Genomics of the Periinfarction Cortex After Focal Cerebral Ischemia *Aigang Lu, *Yang Tang, *Ruiqiong Ran, *Joseph F. Clark, †Bruce J. Aronow, and *Frank R. Sharp *Departments of Neurology, Pediatrics and the Neurosciences Program, University of Cincinnati, and †Division of Molecular Developmental Biology and Informatics, Children’s Hospital Research Foundation, Cincinnati, Ohio, U.S.A. Summary: Understanding transcriptional changes in brain af- ter ischemia may provide therapeutic targets for treating stroke and promoting recovery. To study these changes on a genomic scale, oligonucleotide arrays were used to assess RNA samples from periinfarction cortex of adult Sprague-Dawley rats 24 h after permanent middle cerebral artery occlusions. Of the 328 regulated transcripts in ischemia compared with sham-operated animals, 264 were upregulated, 64 were downregulated, and 163 (49.7%) had not been reported in stroke. Of the functional groups modulated by ischemia: G-protein–related genes were the least reported; and cytokines, chemokines, stress proteins, and cell adhesion and immune molecules were the most highly expressed. Quantitative reverse transcription polymerase chain reaction of 20 selected genes at 2, 4, and 24 h after ischemia showed early upregulated genes (2 h) including Narp, Rad, G33A, HYCP2, Pim-3, Cpg21, JAK2, CELF, Tenascin, and DAF. Late upregulated genes (24 h) included Cathepsin C, Cip-26, Cystatin B, PHAS-I, TBFII, Spr, PRG1, and LPS- binding protein. Glycerol 3-phosphate dehydrogenase, which is involved in mitochondrial reoxidation of glycolysis derived NADH, was regulated more than 60-fold. Plasticity-related transcripts were regulated, including Narp, agrin, and Cpg21. A newly reported lung pathway was also regulated in ischemic brain: C/EBP induction of Egr-1 (NGFI-A) with downstream induction of PAI-1, VEGF, ICAM, IL1, and MIP1. Genes regu- lated acutely after stroke may modulate cell survival and death; also, late regulated genes may be related to tissue repair and functional recovery. Key Words: DNA microarrays— Functional genomics—Cerebral ischemia—Gene expression— Glycerol 3-phosphate dehydrogenase—egr-1—Cathepsins. Recent clinical trials of neuroprotective drugs for the acute treatment of stroke have failed. These included trials of sodium and calcium channel antagonists, N- methyl-D-aspartate receptor antagonists, -amino-butyric acid agonists, free radical scavengers, nitric oxide path- way modulators, blockers of adhesion molecules, and other drug classes (De Keyser et al., 1999; Barber et al., 2001; Albers et al., 2001; Fisher and Schaebitz, 2000). In spite of these failures, there is still optimism that phar- macologic approaches can be developed to treat acute stroke or enhance recovery (White et al., 2000). One new approach to search for targets is to perform genomic studies at different times after stroke to try to identify time-specific gene pathways or gene clusters related to specific injury or recovery processes after stroke. DNA microarrays can assay thousands of transcripts in a single sample (Noordewier and Warren, 2001). The first brain ischemia study used custom-designed 750 gene arrays to examine RNA changes in cortex and stria- tum 3 h after focal ischemia (Soriano et al., 2000). Of the 24 genes regulated more than twofold, most were imme- diate early genes such as c-fos, NGFI-A, NGFI-C, Krox- 20, and Arc (Soriano et al., 2000). Subsequent studies used oligodeoxynucleotide-based or complementary DNA (cDNA) microarrays to study RNA expression in hippocampus of rats subjected to transient global ische- mia (Jin et al., 2001) and in cortex of rats at 6 h or 10 days after focal ischemia (Kim et al., 2002; Keyvani et al., 2002). A recent study combined cDNA array analysis of 74 genes with brain metabolic status studied using positron emission tomography scanning in a baboon fo- cal cerebral ischemia model. A change in the pattern of gene expression when the cerebral metabolic rate for Received November 22, 2002; final version received January 16, 2003; accepted January 20, 2003. This study was supported by NIH grants NS28167, NS38743, NS42774, NS43252, AG19561 and a Bugher award from the American Heart Association. Address correspondence to Dr. Aigang Lu, Department of Neurol- ogy and the Neurosciences Program, University of Cincinnati, Vontz Center for Molecular Studies, Room 2327, 3125 Eden Avenue, Cin- cinnati, OH 45267-0536, U.S.A.; e-mail: [email protected] Journal of Cerebral Blood Flow & Metabolism 23:786–810 © 2003 The International Society for Cerebral Blood Flow and Metabolism Published by Lippincott Williams & Wilkins, Inc., Baltimore 786 DOI: 10.1097/01.WCB.0000062340.80057.06

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Genomics of the Periinfarction Cortex After FocalCerebral Ischemia
*Aigang Lu, *Yang Tang, *Ruiqiong Ran, *Joseph F. Clark, †Bruce J. Aronow, and*Frank R. Sharp
*Departments of Neurology, Pediatrics and the Neurosciences Program, University of Cincinnati, and †Division of MolecularDevelopmental Biology and Informatics, Children’s Hospital Research Foundation, Cincinnati, Ohio, U.S.A.
Summary: Understanding transcriptional changes in brain af-ter ischemia may provide therapeutic targets for treating strokeand promoting recovery. To study these changes on a genomicscale, oligonucleotide arrays were used to assess RNA samplesfrom periinfarction cortex of adult Sprague-Dawley rats 24 hafter permanent middle cerebral artery occlusions. Of the 328regulated transcripts in ischemia compared with sham-operatedanimals, 264 were upregulated, 64 were downregulated, and163 (49.7%) had not been reported in stroke. Of the functionalgroups modulated by ischemia: G-protein–related genes werethe least reported; and cytokines, chemokines, stress proteins,and cell adhesion and immune molecules were the most highlyexpressed. Quantitative reverse transcription polymerase chainreaction of 20 selected genes at 2, 4, and 24 h after ischemiashowed early upregulated genes (2 h) including Narp, Rad,
G33A, HYCP2, Pim-3, Cpg21, JAK2, CELF, Tenascin, andDAF. Late upregulated genes (24 h) included Cathepsin C,Cip-26, Cystatin B, PHAS-I, TBFII, Spr, PRG1, and LPS-binding protein. Glycerol 3-phosphate dehydrogenase, which isinvolved in mitochondrial reoxidation of glycolysis derivedNADH, was regulated more than 60-fold. Plasticity-relatedtranscripts were regulated, including Narp, agrin, and Cpg21.A newly reported lung pathway was also regulated in ischemicbrain: C/EBP induction of Egr-1 (NGFI-A) with downstreaminduction of PAI-1, VEGF, ICAM, IL1, and MIP1. Genes regu-lated acutely after stroke may modulate cell survival and death;also, late regulated genes may be related to tissue repair andfunctional recovery. Key Words: DNA microarrays—Functional genomics—Cerebral ischemia—Gene expression—Glycerol 3-phosphate dehydrogenase—egr-1—Cathepsins.
Recent clinical trials of neuroprotective drugs for theacute treatment of stroke have failed. These includedtrials of sodium and calcium channel antagonists, N-methyl-D-aspartate receptor antagonists, �-amino-butyricacid agonists, free radical scavengers, nitric oxide path-way modulators, blockers of adhesion molecules, andother drug classes (De Keyser et al., 1999; Barber et al.,2001; Albers et al., 2001; Fisher and Schaebitz, 2000). Inspite of these failures, there is still optimism that phar-macologic approaches can be developed to treat acutestroke or enhance recovery (White et al., 2000). One newapproach to search for targets is to perform genomic
studies at different times after stroke to try to identifytime-specific gene pathways or gene clusters related tospecific injury or recovery processes after stroke.
DNA microarrays can assay thousands of transcriptsin a single sample (Noordewier and Warren, 2001). Thefirst brain ischemia study used custom-designed 750gene arrays to examine RNA changes in cortex and stria-tum 3 h after focal ischemia (Soriano et al., 2000). Of the24 genes regulated more than twofold, most were imme-diate early genes such as c-fos, NGFI-A, NGFI-C, Krox-20, and Arc (Soriano et al., 2000). Subsequent studiesused oligodeoxynucleotide-based or complementaryDNA (cDNA) microarrays to study RNA expression inhippocampus of rats subjected to transient global ische-mia (Jin et al., 2001) and in cortex of rats at 6 h or 10days after focal ischemia (Kim et al., 2002; Keyvani etal., 2002). A recent study combined cDNA array analysisof 74 genes with brain metabolic status studied usingpositron emission tomography scanning in a baboon fo-cal cerebral ischemia model. A change in the pattern ofgene expression when the cerebral metabolic rate for
Received November 22, 2002; final version received January 16,2003; accepted January 20, 2003.
This study was supported by NIH grants NS28167, NS38743,NS42774, NS43252, AG19561 and a Bugher award from the AmericanHeart Association.
Address correspondence to Dr. Aigang Lu, Department of Neurol-ogy and the Neurosciences Program, University of Cincinnati, VontzCenter for Molecular Studies, Room 2327, 3125 Eden Avenue, Cin-cinnati, OH 45267-0536, U.S.A.; e-mail: [email protected]
Journal of Cerebral Blood Flow & Metabolism23:786–810 © 2003 The International Society for Cerebral Blood Flow and MetabolismPublished by Lippincott Williams & Wilkins, Inc., Baltimore
786 DOI: 10.1097/01.WCB.0000062340.80057.06

oxygen was reduced by 48% to 66% was suggested toserve as a molecular definition of the penumbra (Chu-quet et al., 2002).
The present study used rat Affymetrix U34A oligo-nucleotide arrays to assess 8,740 transcripts in the peri-infarction cerebral cortex at 24 h after permanent middlecerebral artery (MCA) occlusions in adult rats. Usingvery strict criteria, less than 4% of these transcripts wereregulated, and 49.7% of these had not been reportedpreviously. Real time reverse transcription polymerasechain reaction (RT-PCR) confirmed the expression of 20of these genes and showed two general classes: thoseinduced by 2 h, and hence might be targets for acutestroke therapy; and those induced at later times thatcould be targets for tissue repair and plasticity.
MATERIALS AND METHODS
Animal protocols were approved by the University of Cin-cinnati animal care committee and conform to the NationalInstitutes of Health Guide for Care and Use of LaboratoryAnimals. Male Sprague-Dawley rats weighed approximately300 g to 350 g, had unrestricted access to food and water, andwere housed two per cage with a 12-h light–dark cycle.
Stroke modelThe left MCA was occluded using the intraluminal filament
technique (Rajdev et al., 2000; Schwarz et al., 2002). AdultSprague-Dawley rats (n � 3) were anesthetized with isoflu-rane. During anesthesia, rectal temperature was monitored andbody temperature was maintained at 37 ± 0.2°C with a heatingblanket. The left common carotid artery, external carotid artery,and internal carotid artery were isolated via a ventral midlineincision. To occlude the MCA, a 3–0 monofilament nylon su-ture was inserted into the external carotid artery and advancedinto the internal carotid artery approximately 20 mm from thecarotid bifurcation until mild resistance was felt. The woundwas closed. Once animals recovered, they were returned totheir home cages with food and water available ad libitum. Oneday later (24 h), rats were reanesthetized and killed. Sham-operated animals (n � 3) were treated like ischemic animalsexcept that no suture was inserted into the carotid.
RNA preparationAt 24 h after cerebral ischemia, rats were reanesthetized with
ketamine (100 mg/kg) and xylazine (20 mg/kg) and killed. Theperiinfarction cortex was dissected according to a publishedmethod in rat filament model of unilateral proximal MCA oc-clusion (Ashwal et al., 1998; Schwarz et al., 2002; see thesearticles for diagram of dissected brain region). The brain wasquickly removed and cut coronally into three slices beginning3 mm from the anterior tip of the frontal lobe in a brain matrixin a cold room. A longitudinal cut approximately 2 mm fromthe midline through left hemisphere in the sections was made toavoid medial hemispheric structures, which are supplied pri-marily by the anterior cerebral artery. Then, a transverse di-agonal cut was made at approximately the “2 o’clock” posi-tion—avoiding obvious areas of infarction. The left parietal,periinfarction cortex was dissected. The parietal cerebral cor-texes in ischemic 2- or 4-h rats from the same location werealso dissected. The dissected brain tissues were homogenized ina Teflon–glass homogenizer with TRIzol Total RNA IsolationReagent (Life Technology, Rockville, MD, U.S.A.). TotalRNA was isolated according the manufacturer’s instructions.
Briefly, the brain homogenate was treated with chloroform;RNA was precipitated using isopropyl alcohol and cleaned us-ing a RNAeasy mini kit (Qiagen Inc., Valencia, CA, U.S.A.).
GeneChip expression analysis and database searchGeneChip expression analysis was performed according to
the Affymetrix expression analysis technical manual. Briefly,double-stranded cDNA was synthesized from total RNA with ahigh-performance liquid chromatography–purified oligo-dTprimer. Biotin-labeled complementary RNA (cRNA) was syn-thesized from cDNA using T7 RNA polymerase and biotin-labeled ribonucleotides. The quality of the cRNA was assessedusing gel electrophoresis. The cRNA was hybridized to Af-fymetrix U34A rat arrays (Affymetrix, Santa Clara, CA,U.S.A.). The U34A microarray was scanned with the GeneChipscanner.
The data were analyzed using Affymetrix GeneChip expres-sion analysis software according to the Affymetrix GeneChipAnalysis Suite (Tang et al., 2001). An absolute analysis re-ported the hybridization intensity data (average difference) andwhether transcripts were present, absent, or marginal in thetarget from each probe array. Then, a comparison analysis wasrun. The patterns of change of the whole probe set were used tomake a qualitative call of “Increase,” “Decrease,” “Marginalincrease,” “Marginal decrease,” or “No change.” Three chipswere used for each group (sham-operation and ischemia). Thecross-comparisons were made between sham-operation andischemia groups. Genes were included in the analysis only ifthey met all of the following criteria: they were present in allthree sham or all three ischemia samples; all three ischemiasamples for each gene showed either an “Increase or Decrease”when compared with all three sham samples for each gene; andthe fold change in each of the individual comparisons betweenischemia and sham had to be at least 1.7-fold (Jin et al., 2001).These are stringent criteria that probably eliminated manygenes that were actually regulated by ischemia.
Functional information for the regulated genes was obtainedusing LocusLink, OMIM, GeneCards, PubMed, and referenc-ing gene ontology (Ashburner et al., 2000). By searching Uni-Gene and doing Blast analyses of the GeneseqN database, wedetermined the similarity of the expressed sequence tags(ESTs) on the microarrays with known genes. For genes thatwere represented several times on an array, the alternate genenames are given, and the expression values for that gene wereaveraged. If the ESTs represented known or at least highlyhomologous genes, the known gene name is provided in thetables in the unigene/blast columns (Tables 1–14). Sometimesone EST may blast to short fragments of genes or blast toseveral genes, making these ESTs more difficult to interpret. Adraft rat genome covering more than 90% of the rat genomeis available. Most of the ESTs had known homologues(Butler, 2002).
Real-time quantitative RT-PCRTwenty genes were selected for RT-PCR based on whether
they had important functions in cell death, were previouslyunreported, and had relatively high-fold changes on the micro-arrays. Real-time quantitative RT-PCR was performed on thesegenes (n � 3) using the ABI Prism 5700 Sequence Detectionsystem (Applied Biosystems, Foster City, CA, U.S.A.) (Tanget al., 2001). Primer and probe sequences were selected fromcoding regions of each of the genes with the aid of PrimerExpress 2.0 (Applied Biosystems). All primers and probeswere synthesized using PE Oligofactory (Applied Biosys-tems). Each probe was labeled at the 5�-end with the reporterdye VIC and at the 3�-end with quencher dye TAMRA
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 787
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

