The International Journal of Biochemistry & Cell Biology 44 (2012) 139–149 Contents lists available at SciVerse ScienceDirect The International Journal of Biochemistry & Cell Biology journa l h o me page: www.elsevier.com/locate/biocel Hypersensitivity of A8344G MERRF mutated cybrid cells to staurosporine-induced cell death is mediated by calcium-dependent activation of calpains Guillaume Rommelaere, Sébastien Michel, Jérémy Malaisse, Sophie Charlier, Thierry Arnould, Patricia Renard ∗ Laboratory of Biochemistry and Cell Biology (URBC), NARILIS (NAmur Research Institute for Life Sciences), University of Namur (FUNDP), 61 rue de Bruxelles, 5000 Namur, Belgium a r t i c l e i n f o Article history: Received 6 July 2011 Received in revised form 13 October 2011 Accepted 16 October 2011 Available online 22 October 2011 Keywords: Apoptosis Mitochondria Cybrid MERRF Calpain Caspases a b s t r a c t Mutations in the mitochondrial DNA can lead to the development of mitochondrial diseases such as Myoclonic Epilepsy with Ragged Red Fibers (MERRF) or Mitochondrial Encephalomyopathy, Lactic Aci- dosis and Stroke-like episodes (MELAS). We first show that human 143B-derived cybrid cells harboring either the A8344G (MERRF) or the A3243G (MELAS) mutation, are more prone to undergo apoptosis then their wild-type counterpart, when challenged with various apoptotic inducers such as staurosporine, etoposide and TRAIL. In addition, investigating the mechanisms underlying A8344G cybrid cells hyper- sensitivity to staurosporine-induced cell death, we found that staurosporine treatment activates caspases independently of cytochrome c release in both wild-type and mutated cells. Caspases are activated, at least partly, through the activation of calcium-dependent calpain proteases, a pathway that is more strongly activated in mutated cybrid cells than in wild-type cells exposed to staurosporine. These results suggest that calcium homeostasis perturbation induced by mitochondrial dysfunction could predispose cells to apoptosis, a process that could take part into the progressive cell degeneration observed in MERRF syndrome, and more generally in mitochondrial diseases. © 2011 Elsevier Ltd. All rights reserved. 1. Introduction Several mitochondrial diseases such as the Myoclonic Epilepsy with Ragged Red Fibers (MERRF) and the Mitochondrial myopathy, Encephalopathy, Lactic acidosis, and Stroke-like episodes (MELAS) syndromes are due to defects in mitochondrial tRNA coding genes. Since cells contain multiple copies of the mitochondrial genome, the development and the severity of such mtDNA-associated pathologies depend on the heteroplasmy level of the mutation (i.e. the mutated/wt mtDNA ratio). Clinically, the MERRF syndrome is a neurodegenerative disease characterized by myoclonus, mito- chondrial myopathy, cerebellar ataxia and generalized epilepsy. MELAS patients present intermittent episodes of encephalopathy, Abbreviations: CPEO, Chronic Progressive External Ophthalmoplegia; KSS, Kearns–Sayre Syndrome; LHON, Leber’s Hereditary Optic Neuropathy; MELAS, Mito- chondrial Encephalomyopathy Lactic Acidosis and Stroke-like episodes; MERRF, Myoclonic Epilepsy with Ragged Red Fibers; MILS, Maternally Inherited Leigh Syn- drome; PARP-1, Poly (ADP-Ribose) Polymerase 1; ROS, Reactive Oxygen Species; STS, staurosporine; TRAIL, TNF- Related Apoptosis Inducing Ligand. ∗ Corresponding author. Tel.: +32 81 724128; fax: +32 81 724135. E-mail address: [email protected] (P. Renard). vomiting and migraine-like headache. Histologically, Gomori’s trichrome staining of MERRF or MELAS patients muscle biopsies reveal the presence of the “Ragged Red Fibers” (RRF) due to the subsarcolemmal accumulation of mitochondria (for reviews, see DiMauro and Hirano, 2009; Tuppen et al., 2010; Wallace, 2001). At the molecular level, the MERRF syndrome is mainly asso- ciated with the A8344G transition in the mitochondrial tRNA Lys coding gene, while the A3243G transition in the tRNA Leu is the most common mutation causing the MELAS syndrome. These mutations impair the mitochondrial protein synthesis, leading to a decrease in electron transport chain activity. Ensuing features are a decreased oxidative production of ATP, an increased Reactive Oxygen Species (ROS) production, a decreased mitochondrial membrane potential and modifications in calcium homeostasis (Brini et al., 1999; James et al., 1996; Masucci et al., 1995; Moudy et al., 1995; Vives-Bauza et al., 2006; von Kleist-Retzow et al., 2007). Beside these direct con- sequences of an altered electron transport chain, it has long been suspected that other mitochondria-dependent processes such as apoptosis could be altered in mitochondrial diseases. While sev- eral studies conducted on muscle biopsies of patients presenting MERRF, MELAS or other mitochondrial diseases suggested an ele- vated level of apoptotic markers (Mirabella et al., 2000; Umaki 1357-2725/$ – see front matter © 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.biocel.2011.10.009

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Hsa
GTL6
a
ARRAA
KAMCMCC
1
wEsStptacM
KcMdS
1d
The International Journal of Biochemistry & Cell Biology 44 (2012) 139– 149
Contents lists available at SciVerse ScienceDirect
The International Journal of Biochemistry& Cell Biology
journa l h o me page: www.elsev ier .com/ locate /b ioce l
ypersensitivity of A8344G MERRF mutated cybrid cells totaurosporine-induced cell death is mediated by calcium-dependentctivation of calpains
uillaume Rommelaere, Sébastien Michel, Jérémy Malaisse, Sophie Charlier,hierry Arnould, Patricia Renard ∗
aboratory of Biochemistry and Cell Biology (URBC), NARILIS (NAmur Research Institute for Life Sciences), University of Namur (FUNDP),1 rue de Bruxelles, 5000 Namur, Belgium
r t i c l e i n f o
rticle history:eceived 6 July 2011eceived in revised form 13 October 2011ccepted 16 October 2011vailable online 22 October 2011
eywords:poptosis
a b s t r a c t
Mutations in the mitochondrial DNA can lead to the development of mitochondrial diseases such asMyoclonic Epilepsy with Ragged Red Fibers (MERRF) or Mitochondrial Encephalomyopathy, Lactic Aci-dosis and Stroke-like episodes (MELAS). We first show that human 143B-derived cybrid cells harboringeither the A8344G (MERRF) or the A3243G (MELAS) mutation, are more prone to undergo apoptosis thentheir wild-type counterpart, when challenged with various apoptotic inducers such as staurosporine,etoposide and TRAIL. In addition, investigating the mechanisms underlying A8344G cybrid cells hyper-sensitivity to staurosporine-induced cell death, we found that staurosporine treatment activates caspases
itochondriaybridERRF
alpainaspases
independently of cytochrome c release in both wild-type and mutated cells. Caspases are activated, atleast partly, through the activation of calcium-dependent calpain proteases, a pathway that is morestrongly activated in mutated cybrid cells than in wild-type cells exposed to staurosporine. These resultssuggest that calcium homeostasis perturbation induced by mitochondrial dysfunction could predisposecells to apoptosis, a process that could take part into the progressive cell degeneration observed in MERRFsyndrome, and more generally in mitochondrial diseases.
© 2011 Elsevier Ltd. All rights reserved.
. Introduction
Several mitochondrial diseases such as the Myoclonic Epilepsyith Ragged Red Fibers (MERRF) and the Mitochondrial myopathy,
ncephalopathy, Lactic acidosis, and Stroke-like episodes (MELAS)yndromes are due to defects in mitochondrial tRNA coding genes.ince cells contain multiple copies of the mitochondrial genome,he development and the severity of such mtDNA-associatedathologies depend on the heteroplasmy level of the mutation (i.e.he mutated/wt mtDNA ratio). Clinically, the MERRF syndrome is
neurodegenerative disease characterized by myoclonus, mito-hondrial myopathy, cerebellar ataxia and generalized epilepsy.ELAS patients present intermittent episodes of encephalopathy,
Abbreviations: CPEO, Chronic Progressive External Ophthalmoplegia; KSS,earns–Sayre Syndrome; LHON, Leber’s Hereditary Optic Neuropathy; MELAS, Mito-hondrial Encephalomyopathy Lactic Acidosis and Stroke-like episodes; MERRF,yoclonic Epilepsy with Ragged Red Fibers; MILS, Maternally Inherited Leigh Syn-
rome; PARP-1, Poly (ADP-Ribose) Polymerase 1; ROS, Reactive Oxygen Species;TS, staurosporine; TRAIL, TNF-� Related Apoptosis Inducing Ligand.∗ Corresponding author. Tel.: +32 81 724128; fax: +32 81 724135.
E-mail address: [email protected] (P. Renard).
357-2725/$ – see front matter © 2011 Elsevier Ltd. All rights reserved.oi:10.1016/j.biocel.2011.10.009
vomiting and migraine-like headache. Histologically, Gomori’strichrome staining of MERRF or MELAS patients muscle biopsiesreveal the presence of the “Ragged Red Fibers” (RRF) due to thesubsarcolemmal accumulation of mitochondria (for reviews, seeDiMauro and Hirano, 2009; Tuppen et al., 2010; Wallace, 2001).
