Functional Modulation of Nuclear Steroid Receptors by Tauroursodeoxycholic Acid Reduces Amyloid -Peptide-Induced Apoptosis Susana Sola ´ , Joana D. Amaral, Pedro M. Borralho, Rita M. Ramalho, Rui E. Castro, Ma ´rcia M. Aranha, Cifford J. Steer, and Cecı´lia M. P. Rodrigues Centro de Patoge ´ nese Molecular (S.S., J.D.A., P.M.B., R.M.R., R.E.C., M.M.A., C.M.P.R.), Faculty of Pharmacy, University of Lisbon, Lisbon 1600-083, Portugal; and Departments of Medicine (C.J.S.), and Genetics, Cell Biology, and Development (C.J.S.), University of Minnesota Medical School, Minneapolis, Minnesota 55455 Tauroursodeoxycholic acid (TUDCA) prevents amyloid -peptide (A)-induced neuronal apopto- sis, by modulating both classical mitochondrial pathways and specific upstream targets. In addi- tion, activation of nuclear steroid receptors (NSRs), such as the mineralocorticoid receptor (MR) and the glucocorticoid receptor (GR) differentially reg- ulates apoptosis in the brain. In this study we in- vestigated whether TUDCA, a cholesterol-derived endogenous molecule, requires NSRs for inhibiting A-induced apoptosis in primary neurons. Our re- sults confirmed that TUDCA significantly reduced A-induced apoptosis; in addition, the fluores- cently labeled bile acid molecule was detected dif- fusely in both cytoplasm and nucleus of rat cortical neurons. Interestingly, experiments using small in- terfering RNAs (siRNAs) revealed that, in contrast to GR siRNA, MR siRNA abolished the antiapop- totic effect of TUDCA. A incubation reduced MR nuclear translocation while increasing nuclear GR levels. Notably, pretreatment with TUDCA mark- edly altered A-induced changes in NSRs, includ- ing MR dissociation from its cytosolic chaperone, heat shock protein 90, and subsequent transloca- tion to the nucleus. Furthermore, when a carboxy terminus-deleted form of MR was used, nuclear trafficking of both MR and the bile acid was abro- gated, suggesting that they translocate to the nu- cleus as a steroid-receptor complex. Transfection experiments with wild-type or mutant MR con- firmed that this interaction was required for TUDCA protection against A-induced apoptosis. Finally, in cotransfection experiments with NSR re- sponse element reporter and overexpression con- structs, pretreatment with TUDCA significantly modulated A-induced changes in MR and GR transactivation. In conclusion, these results pro- vide novel insights into the specific cellular mech- anism of TUDCA antiapoptotic function against A-induced apoptosis and suggest targets for po- tential therapeutic intervention. (Molecular Endo- crinology 20: 2292–2303, 2006) A LZHEIMER’S DISEASE (AD) is characterized by a selective damage of synapses and neurons, neu- rofibrillary tangles, activated glia, and presence of se- nile plaques (1). In addition, amyloid -peptide (A) is the major constituent of senile plaques (2), thought to directly induce oxidative stress, inflammation, and neuronal apoptosis (1, 3). Recently, the involvement of apoptosis has been corroborated by studies showing that A alters expression of the Bcl-2 family of apop- tosis-related genes (4), and that survival signaling pathways are required for protection of A-mediated neuronal apoptosis (5). The endogenous hydrophilic bile acid, ursodeoxy- cholic acid (UDCA), and its taurine-conjugated deriv- ative, tauroursodeoxycholic acid (TUDCA), play a unique role in modulating mitochondrial apoptosis (6– 8). They also regulate the expression of Bcl-2 family members by modulating molecular targets upstream of the mitochondrial commitment, in a caspase-inde- pendent manner (9). Curiously, TUDCA is a potent neuroprotective agent not only in pharmacological and transgenic animal models of Huntington’s disease (10, 11), but also for acute ischemic and hemorrhagic stroke (12, 13). Furthermore, TUDCA improved the survival and function of nigral transplants in a rat model of Parkinson’s disease (14) and partially res- cued a Parkinson’s disease model of Caenorhabditis elegans from mitochondrial dysfunction (15). Impor- tantly, TUDCA also prevents A-induced neuronal First Published Online May 25, 2006 Abbreviations: A, Amyloid -peptide; AD, Alzheimer’s disease; CAT, chloramphenicol acetyltransferase; DEVD, N- acetyl-Asp-Glu-Val-Asp; GFP, green fluorescent protein; GR, glucocorticoid receptor; GRE, glucocorticoid response ele- ment; hsp90, heat shock protein 90; LBD, ligand binding domain; MR, mineralocorticoid receptor; MRE, mineralocor- ticoid response element; NBD, nitrobenzoxadiazolyl; NSR, nuclear steroid receptor; PARP, poly(ADP-ribose) polymer- ase; pNA, p-nitroanilide; siRNA, small interfering RNA; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid; Wt, wild type. Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community. 0888-8809/06/$15.00/0 Molecular Endocrinology 20(10):2292–2303 Printed in U.S.A. Copyright © 2006 by The Endocrine Society doi: 10.1210/me.2006-0063 2292

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Functional Modulation of Nuclear Steroid Receptorsby Tauroursodeoxycholic Acid Reduces Amyloid�-Peptide-Induced Apoptosis
Susana Sola, Joana D. Amaral, Pedro M. Borralho, Rita M. Ramalho, Rui E. Castro,Marcia M. Aranha, Cifford J. Steer, and Cecılia M. P. Rodrigues
Centro de Patogenese Molecular (S.S., J.D.A., P.M.B., R.M.R., R.E.C., M.M.A., C.M.P.R.), Faculty ofPharmacy, University of Lisbon, Lisbon 1600-083, Portugal; and Departments of Medicine (C.J.S.),and Genetics, Cell Biology, and Development (C.J.S.), University of Minnesota Medical School,Minneapolis, Minnesota 55455
Tauroursodeoxycholic acid (TUDCA) preventsamyloid �-peptide (A�)-induced neuronal apopto-sis, by modulating both classical mitochondrialpathways and specific upstream targets. In addi-tion, activation of nuclear steroid receptors (NSRs),such as the mineralocorticoid receptor (MR) andthe glucocorticoid receptor (GR) differentially reg-ulates apoptosis in the brain. In this study we in-vestigated whether TUDCA, a cholesterol-derivedendogenous molecule, requires NSRs for inhibitingA�-induced apoptosis in primary neurons. Our re-sults confirmed that TUDCA significantly reducedA�-induced apoptosis; in addition, the fluores-cently labeled bile acid molecule was detected dif-fusely in both cytoplasm and nucleus of rat corticalneurons. Interestingly, experiments using small in-terfering RNAs (siRNAs) revealed that, in contrastto GR siRNA, MR siRNA abolished the antiapop-totic effect of TUDCA. A� incubation reduced MRnuclear translocation while increasing nuclear GRlevels. Notably, pretreatment with TUDCA mark-
edly altered A�-induced changes in NSRs, includ-ing MR dissociation from its cytosolic chaperone,heat shock protein 90, and subsequent transloca-tion to the nucleus. Furthermore, when a carboxyterminus-deleted form of MR was used, nucleartrafficking of both MR and the bile acid was abro-gated, suggesting that they translocate to the nu-cleus as a steroid-receptor complex. Transfectionexperiments with wild-type or mutant MR con-firmed that this interaction was required forTUDCA protection against A�-induced apoptosis.Finally, in cotransfection experiments with NSR re-sponse element reporter and overexpression con-structs, pretreatment with TUDCA significantlymodulated A�-induced changes in MR and GRtransactivation. In conclusion, these results pro-vide novel insights into the specific cellular mech-anism of TUDCA antiapoptotic function againstA�-induced apoptosis and suggest targets for po-tential therapeutic intervention. (Molecular Endo-crinology 20: 2292–2303, 2006)
ALZHEIMER’S DISEASE (AD) is characterized by aselective damage of synapses and neurons, neu-
rofibrillary tangles, activated glia, and presence of se-nile plaques (1). In addition, amyloid �-peptide (A�) isthe major constituent of senile plaques (2), thought todirectly induce oxidative stress, inflammation, andneuronal apoptosis (1, 3). Recently, the involvement of
apoptosis has been corroborated by studies showingthat A� alters expression of the Bcl-2 family of apop-tosis-related genes (4), and that survival signalingpathways are required for protection of A�-mediatedneuronal apoptosis (5).
The endogenous hydrophilic bile acid, ursodeoxy-cholic acid (UDCA), and its taurine-conjugated deriv-ative, tauroursodeoxycholic acid (TUDCA), play aunique role in modulating mitochondrial apoptosis (6–8). They also regulate the expression of Bcl-2 familymembers by modulating molecular targets upstreamof the mitochondrial commitment, in a caspase-inde-pendent manner (9). Curiously, TUDCA is a potentneuroprotective agent not only in pharmacological andtransgenic animal models of Huntington’s disease (10,11), but also for acute ischemic and hemorrhagicstroke (12, 13). Furthermore, TUDCA improved thesurvival and function of nigral transplants in a ratmodel of Parkinson’s disease (14) and partially res-cued a Parkinson’s disease model of Caenorhabditiselegans from mitochondrial dysfunction (15). Impor-tantly, TUDCA also prevents A�-induced neuronal
First Published Online May 25, 2006Abbreviations: A�, Amyloid �-peptide; AD, Alzheimer’s
disease; CAT, chloramphenicol acetyltransferase; DEVD, N-acetyl-Asp-Glu-Val-Asp; GFP, green fluorescent protein; GR,glucocorticoid receptor; GRE, glucocorticoid response ele-ment; hsp90, heat shock protein 90; LBD, ligand bindingdomain; MR, mineralocorticoid receptor; MRE, mineralocor-ticoid response element; NBD, nitrobenzoxadiazolyl; NSR,nuclear steroid receptor; PARP, poly(ADP-ribose) polymer-ase; pNA, p-nitroanilide; siRNA, small interfering RNA;TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholicacid; Wt, wild type.
