Journal of Pathology J Pathol 2003; 200: 448–464. Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/path.1400 Review Article Extracellular matrix remodelling: the role of matrix metalloproteinases Ivan Stamenkovic* Experimental Pathology Division, Institut Universitaire de Pathologie, Universit´ e de Lausanne, Lausanne, Switzerland *Correspondence to: Dr Ivan Stamenkovic, Experimental Pathology Division, Institut Universitaire de Pathologie, Universit´ e de Lausanne, 25 Rue du Bagnon, CH-1011 Lausanne, Switzerland. Abstract Matrix metalloproteinases (MMPs) are a growing family of metalloendopeptidases that cleave the protein components of the extracellular matrix and thereby play a central role in tissue remodelling. For many years following their discovery, MMPs were believed to function primarily as regulators of ECM composition and to facilitate cell migration simply by removing barriers such as collagen. It is becoming increasingly clear, however, that MMPs are implicated in the functional regulation of a host of non-ECM molecules that include growth factors and their receptors, cytokines and chemokines, adhesion receptors and cell surface proteoglycans, and a variety of enzymes. MMPs therefore play an important role in the control of cellular interactions with and response to their environment in conditions that promote tissue turnover, be they physiological, such as normal development, or pathological, such as inflammation and cancer. This review summarizes some of the recent discoveries that have shed new light on the role of MMPs in physiology and disease. Copyright 2003 John Wiley & Sons, Ltd. Keywords: matrix metalloproteinases; remodelling; cancer; inflammation; development Introduction The extracellular matrix (ECM) plays a key role in tis- sue architecture and homeostasis. In most organs, the principal proteinaceous components of the ECM are collagens that are produced and secreted by a variety of stromal cells, although predominantly fibroblasts, and which provide much of the scaffold necessary for the organization of cells that constitute the tis- sue. Numerous other proteins contribute to specialized components of the ECM structure, such as the base- ment membrane, including laminin, entactin, collagen IV, and various growth factors and proteases [1]. A second class of molecules that play an essential role in the composition of the ECM are secreted proteo- glycans whose protein core is covalently bound to high-molecular-weight glycosaminoglycans, including chondroitin, heparan, and keratan sulphate. Proteogly- cans are implicated in numerous processes, two of their major functions including the support of cell adhesion and the binding of a host of latent growth factors [2]. The ECM also contains one major gly- cosaminoglycan, hyaluronan (HA), which is typically not sulphated or bound to a protein core. Hyaluronan is a polymer built of repeating disaccharide units with the structure [D-glucuronic acid (1-β -3) N -acetyl-D- glucosamine (1-β -4)] n , which is synthesized primarily by fibroblasts, although other stromal cells produce HA as well, and deposited in the ECM of virtually all organs. Hyaluronan participates in the regulation of numerous cellular functions, prominent among which are adhesion, trafficking, and signalling [3]. Together, the ECM components thus constitute a structure that in addition to helping maintain tissue integrity regulates cell migration and provides a reservoir of cytokines and growth factors. By its very nature, the ECM is constantly undergoing changes in response to a host of cellular stimuli. These range from dynamic homeosta- sis that typifies resting-state adult organs to full-blown tissue remodelling, as occurs during normal develop- ment, inflammation, wound healing, and cancer, char- acterized by major alterations in both ECM structure and composition. ECM remodelling is the result of multiple concur- rent processes that vary according to the initiating stimulus. Thus, the structural and functional changes within the ECM of a given organ that occur dur- ing an acute inflammatory response differ from those that accompany limb development and tumour inva- sion. Nevertheless, several key mechanistic features of tissue remodelling are common to most aetiologies. Remodelling at the very minimum requires two events: synthesis and deposition of ECM components on the one hand and their proteolytic breakdown on the other. Numerous proteases have been implicated in the prote- olytic degradation of the ECM, most prominent among which are members of the matrix metalloproteinase (MMP) family. Matrix metalloproteinases form a subfamily of the metzincin superfamily of proteases, which constitutes one of several metalloendopeptidase families [4–6]. The metzincins share a conserved structural topology, a consensus motif within the catalytic domain contain- ing three histidines that provide a zinc binding site and Copyright 2003 John Wiley & Sons, Ltd.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Journal of PathologyJ Pathol 2003; 200: 448–464.Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/path.1400
Review Article
Extracellular matrix remodelling: the role of matrixmetalloproteinasesIvan Stamenkovic*Experimental Pathology Division, Institut Universitaire de Pathologie, Universite de Lausanne, Lausanne, Switzerland
*Correspondence to:Dr Ivan Stamenkovic,Experimental Pathology Division,Institut Universitaire dePathologie, Universite deLausanne, 25 Rue du Bagnon,CH-1011 Lausanne, Switzerland.
AbstractMatrix metalloproteinases (MMPs) are a growing family of metalloendopeptidases thatcleave the protein components of the extracellular matrix and thereby play a central rolein tissue remodelling. For many years following their discovery, MMPs were believed tofunction primarily as regulators of ECM composition and to facilitate cell migration simplyby removing barriers such as collagen. It is becoming increasingly clear, however, thatMMPs are implicated in the functional regulation of a host of non-ECM molecules thatinclude growth factors and their receptors, cytokines and chemokines, adhesion receptorsand cell surface proteoglycans, and a variety of enzymes. MMPs therefore play an importantrole in the control of cellular interactions with and response to their environment inconditions that promote tissue turnover, be they physiological, such as normal development,or pathological, such as inflammation and cancer. This review summarizes some of therecent discoveries that have shed new light on the role of MMPs in physiology and disease.Copyright 2003 John Wiley & Sons, Ltd.
