Cent. Eur. J. Phys. • 12(1) • 2014 • 70-79 DOI: 10.2478/s11534-013-0322-1 Central European Journal of Physics First-principles calculations of structural, electronic and thermal properties of Zn 1- Mg S ternary alloys Research Article Samia Lamraoui 1 , Rachid Bensalem 1 , Khadidja Hacini 2 , Hocine Meradji 2* , Sebti Ghemid 2 , Fouad El Haj Hassan 3 1 Laboratoire de Magnétisme et Spectroscopie des Solides, Département de Physique, Faculté des Sciences, Université Badji Mokhtar, Annaba, Algeria 2 Laboratoire de Physique des Rayonnements, Département de Physique, Faculté des Sciences, Université Badji Mokhtar, Annaba, Algeria 3 Laboratoire de physique des matériaux, Faculté des Sciences, Université Libanaise, Elhadath, Beirut, Lebanon Received 8 May 2013; accepted 26 October 2013 Abstract: Structural, electronic and thermal properties of Zn 1- Mg S ternary alloys are studied by using the full potential-linearized augmented plane wave method (FP-LAPW) within the density functional theory (DFT). The Wu-Cohen generalized gradient approximation (WC-GGA) is used in this approach for the exchange- correlation potential. Moreover, the modified Becke-Johnson approximation (mBJ) is adopted for band structure calculations. The dependence of the lattice constant, bulk modulus and band gap on the compo- sition x showed that the first exhibits a small deviation from the Vegard’s law, whereas, a marginal deviation of the second from linear concentration dependence (LCD). The bowing of the fundamental gap versus composition predicted by our calculations agrees well with the available theoretical data. The microscopic origins of the gap bowing are explained by using the approach of Zunger and co-workers. Thermal ef- fects on some macroscopic properties of Zn 1- Mg S alloys are also investigated using the quasi-harmonic Debye model, in which the phononic effects are considered. As, this is the first quantitative theoretical prediction of the thermal properties of Zn 1- Mg S alloys, no other calculated results and furthermore no experimental studies are available for comparison. PACS (2008): 64.75.-q, 65.40.-b, 71.20.-b, 81.90.+c Keywords: FP-LAPW • DFT • structural properties • gap bowing • Debye model © Versita sp. z o.o. * E-mail: [email protected] (Corresponding author) 1. Introduction Binary, ternary and quaternary semiconductor alloys have been lately under increasing investigations both in funda- mental studies and for many potential applications such as electronic and electro-optical devices [1]. Since, it is possible to combine two different elements with different 70

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Cent. Eur. J. Phys. • 12(1) • 2014 • 70-79DOI: 10.2478/s11534-013-0322-1
Central European Journal of Physics
First-principles calculations of structural, electronicand thermal properties of Zn1−xMgxS ternary alloys
Research Article
Samia Lamraoui1, Rachid Bensalem1, Khadidja Hacini2, Hocine Meradji2∗, Sebti Ghemid2,Fouad El Haj Hassan3
1 Laboratoire de Magnétisme et Spectroscopie des Solides,Département de Physique, Faculté des Sciences, Université Badji Mokhtar, Annaba, Algeria
2 Laboratoire de Physique des Rayonnements, Département de Physique,Faculté des Sciences, Université Badji Mokhtar, Annaba, Algeria
3 Laboratoire de physique des matériaux,Faculté des Sciences, Université Libanaise, Elhadath, Beirut, Lebanon
Received 8 May 2013; accepted 26 October 2013
Abstract: Structural, electronic and thermal properties of Zn1−xMgxS ternary alloys are studied by using the fullpotential-linearized augmented plane wave method (FP-LAPW) within the density functional theory (DFT).The Wu-Cohen generalized gradient approximation (WC-GGA) is used in this approach for the exchange-correlation potential. Moreover, the modified Becke-Johnson approximation (mBJ) is adopted for bandstructure calculations. The dependence of the lattice constant, bulk modulus and band gap on the compo-sition x showed that the first exhibits a small deviation from the Vegard’s law, whereas, a marginal deviationof the second from linear concentration dependence (LCD). The bowing of the fundamental gap versuscomposition predicted by our calculations agrees well with the available theoretical data. The microscopicorigins of the gap bowing are explained by using the approach of Zunger and co-workers. Thermal ef-fects on some macroscopic properties of Zn1−xMgxS alloys are also investigated using the quasi-harmonicDebye model, in which the phononic effects are considered. As, this is the first quantitative theoreticalprediction of the thermal properties of Zn1−xMgxS alloys, no other calculated results and furthermore noexperimental studies are available for comparison.
PACS (2008): 64.75.-q, 65.40.-b, 71.20.-b, 81.90.+c
Keywords: FP-LAPW • DFT • structural properties • gap bowing • Debye model© Versita sp. z o.o.
∗E-mail: [email protected] (Corresponding author)1. Introduction
Binary, ternary and quaternary semiconductor alloys havebeen lately under increasing investigations both in funda-mental studies and for many potential applications suchas electronic and electro-optical devices [1]. Since, it ispossible to combine two different elements with different70

Samia Lamraoui et al.
