestin cancer vaccine development following the identification of defined Tcell tumor antigens at the molecular level and the demonstration inmice and humans that T cells of well-defined specificity can mediatethe complete elimination of large solid tumors. Evidence supports thegeneral concept of immune surveillance in experimental mice modelsand in cancer patients [3]. Cancer vaccine development is focusing ondifferent forms of cancer, butmelanomahas been one of themain targetsof research and, thus, most of the immunogenic antigens have been
of interest.livery Systems Group, iMed.UL,Pinto, Edf. E, 1649-003 Lisboa,
rights reserved.
identified in melanoma [4]. In fact, there are strong rational argumentsto expect good outcomes for therapeutic melanoma vaccines, includingdocumented spontaneous remissions, lymphocytic infiltration of tumors,expression of developmental and melanoma-specific antigens on tumortissue, and responses to biological agents, such as interleukin (IL)-2 andinterferon alpha (IFN-α) [5–8]. Several vaccination strategies have beentested in vivo, including whole tumor cells, cell lysates, and proteins orspecific peptide fragments, leading to the generation of tumor-specificT cell responses [9]. Nevertheless, the therapeutic benefit of thesestrategies remains unclear and further well-designed studies are neededto clarify the potential of each approach.
In the last decades, convergence ofmany scientific disciplines explor-ing nanotechnology and nanomedicine has successfully driven the firstgeneration of nanomedicines to the clinic [10]. A great number of prom-ising nano-sized tools for biomedical applications have been emerging.Among them, biodegradable nanoparticles (NPs) are gaining increasedattention for their ability to serve as specialized biocompatible deliverysystems of amultitude ofmolecules, including proteins, peptides, nucleicacids and oligonucleotides that constitute themain vaccine components.There are a considerable number of advantages in using biodegradableNPs as vaccine delivery systems that will be presented throughoutthe manuscript. One aspect that stands out is their unique capacity oftargeting the immune system and co-deliver antigen and adjuvantsimultaneously to the same cell, enabling the generation of an effectiveimmune response [11].
Despite all the challenges in the development of cancer vaccinesand the many mechanisms by which tumors can evade or subvert im-mune responses, immunotherapy is still an attractive strategy for

180 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
cancer therapy and cancer vaccines are one of the most ambitious butpromising tools in this field. In contrast to vaccines that induce pro-tective immunity against viral pathogens associated with the inductionof cancer, such as high risk serotypes of the human papillomavirus(HPV) and the hepatitis B virus (HBV), it is envisioned that general cancervaccines would be administered after the onset or detection of disease.Thus, they would be considered therapeutic rather than prophylacticvaccines. Another critical aspect of cancer vaccines is that they are moreeffective in the adjuvant setting for the treatment of minimal residualdisease than in the treatment of larger bulky primary tumors. Humanleukocyte antigen (HLA) expression can also influence the efficacy ofcancer vaccines, which must, therefore, have sufficient antigen complex-ity to provide broad immunological coverage of the treated patient popu-lation [12]. Most important, cancer vaccines must be constructed takinginto account the desired immune response that will effectively enhancepatient defenses against the tumor and also abrogate the mechanismsof tumor evasion and tolerance.
In this review, the immunological mechanisms behind the develop-ment of effective anti-tumor immune responses will be used as a startingpoint to understand the potential of biodegradable polymeric NPs ascancer vaccine delivery systems. An overview of each of the three essen-tial components of a therapeutic cancer vaccine – antigen, adjuvant anddelivery system – will be provided with special emphasis on the crucialrole of NPs and Toll-like receptor (TLR) ligands in subverting tumor im-munosuppression. Some of the factors that can influence NP uptake bydendritic cells (DCs), such as size, surface charge, surface functionalizationand route of administration, will also be discussed. Throughout thisreview, many examples of the use of NPs as vaccine delivery systems, inparticular those made of biodegradable aliphatic polyesters, such aspoly(lactic-co-glycolic acid) (PLGA) and poly-ε-caprolactone (PCL), willbe presented to support their potential as cancer vaccine deliverysystems.
2. Desired immune response for a cancer vaccine
2.1. Cancer vaccine principles and hurdles
Vaccines have had a significant impact in global health as they per-mitted the control of many infectious diseases and the eradication ofsmallpox [13,14]. After the observation by Edward Jenner in 1796 thatmilkmaids were generally immune to smallpox, he tested a primordialway of vaccination by installing subcutaneously material from milk-maids' lesions containing cowpox in a healthy individual and confirmedprotection against smallpox [15]. Later, Pasteur developed techniques forinactivation of microorganisms and demonstrated for the first time thatthe rationale behind smallpox vaccination could be extended to severalother infectious diseases [16]. Since then, remarkable progresses havebeen made and many tools are now available for the production ofsuccessful vaccines. The idea of employing vaccines to treat cancerarose one century later with the work of Paul Ehrlich and WilliamColey. Ehrlich attempted to generate immunity against cancer byinjecting weakened tumor cells, with no success [17]. Coley developeda mixture of heat-killed Streptococcus pyogenes and Serratia marcescensbacteria (the Coley toxin), and tested it in cancer patients obtainingsuccessful results due to the adjuvant effect of the toxin in the immuneresponse [18]. However, it soon became clear that anticancer vaccineswould have to be therapeutic rather than prophylactic and pour induc-tion of immune responses by tumor cells had to be circumvented [19].
Three critical elements are considered to be essential in the composi-tion of an effective vaccine: antigen, adjuvant, and delivery system [20].Antigens aremolecules (proteins, peptides, lipids, etc.) recognized by theimmune system that are able to induce an adaptive immune response.An adjuvant is a factor that is able to stimulate the innate immunesystem and influence the profile of the elicited immune response whilea delivery system is defined as a platform that insures optimal deliveryof both antigen and adjuvant for the effective activation of both innate
and adaptive immune systems. It has been demonstrated that antigensand adjuvants must act concomitantly on the same antigen-presentingcell (APC) to obtain its full activation [11]. Indeed, the administration ofan isolated antigen can induce a Th2-dominated immune response oreven tolerance to the antigen. As a result, the efficacy of a cancer vaccineformulation can be considerably improved by the development ofdelivery systems able to protect and deliver antigens and adjuvantsconcomitantly [21].
The selection of the appropriate tumor antigens is one of the mainhurdles of cancer vaccine development. In a general way, the primetargets for anti-cancer vaccines must fulfill the following criteria: (i) in-clude peptide sequences that bind to major histocompatibility complex(MHC) class I, (ii) be processed by tumor cells and become availablefor binding toMHC Imolecules, (iii) be recognised by the T cell repertoirein an MHC I-restricted fashion, and (iv) drive the expansion of cytotoxicT lymphocyte (CTL) precursors expressing specific T cell receptors [22].Since the molecular cloning of the first gene reported to encode aCTL-defined human tumor antigen (MAGE-A1), and with the develop-ment of new technologies, many other antigens recognized by T cellson human cancers, the so-called tumor associated antigens (TAA), havebeen identified and characterized [23,24]. This process has clearlybeneficiated from the integration of molecular cloning by screeningtumor-derived cDNA librarieswith autologous tumor-specific T lympho-cyteswith novel strategies such as (i) reverse immunology (epitope pre-diction on the basis of known HLA-binding motifs performed bydedicated software); (ii) biochemical methods associated with chroma-tography and mass spectrometry to fractionate TAA peptides naturallyexpressed on tumor cells in the context of HLA molecules, and (iii)DNA microarray technologies which allow comparison of gene expres-sion profiles in normal and tumor tissues, such as representational differ-ence analysis (RDA), differential display (DD), suppression subtractivehybridization (SSH), and serial analysis of gene expression (SAGE) [24].Apart from the epitopes recognized by CD8+ or CD4+ T cells, othertypes of tumor antigens have been identified or created. These includeanalogs or artificiallymodified epitopes, antigens identified by antibodies(mimotopes), immunogenic fusion proteins (new T-cell epitopes resul-tant from translocation of chromosomes during carcinogenesis resultingin fusion of distant genes), splicing aberrations, point mutations, fusionjunctions in epitopes and antigens detected by the SEREX technology[24–26].
TAA can be broadly classified in two categories: (i) shared tumor anti-gens, and (ii) unique tumor antigens. The first group is expressed bymany tumor types and their expression on normal tissues is either absentor quantitatively and qualitatively different. Examples are the cancer-testis antigens (MAGE, GAGE andNY-ESO1). The second group comprisesantigens that are expressed uniquely by individual tumors and corre-spond to new epitopes that are products of point mutations or splicingalterations [27,28]. An additional group of TAA includes those thatmight be expressed by cancer stem cells (CSC), a minor population oftumor cells which shows the features of stemness and, therefore, mayrepresent an important target for most tumors [29]. TAA can also bedefined as dispensable and indispensable. Indispensable TAA, also calleduniversal TAA, have the remarkable feature of being necessary fortumor growth andprogression. These antigens are overexpressed by neo-plastic and fetal cells, but weakly expressed in a phase-specific way in afew normal cells. Normally they have limited generation of antigen-lossvariants and, therefore, they are attractive targets for cancer vaccines[22]. Examples are the inhibitors of apoptosis proteins (IAP) and thehuman telomerase reverse transcriptase (hTERT) [30,31]. Studies onuniversal TAA demonstrate that such antigens are, more than any otherTAA, frequently expressed in many different human tumors and, there-fore, they apparently provide the most common, neoplasia-related andeasily identifiable immune target. Besides this apparent solution for theselection of antigens for cancer vaccines, universal TAA have severaldrawbacks that obviate this hypothesis. For instance, it remains to beestablished whether they are more immunogenic when compared with

Table 1Pros and cons of universal antigen-based and personalized cancer vaccines.
Universal antigen-based cancer vaccines
Pros Cons
• Frequently expressed in many differenthuman tumors;
• Can be easily identified;• Since they are indispensable for tumorgrowth and progression, the generationof antigen-loss variations is limited oreven absent and the tumor growth con-trol is more efficient.
• Antigen processing-related problems;• Antigen-specific T cells have low fre-quencies;
• Risk of low immunogenicity;• In some cases, these molecules might bemore easily targeted by antibodies ratherthan by antigen-specific T cells as someantigens are normal molecules involvedin migration and cell adhesion;
• Slow and weak primary CTL responses.
Personalized cancer vaccines
Pros Cons
• Generally highly immunogenic antigens;• Memory CTL is able to start rapid andstrong secondary responses;
• Reduced chances of inducing the genera-tion of regulatory T cells (Treg).
• Their production is time consumingand expensive;• The primary response is absent;• The selection of high CTL precursorfrequencies does not ensure tumorregression;• Risk of antigen presentation failure.
181J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
other TAA obtained from the same tumor [32]. Also, the frequency ofpeptide-specific CTL precursors in peripheral blood mononuclear cells(PBMC), which is determinant for the generation of the final number ofeffector T cells, can be very low and not overcome tumor escape fromimmune surveillance [33].
An alternative strategy to overcome the inherent problems of usinguniversal antigens is the application of personalized vaccination. Thisapproach takes into account the immunological diversity of individualpatients by measuring CTL precursors in PBMC prior to vaccination toselect the antigenswith higher CTL precursor frequency [34,35]. Uniquetumor antigens constitute a growing class of T cell-defined epitopes thatexhibit strong immunogenicity. Some of these antigens, which oftenderive from mutation of genes that have relevant biological functions,are less susceptible to immunoselection and may be retained even inadvanced tumors. Immunogenicity and constitutive expression ofunique tumor antigens provide a strong rationale for the design ofnovel patient-tailored therapies that target such determinants [36,37].Pros and cons of both strategies of cancer vaccination are listed inTable 1. The next step in the development of vaccines is to identifybiomarkers of immune response that will help not only in the identifi-cation of vaccine responders but also in new vaccine development. Itis not certain if vaccinologywill lead us to abandon the universal empir-ic vaccine approach but is likely that the future of vaccinology passes byto predict vaccine response, adverse reactions to a vaccine, number ofdoses needed to induce an effective response to a vaccine, and directus toward a science-based directed design/develop paradigm fornovel vaccine candidates [38]. Apart from the problems related to dif-ferent vaccine modalities, cancer vaccines have additional barriersdeeply related to tumor immunology that need to be overtaken. Clinicaltrials of cancer vaccines demonstrated very low overall objectiveresponse rates [39]. Table 2 presents some of the barriers that can con-tribute to explain these disappointing results. These can be either tumorescape mechanisms and/or related to T-cell constraints [40,41]. Suchprocesses can compromise the efficacy of any vaccine targeting sharedor unique tumor antigens. Another set of hurdles in the developmentof therapeutic vaccines as a treatment modality for cancer includesdifficulties in the selection of the optimal dose and schedule and thevalidation ofmethods of evaluation of the efficacy of the vaccine. Obsta-cles in the patentability of many immunotherapeutic strategies, whichhave resulted in a lack of interest and a subsequent lack of funding bythe pharmaceutical industry, have also hampered clinical development[42].
2.2. Cancer vaccines must activate CTL and Th1 cells
Most TAA are not cell surface proteins but rather intracellular proteins.Peptide fragments of these antigens are bound by newly synthesizedMHC class I molecules and shuttled to the cell surface for immunologicalrecognition. Thus, in order to be effective, it is critical that cell-mediatedimmunity, particularly CTLs, is the focus of the immune response elicitedby a cancer vaccine [12]. In the presence of the appropriate stimulation,CD8+ T cells undergo proliferation and differentiate into CTLs [64]. Acti-vated CTLs can migrate to peripheral tissues and exert two main effectorfunctions: direct contact-mediated cytotoxicity and secretion of effectorcytokines, such as IFN-γ and TNF-α, which can mediate local inflamma-tion. Another essential function of activated CD8+T cells is the acquisitionof memory. Memory CD8+ T cells can be maintained for long periods oftime without antigenic stimulation and potentiate a better and faster im-mune response of the host upon relapse or metastasis [65].
In recent years, it has become evident that CD4+ T cells also play acritical role in the development of effective anti-tumor immunity.Indeed, CD8+ lymphocytes can fail to maintain functionality in vivo inthe absence of CD4+ T helper cells. These cells can enhance anti-tumorCTL responses by increasing clonal expansion at the tumor site, promot-ing the generation and maintenance of memory CTL phenotypes,preventing activation-induced cell death (AICD) and functioning as
APCs for CTLs [54]. Classically, effector CD4+ cells are considered to dif-ferentiate into T-helper 1 (Th1) and T-helper 2 (Th2) cells. AlthoughTh1 and Th2 subtypes can both mediate anticancer functions,IFN-γ-secreting Th1 cells are more effective in this role [66]. Becker(2006) reviewed the relationship of the Th1/Th2 imbalance with tumorpresence and prognosis, highlighting that tumor cells induce increasedTh2 cytokine serum levels, namely IL-4, IL-6 and IL-10, which suppressthe production of Th1 cytokines, such as IL-2, IL-12 and IFN-γ, resultingin prevention of activation of CD8+ cytotoxic T cell precursors [67]. Act-ing together in the process of tumor eradication, the combined action ofCD8+ and IFN-γ-secreting Th1 CD4+ T cells starts with activation oftumor-specific CD8+ T cells that can kill tumor cells directly. The survivaland persistence of CD8+ T cells as memory cells are also regulated bytumor-specific CD4+ T cells. Both CD8+ and, especially, Th1 CD4+ Tcells, secrete IFN-γ, which can further sensitize tumor cells to CD8+ Tcells by upregulating MHC class I and other components of theantigen-processing machinery, promoting the recruitment of cells fromthe innate immune system such as natural killer (NK) cells, granulocytesormacrophages, and interferingwith crucial functions of the tumor stro-ma, namely, angiogenesis [68]. A schematic representation of the desiredimmune response that a cancer vaccine must induce is shown in Fig. 1.
2.3. Reaching CTLs and Th1 cells through DCs — the bridge betweenadaptive and innate immune systems
Naïve CD8+ T cells, located in secondary lymphoid organs, constantlysurvey and sample APCs searching for cognate peptide-MHC moleculesfor which the T cell receptor (TCR) has specific affinity. APCs, DCs byexcellence, but alsomacrophages and B cells, are known for their special-ized capacity for antigen presentation to T cells leading to activation ofinnate and adaptive immune responses or tolerance to innocuousantigens [69]. Among the APCs, DCs are considered as the main APCpopulation because of several intrinsic characteristics that are not pres-ent onmacrophages or B cells. DCs take advantage of their strategic loca-tion in peripheral tissues, such as skin and mucosal surfaces, the mostfrequent routes of pathogen entry [70]. Moreover, after internalizationof antigens, DCs are able to migrate toward T cell-rich areas, such assentinel lymph nodes (LNs), where they can activate naïve T cells in anantigen-specific manner, placing a bridge between innate and adaptiveimmune systems. This activation comprises the presentation of antigen-MHC complexes, and the upregulation of costimulatory molecules andsoluble molecules, such as cytokines and chemokines, e.g., IL-12 and
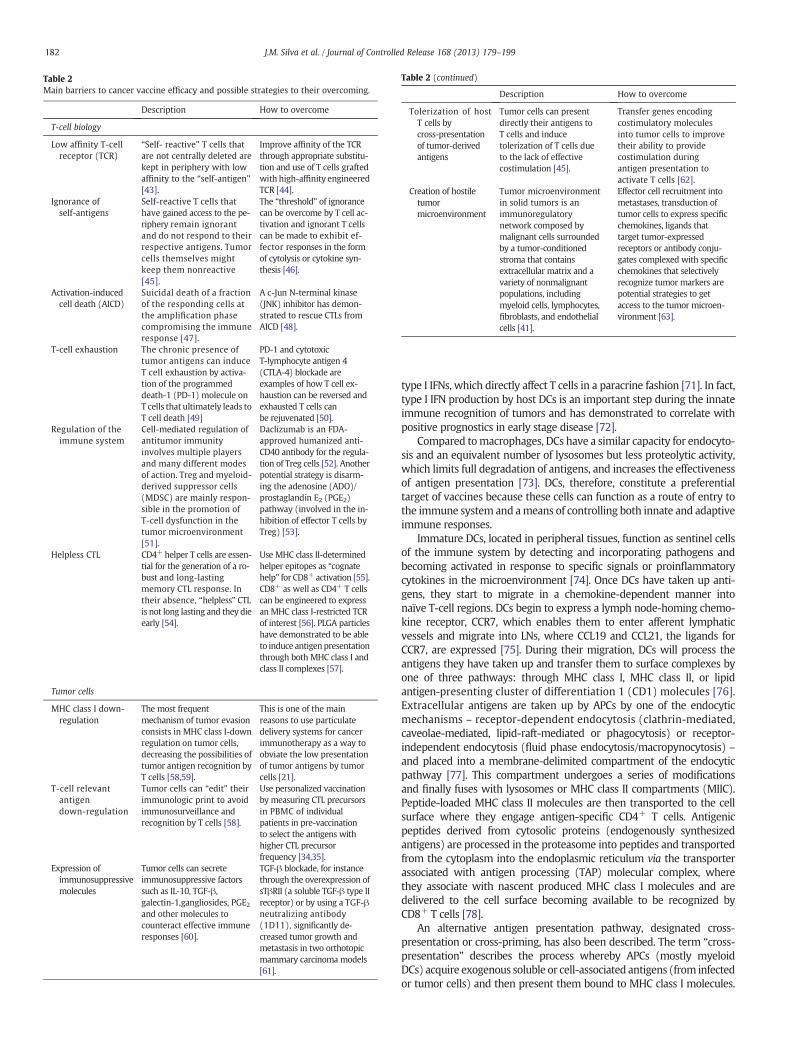
Table 2Main barriers to cancer vaccine efficacy and possible strategies to their overcoming.
Description How to overcome
T-cell biology
Low affinity T-cellreceptor (TCR)
“Self- reactive” T cells thatare not centrally deleted arekept in periphery with lowaffinity to the “self-antigen”[43].
Improve affinity of the TCRthrough appropriate substitu-tion and use of T cells graftedwith high-affinity engineeredTCR [44].
Ignorance ofself-antigens
Self-reactive T cells thathave gained access to the pe-riphery remain ignorantand do not respond to theirrespective antigens. Tumorcells themselves mightkeep them nonreactive[45].
The “threshold” of ignorancecan be overcome by T cell ac-tivation and ignorant T cellscan be made to exhibit ef-fector responses in the formof cytolysis or cytokine syn-thesis [46].
Activation-inducedcell death (AICD)
Suicidal death of a fractionof the responding cells atthe amplification phasecompromising the immuneresponse [47].
A c-Jun N-terminal kinase(JNK) inhibitor has demon-strated to rescue CTLs fromAICD [48].
T-cell exhaustion The chronic presence oftumor antigens can induceT cell exhaustion by activa-tion of the programmeddeath-1 (PD-1) molecule onT cells that ultimately leads toT cell death [49]
PD-1 and cytotoxicT-lymphocyte antigen 4(CTLA-4) blockade areexamples of how T cell ex-haustion can be reversed andexhausted T cells canbe rejuvenated [50].
Regulation of theimmune system
Cell-mediated regulation ofantitumor immunityinvolves multiple playersand many different modesof action. Treg and myeloid-derived suppressor cells(MDSC) are mainly respon-sible in the promotion ofT-cell dysfunction in thetumor microenvironment[51].
Daclizumab is an FDA-approved humanized anti-CD40 antibody for the regula-tion of Treg cells [52]. Anotherpotential strategy is disarm-ing the adenosine (ADO)/prostaglandin E2 (PGE2)pathway (involved in the in-hibition of effector T cells byTreg) [53].
Helpless CTL CD4+ helper T cells are essen-tial for the generation of a ro-bust and long-lastingmemory CTL response. Intheir absence, “helpless” CTLis not long lasting and they dieearly [54].
Use MHC class II-determinedhelper epitopes as “cognatehelp” for CD8+ activation [55].CD8+ as well as CD4+ T cellscan be engineered to expressan MHC class I-restricted TCRof interest [56]. PLGA particleshave demonstrated to be ableto induce antigenpresentationthrough both MHC class I andclass II complexes [57].
Tumor cells
MHC class I down-regulation
The most frequentmechanism of tumor evasionconsists in MHC class I-downregulation on tumor cells,decreasing the possibilities oftumor antigen recognition byT cells [58,59].
This is one of the mainreasons to use particulatedelivery systems for cancerimmunotherapy as a way toobviate the low presentationof tumor antigens by tumorcells [21].
T-cell relevantantigendown-regulation
Tumor cells can “edit” theirimmunologic print to avoidimmunosurveillance andrecognition by T cells [58].
Use personalized vaccinationby measuring CTL precursorsin PBMC of individualpatients in pre-vaccinationto select the antigens withhigher CTL precursorfrequency [34,35].
Expression ofimmunosuppressivemolecules
Tumor cells can secreteimmunosuppressive factorssuch as IL-10, TGF-β,galectin-1,gangliosides, PGE2and other molecules tocounteract effective immuneresponses [60].
TGF-β blockade, for instancethrough the overexpression ofsTβRII (a soluble TGF-β type IIreceptor) or by using a TGF-βneutralizing antibody(1D11), significantly de-creased tumor growth andmetastasis in two orthotopicmammary carcinoma models[61].
Table 2 (continued)
Description How to overcome
Tolerization of hostT cells bycross-presentationof tumor-derivedantigens
Tumor cells can presentdirectly their antigens toT cells and inducetolerization of T cells dueto the lack of effectivecostimulation [45].
Transfer genes encodingcostimulatory moleculesinto tumor cells to improvetheir ability to providecostimulation duringantigen presentation toactivate T cells [62].
Creation of hostiletumormicroenvironment
Tumor microenvironmentin solid tumors is animmunoregulatorynetwork composed bymalignant cells surroundedby a tumor-conditionedstroma that containsextracellular matrix and avariety of nonmalignantpopulations, includingmyeloid cells, lymphocytes,fibroblasts, and endothelialcells [41].
Effector cell recruitment intometastases, transduction oftumor cells to express specificchemokines, ligands thattarget tumor-expressedreceptors or antibody conju-gates complexed with specificchemokines that selectivelyrecognize tumor markers arepotential strategies to getaccess to the tumor microen-vironment [63].
182 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
type I IFNs, which directly affect T cells in a paracrine fashion [71]. In fact,type I IFN production by host DCs is an important step during the innateimmune recognition of tumors and has demonstrated to correlate withpositive prognostics in early stage disease [72].
Compared tomacrophages, DCs have a similar capacity for endocyto-sis and an equivalent number of lysosomes but less proteolytic activity,which limits full degradation of antigens, and increases the effectivenessof antigen presentation [73]. DCs, therefore, constitute a preferentialtarget of vaccines because these cells can function as a route of entry tothe immune system and ameans of controlling both innate and adaptiveimmune responses.
Immature DCs, located in peripheral tissues, function as sentinel cellsof the immune system by detecting and incorporating pathogens andbecoming activated in response to specific signals or proinflammatorycytokines in the microenvironment [74]. Once DCs have taken up anti-gens, they start to migrate in a chemokine-dependent manner intonaïve T-cell regions. DCs begin to express a lymph node-homing chemo-kine receptor, CCR7, which enables them to enter afferent lymphaticvessels and migrate into LNs, where CCL19 and CCL21, the ligands forCCR7, are expressed [75]. During their migration, DCs will process theantigens they have taken up and transfer them to surface complexes byone of three pathways: through MHC class I, MHC class II, or lipidantigen-presenting cluster of differentiation 1 (CD1) molecules [76].Extracellular antigens are taken up by APCs by one of the endocyticmechanisms – receptor-dependent endocytosis (clathrin-mediated,caveolae-mediated, lipid-raft-mediated or phagocytosis) or receptor-independent endocytosis (fluid phase endocytosis/macropynocytosis) –and placed into a membrane-delimited compartment of the endocyticpathway [77]. This compartment undergoes a series of modificationsand finally fuses with lysosomes or MHC class II compartments (MIIC).Peptide-loaded MHC class II molecules are then transported to the cellsurface where they engage antigen-specific CD4+ T cells. Antigenicpeptides derived from cytosolic proteins (endogenously synthesizedantigens) are processed in the proteasome into peptides and transportedfrom the cytoplasm into the endoplasmic reticulum via the transporterassociated with antigen processing (TAP) molecular complex, wherethey associate with nascent produced MHC class I molecules and aredelivered to the cell surface becoming available to be recognized byCD8+ T cells [78].
An alternative antigen presentation pathway, designated cross-presentation or cross-priming, has also been described. The term “cross-presentation” describes the process whereby APCs (mostly myeloidDCs) acquire exogenous soluble or cell-associated antigens (from infectedor tumor cells) and then present them bound to MHC class I molecules.