Molecular Endocrinology is published monthly by TheEndocrine Society (http://www.endo-society.org), theforemost professional society serving the endocrinecommunity.
0888-8809/06/$15.00/0 Molecular Endocrinology 20(10):2292–2303Printed in U.S.A. Copyright © 2006 by The Endocrine Society
doi: 10.1210/me.2006-0063
2292

death by both triggering phosphatidylinositol 3-kinasesurvival signaling (16) and modulating the E2F-1/p53/Bax apoptotic pathway (17).
Nuclear steroid receptors (NSRs) play an heteroge-neous function in apoptosis, which is affected by tis-sue-specific parameters such as alternative initiationsites within nuclear receptor genes, and different ef-fects of comodulators (18, 19). In contrast to its anti-apoptotic effect in the liver, the glucocorticoid recep-tor (GR) up-regulates p53 and proapoptotic Bcl-2members in neuronal and leukemia cells (20–22). In-terestingly, glucocorticoid receptor (GR) activation hasbeen correlated with neuronal pathological conditions,such as AD (23, 24). The hormone-binding domain ofGR is not mutated in AD patients (25), but GR activa-tion enhances oxidative stress-induced cell death inhippocampal neurons (26). Conversely, the mineralo-corticoid receptor (MR) is a potent inhibitor of apopto-sis in the majority of cell types, including neurons (20,27). Although MR and GR share considerable structureand functional homology, MR can form heterodimerswith GR and inhibit its function via the MR N-terminaldomain (28). In neuronal cells, MR decreases p53 lev-els as well as the ratio of pro- relative to antiapoptoticBcl-2 members (20).
Bile acids are natural ligands of NSRs, structurallysimilar to steroid hormones (29). Moreover, UDCA in-teracts with GR ligand binding domain (LBD) (30), per-haps attenuating the interaction between GR and itscoactivators (31). Here, we demonstrate that TUDCAtargets a specific region of MR LBD and dissociatesthe NSR from its cytosolic chaperone, heat shockprotein 90 (hsp90). The bile acid/NSR complex is thentranslocated to the nucleus, thereby modulating NSRtransactivation and ultimately reducing A�-mediatedneuronal apoptosis.
RESULTS
Fluorescent UDCA Is Localized in the Nucleusof Neurons
It has been reported that UDCA and its amino acid-conjugated derivatives prevent neuronal apoptosisfrom A� through inhibition of mitochondrial pathwaysand modulation of upstream targets (16, 17). To furtherunderstand the mechanism by which bile acids mod-ulate apoptosis in neuronal cells, we first investigatedthe subcellular localization of UDCA in primary ratcortical neurons. For this purpose, cells were incu-bated with a fluorescent UDCA molecule (Fig. 1). Un-labeled UDCA did not show any fluorescence at alltime points. In contrast, the 3�-hydroxy-7-nitrobenzo-xadiazolyl (NBD)-UDCA molecule was clearly ob-served in rat cortical neurons. The fluorescent signalwas weak at 5 min; however, it became more intenseat 15 min and at 1 and 2 h of incubation. Moreover,although the green fluorescence was stronger in thecytosol, the signal was diffuse in all compartments of
neurons, including the nucleus. Nuclear localization ofthe NBD-UDCA molecule was confirmed by Hoechstcounterstain (data not shown). Therefore, these resultssuggest that this bile acid may also have a specific rolewithin the nucleus to protect neurons againstapoptosis.
TUDCA Prevents A�-Induced Apoptosis througha NSR-Dependent Mechanism
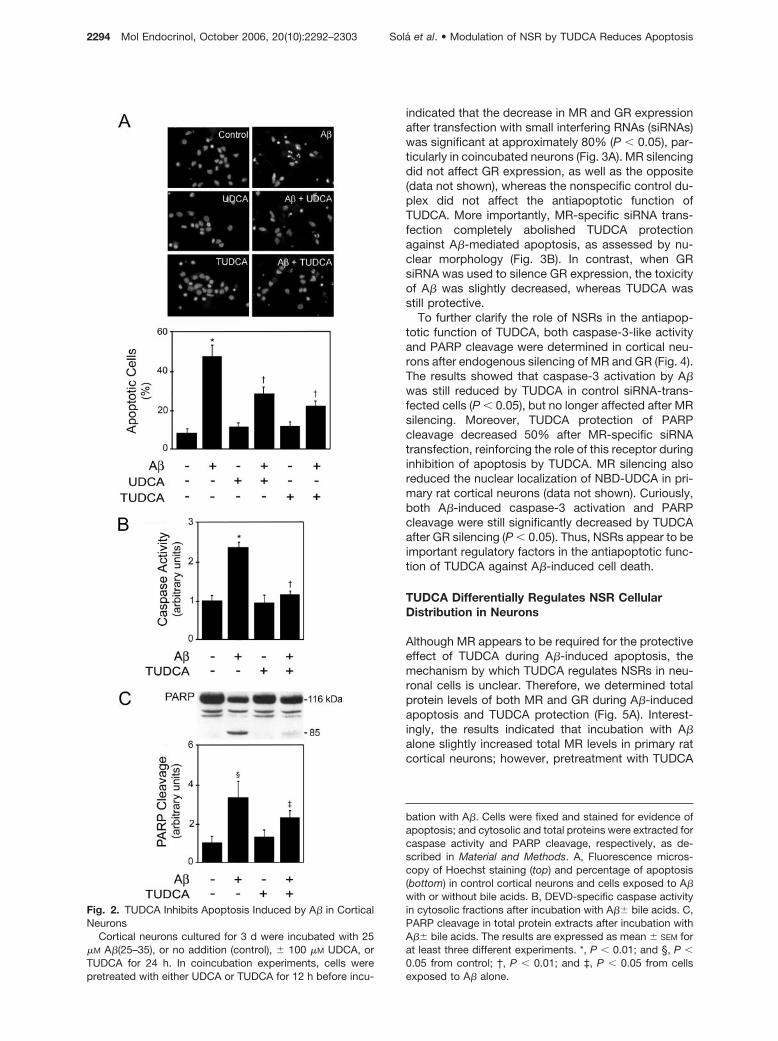
It has been reported that NSRs modulate apoptosis inneuronal cells (20). In addition, we have previouslyshown that UDCA prevents hepatocyte apoptosisthrough an NSR-dependent mechanism (32), and thatGR is crucial for the nuclear translocation of the bileacid and attenuation of apoptosis (30). Therefore, weinvestigated the potential role of NSRs in the protec-tive function of bile acids against A�-induced apopto-sis of neuronal cells. Apoptosis was assessed bychanges in nuclear morphology, caspase activity, andpoly(ADP-ribose) polymerase (PARP) cleavage (Fig. 2).Significant levels of apoptosis were detected in pri-mary rat cortical neurons after incubation with A�(25–35) peptide, with approximately 50% of cell death (P �0.01). Notably, UDCA and TUDCA protected A�-induced nuclear fragmentation by 50% and 65%, re-spectively (P � 0.01). In addition, the active fragmentA�(1–40) resulted in 40% neuronal apoptosis, and thiswas reduced 40% and 50% by UDCA and TUDCA,respectively (data not shown). A�-induced cell deathwas associated with a 2-fold increase in caspase-3-like activation (P � 0.01), whereas TUDCA almostcompletely inhibited this effect (P � 0.01). Further,PARP cleavage was evident during A�-induced celldeath (P � 0.05), but significantly reduced by TUDCA(P � 0.05).
We next determined whether the antiapoptotic ef-fect of TUDCA in rat cortical neurons requires NSRactivation by performing posttranscriptional gene si-lencing experiments for both MR and GR. The results
Fig. 1. Nuclear Localization of UDCA in Cortical NeuronsSubcellular distribution of labeled UDCA in primary rat
cortical neurons. Cortical neurons were cultured for 3 d asdescribed in Materials and Methods and supplemented with100 �M of either NBD-UDCA or unlabeled UDCA for 5, 15,and 30 min and 1 and 2 h. Competition studies demonstratedthe specificity of the NBD-UDCA molecule in primary ratcortical neurons (data not shown). Representative photo-graphs of fluorescence microscopy of at least three differentexperiments.
Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis Mol Endocrinol, October 2006, 20(10):2292–2303 2293
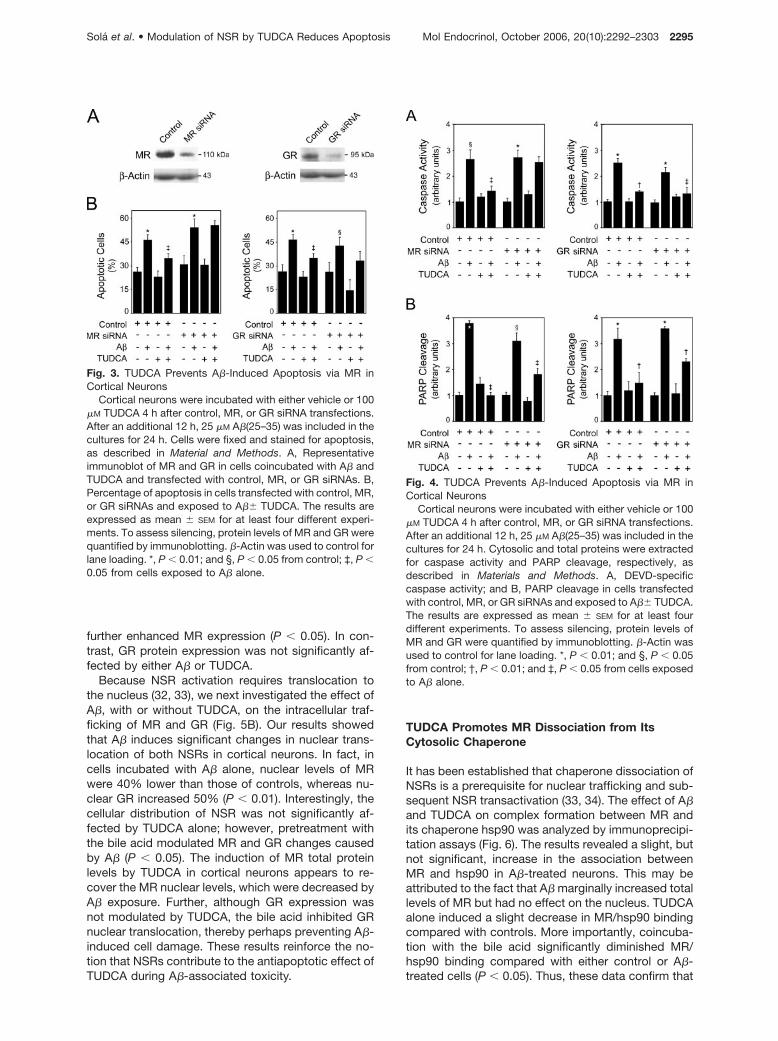
indicated that the decrease in MR and GR expressionafter transfection with small interfering RNAs (siRNAs)was significant at approximately 80% (P � 0.05), par-ticularly in coincubated neurons (Fig. 3A). MR silencingdid not affect GR expression, as well as the opposite(data not shown), whereas the nonspecific control du-plex did not affect the antiapoptotic function ofTUDCA. More importantly, MR-specific siRNA trans-fection completely abolished TUDCA protectionagainst A�-mediated apoptosis, as assessed by nu-clear morphology (Fig. 3B). In contrast, when GRsiRNA was used to silence GR expression, the toxicityof A� was slightly decreased, whereas TUDCA wasstill protective.
