FREQUENCY OF HAEMOGL OF SRI LANKAN PATIENT DESIGN AND IMPLEMEN AND 719A>G POLYMORPH A MATERNALLY INHER TRISOMY 15 (pterq22) IN A C PRANIDH DIS THE UNIV IN PARTIAL FULF MASTER OF GLOBIN BETA GENE (HBB) MUTATIONS I TS REFERRED FOR β THALASSAEMIA S ~ NTATION OF A NEW ASSAY TO GENOTY HISMS IN THE THIOPURINE S-METHYLT (TPMT) GENE ~ RITED PARTIAL TRISOMY 1 (q44qter) AND CHILD WITH SILVER RUSSELL & PART 15q SYNDROME BY HEE BHAGYA JEERASINGHE (B.Sc) FMC/GD/02/2012/02 SSERTATION SUBMITTED TO VERSITY OF COLOMBO, SRI LANKA FILMENT OF THE REQUIREMENTS OF T F SCIENCE IN GENETIC DIAGNOSTICS AUGUST 2014 IN A COHORT SCREENING YPE 460G>A TRANSFERASE D PARTIAL TIAL TRISOMY THE

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

FREQUENCY OF HAEMOGLOBIN BETA GENE (HBB) MUTATIONS IN A COHORT
OF SRI LANKAN PATIENTS REFERRED FOR β THALASSAEMIA SCREENING
~DESIGN AND IMPLEMENTATION OF A NEW ASSAY TO GENOTYPE 460G>A
AND 719A>G POLYMORPHISMS IN THE THIOPURINE S-METHYLTRANSFERASE
(TPMT) GENE
~A MATERNALLY INHERITED PARTIAL TRISOMY 1 (q44qter) AND PARTIAL
TRISOMY 15 (pterq22) IN A CHILD WITH SILVER RUSSELL & PARTIAL TRISOMY
15q SYNDROME
BY
PRANIDHEE BHAGYA JEERASINGHE (B.Sc)
FMC/GD/02/2012/02
DISSERTATION SUBMITTED TO
THE UNIVERSITY OF COLOMBO, SRI LANKA
IN PARTIAL FULFILMENT OF THE REQUIREMENTS OF THE
MASTER OF SCIENCE IN GENETIC DIAGNOSTICS
AUGUST 2014
FREQUENCY OF HAEMOGLOBIN BETA GENE (HBB) MUTATIONS IN A COHORT
OF SRI LANKAN PATIENTS REFERRED FOR β THALASSAEMIA SCREENING
~DESIGN AND IMPLEMENTATION OF A NEW ASSAY TO GENOTYPE 460G>A
AND 719A>G POLYMORPHISMS IN THE THIOPURINE S-METHYLTRANSFERASE
(TPMT) GENE
~A MATERNALLY INHERITED PARTIAL TRISOMY 1 (q44qter) AND PARTIAL
TRISOMY 15 (pterq22) IN A CHILD WITH SILVER RUSSELL & PARTIAL TRISOMY
15q SYNDROME
BY
PRANIDHEE BHAGYA JEERASINGHE (B.Sc)
FMC/GD/02/2012/02
DISSERTATION SUBMITTED TO
THE UNIVERSITY OF COLOMBO, SRI LANKA
IN PARTIAL FULFILMENT OF THE REQUIREMENTS OF THE
MASTER OF SCIENCE IN GENETIC DIAGNOSTICS
AUGUST 2014
FREQUENCY OF HAEMOGLOBIN BETA GENE (HBB) MUTATIONS IN A COHORT
OF SRI LANKAN PATIENTS REFERRED FOR β THALASSAEMIA SCREENING
~DESIGN AND IMPLEMENTATION OF A NEW ASSAY TO GENOTYPE 460G>A
AND 719A>G POLYMORPHISMS IN THE THIOPURINE S-METHYLTRANSFERASE
(TPMT) GENE
~A MATERNALLY INHERITED PARTIAL TRISOMY 1 (q44qter) AND PARTIAL
TRISOMY 15 (pterq22) IN A CHILD WITH SILVER RUSSELL & PARTIAL TRISOMY
15q SYNDROME
BY
PRANIDHEE BHAGYA JEERASINGHE (B.Sc)
FMC/GD/02/2012/02
DISSERTATION SUBMITTED TO
THE UNIVERSITY OF COLOMBO, SRI LANKA
IN PARTIAL FULFILMENT OF THE REQUIREMENTS OF THE
MASTER OF SCIENCE IN GENETIC DIAGNOSTICS
AUGUST 2014

CERTIFICATION
I certify that the contents of this dissertation are my own work and that I have acknowledged the
sources where relevant.
…………………………………………
Signature of the candidate
This is to certify that the contents of this dissertation were supervised by the followingsupervisors:
MUTATION REPORT
……………………………. …………………………….…Dr. U.N.D. Sirisena Prof. V.H.W. Dissanayake
PHARMACOGENOMICS REPORT
……………………………. …………………………….…Dr. K.T. Wettasinghe Prof. V.H.W. Dissanayake
MOLECULAR CYTOGENETICS REPORT
……………………………. …………………………….…Ms. I. Kariyawasam Ms. V. Udalamaththa
…………………………….Prof. V.H.W. Dissanayake

ACKNOWLEDGEMENT
The writing of this dissertation has been one of the significant academic challenges I have faced.
Without the support, patience and guidance of the following people, this dissertation would not
have been completed. It is to them that I owe my deepest gratitude.
I am indebted to Prof Vajira Dissanayake for his continuous encouragement and guidance. He
sets high standards for his students and he encourages and guides them to meet those standards.
Furthermore, Dr. U.N.D. Sirisena, Dr. K.T. Wettasinghe, Ms. I. Kariyawasam, Ms. V.
Udalamaththa who undertook to act as my supervisors for their guidance, wisdom, knowledge
and constant supervision as well as for providing necessary information regarding the
dissertation.
I am also grateful to the molecular laboratory staff at Asiri Centre for Genomics & Regenerative
medicine, Colombo, Sri Lanka for assistance with DNA sequencing and helping me to find
patient data.
I would like to thank all my colleagues for their kind co-operation and encouragement which
helped me throughout in completing this dissertation.
Most importantly, none of this would have been possible without the love and patience of my
family to whom this dissertation is dedicated to. They have been a constant source of love,
concern, support and strength all these years.

MUTATION REPORT
FREQUENCY OF HAEMOGLOBIN BETA
GENE (HBB) MUTATIONS IN A COHORT
OF SRI LANKAN PATIENTS REFERRED
FOR β THALASSAEMIA SCREENING

1
ABSTRACT
Introduction: Thalassaemia is an autosomal recessive disorder commonly seen in the Sri
Lankan population. Majority of mutations seen in the HBB gene leading to β thalassaemia are
single nucleotide substitutions, deletions or insertions of oligonucleotides leading to frame-shift
mutations. Some of these mutations cause an absence of β-chain production and the resulting
disease is called β-zero (B0) thalassaemia, whereas others result in a reduced output of β chains,
β-plus (B+) thalassaemia.
This report presents the frequency of different HBB gene mutations in a cohort of Sri Lankan
patients referred for β thalassaemia screening.
Methods: The mutations that were screened using direct sequencing method in the present study
includes IVS1–5 (G>C), IVS 1–1 (G>A), CD 26 (G>A), CD 6 (A>T), CD15(G>A), IVS1-130
(G>C), CD121 (G>C), IVS1-129 (A>C), IVSII-613 (C>T), CD41/42-TCTT, and 619bp deletion.
Results: Highest frequency was seen in heterozygous carriers having IVS1-5 G>C and IVS1-1
G>A mutations (49.4%) followed by homozygous point mutation IVS1-5 G>C (14.1%).
Conclusion: The findings in this study therefore add to the growing database of HBB mutations
identified in the Sri Lankan population. Screening for the IVS1–5 (G>C), IVS 1–1 (G>A), and
CD 26 (G>A) [HbE] mutations should be carried out for early detection of β thalassaemia
patients as well as carrier/prenatal detection and genetic counseling in order to control and
prevent the severe socio-economic burden of this disease in the country.
Key words: β Thalassaemia, HBB gene, β Globin gene , mutations, Haemoglobinopathies,Sri Lanka

2
Introduction
Haemoglobin Beta (HBB) gene codes for a protein called Beta globin, which is a subunit of
haemoglobin. Haemoglobin normally consists of four protein subunits: two subunits of β-globin
and two subunits of another protein called alpha-globin. More than 180 different mutations of the
β globin gene have been found in patients with β thalassaemia. The majority of these mutations
are single nucleotide substitutions, deletions or insertions of oligonucleotides leading to frame-
shift mutations. An updated list of β-thalassemia mutations is available through the Globin Gene
Server website (http://www.globin.cse.psu.edu). All of us have two HBB genes. One inherited
from the father and the other from the mother. Normal people have two normal genes. Beta
Thalassaemia carriers or heterozygotes have a mutation in one gene. Affected individuals have
mutations in both genes. Individuals who have the same mutation in both genes are known as
homozygotes for that mutation. Individuals who have two different mutations are known as
compound heterozygotes.
These mutations may affect gene function at any level between transcription, processing of the
primary messenger ribonucleic acid (m-RNA) transcript, translation, or post-translational
stability of the gene product. Rarely, β thalassaemia, like alpha thalassaemia, may result from a
partial or complete deletion of the β globin gene. Some of these mutations cause an absence of β-
chain production and the resulting disease is called β-zero (B0) thalassaemia, whereas others
result in a reduced output of β chains, β-plus (B+) thalassemia. Thalassaemia is inherited as a
recessive trait. β-thalassaemia can be classified into: β Thalassemia major, Thalassaemia
intermedia and Thalassaemia minor.
β-thalassemia with associated Hb anomalies: HbC/β-thalassaemia; HbE/β-thalassaemia; HbS/ β-
thalassaemia (a clinical condition which is more similar to sickle cell disease than to