optical and mechanical properties in order to obtain a newcompound with intermediate properties, the II-VI semicon-ductor alloys are used in optoelectronic devices rangingfrom blue to near-ultraviolet spectral region [2] and arealso used to fabricate X-ray and γ-ray detectors [3, 4].Furthermore, large band gaps of Mg containing II-VI semi-conductors make them interesting for opto-electronic de-vices in the whole visible field [5]. Such materials havealso been exploited both for quantum confinement and forachieving waveguides. As a mater of fact Zn1−xMgxS al-loys are promising candidates [6] for their energy gapsand lattice constants which can be varied in a wide rangeof stoichiometry parameter x . Although a number of ex-perimental and theoretical studies of Zn1−xMgxS semicon-ductor alloys have been published [7–12], to the best ofour knowledge there are no available data for their ther-mal properties. Thermodynamic properties for crystallinematerials are very important in many applications involv-ing high pressure and high temperature. Although, ab-initio calculations have successively predicted the elec-tronic and structural properties of various materials, thesecalculations are very often restricted to the 0 K temper-atures. In this work, thermal properties are consideredby the use of the quasi-harmonic Debye model [13], andin order to provide further works on Zn1−xMgxS alloys,the full-potential linearized augmented plane-wave (FP-LAPW) method combined with the quasi-harmonic Debyemodel, is used to determine the structural, electronic, andthermal properties of these alloys. The rest of the paperhas been divided in three parts. In section 2, we brieflydescribe the computational techniques used in this study.The most relevant results of Zn1−xMgxS alloys are pre-sented and discussed in section3. Finally, in section 4 wesummarize the main conclusions of our work.2. Method of calculations
All calculations were performed using the FP-LAPWmethod [14] within the framework of the density functionaltheory (DFT) [15, 16] as implemented in the WIEN2K code[17]. For structural properties, the exchange-correlationpotential was calculated using the generalized gradientapproximation (GGA) in the new form (WC) proposed byWu and Cohen [18] which is an improved form of the mostpopular Perdew-Burke-Ernzerhof (PBE) GGA [19]. In ad-dition, and for electronic properties only, we also appliedthe modified Becke-Johnson (mBJ) exchange potential ap-proximation [20, 21]. In this scheme, crystal unit cell ispartitioned into two regions: non-overlapping Muffin-Tin(MT) spheres centred at the atomic sites and the remain-ing interstitial area. In both regions of unit cell, Kohn-
Sham wave functions, charge density and potential wereexpanded in the different sets of basis functions.In the interstitial region, the basis sets consist of a planewaves. Inside the MT spheres, the basis sets is describedby radial solutions of the one particle Schrödinger equa-tion (at fixed energy) and their energy derivatives mul-tiplied by spherical harmonics. The maximum l quan-tum number for the wave function expansion inside atomicspheres was confined to lmax = 10. The plane wave cut-off parameters were decided by RMTKmax = 8.0 (whereKmax is the largest wave vector of the basis set and RMTis the smallest Muffin-Tin radius in the unit cell) andGmax = 14(Ryd)1/2 for Fourier expansion of potential inthe interstitial region. The RMT of Zn, Mg and S atomshave been chosen to be 2.2, 2, and 2, a.u., respectively.Meshes of 47 special k-points for binary compounds and35 special k-points for alloys were used in the irreduciblewedge of the Brillouin zone. To ensure total energy con-vergence, number of tests has been performed taking dif-ferent RMT values and different sets of special k- points.3. Results and discussions3.1. Structural propertiesThe structural properties of the binary compounds ZnSand MgS in the zinc-blende phase using the WC-GGAscheme were calculated, and the alloys are modeled atsome selected compositions with ordered structures de-scribed in terms of periodically repeated supercells. Forthe compositions x = 0.25, 0.5, and 0.75, the simpleststructure is an eight- atom simple cubic lattice. For thestructures considered, the structural optimization by min-imizing the total energy with respect to the cell param-eters and the atomic positions was performed. The ideaof constructing an alloy by taking a large unit cell (cubiceight atoms) and repeating it three dimensionally for thecalculation of the electronic structure of alloys used byAgrawal et al [22] has been adopted recently by many re-searchers to investigate properties of alloys [23–25]. Forthe compositions x = 0.25 and 0.75 the simplest structureis an eight-atom simple cubic cell (luzonite): the anionswith the lower concentration form a regular simple cubiclattice, but for the composition x = 0.5, the atoms of thesame layer are identical. The calculated total energies atmany different volumes around equilibrium were fitted bythe Murnaghan equation of state [26] in order to deter-mine the ground state properties such as the equilibriumlattice constant a, the bulk modulus B and its pressurederivative B′. The WC-GGA calculated structural param-eters for Zn1−xMgxS at various compositions x are listedin Table 1, where comparison is made with experimental