To further clarify the role of NSRs in the antiapop-totic function of TUDCA, both caspase-3-like activityand PARP cleavage were determined in cortical neu-rons after endogenous silencing of MR and GR (Fig. 4).The results showed that caspase-3 activation by A�was still reduced by TUDCA in control siRNA-trans-fected cells (P � 0.05), but no longer affected after MRsilencing. Moreover, TUDCA protection of PARPcleavage decreased 50% after MR-specific siRNAtransfection, reinforcing the role of this receptor duringinhibition of apoptosis by TUDCA. MR silencing alsoreduced the nuclear localization of NBD-UDCA in pri-mary rat cortical neurons (data not shown). Curiously,both A�-induced caspase-3 activation and PARPcleavage were still significantly decreased by TUDCAafter GR silencing (P � 0.05). Thus, NSRs appear to beimportant regulatory factors in the antiapoptotic func-tion of TUDCA against A�-induced cell death.
TUDCA Differentially Regulates NSR CellularDistribution in Neurons
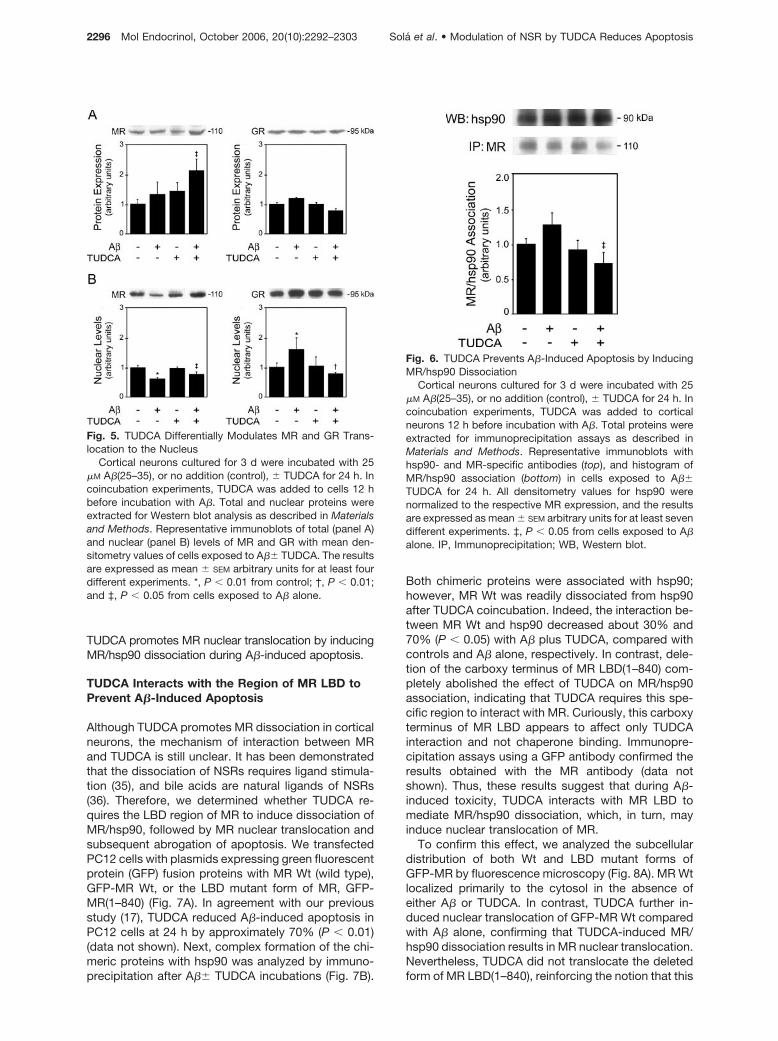
Although MR appears to be required for the protectiveeffect of TUDCA during A�-induced apoptosis, themechanism by which TUDCA regulates NSRs in neu-ronal cells is unclear. Therefore, we determined totalprotein levels of both MR and GR during A�-inducedapoptosis and TUDCA protection (Fig. 5A). Interest-ingly, the results indicated that incubation with A�alone slightly increased total MR levels in primary ratcortical neurons; however, pretreatment with TUDCA
Fig. 2. TUDCA Inhibits Apoptosis Induced by A� in CorticalNeurons
Cortical neurons cultured for 3 d were incubated with 25�M A�(25–35), or no addition (control), � 100 �M UDCA, orTUDCA for 24 h. In coincubation experiments, cells werepretreated with either UDCA or TUDCA for 12 h before incu-
bation with A�. Cells were fixed and stained for evidence ofapoptosis; and cytosolic and total proteins were extracted forcaspase activity and PARP cleavage, respectively, as de-scribed in Material and Methods. A, Fluorescence micros-copy of Hoechst staining (top) and percentage of apoptosis(bottom) in control cortical neurons and cells exposed to A�with or without bile acids. B, DEVD-specific caspase activityin cytosolic fractions after incubation with A�� bile acids. C,PARP cleavage in total protein extracts after incubation withA�� bile acids. The results are expressed as mean � SEM forat least three different experiments. *, P � 0.01; and §, P �0.05 from control; †, P � 0.01; and ‡, P � 0.05 from cellsexposed to A� alone.
2294 Mol Endocrinol, October 2006, 20(10):2292–2303 Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis
further enhanced MR expression (P � 0.05). In con-trast, GR protein expression was not significantly af-fected by either A� or TUDCA.
Because NSR activation requires translocation tothe nucleus (32, 33), we next investigated the effect ofA�, with or without TUDCA, on the intracellular traf-ficking of MR and GR (Fig. 5B). Our results showedthat A� induces significant changes in nuclear trans-location of both NSRs in cortical neurons. In fact, incells incubated with A� alone, nuclear levels of MRwere 40% lower than those of controls, whereas nu-clear GR increased 50% (P � 0.01). Interestingly, thecellular distribution of NSR was not significantly af-fected by TUDCA alone; however, pretreatment withthe bile acid modulated MR and GR changes causedby A� (P � 0.05). The induction of MR total proteinlevels by TUDCA in cortical neurons appears to re-cover the MR nuclear levels, which were decreased byA� exposure. Further, although GR expression wasnot modulated by TUDCA, the bile acid inhibited GRnuclear translocation, thereby perhaps preventing A�-induced cell damage. These results reinforce the no-tion that NSRs contribute to the antiapoptotic effect ofTUDCA during A�-associated toxicity.
TUDCA Promotes MR Dissociation from ItsCytosolic Chaperone
It has been established that chaperone dissociation ofNSRs is a prerequisite for nuclear trafficking and sub-sequent NSR transactivation (33, 34). The effect of A�and TUDCA on complex formation between MR andits chaperone hsp90 was analyzed by immunoprecipi-tation assays (Fig. 6). The results revealed a slight, butnot significant, increase in the association betweenMR and hsp90 in A�-treated neurons. This may beattributed to the fact that A� marginally increased totallevels of MR but had no effect on the nucleus. TUDCAalone induced a slight decrease in MR/hsp90 bindingcompared with controls. More importantly, coincuba-tion with the bile acid significantly diminished MR/hsp90 binding compared with either control or A�-treated cells (P � 0.05). Thus, these data confirm that
Fig. 3. TUDCA Prevents A�-Induced Apoptosis via MR inCortical Neurons
Cortical neurons were incubated with either vehicle or 100�M TUDCA 4 h after control, MR, or GR siRNA transfections.After an additional 12 h, 25 �M A�(25–35) was included in thecultures for 24 h. Cells were fixed and stained for apoptosis,as described in Material and Methods. A, Representativeimmunoblot of MR and GR in cells coincubated with A� andTUDCA and transfected with control, MR, or GR siRNAs. B,Percentage of apoptosis in cells transfected with control, MR,or GR siRNAs and exposed to A�� TUDCA. The results areexpressed as mean � SEM for at least four different experi-ments. To assess silencing, protein levels of MR and GR werequantified by immunoblotting. �-Actin was used to control forlane loading. *, P � 0.01; and §, P � 0.05 from control; ‡, P �0.05 from cells exposed to A� alone.