Fig. 1. Desired immune response elicited by a therapeutic cancer vaccine. Cancer vac-cines must target antigens and “danger signals” or adjuvants to immature dendriticcells (ImDCs) either in the skin or in the lymph nodes (LNs). After processing, antigenswill be presented through major histocompatibility complex (MHC) class I or MHCclass II complexes to CD8+ or CD4+ T lymphocytes, respectively. Simultaneously, “dan-ger signals” promote the activation andmaturation of DCs, which consequently expressco-stimulatory factors and secrete cytokines, such as INF-γ and IL-12, which are crucialin the effective stimulation of T cells and in determining the CD4+ phenotype. CD8+
lymphocytes acquire a cytotoxic phenotype, known as cytotoxic T lymphocytes(CTL), able to cause tumor cell lysis directly. Some of these CD8+ lymphocytes also ac-quire a memory phenotype which is crucial to the maintenance of immunity. T helper 1(Th1) cells enhance the action of CTL by enhancing clonal expansion at the tumor siteand promoting the generation and maintenance of the memory phenotype. Th1 cellssecrete IFN-γ, which can further sensitize tumor cells to CTL action by upregulatingMHC class I and other components of the antigen-processing machinery and promot-ing the recruitment of cells from the innate immune system such as the natural killer(NK) cells, granulocytes or macrophages that also contribute to tumor cell lysis.
183J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
This mechanism insures the activation of CD8+ T cells in response to avirus that does not infect APCs or against tumor cells and is highly rele-vant for anti-tumor vaccines. Cross-presentation seems to occurthrough retrotranslocation of antigens from endosomal compartmentsinto the cytosol, followed by re-import of proteasomally processed pep-tides back into endosomes via TAP, where peptide loading of recycledMHC class I molecules occurs [78,79]. In the absence of stimulus,steady-state APCs constantly present self-antigens to T cells, inducingT cell tolerance to these antigens in order to avoid autoimmune reac-tions. However, in the presence of a foreign antigen, additional stimuli,apart from the simple presentation of the antigen, are necessary toinduce T cell expansion and acquisition of effector functions.
For T cells to be effectively activated byDCs, three signals are required:(i) they need to recognize the processed antigen in the context of MHCclass I (for CD8+ T cells) or MHC class II (for CD4+ T cells); each T cellcarries a unique TCR and only those cells that recognize the antigenspresented by DCs in the form of an MHC/antigen complex will becomeactivated. (ii) During maturation, DCs also upregulate other cell surfacemolecules, such as MHC class I and II, and costimulatory molecules, suchas CD40, the receptor for CD40L expressed by naïve T cells, and alsoCD80 and CD86,which are the ligands for the immunoglobulin superfam-ily member, CD28, also expressed by naïve T cells. This is the reason whyimmature DCs in the periphery cannot efficiently activate T cells. (iii) Tcells need cytokines and chemokines, such as IL-12 and type I IFNs,which are produced by DCs and directly affect T cells in a paracrinefashion [80,81]. These three signals promote T cell expansion, survivaland acquisition of effector functions. Asmentioned, CD8+T cells predom-inantly recognize endogenous intracellular antigens that are presented byMHC class I molecules, which are ubiquitously expressed by the majorityof normal and cancerous tissues. Unfortunately, cancer cells are notori-ously unreliable as antigen presenters. They lack activating costimulatorymolecules, produce immunosuppressive soluble factors, and have unsta-ble genomes that are prone to losing key molecules required for antigenprocessing and presentation [82]. On the other hand, most conventionalvaccines elicit antibody and CD4+ helper T cell responses rather thanCTL responses. For a vaccine to induce CTL responses, the antigen of inter-est needs to reach the cytoplasm of DCs to be cross-presented throughMHC class I to CD8+ T cells. As key players orchestrating immunity, DCs
are a natural focus for immune therapy. Basically, two strategieshave been studied to target these cells and to use them in cancerimmunotherapy: administration of ex vivo tumor-peptide pulsed DCsor in vivo direct delivery of tumor-peptides [76]. For both strategies,there are a number of vaccine delivery systems that are being evaluatedto more efficiently load antigens into DCs, including recombinant viralvectors, virus-like particles, liposomes, micro- and NPs to name a few[12].
2.4. Non-peptide cancer antigens
For a long time, proteins and peptides were seen as the only primarytargets of adaptive immune responses, and so were called T cell-dependent antigens, whereas carbohydrates and lipids were considerednot to be recognized by the complete adaptive machinery and seen as Tcell-independent antigens [83]. Now it is known that lipid antigen recog-nition is also an important component of host defense. APCs also display apathway of antigen presentation for lipid-based antigens based on recog-nition of this class of antigens by members of the MHC I-like CD1 family.The CD1 family, which is constitutively expressed on DCs, macrophagesand B cells, consists of five isoforms that, based on sequence similarity,have been classified into three groups: group 1 (CD1a, CD1b and CD1c)and group 2 (CD1d), both involved in antigen presentation, and group 3(CD1e), involved in lipid processing and trafficking [84,85]. Similar towhat happens with peptide antigens, lipid antigens can be synthesizedoutside APCs by microbes or by other cells and be internalized, or canbe synthesized within APCs as self- or microbial lipids generated byinvading pathogens. Exogenous lipids gain access to the organelles ofthe endocytic pathway after an internalization process facilitated bylipoproteins or by fusion with the plasma membrane [86]. These lipidsare then transported into the cellular compartments where CD1 mole-cules traffic to form antigenic complexes. Group 1 CD1molecules presentlipid antigens to clonally diverse T cells, either CD4+, CD8+ or CD4–CD8double negative T cells, and preferentially release different types of cyto-kines according to their Th1, Th2 or Th17 profile. In contrast, group 2CD1d molecules present lipid antigens to NKT cells, a unique subset of Tlineage cells that coexpress TCRs and receptors of the NK cell lineage[84]. Stimulation of NKT cells induces rapid secretion of large amountsof immune regulatory Th1 and Th2 cytokines, such as IFN-γ and IL-4[87]. A wide range of self and foreign lipid antigens is known to bepresented by CD1 molecules, including lipopeptides, diacylglycerolipids,sphingolipids, mycolates and osphomycoketides [88]. The most exten-sively studied CD1d antigen is alpha-galactosylceramide (α-GalCer).This synthetic glycolipid is an extremely potent stimulatory ligandeliciting the secretion of cytokines by NKT cells, leading these cells toenhance the activation of DCs and up-regulate co-stimulatory andcross-priming of tumor and viral antigens, thus acting as a strongadjuvant for Th1 and CTL responses [89].
Aberrant glycosylation is known to be a hallmark of cancer celltransformation defining stage, direction and tumor progression [90].As such, another class of tumor antigens that is a promising target incancer immunotherapy is the Tumor Associated Carbohydrate Antigens(TACA). A number of TACA have been identified and tested as tumorvaccine targets, including mucin-related (O-linked) (Tn, Sialyl Tn, andThomsen–Friedenreich antigens), blood group Lewis-related (LewisY,Sialyl LewisX, Sialyl LewisA, and LewisX), glycosphingolipids [Globo H,stage-specific embryonic antigen-3 (SSEA-3)], and sialic acid containingglycosphingolipids or gangliosides [GM2, GD2, GD3, fucosyl GM1, theNeuGcGM3 variant of GM3 and the polysialic acid (PSA)] [reviewed in[91]]. A great advantage of targeting TACA is the potential to broadenthe spectrum of antigens recognized by the immune response, andthereby reduce the risk of developing resistant tumors because of lossor mutation of a given protein or lipid antigen. The expression of TACAon normal tissues and the low immunogenicity of the carbohydrate an-tigens are the main drawbacks in the use of TACA as cancer vaccine tar-gets because these effects can result in tolerance or autoimmunity

184 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
responses [92]. Despite the poor immunogenicity of pure carbohydrates,as they seem not to be able to recruit an adaptive immune response, anew generation of TACA-based vaccines that can produce not onlyanti-tumor antibodies but also cellular immune responses is emerging,based on three main approaches: (i) glycoprotein- and glycopeptide-based vaccines to facilitate anti-tumor cellular responses; (ii) targetingof pattern recognition receptors (PRRs) to enhance antigen internaliza-tion and DC activation, as molecules containing carbohydrates (e.g.,lipopolysaccharide) are recognized by some TLRs and C-type lectins;and (iii) substitution of carbohydrate epitopes with protein or peptidesurrogates to overcome the T-cell independent nature of the “anti-carbohydrate” response, for example, by using Carbohydrate MimeticPeptides (CMPs), which can induce anti-tumor antibodies and contrib-ute to cellular immune responses [92,93]. One example of a glycoproteinthat can be modified to yield highly immunogenic TACA is the humanmilk mucin (MUC1), which is overexpressed on tumor cells with an al-tered glycosylation pattern, which makes it one of most promising TAA[94]. Since these endogenous structures are usually tolerated by the im-mune system and owing to the biological microheterogeneity of glyco-proteins, MUC1 isolated from tumor cells is not suitable for vaccination[95]. Synthetic glycopeptide antigens have been conjugated to immunestimulating components in order to generate a sufficient immunogenic-ity. Fully synthetic two-component vaccines either with T-cell peptideepitopes or with TLR ligands or three-component vaccines with boththese stimulants have been synthesized [96]. For instance, MUC1 glyco-peptide antigens have been coupled to tetanus toxoid (TTox), resultingin the induction of a strong and highly selective immune response [97].Later on, it has been demonstrated that bovine serum albumin (BSA)could replace the very expensive TTox as the immune-stimulating carrierprotein in exploratory immunization studies of synthetic MUC1antitumor vaccines [98].
TACA-based cancer vaccines can also be used with nanoparticulatedelivery systems to enhance their efficacy. For example, Mazorra et al.(2008) demonstrated that the conjugation of GM3, a ubiquitous antigenoverexpressed in several epithelial tumor types, to very small sizeproteoliposomes (VSSP) derived from Neisseria meningitides membraneproteins, induced protective anti-tumor antibodies in a mice melanomamodel, whichwere dependent on the activation of CD8+ T cells, showingthe direct involvement of the cellular immune response in the inducedanti-tumor protection [99]. Pejawar-Gaddy et al. (2010) used virus-likeparticles (VLP) decorated with MUC1, and obtained MUC1-specificCD8+ cytotoxic T cells that significantly delayed tumor growth in immu-nized mice [28].
Overall, the selection of a cancer vaccine target antigen should bebased not only on the presence of the antigen in a variety of tumor tissuesbut also on the role that this antigen plays in tumor growth andmetasta-sis. As such, lipid and carbohydrate-based tumor antigensmay be suitablealternatives to peptide tumor antigens.
3. TLR ligands and cancer vaccines
3.1. Potentiating the cancer immune response with TLR ligands
The innate immune system is the first line of defense against micro-bial pathogens. DCs possess a broad spectrum of cell surface receptorsknown as pattern recognition receptors (PRRs), which are involved inthe initiation, promotion and execution of immune responses. PRRs,TLRs, nucleotide oligomerization domain (NOD)-like receptors, scaven-ger receptors, retinoic-acid inducible gene (RIG)-like receptors (RLR)and C-type lectin receptors recognize some conservative elementsexclusively expressed on pathogens, known as pathogen-associatedmolecular patterns (PAMPs), including lipids, lipoproteins, proteins,carbohydrates and nucleic acids [100]. Recognition of PAMPs by PRRsactivates intracellular signaling pathways that induce activation ofDCs, which thus acquire a mature T cell-stimulating phenotype culmi-nating in the induction of inflammatory cytokines, chemokines, IFNs
and upregulation of co-stimulatory molecules [101]. In mammals, thefamily of TLRs is the largest and most extensively studied PRR class.TLRs are mainly expressed on APCs but also on cells of the adaptiveimmune system including several T cell subsets, such as conventionalαβT cells, regulatory T cells, and γδT cells, as well as NKT cells [102].TLR activation can engage not only the innate immune system but canalso induce adaptive immune responses, either through direct engage-ment of TLRs with their ligands on B and T cells, or through indirectmechanisms mediated by TLR-activated DCs.
Mammalian TLRs are classified into several groups based on the typeof PAMP they recognize. TLR1, 2, 4 and 6 recognize lipids, such as lipo-polysaccharide (LPS) from Gram-negative bacteria, lipoteichoic acidfrom Gram-positive bacteria and lipoarabinomannan frommycobacteria.TLR5 senses bacterial flagellin. TLR3, 7, 8 and 9 detect nucleic acids de-rived fromviruses andbacteria. TLR3 has been shown to recognize doublestranded RNA (dsRNA), which is produced by many viruses duringreplication. TLR7 recognizes synthetic imidazoquinoline-like molecules,guanosine analogs such as loxoribine, viral single-stranded RNA(ssRNA) and small interfering RNA (siRNA). Human TLR8, which hashigh homology to mouse TLR7, participates in the recognition ofimidazoquinolines and ssRNA. TLR9 recognizes non-methylated CpGDNA motifs present in bacterial and viral genomes, as well as non-nucleic acids, such as hemozoin from the malaria parasite [103]. TLRscan also be divided into two subfamilies distinguished by their subcellularlocalization. The first subfamily comprises TLR1, 2, 4, 5, 6 and also TLR10in humans and TLR11 in mice, expressed at the cell surface where theydirectly encounter their ligands. The other subfamily of TLRs, whichincludes TLR3, 7, 8 and 9, is localized in a specialized endosomal compart-ment where they detect the presence of viral and bacterial nucleic acids[104].
Recognition of ligands by TLRs induces recruitment of differentcytoplasmic adaptor molecules and can initiate twomajor independentbut complementary signaling pathways: (i) recruitment of the adaptorMyD88on activation of TLR2, 4, 7, 8, and 9 or of Toll-interleukin 1 recep-tor domain (TIR)-containing adaptor molecule (TICAM)-1 on activationof TLR3, both resulting in activation of the nuclear factor-κB (NF-κB)and production of inflammatory cytokines, such as IL-1, IL-6, TNF-α,and IL-12; and (ii) activation of IFN regulatory factor 3 (IRF3) throughTRIF–TICAM-1 recruitment by TLR3 and TLR4 engagement leading totype I IFN production and subsequently induction of IFN-responsivegenes, such as antiviral genes and the CXC–chemokine, IP-10/CXCL10[105,106].The involvement of the humoral branch with production ofantigen-specific antibodies has also been reported [107]. From these ob-servations, one can predict that TLR ligands may act as very potentimmune modulators and vaccine adjuvants and, given the preferentialTh1-profile involved, thesemolecules could have a huge impact on cancerimmunotherapy.
Even that TLR activation is prone to the induction of a Th1-profile,structural differences on TLR agonists seem to induce slightly distinctimmune responses. These differences aremainly related to the inductionof antibody secretion, the activation of NK and CTL and the secretion ofdifferent cytokines [reviewed by Duthie et al. (2011) [108]]. Engagementof TLR2, 4 and 5 seems to be related to the enhancement of the suppres-sive function of Treg which might be related to the expression of thesePRRs on CD4+CD25+ Treg cells, while the other TLRs have a broaderexpression on CD4+ T cells and are related to the reversion of thesuppressive action of Treg [109]. A well known example of thedependence on agonist structure is the case of the TLR9 agonistsCpG oligodeoxynucleotides (ODN), where different classes can bedefined according to their structural characteristics. A-class CpG ODNare generally 20–21 bases in length with a single CpG motif within ahair-pin-forming palindrome phosphodiester region and induce IFN-γproduction by plasmacytoid DCs (pDCs), by NK cells, and monocytematuration into DCs, but are poor inducers of B cell activation. Incontrast, B-class CpG ODN, which incorporate one or more CpG motifswithin a fully phosphorothioate backbone fail, to induce pDC IFN-γ
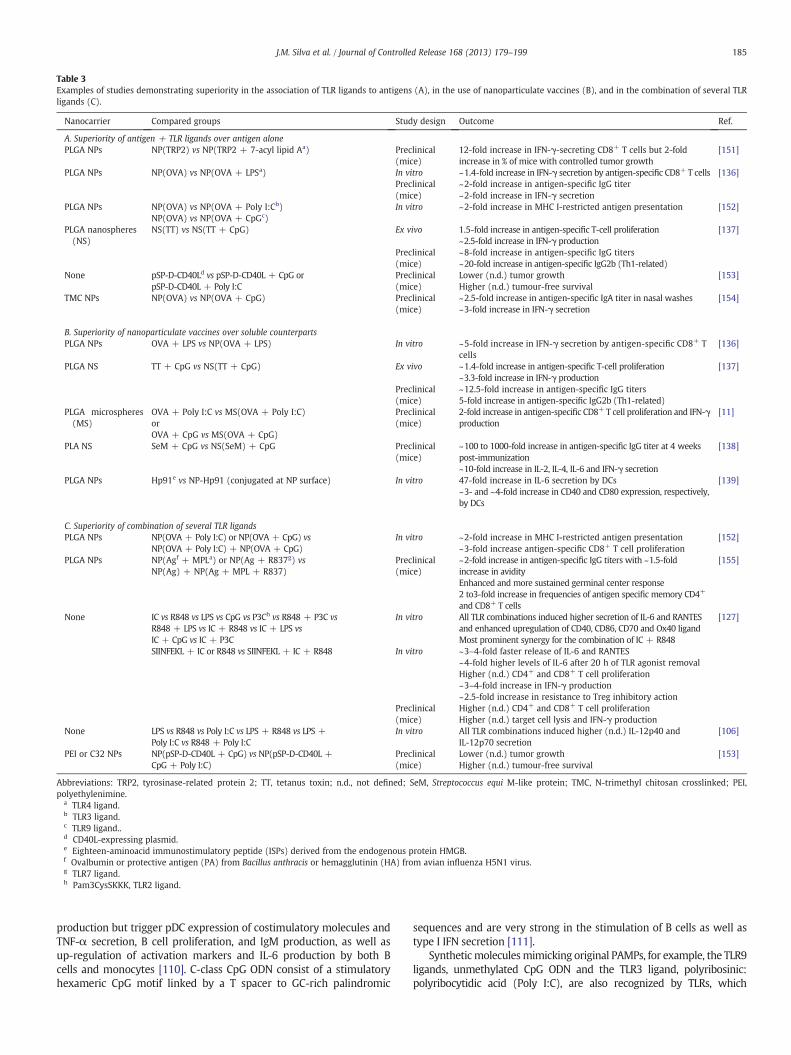
Table 3Examples of studies demonstrating superiority in the association of TLR ligands to antigens (A), in the use of nanoparticulate vaccines (B), and in the combination of several TLRligands (C).
Nanocarrier Compared groups Study design Outcome Ref.
A. Superiority of antigen + TLR ligands over antigen alonePLGA NPs NP(TRP2) vs NP(TRP2 + 7-acyl lipid Aa) Preclinical
(mice)12-fold increase in IFN-γ-secreting CD8+ T cells but 2-foldincrease in % of mice with controlled tumor growth
[151]
PLGA NPs NP(OVA) vs NP(OVA + LPSa) In vitro ~1.4-fold increase in IFN-γ secretion by antigen-specific CD8+ T cells [136]Preclinical(mice)
~2-fold increase in antigen-specific IgG titer~2-fold increase in IFN-γ secretion
PLGA NPs NP(OVA) vs NP(OVA + Poly I:Cb) In vitro ~2-fold increase in MHC I-restricted antigen presentation [152]NP(OVA) vs NP(OVA + CpGc)
PLGA nanospheres(NS)
NS(TT) vs NS(TT + CpG) Ex vivo 1.5-fold increase in antigen-specific T-cell proliferation~2.5-fold increase in IFN-γ production
[137]
Preclinical(mice)
~8-fold increase in antigen-specific IgG titers~20-fold increase in antigen-specific IgG2b (Th1-related)
None pSP-D-CD40Ld vs pSP-D-CD40L + CpG orpSP-D-CD40L + Poly I:C
Preclinical(mice)
Lower (n.d.) tumor growthHigher (n.d.) tumour-free survival
[153]
TMC NPs NP(OVA) vs NP(OVA + CpG) Preclinical(mice)
~2.5-fold increase in antigen-specific IgA titer in nasal washes~3-fold increase in IFN-γ secretion
[154]
B. Superiority of nanoparticulate vaccines over soluble counterpartsPLGA NPs OVA + LPS vs NP(OVA + LPS) In vitro ~5-fold increase in IFN-γ secretion by antigen-specific CD8+ T
cells[136]
PLGA NS TT + CpG vs NS(TT + CpG) Ex vivo ~1.4-fold increase in antigen-specific T-cell proliferation~3.3-fold increase in IFN-γ production
[137]
Preclinical(mice)
~12.5-fold increase in antigen-specific IgG titers5-fold increase in antigen-specific IgG2b (Th1-related)
PLGA microspheres(MS)
OVA + Poly I:C vs MS(OVA + Poly I:C)orOVA + CpG vs MS(OVA + CpG)
Preclinical(mice)
2-fold increase in antigen-specific CD8+ T cell proliferation and IFN-γproduction
[11]
PLA NS SeM + CpG vs NS(SeM) + CpG Preclinical(mice)
~100 to 1000-fold increase in antigen-specific IgG titer at 4 weekspost-immunization~10-fold increase in IL-2, IL-4, IL-6 and IFN-γ secretion
[138]
PLGA NPs Hp91e vs NP-Hp91 (conjugated at NP surface) In vitro 47-fold increase in IL-6 secretion by DCs~3- and ~4-fold increase in CD40 and CD80 expression, respectively,by DCs
[139]
C. Superiority of combination of several TLR ligandsPLGA NPs NP(OVA + Poly I:C) or NP(OVA + CpG) vs
NP(OVA + Poly I:C) + NP(OVA + CpG)In vitro ~2-fold increase in MHC I-restricted antigen presentation
~3-fold increase antigen-specific CD8+ T cell proliferation[152]
PLGA NPs NP(Agf + MPLa) or NP(Ag + R837g) vsNP(Ag) + NP(Ag + MPL + R837)
Preclinical(mice)
~2-fold increase in antigen-specific IgG titers with ~1.5-foldincrease in avidityEnhanced and more sustained germinal center response2 to3-fold increase in frequencies of antigen specific memory CD4+
and CD8+ T cells
[155]
None IC vs R848 vs LPS vs CpG vs P3Ch vs R848 + P3C vsR848 + LPS vs IC + R848 vs IC + LPS vsIC + CpG vs IC + P3C
In vitro All TLR combinations induced higher secretion of IL-6 and RANTESand enhanced upregulation of CD40, CD86, CD70 and Ox40 ligandMost prominent synergy for the combination of IC + R848
[127]
SIINFEKL + IC or R848 vs SIINFEKL + IC + R848 In vitro ~3–4-fold faster release of IL-6 and RANTES~4-fold higher levels of IL-6 after 20 h of TLR agonist removalHigher (n.d.) CD4+ and CD8+ T cell proliferation~3–4-fold increase in IFN-γ production~2.5-fold increase in resistance to Treg inhibitory action
Preclinical(mice)
Higher (n.d.) CD4+ and CD8+ T cell proliferationHigher (n.d.) target cell lysis and IFN-γ production
None LPS vs R848 vs Poly I:C vs LPS + R848 vs LPS +Poly I:C vs R848 + Poly I:C
In vitro All TLR combinations induced higher (n.d.) IL-12p40 andIL-12p70 secretion
[106]
PEI or C32 NPs NP(pSP-D-CD40L + CpG) vs NP(pSP-D-CD40L +CpG + Poly I:C)
Preclinical(mice)
Lower (n.d.) tumor growthHigher (n.d.) tumour-free survival
[153]
Abbreviations: TRP2, tyrosinase-related protein 2; TT, tetanus toxin; n.d., not defined; SeM, Streptococcus equi M-like protein; TMC, N-trimethyl chitosan crosslinked; PEI,polyethylenimine.
a TLR4 ligand.b TLR3 ligand.c TLR9 ligand..d CD40L-expressing plasmid.e Eighteen-aminoacid immunostimulatory peptide (ISPs) derived from the endogenous protein HMGB.f Ovalbumin or protective antigen (PA) from Bacillus anthracis or hemagglutinin (HA) from avian influenza H5N1 virus.g TLR7 ligand.h Pam3CysSKKK, TLR2 ligand.
185J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
production but trigger pDC expression of costimulatory molecules andTNF-α secretion, B cell proliferation, and IgM production, as well asup-regulation of activation markers and IL-6 production by both Bcells and monocytes [110]. C-class CpG ODN consist of a stimulatoryhexameric CpG motif linked by a T spacer to GC-rich palindromic
sequences and are very strong in the stimulation of B cells as well astype I IFN secretion [111].
Syntheticmoleculesmimicking original PAMPs, for example, the TLR9ligands, unmethylated CpG ODN and the TLR3 ligand, polyribosinic:polyribocytidic acid (Poly I:C), are also recognized by TLRs, which