At the molecular level, the MERRF syndrome is mainly asso-ciated with the A8344G transition in the mitochondrial tRNALys
coding gene, while the A3243G transition in the tRNALeu is the mostcommon mutation causing the MELAS syndrome. These mutationsimpair the mitochondrial protein synthesis, leading to a decrease inelectron transport chain activity. Ensuing features are a decreasedoxidative production of ATP, an increased Reactive Oxygen Species(ROS) production, a decreased mitochondrial membrane potentialand modifications in calcium homeostasis (Brini et al., 1999; Jameset al., 1996; Masucci et al., 1995; Moudy et al., 1995; Vives-Bauzaet al., 2006; von Kleist-Retzow et al., 2007). Beside these direct con-sequences of an altered electron transport chain, it has long beensuspected that other mitochondria-dependent processes such as
apoptosis could be altered in mitochondrial diseases. While sev-eral studies conducted on muscle biopsies of patients presentingMERRF, MELAS or other mitochondrial diseases suggested an ele-vated level of apoptotic markers (Mirabella et al., 2000; Umaki
1 l of Bi
e2fiwsi(aplm
omiaeraYeasrecW2
mCbs2WhtatsWcdsrbb1
fotrLaticbcctctos
40 G. Rommelaere et al. / The International Journa
t al., 2002), these findings have been controversial (Ikezoe et al.,002) until a large-scale study conducted on individual musclebers from patients suffering from various mitochondrial diseasesas published by the group of A. Lombes (Aure et al., 2006). This
tudy clearly highlighted the presence of apoptotic features specif-cally in muscle fibers presenting an accumulation of mitochondriaRRF) and harboring a high percentage of mutation load. Whilen increased apoptosis in muscle fibers could contribute to therogressive muscle loss observed in these patients, the molecu-
ar mechanisms underlying increased apoptosis in the context ofitochondrial mutations are still unclear.Apoptosis has been evidenced in several in vitro cellular models
f mitochondrial diseases. In �0 cells, which are totally depleted oftDNA, we and others have shown a hypersensitivity to apoptosis
nduced by various inducers such as staurosporine, UV, cisplatinnd H2O2 when compared with WT cells (Cardoso et al., 2004; Liut al., 2004; Rommelaere et al., 2011; Yen et al., 2005) while otheresearch groups have shown an increased resistance to inducedpoptosis (Biswas et al., 2005; Ferraresi et al., 2008; Lee et al., 2004;u et al., 2009). These contradictory results may be due to the differ-ntial expression of Bcl-2 proteins between WT and depleted cellsccording to the cell model used. For example our group recentlyhowed that mtDNA depleted 143B cells were sensitive to stau-osporine due to the decreased expression of Bcl-2 (Rommelaeret al., 2011) while in mtDNA-depleted mouse myoblasts C2C12ells, the same protein is overexpressed when compared to
T C2C12, protecting cells from staurosporine (Biswas et al.,005).
Apoptosis has also been widely studied in cybrid cells harboringtDNA mutations responsible for many mitochondrial pathologies.
ybrid cells with mtDNA mutations have mostly been reported toe more sensitive to apoptosis triggered by many inducers such astaurosporine, UV exposure, Fas or H2O2 exposure (Carrozzo et al.,004; Danielson et al., 2002; Liu et al., 2007; Schoeler et al., 2005;ong and Cortopassi, 1997). To our knowledge, only one study
as shown resistance of cybrid cells harboring the A8344G muta-ion to apoptosis induced by staurosporine, etoposide, thapsigarginnd tunicamycin (Kwong et al., 2007). These authors suggest thathis resistance is caused by an increase in Bcl-2 protein expres-ion, but they used parental 143B cells as a reference instead of
T cybrid cells. Regarding the A8344G and the A3243G cybridells derived from 143B osteosarcoma cells, two other indepen-ent studies have shown that these cells are more sensitive totaurosporine or H2O2-induced apoptosis than WT cybrid cells, asevealed by higher caspase-3 activation and DNA fragmentation,ut the molecular mechanisms leading to apoptosis have nevereen fully investigated (Liu et al., 2004; Wong and Cortopassi,997).
In this study, we first confirmed that cybrid cells derivedrom 143B human osteosarcoma cells and harboring the A8344Gr the A3243G mutation are more sensitive than their wildype (WT) counterpart to several apoptosis inducers: stau-osporine, etoposide and TRAIL (TNF-� Related Apoptosis Inducingigand). The second objective was to study the molecular mech-nisms underlying the hypersensitivity of MERRF cybrid cellso staurosporine-induced apoptosis. Unexpectedly, staurosporinenduces the activation of effector caspases without any cytochrome
release from mitochondria. Actually, in these cells caspases woulde activated, at least partly, by the calcium-activated proteasesalpains, a pathway that is more strongly activated in mutatedybrid cells than in wild type counterpart. These data suggesthat impaired calcium homeostasis observed in cells with mito-
hondrial disorders could be responsible for their hypersensitivityo induced apoptosis and could contribute to the developmentf mitochondrial diseases such as the MERRF and the MELASyndrome.ochemistry & Cell Biology 44 (2012) 139– 149
2. Material and methods
2.1. Cell culture and induction of apoptosis
The homoplasmic WT and A8344G MERRF mutated cybrid cellswere a gift from Prof. Andreu (University Hospital Vall d’Hebron,Barcelona, Spain) and were originally obtained by fusion of patientplatelets containing the A8344G mtDNA mutation with humanosteosarcoma cells devoid of mtDNA (�0143B cells) (Vives-Bauzaet al., 2006). Cells were cultured in 4.5 g glucose-containing DMEMsupplemented with 10% FBS, containing 110 mg/L pyruvate andcontaining 50 �g/mL uridine for �0143B cells. Cells were plated48 h before experiments at a density of 1,500,000 cells/75 cm2
flask for mutated cybrid cells and 1,000,000 cells/75 cm2 flask forWT cybrid cells as population doubling time is slightly differentbetween these cell lines. Cybrid cells were then incubated with500 nM staurosporine (Sigma) to induce apoptosis. When inhibitorswere used, cells were pre-incubated for 60 min with 20 �M zVAD-fmk (BD Pharmingen), 10 �M ALLN (Sigma), 50 �M calpeptin(Calbiochem), 50 �M of PD150606 (Calbiochem) or 20 �M LEVD-CHO (Calbiochem) before staurosporine addition. At the end ofthe incubations, adherent and floating cells were collected foranalyses.
2.2. Protein extraction
At the end of the incubations, cells were rinsed once with PBSand scraped in lysis buffer (20 mM Tris, pH 7.4, 150 mM KCl, 1 mMEDTA, 1% Triton X-100) complemented with phosphatase and pro-tease inhibitors. Floating cells and rinsing PBS were harvestedand centrifuged 5 min at 1000 rpm. The pellet was resuspendedin lysis buffer and added to the cell lysate. After 5 min on ice,the cell lysates were centrifuged for 10 min at 13,000 rpm at4 ◦C. The supernatants referred as “clear cell lysates” were col-lected, protein content was quantified with the BioRad proteinassay.
2.3. Western blotting analysis
The proteins were resolved by electrophoresis on 12% Tricinegels, on 4–12% bis–Tris gel or on 3–8% Tris-Acetate gels (NuPage,Invitrogen). After semi-dry transfer onto PVDF (polyvinylidenefluoride) membranes, Western blotting analysis was performedwith primary antibodies against the following human proteins:Poly (ADP-Ribose) Polymerase-1 (PARP-1, Pharmingen; dilution1:5000), caspase-9 (Cell Signaling; 1:3000), cleaved caspase-3 (CellSignaling; 1:1000), caspase-7 (Cell Signaling; 1:1000), �-fodrin(Biomol; 1:10,000), cytochrome c (BD Pharmingen; 1:5000). West-ern blot analysis was performed either by chemoluminescence (ECLadvanced, Amersham Biosciences) using a HRP-conjugated per-oxidase secondary antibody (dilution 1:100,000 or a 1:300,000)or by infrared fluorescence (Odyssey scanner, Li-Cor) using asecondary antibody coupled to Infrared dyes (Li-Cor; 1:10,000).Protein loadings for cleared cell lysates or cytosolic fractionswere controlled by the immunodetection of �-tubulin (Sigma;1:20,000) or �-actin (Sigma, 1:20,000). Protein loading for“mitochondria-enriched fractions” was controlled by the immun-odetection of TOM-20 (Transporter of Outer Membrane-20; BDBioscience; 1:20,000). Molecular weight markers used wereSeeBlue Plus2 Protein Standard (Invitrogen) and Odyssey Two-Color Molecular Weight Markers (Li-Cor) for the revelation by
chemoluminescence and by infrared fluorescence, respectively.For Tris-Acetate gels, the HiMark Pre-Stained Protein Stan-dard (Invitrogen) was also used for one experiment presentedin Fig. 3B.
l of Biochemistry & Cell Biology 44 (2012) 139– 149 141
2
ARcsFotaaAt(oaw
2
d(su
2
t(fe
2
Dr
2
attbo
3
3a
tcimmm(aca
Fig. 1. Effect of staurosporine on the key markers of apoptosis in WT and MERRFmutated cybrid cells. Wild-type (white) and MERRF mutated cybrid cells (black)were incubated for the indicated times with 500 nM staurosporine (STS) beforecollected for the analysis of DEVDase activity using the fluorescent substrate (Ac-DEVD-AFC) (A), for the Western blotting analysis of caspase-3 (B), caspase-7 (C)and PARP-1 (D) (protein loading being controlled by the immunodetection of �-tubulin), and for the analysis of DNA fragmentation, using an ELISA (E). Theseresults represent means ± 1 S.D. for n = 3. *Significantly different from untreatedcontrols as determined by a Student–Newman–Keuls test with p < 0.05 or less.+Significantly different from the corresponding treated WT cybrid cells as deter-
G. Rommelaere et al. / The International Journa
.4. Flow cytometry
Cytosolic calcium concentration was evaluated using Fluo-3-M and FuraRed fluorescent probes as described (Novak andabinovitch, 1994), allowing a ratiometric determination of cal-ium concentration. Cybrid cells were treated with 500 nMtaurosporine for 1 or 2 h before the addition of Fluo-3-AM anduraRed for an extra 30 min incubation at a final concentrationf 2 �M and 5 �M, respectively. Cells were then rinsed with PBS,rypsinised, rinsed with PBS and resuspended in HBSS (Hank’s Bal-nced Salt Solution). Cells were analyzed by flow cytometry with
FACScalibur (BD Biosciences), using FL1-H channel for Fluo-3-M and FL3-H channel for FuraRed. Data were processed using
he Cell Quest Pro software. Mean Channel Fluorescence RatiosMCFRs) were calculated for each condition by dividing Fluo-3-AMr FuraRed mean values by the mean values of their respective neg-tive control (same treatment with unlabelled cells). Finally, resultsere expressed as Fluo-3-AM MCFR normalized by FuraRed MCFR.
