Gene expression Gene expression Terry Speed Lecture 4, December 18, 2001

Gene expression Terry Speed Lecture 4, December 18, 2001.
Dec 20, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Gene expressionGene expression
Terry Speed
Lecture 4, December 18, 2001

Thesis:Thesis: the analysis of gene the analysis of gene expression data is going to be big expression data is going to be big
in 21st century statisticsin 21st century statistics
Many different technologies, including
High-density nylon membrane arrays
Serial analysis of gene expression (SAGE)
Short oligonucleotide arrays (Affymetrix)
Long oligo arrays (Agilent)
Fibre optic arrays (Illumina)
cDNA arrays (Brown/Botstein)*
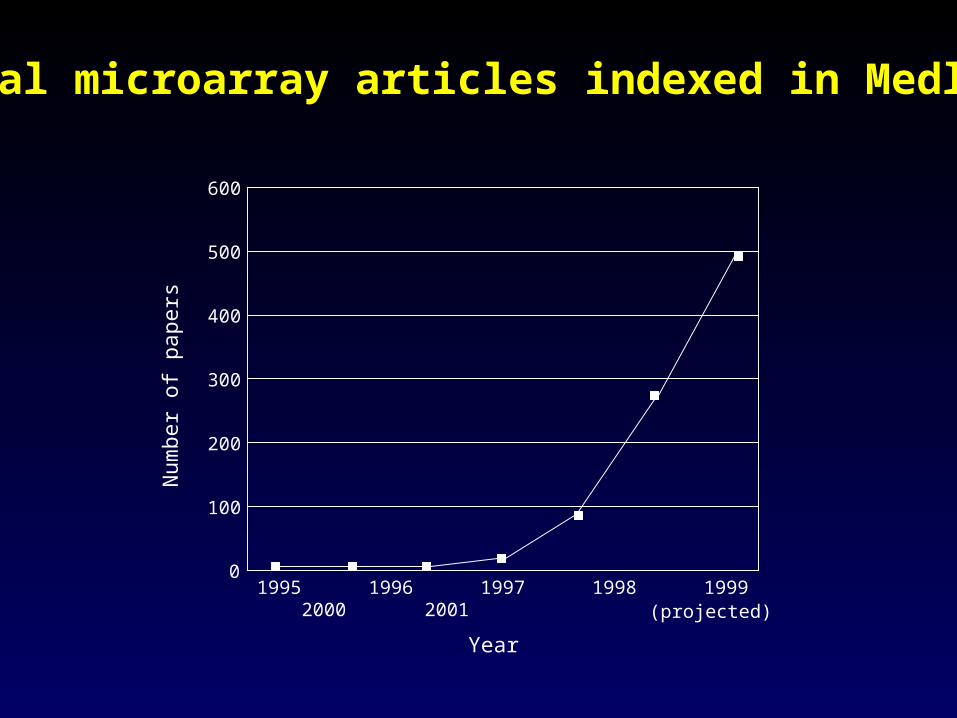
1995 1996 1997 1998 1999 2000 2001
0
100
200
300
400
500
600
(projected)
Year
Num
ber
of
papers
Total microarray articles indexed in Medline