3
thalassaemia major or intermedia); HbD/β-thalassaemia (a clinical condition which has
manifestations ranging from mild to moderate disease, resembling either thalassemia minor or
thalassemia intermedia) (12).
β thalassaemia major patients require regular red blood cell transfusions and are usually detected
within the first two years of life. Unlike thalassaemia major, thalassaemia intermedia patients do
not require regular transfusions and may be detected later in life. Except in the rare dominant
forms, heterozygous β-thalassaemia usually results in the clinically silent carrier state. HbE/β-
thalassaemia and HbC/β-thalassaemia exhibit a wide range in terms of diversity of phenotypes
and spectrum of severity (1). Clinical features of thalassaemia major includes severe anaemia,
feeding problems, diarrhea, irritability, recurrent bouts of fever, and progressive enlargement of
the abdomen caused by spleen and liver enlargement. Individuals with thalassemia intermedia
present with milder anemia and do not require or only occasionally require blood transfusions. In
some of the severe cases, patients between the ages of 2 and 6 years survive without regular
blood transfusions, but growth retardation and developmental delay can occur. They also
frequently develop leg ulcers and have an increased predisposition to thrombosis. Thalassaemia
minor sometimes manifests mild anemia but are usually asymptomatic (1).
The thalassaemias occur at a particularly high frequency in a band stretching from the
Mediterranean region, through the Middle East and Indian subcontinent into South East Asia (2).
The total annual incidence of symptomatic individuals is estimated at 1 in 100,000 throughout
the world and 1 in 10,000 people in the European Union (1). South Asia as a whole is a region
with a high prevalence of β thalassaemia with an estimated 17 million β thalassaemia carriers in
India, ~ 8 million carriers in Pakistan, 3 million carriers in Bangladesh, and ~ 0.5 million carriers
in Sri Lanka. In Sri Lanka nearly 50 per cent of transfusion-dependent thalassaemic children

4
were found to have β-thalassaemia/Hb E (3, 4). In Southeast Asia, β°-thalassaemia far exceeds
β+-thalassaemias (5).
The HBB gene maps in the short arm of chromosome 11p15.5 which is a region also containing
the delta globin gene, the embryonic epsilon gene, the fetal A-gamma and G-gamma genes, and a
pseudo gene (ψB1). The order of developmental expression of the five functional globin genes
is: HBE1, HBG2, HBG1, HBD, and HBB. The HBB gene, which spans 1.6 Kb, contains three
exons and both 5′ and 3′ un-translated regions (UTRs) (1). Some of the major β thalassaemia
mutations and their frequencies in Sri Lanka are shown in Table 1 (6). Many techniques have
been applied to the molecular analysis and prenatal diagnosis of β-thalassemia. These include
amplification refractory mutation system (ARMS), denaturing gradient gel electrophoresis,
restriction fragment length polymorphism analysis, dot-blot hybridization with allele-specific
oligonucleotides, reverse dot-blot hybridization, direct DNA sequencing, and multiplex mini
sequencing (15).
In this paper, the frequency of common HBB gene mutations in a cohort of Sri Lankan patients
referred for β thalassaemia screening are described.
Materials And Methods
HBB mutation analysis reports of 124 patients who were referred to the Asiri Surgical Hospital,
Colombo, Sri Lanka mainly for prenatal testing, carrier testing, and diagnostic testing of
transfusion dependent cases during the period 2007 to 2014 were analyzed. The diagnosis of β
thalassaemia was made based on clinical and hematological indices. Ninety five (95) peripheral
blood samples, four (04) cord blood samples and twenty five (25) amniotic fluid samples were
collected from the individuals after obtaining written informed consent. The cohort consisted of

5
61 symptomatic individuals for diagnostic testing, 34 individuals for carrier testing, and 29
individuals for prenatal testing.
Genomic DNA was extracted from peripheral blood, amniotic fluid, and cord blood using
Promega DNA extraction kit. Eleven mutations; IVS1–5 (G>C), IVS 1–1 (G>A), CD 26 (G>A),
CD 6 (A>T), CD15 (G>A), IVS1-130 (G>C), CD121 (G>C), IVS1-129 (A>C), and IVSII-
613(C>T), IVS2-1G>C, CD 41/42 (–TTCT)–deletion were screened by mini sequencing and
∆619bp deletion was screened by gap-PCR. Both these techniques were performed according to
the methodologies originally described by Wen Wang et al (15).
Results
Out of 124 individuals that were screened, 99 individuals showed the presence of one of the
above mutations. The frequencies of different HBB mutations detected in this cohort are shown
in Table 2.
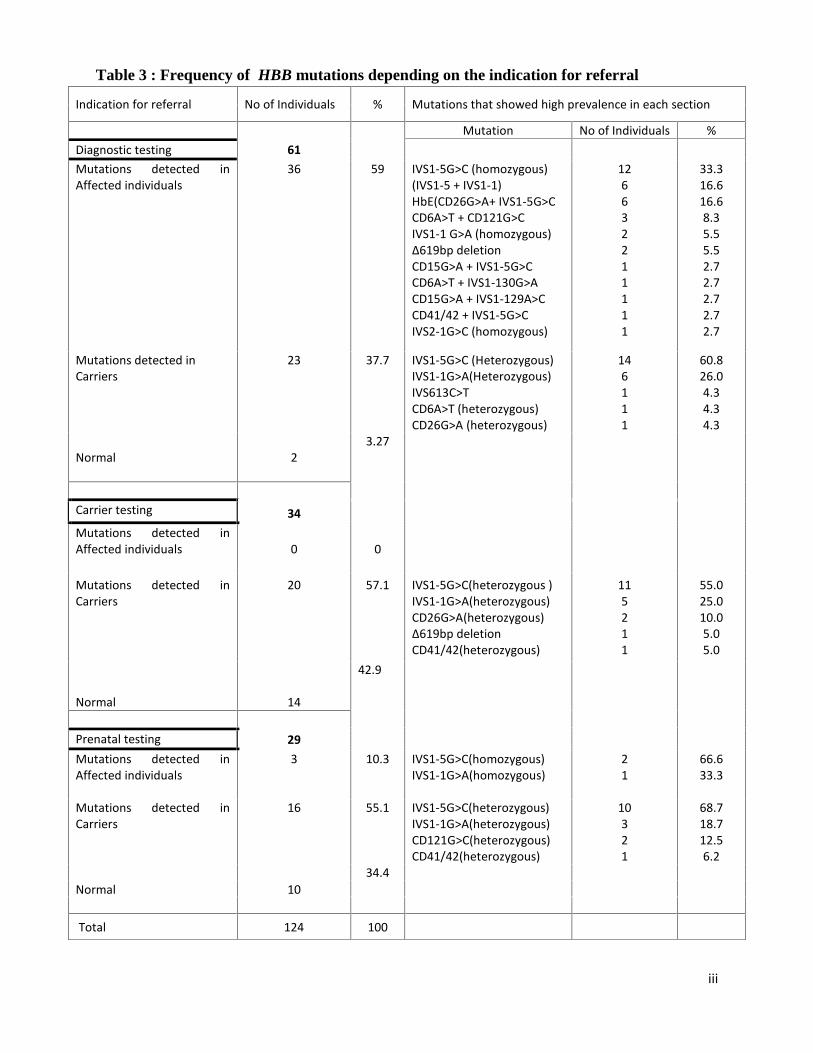
Table 3 gives elaborated figures of different mutations present in the cohort based on the
indication for referral. Overall, highest prevalence was seen in the heterozygous carriers having
IVS1-5 G>C and IVS1-1 G>A mutations (49.4%) followed by the homozygous point mutation
IVS1-5 G>C (14.1%). In a homozygous 619bp deletion, 1671-bp fragment was amplified and in
individuals heterozygous for the 619bp allele, both 1671bp & 1457bp fragments were amplified
and easily discriminated by simple agarose gel electrophoresis. In addition we also found three
intragenic single nucleotide polymorphisms (SNPs) [rs10768683-IVSII 16 G>C, rs7480526-
IVSII74 A>C, rs713040-CD2 T>C] in 12 patients (Table 04). Figure 1 and 2 illustrate the DNA
sequences of IVS1-1 G>A & IVS1-5 G>C heterozygote mutations, respectively. Figure 3
illustrates the DNA sequence of a IVSI-5 G>C homozygote mutation in a patient with
thalassaemia major. Figure 4 illustrates the DNA sequence of a compound heterozygote for CD