.5. Caspase activity assay
Caspase-3/-7 and caspase-4 activity assays were performed asescribed (Rommelaere et al., 2011) using 10 �g/mL Ac-DEVD-AFCBD Pharmingen) or 40 �M Ac-LEVD-AFC (Alexis) fluorescent sub-trates, respectively. Results are expressed in arbitrary fluorescencenits normalized for protein content used for the assay.
.6. Subcellular fractionation by differential centrifugation
Subcellular fractionation was performed by differential cen-rifugation as previously described for 143B and �0143B cellsRommelaere et al., 2011) in order to analyze cytochrome c releaserom MLP fractions (mitochondrial, lysosomal and microsomalnriched fractions) into cytosolic fractions.
.7. DNA fragmentation assays
Quantitative DNA fragmentation was assessed with the Celleath Detection ELISA kit (Roche) following the manufacturer’s
ecommendations.
.8. Statistical analysis
Data from at least three independent experiments are presenteds means ± S.D. and were analyzed with the appropriate statisticalest such as ANOVA 2 or ANOVA on ranks followed by a Holm–Sidakest or a Student–Newman–Keuls test, respectively, as determinedy the software SigmaStat 3.1. Differences between means werenly considered as statistically significant when p < 0.05 or less.
. Results
.1. MERRF mutated cybrid cells are more sensitive to inducedpoptosis.
In order to study the impact of a mitochondrial disorder onhe apoptotic process, we used homoplasmic transmitochondrialybrid cells derived from 143B osteosarcoma cells and harbor-ng (or not for WT control cells) the A8344G transition in the
tDNA, responsible for the MERRF syndrome. WT and MERRFutated cybrid cells were treated with 500 nM staurosporine, aolecule described to induce mitochondria-mediated apoptosis
Diwakarla et al., 2009), for 1–16 h before evaluating caspase-3/-7ctivity through DEVDase activity assay, assessed with a fluores-ent substrate. As seen in Fig. 1A, staurosporine induces DEVDasectivity in both cell lines but more rapidly and more intensively in
mined by a Student–Newman–Keuls with p < 0.05 or less.
MERRF mutated cybrid cells. As both caspases-3 and -7 are able tocleave the fluorescent substrate, we next searched for caspases-3 and -7 cleavages in WT and mutated cybrid cells by Westernblot, using antibodies specific for the active cleaved forms of bothcaspases (Fig. 1B and C). The active fragments of both proteasesare already detected after 4 h of 500 nM staurosporine treatmentin mutated cybrid cells and their abundance increases in a time-dependent manner while in WT cybrid cells, the cleaved fragmentsare only barely detectable after 6 h of staurosporine treatment andare always less abundant than in MERRF mutated cybrid cells,for both caspases-3 and -7. These kinetics are correlated to the
detection of the cleaved form of PARP-1, a well-known endoge-nous substrate of caspases (Fig. 1D). Finally, internucleosomal DNAcleavage, a final step of the apoptotic process was quantified. A
1 l of Biochemistry & Cell Biology 44 (2012) 139– 149
shWfsttMma(tiTtstuesacDwmtb
3m
hrartcwWcccimsfiwdtscDdm
wAioascic
Fig. 2. Non-canonical mechanisms are triggered by staurosporine. Wild-type andMERRF mutated cybrid cells were incubated for the indicated time with 500 nMstaurosporine (STS) before collected for the Western blotting analysis of caspase-9 (A). Western blotting analysis of cytochrome c performed on cytosolic and onMLP (mitochondrial, lysosomal and microsomal enriched) fractions of �0143B cellstreated with 2 �M staurosporine (STS) for 6 h or of WT and mutated MERRF cybridcells treated with or without 500 nM staurosporine for 6 h (STS) (B). WT (white)and MERRF mutated cybrid cells (black) were incubated with or without 500 nMstaurosporine (STS) for 6 h in the presence or in the absence of 20 �M zVAD-fmkor 10 �M ALLN. After the incubations: Clear cell lysates were prepared for theWestern blotting analysis of PARP-1. For a clearer presentation, two nonessentialconditions have been sliced from the original blot. Band cut is indicated by theblack lines (C). Cells were processed for the colorimetric DNA fragmentation assayas described in Section 2. The results are expressed as optical density (O.D.) nor-malized for protein content (O.D./�g protein) and represent means ± 1 S.D. for n = 3(D). Protein loadings for Western blotting were controlled by the immunodetec-tion of �-tubulin and TOM 20 for S fraction and MLP fractions, respectively and bythe immunodetection of �-tubulin and �-actin for clear cell lysates. *Significantlydifferent from untreated controls as determined by a Student–Newman–Keulstest with p < 0.05 or less. +Significantly different from the corresponding treated
42 G. Rommelaere et al. / The International Journa
ignificant increase in DNA cleavage is already detected after a 4--treatment in MERRF mutated cybrid cells and only after 6 h inT cybrid cells (Fig. 1E). As observed for caspase activation, DNA
ragmentation increases with incubation time in the presence oftaurosporine and is always stronger in mutated cybrid cells. Takenogether, these results show that the mitochondrial A8344G muta-ion induces a hypersensitivity to staurosporine-induced cell death.
oreover, this phenomenon was not limited to this particulartDNA mutation since hypersensitivity to staurosporine-induced
poptosis was similarly evidenced in A3243G MELAS cybrid cellsFig. S1). Indeed, caspase-3/-7 activity as well as DNA fragmen-ation induced by staurosporine increase more rapidly and morentensively in MELAS mutated cybrid cells than in WT cybrid cells.hese observations confirm that two different point mtDNA muta-ions leading to impaired mitochondrial protein synthesis increaseensitivity to cell death. Finally, other apoptotic inducers, known torigger apoptosis by other pathways than staurosporine, were alsosed to challenge A8344G MERRF cybrid cells. TRAIL, triggering thextrinsic apoptosis pathway (Griffith and Lynch, 1998) and etopo-ide, a DNA damage-inducing agent (Walker et al., 1991), were alsoble to trigger a more pronounced apoptotic response in A8344Gybrid cells when compared with WT cybrid cells as evidenced byEVDase activity and DNA fragmentation (Fig. S2). In summary,hatever the inducer (staurosporine, etoposide or TRAIL) or thetDNA mutation is (MERRF or MELAS), the mitochondrial dysfunc-
ion always sensitizes the cell line to induced apoptosis as showny caspase activation and DNA fragmentation analyses.
.2. Staurosporine-induced apoptosis involves non classicalechanisms
To decipher the molecular mechanisms responsible for theypersensitivity of the mutated MERRF cybrid cells to stau-osporine, the key components of the intrinsic pathway were firstnalyzed. We looked for caspase-9 processing and cytochrome celease in the cytosol of both cell lines after a 500 nM staurosporinereatment. As shown in Fig. 2A, staurosporine induces caspase-9leavage, appearing after 4 h of treatment in mutated cybrid cellshile the cleavage is only barely detected after 6 h of incubation inT cybrid cells and is always less abundant than in MERRF mutated
ybrid cells. This result confirms the hypersensitivity of mutatedybrid cells observed in Fig. 1 and suggests that caspase cascadeould be activated by the well described mitochondria-dependentntrinsic pathway involving outer mitochondrial membrane per-
eabilization. Cytochrome c abundance was then analyzed inubcellular fractions of both cell lines treated with staurosporineor 6 h. Unexpectedly, we could not find any cytochrome c releasen the cytosol of WT and MERRF mutated cybrid cells incubated
ith staurosporine (Fig. 2B), while cytosolic cytochrome c could beetected in �0143B challenged with 2 �M staurosporine, a condi-ion used as a positive control (Rommelaere et al., 2011). Moreover,taurosporine does not induce cytochrome c release in the MELASybrid cells harboring the A3243G mutation in the mitochondrialNA (Fig. S3), suggesting that the mechanisms that mediate celleath in MERRF and MELAS cybrid cells treated with staurosporineay be similar.As we recently showed that staurosporine-induced cell death
as inhibited by the use of the calpain–cathepsin B inhibitorLLN in mtDNA-depleted �0143B, this inhibitor was then tested
n WT and MERRF mutated cybrid cells to look for a contributionf non-caspase proteases to staurosporine-induced PARP-1 cleav-ge (Fig. 2C) and DNA fragmentation (Fig. 2D). ALLN decreases
taurosporine-induced PARP-1 cleavage in MERRF mutated cybridells and in WT cybrid cells. DNA fragmentation was quantifiedn the same conditions as in the presence zVAD-fmk, a pan-aspase inhibitor. While none of the inhibitors used induces aWT cybrid cells as determined by a Student–Newman–Keuls with p < 0.05 or less.§Significantly different from corresponding staurosporine treated cells as deter-mined by a Student–Newman–Keuls test with p < 0.05 or less.