Fig. 4. TUDCA Prevents A�-Induced Apoptosis via MR inCortical Neurons
Cortical neurons were incubated with either vehicle or 100�M TUDCA 4 h after control, MR, or GR siRNA transfections.After an additional 12 h, 25 �M A�(25–35) was included in thecultures for 24 h. Cytosolic and total proteins were extractedfor caspase activity and PARP cleavage, respectively, asdescribed in Materials and Methods. A, DEVD-specificcaspase activity; and B, PARP cleavage in cells transfectedwith control, MR, or GR siRNAs and exposed to A�� TUDCA.The results are expressed as mean � SEM for at least fourdifferent experiments. To assess silencing, protein levels ofMR and GR were quantified by immunoblotting. �-Actin wasused to control for lane loading. *, P � 0.01; and §, P � 0.05from control; †, P � 0.01; and ‡, P � 0.05 from cells exposedto A� alone.
Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis Mol Endocrinol, October 2006, 20(10):2292–2303 2295
TUDCA promotes MR nuclear translocation by inducingMR/hsp90 dissociation during A�-induced apoptosis.
TUDCA Interacts with the Region of MR LBD toPrevent A�-Induced Apoptosis
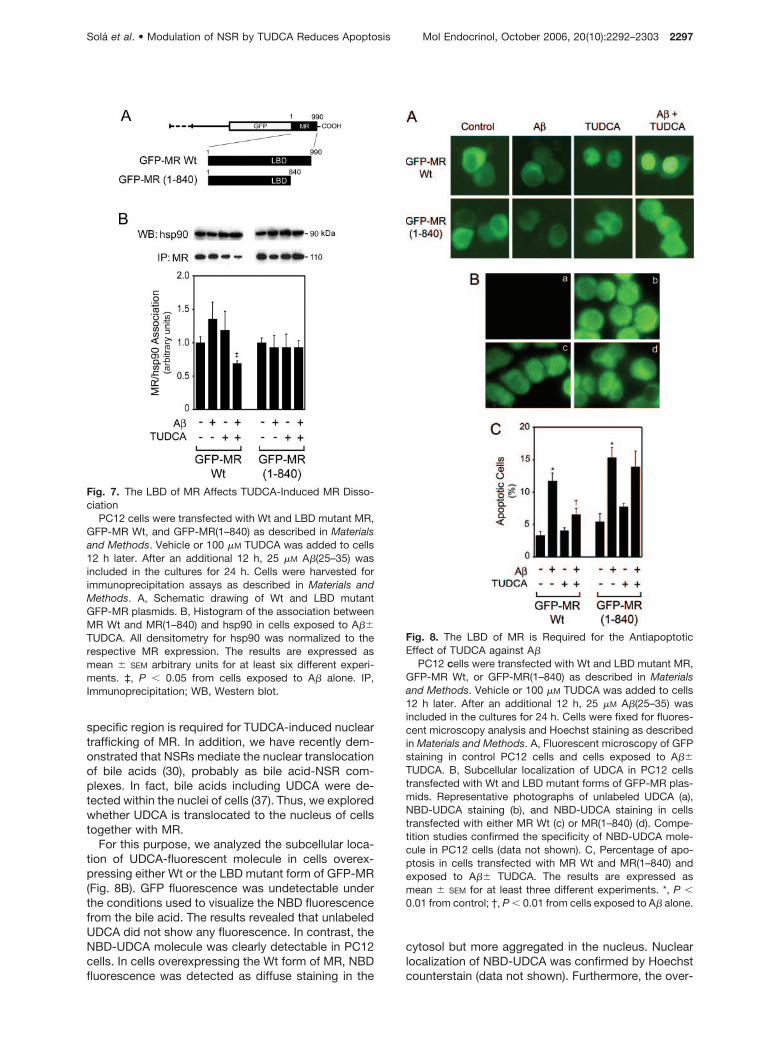
Although TUDCA promotes MR dissociation in corticalneurons, the mechanism of interaction between MRand TUDCA is still unclear. It has been demonstratedthat the dissociation of NSRs requires ligand stimula-tion (35), and bile acids are natural ligands of NSRs(36). Therefore, we determined whether TUDCA re-quires the LBD region of MR to induce dissociation ofMR/hsp90, followed by MR nuclear translocation andsubsequent abrogation of apoptosis. We transfectedPC12 cells with plasmids expressing green fluorescentprotein (GFP) fusion proteins with MR Wt (wild type),GFP-MR Wt, or the LBD mutant form of MR, GFP-MR(1–840) (Fig. 7A). In agreement with our previousstudy (17), TUDCA reduced A�-induced apoptosis inPC12 cells at 24 h by approximately 70% (P � 0.01)(data not shown). Next, complex formation of the chi-meric proteins with hsp90 was analyzed by immuno-precipitation after A�� TUDCA incubations (Fig. 7B).
Both chimeric proteins were associated with hsp90;however, MR Wt was readily dissociated from hsp90after TUDCA coincubation. Indeed, the interaction be-tween MR Wt and hsp90 decreased about 30% and70% (P � 0.05) with A� plus TUDCA, compared withcontrols and A� alone, respectively. In contrast, dele-tion of the carboxy terminus of MR LBD(1–840) com-pletely abolished the effect of TUDCA on MR/hsp90association, indicating that TUDCA requires this spe-cific region to interact with MR. Curiously, this carboxyterminus of MR LBD appears to affect only TUDCAinteraction and not chaperone binding. Immunopre-cipitation assays using a GFP antibody confirmed theresults obtained with the MR antibody (data notshown). Thus, these results suggest that during A�-induced toxicity, TUDCA interacts with MR LBD tomediate MR/hsp90 dissociation, which, in turn, mayinduce nuclear translocation of MR.
To confirm this effect, we analyzed the subcellulardistribution of both Wt and LBD mutant forms ofGFP-MR by fluorescence microscopy (Fig. 8A). MR Wtlocalized primarily to the cytosol in the absence ofeither A� or TUDCA. In contrast, TUDCA further in-duced nuclear translocation of GFP-MR Wt comparedwith A� alone, confirming that TUDCA-induced MR/hsp90 dissociation results in MR nuclear translocation.Nevertheless, TUDCA did not translocate the deletedform of MR LBD(1–840), reinforcing the notion that this
Fig. 5. TUDCA Differentially Modulates MR and GR Trans-location to the Nucleus
Cortical neurons cultured for 3 d were incubated with 25�M A�(25–35), or no addition (control), � TUDCA for 24 h. Incoincubation experiments, TUDCA was added to cells 12 hbefore incubation with A�. Total and nuclear proteins wereextracted for Western blot analysis as described in Materialsand Methods. Representative immunoblots of total (panel A)and nuclear (panel B) levels of MR and GR with mean den-sitometry values of cells exposed to A�� TUDCA. The resultsare expressed as mean � SEM arbitrary units for at least fourdifferent experiments. *, P � 0.01 from control; †, P � 0.01;and ‡, P � 0.05 from cells exposed to A� alone.
Fig. 6. TUDCA Prevents A�-Induced Apoptosis by InducingMR/hsp90 Dissociation
Cortical neurons cultured for 3 d were incubated with 25�M A�(25–35), or no addition (control), � TUDCA for 24 h. Incoincubation experiments, TUDCA was added to corticalneurons 12 h before incubation with A�. Total proteins wereextracted for immunoprecipitation assays as described inMaterials and Methods. Representative immunoblots withhsp90- and MR-specific antibodies (top), and histogram ofMR/hsp90 association (bottom) in cells exposed to A��TUDCA for 24 h. All densitometry values for hsp90 werenormalized to the respective MR expression, and the resultsare expressed as mean � SEM arbitrary units for at least sevendifferent experiments. ‡, P � 0.05 from cells exposed to A�alone. IP, Immunoprecipitation; WB, Western blot.
2296 Mol Endocrinol, October 2006, 20(10):2292–2303 Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis
specific region is required for TUDCA-induced nucleartrafficking of MR. In addition, we have recently dem-onstrated that NSRs mediate the nuclear translocationof bile acids (30), probably as bile acid-NSR com-plexes. In fact, bile acids including UDCA were de-tected within the nuclei of cells (37). Thus, we exploredwhether UDCA is translocated to the nucleus of cellstogether with MR.
For this purpose, we analyzed the subcellular loca-tion of UDCA-fluorescent molecule in cells overex-pressing either Wt or the LBD mutant form of GFP-MR(Fig. 8B). GFP fluorescence was undetectable underthe conditions used to visualize the NBD fluorescencefrom the bile acid. The results revealed that unlabeledUDCA did not show any fluorescence. In contrast, theNBD-UDCA molecule was clearly detectable in PC12cells. In cells overexpressing the Wt form of MR, NBDfluorescence was detected as diffuse staining in the
cytosol but more aggregated in the nucleus. Nuclearlocalization of NBD-UDCA was confirmed by Hoechstcounterstain (data not shown). Furthermore, the over-
Fig. 7. The LBD of MR Affects TUDCA-Induced MR Disso-ciation
PC12 cells were transfected with Wt and LBD mutant MR,GFP-MR Wt, and GFP-MR(1–840) as described in Materialsand Methods. Vehicle or 100 �M TUDCA was added to cells12 h later. After an additional 12 h, 25 �M A�(25–35) wasincluded in the cultures for 24 h. Cells were harvested forimmunoprecipitation assays as described in Materials andMethods. A, Schematic drawing of Wt and LBD mutantGFP-MR plasmids. B, Histogram of the association betweenMR Wt and MR(1–840) and hsp90 in cells exposed to A��TUDCA. All densitometry for hsp90 was normalized to therespective MR expression. The results are expressed asmean � SEM arbitrary units for at least six different experi-ments. ‡, P � 0.05 from cells exposed to A� alone. IP,Immunoprecipitation; WB, Western blot.