6
26 (G>A) and IVS1-5 (G>C) mutations in a patient with HbE/β-Thalassaemia. Figure 5
illustrates the deletion analysis result of homozygous ∆619bp deletion in a patient with β-
Thalassaemia.
Discussion
β thalassaemia is one of the most common autosomal recessive genetic diseases in Sri Lanka.
Many published data indicate that there is a high prevalence of β thalassaemia mutations in many
populations around the world (4).
Based on previously published data, the mutations that account for the highest prevalence in Sri
Lanka are IVSI-5 (G>C), which was observed at the highest frequency (56.2%), followed by
IVSI-1 (G>A), found at a frequency of 15.2%, and Hb E/β thalassaemia which accounted for
26.3% (6). In our study, the highest frequency was found in β thalassaemia patients who were
heterozygous carriers of IVSI-5 G>C & IVSI-1 G>A showing 49.4%. In patients who were
homozygous for IVSI-1(G>C), IVSI-1(G>A) and HbE CD26 (G>A) mutations, the frequencies
were found to be 14.1%, 3% and 10.1%, respectively. The results obtained in this study are in
agreement with other studies across the Indian subcontinent which show the IVSI-5 mutation as
being the commonest mutation (6)(8)(9). IVSI-1 (G>A), which was found at a frequency of 3%,
occurs at a very low frequency in north India (9) but is more commonly reported in the
Mediterranean region, Portugal, Spain and Middle East (7)(16). The data reported from the
Mediterranean region indicates that IVSI-1 (G>A), IVSI-110 (G>A), and IVSI-6 (T>C) are the
predominant β thalassaemia mutations while -101 (C>T) mutation is one of the most prevalent
silent β thalassaemia mutations (10)(11). Haemoglobin E occurs at a high frequency in parts of
North-east India and throughout South-east Asia (12). According to a previous study done in Sri

7
Lanka (6), HbE/ β-thalassaemia (CD26 G>A) mutation was observed at a frequency of 26.3%
(6). Both these findings are compatible with the comparatively high frequency of HbE/ β-
thalassaemia (CD26 G>A) mutation which accounted for 10.1% in our study. The other β-
thalassaemia mutations observed in this cohort, comprising 22% of the total, included:
CD15G>A, IVS1-129 A>C, CD 41/42, IVSII-613 C>T, CD6 A>T, IVS I-130 G>C, CD121
G>C, ∆619bp deletion, IVS2-1 G>C. These mutations have previously been reported in Sri
Lanka, India, Middle East and the Mediterranean region (6)(16). The structural haemoglobin
variants that were encountered, haemoglobins S and D appeared to occur at low frequencies
(total 7%) which agrees with the previous study done in Sri Lanka by Fisher C.A et.al in 2003
(6).
In addition to these however, we also found three intragenic SNPs [rs10768683- IVSII 16 G>C,
rs7480526-IVSII74 A>C, rs713040-CD2 T>C] that were used for haplotyping of the beta globin
gene in previous studies worldwide. These three SNPs have been used in one of the most
common haplotypes used for screening HBB mutations in diagnostic laboratories and in linkage
studies, in order to obtain useful information about the origin of the beta gene mutations and to
elaborate the clinical heterogeneity in β thalassaemia as well as the gene flow among countries
(17,18).
The findings in this study therefore add to the growing database of HBB mutations identified in
the Sri Lankan population. Screening for the IVS1–5 (G>C), IVS 1–1 (G>A), and CD 26 (G>A)
[HbE] mutations should be carried out for early detection of β thalassaemia patients as well as
carrier/prenatal detection and genetic counseling in order to control and prevent the severe socio-
economic burden of this disease in the country.

8
REFERENCES
(1). Galanello R, Origa R. Β-thalassemia. Orphanet J Rare Dis 2010;5:11.
(2). Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the
thalassaemias. Nat Rev Genet 2001;2:245-55.
(3). Olivieri NF, Thayalsuthan V, O'Donnell A, Premawardhena A, Rigobon C, Muraca G, et al.
Emerging insights in the management of hemoglobin E β thalassemia. Ann N Y Acad Sci
2010;1202:155-7.
(4). Premawardhena A, Fisher CA, Olivieri NF, de Silva S, Arambepola M, Perera W, et al.
Haemoglobin E β thalassaemia in Sri Lanka. Lancet 2005;366:1467-70.
(5). Fucharoen S, Winichagoon P. Haemoglobinopathies in southeast Asia. Indian J Med Res
2011;134:498-506.
(6). Fisher CA, Premawardhena A, de Silva S, Perera G, Rajapaksa S, Olivieri NA, et al. The
molecular basis for the thalassaemias in Sri Lanka. Br J Haematol 2003;121:662-71.
(7). Black ML, Sinha S, Agarwal S, Colah R, Das R, Bellgard M, et al. A descriptive profile of
β-thalassaemia mutations in India, Pakistan and Sri Lanka. J Community Genet 2010;1:149-
57.
(8). Garewal G, Das R, Awasthi A, Ahluwalia J, Marwaha RK. The clinical significance of the

9
spectrum of interactions of CAP+1 (A-->C), a silent β-globin gene mutation, with other β-
thalassemia mutations and globin gene modifiers in north Indians. Eur J Haematol
2007;79:417-21.
(9). Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of β-
thalassaemia mutations on the Indian subcontinent: the basis for prenatal diagnosis. Br J
Haematol 1991;78:242-7.
(10). Maragoudaki E, Kanavakis E, Traeger-Synodinos J, Vrettou C, Tzetis M, Metaxotou-
Mavrommati A, et al. Molecular, haematological and clinical studies of the -101 C --> T
substitution of the β-globin gene promoter in 25 β-thalassaemia intermedia patients and 45
heterozygotes. Br J Haematol 1999;107:699-706.
(11). Saeed Akhtar Khan Khattak SA, Nadir Ali ,Jaleel Anwar Kashif Hafeez Shaikh
Prevalence of various mutations in β thalassaemia and its association with haematological
parameters. Journal of Pakistan Medical Association 2012;62.
(12). Weatherall DJ, Glegg JB. The Thalassemia Syndromes. 4th ed. Oxford (UK): Blackwell
Science, 2001.
(13). Agarwal S, Pradhan M, Gupta UR, Sarwai S, Agarwal SS. Geographic and ethnic
distribution of beta-thalassaemia mutations in Uttar Pradesh. Hemoglobin. 2000a;24:89–
97. http://dx.doi.org/10.3109/03630260009003427. [PubMed]

10
(14). Agarwal S, Tamhankar PM, Kumar R, Dalal A. Clinical and haematological features in a compound
heterozygote (HBB:c.92 + 5G > C/HBB:c.93-2A > C) case of thalassaemia major. Int J Lab
Hematol. 2010;32(3):369–7. http://dx.doi.org/10.1111/j.1751-553X.2009.01157.x. [PubMed]
(15). Wang W, Kham SK, Yeo GH, et al. Multiplex minisequencing screen for common
Southeast Asian and Indian beta-thalassemia mutations. Clin Chem. 2003;49:209–218.
(16). Huisman, T.H.J., Carver, M.F.H. & Baysal, E. (1997) A Syllabus of Thalassemia
Mutations. The Sickle Cell Anemia Foundation, Augusta. 309.
(17). Beta Globin Frameworks in Thalassemia Major Patients from North Iran. Haleh Akhavan-
Niaki, Ali Banihashemi, and Mandana Azizi.
(18). The association between intragenic SNP haplotypes and mutations of the beta globin gene
in a Turkish population. Turker Bilgen, Yunus Arikan, Duran Canatan, Akif Yeşilipek,
Ibrahim Keser.

i
Table 1: Frequency of the common β thalassaemia mutations in Sri Lanka
(Fisher C.A. et al 2003) (6)
Mutation Allele Number Frequency (%)
IVS1-5(G>C) 697 56.2
IVS1-1(G>A) 189 15.2
HbE (CD26 G>A) 162 13.1
CD41/42 (-TCTT) 38 3.1
CD15 (G>A) 27 0.6
HbS (CD 6 A>T) 05 0.4
619bp deletion 01 0.1