71
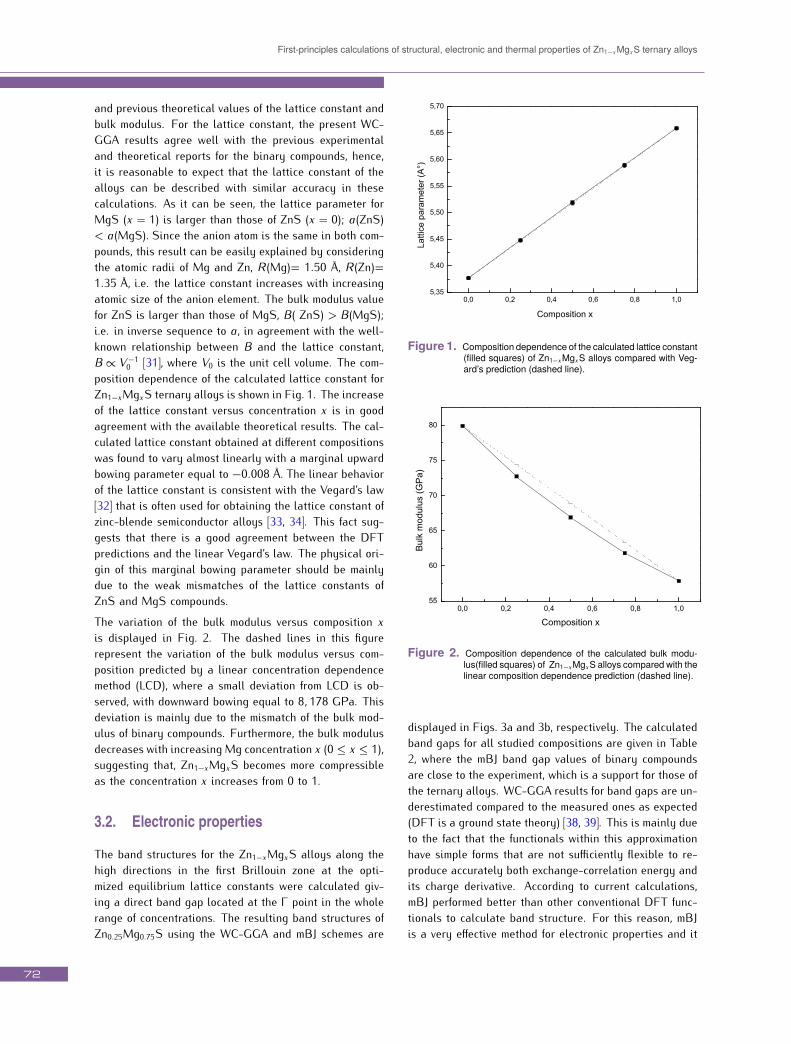
First-principles calculations of structural, electronic and thermal properties of Zn1−xMgxS ternary alloys
and previous theoretical values of the lattice constant andbulk modulus. For the lattice constant, the present WC-GGA results agree well with the previous experimentaland theoretical reports for the binary compounds, hence,it is reasonable to expect that the lattice constant of thealloys can be described with similar accuracy in thesecalculations. As it can be seen, the lattice parameter forMgS (x = 1) is larger than those of ZnS (x = 0); a(ZnS)< a(MgS). Since the anion atom is the same in both com-pounds, this result can be easily explained by consideringthe atomic radii of Mg and Zn, R (Mg)= 1.50 Å, R (Zn)=1.35 Å, i.e. the lattice constant increases with increasingatomic size of the anion element. The bulk modulus valuefor ZnS is larger than those of MgS, B( ZnS) > B(MgS);i.e. in inverse sequence to a, in agreement with the well-known relationship between B and the lattice constant,B ∝ V −10 [31], where V0 is the unit cell volume. The com-position dependence of the calculated lattice constant forZn1−xMgxS ternary alloys is shown in Fig. 1. The increaseof the lattice constant versus concentration x is in goodagreement with the available theoretical results. The cal-culated lattice constant obtained at different compositionswas found to vary almost linearly with a marginal upwardbowing parameter equal to −0.008 Å. The linear behaviorof the lattice constant is consistent with the Vegard’s law[32] that is often used for obtaining the lattice constant ofzinc-blende semiconductor alloys [33, 34]. This fact sug-gests that there is a good agreement between the DFTpredictions and the linear Vegard’s law. The physical ori-gin of this marginal bowing parameter should be mainlydue to the weak mismatches of the lattice constants ofZnS and MgS compounds.The variation of the bulk modulus versus composition xis displayed in Fig. 2. The dashed lines in this figurerepresent the variation of the bulk modulus versus com-position predicted by a linear concentration dependencemethod (LCD), where a small deviation from LCD is ob-served, with downward bowing equal to 8, 178 GPa. Thisdeviation is mainly due to the mismatch of the bulk mod-ulus of binary compounds. Furthermore, the bulk modulusdecreases with increasing Mg concentration x (0 ≤ x ≤ 1),suggesting that, Zn1−xMgxS becomes more compressibleas the concentration x increases from 0 to 1.3.2. Electronic properties
The band structures for the Zn1−xMgxS alloys along thehigh directions in the first Brillouin zone at the opti-mized equilibrium lattice constants were calculated giv-ing a direct band gap located at the Γ point in the wholerange of concentrations. The resulting band structures ofZn0.25Mg0.75S using the WC-GGA and mBJ schemes are
Figure 1. Composition dependence of the calculated lattice constant(filled squares) of Zn1−xMgxS alloys compared with Veg-ard’s prediction (dashed line).
Figure 2. Composition dependence of the calculated bulk modu-lus(filled squares) of Zn1−xMgxS alloys compared with thelinear composition dependence prediction (dashed line).
displayed in Figs. 3a and 3b, respectively. The calculatedband gaps for all studied compositions are given in Table2, where the mBJ band gap values of binary compoundsare close to the experiment, which is a support for those ofthe ternary alloys. WC-GGA results for band gaps are un-derestimated compared to the measured ones as expected(DFT is a ground state theory) [38, 39]. This is mainly dueto the fact that the functionals within this approximationhave simple forms that are not sufficiently flexible to re-produce accurately both exchange-correlation energy andits charge derivative. According to current calculations,mBJ performed better than other conventional DFT func-tionals to calculate band structure. For this reason, mBJis a very effective method for electronic properties and it72

Samia Lamraoui et al.
Table 1. Calculated equilibrium lattice constant and bulk modulus of zinc-blende ZnS and MgS compounds and their ternary alloys.