Fig. 8. The LBD of MR is Required for the AntiapoptoticEffect of TUDCA against A�
PC12 cells were transfected with Wt and LBD mutant MR,GFP-MR Wt, or GFP-MR(1–840) as described in Materialsand Methods. Vehicle or 100 �M TUDCA was added to cells12 h later. After an additional 12 h, 25 �M A�(25–35) wasincluded in the cultures for 24 h. Cells were fixed for fluores-cent microscopy analysis and Hoechst staining as describedin Materials and Methods. A, Fluorescent microscopy of GFPstaining in control PC12 cells and cells exposed to A��TUDCA. B, Subcellular localization of UDCA in PC12 cellstransfected with Wt and LBD mutant forms of GFP-MR plas-mids. Representative photographs of unlabeled UDCA (a),NBD-UDCA staining (b), and NBD-UDCA staining in cellstransfected with either MR Wt (c) or MR(1–840) (d). Compe-tition studies confirmed the specificity of NBD-UDCA mole-cule in PC12 cells (data not shown). C, Percentage of apo-ptosis in cells transfected with MR Wt and MR(1–840) andexposed to A�� TUDCA. The results are expressed asmean � SEM for at least three different experiments. *, P �0.01 from control; †, P � 0.01 from cells exposed to A� alone.
Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis Mol Endocrinol, October 2006, 20(10):2292–2303 2297

expression of GFP-MR(1–840) resulted in an approx-imately 30% decrease of UDCA nuclear aggregates,indicating that the interaction between the bile acidand the LBD region is required for nuclear uptake ofthe bile acid via MR. We next determined whethernuclear migration of the bile acid-MR complex wasindeed critical for TUDCA antiapoptotic functionagainst A�. The results showed that unlike MR Wt, MRLBD(1–840) completely abolished TUDCA protectionof apoptosis in A�-treated cells (Fig. 8C). Thus,TUDCA appears to interact with a specific region ofMR LBD to induce MR dissociation, followed by nu-clear translocation of the bile acid-MR complex, andinhibition of apoptosis induced by A�.
TUDCA Modulates NSR Transactivation toPrevent A�-Induced Apoptosis
We recently reported that GR translocation of TUDCAwas required to reduce TGF-�1-induced hepatocyteapoptosis but did not require an increase in NSRtransactivation (30). Therefore, we investigatedwhether the antiapoptotic function of TUDCA againstA� peptide is also NSR transactivation dependent.Cells were cotransfected with a GR/MR-responsiveelement-reporter construct, pGRE/MRE-luciferase,and either MR (pRShMR) or GR (pRShGR), overex-pression plasmids. Surprisingly, our results revealedthat TUDCA differentially regulates NSR transactiva-tion during A�-induced cell death (Fig. 9). In fact, whencells were cotransfected with MR overexpressionplasmid, pRShMR, A� significantly decreased MR ac-tivity (P � 0.01), whereas the bile acid alone increasedMR transactivation by approximately 30% (P � 0.05).Notably, coincubation with TUDCA resulted in approx-imately 40% greater MR response element activation,as compared with A� alone (P � 0.05). Nevertheless,when cells were cotransfected with GR overexpres-sion plasmid, pRShGR, A� induced GR activity byapproximately 50% (P � 0.01), whereas TUDCA pre-treatment completely inhibited A�-induced GR trans-activation (P � 0.05). Interestingly, the results showthat without MR/GR overexpression, A� has an effectthat is more MR- than GR-like. It is possible that otherNSRs may account for the modulation of MRE/GRE-dependent reporter activity in PC12 cells, particularlybecause PC12 cells do not express endogenous MR.Also, in cells cotransfected with MR overexpressionplasmid, 10 �M corticosterone diminished TUDCA-induced GRE/MRE transactivation from A� by approx-imately 30% (P � 0.05). Interestingly, this effect wasgreater with 20 �M corticosterone. The competitioneffect of corticosterone for nuclear translocation ofUDCA through MR was also confirmed by fluorescentmicroscopy in PC12 cells overexpressing GFP-MR Wt(data not shown). Collectively, these results indicatethat during A�-induced apoptosis, TUDCA pretreat-ment results in differential cellular distribution of NSRsthat ultimately affects their transactivation. Thus, mod-ulation of NSR transactivation by TUDCA may be re-
quired for bile acid protection against A� peptide-induced apoptosis.
DISCUSSION
The accumulation of A� in the brain has been impli-cated as a potential cause for the neuronal loss thatoccurs in AD. Indeed, cell death from A�-inducedtoxicity is a complex process involving a number ofdifferent pathways, including oxidative stress, pertur-bation of calcium homeostasis, mitochondrial dys-function, and caspase activation (1).
In contrast to hydrophobic bile acids, UDCA andTUDCA are nontoxic species and can act as antiapop-totic agents (6, 38). They directly inhibit reactive oxy-gen species production, collapse of the transmem-brane potential, and disruption of the outermitochondrial membrane (39). We have previously re-ported that TUDCA is a pleiotropic agent that preventsA�-induced apoptosis by both inhibiting the mito-chondrial pathway of cell death (16) and modulatingthe E2F-1/p53/Bax pathway (17). Our results providean extended mechanism of action for TUDCA, in which
Fig. 9. TUDCA Prevents A�-Induced Apoptosis by Regulat-ing NSR Transactivation
PC12 cells were cotransfected with a GR/MR-responsiveelement-reporter construct, pGRE/MRE-luciferase, and ei-ther MR (pRShMR), or GR (pRShGR) overexpression plas-mids, as described in Materials and Methods. Vehicle or 100�M TUDCA was added to cells 12 h later. After an additional12 h, 25 �M A�(25–35) was included in the cultures, and cellswere harvested for luciferase assays. Transfection efficien-cies were determined using a reporter plasmid expressing�-galactosidase. Based on this control, transfection efficien-cies were approximately 70% and did not differ betweenreporter and expression plasmids. Histogram of the GREtransactivation in cells exposed to A� for 24 h � TUDCA, withor without NSR overexpression. Luciferase activity (relativelight units/mg protein) was normalized to control PGL3-CATexpression, and the results are expressed as mean � SEM foreight different experiments. *, P � 0.01; and §, P � 0.05 fromcontrol; †, P � 0.01; and ‡, P � 0.05 from cells exposed toA� alone.
2298 Mol Endocrinol, October 2006, 20(10):2292–2303 Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis

the bile acid binds to MR and is translocated as acomplex to the nucleus of neuronal cells where itprotects against A�-induced apoptosis.
Recent studies have identified an increasing numberof ligands for nuclear receptors, including certain bileacids (40–42). It has been demonstrated that UDCAactivates GR in liver cells, in the absence of ligandbinding (31, 43), suggesting a modulatory effect forthis bile acid. However, UDCA-induced GR modula-tion has been correlated primarily with its antiinflam-matory properties, but not with UDCA’s antiapoptoticfunction. In fact, nuclear receptors act as ligand-acti-vated transcription factors that regulate expression oftarget genes to affect a variety of cellular processes,including apoptosis.
It has been demonstrated that a number of apopto-sis-related proteins such as p53, Bax, Bcl-2, Bcl-xL,and the cellular inhibitor of apoptosis 2 (c-IAP2) arestrongly regulated by NSRs in several cell types (20,44, 45). Recently, we showed that UDCA modulateshepatocyte apoptosis through a NSR-dependentmechanism (32). However, the modulatory effect ofNSRs is cell specific, often involving the expressionand attenuation of the same gene between differenttissues.
In the present study, using a fluorescently labeledUDCA molecule, we demonstrated that UDCA reachesthe nucleus of primary rat cortical neurons. This cor-roborates previous observations in liver cells (30, 37)and suggests that UDCA may also have specific func-tions in the nucleus of neuronal cells. In addition, ourresults clearly showed that NSRs are involved in TUD-CA’s antiapoptotic action, because MR silencing re-sulted in significant blockade of its protective effect inneurons exposed to A�. A� significantly increased GRnuclear levels, while slightly increasing total MR ex-pression and markedly reducing nuclear MR. This isconsistent with previous studies showing high levels ofgr mRNA in the brain of patients with AD (23). Further-more, GR and MR appear to have opposing effects, invarious cell types, inhibiting each other’s transcrip-tional function, and thus differentially influencing theultimate fate of the cell (20, 28). Notably, in contrastwith UDCA and TUDCA in liver (32), pretreatment withTUDCA differentially modulates MR and GR nucleartranslocation in neuronal cells. It has been shown thatMR and GR share a significant sequence identity, of-ten binding the same ligands (46). Nevertheless, theeffect of TUDCA is not surprising, because NSR li-gands mediate effects that are receptor- rather thanligand-specific (47). In addition, ligand-induced nu-clear translocation of MR and GR exhibits differentialpatterns depending on the cell type (48). Curiously, itwas demonstrated that the presence or absence ofcortisol in the same cell type determines the activationof either GR and MR, or only MR by the same ligand(47). Thus, it is possible that specific comodulators inneuronal cells may somehow induce TUDCA-medi-ated MR nuclear translocation, rather than GR nucleartraffic. Others have demonstrated that the ability of
corticosteroids to stimulate receptor transactivationfunction is also dependent on the stability of the ste-roid-receptor complexes (49).
To investigate a possible interaction betweenTUDCA and MR, and having shown that UDCA bindsto a specific GR LBD region in hepatic cells (30), weevaluated the association between TUDCA and MRLBD during A�-mediated apoptosis. Our resultsshowed that, in the presence of A�, TUDCA appearsto interact with a specific region of MR LBD. Thispromotes MR dissociation from its cytosolic chaper-one, hsp90, and translocation to the nucleus possiblyas a bile acid-receptor complex. By competition stud-ies, corticosterone diminished both TUDCA-inducedGRE/MRE transactivation and nuclear translocation ofthe bile acid. In fact, the deleted form of MR(1–840)lacks an important region of the steroid-bindingpocket, which is thought to be necessary for the hy-drophobic contact between ligands and NSRs (50).This mutation affected nuclear translocation of bothMR and the bile acid. In fact, MR LBD appears to be aprerequisite for the antiapoptotic function of TUDCAduring A�-induced apoptosis. MR may be required asthe nuclear transporter molecule for TUDCA, and/ormay also be transcriptionally modulated by the bileacid to prevent apoptosis. Indeed, the antiapoptoticaction of MR has been extensively described in neu-ronal cells (20, 51–53). Interestingly, it has been shownthat the increase of neuronal survival is reversed by aMR antagonist (27). Thus, to determine whetherTUDCA does require modulation of NSR activity toprotect A�-induced apoptosis, we measured both MRand GR transactivation. In contrast to our results inprimary rat hepatocytes (30), TUDCA significantly al-tered MR and GR transactivation in PC12 cells; thetranscriptional activity of MR was increased, whereasGR transactivation was reduced by TUDCA pretreat-ment. It has been postulated that although MR inhibitsGR transactivation in lymphocytes, it does not preventtranslocation of GR into the nucleus with hormonebinding (28). Nevertheless, during A�-mediated neu-ronal apoptosis, repression of GR activity by TUDCAmay be attributed, in part, to the significant decreasein GR nuclear translocation. In addition, it is importantto note that the context of this study is therapeutic anddoes not immediately apply to physiological modula-tion of NSR function. In fact, the levels of bile acidsused are pharmacological, rather than (patho)physio-logical, concentrations.
