GROUP-CONTRIBUTION METHODS IN ESTIMATING LIQUID-LIQUID DISTRIBUTION COEFFICIENTS by CHE KEUNG LIU, B.A. A THESIS IN CHEMICAL ENGINEERING Submitted to the Graduate Faculty of Texas Tech University in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE IN CHEMICAL ENGINEERING Approved May, 1981

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

GROUP-CONTRIBUTION METHODS IN ESTIMATING
LIQUID-LIQUID DISTRIBUTION COEFFICIENTS
by
CHE KEUNG LIU, B.A.
A THESIS
IN
CHEMICAL ENGINEERING
Submitted to the Graduate Faculty of Texas Tech University in
Partial Fulfillment of the Requirements for
the Degree of
MASTER OF SCIENCE
IN
CHEMICAL ENGINEERING
Approved
May, 1981

/ '
ACKNOWLEDGMENTS
To Dr. L. Davis Clements, chairman of my committee, I extend my
deepest appreciation for his helpful and expert advice, guidance, and
encouragement throughout this project. I would also like to thank the
rest of the committee. Dr. S. R. Beck and Dr. H. R. Heichelheim, for
their suggestions and criticisms.
Finally, my special thanks go to Ms. Sue Willis for her assistance
in preparing this thesis.
n

TABLE OF CONTENTS
PAGE
ACKNOWLEDGMENTS ii
LIST OF TABLES v
LIST OF FIGURES vi
CHAPTER I INTRODUCTION 1
CHAPTER II LITERATURE REVIEW 4
Uses of Distribution Coefficient 4
History 7
Thermodynamic Derivation of Distribution Law . . . 9
Activity Coefficient Models 11
Modifications of Wilson's Equation 12
NRTL (Non-random Two-liquid Equation) 13
UNIQUAC (Universal Quasi-Chemical) Equation. . . . 14
Group-Contribution Models 16
ASOG 18
UNIFAC Method (UNIQUAC Functional-Group
Activity Coefficients) 20
Empirical Correlation Methods 21
CHAPTER III DISTRIBUTION COEFFICIENTS OF HOMOLOGUES 23
Introduction 23
Relation of the Distribution Coefficients of Homologues to Molecular Structure 23 Derivation to Free-Energy Equations of Partitioning Process 32 Determination of Free Energy of Transfer of Methylene Group 40
. • •

PAGE
CHAPTER IV ESTIMATION OF LIQUID-LIQUID DISTRIBUTION COEFFICIENTS FROM GROUP CONTRIBUTIONS 43
Reasons for New Model Development 43
Data Collection 43
Group Contributions Model 44
Method 47
Predictivity of the Model 50
CHAPTER V EVALUATION OF FACTORS INFLUENCING LIQUID-LIQUID DISTRIBUTION COEFFICIENTS 61
Introduction 61
Effect of pH on Distribution Coefficient 61
Temperature Effect on Distribution Coefficient 68
Effect of Concentration on Distribution Coefficient 76
Discussion 79
CHAPTER VI CONCLUSIONS AND RECOMMENDATIONS FOR
FUTURE STUDIES 81
Summary of the Research 81
Conclusions 81
Recommendation for Future Studies 82
LIST OF REFERENCES 83 NOMENCLATURE 90
TV

LIST OF TABLES
PAGE
Table 1 RELATION OF DISTRIBUTION COEFFICIENTS TO THE NUMBER OF CARBON NUMBER AND METHYLENE NUMBER 25
Table 2 DISTRIBUTION COEFFICIENTS OF ACIDS OBTAINED FROM EXPERIMENTS AND CORRELATIONS 31
Table 3 FREE ENERGY CHANGES (AT 25°C) TO TRANSFER
A MOLE OF METHYLENE 42
Table 4 GROUP CONTRIBUTIONS FOR THE FOUR SYSTEMS 49
Table 5 EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-OCTANOL-WATER SYSTEM 51
Table 6 EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-DIETHYL ETHER-WATER SYSTEM 55
Table 7 EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-CHLOROFORM-WATER SYSTEM. . . . 57
Table 8 EXPERIMENTAL AND CALCULATED DISTRIBUTION
COEFFICIENTS OF SOLUTE-BENZENE-WATER SYSTEM 59
Table 9 SHIFT OF DISTRIBUTION COEFFICIENT WITH pH 62
Table 10 SHIFT OF DISTRIBUTION COEFFICIENTS WITH pH 65
Table 11 EFFECT OF TEMPERATURE UPON DISTRIBUTION
COEFFICIENT 70 Table 12 SHIFT OF DISTRIBUTION COEFFICIENTS OF
STRONG ACIDS WITH CONCENTRATION 77

LIST OF FIGURES
PAGE
Figure 1. Relation Of The Distribution Coefficient To The (Number of Methylene Group) Number Of Carbon Atoms 28
Figure 2. Relation Of The Distribution Coefficient To The (Number of Methylene Group) Number Of Carbon Atoms 29
Figure 3. Relation Of The Distribution Coefficient To The (Number of Methylene Group) Number Of Carbon Atoms 30
Figure 4. Relation Of The Distribution Coefficient To The (Number Of Methylene Group) Number Of Carbon Atoms 33
Figure 5. Relation Of The Distribution Coefficient To The (Number Of Methylene Group) Number Of Carbon Atoms 34
Figure 6. Relation Of The Distribution Coefficient To The (Number of Methylene Group) Number Of Carbon Atoms 35
Figure 7. Relation Of The Distribution Coefficient To The (Number of Methylene Group) Number Of Carbon Atoms 36
Figure 8. Relation Of The Distribution Coefficient To
The Number Of Carbon Atoms 37
Figure 9. Shift Of K With pH 63
Figure 10. Shift Of K With pH 66 Figure 11. Effect Of Temperature Upon Distribution
Coefficient 72
Figure 12. Effect Of Temperature Upon Distribution Coefficient 73
Figure 13. Effect Of Temperature Upon Distribution Coefficient 74
VI

PAGE
Figure 14. Effect Of Temperature Upon Distribution Coefficient 75
Figure 15. Shift Of Distribution Coefficient With Concentration 78
v n

CHAPTER I
INTRODUCTION
A large part of chemical engineering design is concerned with sepa
ration processes such as distillation, absorption, and extraction. For
design of a liquid-liquid extraction system, one needs to know the rela
tive distribution of the solutes between the two liquid phases. The key
to carrying out an effective extraction depends on the choice of a suit
able solvent. The distribution coefficient is one of the important fac
tors in the choice of solvents. According to Henley and Staffin (1),
the liquid-liquid distribution coefficient of a good solvent should be
at least 5, and perhaps as much as 50. At the early stages of process
development we can save money and time if we eliminate solvents which
appear least useful and work on those which are most promising. However,
the choice of solvent requires either a large base of experimental data,
or accurate prediction methods.
Since the number of different mixtures in chemical technology is
extremely large, satisfactory experimental equilibrium data are seldom
available for the conditions in a particular design problem. It is
therefore necessary to be able to predict liquid-liquid distribution co
efficients from the available experimental data based on thermodynamics
and empirical models for the mixture. However, when suitable data are
lacking, we have to predict the coefficients from some generalized method,
For many years group-contribution methods have been used to predict
properties of pure substances with great success. For example, Souders
1

and Matthews (2) used group-contribution methods for the prediction of
heat capacity and heat contents of hydrocarbons in the ideal state.
During the last 15 years, group-contribution methods have also been
developed for the prediction of thermodynamic properties of liquid mix
tures. These correlation methods are based on the premise that thermo
dynamic functions within a molecule are additive. Thus, the values for
the liquid molecules can be built up from an assignment of contributions
to the various groups which make up the molecule under consideration.
The group-contribution method has two advantages. First, i t enables
systematic interpolation and extrapolation of liquid-liquid equilibrium
data for many chemically related mixtures. Secondly, i t provides a rea
sonable method for predicting the properties of a wery large number of
mixtures in terms of a relatively small number of group parameters.
There is a need for more work on methods for the correlation and
prediction of liquid-liquid distribution coefficients. Today, the goal
of predicting the compositions of the two liquid phases in equilibrium
has remained elusive. Several attempts have been made to use the ASOG
(Analytical Solution of Groups) and UNIFAC (UNIQUAC Functional-group
Activity Coefficients) group-contribution methods to predict the liquid-
liquid equilibrium compositions. The agreement between experimental and
calculated equilibrium composition is not too convincing. Also, these
models are based on parameters estimated from experimental phase-
equilibrium data for the mixtures of interest and are not readily gen
eralized to systems for which there are no data.
The purpose of this study was to investigate the properties of
liquid-liquid distribution coefficients in order to determine i f

development of a systematic group-contribution method to predict the
value of liquid-liquid distribution coefficients based on a knowledge
of the structural formula of the compound only is possible.

CHAPTER II
LITERATURE REVIEW
Uses of Distribution Coefficient
Solvent extraction is an important industrial separation method.
Industrial application of solvent extraction has increased rapidly over
the past two decades. The unique ability of solvent extraction to achieve
separation according to chemical type rather than according to physical
characteristics allows application to a great variety of processes. Ap
plications range from nuclear-fuel enrichment and reprocessing to ferti
lizer manufacture, and from petroleum refining to food processing.
The choice of solvents for extraction depends upon several factors,
including cost, availability, stability, selectivity for the desired
component and the distribution coefficient. In evaluating a solvent for
use in an extraction operation, it is of prime importance to know the
relative distribution of the material being extracted between the solvent
and raffinate phases. Both the quantity of solvent needed and the number
of extraction stages required to obtain the desired separation depend on
the value of distribution coefficient.
The distribution coefficient, K, is the ratio of the composition of
the solute in each liquid phase in equilibrium:
Y. <
'a K = -y^ at equilibrium (1) a A.
where Y = composition of solute a in the extract phase a
X^ = composition of solute a in the raffinate phas( a

The value of K is one of the main parameters used to establish the
minimum sol vent-feed ratio that can be used in an extraction process.
Consider the case that solvent S and carrier solvent R are immiscible.
The minimum solvent to feed ratio can be estimated from the liquid-liquid
distribution coefficient.
From the definition of the distribution coefficient, we have
B£/(S + B ) D = B /(R + Bj ) (2)
where Br = quantity of solute B in extract phase
Bn = quantity of solute B in raffinate phase
S = quantity of solvent
R = quantity of carrier solvent
If the concentration of solute B is low, then S is much bigger than B^
and R is much bigger than B, and the distribution coefficient can be
expressed as
Bp/S
S^VF ( )
where B = total quantity of B
F = quantity of feed (F = B+ R)
Rearranging the terms, the solvent to feed ratio, S/F, is
i=L.!i (4)
For 99% extraction, the values of b^ and B are 0.01 and 0.99, respec
tively. Thus, i f the distribution coefficient is 4, 24.75 pounds of

solvent would be required to remove 99% of the solute from 1 pound of
feed.
The distribution coefficient also influences the selectivity. It
is desirable in any extraction process that the solvent used dissolve a
large portion of the solute, while at the same time the amount of non-
consolute component transferred be as low as possible. A measure of
this separation is the selectivity of the solvent.
The selectivity a^^ of solvents for solute C from a mixture of A
and C is
"ca K " \ ( Y j a a (5)
For separation to be possible, a should be larger than 1. The
larger a , the better the extraction process, since as a , increases, " ca
fewer stages- are required for a given separation and capital costs are
lower.
For extraction of solute C from a mixture of A and C, since
X^/Y^ > 1, then for solvent to have a high selectivity for the desired
component, the distribution coefficient, K , should have a favorable
value (K » 1). Thus the distribution coefficient is an important
property of the solvent in that it influences the selectivity.
Distribution coefficients have been used as an aid in identifica
tion of extremely small amounts of organic compounds (3). From the
rate of migration of a substance in the countercurrent distribution ap
paratus, its distribution coefficient may be calculated. It is thus pos
sible to characterize an unknown substance, never before isolated, in
terms of its distribution constants in mixtures of different solvents.

7
The distribution coefficient is one of the fundamental properties
of an emulsifier. Davies (4), who studied the kinetics of coalescence
in emulsion systems, has related the HLB (hydrophile-lipophile balance)
system to the distribution coefficient. Thus, the purely empirical HLB
system is related to the distribution coefficient, which is in turn based
firmly on thermodynamics. This relationship makes it possible to deter
mine the molecule's ability to function as a wetting agent, detergent,
or defoamer.
Many equilibrium constants have been determined by distribution
measurements. Vandalen (5) measured the equilibrium constants for metal
ion complexing agents among water and organic phases. Other types of
equilibrium studied by distribution measurements are those which have
dimer dissociation and those which form Schiff bases (6, 7).
History
As early as 1870, Berthelot and Jungfleisch (8) investigated the
distribution of I^ and Br^ between CSp and water. They also studied the
partition of various organic acids between ethyl ether and water. They
showed that the ratio of the concentrations of solute distributed between
two immiscible solvents was a constant at constant temperature. From
these early investigations, they also observed that the liquid-liquid
distribution coefficient varies with temperature.
In 1891, Nernst (9) developed the distribution law. The distribu
tion law stated that a solute will distribute between two essentially
immiscible solvents in such a manner that, at equilibrium, the ratio of
the concentrations of the solute in the two phases at a particular

8
temperature will be a constant, provided the solute has the same molecu
lar weight in each phase. For a solute B distributing between solvents
1 and 2, we have
B^^B^
K = [ByCB]^ (6)
where K is the distribution coefficient, a constant independent of total
solute concentration, and [B]^ is the concentration of solute in solvent
phase 2, and [B]^ is the concentration of solute in solvent phase 1.
Although the distribution law is a useful approximation, it has two
shortcomings. First, the distribution law does not consider chemical re
actions such as association and dissociation of solute in different
phases. Secondly, the distribution law is not thermodynamically rigor
ous.
Chemical interactions of the distributing species with the other
components in different phases affect the concentrations of the distri
buting species. If all the significant interactions of the distributing
species are known, we may evaluate a distribution ratio which describes
the stoichiometric ratio including all species of the same component in
the respective phases. We may express the distribution ratio, D, as
Pj _ Total concentration in organic phase , . Total concentration in aqueous phase ^''
Lacroix (10) illustrated the situation of ionization of solute in
the aqueous phase. He showed that the distribution ratio of 8-quinolinol

between chloroform and water with the problem of ionization of 8-quinolinol
could be related to distribution coefficient as
lM^+1 + ^ • 1 [H^]
where K, = first ionization constant
K2 = second ionization constant
K = distribution coefficient
Thermodynamic Derivation Of Distribution Law
The distribution law could be explained by classical thermodynamics
under the conditions that the solvent systems are almost completely im
miscible and the solute concentration is wery low. Treybal (11) has pre
sented a discussion of the thermodynamic derivation of distribution law.
The following discussion relies heavily on his summary.
At constant temperature and pressure, equilibrium is attained when
the chemical potentials, y, of the solute in each phase are equal. Thus
where 1 = solute phase 1
2 = solute phase 2
Consider the solution to be represented by an ideal molar solution,
and we have
y^^ + RT In m^ + RT In y^ = M^^ + RT In m^ + RT In y^ (10)

10
where m = solute concentration in molarity
Y = molar activity coefficient
y-j** = chemical potential of solute in a hypothetical ideal molar
solution.
From the above expression, the molar distribution coefficient, K,
can be expressed as
mp Yi '{\i^° - yi°)/RT
m Y2 . ^ '
If the presence of the solute does not significantly affect the
mutual solubilities of the two solvents, the y°'s become constant. Then
we have
mp Y^ K = ir = 7-C (12)
n ^2
where C is a constant for the system at constant temperature.
Thus, the distribution coefficient varies with the variations in the
activity coefficients in each of the phases. When solute concentration
is yery low, the distribution coefficient becomes constant as the acti
vity coefficients approach unity. For example, Grahame and Seaborg (12)
found that the distribution coefficient of gallium chloride between
ethyl ether and 6 m hydrochloric acid remains essentially constant (with-
-12 -3 in 5%) over a concentration range from 10 to 2 x 10 m gallium
chloride.

n
Act iv i ty Coefficient Models
Unfortunately, the simplif ied approach of assuming that the rat io
of the ac t iv i t y coefficients of any component in the coexisting phases
approaches unity does not apply to the whole concentration range. For
calculation of distr ibut ion coefficients i t is necessary, in pr inc ip le,
to know the ac t iv i ty coefficients of solute in the equilibrium phases
over the concentration range of interest. Once a reasonable method for
predicting ac t iv i ty coefficients is available, one may use the ac t iv i ty
coeff icient of solute in each phase to calculate distr ibut ion coef f i c i
ents.
Tradi t ional ly, two dif ferent categories of models have been used in
correlating ac t iv i ty coeff icients. The f i r s t type of model uses empiri
cal relations for interpolat ion. The second type of model is a direct
application of the thermodynamics of fluid-phase equi l ibr ia .
P ie ro t t i , Deal and Derr (13) have correlated in f in i te -d i lu t ion ac
t i v i t y coefficients with the molecular structure of the components in
volved. In binary systems where the in f in i te -d i lu t ion act iv i ty coef f i
cients are quite large (larger than 25), so that the mutual so lub i l i t ies
are small, good estimates of the mutual so lubi l i t ies can be made on the
basis of the in f i n i te -d i l u t i on act iv i ty coeff icients. However, in sys
tems with smaller immiscibi l i ty gaps and in multicomponent systems, the
l iqu id - l iqu id equilibrium compositions cannot be predicted with s u f f i
cient accuracy from in f in i te -d i l u t i on act iv i ty coefficients alone.
Hildebrand and Scott's regular solution theory (14) offers a pos
s i b i l i t y of calculating ac t iv i ty coefficients without any use of mixture
experimental information. Al l one keeds to know is the "so lub i l i t y

12
parameter" for each pure component. However, regular solution theory
does not apply to even moderately polar substances. This is a serious
disadvantage, since water is present in most of the liquid-liquid systems
of interest. In situations where regular solution theory holds, i t may,
at most, be expected to yield predictions which are in qualitative agree
ment with experiment.
The modifications of Wilson's equation, NRTL and UNIQUAC equations
are widely used today for calculating liquid phase activity coefficients.
These models were derived by various researchers by using Wilson's "local
composition" concept for representation of excess Gibbs energies of
liquid mixtures. The local composition concept provides a convenient
method for introducing nonrandomness into the liquid-mixture model. The
central idea of the local composition concept is that when viewed micro
scopically, a liquid mixture is not homogeneous; the composition at one
point in the mixture is not necessarily the same as that at another point.
In engineering applications and in typical laboratory work only the
average, overall composition matters. However, for constructing liquid-
mixture models, i t appears that the local composition, rather than the
average composition is a more realistic primary variable.
Modifications of Wilson's Equation
Wilson's equation is not applicable to liquid mixtures which are
only partially miscible. The most used modification of Wilson's equa
tion was introduced by Tsuboka and Katayama (15). They modified Wilson's
equation by introducing the parameter A.. which is defined as the

13
probability of finding a molecule of type j, next to a molecule of
type i.
The Wilson equation modified by Tsuboka and Katayama for the excess
Gibbs energy is:
G^ ^ , " J |y = - Z X. (In Z X. A.. + In E x. p. .) (13)
Pij = V./Vj. ; i, j = 1,2, . . .N (14)
The modified Wilson's equation uses two parameters per binary and
correlates binary and ternary data well. Prediction of ternary liquid-
liquid equilibrium from binary data is qualitatively satisfactory.
NRTL (Non-random Two-liquid Equation)
The NRTL is the most extensively used model for liquid-liquid equil
ibrium to date. The equation was developed by Renon and Prausnitz (16).
To take into account nonrandomness in liquid mixtures, Renon modified
Wilson's equation by adding a term 3-]2» which is characteristic of the
nonrandomness of the mixture. Also, he introduced the two-liquid theory
of Scott (17), which assumes that there are two kinds of cells in a
binary mixture: one with molecule 1 at the center surrounded by 1 and 2
and the other, with molecule 2 at the center.
Renon defined the molar excess Gibbs energy for a binary solution
as the sum of two changes in residual Gibbs energy: f i rs t , that of
transferring n-. molecules from a cell of the pure liquid 1 into a cell 1
of the solution, and second, that of transferring n molecules from a
cell of the pure liquid 2 into a cell 2 of the solution.