Common themes themes
• Parallel approach to collection of very large amounts of data (by biological standards)
• Sophisticated instrumentation, requires some understanding
• Systematic features of the data are at least as important as the random ones
• Often more like industrial process than single investigator lab research
• Integration of many data types: clinical, genetic, molecular…..databases
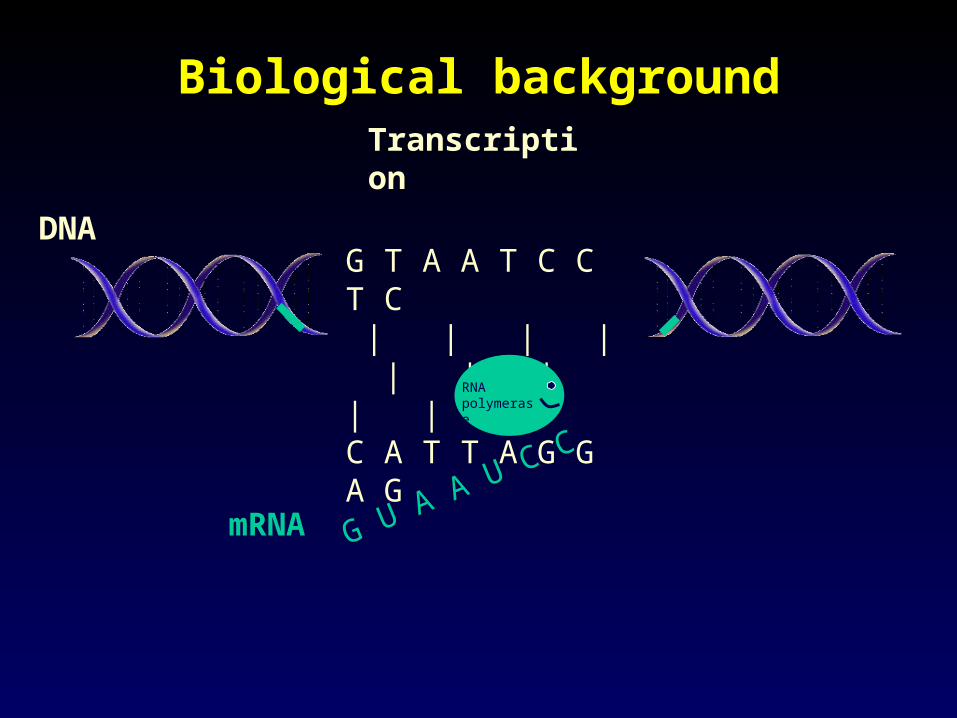
Biological backgroundBiological background
G T A A T C C T C | | | | | | | | | C A T T A G G A G
DNA
G U A A U C C
RNA polymerase
mRNA
Transcription
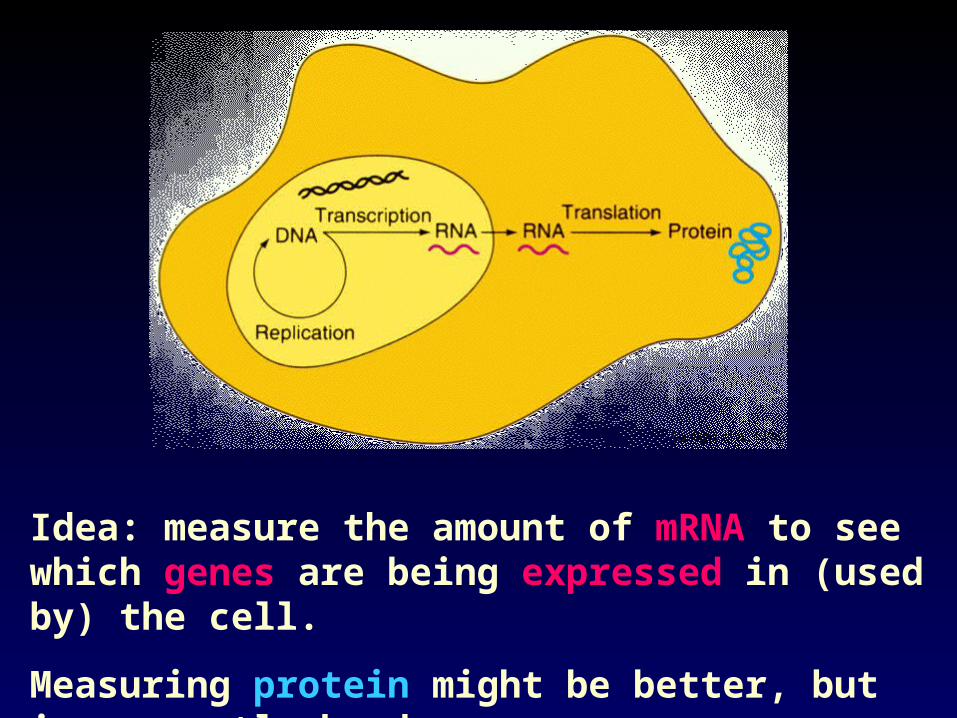
Idea: measure the amount of mRNA to see which genes are being expressed in (used by) the cell.
Measuring protein might be better, but is currently harder.

Reverse transcriptionReverse transcriptionClone cDNA strands, complementary to the mRNA
G U A A U C C U C
Reverse transcriptase
mRNA
cDNA
C A T T A G G A G C A T T A G G A G C A T T A G G A G C A T T A G G A G
T T A G G A G
C A T T A G G A G C A T T A G G A G C A T T A G G A G
C A T T A G G A G
C A T T A G G A G

cDNA microarray experimentscDNA microarray experiments
mRNA levels compared in many different contexts
Different tissues, same organism (brain v. liver) Same tissue, same organism (ttt v. ctl, tumor v. non-tumor) Same tissue, different organisms (wt v. ko, tg, or mutant)
Time course experiments (effect of ttt, development)
Other special designs (e.g. to detect spatial patterns).

cDNA microarrayscDNA microarrays
cDNA clones

cDNA microarrayscDNA microarraysCompare the genetic expression in two samples of cells
cDNA from one gene on each spot
SAMPLES
cDNA labelled red/green
e.g. treatment / control
normal / tumor tissue

HYBRIDIZE
Add equal amounts of labelled cDNA samples to microarray.
SCAN
Laser Detector

Biological questionDifferentially expressed genesSample class prediction etc.
Testing
Biological verification and interpretation
Microarray experiment
Estimation
Experimental design
Image analysis
Normalization
Clustering Discrimination
R, G
16-bit TIFF files
(Rfg, Rbg), (Gfg, Gbg)

Some statistical questionsSome statistical questions
Image analysis: addressing, segmenting, quantifying Normalisation: within and between slides
Quality: of images, of spots, of (log) ratios
Which genes are (relatively) up/down regulated?
Assigning p-values to tests/confidence to results.

Some statistical questions, ctdSome statistical questions, ctd
Planning of experiments: design, sample size
Discrimination and allocation of samples
Clustering, classification: of samples, of genes
Selection of genes relevant to any given analysis
Analysis of time course, factorial and other special experiments…..…...& much more.

Some bioinformatic questionsSome bioinformatic questions
Connecting spots to databases, e.g. to sequence, structure, and pathway databases
Discovering short sequences regulating sets of genes: direct and inverse methods
Relating expression profiles to structure and function, e.g. protein localisation
Identifying novel biochemical or signalling pathways, ………..and much more.