Finally, it remains to be determined which moleculartargets are directly affected by TUDCA-mediated reg-ulation of NSR transactivation during A�-induced ap-optosis. The members of both the phosphatidylinositol3-kinase survival signaling and the E2F-1/p53/Bax ap-optotic pathway are good candidates, because wehave previously shown that both pathways are mod-ulated by TUDCA during A�-mediated cell death (16,17).
Collectively, our studies further expand the anti-apoptotic mechanism for TUDCA during A�-induced
Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis Mol Endocrinol, October 2006, 20(10):2292–2303 2299

cell injury. The results demonstrate that TUDCA pref-erentially targets MR in neural cells, rather than GR,interacting with a specific region of MR LBD and dis-sociating MR from its cytosolic chaperone, hsp90.TUDCA is then translocated into the nucleus togetherwith MR, modulating NSR transactivation and thusinhibiting A�-induced apoptosis. Finally, a completeelucidation of the molecular targets affected by theTUDCA-mediated regulation of NSRs during A�-in-duced apoptosis would promise to accelerate devel-opment of neuroprotective strategies for the treatmentof AD.
MATERIALS AND METHODS
Cell Cultures
Primary cultures of rat cortical neurons were prepared fromfetuses of 17- to 18-d pregnant Wistar rats as previouslydescribed (16). Briefly, after trypsinization, cells were washedtwice in Hank’s balanced salt solution without Ca2� andMg2� (HBSS-2), containing 10% fetal calf serum, and resus-pended in Neurobasal medium supplemented with 0.5 mM
L-glutamine, 25 �M L-glutamic acid, 2% B-27 supplement,and 12 mg/ml gentamicin (Invitrogen, San Diego, CA). Neu-rons were then plated on tissue culture plates precoated withpoly-D-lysine at 2 � 106 cells/ml and maintained at 37 C in ahumidified atmosphere of 5% CO2. All experiments wereperformed using cells cultured for 2–3 d in fresh Neurobasalmedium without glutamic acid and B-27 supplement. Neu-rons were characterized by phase contrast microscopy andindirect immunocytochemistry for neurofilaments and glialfibrillary acidic protein. Neuronal cultures were more than95% pure. All animals received humane care according to thecriteria outlined in the Guide for the Care and Use of Labo-ratory Animals prepared by the national Academy of Sci-ences and published by the National Institutes of Health (NIH)(NIH publication 86–23, revised 1985). In DNA plasmid trans-fection experiments, PC12 cells were grown in RPMI-1640medium (Sigma Chemical Co., St. Louis, MO) supplementedwith 10% heat-inactivated horse serum (Sigma ChemicalCo.), 5% fetal bovine serum (Invitrogen), and 1% penicillin/streptomycin, and maintained at 37 C in a humidified atmo-sphere of 5% CO2. Cells were plated on tissue culture platesprecoated with poly-D-lysine at 4 � 105 cells/ml.
Intracellular Distribution of UDCA
Cellular distribution of UDCA was visualized by incubatingcells with NBD-UDCA fluorescent molecule, with transportproperties similar to those of natural bile acids (54). Cellswere incubated in fresh medium supplemented with 100 �M
of either NBD-UDCA or unlabeled UDCA for 5, 15, and 30 minand 1 and 2 h. Competition studies were performed by pre-treating cells with 500 �M unlabeled UDCA for 5 min, wash-ing, and replacing the medium with 100 �M NBD-UDCA for 1and 2 h. Attached cells were washed three times with PBSand fixed with 4% paraformaldehyde in PBS, pH 7.4, for 10min at room temperature. After additional washes, sampleswere mounted using Fluoromount-G, and fluorescence wasvisualized using an Axioskop fluorescence microscope (CarlZeiss GmbH, Jena, Germany). Nuclear localization of theNBD-UDCA molecule was confirmed by Hoechstcounterstain.
Induction of Apoptosis
Cells were cultured as described above and then incubatedwith either 25 �M A�(25–35) active fragment (Bachem AG,Bubendorf, Switzerland), or no addition (control) for 24 h, withor without UDCA or TUDCA (Sigma Chemical Co.). A�(1–40)(Bachem AG) at 10 �M was also tested in a subset of exper-iments. In coincubation studies, cells were pretreated with100 �M of either UDCA or TUDCA for 12 h before incubationwith A� peptides. Although these bile acid concentrations aregreater than physiological levels, they can be achieved afterexogenous administration (11). In parallel competition exper-iments, cells were pretreated with 10 �M of corticosterone(Sigma Chemical Co.), with or without bile acids, 12 h beforeincubation with A� peptides. Attached and floating cells werecombined to extract cytosolic, nuclear, and total proteins forcaspase activity assays, as well as immunoblot and immu-noprecipitation analysis. In parallel experiments, cells werealso fixed for morphological examination.
Morphological Evaluation of Apoptosis and CaspaseActivation
Hoechst labeling of cells was used to detect apoptotic nuclei(9). General caspase-3-like activity was determined by enzy-matic cleavage of chromophore p-nitroanilide (pNA) fromthe substrate N-acetyl-Asp-Glu-Val-Asp-pNA (DEVD-pNA;Sigma Chemical Co.) in cytosolic protein extracts (Complete;Roche Applied Science, Mannheim, Germany) (9). Caspase-3activation was confirmed by cleavage of PARP, a specificcaspase-3 endogenous substrate, in total protein extractsusing immunoblot analysis.
Transfections with siRNAs and GFP-MRChimeric Proteins
siRNA sequences targeting mr (GenBank Accession no.M36074) and gr (GenBank accession no. NM012576) mRNAcorresponded to the coding regions 523–543 (GGCGCTG-GAGTCAAGTGTCTC) and 129–149 (GGCCAAGGGAGGGG-GAGCGTA), respectively. The location of targeting se-quences was 51 and 50 nucleotides downstream, relative tothe first nucleotide of the start codon for mr and gr, respec-tively. The 23-nucleotide double-stranded RNAs were pre-pared by annealing both forward and reverse sequences ofMR and GR targeting regions; mr forward 5�-rGrGrCrGrCrU-rGrGrArGrUrCrArArGrUrGrUrCrUrCTT-3�; mr reverse 5�-rGrArGrArCrArCrUrUrGrArCrUrCrCrArGrCrGrCrCTT-3�; grforward 5�-rGrGrCrCrArArGrGrGrArGrGrGrGrGrArGrCrGrUr-ATT-3�; and gr reverse 5�-rUrArCrGrCrUrCrCrCrCrCrUrCrC-rCrUrUrGrGrCrCTT-3�. A nonspecific duplex was used ascontrol (5�-rCrArGrUrGrGrArGrArUrCrArArCrGrUrGrCrArAr-GUU-3�), which did not significantly affect MR and GR proteinlevels relative to the untransfected controls. Twelve hoursafter plating, primary rat cortical neurons at 40% confluencewere transfected with approximately 6 �g of siRNA usingJetSI Transfection Reagent for siRNA (Polyplus-Transfection,Illkirch, France), according to the manufacturer’s instructions.To assess gene silencing, protein levels of MR and GR weredetermined by immunoblotting. In addition, PC12 cells weretransiently transfected with the expression plasmids for thechimeric proteins of GFP and human MR, including the Wtform, GFP-MR Wt, and a carboxy terminus-deleted form ofMR LBD, GFP-MR(1–840). GFP-MR Wt gene expression wasunder cytomegalovirus-IEP T7 enhancer/promoter control(55, 56). To generate the chimeric plasmid GFP-MR(1–840),the insert hMR-GFP was removed from the pCMX vector anddigested with BfrBI and PmlI to delete the carboxy terminusof MR LBD (amino acids 840–990). Upon ligation of the insert,the fragment was recloned into HindIII and XhoI sites of thepCMX original vector. For transfection assays, PC12 cellswere washed with PBS to remove dead cells and incubated
2300 Mol Endocrinol, October 2006, 20(10):2292–2303 Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis

in RPMI-1640 medium supplemented with 1% heat-inacti-vated horse serum. At 24 h after plating, cells at approxi-mately 40% confluence were transfected with 5 �g of eachconstruct using Lipofectamine 2000 (Invitrogen), accordingto the manufacture’s instructions.
Immunoblotting
Steady-state levels of MR, GR, GFP, and hsp90 proteins, aswell as NSR distribution and PARP cleavage, were deter-mined by Western blot, using primary rabbit polyclonal anti-bodies reactive to MR, GR, hsp90, and PARP (Santa CruzBiotechnology, Inc., Santa Cruz, CA) or primary mousemonoclonal antibody to GFP (BD Biosciences, Palo Alto, CA),as well as secondary antibodies conjugated with horseradishperoxidase (Bio-Rad Laboratories, Hercules, CA). Mem-branes were processed for protein detection using SuperSignal substrate (Pierce Chemical Co., Rockford, IL). �-Actinwas used as a loading control. Protein concentrations weredetermined using the Bio-Rad protein assay kit according tothe manufacturer’s specifications.