ii
Table 2 . Frequency of the different HBB mutations in a cohort of Sri Lankan patients
referred for β thalassaemia screening
Type of HBB Mutation DetectedNo. of
samples%
β- Thalassaemia - Heterozygous carrierIVS1-5 G>CIVS1-1 G>A
493613
49.436.313.1
β- Thalassaemia Affected - HomozygousIVSI-5 G>C
14 14.1
β- Thalassaemia Affected- HomozygousIVSI-I G>A
3 3.0
β- Thalassaemia Affected- CompoundHeterozygousIVS1-5 G>C + IVS1-1G>AIVS1-5 G>C + CD15G>AIVS1-5 G>C+ CD 41/42CD15 G>A + IVS1-129 A>C
85111
8.05.01.01.01.0
β- Thalassaemia Carrier - HeterozygousIVSII-613 C>T
1 1.0
HbE/ β- ThalassaemiaCD26 G>ACD26 G>A + IVS I-5 G>C
1046
10.14.06.0
HbS/ β- ThalassaemiaCD6 A>T + IVS I-130 G>CHbS (Sicle cell carrier) CD6A>T
211
2.01.01.0
β- Thalassaemia Carrier - HeterozygousCD15 G>ACD41/42 –TCTTCD121 G>C
5122
5.11.02.02.0
β- ThalassaemiaHomozygous Affected ∆619bp deletionHeterozygous carrier ∆619bp deletion
321
3.02.01.0
β- Thalassaemia Affected -CompoundheterozygousCD6 A>T + CD121 G>C
3 3.0
β- Thalassaemia Carrier - HeterozygousIVS2-1 G>C
1 1.0
No mutations detected 25 20.16
Total 124 100
iii
Table 3 : Frequency of HBB mutations depending on the indication for referral
Indication for referral No of Individuals % Mutations that showed high prevalence in each section
Mutation No of Individuals %Diagnostic testing 61Mutations detected inAffected individuals
36 59 IVS1-5G>C (homozygous)(IVS1-5 + IVS1-1)HbE(CD26G>A+ IVS1-5G>CCD6A>T + CD121G>CIVS1-1 G>A (homozygous)∆619bp deletionCD15G>A + IVS1-5G>CCD6A>T + IVS1-130G>ACD15G>A + IVS1-129A>CCD41/42 + IVS1-5G>CIVS2-1G>C (homozygous)
126632211111
33.316.616.68.35.55.52.72.72.72.72.7
Mutations detected inCarriers
23 37.7 IVS1-5G>C (Heterozygous)IVS1-1G>A(Heterozygous)IVS613C>TCD6A>T (heterozygous)CD26G>A (heterozygous)
146111
60.826.04.34.34.3
Normal 23.27
Carrier testing 34Mutations detected inAffected individuals 0 0
Mutations detected inCarriers
20 57.1 IVS1-5G>C(heterozygous )IVS1-1G>A(heterozygous)CD26G>A(heterozygous)∆619bp deletionCD41/42(heterozygous)
115211
55.025.010.05.05.0
Normal 14
42.9
Prenatal testing 29Mutations detected inAffected individuals
3 10.3 IVS1-5G>C(homozygous)IVS1-1G>A(homozygous)
21
66.633.3
Mutations detected inCarriers
16 55.1 IVS1-5G>C(heterozygous)IVS1-1G>A(heterozygous)CD121G>C(heterozygous)CD41/42(heterozygous)
10321
68.718.712.56.2
Normal 1034.4
Total 124 100

iv
Table 4. Data on HBB gene mutations found with the presence of three single nucleotide
polymorphisms in 12 patients
HBB gene mutation HBB Gene SNPs GeneticIVSII 16
C>GIVSII 74
A>CCD2A>C
Diagnosis
rs10768683
rs7480526 rs713040
CD26 G>A + IVS I-5 G>C CG AA AC HbE/ β thalassemia
IVSI-5 G>C Homozygous CC AA AA βthalassaemia Major
IVSI-5 G>C Homozygous GG AA AA βthalassaemia Major
IVSI-5 G>C Heterozygous CG AC AC Carrier
IVSI-5 G>C Heterozygous CG AC AC Carrier
CD15 G>A Heterozygous CG AC AC Carrier
IVSI-5 G>C + CD15 G>A CC AA AA β thalassaemia Major
∆619bp Homozygous
deletion
CC AA AA β thalassemia Major
IVSI-1 G>A Heterozygous CG AA AC Carrier
CD6 A>T + IVS1-130 G>A CG AA AC HbS/β thalassemia
IVSI-5 G>C Heterozygous CG AC AC Carrier
IVSI-5 G>C + IVSI-1 G>A CC AA AA β thalassemia Major

v
Figure 1: IVS 1-1 (G>A) heterozygous mutation in a patient with Beta Thalassaemia
v
Figure 1: IVS 1-1 (G>A) heterozygous mutation in a patient with Beta Thalassaemia
v
Figure 1: IVS 1-1 (G>A) heterozygous mutation in a patient with Beta Thalassaemia

vi
Figure 2: IVS1-5 (G>C) heterozygous mutation in a patient with Beta Thalassaemia
vi
Figure 2: IVS1-5 (G>C) heterozygous mutation in a patient with Beta Thalassaemia
vi
Figure 2: IVS1-5 (G>C) heterozygous mutation in a patient with Beta Thalassaemia

vii
Figure 3: IVS1-5 (G>C) homozygous mutation in a patient with Beta Thalassaemia
IVSI-5 G>C
vii
Figure 3: IVS1-5 (G>C) homozygous mutation in a patient with Beta Thalassaemia
IVSI-5 G>C
vii
Figure 3: IVS1-5 (G>C) homozygous mutation in a patient with Beta Thalassaemia
IVSI-5 G>C

viii
Figure 4: Compound heterozygosity for CD 26 (G>A) and IVS1-5 (G>C) mutations in a
patient with HbE/β-Thalassaemia
IVS1-5 G>CCD26 G>A
viii
Figure 4: Compound heterozygosity for CD 26 (G>A) and IVS1-5 (G>C) mutations in a
patient with HbE/β-Thalassaemia
IVS1-5 G>CCD26 G>A
viii
Figure 4: Compound heterozygosity for CD 26 (G>A) and IVS1-5 (G>C) mutations in a
patient with HbE/β-Thalassaemia
IVS1-5 G>CCD26 G>A

ix
Figure 5: Homozygous deletion for ∆619bp in a patient with β-Thalassaemia
Blank

PHARMACOGENOMICS REPORT
DESIGN AND IMPLEMENTATION OF A
NEW ASSAY TO GENOTYPE 460G>A
AND 719A>G POLYMORPHISMS IN THE
THIOPURINE S-METHYLTRANSFERASE
(TPMT) GENE

1
ABSTRACT
Introduction: Thiopurine S-methyltransferase (TPMT) is a cytosolic enzyme which catalyses
the S-methylation of thiopurine drugs that are commonly used to treat a wide range of
conditions. Variation in sensitivity to these drugs among patients has been detected due to the
presence of point mutations in the TPMT gene. Studies have shown that TPMT*3C is known to be
the predominant mutant allele reported in Asian and African populations whereas TPMT*3A is
the predominant mutant allele found in Caucasians. The tetra-primer Amplification Refractory
Mutation System ARMS assay described here provides genotyping for two common TPMT
mutations 460G>A, and 719A>G seen in the South Asian region, and offers a simple, cost
effective, precise and rapid option for screening of patients in clinical settings. The aim of this
study was to design and implement new assay to genotype 460G>A, and 719A>G
polymorphisms in the TPMT gene.
Method: We designed a tetra-primer ARMS-Polymerase Chain Reaction PCR to genotype
polymorphisms in the TPMT gene.
Results: Out of the 30 samples used, none were found to be heterozygous or homozygous for
both mutations. All were of the wild type genotype (TPMT*3C – AA and TPMT*3B – GG).
Conclusion: This assay can be used to detect and analyze more variants of the TPMT gene by
designing tetra primers for each polymorphism in order to identify patients who are at risk of
developing hematotoxicity in response to treatment with thiopourine drugs.
Keywords: Thiopurine S-methyltransferase gene; Drug metabolism; Pharmacogenetics

2
Introduction
Thiopurinemethyltransferase or thiopurine S-methyltransferase (TPMT) is a cytosolic enzyme in
humans that is encoded by the TPMT gene located on chromosome 6p22.3 and is approximately
27Kb in size consisting of 9 exons (1). TPMT enzyme is involved in S-methylation of aromatic
and heterocyclic sulfhydryl compounds including the anticancer agents, 6-mercaptopurine, 6-
thioguanine and the immunosuppressant, azathioprine (2). Such thiopurine drugs are prescribed
for the treatment of many diseases including hematologic malignancies, rheumatoid arthritis,
inflammatory bowel disease (IBD) and as immunosuppressants in solid organ transplants (3).
These thiopurine drugs are metabolized to nucleotide intermediates by hypoxanthine-guanine
phosphoribosyl transferase and further metabolized to thioguanine nucleotides (TGN).
Alternatively, TPMT enzyme metabolizes thiopurine to inactive S-methylated metabolites and 6-
thiouric acid by xanthine oxidase (XO). This causes decrease in the amount of drug available for
activation to TGN. Negative correlation between the activity of TPMT in erythrocytes and
intracellular concentration of TGN has been reported (1). Thus, people who are having
intermediate or low activity of TPMT enzyme shows an increased risk of hematopoietic toxicity
when treated with conventional doses of drugs which are metabolized by TPMT enzyme (4).
TPMT enzyme deficiency is inherited in an autosomal recessive manner (4). Previous studies in
Caucasians and African-Americans have shown that most of them possess high activity ~90%,
10% shows intermediate activity and 0.3% low activity. Those who show high activity have the
TPMT*1/*1 (wild-type) genotype. Those who exhibit intermediate activity have the
heterozygous TPMT genotype and possess one TPMT variant allele. They may experience
moderate to severe myelosuppression. Finally the ones who show low activity of the enzyme
have the homozygous TPMT variant genotype and they have the highest risk of developing severe