Part of the image of one channel false-coloured on a white (v. high) red (high) through yellow and green (medium) to blue (low) and black scale

Does one size fit all?

Segmentation: limitation of the Segmentation: limitation of the fixed circle methodfixed circle method
SRG Fixed Circle
Inside the boundary is spot (foreground), outside is not.

Some local backgroundsSome local backgrounds
We use something different again: a smaller, less variable value.
Single channelgrey scale

Quantification of expressionQuantification of expression
For each spot on the slide we calculate
Red intensity = Rfg - Rbg
fg = foreground, bg = background, and
Green intensity = Gfg - Gbg
and combine them in the log (base 2) ratio
Log2( Red intensity / Green intensity)

Gene Expression DataGene Expression Data On p genes for n slides: p is O(10,000), n is O(10-100), but growing,
Genes
Slides
Gene expression level of gene 5 in slide 4
= Log2( Red intensity / Green intensity)
slide 1 slide 2 slide 3 slide 4 slide 5 …
1 0.46 0.30 0.80 1.51 0.90 ...2 -0.10 0.49 0.24 0.06 0.46 ...3 0.15 0.74 0.04 0.10 0.20 ...4 -0.45 -1.03 -0.79 -0.56 -0.32 ...5 -0.06 1.06 1.35 1.09 -1.09 ...
These values are conventionally displayed on a red (>0) yellow (0) green (<0) scale.


The red/green ratios can be spatially biasedThe red/green ratios can be spatially biased
• .Top 2.5%of ratios red, bottom 2.5% of ratios green
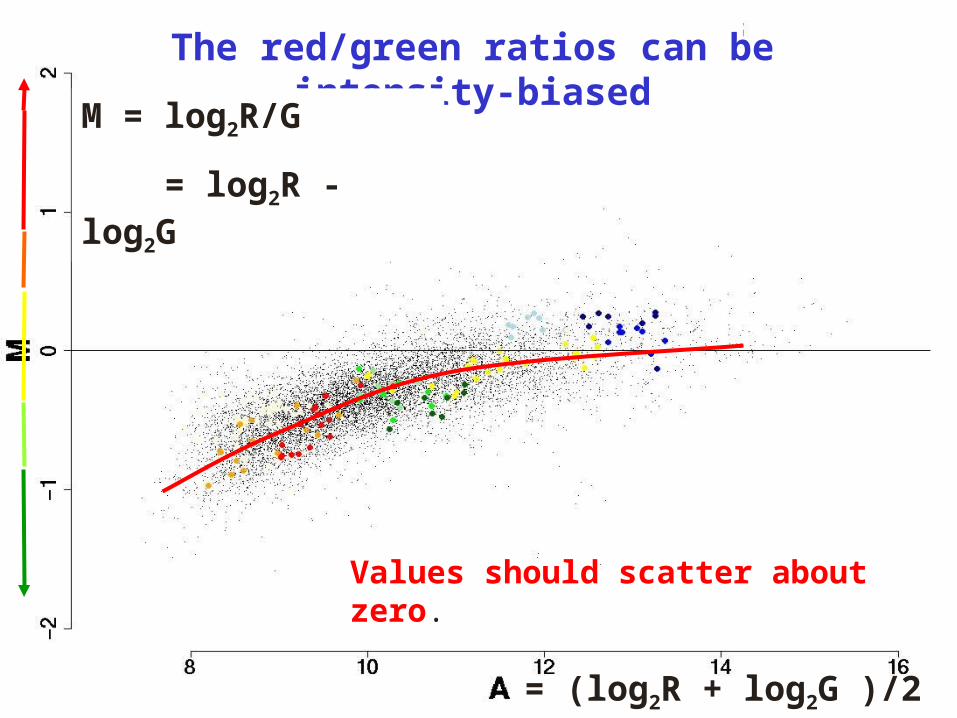
The red/green ratios can be intensity-biased
M = log2R/G
= log2R - log2G
= (log2R + log2G )/2
Values should scatter about zero.

Yellow: GAPDH, tubulin Light blue: MSP pool / titration
Orange: Schadt-Wong rank invariant set Red line: lowess smooth
Normalization: how we “fix” the previous problem
The curved line becomes the new zero line
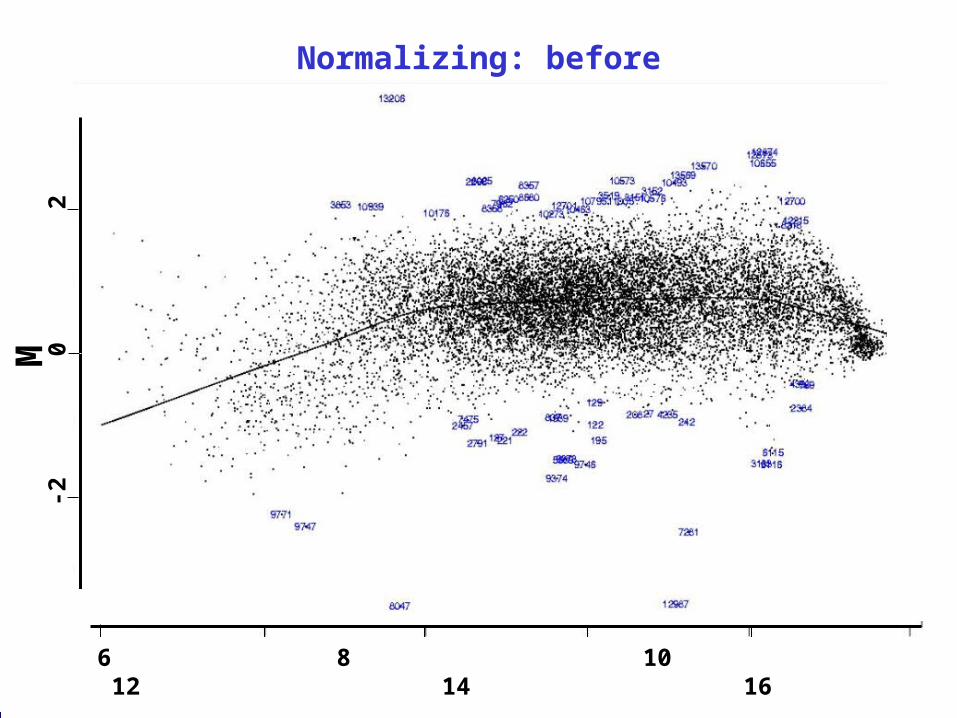
Normalizing: before2
0-2
-4
6 8 10 12 14 16
M