14
In a multicomponent mixture, the NRTL equation for the excess Gibbs
energy and the activity coefficient are:
N
gE N .?, ji ji ^j fp= Z x.^ ; l,j,p = 1,2, ..., N (15)
^ ki \ k=l ^^ ^
N N Zr.. G..X. .1 „ r Z x F . G . .
'" i N ._, N ^Mj N ; Ub; ^ S. . X. "" Z G, . X, Z G, . X,
k=l ^ ^ k=l ^J ^ k=l 'J ^
There are three parameters per binary: r . . , r.. and 3^-. The para-
meters are calculated from experimental compositions of the two equili
brated liquid phases. The NRTL equation appears to be applicable to a
wide variety of mixtures for calculating vapor-liquid and liquid-liquid
equilibria. The NRTL equation often correlates binary and ternary
liquid-liquid equilibria quantitatively correctly. Prediction of ternary
liquid-liquid equilibria from binary data is often qualitatively correct.
UNIQUAC (Universal Quasi-Chemical) Equation
The UNIQUAC equation was developed by Abrams and Prausnitz (18) in
1975 and modified by Anderson and Prausnitz in 1978 (19). The UNIQUAC
equation generalized the theory of Guggenheim to mixtures containing
molecules of different size and shape by utilizing the local composition
concept. The original Guggenheim quasi-chemical lattice model is re
stricted to small molecules of essentially the same size. The effect of
molecular size and shape are introduced through structural parameters

15
obtained from pure-component data and through use of Staverman's combi
natorial entropy as a boundary condition for athermal mixtures.
In a multicomponent mixture, the UNIQUAC equation for the activity
coefficient of component i is
In Y . = In Y.^ + In Y ^ (17)
combinatorial residual
The UNIQUAC model has only two adjustable parameters, r and r . ,
per binary. These parameters must be evaluated from experimental phase-
equilibrium data. No ternary parameters are required for systems con
taining three or more components. Since i t often happens that binary-
parameter sets cannot be determined uniquely, ternary data should then
be used to fix the best binary sets from the ranges obtained from the
binary data.
For a few systems, Abrams and Prausnitz (18) show that UNIQUAC per
forms reasonably well, both in predicting ternary diagrams from binary
information only and in correlating ternary diagrams. Anderson and Praus
nitz (19) show that UNIQUAC predicts ternary diagrams very well from bi
nary information when binary vapor-liquid and liquid-liquid equilibrium
data are correlated simultaneously with only a few ternary tie lines.
Comparing these models for correlating liquid-liquid equilibrium,
Fredenslund, et a l . (20) concluded that UNIQUAC is as good as or better
than NRTL and modifications of Wilson's equation by Tsuboka and Katayama.
However, these local composition models were developed for vapor-liquid
equilibrium and are not fully successful for liquid-liquid equilibrium.
I t is often not possible to represent the solute distribution coeffici
ents with sufficient accuracy for extraction design purposes. Freden
slund, et a l . (20) reported that the predicted value of distribution

16
coefficients by UNIQUAC using four parameters may be in error by more
than a factor of two.
Group-Contribution Models
The NRTL and UNIQUAC models are widely used for correlating liquid-
liquid equilibrium data. The parameters in these models are estimated
from experimental phase equilibrium data. For systems for which little
or no experimental information is available one needs prediction methods.
In recent years the group contribution approach has become a valuable
tool for such predictions. Notable in this development are the pioneer
ing work by Pierotti, Deal and Derr (13), Wilson and Deal (21), and sub
sequent contributions by Scheller (22), Rateliff and Chao (23), Derr and
Deal (24), and Fredenslund, Jones and Prausnitz (25).
In 1962, Wilson and Deal (21) presented the solution of groups con
cept to calculate activity coefficients on the basis of solute and sol
vent structures. The four assumptions they used became the basis for
most group-contribution methods used for the estimation of activity
coefficients.
The four assumptions are:
Assumption 1. The liquid solution can be treated as a solution of groups
which make up the components of the mixture. The "groups" are any con
venient structural units such as -CH^, -OH, and -CH2OH.
Assumption 2. The partial molar excess free energy, or, simply, the
logarithm of the activity coefficient of a component is assumed to be
the sum of two contributions - one associated with differences in molecu
lar size and shape and the other with energetic interactions between the

17
groups. For molecular solute i in any solution:
In Yi = In yS' + In Y - ^ (18)
C R where Y - is the combinatorial or size or entropy part and Y,- is the
residual or interaction or enthalpy part.
Assumption 3. The contribution from interactions of molecular "groups"
is assumed to be the sum of the individual contributions of each solute
"group" in the solution, less the sum of the individual contributions in
the conventional standard state environment. For molecular solute i,
containing groups K:
In y.^ = Z v^[ln r^ - In r^^^'^ (19)
K = 1, 2 ... N, where N is the number of different groups in the mixture,
r. is the residual activity coefficient of group K in a solution; r. ^
is the residual activity coefficient of group K in a reference solution
containing only molecules of type i; ^^ is the number of "interaction"
groups of kind K in molecule i. The standard state for the group resi
dual activity coefficient need not be defined due to cancellation of
terms.
Assumption 4. The individual group contributions in any environment con
taining groups of given kinds are assumed to be only a function of group
concentrations.
= F(x^, X2 . . .x^) (20)

18
(i) The same function is used to represent r. and T.^ \ The group fraction
F is defined by:
Fu = ^-:^ (21) Z Z v.^^^x.
i = 1, 2 . . .M (number of components)
j = 1, 2 . . .N (number of groups)
The assumption that individual group contributions are functions
only of group concentrations permits experimental data for one system to
be applied to a second system involving the same groups.
The pioneering work of Wilson and Deal lead to the development of
various group-contribution methods as stated previously. The difference
between the various group-contribution methods is essentially due to the
differences in the definition of functional groups and in the equations
used for calculating the combinatorial or size activity coefficient and
the group activity coefficient.
ASOG and UNIFAC models are based on the solution of groups concept.
These models may be used to compute the liquid phase activity coeffi
cients by the properties of the groups. Hence, liquid-liquid distribu
tion compositions at equilibrium may be predicted in the absence of ex
perimental information for the mixture of interest.
ASOG
The "Analytical Solutions of Groups" (ASOG) method was developed
by Derr and Deal from previous work on group-contribution theory by
Wilson (21) and Pierotti, Deal and Derr (13). The Flory-Huggins relation

19
was used for calculating the size term
In Y - = In r. + 1 - r. (22)
Here r is defined as the ratio of solute groups to the total number of
groups in the average liquid molecule:
r, = j-^ (23)
Z S. X. J J
here S. and S. are the number of "s ize" groups in each of the molecular
species in the so lu t ion.
Predict ion of l i q u i d - l i q u i d equ i l i b r i a has been based on the ca l
culat ion of mole f rac t ion concentrations x. , x. which sat is fy the
l i q u i d - l i q u i d equ i l i b r i a condit ions.
(Yi x . )^ = {y. x . )^^ (24)
Z x.^ = 1 ; Z x,^^ = 1 (25) i= l ^ i= l ^
Tochigi and Kojima (26) discuss the predict ion of l i q u i d - l i q u i d
equi l ibr ium by ASOG for 9 ternary systems make up of CH2(=CH2), OH and
CO groups at 25°C and 37.8°C. The predicted values and the observed
ones fo r the 9 ternary systems only agree semi-quant i tat ively. The pre
dicted values are not in agreement with the observed ones around the
p l a i t po in t .
Sugi and Katayama (27) measured l i q u i d - l i q u i d equi l ibr ium data for
three d i f f e ren t aqueous alcohol solut ions. They determined the group-

20
interaction parameters based on the data for the mutual solubility of
water and 1-butanol at 25**C. These parameters were then used for the
prediction of liquid-liquid equilibria for al l the other measured systems
Their results were in qualitative, but not quantitative, agreement with
experiment.
UNIFAC f^thod (UNIQUAC Functional-Group Activity Coefficients)
The UNIFAC method was originally developed by Fredenslund, et a l .
(25) in 1975 based on the UNIQUAC model for liquid mixtures. In the
UNIFAC method, the combinatorial term of the activity coefficient takes
into account not only the differences in molecular sizes as given by
group volumes but also the differences in molecular forms as presented
by group surface areas. The method was later revised and detailed des
criptions of the method have been presented by Fredenslund, et a l . (28).
The UNIFAC equations for the calculation of activity coefficients
are:
In y- = In y.^ + In Y / (26)
combinatorial residual
In Y - = (In c^/x. + 1 - 4)^./x.) - 1/2 Z q.(ln (D./e. + 1 - <t)./9.) (27)
Is
The parameters needed for the use of UNIFAC are group volumes (R,^),
group surface areas (Qj ) and group interaction parameters (A^^ and A^^ ).
The group interaction parameters must be evaluated from phase equilibri urn

21
data. Extensive tables with parameters are given by Fredenslund, et
a l . (28).
Recently, Magnussen, e t a l . (29) decided to develop a new parameter
table e x p l i c i t l y for l i q u i d - l i q u i d equi l ibr ium at 25^C. These new para
meter may well lead to s ign i f i can t improvements. However, the d i s t r i b u
t ion coef f ic ients cannot be expected to be predicted well since small
concentrations may be a f f l i c t e d with large re la t ive er rors .
Empirical Correlat ion Methods
Both the ASOG and UNIFAC approaches to estimating l i q u i d - l i q u i d
equi l ibr ium behavior have a common thermodynamic basis, and both depend
upon estimation of solute a c t i v i t y coef f ic ients to calculate a d i s t r i b u
t ion coe f f i c ien t . Soviet authors have taken a much more s imp l is t i c ap
proach to corre la t ion of l i q u i d - l i q u i d d is t r ibu t ion coe f f i c ien ts .
Korenman, et a l . (30) found a l inear re la t ion between the number of
carbon atoms in the extractable molecule and the logarithm of the cor
responding d i s t r i bu t i on coef f ic ients in n-alkanols, a l iphat ic amines,
and 2-alkoxyethanols homologues.
Abramzon, et a l . (31) observed that the re lat ion of the d i s t r i b u
t ion coe f f i c ien t values of amines in a homologous series to the number
of hydrogen atoms in the hydrocarbon chain is l inear and the graphs for
a homologous series of amines in d i f fe ren t solvents are p a r a l l e l . They
concluded that from the re la t ion of the d is t r ibu t ion coef f i c ien t to the
interphase tension and the size of the molecule undergoing p a r t i t i o n ,
i t i s possible to predict the d i s t r i bu t i on coef f ic ients of primary
amines in various l i q u i d - l i q u i d systems.

22
Nys and Rekker (32) estimated liquid-liquid distribution coeffi
cients of solutes of different structures in octanol-water system.
Their calculations are based on the following equation:
n log K = Z a f (29)
where f is the group contribution, n is the structure type and a is the
number of times a given group occurs in the structure.
By means of regression analysis, Rekker, et al. obtained the group
values of 11 type of structure. The overall accuracy of the estimating
distribution coefficients is extremely good. The average absolute percent
error is 9.4%.

CHAPTER I I I
DISTRIBUTION COEFFICIENTS OF HOMOLOGUES
Introduction
The works of Korenman, et a l . (30) and Abramzon, et a l . (31) with
a limited number of homologous series of solute materials suggests an
interesting possibility. I f the linear relationship between distribu
tion coefficient and carbon number in a homologous series is a general
relation, then one should be able to estimate the distribution coeffi
cient of larger molecules in a series based solely upon values for the
smaller molecules of a series. Further, there exists the opportunity
of predicting, a priori, values for liquid-liquid distribution coeffi
cients at infinite dilution based solely upon molecular structure, in
a manner similar to prediction of ASOG or UNIFAC parameters.
The work of this chapter has two purposes. First, the relation of
the distribution coefficients of several homologues to the number of
carbon atoms in the molecule is studied in order to verify the property
of log -linear distribution coefficients for homologues. Second, free-
energy equations are used to determine the free energy of the methylene
group, as a test to see i f other group-free-energy contributions may be
determined.
Relation of the Distribution Coefficients of Homologues to Molecular Structure
The liquid-liquid distribution coefficients of several homologues-
alkylamines, n-alkanols and carboxylic acids in octanol-water system.
23