3
myelosuppression. Such patients should not be treated with a thiopurine drug or the drug dose
should be reduced (5, 6).
TPMT displays genetic polymorphisms including 10 different allelic variants to date, of which
the most commonly studied are TPMT *2, *3A, *3B, *3C, and *4 (3).The TPMT*3A allele
comprises two transitions, 460G>A and 719A>G, which in turn produce the amino acid
substitutions Ala154Thr and Tyr240Cys, respectively, where as the TPMT*3B or TPMT*3C
comprises only the 460G>A or 719A>G transition (7).
According to the genotype analysis done in previous studies, the TPMT *3A mutation is known
to be the TPMT ancestral mutant allele as it is found in the African, Caucasian and Asian
populations (8). Further studies have shown that TPMT*3C is known to be the predominant
mutant allele reported in Asian and African populations whereas TPMT*3A is the predominant
mutant allele found in Caucasians (9). In the present study we describe the implementation and
development of a Tetra-primer Amplification refractory mutation system (ARMS) assay for the
detection of TPMT*3C (719A>G) rs1142345 and TPMT*3B (460G>A) rs1800460 single
nucleotide polymorphisms (SNPs) in order to study the predominant polymorphisms of TPMT
gene in the Sri Lankan population.
Method
Genomic DNA was extracted from 30 peripheral blood samples from a general Sri Lankan
population (10 samples each from three ethnic groups – Sinhalese, Moor and Tamil) using
QIAamp DNA Mini Kit and both the TPMT polymorphisms were genotyped in each sample.
PCR amplification was performed using primer sets for each SNP (pair of common outer primers
and a pair of inner/allele specific primers) (Table 1). Primers were designed using Primer 1
(primer design for tetra-primer ARMS-PCR) tool. Allele specificity was maximized by
introducing a 3` mismatch in each of the allele specific primers. Both SNPs were amplified

4
separately. Annealing temperatures for each allele specific primer were optimized and then
combined. Reactions consisted of a total volume of 25 µl containing , 12.4 µL of double distilled
water, 5 µl of 5X buffer, 0.5 µl of 10 mM dNTPS, 0.5 µl of each primer, 3 µL of 25 mM MgCl2,
0.1 µl of 5U/µL Taq DNA polymerase (Promega Corp,USA) and 2 µl of genomic DNA. An
initial denaturation step at 94°C for 5 min was followed by 30 cycles consisting of denaturation
step at 94°C for 45s, annealing for 45s at 59°C, and extension at 72°C for 45s. The final
extension was subsequently performed at 72°C for 10 min. The DNA fragments were
subsequently analyzed using a 3% agarose gel electrophoresis and stained with ethidium bromide
(0.03g/ml). The identity of positive control was confirmed by automated DNA sequencing on an
ABI Prism 3100-Avant Genetic Analyzer (Applied Biosystems, CA, USA), using Big Dye
Terminator chemistry version 3.1 (Applied Biosystems).
Results
Tetra-primer ARMS-PCR, which employs two primer pairs to amplify the two different alleles
of a SNP in a single PCR reaction was designed and implemented in order to determine the
predominant polymorphisms of TPMT gene in the Sri Lankan population. Agarose gel
electrophoresis images showing the genotyping results are given in Figure 1. Out of the 30
samples used, none were found to be heterozygous or homozygous for both mutations. All were
of the wild type genotype (TPMT*3C – AA and TPMT*3B – GG).

5
Table 1- Oligonucleotide primers used in this assay
TMPT*3C TPMT*3B
Figure1. Images of PCR amplicons containing SNP loci of TPMT*3C (NM_0003672:c.719A>G) andTPMT*3B (NM_000367.2:c.460G>A) after agarose gel electrophoresis. Sizes of the amplicons areindicated by arrows. Lane 1 shows the 500bp DNA ladder, Lane 2-3: samples showing wild typegenotype of TPMT*3C (Homozygous wild - AA) consisting of 475bp and 191bp amplicons. Lane 6-7:samples showing wild type genotype of TPMT*3B (Homozygous wild - GG) consisting of 506bp and263bp amplicons. Lanes 4 and 8 : Control sample.
# Primer name Sequence1 TPMT*3C-Forward (Inner Primer -A allele)
rs1142345 5’ GAATTGACTGTCTTTTTGAAAAGTTCTA 3’
2 TPMT*3C-Reverse (Inner primer -G allele)rs1142345 5’TGTCTCATTTACTTTTCTGTAAGTATAC 3’
3 TPMT*3C-Forward (Outer primer-(5' - 3'))rs1142345 5’TACCCAGCTCATTTTGTATTTTTAGTA3’
4 TPMT*3C-Reverse (Outer Primer- (5' - 3'))rs1142345 5’TACTAAAAAGCCATTTTTAGTAAAGATC3’
5 TPMT*3A/B-Forward (Inner Primer-G allele)rs1800460 5’GCAAATTTGACATGATTTGGGATAGAGTAG3’
6 TPMT*3A/B-Reverse (Inner Primer-A allele)rs1800460 5’ATCACCTGGATTGATGGCAACTAAGGT3’
7 TPMT*3A/B-Forward (Outer Primer- (5' - 3'))rs1800460 5’ATGTCCCCAAATCATAACAGAGTGGG3’
8 TPMT*3A/B-Reverse (Outer Primer- (5' - 3'))rs1800460 5’TAGCCTTACACCCAGGTCTCTGTAGTCA3’
50bpladder
506bp263bp
500bp
200bp100bp50bp
Blank
Control
Sample 1
Sample 2
Control
Sample 1
Sample 2
475bp191bp
1 2 3 4 5 6 7 8

6
Discussion
Many published data have demonstrated convincingly the relationship between TPMT enzyme
deficiency and the occurrence of thiopurine-induced toxicity in leukemia patients and recipients
of organ transplants (10). It is important to have reliable tests for TPMT alleles that can be
readily applied in diagnostic testing and research settings. The tetra-primer ARMS assay
described here provides genotyping for two common TPMT mutations seen in the South Asian
region, and offers a simple, cost effective, precise and rapid option for screening of patients in
clinical settings.
Studies have been developed using the multiplexed ARMS assay using three primers which
simultaneously detects TPMT*2, TPMT*3A, TPMT*3B, and TPMT*3C alleles (4) and many
others using sequencing and PCR-RFLP (Restriction Fragment Length Polymorphism)
techniques. Mostly research has been carried out using real-time PCR or RFLP technique.
However, reagent and equipment costs of those methods have limited the implementation of
those assays in many research and clinical laboratories especially in developing countries. The
assay we have designed has the added advantage that it can be carried out without the need for
costly specialized equipment.
According to the genotype analysis done by previous studies, the TPMT *3A (719A>G)
mutation is known to be the TPMT ancestral mutant allele as it is found in the African, Caucasian
and Asian populations (8). Further studies have shown that TPMT*3C is known to be the
predominant mutant allele reported in Asian and African populations whereas TPMT*3A is the
predominant mutant allele found in Caucasians (9). Hence, in this assay we chose the two most
prevalent TPMT polymorphisms (460G>A, and 719A>G) seen in the Asian and South East
Asian populations based on previously published data.

7
To date, no tests have been designed in Sri Lanka to determine the frequencies of the TPMT
alleles that are important in the metabolism of the thiopurine drugs.
Although none of the samples we used were positive for both mutations, our results indicated
that both 460G>A, and 719A>G TPMT polymorphisms can be detected using the tetra primer
ARMS assay efficiently. We carried out several optimizations in order to multiplex this assay
and detect both the variants simultaneously in one reaction mixture. However, the control bands
of each polymorphism failed to separate in gel electrophoresis when amplified in one reaction
mixture. Thus, both polymorphisms can be detected clearly when amplified in separate reaction
mixtures.
This assay can be used to detect and analyze more variants of the TPMT gene by designing tetra
primers for each polymorphism in order to identify patients who are at risk of developing
hematotoxicity in response to treatment with thiopurine drugs.
Acknowledgement
We thank Asiri Centre for Genomics & Regenerative medicine, Colombo, Sri Lanka for
assistance with DNA sequencing.
References
1. Seki T, Tanaka T, Nakamura Y. Genomic structure and multiple single-nucleotide
polymorphisms (SNPs) of the thiopurine S-methyltransferase (TPMT) gene. Journal of
human genetics. 2000;45(5):299-302.
2. Srimartpirom S, Tassaneeyakul W, Kukongviriyapan V, Tassaneeyakul W. Thiopurine S-
methyltransferase genetic polymorphism in the Thai population. British journal of clinical
pharmacology. 2004;58(1):66-70.
3. Nguyen CM, Mendes MA, Ma JD. Thiopurine methyltransferase (TPMT) genotyping to

8
predict myelosuppression risk. PLoS currents. 2011;3:RRN1236.
4. Roberts RL, Barclay ML, Gearry RB, Kennedy MA. A multiplexed allele-specific
polymerase chain reaction assay for the detection of common thiopurine S-
methyltransferase (TPMT) mutations. Clinica chimica acta; international journal of
clinical chemistry. 2004;341(1-2):49-53.
5. Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic
inheritance of erythrocyte thiopurine methyltransferase activity. American journal of
human genetics. 1980;32(5):651-62.
6. Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, et al.
Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine
methyltransferase genotype and thiopurine dosing. Clinical pharmacology and
therapeutics. 2011;89(3):387-91.
7. Tai HL, Krynetski EY, Yates CR, Loennechen T, Fessing MY, Krynetskaia NF, et al.
Thiopurine S-methyltransferase deficiency: two nucleotide transitions define the most
prevalent mutant allele associated with loss of catalytic activity in Caucasians. American
journal of human genetics. 1996;58(4):694-702.
8. Ameyaw MM, Collie-Duguid ES, Powrie RH, Ofori-Adjei D, McLeod HL. Thiopurine
methyltransferase alleles in British and Ghanaian populations. Human molecular genetics.
1999;8(2):367-70.
9. Otterness D, Szumlanski C, Lennard L, Klemetsdal B, Aarbakke J, Park-Hah JO, et al.
Human thiopurine methyltransferase pharmacogenetics: gene sequence polymorphisms.
Clinical pharmacology and therapeutics. 1997;62(1):60-73.
10. McLeod HL, Siva C. The thiopurine S-methyltransferase gene locus -- implications for
clinical pharmacogenomics. Pharmacogenomics. 2002;3(1):89-98.