significant decrease in STS-induced DNA cleavage in WT cybridcells, the situation is different in mutated cybrid cells. Indeed, asfor PARP-1 cleavage, ALLN but also zVAD-fmk cause a decrease
in staurosporine-induced DNA fragmentation in MERRF mutatedcybrid cells. As ALLN is not a highly specific inhibitor (it inhibitscalpains but also cathepsin B), this suggests that in addition to cas-pases, calpains or cathepsin B could also be involved in the induced
G. Rommelaere et al. / The International Journal of Biochemistry & Cell Biology 44 (2012) 139– 149 143
Fig. 3. Staurosporine induces calcium concentration increase and calpain activation. (A) WT (white) and mutated (black) MERRF cybrid cells were treated or not (CTL) with500 nM staurosporine (STS) for 1 or 2 h before cells were stained for 30 min with Fluo-3-AM and FuraRed in the presence of staurosporine. Fluorescence was analyzed by flowcytometry as described in Section 2. Results are expressed as Fluo-3 MCFR (Mean Channel Fluorescence Ratio) normalized by FuraRed MCFR and represent means ± 1 S.D.for n = 4. **, ***Significantly different from corresponding untreated control cells as determined by a Holm–Sidak test with p < 0.01 and p < 0.001, respectively. +++Significantlydifferent from corresponding WT cybrid cells as determined by a t-test with p < 0.001. N.S.: non significant. (B and C) WT and mutated MERRF cybrid cells were incubated for6 h or 16 h with or without 500 nM staurosporine (STS) in the presence or in the absence of: (B) 20 �M zVAD-fmk or 10 �M ALLN or (C) 50 �M calpeptin or 50 �M PD150606.Clear cell lysates were prepared and the abundance of �-fodrin breakdown products was monitored by Western blotting analysis. Protein loading was controlled by thei
pro2owncatcmqc
3t
tlcTrt
mmunodetection of �-tubulin.
rogrammed cell death. As we previously showed that stau-osporine could induce cathepsin B release in the cytosol of �0143Bsteosarcoma cells to contribute to cell death (Rommelaere et al.,011), we analyzed the localization of cathepsin B by immunoflu-rescence staining in WT and mutated cybrid cells treated or notith 500 nM staurosporine for 6 h. As seen in Fig. S4, cathepsin B isot released in the cytosol after staurosporine treatment. Indeed,athepsin B staining in MERRF cybrid cells remains very punctu-ted and without any diffused fluorescence in the cytosol contraryo what is observed in staurosporine-treated �0143B osteosarcomaells. We thus can assume that the effect of ALLN on cell death inutated cybrid cells is not due to cathepsin B inhibition. Conse-
uently, we looked for calpain activation in WT and mutated cybridells treated with staurosporine.
.3. Calpains are activated by staurosporine and participate inhe apoptotic response
Calpains are calcium-activated proteases which can participateo cell death (for a review, see Goll et al., 2003). First, free cytoso-ic calcium concentration was analyzed by flow cytometry in cells
hallenged with staurosporine using Fluo-3 and Fura Red probes.he simultaneous use of this couple of probes allows a ratiomet-ic measure of cytosolic calcium concentration, thereby enablinghe comparison of calcium concentration (basal and in responseto staurosporine) between two cell lines susceptible to differ-entially incorporate the probes (Novak and Rabinovitch, 1994).As shown in Fig. 3A, MERRF mutated cybrid cells respond tostaurosporine treatment by a 2-fold increase in calcium concen-tration after 1 h while it takes 2 h in WT cybrid cells. As calciumconcentration increases rapidly upon staurosporine treatment,calpain activity was then monitored by analyzing the �-fodrinbreakdown products. This 240 kDa protein can be cleaved byboth caspases and calpains, but cleavage by caspases generates145/120 kDa �-fodrin fragments while proteolysis by calpains leadsto 150/145 kDa fragments (Zhang et al., 2009). We found that�-fodrin is cleaved after a 6-h-treatment with staurosporine inboth cybrid cell lines but the cleavage products are clearly moreabundant in mutated cybrids, confirming the hypersensitivity ofthis cell line to staurosporine (Fig. 3B). However, as the 145and 150 kDa products cannot be distinguished on the blots, theactivation of calpains is difficult to assess. To look for a putativeactivation of calpains, both cell lines were challenged with stau-rosporine in the presence of 10 �M ALLN or 20 �M zVAD-fmk.As the calpain inhibitor (ALLN) slightly decreases the 145/150 kDaproducts abundance while the caspase inhibitor (zVAD-fmk) only
decreases the 120 kDa product abundance, these data suggest thatstaurosporine induces calpain activity in both cell lines, even if itis less pronounced in the WT cybrid cells. As ALLN is known toinhibit other proteases such as cathepsin B, more specific inhibitors
144 G. Rommelaere et al. / The International Journal of Bi
Fig. 4. Effect of calpeptin on staurosporine-induced apoptosis markers. WT (white)and MERRF mutated (black) cybrids cells were incubated for 6 h with or without500 nM staurosporine (STS) in the presence or in the absence of 50 �M calpeptin.Cells were then collected for the analysis of DEVDase activity using the fluores-cent substrate (Ac-DEVD-AFC) (A), for the Western blotting analysis of caspase-3(B), caspase-7 (C) and PARP-1 (D). Caspase-3 and -7 abundances were analyzedin mutated cybrid cells only and PARP-1 cleavage was also analyzed after 16 h oftreatment. For a clearer presentation, one nonessential condition has been slicedfrom the original blots (Fig. 4D). Band cut is indicated by the black lines. Proteinloadings were controlled by the immunodetection of �-tubulin. DEVDase activityresults are expressed as fluorescence arbitrary units normalized for protein con-tent (fluorescence/�g protein) and represent means ± 1 S.D. for n = 3. *Significantlydifferent from untreated controls as determined by a Student–Newman–Keuls testwith p < 0.05 or less. +Significantly different from the corresponding treated WTcybrid cells as determined by a Student–Newman–Keuls test with p < 0.05 or less.N
wic1tcb�a
pitdeicarPbc
death. It is thus only when inhibitors are used in combination thatPARP cleavage and DNA fragmentation induced by staurosporine
.S.: non significant.
ere tested on the cleavage of �-fodrin induced by staurosporinen both cell lines. Both calpeptin and PD150606, two calpain spe-ific inhibitors (Tsujinaka et al., 1988; Wang et al., 1996), reduce the45/150 kDa products in MERRF mutated cybrid cells, confirminghe activation of calpains by staurosporine treatment. In WT cybridells, only calpeptin decreases the cleavage of �-fodrin inducedy staurosporine (Fig. 3C). As calpeptin inhibits the cleavage of-fodrin more efficiently than PD150606, calpeptin was kept as
specific calpain inhibitor for following experiments.We thus next used calpeptin to evaluate the contribution of cal-
ains in stauroporine-induced cell death in cybrid cells. As shownn Fig. 4A, calpeptin has no significant effect on the cleavage ofhe DEVDase fluorescent substrate induced by staurosporine butecreases the activating cleavage of both caspase-3 and to a lesserxtend of caspase-7 (Fig. 4B and C, respectively). As calpeptinnhibitory effect is far from being complete on the cleavage of theseaspases, the lack of significant effect of calpeptin on DEVDasectivity induced by staurosporine can probably be explained by theesidual activity of these enzymes. Finally, staurosporine-inducedARP-1 cleavage was also decreased in the presence of calpeptin in
oth cell lines (Fig. 4D). Taken together, these results suggest thatalpains are activated by staurosporine in 143B-derived cybrid cellsochemistry & Cell Biology 44 (2012) 139– 149
and participate to effector caspases-3 and -7 activation and PARP-1cleavage.
3.4. Caspase-4 is activated following staurosporine treatmentand is involved in cell death
Calpain-dependent activation of caspases has been reported inmice models where it induces caspase-12 activation, triggeringcaspase cascade activation (Nakagawa and Yuan, 2000). However,human caspase-12 is truncated and does not show any proteaseactivity and caspase-4 has been postulated to play the role ofcaspase-12 in humans (Matsuzaki et al., 2010). The activity of thisenzyme was thus measured with a specific fluorescent substrate,ac-LEVD-AFC (Fig. 5A) in cybrid cells treated with 500 nM stau-rosporine for 6 or 12 h. Caspase-4 is activated upon staurosporinetreatment after 6 and 12 h in mutated cybrid cells. The activity of theprotease triggered by the staurosporine treatment is also higher inMERRF mutated cybrid cells than in WT cells. To check if caspase-4 activation could be dependent on calpain activation, caspase-4activity was next assayed after a treatment with staurosporinefor 6 h in the presence or in the absence of the calpain inhibitors,ALLN or calpeptin. As shown in Fig. 5B, even if not significant, bothmolecules slightly prevent caspase-4 activation in both cell lines,suggesting that calpains may be, at least partly, involved in theactivation of this protease. Moreover, using a caspase-4 inhibitor(LEVD-CHO), we also showed that caspase-4 is involved in DEVDaseactivity induced by staurosporine (Fig. 5C).