(6-carboxytetramethyl-rhodamine) and was phosphate blockedat the 3�-end to prevent extension by AmpliTaq Gold DNApolymerase. One-step RT-PCR was performed according to theTaqman One-Step RT-PCR Master Mix Reagent kit protocol(Applied Biosystems). Fifty to 100-ng total RNA, 900-nmol/Lprimer and 250-nmol/L probe were added for the selectedgenes. Thermal cycling was carried out as follows. Reversetranscription: 48°C for 30 minutes; activation of hot startedAmpliTaq Gold DNA polymerase: 95°C for 10 minutes; ther-mal cycling: 95°C for 15 seconds, and 60°C for 1 minute for 40cycles. The amplified transcripts were quantified with the rela-tive standard curve method and using GAPDH as a loadingcontrol.
RESULTS
Total number of regulated genesA significant number of genes were regulated at 24 h
after permanent focal ischemia (Figs. 1 and 2; Tables1–15). Of the 328 transcripts (from 8,740 on the micro-arrays) that differed from the sham-operation group us-ing the criteria above (all present and fold change >1.7for all comparisons), 264 genes and ESTs were upregu-lated, and 64 genes and ESTs were downregulated. Fig-ure 1 shows a scatter plot of increased expression (brownand red) and decreased expression (green) of transcriptsin the 24-h ischemic samples compared with sham-operation controls. It is notable that many genes showedgreater than 10-fold increases of expression. Figure 2shows that the 328 regulated genes cluster into twogroups: those that increase in animals with stroke andthose that decrease in animals with stroke (Fig. 2). Thisunsupervised cluster also shows the relative consistencyof expression of upregulated (red, threefold increase) anddownregulated genes (blue, threefold decrease) in thethree stroke animals compared to the three sham-operated animals (Fig. 2).
By searching PubMed and performing Blast analyses,it was estimated that 165 (50%) of the 328 regulatedgenes had been reported in previous stroke studies. Ofnote, 147 known genes (45%) had not been reported tobe regulated after stroke or other type of ischemia, and16 (5%) were unknown ESTs. Thus, half (49.7%) of thetranscripts reported in this study have not been reportedin previous stroke or ischemia studies (Fig. 3).
Functional categories of differential expressed genesBy searching LocusLink, OMIM, GeneCards, and
PubMed and reference gene ontology, we divided theregulated genes into 14 different functional categories(Fig. 4, Tables 1–15). Although some of the categoriesare somewhat artificial because some genes fall into sev-eral categories, these categories help in assessing thelarge amount of data (Figs. 4A–C).
Transcription factors and metabolism- and signaltransduction–related categories had the most numbers ofregulated transcripts (Fig. 4A). The genes showing thehighest fold changes (more than fivefold) included thecytokines and chemokines, cell adhesion, motility and
immune response–related genes, stress proteins, andtranscriptional factors (Fig. 4B). Stress proteins, growthfactors, cytokines and chemokines, cell adhesion, motil-ity and immune response–related genes, and enzymesand enzyme inhibitors had been the most reported,whereas many of the G-protein–related genes and me-tabolism-related genes shown to be induced by stroke inthis study had not been previously reported (Fig. 4C).
Enzymes and inhibitors. Several enzymes and pro-teases well known in cerebral ischemia were detectedusing the microarrays including the ICE-like cysteineprotease, calpain and cathepsin L (cysteine protease),gelatinase B (collagenase), and TIM1 (metalloproteinaseinhibitor). Additional upregulated genes includedcathepsin K and C (cysteine protease) and PS-PLA1(phospholipase); enzyme inhibitors, such as cystatin B(cysteine protease inhibitor); contrapsin-like protease in-hibitor–related protein (serine protease inhibitor); and aribonuclease inhibitor (Table 1).
Metabolism-related genes. Several genes in Table 2known to be induced by stroke included GLUT1 (glucosetransporter), ornithine decarboxylase (rate-limiting en-zyme of polyamine biosynthesis), and PCNA/cyclin(DNA replication and repair). Additional regulated genesincluded glycerol 3-phosphate dehydrogenase (GPDH)that was induced more than 60-fold, PHAS-I (translationnegative regulation), PKBS (benzodiazepine receptor,flow of cholesterol into mitochondria), and TBFII (RNAbinding, splicing, and processing) (Table 2).
Stress response proteins. The transcripts of almost allheat shock proteins (Hsp) present on the microarrayswere induced. Hsp27, Hsp70, and heme oxygenase werehighly expressed as previously reported (Sharp et al.,2000). The DNA damage response gene GADD45 andglutathione peroxidase were also upregulated. The pre-viously unreported antioxidant protein 2 transcript wasdecreased (Table 3).
Neurotransmitter and hormone–related genes. Un-reported stroke-inducible genes, such as Ania-3 (metabo-tropic glutamate receptor signal pathway) and �-typecalcitonin gene–related peptide (vasodilator) werehighly expressed. Retinoic acid–related genes were sig-nificantly regulated, and the angiotensinogen transcriptdecreased. Reported stroke-inducible genes, such asNarp (extracellular aggregating for �-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid [AMPA] receptor),�-type calcitonin gene-related peptide (vasodilator), cy-clooxygenase-2 (rate-limiting enzyme in the conversionof arachidonic acid to prostaglandins), and thyrotropin-releasing hormone were also highly expressed (Table 4).
Growth factor–related genes. Several growth factorsinduced by ischemia were detected including brain-derived neurotrophic factor, transforming growth factor-�-1, heparin binding epidermal growth factor-likegrowth factor and vascular endothelial growth factor
A. LU ET AL.788
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

FIG. 1. Scatter plot showing the av-erage hybridization signal intensity ofthe genes in the ischemic (n = 3)compared with the sham surgery (n =3) group. The diagonal lines indicatetwofold change in the ischemic groupcompared with the sham group (two-fold increase, upper line; twofold de-crease, lower line). Significantly up-regulated transcripts in the ischemiccompared with sham groups areshown in red–brown; significantlydownregulated transcripts in the ische-mic compared to the sham groups areshown in green.
FIG. 2. Hierarchical clustering ofregulated genes in the ischemicgroup compared with sham group.Red represents threefold increases ofexpression compared with themeans, and purple–blue indicatesthreefold decreases of expressioncompared with the means. The ex-pression of the three sham animals isshown in the top three rows, and theexpression of the three ischemic ani-mals is shown in the bottom threerows. The expression of individualgenes is shown in thin vertical col-umns. Note that a great many genesare induced in the three ischemic ani-mals compared with the shams (leftthree fourths of the cluster), and asmaller set of genes are decreased inthe three ischemic animals comparedwith the shams (right one fourth of thecluster).
FIG. 3. Pie chart showing the per-centages of genes identified as regu-lated by ischemia in the periinfarctioncerebral cortex in this study. The pro-portion of genes reported in previousischemic stroke studies (blue), not re-ported in previous ischemic strokestudies (lavender), and the percent-ages of expressed sequence tags(ESTs) (yellow) are shown.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 789
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

TABLE 1. Enzymes and inhibitors
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chip
RNAchangein paper Unigene/blast
Percent identity,aligned region
Oxidoreductase EST220459 Oxidoreductase, drug metabolismand synthesis of cholesterol,steroids, and other lipids. Eyemorphogenesis
7.8 AI176856 I I(CYP2E) CP1B ratcytochromeP450 1B1
100% 542 aa
Cytochrome P450(CYP1B1)
Oxidoreductase, drug metabolismand synthesis of cholesterol,steroids, and other lipids. Eyemorphogenesis
6.4 U09540 I I(CYP2E)
Cysteine proteasesand inhibitor
Calpain II 80 kDasubunit
Calcium-activated neutral proteases,nonlysosomal, intracellularcysteine proteases, catalyzinglimited proteolysis of substratesinvolved in cytoskeletalremodeling and signal tranduction.
1.9 L09120 I I
Cyclic Protein-2� cathepsinL proenzyme
Lysosomal cysteine (thiol) protease 1.8 S85184 I I
EST Lysosome cysteine-type peptidase 2.8 AA925246 I X Cathepsin Kprecursor
100% 328 aa
Cathepsin C Lysosomal cysteine (thiol) protease 3.2 D90404 I IICE-like cysteine
proteaseCysteine (thiol) protease, induction
of apoptosis3.8 U49930 I I
EST203339 Cysteine protease inhibitor, inhibitspapain (cathepsins L, h and b)
2.1 AI008888 I I Cystatin B 100% 97 aa
Major acute phasealpha-1
Inhibitors of thiol proteases,releasing bradykinin
3.8 K02814 I X
Serine proteaseand inhibitor
Tissue-typeplasminogenactivator (t-PA)
Serine protease, converts inactiveplasminogen to plasmin
2.6 M23697 I I
Plasminogenactivatorinhibitor-1(PAI-1)
Member of the serpin family ofserine protease inhibitors, “bait”for tissue plasminogen activator,urokinase, and protein c,regulation of fibrinolysis
17 M24067 I I
EST189815 Serine protease inhibitor, inhibitionof complement activation
2.1 AA800318 I X Complement C1inhibitorprecursor
81% 171 aa
Contrapsin-likeproteaseinhibitor relatedprotein (CPi-26)
Serine protease inhibitor, inhibitingneutrophil cathepsin g and mastcell chymase
∼71.6 D00753 I I
Metalloproteinaseand inhibitor
EST202002 Component of the neutrophilgelatinase complex, modulator ofinflammation, apoptosis
∼21 AA94650 I I NGAL ratneutrophilgelatinase-associatedlipocalinprecursor
100% 197 aa
Gelatinase B(GelB)
Collagenase, degrades type IV and Vcollagens
∼17.4 U24441 I I
EST215162 Inhibitors of the matrixmetalloproteinases, known to act onmmp-1, mmp-2, mmp-3, mmp-7,mmp-8, mmp-9, mmp-10, mmp-11,mmp-12, mmp-13 and mmp-16.Does not act on mmp-14.
∼21 AI169327 I I TIM1 rat metallo-proteinaseinhibitor 1precursor
100% 216 aa
Phospholipaseand inhibitor
PS-PLA1 Phospholipase A1,phosphatidylserine metabolism
∼13.1 D88666 I X
EST217956 Ca2+-dependentphospholipid-binding protein,inhibiting phospholipase A2 andantiinflammation
4.3 AI171962 I I Annexin I 100% 345 aa
Annexin II Calcium-dependentphospholipid-binding protein,regulation of cellular growth, andsignal transduction
5.1 L13039 I I
RNA helicase Nuclear RNAhelicase
ATP dependent RNA helicase,down-regulation of acute phasecytokine production (TNFalpha,IL-1, and IL-6)
2.7 AF063447 I X
Ribonucleaseinhibitor
Ribonucleaseinhibitor
Ribonuclease inhibitor 2.6 X62528 I X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.790
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