Keywords: matrix metalloproteinases; remodelling; cancer; inflammation; development
Introduction
The extracellular matrix (ECM) plays a key role in tis-sue architecture and homeostasis. In most organs, theprincipal proteinaceous components of the ECM arecollagens that are produced and secreted by a varietyof stromal cells, although predominantly fibroblasts,and which provide much of the scaffold necessaryfor the organization of cells that constitute the tis-sue. Numerous other proteins contribute to specializedcomponents of the ECM structure, such as the base-ment membrane, including laminin, entactin, collagenIV, and various growth factors and proteases [1]. Asecond class of molecules that play an essential rolein the composition of the ECM are secreted proteo-glycans whose protein core is covalently bound tohigh-molecular-weight glycosaminoglycans, includingchondroitin, heparan, and keratan sulphate. Proteogly-cans are implicated in numerous processes, two oftheir major functions including the support of celladhesion and the binding of a host of latent growthfactors [2]. The ECM also contains one major gly-cosaminoglycan, hyaluronan (HA), which is typicallynot sulphated or bound to a protein core. Hyaluronanis a polymer built of repeating disaccharide units withthe structure [D-glucuronic acid (1-β-3) N -acetyl-D-glucosamine (1-β-4)]n , which is synthesized primarilyby fibroblasts, although other stromal cells produceHA as well, and deposited in the ECM of virtually allorgans. Hyaluronan participates in the regulation ofnumerous cellular functions, prominent among whichare adhesion, trafficking, and signalling [3]. Together,
the ECM components thus constitute a structure that inaddition to helping maintain tissue integrity regulatescell migration and provides a reservoir of cytokinesand growth factors. By its very nature, the ECM isconstantly undergoing changes in response to a host ofcellular stimuli. These range from dynamic homeosta-sis that typifies resting-state adult organs to full-blowntissue remodelling, as occurs during normal develop-ment, inflammation, wound healing, and cancer, char-acterized by major alterations in both ECM structureand composition.
ECM remodelling is the result of multiple concur-rent processes that vary according to the initiatingstimulus. Thus, the structural and functional changeswithin the ECM of a given organ that occur dur-ing an acute inflammatory response differ from thosethat accompany limb development and tumour inva-sion. Nevertheless, several key mechanistic featuresof tissue remodelling are common to most aetiologies.Remodelling at the very minimum requires two events:synthesis and deposition of ECM components on theone hand and their proteolytic breakdown on the other.Numerous proteases have been implicated in the prote-olytic degradation of the ECM, most prominent amongwhich are members of the matrix metalloproteinase(MMP) family.
Matrix metalloproteinases form a subfamily of themetzincin superfamily of proteases, which constitutesone of several metalloendopeptidase families [4–6].The metzincins share a conserved structural topology,a consensus motif within the catalytic domain contain-ing three histidines that provide a zinc binding site and
Copyright 2003 John Wiley & Sons, Ltd.

Extracellular matrix remodelling 449
a conserved ‘Met-turn’ motif that resides beneath theactive site zinc ion [5]. Based on structural similarity,the metzincins are subdivided into four subfamilies:the serralysins, adamalysins, astracins, and matrixins(MMPs).
The first insight into the function of MMPs wasobtained as a result of the observation that their pro-teolytic activity was responsible for the dissolutionof the tadpole tail [7]. It was subsequently recog-nized that the >20 human MMPs and homologuesfrom other species could cleave practically all of theprotein components of the ECM. Based on their per-ceived specificity for ECM proteins, MMPs have beendivided into collagenases, gelatinases, stromelysins,and matrilysins. However, the high degree of over-lap among MMP substrate specificities and the notionthat MMPs can cleave a growing list of substrates thatare not part of the ECM [8,9] render this nomenclatureimprecise at best. MMPs are assigned a number thatcorresponds to the chronology of their identification,but a more rational approach as to their classificationbased on their structure is currently being adopted [9].Thus several subclasses of MMPs have been identified,some of which are secreted whereas others are mem-brane bound (Figure 1). All MMPs are produced aszymogens containing a secretory signal sequence anda propeptide whose proteolytic cleavage is requiredfor MMP activation. The propeptide is followed bythe catalytic domain that contains the consensus zinc-binding motif HEBXHXBGBXH, where X is a vari-able residue and B is a bulky hydrophobic residue. Atleast two MMPs (MMP-7 and MMP-26) are composedonly of the signal peptide, propeptide, and catalyticdomain, and are known as minimal domain MMPs(Figure 1). Most of the remaining MMPs contain ahaemopexin-like domain that is thought to confer somedegree of substrate specificity, and several have addi-tional features, such as fibronectin-like repeats or ser-ine protease recognition motifs (Figure 1). Finally,a subclass of MMPs contains a transmembrane andintracellular domain and are often referred to as MT-MMPs.
Regulation of MMP activationand proteolytic activity
MMP activity is controlled at least at three levels:transcription, proteolytic activation of the zymogenform, and inhibition of the active enzyme by a hostof natural inhibitors. Most MMPs are expressed atlow levels or not at all in resting-state adult tissues.However, numerous cytokines and growth factors aswell as physical cellular interactions provide stimulithat can rapidly induce MMP expression (reviewed in[10] and references therein).
The propeptide serves to maintain pro-MMPs inan inactive state. A cysteine-sulphydryl group withinthe distinctive consensus PRCGXPDV motif in thepropeptide domain binds to the active site zinc atom,
thereby preventing activation. Disruption of this inter-action by physical or chemical means constitutes thefirst step in MMP activation, which is followed by pro-teolytic cleavage of the COOH-terminal side of thePRCGXPDV site with irreversible loss of the cys-teine residue [6,11,12]. Whereas activation of mostMMPs occurs outside the cell, several MMPs, includ-ing MMP-11 and -28 and membrane-bound MMPs,contain furin protease cleavage sites and may be acti-vated by intracellular furin-like serine proteases beforereaching the cell surface [12]. Most MMPs can be acti-vated by other MMPs (Table 1) and a variety of serineproteases in vitro [11]. However, with one notableexception, the mechanisms of physiological extracel-lular activation of MMPs remain to be elucidated.The exception is activation of MMP-2. It has beendemonstrated that MMP-14 binds the tissue inhibitorof metalloproteinases 2 (TIMP-2) on the cell surfaceand recruits MMP-2, which binds to the immobilizedTIMP-2 through its haemopexin-like domain [13,14].A TIMP-free MMP-14 is then recruited to the com-plex and cleaves the MMP-2 propeptide, resulting inpartial MMP-2 activation, which is completed by anadjacent active MMP-2 [15].