Immunoprecipitation
Binding of MR to hsp90 was detected by immunoprecipita-tion analysis. In brief, whole-cell extracts were prepared bylysing cells in M-PER (Mammalian Protein Extraction Re-agent) (Pierce). Immunoprecipitation experiments were car-ried out using the antibody to hsp90 and the Ezview RedProtein G Affinity Gel (Sigma Chemical Co.). Typically, 200 �gof lysate was incubated with 1 �g of primary mouse mono-clonal antibody to hsp90 overnight at 4 C. Immunoblots werethen probed with the rabbit polyclonal MR antibody. Hsp90expression was determined in the same membrane afterstripping off the immune complex for the detection of MR. Inparallel, 20 �g of whole-cell extract was independently usedfor immunodetection of MR and hsp90. Immunoprecipitationassays using high-detergent conditions as well as Westernblot analysis showed absence of nonspecific binding of thehsp90 antibody to MR. In addition, immunoprecipitation as-says using the mouse monoclonal antibody reactive to GFPwere performed to confirm the results obtained with the MRantibody. Finally, immunoprecipitation assays using themouse monoclonal antibody reactive to �-actin showed nodetectable association with either hsp90 or MR.
Subcellular Localization of GFP Fusion Proteins
Analysis of intracellular trafficking of GFP-MR in cells wasperformed by using PC12 cells transiently transfected withchimeric proteins GFP-MR Wt or GFP-MR(1–840). For detec-tion of GFP fluorescence, transfected cells were incubated at30 C for 4 h, fixed with 4% paraformaldehyde in PBS, pH 7.4,at room temperature for 10 min, washed, and mounted usingFluoromount-G. Fluorescence was visualized using an Axios-kop fluorescence microscope (Carl Zeiss GmbH). Nuclearlocalization of the GFP-MR molecule was confirmed byHoechst counterstain.
Transactivation of NSRs
Transactivation of NSRs was investigated by cotransfectingPC12 cells with a GR/MR-responsive reporter plasmid,pGRE/MRE-luciferase, and the Wt human MR (pRShMR) orGR (pRShGR). The pGRE/MRE plasmid consisted of the en-tire human GR/MR-responsive promoter fused to the lucif-erase gene (57); pRShMR and pRShGR overexpression plas-mids were placed under simian virus 40 enhancer/promotercontrol (55). Twelve hours after plating, cells at approximately40% confluence were transfected with 3 �g of pGRE/MRE-
luciferase plasmid and 10 ng of the receptor expressionplasmids, pRShMR or pRShGR. For normalization, cells werecotransfected with the chloramphenicol acetyltransferase(CAT) reporter construct, PGL3-Control vector (PromegaCorp., Madison, WI). Transfection efficiencies of approxi-mately 70% were determined in cortical neurons using areporter plasmid expressing �-galactosidase and did not dif-fer between reporter and expression plasmids (data notshown). The cells were harvested for luciferase assays (Pro-mega Corp.) and CAT ELISA (Roche Applied Science, India-napolis, IN), according to the manufacturer’s recommendation.
Densitometry and Statistical Analysis
The relative intensities of protein bands were analyzed usingthe ImageMaster 1D Elite densitometric analysis program(Amersham Biosciences, Piscataway, NJ). Statistical analysiswas performed using GraphPad InStat version 3.00 for Win-dows 95 (GraphPad Software, San Diego, CA) for the ANOVAand Bonferroni’s multiple comparison tests. Values of P �0.05 were considered statistically significant.
Acknowledgments
We thank Dr. Alan F. Hoffman, University of California, SanDiego, CA, for the generous gift of NBD-UDCA and Dr. Hiro-toshi Tanaka, University of Tokyo, Tokyo, Japan, for provid-ing the pGRE/MRE reporter plasmid, as well as the pRShMR,pRShGR, and GFP-MR Wt overexpression plasmids. We arealso deeply grateful to Dr. Elsa Rodrigues, University of Lis-bon, Lisbon, Portugal, for her help with the construction ofthe carboxy terminus-deleted form of MR LBD overexpres-sion plasmid, GFP-MR(1-840).
Received February 6, 2006. Accepted May 10, 2006.Address all correspondence and requests for reprints to:
Dr. Cecılia M. P. Rodrigues, Avenida das Forcas Armadas,1600–083 Lisbon, Portugal. E-mail: [email protected].
This work was supported by POCTI/BCI/44929/2002 fromFundacao para a Ciencia e a Tecnologia, and L-V-595/2004from Fundacao Luso-Americana, Lisbon, Portugal (toC.M.P.R.). S.S. was the recipient of postdoctoral fellowship(SFRH/BPD/20834/2004). J.D.A., P.M.B., R.M.R. and R.E.C.were recipients of Ph.D. fellowships (BD/17799/2004, BD/24165/2005, BD/12641/2003, and BD/12655/2003, respec-tively) from Fundacao para a Ciencia e a Tecnologia.
Disclosure of potential conflicts of interest: S.S., J.D.A.,P.M.B., R.M.R., R.E.C., M.M.A., C.J.S., and C.M.P.R. havenothing to declare.
REFERENCES
1. Selkoe DJ 2001 Alzheimer’s disease: genes, proteins,and therapy. Physiol Rev 81:741–766
2. Haass C, Selkoe DJ 1993 Cellular processing of �-amy-loid precursor protein and the genesis of amyloid �-pep-tide. Cell 75:1039–1042
3. Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, GuoZ, Greig NH, Mattson MP 2001 A synthetic inhibitor ofp53 protects neurons against death induced by ischemicand excitotoxic insults, and amyloid �-peptide. J Neuro-chem 77:220–228
4. Yao M, Nguyen TV, Pike CJ 2005 �-Amyloid-inducedneuronal apoptosis involves c-Jun N-terminal kinase-de-pendent downregulation of Bcl-w. J Neurosci 25:1149–1158
Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis Mol Endocrinol, October 2006, 20(10):2292–2303 2301

5. Watson K, Fan GH 2005 Macrophage inflammatory pro-tein 2 inhibits �-amyloid peptide (1–42)-mediated hip-pocampal neuronal apoptosis through activation of mi-togen-activated protein kinase and phosphatidylinositol3-kinase signaling pathways. Mol Pharmacol 67:757–765
6. Rodrigues CMP, Fan G, Ma X, Kren BT, Steer CJ 1998 Anovel role for ursodeoxycholic acid in inhibiting apoptosisby modulating mitochondrial membrane perturbation.J Clin Invest 101:2790–2799
7. Rodrigues CMP, Stieers CL, Keene CD, Ma X, Kren BT,Low WC, Steer CJ 2000 Tauroursodeoxycholic acid par-tially prevents apoptosis induced by 3-nitropropionicacid: evidence for a mitochondrial pathway independentof the permeability transition. J Neurochem 75:2368–2379
8. Rodrigues CMP, Sola S, Brites D 2002 Bilirubin inducesapoptosis via the mitochondrial pathway in developingrat brain neurons. Hepatology 35:1186–1195
9. Sola S, Ma X, Castro RE, Kren BT, Steer CJ, RodriguesCMP 2003 Ursodeoxycholic acid modulates E2F-1 andp53 expression through a caspase-independent mecha-nism in transforming growth factor �1-induced apoptosisof rat hepatocytes. J Biol Chem 278:48831–48838
10. Keene CD, Rodrigues CMP, Eich T, Linehan-Stieers C,Abt A, Kren BT, Steer CJ, Low WC 2001 A bile acidprotects against motor and cognitive deficits and re-duces striatal degeneration in the 3-nitropropionic acidmodel of Huntington’s disease. Exp Neurol 171:351–360
11. Keene CD, Rodrigues CMP, Eich T, Chhabra MS, SteerCJ, Low WC 2002 Tauroursodeoxycholic acid, a bileacid, is neuroprotective in a transgenic animal model ofHuntington’s disease. Proc Natl Acad Sci USA 99:10671–10676
12. Rodrigues CMP, Spellman SR, Sola S, Grande AW, Line-han-Stieers C, Low WC, Steer CJ 2002 Neuroprotectionby a bile acid in an acute stroke model in the rat. J CerebBlood Flow Metab 22:463–471
13. Rodrigues CMP, Sola S, Nan Z, Castro RE, Ribeiro PS,Low WC, Steer CJ 2003 Tauroursodeoxycholic acid re-duces apoptosis and protects against neurological injuryafter acute hemorrhagic stroke in rats. Proc Natl AcadSci USA 100:6087–6092
14. Duan WM, Rodrigues CMP, Zhao LR, Steer CJ, Low WC2002 Tauroursodeoxycholic acid improves the survivaland function of nigral transplants in a rat model of Par-kinson’s disease. Cell Transplant 11:195–205
15. Ved R, Saha S, Westlund B, Perier C, Burnam LG, SluderA, Hoener M, Rodrigues CMP, Alfonso A, Steer C, Liu L,Przedborski S, Wolozin B 2005 Similar patterns of mito-chondrial vulnerability and rescue induced by geneticmodification of �-synuclein, parkin and DJ-1 in C. el-egans. J Biol Chem 280:42655–42668
16. Sola S, Castro RE, Laires PA, Steer CJ, Rodrigues CMP2003 Tauroursodeoxycholic acid prevents amyloid-�peptide-induced neuronal death via a phosphatidylinosi-tol 3-kinase-dependent signaling pathway. Mol Med9:226–234
17. Ramalho RM, Ribeiro PS, Sola S, Castro RE, Steer CJ,Rodrigues CMP 2004 Inhibition of the E2F-1/p53/Baxpathway by tauroursodeoxycholic acid in amyloid �-pep-tide-induced apoptosis of PC12 cells. J Neurochem 90:567–575
18. McCormick JA, Lyons V, Jacobson MD, Noble J, DiorioJ, Nyirenda M, Weaver S, Ester W, Yau JL, Meaney MJ,Seckl JR, Chapman KE 2000 5�-Heterogeneity of glu-cocorticoid receptor messenger RNA is tissue specific:differential regulation of variant transcripts by early-lifeevents. Mol Endocrinol 14:506–517