TABLE 2. Genes related to metabolism 1
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Carbohydratemetabolism
GLUT1�glucosetransporter 1,Rat brainglucose-transporter
Glucose transporter,transport ofdehydroascorbic acid
2.9 S68135, M13979
I I
EST189687 Glycogen catabolism −2.1 AA800190 D X Glycogen phosphorylase,brain form
99%, 137 aa
HK2 gene fortype IIhexokinase
Phosphorylating glucose toproduce glucose-6-phosphate, glycolysis
∼11 D26393 I X
6-phosphofructo-2-kinase/fructose-2,6bisphosphatase
Regulation of theconcentration offructose-2,6-bisphosphate,glycolysis
∼8.2 AB006710 I X
Glycerol3-phosphatedehydrogenase
Glycolysis, oxidoreductase ∼61.7 AB002558 I X
Amino acid,proteinmetabolism
EST226923 Serine synthesis −2.1 AI230228 D X Human phosphoserinetransaminase
54%, 108 aa
EST212157 Serine synthesis −2.5 AI102868 D X Phosphoserinetransaminase
54%, 108 aa
Cystathioninegamma-lyase
Cysteine metabolism ∼3.1 D17370 I X
L-cysteine oxygenoxidoreductase
Degradation of cysteine,Taurine and hypotaurinemetabolism, electrontransporter
2.9 E03229 I X
EST198184 Degradation of cysteine,Taurine and hypotaurinemetabolism, electrontransporter
2.5 AA942685 I X CYDX rat cysteinedioxygenase
100%, 199 aa
S-adenosyl-methioninesynthetase
Amino acid metabolism,biosynthesis ofpolyamines
2 J05571 I X
Ornithinedecarboxylase(ODC)
Rate-limiting enzyme of thepolyamine biosynthesis
2.8 J04791, J04792,X07944
I I
EST Mitochondrial ribosome 28Ssubunit protein
2.1 AI639387 I X Mitochondrial ribosomalprotein S6 (MRPS6)
Ribosomal proteinS27
Cytosolic small ribosomal(40S)-subunit
2.7 X59375 I I
PHAS-I Binding EIF4E, translationalnegative regulation
∼10.1 U05014 I X
EST195768 Fructosamine-3-kinase,deglycation
−2.8 AA891965 D X Fructosamine-3-kinase(FN3K gene)
EST197246 Nuclear and cytoplasmicprotein glycosylation
1.8 AA893443 I X UDP-N-acetylglu-cosamine-peptideN-acetylglucos-aminyltransferase 110Kda subunit (OGT1)
100%, 1035 aa
EST196352 N-glycosylation 2 AA892549 I X Mannosyl-oligosaccharidealpha-1,2-mannosidase
EST196578 Hydrolysis andtransglycosylation
2.9 AA892775 I I Rat lysozyme c, type 1precursor
100%, 146 aa
Lipidmetabolism
EST Glycolipids catabolism −2.5 AA859911 D X Cmp-n-acetyl-neuraminate-beta-galactosamide-alpha-2,3-sialyltransferase(alpha 2,3-st)(gal-nac6s) (st3gala.2)(siat4-b)
100%, 349 aa
Fatty acidtransportprotein
Membrane transporter forlong-chain fatty acids
−2.6 U89529 D X
Low molecularweight fattyacid bindingprotein
Lipid transporter, tumorsuppressor
−2.2 J02773 D X
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 791
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

(VEGF) (Lipton, 1999). Other factors, not previouslydescribed in ischemic stroke, were also induced by is-chemia: PT N1 (negative regulation of insulin signaling),DOC-2 p59 isoform (CSF1 signal transduction pathway),and GRB2 (linking EGFR and PDGFRB to Ras and Racpathway). Of note, the Noggin transcript (binding andinactivating members of TGF-� superfamily signal pro-tein) decreased (Table 5).
Cytokine and chemokine–related gene. Ischemia isknown to induce many of these genes including tumornecrosis factor (TNF) receptor, interleukin (IL)-1�, IL-1
receptor antagonist, IL-6, CC chemokine ST38 precur-sor, MIP-1, and interferon-induced messenger RNA.Other inducible genes in this class included HCYP2(serine/threonine kinase, interacting with TNFR1and TRAF2), PRG1 (protection of cells from Fas orTNF�-induced apoptosis), gro, calgranulin, and IL-18(Table 6).
Signal transduction–related genes. GTP cyclohydro-lase I (NO-synthesis related), protein kinase C-�, oxida-tive stress-inducible protein tyrosine phosphatase,MKP-3 (inactivation of MAPK), and SOCS-3 (negative
TABLE 2—(Continued)
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Lipidmetabolism
C-FABP �cutaneous fattyacid-bindingprotein
Lipid binding,differentiation
2.4 S69874 I I
EST218294 Cholesterol biosynthesis 1.9 AI172293 I X RANP-1 100%, 292 aaEST Oxidoreductase,
cholesterol biosynthesis3.1 E12625 I X
Isopentenyldiphosphate-dimethylallyldiphosphateisomerase
Isoprenoid biosynthesis,steroids synthesis
3.5 AF003835 I X /C-4 methyl steroloxidase
Peripheral-typebenzodiazepinereceptor (PKBS)
Benzodiazepine receptor,involving in the flow ofcholesterol intomitochondria
11.3 J05122 I I
EST LDL receptor, proteolysisand peptidolysis
∼5.6 AI071531 I I JE0111 lectin-likeoxidized LDLreceptor-mouse
78%, 360 aa
Aminopeptidase-B Exopeptidase, eukotrieneA4 hydrolase
−1.8 D87515 D X
Nucleotidemetabolism
Adenosine kinase Conversion of adenosineto AMP, ribonucleosidemonophosphatebiosynthesis
−2 U57042 D X
Proliferating cellnuclear antigen(PCNA/cyclin)
Delta-DNA polymerasecofactor, DNAreplication and repair,cell cycle control
2.4 M24604 I I
EST Production of thepyrimidine nucleosidetriphosphates
∼11.8 AA859827 I X URK1 mouse uridinekinase
78%, 184 aa
Phosphodiesterase I Nucleotidepyrophosphatase, broadspecificity
−2.7 D28560 D X
Phosphodiesterase I Phosphodiesterase I.Nucleotidepyrophosphatase
∼6.2 D30649 I X
TBFII mRNA forpolypyrimidinetract bindingprotein
RNA binding, mRNAsplicing, mRNAprocessing
∼11.8 X74565 I X
EST189008 pre-mRNAs binding,pre-mRNA processing,and other aspects ofmRNA metabolism andtransport
2 AA799511 I X ROA2_humanheterogeneous nuclearribonucleoproteinsA2/B1
97%, 41 aa
EST SnoRNP assembly, pre-18srRNA processing
3 AF069782 I X /nucleolar protein 5(NOP5)
Gas-5 growth arresthomolognon-translatedmRNA sequence
Processing of ribosomalRNA
3 U77829 I X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.792
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

regulator in the JAK-STAT pathway) are induced byischemia as previously reported. Unreported genes in-cluded Ssecks 322 (PKA anchoring protein), Cpg21 (in-activating ERK1), JAK2 (JAK-STAT cascade), and sev-eral genes related to NO metabolism (Table 7).
G-protein–related genes. This was the most surpris-ing group of genes because ischemia was not generallyknown to regulate them. Highly expressed transcriptsincluded Rad (small monomeric GTPase) and G33A
(Rho GTPase binding and activator) (Table 8). G-protein–coupled receptors, GTPase binding molecules,and GTP signaling are remarkably regulated in the peri-infarction cortex after ischemia (Table 8).
Transcriptional regulatory proteins. Many tran-scription factors are known to be induced by ischemiaand were also regulated on the microarrays includingc-fos, Fra-1, c-jun, jun-B, c-myc, NGFI-A, krox20, andHIF1. Other highly regulated transcriptional factors de-
TABLE 3. Stress response genes
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Oxidativestress
EST211851 Heavy metal binding,detoxifing metals,antioxidation
8.4 AI102562 I I MT1 ratmetallothionein-1
100%, 60 aa
Metallothionein-2metallothionein-1
Heavy metal binding,detoxifing metals,antioxidation
8 M11794 I I
EST220041 Storage of trace-metals, cellstress
2.9 AI176456 I I Rat metallothionein-II 100%, 60 aa
GlutathioneS-transferaseYc1 subunit
Conjugation of reducedglutathione to a widenumber of hydrophobicelectrophiles
−2.2 S72505 D D
GlutathioneS-transferase Ycsubunit
Glutathione transferase −2.3 K01932 D D
EST195844 Peroxidase and antioxidantactivity
−2 AA892041 D X Antioxidant protein 2 100%, 223 aa
EST 190084 Selenium-dependentglutathione peroxidase,reduction of hydrogenperoxide, organichydroperoxide, and lipidperoxides
∼9.6 AA800587 I I A45207 glutathioneperoxidase
94%, 166 aa
Heat shockprotein
Heat shock protein70
Heat shock protein ∼103.7 L16764, Z27118 I I
EST, EST191323 Heat shock protein ∼91.6 AA818604,AA848563
I I Rat heat shock 70 Kdprotein 1/2
100%, 640 aa
EST Heat shock protein 2 AA875620 I I Heat shock 70 Kd protein3 (HSP70.3)
100%, 640 aa
Hsp70.2 Heat shock protein 52.6 Z75029 I IHeat shock protein
(Hsp27)Heat shock protein ∼358.7 M86389 I I
EST Heat shock protein 53 AA998683 I I Heat shock 27protein—rat
100%, 204 aa
EST220250 Heat shock protein 11.2 AI176658 I I Heat shock 27protein—rat
100%, 204 AA
Collagen-bindingprotein (gp46)
Heat shock protein 3.2 M69246 I I
EST216547 Heat shock protein 2 AI170613 I I 10 Kd heat shock protein,mitochondrial
99%, 101 aa
EST0020 Heat shock protein 2.8 AA108277 I I /Hsp105Alpha-crystallin B
chainMolecular chaperone 3.2 M55534, X60351 I I/−
EST223333 Heat shock protein 15.1 AI179610 I I A Chain A, Crystalstructure of rat hemeoxygenase-1 incomplex with heme
100%, 288 aa
Heme oxygenasegene
Heme oxygenase ∼88.5 J02722 I I
DNA damageresponse
Progressionelevated gene 3protein(Gadd34)
DNA damage response,initiation of proteintranslation, proapoptoticfunction
3.2 AF020618 I I
EST DNA damage response,inducing p38/JNKactivation and
7.8 AI070295 I I Growth arrest anddna-damage-inducibleprotein GADD45
100%, 164 aa
GADD45 DNA damage response,inducing p38/JNKactivation and
17.4 L32591 I I
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 793
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