MMP activity is tightly controlled by severalendogenous inhibitors. In tissue fluids, the principalMMP inhibitor is α2-macroglobulin, which binds toMMPs and creates a complex that is itself bound andirreversibly cleared by scavenger receptors [16,17].The most thoroughly studied MMP inhibitors, how-ever, are TIMPs. The four TIMPs that have been char-acterized thus far are small molecules of 21–28 kDathat bind to MMPs in a 1 : 1 stoichiometric ratio andreversibly block MMP activity (reviewed in [18]).TIMPs differ in their expression patterns and affin-ity for the various MMPs. Thus, TIMP-1 and TIMP-2 inhibit the activity of a broad range of MMPsand, by analogy to the TIMP-2/pro-MMP-2 interac-tion, TIMP-1 may form a complex with pro-MMP-9[18,19]. The TIMP-1/pro-MMP-9 complex is thoughtto recruit MMP-3, forming a stable ternary com-plex that causes MMP-3 inactivation [20]. However,the physiological relevance of this putative com-plex remains to be established. TIMP-3 preferen-tially inhibits the activity of MMP-1, -3, -7, and -13,and unlike other TIMPs blocks the activity of themore distantly related metalloproteinase ADAM-17[21]. TIMP-4 has a more restricted inhibitory activ-ity that primarily blocks MMP-2 and -7 and to alesser extent MMP-1, -3, and -9 [22]. Recent evi-dence suggests that TIMPs may have additional func-tions to those of MMP inhibition [18]. A case inpoint is the observation that an important physiologi-cal function of TIMP-2 is the activation of MMP-2[23]. Among other molecules capable of regulatingMMP proteolytic activity are thrombospondin andRECK. Thrombospondin-2 binds MMP-2 in a com-plex that facilitates scavenger receptor-mediated endo-cytosis and clearance [17], whereas thrombospondin-1binds to proMMP-2 and -9 and directly blocks their
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

450 I Stamenkovic
MM
P st
ruct
ure
Min
imal
dom
ain:
MM
P-7
MM
P-26
Sim
ple
hem
opex
in d
omai
n: M
MP-
1 M
MP-
3M
MP-
8 M
MP-
10 M
MP-
12 M
MP-
13 M
MP-
19M
MP-
20 M
MP-
27
Typ
e II
fib
rone
ctin
-lik
e re
peat
s: M
MP-
2 M
MP-
9
Sim
ple
hem
opex
in d
omai
n, f
urin
-act
ivat
ed:
MM
P-11
MM
P-28
Tra
nsm
embr
ane
MM
Ps: M
MP-
14 M
MP-
15M
MP-
16 M
MP-
24
GPI
-anc
hore
d M
MPs
: MM
P-17
MM
P-25
tran
smem
bran
e do
mai
n
cyto
plas
mic
dom
ain
GPI
anc
hor
sign
al s
eque
nce
pro-
dom
ain
furi
n re
cogn
ition
mot
if
Typ
e II
fib
rone
ctin
rep
eats
Zin
c bi
ndin
g do
mai
n
cata
lyti
c do
mai
n
Hin
ge r
egio
nH
emop
exin
-lik
e do
mai
n
S
S
S SS
SS
SS
S
S
S
Fig
ure
1.Pr
otei
nst
ruct
ure
ofM
MPs
.The
prin
cipa
lstr
uctu
rals
ubcl
asse
sof
MM
Psar
esh
own
and
the
diffe
rent
dom
ains
indi
cate
d.In
divi
dual
MM
Psth
atbe
long
toea
chst
ruct
ural
subc
lass
are
liste
d
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

Extracellular matrix remodelling 451
Table 1. Non-ECM substrates of matrix metalloproteinases
MMPGrowth factor precursors
and binding proteinsCytokines
chemokines Adhesion receptors Miscellany
1 IGFBP, perlecan CXCL12, proTNF-α α1-Antichymotrypsinα1-Proteinase inhibitorproMMP-1, 2
2 Decorin, proTGF-β , IGF-BP,FGFR1
proTNFα, MCP-3 (CCL7)proIL-1β
proMMP-1, -2, -13
3 IGFBP, perlecan, decorin,proHB-EGF
proTNF-αproIL-1β
CXCL12
E-cadherin Plasminogenα1-Antichymotrypsinα1-Proteinaseinhibitor proMMP-1,-3, -7, -9, -13
7 Decorin, proHB-EGF FasL,proTNF-α
E-cadherinβ4-IntegrinSyndecan-1
PlasminogenPro-α-defensinproMMP-7
8 α1-Antichymotrypsinα1-Proteinase inhibitorproMMP-8
9 ProTGF-β ProIL-1β
IL-2Rα
proTNF-αCXCL7, CXCL8CXCL1, CXCL12KitL
ICAM-1 Plasminogenα1-Proteinase inhibitor
10 proMMP-1, -1011 IGFBP α1-Proteinase inhibitor12 Plasminogen13 CXCL12 proMMP-9, -1314 CXCL12 CD44
αv integrinCell surface tissueTransglutaminase (tTG)pro-MMP-2
15 tTG16 proMMP-217 proMMP-2
activation [24,25]. Interestingly, thrombospondin-1has also been reported to increase MMP-2 and -9activation [26]. A seemingly potent MMP inhibitor isthe cell surface receptor known as reversion-inducingcysteine-rich protein with kazal motifs (RECK). It isthe only cell surface MMP inhibitor characterized thusfar [27].
A recently identified mechanism that controls MMPactivity is cell surface localization. It has long beensuggested that MMPs function at or in the vicin-ity of the cell surface [28]. Accumulating evidencenow suggests that cell surface association may becritical for optimal MMP function. MT-MMPs havebeen shown to localize to invadopodia — specializedplasma membrane protrusions believed to constitutethe cellular structures that direct invasion[29,30] — and to lose their proteolytic activity whenexpressed in secreted form [29,31]. Moreover, anincreasing number of secreted MMPs are now recog-nized to at least transiently localize to the cell surface,most often in association with adhesion receptors orcell surface proteoglycans. Thus, MMP-1 associateswith integrins [32] and EMMPRIN, an immunoglob-ulin superfamily member that induces MMP expres-sion [33,34]. In addition to the MMP-14/TIMP-2complex, MMP-2 interacts with αvβ3-integrin via its
haemopexin-like domain [35]. MMP-7 binds cell sur-face heparan sulphate proteoglycans (HSPG), includ-ing HSPG isoforms of the hyaluronan receptor CD44[36] (Figure 2), and MMP-9 binds several cell surfacereceptors including CD44 [37,38], ICAM-1 [39], andintegrins (Lan and Stamenkovic, unpublished). MMP-9 has also been suggested to use cell surface-boundcollagen IV chains as a docking mechanism [40].MMP-13 binds HSPG (Yu and Stamenkovic, unpub-lished) and MMP-19 localizes to the cell surface by amechanism that is as yet undefined [41].