186 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
increases their potential for clinical investigation [112].Molecular dynam-ic simulation using computational approaches has been demonstrated tobe an important tool to improve understanding of the interactionsbetween TLRs and their ligands and to identify key structural issues thatpotentiate the rational design of newmolecules with pre-defined agonistor antagonist action on these receptors [113,114]. Some examples ofstudies demonstrating superiority in immune responses obtained withthe association of TLR ligands over the antigen used alone are presentedin Table 3A. Several TLR ligands have been tested as adjuvants of cancervaccines in clinical trials, including for melanoma [115], lymphoma[116], glioblastoma [117], breast, prostate, ovarian and lung cancers[118], with quite promising results, supporting the broad conclusionthat they can be safe and effective vaccine adjuvants [also reviewed byEngel et al. (2011) [119]].
In spite of the focus of this review on the use of PRR agonists, mainlyfor TLRs, as cancer vaccine adjuvants, there are other approaches thathave been tested for that purpose [120]. For several years, aluminum-based compounds (mainly aluminum hydroxide or phosphate) werethe only approved vaccine adjuvants and despite their relatively stronghumoral-inducing capacity, their weak adjuvant action in inducingcellular immune responses has driven intense research for alternatives[121]. For example, stimulation of cells of the innate immune systemthat engage in intimate interactions with DCs seems to be a promisingmethodology. Invariant natural killer T (iNKT) cells represent such a celltype, which holds substantial promise as a target for the developmentof vaccine adjuvants. As mentioned in Section 2.4, iNKT cells recognizeglycolipids presented by DCs and their stimulation induces rapidsecretion of large amounts of immune regulatory Th1 and Th2 cytokines,such as IFN-γ and IL-4, respectively [87]. Co-administration of theglycolipid α-GalCer along with a peptide antigen led to enhanced CD4+
and CD8+ T cell responses, which were shown to be dependent on DCconditioning by activated iNKT cells [122]. By activating various othereffector cells, such as NK cells and neutrophils, in the innate immune sys-tem, iNKT cells link the two arms of the immune system, forming a bridgebetween innate and acquired immunity.
3.2. Using TLR ligands to reverse tumor immunosuppression
One of the main hurdles in cancer immunotherapy is the difficultyto abrogate the immunosuppressive tumor environment. There area number of mechanisms by which tumors may actively evade or sup-press an immune response: deletion of immune effector cell by ex-pression of death-inducing ligands by tumor cells; induction oftolerance of tumor-reactive T cells induced by tumor cells; suppressionof tumor-reactive T cells by Treg cells or myeloid-derived suppressorcells (MDSCs); ignorance of tumor as a result of spatial separation of Tand tumor cells; and induction of tolerance of host T cells by cross-presentation of tumor-derived antigens [45,123].
Despite the vital importance of functional Treg in balancing inflamma-tory responses and preventing autoimmune diseases, these cells havebeen frequently found to comprise the majority of tumor-infiltratinglymphocytes and to correlate with poor survival in cancer patients[124]. Regulating the powerful immune-modulating activity of Treg isanticipated to become an important tool in cancer immunology and hasbeen demonstrated to increase T-cell activation and infiltration in largetumors [125]. Along with costimulatory molecules and cytokines, TLRligands are one of the major classes of molecules that regulate Treg andalter their function, through direct or indirect effects. Indirect effectsresult from the activation of DCs by TLR ligands. TLR-activatedDCs secretecytokines,mainly IL-6, IFN-γ and IL-4,whichmake T cells refractory to thesuppressive actions of Treg [126].Warger et al. (2006) demonstrated thatdistinct combinations of two TLR ligands resulted in synergistic activationof DCs that converted to superior CD4+ and CD8+ T-cell responses whileabrogating Treg-mediated suppression [127]. However, in the absence ofTLR stimulus, antigen-loadedDCswere shown to induce Treg cell prolifer-ation with retention of their immunosuppressive activity, demonstrating
that the capacity of DCs to reverse Treg immunosuppressive function ishighly dependent on TLR-mediated activation [128]. Treg cells havebeen shown to selectively express a multitude of TLR, namely TLR-4, -5,-7 and -8, activation which induces upregulation of several activationmarkers and enhances their survival/proliferation, thus reversing theirimmunosuppressive function [129]. On the other hand, TLR2 engagementon Treg has been shown to augment proliferation and survival, enhancingtheir suppressive function [130]. Olivier et al. (2011) tested a large panelof TLR ligands in combination with a model antigen covalently linked toan inert delivery system and observed that the negative modulation ofeffector CD4+ T cells exerted by Tregs could not be circumvented, which-ever TLR ligand was used as adjuvant [131]. Given the contradictoryresults obtained for the role of TLR engagement on Treg, it has beenhypothesized that TLR ligands can regulate T cell-mediated immuneresponses by multiple approaches, possibly including: (i) TLR-mediatedactivation of DCs resulting in secretion of cytokines and costimulatoryfactors that stimulate effector T cells; (ii) direct effects on effector T cellsenhancing their functions and clonal expansion rendering them resistantto Treg suppressive actions; and (iii) expansion of the Treg cell populationwith a transient loss of immunosuppressive function in the early responsestage, and reacquisition at the late stage of immune response to regulatethe expanded effector T cells following clearance of the TLR ligands. At thisphase, TLR-mediated enhanced Treg function can then play a critical rolein the prevention of immune pathology or in maintaining low levels ofpathogens, effects that are needed for maintenance of immunologicmemory [130]. In some types of cancers, intratumoral Treg infiltrationhas been shown to correlate with a better control of the tumor, possiblythrough the production of pro-inflammatory cytokines, and becauseintratumoral accumulation of Tregs is often associated with that ofother immune cells, and hencemay reflect the overall level of tumor infil-tration by immune cells, including CTL [132].
Another important action of TLR ligands in reversing tumor immuno-suppression is exerted on MDSC. MDSC accumulates in tumor-bearinghosts and cancer patients and plays a major role in tumor-related immu-nosuppression, hampering successful immunotherapy through inhibitionof CD4+andCD8+Tcell function and control ofNK cell cytotoxicity [123].Zoglmeier et al. (2011) showed for the first time that short-termactivation with the TLR9 ligand, CpG, blocked the suppressive functionof MDSC in tumor-bearing mice, promoting MDSC differentiation andrestoring the disturbed balance of mature and immature myeloid cellsthrough the induction of IFN-α [133].
In summary, developing effective immunotherapy for cancer requirestwo steps: targeted, antigen-specific immune stimulation througheffective DC activation; and concurrent reversal of tumor-driven immunesuppression. TLR ligands have been shown to play important roles in bothsteps and promise to become essential tools for effective cancerimmunotherapy.
4. Reaching the immune system with nanoparticles forcancer vaccines
Peptide immunization or subunit vaccines elicit poorly protectiveCD8+ T cell responses, whereas virulent acute pathogens and livevaccines generally induce excellent effector andmemory cell formation.It can thus be anticipated that mimicking the signals that are opera-tional during challenge with virulent microbes by inducing similarsignals in a comparably timed manner should significantly improvepeptide vaccination regimens [134].
Uptake of antigens by DCs depends on several antigen-associatedproperties, such as size, shape, surface charge, hydrophobicity/hydrophilicity, as well as receptor interactions. Particulate vaccines,such as whole cell vaccines, virosomes, virus-like particles or antigensformulated in particulate adjuvants, such as liposomes, micro- and NPs,have large surfaces, which present electrostatic or receptor-interactingproperties making possible a better interaction with DCs compared withsoluble antigens [135]. Several studies have demonstrated that particulate

187J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
delivery systems could enhance the uptake of antigens and adjuvants byDCs and that these systems are associated with better immune re-sponses than those obtained with the soluble counterparts (Table 3B)[11,136–139]. Use of either PLGA NPs ormicroparticles (MPs) to encap-sulate a synthetic analog of the glycolipid, α-GalCer, has been demon-strated to drive activation from iNKT in vivo and in vitro [140]. Apartfrom the higher uptake by DCs, nanocarriers also provide protectionto the encapsulated agents, including proteins, peptides, and nucleicacids, which would otherwise suffer immediate degradation whenadministered [141]. TLR ligands also benefit from nanocarrier encapsula-tion, sincemolecules, such as viral and bacterial nucleic acids, are alsoeasy targets of degradation. Some TLR ligands can cause a life-threatening septic shock-like state and may contribute to organ dys-function remote from the site where beneficial effects are desired[142]. As such, these molecules not only benefit from protection bynanocarriers but their safety is also improved because lower doses areneeded and their systemic distribution is restricted [143]. Moreover,nanocarriers can provide direct intracellular access, facilitating engage-ment of the intracellular TLR3, 7, 8 and 9 by their ligands, improvingtheir efficacy as vaccine adjuvants [144].
Nanocarriers composed of biodegradable polymers, such as amphi-philic polyesters, are able to confer a controlled release profile to theencapsulated agents, the rate of release of which can be regulated bythe manipulation of the polymers' physicochemical characteristics,such as molecular weight, in the formulation [145]. A sustained releaseof adjuvants, such as TLR ligands, is essential to properly stimulate DCsfor long-lasting proinflammatory cytokine production and to reversethe immunosuppressive action of Treg. It has been shown that onceTLR ligands are not present, Treg are able to return to the resting stateand recover their suppressive capacities [146]. Similarly, Yang et al.(2004) demonstrated that DCs that were no longer exposed to LPSsecreted no IL-12, and the production of IL-1 and IL-6 was reduced bymore than 90% compared with that of DCs that were cultured continu-ously in the presence of LPS [128].
Additionally, some particulate systems, particularly those constitutedby the polyester polymer, PLGA, have been shown to escape from earlyendosomes to the cytoplasm potentiating the proteasome-dependentprocessing of encapsulated antigens and cross-presentation throughMHC class I [147,148]. Finally, particulate formulation of different sizescan deliver antigens to separate subsets of DCs, inducing Th1 and/orTh2 responses as required [149].
A significant shift in the isotype of the immune response can beachieved by co-administering antigen and Th1-inducing adjuvants,such as TLR ligands. A delivery system that can package and transportantigen(s) and adjuvant(s) to the targeted site of action is thus essentialfor an ideal cancer vaccine formulation [150]. Antigens and adjuvantshave been successfully encapsulated together into the same particulatedelivery system (Table 3A) [136,137,151–154]. Schlosser et al.(2008) demonstrated that the efficiency of cross-priming and inductionof CTL responses in mice after vaccination with biodegradable PLGA MSwas enhanced when ovalbumin was coencapsulated together with ei-ther a CpG ODN or Poly I:C as compared to co-inoculation ofovalbumin-bearing MS with soluble or separately encapsulated adju-vants [11]. Furthermore, coencapsulation of glycolipids, such asα-GalCer, along with peptide antigens can potentiate the activation ofthe iNKT cell-DC loop as it has been demonstrated that simultaneouspresentation of α-GalCer and antigen by DCs enhances T cell responsesthrough the conditioning of DCs by stimulated iNKT cells [122].
In addition to the advantage of delivering antigens and adjuvantssimultaneously in the same particulate delivery system, the combina-tion of several adjuvants can also have a positive effect on the activationof the immune system. Combined TLR ligation on DCs has been demon-strated to trigger distinct signaling pathways that resulted in activationof these cells in a synergistic manner in terms of proinflammatory cyto-kine and chemokine production and also expression of costimulatoryfactors (Table 3C) [106,127,152,153,155]. For example, Warger et al.
(2006) observed that DCs activated by the combined use of TLR7 andTLR3 ligands induced CD4+ and CD8+ T cells that were insensitive tothe inhibitory effects of Treg. These DCs demonstrated a faster andmore sustained secretion of proinflammatory cytokines and expressionof costimulatory molecules, resulting in a marked increase in CTLeffector functions in wild-type mice in vivo [127]. Gautier et al. (2005)demonstrated that the different TLR signaling pathways cooperate inthe secretion of IL-12p70 through an autocrine loop of type I IFN, andthat different TLR ligands are likely to be necessary for inducing anoptimal response to pathogens [106]. Kasturi et al. (2011) demonstrat-ed that immunization of mice with synthetic NPs containing antigensand ligands for TLR4 (MPL) and TLR7 (R837) induced synergisticincreases in antigen-specific neutralizing antibodies compared toimmunization with a single TLR ligand. Moreover, these investigatorsdemonstrated that direct triggering of TLRs on B cells was essential forantibody responses and that co-activation of both TLRs on the same Bcell was needed [155]. The synergistic effect observedwith the simulta-neous stimulation of MyD88-independent and MyD88-dependentpathways by distinct TLR ligands can be explained by an enhanced acti-vation of DCs in terms of proinflammatory cytokine and chemokineproduction and also expression of costimulatory molecules resultingfrom the greater induction of gene expression because two indepen-dent signaling pathways are activated in parallel [156].
The optimal size for NP uptake by APCs is still a matter of debate. It islikely that APCs have evolved to effectively process any antigen withdimensions that are similar to pathogens, ranging from viruses(20–100 nm) to bacteria and even cells (in the micrometer range)[135]. DCs and macrophages have been shown to optimally phagocyteparticles up to 10 and 30 μm in diameter, respectively [157]. Fogedand colleagues (2005) investigated DC uptake of model fluorescentpolystyrene particles with a broad size range and variable surface prop-erties. The optimal particle diameter for fast and efficient acquisition bya substantial percentage of the DCswas 0.5 μmor less [158]. Apart fromthe fact that peripheral DCs can capture NPs at peripheral sites, such asepidermis and dermis, it seems advantageous to directly target NPsto the LNs. In this case, additional size aspects must be taken intoaccount. Initial lymph vessels have diameters around 10–60 μmand the sinusoid in the spleen varies from 150 to 200 nm [159].Only molecules of 20–200 nm can effectively enter in lymphaticsystem. Particles larger than 200–500 nm do not enter the lymphaticsystem unless they are associatedwith DCs. NPs of less than 200 nm can,therefore, reach the lymphoid organs directly within hours after injec-tion, whereas particles larger than 200–500 nm require DCs, which cansqueeze through openings of overlapping endothelial cells and willtake approximately 24 h to arrive in the LNs [135]. The size of the NPsalso seems to influence their cellular uptake mechanism (endocytosis,macropinocytosis, phagocytosis, clathrin-dependent and/or caveloae-mediated) and intracellular pathway. Each endocytic pathway is alsodefined by a specific size range of the engulfed soluble or particulatematerial. In general, virus sized particles (20–200 nm) are usually takenup via classic receptor-mediated endocytosis (clathrin-dependent)(particles b150 nm) or endocytosis through caveolae (particles of50–80 nm) and tend to generate a virus-like immune responsewith activation of CTL and Th1. Endocytosis of larger sized particles(>0.5 μm) occurs mainly via macropinocytosis and phagocytosisand is restricted to a few, specialized cells, such as macrophagesand Langerhans cells (LCs) in the skin, and preferentially generates abacteria-like immune responsewith activation of Th2 cells and antibodies[160]. The size range of each endocytic pathway is probably subject ofsome degree of flexibility and dependent on other factors. For instance,Cureton et al. (2009) demonstrated how physical properties of theparticle dictate the mechanism of virus particles internalization bydescribing that vesicular stomatitis virus (VSV), an enveloped virus withbullet-shaped virions measuring 70 × 200 nm, enters cells through apartial clathrin coat that require local actinfilament assembly to completevesicle budding and internalization, probably because it exceeds the