i
SOP : TPMT genotyping
Title : Detection of TPMT*3C (460G>A), and TPMT*3B (719A>G) polymorphisms in the(Thiopurine S-methyltransferase) TPMT gene
Last Revised : June 2014
Test : Molecular Genetic Test
Purpose : To detect TPMT gene mutations for prediction of response to thiopurine drugs in
patients at risk of developing toxicity during thiopurine drug therapy in Sri Lanka
Method : Tetra-primer Allele- Specific PCR (AS-PCR)
1. DNA extraction
2. Tetra-primer Allele specific PCR
3. Agarose gel electrophoresis
Primer reconstitution (SOP003).
# Primer name Sequence
1 TPMT*3C-Forward (Inner Primer -A allele)rs1142345
5’ GAATTGACTGTCTTTTTGAAAAGTTCTA 3’
2 TPMT*3C-Reverse (Inner primer -G allele)rs1142345
5’TGTCTCATTTACTTTTCTGTAAGTATAC 3’
3 TPMT*3C-Forward (Outer primer-(5' - 3'))rs1142345
5’TACCCAGCTCATTTTGTATTTTTAGTA3’
4 TPMT*3C-Reverse (Outer Primer- (5' - 3'))rs1142345
5’TACTAAAAAGCCATTTTTAGTAAAGATC3’
5 TPMT*3A/B-Forward (Inner Primer-G allele)rs1800460
5’GCAAATTTGACATGATTTGGGATAGAGTAG3’
6 TPMT*3A/B-Reverse (Inner Primer-A allele)rs1800460
5’ATCACCTGGATTGATGGCAACTAAGGT3’
7 TPMT*3A/B-Forward (Outer Primer- (5' - 3'))rs1800460
5’ATGTCCCCAAATCATAACAGAGTGGG3’
8 TPMT*3A/B-Reverse (Outer Primer- (5' - 3'))rs1800460
5’TAGCCTTACACCCAGGTCTCTGTAGTCA3’
Step 1 : DNA extraction using peripheral blood leucocytes (Ref.SOP001/SOP002)
Mutated gene (V617F)

ii
Step 2 : Set-up PCR.Master mix 1 – For TPMT*3C25µL Master mix contains;
Component Volume
dH2O 12.4 µl
MgCl2 (25mM) 3 µl
5X PCR Buffer 5 µl
dNTP’s (10mM mix)(Ref.SOP007) 0.5 µl
Total Primer volume – 4 primers (per primer – 0.5 µl) 2µl
Taq polymerase 0.1 µl
DNA 2 µl
Master mix 1 – For TPMT*3BComponent Volume
dH2O 12.4 µl
MgCl2 (25mM) 3 µl
5X PCR Buffer 5 µl
dNTP’s (10mM mix)(Ref.SOP007) 0.5 µl
Total Primer volume – 4 primers (per primer – 0.5 µl) 2µl
Taq polymerase 0.1 µl
DNA 2 µl
Step 3:
Run PCR.
PCR cycle conditions;
Initial denaturation at 95˚c for 5 min.
Denaturation at 95 ˚c for 30 sec.
Annealing at 59 ˚c for 45 sec. 30 cycles
Extension at 72 ˚c for 1 min.
Final extension at 72 ˚c for 7 min.
Cooling at 10 ˚c ∞

iii
Step 4:
Analysis by gel electrophoresis (Ref:SOP004)
Run 10µl of PCR product at 60v in 3% gel for 30 min.
Gel
Interpretation
Results
Interpretation
:
:Lane No Size of bands Genotype
1 500bp ladder2 Sample 1 (TPMT*3C) 475bp, 191bp AA (Homozygous Wild)3 Sample 2 (TPMT*3C) 475bp, 191bp AA (Homozygous Wild)4 Control (TPMT*3C) 475bp, 191bp AA (Homozygous Wild)5 Blank6 Sample 1 (TPMT*3B) 506bp, 263bp GG(Homozygous Wild)7 Sample 2 (TPMT*3B) 506bp, 263bp GG(Homozygous Wild)8 Control (TPMT*3B) 506bp, 263bp GG(Homozygous Wild)
Genotype Expected bandsTPMT*3C (AA) homozygous wild 475bp, 191bpTPMT*3C (AG) heterozygous mutant 475bp, 191bp, 340bpTPMT*3C (GG) homozygous mutant 475bp, 340bpTPMT*3B (GG) homozygous wild 506bp, 263bpTPMT*3B (GA) heterozygous mutant 506bp, 263bp, 300bpTPMT*3B (AA) homozygous mutant 506bp, 300bp
1 2 3 4 5 6 7 8

iv
Reference : Roberts RL, Barclay ML, Gearry RB, Kennedy MA. A multiplexed allele-specificpolymerase chain reaction assay for the detection of common thiopurine S-methyltransferase (TPMT) mutations. Clinica chimica acta; international journal ofclinical chemistry. 2004;341(1-2):49-53.

v
Thiopurine S-methyltransferase, encodedby the TPMT gene, is the major enzymein hematopoietic cells responsible for theinactivation of thiopurines. Thiopurinedrugs are used in transplantation and thetreatment of several disorders includingchronic inflammatory diseases andhematological malignancies. Thiopurinedrugs have a narrow therapeutic indexand can cause life-threatening toxicity,which has been associated with anaccumulation of TPMT’s substrates, 6-thioguanine.Individuals with intermediateenzyme activity are heterozygous for onevariant in TPMT while those with low orno activity are homozygous or compoundheterozygous for variants in TPMT.
Reasons for Referral:
Individuals with conditions requiringtreatment with thiopurine drugs
Limitations:
Not all variants with known impact on enzymeexpression and activity are tested in this assay.Rare genetic alterations at primer binding sitesmay result in diagnostic errors.
Testing Methodology:
DNA amplification by Tetra-primer allelespecific PCR and analyzing PCR products byagrose gel electrophoresis.
Variant allele tested:
TPMT*3C (719A>G) rs1142345 TPMT*3B (460G>A) rs1800460
Specimen Requirement:
3cc of blood collected in EDTA (purple -top)tubes
Requisition must accompany specimen. Prior toany genetic testing we request that the subjectsign our consent form and submit it with thesample. To receive our forms, additionalinformation, please contact our unit.
Turnout time: 2 weeks
References
Seki T, Tanaka T, Nakamura Y. Genomic structureand multiple single-nucleotide polymorphisms(SNPs) of the thiopurine S-methyltransferase(TPMT) gene. Journal of human genetics.2000;45(5):299-302.
Srimartpirom S, Tassaneeyakul W, KukongviriyapanV, Tassaneeyakul W. Thiopurine S-methyltransferasegenetic polymorphism in the Thai population. Britishjournal of clinical pharmacology. 2004;58(1):66-70.
Performing Laboratory:Human Genetics UnitFaculty of MedicineUniversity of ColomboKynsey Road, Colombo 8, Sri LankaPhone (94-011) 2695 300, 2689 545 (Direct)Fax (94-011) [email protected]://www.hgucolombo.or
Thiopurine-S-Methyltransferase (TPMT) Genotyping
Human Genetics Unit, Faculty of Medicine, University Colombo

vi
Confidential Molecular Genetic Laboratory Test Report Date:
Human Genetics UnitFaculty of Medicine
University of ColomboKynsey Road, Colombo 8, Sri Lanka
Phone (94-011) 2695300, 2689545 (Direct)Fax (94-011) 2689979
[email protected]://www.hgucolombo.org
PatientIdentification:
Name: Age: Sex:
\
Lab Reference:
Indication:
Material Tested: EDTA Blood
Test: TPMT Genotyping
AnalysisPerformed:
The following mutations in the TPMT gene were genotyped by Tetra-primer AlleleSpecific PCR: TPMT*3B (460 G>A) and TPMT*3C (719 A>G)
Result: TPMT*3C or TPMT*3B mutations detected
TPMT*3C
Polymorphism Genotype TPMT Enzyme Activity
719A>G AG IntermediateHeterozygous mutation
719A>G GG Low/DeficientHomozygous Mutation
TPMT*3B
Polymorphism Genotype TPMT Enzyme Activity
460G>A GA IntermediateHeterozygous mutation
460G>A AA Low/DeficientHomozygous Mutation
719A>G & 460G>A AG, GACompound Heterozygote Low/Deficient

vii
Analysis Requested by:
Because of their complexity and their potential implications for other family members, all genetic tests should beaccompanied by genetic counseling.
Prof. Rohan W. Jayasekara MBBS (Ceylon), PhD (Newcastle), C.Biol., MSB (London) – Medical Geneticist and DirectorProf. Vajira H. W. Dissanayake MBBS (Colombo), PhD (Nottingham) – Medical Geneticist
Remarks: Based on this individual’s Genetic Result:
If heterozygous for one mutation – indicates an intermediate metabolizer ofthiopurine drugs. Hence this patient is at a risk of developing side effects orhematologic toxicity, and may require a lower dosage.
If homozygous/compound heterozygous - indicates a poor metabolizer ofthiopurine drugs and is at high risk for life-threatening hematologic toxicity ifgiven full doses of thiopurine drugs. Alternative therapy or greatly reduced dosageshould be considered for this patient.
If no mutations are detected –Since the above two mutations were not detected, this patient is having the wildtype genotype which is consistent with normal TPMT enzyme activity. Standarddoses of thiopurine drugs are less likely to be toxic in individuals with thisgenotype
Prof. Vajira H. W. Dissanayake MBBS, PhD, FNASSLMedicalGeneticist
………………………..
Analysis Performed by: ………………………..