Normalizing: after2
0-2
-4
M n
orm
alis
ed
6 8 10 12 14 16

SCIENTIFIC: To determine which genes are differentially expressed between two sources of mRNA (trt, ctl).
STATISTICAL: To assign appropriately adjusted p-values to thousands of genes.
A basic problemA basic problem

• 8 treatment mice and 8 control mice
• 16 hybridizations: liver mRNA from each of the 16 mice (Ti , Ci ) is labelled with Cy5, while pooled liver mRNA from the control mice (C*) is labelled with Cy3.
• Probes: ~ 6,000 cDNAs (genes), including 200 related to lipid metabolism.
Goal. To identify genes with altered expression in the livers of Apo AI knock-out mice (T) compared to inbred C57Bl/6 control mice (C).
Apo AI experiment (Callow Apo AI experiment (Callow et alet al 2000, LBNL) 2000, LBNL)

Leukemia experiments (Golub Leukemia experiments (Golub et alet al 1999,WI) 1999,WI)
Goal. To identify genes which are differentially expressed in acute lymphoblastic leukemia (ALL) tumours in comparison with acute myeloid leukemia (AML) tumours.
• 38 tumour samples: 27 ALL, 11 AML.• Data from Affymetrix chips, some pre-processing.• Originally 6,817 genes; 3,051 after reduction.
Data therefore a 3,051 38 array of expression values.

Univariate hypothesis testingUnivariate hypothesis testing
Initially, focus on one gene only.
We wish to test the null hypothesis H that the gene is not differentially expressed.
In order to do so, we use a two sample t-statistic:
€
t=averofn1 trtx − averofn2ctlx
[1n1
(SDofn1trtx)2 +
1n1
(SDofn1ctlx)2]

Single-step adjustments of Single-step adjustments of pi
• Bonferroni: min (mpi, 1), m= #genes
•Sidák: 1 - (1 - pi)m
minP method of Westfall and Young:
Pr( min Pl ≤ pi | H)
1≤l≤m
• maxT method of Westfall and Young:
Pr( max |Tl | ≥ | ti | | H0C )
1≤l≤m

More powerful methods: More powerful methods: step-down adjustmentsstep-down adjustments
The idea: S Holm’s modification of Bonferroni.
Also applies to Sidák, maxT, and minP.
We illustrate this last adjustment.

Step-down adjustment of Step-down adjustment of minPminP
Initialization: Order the unadjusted p-values such that pr1 ≤ pr2 ≤ ≤ prm. The indices r1, r2, r3,.. are fixed for given data.
Step-down adjustment:
1. Compare min {Pr1, , Prm} with pr1 ;
2. Compare min {Pr2, , Prm} with pr2 ;
3 Compare min {Pr3 , Prm} with pri3 …….
m. Compare Prm with prm
Enforce the monotonicity on the adjusted pri


gene t unadj. p minP plower maxT
index statistic (104) adjust. adjust.
2139 -22 1.5 .53 8 10-5 2 10-4
4117 -13 1.5 .53 8 10-5 5 10-4
5330 -12 1.5 .53 8 10-5 5 10-4
1731 -11 1.5 .53 8 10-5 5 10-4
538 -11 1.5 .53 8 10-5 5 10-4
1489 -9.1 1.5 .53 8 10-5 1 10-3
2526 -8.3 1.5 .53 8 10-5 3 10-3
4916 -7.7 1.5 .53 8 10-5 8 10-3
941 -4.7 1.5 .53 8 10-5 0.65
2000 +3.1 1.5 .53 8 10-5 1.00
5867 -4.2 3.1 .76 0.54 0.90
4608 +4.8 6.2 .93 0.87 0.61
948 -4.7 7.8 .96 0.93 0.66
5577 -4.5 12 .99 0.93 0.74

Apo AI. Histogram & Q-Q plotApo AI. Histogram & Q-Q plot
ApoA1


Brief discussion
Not mentioned: strong vs weak control of Type 1 error.
The minP adjustment seems more conservative than the maxT adjustment, but is essentially model-free.
The adjusted minP values are very discrete; it seems that 12,870 permutations are not enough for 6,000 tests.
Extends to other statistics: Wilcoxon, paired t, F, blocked F..
Major question in practice: minP, maxT or something else?
Wanted are guidelines for use of minP in terms of sample sizes and number of genes.
Other approaches: False Discovery Rate (V/R), Bayes.