188 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
capacity of typical clathrin-coated vesicles [161]. On the other hand,shorter VSV (75 nm long) defective interfering particle (DI-T) internaliza-tion occurs through complete clathrin-coated vesicles and does notrequire actin polymerization [162].
Surface charge has also a profound effect on internalization capability.This effect is partly due to the fact that the cell membrane is negativelycharged and will have a greater affinity for positively charged molecules[163]. Intracellular trafficking studies indicate that some of positivelycharged NPs could escape from lysosomes after being internalized andexhibit perinuclear localization, whereas the negatively and neutrallycharged NPs prefer to colocalize with lysosomes [164,165]. For over halfa century the measurement of zeta potential by determining electropho-retic mobility by electrophoretic light scattering has become essential forNP characterization and research. However, the interpretation of NPelectrophoretic mobility can be complex. NP surface charged can beshielded by a diffuse layer of solvated counterions and consequently thezeta potential is highly dependent on immediate environmental factorssuch as, ionic composition and ionic strength of the dispersant, the pHof the dispersant and the mathematical models applied to derivitizedzeta potential fromelectrophoreticmobility [166]. Particle shape is anoth-er parameter of great impact in the context of biodistribution, cellularuptake and toxicity. Venkataraman et al. (2011) provide a comprehensivesummary on current understanding of the influence of nanostructureswith different shapes on important biological processes in drug delivery.Non-spherical particles possess drug loading capacities and biologicalbehaviors that frequently deviate from their historically well-studiedspherical counterparts, which have been shown to be advantageous forimproving blood circulation time, organ distribution and avoiding prema-ture clearance by phagocytosis in various situations [167]. Nonsphericalparticles showed lower uptake by cells than their spherical counterpartswith a negative correlation between aspect ratio and uptake rate. This isattributed to the larger average curvature radius of adsorbed non-spherical particles experienced by the cells [168].
It iswell known that, upon contactwith a biologicalfluid (e.g.plasma),NPs tend to associate with a range of biopolymers, especially proteins,organized into the so-called “protein corona”. The composition of the“protein corona” at any given time is determined by the concentrationsof the over 3700 proteins in plasma, and the equilibrium bindingconstants of each protein for the particular NP. Themain class of proteinsthat are known to adsorb at NP surface is the opsonins (mainly comple-ment proteins such as C3, C4, and C5 and immunoglobulins such as IgGand IgA) [169]. The nature and the organization of the adsorbed proteinson the surface ofNPs, and any subsequent colloidal instability of either theNPs (e.g. particle aggregation, flocculation, and precipitation) determinesthe initial biological responses to the presence of NPs and can influencetheir biodistribution and biocompatibility [170,171]. For instance,the presence of opsonins promotes rapid clearance from the blood-stream by phagocytosis by the mononuclear phagocyte system (MPS) inthe liver and spleen [172]. The complexity of the plasma corona and itsphysicochemical regulation is still far from being fully understood. Somestudies demonstrated that both size and surface properties play a verysignificant role in determining the composition of the “protein corona”on the surface of different NPs [173,174]. Greater knowledge of how pro-teins bind to NPs and the factors that determine affinity is necessary.
In addition to the factors that can influence passive targeting of NPs,several strategies are available to functionalize the NP surface to active-ly target DCs. One such approach will be covered in the next chapter ofthis review.
5. Functionalizing nanoparticles for active targeting to DCs
Surface functionalization of NPs can be used to increase the resi-dence time in the blood, reduce nonspecific distribution, and in somecases, target tissues or specific cell surface antigens [172]. A multitudeof ligands are currently being assessed to functionalize nanocarriers,including peptides, antibodies, proteins, polysaccharides, glycolipids,
glycoproteins, and lectins. Some ligands make use of mononuclearphagocyte characteristic receptor expression and phagocytic in-nate processes [175]. PEGylation – the introduction of the hydro-philic poly(ethylene glycol) (PEG) chains by grafting, conjugationor adsorption to the NP surface – is normally used to provide stericstabilization and “stealth” properties, such as prevention of theadsorption of proteins at their surface. Several studies in rats and micehave demonstrated that pegylated PLGA NPs exhibit significantly in-creased blood circulation time and decreased liver uptake comparedwith non-pegylated NPs [176]. Notwithstanding, pegylation of NPs isalso a highly suited platform for the introduction of functional ligandsas they can readily be attached to the terminal ends of PEG chains,using standard protocols [177].
Two specific goals may be achieved by the introduction of suchligands. First, introduction of ligands for defined cell surface receptorsmay enhance cellular uptake or may be used to target defined celltypes (for example DCs). Second, the introduction of TLR ligands orligands for other PRRs on the surface of NPs is an attractive option tofurther enhance the immunogenicity of nanoparticulate vaccines, byproviding an intrinsic danger signal [178,179].
Among the PRRs expressed by APCs are the C-type lectin receptors(CLRs), which are characterized by the presence of carbohydrate-binding domains either produced as transmembrane proteins or secretedas soluble proteins [180]. CLRs are involved in the recognition and inter-nalization of many glycosylated self-antigens and pathogens, such asviruses, bacteria, and fungi. CLRs are highly specific internalizationreceptors that facilitate antigen uptake and processing and influenceantigen-routing and loading on MHC class I and II. Depending on theCLR targeted, either CD4+ or CD8+ T-cell responses can be specificallyinduced [181]. Some membrane-bound forms of the C-type lectins areexpressed on DCs. The mannose receptor (MR/CD206), the prototype ofthis family, recognizes sugars terminating in D-mannose, L-fucose orN-acetyl glucosamine, not only on a range of bacteria, viruses, and yeasts,but also on certain endogenous glycoproteins, such as lysosomal hydro-lases [182]. Antigens targeted to the MR seem to be directed to distinctearly endosomes that do not mature, acidify, or fuse with lysosomes,but instead remain stable over extended periods within APCs, finallybeing presented exclusively to CD8+ T cells by the cross-presentationpathway through MHC class I [183].
Targeting antigens to endocytic receptors on DCs represents anattractive strategy to enhance the efficacy of vaccines. A schematic repre-sentation of the targeting of DCs with NPs designed for cancer vaccinescan be seen in Fig. 2. Several studies have demonstrated the successfuluse of mannose-grafted nanocarriers and the increase in the uptake ofthese carriers by DCs. PLGA NPs decorated with mannan, a naturalpolymannose isolated from the cell wall of Saccharomyces cerevisae,have been shown to have a stronger binding affinity and increasedCD4+ and CD8+ T cell responses compared to non-decorated NPs [184].Other particulate vaccines have been successfully developed to spe-cifically target the MR, such as mannose–PEG3–NH2-decorated PLGAMS [185], mannosylated-gelatin NPs [186] and liposomes containingmannosyl lipid derivatives [187]. In vivo, it has been demonstratedthat targeting antigens to CLRs can result in enhanced humoral andcellular responses sufficient to induce immunity against tumors[188,189]. Targeting CLRs with anti-lectin antibodies instead ofusing one of their ligands has also been successful [190].
6. Using aliphatic polyesters to formulate biodegradablepolymeric nanoparticles
Different classes of nanocarriers have been tested for cancer immuno-therapy applications, as reviewed by Krishnamachari et al. (2011) [21].Among these classes, the thermoplastic aliphatic polyesters, includingpoly-lactic acid (PLA), poly-glycolic acid (PGA) and especially the copoly-mer PLGA, have a long history of use for biomedical applications[reviewed in [191]]. Because of their excellent biocompatibility and

189J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
biodegradability, they are used since 1970s as biodegradable sutures andare approved by theUS Food andDrugAdministration (FDA) and Europe-an Medicine Agency (EMA) for drug delivery [192].
The aliphatic polyesters mainly used for biomedical applications arethose derived from the monomers glycolide/glycolic acid (GA), lactide/lactid acid (LA), β-butyrolactone (β-BL), ε-caprolactone (ε-CL), 1,5-dioxepan-2-one (DXO) and trimethylene carbonate (TMC) [193]. Threemajor routes have beendescribed for the synthesis of aliphatic polyesters:(i) polycondensation of a hydroxy acid or a diol and a diacid, a methodthat normally drives to a low degree of polymerization resulting in lowmolecularweight polymers and the impossibility of synthesis of block co-polymers [194]; (ii) ring opening polymerization (ROP) of lactones andother cyclic diesters such as lactide and glycolide which under properconditions can result in polyesters with highmolecularweights with lim-ited side reactions [193,195]; and (iii) enzymatic polymerizationwhich iscarried out under mild conditions avoiding the use of toxic reagents andallowing catalyst recycle, but yielding relatively low molecular weightpolymers [196,197]. Aliphatic polyesters can be classified as homopoly-mers or copolymers depending on their homogenous or heterogenouscomposition on repeating units, respectively. Table 4 presents the molec-ular structures of the mainly used aliphatic polyester homo- and copoly-mers and their respective monomers.
Random, block, and graft copolymerization can yield products withsignificantly different properties such as glass transition temperature(Tg), melting temperature (Tm), and crystallinity and be used to obtaina product with a particular combination of desirable properties [208].For instance, in the 1970s it had already been recognized that PCL wasparticularly amenable to blending and polymer blends [209]. Completelyamorphous polymers like poly(1,5-dioxepan-2-one) (PDXO) are alsovery valuable for copolymerization to improve the elasticity of a polymer[210]. PLA is also a good candidate for copolymerization due to itstoughness [211]. The reduced stability of poly(3-hydroxybutyrate)(P3HB) justifies its use as a copolymer with 3-hydroxyvalerate to formthe less crystalline poly(3-hydroxybutyrate-co-3-hydroxyvalerate)(PHBV), widely used not only to tissue engineering, [212,213] butalso to produce particulate drug delivery systems (DDS) [214,215].
Random copolymers can be easily synthesized by polymerization ofdifferent monomers through the synthesis methods referred in Table 4.The final copolymer composition depends on the composition of themonomer feed as well as the reactivity ration of the monomers. Copoly-mers canbe representedby “AB”,whereAandB represent the constituentblocks of different natures. PLGA, the copolymer of the lactide/lactic acidand glycolide/glycolic acid monomers is by far the most studied randompolyester copolymer [191]. PLGA can be producedwith different stoichio-metric ratios of itsmonomers and severalmolecularweights, two param-eters that are of determining influence on the hydrophobicity and releasekinetics of the polymer. The forms of PLGA are usually identified by themonomers' ratio used. Accordingly, PLGA 50:50 identifies a copolymerwith 50% lactic acid and 50% glycolic acid, which is by far themost widelyused PLGA copolymer in nanotechnology [216]. PLGAhas been extensive-ly applied in the formulation development of different biodegradabledevices, such as MPs and NPs, implants and miscellaneous devices, aswell as in situ-formed devices [217]. Due to the protection providedfrom the aggressive in vivo environment, PLGA particles have been inves-tigated for sustained and targeted/localized delivery of various agents,including hydrophilic and hydrophobic drugs, proteins and peptides,plasmid DNA and siRNA [218,219]. It has also been extensively tested asvaccine particulate delivery systems to encapsulate a variety of antigenssuch as proteins, peptides, lipopeptides, cell lysates, viruses or plasmidDNA [139,220–224]. Some advantages and disadvantages of PLGA as aparticulate vaccine delivery system, some of which transversal to allaliphatic polyester-based vaccine particulate delivery systems, arepresented in Table 5.
The success of the pharmaceutical applications of PLA, PGA and PLGAcopolymers has led to the evaluation of other aliphatic polyesters, such asPCL. PCL has attracted attention because of its low cost, good safety profile
and compatibility with a variety of polymers. Moreover, PCL has severalother advantages, including high permeability to small drug moleculesand an exceptional ability to form blends with other polymers.PCL-based systems have a great potential for protein delivery mainlybecause, during particle degradation, the decrease inmicroenvironmentalpH is not so pronounced and protein integrity is better preserved. In fact,PCL degrades very slowly and, unlike PLGA, does not generate an acidicenvironment that could adversely affect the antigenicity of the vaccine;hence, PCL is a promising vaccine carrier [229].
Synthetic polymer chemistry evolution has brought almost unlimitedstrategies for producing block copolymers beyond the established AB,with higher numbers and types of repeating units. Combining morechemically distinct blocks and block types in ABA or BAB triblock copoly-mers, multiblock, star-block and graft copolymers offer unparalleledopportunities for designing new nanostructured materials with diversi-fied functionalities and desirable properties [230,231]. AB-diblock andmultiblock copolymers are also known as amphiphilic block copolymers(ABCs) as they possess amphiphilic properties due to their two regionsof distinct chemical nature that undergo phase separation as a result ofchain association in solvents that selectively dissolve one of the blocks.This process results in the formation of nanoscopic supramolecularcore/shell structures – polymeric micelles – by the relative interactionsbetween the hydrophilic and hydrophobic blocks with one another andthe surrounding medium. The chemical nature of the core-formingblockmay allowdrug incorporation via chemical, physical, and/or electro-static means [232]. These systems offer several advantages for drugdelivery as it may be possible to choose appropriate block copolymersfor specific purposes, such as prolonging circulation time, introductionof targeting moieties, and modification of the drug-release profile [233].Poly(ethylene glycol)-block-poly(ester) is one class of the most frequentABCs used to polymericmicelle formation. Triblock copolymermicelles ofPLA–PEG–PLA and PLGA–PEG–PLGA were used to encapsulate plasmidDNA encoding for a multiple-epitope antigen gene of the hepatitis Cvirus (HCV). As controlled-released gene coatings, these triblock micelleshad shown high DNA loading efficiency, low cytotoxicity, a sustained-release profile and achieved satisfying immune responses in vivo [234].A new poly(lactide-co-glycolide-co-ε-caprolactone)-graft mannosylatedpoly(ethylene oxide) copolymer was synthesized by combination of“Clip” and “Click” chemistries and demonstrated potential to formulatemicelles with DC- and macrophage-targeting capacity [177].
In addition to the amphiphilic structure, block copolymers can also beproduced by stimuli responsive copolymers by combining degradablehydrophobic blocks with functional hydrophilic ones, allowing the ad-ministration of polymer–drug conjugates in block copolymer-basedformulations where degradation of the amphiphilic block copolymerleads to a destabilization of aggregates. Barz et al. (2010) combined aN-(2-hydroxypropyl)-methacrylamide (HPMA)with PLA to synthesizednovel P(HPMA)-block-P(DLLA) copolymers combining ROP, reversibleaddition fragmentation chain transfer (RAFT) polymerization as well asthe activated ester approach [235]. Because the lactide/lactic acid mono-mer exists in three diastereomeric forms, PLA possesses chiral propertiesand can be found in four forms: poly(L-lactic acid) (PLLA), poly(D-lacticacid) (PDLA), poly(D,L-lactic acid) (PDLLA) – a racemic mixture of PLLAand PDLA – and meso-poly(lactic acid). When polymerized, atactic,heterotactic and isotactic chains exhibit strikingly different crystallinity,thermal properties and degradability. Considering its use in biomedicalresearch, only PLLA and PDLLA have shown promise and thus havebeen extensively studied [236]. Barz et al. (2012) also verified that poly-mer tacticity and stereoregularity represent a key feature ondeterminingcellular uptake and efficiency for the P(HPMA)-block-P(DLLA) copolymerdrug formulations [237]. DDS based on PEG-aliphatic polyester functionalblock copolymers can also exploit both the thick membrane of the blockcopolymer vesicles and their aqueous lumen for drug delivery as well aspH-triggered release within endolysosomes [238].
Aliphatic polyesters can also form star polymers by means of ROP ofcyclic esters in a “core-first” approach to polymer synthesis [239]. Star

190 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
polymers are branched, multi-armed polymeric structures in which thebranches radiate from a central core. They offer an increased concentra-tion of functional end groups for polymers of equal molecular weight,have improved solubility and often have lower melt viscosities, differentthermal properties and improved physical processing. Aliphatic biode-gradable star polymers have been prepared from the monomers lactide,caprolactone, glycolide, b-butyrolactone and trimethylene carbonate aswell as from their copolymers [240]. They have found particular utilityas controlled release DDSs andmolecular imaging tools. Their hydropho-bic cores and hydrophilic coronas can accommodate hydrophobic drugswithin their interior and water-soluble molecules in their hydrophilicarms, providing transport of the payload through aqueousmedia controlthe rate of drug release [241–243]. It is noteworthy that antigens and ad-juvants can be either hydrophilic or hydrophobicmolecules, making starpolymers promising tools for vaccine delivery systems. Unfortunately,these structures are still under-explored as vaccine delivery systems.
Apart from the biodegradability, the biocompatibility of a polymer isdependent on the elimination of its degradation products from the body,a property known as bioresorbability [244]. The homopolymers PLA, PGAand PCL and their copolymers are known to be bioresorbable [193,245].Aliphatic polyesters are bulk-eroding polymers, meaning that erosionhappens throughout the polymer bulk and not exclusively from thesurface inwards as happens with surface-eroding polymers such aspolyanhydrides [246]. The in vivo degradation of PLGA (and also otherpolyesters) happens by a chemical hydrolysis reaction of ester bonds inits backbone resulting in the production of the originalmonomers, (lacticacid and glycolic acid in the case of PLGA) [247]. These resulting productsof PLGA degradation are easily metabolized in the body via the Krebscycle and thus physiologically eliminated [248]. The reaction can beacid- or base-catalyzed, where the acid-catalyzed is the most relevant.The source of the catalyst can be external (non-autocatalytic reaction)or internal (autocatalytic reaction) when it happens by the carboxylic
Fig. 2. Immune system targeting by nanoparticles for cancer vaccines. Biodegradablepolymeric nanoparticles (NPs) can entrap tumor antigens along with adjuvants, suchas the Toll-like receptor ligands (TLRs), and be targeted to specific receptors expressedon dendritic cells (DCs). After uptake, polymeric NPs start to degrade and release theantigens and adjuvants. Binding of adjuvants to their cognate receptors will providestimulus to the initiation of DC maturation. During this process, DCs upregulate the ex-pression of cell surface molecules, such as MHC class I and class II, and costimulatorymolecules, such as CD40, the receptor for CD40L expressed by naïve T cells, and alsoCD80 and CD86, which are the ligands for the immunoglobulin superfamily memberCD28, also expressed by naïve T cells. Mature DCs also release cytokines, such as typeI IFN and IL-12, which act on T cells in a paracrine way. This process culminates withthe presentation of antigens through MHC-peptide complexes to T cells. T cells under-go clonal expansion and proceed to tumor destruction, a process that involves the ac-tion of cytotoxic T lymphocytes (CTL) along with T helper 1 cells (Th1) as well as othereffector cells, such as macrophages (Mph), natural killer T cells (NK) and granulocytes(Gran).
acid end groups of the polymer chain [249]. The degradation rate ofPLGA is related to the monomer ratio used in production: glycolic acidis slightly more hydrophilic than lactic acid, and its higher contentleads to higher hydrolysis rates [250]. From the several monomer ratiocombinations available in the market (50:50, 65:35, 75:25 and 80:20)PLGA 50:50 is a fairly hydrophilic polymer and has the fastest biodegra-dation rate of the PLGA polymers [251]. For instance, PLGA 50:50 of14 kDa is almost completely degraded within 30 days in water [252].PCL degradation is a homogenous process that happens in two stages:(i) the non-enzymatic hydrolytic cleavage of ester groups, and (ii) intra-cellular degradation for more crystalline polymers and of lowmolecularweight (less than 3000 kDa) the polymer is shown to undergo intracel-lular degradation [209]. The liberated carboxylic acid end groups catalyzethe hydrolysis, i.e., the cleavage of additional ester groups.
There is no consensus opinion as towhether aliphatic polyester-basedvaccine delivery systems are immunomodulatory by themselves(i.e., without the addition of an immunomodulator molecule) orcompletely inert systems once inside the body [253]. Some syntheticpolymeric NPs have demonstrated to have immunomodulatoryproperties. For instance, Frick et al. (2012) showed that sulfonate- andphosphonate-functionalized polystyrene NPs enhanced DC maturationand T-cell stimulatory capacity [254]. A study on PLGA MPs and NPsdemonstrated potent inflammatory stimulus with NF-κB translocationto cell nucleus and further pro-inflammatory cytokine production,mainly by MP-stimulated cells [255]. On the other hand, it has beendemonstrated that empty PLGAMPs with or without polycationic coat-ings did not induced significantmaturation and did not affect the capac-ity of DCs to respond to a maturation-inducing cocktail afterwards[256]. Moreover, at least some features related to their physicochemicalproperties and the passive targeting of the immune system can be men-tioned as an intrinsic adjuvant effect [21]. These factors are enumeratedin Table 6. It is well known that non-functionalized polymeric particlescan be rapidly removed from the blood stream by the macrophages ofthe MPS. However, it is not clear if aliphatic polyester particles are ableto induce complement activation during this process. It seems that hy-drophobic surfaces and chemical groups such as –NH3, –OH or –COOHmight induce the activation of complement [257]. Also, other parameterssuch as chain length, architecture (e.g., linear, branched, star-shaped,di-block and tri-block co-polymers, dendrimers), and solution properties(monomer versus micellar forms) may affect complement activationdifferently and through different initiation pathways [258,259].
The “double emulsion solvent evaporation” method has been widelyapplied to produce aliphatic polyester-based NPs [138,260,261]. Thismethod allows the production of water-in–oil-in–water nano-emulsionsthat originate NP suspensions after the evaporation of organic phase[262]. The main particularity of nano-emulsions is their great stability ofdroplet suspension and absence of the flocculation phenomenon innano-emulsions, naturally prevented by steric stabilization, essentiallydue to the sub-micrometric droplet size [263]. Negative aspects arefrequently pointed to this method as the contact with organic solvents,the use of high-shear devices and the need of surfactants to stabilizeemulsion droplets. Even though, the maintenance of integrity andantigenicity of proteins and peptides encapsulated into NPs using thedouble-emulsion solvent evaporation method has been confirmed byour group and others using SDS-PAGE and Western blotting techniques,respectively [220,222,227,264]. Also, the residual amount of organicsolvent on PCL MS has been quantified by nuclear magnetic resonance(NMR) spectroscopy technique and demonstrated to be very low [265].The permanence of surfactant molecules at NP surface remains a concernsince these compounds are normally associated with toxic effects. Nor-mally, NP formulation comprises washing procedures such as repetitivecentrifugation, filtration or dialysis to remove the excess of surfactantfrom NP surface. Even though, a fraction of the surfactant remains associ-ated with the NP due to the interconnected network with the polymer atthe interface [266]. It seems that some polymers and surfactants that re-main associated with aliphatic polyester NPs can have an impact on
Table 4Examples of synthetic aliphatic polyesters and their synthesis methods.
Monomer molecular structure Examples of synthesis methods Polymer molecular structure
HomopolymersROP of glycolidePolycondensation of glycolic acid [198]
Polyglycolide or polyglycolic acid (PGA)
ROP of lactidePolycondensation of lactic acid [199,200]
Polylactide or polylactic acid (PLA)
ROP of caprolactoneFree radical ROP of 2-methylene-1-3-dioxepanePolycondensation of 6-hydroxyhexanoic acid[201,202]
Polycaprolactone (PCL)
ROP of 1,5-dioxepan-2-one [193]
Poly(1,5-dioxepan-2-one) (PDXO)
ROP of trimethylene carbonate [193]
Poly(trimethylene carbonate) (PTMC)
ROP of β-butyrolactonePolycondensation of 3-hydroxybutyric acid[203]
Poly(3-hydroxybutyrate) (P3HB)
Combination of propylene glycol and fumarylchloride to form an intermediate, fumaric diester,which is then transesterified [204]
Poly(propylene fumarate) (PPF)
Esterification and polycondensation of adipicacid and ethylene glycol [205]
Polyethylene adipate (PEA)
CopolymersRing-opening copolymerization of glycolideand lactide [206]
Poly(lactic-co-glycolide) (PLGA)
ROP of butyrolactone and valerolactone (oligomericaluminoxane as a catalyst) orcopolymerization of 3-hydroxybutyric acidand 3-hydroxypentanoic acid[207]
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)
(continued on next page)
191J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199