24
diethyl ether-water system, chloroform-water system and benzene-water
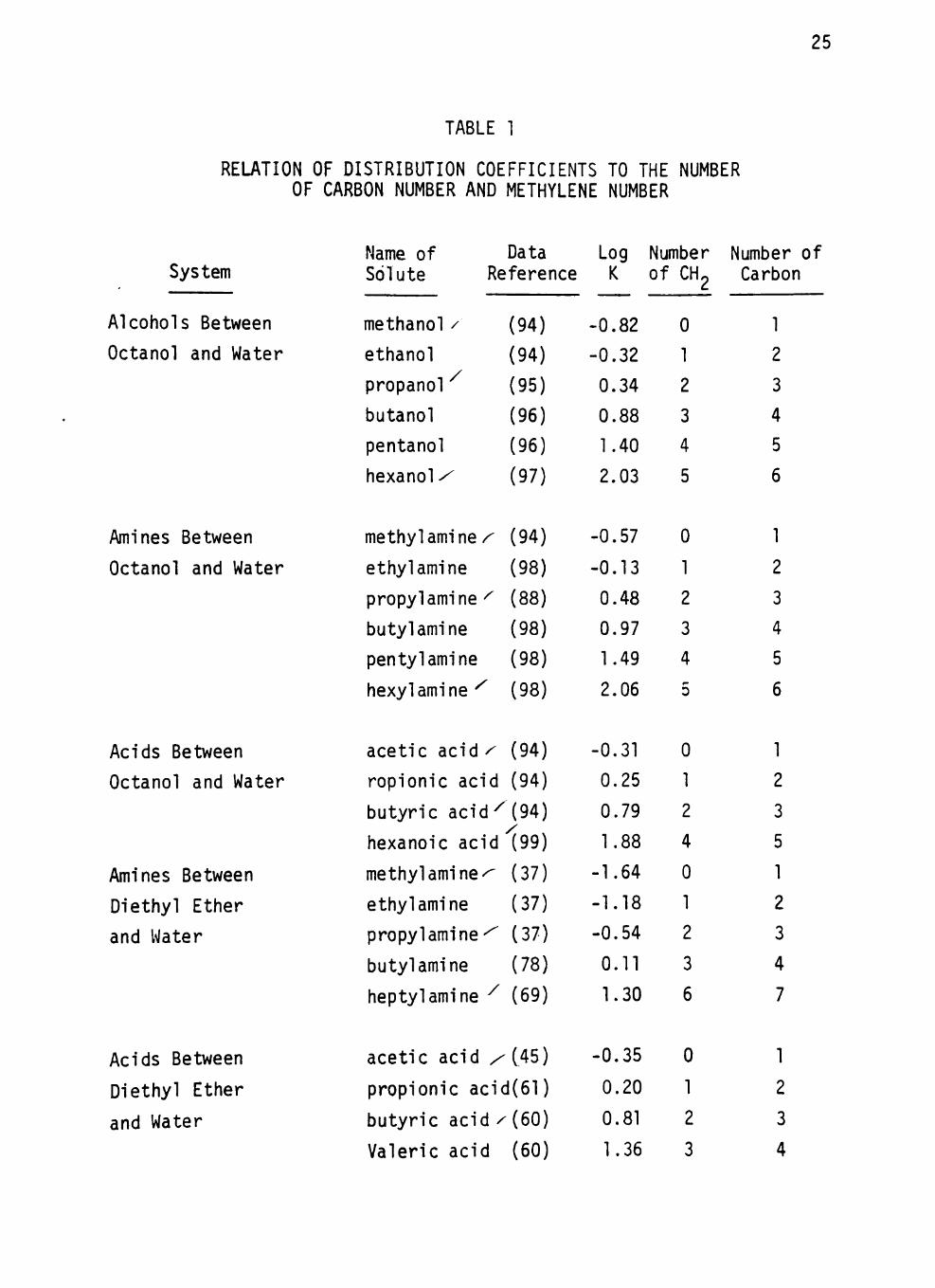
system have been tabulated from the l i terature in Table 1. I t is obvious
that the distr ibut ion coefficients of these homologues increase with an
increase in the number of carbon atoms in the molecule.
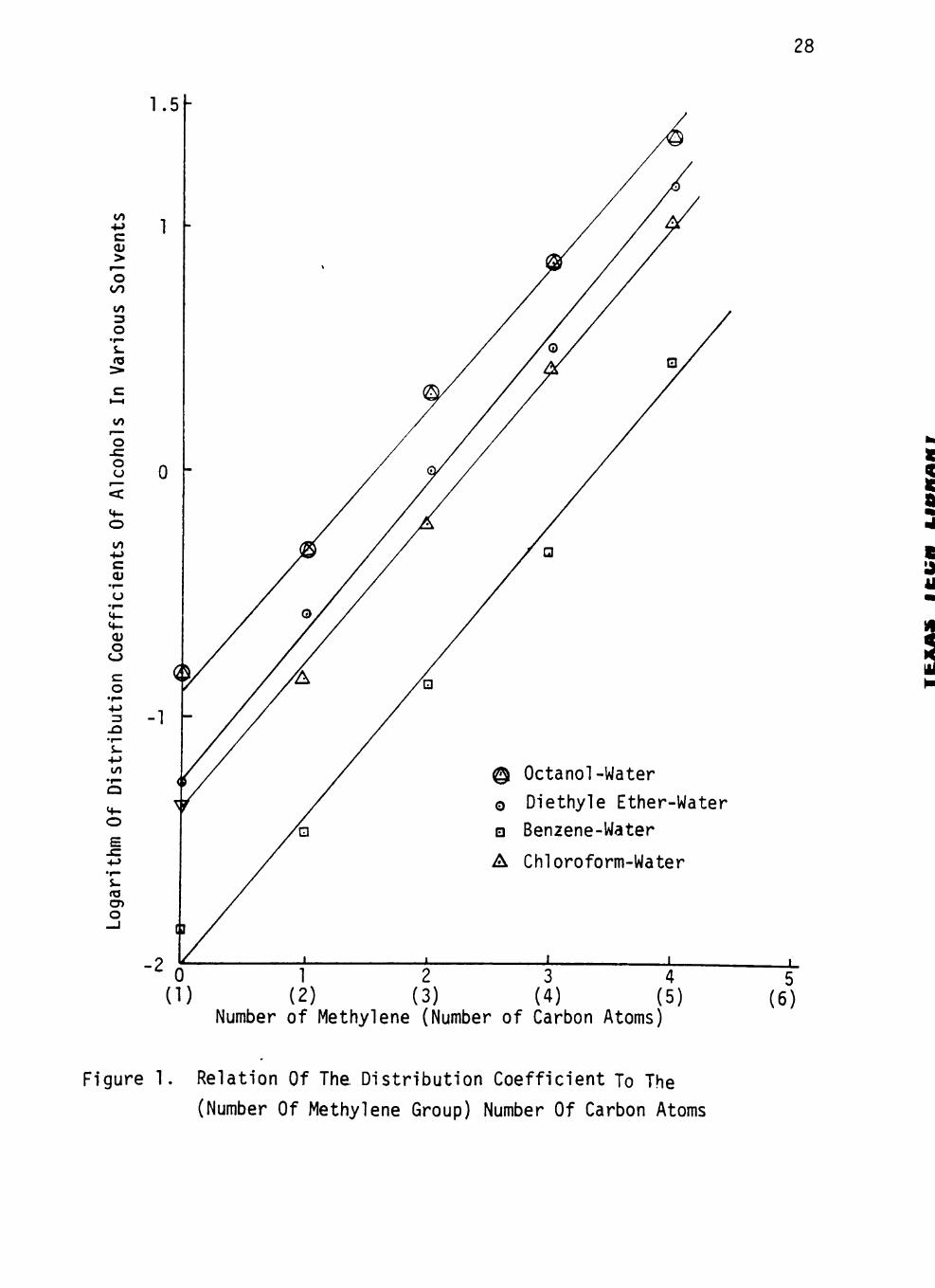
The logarithms of distr ibut ion coefficients of the homologous series
of solutes in various systems are plotted against solute carbon number
in the hydrocarbon chain in Figures 1 to 3. In a l l cases a l inear rela
t ion is observed between the number of carbon atoms in the hydrocarbon
chain of the solute and the logarithm of the corresponding distr ibut ion
coeff ic ients. The correlation coefficients of these relations are close
to unity.
The log-l inear behavior may be of practical value for predicting
the distr ibut ion coefficients of certain compounds. Table 2 gives an
example of the results obtained from correlation of acids in four d i f
ferent systems together with the experimental values. Good mutual agree
ment is evident.
The homologous difference is given by the slope of the straight
l ines, and depends in general on the nature of the solvent (Figures 1 to
3). Comparing the distr ibut ion coefficients of the same solute in d i f
ferent solvents, i t appears that the distr ibut ion coefficients decrease
with an increase in solvent polar i ty. Acids in chloroform-water system
and in the benzene-water system give exactly the same slope by s t a t i s t i
cal analysis. Also, acids in octanol-water and in diethyl ether-water
system give approximately the same slope (0.57 vs. 0.55). This regular
i t y can be explained by the properties of solvents. Chloroform and
benzene are classif ied as polar solvents and proton donors; while
25
TABLE 1
RELATION OF DISTRIBUTION COEFFICIENTS TO THE NUMBER OF CARBON NUMBER AND METHYLENE NUMBER
System
Alcohols Between
Octanol and Water
Amines Between
Octanol and Water
Acids Between
Octanol and Water
Amines Between
Diethyl Ether
and Water
Acids Between
Diethyl Ether
and Water
Name of Data Solute Reference
methanol / (
ethanol (
propanol (
butanol (
pentanol (
hexanol^ (
methyl amines (
ethyl amine i
propylamine'^ i
butyl amine i
pentylamine i
hexylamine ^ i
acetic acid ^ '
ropionic acid
butyric acid^
hexanoic acid
methyl amines (
ethyl amine
propylamine'^
butyl amine
heptylamine ^
acetic acid x
propionic acid
butyric acid ^
Valeric acid
94)
94)
95)
96)
96)
'97)
,94)
.98)
;88)
[98)
[98)
[98)
[94)
[94)
[94)
[99)
[37)
[37)
[37)
[78)
(69)
[45)
(61)
(60)
(60)
Log K
-0.82
-0.32
0.34
0.88
1.40
2.03
-0.57
-0.13
0.48
0.97
1.49
2.06
-0.31
0.25
0.79
1.88
-1.64
-1.18
-0.54
0.11
1.30
-0.35
0.20
0.81
1.36
Number of CH^
0
1
2
3
4
5
0
1
2
3
4
0
0
1
2
4
0
1
2
3
6
0
1
2
3
Number of Carbon
1
2
3
4
5
6
1
2
3
4
5
6
1
2
3
5
1
2
3
4
7
1
2
3
4
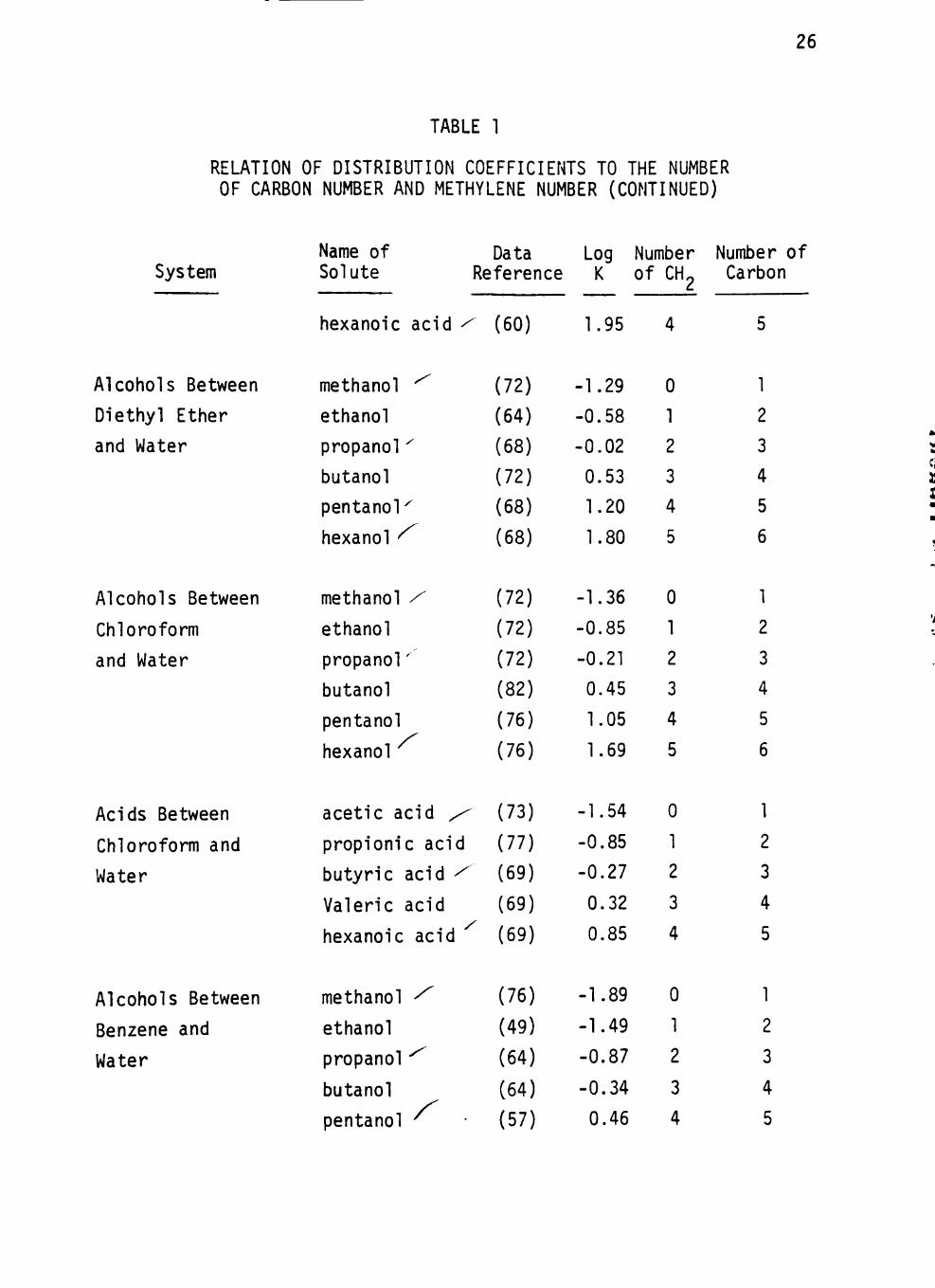
26
TABLE 1
RELATION OF DISTRIBUTION COEFFICIENTS TO THE NUMBER OF CARBON NUMBER AND METHYLENE NUMBER (CONTINUED)
System Name of Solute
Data Log Number Number of Reference K of CH,
hexanoic acid - (60) 1.95
Carbon
Alcohols Between
Diethyl Ether
and Water
Alcohols Between
Chloroform
and Water
Acids Between
Chloroform and
Water
Alcohols Between
Benzene and
Water
methanol ^
ethanol
propanol^
butanol
pentanoK
hexanol ^
methanol /
ethanol
propanol'
butanol
pentanol
hexanol
acetic acid ^
propionic acid
butyric acid ^
Valeric acid
hexanoic acid
methanol ^
ethanol
propanol ^
butanol
pentanol ^
(72)
(64)
(68)
(72)
(68)
(68)
(72)
(72)
(72)
(82)
(76)
(76)
(73)
(77)
(69)
(69)
(69)
(76)
(49)
(64)
(64)
(57)
-1.29
-0.58
-0.02
0.53
1.20
1.80
-1.36
-0.85
-0.21
0.45
1.05
1.69
-1.54
-0.85
-0.27
0.32
0.85
-1.89
-1.49
-0.87
-0.34
0.46
0
1
2
3
4
5
0
1
2
3
4
5
0
1
2
3
4
0
1
2
3
4
1
2
3
4
5
6
1
2
3
4
5
6
1
2
3
4
5
1
2
3
4
5
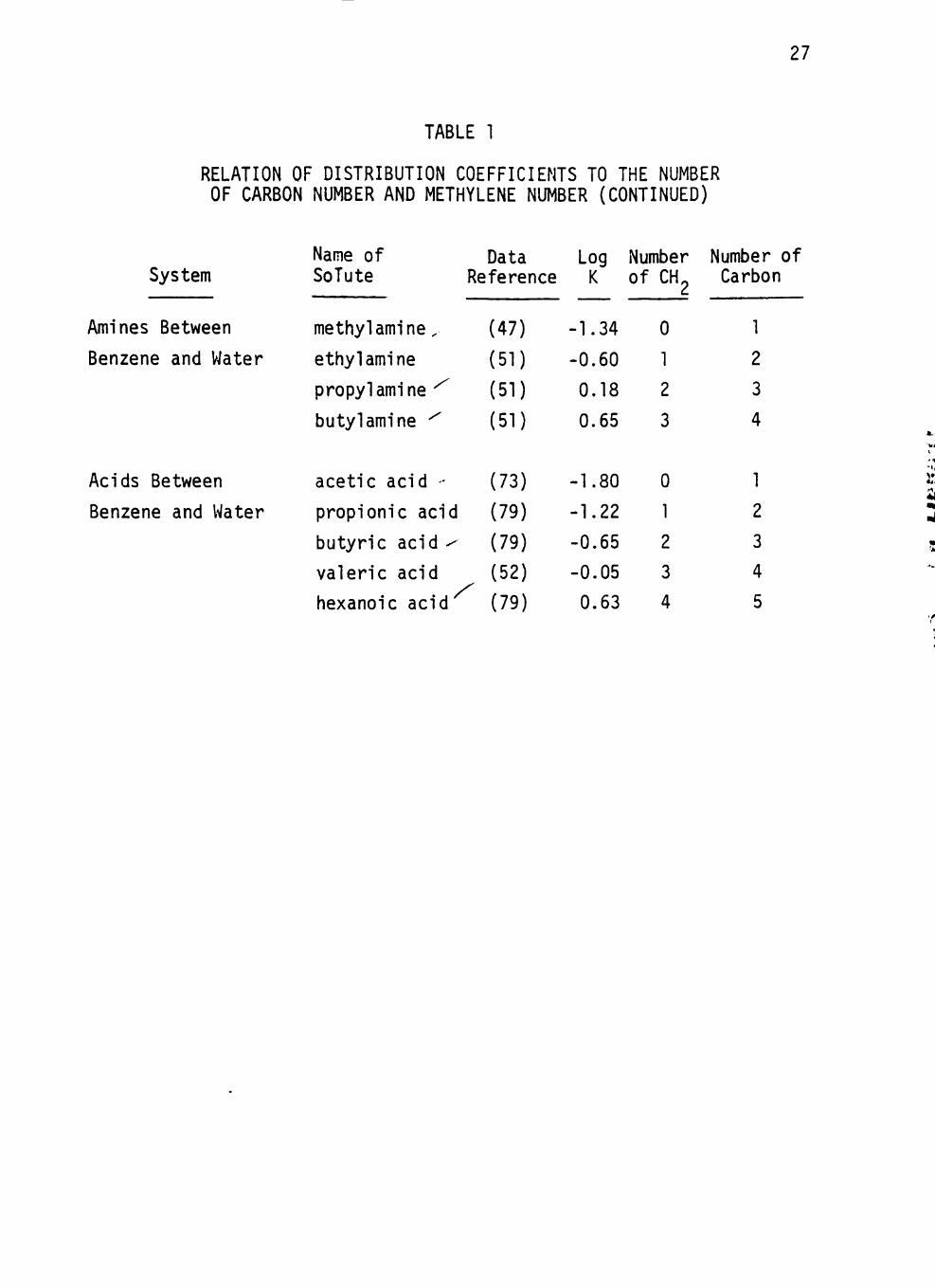
27
TABLE 1
RELATION OF DISTRIBUTION COEFFICIENTS TO THE NUMBER OF CARBON NUMBER AND METHYLENE NUMBER (CONTINUED)
System
Amines Between
Benzene and Water
Acids Between
Benzene and Water
Name of Data SoTute Reference
methyl amine.
ethyl amine
propylamine ^
butyl amine ^
acetic acid -
propionic acid
butyric acid ^
valeric acid
hexanoic acid
(47)
(51)
(51)
(51)
(73)
(79)
(79)
(52)
(79)
Log K
-1.34
-0.60
0.18
0.65
-1.80
-1.22
-0.65
-0.05
0.63
Number of CH^
0
1
2
3
0
1
2
3
4
Number of Carbon
1
2
3
4
1
2
3
4
5
28
4 0
>
o 0 0
CO 3 O
z. ns
to
'o o u
o 10
+J c
o
0)
o
3
4->
to
5 o E
o
«
s 8
41
1 2 3 4 (2) (3) (4) (5)
Number of Methylene (Number of Carbon Atoms)
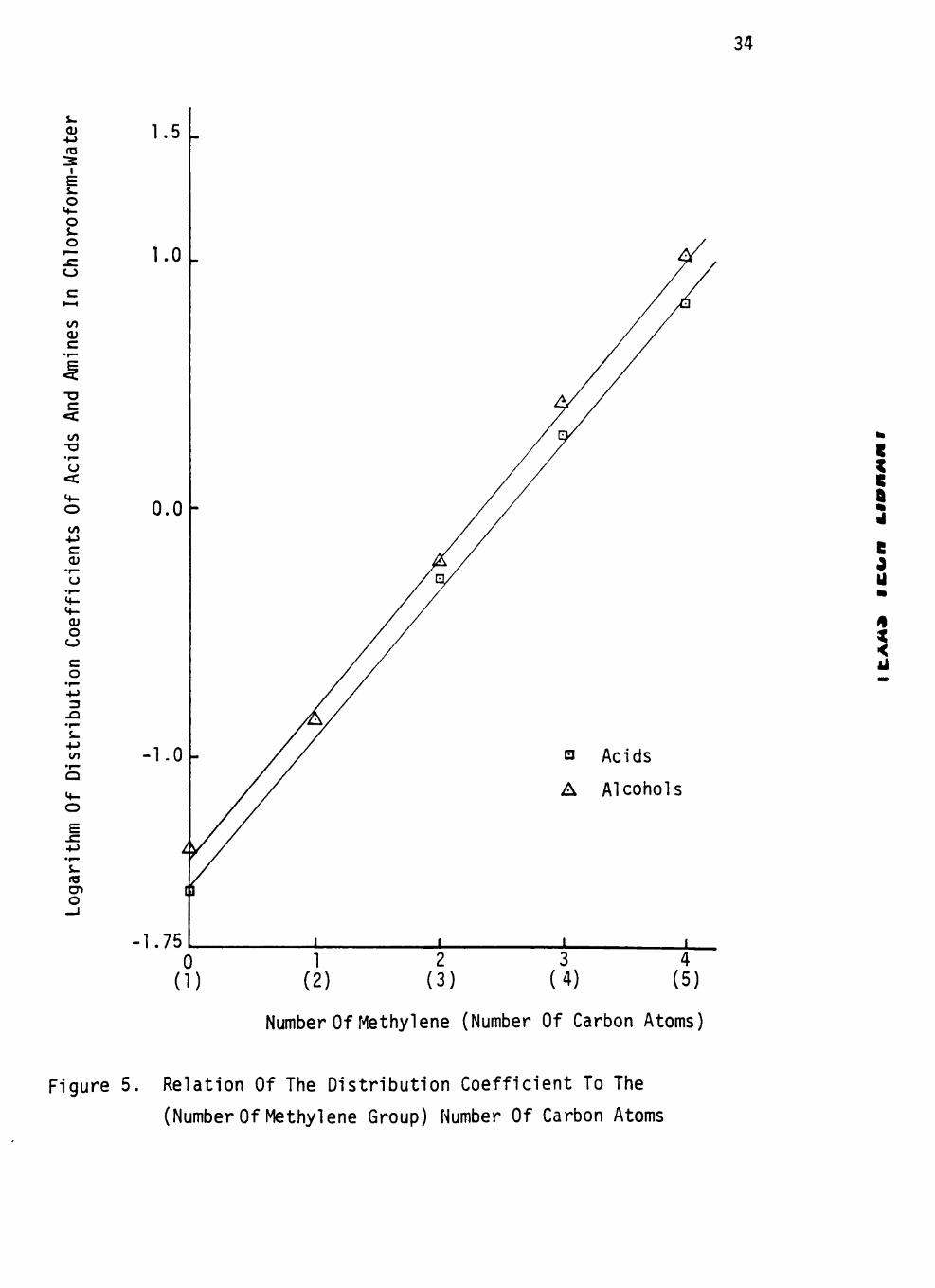
Figure 1. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms

29
to 4 J C (U
>
o CO to 3
o z. n3
to
•r— u
<:
to +J c a>
•r— O
•r -
^-
o CJ c o 3
.o •r— +-> to
4 -O
CD O
Number Of Methylene (Number Of Carbon Atoms)
Figure 2. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms

30
to +J c a; >
o oo to 3 o
•r -& .
to
d
'i
to 4-> C <u
• I —
o
0) o CJ
• M to
•r—
a
o o Diethyl Ether-Water
a Octanol-Water
A Benzene-Water
-1.75 0 1 2 3 4 (1) (2) (3) (4) (5)
Number Of Methylene (Number Of Carbon Atoms)
Figure 3. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms