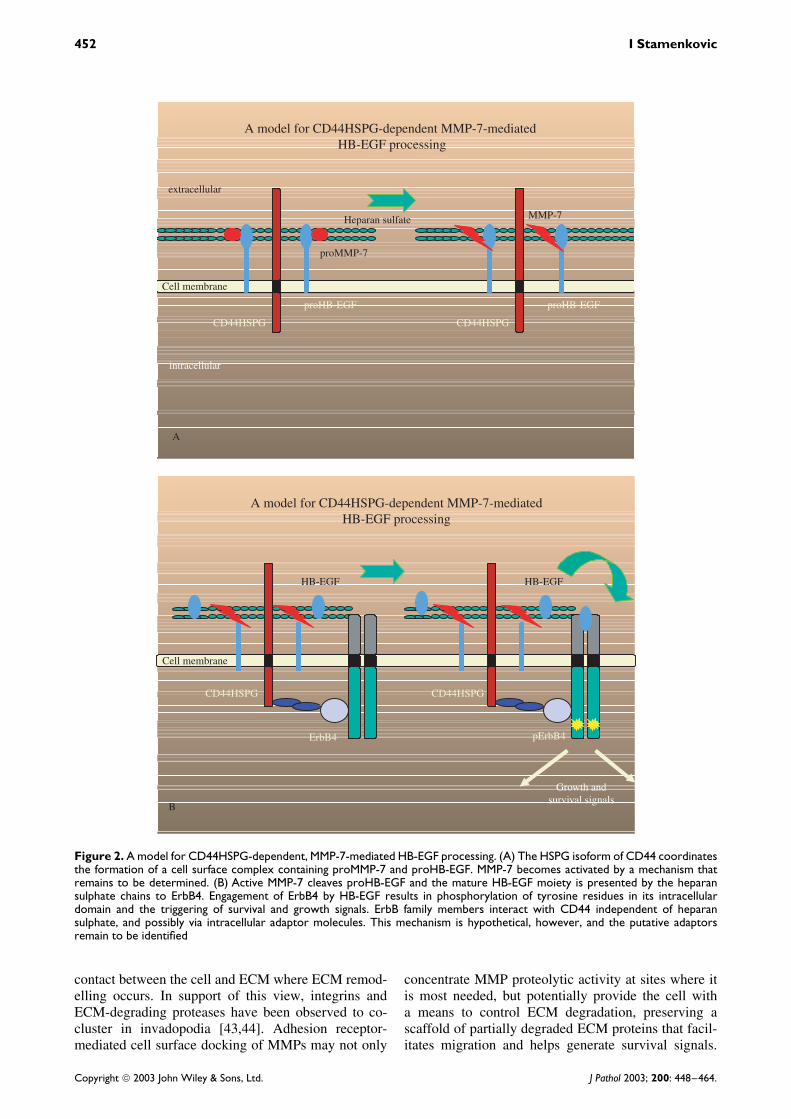
The potential functional importance of cell surfacedocking of secreted MMPs has recently been high-lighted by several studies. Inhibition of cell surfacelocalization of MMP-9 in a mouse mammary carci-noma cell line led to the loss of its invasive andmetastatic properties that could be rescued by con-stitutive cell surface expression of an MMP-9 fusionprotein [37,42]. Similarly, disruption of the interactionbetween CD44HSPG and MMP-7 was observed toresult in MMP-7 relocation from the apical to thebasal cell surface in postpartum uterine and lactatingmammary epithelia that was associated with increasedepithelial cell death and inappropriate tissue remod-elling in vivo [36] (Figure 2).
Cell surface docking may provide a mechanismto tether MMP activity to the regions of physical
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.
452 I Stamenkovic
A model for CD44HSPG-dependent MMP-7-mediated HB-EGF processing
A
CD44HSPG CD44HSPG
extracellular
MMP-7Heparan sulfate
proMMP-7
Cell membrane
proHB-EGF proHB-EGF
intracellular
A model for CD44HSPG-dependent MMP-7-mediatedHB-EGF processing
B
CD44HSPG CD44HSPG
HB-EGF HB-EGF
Cell membrane
ErbB4 pErbB4
Growth andsurvival signals
Figure 2. A model for CD44HSPG-dependent, MMP-7-mediated HB-EGF processing. (A) The HSPG isoform of CD44 coordinatesthe formation of a cell surface complex containing proMMP-7 and proHB-EGF. MMP-7 becomes activated by a mechanism thatremains to be determined. (B) Active MMP-7 cleaves proHB-EGF and the mature HB-EGF moiety is presented by the heparansulphate chains to ErbB4. Engagement of ErbB4 by HB-EGF results in phosphorylation of tyrosine residues in its intracellulardomain and the triggering of survival and growth signals. ErbB family members interact with CD44 independent of heparansulphate, and possibly via intracellular adaptor molecules. This mechanism is hypothetical, however, and the putative adaptorsremain to be identified
contact between the cell and ECM where ECM remod-elling occurs. In support of this view, integrins andECM-degrading proteases have been observed to co-cluster in invadopodia [43,44]. Adhesion receptor-mediated cell surface docking of MMPs may not only
concentrate MMP proteolytic activity at sites where itis most needed, but potentially provide the cell witha means to control ECM degradation, preserving ascaffold of partially degraded ECM proteins that facil-itates migration and helps generate survival signals.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

Extracellular matrix remodelling 453
Growth factor precursor processing on the cell sur-face, in proximity to the corresponding receptors, mayprovide MMPs with the ability to optimize the effec-tiveness of growth factor-mediated stimulation. It isalso conceivable that cell surface localization may playa part in the control of MMP activation, as has beenshown to be the case for MMP-2. Finally, the apparentpersistent activity of cell membrane-anchored MMP-7and MMP-9 [36,37,42] raises the possibility that thecell surface may provide a protective niche for MMPsfrom natural inhibitors. However, there is no indica-tion at present as to the mechanisms that may underliesuch protection.
MMP substrates
MMPs have long been thought to primarily degradeECM proteins and their ECM substrate specificityhas been extensively reviewed [8–10]. Facilitation ofcell migration was believed to be the principal effectof the MMP-mediated breakdown of ECM barriers.However, the effects of ECM degradation on cellbehaviour are likely to be considerably more complex,due, at the very least, to a combination of changes inthe nature of physical interactions between the celland the degraded ECM proteins on the one hand, andthe release of a host of ECM-sequestered cytokineson the other. Several ECM degradation products havebeen shown to display unique biological properties atleast in part by exposure of new recognition sites forcell surface ECM receptors that can trigger a varietyof cellular signals. For example, cleavage of collagenIV and laminin-5 generates exposure of cryptic sitesthat promote migration [45,46].