3.5. The interconnected calpains–caspases pathway is not theonly pathway mediating cell death induced by staurosporinein WT and MERRF mutated cybrid cells
Altogether these results indicate that caspase-4 is involved instaurosporine-induced cell death, making directly or indirectly, thelink between calpains and executioner caspases-3 and -7 lead-ing to cell death. However, the caspase inhibitor zVAD-fmk andthe calpain inhibitors (ALLN and calpeptin) do not have completeinhibitory effect on cell death markers and/or effectors (caspaseactivity, PARP cleavage, DNA fragmentation) when used separately(Figs. 2, 4 and 5). We thus wondered whether calpain and cas-pase activities were exclusively connected to each other or if eachproteases family could also act independently from the other. Toaddress this question, cybrid cells were treated with staurosporinefor 6 h in the presence of zVAD-fmk, calpeptin or both to look for aneventual additive or synergistic effect of these inhibitors on DEV-Dase activity, PARP-1 cleavage and DNA fragmentation. As shownin Fig. 6A, concerning DEVDase activity, calpeptin alone inducesa slight and non significant inhibitory effect when comparedwith staurosporine-treated cells (as already observed in Fig. 4A)while as expected, zVAD-fmk completely inhibits the activity ofcaspases-3 and -7 induced by staurosporine. However, regardingstaurosporine-induced PARP-1 cleavage and DNA fragmentation,the combination of both inhibitors induces a stronger effect thanwhen inhibitors are used separately (Fig. 6B and C). These resultssuggest that when calpains are inhibited (by calpeptin alone), cas-pases are still activated and contribute to cell death. Conversely, celldeath still occurs via calpains-dependent mechanisms when cas-pases are inhibited (by zVAD-fmk alone). Altogether these resultssuggest that even if calpains and caspases are connected throughcaspase-4 activation upon staurosporine treatment (Fig. 5), theycould also take part in independent pathways that contribute to cell
are very low as main effectors activated by staurosporine areinhibited.

G. Rommelaere et al. / The International Journal of Biochemistry & Cell Biology 44 (2012) 139– 149 145
Fig. 5. Involvement of caspase-4 in staurosporine-induced apoptosis. (A) WT(white) and mutated MERRF cybrid cells (black) were treated with or without (CTL)500 nM staurosporine (STS) for 6 or 12 h (A) or with 500 nM staurosporine for 6 h inthe presence or in the absence of 10 �M ALLN or 50 �M calpeptin (B). Caspase-4 activity was then assayed using the fluorescent substrate (Ac-LEVD-AFC). (C)WT (white) and mutated MERRF cybrid cells (black) were incubated with 500 nMstaurosporine (STS) for 6 h in the presence or in the absence of 20 �M LEVD-CHO(caspase-4 inhibitor). DEVDase activity was then assayed using the fluorescentsubstrate Ac-DEVD-AFC. The results are expressed as fluorescence arbitrary unitsnormalized for protein content (fluorescence/�g protein) and represent means ± 1S.D. for n = 3. *Significantly different from untreated controls as determined by aStudent–Newman–Keuls test with p < 0.05 or less. +Significantly different from thecorresponding treated WT cybrid cells as determined by a Student–Newman–Keulstest with p < 0.05 or less. §Significantly different from corresponding staurosporinetN
4
c2ec
Fig. 6. Synergic inhibitory effect of zVAD-fmk and calpeptin on staurosporine-induced apoptosis. WT (white) and mutated MERRF cybrid cells (black) wereincubated with 500 nM staurosporine (STS) for 6 h with or without 50 �M calpeptin,20 �M zVAD-fmk or both. At the end of the incubations: (A) Cell lysates wereprepared and DEVDase activity was assayed with the fluorescent substrate (Ac-DEVD-AFC). The results are expressed as fluorescence arbitrary units normalizedfor protein content (fluorescence/�g protein) and represent means ± 1 S.D. for n = 3.(B) A Western blotting analysis of PARP-1 cleavage was performed on clear celllysates. Protein loading was controlled by the immunodetection of �-actin. (C) Cellswere processed for the colorimetric DNA fragmentation assay as described in Sec-tion 2. The results are expressed as optical density (O.D.) normalized for proteincontent (O.D./�g protein) and represent means ± 1 S.D. for n = 3. *Significantly dif-ferent from untreated controls as determined by a Student–Newman–Keuls testwith p < 0.05 or less. +Significantly different from the corresponding treated WT
(Aure et al., 2006). In the particular case of the MERRF syndrome,
reated cells as determined by a Student–Newman–Keuls test with p < 0.05 or less..S.: non significant.
. Discussion
Even if some controversy does exist about whether or not mito-hondrial dysfunction can sensitize cells to apoptosis (Biswas et al.,
005; Danielson et al., 2002; Ferraresi et al., 2008; Rommelaeret al., 2011), it has been extensively demonstrated that mito-hondrial dysfunction induced either chemically by the use ofcybrid cells as determined by a Student–Newman–Keuls test with p < 0.05 or less.§Significantly different from corresponding staurosporine treated cells as deter-mined by a Student–Newman–Keuls test with p < 0.05 or less.
respiratory chain inhibitors (Antimycin A, rotenone,. . .) or uncou-plers (FCCP,. . .; Sapkota et al., 2011; Tiwari et al., 2011; You andPark, 2010), or genetically by mtDNA depletion or mutations, caninduce or sensitize cells to apoptosis (Liu et al., 2004; Rommelaereet al., 2011). Getting more knowledge about the sensitivity toapoptosis of cells that display mitochondrial dysfunction is of par-ticular interest in the context of mitochondrial myopathies, sincean increased apoptosis in muscle tissue could contribute to the pro-gressive muscle loss observed in most mitochondrial myopathies
mainly caused by the A8344G point mutation in the tRNALys cod-ing gene, elevated apoptosis markers have been evidenced in vivo,in muscle biopsies (Mirabella et al., 2000), as well as in vitro, in

1 l of Bi
1C
adMsiMiidMKriiFmaamtp
smtrscfcoppssltoR
drocdtocmhs(2rcwcActcce
46 G. Rommelaere et al. / The International Journa
43B-derived cybrid cells challenged with H2O2 (Wong andortopassi, 1997) or staurosporine (Liu et al., 2004).
In this study, by analyzing PARP-1 cleavage, effector caspasectivity and DNA fragmentation, we showed that cybrid cellserived from 143B osteosarcoma cells harboring the A8344GERRF mutations in the mitochondrial DNA are more sensitive to
taurosporine, etoposide and TRAIL. In addition, this phenomenons not limited to this particular mtDNA mutation since the A3243G
ELAS cybrid cells are also more sensitive to staurosporine-nduced cell death than their WT counterpart. These results aren agreement with several studies showing that mutations oreletions responsible for mitochondrial diseases such as MELAS,ERRF, Chronic Progressive External Ophthalmoplegia (CPEO),
earns–Sayre Syndrome (KSS), Leber’s Hereditary Optic Neu-opathy (LHON) or Maternally Inherited Leigh Syndrome (MILS)ncrease cell sensitivity to apoptosis induced by various apoptoticnducers such as staurosporine, UV exposure, H2O2, TRAIL andAS (for a review, see Liu et al., 2009). It has been reported thatutated cybrid cells are hypersensitive to H2O2-induced apoptosis,
process mediated by calcium and the opening of the perme-bility transition pore (Wong and Cortopassi, 1997). However, theechanisms involved in the hypersensitivity of cells harboring
he A8344G MERRF mtDNA mutation to induced-apoptosis remainoorly understood.
As mitochondria are the main integrator of the canonical intrin-ic apoptosis pathway, we first hypothesized that A8344G mtDNAutation could alter this pathway. In the commitment of apoptosis
hrough the intrinsic pathway, caspase-9 is cleaved following theelease of cytochrome c from the mitochondrial intermembranepace into the cytosol leading to the formation of the multimericomplex apoptosome (for a review, see Bratton et al., 2001). Weound that even if caspase-9 is cleaved in staurosporine-treatedybrid cells, cytochrome c release from the mitochondria was neverbserved in these conditions, meaning that the activation of cas-ase cascade might be initiated by an apoptosome-independentathway. It is to note that caspase-9 cleavage might happen down-tream of effector caspase activation (for a review on caspases,ee Pop and Salvesen, 2009). The absence of cytochrome c cytoso-ic release was not due to technical limitations, as we were ableo detect the release of cytochrome c in mtDNA devoid 143B �0
steosarcoma cells treated with 2 �M staurosporine (Fig. 2B andommelaere et al., 2011).