31
TABLE 2
DISTRIBUTION COEFFICIENTS OF ACIDS OBTAINED FROM EXPERIMENTS AND CORRELATIONS
System
Acids between
Octanol and Water
Name of Solute
Distribution Coefficients
From Experiments From Correlation
acetic acid 0.49 0.50
propoinic acid 1.78 1.74
butyric acid 6.17 6.17
hexanoic acid 75.86 75.86
Acids between
Diethyl-ether and
Water
Acids between
Chloroform and
Water
Acids between
Benzene and
Water
acetic acid
propoinic acid
butyric acid
valeric acid
acetic acid
propoinic acid
butyric acid
valeric acid
hexanoic acid
acetic acid
propoinic acid
butyric acid
valeric acid
hexanoic acid
0.45
1.58
6.46
22.91
0.03
0.14
0.54
2.09
7.08
0.02
0.06
0.22
0.89
4.27
0.44
1.66
6.17
23.44
0.03
0.13
0.50
2.00
7.76
0.02
0.06
0.24
0.95
3.89
c t t 0
u
5

32
octanol and diethyl ether are classified as non-polar solvents and pro
ton acceptors (33).
In order to study the effect of the nature of solute on distribu
tion coefficients for extraction by a given solvent, the logarithms of
distribution coefficients of alcohols, acids and amines in same liquid-
liquid systems are plotted against solute carbon number in the hydrocar
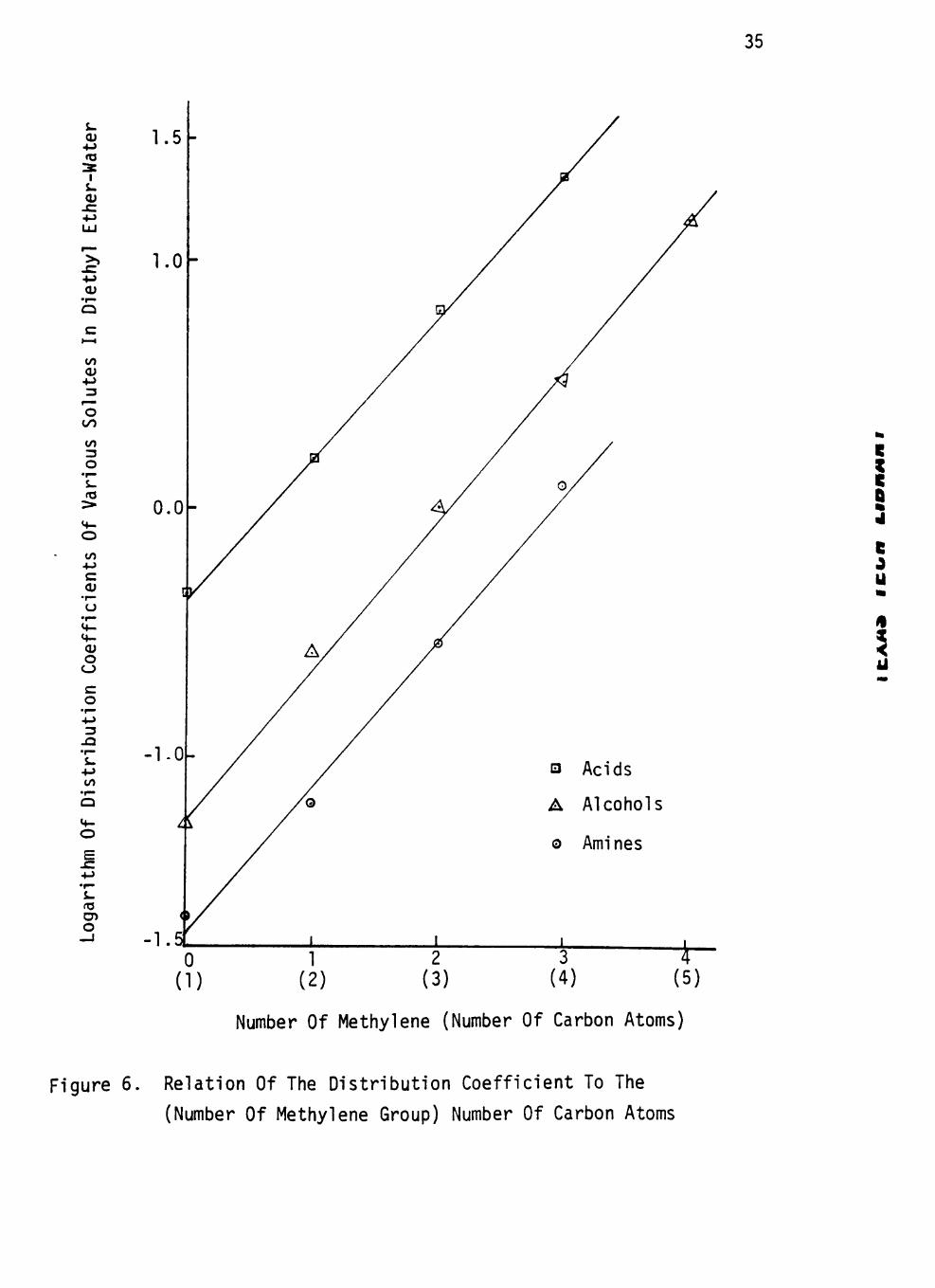
bon chain in Figures 4 to 7. The slopes of amines, alcohols and acids
in same solvent systems are different except for that of the chloroform-
water system. It seems that the nature of the extractable compounds also
affects the size of the homologous difference for extraction by a given J
solvent.
c
0
e Figure 8 shows plots that form a series of parallel lines. The J
u
se lect iv i ty of a solvent for separating two components A and B is deter-
mined by the difference between the logarithms of the distr ibut ion coef- ^
f ic ients of A and B. Therefore, the vertical displacement between 2 par- "*
a l l e l l ines of the same solvent pair determines the select iv i ty of the
solvent pair. Also, the horizontal displacement between 2 parallel lines
of the same solvent pair can be taken as a measure of the ab i l i t y of the
solvent pair to separate broad-range mixtures. Therefore, the se lect i
v i ty and a b i l i t y of the solvent pair, chloroform-water in separating
broad-range mixtures can be determined by the vertical and horizontal
displacement between the two parallel l ines.
Derivation to Free-Energy Equations of Parti t ioning Process
Cratin (34) and Everett (35) have given an analysis of the thermo
dynamics of the distr ibut ion process. The following mathematical develop
ment is patterned after Cratin and Everett.

33
s.
+J
I
o c m
• M u o
to
o 00
to 3
o Z.
O
to
CU
4 -<U O
CJ
c o
•r— 4J 3
X i
&-• M to
•f— O
E x :
(a en o
C
e
e u
4
1 2 3 4 (2) (3) (4) (5)
Number Of Methylene (Number Of Carbon Atoms)
Figure 4. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms
34
•M
I
E O
H-O L. O
CJ
c
to
c
T3 C
< :
to
-o <
to
<u
CU o CJ
c o
3
.a •M to
O
C 0
e u
i i
-1.75
Number Of Methylene (Number Of Carbon Atoms)
Figure 5. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms
35
QJ 4->
<a
I <L)
(U
Q
C
to CD
O OO
to 3 O
s _ (T3
to
o CJ
o
4J to
o E
(T3 cn o
Z c 0
e u
Number Of Methylene (Number Of Carbon Atoms)
Figure 6. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms

36
4-> «T3
CU C Q) N C
<u CO
to (U
o oo to 3 o
4-O I/)
CU
( J • r -«4-4 -CU
o CJ e o
4-> 3
J 3
S-4 J to
•r -O
O
E 4 0
&.
o
Number Of Methylene (Number of Carbon Atoms)
Figure 7. Relation Of The Distribution Coefficient To The
(Number Of Methylene Group) Number Of Carbon Atoms

37
1.75 •
to C <u CJ
CU o C_J
3
.a •r -i-4-> to
O E sz
O
e t e
6
Acids In Chloroform-Water
Alcohols In Chloroform-Water
Alcohols In Diethyl Ether-Water
Acids In Benzene-Water
-1.9
Figure 8,
2 3 4 5 Number Of Carbon Atoms In The Hydrocarbon Chain
Relation Of The Distribution Coefficient To The
Number Of Carbon Atoms

38
An ideal solution is defined as one in which each component follows
the equation:
y.(T,P,X) = y.°(T,P) + RT In x. (30)
where y.** is the chemical potential of pure "i" in the solution at a
specified temperature and pressure, and x . is its mole fraction. y.° is
not the actual chemical potential of pure "i" but the value it would have
if the solution remained ideal up to x.=l.
The molar concentration of the ith component, C., is defined as
C i = ^ (31)
where N- is the number of moles of component i, and V is the volume of
solution.
If the solution is sufficiently dilute,
V = N3 \ ° (32)
o
in which N3 and V are the number of moles and molar volume, respec
t ive ly , of solvent in the solution, Thus, N.
(33)
Furthermore,
L J - " 0 N V s s
N. 1
- i N. + N h h
(34)
since N^ » N^.
By substituting x^ for N./N^ in Equation 33 we obtain
C, = 0 (35)

39
Rearranging Equation 35 and taking logarithms will give
In X. = In C. + In V ° (36)
l i s
Finally, Equation 30 may be written for component i in the follow
ing manner:
y^-(T,P,X) = u.°(T,P) + RT In YJ + RT In C- (37)
Equation 37 shows that, for dilute solutions, the chemical potential
based upon mole fraction is larger than that based upon the molar concen-o
tration by RT In V .
Assume that the total free energy of a molecule, y., made up of the
contributions from a lipophilic group (y, ) and "n" hydrophilic groups
(yn) fnay be represented by the equation
y^(W) = M^W + n y^(W) (38)
and
y^(0) = M^{Q) + n y^(0) (39)
where (W) and (0) refer to the aqueous and nonaqueous phases.
For ideal solutions we may write
M^W = yJ(W) + n yj °(W) + RT In x(W) (40)
\i^(0) = yL°(0) + n y^°(0) + RT ]n x{0) (41)
Introducing Equation 36 into Equations 40 and 41, we obtain
y^(W) = yL°(W) + n y^°(W) + RT In V^W) + RT In C(W) (42)

40
and
y^(0) = y|_°(0) + n y^°(0) + RT In V°(0) + RT In C(0) (43)
When equilibrium is established between phases, y.(W) = y . (0) , we
may equate Equations 42 and 43 collect terms and replace C(0)/C(W) by
distribution coefficient, K, to obtain
l\°W ' yL°(0)] + RT ln[¥<'(W)/r(0)] + n[y^°(W) - y^°(0)] = RTlnK (44)
To simplify Equation 44, let us put
Ay° = y°(W) - y°(0) (45)
and, n Ay ^ Ay °
^°9 K = 273"^ " 273Tr •" Tog[V°(W)/V°(0)] (46)
According to Equation 46, a plot of log K vs. n will be linear with a
slope equal to AyL|°/2.3 RT, and with an intercept of Ay. °/2.3 RT + log
[V°(W)/V°(0)].
Determination of Free Energy of Transfer of Methylene Group
The validity of Equation 46 is tested by plotting methylene number
vs. log K for different solutes in different systems from data tabulated
in Table 1. The graphs are the. same as Figures 1 to 7. A good linear
relation was observed for all except amines in the diethyl ether-water
system.
The linear relationship between number of methylene of homologous
alkanols in chloroform-water system can be expressed as

41
log K = -0.61 n + 1.42 (47)
This corresponds to a standard free energy change (at 25°C) for transfer
of a mole of methylene from chloroform to water of -0.83 k cal. The
negative sign indicates that the transfer process is spontaneous.
Results of free energy changes (at 25*'C) to transfer a mole of
methylene for a variety of solutes in different solvent systems are sum
marized in Table 3. I t indicates that the range of the free energy of
transfer of a mole of methylene is from 0.68 k cal to 0.93 k cal. From
Table 3, i t can be observed that the standard free energy required to
transfer a mole of methylene for homologous series of alcohols in ether-
water and chloroform-water systems are the same. A similar situation is
observed for acids in chloroform-water and benzene-water system. No
useful generalizations are generated due to insufficient data. For dif
ferent solutes in the same liquid-liquid system, the standard free ener
gies of transfer per mole of methylene are different except in chloroform-
water system. I t seems that the standard free energy for transferring
also depends on solute molecular weight and type.
The validity of Cratin's free energy equation suggests that free-
energy values contributing to distribution coefficients may be group ad
ditive. The equation indicates the possibility of determining free
energies of other groups, provided that data of two homologous series of
compounds, both having the same lipophile, but different hydrophiles,
are available.

42
TABLE 3
FREE ENERGY CHANGES (AT 25°C) TO TRANSFER A MOLE OF METHYLENE
AyCH^ of AcidS' AyCHp of Alcohols AyCHp of Amines
Solvents (kcal/mole) (kcal/mole) (kcal/mole)
Octanol and Water 0.75 0.78 0.72
Diethyl Ether .... 0.79 0.83 0.68 and Water
Chloroform and 0.83 0.83 Water
Benzene and Water 0.83 0.80 0.93

CHAPTER IV
ESTIMATION OF LIQUID-LIQUID DISTRIBUTION COEFFICIENTS FROM GROUP CONTRIBUTIONS
Reasons for New Model Development
The activity-coefficient models have not lived up to their early
expectation for predicting liquid-liquid distribution coefficients. I t
is often not possible to represent the solute distribution coefficients
with sufficient accuracy for extraction design purposes. There is a
strong need for more work on methods for the correlation and prediction
of liquid-liquid distribution coefficients.
In the meantime, we are in need of a practical procedure for esti
mating liquid-liquid distribution coefficients. The model should be
fairly simple and easily applied. The results should have sufficient
accuracy to serve in preliminary design calculations and for screening
solvents.
The present work is intended as a demonstration of a simple group-
contribution technique. I t is intended to provide a way to estimate
liquid-liquid distribution coefficients for a wide variety of solutes
using the idea of structural group-contributions.
Data Collection
In spite of a large amount of work on liquid-liquid equilibrium, no
extensive l is t of liquid-liquid distribution coefficients has appeared
in the literature. Even the latest compilations are quite old. Some of
them are: Seidel, et a l . (36), Collandar (37), Von Metzsch (38), and
International Critical Tables (39).
43 .

44
From an intensive literature survey, liquid-liquid distribution co
efficients for ternary mixtures have been collected. The kinds of sys
tems considered are solutes with octanol-water, diethyl ether-water,
chloroform-water and benzene-water. The temperature range is roughly
15-35°C; and the pressure is atmospheric.
All available data were used for the model development except those
which are obviously erroneous such as the case that the summation of
mole fraction does not equal unity. No thermodynamic equation such as
Gibbs-Duhem equation was used for testing thermodynamic consistency.
Group Contributions Model
The group-contribution method is based on the premise that thermo
dynamic functions for structural components of a molecule are additive.
Molecular structure groups have the same contribution to the thermodynamic
function no matter what molecule they appear in. Thus, the value of a
thermodynamic function can be built up from an assignment of specific
contributions to the various groups which make up the molecule.
Three assumptions were made in developing this group-contribution
method:
Assumption 1. The solute can be treated as mixtures of groups (CH^-,
-CHp- , -OH, etc.) which make up the molecular species present.
Assumption 2. The logarithm of the liquid-liquid distribution coeffici
ent of a component is assumed to be the sum of two contributions. One
associated with the difference in free energy of the solute between the
two liquid phases and the other with the energy required to transfer
solute through the liquid-liquid interface.

45
For molecular solute j in any solution:
log K. = log K { + log K^ (48)
where K . is the part associated with the difference in free energy of the
solute between the two liquid phases and K. is the residue part associated
with the energy required to transfer solute through the liquid-liquid in-
terface.
The excess Gibbs energy G and the activity coefficient Y- are in
terrelated by the following expressions:
G"" = RT Z X. In Y . (49)
and
"T 1" i = ^H^h,P,u, (50)
Since the activity coefficient is taken to be a function of tempera
ture and liquid composition, the activity coefficient can be calculated
once G is expressed as a function of composition and temperature.
The excess Gibbs energy is related to the excess enthalpy and the
excess entropy by the following relationship:
G^ = H^ - TS^ (51)
The condition for equilibrium between two liquid phases I and II
is:
x / Y / = X.^^ y.^^ i = 1, 2 . . .M (52)

46
The liquid-liquid distribution coefficient K. for component i is
then defined as:
K.--ljj-.^ (53)
N i
Thus it is natural to assume the excess free energy, or, the liquid-
liquid distribution coefficient to be the sum of two contributions. The
model has a contribution to the distribution coefficient, associated with
the difference in free energy of the solute between the liquid phases and
a residual contribution, essentially associated with the extra energy re
quired to transfer solute through the liquid-liquid interphase.
Assumption 3. The contribution associated with the difference in free
energy of the solute between the two liquid phases is assumed to be the
sum of the individual contributions of each solute group in the solution.
For molecular solute j containing group k:
log K^ = ^(f^kj^^^k^ ' ^
where: N. . = number of groups of type k in solute component j
r. = distribution coefficient of group k in the solution
environment
Substituting log KT by Equation 54, Equation 48 becomes
k , log K. = 2:(N^j)(r^) + log K^ (55)
The assumptions used for the new model are basically the same as
those used for the ASOG and the UNIFAC models. The new model uses the
term free energy difference to account for the contributions from the

47
enthalpy (interaction) part. A new term K. is added to the new model to
deal with the extra energy required for the transfer process due to the
nature of the liquid-liquid interface.
The main difference of the three models are the assumptions used
for calculating the entropy (size) part and the enthalpy (interaction)
part. The ASOG and UNIFAC models are theoretically based. The ASOG
model assumes that the combinatorial contribution of the excess Gibbs
energy of a mixture can be expressed by Flory-Huggins equation. The
UNIFAC model assumes that a semi-theoretical equation for the combina
torial contribution of the excess Gibbs energy of a liquid mixture can
be obtained through generalization of Guggenheim quasi-chemical theory.
The new model uses an empirical approach to correlate the group distri
bution coefficients and assumes the contribution from the difference in
free energy of the solute between the liquid phases depending upon the
numbers and kinds of structural groupings of the solute.
Method
The liquid-liquid distribution coefficients of any compound can be
determined by Equation 55, given appropriate structural parameters.
It is possible to obtain values of distribution coefficient from
published data on appropriate compounds, and to fit an equation of the
form denoted by Equation 55. The values of each of these groups can be
obtained by using multiple regression analysis to fit a set of equations
of the form denoted by Equation 55. The numbers of group k in solute
component i, N. ., are introduced as independent parameters and the logar
ithm of the distribution of the compound, log K., as a dependent

48
parameter. The group-distribution coefficients, r. , can be obtained from
the regression coefficients and the extra energy part, log K. from the
intercept of the output.
Calculations were performed on NAS AS/6 computing system. The Sta-
tistical Analysis System (SAS) program, maximum R improvement technique
developed by James H. Goodnight was used to find r. and log K.. K J
It would be ideal to have a well balanced distribution of the groups
among data. Ten to eleven most populated structure types were selected
for group-contribution determination. Among them are: CH^-, -CH2-,
-CH-, NH2-, -NH-, -N-, CgHg-, H0-, -0-, HOOC-, and -CO-.
The calculated values for these groups are summarized in Table 4.
As an example, the liquid-liquid distribution coefficient of methoxyeth-
anol between octanol and water is calculated by the group-contribution
method adopted in this study: structure CH2-O-CH2-CH2-OH. The molecule
contains one CH^- group, two -CH2- groups, one -0- group and one -OH
group. From the group-contribution values and the log K. values pre-
sented in Table 4, the log K value for methanoxyethanol is calculated
as follows:
log K = (1 X r , ) + (1 X r.Q_) + (2 X r.cH2'
+ (1 X r_Qj^) + log K^
= (0.705) + (-1.142) + (2 X 0.505)
+ (-1.077) + (-0.213)
= -0.72
K = 0.19
The experimental value reported by Korenman, et a l . (40) is 0.17
and was not utilized for the establishment of group-contribution values
presented in Table 4. Yet the percent error was found to be 11.7%.