TABLE 4. Neurotransmitter and hormone-related genes
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Glutamaterelated
Glutamatereceptor(GluR-B)
Subunit 2 of AMPA subtypeof ionotropic glutamatereceptor for synaptictransmission
−2 M38061 D D
Metabotropicglutamatereceptor 3
Metabotropic glutamatereceptor
−2.7 M92076 D D
Narp � neuronalactivityregulatedpentraxin(NPTX2)
Transportor, extracellularaggregating factor forAMPA receptors,synapses plasticity anduptake of extracellularmaterial
∼101.5 S82649 I I
AMPA receptorinteractingprotein GRIP
Adapter protein, lustering ofAMPA receptors
∼3.1 U88572 I X
VesI Modulation of metabotropicglutamate and IP3receptors
3 AB003726 I X
Activity andneurotrans-mitter-inducedearly gene 3(ania-3)
Metabotropic glutamatereceptor signalingpathway, neuronalimmediate early gene
∼6.0 AF030088 I X
GABA related EST225364 GABA transporter −2.1 AI228669 D X Sodium- and chloridedependent GABAtransporter 1
100%, 598 aa
GABA-A receptorgamma-2subunit
Benzodiazepine receptor,gamma-aminobutyricacid-inhibited chloridechannel
−2.2 L08497 D D/I
Angiotensinogen Angiotensinogen Angiotensinogen, hormone −5 M12112 D XEndothelin related Endothelin-
convertingenzyme
Endothelin-convertingenzyme,metalloendopeptidase
1.8 D29683 I X
EST Endothelin receptor, Gprotein-coupled receptor
3.3 AA818970 I I Rat endothelin Breceptor precursor
100%, 441 aa
Neuropeptide Neuropeptide Neuropeptide Y,neuropeptide hormone andinhibitory neuromodulator
2.3 M15880 I I
Alpha-typecalcitoningene-relatedpeptide
Ligands for receptors foundin nervous system- andperipheral tissues,inducing vasodilatation
∼9.5 M11597 I X
Beta-typecalcitoningene-relatedpeptide
Neuropeptide hormone,calcium ion homeostasis,vasodilatation
∼10.3 M11596 I I
Adrenomedullinprecurosr
A hypotensive peptide andvasodilatator agent
∼6.4 D15069 I I
Galanin (aneuropeptide)
Neuropeptide 5.5 J03624 I I
Prostaglandinrelated
Cyclooxygenaseisoform COX-2
Prostaglandin-endoperoxidesynthase, the firstrate-limiting step in theconversion of arachidonicacid to prostaglandins
7.6 S67722,L25925
I I
Thyrotropinreleasinghormone
Thyrotropinreleasinghormone (TRH)
A regulator of thebiosynthesis of TSHand as aneurotransmitter/neuromodulatorin the central andperipheral nervoussystems
∼11 M23643,M36317
I I
Retinoic acidrelated
Cellular retinoicacid-bindingprotein II(CRABP II)
Retinoic acid-binding protein 2.2 U23407 I X
Cytosolicretinol-bindingprotein (CRBP)
Retinoid binding andtransport
3.9 M19257 I I
Aldehydedehydrogenase(ALDH)
Aldehyde dehydrogenase −2.5 AF001898 D X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.794
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

tected on the microarrays included RL/IF-1 (I�B), reti-noic acid receptor �1, chromosomal protein HMG2,transcriptional repressor CREM, C/EBP-related tran-scription factor, and CELF (Tables 9 and 10).
Extracellular matrix. Osteopontin, a ligand of inte-grin and CD44, was highly expressed along with decorin(binding fibronectin and TGF-�), tenascin (ligand of in-tegrins), syndecan (receptor of extracellular matrix),and lysyl oxidase (protein modification of extracel-lular matrix) (Table 11). Other genes related to fibronec-tin and procollagen were also regulated as previouslyreported.
Cytoskeletal protein–related genes. Glial fibrillaryacidic protein � and � (intermediate filament) and tubu-
lin �-1 chain (microtubule protein) were regulated byischemia as expected. Other stroke-regulated genes in-cluded moesin (cross-linking actin to membranes), talin(linking cytoskeleton to extracellular matrix receptor),and spr (intermediate filament) (Table 12).
Genes-related ion, vesicular transport, and synap-tic transmission. Brain digoxin carrier protein (trans-porter of organic anions), potassium channel �-subunit,neuronal pentraxin I, and agrin expression decreased.Transferrin receptor and ceruloplasmin (ferroxidase,iron efflux) increased (Table 13).
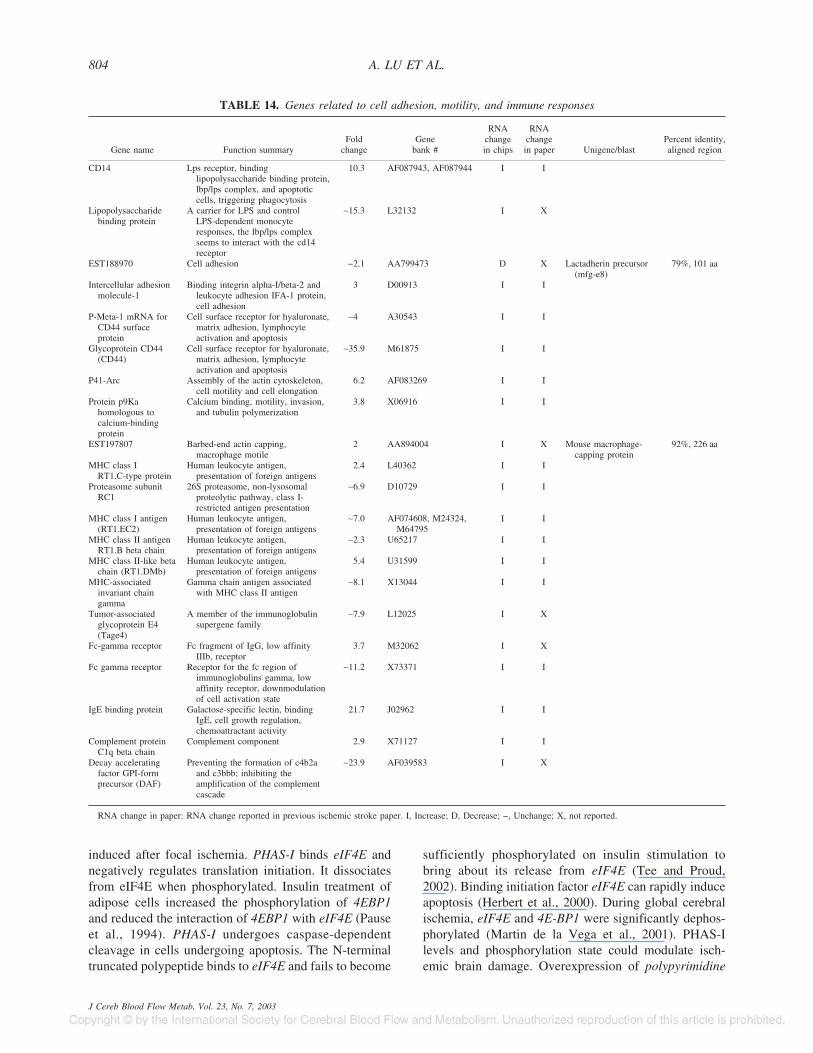
Cell adhesion, motility, and immune response–related genes. CD14 (binding apoptotic cell, triggeringphagocytosis), CD44 (cell surface receptor, matrix adhe-
TABLE 5. Growth factor-related genes
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
IGF-related Insulin-likegrowth factor II
Insulin-like growth factorreceptor ligand
−2.5 X17012 D I
EST Negative regulation ofinsulin signaling
2.2 AI113289 I X Protein-tyrosinephosphatase,non-receptor type 1(PTP-1b) (PTN1)
100%, 431 aa
EST203856 Insulin-like growth factorreceptor binding protein
∼13.4 AIl009405 I I IBP3 rat insulin-likegrowth factor bindingprotein 3
100%, 291 aa
Insulin-likegrowthfactor-bindingprotein(IGF-BP3)
Insulin-like growth factorreceptor binding protein
∼11.4 M31837 I I
BDNF EST Growth factor 5.1 AI030286 I I Brain-derivedneurotrophic factorprecursor
100% 248 aa
BDNF �brain-derivedneurotrophicfactor
Growth factor, neurogenesis,neuronal development
12, 7.3 S76758, D10938 I I
TGF related Transforminggrowth factorbeta 1 (TGFB1)
Ligand of TGF receptor, cellproliferation,differentiation, andapoptosis. Fibrosis.
∼29.1 X52498 I I
EST Binds and inactivatesmembers of TGF-betasuperfamily signalingproteins
−4.1 AA859752 D X Noggin 100%, 143 aa
Furin Processes hormoneprecursors: PTH, TGFb
2.2 X55660 I I
CSF related DOC-2 p59isoform
Csf-1 signal transductionpathway, tumor suppressor
2.7 U95178 I X
EGF related Heparin bindingEGF-likegrowth factor
Epidermal growth factorreceptor ligand, positivecontrol of cellproliferation
17.7 L05489 I I
EST216715 Linking EGFR andPDGFRB to Ras and Racsignaling pathways
1.9 AI170776 I X Growth factorreceptor-bound protein2 (GRB2)
100%, 202 aa
VEGF related EST193502 Vascular endothelial growthfactor I
2.3 AA850734 I I Mouse vascularendothelial growthfactor precursor
98%, 213 aa
Vascularendothelialgrowth factorform 3
Vascular endothelial growthfactor receptor ligand,induces endothelial cellproliferation and vascularpermeability
∼11.1 L20913 I I
FGF receptor Fibroblast growthfactor receptor1 beta-isoform
Fibroblast growth factorreceptor
4 S54008 I I
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 795
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

sion, and apoptosis), and immunoglobulin E binding pro-tein (galactose-specific lectin) increased as previouslyreported in other cerebral ischemia studies. Other regu-lated transcripts included lipopolysaccharide bindingprotein (carrier for lipopolysaccharide, interacting with
CD14), proteasome subunit RC1 (26S proteasome, classI-restricted antigen presentation), and DAF (inhibitingamplification of complement cascade) (Table 14).
Others. Urinary plasminogen activator receptor 1,serine threonine kinase (pim-3), intracellular calcium–
TABLE 6. Genes related to cytokines and chemokines
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
TNF related Tumor necrosisfactor receptor(TNF receptor)
Tumor necrosis factorreceptor, apoptosis andnf-kappa b signaling
4.2 M63122 I I
Homocysteinerespondentprotein HCYP2
Serine-threonine kinase,interacting with TNFR1and TRAF2 and cativtingcaspase
∼6.4 AF036537 I I
EST190110 Zinc finger protein,single-stranded RNAbiding, inhibiting,macrophage TNF-alphaproduction
5.2 AA800613 I I Rat tristetraproline 100%, 319 aa
PRG1(PACAP-responsive gene1)
Protection of cells from Fas-or tumor necrosis factoralpha-induced apoptosis
5.5 X96437 I X
Osteoprotegerin(OPG)
Decoy receptor for osteoclastdifferentiation factor
5 U94330 I X
IL related Interleukin 1-beta Interleukin-1 beta, apoptosis 3 M98820 I IInterleukin 1
receptorantagonist
Interleukin-1 receptorantogonist, inhibiting thebinding of IL-1-alpha andIL 1-beta
∼4.4 M63101 I I
Interleukin 6 (IL6) IL6R ligand growth factor ∼18 M26744,M26745
I I
Interleukin 18(IL-18)
Cytokine, immune response 2.9 AJ222813 I I
Chemokine CC chemokineST38 precursor
C-C chemokine, chemotaxis 22.3 AF053312 I I
EST201236 CXC chemokine receptor,mediating intracellularcalcium flux
∼3.2 AA945737 I X Chemokine receptorLCR1
100%, 331 aa
Macrophageinflammatoryprotein-1alpha
Chemokine cc,inflammatory, andchemokinetic properties
∼20.4 U22414 I I
Macrophageinflammatoryprotein-1 beta(MIP-1 beta)
Chemokine, inflammatory,and chemokineticproperties
∼6.5 U06434 I I
Immediate-earlyserum-responsiveJE gene
Chemotactic factor formonocyes, binding to ccr2and ccr4
61.5 X17053 I I
Gro Chemokine, inflammatoryprocesses
∼9.2 D11445 I I
Interferonrelated
Interferon inducedmRNA
Cell surface receptor linkedsignal transduction, thetransduction ofantiproliferative andhomotypic adhesionsignals
5.6 X61381 I I
EST207718 Regulating gene activity inthe proliferative and/ordifferentiative pathwaysinduced by ngf, may bean autocrine factor
5 AI014163 I X Interferon-related proteinPC4
100%, 448 aa
Others Platelet-activatingfactoracetylhydrolasealpha 1 subunit(PAF-AH alpha1)
Inactivates PAF 6.6 AF016047 I I
EST A member of the S100family of proteins,inhibition of casein kinaseand as a cytokine
∼3.4 AA957003 I X S108 RAT calgranulin A 100%, 88 aa
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.796
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