Lattice constant a (Å) Bulk Modulus B (GPa)This work Exp Other calculations This work Exp Other calculationsAlloy x WC-GGA WC-GGA0 5.376 5.409a 5.465e, 5.427d , 5.458f 79.904 76.9g 77.4c , 69.62e, 69.30f0.25 5.448 - 5.520e 72.747 - 65.14eZn1−xMgxS 0.5 5.519 - 5.583e 66.905 - 61.12e0.75 5.589 - 5.644e 61.815 - 57.66e1 5.658 5.62b 5.584c , 5.635d , 5.703e, 5.708f 57.860 - 57.5c , 55.46e, 55.59faRef. [23], bRef. [24], cRef. [25], dRef. [6], eRef. [12], fRef. [11], gRef. [26]
can be used for a wide range of semiconductors.Fig. 4 shows the composition dependence of the calculatedband gaps using WC-GGA and mBJ schemes, where, theband gap varies non-linearly with the composition x pro-viding a band gap bowing which is calculated by fittingthe non-linear variation of the calculated band gap versusconcentration with quadratic function. The results obeythe following variations:EWG−GGAg = 2.062 + 0.738 + 0.434x2, (1)EmBJg = 3.762 + 0.85x + 1.46x2, (2)
where the quadratic terms express the band gap bowingparameters.The main influence of the band gap energy is due to thelattice constant and the electronegativity mismatch of theparent atoms.The physical origins of the band gap bowing in these al-loys are better understood by following the procedure ofBernard and Zunger [40], in which the bowing parameter(b) is decomposed into three physically distinct contribu-tions. In fact the overall band gap bowing coefficient atx = 0.50 measures the change in the band gap accordingto the reaction:
AB(aAB) + AC (aAC ) −→ AB0.5C0.5(aeq), (3)which can be decomposed into the following three steps:
AB(aAB) + AC (aAC ) VD−→ AB(a) + AC (a), (4)AB(a) + AC (a) CE−→ AB 0.5C 0.5 (a), (5)AB 0.5C 0.5 (a) SR−→ AB 0.5C 0.5 (aeq ), (6)
where aAB and aAC are the equilibrium lattice constantsof the binary compounds AB and AC, respectively; aeq isthe alloy equilibrium lattice constant.
The first step measures the volume deformation (VD) ef-fect on the bowing. The corresponding contribution to thetotal gap bowing parameter bVD represents the relativeresponse of the band structure of the binary compoundsAB and AC to hydrostatic pressure, which arises from thechange of their individual equilibrium lattice constants tothe alloy equilibrium lattice constant value a(x) (Vegard’srule). The second contribution, the charge exchange (CE)contribution bCE , reflects a charge transfer effect whichis due to the different (averaged) bonding behavior at thelattice constant a. The final step measures change dueto structural relaxation (SR) in passing from unrelaxed tothe relaxed alloy. Consequently, the total gap bowingparameter is defined as:b = bVD + bCE + bSR , (7)
wherebVD = 2[εAB(aAB)− εAB(a) + εAC (aAC )− εAC (a)], (8)
bCE = 2[εAB(a) + εAC (a)− 2εABC (a)], (9)bSR = 2[εABC (a)− εABC (aeq)], (10)
where ε is the energy gap calculated for the indicatedatomic structures and lattice constants. We calculatedthe bowing contributions at composition x = 0.5 basedon Eqs. (8)–(10) with self-consistent band structure FP-LAPW within both WC-GGA and mBJ approaches. Theresults are given in Table 3 together with the bowing ob-tained using a quadratic variation of the band gap energyversus composition x and compared with available theo-retical results.The calculated quadratic parameters (gap bowing) are inreasonable agreement with the values found from Bernardand Zunger’s approach. The charge transfer contribu-tion bCE dominates the total band gap bowing parameter73

First-principles calculations of structural, electronic and thermal properties of Zn1−xMgxS ternary alloys
in the studied alloys; this is related to electronegativitymismatch between the constituting atoms: Zn (1.65), Mg(1.31) and S (2.58). Furthermore, the contribution of bVDto b is negative, i.e. it decreases the value of b. The con-tribution of the structural relaxation bSR is small in thesealloys. Finally, the mBJ values for bowing parameters arelarger than the corresponding values within WC-GGA.3.3. Thermal propertiesThe study of of Zn1−xMgxS alloy thermal properties, wasdone within the quasi-harmonic Debye model [13] andcombined with ab-initio calculations of the static to-tal energy, in which the non equilibrium Gibbs functionG∗(V ;P, T ) can be written in the form:
G∗(V ;P, T ) = E(V ) + PV + AV ib[θ(V );T ], (11)where E(V ) is the total energy per unit cell, PV cor-responds to the constant hydrostatic pressure condition,θ(V ) is the Debye temperature, and AV ib is the vibrationalterm, which can be written using the Debye model of thephonon density of states as [41, 42]:AV ib = nkBT
[ 9θ8T + 3 ln (1− e−θ/T )−D(θ/T )] , (12)where D(θ/T ) represents the Debye integral, n is thenumber of atoms per formula unit.For an isotropic solid, θ is expressed as [41]:
θD = ~kB [6π2V 1/2n]1/3f (σ )√BS
M , (13)M being the molecular mass per unit cell and BS theadiabatic bulk modulus, approximated by the static com-pressibility [13]
Bs ∼= B(V ) = V{d2E(V )dV 2
} (14)f (σ ) is given by:
f (σ ) =3[2( 21 + σ31− 2σ
)3/2 + (11 + σ31− σ)3/2]−1
1/3.