Although ECM degradation remains an impor-tant physiological function of MMPs, recent obser-vations provide convincing evidence that MMP sub-strates include a host of non-ECM molecules rangingfrom growth factor precursor and binding proteinsto cell surface adhesion receptors (Table 1). Thesenewly recognized properties suggest a more extensiveinvolvement of MMPs in a variety of physiologicaland pathological processes than had previously beenappreciated.
Degradation of insulin-like growth factor bind-ing protein (IGF-BP) by several MMPs releasesIGFs [47,48], cleavage of perlecan releases fibroblastgrowth factors, [49] and proteolysis of latent TGF-β binding proteins, including decorin, augments thebioavailability of latent TGF-β. Latent TGF-β 1 and2 can be proteolytically activated by MMP-2 andMMP-9 [42], and recent evidence suggests that activeTGF-β can be released my MMP-14 from cell surfacecomplexes involving αvβ8-integrin [50]. In addition,growth factor precursors can be activated and releasedby MMP-mediated proteolytic cleavage. Cell surfacegrowth factor precursors that undergo MMP-mediatedcleavage identified thus far include members of the
EGF family, particularly heparin-binding EGF (HB-EGF) precursor, cleaved and activated by MMP-3 [51]and MMP-7 [36].
Indirect evidence suggests that several growth fac-tor receptors are MMP substrates. Shedding of mem-bers of the EGF receptor family, including ErbB2and ErbB4, is blocked by TIMP-1 [52,53], whereasthe release of hepatocyte growth factor/scatter factorreceptor c-Met [54] is blocked by TIMP-3. Fibroblastgrowth factor receptor 1 is proteolytically cleaved byMMP-2 [55]. In all cases, cleavage occurs within theextracellular domain.
Cell surface adhesion receptors and proteoglycansare also subject to MMP-mediated cleavage and shed-ding. E-cadherin is cleaved by MMP-3 and -7 [56]whereas the extracellular domain of the hyaluronan(HA) receptor CD44 is cleaved by MMP-14 [57]. Inte-grins have been observed to serve as MMP substrates,as exemplified by the cleavage of αv integrin chain byMMP-14 [58], and that of β4-integrin by MMP-7 [59].Shedding of ICAM-1 has recently been shown to beMMP-9 mediated [39], and MMP-7 has been found tocleave the proteoglycan syndecan-1 [60]. Syndecan-1associates with the CXC chemokine KC, and releaseof the syndecan/KC complex from the cell surface asa result of MMP-7-mediated proteolysis is reported toplay an important role in regulating neutrophil influxto sites of injury [60].
Finally, a variety of cytokines, cytokine receptors,and chemokines have been observed to undergo MMP-mediated cleavage. Tumour necrosis factor α (TNFα)is released from the cell surface by ADAM-17 andMMP-1, -3, and -7 [8,61], while Fas ligand (FasL)is shed as a result of MMP-7-mediated proteolysis[62,63]. Interleukin-2 receptor α has been proposedto undergo MMP-9-dependent downregulation [64].MMP-2 cleaves and inactivates monocyte chemoat-tractant protein-3 (MCP-3, also known as CCL7[65]). MMP-9 cleaves the neutrophil chemoattractantinterleukin-8 (IL-8, also known as CXCL8), stronglyincreasing its activity [66]. By contrast, MMP-9 prote-olytically inactivates connective tissue-activating pep-tide III (CTAP-III, also known as CXCL7), plateletfactor 4 (PF4, also known as CXCL4), and growth-related oncogene α (GROα, also known as CXCL1[66]). Stromal cell-derived factor (SDF-1 also knownas CXCL12) undergoes inactivating cleavage by sev-eral MMPs, including MMP-1, -3, -9, -13, and -14[67]. Interestingly, induction of SDF-1 expressionin the context of bone marrow ablation upregulatesMMP-9 expression, resulting in the proteolytic cleav-age of Kit ligand (KitL) and the recruitment of cKit+stem cells [68].
MMPs and development
The nature and variety of MMP substrates predictthat a major physiological role of MMPs should liein tissue morphogenesis and organization. Indeed,
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

454 I Stamenkovic
numerous experimental approaches have shown thatMMPs help control processes that range from cellaggregation in vitro to branching morphogenesis ofseveral organs and hormone-dependent tissue cycling.Adipocytes cultured on basement membrane organizeinto large multicellular clusters that secrete MMP-2. Inhibition of MMP activity blocks migration ofthese cells and three-dimensional cluster organization[69]. Pancreatic islet cell morphogenesis also appearsto depend on MMP activity. In vitro, embryonicpancreatic islet epithelial cells cultured on collagengels differentiate and organize into aggregates thatresemble the islets of Langerhans and secrete MMP-2.Inhibition of MMP activity abrogates cell organizationinto islet type structures without affecting endocrinecell differentiation [70].
MMPs are implicated in branching morphogenesis,although the mechanisms appear to vary accordingto the type of tissue. Culture of day 10 embryonickidney explants results in ureteric bud developmentby branching morphogenesis that is inhibited by block-ing anti-MMP-9 antibody [71]. This process may alsoimplicate MMP-14, and can be enhanced by inhibitionof TIMP-2 expression [72]. In contrast, branching ofcultured salivary glands is enhanced by MMP inhibi-tion [73]. This dichotomy may be explained by differ-ences in the mechanisms that underlie branching of thetwo epithelia. Whereas kidney epithelium branches bybudding and growing into the mesenchyme, a processthat requires ECM degradation, salivary gland epithe-lium branches by the formation of clefts composedof collagen bundles in the epithelium in parallel togrowth into the surrounding mesenchyme. It followsthat in the kidney ECM degradation would be pre-dicted to enhance branching by reducing the resistanceof the ECM barrier, whereas in salivary glands MMP-mediated degradation of collagen would be expectedto destabilize the clefts and reduce branching.