49
to
3 O %. CD
(U 3 to
i-rO OH O
TABLE 4
GROUP CONTRIBUTIONS FOR THE FOUR SYSTEMS
Systems
CH3-
-CH2-
-CH-1
^6^5-
-coo-
-COOH
- 0 -
-OH
NH2-
-NH-
-N-1
c=o
Octanol and Water
0.705
0.505
0.182
1.935
-1.027
-0.681
-1,142
-1.077
-1.094
-1.560
-1.734
—
Diethyl Ether and Water
0.823
0.246
-0,218
2.470
0.174
-0.094
-0.839
-1.057
-1.760
-2.228
-1.153
—
Chloroform and Water
1,517
0,545
-0,403
3.173
—
-1.244
-0,002
-0.800
-0.641
-1.637
—
-0.407
Benzene and Water
2,149
0.544
-1.328
4.020
-0,690
-0.656
-1,316
-0.809
-0.019
-2,095
-3,667
-0.430
Log K}- -0.213 -0.112 -1.767 -3.177
50
Predictivity of the Model
All of the correlations are quite good, especially when one consid
ers that the correlations presented cover a broad range of distribution
coefficients. Also the data are taken from the work of many investiga
tors whose results are obtained by different techniques on compounds of
various degrees of purity over a temperature range of 15-35°. The data
are necessarily of limited accuracy. A realistic idea of the accuracy
of the correlations and predictions can only be obtained by a detailed
review of the cases treated. However, a rough idea of the predictive
performance of the model has been obtained by comparing with data in
cluded in the set of reference systems used to evaluate the group
parameters. Results of experimental distribution coefficients and cal
culated distribution coefficients of the four systems studied are tabu
lated in Tables 5 to 8. The average % error and average absolute % er
ror for the four analysis with the new model are summarized as follows:
Octanol- Diethylether- Chloroform- Benzene-Water Water Water Water
average •,« p absolute Jn.x* 32.2 26.2 15.8 0/ % error
(9.4)
average -2.3 13.6 -2.9 0.5 % error (-0.3)*
*Nys and Rekker's result.
Comparing the predictivity of the new model with Nys and Rekker's model
in octanol-water system, we find out that Nys and Rekker's model yields
3.5% less in average absolute % error (12.8% vs. 9.4%). However, Nys
and Rekker's model cannot be applied to chloroform-water system and
benzene-water system with accuracy.
51
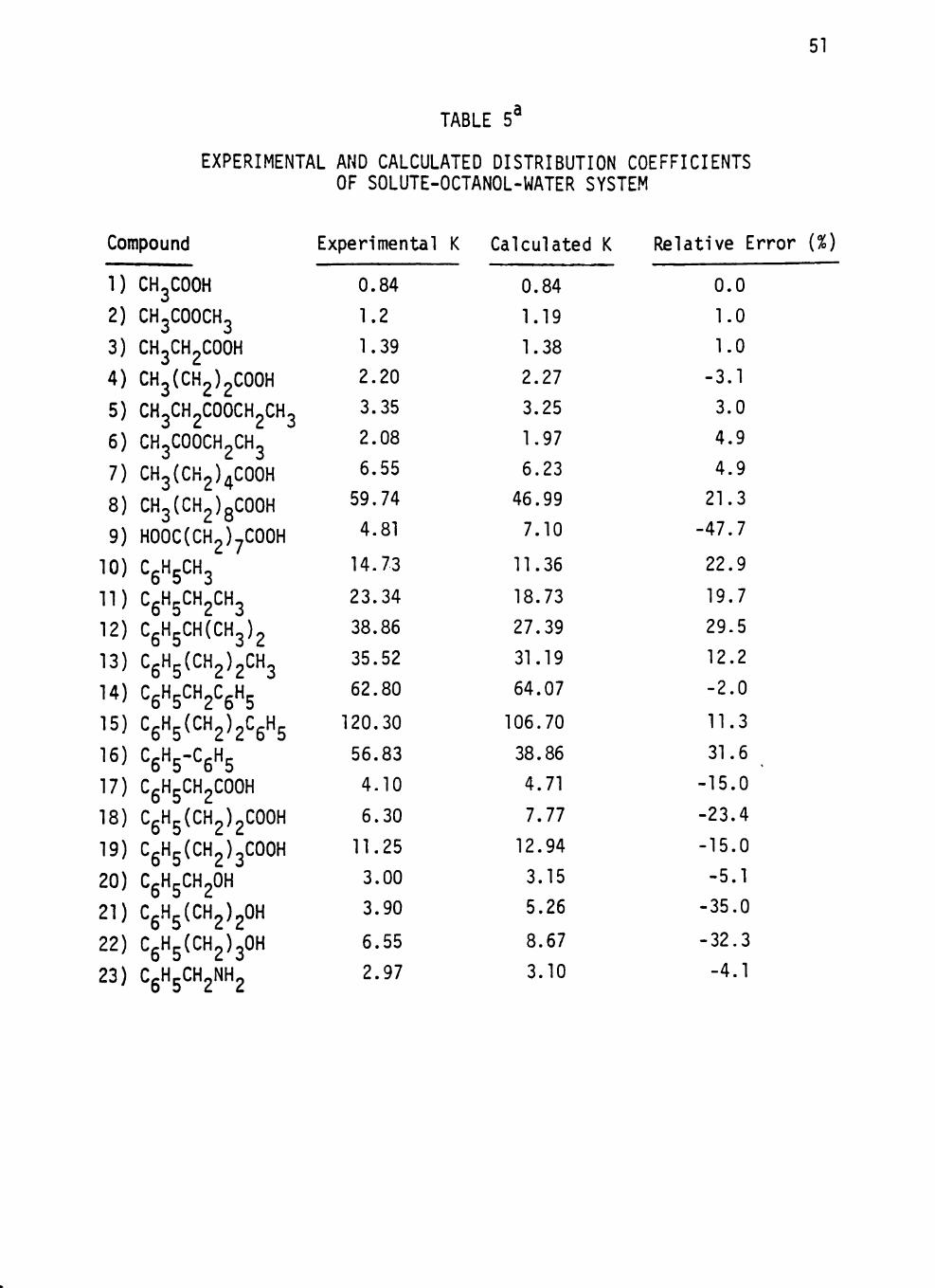
TABLE S"*
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-OCTANOL-WATER SYSTEM
Cc
1)
2]
3]
4]
5]
6]
1]
8 ;
9;
10^
11
12'
13
14
15
16.
17'
18'
19'
20:
21.
22;
23;
impound
CH3COOH
CH3COOCH3
CH3CH2COOH
1 CH3(CH2)2C00H
1 CH3CH2COOCH2CH3
) CH3COOCH2CH3
) CH3(CH2)4C00H
1 CH3(CH2)3C00H
) H00C(CH2)7C00H
) CgH5CH3
) CgH5CH2CH3
) CgH5CH(CH3)2
1 CgH5(CH2)2CH3
* ^6"5^"2^6"5 ) CgH5(CH2)2C5H5
' ^e^s'^e'^s ) CgH5CH2C00H
) CgHg(CH2)2C00H
) CgHg(CH2)3C00H
) CgHgCH20H
) CgHg(CH2)20H
) CgHg(CH2)30H
1 CgH5CH2NH2
Experimental K
0.84
1.2
1.39
2.20
3.35
2.08
6.55
59.74
4.81
14.73
23.34
38.86
35.52
62.80
120.30
56.83
4.10
6.30
11.25
3.00
3.90
6.55
2.97
Calculated K
0.84
1.19
1.38
2.27
3.25
1.97
6.23
46.99
7.10
11.36
18.73
27.39
31.19
64.07
106.70
38.86
4.71
7.77
12.94
3.15
5.26
8.67
3.10
Relative Error (%)
0.0
1.0
1.0
-3.1
3.0
4.9
4.9
21.3
-47.7
22.9
19.7
29.5
12.2
-2.0
11.3
31.6
-15.0
-23.4
-15.0
-5.1
-35.0
-32.3
-4.1

52
TABLE 5^
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-OCTANOL-WATER SYSTEM (CONTINUED)
Compound Experimental K Calculated K Relative Error (%)
24) CgHg(CH2)2NH2
25) CgHgCH2C00CH3
26) CH3C00CH2CgHg
27) CgHg(CH2)2C00CH3
28) CgHg(CH2)3C00CH3
29) CgH5(CH2)30CH3
30) CgHgCHOHCgHg
31) (CgHg)2CH.0.(CH2
32) pH2- ..^ CgHc-CH-C00-CH^CH« N-CH,
0 3 I I 2 3 CHoOH \ ruj
4.10
6.23
7.10
10.18
15.96
14.88
14.44
)2N(CH3)2
CH
26.
6.
31
23
5.16
6.75
6.75
11.13
18.54
16.44
15.80
29.37
6.37
-25.8
-8.3
4.9
-9.4
-16.2
-10.5
-9.4
-11.6
-2.0
i2un . CH 2
33;
34
35;
36;
37;
38]
39;
40;
41;
42;
43;
44;
CH2-
) CgH5CH2N(CH3)2
) CH3NH2
) CH3CH2NH2
) CH3(CH2)2NH2
) CH3(CH2)3NH2
1 CH3(CH2)4NH2
) CH3(CH2)5NH2
1 CH3(CH2)gNH2
I (CH3)2CHCH2NH2
> CH3CH2CHCH3NH2
1 CH3(CH2)4CH(CH2CF
1 CHo ~ CH« / Z / Z CH« CH - NH« Z s Z ^ CH2 - CH2
CH
6.75
0.57
0.88
1.62
2.41
4.44
7.24
13.07
2.41
2.10
l3)NH2 16.78
4.44
6.69
0.55
0.90
1.51
2.48
4.14
6.82
11.36
2.20
2.20
16.61
4.06
1.0
2.9
-2.3
6.7
-3.1
6.8
5.8
13.1
8.6
-4.8
1.0
8.6

53
TABLE 5^
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-OCTAHOL-WATER SYSTEM (CONTINUED)
Compound Exp
45) (CH3)2CHNH2
46) CH3CH2NHCH3
47) (CH3CH2CH2)2NH
48) (CH3CH2CH2CH2)2NH
49) (CH3CHp)2NH
erimental K
1.30
1.16
5.31
14.59
1.79
50) CH3(CH2)2NH(CH2)3CH3 8.33
51) CH3(CH2)3NHCH3
52) ^CH2 - CH2
CH« NH
CH2 - CH2
53) CH3CH2NHCH(CH3)2
54) CH3CH2CHCH3NH(CH2)
3.78
2.25
2.53
2CH3 6.75
55) CH3(CH2)2NHCH2CH(CH3)2 7.92
56) (CH3)3N
57) (CH3)2N(CH2)3CH3
58) ^ C H 2 - C^2
CH, CH - OH
CHp •" CHp
59) CH3OH
60) CH3CH2OH
61) CH3(CH2)20H
62) CH3(CH2)30H
63) CH3(CH2)40H
64) CH3(CH2)50H
65) CH3(CH2)70H
66) CH3CH2OCH2CH3
1.31
5.47
3.42
0.52
0.73
1.40
2.41
4.06
7.61
23.34
2.16
Calculated K
1.34
1.15
5.26
14.44
1.93
8.67
3.19
2.12
2.80
7.69
7.69
1.46
5.37
4.14
0.56
0.92
1.54
2.53
4.22
6.96
19.11
2.92
Relative Error (%)
-3.0
1.0
1.0
1.0
-8.3
-4.1
15.6
5.8
-10.5
-13.9
3.0
-11.6
2.0
-20.9
-8.3
-27.0
-9.5
-5.1
-4.1
8.6
18.1
-35.0
67) CH3(CH2)20(CH2)2CH3 7.61 8.00 -5.1

54
TABLE 5^
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-OCTANOL-WATER SYSTEM (CONTINUED)
Compound Experimental K Calculated K Relative Error (%)
68) CH3CH20(CH2)3CH3
69) (CH3)2CHCH20H
70) CH3CH2CH(CH3)0H
71) (CH3)2CH(CH2)20H
72) CH3CH(0H)CH(0H)CH3
73) CH3CH20(CH2)20H
74) (CgH5)2CHC00H
75) (CH3CH2)3N
76) HOCH2COOH
77) CH3CHOHCOOH
78) CgHgCHOHCOOH
79) CH3CH2OCH2OCH2CH3
80) H00C(CH2)2C00H
81) C.Hc CH-CH-NH-CH, ' 6 5 I \ 3 OH CH3
82) OHCHpCHpNHp ^ Cm Cm
83) CHp - CHp
0 0 1 1 CH2 - CH2
84) (H0CHCH2)2NH
CH3
^Reference (32)
7.61
1.92
1.84
3.19
0.40
0.58
21.12
4.22
0.33
0.54
1.75
2.32
0.55
2.53
0.27
0.66
. 0.44
8.00
2.25
2.25
3.71
0.55
0.81
23.57
5.37
0.29
0.34
1.16
1.54
0.57
2.36
0.25
0.62
0.32
-5.1
-21.2
-22.2
-16.2
-39.0
-39.3
-11.6
-27.1
13.0
36.7
33.7
33.6
-3.0
6.8
5.8
5.8
27.4

55
TABLE 6
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-DIETHYL ETHER-WATER SYSTEM
1
2
3
4
5
6
7
8
9
10
11
C>0 CO
14;
is; 16;
17;
is; 19]
20]
21]
22]
23]
24]
25)
26]
Compound
) HOOCCHOHCH2COOH
) (CH0HC00H)2
) CH3(CH2)2C00H
) CH3(CH2)4C00H
) (CH2)7(C00H)2
) (CH2)3(C00H)2
) CH3NH2
) (CH3)2NH
) C2H5NH2
) CH3(CH2)2NH2
) (CH3CH2)2NH
Experimental K
0.16
0.088
2.24
6.55
3.32
5.81
0.19
0.30
0.31
0.58
0.76
) CgHg(CH2)3NH2 3.40
) CgHgCH2CH3(CH2)2NH 4.44
) CH3OH
) CH3CH2OH
) CH3(CH2)20H
1 CH0H(CH20H)2
) CH3(CH2)30H
1 CH3(CH2)2CH(0H)2
1 (CH3)2(CH0H)2
CH3(CH2)2CH(CH)2
CH3CH20(CH2)20H
CH3(CH2)40H
» CgHgOH
CH3(CH2)50H
C2H5OC2H5
0.43
0.61
1.32
0.52
2.34
0.25
0.21
0.25
0.50
3.32
4.86
6.05
2.72
Data Calculated Relative Reference K Error(%)
(58
(59
(60
(61
(62
(62
(37
(37
(37
(37
(37
(65
(65
(37
(37
(37
(66
(37
(67
(37
(67
(67
(68
(63
(68
f37
0.26
0.058
3.03
4.60
4.14
5.31
0.34
0.49
0.45
0.57
0.82
3.82
5.41
0.70
0.90
1.16
0.11
1.48
0.32
0.36
0.32
0.63
1.90
3.67
2.41
3.29
-67.0
34.1
-35.1
29.8
-24.6
8.6
-78.9
-63.3
-45.2
1.7
-7.9
-12.4
-21.9
-62.8
-47.5
12.2
78.8
36.8
-28.0
-69.6
-28.0
-26.0
42.8
24.4
60.1
-20.9

56
TABLE 6
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-DIETHYL ETHER-WATER SYSTEM (CONTINUED)
27)
28)
29)
30)
31)
32)
33)
34)
35)
36)
37)
38)
39)
40)
41)
42)
43)
44)
45)
46)
47)
48)
Experimental Compound
CH30(CH2)2CH(0H)2
NH2COOCH3
NH2(CH2)20H
CH3CHOHCOOH
HOCH2CHOHCOOH
CH30(CH2)20H
CH3CHOHCH2COOH
CH2CHCH(CH20CH3)0H
HN(CH2CH20H)2
CH3(CH2)3C00H
CH(CH2)20(CH2)20CH3
CgH5NH2
(C00H)3(CH2)20H
HO(CH2)gOH
(CH2)5CH(0H)3
(H0CH2CH20CH2)2
N(CH2CH20H)3
CgHgCOOH
CH30CgHg
{(^zhh^^^h^^h CgHgCOOH
C^HcCHOHCHCH^^NHCH^
K
0.18
0.43
0.056
0.53
0.13
0.30
0.67
0.18
0.024
3.90
• 0.24
2.34
0.27
0.40
0.081
0.081
0.052
6.62
11.70
0.63
6.42
1.35
Data Reference
(37)
(37)
137)
(69)
(37)
(37)
(69)
(37)
(37)
(60)
(37)
(70)
(61)
(37)
(67)
(37)
(37)
(37)
(63)
(37)
(37)
(37)
Calculated K
0.14
0.42
0.087
0.52
0.10
0.50
0.66
0.14
0.031
3.86
0.35
1.82
0.38
0.47
0.103
0.088
0.090
9.58
10.40
0.58
9.58
1.32
Relative Error(%)
22.0
2.3
-55.4
1.9
23.1
-66.6
1.5
22.0
-29.2
1.0
-45.8
22.2
-40.9
-17.5
-27.2
-8.4
-73.1
-44.7
11.1
7.9
-49.2
2.0