TABLE 7. Genes related to signal transduction 1
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Calciumrelated
Plasma membraneCA2+ ATPase-isoform 2
Calcium-transportingATPase, transport ofcalcium out of the cell
−2.3 J03754 D D
EST199283 Calcium-transportingATPase, transport ofcalcium out of the cell
−2.1 AA943784 D D Calcium-transportingatpase plasmamembrane, brainisoform 2
100%, 1197 aa
Calmodulin-sensitive plasmamembraneCa2+-transportingATPase (PMCA3)
Calcium-transportingATPase, transport ofcalcium out of the cell
−2.1 J05087 D D
EST Calcium-binding,actin-cytoskeletonassociated
−2.6 AI639532 D X Human troponin C, fastskeletal muscle
97%, 133 aa
Activity andneurotransmitter-induced earlygene 4 (ania-4)
Calcium/calmodulin-dependent proteinkinase-related
∼4 AF030089 I X
Kinase Arelated
cAMPphosphodiesterase
cAMP-specificphosphodiesterase
4.1 J04563 I I
cAMPphosphodiesterase(PDE4)
cAMP-specificphosphodiesterase
2.5 M25350 I I/D
Ssecks 322, PKCbinding protein,and substratemRNA
Protein kinase A anchoringprotein
∼6.6 U75404,U41453,U23146
I X
NO-kinaseG related
EST191798 Guanylate cyclase, NOmediated signaltransduction
−6.7 AA849036 D X Guanylate cyclasesoluble, alpha-1 chain
100%, 689 aa
Cyclic nucleotidephosphodiesterase(CaM-PDE)
CaM-dependentcyclic-nucleotidephosphodiesterase,hydrolase
−2.6 M94537 D X
EST NO mediated signaltransduction
2.1 AI058941 I X N(G),N(G)-dimethylargininedimethylaminohydrolase1
100%, 284 aa
GTP cyclo-hydrolase I
Synthesis oftetrahydrobiopterin,cofactor in aromaticamino acid hydroxylation,and nitric oxide synthesis
7.2 M58364 I I
Membranereceptortyrosinekinase
EST Transmembrane receptorprotein tyrosine kinase,neurogenesis
−1.7 M59814 D X Ephrin type-b receptor 1precursor (elk)
100%, 983 aa
Sky Transmembrane receptorsignaling protein tyrosinekinase, cell adhesion,particularly in CNS
−2.2 D37880 D X
EST Receptor tyrosine kinase,neural crest development
5.2 AI639318 I I Proto-oncogenetyrosine-protein kinasereeptor RET precursor
92%, 1115 aa
Inositolphosphatemetabolism/PKC
Phospholipase C-1 Inositol phosphatemetabolism, producingIP3 and DAG.Translocation of tubby
−2.7 M20636 D I/D/− /cornichon-like protein
PhospholipaseC-beta1b
Inositol phosphatemetabolism
−2.1 L14323 D I/D/−
Inositol1,4,5-triphosphate3-kinase
Inositol phosphatemetabolism, convertingIP3 to Ins(1, 3, 4, 5)P4
−2.2 X56917 D D/−
Protein kinase Cdelta subspecies
Diacylglycerol-activated/phosholipid dependentprotein kinase C
∼15.5 M18330 I I
MAPkinaserelated
Oxidativestress-inducibleprotein tyrosinephosphatase
Inactivation of MAP kinase(erk1/2), oxidative stressresponse
1.8 S81478 I I
Protein tyrosinephosphatase
Inactivation of MAPkinase(erk1/2), oxidativestress response
3.5 S74351U02553
I I
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 797
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

binding protein, the major vault protein—which is in-volved in nucleo-cytoplasmic transport—and neuro-transmitter-induced early gene 6 were all regulated byischemia (Table 15).
Real-time RT-PCRReal-time RT-PCR was performed on 20 selected
genes at 2, 4, and 24 h after ischemia (Fig. 5). The earlytimes were chosen becausethese would be times when
TABLE 8. G protein-related genes
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
EST G protein −2.9 AA925506 D D Guaninenucleotide-bindingprotein G(i)/G(s)/G(o)gamma-7 subunit
100%, 68 aa
N-chimaerin GTPase activator,cooperating with Rac1and Cdc42Hs
−1.5 X67250 D X
EST19812 GTPase activator −2.2 AA894317 D X N-CHIMAERIN/chimerin(chimaerin) 2 (CHN2)
100%, 333 aa
EST196299 GTPase activator −2.9 AA892496 D XSPA-1 like
proteinGTPase activator,
stimulating Rsr1GTPase
−1.8 AF026504 D X
Ras-related rab1Bprotein
Small GTP-bindingprotein
−1.7 X13905 D X
Ras-relatedprotein (rad)
Small monomericGTPase, member ofthe ras family of GTPbinding proteins
14.8 U12187 I I
G33A Rat mRNAfor gene 33polypeptide
RHO GTPase bindingand activator, stressresponse
∼37.2 X07266 I X
EST215655 RHO GTPase bindingand activator, stressresponse
3.8 AI169756 I X Gene 33 polypeptide-rat 97%, 136 aa
EST195743 RHO small monomericGTPase, ras-related,reorganization of theactin cytoskeleton
2.7 AA891940 I X RhoA:1 humanGTP-binding proteinrhoC 6
96%, 32 aa
Isoprenylated 67kDa protein
GTP-bindingprotein/GTPase,immune response
∼3.7 M80367 I X
Putative G-proteincoupledreceptor RA1c
G-protein coupledreceptor
−5 AF079864 D X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
TABLE 7—(Continued)
Category Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
MAP kinaserelated
Dual-specificity proteintyrosine phosphatase(rVH6)
Inactivation of ERK2 3.5 U42627 I I
Dual specificityphosphatase, MKP-3
Inactivation of MAPK 6.3 X94185 I I
MAP-kinase phosphatase(cpg21)
Inactivates ERK1 10.7 AF013144 I X
JAK-STATcascade
Protein-tyrosine kinase(JAK2)
Protein tyrosine kinase,JAK-STAT cascade
∼6.0 U13396,AJ000557
I X
Stat3 protein JAK-STAT cascade,transcription factor
3.2 X91810 I I
Suppressor of cytokinesignaling-3 (SOCS-3)
SH2-containing protein,negative regulator in theJAK-STAT pathway
∼25.7 AF075383 I I
Other EST Signaling role during T-cellactivation
−2.2 AA866291 D X /cornichon-like protein
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.798
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

acute stroke treatment might be initiated. The 24-h timewas chosen because this would be the time when infarctsize was nearly stable but tissue repair and plasticityresponses would likely have been initiated. The geneswere chosen based on their high fold induction or theirfunctions or functional classes.
The PCR results showed good agreement with the cor-responding microarray data in terms of increase and de-crease of expression. Early upregulated genes included
glutamate receptor–related gene (Narp), G-protein–related genes (Rad and G33A), kinases and phosphatases(HYCP2, Pim-3, Cpg21, JAK2), and the transcriptionalfactor CELF (Figs. 5A and 5B). Two other genes showedmodest regulation at early times and greater increases at24h including the extracellular matrix gene Tenascin andthe complement inhibitor, DAF (Fig. 5A). Early upregu-lated genes might be involved in acute neuronal injurycaused by stroke.
TABLE 9. Transcriptional Regulation 1
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
C-fos Transcription factor 15.9 X06769 I IFBR-murine
osteosarcomaprovirus genome
Transcription factor 14.3 X03347 I I /c-fos
Fos-related antigen(Fra-1)
Transcription factor,similar to Fos
∼21 M19651 I I
EST Transcription factor 3.5 AA875032 I I /fos-related antigen-2(fra-2 gene)
c-jun oncogene Transcription factor ∼17.6 X17163 I IEST201366 Transcription factor 2.3 AA945867 I I /C-junEST219534 Transcription factor 2.8 AI175959 I I /C-junEST194844 Transcription factor,
growth factor response6.4 AA891041 I I Rat transcription factor
jun-B100%, 343 aa
pJunB Transcription factor,growth factor response
7.1 X54686 I I
c-myc Transcription factor,activating and repressingexpression of targetgenes
∼10.4 Y00396 I I
EST191797 Transcriptional regulatoryprotein, cell growth,and/or maintenance
−2.1 AA849035 D X MYCB 100%, 177 aa
Nerve growthfactor-induced(NGFI-A) gene(egr1)
Transcription factor,mitogenesis, anddifferentiation
2.1 M18416 I I
Immediate early genetranscription factorNGFI-B
Transcriptional factor,orphan nuclear receptor,steroid receptor family
2.3 U17254 I I
NGFI-C3E exon#1-2 Transcription factor,positive control of cellproliferation
2.5 M92433 I I
krox20 Transcription factor,nervous systemdevelopment
7.2 U78102 I I
EST Transcription factor,repression oftranscription, negativecontrol of cellproliferation
3.6 AI071299 I X Mouse transforminggrowth factor-beta-inducible early growthresponse protein 1(MGIF)
93%, 479 aa
Hypoxia-induciblefactor 1
Transcription factor, stressresponse
2.7 Y09507 I I
RL/IF-1 Transcription factorbinding and cytoplasmicsequestering
∼16.4 X63594 I I
NGF-inducibleanti-proliferativeputative secretedprotein (PC3)
Transcription factor, DNArepair, negative controlof cell proliferation
4.1 M60921 I I
EST199655 Transcription factor, DNArepair, negative controlof cell proliferation
2.6 AA944156 I I BTG2 rat btg2 proteinprecursor
100%, 157 aa
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 799
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

Late upregulated transcripts included enzymes and inhibi-tors (Cathepsin C, Cip-26, Systatin B), metabolism-relatedgenes (PHAS-I, TBFII), cytoskeletal proteins (Spr), andthe lipopolysaccharide binding protein (Figs. 5C and 5D).These genes may be related to tissue immune and repair re-sponses or possibly to some aspect of plasticity after thestroke. Several transcripts were also downregulated on themicroarrays (Tables 1–15). The expression of two of these
transcripts, brain digoxin carrier protein and a potassiumchannel, was confirmed by RT-PCR (Fig. 5E).
DISCUSSION
Various members of virtually every class of genesthroughout the entire genome appear to be regulated in
TABLE 10. Trancriptional Regulation 2
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
FSH-regulatedprotein
Transcription factor,negative control of cellproliferation
∼5.5 L26292 I X /Kruppel-like factor 4(KLF4)
RYB-a Cell proliferation and DNArepair, repression oftranscription
3 D28557 I X
Sry-relatedHMG-box proteinSox11
Transcription activatingfactor, neurogenesis
∼5.0 AJ004858 I X
EST198080 Transcription factor, neuraldevelopment
−3.1 AA894277 D X /Soggy precursor andTEAD-2
BHF-1 Differentiation factor duringneurogenesis
∼−7.7 D82074 D X
EST Transcription factor,differentiation factorduring neurogenesis
∼−5.8 AI639109 D X /Neurogenic differentiation(NEUROD1, BHF-1,NIDDM)
LIM homeodomainprotein (LH-2)
Transcription factor, celldifferentiation
−2 L06804 D X
Retinoic acidreceptor alpha1
Nuclear hormone receptor,differentiation
∼9.8 AJ002940 I X
Inhibitor ofDNA-binding,splice variantId1.25
Transcription factor,inhibiting dna binding,negative regulation of celldifferentiation
1.7 L23148 I X
High MobilityGroup Protein I(Y)
AT DNA binding,transcription regulation
2.3 X62875 I X
Chromosomalprotein HMG2
DNA-binding protein,affecting transcription andcell differentiation, DNArepairing
5.2 D84418 I X
Leucine zipperprotein
Transcription factor,transcription co-repressor,cAMP responsive elementbinding
2.6 M63282 I X
Transcriptionalrepressor CREM
Regulator of thetranscription ofcAMP-inducible genes
∼17.1 S66024 I I
Interferon regulatoryfactor 1 (IRF-1)
Transcription activatingfactor, regulatingapoptosis andtumor-suppression
2.6 M34253 I I
EST, EST234097 Transcription factor 2.2 U53184, AI237535 I X Human LPS-inducedTNF-alpha factor
80%, 134 aa
rNFIL-6�C/EBP-relatedtranscriptionfactor
Transcription factor, immuneand inflammatoryresponses, antiapoptosis
∼14.2 S77528 I X
Sfb mRNA forsilencer factor B
Transcription factor 6.2 X60769 I I
CELF Transcription factors,immune and inflammatoryresponses
14 M65149 I I
EST Transcription activatingfactor, immune response
∼43.8 AI045030 I X Rat CCAAT/enhancerbinding protein delta
100%, 267 aa
Zinc finger protein Transcriptional activator,b-cell growth anddevelopment, fibrogenesis
∼15.4 AF001417 I X
EST Transcription factor,activator
6.5 AA900476 I X MRG1 mouse MSG-relatedprotein 1
98%, 268 aa
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.800
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