Table 4 (continued)
Monomer molecular structure Examples of synthesis methods Polymer molecular structure
Ring-opening copolymerization of α-cloro-ε-caprolactone,glycolide and lactideThe chloride can be then substituted to furtherfunctionalization [177]
Poly(lactide-co-glycolide-co-α-cloro-ε-caprolactone)(PLGA-α-Cl-ε-PCL)
n, number of repeated units; x, number of units of lactic acid; y, number of units of glycolic acid; k, number of units of 3-hydroxybutyric acid; z, number of units of3-hydroxypentanoic acid; r, number of units of α-cloro-ε-caprolactone.
192 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
NP-cell interaction process and even increment their immunogenicity[267]. For instance, the addition of poloxamer 188 (an amphiphilictriblock co-polymer with PEG chains linked to a more hydrophobic poly-propylene glycol backbone) to PLGA NPs increased in vivo intranasal up-take and the ability of the particle targeting peptide to interact with themucosal epithelial membrane without specifically increasing particle ad-hesion [268]. Another study comparing the effect of coating PLGA NPswith either Pluronic F127 (non-ionic bifunctional triblock copolymersurfactant) and the commonly used polyvinyl alcohol (PVA) on PLGANPs properties demonstrated that the first surfactant provides higher in-ternalization of NPs by prostate and breast cancer cells [269]. It is possibleto quantify the remaining surfactant on NP suspensions and optimize NPformulation development providing insight into the mechanisms of NPstabilization [270]. It is also possible to formulate surfactant-free PLGANPs by using alternative stabilizers such as sucrose [271].
The non-specific nature of the phagocytosis by the MPS representsone of the main hurdles that need to be overcome in order to achieveselective targeting of particulates to selected cells. It is known that alonger exposure to smaller amounts of antigen provides more effectiveimmune responses with proper clonal expansion of CD8+ T cells andacquisition of memory [272]. In a study in which pegylated PLGA NPswere used as a strategy for the delivery of recombinant hepatitis Bsurface antigen (HBsAg), it was demonstrated that the presence of PEGon the surface of NPs delayed their uptake by DCs and a gradual increasein uptake was observed at longer incubation times. This study furtherdemonstrated the in vivo production of anti-HBs antibodies after immu-nization of mice with this particulate vaccine [176]. In a dermal DNAvaccine approach, immobilization of NPs in the extracellular matrix,possibly caused by their electrostatic interactions with negativelycharged extracellular matrix components, was resolved by shieldingtheir surface by pegylation, which improved in vivo antigen expressionmore than 55-fold. These observations suggest that charge shielding pro-vides a generally applicable strategy for the development of dermal ap-plied vaccine formulations [178]. Another potential advantage providedby hydrophilic PEG is the improvement in biocompatibility of the deliv-ery vehicle. This effect occurs because most of the biological environ-ment is hydrophilic in nature and biocompatibility appears to becorrelated directly with the degree of hydrophilicity that a surface ex-hibits [273]. Moreover, pegylated NPs tend to present slightly smaller di-ameters with lower polydispersities than those prepared fromnon-pegylated polymers [274].
7. Rational choice of the route of administration for cancer vaccines
DCs can be found in considerable numbers in the epidermis and der-mis, where they continuously sample antigens and rapidly start immuneresponses upon the presence of a pathogenic stimulus. As such, the skin isan attractive organ to be used as an immunization route [275]. LCs are theonly DCs that are found in the epidermis during the steady-state. LCsextend dendritic processes that allow them to sample a large area of theskin and to have access not only to antigens from pathogens that invadethe epidermis but also to self-antigens, as well as tumor antigens fromepidermal neoplasias [276]. Similar to all peripheral DCs, LCs migrate toregional LNs after acquisition of the antigen in the periphery where they
can present antigen to naïve and memory T cells. Upon an inflammatorystimulus, LCs arrive in the LNswith a relatively long delay (~72 h), whichreflects the time needed to cross the basement membrane that separatesthe epidermis and dermis. In the dermis, during the steady-state, a varietyof immune cells can be found, including memory T cells, mast cells, anddermal DCs. Like LCs, dermal DCs migrate to skin-draining LNs, both inthe steady-state and in response to inflammation. They are also able topresent antigen acquired in the periphery to T cells. Unlike LCs, they arriveat LNs more quickly and can be identified within 18 h after stimulation[277].
Another primary reason for considering the intradermal route is thepotential for safe immune stimulation, because this route avoids directcontact between potent adjuvants and the general circulation. Moreover,the most abundant epidermal cells, keratinocytes, also play an importantrole in innate immunity on the skin as they produce a wide range ofcytokines, chemokines, and antimicrobial peptides, and they expressseveral TLRs [278]. As a result, these cells are able to kill invading patho-gens and recruit immune cells, such as DCs, helping tomaintain an appro-priate balance between reactivity and tolerance of the immune system. Inaddition, keratinocytes have been reported to function as accessory cells,able to prime naïve skin reactive T cells [279].
As mentioned earlier, when immature DCs located in peripheraltissues encounter activatory/inflammatory signals present in pathogens,they incorporate them and become activated. Activated DCs migrate to-ward the sentinel LNs where they present the processed antigens tonaïve T cells, stimulating them to proliferate and differentiate, and initi-ating an adaptive immune response [75]. Duringmaturation, DCs acquirea specific phenotype, which is associated with several coordinatedevents, such as loss of endocytic and phagocytic receptors, high-levelexpression of costimulatory molecules, changes in morphology, andactivation of the Ag-processingmachinery, including a shift in lysosomalcompartments [280]. Mature DCs are highly capable of presenting theprocessed antigens but their capacity to subsequently uptake andprocess newly encountered antigens is much reduced [281]. This factmay lead to the conclusion that targeting DCs at peripheral tissues,where they have an immature phenotype, is a much more logicalapproach than targeting DCs at secondary lymphoid organs. Neverthe-less, preclinical and clinical studies in which DCs were targeted directlyat the LNs through direct vaccine injections into this organ have showngood responses [282–284].
When injection routes were compared in preclinical studies with thegoal of optimizing DNA vaccination, administration of a plasmid DNAvaccine directly into lymphoid tissues was more effective for generationof CTL immune responses by at least three orders of magnitude com-pared to intradermal or intramuscular routes [285]. Another study withnaked-antigen-encoding RNA vaccination showed that, compared toother application routes, the intranodal route generated stronger T-cellresponses and superior antitumor immunity because of the bioavailabil-ity and inherent adjuvant effects of RNA in the LN microenvironment[286]. This apparent paradox can be explained by recent findings show-ing that although mature DCs markedly downregulate their capacity forphagocytosis and general depletion of MHC II from late endocytic com-partments, they continue to capture, process, and present antigens inter-nalized via endocytic receptors, suggesting that they may continuously

Table 5Main advantages and disadvantages of PLGA as a particulate vaccine delivery systemcomponent.
Advantages
• Biodegradability and biocompatibility;• Approval for parenteral use by regulatory authorities around the world;• Commercially available with different physico-chemical properties (severalmolecular weights and lactide:glycolide ratios);
• Drug release profile can be tailored by selecting PLGA polymers with the appropriateproperties. A pulsatile release is also possible, which provides more effective immuneresponses as it can avoid the risk of tolerance and substitute the need of severalboosting administrations typically required to induce protective immunity [225];
• Protection of encapsulated biomolecules from in vivo degradation in the bloodstream, gastro-intestinal tract and nasal mucosa [226,227];
• Delivery of antigens to be presented by both MHC class I andMHC class II pathways;• Transportation of more than one molecule concomitantly in the same particulatedelivery system (e.g., several antigens or co-encapsulation of antigen and adjuvant)increasing the probability of coordinate and synergistic action;
• Capacity to induce strong T cell responses containing very low doses of antigens andadjuvants, allowing the use of lower doses of these molecules minimizing thepotential side effects often associated with the use of adjuvants [228];
• Possibility of blending or co-polymerization with other materials;• Can be stably stored and easily scaled up for pharmaceutical manufacture.
Disadvantages
• Environment acidification upon degradation compromising cargo-stability;• Risk of denaturation of the incorporated biomacromolecules by the organic solventsand/or shear stress used during particle formulation;
• Low encapsulation efficiencies of some macromolecules;• Lack of suitable functional groups for efficient covalent bioconjugation of bioactiveligands or the adsorption of negatively charged therapeutics such as nucleic acids;
• Required sterilization for clinical use;• Potential expensive DDS.
Table 6Inherent adjuvanticity of aliphatic polyester particulate delivery systems for vaccines.
• Ability to protect the immunogens from rapid degradation and clearance;• Efficient internalization by APCs;• Extracellular reservoirs of immunogens due to sustained release occurring for lengthyperiods (depot effect);
• Intracellular reservoirs of immunogens in APCs after particle uptake so thatintracellularly released antigens can be presented by the APCs to specific T cells overextended periods of time;
• Capacity to directly migrate to lymph nodes after peripheral administration (forparticles b200 nm);
• Codelivery of several immunogens, being either several antigens or antigens andadjuvant molecules;
• Capacity to induce presentation of antigens through bothMHC class I andMHC class IIcomplexes.
193J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
initiate responses to newly encountered antigens during the course ofinfection [287]. It has also been found that the lymphoid organs containa large cohort of immature DCs, most likely to maintain peripheraltolerance, which can respond to infections that reach these organs andmature in situ [288]. The fact that DCs exist in high concentrations inthe LNs and a substantial fraction of them are phenotypically and func-tionally immature and thus able to process new antigens, may explainthe good responses obtainedwith the administration of antigens directlyin the LNs [289]. Hence, targeting anti-tumor vaccines to LNs throughdirect intra-nodal administration or by using particulate deliverysystems that can travel through the lymphatic vessels and reach theLNs seems to be an attractive option. Refer to Section 4 for detailed infor-mation regarding particle size requirements to target lymphoid organs.Regarding future applications in clinical trials, injection into a humansubcutaneous LNs is readily feasible with the use of ultrasound guidanceand is a simple procedure that takes only a few minutes [290].
8. Regulatory issues and concluding remarks
The ultimate goal of the current review was to highlight thepotential of NPs, in particular those constituted by biodegradablealiphatic polyesters, for use as cancer vaccines to target tumor antigensand adjuvants to DCs for priming antigen-specific T cell responses. Im-proved understanding of the role of the immune system in controllingtumor growth and recent developments in the field of nanoparticulatecancer vaccines have created a degree of knowledge that promises tolead to favorable results with this approach in the near future. Never-theless, several questions remain unanswered and difficulties need tobe overcome. Methods to clinically evaluate cancer vaccine responsesneed to be validated. More information about the most relevanttumor antigens and epitopes, the optimal patient population and opti-mal doses and schedules need to be collected.
Regulatory issues regarding this area deal with two sets of issues: thematerials involved and the cancer vaccine approach in itself. From thematerials side, certainly a number of polymeric and macromolecular
carriers havemade theway to clinical practice and being used for paren-teral administration will have a facilitated access to regulatory appraisal.But in any specific case a full assessment will have to be considered andthe nonclinical requirements to clinical translation will vary accordingalso to disease pathophysiology and its impact (together with materialproperties and administration routes) on overall clinical benefit-riskassessment [10,291–293]. A number of activities particularly dealingwith vaccines have a growing place in the European Medicines Agencyactivities. Even though, current priorities go further than specific anti-infectious vaccines and even consider DNA vaccines meanwhile theylack a strong commitment on the Cancer Therapeutic Vaccines area[294]. Current regulatory compliance for clinical development ofvaccines applicable also to cancer vaccines, in Europe has long timedealt both with Note for guidance on the clinical evaluation of vaccines[295] and also Guideline on adjuvants in vaccines for human use [296]as well as other relevant documents for vaccines in general. Meanwhile,FDA as issued in October 2011 a relevant guidance document preparedby the Office of Cellular, Tissue and Gene Therapies in FDA's Center forBiologics Evaluation and Research. The guidance on “Clinical Consider-ations for Therapeutic Cancer Vaccines” aims at providing guidance to“sponsors who wish to submit an Investigational New Drug application(IND) for a therapeutic cancer vaccine with recommendations on criticalclinical considerations for investigational studies of these products” [297].This will have amajor impact in present and future plans tomove CancerTherapeutic Vaccines.
Tumor vaccines might have the capacity to combat alreadyestablished malignant disease, but it must be kept in mind thatthe success of immunological strategies will correlate with thestage of the disease. The ideal scenario for the application of a therapeuticcancer vaccine might be minimal residual disease with low numbers ofaberrant or cancer stem cells, where the actively induced immunemech-anisms would have a fair chance to prevent tumor relapse throughreduction of circulating tumor cells and micrometastases. In addition,prophylactic TAA-based vaccines may be conceivable in cases of heredi-tary predisposition.
Notwithstanding these remaining issues, the development ofnanoparticulate cancer vaccines has received great impetus and effortsare being applied to the development of novel vaccine adjuvants andnanoparticulate delivery systems, moving from the understanding ofthe inherent cellular mechanisms to the evaluation of therapeutic po-tential in clinical practice.
Acknowledgments
The authors would like to acknowledge the institutional affiliationsthat supported the construction of the present manuscript, namely,iMed.UL— Research Institute for Medicines and Pharmaceutical Sciences,from the Faculty of Pharmacy of the University of Lisbon, Portugal, andLouvain Drug Research Institute from the Université Catholique de Lou-vain, Brussels, Belgium. Also, the authors would like to acknowledge

194 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
Fundação para a Ciência e Tecnologia, Ministério da Ciência e Tecnologia,Portugal (SFRH/BD/64295/2009 and PTDC/SAU-FAR/119389/2010) andFonds de la Recherche Scientifique Médicale (Belgium) for supportingJoana M. Silva.
References
[1] J. Begley, A. Ribas, Targeted therapies to improve tumor immunotherapy, Clin.Cancer Res. 14 (14) (2008) 4385–4391.
[2] P.M. Dimberu, R.M. Leonhardt, Cancer immunotherapy takes a multi-faceted ap-proach to kick the immune system into gear, Yale J. Biol.Med. 84 (4) (2011) 371–380.
[3] D.E. Speiser, P. Romero, Molecularly defined vaccines for cancer immunothera-py, and protective T cell immunity, Semin. Immunol. 22 (3) (2010) 144–154.
[4] H. Benlalam, N. Labarrière, B. Linard, L. Derré, E. Diez, M.C. Pandolfino, M.Bonneville, F. Jotereau, Comprehensive analysis of the frequency of recognitionof melanoma associated antigen (MAA) by CD8 melanoma infiltrating lympho-cytes (TIL): implications for immunotherapy, Eur. J. Immunol. 31 (7) (2001)2007–2015.
[5] R. Barnetson, G. Halliday, Regression in skin tumours: a common phenomenon,Australas. J. Dermatol. 38 (Suppl. 1) (1997) S63–S65.
[6] U.N. Rao, S.J. Lee, W. Luo, M.C. Mihm, J.M. Kirkwood, Presence of tumor-infiltratinglymphocytes and a dominant nodule within primary melanoma are prognosticfactors for relapse-free survival of patients with thick (t4) primary melanoma:pathologic analysis of the e1690 and e1694 intergroup trials, Am. J. Clin. Pathol.133 (4) (2010) 646–653.
[7] H. Yaziji, A.M. Gown, Immunohistochemical markers of melanocytic tumors, Int.J. Surg. Pathol. 11 (1) (2003) 11–15.
[8] C. Nicholas, G.B. Lesinski, Immunomodulatory cytokines as therapeutic agentsfor melanoma, Immunotherapy 3 (5) (2011) 673–690.
[9] L. Vujanovic, L.H. Butterfield, Melanoma cancer vaccines and anti-tumor T cellresponses, J. Cell. Biochem. 102 (2) (2007) 301–310.
[10] R. Duncan, R. Gaspar, Nanomedicine(s) under the microscope, Mol. Pharm. 8 (6)(2011) 2101–2141.
[11] E. Schlosser, M. Mueller, S. Fischer, S. Basta, D.H. Busch, B. Gander, M. Groettrup,TLR ligands and antigen need to be coencapsulated into the same biodegradablemicrosphere for the generation of potent cytotoxic T lymphocyte responses,Vaccine 26 (13) (2008) 1626–1637.
[12] R.A. Henderson, S. Mossman, N. Nairn, M.A. Cheever, Cancer vaccines and immu-notherapies: emerging perspectives, Vaccine 23 (17–18) (2005) 2359–2362.
[13] P.H. Verardi, A. Titong, C.J. Hagen, A vaccinia virus renaissance: new vaccine and im-munotherapeutic uses after smallpox eradication, Hum. Vaccin. Immunother. 8 (7)(2012) 961–970.
[14] P. Ravanfar, A. Satyaprakash, R. Creed, N. Mendoza, Existing antiviral vaccines,Dermatol. Ther. 22 (2) (2009) 110–128.
[15] S. Riedel, Edward Jenner and the history of smallpox and vaccination, Proc.(Bayl. Univ. Med. Cent.) 18 (1) (2005) 21–25.
[16] S.A. Plotkin, History of Vaccine Development, in: S.A. Plotkin (Ed.), Springer NewYork, New York, NY, 2011.
[17] T.A. Waldmann, Immunotherapy: past, present and future, Nat. Med. 9 (3) (2003)269–277.
[18] J.I. Lundin, H. Checkoway, Endotoxin and cancer, Environ. Health Perspect. 117(9) (2009) 1344–1350.
[19] E. Vacchelli, I. Martins, A. Eggermont, W.H. Fridman, J. Galon, C. Sautès-Fridman, E.Tartour, L. Zitvogel, G. Kroemer, L. Galluzzi, Trial watch: peptide vaccines in cancertherapy, Oncoimmunology 1 (9) (2012) 1557–1576.
[20] P. Malyala, D.T. O'Hagan, M. Singh, Enhancing the therapeutic efficacy of CpGoligonucleotides using biodegradable microparticles, Adv. Drug Deliv. Rev. 61(3) (2009) 218–225.
[21] Y. Krishnamachari, S.M. Geary, C.D. Lemke, A.K. Salem, Nanoparticle deliverysystems in cancer vaccines, Pharm. Res. 28 (2) (2011) 215–236.
[22] L. Matera, The choice of the antigen in the dendritic cell-based vaccine therapyfor prostate cancer, Cancer Treat. Rev. 36 (2) (2010) 131–141.
[23] P. Van der Bruggen, C. Traversari, P. Chomez, C. Lurquin, E. De Plaen, B.J. Van denEynde, A. Knuth, T. Boon, A gene encoding an antigen recognized by cytolytic Tlymphocytes on a human melanoma, J. Immunol. 178 (5) (2007) 2617–2621.
[24] L. Novellino, C. Castelli, G. Parmiani, A listing of human tumorantigens recognizedbyTcells: March 2004 update, Cancer Immunol. Immunother. 54 (3) (2005) 187–207.
[25] L. Zhao, Z. Liu, D. Fan, Overview of mimotopes and related strategies in tumorvaccine development, Expert Rev. Vaccines 7 (10) (2008) 1547–1555.
[26] G. Parmiani, C. Castelli, P. Dalerba, R. Mortarini, L. Rivoltini, F.M. Marincola, A.Anichini, Cancer immunotherapy with peptide-based vaccines: what have weachieved? Where are we going? J. Natl. Cancer Inst. 94 (11) (2002) 805–818.
[27] J.P. Higgins, M.B. Bernstein, J.W. Hodge, Enhancing immune responses totumor-associated antigens, Cancer Biol. Ther. 8 (15) (2009) 1440–1449.
[28] S. Pejawar-Gaddy, Y. Rajawat, Z. Hilioti, J. Xue, D.F. Gaddy, O.J. Finn, R.P. Viscidi, I.Bossis, Generation of a tumor vaccine candidate based on conjugation of aMUC1 pep-tide to polyionic papillomavirus virus-like particles, Cancer Immunol. Immunother. 59(11) (2010) 1685–1696.
[29] P. Dalerba, R.W. Cho, M.F. Clarke, Cancer stem cells: models and concepts, Annu. Rev.Med. 58 (2007) 267–284.
[30] M.H. Andersen, I.M. Svane, J.C. Becker, P.T. Straten, The universal character ofthe tumor-associated antigen survivin, Clin. Cancer Res. 13 (20) (2007)5991–5994.
[31] Z.-L. Liao, X.-D. Tang, M.-H. Lü, Y.-Y. Wu, Y.-L. Cao, D.-C. Fang, S.-M. Yang, H. Guo,Antitumor effect of new multiple antigen peptide based on HLA-A0201-restrictedCTL epitopes of human telomerase reverse transcriptase (hTERT), Cancer Sci. 103(11) (2012) 1920–1928.
[32] G. Parmiani, V. Russo, A. Marrari, G. Cutolo, C. Casati, L. Pilla, C. Maccalli, L. Rivoltini,C. Castelli, Universal and stemness-related tumor antigens: potential use in cancerimmunotherapy, Clin. Cancer Res. 13 (19) (2007) 5675–5679.
[33] A. Anichini, A. Molla, R. Mortarini, G. Tragni, I. Bersani, M. Di Nicola, A.M. Gianni, S.Pilotti, R. Dunbar, V. Cerundolo, G. Parmiani, An expanded peripheral T cell popula-tion to a cytotoxic T lymphocyte (CTL)-defined, melanocyte-specific antigen inmetastatic melanoma patients impacts on generation of peptide-specific CTLs butdoes not overcome tumor escape from immune surveillance in metastatic lesions,J. Exp. Med. 190 (5) (1999) 651–667.
[34] Y. Maeda, N. Hida, F. Niiya, K. Katagiri, M. Harada, H. Yamana, T. Kamura, M.Takahashi, Y. Sato, S. Todo, K. Itoh, Detection of peptide-specific CTL-precursorsin peripheral blood lymphocytes of cancer patients, Br. J. Cancer 87 (7) (2002)796–804.
[35] M. Noguchi, T. Sasada, K. Itoh, Personalized peptide vaccination: a new approachfor advanced cancer as therapeutic cancer vaccine, Cancer Immunol. Immunother.(2012), http://dx.doi.org/10.1007/s00262-012-1379-1.
[36] M. Sensi, A. Anichini, Unique tumor antigens: evidence for immune control ofgenome integrity and immunogenic targets for T cell-mediated patient-specificimmunotherapy, Clin. Cancer Res. 12 (17) (2006) 5023–5032.
[37] K. Itoh, A. Yamada, Personalized peptide vaccines: a new therapeutic modalityfor cancer, Cancer Sci. 97 (10) (2006) 970–976.
[38] G.A. Poland, R.B. Kennedy, I.G. Ovsyannikova, Vaccinomics and personalizedvaccinology: is science leading us toward a new path of directed vaccine devel-opment and discovery? G.F. Rall (Ed.), PLoS Pathog. 7 (12) (2011) e1002344.
[39] S.A. Rosenberg, J.C. Yang, N.P. Restifo, Cancer immunotherapy: moving beyondcurrent vaccines, Nat. Med. 10 (9) (2004) 909–915.
[40] J. Crespo, H. Sun, T.H. Welling, Z. Tian, W. Zou, T cell anergy, exhaustion, senes-cence, and stemness in the tumor microenvironment, Curr. Opin. Immunol. 25(2013) 1–8.
[41] T.F. Gajewski, Y. Meng, C. Blank, I. Brown, A. Kacha, J. Kline, H. Harlin, Immune resis-tance orchestrated by the tumor microenvironment, Immunol. Rev. 213 (2006)131–145.
[42] W.J. Lesterhuis, J.B. Haanen, C.J. Punt, Cancer immunotherapy—revisited, Nat.Rev. Drug Discov. 10 (8) (2011) 591–600.
[43] M. Aleksic, N. Liddy, P.E. Molloy, N. Pumphrey, A. Vuidepot, K.-M. Chang, B.K.Jakobsen, Different affinity windows for virus and cancer-specific T-cell recep-tors: implications for therapeutic strategies, Eur. J. Immunol. 42 (12) (2012)3174–3179.
[44] M. Hebeisen, L. Baitsch, D. Presotto, P. Baumgaertner, P. Romero, O. Michielin,D.E. Speiser, N. Rufer, SHP-1 phosphatase activity counteracts increased T cellreceptor affinity, J. Clin. Invest. 123 (3) (2013) 1044–1056.
[45] M.Y. Mapara, M. Sykes, Tolerance and cancer: mechanisms of tumor evasion andstrategies for breaking tolerance, J. Clin. Oncol. 22 (6) (2004) 1136–1151.
[46] S. Ray, A. Chhabra, S. Mehrotra, N.G. Chakraborty, A. Ribas, J. Economou, B.Mukherji, Obstacles to and opportunities for more effective peptide-based ther-apeutic immunization in human melanoma, Clin. Dermatol. 27 (6) (2009)603–613.
[47] H. Prado-Garcia, S. Romero-Garcia, D. Aguilar-Cazares, M. Meneses-Flores, J.S.Lopez-Gonzalez, Tumor-induced CD8+ T-cell dysfunction in lung cancer patients,Clin. Dev. Immunol. 2012 (2012) 741741.
[48] S. Mehrotra, A. Chhabra, S. Chattopadhyay, D.I. Dorsky, N.G. Chakraborty, B.Mukherji, Rescuing melanoma epitope-specific cytolytic T lymphocytes fromactivation-induced cell death, by SP600125, an inhibitor of JNK: implicationsin cancer immunotherapy, J. Immunol. 173 (10) (2004) 6017–6024.
[49] S. Mumprecht, C. Schürch, J. Schwaller, M. Solenthaler, A.F. Ochsenbein,Programmed death 1 signaling on chronicmyeloid leukemia-specific T cells resultsin T-cell exhaustion and disease progression, Blood 114 (8) (2009) 1528–1536.
[50] C. Zielinski, S. Knapp, C. Mascaux, F. Hirsch, Rationale for targeting the immunesystem through checkpoint molecule blockade in the treatment of non-small-cell lung cancer, Ann. Oncol. (2013), http://dx.doi.org/10.1093/annonc/mds647,(Electronic publication ahead of print).
[51] D. Lindau, P. Gielen, M. Kroesen, P. Wesseling, G.J. Adema, The immunosuppres-sive tumour network: myeloid-derived suppressor cells, regulatory T cells andnatural killer T cells, Immunology 138 (2) (2013) 105–115.
[52] R.Morita, Y.Hirohashi, N. Sato, Depletion of Tregs in vivo: a promising approach to en-hance antitumor immunity without autoimmunity, Immunotherapy 4 (11) (2012)1103–1105.
[53] T.L. Whiteside, Disarming suppressor cells to improve immunotherapy, CancerImmunol. Immunother. 61 (2) (2012) 283–288.
[54] R. Kennedy, E. Celis, Multiple roles for CD4+T cells in anti tumor immune responses,Immunol. Rev. 222 (1) (2008) 129–144.
[55] H.M. Zarour, J.M. Kirkwood, L.S. Kierstead,W. Herr, V. Brusic, C.L. Slingluff, J. Sidney,A. Sette, W.J. Storkus, Melan-A/MART-1(51-73) represents an immunogenicHLA-DR4-restricted epitope recognized by melanoma-reactive CD4(+) T cells,Proc. Natl. Acad. Sci. U. S. A. 97 (1) (2000) 400–405.
[56] S.P. Kerkar, L. Sanchez-Perez, S. Yang, Z. Borman, P. Muranski, Y. Ji, D.Chinnasamy, A.D.M. Kaiser, C. Hinrichs, C.A. Klebanoff, C. Scott, L. Gattinoni,R.A. Morgan, S.A. Rosenberg, N.P. Restifo, Genetic engineering of murineCD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies, J.Immunother. 34 (4) (2011) 343–352.
[57] H. Shen, A.L. Ackerman, V. Cody, A. Giodini, E.R. Hinson, P. Cresswell, R.L. Edelson,W.M. Saltzman, D.J. Hanlon, Enhanced and prolonged cross presentation following