Olfactory Epithelium
VomeroNasal Organ
Main (Auxiliary)Olfactory Bulb
From Buck (2000)
From a study of the mouse olfactory system

Axonal connectivity between the nose and the mouse olfactory bulb
>2M, ~1,800 types
Two principles: “zone-to-zone projection”, and “glomerular convergence”
Neocortex

Of interest: the hardwiring of the Of interest: the hardwiring of the vertebrate olfactory systemvertebrate olfactory system
• Expression of a specific odorant receptor gene by an olfactory neuron.
• Targeting and convergence of like axons to specific glomeruli in the olfactory bulb.

The biological question in this caseThe biological question in this case
Are there genes with spatially restricted expression patterns within
the olfactory bulb?
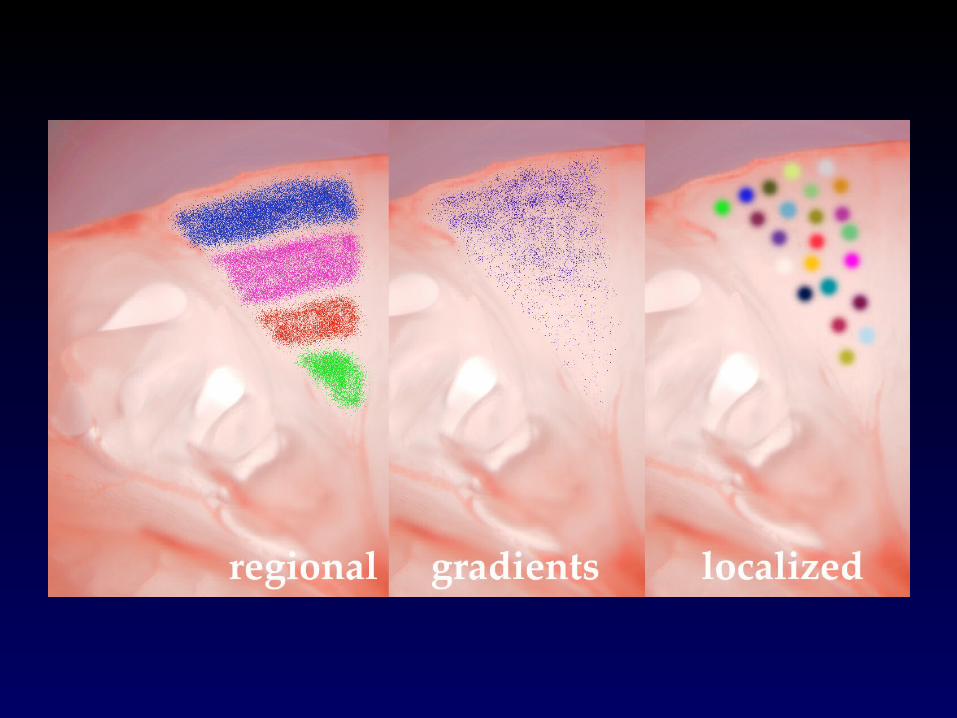

Layout of the cDNA MicroarraysLayout of the cDNA Microarrays
• Sequence verified mouse cDNAs• 19,200 spots in two print groups of 9,600 each
– 4 x 4 grid, each with 25 x24 spots– Controls on the first 2 rows of each grid.
77
pg1 pg2

Design: How We Sliced Up the BulbDesign: How We Sliced Up the Bulb
A
P D
V
M
L

Design: Two Ways to Do the Design: Two Ways to Do the ComparisonsComparisons
Goal: 3-D representation of gene expression
P
D
MA
V
L
R
Compare all samples to a common reference sample (e.g., whole bulb)
P
D
MA
V
L
Multiple direct comparisons between different samples (no common reference)

An Important Aspect of Our DesignAn Important Aspect of Our Design
Different ways of estimating the same contrast:
e.g. A compared to P
Direct = A-P
Indirect = A-M + (M-P) or
A-D + (D-P) or
-(L-A) - (P-L)
How do we combine these?
LL
PPVV
DD
MM
AA

Analysis using a linear model
Define a matrix X so that E(M)=X
Use least squares estimates for A-L, P-L, D-L, V-L, M-LIn practice, we use robust regression.
Estimates for other estimable contrasts follow in the usual way.
€
E
m1
m2
M
mn
⎛
⎝
⎜ ⎜ ⎜
⎞
⎠
⎟ ⎟ ⎟ =
0 0 0 −1 1
−1 0 0 0 0
M O M
−1 1 0 0 0
⎛
⎝
⎜ ⎜ ⎜
⎞
⎠
⎟ ⎟ ⎟ •
A−L
P −L
D−L
V −L
M−L
⎛
⎝
⎜ ⎜ ⎜ ⎜
⎞
⎠
⎟ ⎟ ⎟ ⎟
ˆ = X' X( )−1X'M