57
TABLE 7
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-CHLOROFORM-WATER SYSTEM
1)
2)
3)
4)
5)
6)
7]
8]
9]
10]
n; 12;
13;
14;
15;
16;
17'
18'
19
20'
21
22
23
24
25
26
27
Compound
CH3NH2
HONH2
CH3COOH
CgHgOH
CgHgNH2
CH3(CH2)4C00H
CH30CgH5
CgHgCH3CH2
CgH5CH2NH2
1 CH3(CH2)gOH
) CH3OCH2COOH
I CH3OH
) CH3CH2OH
) CH3(CH2)30H
) CH3(CH2)40H
) CH3(CH2)50H
) CH3CH2COOH
) CH3CHOHCOOH
) CH3(CH2)3NH2
) (CH3CH2)2NH
) CgHgCH3
) CgHgCHOHCOOH
) CH3CH20CgHg
) CH3CH(CgH5)NH2
) CH3CHCgHgC00H
) (CH2)7(C00H)2
) CgHg(CH2)3C00H
Experimental K
0.41
0.076
0.021
1.55
3.42
2.34
22.75
39.65
3.25
11.13
0.27
0.28
0.43
1.57
2.86
5.42
0.43
0.11
2.69
2.25
30.27
0.28
37.34
4.02
3.00
0.56
5.64
Data Reference
(71)
(72)
(73)
(74)
(75)
(69)
(68)
(68)
(69)
(76)
(77)
(76)
(76)
(76)
(76)
(76)
(77)
(77)
(78)
(78)
(68)
(79)
(68)
(80)
(69)
(81)
(82)
Calculated K
0.41
0.040
0.023
1.84
2.16
1.99
18.54
32.14
3.71
9.21
0.38
0.35
0.61
1.80
3.10
5.37
0.39
0.067
2.12
2.10
18.54
0.35
32.14
6.55
3.60
0.64
6.05
Relative Error(%)
0.0
46.6
-9.5
-18.6
36.9
14.8
18.1
18.9
-13.9
17.3
-42.3
•-25.0
-41.8
-14.7
-8.32
0.09
9.30
39.1
21.2
6.7
38.7
-25.0
13.9
-62.9
-19.8
-14.3
-7.3

58
TABLE 7
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-CHLOROFORM-WATER SYSTEM (CONTINUED)
Compound
28) CH3COCOOH
29) CH3COCH3
30) CH3CH2CONH2
31) (CH3C0)2CH2
32) CH3C0(CH2)2C00H
33) CH3C0CgHg
34) CgHgNHC0CH3
35) CgHgC0CH2C0CH3 .
Experimental K
0.11
2.05
0.25
2.16
0.30
16.28
2.44
36.60
Data Reference
(77)
(74)
(83)
(84)
(77)
(68)
(85)
(86)
Calculated K
0.14
2.36
0.47
2.72
0.44
12.43
2.41
14.30
Relative Error(%)
-27.3
-15.1
-88.0
-25.9
-46.7
23.7
1.0
60.9

59
TABLE 8
EXPERIMENTAL AND CALCULATED DISTRIBUTION COEFFICIENTS OF SOLUTE-BENZENE-WATER SYSTEM
1)
2)
3)
4)
5)
6)
7]
8]
9]
lo ;
11-
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
Compound
CH3OH
CH3NH
CH3COOH
CH3CH2OH
(CH3)2NH
CH3CH2NH2
CH3CH2COOH
1 CH3(CH2)20H
> C2HgC00CH3
1 CH3(CH2)2C00H
) CH3(CH2)30H
) CH3CH20(CH2)20H
) CH3(CH2)3NH2
) (CH3C0)2CH2
) CH3(CH2)2C00CH3
) CH3(CH2)3C00H
) CH3(CH2)40H
) CgH5NH2
) CH3(CH2)4C00H
) (C3H7)2NH
) (C2H5)3N
) CgHgCOOH
) CgH5CH2NH2
) CH3(CH2)gOH
) CH3C0CgH5
) CgHgCHOHCOOH
) CcH.COCH,COCH, ' 6 D Z 3 ) (C.H,CO)pCHp
Experimental K
0.16
0.26
0.18
0.26
0.44
0.55
0.31
0.50
2.75
0.52
0.83
0.22
1.92
2.14
4.57
0.95
1.58
2.72
1.88
2.86
3.10
1.20
1.84
6.75
9.03
0.14
20.70
208.5
Data Reference
(46)
(47)
(48)
(46j
(50)
(51)
(52)
(76)
(53)
(52)
(54)
(55)
(51)
(56)
(53)
(52)
(57)
(87)
(79)
(47)
(88)
(89)
(90)
(76)
(83)
(91)
(92)
(93)
Calculated K
0.16
0.35
0.19
0.28
0.38
0.61
0.32
0.47
2.66
0.55
0.82
0.22
1.80
2.25
4.57
0.95
1.40
2.27
1.63
3.32
3.46
1.21
3.90
4.18
12.94
0.14
14.59
94.6
Relative Error(%)
0.0
-33.9
-6.0
-5.2
13.9
-10.5
-2.0
4.8
3.0
-5.2
1.0
0.0
6.0
-5.1
0.0
0.0
11.3
16.5
13.1
-16.2
-11.6
-1.0
-111.7
38.1
-43.3
0.0
29.5
54.6

60
It is difficult to compare the predictivity of the new model with
that of the UNIFAC and ASOG model. The ASOG and UNIFAC models are not
thoroughly tested in correlating liquid-liquid distribution coefficients
and not many results are available in the open literature. We think the
new model has better accuracy in correlating liquid-liquid distribution
coefficients than the ASOG and UNIFAC models at the present time. The
reason is based on the performance of the UNIFAC model carried out by
Fredenslund, et al. (28) for several ternary systems and the performance
of the ASOG model by Tochigi and Kojima (26) for 9 ternary systems. The
predicted liquid compositions at equilibrium are not quantitatively ac
ceptable for the design purpose in either case. The advantage of ASOG
and UNIFAC models is that these models can be applied to e\/ery system
and are not specific to one system. However, the predicted liquid-liquid
distribution coefficients are usually of semi-quantitative values. The
new model is an empirical approach and correlates specific systems so
that quantitative results are obtained.

CHAPTER V
EVALUATION OF FACTORS INFLUENCING LIQUID-LIQUID DISTRIBUTION COEFFICIENTS
Introduction
Method of Chapter IV allows estimation of liquid-liquid distribu
tion coefficients at single temperature, infinite dilution. It is de
sirable to expand the application for other conditions. Several factors
which can affect the distribution of solute between two different phases
include pH of the aqueous phase, temperature, pressure, original concen
tration of solute and structure of solute. For liquid mixtures at ordi
nary pressure, the effect of pressure is negligible. In this chapter,
the evaluation of the effects of pH, temperature and concentration upon
liquid-liquid distribution coefficients is presented.
Effect of pH on Distribution Coefficient
For ionizable solutes, distribution coefficients will vary with pH.
Generally, dependence of distribution coefficients upon pH is not linear.
Data from Colaizzi and Klink (41) show the distribution behavior of metha-
cycline hydrochloride between n-octyl alcohol and water. Methacycline
hydrochloride is ionized at all pH values. The variation of distribu
tion coefficient with pH is summarized in Table 9. The shift of log K
with change of pH is expressed graphically in Figure 9. The increase
in distribution coefficient observed with pH increases from 2.1 to
8.5 were nonuniform. The maximum distribution coefficient occurred at
pH equal to 5.6.
61

62
TABLE 9^
SHIFT OF DISTRIBUTION COEFFICIENT WITH pH
System pH
Mathacylcine Hydrochloride In
N-Octyl alcohol-Water
Reference (41)
2.1
3,0
3,9
5.6
6,6
7,5
8,5
0,69
0.72
0,82
0.91
0,82
0,43
0,16
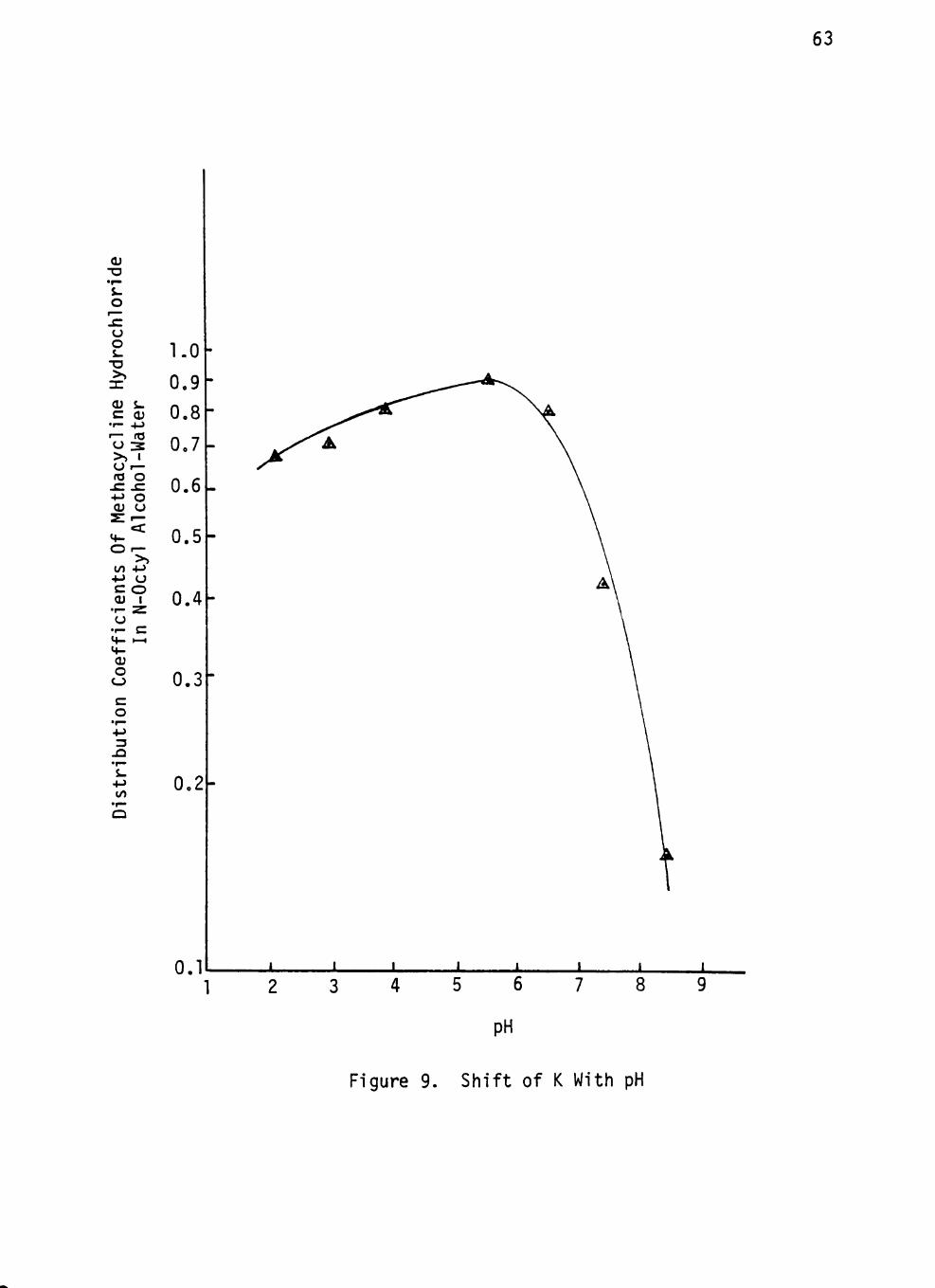
63
CU
•r— &-o
j r o o &--o
CU S -c <u
• I - + j >— ro O S >> I O f— ro O
. C JO. •M O CU O
O r ->>
in 4-> -> CJ C O (U I
•r- 2 1 U
•r- C
V « -cu O
o
3
s-• » - )
I/)
o
1.0 0,9 0,8 0,7
0.6
0.5
0.4
0.3
0,2
0.1 8
pH
Figure 9. S h i f t o f K With pH

64
A wide variety of phenolic compounds are potentially available in
huge amounts from coal-hydrogenation oils. Such mixtures are composed
largely of homologous and isomeric phenols, and separation by the usual
procedures of precise fractional distillation and fractional crystalliza
tion is difficult. pH adjustment is used to increase the distribution
coefficients among groups of closely related phenols and thus separation
of isomeric phenols can be achieved.
Golumbic, et al. (42) studied the distribution of phenol between a
non-ionizing organic phase and an immiscible aqueous phase. Data for
distribution coefficient at different pH values are tabulated in Table
10. The results are shown in Figure 10. In Figure 10, the observed
distribution coefficient of 4 phenols, distributed in the system cyclo-
hexane - 0.5 m phosphate buffer, are plotted as a function of pH. A
straight line occurred in all cases. Three factors affect the distribu
tion of a phenol between the organic and aqueous phase: pH of the aque
ous phase, ionization constant of the phenol, and distribution coeffici
ent of the unionized phenol.
Golumbic, et al., derived the relationship between distribution co
efficient, pH and ionization constant of a weak acid, such as phenol be
tween a non-ionizing organic phase and an immiscible aqueous phase.
When no association occurs in either phase, the observed distribution
coefficient, K', is given as the resultant of two equilibria: (a) the
distribution of the undissociated acid between the immiscible phases
and (b) the ionization of the acid in the aqueous phase.
[HA]o

65
TABLE 10^
SHIFT OF DISTRIBUTION COEFFICIENTS WITH pH
System pH Log K
P-Cyclohexylhenol In Cyclohexane-Phosphate 11.67 1.07
11.92 0.78
12.55 -0.04
0-Ethylphenol In Cyclohexane-Phosphate 11.11 0.17
11,43 -0,17
11,92 -0.63
12,55 -1,23
2,5 - Xylenol In Cyclohexane-Phosphate 11,12 -0,03
11.43 -0.32
11.92 -0.80
12.30 -1,13
M-Cresol In Cyclohexane-Phosphate 11.11 -1.03
11,43 -1,34
12.30 -1,99
^Reference (42)

66
1.1
0.8
0.4
CO +J c CU
o • I - O)
M- ro CU x : O CL
CJ to o
C J C O CL.
• I - I 40 OJ 3 C
.a ro • I - X S - CU
4 J x : CO O
•O CJ CU > c CU CO CO
.a 1 — o o M - CU o sz a. E
sz 4-+J o •r—
s -(O cn o
0.0
-0.4
-0.8
i 12.4 12,6
Figure 10. Shift of K With pH

67
The ionization constant of the acid, k, is defined as
^ _ [H^lwrA'lw ^ [HAJw in water (57)
The distribution coefficient, K, of the unnonized acid is given as
K = I . . . •
HA w (58)
From Equation 58 [HA]o = K[HA]w, and Equation 56 becomes
K' = K ' [HA]w
([HA]w + [ A ' ] w ) (59)
or
K' = K
(1 + A-HA < ^ )
(60)
w
From Equation 57, [A']w/[HA]w = k/[H ]w, so Equation 60 can be
written as
K' = K
(1 + - ^ ) (61)
When the aqueous phase contains a buffer of sufficient alkalinity
so that [H ] « k. Equation 61 can be approximated by
^ k (62)
or
log K' = log K - pH + pK (63)
According to Equation 63, a plot of log K' versus pH would be a
straight line with a slope of -1. Experimental results, represented

68
graphically in Figure 10, verified this relation. This observation also
indicates that there was no significant association in the organic phase.
Temperature Effect on Distribution Coefficient
The distribution coefficient of a solute between two solvent layers
is closely related to the solubility of the solute in either of the sol
vents. In the condition in which the solute concentration is very low,
the distribution coefficient can be considered as a thermodynamic pro
perty.
From thermodynamics, the standard free energy of transfer of solute
in the distribution process is given by
AG^^ = Ail = RT In K^ (64)
With the assumption that the molar heat of transfer of solute be
tween two solvent layers is not temperature dependent over the range
studied, we get
d log K. = - ^ ^ dT (65)
^ 2.3 RT"
where AHs is the molar heat of transfer of solute between two solvents.
Equation 65 shows the expected manner for the distribution coeffi
cient to vary with temperature. When Equation 65 is integrated, we have
When log K^ is plotted against 1/T, a straight line with (-AHs/2.3 R)
as slope is expected.

69
From the Clausius-Clapeyron equation, at the same temperature,
d log P' = ^' p dT (67) 2.3 RT
If Equation 65 is divided by the Clausius-Clapeyron equation, always at
the same temperature, we get
' ^^g 'D . AHs I,.. d log P' " H' ( ^
• Assuming AHs/AH' to remain constant and integrating the above equa
tion, yields approximately
log Kj3 = -^ log P' + C (69)
where P' is vapor pressure of the reference liquid, H', the molar latent
heat of vaporization of the reference liquid, is assumed to remain nearly
constant.
It is obvious that the Othmer and Thaker plot (43), a plot of dis
tribution coefficient versus the vapor pressures of the reference liquid
at the same temperature, will give a straight line on logarithm paper.
The slope of the line, AHs/H' represents the ratio of the molar heat of
transfer for the solute from one solvent layer to the other to the molar
latent heat of vaporization of the reference liquid. Examples of Othmer
and Thaker plots for several systems are given in Table 11 and plotted
in Figures 11 to 14.
The plot of distribution coefficient versus the vapor pressure of
the reference liquid has a more practical advantage. First, the lines
so obtained are more nearly straight on the logarithmic plot than on
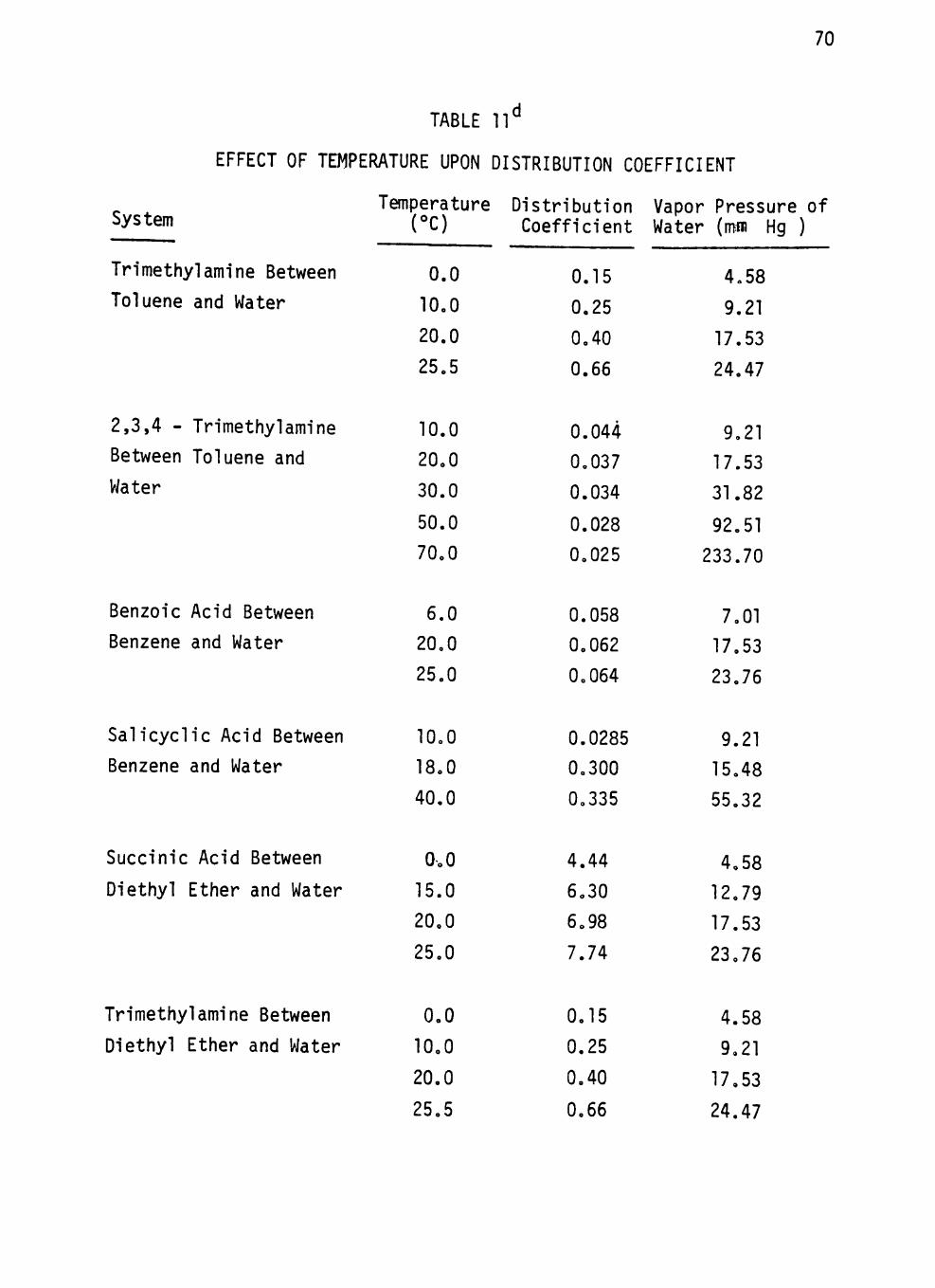
70
TABLE n ^
EFFECT OF TEMPERATURE UPON DISTRIBUTION COEFFICIENT
^ ^ Temperature Distribution Vapor Pressure of ^y^^^^ iX) Coefficient Water (mm Hg )
Trimethyl amine Between Toluene and Water
2,3,4 - Trimethylamine Between Toluene and Water
Benzoic Acid Between Benzene and Water
Salicyclic Acid Between Benzene and Water
Succinic Acid Between
Diethyl Ether and Water
Trimethylamine Between Diethyl Ether and Water
0.0
10,0
20.0
25,5
10.0
20,0
30.0
50.0
70,0
6.0 20,0
25,0
10,0
18.0
40.0
0.0
15.0
20,0
25,0
0.0
10.0
20.0
25,5
0.15
0.25
0.40
0.66
0.044 0.037
0.034
0.028 0,025
0.058 0,062
0,064
0.0285
0,300
0,335
4.44
6,30
6.98
7.74
0.15
0.25
0.40
0,66
4.58
9.21
17.53
24,47
9,21 17.53
31.82
92,51
233.70
7,01 17.53
23.76
9.21
15,48
55,32
4,58
12,79
17,53
23.76
4.58
9,21
17,53
24.47