195J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
endosomal escape of exogenous antigens encapsulated in biodegradablenanoparticles, Immunology 117 (1) (2006) 78–88.
[58] J. Bukur, S. Jasinski, B. Seliger, The role of classical and non-classical HLA class Iantigens in human tumors, Semin. Cancer Biol. 22 (4) (2012) 350–358.
[59] J. Bubeník, MHC class I down-regulation: tumour escape from immune surveil-lance? (Review), Int. J. Oncol. 25 (2) (2004) 487–491.
[60] G.A. Rabinovich, D. Gabrilovich, E.M. Sotomayor, Immunosuppressive strategiesthat are mediated by tumor cells, Annu. Rev. Immunol. 25 (2007) 267–296.
[61] J. Liu, S. Liao, B. Diop-Frimpong, W. Chen, S. Goel, K. Naxerova, M. Ancukiewicz, Y.Boucher, R.K. Jain, L. Xu, TGF-β blockade improves the distribution and efficacy oftherapeutics in breast carcinoma by normalizing the tumor stroma, Proc. Natl. Acad.Sci. U. S. A. 109 (41) (2012) 16618–16623.
[62] S. Nawrocki, A. Mackiewicz, Genetically modified tumour vaccines—where weare today, Cancer Treat. Rev. 25 (1) (1999) 29–46.
[63] M.L. Taddei, E. Giannoni, G. Comito, P. Chiarugi, Microenvironment and tumorcell plasticity: an easy way out, Cancer Lett. (2013), http://dx.doi.org/10.1016/j.canlet.2013.01.042.
[64] Y. Huang, S. Shah, L. Qiao, Tumor resistance to CD8+ T cell-based therapeuticvaccination, Arch. Immunol. Ther. Exp. (Warsz.) 55 (4) (2007) 205–217.
[65] J.D. Ahlers, I.M. Belyakov, Memories that last forever: strategies for optimizingvaccine T-cell memory, Blood 115 (9) (2010) 1678–1679.
[66] T. Nishimura, K. Iwakabe, M. Sekimoto, Y. Ohmi, T. Yahata, M. Nakui, T. Sato, S.Habu, H. Tashiro, M. Sato, A. Ohta, Distinct role of antigen-specific T helpertype 1 (Th1) and Th2 cells in tumor eradication in vivo, J. Exp. Med. 190 (5)(1999) 617–627.
[67] Y. Becker,Molecular immunological approaches to biotherapy of human cancers—areview, hypothesis and implications, Anticancer Res. 26 (2A) (2006) 1113–1134.
[68] E. Gilboa, The promise of cancer vaccines, Nat. Rev. Cancer 4 (5) (2004) 401–411.[69] M. Rossi, J.W. Young, Human dendritic cells: potent antigen-presenting cells at the
crossroads of innate and adaptive immunity, J. Immunol. 175 (3) (2005) 1373–1381.[70] M. Merad, M.G. Manz, Dendritic cell homeostasis, Blood 113 (15) (2009) 3418–3427.[71] O. Takeuchi, S. Akira, Pattern recognition receptors and inflammation, Cell 140
(6) (2010) 805–820.[72] T.F. Gajewski, M.B. Fuertes, S.-R. Woo, Innate immune sensing of cancer: clues
from an identified role for type I IFNs, Cancer Immunol. Immunother. 61 (8)(2012) 1343–1347.
[73] L. Delamarre, M. Pack, H. Chang, I. Mellman, E.S. Trombetta, Differential lyso-somal proteolysis in antigen-presenting cells determines antigen fate, Science307 (5715) (2005) 1630–1634.
[74] N. Kadowaki, Dendritic cells: a conductor of T cell differentiation, Allergol. Int.56 (3) (2007) 193–199.
[75] A. Martín-Fontecha, S. Sebastiani, U.E. Höpken, M. Uguccioni, M. Lipp, A.Lanzavecchia, F. Sallusto, Regulation of dendritic cell migration to the draininglymph node, J. Exp. Med. 198 (4) (2003) 615–621.
[76] A.K. Palucka, H. Ueno, J. Fay, J. Banchereau, Dendritic cells: a critical player incancer therapy? J. Immunother. 31 (9) (2008) 793–805.
[77] H. Hillaireau, P. Couvreur, Nanocarriers' entry into the cell: relevance to drugdelivery, Cell. Mol. Life Sci. 66 (17) (2009) 2873–2896.
[78] J.A. Hubbell, S.N. Thomas, M.A. Swartz, Materials engineering for immunomodulation,Nature 462 (7272) (2009) 449–460.
[79] M. Raghavan, N. Del Cid, S.M. Rizvi, L.R. Peters, MHC class I assembly: out andabout, Trends Immunol. 29 (9) (2008) 436–443.
[80] I. Hwang, D.L. Ki, Receptor-mediated T cell absorption of antigen presentingcell-derived molecules, Front. Biosci. 16 (2011) 411–421.
[81] S. Takeuchi, M. Furue, Dendritic cells: ontogeny, Allergol. Int. 56 (3) (2007) 215–223.[82] P. Muranski, N.P. Restifo, Adoptive immunotherapy of cancer using CD4+ T
cells, Curr. Opin. Immunol. 21 (2) (2009) 200–208.[83] B.A. Cobb, D.L. Kasper, Coming of age: carbohydrates and immunity, Eur. J.
Immunol. 35 (2) (2005) 352–356.[84] G. De Libero, L. Mori, How the immune system detects lipid antigens, Prog. Lipid
Res. 49 (2) (2010) 120–127.[85] F. Facciotti, M. Cavallari, C. Angénieux, L.F. Garcia-Alles, F. Signorino-Gelo, L.
Angman, M. Gilleron, J. Prandi, G. Puzo, L. Panza, C. Xia, P.G. Wang, P.Dellabona, G. Casorati, S.A. Porcelli, H. Salle, L. Mori, G. De Libero, Fine tuning byhuman CD1e of lipid-specific immune responses, Proc. Natl. Acad. Sci. U. S. A. 108(34) (2011) 14228–14233.
[86] G. De Libero, A. Collmann, L. Mori, The cellular and biochemical rules of lipid an-tigen presentation, Eur. J. Immunol. 39 (10) (2009) 2648–2656.
[87] D.I. Godfrey, D.G. Pellicci, O. Patel, L. Kjer-Nielsen, J. McCluskey, J. Rossjohn, An-tigen recognition by CD1d-restricted NKT T cell receptors, Semin. Immunol. 22(2) (2010) 61–67.
[88] A. Schiefner, I.A. Wilson, Presentation of lipid antigens by CD1 glycoproteins,Curr. Pharm. Des. 15 (28) (2009) 3311–3317.
[89] S.-I. Fujii, K. Shimizu, C. Smith, L. Bonifaz, R.M. Steinman, Activation of natural killer Tcells by alpha-galactosylceramide rapidly induces the fullmaturation of dendritic cellsin vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity toa coadministered protein, J. Exp. Med. 198 (2) (2003) 267–279.
[90] S. Hakomori, Tumor malignancy defined by aberrant glycosylation andsphingo(glyco)lipid metabolism, Cancer Res. 56 (23) (1996) 5309–5318.
[91] J. Heimburg-Molinaro, M. Lum, G. Vijay, M. Jain, A. Almogren, K. Rittenhouse-Olson,Cancer vaccines and carbohydrate epitopes, Vaccine 29 (48) (2011) 8802–8826.
[92] A. Pashov, B. Monzavi-Karbassi, T. Kieber-Emmons, Glycan mediated immuneresponses to tumor cells, Hum. Vaccin. 7 (2011) 156–165, (Suppl.).
[93] B. Monzavi-Karbassi, A. Pashov, F. Jousheghany, C. Artaud, T. Kieber-Emmons,Evaluating strategies to enhance the anti-tumor immune response to a carbohy-drate mimetic peptide vaccine, Int. J. Mol. Med. 17 (6) (2006) 1045–1052.
[94] M.A. Cheever, J.P. Allison, A.S. Ferris, O.J. Finn, B.M. Hastings, T.T. Hecht, I.Mellman, S.A. Prindiville, J.L. Viner, L.M. Weiner, L.M. Matrisian, The prioritiza-tion of cancer antigens: a national cancer institute pilot project for the acceler-ation of translational research, Clin. Cancer Res. 15 (17) (2009) 5323–5337.
[95] A.M. Vlad, J.C. Kettel, N.M. Alajez, C.A. Carlos, O.J. Finn, MUC1 immunobiology:from discovery to clinical applications, Adv. Immunol. 82 (2004) 249–293.
[96] N. Gaidzik, U. Westerlind, H. Kunz, The development of synthetic antitumourvaccines from mucin glycopeptide antigens, Chem. Soc. Rev. (2013), http://dx.doi.org/10.1039/C3CS35470A, (Electronic publication ahead of print).
[97] A. Kaiser, N. Gaidzik, U.Westerlind, D. Kowalczyk, A. Hobel, E. Schmitt, H. Kunz, A syn-thetic vaccine consisting of a tumor-associated sialyl-T(N)-MUC1 tandem-repeat gly-copeptide and tetanus toxoid: induction of a strong and highly selective immuneresponse, Angew. Chem. Int. Ed Engl. 48 (41) (2009) 7551–7555.
[98] H. Cai, Z.-H. Huang, L. Shi, Z.-Y. Sun, Y.-F. Zhao, H. Kunz, Y.M. Li, Variation of theglycosylation pattern in MUC1 glycopeptide BSA vaccines and its influence onthe immune response, Angew. Chem. Int. Ed Engl. 51 (7) (2012) 1719–1723.
[99] Z. Mazorra, C. Mesa, A. Fernández, L.E. Fernández, Immunization with a GM3 gan-glioside nanoparticulated vaccine confers an effector CD8(+) T cells-mediatedprotection against melanoma B16 challenge, Cancer Immunol. Immunother. 57(12) (2008) 1771–1780.
[100] H. Kumar, T. Kawai, S. Akira, Pathogen recognition in the innate immune response,Biochem. J. 420 (1) (2009) 1–16.
[101] B. Beutler, Microbe sensing, positive feedback loops, and the pathogenesis of in-flammatory diseases, Immunol. Rev. 227 (1) (2009) 248–263.
[102] D. Wesch, C. Peters, H.-H. Oberg, K. Pietschmann, D. Kabelitz, Modulation of γδ Tcell responses by TLR ligands, Cell. Mol. Life Sci. 68 (14) (2011) 2357–2370.
[103] G. Schreibelt, J. Tel, K.H. Sliepen, D. Benitez-Ribas, C.G. Figdor, G.J. Adema, I.J.de Vries, Toll-like receptor expression and function in human dendritic cellsubsets: implications for dendritic cell-based anti-cancer immunotherapy,Cancer Immunol. Immunother. 59 (10) (2010) 1573–1582.
[104] S.S. Diebold, Activation of dendritic cells by Toll-like receptors and C-typelectins, Handb. Exp. Pharmacol. 188 (2009) 3–30.
[105] K. Takeda, T. Kaisho, S. Akira, Toll-like receptors, Annu. Rev. Immunol. 21 (1) (2003)335–376.
[106] G. Gautier, M. Humbert, F. Deauvieau, M. Scuiller, J. Hiscott, E.M. Bates, G.Trinchieri, C. Caux, P. Garrone, A type I interferon autocrine–paracrine loopis involved in Toll-like receptor-induced interleukin-12p70 secretion bydendritic cells, J. Exp. Med. 201 (9) (2005) 1435–1446.
[107] C.A. Sariol, M.I. Martínez, F. Rivera, I.V. Rodríguez, P. Pantoja, K. Abel, T. Arana, L.Giavedoni, V. Hodara, L.J. White, Y.I. Angleró, L.J. Montaner, E.N. Kraiselburd, De-creased dengue replication and an increased anti-viral humoral response with theuse of combined Toll-like receptor 3 and 7/8 agonists in macaques, PLoS One 6 (4)(2011) e19323.
[108] M.S. Duthie, H.P. Windish, C.B. Fox, S.G. Reed, Use of defined TLR ligands as ad-juvants within human vaccines, Immunol. Rev. 239 (1) (2011) 178–196.
[109] G. Liu, L. Zhang, Y. Zhao, Modulation of immune responses through direct activa-tion of Toll like receptors to T cells, Clin. Exp. Immunol. 160 (2) (2010) 168–175.
[110] A.M. Avalos, E. Latz, B. Mousseau, S.R. Christensen, M.J. Shlomchik, F. Lund, A.Marshak-Rothstein, Differential cytokine production and bystander activation ofautoreactive B cells in response to CpG-A and CpG-B oligonucleotides, J. Immunol.183 (10) (2009) 6262–6268.
[111] J. Vollmer, R. Weeratna, P. Payette, M. Jurk, C. Schetter, M. Laucht, T. Wader, S. Tluk,M. Liu, H.L. Davis, A.M. Krieg, Characterization of three CpG oligodeoxynucleotideclasses with distinct immunostimulatory activities, Eur. J. Immunol. 34 (1) (2004)251–262.
[112] S. Agrawal, E. Kandimalla, Synthetic agonists of Toll-like receptors 7, 8 and 9,Biochem. Soc. Trans. 35 (6) (2007) 1461–1467.
[113] T. Barata, I. Teo, S. Lalwani, E. Simanek, M. Zloh, S. Shaunak, Computationaldesign principles for bioactive dendrimer based constructs as antagonists ofthe TLR4-MD-2-LPS complex, Biomaterials 32 (33) (2011) 8702–8711.
[114] T.S. Barata, I. Teo, S. Brocchini, M. Zloh, S. Shaunak, Partially glycosylateddendrimers block MD-2 and prevent TLR4–MD-2–LPS complex mediated cyto-kine responses, PLoS Comput. Biol. 7 (6) (2011) e1002095.
[115] S.M. Goldinger, R. Dummer, P. Baumgaertner, D. Mihic-Probst, K. Schwarz, A.Hammann-Haenni, J. Willers, C. Geldhof, J.O. Prior, T.M. Kündig, O. Michielin,M.F. Bachmann, D.E. Speiser, Nano-particle vaccination combined with TLR-7and -9 ligands triggers memory and effector CD8+ T-cell responses in melano-ma patients, Eur. J. Immunol. 42 (11) (Novembro de 2012) 3049–3061.
[116] B.K. Link, Z.K. Ballas, D. Weisdorf, J.E. Wooldridge, A.D. Bossler, M. Shannon, W.L.Rasmussen, A.M. Krieg, G.J. Weiner, Oligodeoxynucleotide CpG 7909 deliveredas intravenous infusion demonstrates immunologic modulation in patientswith previously treated non-Hodgkin lymphoma, J. Immunother. 29 (5)(2006) 558–568.
[117] A. Carpentier, F. Laigle-Donadey, S. Zohar, L. Capelle, A. Behin, A. Tibi, N.Martin-Duverneuil, M. Sanson, L. Lacomblez, S. Taillibert, L. Puybasset, R. VanEffenterre, J.Y. Delattre, A.F. Carpentier, Phase 1 trial of a CpGoligodeoxynucleotide for patients with recurrent glioblastoma, Neuro Oncol. 8(1) (2006) 60–66.
[118] C. Manegold, D. Gravenor, D. Woytowitz, J. Mezger, V. Hirsh, G. Albert, M.Al-Adhami, D. Readett, A.M. Krieg, C.G. Leichman, Randomized phase II trial ofa Toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combinationwith first-line taxane plus platinum chemotherapy for advanced-stagenon-small-cell lung cancer, J. Clin. Oncol. 26 (24) (2008) 3979–3986.
[119] A.L. Engel, G.E. Holt, H. Lu, The pharmacokinetics of Toll-like receptor agonistsand the impact on the immune system, Expert Rev. Clin. Pharmacol. 4 (2)(2011) 275–289.