(15)The Poisson ratio σ is taken as 0.25 [43]. Therefore, thenon-equilibrium Gibbs function G∗(V ;P, T ) as a functionof (V ;P, T ) can be minimized with respect to volume V :[∂G∗(V ;P, T )
∂V
]P,T
= 0. (16)
By solving Eq. (16), one can get the thermal equationof state (EOS)V (P, T ). The isothermal bulk modulus BT ,the heat capacity CV and the thermal expansion coefficientα are given by [43]:
BT (P, V ) = V[δ2G∗(V ;P, T )
δV 2], (17)
CV = 3nkB [4D(θ/T )− 3θ/Teθ/T − 1
], (18)
α = γCVBTV
. (19)Where γ is the Grüneisen parameter, which is defined as:
γ = −d lnθ(V )d lnV . (20)
Through the quasi-harmonic Debye model, one could cal-culate the thermodynamic quantities of any temperaturesand pressures of compounds from the calculated E − Vdata at T = 0 and P = 0. The thermal properties are de-termined in the temperature and pressure ranges 0−900 Kand 0− 6 GPa, respectively. Since, to the best of knowl-edge no experimental nor theoretical data are availablefor comparison, this work is the first theoretical predic-tion for these thermal quantities. A similar behaviour isobtained for all concentrations, therefore, only the resultsrelated to the concentration 0.75, are presented.The variation of the lattice constant a of Zn0.25Mg0.75Salloy versus temperature at several pressures is shown inFig. 5. When T is smaller than 100 K, a is nearly constantfor all applied pressures, but, when T>100 K, the latticeconstant increases with increasing temperature at a givenpressure. On the other side, as the pressure increases,the lattice constant decreases at a given temperature. Therate of increase of the lattice constant with temperaturedecreases with increasing pressure. This effect of increas-ing pressure on Zn0.25Mg0.75S is just the same as that ofdecreasing temperature on this material.The bulk modulus B is related to interatomic potentialsand can be obtained by the second derivative of the in-ternal energy with respect to strain. The temperature de-pendence of B partially reflects the anharmonic interac-tions since for a purely harmonic crystal, the bulk moduluswould be independent of temperature. The bulk modulusB of Zn0.25Mg0.75S alloy as a function of temperature upto 900 K at several pressures is plotted in Fig. 6. B re-mains constant for T < 100 K, but for T > 100 K, B de-creases dramatically as T increases. This indicates thatthe volume of Zn0.25Mg0.75S alloy varies significantly asthe temperature increases when T > 100 K. However, the
74

Samia Lamraoui et al.
Table 2. Direct band gap energy Eg (eV) of Zn1-xMgxS alloys at various x compositions.
Eg (eV) Γ− ΓAlloy This work Exp Other calculationsx WC-GGA mBJ0 1.993 3.722 3.68a, 3.80g, 2.37c , 1.953d , 2.792d , 1.97e, 2.823e0.25 2.429 3.950 2.263d , 3, 114dZn1−xMgxS 0.5 2.491 4.167 2.509d , 3.377d0.75 2.771 4.441 2.817d , 3.715d1 3.288 5.141 4.45a, 4.50b 4.48f , 3.42c , 3.327d , 4.385d , 3.333e, 4.418eaRef. [6], bRef. [24], cRef. [25], dRef. [12], eRef. [11], fRef. [31] , gRef. [32]
Table 3. Decomposition of bowing parameter into volume deformation (VD), Charge exchange (CE), and structural relaxation (SR) contributionscompared with that obtained by a quadratic fit and other predictions (all values are in eV).
This work Other calculationsAlloy Zunger approach Quadratic fitsmBJ WC-GGA mBJ WC-GGAbVD −0.226 −0.103 −0.099a, −0.657bbCE 1.181 0.689 0.502a, 1.509bZn1−xMgxS bSR 0.103 0.013 0.123a, −0.007bb 1.058 0.599 1.146 0.434 0.526a, 0.845baRef. [12] using PBE-GGA approximation