The Olfactory Bulb ExperimentsThe Olfactory Bulb Experiments completed so farcompleted so far
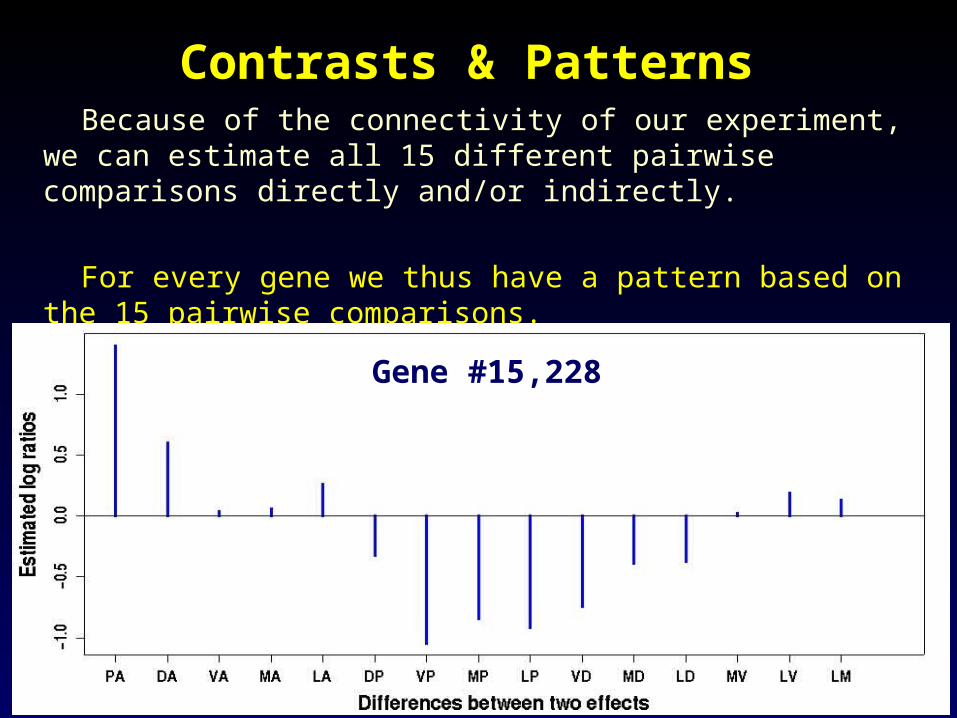
Contrasts & PatternsContrasts & Patterns Because of the connectivity of our experiment, we can estimate
all 15 different pairwise comparisons directly and/or indirectly.
For every gene we thus have a pattern based on the 15 pairwise comparisons.
Gene #15,228

Contrasts & patterns:another wayContrasts & patterns:another way Instead of estimating pairwise comparisons between each of the six
effects, we can come closer to estimating the effects themselves by doing so subject to the standard zero sum constraint (6 parameters, 5 d.f.).
What we estimate for A, say, subject to this constraint, is in reality an estimate of
A - 1/6(A + P + D + V + M + L).
This set of parameter estimates gives results similar to, but better than, the ones we would have obtained had we carried out the experiments with whole-bulb reference tissue.
In effect we have created the whole-bulb reference in silico.

Alternative pattern representationAlternative pattern representation
Gene #15,228 once again.

Reconstruction of the Bulb as a Cube:Reconstruction of the Bulb as a Cube:Expression of Gene # 15,228Expression of Gene # 15,228
ExpressionLevel
High
Low

Patterns, More Globally...Patterns, More Globally...
1. Find the genes whose expression fits specific, predefined patterns.
2. Perform cluster analysis - see what expression patterns emerge.
Can we identify genes with interesting patterns of expression across the bulb?
Two approaches:

Clustering procedureClustering procedure
Start with a sets of genes exhibiting some minimal level of differential expression across the bulb; here ~650 were chosen from all 15 contrasts.
Carry out hierarchical clustering, building a dendrogram: Mahalanobis distance and Ward agglomeration (minimum variance) were used.
Now consider all clusters of 2 or more genes in the tree. Singles are added separately.
Measure the heterogeneity h of a cluster by calculating the 15 SDs
across the cluster of each of the pairwise effects, and taking the largest.
Choose a score s (see plots) and take all maximal disjoint clusters with
h < s. Here we used s = 0.46 and obtained 16 clusters.
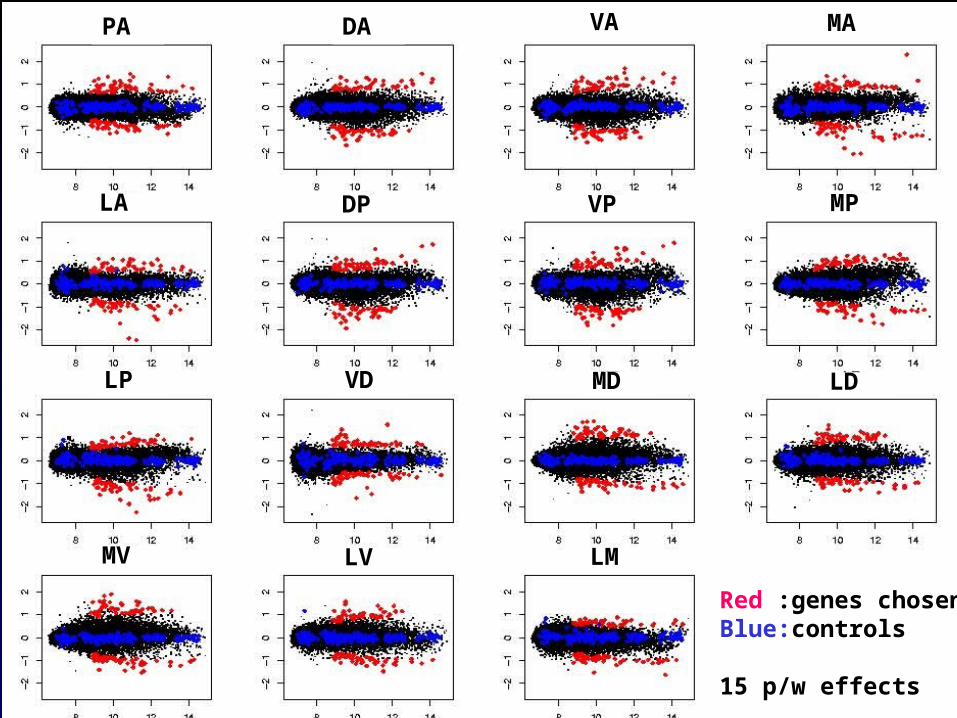
Red :genes chosenBlue:controls
15 p/w effects
PA DA VA
LA DP VPLA MP
MA
LP VD MD
LA LV LMMV
LD