The ECM of the mammary gland is subject to majorremodelling during development, lactation, and wean-ing, and several lines of evidence suggest that MMPsare implicated in each of these events. Mammary glandformation during embryonic development begins bythe budding of an epithelial tubular structure into themammary fat pad. Extensive growth and branchingat the terminal end bud follow to form the epithelialducts. These ducts end in structures that differenti-ate into alveoli, which constitute the milk-secretingunits during lactation. Alveolar development duringpregnancy and lactation dramatically alters mammarygland architecture, which regains its pre-pregnancystate during weaning by a process known as invo-lution. Expression of an active form of MMP-3 inthe mammary gland alters gland development [74,75].Virgin female mice that express a low level of thetransgene in mammary epithelia display abnormalgland morphology characterized by excessive branch-ing of the primary ducts and early alveolar devel-opment with β-casein expression at levels compa-rable to those observed in early to mid-pregnancy
glands. Lactating glands have high levels of trans-gene expression and show loss of basement membraneintegrity with depletion of laminin and collagen IV.This is accompanied by a reduction in alveolar sizeand premature alveolar epithelial apoptosis during latepregnancy. The extracellular MMP activity correlateswith increased cleavage of the basement membranemolecule entactin in the vicinity of cells undergoingapoptosis. Alveolar epithelial cells are rescued fromapoptosis when mice overexpressing MMP-3 in themammary epithelium are crossed with mice overex-pressing TIMP-1 [76].
Interestingly, mice deficient in CD44, which pro-vides a docking receptor for MMP-7 on the epithe-lial cell surface, display a similar mammary glanddefect [36]. In these mice MMP-7 activity is mis-localized, such that instead of being confined to theapical epithelial surface, it diffuses to the basementmembrane. Alveolar epithelial cells undergo prematureapoptosis, presumably because of impaired MMP-7-mediated proHB-EGF processing [36], and the glandsundergo premature involution that may be due, at leastin part, to ECM degradation resulting from inappropri-ate localization of MMP-7 proteolytic activity. Clearly,MMP activity can have a potent effect on mammarygland development and function, although the fullextent of its involvement remains to be elucidated.
MMP-7 has recently been found to play a rolein postpartum uterine involution. It has been knownthat in the rat MMP-7 expression is induced inthe uterus at the time of delivery and reaches amaximum at about 48 h postpartum, following whichit decreases rapidly, becoming undetectable at 5 days[77]. In CD44-deficient mice, expression of MMP-7 in postpartum uterus is comparable to that inwild-type tissue but the cellular distribution of theenzyme is altered. Instead of localizing to the apicalcell surface, as in postpartum uteri of wild-typemice, MMP-7 diffuses to the basal compartmentand is released into the basement membrane. Thisrelocalization is accompanied by degradation of theECM and accelerated involution compared to uteri ofwild-type animals [36]. These observations support thenotion that location plays a major role in the effect ofMMP activity.
MMPs in bone development
There is increasing evidence that MMPs are impli-cated in bone development. Two types of ossifica-tion underlie skeletal development: intramembranousand endochondral [78]. Intramembranous ossification,restricted to the skull and clavicles, results from directdifferentiation of the mesenchymal condensations thatprecede the future skeletal elements into osteoblasts.Endochondral ossification is the process whereby therest of the future skeleton develops, characterized bythe differentiation of the mesenchymal condensationsinto chondrocytes, which form the cartilage anlagen of
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

Extracellular matrix remodelling 455
the future bones. In the periphery of the anlage, cellsfrom the perichondrium differentiate into osteoblasts,whereas the periphery of the anlage becomes hyper-trophic as a result of chondrocyte proliferation andmaturation. Hypertrophic cartilage is progressivelyresorbed by the action of chondroclasts that invade andpenetrate the cartilage and replaced by bone matrix fol-lowing the invasion of capillaries. The bone matrix isdeposited by osteoblasts that follow capillary invasion,and is remodelled by the action of osteoclasts. Home-ostasis in adult bone is maintained by the balance ofosteoblast and osteoclast activity. Thus far, at least twoMMPs have been implicated in the regulation of bonedevelopment: MMP-9 and -14.
Mice in which the MMP-9 gene has been inacti-vated display a specific defect in endochondral boneformation, characterized by an accumulation of hyper-trophic cartilage at the skeletal growth plates [79].This inappropriate cartilage accumulation appears tobe due to impaired vascular invasion into the hyper-trophic cartilage zone, which leads to inhibition ofendochondral ossification. In addition, apoptosis ofterminal hypertrophic chondrocytes is delayed, sug-gesting that death of these cells is related to capillaryinvasion. This notion is supported by the observationthat chondrocytes proximal to the capillaries are theones that undergo cell death. Although it is not clearhow MMP-9 activity regulates endochondral ossifica-tion, one possibility may be that MMP-9 helps releaseone or several angiogenic factors from the hyper-trophic cartilage ECM [79]. This would explain theobserved defect in angiogenesis in the absence ofMMP-9, along with the inhibition of the processes thatare dependent on it. In support of this notion, VEGFhas been shown to be a major angiogenic factor at thegrowth plate, and its inhibition by recombinant solubleVEGF receptor (VEGFR) leads to abrogation of vas-cular ingrowth into hypertrophic cartilage, resulting ina phenotype that is similar to but more severe than thatobserved in MMP-9 null mice [80]. Expression of sol-uble VEGFR, however, has additional effects on bonedevelopment, including a decrease in chondroclast andosteoblast recruitment. It seems therefore that MMP-9 may regulate bone formation by a partial effect onVEGF activity or on the activity of other angiogenicfactors that cooperate with VEGF. It is noteworthy thatimpaired endochondral ossification is the only devel-opmental defect in MMP-9 null mice, suggesting thatthe non-redundant functions of MMP-9 during devel-opment are highly restricted.
In contrast to MMP-9 null mice, MMP-14 null ani-mals display numerous skeletal defects [81]. In addi-tion to craniofacial dysmorphisms caused by impairedintramembranous bone formation, these mice displaydwarfism that may reflect defective endochondral ossi-fication as well as osteopenia, arthritis, and fibrosisof soft tissues [81]. The cranial abnormalities are duein part to impaired removal of the calvarial cartilageprimordia, which persist and transform into a fibroticvestige. This may interfere with the normal formation
of the calvarial bones and suture closure. In the longbones, there is defective ossification of the epiphysis.Under normal conditions, hypertrophic cartilage isformed in the centre of the epiphysis from the inwardmaturation of chondrocytes, and undergoes ossifica-tion by perichondral vessel invasion through vascu-lar canals formed by the perichondrium. In MMP-14null mice, no vascular canal is formed. Ossificationis therefore delayed and occurs only when the hyper-trophic cartilage has expanded into the perichondriumby direct invasion of perichondrial vessels. Delayedepiphyseal ossification in these mice is associated withgrowth plate disorganization and reduced chondrocyteproliferation, which may contribute to the observeddwarfism. Progressive fibrosis of the periskeletal softtissues may also contribute to dwarfism and, inter-estingly, similar defects have been described in micewith a mutation in the collagenase cleavage site oftype I collagen [82]. Bone marrow stromal cells andfibroblasts isolated from MMP-14 null mice displayimpaired collagenolytic activity in vitro, suggestingthat the observed developmental defects may be due,at least in part, to inadequate remodelling of the skele-tal matrix and periskeletal soft-tissue ECM. Decreasedosteogenesis from bone marrow stromal cells in MMP-14 null mice suggests that differentiation or functionof osteoblasts may be impaired, while the observedincrease in the osteoclast number suggests that MMP-14 may play a regulatory role in the development ofthese cells as well.