bRef. [12] using Engel-Vosko EV-GGA approximation.variation of the cell volume is very small when T < 100K. For a given temperature, the bulk modulus increaseswith increasing pressure. The hardness of this materialdecreases with increasing temperature and increases withapplied pressure.The heat capacity of a substance not only provides es-sential insight into its vibrational properties but is alsomandatory for many applications. Two famous limitingcases are correctly predicted by the standard elastic con-tinuum theory [44]. At high temperature, the constant vol-ume heat capacity Cv does not depend much on temper-ature and tends to the Dulong-Petit limit (Cv (T ) ≈ 3Rfor mono-atomic solids) [45], which is true to all solidsat high temperatures. At sufficiently low temperature, Cvis proportional to T 3 [44]. At intermediate temperatures,however, the temperature dependence of Cv is governed bythe details of vibrations of the atoms and for a long timecould only be determined from experiments. Fig. 7. dis-plays the dependence of Cv on temperature and pressurefor the Zn0.25Mg0.75S alloy. It is shown that for T < 500K,the heat capacity depends on both temperature and pres-sure. At high temperatures, (T > 500 K) Cv tends tothe Dulong-Petit limit, Cv tends to 48, 97 Jmol−1K−1 athigh temperatures. The variation of Cv with respect totemperature under low pressure is great than those un-der higher pressure. The variation of the heat capacityat constant pressure Cp versus temperature at 0, 2, 4 and
6 GPa pressures for the Zn0.25Mg0.75S alloy is shown inFig. 8. The variation features of Cp at low temperature aresimilar to the behavior of Cv . However, at high- tempera-ture, Cp exhibits apparently different features. Cp valuesdecrease with increasing pressures and do not convergeto a constant value. At room temperature and zero pres-sure, the value of Cp is about 42.07 J mol−1K−1. Fig. 9.shows the variation of the thermal expansion coefficientα with respect to the temperature at several pressuresfor the Zn0.25Mg0.75S alloy. As the thermal expansion co-efficients have anharmonic behaviors, no linear variationwith temperature T is expected. α increases with T 3 atlow temperatures and gradually approaches a linear in-crease with enhanced temperature and the propensity ofincrement becomes moderate, which means that the tem-perature dependence of α is very small at high tempera-ture. For a given temperature, α decreases dramaticallywith increasing pressure. With increasing pressure, theincrease in α is weakened. The calculated properties atdifferent temperatures are very sensitive to the vibrationaleffects. In the quasi-harmonic Debye model, the Debyetemperature is a key parameter. The temperature depen-dence of the Debye temperature θD at several pressuresfor the Zn0.25Mg0.75S alloy is plotted in Fig. 10. θD isnearly constant from 0 to 100 K and decreases almostlinearly with increasing temperature for T > 100 K. It isalso shown that θD increases with applied pressure at a
75
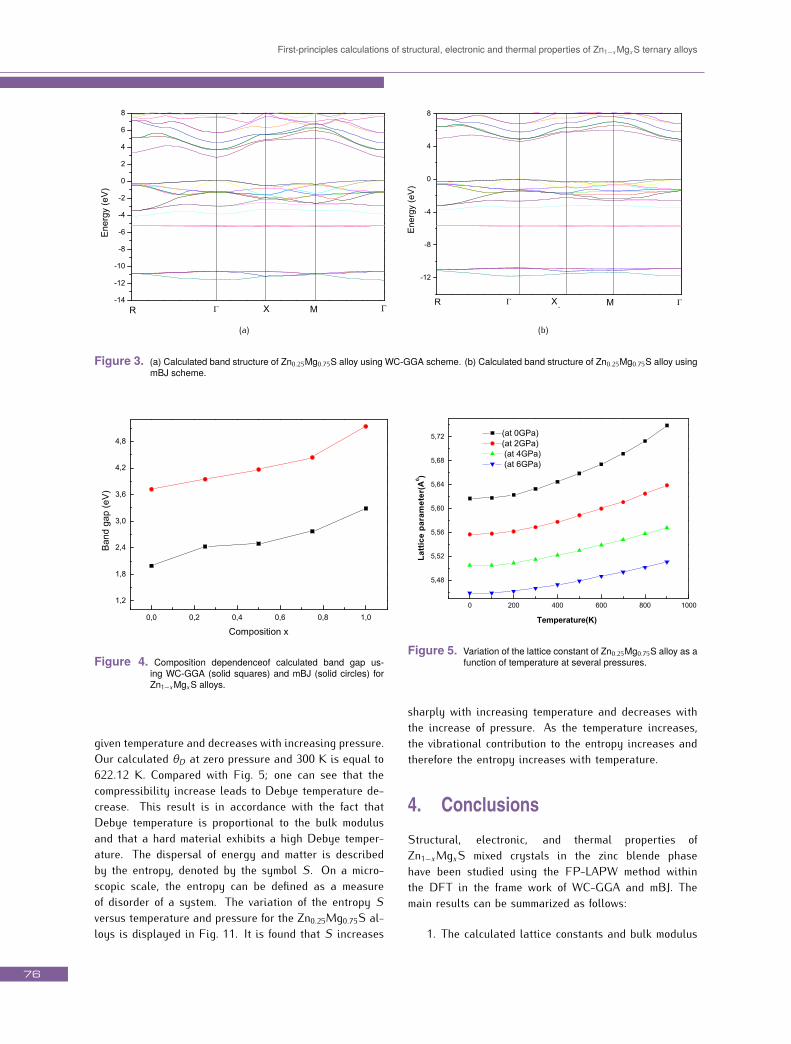
First-principles calculations of structural, electronic and thermal properties of Zn1−xMgxS ternary alloys
(a) (b)Figure 3. (a) Calculated band structure of Zn0.25Mg0.75S alloy using WC-GGA scheme. (b) Calculated band structure of Zn0.25Mg0.75S alloy using
mBJ scheme.
Figure 4. Composition dependenceof calculated band gap us-ing WC-GGA (solid squares) and mBJ (solid circles) forZn1−xMgxS alloys.
given temperature and decreases with increasing pressure.Our calculated θD at zero pressure and 300 K is equal to622.12 K. Compared with Fig. 5; one can see that thecompressibility increase leads to Debye temperature de-crease. This result is in accordance with the fact thatDebye temperature is proportional to the bulk modulusand that a hard material exhibits a high Debye temper-ature. The dispersal of energy and matter is describedby the entropy, denoted by the symbol S. On a micro-scopic scale, the entropy can be defined as a measureof disorder of a system. The variation of the entropy Sversus temperature and pressure for the Zn0.25Mg0.75S al-loys is displayed in Fig. 11. It is found that S increases
Figure 5. Variation of the lattice constant of Zn0.25Mg0.75S alloy as afunction of temperature at several pressures.