71
TABLE 11^
EFFECT OF TEMPERATURE UPON DISTRIBUTION COEFFICIENT (CONTINUED)
Temperature Distribution Vapor Pressure of System •(°C) Coefficient Water (mm Hg.)
Oxalic Acid Between
Diethyl Ether and Water
Benzoic Acid Between
Chloroform and Water
Salicyclic Acid Between
Chloroform and Water
15.0 25,0
27.0
10,0
25,0
40.0
10.0
25.0
40,0
14,00
16.80 17,00
0.215
0,240
0.277
0,47
0,60
0.695
12.79 23,76 26,74
9,21 23,76
55,32
9,21
23,76
55.32
^Reference (39)

72
Temperature (°C)
20 30 40
CU +J ro
"T CU c CU
CO (U c
CO 4J £ (U
CJ
(U o CJ
3 j Q •r—
S . 4-> CO
0.1 • Trimethylamine
A 2,3,4 - trimethypyridine
0.01 100
Figure 11.
10 20 30 40 50 Vapor Pressure (mm Hg)
Effect Of Temperature Upon Distribution Coefficient
200 300 400

73
Temperature (°C)
10 18 20 25
0.4 -
s-CU
4-> ro 1
<U c CU N c CU
CO
c 1—1
T 3 •^ o
< vt-O
to +J
c (U • r -
o •r— H -<+-(U
o CJ
c: o
• r " 4-> 3
.a •r—
s.. + j CO
o
0,
0.
0.
0,
0.
0.
2
.1
08
.06
.04
02
0,01
•*r T—r
• Benzoic Acid
A Salicyclic Acid
± ± 10 20 30 40 50 60
Vapor Pressure Of Water (mm Hg)
Figure 12, Effect Of Temperature Upon Distribution Coefficient
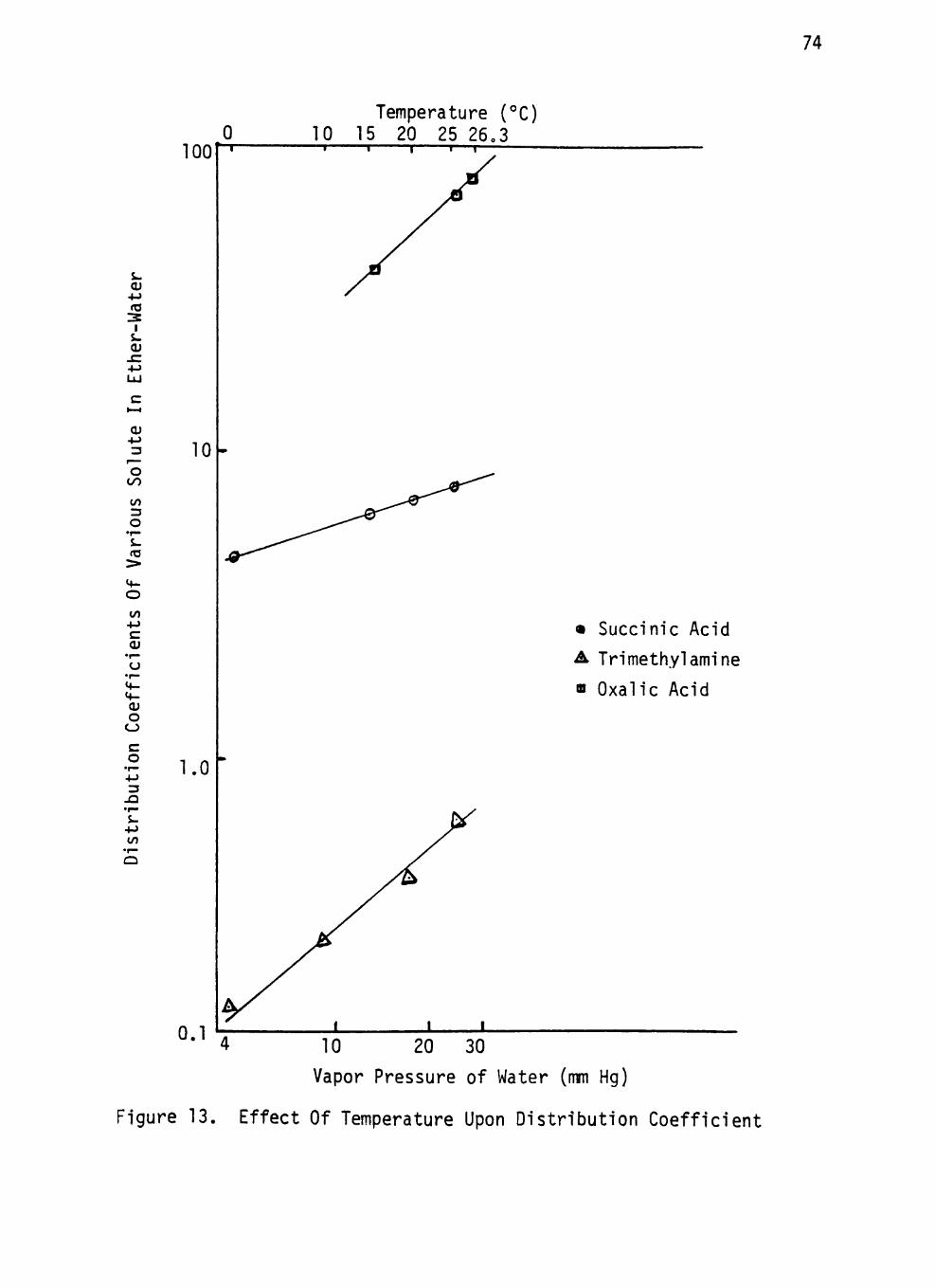
74
CU • M ro
CU
CU
3
'o
to 3 O
•r -&. ro
>
vv-O
to 4 J
CU • I —
CJ
CU o
CJ
c: o
3
•r-s -
4-) CO
o
100 0 10
10
1.0
0.1
Temperature (°C) 15 20 25 26,3
• Succinic Acid
A Trimethyl amine
n Oxalic Acid
10 20 30
Vapor Pressure of Water (mm Hg)
Figure 13. Effect Of Temperature Upon Distribution Coefficient

75
Temperature (°C)
CU 4-i ro
I
O M-O &. O
x: CJ
CO " O •r -CJ
<:
O
CO
CU
CJ
CU o
CJ
c o
3 .a s.
4-> CO
O
10
1.0
^ 25 40
—r
< Benzoic Acid
a Salicyclic Acid
5 10 30 50 70 90
Vapor Pressure Of Water (mm Hg)
Figure 14. Effect Of Temperature Upon Distribution Coefficient

76
the reciprocal temperature plot. AHs/H' is more nearly constant with a
variation of temperature than AHs alone, since the numerator and the de
nominator of the fractions are both decreasing with increasing tempera
ture. Secondly, using the known molar latent heat of vaporization of
reference liquid as standard, the amount of heat needed to transfer a
certain quantity of solute between two solvents can be estimated.
From Figure 11, the effect of temperature upon the distribution co
efficient of 2,3,4-trimethylpyridine between toluene and water in the
logarithmic plot can be represented by the equation:
log K^ = -0.17 log P'- 1.21 (70)
Assuming the molar latent heat for pure water to be 10,330 cal. per mole
at the mean temperature of the range (40°C), -1756 cal. per mole would
be required for the molar heat of transfer of 2,3,4-trimethylpyridine as
estimated from the slope of the logarithmic plot. Therefore, l i t t l e
difficulty should be encountered in maintaining practically constant
temperature in an extraction column.
Effect of Concentration on Distribution Coefficient
Effect of concentration on distribution coefficient is usually not
yery marked. The distribution coefficient tends to reach a constant
value at high dilutions. Data from Carpenter, et a l . (44) indicate the
variation of distribution of several acids with change of concentration
(Table 12). The results are represented graphically in Figure 15. The
shift of distribution coefficient with change of concentration behaves
differently among the acids. The decrease of the distribution

77
•a:
CJ
o CJ
CU CM
OQ ?a:
in Q t—(
CJ < CD
O CC I — C>0
o lyO
I — UJ l-H CJ
UJ
o CJ
o
o • r —
o
o o s..
o <:
o •r—
o in CM
vo cn CM
CO vo CO
cn
CM
CO 00 CM
00 00 CM
o o o m en o I— I CM CO ro CO
CO vo CO
u r*. CO
CO cn 00 r- CM CO "5J- «?r *;r
o cn '^
CO
vo * ; * •
O r>>. ^
in r-* «:3-
00 00 «;r
OQ 1—4
ai f-co
o
C>0
< :
o
o
>^
zc
(J <:
u &. o
CJ &-CU
a.
cn CM CO
00 CO CO
o cn CO
CO CO CO
O r>*. CO
CO r»« CO
00 CO
00 CO CO
vo CT» CO
o o
o cn in
o CM vo
o vo vo
o cn vo
o
r>. ,
o
o o cn r^
o vo cn O
ro f— S- ro •M E e s. CU o u z o CJ
o
o CM • o
CO
C3
*a-O
cn o
vo o
o
r •
o
CO
o cn •
o
o 0
^ «!»•
CJ S= CU
CU c*-CU
oc CU
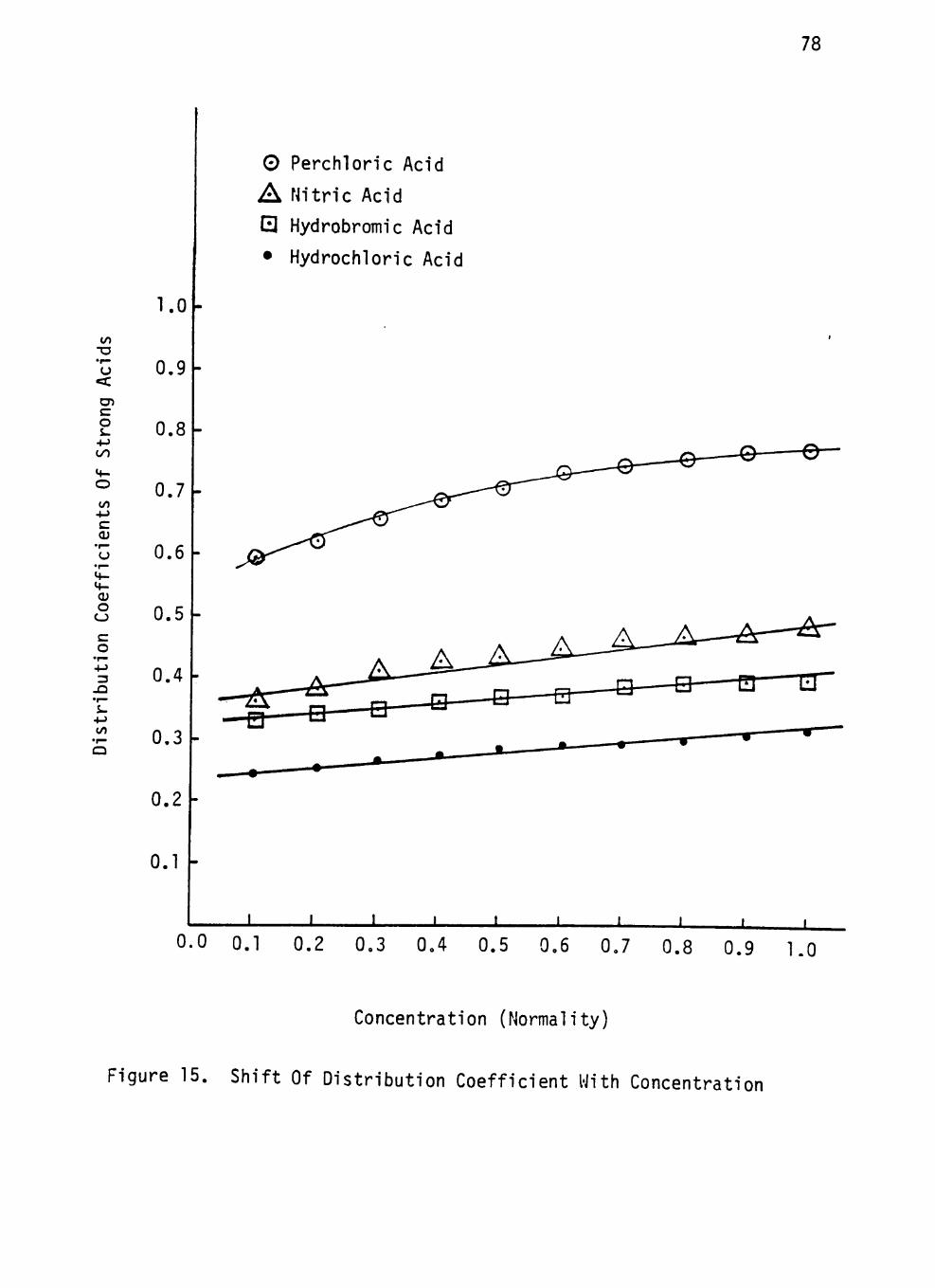
78
CO • o •r -
U
cn c o s.
4 J LT) Vf-O CO
+ J c CU
•r-CJ
^
CU O
CJ
e o
•r— +J 3
JZX •r— S-
^ - > CO
o
1,0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0 Perchloric Acid
A Nitric Acid
Q Hydrobromic Acid
• Hydrochloric Acid
0.0 0.1 0.2 0.3 0.4 0.5 0,6 0.7 0.8 0.9 1.0
Concentration (Normality)
Figure 15. Shift Of Distribution Coefficient With Concentrati on

79
coefficients with dilution becomes increasingly greater from hydrobromic,
to nitric, to perchloric acids. The amount and rate of change appears
to be correlated with the size of the distribution coefficients of these
aci ds.
Discussion
Variation of liquid-liquid distribution coefficients with solute
concentration in dilute region is usually small and can be neglected.
In a system with a highly concentrated solution of polar compounds, the
composition of the solute in the equilibrium phases is a complex func
tion of association, dissociation and other processes. Therefore, it
is not feasible to correlate liquid-liquid distribution coefficients at
high concentration.
For ionizable solutes, liquid-liquid distribution coefficients vary
with pH. Generally, the degree to which the liquid-liquid distribution
coefficients are shifted with pH is not linear. Also, the direction of
the change in distribution coefficients depends on the nature of the
solutes. Therefore, it is not possible to predict the effect of pH on
distribution coefficients except by experimental determinations.
Temperature has a marked effect on liquid-liquid distribution coef
ficients. At infinite dilution, with the assumption that the standard
molar enthalpy change for the transfer process is not temperature de
pendent in the range studied, it is possible to estimate the distribu
tion coefficients at different temperature from
, " Dl _ -AH , 1 1 X ,^.^ Np2 K 2 'l

80
However, the predicted liquid-liquid distribution coefficients are
not precise because the solvent molar volume and the solvents miscibili-
ties vary with temperature. Also, the necessary enthalpies of transfer,
AH, are rarely available.
The present status of the new model is that it is not capable of
correlating liquid-liquid distribution coefficients at temperatures other
than 25°C. There are insufficient data to establish a general expression
to account for the temperature dependence of distribution coefficients.
Most of published liquid-liquid equilibrium data are within 10°C of room
temperature. In order to extrapolare liquid-liquid distribution coeffi
cients with temperature, we need an improved model with temperature para
meters and more liquid-liquid distribution coefficients outside this
temperature range.

CHAPTER VI
CONCLUSIONS AND RECOMMENDATIONS FOR FUTURE STUDIES
Summary of the Research
1. A group contribution method was used in estimating the liquid-
liquid distribution coefficients of solute-octanol-water, solute-
diethyl ether-water, solute-chloroform-water and solute-benzene-
water system.
2. The properties of liquid-liquid distribution coefficients of
homologues were related to the number of carbon atoms in the
hydrocarbon chain.
3. The effect of temperature, pH and solute concentration on liquid-
liquid distribution coefficients were studied.
Conclusions
From the results of study, the following conclusions can be drawn:
1. The group contribution method proposed by this study provides
a rapid means to estimate distribution coefficients which are
difficult to obtain experimentally and select proper solvents.
The advantage of this method is that no physical properties are
required and distribution coefficients can be calculated from
structure considerations alone.
2. The log K versus number of carbon atoms plots give very good
correlation coefficients and are useful for predicting the dis
tribution coefficients of certain compounds.
3. Variation of distribution coefficients with solute concentration
is usually small. Distribution coefficients tend to reach a
81

82
constant value at high dilutions. In some cases, the shift of
distribution coefficients with solute concentration is related
to association in one or both of the phases.
4. Often, the effect of temperature is not large when consideration
is restricted to a moderate temperature range. The molar heat of
transfer of solute can be estimated from the slope of the plot of
distribution coefficient versus the vapor pressure of the refer
ence liquid, provided that the molar latent heat of vaporization
of the reference liquid is known.
5. For ionizable solutes, distribution coefficients vary with pH.
Generally, the degree to which the distribution coefficients
are shifted with change in pH is not linear. pH adjustment is
useful in increasing the distribution coefficients among groups
of closed related compounds and thus separation of isomeric
compounds can be achieved.
Recommendation for Future Studies
For better prediction of distribution coefficient, it is recommended
that:
1. The liquid-liquid distribution coefficients of other systems and
outside the room temperature range need to be collected in order
to correlate the distribution coefficients to other solvent
systems and at other temperature.
2. More research should be done to understand the effect of inter-
molecular interaction of the liquid-liquid interphase on liquid-
liquid distribution coefficients.