196 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
[120] R. Brunner, E. Jensen-Jarolim, I. Pali-Schöll, The ABC of clinical and experimentaladjuvants—a brief overview, Immunol. Lett. 128 (1) (2010) 29–35.
[121] N. Petrovsky, J.C. Aguilar, Vaccine adjuvants: current state and future trends,Immunol. Cell Biol. 82 (5) (2004) 488–496.
[122] I.F. Hermans, J.D. Silk, U. Gileadi, M. Salio, B. Mathew, G. Ritter, R. Schmidt, A.L.Harris, L. Old, V. Cerundolo, NKT cells enhance CD4+ and CD8+ T cell re-sponses to soluble antigen in vivo through direct interaction with dendriticcells, J. Immunol. 171 (10) (2003) 5140–5147.
[123] S. Kusmartsev, D.I. Gabrilovich, Role of immature myeloid cells in mechanismsof immune evasion in cancer, Cancer Immunol. Immunother. 55 (3) (2006)237–245.
[124] Q. Gao, S.-J. Qiu, J. Fan, J. Zhou, X.-Y. Wang, Y.-S. Xiao, Y. Xu, Y.W. Li, Z.Y. Tang,Intratumoral balance of regulatory and cytotoxic T cells is associated with prog-nosis of hepatocellular carcinoma after resection, J. Clin. Oncol. 25 (18) (2007)2586–2593.
[125] Y. Zhang, F. Luo, Y. Cai, N. Liu, L. Wang, D. Xu, Y. Chu, TLR1/TLR2 agonist inducestumor regression by reciprocal modulation of effector and regulatory T cells,J. Immunol. 186 (4) (2011) 1963–1969.
[126] H.-H. Oberg, M. Juricke, D. Kabelitz, D. Wesch, Regulation of T cell activation byTLR ligands, Eur. J. Cell Biol. 90 (6–7) (2011) 582–592.
[127] T. Warger, P. Osterloh, G. Rechtsteiner, M. Fassbender, V. Heib, B. Schmid, E.Schmitt, H. Schild, M.P. Radsak, Synergistic activation of dendritic cells by com-bined Toll-like receptor ligation induces superior CTL responses in vivo, Blood108 (2) (2006) 544–550.
[128] Y. Yang, C.T. Huang, X. Huang, D.M. Pardoll, Persistent Toll-like receptor signalsare required for reversal of regulatory T cell-mediated CD8 tolerance, Nat.Immunol. 5 (5) (2004) 508–515.
[129] I. Caramalho, T. Lopes-Carvalho, D. Ostler, S. Zelenay, M. Haury, J. Demengeot,Regulatory T cells selectively express Toll-like receptors and are activated by li-popolysaccharide, J. Exp. Med. 197 (4) (2003) 403–411.
[130] Q. Chen, T.S. Davidson, E.N. Huter, E.M. Shevach, Engagement of TLR2 does notreverse the suppressor function of mouse regulatory T cells, but promotestheir survival, J. Immunol. 183 (7) (2009) 4458–4466.
[131] A. Olivier, A. Sainz-Perez, H. Dong, T. Sparwasser, L. Majlessi, C. Leclerc, The ad-juvant effect of TLR agonists on CD4(+) effector T cells is under the indirect con-trol of regulatory T cells, Eur. J. Immunol. 41 (8) (2011) 2303–2313.
[132] C. Tanchot, M. Terme, H. Pere, T. Tran, N. Benhamouda, M. Strioga, C. Banissi, L.Galluzzi, G. Kroemer, E. Tartour, Tumor-infiltrating regulatory T cells: pheno-type, role, mechanism of expansion in situ and clinical significance, CancerMicroenviron. (2012), http://dx.doi.org/10.1007/s12307-012-0122-y, (Elec-tronic publication ahead of print).
[133] C. Zoglmeier, H. Bauer, D. Nörenberg, G. Wedekind, P. Bittner, N. Sandholzer, M.Rapp, D. Anz, S. Endres, C. Bourquin, CpG blocks immunosuppression bymyeloid-derived suppressor cells in tumor-bearing mice, Clin. Cancer Res. 17(7) (2011) 1765–1775.
[134] R. Arens, S.P. Schoenberger, Plasticity in programming of effector and memoryCD8+ T cell formation, Immunol. Rev. 235 (1) (2010) 190–205.
[135] M.F. Bachmann, G.T. Jennings, Vaccine delivery: amatter of size, geometry, kineticsand molecular patterns, Nat. Rev. Immunol. 10 (11) (2010) 787–796.
[136] S.L. Demento, S.C. Eisenbarth, H.G. Foellmer, C. Platt, M.J. Caplan, W.M. Saltzman, I.Mellman, M. Ledizet, E. Fikrig, R.A. Flavell, T.M. Fahmy, Inflammasome-activatingnanoparticles as modular systems for optimizing vaccine efficacy, Vaccine 27(23) (2009) 3013–3021.
[137] M. Diwan, M. Tafaghodi, J. Samuel, Enhancement of immune responses byco-delivery of a CpG oligodeoxynucleotide and tetanus toxoid in biodegradablenanospheres, J. Control. Release 85 (1–3) (2002) 247–262.
[138] H.F. Florindo, S. Pandit, L. Gonçalves, M. Videira, O. Alpar, A.J. Almeida, Antibodyand cytokine-associated immune responses to S. equi antigens entrapped in PLAnanospheres, Biomaterials 30 (28) (2009) 5161–5169.
[139] C. Clawson, C.-T. Huang, D. Futalan, D.M. Seible, R. Saenz, M. Larsson, W. Ma, B.Minev, F. Zhang, M. Ozkan, C. Ozkan, S. Esener, D. Messmer, Delivery of a peptidevia poly (d, l-lactic-co-glycolic) acid nanoparticles enhances its dendriticcell-stimulatory capacity, Nanomedicine 6 (5) (2010) 651–661.
[140] E. Macho Fernandez, J. Chang, J. Fontaine, E. Bialecki, F. Rodriguez, E.Werkmeister, V. Krieger, C. Ehreti, B. Heurtault, S. Fournel, B. Frisch, D.Betbeder, C. Faveeuw, F. Trottein, Activation of invariant Natural Killer T lym-phocytes in response to the α-galactosylceramide analogue KRN7000 encapsu-lated in PLGA-based nanoparticles and microparticles, Int. J. Pharm. 423 (1)(2012) 45–54.
[141] J. Panyam, V. Labhasetwar, Biodegradable nanoparticles for drug and gene deliv-ery to cells and tissue, Adv. Drug Deliv. Rev. 55 (3) (2003) 329–347.
[142] J. Damm, F. Wiegand, L.M. Harden, R. Gerstberger, C. Rummel, J. Roth, Fever,sickness behavior, and expression of inflammatory genes in the hypothalamusafter systemic and localized subcutaneous stimulation of rats with the Toll-likereceptor 7 agonist imiquimod, Neuroscience 201 (2012) 166–183.
[143] P.J. Tacken, I.S. Zeelenberg, L.J. Cruz, M.A. van Hout-Kuijer, G. van de Glind, R.G.Fokkink, A.J. Lambeck, C.G. Figdor, Targeted delivery of TLR ligands to humanand mouse dendritic cells strongly enhances adjuvanticity, Blood 118 (26)(2011) 6836–6844.
[144] D.N. Nguyen, K.P. Mahon, G. Chikh, P. Kim, H. Chung, A.P. Vicari, K.T. Love, M.Goldberg, S. Chen, A.M. Krieg, J. Chen, R. Langer, D.G. Anderson, Lipid-derivednanoparticles for immunostimulatory RNA adjuvant delivery, Proc. Natl. Acad.Sci. U. S. A. 109 (14) (2012) E797–E803.
[145] R.A. Jain, C.T. Rhodes, A.M. Railkar, A.W. Malick, N.H. Shah, Controlled release ofdrugs from injectable in situ formed biodegradable PLGA microspheres: effect ofvarious formulation variables, Eur. J. Pharm. Biopharm. 50 (2) (2000) 257–262.
[146] R.P.M. Sutmuller, M.E. Morgan, M.G. Netea, O. Grauer, G.J. Adema, Toll-like re-ceptors on regulatory T cells: expanding immune regulation, Trends Immunol.27 (8) (2006) 387–393.
[147] K. Park, Mechanism of cross-presentation of microencapsulated antigen, J. Con-trol. Release 151 (3) (2011) 219.
[148] J. Panyam, W. Zhou, S. Prabha, S.K. Sahoo, V. Labhase, Rapid endo-lysosomalescape of poly (DL-lactide-co-glycolide) nanoparticles: implications for drugand gene delivery, FASEB J. 16 (10) (2002) 1217–1226.
[149] A. Gamvrellis, D. Leong, J.C. Hanley, S.D. Xiang, P. Mottram, M. Plebanski, Vaccinesthat facilitate antigen entry into dendritic cells, Immunol. Cell Biol. 82 (5) (2004)506–516.
[150] Y. Krishnamachari, A.K. Salem, Innovative strategies for co-delivering antigensand CpG oligonucleotides, Adv. Drug Deliv. Rev. 61 (3) (2009) 205–217.
[151] S. Hamdy, O. Molavi, Z. Ma, A. Haddadi, A. Alshamsan, Z. Gobti, S. Elhasi, J.Samuel, A. Lavasanifar, Co-delivery of cancer-associated antigen and Toll-like re-ceptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediatedanti-tumor immunity, Vaccine 26 (39) (2008) 5046–5057.
[152] Y.R. Lee, Y.H. Lee, S.A. Im, I.H. Yang, G.W. Ahn, K. Kim, C.K. Lee, Biodegradablenanoparticles containing TLR3 or TLR9 agonists together with antigen enhanceMHC-restricted presentation of the antigen, Arch. Pharm. Res. 33 (11) (2010)1859–1866.
[153] G.W. Stone, S. Barzee, V. Snarsky, C. Santucci, B. Tran, R. Langer, G.T. Zugates, D.G.Anderson, R.S. Kornbluth, Nanoparticle-delivered multimeric soluble CD40L DNAcombined with Toll-like receptor agonists as a treatment for melanoma, PLoS One 4(10) (2009) e7334.
[154] B. Slütter, W. Jiskoot, Dual role of CpG as immune modulator and physicalcrosslinker in ovalbumin loaded N-trimethyl chitosan (TMC) nanoparticles fornasal vaccination, J. Control. Release 148 (1) (2010) 117–121.
[155] S.P. Kasturi, I. Skountzou, R.A. Albrecht, D. Koutsonanos, T. Hua, H. Nakaya, R.Ravindran, S. Stewart, M. Alam, M. Kwissa, F. Villinger, N. Murthy, J. Steel, J.Jacob, R.J. Hogan, A. García-Sastre, R. Compans, B. Pulendran, Programming themagnitude and persistence of antibody responses with innate immunity, Nature470 (7335) (2011) 543–547.
[156] D. Tross, L. Petrenko, S. Klaschik, Q. Zhu, D.M. Klinman, Global changes in geneexpression and synergistic interactions induced by TLR9 and TLR3, Mol. Immunol.46 (13) (2009) 2557–2564.
[157] A. Sapin, E. Garcion, A. Clavreul, F. Lagarce, J.P. Benoit, P. Menei, Development ofnew polymer-based particulate systems for anti-glioma vaccination, Int. J. Pharm.309 (1-2) (17 de Fevereiro de 2006) 1–5.
[158] C. Foged, B. Brodin, S. Frokjaer, A. Sundblad, Particle size and surface charge affectparticle uptake by human dendritic cells in an in vitro model, Int. J. Pharm. 298(2) (2005) 315–322.
[159] K. Cho, X. Wang, S. Nie, Z.G. Chen, D.M. Shin, Therapeutic nanoparticles for drugdelivery in cancer, Clin. Cancer Res. 14 (5) (2008) 1310–1316.
[160] S.D. Xiang, A. Scholzen, G. Minigo, C. David, V. Apostolopoulos, P.L. Mottram, M.Plebanski, Pathogen recognition and development of particulate vaccines: doessize matter? Methods 40 (1) (2006) 1–9.
[161] D.K. Cureton, R.H. Massol, S. Saffarian, T.L. Kirchhausen, S.P.J. Whelan, Vesicularstomatitis virus enters cells through vesicles incompletely coated with clathrinthat depend upon actin for internalization, PLoS Pathog. 5 (4) (2009) e1000394.
[162] D.K. Cureton, R.H. Massol, S.P.J. Whelan, T. Kirchhausen, The length of vesicularstomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis, J.A.T. Young (Ed.), PLoS Pathog. 6 (9) (2010) e1001127.
[163] K.T. Thurn, E.M. Brown, A. Wu, S. Vogt, B. Lai, J. Maser, T. Paunesku, G.E.Woloschak, Nanoparticles for applications in cellular imaging, Nanoscale Res.Lett. 2 (9) (2007) 430–441.
[164] J. Liu, H. Bauer, J. Callahan, P. Kopecková, H. Pan, J. Kopecek, Endocytic up-take of a large array of HPMA copolymers: elucidation into the dependenceon the physicochemical characteristics, J. Control. Release 143 (1) (2010)71–79.
[165] Z.G. Yue,W.Wei, P.P. Lv, H. Yue, L.Y.Wang, Z.G. Su, G.H.Ma, Surface charge affectscellular uptake and intracellular trafficking of chitosan-based nanoparticles,Biomacromolecules 12 (7) (2011) 2440–2446.
[166] T.L. Doane, C.-H. Chuang, R.J. Hill, C. Burda, Nanoparticle ζ-potentials, Acc. Chem.Res. 45 (3) (2012) 317–326.
[167] S. Venkataraman, J.L. Hedrick, Z.Y. Ong, C. Yang, P.L. Ee, P.T. Hammond, Y.Y. Yang,The effects of polymeric nanostructure shape on drug delivery, Adv. Drug Deliv.Rev. 63 (14–15) (2011) 1228–1246.
[168] L. Florez, C. Herrmann, J.M. Cramer, C.P. Hauser, K. Koynov, K. Landfester, D.Crespy, V. Mailänder, How shape influences uptake: interactions of anisotropicpolymer nanoparticles and human mesenchymal stem cells, Small 8 (14)(2012) 2222–2230.
[169] D.E. Owens, N.A. Peppas, Opsonization, biodistribution, and pharmacokinetics ofpolymeric nanoparticles, Int. J. Pharm. 307 (1) (2006) 93–102.
[170] I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse, K.A. Dawson, Thenanoparticle–protein complex as a biological entity; a complex fluids and sur-face science challenge for the 21st century, Adv. Colloid Interface Sci. 134-135(2007) 167–174.
[171] P. Aggarwal, J.B. Hall, C.B. McLeland, M.A. Dobrovolskaia, S.E. McNeil, Nanoparti-cle interaction with plasma proteins as it relates to particle biodistribution, bio-compatibility and therapeutic efficacy, Adv. Drug Deliv. Rev. 61 (6) (2009)428–437.
[172] F. Alexis, E. Pridgen, L.K.Molnar, O.C. Farokhzad, Factors affecting the clearance andbiodistribution of polymeric nanoparticles, Mol. Pharm. 5 (4) (2008) 505–515.
[173] M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall, K.A. Dawson, Nanoparticlesize and surface properties determine the protein corona with possible

197J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
implications for biological impacts, Proc. Natl. Acad. Sci. U. S. A. 105 (38) (2008)14265–14270.
[174] S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S.K. Knauer, C.Bantz, T. Nawroth, C. bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M.Maskos, H. Schild, R.H. Stauber, Nanoparticle size is a critical physicochemicaldeterminant of the human blood plasma corona: a comprehensive quantitativeproteomic analysis, ACS Nano 5 (9) (2011) 7155–7157.
[175] C. Kelly, C. Jefferies, S.A. Cryan, Targeted liposomal drug delivery to monocytesand macrophages, J. Drug Deliv. 2011 (2011) 727241.
[176] D.J. Bharali, V. Pradhan, G. Elkin, W. Qi, A. Hutson, S.A. Mousa, Y. Thanavala,Novel nanoparticles for the delivery of recombinant hepatitis B vaccine,Nanomedicine 4 (4) (2008) 311–317.
[177] H. Freichels, V. Pourcelle, R. Auzély-Velty, J. Marchand-Brynaert, C. Jérôme,Synthesis of poly(lactide-co-glycolide-co-ε-caprolactone)-graft-mannosylatedpoly(ethylene oxide) copolymers by combination of «clip» and «click» chemistries,Biomacromolecules 13 (3) (2012) 760–768.
[178] J.H. Van den Berg, K. Oosterhuis, W.E. Hennink, G. Storm, L.J. Van der Aa, J.F.Engbersen, J.B. Haanenc, J.H. Beijnena, T.N. Schumacherc, B. Nuijena, Shieldingthe cationic charge of nanoparticle-formulated dermal DNA vaccines is essentialfor antigen expression and immunogenicity, J. Control. Release 141 (2) (2010)234–240.
[179] E. Fernandez-Megia, R.Novoa-Carballal, E. Quiñoá, R. Riguera, Conjugation of bioac-tive ligands to PEG-grafted chitosan at the distal end of PEG, Biomacromolecules 8(3) (2007) 833–842.
[180] Y. Van Kooyk, C-type lectins on dendritic cells: key modulators for the inductionof immune responses, Biochem. Soc. Trans. 36 (Pt 6) (2008) 1478–1481.
[181] W.W.J. Unger, Y. Van Kooyk, «Dressed for success» C-type lectin receptors for the de-livery of glyco-vaccines to dendritic cells, Curr. Opin. Immunol. 23 (1) (2010)131–137.
[182] U. Gazi, L. Martinez-Pomares, Influence of the mannose receptor in host immuneresponses, Immunobiology 214 (7) (2009) 554–561.
[183] S. Burgdorf, A. Kautz, V. Böhnert, P.A. Knolle, C. Kurts, Distinct pathways of anti-gen uptake and intracellular routing in CD4 and CD8 T cell activation, Science316 (5824) (2007) 612–616.
[184] S. Hamdy, A. Haddadi, A. Shayeganpour, J. Samuel, A. Lavasanifar, Activation ofantigen-specific T cell-responses by mannan-decorated PLGA nanoparticles,Pharm. Res. 28 (9) (2011) 2288–3301.
[185] N. Brandhonneur, F. Chevanne, V. Vié, B. Frisch, R. Primault, M.F. Le Potier, P. LeCorre, Specific and non-specific phagocytosis of ligand-grafted PLGA micro-spheres by macrophages, Eur. J. Pharm. Sci. 36 (4–5) (2009) 474–485.
[186] G.K. Saraogi, B. Sharma, B. Joshi, P. Gupta, U.D. Gupta, N.K. Jain, G.P. Agrawal,Mannosylated gelatin nanoparticles bearing isoniazid for effective managementof tuberculosis, J. Drug Target. 19 (3) (2011) 219–227.
[187] S. Espuelas, C. Thumann, B. Heurtault, F. Schuber, B. Frisch, Influence of ligandvalency on the targeting of immature human dendritic cells by mannosylatedliposomes, Bioconjug. Chem. 19 (12) (2008) 2385–2393.
[188] L.Z. He, A. Crocker, J. Lee, J. Mendoza-Ramirez, X.T. Wang, L.A. Vitale, T. O'Neill, C.Petromilli, H.F. Zhang, J. Lopez, D. Rohrer, T. Keler, R. Clynes, Antigenic targetingof the human mannose receptor induces tumor immunity, J. Immunol. 178 (10)(2007) 6259–6267.
[189] V. Ramakrishna, J.F. Treml, L. Vitale, J.E. Connolly, T. O'Neill, P.A. Smith, C.L. Jones,L.Z. He, J. Goldstein, P.K. Wallace, T. Keler, M.J. Endres, Mannose receptor targetingof tumor antigen pmel17 to human dendritic cells directs anti-melanoma T cell re-sponses via multiple HLA molecules, J. Immunol. 172 (5) (2004) 2845–2852.
[190] V. Flacher, C.H. Tripp, P. Stoitzner, B.Haid, S. Ebner, B.Del Frari, F. Koch, C.G. Park, R.M.Steinman, J. Idoyaga, N. Romani, Epidermal Langerhans cells rapidly capture andpresent antigens from C-type lectin-targeting antibodies deposited in the dermis, J.Invest. Dermatol. 130 (3) (2009) 755–762.
[191] F. Danhier, E. Ansorena, J.M. Silva, R. Coco, A. Le Breton, V. Préat, PLGA-basednanoparticles: an overview of biomedical applications, J. Control. Release 161(2) (2012) 505–522.
[192] K.A. Athanasiou, G.G. Niederauer, C. Agrawal, Sterilization, toxicity, biocompati-bility and clinical applications of polylactic acid/polyglycolic acid copolymers,Biomaterials 17 (2) (1996) 93–102.
[193] A.-C. Albertsson, I.K. Varma, Recent developments in ring opening polymerization oflactones for biomedical applications, Biomacromolecules 4 (6) (2003) 1466–1486.
[194] S.H. Hyon, K. Jamshidi, Y. Ikada, Synthesis of polylactides with different molecu-lar weights, Biomaterials 18 (22) (1997) 1503–1508.
[195] C.M. Thomas, Stereocontrolled ring-opening polymerization of cyclic esters:synthesis of new polyester microstructures, Chem. Soc. Rev. 39 (1) (2010)165–173.
[196] S. Kobayashi, H. Uyama, S. Kimura, Enzymatic polymerization, Chem. Rev. 101(12) (2001) 3793–3818.
[197] J.S. Dordick, Enzymatic and chemoenzymatic approaches to polymer synthesis,Trends Biotechnol. 10 (8) (1992) 287–290.
[198] O. Dechy-Cabaret, B. Martin-Vaca, D. Bourissou, Controlled ring-opening polymeriza-tion of lactide and glycolide, Chem. Rev. 104 (12) (2004) 6147–6176.
[199] R. Platel, L. Hodgson, C. Williams, Biocompatible initiators for lactide polymerization,Polym. Rev. 48 (1) (Janeiro de 2008) 11–63.
[200] H.R. Kricheldorf, Syntheses and application of polylactides, Chemosphere 43 (1)(2001) 49–54.
[201] M. Labet, W. Thielemans, Synthesis of polycaprolactone: a review, Chem. Soc.Rev. 38 (12) (2009) 3484–3504.
[202] H. Huatan, J.H. Collett, D. Attwood, C. Booth, Preparation and characterization ofpoly(epsilon-caprolactone) polymer blends for the delivery of proteins, Bioma-terials 16 (17) (1995) 1297–1303.
[203] H. Abe, Y. Doi, Structural effects on enzymatic degradabilities for poly[(R)-3-hydroxybutyric acid] and its copolymers, Int. J. Biol. Macromol. 25 (1-3)(1999) 185–192.
[204] S.J. Peter, M.J. Yaszemski, L.J. Suggs, R.G. Payne, R. Langer, W.C. Hayes, M.R.Unroe, I.B. Alemany, P.S. Engel, A.G. Mikos, Characterization of partially saturat-ed poly(propylene fumarate) for orthopaedic application, J. Biomater. Sci.Polym. Ed. 8 (11) (1997) 893–904.
[205] J. Liu, P. Chen, J. Li, S.-H. Jiang, Z.-Q. Jiang, Q. Gu, Synthesis of poly(ethyleneadipate-co-l-lactic acid) copolymers via ring opening polymerization, Polym.Bull. 66 (2) (2011) 187–197.
[206] H. Qian, A.R. Wohl, J.T. Crow, C.W. Macosko, T.R. Hoye, A strategy for control of«random» copolymerization of lactide and glycolide: application to synthesisof PEG-b-PLGA block polymers having narrow dispersity, Macromolecules 44(18) (2011) 7132–7140.
[207] C.W. Lee, R. Urakawa, Y. Kimura, Copolymerization of γ-valerolactone andβ-butyrolactone, Eur. Polym. J. 34 (1) (1998) 117–122.
[208] S. Malberg, A. Hoglund, A.-C. Albertsson, Macromolecular design of aliphaticpolyesters with maintained mechanical properties and a rapid, customized deg-radation profile, Biomacromolecules 12 (6) (2011) 2382–2388.
[209] M.A. Woodruff, D.W. Hutmacher, The return of a forgotten polymer—polycaprolactone in the 21st century, Prog. Polym. Sci. 35 (10) (2010) 1217–1256.
[210] M. Ryner, A.-C. Albertsson, Resorbable and highly elastic block copolymers from1,5-dioxepan-2-one and L-lactide with controlled tensile properties and hydro-philicity, Biomacromolecules 3 (3) (2002) 601–608.
[211] X. Pang, X. Zhuang, Z. Tang, X. Chen, Polylactic acid (PLA): research, develop-ment and industrialization, Biotechnology 5 (11) (2010) 1125–1136.
[212] G.T. Köse, H. Kenar, N. Hasirci, V. Hasirci, Macroporous poly(3-hydroxybutyrate-co-3-hydroxyvalerate) matrices for bone tissue engineering, Biomaterials 24 (11)(2003) 1949–1958.
[213] K.S. Jack, S. Velayudhan, P. Luckman, M. Trau, L. Grøndahl, J. Cooper-White, The fabri-cation and characterization of biodegradable HA/PHBV nanoparticle–polymer com-posite scaffolds, Acta Biomater. 5 (7) (2009) 2657–2667.
[214] G. Khang, S.W. Kim, J.C. Cho, J.M. Rhee, S.C. Yoon, H.B. Lee, Preparation and charac-terization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) microspheres for thesustained release of 5-fluorouracil, Biomed. Mater. Eng. 11 (2) (2001) 89–103.
[215] J.B.E. Mendes, M.K. Riekes, V.M. De Oliveira, M.D. Michel, H.K. Stulzer, N.M.Khalil, S.F. Zawadzki, R.M. Mainardes, P.V. Farago, PHBV/PCL microparticles forcontrolled release of resveratrol: physicochemical characterization, antioxidantpotential, and effect on hemolysis of human erythrocytes, Scientific World Jour-nal 2012 (2012) 542937.
[216] J.M. Lü, X. Wang, C. Marin-Muller, H. Wang, P.H. Lin, Q. Yao, C. Chen, Current ad-vances in research and clinical applications of PLGA-based nanotechnology, Ex-pert Rev. Mol. Diagn. 9 (4) (2009) 325–341.
[217] R.A. Jain, The manufacturing techniques of various drug loaded biodegradablepoly (lactide-co-glycolide)(PLGA) devices, Biomaterials 21 (23) (2000)2475–2490.
[218] R.G. Soderquist, E.M. Sloane, L.C. Loram, J.A. Harrison, E.C. Dengler, S.M. Johnson,L.D. Amer, C.S. Young, M.T. Lewis, S. Poole, M.G. Frank, L.R. Watkins, E.D.Milligan, M.J. Mahoney, Release of plasmid DNA-encoding IL-10 from PLGA mi-croparticles facilitates long-term reversal of neuropathic pain following a singleintrathecal administration, Pharm. Res. 27 (5) (2010) 841–854.
[219] D. Cun, D.K. Jensen, M.J. Maltesen, M. Bunker, P. Whiteside, D. Scurr, C. Foged,H.M. Nielsen, High loading efficiency and sustained release of siRNA encapsulat-ed in PLGA nanoparticles: quality by design optimization and characterization,Eur. J. Pharm. Biopharm. 77 (1) (2010) 26–35.
[220] C. Solbrig, J. Saucier-Sawyer, V. Cody, W. Saltzman, D. Hanlon, Polymernanoparticles for immunotherapy from encapsulated tumor-associated antigensand whole tumor cells, Mol. Pharm. 4 (1) (2007) 47–57.
[221] M. Diwan, P. Elamanchili, H. Lane, A. Gainer, J. Samuel, Biodegradable nano-particle mediated antigen delivery to human cord blood derived dendriticcells for induction of primary T cell responses, J. Drug Target. 11 (8–10)(2003) 495–507.
[222] S. Prasad, V. Cody, J.K. Saucier-Sawyer, W.M. Saltzman, C.T. Sasaki, R.L. Edelson,M.A. Birchall, D.J. Hanlon, Polymer nanoparticles containing tumor lysates as an-tigen delivery vehicles for dendritic cell-based antitumor immunotherapy,Nanomedicine 7 (1) (2011) 1–10.
[223] C.M. Thomas, A. Rawat, L.J. Hope-Weeks, F. Ahsan, Aerosolized PLA and PLGAnanoparticles enhance humoral, mucosal and cytokine responses to hepatitis Bvaccine, Mol. Pharm. 27 (2010) 905–919.
[224] J. Tian, J. Yu, Cell biology of antigen processing in vitro and in vivo, Fish ShellfishImmunol. 30 (1) (2010) 109–117.
[225] W. Jiang, R.K. Gupta, M.C. Deshpande, S.P. Schwendeman, Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine anti-gens, Adv. Drug Deliv. Rev. 57 (3) (2005) 391–410.
[226] M. Garinot, V. Fiévez, V. Pourcelle, F. Stoffelbach, A. Des Rieux, L. Plapied, I.Theate, H. Freichels, C. Jérôme, J. Marchand-Brynaert, Y.J. Schneider, V. Préat,PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination,J. Control. Release 120 (3) (2007) 195–204.
[227] H.F. Florindo, S. Pandit, L.M.D. Gonçalves, H.O. Alpar, A.J. Almeida, New approachon the development of a mucosal vaccine against strangles: systemic and muco-sal immune responses in a mouse model, Vaccine 27 (8) (2009) 1230–1241.
[228] M. Diwan, P. Elamanchili, M. Cao, J. Samuel, Dose sparing of CpGoligodeoxynucleotide vaccine adjuvants by nanoparticle delivery, Curr.Drug Deliv. 1 (4) (2004) 405–412.
[229] V. Sinha, K. Bansal, R. Kaushik, R. Kumria, A. Trehan, Poly-[epsilon]-caprolactonemicrospheres and nanospheres: an overview, Int. J. Pharm. 278 (1) (2004) 1–23.