Very interestingly, cytochrome c release could neither beetected in A3243G MELAS cybrid cells treated with 2 �M stau-osporine for 6 h. As osteosarcoma-derived cybrid cells (MERRFr MELAS) incubated with staurosporine do not undergo mito-hondrial outer membrane permeabilization while osteosarcoma-erived �0 cells do (Rommelaere et al., 2011), this suggests thathe mechanisms that mediate apoptotic cell death could dependn the origin of the mitochondrial dysfunction. However, in someircumstances, mtDNA mutated cybrid cells could also undergoitochondrial outer membrane permeabilization as other studies
ave shown that cybrid cells harboring mtDNA mutations respon-ible for LHON and MILS syndrome are hypersensitive to galactoseGhelli et al., 2003) or to staurosporine and TNF-� (Carrozzo et al.,004), respectively, through pathways involving cytochrome celease. Interestingly in LHON cybrid cells treated with galactose,aspases are not activated even if cytochrome c release is observed,hich is probably due to the lack of energy in these cells. In these
onditions, the authors suggest that Endo G (Endonuclease G) andIF (Apoptosis Inducing Factor) mediate nuclear fragmentation andell death following mitochondrial outer membrane permeabiliza-
ion (Zanna et al., 2005). Liu and colleagues have also reported thatytochrome c is released from mitochondria to the cytosol in cybridells that display the 4366-bp deletion responsible for CPEO whenxposed to UV light (Lee et al., 2005; Liu et al., 2004, 2007). Whileochemistry & Cell Biology 44 (2012) 139– 149
the same authors have also described the hypersensitivity of MERRFcybrid cells to staurosporine, they did not check for cytochrome crelease. Altogether, these results indicate that apoptosis induced incybrid cells could be mediated by cytochrome c release dependingon the apoptotic inducer used and on the origin of the mitochon-drial dysfunction but, to our knowledge, cytochrome c release inthe cytosol has never been described for A8344G mutated cybridcells subjected to induced apoptosis.
Although unusual, cell death mediated by caspases indepen-dently of cytochrome c release has already been observed (Chauhanet al., 2001; Nakashima et al., 2003). For example, the P39human myelomonocytoid cell line treated with etoposide undergocaspase-dependent apoptosis without cytochrome c release in thecytosol (Hishita et al., 2001). In theses conditions, caspase activa-tion was dependent on the release of lysosomal enzymes such ascathepsin L.
In this study, using caspase and calpain inhibitors we showedthat these proteases are directly or indirectly involved in PARP-1cleavage and DNA fragmentation induced by staurosporine (Fig. 2).This result is very interesting since we recently reported that, in�0143B that completely lack mtDNA, another protease, cathepsinB, was released in the cytosol in response to a 2 �M staurosporinetreatment and participates to cell death (Rommelaere et al., 2011).However, in MERRF cybrid cells challenged with 500 nM stau-rosporine, cathepsin B seems to remain in lysosomes (Fig. S4),confirming that, as observed for cytochrome c release, cell deathpathways involved in �0143B cells and in A8344G MERRF cybridcells treated with staurosporine are not the same.
As the calpain inhibitor (ALLN) inhibited PARP-1 cleavageand DNA fragmentation induced by staurosporine in cybrid cells,calcium concentration and calpain activation were analyzed. Stau-rosporine treatment triggers an increase in the free cytosoliccalcium concentration in both cell lines, but this increase is fasterand stronger in MERRF mutated cybrid cells. This data correlatedwith a stronger activation of calpains found in mutated cybrid cells,when compared with their wild-type counterpart (Fig. 3B). Theseresults are in agreement with previous studies showing that stau-rosporine is able to increase cytosolic calcium concentration (Liuet al., 2004; Morales et al., 2011; Norberg et al., 2010) as well as totrigger calpain activation (Janssens et al., 2009; Zhang et al., 2009).
While an elevated intracellular calcium concentration has beenreported in numerous cases of mitochondrial dysfunction, both inprimary cells from patients suffering from a mitochondrial diseases,like MELAS (Moudy et al., 1995) or Leigh’s disease (Menzies et al.,2009), and in mtDNA-depleted or cybrid cell lines (Arnould et al.,2002) its role in cell death and sensitivity to apoptosis has neverbeen deeply investigated. To confirm that calcium concentrationincrease could lead to hypersensitivity of cells with mitochondrialdysfunction, WT and MERRF mutated cybrid cells were treated for24 h with 1 �M thapsigargin, a molecule known to increase calciumconcentration and cell death (Furuya et al., 1994). PARP-1 cleavagewas then analyzed and as shown in Fig. S5, MERRF mutated cybridcells are more sensitive to that calcium stressor than the WT cybridcells. Cell death could thus be another cell response modulated bycalcium in the context of mitochondrial dysfunction.
Indeed, in mtDNA-depleted cells, we previously showed thatelevated calcium concentration participates, through the activationof a Ca++–CaMKIV–CREB axis (Arnould et al., 2002), to the modula-tion of gene expression leading (1) to the enhanced expression ofmtCLIC, a mitochondrial chloride channel contributing to maintaina mitochondrial membrane potential (Arnould et al., 2003) and (2)to control the mitochondrial biogenesis in mtDNA-depleted cells
(Mercy et al., 2005). Mitochondria dysfunction also impairs cal-cium homeostasis and calcium buffering capacity of mitochondria,as reported in MERRF cybrids harboring the A8356T mutation onthe tRNALys (Brini et al., 1999) as well as in A3243G MELAS cybrids
l of Biochemistry & Cell Biology 44 (2012) 139– 149 147
(scea
ieiwwcwtt1etiiim
idcdsdahtDwtaobceptaf
iMpcaiffzic“wa
pmeiel
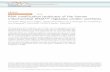
STS
Ca++ Cytochrome c-independen talter nat ive pathw ay
Calpains
p y
2
3
Caspase -41
Caspase-3/7
DNA FragmentationCell death
Fig. 7. Hypothetical model for staurosporine-induced apoptosis in WT and MERRFmutated cybrid cells. Staurosporine (STS) induces a rapid increase in intracellularcalcium concentration that, in turn activates calpain proteases. These calcium-activated proteases can actively participate to apoptosis by the cleavage oftheir numerous substrates but can also contribute to effector caspase activation(caspases-3 and -7) through the activation of caspase-4 (plain arrows). Althoughthis calcium-dependent activation of proteases participates in the induction of apo-ptosis hallmarks, like PARP-1 cleavage and DNA fragmentation, alternative pathwaycould also be involved in cell death. When caspases are inhibited by the use ofzVAD-fmk, calpains are still activated and mediate the remaining PARP-1 cleav-age and DNA fragmentation (intermittent arrow #1). Inversely, when calpainsare inhibited by calpeptin, caspases are still partly activated by an unidentifiedcytochrome c-independent pathway, allowing cell death to occur (intermittentarrow #2). Finally, the fact that PARP cleavage and DNA fragmentation can still beslightly observed when both caspases and calpains are inhibited by zVAD-fmk and
G. Rommelaere et al. / The International Journa
von Kleist-Retzow et al., 2007). Altogether these studies demon-trate that calcium homeostasis and concentration are perturbed inells with impaired mitochondria, which seems to modulate sev-ral cell responses such as mitochondrial biogenesis or cell deaths shown by our results.
As caspase-4, an initiator caspase, can be activated by calpainsn a context of endoplasmic reticulum stress (Das et al., 2010; Jiangt al., 2009; Matsuzaki et al., 2010), and as caspase-4 is activatedn Jurkat cells treated with staurosporine in an apoptotic path-
ay occurring without cytochrome c release (Cusinato et al., 2006),e hypothesized that staurosporine-induced apoptotic response in
ybrid cells could be mediated by a Ca++–calpains–caspases path-ay. Indeed, caspase-4 is activated in response to a staurosporine
reatment, partly in a calpain-dependent manner, as shown byhe use of calpeptin, a specific calpain inhibitor (Tsujinaka et al.,988). In addition, the inhibition of caspase-4, by LEVD-CHO (Duant al., 2004), decreases caspase-3/-7 activity (Fig. 5), known to leado DNA fragmentation and cell death. While this pathway occursn both cell lines, it develops more rapidly and more intensivelyn MERRF mutated cybrid cells. This may be caused by the rapidncrease in free cytosolic calcium concentration observed in MERRF
utated cybrid cells following staurosporine exposure (Fig. 3A).We thus have shown that staurosporine-induced apoptosis
nvolves the activation of caspases by a calcium-activated calpain-ependent mechanism. However, inhibiting calpains does notompletely inhibit caspase activation and inhibition of caspasesoes not totally prevent DNA fragmentation in cells incubated withtaurosporine. This suggests that even if connected by the calpain-ependent cleavage of caspase-4, the two groups of proteases couldlso be activated and/or act independently of each other. Thisypothesis was tested by using calpeptin and zVAD-fmk simul-aneously. Analyzing caspase-3/-7 activity, PARP-1 cleavage andNA fragmentation, we have shown that inhibitors used togetherere more effective than each inhibitor used alone. This means
hat when a group of proteases is inhibited, the other still shows residual activity allowing cell death to occur. A poor efficiencyf the inhibitors used could have also caused this observation,ut as shown in Fig. 6A, zVAD-fmk completely inhibits effectoraspase-3/-7 activation while calpeptin was shown to be the mostfficient calpain inhibitor (Fig. 3C). One can assume that even ifartly activated by calpains, staurosporine-induced caspase activa-ion is rather due to other cytochrome c-independent mechanisms,s calpeptin does not completely prevent the onset of cell deatheatures.