LIST OF REFERENCES
1. Henley, E. J., and Staffin, K., Stagewise Process Design, John Wiley and Sons, New York, 53 (197^)7
2. Souders, M.. Jr., Matthews, C. S. and Hurd, C. 0., "Relationship of Thermodynamic Properties to Molecular Structure," Ind. Eng. Chem., 41' 1037-1047 (1949).
3. Craig, L. C., "Identification of Small Amounts of Organic Compounds by Distribution Studies," J. Biol. Chem., 155, 519-534 (1944).
4. Davies, J. T., Rideal, E. K., Interfacial Phenomena, Academic Press, New York, 343 (1961).
5. Bait, S., and Vandaler, E., "Extraction Dissociation Constants of the Carbazine Complexes," Anal. Chim. Acta., 30, 434-442 (1964).
6. Moelwyn-Hughes, S., Physical Chemistry, 2nd Edition, Pergamon Press, New York, 1077-1085 (1961).
7. Green, R., and Alexander, P., "Schiff Base Equilibria," Aust. J. Chem., 2^, 329-336 (1965).
8. Berthelot and Jungfleisch, "Sur Les Chlorures D'Acetylene Et Sur La Synthese du Chlorure de Jul in," Ann. Chim. Phys., 4 , 26-30 (1872).
9. Nernst, W., "Verteilung eines stoffes zweischen zwei ISsungsmitteln und Zwischen Lbsungsmittel und Dampftaum," Z. Physik. Chem., 8, 110-139 (1891).
10. Lacroix, S., "Separation Du Gallium Par Extraction Par Le Chloro-forme," Anal. Chim. Acta., 1, 260-267 (1947).
11. Treybal, R. E., Liquid Extraction, 2nd Edition, McGraw-Hill, New York, 64-76 (19281T
12. Grahame, D. C., and Seaborg, G. T., "The Distribution of Minute Amounts of Material Between Liquid Phases," J. Am. Chem. Soc, 60, 2524-2528 (1938).
13. Pierotti, G. J., Deal, C. H. and Derr, E. L., "Activity Coefficients and Molecular Structure," Ind. Eng. Chem., 51, 95-102 (1959).
14. Hildebrand, J. H., and Scott, R. L., The Solubility of Nonelec-trolytes, Reinhold, New York (1950).
83

84
15. Tsuboka, T., and Katayame, T., "Correlations Based on Local Fraction Model between New Excess Gibbs Energy Equations," J. Chem. Eng. Japan, 8 , 404-418 (1975).
16. Renon, H., and Prausnitz, J. M., "Local Compositions in Thermodynamic Excess Functions for Liquid Mixtures," A.I.Ch.E. J., 14, 135-144 (1968). —
17. Scott, R. L., "Corresponding States Treatment of Nonelectrolyte Solutions," J. Chem. Phys., ^ , 193-205 (1956).
18. Abrams, D. S., and Prausnitz, J. M., "Statistical Thermodynamics of Liquid Mixtures: A New Expression for the Excess Gibbs Energy of Partly or Completely Miscible Systems," A.I.Ch.E. J., 21. 116-128 (1975). —
19. Anderson, T. F., and Prausnitz, J. M., "Application of the UNIQUAC Equation to Calculation of Multicomponent Phase Equilibria," Ind. Eng. Chem., 15, 561-567 (1978).
20. Sorensen, J. M., Magnussen, T., Rasmussen, P. and Fredenslund, Aa., "Liquid-Liquid Equilibrium Data: Their Retrieval, Correlation and Prediction. Part II: Correlation," Fluid Phase Equilibria, 1, 47-82 (1978).
21. Wilson, G. M., and Deal, C. H., "Activity Coefficients and Molecular Structure," Ind. Eng. Chem. Fundamentals, 1, 20-33 (1962).
22. Scheller, W. A., "Group Contributions To Activity Coefficients," Ind. Eng. Chem. Fundamentals, 4 , 459-462 (1965).
23. Rateliff, G. A., and Chao, K. C , "Prediction of Thermodynamic Properties of Polar Mixtures by a Group Solution Model," Can. J. Chem. Eng., 47, 148-153 (1969).
24. Derr, E. L., and Deal, C. H., "Group Contributions in Mixtures," Ind. Eng. Chem., 60, 28-38 (1968).
25. Fredenslund, A., Jones, R. L., and Prausnitz, J. M., "Group-Contribution Estimation of Activity Coefficients in Nonideal Liquid Mixtures," A.I.Ch.E. J., 21, 1086-1975 (1975).
26. Tochigi, K., and Kojima, J., "Prediction of Liquid-Liquid Equilibria by an Analytical Solutions of Groups," J. Chem. Eng. Japan, 10, 61-63 (1977).
27. Sugi, H., and Katayama, T., "Liquid-Liquid Equilibrium Data for Three Ternary Systems of Aqueous Alcohol Solutions and Applicability of the Analytical Solutions of Groups," J. Chem. Eng. Japan, 10, 400-402 (1977).

85
28. Fredenslund, Aa., Gmehling, J., and Rasmussen, P., Vapor-Liquid Equilibria Using UNIFAC, Elsevier, Amsterdam (1977).
29. Magnussen, T., Sorensen, J. M., Rasmussen, P., and Fredenslund, Aa., "Liquid-Liquid Equilibrium Data: Their Retrieval, Correlation and Prediction. Part III: Prediction," Fluid Phase Equilibria,, 4, 151-163 (1979). ^
30. Korenman, I. M., and Others, "Extraction of Homologues," Zhur. Fiz. Khim.. IE, 177-178 (1974).
31. Abramzon, A. A., and Ovchatova, A. D., "Relation of the Partition Coefficients of Amines in Liquid-Liquid Systems to Molecular Structure," Zhur. Obs. Khim., £, 1347-1350 (1974).
32. Nys, G. C , and Rekker, R. F., "Statistical Analysis of a Series of Partition Coefficients with Special Reference to the Predictability of Folding of Drug Molecules," Chim. Therap., 8, 521-535 (1973). -
33. Snyder, L. R., "Solutions to Solution Problems," Chem. Tech., 10, 191-194 (1980). •—
34. Cratin, P. D., "Partitioning at the Liquid-Liquid Interface," Ind. Eng. Chem., 60, 14-19 (1968).
35. Everett, D. M., Chemical Thermodynamics, Logmans, London, 54-90 (1959).
36. Seidell, A., Solubility of Organic Compounds, Vol. II, 3rd Edition, Van Nostrand, Princeton, New Jersey (1941).
37. Col lander, R., "Die Verteilung Organischer Verbindungen Zwischen Ather und Wasser," Acta Chem. Scand., 3, 717-747 (1949).
38. Metzsch, F. Von., "Wahl der Losungsmittel fur die Verteilung Zwischen Zwei Flussigen Phasen," Angew. Chem., 65 , 586-598 (1953).
39. Washburn, E. W., International Critical Tables of Numerical Data, Physics, Chemistry and Technology," Vol. Ill, McGraw-Hill, New York (1928).
40. Korenman, I. M., and Dobromyslova, T. M., "Distribution of Cello-solves between Organic Solvents and Water," Zh. Prikl. Khim., 48, 2711-2714 (1975).
41. Colaizzi, J., and Klink, P., "pH-Partition Behavior of Tetracyclines," J. Pharm. Sci., 58, 1184-1189 (1969).

86
42. Golumbic, C , Orchin, M., and Weller, S., "Relationship between Partition Coefficient and Ionization Constant," J. Amer. Soc., 71, 2624-2627 (1949).
43. Othmer, D. F., and Thaker, M. S., "Correlating Solubility and Distribution Coefficient Data," Ind. Eng. Chem., 44, 1654-1656 (1952).
44. Carpenter, F. H., McGregor, W. H., and Close, J. A., "A Relationship Between Charge and Distribution Constants of Compounds," J. Amer. Soc, 81, 849-855 (1959).
45. Archibald, R., "Distribution of Acids Between Water and Several Immiscible Solvents," J. Am. Chem. Soc, 54, 3178-3185 (1932).
46. Tanaka, M., and Kojima, I., "Synergic Extraction of Vandium 8-Quinolinolate in the Presence of Alcohol, as Explained by the Esterification," J. Inorg. Nucl. Chem., 2^, 1769-1797 (1967).
47. Herz, W., and Stanner, E., "Uber Verteilungskoeffizienten und ihre Beeinflussung durch Salzzusatze," Z. Physik. Chem., 128, 399-411 (1927).
48. Hantzsch, A., and Sebalt, F., "Veber den Zustand Wasseriger Ammoniak und Aminlbsungen," Z. Physik. Chem., 30, 258-299 (1899).
49. Hutchinson, E., "Diffusion Across Oil-Water Interfaces," J. Phys. Chem., 52, 897-907 (1948).
50. Greenfield, B., and Hardy, C , "Studies on Mono- and Di-n-butylphosphoric Acids," J. Inorg. Nucl. Chem., 21, 359-365 (1961).
51. Korenman, I., Gurevich, N. and Kulagina, T., "Extraction of Pri-marv Aliphatic Amines at Different Temperatures," Russ. J. Phys. Chem., 46, 1523EE (1972).
52. Brown, F., and Bury, C , "The Distribution of Normal Fatty Acids between Water and Benzene," J. Chem. Soc, 123, 2430-2434 (1923).
53. Traube, J., "Theorie der Osmose und Narkose," Archiv. Fur die Gesammte Physiologie, 105, 541-558 (1904).
54. Meeussen, E., and Huyskens, P., "'Etude De La Structure Du Butanol-n En Solution Par Les Coefficients De Partage," J. Chim. Phys., 62, 845-854 (1966).
55. Korenman, I. M., and Dobromyslova, T. M., "Distribution of Cello-solves between Organic Solvents and Water," Zh. Prikl. Khim., 48, 2711-2714 (1975).

87
56. Rydberg, J., Svensk, T., "The Distribution of Acetylacetone between Benzene and Water," Kern. Tid., 62, 179-184 (1950).
57. Dyrssen, D., "Studies on the Extraction of Metal Complexes," Acta. Chem. Scand., U , 1771-1786 (1957).
58. Pinnow, J., "Verteilungs Koeffizient und Extraktionsgeschwin-digkeit einger Organischer SSuren," Z. Anal. Chem., 54, 321-345 (1915).
59. Dermer, 0., "Partition Ratios of Some Organic Acids between Water and Ethyl Ether," J. Am. Chem. Soc, 63_, 3524-3525 (1941).
60. Behrens, W., "Quantitative Analyse von Gemischen flUchtiger Fettsauren durch Verteilung Zwischen Athyeather und Wasser," Z. Anal. Chem., 69., 97-107 (1926).
61. Dermer, 0. and Dermer, V., "Identification of Organic Acids by Partition between Ethyl Ether and Hater," J. Am. Chem. Soc., 65, 1653-1656 (1943).
62. Chandler, E., "The Ionization Constants of the Second Hydrogen Ion of Dibasic Acids," J. Am. Chem. Soc., 30, 696-713 (1908).
63. Lindberg, J., "The Relation between the Distribution in Ether and Water and the Infrared Spectrum of Several Guaiacyl and Syringyl Compounds," Chem. Abs., 54, 14877 (1960).
64. Meyer, K., and Hemmi, H., "Beitrage zur Theorie der Narkose," Biochem. Z., 277, 39-76 (1935).
65. Thies, H., and Ermer, E., "Verteilungskoeffizienten und Konsti-tution bei Weckaminen und Verwandten Arylalky-laminderivaten," Naturwissen., 49, 37-40 (1962).
66. Ross, E., "The Transfer of Non-Electrolytes across the Blood-Aqueous Barrier," J. Physiol., 112, 299-237 (1951).
67. Collander, R., "The Permeability of Nitella Cells to Non-electrolytes," Phys. Plant., 7., 420-445 (1954).
68 Bowen, C , "Distribution of Anabasine between Certain Organic Solvents and Water," Ind. Eng. Chem., 41, 1295-1296 (1949).
69. Smith, H., "The Nature of Secondary Valence," J. Phys. Chem., 25 , 204-263 (1921).
70 Gier, T., and Hougen, J., "Concentration Gradients in Spray and Packed Extraction Column," Ind. Eng. Chem., 45, 1362-1370 (1953).

88
71. Moore, T., and Winmill, T., "The State of Amines in Aqueous Solution," J. Chem. Soc, 101, 1635-1676 (1912).
72. Sandell, K., "Uber das Wasserstoffbindungsvermbgen von Sauren," Naturwissen., 51, 336-338 (1964).
73. Ivanov, B., and Makeikina, V., "Distribution Coefficients in the Systems Water-Phenol-Organic Solvent," Chem. Abs., 62, 15896 B (1965). ~
74. Herz, W., and Rathman, W., "Anwendungen des Verteilungssatzes," Z. Electrochem., 1^, 552-553 (1913).
75. Mayer, S., Maickel, R., and Brodie, B., "Kinetics of Penetration of Drugs and Other Foreign Compounds into Cerebrospinal Fluid and Brain," J. Pharm. and Expl. Ther., 127, 205-211 (1959).
76. Korenman, I., and Chernorukova, Z., "Distribution of Normal Alcohols between Organic Solvents and Water," Zh. Prikl. Khim., 47, 2595-2597 (1974). "~
77. Sandell, K., "Uber das Wasserstoff-Bindungsvermbgen und die Struktur von Thioessigsaure und Thioacetamid," Naturwissen., 53, 330-331 (1966).
78. Sandell, K., "Verteilung Aliphatischer Amine Zwischen Wasser und Mischungen von Organischen Lbsungsmitteln," Naturwissen., 49, 12-14 (1962).
79. Smith, H., and White, T., "The Distribution Ratios of Some Organic Acids between Water and Organic Liquids," J. Phys. Chem., 33, 1953-1974 (1929).
80. Knuosen, L., and Grove, D., "A Graphic Method of Studying the Separation of Mixtures bv Immiscible Solvents," Ind. Eno. Chem. Anal. Ed., ]±, 556-557, "(1942).
81. Marvel, C , and Richards, J., "Separation of Polybasic Acids by Fractional Extraction," J. Anal. Chem., 21, 1480-1482 (1949).
82. Gordon, N., and Reid, E., "The Solubility of Liquids in Liquids," J. Phys. Chem., ^ , 773-789 (1922).
83. Carstensen, H., "Separation of Corticusteroids by Countercurrent Distribution," Acta Chem. Scand., £, 1026-1028 (1955).
84. Stevancevic, D., and Antonijevic, V., "Extraction of Copper and Uranium with 2-Methyl-2,4-heptane-dione and 2,4-hexane-dione," Chem. Ab., 63, 17215 (1965).

89
85. Marden, J . , "A Study of the Methods for Extractions by means of Immiscible Solvents from the Point of View of the Distribution Coefficients," Ind. Eng. Chem., i , 315-320 (1914).
86. Stary, I . , and Rudenko, N., "The Dissociation Constant of Benzoyl acetone and its Distribution Coefficients between Some Organic Solvents and the Aqueous Phase," Chem. Ab., S3_, 5828 (1959).
87. Farmer, R., and Warth, F., "The Affinity Constants of Aniline and i ts Derivatives," J. Chem. Soc , 85, 1713-1715 (1904).
88. Greene, R. and Black, A., "The Preparation of Pure d-Riboflavin from Natural Sources," J. Am. Chem. Soc , 5^, 1820-1822 (1937).
89. Huq, A., and Lodhi, S. , "Distribution of Benzoic Acid between Benzene and Water and Dimerization of Benzoic Acid in Benzene," J. Phys. Chem., 70, 1354-1364 (1966).
90. Williams, G., and Soper, F., "The Ionization Constants of Some Chloro and Nitro-anilines by the Partition Method," J. Chem. Soc., 132, 2469-2474 (1930).
91. Schilow, N., and Lepin, L., "Studien liber die Verteilung von Stoffen Zwischen Zwei Lbsungsmitteln," Z. Physik. Chem., 101, 353-402 (1922).
92. Korenman, I . , and Graznova, M., "Distribution of 3-Diketones between Organic Solvents and Water," Zh. Anal. Khim., 29., 964-965 (1974).
93. Kuznetsova, E., "Theory of the Extraction of Organic Molecules with a High Dipole Moment into a Non-polar Solvent," Zh. Fizich. Khim., 49, 408 (1975).
94. Collander, R., "The Partition of Organic Compounds between Higher Alcohols and Water," Acta Chem. Scand., 5., 774-780 (1951).
95. Dillingham, E. 0 . , Mast, R. W., Bass, G. E., and Autian, J . , "Toxicity of Methyl and Halogen-Substituted Alcohols in Tissue Culture Relative to Structure-Activity Models and Acute Toxicity in Mice," J. Pharm. Sc i . , 62., 22-30 (1973).
96. Rogers, K., and Cammarata, A., "Superdelocalizability and Charge Density," J. Med. Chem., U, 692-694 (1969).
97. Hansch, C , and Anderson, S. , "The Effect of Intramolecular Hydrophobic Bonding on Partition Coefficients," J. Org. Chem., 32, 2583-2586 (1967).
98. Kurihara, N., and Fuji ta, T . , "Studies on BHC Isomers and Related ' mpounds," Pest. Biochem. and Physiol., 2, 383-390 (1973).
99. Korenman, Y., "Phenol Solvates in Organic Solvents," Russ. J. Phys. Chem., 46, 42-43 (1972).

NOMENCLATURE
a = number of occurrences
A = interaction parameter
B = total quantity of B
B^ = quantity of solute B in extract phase
Bf = quantity of solute B in raffinate phase
D = distribution ratio
F = quantity of feed
G = free energy
G = excess Gibbs energy
G.. = coefficient of NRTL equation
H = enthalpy
H' = molar latent of vaporization
H^ = molar heat of transfer
k = ionization constant
K' = observed liquid-liquid distribution coefficient
K = liquid-liquid distribution coefficient (molar basis)
Kp = liquid-liquid distribution coefficient (mass basis)
m = molarity
n = number
0 = non-aqueous phase
P' = vapor pressure
r = ratio of solute groups to the total number of groups
R = universal gas constant
R' = quantity of carrier solvent
90

91
S
T
V
W
x
X,Y
Greek
a
3
Y
U
r
9
0
V
A
=
=
=
s
=
=
quantity of solvent
absolute temperature
volume
aqueous phase
mole fraction
composition
Symbols
=
=
=
=
=
=
=
=
=
Subscripts
selectivity
non-randomness param
activity coefficient
chemical potential
group contribution
area fraction
volume fraction
number
difference
1,2,3, = components
a,c,i,j,k,l,n,m,p = components
E = extract phase
H
L
R
s
t
s
=
=
=
=
hydrophilic group
lipophilic group
raffinate phase
solvent
total

92
Superscripts
^»II = phases
^ = combinatorial
L = part associated with the energy required to transfer solute through liquid-liquid interface
^ = residue
T = part associated with the differences in free energy of the solute between the two liquid phases
Related Documents