response to cerebral ischemia. The challenge will be todevelop strategies for identifying the best targets to treatacute stroke or to promote recovery and repair. Our pre-vious studies identified groups of molecules that wereregulated in common by several types of injuries thatmight provide such targets (Tang et al., 2002). The pre-sent study focuses on genes with high expression levelsor on groups of genes that appear to signify activation ofspecific pathways. The multiplicity of genes induced af-ter focal ischemia would suggest that pharmacologic ap-proaches that targeted multiple pathways simultaneouslymight be more successful than single gene or even singlegene pathway approaches. High-throughput drug screen-ing might be designed to detect single compounds thatmodulated several different but critical injury or recov-ery pathways.
RNAs for calpain, ICE-like cysteine protease, cathep-sin L, cathepsin K, cathepsin C, cystatin B, and contrap-sin-like protease inhibitor–related protein (Cpi-26) wereall induced after stroke. The ICE-like cysteine protease ishomologous to caspase-3 (Juan et al., 1996). Caspase-3,a central executioner in mitochondrial apoptotic path-ways (Hengartner, 2000), is induced along withcaspase-9 in the penumbra of some (Benchoua et al.,2001; Namura et al., 1998) but not all focal ischemiastudies of adult brain (Gill et al., 2002; Loetscher et al.,2001). The present results support the induction of sev-eral caspase-like RNAs by focal ischemia (Dirnagl et al.,1999). However, it is likely that regulation of mostcaspases occurs by cleavage-induced activation of theirproteolytic domains rather than being transcriptionallyregulated (Phan et al., 2002; Dirnagl et al., 1999).
TABLE 11. Extracellular matrix-related genes
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Fibronectin gene 3 end Extracellular matrix, celladhesion and migration,signal transduction
1.7 X05834 I I
Brevican core protein Chondroitin sulfateproteoglycan, extracellularmatrix, differentiation,gliotic response
−2.3 U37142 D X
Dermatan sulfateproteoglycan-II(decorin)
Small proteoglycan, bindingcollagen, fibronectin, andtgf-beta. Suppressing thegrowth of various tumorcell lines
∼10.6 Z12298 I X
EST206481 Calcium-binding of theextracellular matrix;inhibitor of calcification
5 AI012030 I I MGP rat matrix gla-proteinprecursor
100%, 102 aa
Osteopontin Ligand for integrinalpha-v/beta-3 and CD44
60.9 M14656 I I
Tenascin Extracellular matrixglycoprotein, a ligand forintegrins
∼7.7 U09401, U15550 I X
Syndecan Cell surface proteoglycanand receptor for theextracellular matrix, celladhesion, action of growthfactors
5.2 X60651, S61865 I I
Ryudocan�heparansulfate proteoglycancore protein
Cell surface proteoglycan,neutrophil migration,receptors or coreceptors
2.2 S61868 I I
Neuroglycan C precursor Chondroitin sulfateproteoglycan,CNS-specific functions
−2.3 U33553 D X
UDP-glucosedehydrogenase
Excellular matrixbiosynthesis
1.7 AB013732 I X
Protein disulphideisomerase (PDI)
Procollagen-prolinehydroxylation,extracellular matrixmaintenance
2.3 X02918 I I
Iodothyronine5-monodeiodinase(5-MD)
Procollagen-prolinehydroxylation,extracellular matrixmaintenance
3.5 M21476 I I
EST230748 Protein modification ofextracellular matrix, tumorsuppressor
1.8 AI234060 I X Protein-lysine 6-oxidaseprecursor
100%, 410 aa
Lysyl oxidase Protein modification ofextracellular matrix, tumorsuppression
∼12.6 S77494, S66184 I X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 801
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

Calpain is a calcium-activated nonlysosomal, intracel-lular cysteine protease that is induced and activated dur-ing cerebral ischemia (Neumar et al., 2001; Wang,2000). Calpains can compromise lysosomal membraneintegrity and lead to cathepsin B and L activation. Duringcerebral ischemia and neuronal apoptosis, cathepsin Band L were activated (Islekel et al., 1999; Yoshida et al.,2002). Cathepsin B activates caspase-3 via caspase-11during liver cell apoptosis, and cathepsin L activates a
caspase-3–like protease in liver cells. Calpain can alsodirectly activate caspase-12 and cause endoplasmic re-ticulum stress. Calpain and caspase-3 cleave severalcommon cytoskeletal, cytosolic, and nuclear protein sub-strates (Huang and Wang, 2001; Yamashima, 2000;Wang, 2000). It is not known whether caspase-11 or 12are activated after cerebral ischemia, but it is likely thatcalpain and some caspase-related proteases are effectorsof injury and tissue repair.
TABLE 12. Genes related to cytoskeletal proteins
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Lamina associatedpolypeptide 2 (LAP2)
Binding lamin B1 andchromosomes, regulationof nuclear architecture
1.7 U18314 I X
EST Lamin B receptor,lamin/chromatin binding
AI145490 I X Rat NBP60 100%, 619 aa
EST189270 Crosslinking actin tomembrane glycoproteins,transducing ligand-receptor binding to actinreorganization, neuronalmigration
∼13.9 AA799773 I X ABP2_human endothelialactin-binding protein
60%, 124 aa
EST Cross-linkers betweenplasma membranes andactin-based cytoskeletons,cell-cell recognition andsignaling and cellmovement
∼15.3 AF004811 I I Mouse moesin 99%, 576 aa
EST190459 Cytoskeletal protein, linkingcytoskeleton toextracellular matrixreceptors
∼7.1 AA800962 I X TALI_human talin 96%, 113 aa
Growth factor (Arc) Actin binding, endodermdevelopment
2.9 U19866 I I
Myosin regulatory lightchain (RLC)
Cytoskeleton organizationand biogenesis
2.1 X05566 I X
RLC-A gene for myosinregulatory light chain,exon 4
Cytoskeleton organizationand biogenesis
2.5 X54617 I X
SM22 Actin-binding proteins,muscle development
∼21 M83107 I X
Tropmyosin (TM-4) Binds actin filament 3 J02780 I XEST Tropomyosin, cytoskeletal
type, binds actin filament5 AA859305 I X Tropomyosin isoform 6-rat 100%, 247 aa
CLP36 (clp36) Cytoskeletal protein, adapter,actin binding
2.6 U23769 I X
Striated musclealpha-tropomyosin
Actin-binding andtroponin-binding proteins
1.9 X02412 I X
EST199921 Actin-binding proteins,inhibiting actomyosinMg(2+)-ATPase
2.4 AA944422 I I Calponin, acidic isoform 100%, 329 aa
Nestin Intermediate filament,neurogenesis
1.9 M34384 I I
Vimentin Intermediate filament,cytoskeletal protein
3.2 X62952 I I
Glial fibrillary acidic proteinalpha (GFAP) and glialfibrillary acidic proteindelta (GFAP)
Intermediate filament,cell-specific marker
∼22.6 AF028784
Small proline-rich protein(spr)
Intermediate filament, cellshape and cell size control
5.6 L46593 I I
EST195714 Member of the cornifiedenvelope precursor proteinfamily, structural protein,epidermal differentiation
13.5 AA891911 I X CORA rat cornifin alpha 100%, 151 aa
EST196136 Microtubule, cytoskeletalstructural protein
16.8 AA892333 I D Tubulin alpha-1 chain 100%, 450 aa
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.802
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

Cystatin B is an inhibitor of cathepsins B, H, and L(Cimerman et al., 2001). Cystatin B–deficient mice haveincreased expression of apoptosis/glial activation genes(Lieuallen et al., 2001), suggesting that Cystatin B mayprotect. Cathepsin C, a lysosomal cysteine protease, pro-cesses granzyme A and B. Granzymes are expressed inthe granules of activated cytotoxic lymphocytes, thegranules entering target cells via perforin after cytotoxiclymphocytes synapse with the target cells. Granzyme Bcan act upstream and downstream of caspases. GranzymeA causes apoptosis via a caspase-independent pathway(Podack, 1999). Since lymphocytes infiltrate ischemicbrain (Jander et al., 1995), these cells may mediate ap-optosis of target ischemic brain cells via these genes. It isnot known whether cystatins, cathepsins, and granzymesplay a role in neurons or glia after ischemia, althoughkeratinocytes can express both perforin and granzymes(Berthou et al., 1997).
Contrapsin-like protease inhibitor related protein (Cpi-26), a member of the ‘serpins‘ subfamily, is homologousto antichymotrypsin (Ohkubo et al., 1991). Antichymo-trypsin can inhibit caspase activity and is antiapoptotic(Ikari et al., 2001). If Cpi-26 functions like antichymo-trypsin, it may protect against focal cerebral ischemia.
Glycerol 3-phosphate dehydrogenase expression in-creased more than 60-fold at 1 d after focal ischemia.Glycerol 3-phosphate dehydrogenase is localized to mi-tochondria and is involved in mitochondrial hydrogenshuttles necessary for the reoxidation of glycolysis-derived NADH. Activated GPDH increases intracellularlevels of NADH and increases cellular resistance againstH2O2 injury (Hwang et al., 1999). It is possible thatGPDH and PARP interact to modulate NADH/NADPHlevels and cell survival and death.
Translation-related transcripts, including PHAS-I andpolypyrimidine track-binding protein (TBFII), were also
TABLE 13. Ion, vesicular transport, and synaptic transmission
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
Brain digoxin carrier protein Na(+)-independenttransporter of organicanions
∼−14.7 U88036 D I
EST Glycine-inhibited chloridechannel
−2 AI145044 D D Glycine receptor alpha-2chain variant A precursor
100%, 451 aa
Potassium channel, alphasubunit
Potassium channel regulator ∼−11 Y17606 D X
Serum and glucocorticoid-regulated kinase (sgk)
Protein serine/threoninekinase, sodium transport
2.2 L01624 I I
Sodium myo-inositoltransporter (SMIT)
Small molecule transport,regulation of membranepotential and tissueosmolyte
2.9 AJ001290 I I
Na-K-2Cl cotransporter(Nkcc1)
Sodium:chloride/potassium:chloridesymporter
5 AF086758 I X
Transferrin receptor Iron transport 2.9 M58040 I XCeruloplasmin Ferroxidase, iron efflux 5 L33869 I IEST Ferroxidase, iron efflux ∼5.1 AA817854 I X CERU rat ceruloplasmin
precursor100%, 1058 aa
Complexin II Docking protein, exocytosis,neurotransmitter release
−1.8 U35099 D X
Alpha-soluble NSFattachment protein
Membrane fusion,exocytosis, andintra-Golgi transport
−1.8 X89968 D X
B/K protein Calcium-dependentphospholipid binding
−3.6 U30831 D X
EST Membrane fusion, bindsphospholipid vesicles
−3.4 AI639118 D X Calcium-dependentactin-binding protein
80%, 41 aa
Synaptotagmin IV homolog Ca2+-dependent vesiculartrafficking and exocytosis
1.9 U14398 I I
S-100 related protein Calcium binding, annexin IIligand, exocytosis andendocytosis
5 J03627 I I
Clathrin assembly proteinshort form (CALM)
Retrieving synaptic vesicles 2.3 AF041373 I X
EST Synaptic transmission,synaptic uptake ofextracellular material
−2 AI072943 D D Neuronal pentraxin Iprecursor (np-I) (np1)(47 kd taipoxin-bindingprotein)
100%, 431 aa
Secretogranin II Packaging or sorting ofpeptide hormones andneuropeptides intosecretory vesicles
2.2 M93669 I I
Agrin Synaptogenesis, extracellularmatrix
−2 M64780 D X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 803
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003
induced after focal ischemia. PHAS-I binds eIF4E andnegatively regulates translation initiation. It dissociatesfrom eIF4E when phosphorylated. Insulin treatment ofadipose cells increased the phosphorylation of 4EBP1and reduced the interaction of 4EBP1 with eIF4E (Pauseet al., 1994). PHAS-I undergoes caspase-dependentcleavage in cells undergoing apoptosis. The N-terminaltruncated polypeptide binds to eIF4E and fails to become
sufficiently phosphorylated on insulin stimulation tobring about its release from eIF4E (Tee and Proud,2002). Binding initiation factor eIF4E can rapidly induceapoptosis (Herbert et al., 2000). During global cerebralischemia, eIF4E and 4E-BP1 were significantly dephos-phorylated (Martin de la Vega et al., 2001). PHAS-Ilevels and phosphorylation state could modulate isch-emic brain damage. Overexpression of polypyrimidine
TABLE 14. Genes related to cell adhesion, motility, and immune responses
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
CD14 Lps receptor, bindinglipopolysaccharide binding protein,lbp/lps complex, and apoptoticcells, triggering phagocytosis
10.3 AF087943, AF087944 I I
Lipopolysaccharidebinding protein
A carrier for LPS and controlLPS-dependent monocyteresponses, the lbp/lps complexseems to interact with the cd14receptor
∼15.3 L32132 I X
EST188970 Cell adhesion −2.1 AA799473 D X Lactadherin precursor(mfg-e8)
79%, 101 aa
Intercellular adhesionmolecule-1
Binding integrin alpha-I/beta-2 andleukocyte adhesion IFA-1 protein,cell adhesion
3 D00913 I I
P-Meta-1 mRNA forCD44 surfaceprotein
Cell surface receptor for hyaluronate,matrix adhesion, lymphocyteactivation and apoptosis
∼4 A30543 I I
Glycoprotein CD44(CD44)
Cell surface receptor for hyaluronate,matrix adhesion, lymphocyteactivation and apoptosis
∼35.9 M61875 I I
P41-Arc Assembly of the actin cytoskeleton,cell motility and cell elongation
6.2 AF083269 I I
Protein p9Kahomologous tocalcium-bindingprotein
Calcium binding, motility, invasion,and tubulin polymerization
3.8 X06916 I I
EST197807 Barbed-end actin capping,macrophage motile
2 AA894004 I X Mouse macrophage-capping protein
92%, 226 aa
MHC class IRT1.C-type protein
Human leukocyte antigen,presentation of foreign antigens
2.4 L40362 I I
Proteasome subunitRC1
26S proteasome, non-lysosomalproteolytic pathway, class I-restricted antigen presentation
∼6.9 D10729 I I
MHC class I antigen(RT1.EC2)
Human leukocyte antigen,presentation of foreign antigens
∼7.0 AF074608, M24324,M64795
I I
MHC class II antigenRT1.B beta chain
Human leukocyte antigen,presentation of foreign antigens
∼2.3 U65217 I I
MHC class II-like betachain (RT1.DMb)
Human leukocyte antigen,presentation of foreign antigens
5.4 U31599 I I
MHC-associatedinvariant chaingamma
Gamma chain antigen associatedwith MHC class II antigen
∼8.1 X13044 I I
Tumor-associatedglycoprotein E4(Tage4)
A member of the immunoglobulinsupergene family
∼7.9 L12025 I X
Fc-gamma receptor Fc fragment of IgG, low affinityIIIb, receptor
3.7 M32062 I X
Fc gamma receptor Receptor for the fc region ofimmunoglobulins gamma, lowaffinity receptor, downmodulationof cell activation state
∼11.2 X73371 I I
IgE binding protein Galactose-specific lectin, bindingIgE, cell growth regulation,chemoattractant activity
21.7 J02962 I I
Complement proteinC1q beta chain
Complement component 2.9 X71127 I I
Decay acceleratingfactor GPI-formprecursor (DAF)
Preventing the formation of c4b2aand c3bbb; inhibiting theamplification of the complementcascade
∼23.9 AF039583 I X
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
A. LU ET AL.804
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