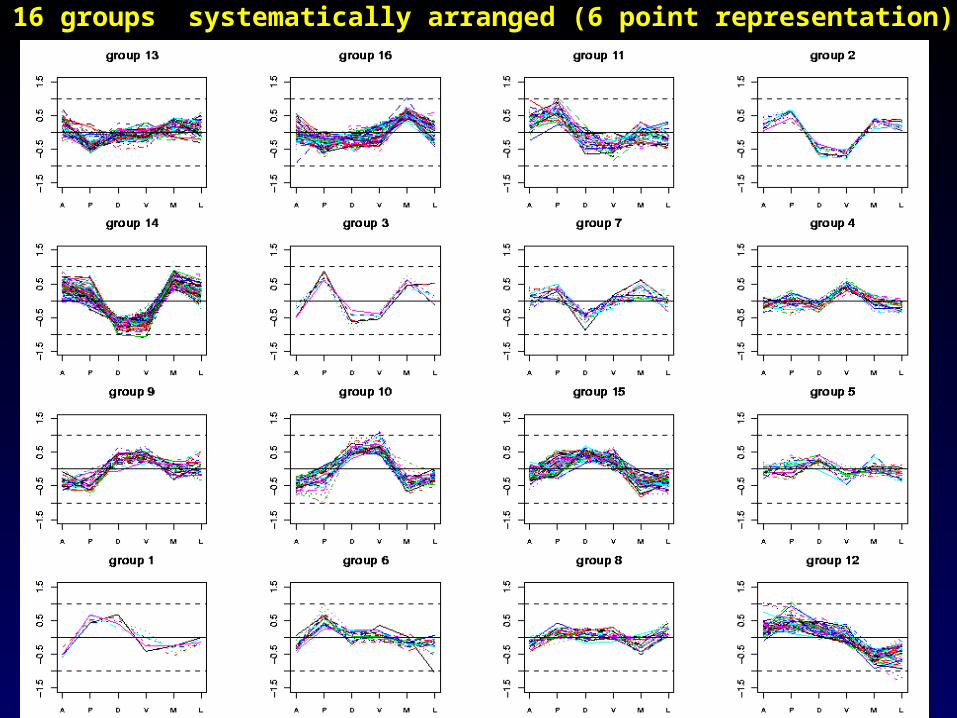
The 16 groups systematically arranged (6 point representation)