MMPs in physiological invasion and woundhealing
There is strong evidence that MMPs participate in nor-mal cell invasion, migration, and process outgrowth inphysiological settings. ‘Physiologically’ invasive cellsinclude, among others, trophoblasts and osteoclasts,whereas neurons require mechanisms that control neu-rite extension. At the implantation site, trophoblastsmust penetrate into the maternal decidua. Trophoblastsexpress high levels of MMP-9, and blocking anti-MMP-9 antibody can abrogate their invasion of ECMin vitro [83,84]. Osteoclasts are recruited to bone sur-faces during bone remodelling, and their migrationthrough collagen gels is blocked by MMP inhibitors[85]. In a model of neuronal differentiation, the neu-roblastoma cell line SKSNBE displays extensive neu-rite outgrowth in response to retinoic acid. MMP-9 isinduced and expressed in the neurites, suggesting itsparticipation in the outgrowth [86]. These observationsare supported by studies showing that oligodendro-cytes in culture express active MMPs at the tip oftheir processes and that blocking anti-MMP-9 antibodyinhibits extension of the processes [87]. Oligoden-drocytes from MMP-9-deficient mice fail to displayprocess outgrowth in culture.
Wound healing relies on processes that are simi-lar to those required to promote physiological devel-opment and malignant tumour progression, including
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

456 I Stamenkovic
migration, ECM degradation, and invasion. In cul-ture, keratinocytes migrate on collagen-1 in a man-ner that requires specific cleavage of collagen-1 byMMP-1. MMP inhibitors can block this migrationand epithelial cells that do not express MMP-1 donot migrate on collagen type I. Consistent with thisobservation, keratinocytes do not migrate on col-lagen type I that bears a mutation in the MMP-1 cleavage site [88]. Keratinocytes at the edge ofthe wound must migrate to cover, or re-epithelializethe wound surface. The fibrin-rich provisional matrixthat is deposited following wounding must then beremoved and the dermis contracts to facilitate woundclosure. In wound-healing models, MMP inhibitorsabrogate keratinocyte migration and delay wound heal-ing in vivo [89]. Similar wound-healing impairmentis observed in plasminogen-deficient mice, suggest-ing that both plasmin and MMPs play a signifi-cant role in keratinocyte migration during woundhealing [90]. Keratinocyte migration is completelyblocked in plasminogen-deficient mice treated withMMP inhibitors, indicating synergism between thesetwo classes of enzymes [89]. Wound healing is delayedin MMP-3 null mice, although by a mechanism thatdoes not appear to implicate impaired keratinocytemigration. MMP-3 activity appears to be importantin fibroblast-mediated wound contraction as MMP-3deficient fibroblasts have a reduced ability to contractcollagen gels [91,92].
MMPs in cancer
Most of the evidence that MMPs are implicatedin cancer development and progression stems fromexperimental models in which expression of selectedMMPs in tumour cells has been observed to promotetumour growth, invasion, and metastasis, whereasnatural and synthetic MMP inhibitors have been shownto reduce and even abrogate tumour development(reviewed in [93]). In virtually all human cancers,MMP expression and activity has been found tobe increased and to correlate with invasiveness andpoor prognosis, supporting the experimental data andthe notion that MMPs play a role in human cancerprogression.
Despite the conceptually straightforward relation-ship between MMP activity and tumour aggressive-ness, there are instances where MMP expressionappears to be associated with a more favourable prog-nosis. In colon cancer, expression of MMP-12 bythe tumour cells and MMP-9 expression by infiltrat-ing macrophages have been reported to correlate withreduced metastatic proclivity [94,95]. To what extentMMP activity is the driving force behind the biologi-cal behaviour of these tumours remains to be clarified.The notion that MMP activity correlates with tumouraggressiveness would imply an inverse relationshipbetween MMP inhibitor expression and tumour pro-gression. However, tumours with poor prognosis have
been observed to express elevated levels of TIMP-1and -2 (reviewed in [18]). One possible explanationmay be that these tumours display high levels of MMPactivity and that the seemingly elevated TIMPs reflectan ineffective attempt of the host tissue to controlMMP-mediated proteolysis. However, several obser-vations suggest that TIMPs may have functions otherthan the ability to block MMPs. First, as discussedabove, TIMP-2 is a key component in MMP-2 activa-tion. In addition, at least under some circumstances,TIMPs can induce VEGF expression and promotetumour angiogenesis [96], tumour cell survival [97],and tumour growth [98]. Thus the balance betweenMMP and TIMP expression may have effects thatreach beyond the regulation of MMP activity alone.