198 J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
[230] F.S. Bates, M.A. Hillmyer, T.P. Lodge, C.M. Bates, K.T. Delaney, G.H.Fredrickson, Multiblock polymers: panacea or Pandora's box? Science 336(6080) (2012) 434–440.
[231] T. Kissel, Y. Li, F. Unger, ABA-triblock copolymers from biodegradable polyesterA-blocks and hydrophilic poly(ethylene oxide) B-blocks as a candidate for insitu forming hydrogel delivery systems for proteins, Adv. Drug Deliv. Rev. 54(1) (2002) 99–134.
[232] X.-B. Xiong, A. Falamarzian, S.M. Garg, A. Lavasanifar, Engineering of amphiphilicblock copolymers for polymeric micellar drug and gene delivery, J. Control.Release 155 (2) (2011) 248–261.
[233] M.L. Adams, A. Lavasanifar, G.S. Kwon, Amphiphilic block copolymers for drugdelivery, J. Pharm. Sci. 92 (7) (2003) 1343–1355.
[234] Y. Yang, Y. Kuang, Y. Liu, W. Li, Z. Jiang, L. Xiao, M. Li, Immunogenicity ofmultiple-epitope antigen gene of HCV carried by novel biodegradable polymers,Comp. Immunol. Microbiol. Infect. Dis. 34 (1) (2011) 65–72.
[235] M. Barz, F.K. Wolf, F. Canal, K. Koynov, M.J. Vicent, H. Frey, R. Zentel, Synthesis,characterization and preliminary biological evaluation of P(HPMA)-b-P(LLA)copolymers: a new type of functional biocompatible block copolymer,Macromol. Rapid Commun. 31 (17) (2010) 1492–1500.
[236] J. Shao, J. Sun, X. Bian, Y. Cui, G. Li, X. Chen, Investigation of poly(lactide)stereocomplexes: 3-armed poly(L-lactide) blended with linear and 3-armed en-antiomers, J. Phys. Chem. B 116 (33) (2012) 9983–9991.
[237] M. Barz, A. Armiñán, F. Canal, F. Wolf, K. Koynov, H. Frey, R. Zentel, M.J. Vicent,P(HPMA)-block-P(LA) copolymers in paclitaxel formulations: polylactide ste-reochemistry controls micellization, cellular uptake kinetics, intracellular local-ization and drug efficiency, J. Control. Release 163 (1) (2012) 63–74.
[238] F. Ahmed, R.I. Pakunlu, G. Srinivas, A. Brannan, F. Bates, M.L. Klein, T. Minko, D.E.Discher, Shrinkage of a rapidly growing tumor by drug-loaded polymersomes:pH-triggered release through copolymer degradation, Mol. Pharm. 3 (3)(2006) 340–350.
[239] A. Blencowe, J.F. Tan, T.K. Goh, G.G. Qiao, Core cross-linked star polymers viacontrolled radical polymerisation, Polymer 50 (1) (2009) 5–32.
[240] D.J.A. Cameron, M.P. Shaver, Aliphatic polyester polymer stars: synthesis, prop-erties and applications in biomedicine and nanotechnology, Chem. Soc. Rev. 40(3) (2011) 1761–1776.
[241] J. Jansen, M.P. Tibbe, G. Mihov, J. Feijen, D.W. Grijpma, Photo-crosslinked net-works prepared from fumaric acid monoethyl ester-functionalized poly(D, L-lac-tic acid) oligomers and N-vinyl-2-pyrrolidone for the controlled and sustainedrelease of proteins, Acta Biomater. 8 (10) (2012) 3652–3659.
[242] J. Zotzmann, A. Alteheld, M. Behl, A. Lendlein, Amorphous phase-segregatedcopoly(ether)esterurethane thermoset networks with oligo(propylene glycol)and oligo[(rac-lactide)-co-glycolide] segments: synthesis and characterization,J. Mater. Sci. Mater. Med. 20 (9) (2009) 1815–1824.
[243] K. Nagahama, T. Saito, T. Ouchi, Y. Ohya, Biodegradable nano-aggregates ofstar-shaped 8-arm PEG -PLLA block co-polymers for encapsulation ofwater-soluble macromolecules, J. Biomater. Sci. Polym. Ed. 22 (1–3)(2011) 407–416, (10).
[244] M. Vert, S.M. Li, G. Spenlehauer, P. Guerin, Bioresorbability and biocompatibilityof aliphatic polyesters, J. Mater. Sci. Mater. Med. 3 (6) (1992) 432–446.
[245] N.A. Weir, F.J. Buchanan, J.F. Orr, G.R. Dickson, Degradation of poly-L-lactide. Part1: in vitro and in vivo physiological temperature degradation, Proc. Inst. Mech.Eng. H 218 (5) (2004) 307–319.
[246] A. Göpferich, J. Tessmar, Polyanhydride degradation and erosion, Adv. DrugDeliv. Rev. 54 (7) (2002) 911–931.
[247] S. Li, Hydrolytic degradation characteristics of aliphatic polyesters derived fromlactic and glycolic acids, J. Biomed. Mater. Res. 48 (3) (1999) 342–353.
[248] S.C. Loo, C.P. Ooi, S.H. Wee, Y.C. Boey, Effect of isothermal annealing on the hy-drolytic degradation rate of poly (lactide-co-glycolide)(PLGA), Biomaterials 26(16) (2005) 2827–2833.
[249] A.N. Ford Versypt, D.W. Pack, R.D. Braatz, Mathematical modeling of drug deliv-ery from autocatalytically degradable PLGA microspheres — a review, J. Control.Release 165 (1) (2013) 29–37.
[250] M.L. Houchin, E.M. Topp, Chemical degradation of peptides and proteins inPLGA: a review of reactions and mechanisms, J. Pharm. Sci. 97 (7) (2008)2395–2404.
[251] R.C.Mundargi, V.R. Babu,V. Rangaswamy, P. Patel, T.M.Aminabhavi, Nano/micro tech-nologies for delivering macromolecular therapeutics using poly (D, L-lactide-co-glycolide) and its derivatives, J. Control. Release 125 (3) (2008) 193–209.
[252] Y. Waeckerle-Men, M. Groettrup, PLGA microspheres for improved antigendelivery to dendritic cells as cellular vaccines, Adv. Drug Deliv. Rev. 57 (3) (2005)475–482.
[253] S.K. Mallapragada, B. Narasimhan, Immunomodulatory biomaterials, Int. J. Pharm.364 (2) (2008) 265–271.
[254] S.U. Frick, N. Bacher, G. Baier, V. Mailänder, K. Landfester, K. Steinbrink, Functional-ized polystyrene nanoparticles trigger humandendritic cellmaturation resulting inenhanced CD4+ T cell activation, Macromol. Biosci. 12 (12) (2012) 1637–1647.
[255] R. Nicolete, D.F. dos Santos, L.H. Faccioli, The uptake of PLGA micro ornanoparticles by macrophages provokes distinct in vitro inflammatory re-sponse, Int. Immunopharmacol. 11 (10) (2011) 1557–1563.
[256] S. Fischer, E. Uetz-von Allmen, Y. Waeckerle-Men, M. Groettrup, H.P. Merkle, B.Gander, The preservation of phenotype and functionality of dendritic cells uponphagocytosis of polyelectrolyte-coated PLGA microparticles, Biomaterials 28 (6)(2007) 994–1004.
[257] B. Nilsson, K.N. Ekdahl, T.E. Mollnes, J.D. Lambris, The role of complement inbiomaterial-induced inflammation, Mol. Immunol. 44 (1–3) (2007) 82–94.
[258] S.M. Moghimi, A.J. Andersen, D. Ahmadvand, P.P. Wibroe, T.L. Andresen, A.C.Hunter, Material properties in complement activation, Adv. Drug Deliv. Rev. 63(12) (2011) 1000–1007.
[259] V.C. Mosqueira, P. Legrand, A. Gulik, O. Bourdon, R. Gref, D. Labarre, G. Barratt,Relationship between complement activation, cellular uptake and surface phys-icochemical aspects of novel PEG-modified nanocapsules, Biomaterials 22 (22)(2001) 2967–2979.
[260] Q. He, J. Liu, X. Sun, Z.R. Zhang, Preparation and characteristics of DNA-nanoparticlestargeting to hepatocarcinoma cells, World J. Gastroenterol. 10 (5) (2004) 660–663.
[261] S. Malathi, S. Balasubramanian, Synthesis of biodegradable polymeric nanoparticlesand their controlled drug delivery for tuberculosis, J. Biomed. Nanotechnol. 7 (1)(2011) 150–151.
[262] A. Des Rieux, V. Fievez, M. Garinot, Y.J. Schneider, V. Préat, Nanoparticles aspotential oral delivery systems of proteins and vaccines: a mechanistic approach,J. Control. Release 116 (1) (2006) 1–27.
[263] N. Anton, J.P. Benoit, P. Saulnier, Design and production of nanoparticles formu-lated from nano-emulsion templates—a review, J. Control. Release 128 (3)(2008) 185–199.
[264] M. Estevan, J.M. Irache, M.J. Grillo, J.M. Blasco, C. Gamazo, Encapsulation of anti-genic extracts of Salmonella enterica serovar: Abortusovis into polymeric sys-tems and efficacy as vaccines in mice, Vet. Microbiol. 118 (1–2) (2006) 124–132.
[265] H. Florindo, S. Pandit, L. Goncalves, H. Alpar, A. Almeida, S. equi antigens adsorbedonto surface modified poly-epsilon-caprolactone microspheres induce humoraland cellular specific immune responses, Vaccine 26 (33) (2008) 4168–4177.
[266] S.K. Sahoo, J. Panyam, S. Prabha, V. Labhasetwar, Residual polyvinyl alcohol asso-ciated with poly (D, L-lactide-co-glycolide) nanoparticles affects their physicalproperties and cellular uptake, J. Control. Release 82 (1) (2002) 105–114.
[267] T. Storni, T. Kundig, G. Senti, P. Johansen, Immunity in response to particulateantigen-delivery systems, Adv. Drug Deliv. Rev. 57 (3) (2005) 333–355.
[268] T.E. Rajapaksa, K.M. Bennett, M. Hamer, C. Lytle, V.G.J. Rodgers, D.D. Lo, Intrana-sal M cell uptake of nanoparticles is independently influenced by targetingligands and buffer ionic strength, J. Biol. Chem. 285 (31) (2010) 23739–23746.
[269] J.U. Menon, S. Kona, A.S. Wadajkar, F. Desai, A. Vadla, K.T. Nguyen, Effects of surfac-tants on the properties of PLGA nanoparticles, J. Biomed. Mater. Res. A 100A (8)(2012) 1998–2005.
[270] A.M. Cerdeira, I.A.Werner, M. Mazzotti, B. Gander, Simultaneous quantificationof polymeric and surface active stabilizers of nanosuspensions by usingnear-infrared spectroscopy, Drug Dev. Ind. Pharm. 38 (11) (2012) 1360–1370.
[271] Y.-C. Wang, Y.-T. Wu, H.-Y. Huang, C.-S. Yang, Surfactant-free formulation ofpoly(lactic/glycolic) acid nanoparticles encapsulating functional polypeptide: atechnical note, AAPS PharmSciTech 10 (4) (2009) 1263–1267.
[272] M. Zanetti, G. Franchini, T cell memory and protective immunity by vaccination:is more better? Trends Immunol. 27 (11) (2006) 511–517.
[273] S. Moffatt, R.J. Cristiano, Uptake characteristics of NGR-coupled stealth PEI/pDNAnanoparticles loadedwith PLGA–PEG–PLGA tri-block copolymer for targeted deliveryto humanmonocyte-derived dendritic cells, Int. J. Pharm. 321 (1–2) (2006) 143–154.
[274] X. Shan, Y. Yuan, C. Liu, X. Tao, Y. Sheng, F. Xu, Influence of PEG chain on thecomplement activation suppression and longevity in vivo prolongation of thePCL biomedical nanoparticles, Biomed. Microdevices 11 (6) (2009) 1187–1194.
[275] N. Romani, M. Thurnher, J. Idoyaga, R.M. Steinman, V. Flacher, Targeting of anti-gens to skin dendritic cells: possibilities to enhance vaccine efficacy, Immunol.Cell Biol. 88 (4) (2010) 424–430.
[276] M. Peiser, J. Koeck, C.J. Kirschning, B. Wittig, R. Wanner, Human Langerhans cellsselectively activated via Toll-like receptor 2 agonists acquire migratory andCD4+ T cell stimulatory capacity, J. Leukoc. Biol. 83 (5) (2008) 1118–1127.
[277] D.H. Kaplan, In vivo function of Langerhans cells and dermal dendritic cells,Trends Immunol. 31 (12) (2010) 446–451.
[278] F.O. Nestle, P. Di Meglio, J.Z. Qin, B.J. Nickoloff, Skin immune sentinels in healthand disease, Nat. Rev. Immunol. 9 (10) (2009) 679–691.
[279] B.S. Kim, F. Miyagawa, Y.H. Cho, C.L. Bennett, B.E. Clausen, S.I. Katz, Keratinocytesfunction as accessory cells for presentation of endogenous antigen expressed inthe epidermis, J. Invest. Dermatol. 129 (12) (2009) 2805–2817.
[280] K. Sato, S. Fujita, Dendritic cells: nature and classification, Allergol. Int. 56 (3)(2007) 183–191.
[281] C. Winzler, P. Rovere, M. Rescigno, F. Granucci, G. Penna, L. Adorini, V.S.Zimmermann, J. Davoust, P. Ricciardi-Castagnoli, Maturation stages of mousedendritic cells in growth factor-dependent long-term cultures, J. Exp. Med.185 (2) (1997) 317–328.
[282] M. Adamina, R. Rosenthal, W.P. Weber, D.M. Frey, C.T. Viehl, M. Bolli, R.W. Huegli,A.L. Jacob, M. Heberer, D. Oertli, W. Marti, G.C. Spagnoli, P. Zajac, Intranodal immu-nization with a vaccinia virus encoding multiple antigenic epitopes andcostimulatory molecules in metastatic melanoma, Mol. Ther. 18 (3) (2009)651–659.
[283] K.A. Smith, B.L. Meisenburg, V.L. Tam, R.R. Pagarigan, R. Wong, D.K. Joea, L.Lantzy, M.A. Carrillo, T.M. Gross, U.M. Malyankar, C.S. Chiang, D.M. Da Silva,T.M. Kündig, W.M. Kast, Z. Qiu, A. Bot, Lymph node-targeted immunotherapymediates potent immunity resulting in regression of isolated or metastatichuman papillomavirus-transformed tumors, Clin. Cancer Res. 15 (19) (2009)6167–6176.
[284] S.T. Reddy, A.J. van der Vlies, E. Simeoni, V. Angeli, G.J. Randolph, C.P. O'Neil, L.K.Lee, M.A. Swartz, J.A. Hubbell, Exploiting lymphatic transport and complementactivation in nanoparticle vaccines, Nat. Biotechnol. 25 (10) (2007) 1159–1164.
[285] K.J. Maloy, I. Erdmann, V. Basch, S. Sierro, T.A. Kramps, R.M. Zinkernagel, S.Oehen, T.M. Kündig, Intralymphatic immunization enhances DNA vaccination,Proc. Natl. Acad. Sci. U. S. A. 98 (6) (2001) 3299–3303.

199J.M. Silva et al. / Journal of Controlled Release 168 (2013) 179–199
[286] S. Kreiter, A. Selmi, M. Diken, M. Koslowski, C.M. Britten, C. Huber, Ö. Türeci, U.Sahin, Intranodal vaccinationwith naked antigen-encoding RNA elicits potent pro-phylactic and therapeutic antitumoral immunity, Cancer Res. 70 (22) (2010)9031–9040.
[287] C.D. Platt, J.K. Ma, C. Chalouni, M. Ebersold, H. Bou-Reslan, R.A. Carano, I.Mellman, L. Delamarre, Mature dendritic cells use endocytic receptors to cap-ture and present antigens, Proc. Natl. Acad. Sci. U. S. A. 107 (9) (2010)4287–4292.
[288] N.S. Wilson, D. El-Sukkari, G.T. Belz, C.M. Smith, R.J. Steptoe, W.R. Heath, K.Shortman, J.A. Villadangos, Most lymphoid organ dendritic cell types are pheno-typically and functionally immature, Blood 102 (6) (2003) 2187–2194.
[289] J.A. Villadangos, W.R. Heath, Life cycle, migration and antigen presenting functionsof spleen and lymph node dendritic cells: limitations of the Langerhans cellsparadigm, Semin. Immunol. 17 (4) (2005) 262–272.
[290] S.T. Tagawa, P. Lee, J. Snively, W. Boswell, S. Ounpraseuth, S. Lee, B.Hickingbottom, J. Smith, D. Johnson, J.S. Weber, Phase I study of intranodal de-livery of a plasmid DNA vaccine for patients with stage IV melanoma, Cancer98 (1) (2003) 144–154.
[291] R. Gaspar, Regulatory issues surrounding nanomedicines: setting the scene for thenext generation of nanopharmaceuticals, Nanomedicine (Lond.) 2 (2) (2007)143–147.
[292] R. Gaspar, R. Duncan, Polymeric carriers: preclinical safety and the regulatory im-plications for design and development of polymer therapeutics, Adv. Drug Deliv.Rev. 61 (13) (2009) 1220–1231.
[293] D. Bowman, A. Maynard, R. Gaspar, Chapter 14: therapeutic products: regulatingdrugs and medical devices, International Handbook on Regulating Nanotechnol-ogies, Edward Elgar Pub, Northampton, MA, 2010. 291–320.
[294] European Medicines Agency C, Vaccines Working Party — «Workplan for 2013»,http://www.ema.europa.eu/docs/en_GB/document_library/Work_programme/2010/02/WC500073590.pdf2013.
[295] EuropeanMedicinesAgency C, Note for guidance on the clinical evaluation of vaccines[Internet], EMEA/CHMP/VWP/164653/2005, 2005. (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003875.pdf).
[296] European Medicines Agency C, Guideline on adjuvants in vaccines for humanuse [Internet], EMEA/CHMP/VEG/134716/2004, 2005. (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003809.pdf).
[297] Food and Drugs Administration O of C, Tissue and Gene Therapies. CLINICAL CON-SIDERATIONS FOR THERAPEUTIC CANCER VACCINES [Internet]. FDA's Center forBiologics Evaluation and Research, 2011. (http://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/vaccines/ucm278673.pdf).
Related Documents