MOLECULAR CYTOGENETICS REPORT
A MATERNALLY INHERITED PARTIAL
TRISOMY 1q (q44qter) AND PARTIAL
TRISOMY 15 (pterq22) IN A CHILD WITH
SILVER RUSSELL & PARTIAL TRISOMY
15q SYNDROME

1
ABSTRACT
A male infant with partial trisomy 1q syndrome and partial trisomy 15q syndrome
47,XY,+der(15)t(1;15)(q44;q22) is described. The baby presented with feeding difficulty,
developmental delay and dysmorphic features including macrocephaly, triangular face, high
nasal bridge, low set ears with a simple and malformed left ear, long philtrum, bilateral single
palmer creases, bilateral syndactyly and Atrial Septal Defect. Cytogenetic analysis (GTG
banding and karyotyping) and subsequent Fluorescence in situ Hybridization (FISH) showed a
derivative chromosome 15 as a result of maternal translocation involving chromosome 1 and 15.
According to our knowledge this is the first report of a patient with maternally inherited 1q
trisomy showing Silver Russell phenotype. We conclude that this patient shows both features of
Silver Russell Syndrome and partial trisomy 15q.
Keywords: Partial Trisomy 1q, Partial Trisomy 15q, Unbalanced Translocation, FISH,
Silver Russell Syndrome

2
Introduction
The inheritance of a derivative unbalanced chromosome creates a partial trisomy or partial
monosomy creating an unbalanced genotype that results in abnormal phenotypic features. Partial
trisomy 1q syndrome is a rare chromosomal abnormality, arising in most cases with de novo
translocation, duplication or insertion [1]. Two major partial trisomy 1q syndromes with regard
to the breakpoint localization have been described, one being a proximal partial trisomy ( 1q32-
qter) and the other as distal partial trisomy (1q42-qter) [2, 3]. Distal partial trisomy 1q syndrome
is often accompanied by other chromosome aberrations, which makes the definition of a
phenotype difficult [4].
A number of individuals have been reported with duplications of the proximal portion of 15q22
having 47 chromosomes, with the extra chromosome being a de novo bisatellited chromosome
15 [5] . Partial trisomy of the proximal part of the long arm of chromosome 15 can arise from a
balanced parental translocation, as a result of parental mosaicism or de novo which in turn leads
to either partial monosomy, or partial trisomy for different autosomes [6].
In this study, we report the results of standard cytogenetics and Fluorescence in situ
Hybridization (FISH). The phenotype of the child is also described and compared with other
previous prenatal cases reported in the literature.
Case report
We report a male infant who was examined at the age of 1 ½ months and died at the age of 8
months. He was the only child of non consanguineous parents with a history of a previous first
trimester miscarriage. He was born at 36 weeks of gestation by cesarean section due to lack of
progression and fetal distress with a birth weight of 2.1Kg (<10th percentile) and length of
34cm.( At birth the only dysmorphic feature which was noted was bilateral syndactyly of the 2nd

3
and 3rd toes. After 4 days the baby was admitted to the hospital due to poor sucking and
diagnosed with pyloric stenosis, jaundice due to lactation failure and weight loss (weight –
1.9kg), though there was no sign of fever. Ramstead Pyloromyotomy was performed and he was
managed in the PBU (Premature Babies Unit) and referred for genetic analysis.
The baby presented with feeding difficulty, developmental delay and dysmorphic features;
macrocephaly, triangular face, high nasal bridge, low set ears with a simple and malformed left
ear, long philtrum, bilateral single palmer creases and bilateral syndactyly. Echocardiographydetected Atrial Septal Defect and ultrasound scanning of abdomen and brain showed no
abnormalities. At the age of 4 months his weight was 3.65Kg. Figure 1 shows some of the
clinical features of the patient.

4
A B
C
Figure 1: Some clinical features of the Patient: A- Large head, Triangular face, Long Philtrum, low set
ears, B-B/L (Bi-lateral) Syndactyly of toes, C- Single palmer crease
Methods
The study was approved by the Ethics Review Committee, Faculty of Medicine, University of
Colombo and written informed consent was obtained from both parents.
Karyotyping
Chromosome culture and karyotyping was performed on peripheral blood lymphocytes of the
baby and both the parents according to the standard procedures. Metaphase chromosome spreads
4
A B
C
Figure 1: Some clinical features of the Patient: A- Large head, Triangular face, Long Philtrum, low set
ears, B-B/L (Bi-lateral) Syndactyly of toes, C- Single palmer crease
Methods
The study was approved by the Ethics Review Committee, Faculty of Medicine, University of
Colombo and written informed consent was obtained from both parents.
Karyotyping
Chromosome culture and karyotyping was performed on peripheral blood lymphocytes of the
baby and both the parents according to the standard procedures. Metaphase chromosome spreads
4
A B
C
Figure 1: Some clinical features of the Patient: A- Large head, Triangular face, Long Philtrum, low set
ears, B-B/L (Bi-lateral) Syndactyly of toes, C- Single palmer crease
Methods
The study was approved by the Ethics Review Committee, Faculty of Medicine, University of
Colombo and written informed consent was obtained from both parents.
Karyotyping
Chromosome culture and karyotyping was performed on peripheral blood lymphocytes of the
baby and both the parents according to the standard procedures. Metaphase chromosome spreads

5
were digested and stained using GTG banding technique. Chromosome spreads were observed
under light microscope (BX 61) and analyzed using Cytovision 3.1 soft ware. Out of 26
metaphase spreads, 14 were analyzed and 8 were karyotyped under a banding resolution of 450.
FISH (Fluorescence in situ Hybridization)
A preliminary FISH experiment was performed on the harvested whole blood lymphocyte cell
suspension of the baby using commercial fluorescent labeled probes of chromosomes 1 (BAC
clone: RP11-624F6, gene- HNRNPU, 1q44 (chr1:245,003,602-245,037,844), color- Green) and
chromosome 15 (BAC Clone: RP11-1059N24, gene- IGF1R 15q26.3 (chr15: 97,010,283 -
97,325,282), color- Red) according to the protocols and procedures described by Empire
Genomics LLC, NY, US (2014). According to the GTG banding technique we concluded that the
translocated regions of both chromosome 1 & 15 were present in the derivative chromosome 15
as 1q44-qter and 15pter-q22 respectively. FISH probes were chosen by selecting a gene (Figure
2) in each of these regions which correlated with the clinical phenotype of the patient. Though
RP11-1059N24 is not located within the 15pter-15q22 region, this probe was used to detect the
presence of derivative chromosome 15.
Initially, the cell culture was harvested using standard cytogenetics protocol. The fixative
(Carnoy’s 3:1 methanol: acetic acid) in cell suspension tube was changed until supernatant was
colourless and then re-fixed in fresh fixative prior to slide preparation. The slides were cleaned
by placing in a coplin jar with 70% alcohol for 5 minutes and then placing in a coplin jar with
fresh 3:1 acetic acid:methanol fixative. Three drops of cell suspension were added on to one slide
in a vertical angle. Then the slide was gently rotated, tipping slightly after ~15 seconds to drain
excess suspension. The slides were kept horizontally until a grainy appearance was observed and
the edges of the slide were dried. 10μl of each probe mixture was added on to two slides (2ul

6
probe + 8ul hybridization buffer) separately. A clean 22 x 22 cover slip was applied on to each
slide and the edges of the cover slip were sealed using rubber cement. The probes were
hybridized with metaphase chromosomes in a StatSpin®ThermoBrite® Hybridizer (Abbot
molecular); denaturation at 73◦C for 2 minutes/Hybridize at 37◦C for 16 hours. After
hybridization the slides were taken and the cover slips were removed. A pre-warmed WS1
(0.4xSSC/0.3% NP-40) at 73◦C was used to wash the slides for 2 min and transferred to WS2
(2xSSC/0.1% NP-40) at room temp/1min. The slides were dried in a dark room. Chromosomes
were counterstained with 10μl of 4’, 6-diamidino-2-phenylindole (DAPI) and covered with 22 x
22 cover slips. After 15-30 minutes the slides were visualized and the images were captured
using Olympus BX61 epifluorescence microscope (Olympus, Tokyo, Japan), ×100/1.3
magnification objective with CCD camera model ER-3339 (Applied Imaging, Newcastle, UK)
and analyzed using GenASIs software (Applied Spectral Imaging, USA).
A
B
Figure 2: A- Chromosomal region of HNRNPU gene (1q44); B- Chromosomal region of IGF1R
gene (15q26.3) (Source: UCSC genome browser)
Results
Chromosomal culture and karyotyping showed a marker chromosome resulting in an unbalanced
structural abnormality (Figure 2). Further analysis by parental screening showed a balanced
translocation between chromosome 1 and 15 in the mother; 46, XX,t(1;15)(q44;q22) and the
father having a karyotype with no structural and numerical abnormalities (46,XY). Therefore the