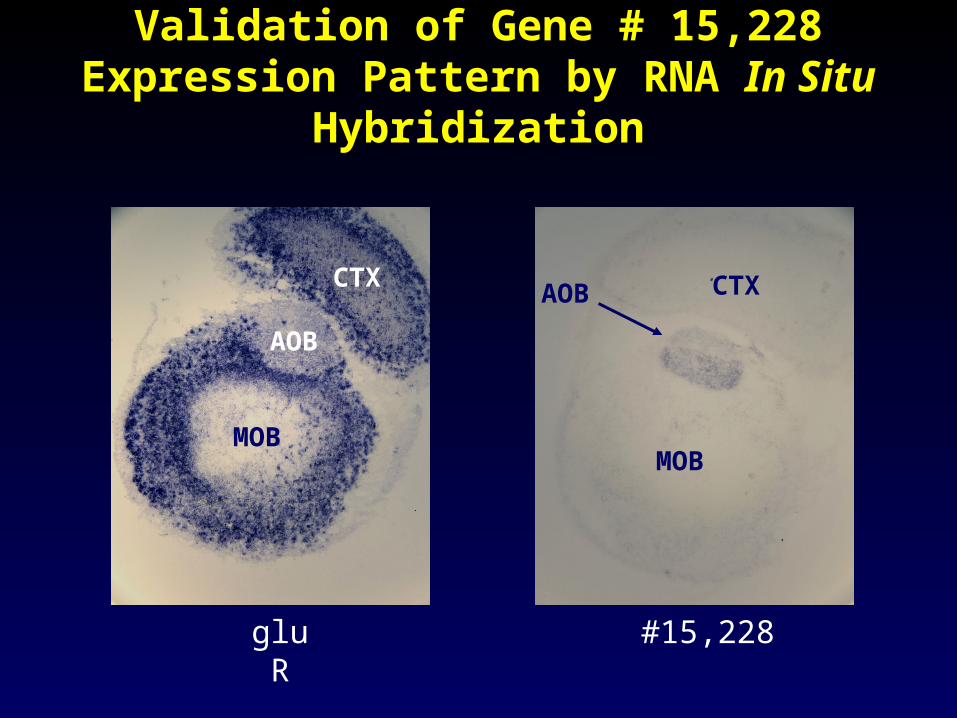
Validation of Gene # 15,228 Expression Validation of Gene # 15,228 Expression Pattern by RNA Pattern by RNA In SituIn Situ Hybridization Hybridization
gluR
CTX
MOB
AOB
#15,228
CTXAOB
MOB
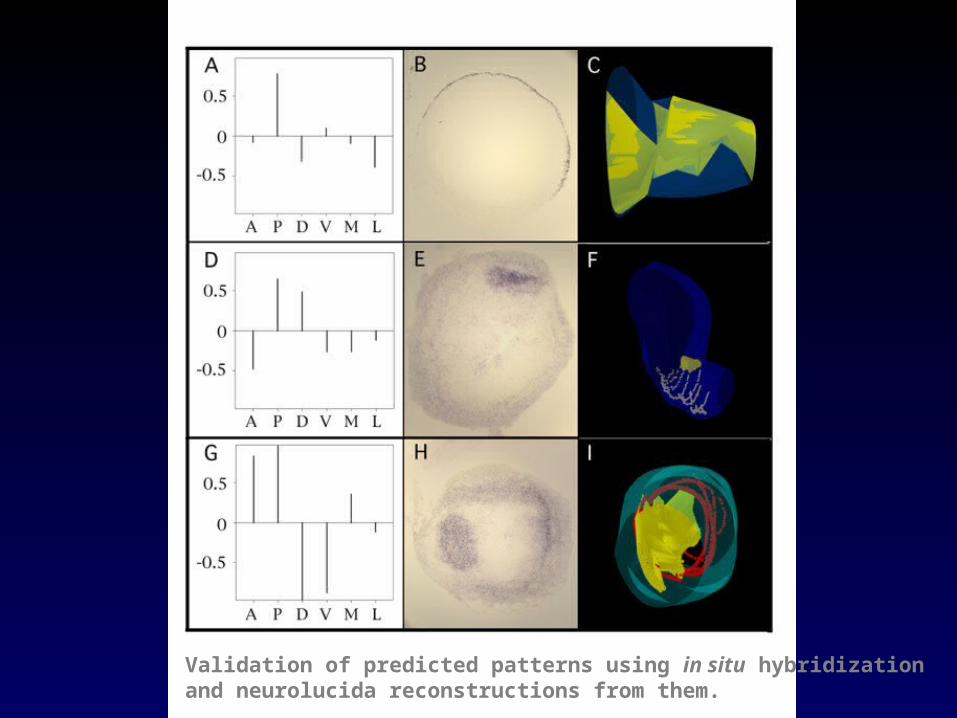
Validation of predicted patterns using in situ hybridizationand neurolucida reconstructions from them.

Some statistical research stimulated Some statistical research stimulated by microarray data analysisby microarray data analysis
Experimental design : Churchill & Kerr
Image analysis: Zuzan & West, ….
Data visualization: Carr et al
Estimation: Ideker et al, ….
Multiple testing: Westfall & Young , Storey, ….
Discriminant analysis: Golub et al,…
Clustering: Hastie & Tibshirani, Van der Laan, Fridlyand & Dudoit, ….
Empirical Bayes: Efron et al, Newton et al,…. Multiplicative models: Li &Wong
Multivariate analysis: Alter et al
Genetic networks: D’Haeseleer et al and more

AcknowledgmentsAcknowledgmentsStatistical collaboratorsStatistical collaboratorsYee Hwa Yang (Berkeley)Yee Hwa Yang (Berkeley)Sandrine Dudoit (Berkeley)Sandrine Dudoit (Berkeley)Ingrid Lönnstedt (Uppsala)Natalie Thorne (WEHI)Natalie Thorne (WEHI)Mauro Delorenzi (WEHI)
CSIRO Image Analysis GroupMichael BuckleyMichael BuckleyRyan Lagerstorm
WEHIGlenn BegleySuzie GrantRob Good
PMCIChuang Fong Kong
Ngai Lab (Berkeley)Cynthia DugganJonathan ScolnickDave Lin Vivian Peng Percy LuuElva DiazJohn Ngai
LBNLMatt Callow
RIKEN Genomic Sciences CenterRIKEN Genomic Sciences CenterYasushi OkazakiYoshihide Hayashizaki

Some web sites:
Technical reports, talks, software etc.
http://www.stat.berkeley.edu/users/terry/zarray/Html/
Statistical software R “GNU’s S” http://lib.stat.cmu.edu/R/CRAN/
Packages within R environment:
-- Spot http://www.cmis.csiro.au/iap/spot.htm
-- SMA (statistics for microarray analysis) http://www.stat.berkeley.edu/users/terry/zarray/Software /smacode.html
Related Documents