Altogether, our results prompted us to propose a hypothet-cal model of cell death induced by staurosporine in WT and
ERRF mutated cybrid cells (Fig. 7). Staurosporine induces a firstathway, marked by an increase in the free cytosolic calcium con-entration leading to the activation of calpains, followed by thectivation of caspase-4 and executioner caspases-3 and -7, induc-ng cell death. Calpains could also be involved in cell death and DNAragmentation independently of caspase activation as both DNAragmentation and PARP-1 cleavage still occur in the presence ofVAD-fmk. However, we cannot rule out that cell death occurringn the presence of zVAD-fmk is mediated by other mechanisms thanaspases and calpains. In addition, beside the calpains–caspasesaxis”, staurosporine could also stimulate an alternative pathwayhich induces the activation of caspases independently of calpains
nd cytochrome c release.According to this model and experimental data, the cell death
athway induced by staurosporine would be mediated by the sameechanisms in both WT and mutated MERRF cybrid cells. How-
ver, this pathway is triggered more rapidly and more intensivelyn cells with the mitochondrial defect, which could be due to differ-nces in free cytosolic calcium concentration between the two cellines: the basal calcium concentration is already slightly even if non
calpeptin, respectively, suggests that staurosporine can trigger cell death through acytochrome c-independent pathway, independently of caspases and calpains (inter-mittent arrow #3).
significantly higher in MERRF mutated cybrid cells, and is morerapidly increased in response to staurosporine when comparedwith WT cybrid cells (Fig. 3A). This data strongly suggeststhat impaired calcium homeostasis could be responsible for anincreased apoptosis in a cell model of a mitochondrial myopathy.By using medium calcium depletion and cyclosporine A, Cortopassiand Wong had previously suggested that H2O2-mediated cell deathin cybrid cells harboring the MERRF, MELAS and LHON mutationswas mediated by calcium and by the PTP opening (Wong andCortopassi, 1997). The data presented in this paper confirm animportant role of calcium in staurosporine-induced cell death inMERRF cybrid cells and propose calpain activation as a molecularlink between higher calcium concentration and hypersensitivity tocell death.
We have to mention that calcium homeostasis deregulation andcalpain activation are also known to be involved in the developmentof others neurodegenerative diseases such as Parkinson’s (PD) andAlzheimer’s (AD) diseases in which mitochondrial dysfunction is acommon feature (Vosler et al., 2008). It has been shown for examplethat in cybrid cells containing impaired mitochondria from a PDpatient, there is an increase in cytosolic calcium concentration and
in calpain activity which could participate to the pathology (Esteveset al., 2010).Finally, we have to keep our results in perspective in a physio-logical point of view. Indeed, MERRF cybrid cells are useful models

1 l of Bi
ahaMcdtewamchMR(bemmicscits
A
sl–aafcr
A
t
R
A
A
A
B
B
B
C
C
48 G. Rommelaere et al. / The International Journa
llowing to easily compare WT versus A8344G mutated mtDNAarboring cells, but they are derived form cancer cells in whichpoptosis is known to be perturbed in comparison to normal cells.oreover, cybrid cells are highly dividing cells, and this is not the
ase of muscles cells in which apoptosis features have been evi-enced (Mirabella et al., 2000). Our findings would have thus to beested in more relevant models even if several studies in such mod-ls already support our data. For example, the “mutator” mice, inhich mtDNA mutations induce mitochondrial dysfunction, show
n increased apoptosis in skeletal muscles cells compared to WTice (Hiona et al., 2010), a feature also observed in MERRF cybrid
ells treated with staurosporine. In addition, cell death markersave been observed in muscles cells originating from MELAS andERRF patients (Mirabella et al., 2000), and particularly in Ragged
ed Fibers (RRF) in which mitochondrial abundance is increasedAure et al., 2006). As calcium is known to promote mitochondrialiogenesis (for a review on mitochondrial abundance, see Michelt al., 2011) and as increased calcium concentrations have beeneasured in primary cells of patients with mitochondrial DNAutations (Menzies et al., 2009; Moudy et al., 1995), it is tempt-
ng to speculate that in muscle fibers with a high level of mutation,alcium could induce mitochondrial biogenesis (RRF) as well as sen-itization to cell death. In regard to these results, the analysis ofalcium concentration and/or of calpain activity directly measuredn muscles from patients with mitochondrial diseases would cer-ainly be of great interest to better understand the development ofuch pathologies.
cknowledgements
G. Rommeleare and S. Michel are recipients of doctoral fellow-hips from the FRIA (Fonds pour la Recherche dans l’Industrie et’Agriculture). This work was supported by the FRFC (no. 2.4650.06)
Fonds National de la Recherche Scientifique (FRS-FNRS), Brussels;nd by the Belgian Association for Muscular Diseases (ABMM). Theuthors especially thank Prof. Andreu, Dr. Janssen and Prof. Attardior the gift of the A8344G MERRF cybrid cells, the A3243G MELASybrid cells and the mtDNA-depleted 143B �0 osteosarcoma cells,espectively.
ppendix A. Supplementary data
Supplementary data associated with this article can be found, inhe online version, at doi:10.1016/j.biocel.2011.10.009.
eferences
rnould T, Mercy L, Houbion A, Vankoningsloo S, Renard P, Pascal T, et al. mtCLICis up-regulated and maintains a mitochondrial membrane potential in mtDNA-depleted L929 cells. FASEB J 2003;17:2145–7.
rnould T, Vankoningsloo S, Renard P, Houbion A, Ninane N, Demazy C, et al. CREBactivation induced by mitochondrial dysfunction is a new signaling pathwaythat impairs cell proliferation. EMBO J 2002;21:53–63.
ure K, Fayet G, Leroy JP, Lacene E, Romero NB, Lombes A. Apoptosis in mitochondrialmyopathies is linked to mitochondrial proliferation. Brain 2006;129:1249–59.
iswas G, Anandatheerthavarada HK, Avadhani NG. Mechanism of mitochondrialstress-induced resistance to apoptosis in mitochondrial DNA-depleted C2C12myocytes. Cell Death Differ 2005;12:266–78.
ratton SB, Walker G, Srinivasula SM, Sun XM, Butterworth M, Alnemri ES, et al.Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apopto-some and associated XIAP complexes. EMBO J 2001;20:998–1009.
rini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signalingdefect in the pathogenesis of a mitochondrial DNA inherited oxidative phospho-rylation deficiency. Nat Med 1999;5:951–4.
ardoso SM, Rego AC, Penacho N, Oliveira CR. Apoptotic cell death induced by hydro-
gen peroxide in NT2 parental and mitochondrial DNA depleted cells. NeurochemInt 2004;45:693–8.arrozzo R, Rizza T, Stringaro A, Pierini R, Mormone E, Santorelli FM, et al.Maternally-inherited Leigh syndrome-related mutations bolster mitochondrial-mediated apoptosis. J Neurochem 2004;90:490–501.
ochemistry & Cell Biology 44 (2012) 139– 149
Chauhan D, Hideshima T, Rosen S, Reed JC, Kharbanda S, Anderson KC. Apaf-1/cytochrome c-independent and Smac-dependent induction of apoptosis inmultiple myeloma (MM) cells. J Biol Chem 2001;276:24453–6.
Cusinato F, Pighin I, Luciani S, Trevisi L. Synergism between staurosporine anddrugs inducing endoplasmic reticulum stress. Biochem Pharmacol 2006;71:1562–9.
Danielson SR, Wong A, Carelli V, Martinuzzi A, Schapira AH, Cortopassi GA. Cellsbearing mutations causing Leber’s hereditary optic neuropathy are sensitizedto Fas-Induced apoptosis. J Biol Chem 2002;277:5810–5.
Das A, Banik NL, Ray SK. Flavonoids activated caspases for apoptosis in humanglioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer2010;116:164–76.
DiMauro S, Hirano M. Pathogenesis and treatment of mitochondrial disorders. AdvExp Med Biol 2009;652:139–70.
Diwakarla S, Nagley P, Hughes ML, Chen B, Beart PM. Differential insult-dependentrecruitment of the intrinsic mitochondrial pathway during neuronal pro-grammed cell death. Cell Mol Life Sci 2009;66:156–72.
Duan L, Aoyagi M, Tamaki M, Yoshino Y, Morimoto T, Wakimoto H, et al. Impair-ment of both apoptotic and cytoprotective signalings in glioma cells resistant tothe combined use of cisplatin and tumor necrosis factor alpha. Clin Cancer Res2004;10:234–43.
Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM. Dysfunctionalmitochondria uphold calpain activation: contribution to Parkinson’s diseasepathology. Neurobiol Dis 2010;37:723–30.
Ferraresi R, Troiano L, Pinti M, Roat E, Lugli E, Quaglino D, et al. Resistance of mtDNA-depleted cells to apoptosis. Cytometry A 2008;73:528–37.
Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT. The role of calcium, pH,and cell proliferation in the programmed (apoptotic) death of androgen-independent prostatic cancer cells induced by thapsigargin. Cancer Res 1994;54:6167–75.
Ghelli A, Zanna C, Porcelli AM, Schapira AH, Martinuzzi A, Carelli V, et al.Leber‘s hereditary optic neuropathy (LHON) pathogenic mutations inducemitochondrial-dependent apoptotic death in transmitochondrial cells incu-bated with galactose medium. J Biol Chem 2003;278:4145–50.
Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev2003;83:731–801.
Griffith TS, Lynch DH. TRAIL: a molecule with multiple receptors and control mech-anisms. Curr Opin Immunol 1998;10:559–63.
Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, et al. Mitochondrial DNAmutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skele-tal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010;5:e11468.
Hishita T, Tada-Oikawa S, Tohyama K, Miura Y, Nishihara T, Tohyama Y, et al.Caspase-3 activation by lysosomal enzymes in cytochrome c-independentapoptosis in myelodysplastic syndrome-derived cell line P39. Cancer Res2001;61:2878–84.
Ikezoe K, Nakagawa M, Yan C, Kira J, Goto Y, Nonaka I. Apoptosis is sus-pended in muscle of mitochondrial encephalomyopathies. Acta Neuropathol2002;103:531–40.