sharply with increasing temperature and decreases withthe increase of pressure. As the temperature increases,the vibrational contribution to the entropy increases andtherefore the entropy increases with temperature.4. ConclusionsStructural, electronic, and thermal properties ofZn1−xMgxS mixed crystals in the zinc blende phasehave been studied using the FP-LAPW method withinthe DFT in the frame work of WC-GGA and mBJ. Themain results can be summarized as follows:
1. The calculated lattice constants and bulk modulus76
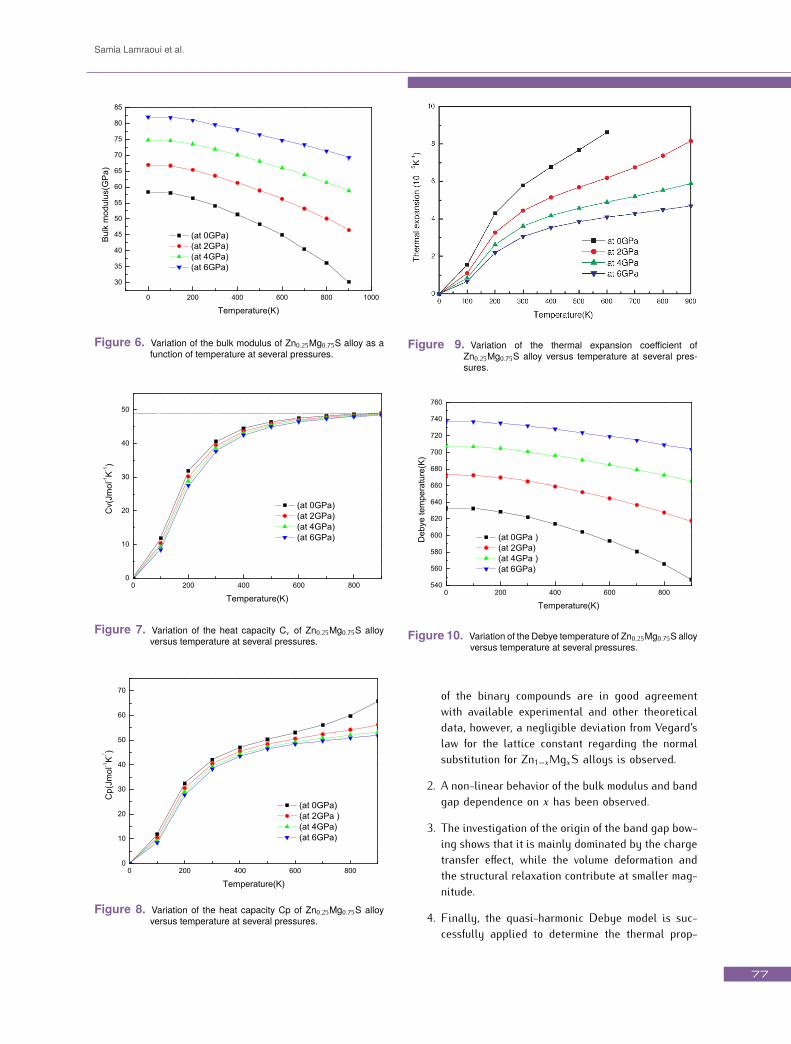
Samia Lamraoui et al.
Figure 6. Variation of the bulk modulus of Zn0.25Mg0.75S alloy as afunction of temperature at several pressures.
Figure 7. Variation of the heat capacity Cv of Zn0.25Mg0.75S alloyversus temperature at several pressures.
Figure 8. Variation of the heat capacity Cp of Zn0.25Mg0.75S alloyversus temperature at several pressures.
Figure 9. Variation of the thermal expansion coefficient ofZn0.25Mg0.75S alloy versus temperature at several pres-sures.
Figure 10. Variation of the Debye temperature of Zn0.25Mg0.75S alloyversus temperature at several pressures.
of the binary compounds are in good agreementwith available experimental and other theoreticaldata, however, a negligible deviation from Vegard’slaw for the lattice constant regarding the normalsubstitution for Zn1−xMgxS alloys is observed.2. A non-linear behavior of the bulk modulus and bandgap dependence on x has been observed.3. The investigation of the origin of the band gap bow-ing shows that it is mainly dominated by the chargetransfer effect, while the volume deformation andthe structural relaxation contribute at smaller mag-nitude.4. Finally, the quasi-harmonic Debye model is suc-cessfully applied to determine the thermal prop-
77
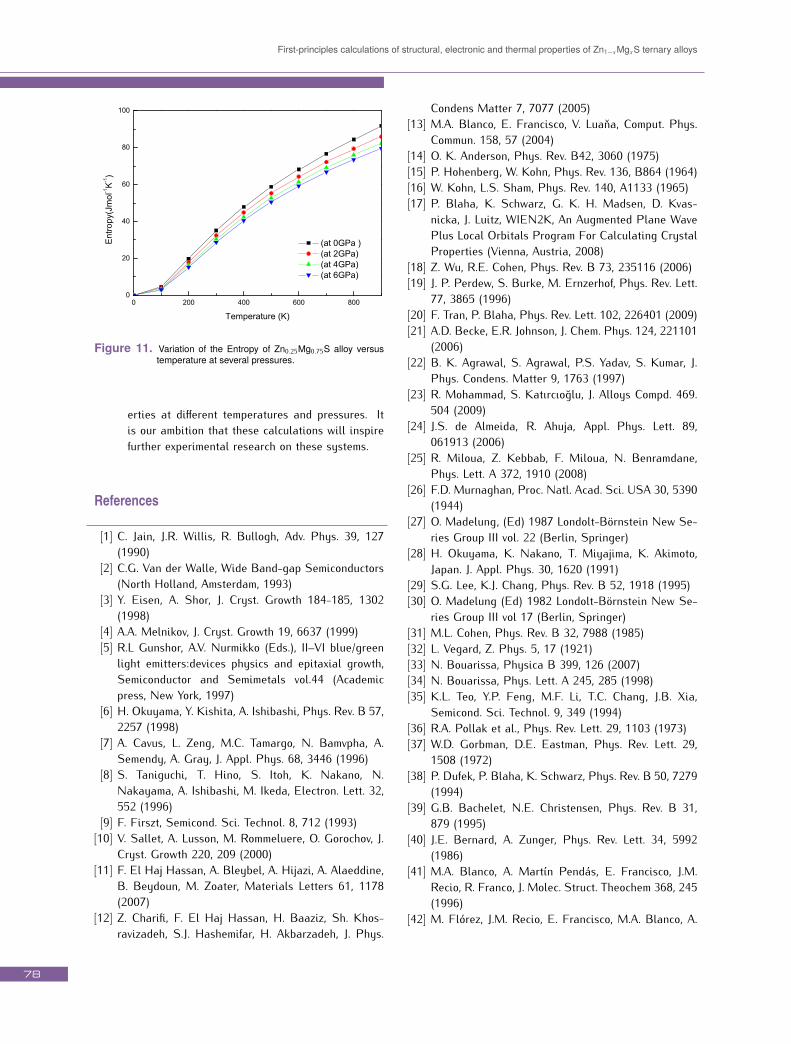
First-principles calculations of structural, electronic and thermal properties of Zn1−xMgxS ternary alloys
Figure 11. Variation of the Entropy of Zn0.25Mg0.75S alloy versustemperature at several pressures.