track-binding protein stimulates Apaf-1 internal ribo-some entry site function (Mitchell et al., 2001). Polypy-rimidine track-binding protein also modulates alternativesplicing of caspase-2 (Cote et al., 2001), although therole for this gene in ischemic brain injury is unclear.
Narp expression, increased by 2- to 5-fold at 2 to 4 hof ischemia, increased more than 25-fold at 24 h afterischemia. Narp is a secreted immediate-early gene in-duced by a wide variety of stimuli. Since it is an extra-cellular aggregating factor for AMPA receptors (O’Brien
et al., 1999), it might exacerbate early ischemic excito-toxicity. Agrin, another aggregating factor, was de-creased after cerebral ischemia. Agrin is an extracellularheparan sulfate proteoglycan that signals AChR cluster-ing, particularly during the formation of the neuromus-cular junction. Agrin is also widely expressed in the cen-tral nervous system (Fong and Craig, 1999), althoughany effects of decreased agrin expression on central ace-tylcholine neurotransmission after ischemia are uncer-tain. The chronic changes of Narp and agrin and effects
TABLE 15. Others
Gene name Function summaryFold
changeGene
bank #
RNAchangein chips
RNAchangein paper Unigene/blast
Percent identity,aligned region
MG87 Iron-sulfur protein −1.8 AF095741 D X Probable iron-sulfur proteinb0947
32%, 279 aa
EST196401 Nucleotide binding 2.4 AA892598 I X /Putative nucleotide bindingprotein, estradiol-induced
EST221135 Compaction of DNA 2.1 AI177503 I X Human histone H3.3 100%, 135 aaEST Probably binding Ca2+ and an
unknown ligand2.6 AI639058 I X Human protein c18ORF1 73%, 141 aa
CD9 CD9 antigen, platelet activationand aggregation
2.1 X76489 I X
ad1-antigen Antigens, mast cell marker,platelet activation marker
2.9 X61654 I X
Urinary plasminogenactivator receptor 1(uPAR-1)
Receptor for urokinaseplasminogen activator,localizing and promotingplasmin formation,proteolysis-independentsignal transduction
∼16.3 X71898 I X
EST189300 Egf-like module 2.1 AA799803 I X Structure of the Egf-likemodule of human C1r, Nmr,19 structures
76%, 134 aa
Major vault protein Nucleocytoplasmic transport 3.1 U09870 I XSerine threonine kinase
(pim-3)Protein serine-threonine kinase,
developmental processes5.3 AF086624 I X
EST105188 Similar to protein kinases ∼4.1 H31287 I X GS3955 61%, 85aaIntracellular calcium-
binding protein(MRP14)
Inhibition of casein kinase ∼16.2 L18948 I X
EST229907 The domain of insulin growthfactor-binding proteinhomologues
∼5.2 AI233219 I X MEGF6 protein-rat 37%, 78 aa
Epithelial celltransmembraneprotein antigenprecursor (RTI40)
An apical integral plasmamembrane protein expressedin type I but not type IIalveolar epithelial cells
10.6 U92081 I I
EST197083 Globule surface membranematerial, indication ofadipocyte differentiation
12.3 AA893280 I X Mouse adipose differentiation-related protein
86%, 180 aa
Epithelial membraneprotein-1
Membrane glycoproteins, celldifferentiation
∼8.9 Z54212 I X
ASM15 Encoding an untranslated RNA,lies at the end of a cluster ofimprinted genes; tumorsuppressor activity
∼40.3 X59864 I I
Ri1 A LIM domain protein,LIM/double zinc fingerdomains
∼31.2 X76454 I X
Activity andneurotransmitter-induced early gene 6(ania-6)
Unclear; low similarity tocyclins
∼6.8 AF030091 I X
EST196323 Unclear 3.5 AA892520 I X /Membrane protein ofcholinergic synaptic vesicles(VATI), human BRCA1,Rho7 and vatI genes,complete cds, and ipf35gene, partial cds
RNA change in paper: RNA change reported in previous ischemic stroke paper. I, Increase; D, Decrease; −, Unchange; X, not reported.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 805
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

FIG. 4. (A) Percentages of the 328 genesregulated by ischemia in this study thatwere in each functional category. (B) Per-centages of the genes in each functionalcategory that were regulated more thanfivefold. (C) Percentages of genes in eachfunctional category that had not been re-ported in previous stroke studies. EST, ex-pressed sequence tag.
A. LU ET AL.806
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

on glutamate and acetylcholine neurotransmission mightalso play important roles in synaptic plasticity and re-covery after stroke.
Brain-derived neurotrophic factor RNA upregulationon the microarrays confirms many previous cerebralischemia studies. Brain-derived neurotrophic factor mayprotect against cerebral ischemia in part by phosphory-lation of ERK1/2 (Han and Holtzman, 2000; Encinas etal., 1999). Ischemia also induced the MAP-kinase phos-
phatase (cpg21) that inactivates ERK1 (Ishibashi et al.,1994). This might decrease the protective effects ofbrain-derived neurotrophic factor. However, ischemia-induced glutamate accumulation activates the P44/42MAP-kinase pathway. Since inhibition of the MAP-kinasepathways protected neurons against glutamate excitotox-icity (Grant et al., 2001), induction of the cpg21 MAP-kinase phosphatase may also protect against brain isch-emia. It is notable that cpg21 is induced more than 20-
FIG. 5. Real-time quantitative reverse transcription polymer-ase chain reaction showing the expression of selected genes(y-axis) at 2, 4, and 24 h after cerebral ischemia in the peri-infarction cerebral cortex. (A and B) Early upregulated genesthat show a twofold or greater increase of expression at 2 or4 h after cerebral ischemia compared with the sham-controls.(C and D) Late upregulated genes that showed a twofold orgreater increase of expression at 24 h after cerebral ischemiacompared with the sham-controls. (E) Downregulated genes.LPS, lipopolysaccharide; TBF, track-binding protein; DAF, de-cay accelerating factor.
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 807
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