19. Bastian LS, Nordeen SK 1991 Concerted stimulation oftranscription by glucocorticoid receptors and basal tran-scription factors: limited transcriptional synergism sug-gests mediation by coactivators/adaptors. Mol Endocri-nol 5:619–627
20. Almeida OF, Conde GL, Crochemore C, Demeneix BA,Fischer D, Hassan AH, Meyer M, Holsboer F, MichaelidisTM 2000 Subtle shifts in the ratio between pro- andantiapoptotic molecules after activation of corticosteroidreceptors decide neuronal fate. FASEB J 14:779–790
21. Crochemore C, Michaelidis TM, Fischer D, Loeffler JP,Almeida OF 2002 Enhancement of p53 activity and inhi-bition of neural cell proliferation by glucocorticoid recep-tor activation. FASEB J 16:761–770
22. Wang Z, Garabedian MJ 2003 Modulation of glucocorti-coid receptor transcriptional activation, phosphorylation,and growth inhibition by p27Kip1. J Biol Chem 278:50897–50901
23. Wetzel DM, Bohn MC, Kazee AM, Hamill RW 1995 Glu-cocorticoid receptor mRNA in Alzheimer’s diseased hip-pocampus. Brain Res 679:72–81
24. Rasmuson S, Andrew R, Nasman B, Seckl JR, WalkerBR, Olsson T 2001 Increased glucocorticoid productionand altered cortisol metabolism in women with mild tomoderate Alzheimer’s disease. Biol Psychiatry 49:547–552
25. Tsolakidou AF, Coulocheri SA, Trikkas G, Moutsatsou P2004 Gene analysis of the glucocorticoid receptor � inAlzheimer’s disease. Clin Chim Acta 349:167–172
26. Behl C, Lezoualc’h F, Trapp T, Widmann M, Skutella T,Holsboer F 1997 Glucocorticoids enhance oxidativestress-induced cell death in hippocampal neurons invitro. Endocrinology 138:101–106
27. Macleod MR, Johansson IM, Soderstrom I, Lai M, GidoG, Wieloch T, Seckl JR, Olsson T 2003 Mineralocorticoidreceptor expression and increased survival followingneuronal injury. Eur J Neurosci 17:1549–1555
28. Planey SL, Derfoul A, Steplewski A, Robertson NM,Litwack G 2002 Inhibition of glucocorticoid-induced apo-ptosis in 697 pre-B lymphocytes by the mineralocorticoidreceptor N-terminal domain. J Biol Chem 277:42188–42196
29. Mendoza ME, Monte MJ, El-Mir MY, Badia MD, Marin JJ2002 Changes in the pattern of bile acids in the nuclei ofrat liver cells during hepatocarcinogenesis. Clin Sci(Lond) 102:143–150
30. Sola S, Amaral JD, Castro RE, Ramalho RM, BorralhoPM, Kren BT, Tanaka H, Steer CJ, Rodrigues CMP 2005Nuclear translocation of UDCA by the glucocorticoid re-ceptor is required to reduce TGF-�1-induced apoptosisin rat hepatocytes. Hepatology 42:925–934
31. Miura T, Ouchida R, Yoshikawa N, Okamoto K, Makino Y,Nakamura T, Morimoto C, Makino I, Tanaka H 2001Functional modulation of the glucocorticoid receptor andsuppression of NF-�B-dependent transcription by ur-sodeoxycholic acid. J Biol Chem 276:47371–47378
32. Sola S, Castro RE, Kren BT, Steer CJ, Rodrigues CMP2004 Modulation of nuclear steroid receptors by ursode-oxycholic acid inhibits TGF-�1-induced E2F-1/p53-me-diated apoptosis of rat hepatocytes. Biochemistry 43:8429–8438
33. Pratt WB, Toft DO 1997 Steroid receptor interactionswith heat shock protein and immunophilin chaperones.Endocr Rev 18:306–360
34. Prima V, Depoix C, Masselot B, Formstecher P, LefebvreP 2000 Alteration of the glucocorticoid receptor subcel-lular localization by non steroidal compounds. J SteroidBiochem Mol Biol 72:1–12
35. Bohen SP, Kralli A, Yamamoto KR 1995 Hold ’em andfold ’em: chaperones and signal transduction. Science268:1303–1304
36. Whitfield GK, Jurutka PW, Haussler CA, Haussler MR1999 Steroid hormone receptors: evolution, ligands, andmolecular basis of biologic function. J Cell BiochemSuppl 32–33:110–122
37. Setchell KD, Rodrigues CMP, Clerici C, Solinas A, MorelliA, Gartung C, Boyer J 1997 Bile acid concentrations in
2302 Mol Endocrinol, October 2006, 20(10):2292–2303 Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis

human and rat liver tissue and in hepatocyte nuclei.Gastroenterology 112:226–235
38. Rodrigues CMP, Ma X, Linehan-Stieers C, Fan G, KrenBT, Steer CJ 1999 Ursodeoxycholic acid prevents cyto-chrome c release in apoptosis by inhibiting mitochondrialmembrane depolarization and channel formation. CellDeath Differ 6:842–854
39. Rodrigues CMP, Sola S, Brito MA, Brondino CD, BritesD, Moura JJ 2001 Amyloid �-peptide disrupts mitochon-drial membrane lipid and protein structure: protectiverole of tauroursodeoxycholate. Biochem Biophys ResCommun 281:468–474
40. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Con-sler TG, Kliewer SA, Stimmel JB, Willson TM, ZavackiAM, Moore DD, Lehmann JM 1999 Bile acids: naturalligands for an orphan nuclear receptor. Science 284:1365–1368
41. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM,Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B 1999Identification of a nuclear receptor for bile acids. Science284:1362–1365
42. Wang H, Chen J, Hollister K, Sowers LC, Forman BM1999 Endogenous bile acids are ligands for the nuclearreceptor FXR/BAR. Mol Cell 3:543–553
43. Tanaka H, Makino Y, Miura T, Hirano F, Okamoto K,Komura K, Sato Y, Makino I 1996 Ligand-independentactivation of the glucocorticoid receptor by ursodeoxy-cholic acid. Repression of IFN-�-induced MHC class IIgene expression via a glucocorticoid receptor-depen-dent pathway. J Immunol 156:1601–1608
44. Yamamoto M, Fukuda K, Miura N, Suzuki R, Kido T,Komatsu Y 1998 Inhibition by dexamethasone of trans-forming growth factor �1-induced apoptosis in rat hep-atoma cells: a possible association with Bcl-xL induction.Hepatology 27:959–966
45. Webster JC, Huber RM, Hanson RL, Collier PM, HawsTF, Mills JK, Burn TC, Allegretto EA 2002 Dexametha-sone and tumor necrosis factor-� act together to inducethe cellular inhibitor of apoptosis-2 gene and preventapoptosis in a variety of cell types. Endocrinology 143:3866–3874
46. Rogerson FM, Dimopoulos N, Sluka P, Chu S, Curtis AJ,Fuller PJ 1999 Structural determinants of aldosteronebinding selectivity in the mineralocorticoid receptor.J Biol Chem 274:36305–36311
47. Lim-Tio SS, Fuller PJ 1998 Intracellular signaling path-ways confer specificity of transactivation by mineralocor-ticoid and glucocorticoid receptors. Endocrinology 139:1653–1661
48. Nishi M, Ogawa H, Ito T, Matsuda KI, Kawata M 2001Dynamic changes in subcellular localization of mineralo-corticoid receptor in living cells: in comparison with glu-cocorticoid receptor using dual-color labeling with greenfluorescent protein spectral variants. Mol Endocrinol 15:1077–1092
49. Hellal-Levy C, Couette B, Fagart J, Souque A, Gomez-Sanchez C, Rafestin-Oblin M 1999 Specific hydroxyla-tions determine selective corticosteroid recognition byhuman glucocorticoid and mineralocorticoid receptors.FEBS Lett 464:9–13
50. Stevens A, Garside H, Berry A, Waters C, White A, Ray D2003 Dissociation of steroid receptor coactivator 1 andnuclear receptor corepressor recruitment to the humanglucocorticoid receptor by modification of the ligand-receptor interface: the role of tyrosine 735. Mol Endocri-nol 17:845–859
51. Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F,Almeida OF 1996 Exacerbation of apoptosis in the den-tate gyrus of the aged rat by dexamethasone and theprotective role of corticosterone. Exp Neurol 140:43–52
52. Hassan AH, Patchev VK, von Rosenstiel P, Holsboer F,Almeida OF 1999 Plasticity of hippocampal corticoste-roid receptors during aging in the rat. FASEB J 13:115–122
53. McCullers DL, Herman JP 1998 Mineralocorticoid recep-tors regulate bcl-2 and p53 mRNA expression in hip-pocampus. Neuroreport 9:3085–3089
54. Holzinger F, Schteingart CD, Ton-Nu HT, Eming SA,Monte MJ, Hagey LR, Hofmann AF 1997 Fluorescent bileacid derivatives: relationship between chemical structureand hepatic and intestinal transport in the rat. Hepatol-ogy 26:1263–1271
55. Yoshikawa N, Makino Y, Okamoto K, Morimoto C,Makino I, Tanaka H 2002 Distinct interaction of cortivazolwith the ligand binding domain confers glucocorticoidreceptor specificity: cortivazol is a specific ligand for theglucocorticoid receptor. J Biol Chem 277:5529–5540
56. Yoshikawa N, Yamamoto K, Shimizu N, Yamada S, Mo-rimoto C, Tanaka H 2005 The distinct agonist propertiesof the phenylpyrazolosteroid cortivasol reveal interdo-main communication within the glucocorticoid receptor.Mol Endocrinol 19:1110–1124
57. Makino Y, Yoshikawa N, Okamoto K, Hirota K, Yodoi J,Makino I, Tanaka H 1999 Direct association with thiore-doxin allows redox regulation of glucocorticoid receptorfunction. J Biol Chem 274:3182–3188
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremostprofessional society serving the endocrine community.
Sola et al. • Modulation of NSR by TUDCA Reduces Apoptosis Mol Endocrinol, October 2006, 20(10):2292–2303 2303
Related Documents