James AM, Wei YH, Pang CY, Murphy MP. Altered mitochondrial function in fibrob-lasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem J1996;318(Pt 2):401–7.
Janssens V, Derua R, Zwaenepoel K, Waelkens E, Goris J. Specific regulation of pro-tein phosphatase 2A PR72/B subunits by calpain. Biochem Biophys Res Commun2009;386:676–81.
Jiang CC, Mao ZG, Avery-Kiejda KA, Wade M, Hersey P, Zhang XD. Glucose-regulatedprotein 78 antagonizes cisplatin and adriamycin in human melanoma cells. Car-cinogenesis 2009;30:197–204.
Kwong JQ, Henning MS, Starkov AA, Manfredi G. The mitochondrial respiratory chainis a modulator of apoptosis. J Cell Biol 2007;179:1163–77.
Lee CF, Liu CY, Chen SM, Sikorska M, Lin CY, Chen TL, et al. Attenuation of UV-inducedapoptosis by coenzyme Q10 in human cells harboring large-scale deletion ofmitochondrial DNA. Ann N Y Acad Sci 2005;1042:429–38.
Lee MS, Kim JY, Park SY. Resistance of rho(0) cells against apoptosis. Ann N Y AcadSci 2004;1011:146–53.
Liu CY, Lee CF, Hong CH, Wei YH. Mitochondrial DNA mutation and depletionincrease the susceptibility of human cells to apoptosis. Ann N Y Acad Sci2004;1011:133–45.
Liu CY, Lee CF, Wei YH. Quantitative effect of 4977 bp deletion of mitochondrial DNAon the susceptibility of human cells to UV-induced apoptosis. Mitochondrion2007;7:89–95.
Liu CY, Lee CF, Wei YH. Role of reactive oxygen species-elicited apoptosis in thepathophysiology of mitochondrial and neurodegenerative diseases associatedwith mitochondrial DNA mutations. J Formos Med Assoc 2009;108:599–611.
Masucci JP, Davidson M, Koga Y, Schon EA, King MP. In vitro analysis of muta-tions causing myoclonus epilepsy with ragged-red fibers in the mitochondrialtRNA(Lys)gene: two genotypes produce similar phenotypes. Mol Cell Biol1995;15:2872–81.
Matsuzaki S, Hiratsuka T, Kuwahara R, Katayama T, Tohyama M. Caspase-4 is par-tially cleaved by calpain via the impairment of Ca2+ homeostasis under the ERstress. Neurochem Int 2010;56:352–6.
Menzies KJ, Robinson BH, Hood DA. Effect of thyroid hormone on mitochondrial
properties and oxidative stress in cells from patients with mtDNA defects. Am JPhysiol Cell Physiol 2009;296:C355–62.Mercy L, Pauw A, Payen L, Tejerina S, Houbion A, Demazy C, et al. Mitochondrialbiogenesis in mtDNA-depleted cells involves a Ca2+-dependent pathway and areduced mitochondrial protein import. FEBS J 2005;272:5031–55.

l of Bi
M
M
M
M
N
N
N
N
P
R
S
S
T
T
G. Rommelaere et al. / The International Journa
ichel S, Wanet A, De Pauw A, Rommelaere G, Arnould T, Renard P. Crosstalkbetween mitochondrial (dys)function and mitochondrial abundance. J Cell Phys-iol 2011., doi:10.1002/jcp.23021.
irabella M, Di Giovanni S, Silvestri G, Tonali P, Servidei S. Apoptosis in mito-chondrial encephalomyopathies with mitochondrial DNA mutations: a potentialpathogenic mechanism. Brain 2000;123(Pt 1):93–104.
orales AP, Carvalho AC, Monteforte PT, Hirata H, Han SW, Hsu YT, et al. Endoplas-mic reticulum calcium release engages Bax translocation in cortical astrocytes.Neurochem Res 2011;36:829–38.
oudy AM, Handran SD, Goldberg MP, Ruffin N, Karl I, Kranz-Eble P, et al. Abnormalcalcium homeostasis and mitochondrial polarization in a human encephalomy-opathy. Proc Natl Acad Sci USA 1995;92:729–33.
akagawa T, Yuan J. Cross-talk between two cysteine protease families. Activationof caspase-12 by calpain in apoptosis. J Cell Biol 2000;150:887–94.
akashima S, Hiraku Y, Tada-Oikawa S, Hishita T, Gabazza EC, Tamaki S, et al. Vac-uolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in thehuman gastric cancer cell line MKN-1. J Biochem 2003;134:359–64.
orberg E, Karlsson M, Korenovska O, Szydlowski S, Silberberg G, Uhlen P, et al.Critical role for hyperpolarization-activated cyclic nucleotide-gated channel 2in the AIF-mediated apoptosis. EMBO J 2010;29:3869–78.
ovak EJ, Rabinovitch PS. Improved sensitivity in flow cytometric intracellular ion-ized calcium measurement using fluo-3/Fura Red fluorescence ratios. Cytometry1994;17:135–41.
op C, Salvesen GS. Human caspases: activation, specificity, and regulation. J BiolChem 2009;284:21777–81.
ommelaere G, Michel S, Mercy L, Fattaccioli A, Demazy C, Ninane N, et al.Hypersensitivity of mtDNA-depleted cells to staurosporine-induced apopto-sis: roles of Bcl-2 downregulation and cathepsin B. Am J Physiol Cell Physiol2011;300:C1090–106.
apkota K, Kim S, Park SE, Kim SJ. Detoxified extract of Rhus verniciflua stokesinhibits rotenone-induced apoptosis in human dopaminergic cells, SH-SY5Y.Cell Mol Neurobiol 2011;31:213–23.
choeler S, Szibor R, Gellerich FN, Wartmann T, Mawrin C, Dietzmann K, et al. Mito-chondrial DNA deletions sensitize cells to apoptosis at low heteroplasmy levels.Biochem Biophys Res Commun 2005;332:43–9.
iwari M, Lopez-Cruzan M, Morgan WW, Herman B. Loss of caspase-2-dependent
apoptosis induces autophagy after mitochondrial oxidative stress in primarycultures of young adult cortical neurons. J Biol Chem 2011;286:8493–506.sujinaka T, Kajiwara Y, Kambayashi J, Sakon M, Higuchi N, Tanaka T, et al. Synthesisof a new cell penetrating calpain inhibitor (calpeptin). Biochem Biophys ResCommun 1988;153:1201–8.
ochemistry & Cell Biology 44 (2012) 139– 149 149
Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations andhuman disease. Biochim Biophys Acta 2010;1797:113–28.
Umaki Y, Mitsui T, Endo I, Akaike M, Matsumoto T. Apoptosis-related changes inskeletal muscles of patients with mitochondrial diseases. Acta Neuropathol2002;103:163–70.
Vives-Bauza C, Gonzalo R, Manfredi G, Garcia-Arumi E, Andreu AL. Enhanced ROSproduction and antioxidant defenses in cybrids harbouring mutations in mtDNA.Neurosci Lett 2006;391:136–41.
von Kleist-Retzow JC, Hornig-Do HT, Schauen M, Eckertz S, Dinh TA, Stassen F, et al.Impaired mitochondrial Ca2+ homeostasis in respiratory chain-deficient cellsbut efficient compensation of energetic disadvantage by enhanced anaerobicglycolysis due to low ATP steady state levels. Exp Cell Res 2007;313:3076–89.
Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronalinjury and neurodegeneration. Mol Neurobiol 2008;38:78–100.
Walker PR, Smith C, Youdale T, Leblanc J, Whitfield JF, Sikorska M. TopoisomeraseII-reactive chemotherapeutic drugs induce apoptosis in thymocytes. Cancer Res1991;51:1078–85.
Wallace DC. Mitochondrial defects in neurodegenerative disease. Ment Retard DevDisabil Res Rev 2001;7:158–66.
Wang KK, Nath R, Posner A, Raser KJ, Buroker-Kilgore M, Hajimohammadreza I,et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci USA1996;93:6687–92.
Wong A, Cortopassi G. mtDNA mutations confer cellular sensitivity to oxidant stressthat is partially rescued by calcium depletion and cyclosporin A. Biochem Bio-phys Res Commun 1997;239:139–45.
Yen HC, Tang YC, Chen FY, Chen SW, Majima HJ. Enhancement of cisplatin-inducedapoptosis and caspase 3 activation by depletion of mitochondrial DNA in ahuman osteosarcoma cell line. Ann N Y Acad Sci 2005;1042:516–22.
You BR, Park WH. The effects of antimycin A on endothelial cells in cell death, reactiveoxygen species and GSH levels. Toxicol In Vitro 2010;24:1111–8.
Yu M, Shi Y, Wei X, Yang Y, Zang F, Niu R. Mitochondrial DNA depletion promotesimpaired oxidative status and adaptive resistance to apoptosis in T47D breastcancer cells. Eur J Cancer Prev 2009;18:445–57.
Zanna C, Ghelli A, Porcelli AM, Martinuzzi A, Carelli V, Rugolo M. Caspase-independent death of Leber’s hereditary optic neuropathy cybrids is driven
by energetic failure and mediated by AIF and Endonuclease G. Apoptosis2005;10:997–1007.Zhang Z, Larner SF, Liu MC, Zheng W, Hayes RL, Wang KK. Multiple alphaII-spectrinbreakdown products distinguish calpain and caspase dominated necrotic andapoptotic cell death pathways. Apoptosis 2009;14:1289–98.
Related Documents