fold after 2 h of ischemia and more than 120-fold by 24h of ischemia, suggesting an important but unstudied rolefor this gene.
Interleukin-6, Jak2, SOCS-3, and Pim-3 RNAs werealso regulated by cerebral ischemia. Interleukin-6 in-duces gp130-homodimerization and activates Jak1, Jak2,Tyk2, STAT1, and STAT3. STAT1 and STAT3 translocateto the nucleus to activate transcription (Heinrich et al.,1998). Interleukin-6 protects the brain against focal andglobal ischemia (Loddick et al., 1998; Matsuda et al.,1996). SOCS-3 binds all four JAK kinases and inhibitstheir activity (Chen et al., 2000), possibly suggesting thatSOCS-3 might counter the protective effects of IL-6. Co-expression of Pim kinases with Socs-1 results in phos-phorylation and stabilization of Socs-1 protein (Chen etal., 2002). Lymphocytes from SOCS-1−/− mice undergoaccelerated apoptosis associated with increased Bax, andmurine embryonic fibroblasts lacking SOCS-1 are moresensitive to TNF-induced cell death (Chen et al., 2000).Some Pim kinases prolong cell survival and inhibit ap-optosis activity (Lilly et al., 1999). Thus, although therole of SOCS-3 is uncertain, IL-6, Pim kinases, andSOCS-1 may protect against cerebral ischemia.
rNFIL-6 (C/EBP �) and CELF (C/EBP �) were highlyinduced. C/EBP plays a role in regulating other genes,since Egr-1 (NGFI-A) and MKP-1 mRNA were signifi-cantly reduced in liver of C/EBP knockout mice (Green-baum et al., 1998). Our data show that, after stroke, bothEgr-1 and oxidative stress-inducible protein tyrosinephosphatase (MKP-1) RNAs increased. The target genesof Egr-1, including PAI-1, VEGF, ICAM-1, IL1-�, andimmediate-early serum-responsive JE gene (MIP-1)were also induced. Recent lung studies show that Egr-1regulates many downstream genes and could have a cen-tral role in the pathogenesis of ischemic lung tissue dam-age (Yan et al., 2000). The present data support the pos-sibility that C/EBP regulation of Egr-1 (NGFI-A) and itsdownstream genes including PAI-1, VEGF, ICAM-1, IL-1�, and MIP-1 could be an important signaling pathwayin cerebral ischemia as well.
Transcripts for CD14, IgE binding protein (lectin),lectin-like oxidized LDL receptor (LOX-1), complementprotein C1q, PS-PLA1 (serine-phospholipid-selectivephospholipase A), and lipopolysaccharide binding pro-tein were all increased after cerebral ischemia. CD14,lectin, and LOX-1 are phagocyte transmembrane mol-ecules. CD14 and LOX-1 can bind phosphatidylserine(PtdSer) and other surface molecules to initiate apopto-sis. C1q is an adhesive “bridging” molecule betweenapoptotic cells and phagocytes. These molecules cantether apoptotic cells and shuttle them to the phagocyticmachinery (Savill and Fadok, 2000; Oka et al., 1998).PS-PLA1 hydrolyzes a fatty acyl residue at the sn-1 po-sition of lysophosphatidylserine and phosphatidylserine(Sato et al., 1997). Lipopolysaccharide binding protein
can bind CD14 (Gutsmann et al., 2001). The increase ofthe latter molecules can decrease phagocytic recognitionof apoptotic cells, which can enhance cell survival whencells are subjected to weak proapoptotic signals (Hoepp-ner et al., 2001). During cerebral ischemia, microglia–macrophages are markedly activated (Stevens et al.,2002). Although inhibition of microglial activation is re-ported to protect against global brain ischemia (Yrjan-heikki et al., 1998), macrophage and microglial engulf-ment of apoptotic cells can suppress the secretion ofproinflammatory mediators such as tumor necrosis fac-tor-� and suppress the inflammatory response to isch-emia (Savill and Fadok, 2000). There seems to be abalance of the detrimental and neuroprotective effects ofactivated microglia and macrophages that may vary overtime and with the severity of injury (Stoll et al., 1998).Exactly how these pathways influence acute injury orrecovery after stroke is still unclear.
The focus of this study was to examine the periinfarc-tion cortex at 24 h after a permanent MCA occlusion, ata time well beyond that when any treatment for acutestroke would likely be useful. The changes in many ofthe 328 transcripts may be related to the onset of tissuerepair and the beginning of recovery. As many as two tothree times as many molecules may be identified oncethe entire rat and human genomes are studied as a func-tion of time and area of brain injured after stroke. Futureapproaches at defining those pathways crucial for recov-ery might include looking for genes shared with toler-ance models, models where recovery is improved or ispoor, and other approaches that will help refine thesearch for clinically important pathways. We have pre-viously reported genes that are induced in common byseveral types of injury in brain and in blood (Tang et al.,2001, 2002). These common genes may relate to com-mon mechanisms of injury and could represent commontargets for a common therapy.
Acknowledgments: The authors thank Melinda Reilly forexcellent technical assistance, and regret the inability to refer-ence all of the publications related to the regulated genes in thisstudy.
REFERENCES
Albers GW, Goldstein LB, Hall D, Lesko LM (2001) Aptiganel hy-drochloride in acute ischemic stroke: a randomized controlled trial.JAMA 286:2673–2682
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM,Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP,Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE,Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: toolfor the unification of biology. The Gene Ontology Consortium. NatGenet 25:25–29
Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ (1998) Core andpenumbral nitric oxide synthase activity during cerebral ischemiaand reperfusion. Stroke 29:1037–1046
Barber PA, Auer RN, Buchan AM, Sutherland GR (2001) Understand-ing and managing ischemic stroke. Can J Physiol Pharmacol79:283–296
A. LU ET AL.808
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D,Onteniente B (2001) Specific caspase pathways are activated in thetwo stages of cerebral infarction. J Neurosci 21:7127–7134
Berthou C, Michel L, Soulie A, Jean-Louis F, Flageul B, Dubertret L,Sigaux F, Zhang Y, Sasportes M (1997) Acquisition of granzymeB and Fas ligand proteins by human keratinocytes contributes toepidermal cell defense. J Immunol 159:5293–5300
Butler D (2002) Piecing it all together. Nature 420:460Chen XP, Losman JA, Rothman P (2000) SOCS proteins, regulators of
intracellular signaling. Immunity 13:287–290Chen XP, Losman JA, Cowan S, Donahue E, Fay S, Vuong BQ,
Nawijn MC, Capece D, Cohan VL, Rothman P (2002) Pimserine/threonine kinases regulate the stability of Socs-1 protein.Proc Natl Acad Sci U S A 99:2175–2180
Chuquet J, Benchenane K, Liot G, Fernandez-Monreal M, Toutain J,Blanchet S, Eveno E, Auffray C, Pietu G, Buisson A, Touzani O,MacKenzie ET, Vivien D (2002) Matching gene expression withhypometabolism after cerebral ischemia in the nonhuman primate.J Cereb Blood Flow Metab 22:1165–1169
Cimerman N, Mesko Brguljan P, Krasovec M, Suskovic S, Kos J(2001) Serum concentration and circadian profiles of cathepsins B,H and L, and their inhibitors, stefins A and B, in asthma. Clin ChimActa 310:113–122
Cote J, Dupuis S, Wu JY (2001) Polypyrimidine track-binding proteinbinding downstream of caspase-2 alternative exon 9 represses itsinclusion. J Biol Chem 276:8535–8543
De Keyser J, Sulter G, Luiten PG (1999) Clinical trials with neuropro-tective drugs in acute ischemic stroke: are we doing the right thing?Trends Neurosci 22:535–540
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ische-mic stroke: an integrated view. Trends Neurosci 22:391–397
Encinas M, Iglesias M, Llecha N, Comella JX (1999) Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved inbrain-derived neurotrophic factor-mediated survival and neurito-genesis of the neuroblastoma cell line SH-SY5Y. J Neurochem73:1409–1421
Fisher M, Schaebitz W (2000) An overview of acute stroke therapy:past, present, and future. Arch Intern Med 160:3196–3206
Fong DK, Craig AM (1999) The Narp hypothesis? Neuron 23:195–197Gill R, Soriano M, Blomgren K, Hagberg H, Wybrecht R, Miss MT,
Hoefer S, Adam G, Niederhauser O, Kemp JA, Loetscher H (2002)Role of caspase-3 activation in cerebral ischemia-induced neuro-degeneration in adult and neonatal brain. J Cereb Blood FlowMetab 22:420–430
Grant ER, Errico MA, Emanuel SL, Benjamin D, McMillian MK,Wadsworth SA, Zivin RA, Zhong Z (2001) Protection againstglutamate toxicity through inhibition of the p44/42 mitogen-activated protein kinase pathway in neuronally differentiated P19cells. Biochem Pharmacol 62:283–296
Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V,Taub R (1998) CCAAT enhancer-binding protein � is required fornormal hepatocyte proliferation in mice after partial hepatectomy.J Clin Invest 102:996–1007
Gutsmann T, Muller M, Carroll SF, MacKenzie RC, Wiese A, SeydelU (2001) Dual role of lipopolysaccharide (LPS)-binding protein inneutralization of LPS and enhancement of LPS-induced activationof mononuclear cells. Infect Immun 69:6942–6950
Han BH, Holtzman DM (2000) BDNF protects the neonatal brain fromhypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci20:5775–5781
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L(1998) Interleukin-6-type cytokine signaling through thegp130/Jak/STAT pathway. Biochem J 334:297–314
Hengartner MO (2000) The biochemistry of apoptosis. Nature407:770–776
Herbert TP, Fahraeus R, Prescott A, Lane DP, Proud CG (2000) Rapidinduction of apoptosis mediated by peptides that bind initiationfactor eIF4E. Curr Biol 10:793–796
Hoeppner DJ, Hengartner MO, Schnabel R (2001) Engulfment genescooperate with ced-3 to promote cell death in Caenorhabditis el-egans. Nature 412:202–206
Huang Y, Wang KK (2001) The calpain family and human disease.Trends Mol Med 7:355–362
Hwang K, Jeong DW, Lee JW, Kim IH, Chang HI, Kim HJ, Kim IY(1999) Alteration of the NAD+/NADH ratio in CHO cells by stabletransfection with human cytosolic glycerol-3-phosphate dehydro-genase: resistance to oxidative stress. Mol Cells 9:429–435
Ikari Y, Mulvihill E, Schwartz SM (2001) Alpha 1-Proteinase inhibitor,alpha 1-antichymotrypsin, and alpha 2-macroglobulin are the an-tiapoptotic factors of vascular smooth muscle cells. J Biol Chem276:11798–11803
Ishibashi T, Bottaro DP, Michieli P, Kelley CA, Aaronson SA (1994)A novel dual specificity phosphatase induced by serum stimulationand heat shock. J Biol Chem 269:29897–29902
Islekel H, Islekel S, Guner G, Ozdamar N (1999) Evaluation of lipidperoxidation, cathepsin L and acid phosphatase activities in ex-perimental brain ischemia-reperfusion. Brain Res 843:18–24
Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G (1995) Lym-phocytic infiltration and expression of intercellular adhesion mol-ecule-1 in photochemically induced ischemia of the rat cortex. JCereb Blood Flow Metab 15:42–51
Jin K, Mao XO, Eshoo MW, Nagayama T, Minami M, Simon RP,Greenberg DA (2001) Microarray analysis of hippocampal geneexpression in global cerebral ischemia. Ann Neurol 50:93–103
Juan TS, McNiece IK, Jenkins NA, Gilbert DJ, Copeland NG, FletcherFA (1996) Molecular characterization of mouse and rat CPP32 �gene encoding a cysteine protease resembling interleukin-1� con-verting enzyme and CED-3. Oncogene 13:749–755
Keyvani K, Witte OW, Paulus W (2002) Gene expression profiling inperilesional and contralateral areas after ischemia in rat brain. JCereb Blood Flow Metab 22:153–160
Kim YD, Sohn NW, Kang C, Soh Y (2002) DNA array reveals alteredgene expression in response to focal cerebral ischemia. Brain ResBull 58:491–498
Lieuallen K, Pennacchio LA, Park M, Myers RM, Lennon GG (2001)Cystatin B-deficient mice have increased expression of apoptosisand glial activation genes. Hum Mol Genet 10:1867–1871
Lilly M, Sandholm J, Cooper JJ, Koskinen PJ, Kraft A (1999) ThePIM-1 serine kinase prolongs survival and inhibits apoptosis-related mitochondrial dysfunction in part through a bcl-2-dependent pathway. Oncogene 18:4022–4031
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev79:1431–1568
Loddick SA, Turnbull AV, Rothwell NJ (1998) Cerebral interleukin-6is neuroprotective during permanent focal cerebral ischemia in therat. J Cereb Blood Flow Metab 18:176–179
Loetscher H, Niederhauser O, Kemp J, Gill R (2001) Is caspase-3inhibition a valid therapeutic strategy in cerebral ischemia? DrugDiscov Today 6:671–680
Martin de la Vega C, Burda J, Nemethova M, Quevedo C, Alcazar A,Martin ME, Danielisova V, Fando JL, Salinas M (2001) Possiblemechanisms involved in the down-regulation of translation duringtransient global ischemia in the rat brain. Biochem J 357:819–826
Matsuda S, Wen TC, Morita F, Otsuka H, Igase K, Yoshimura H,Sakanaka M (1996) Interleukin-6 prevents ischemia-induced learn-ing disability and neuronal and synaptic loss in gerbils. NeurosciLett 204:109–112
Mitchell SA, Brown EC, Coldwell MJ, Jackson RJ, Willis AE (2001)Protein factor requirements of the Apaf-1 internal ribosome entrysegment: roles of polypyrimidine tract binding protein and up-stream of N-ras. Mol Cell Biol 21:3364–3374
Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, YuanJ, Moskowitz MA (1998) Activation and cleavage of caspase-3 inapoptosis induced by experimental cerebral ischemia. J Neurosci18:3659–3668
Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA,Siman R (2001) Calpain activity in the rat brain after transientforebrain ischemia. Exp Neurol 170:27–35
Noordewier MO, Warren PV (2001) Gene expression microarrays andthe integration of biological knowledge. Trends Biotechnol19:412–415
O’Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P(1999) Synaptic clustering of AMPA receptors by the extracellularimmediate-early gene product Narp. Neuron 23:309–323
Ohkubo K, Ogata S, Misumi Y, Takami N, Ikehara Y (1991) Molecular
GENOMICS OF PERIINFARCTION CORTEX AFTER ISCHEMIA 809
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003

cloning and characterization of rat contrapsin-like protease inhibi-tor and related proteins. J Biochem (Tokyo) 109:243–250
Oka K, Sawamura T, Kikuta K, Itokawa S, Kume N, Kita T, Masaki T(1998) Lectin-like oxidized low-density lipoprotein receptor 1 me-diates phagocytosis of aged/apoptotic cells in endothelial cells.Proc Natl Acad Sci U S A 95:9535–9540
Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC Jr,Sonenberg N (1994) Insulin-dependent stimulation of protein syn-thesis by phosphorylation of a regulator of 5�-cap function. Nature371:762–767
Phan TG, Wright PM, Markus R, Howells DW, Davis SM, Donnan GA(2002) Salvaging the ischemic penumbra: more than just reperfu-sion? Clin Exp Pharmacol Physiol 29:1–10
Podack ER (1999) How to induce involuntary suicide: the need fordipeptidyl peptidase I. Proc Natl Acad Sci U S A 96:8312–8314
Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, Weinstein PR,Sharp FR (2000) Mice overexpressing rat heat shock protein 70 areprotected against cerebral infarction. Ann Neurol 47:782–791
Sato T, Aoki J, Nagai Y, Dohmae N, Takio K, Doi T, Arai H, Inoue K(1997) Serine phospholipid-specific phospholipase A that is se-creted from activated platelets: a new member of the lipase family.J Biol Chem 272:2192–2198
Savill J, Fadok V (2000) Corpse clearance defines the meaning of celldeath. Nature 407:784–788
Schwarz DA, Barry G, Mackay KB, Manu F, Naeve GS, Vana AM,Verge G, Conlon PJ, Foster AC, Maki RA (2002) Identification ofdifferentially expressed genes induced by transient ischemicstroke. Brain Res Mol Brain Res 101:12–22
Sharp FR, Lu A, Tang Y, Millhorn DE (2000) Multiple molecularpenumbras after focal cerebral ischemia. J Cereb Blood FlowMetab 20:1011–1032
Soriano MA, Tessier M, Certa U, Gill R (2000) Parallel gene expres-sion monitoring using oligonucleotide probe arrays of multipletranscripts with an animal model of focal ischemia. J Cereb BloodFlow Metab 20:1045–1055
Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP(2002) The use of flow cytometry to evaluate temporal changes in
inflammatory cells following focal cerebral ischemia in mice.Brain Res 932:110–119
Stoll G, Jander S, Schroeter M (1998) Inflammation and glial responsesin ischemic brain lesions. Prog Neurobiol 56:149–171
Tang Y, Lu A, Aronow BJ, Sharp FR (2001) Blood genomic responsesdiffer after stroke, seizures, hypoglycemia, and hypoxia: bloodgenomic fingerprints of disease. Ann Neurol 50:699–707
Tang Y, Lu A, Aronow BJ, Wagner KR, Sharp FR (2002) Genomicresponses of the brain to ischemic stroke, intracerebral hemor-rhage, kainate seizures, hypoglycemia, and hypoxia. Eur J Neuro-sci 15:1937–1952
Tee AR, Proud CG (2002) Caspase cleavage of initiation factor 4E-binding protein 1 yields a dominant inhibitor of cap-dependenttranslation and reveals a novel regulatory motif. Mol Cell Biol22:1674–1683
Wang KK (2000) Calpain and caspase: can you tell the difference?Trends Neurosci 23:20–26
White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Gross-man LI, Rafols JA, Krause GS (2000) Brain ischemia and reper-fusion: molecular mechanisms of neuronal injury. J Neurol Sci179:1–33
Yamashima T (2000) Implication of cysteine proteases calpain, cathep-sin and caspase in ischemic neuronal death of primates. Prog Neu-robiol 62:273–295
Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ,Stern DM (2000) Egr-1, a master switch coordinating upregulationof divergent gene families underlying ischemic stress. Nat Med6:1355–1361
Yoshida M, Yamashima T, Zhao L, Tsuchiya K, Kohda Y, TonchevAB, Matsuda M, Kominami E (2002) Primate neurons show dif-ferent vulnerability to transient ischemia and response to cathepsininhibition. Acta Neuropathol (Berl) 104:267–272
Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J (1998)Tetracyclines inhibit microglial activation and are neuroprotectivein global brain ischemia. Proc Natl Acad Sci U S A 95:15769–15774
A. LU ET AL.810
J Cereb Blood Flow Metab, Vol. 23, No. 7, 2003
Related Documents