7
marker chromosome that was present in the child was the derivative chromosome 15 inherited
from the mother.
The origin of the additional material on chromosome 15 from the long arm of chromosome 1,
suggested by GTG banding pattern, was confirmed by FISH results (Figure 3). The FISH probe
on chromosome 1 (RP11-624F6) was localized in 1q44 and the signals were observed on both
normal chromosomes 1 and on the derivative chromosome 15. The FISH probe on chromosome
15 (RP11-1059N24) was localized in 15q26 and both the normal chromosomes 15 showed two
signals but it was absent on the derivative chromosome 15.
A B
C D
Figure 2. A-Karyogram of the proband bearing a derivative chromosome 15 inherited from mother, B-Ideogram of trisomy 15 with the derivative chromosome 15 illustrating the maternal translocationbetween chromosome 1 and 15, C - Karyogram of the mother bearing a balanced translocation betweenchromosome 1 and 15 : 46,XX,t(1;15)(q44;q22), D- Ideogram of maternal t((1;15)(q44;q22).
Derivative chromosome 15:Trisomy 15(pter-q22)Trisomy 1(q44-qter)
7
marker chromosome that was present in the child was the derivative chromosome 15 inherited
from the mother.
The origin of the additional material on chromosome 15 from the long arm of chromosome 1,
suggested by GTG banding pattern, was confirmed by FISH results (Figure 3). The FISH probe
on chromosome 1 (RP11-624F6) was localized in 1q44 and the signals were observed on both
normal chromosomes 1 and on the derivative chromosome 15. The FISH probe on chromosome
15 (RP11-1059N24) was localized in 15q26 and both the normal chromosomes 15 showed two
signals but it was absent on the derivative chromosome 15.
A B
C D
Figure 2. A-Karyogram of the proband bearing a derivative chromosome 15 inherited from mother, B-Ideogram of trisomy 15 with the derivative chromosome 15 illustrating the maternal translocationbetween chromosome 1 and 15, C - Karyogram of the mother bearing a balanced translocation betweenchromosome 1 and 15 : 46,XX,t(1;15)(q44;q22), D- Ideogram of maternal t((1;15)(q44;q22).
Derivative chromosome 15:Trisomy 15(pter-q22)Trisomy 1(q44-qter)
7
marker chromosome that was present in the child was the derivative chromosome 15 inherited
from the mother.
The origin of the additional material on chromosome 15 from the long arm of chromosome 1,
suggested by GTG banding pattern, was confirmed by FISH results (Figure 3). The FISH probe
on chromosome 1 (RP11-624F6) was localized in 1q44 and the signals were observed on both
normal chromosomes 1 and on the derivative chromosome 15. The FISH probe on chromosome
15 (RP11-1059N24) was localized in 15q26 and both the normal chromosomes 15 showed two
signals but it was absent on the derivative chromosome 15.
A B
C D
Figure 2. A-Karyogram of the proband bearing a derivative chromosome 15 inherited from mother, B-Ideogram of trisomy 15 with the derivative chromosome 15 illustrating the maternal translocationbetween chromosome 1 and 15, C - Karyogram of the mother bearing a balanced translocation betweenchromosome 1 and 15 : 46,XX,t(1;15)(q44;q22), D- Ideogram of maternal t((1;15)(q44;q22).
Derivative chromosome 15:Trisomy 15(pter-q22)Trisomy 1(q44-qter)

8
A B
Figure 3 : A-FISH image of the BAC clone RP11-1059N24 (15q26.3) showing a double signal on bothnormal chromosomes 15, B- FISH image of the BAC clone RP11-624F6 (1q44-ter) showing a triplesignal on both normal chromosomes 1 and the derivative chromosome 15.
Discussion
The clinical phenotypes of partial trisomy 1q syndrome vary widely, due to the different
breakpoints on chromosome 1q and the extent of aberrations involved in other autosomes. Partial
trisomy 1q syndrome can be classified according to breakpoint position as 1q32-qter or 1q42-
qter [2, 3]. Duplication of 1q42-qter with no other involved chromosomal abnormality usually
presents as a mild phenotype, which may include macrocephaly with wide fontanelles, flat nasal
bridge, low-set ears, facial capillary nevi, growth retardation, and developmental delay [3, 7].
The clinical findings and cytogenetic results of a patient with pure trisomy 1q32.1-q42.1 describe
the characteristics of Silver-Russell syndrome [8]. According to previously published data,
authors had considered the diagnosis of Silver-Russell syndrome in a patient with pure trisomy
1q. In that particular case, however, the trisomic region was limited to 1q42-qter [9] which
correlate with the clinical findings of our patient. In our study FISH mapping confirmed the
presence of 1q44-qter region in the derivative chromosome 15 (BAC clone- RP11-624F6).
According to our knowledge, this is the first report of a patient with maternally inherited 1q
duplication localized at 1q44-ter region showing Silver Russell phenotype. The partial 1q
trisomy in this study cannot be defined as ‘pure’, because it is associated with partial 15q
1q
1q
der(15q)
15q15q

9
trisomy. Since most of these symptoms are commonly seen in patients with other chromosome
aberrations, it is impossible to delineate a pure trisomy 1q syndrome or Silver Russell Syndrome
solely based on these features.
In our study, we could not find a gene located within the 15pter-15q22 region that correlated
with the clinical findings of our patient. Hence, we chose the probe RP11-1059N24 located in
15q26.3 to detect the presence of the distal arm of chromosome 15. This probe locates the IGF1R
gene region which correlated with the clinical phenotype of the patient. The absence of this
probe signal on the derivative chromosome 15 indicates that the breakpoint of 15q, mapped by
FISH experiments must be located below 15q22 region (in the distal q arm of chromosome 15).
Distal 15q trisomy includes many of the common characteristics of unbalanced autosomal
abnormalities, such as growth and mental retardation with microcephaly, congenital heart disease
and seizures, facial asymmetry, often with torticollis; down-slanting, narrow palpebral fissures;
ptosis; a prominent nose with a broad nasal root; a long, well-defined philtrum; a down-turned
mouth with a midline V-shaped crease in the lower lip; puffy cheeks; and micrognathia [5].
However, the facial asymmetries, prominent nose, long philtrum, atrial septal defect seen in our
patient are features of the more characteristic phenotype of partial distal trisomy 15q [6]. Table
1 presents the clinical features compared with few of the previously published data. Since the
derivative chromosome 15 in our patient consisted of the distal part of chromosome 15 and distal
part of chromosome 1 and the clinical features are also similar to previously reported cases in the
literature, we conclude that this patient shows features of both Silver Russell Syndrome and
partial distal trisomy 15q.

10
Table 1. Clinical Features of our case with previously published data [8] [10]
Feature Our patient Haelst et al SRS features
Post natal growth retardation + + +
Asymmetry - + +
Relative macrocephaly + + +
Wide Forehead + + +
Downturned mouth corners + + +
Triangular face + + +
Broad/flat nasal bridge + + ?
Low set ears/ ear anomalies + - +
Abnormal Finger/toes + + +
Mental retardation + + +
Spine defects ? + +
References
1. Rasmussen SA, Frias JL, Lafer CZ, Eunpu DL, Zackai EH. Partial duplication 1q: report
of four patients and review of the literature. Am J Med Genet. 1990; 36: 137-43.
2. DuPont BR, Huff RW, Ridgway LE, Stratton RF, Moore CM. Prenatal diagnosis of
partial trisomy 1q using fluorescent in situ hybridization. Am J Med Genet. 1994; 50: 21-
7.

11
3. Emberger W, Petek E, Kroisel PM, Zierler H, Wagner K. Clinical and molecular
cytogenetic characterization of two patients with partial trisomy 1q41-qter: further
delineation of partial trisomy 1q syndrome. Am J Med Genet. 2001; 104: 312-8.
4. Chia NL, Bousfield LR, Poon CC, Trudinger BJ. Trisomy (1q)(q42----qter): confirmation
of a syndrome. Clin Genet. 1988; 34: 224-9.
5. Schnatterly P, Bono KL, Robinow M, Wyandt HE, Kardon N, Kelly TE. Distal 15q
trisomy: phenotypic comparison of nine cases in an extended family. Am J Hum Genet.
1984; 36: 444-51.
6. Herweijer TJ, Oorthuys JW, Leschot NJ. De novo partial trisomy 15q (proximal type). J
Med Genet. 1988; 25: 260-2.
7. Concolino D, Cinti R, Ferraro L, Moricca MT, Strisciuglio P. Partial trisomy 1(q42--
>qter): a new case with a mild phenotype. J Med Genet. 1998; 35: 75-7.
8. van Haelst MM, Eussen HJ, Visscher F, de Ruijter JL, Drop SL, Lindhout D, et al.
Silver-Russell phenotype in a patient with pure trisomy 1q32.1-q42.1: further delineation
of the pure 1q trisomy syndrome. J Med Genet. 2002; 39: 582-5.
9. Kennerknecht I, Barbi G, Rodens K. Dup(1q)(q42-->qter) syndrome: case report and
review of literature. Am J Med Genet. 1993; 47: 1157-60.
10. Utine GE, Aktas D, Alanay Y, Gucer S, Tuncbilek E, Mrasek K, et al. Distal partial
trisomy 1q: report of two cases and a review of the literature. Prenat Diagn. 2007; 27:
865-71.
Related Documents