erties at different temperatures and pressures. Itis our ambition that these calculations will inspirefurther experimental research on these systems.References
[1] C. Jain, J.R. Willis, R. Bullogh, Adv. Phys. 39, 127(1990)[2] C.G. Van der Walle, Wide Band-gap Semiconductors(North Holland, Amsterdam, 1993)[3] Y. Eisen, A. Shor, J. Cryst. Growth 184-185, 1302(1998)[4] A.A. Melnikov, J. Cryst. Growth 19, 6637 (1999)[5] R.L Gunshor, A.V. Nurmikko (Eds.), II–VI blue/greenlight emitters:devices physics and epitaxial growth,Semiconductor and Semimetals vol.44 (Academicpress, New York, 1997)[6] H. Okuyama, Y. Kishita, A. Ishibashi, Phys. Rev. B 57,2257 (1998)[7] A. Cavus, L. Zeng, M.C. Tamargo, N. Bamvpha, A.Semendy, A. Gray, J. Appl. Phys. 68, 3446 (1996)[8] S. Taniguchi, T. Hino, S. Itoh, K. Nakano, N.Nakayama, A. Ishibashi, M. Ikeda, Electron. Lett. 32,552 (1996)[9] F. Firszt, Semicond. Sci. Technol. 8, 712 (1993)[10] V. Sallet, A. Lusson, M. Rommeluere, O. Gorochov, J.Cryst. Growth 220, 209 (2000)[11] F. El Haj Hassan, A. Bleybel, A. Hijazi, A. Alaeddine,B. Beydoun, M. Zoater, Materials Letters 61, 1178(2007)[12] Z. Charifi, F. El Haj Hassan, H. Baaziz, Sh. Khos-ravizadeh, S.J. Hashemifar, H. Akbarzadeh, J. Phys.
Condens Matter 7, 7077 (2005)[13] M.A. Blanco, E. Francisco, V. Luaňa, Comput. Phys.Commun. 158, 57 (2004)[14] O. K. Anderson, Phys. Rev. B42, 3060 (1975)[15] P. Hohenberg, W. Kohn, Phys. Rev. 136, B864 (1964)[16] W. Kohn, L.S. Sham, Phys. Rev. 140, A1133 (1965)[17] P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvas-nicka, J. Luitz, WIEN2K, An Augmented Plane WavePlus Local Orbitals Program For Calculating CrystalProperties (Vienna, Austria, 2008)[18] Z. Wu, R.E. Cohen, Phys. Rev. B 73, 235116 (2006)[19] J. P. Perdew, S. Burke, M. Ernzerhof, Phys. Rev. Lett.77, 3865 (1996)[20] F. Tran, P. Blaha, Phys. Rev. Lett. 102, 226401 (2009)[21] A.D. Becke, E.R. Johnson, J. Chem. Phys. 124, 221101(2006)[22] B. K. Agrawal, S. Agrawal, P.S. Yadav, S. Kumar, J.Phys. Condens. Matter 9, 1763 (1997)[23] R. Mohammad, S. Katırcıoğlu, J. Alloys Compd. 469.504 (2009)[24] J.S. de Almeida, R. Ahuja, Appl. Phys. Lett. 89,061913 (2006)[25] R. Miloua, Z. Kebbab, F. Miloua, N. Benramdane,Phys. Lett. A 372, 1910 (2008)[26] F.D. Murnaghan, Proc. Natl. Acad. Sci. USA 30, 5390(1944)[27] O. Madelung, (Ed) 1987 Londolt-Börnstein New Se-ries Group III vol. 22 (Berlin, Springer)[28] H. Okuyama, K. Nakano, T. Miyajima, K. Akimoto,Japan. J. Appl. Phys. 30, 1620 (1991)[29] S.G. Lee, K.J. Chang, Phys. Rev. B 52, 1918 (1995)[30] O. Madelung (Ed) 1982 Londolt-Börnstein New Se-ries Group III vol 17 (Berlin, Springer)[31] M.L. Cohen, Phys. Rev. B 32, 7988 (1985)[32] L. Vegard, Z. Phys. 5, 17 (1921)[33] N. Bouarissa, Physica B 399, 126 (2007)[34] N. Bouarissa, Phys. Lett. A 245, 285 (1998)[35] K.L. Teo, Y.P. Feng, M.F. Li, T.C. Chang, J.B. Xia,Semicond. Sci. Technol. 9, 349 (1994)[36] R.A. Pollak et al., Phys. Rev. Lett. 29, 1103 (1973)[37] W.D. Gorbman, D.E. Eastman, Phys. Rev. Lett. 29,1508 (1972)[38] P. Dufek, P. Blaha, K. Schwarz, Phys. Rev. B 50, 7279(1994)[39] G.B. Bachelet, N.E. Christensen, Phys. Rev. B 31,879 (1995)[40] J.E. Bernard, A. Zunger, Phys. Rev. Lett. 34, 5992(1986)[41] M.A. Blanco, A. Martín Pendás, E. Francisco, J.M.Recio, R. Franco, J. Molec. Struct. Theochem 368, 245(1996)[42] M. Flórez, J.M. Recio, E. Francisco, M.A. Blanco, A.78

Samia Lamraoui et al.
Martín Pendás, Phys. Rev. B 66, 144112 (2002)[43] J. P. Poirier, Introduction to the Physics of the Earth’sInterior (Oxford, Cambridge University Press, 2000)p. 39[44] P. Debye, Ann. Phys. 39, 789 (1912)[45] A.T. Petit, P.L. Dulong, Ann. Chim. Phys. 10, 395(1819)
79
Related Documents