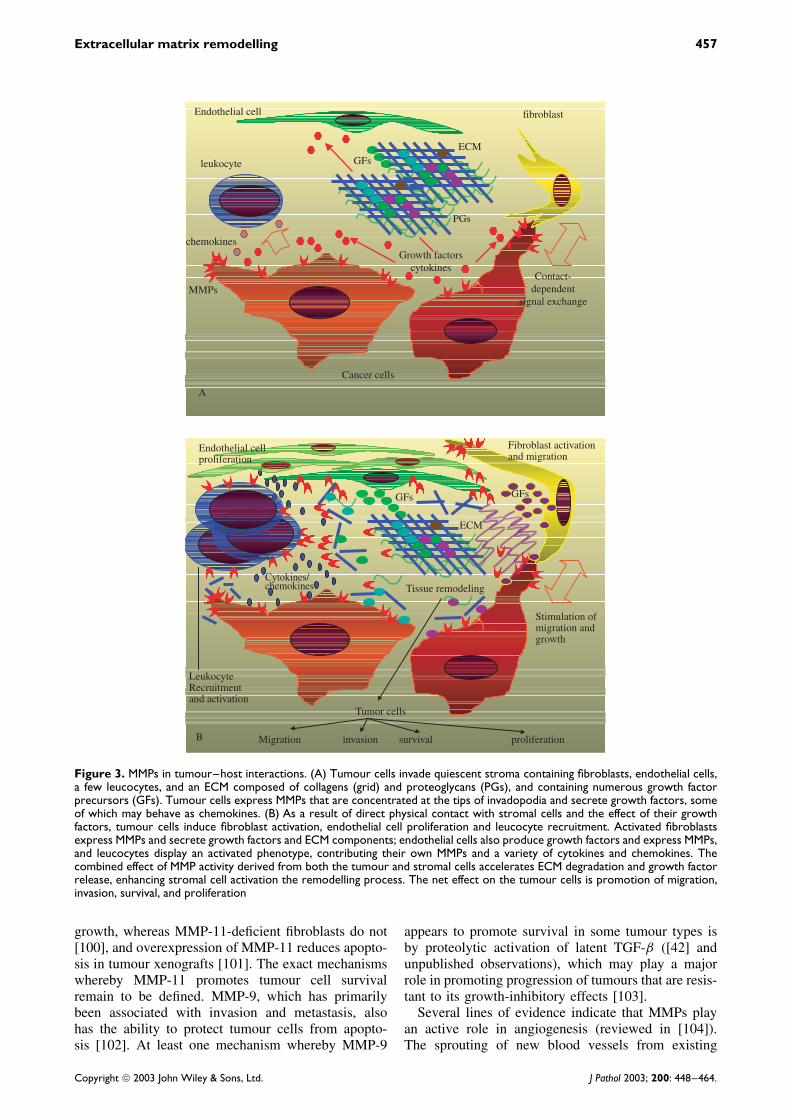
Although it was initially believed that tumour-derived MMPs play the principal role in tumourprogression, recent evidence from transgenic mousemodels indicates that stromal cell-derived MMPs mayplay an equally important role. Moreover, whereassome MMPs such as MMP-7 are primarily expressedby tumour cells, others, including MMP-2, -3, and -9,are expressed by stromal cells, sometimes predomi-nantly so. Stromal cell expression of MMPs may beinduced by tumour cell infiltration, by direct cell–cellcontact, in paracrine manner via secretion of tumour-derived growth factors, or as a result of stimula-tion by growth factors released from degraded ECM(Figure 3).
Until recently, MMP activity has been associatedprimarily with tumour invasion and metastasis. Itis now becoming clear, partly owing to transgenicmouse models (Table 2), that MMPs may play acritical role in early events in tumour development.Several lines of evidence indicate that MMPs regulatecell growth and survival. By activating cell surfacegrowth factor precursors, releasing and activatinglatent growth factors sequestered in the ECM andaltering the structure of essential ECM components,MMPs directly participate in the generation of signalsthat induce tumour cell proliferation. At least twoclasses of growth factor precursors that promote cellproliferation, TGF-α and IGFs, are known to bereleased and activated by MMPs [47,48].
Several mediators of tumour cell survival are alsoregulated by MMPs. It has been suggested that MMP-7promotes carcinoma cell survival rather than invasive-ness [99]. At least two mechanisms support this notion.First, MMP-7 cleaves Fas ligand (FasL) from the cellsurface, yielding a soluble form whose effectivenessin triggering cell death by engaging its receptor Fasis significantly reduced in some settings [64], eventhough it may enhance cell death in others [63]. Sec-ond, MMP-7 cleaves the heparin-binding EGF pre-cursor from the cell surface, releasing the matureactive form that binds ErbB1 and ErbB4 receptors,generating signals that confer protection from apop-tosis [36] (Figure 2). Another metalloproteinase thatpromotes tumour cell survival is MMP-11. Mousefibroblasts expressing MMP-11 support carcinoma cell
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.
Extracellular matrix remodelling 457
Endothelial cell
Cancer cells
Growth factorscytokines
chemokines
PGs
GFs
MMPs
A
ECM
leukocyte
Contact-dependent
signal exchange
fibroblast
Tissue remodeling
Tumor cells
Endothelial cellproliferation
LeukocyteRecruitmentand activation
Fibroblast activationand migration
ECM
GFsGFs
Cytokines/chemokines
Stimulation ofmigration andgrowth
B Migration invasion survival proliferation
Figure 3. MMPs in tumour–host interactions. (A) Tumour cells invade quiescent stroma containing fibroblasts, endothelial cells,a few leucocytes, and an ECM composed of collagens (grid) and proteoglycans (PGs), and containing numerous growth factorprecursors (GFs). Tumour cells express MMPs that are concentrated at the tips of invadopodia and secrete growth factors, someof which may behave as chemokines. (B) As a result of direct physical contact with stromal cells and the effect of their growthfactors, tumour cells induce fibroblast activation, endothelial cell proliferation and leucocyte recruitment. Activated fibroblastsexpress MMPs and secrete growth factors and ECM components; endothelial cells also produce growth factors and express MMPs,and leucocytes display an activated phenotype, contributing their own MMPs and a variety of cytokines and chemokines. Thecombined effect of MMP activity derived from both the tumour and stromal cells accelerates ECM degradation and growth factorrelease, enhancing stromal cell activation the remodelling process. The net effect on the tumour cells is promotion of migration,invasion, survival, and proliferation
growth, whereas MMP-11-deficient fibroblasts do not[100], and overexpression of MMP-11 reduces apopto-sis in tumour xenografts [101]. The exact mechanismswhereby MMP-11 promotes tumour cell survivalremain to be defined. MMP-9, which has primarilybeen associated with invasion and metastasis, alsohas the ability to protect tumour cells from apopto-sis [102]. At least one mechanism whereby MMP-9
appears to promote survival in some tumour types isby proteolytic activation of latent TGF-β ([42] andunpublished observations), which may play a majorrole in promoting progression of tumours that are resis-tant to its growth-inhibitory effects [103].
Several lines of evidence indicate that MMPs playan active role in angiogenesis (reviewed in [104]).The sprouting of new blood vessels from existing
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

458 I Stamenkovic
Table 2. Tumour development in MMP transgenic and knockout mice
MMP PromoterAdditionalstimulus Phenotype Reference
MMP overexpression1 Haptoglobin None Hyperkeratosis [136]
DMBA + PMA Increased skin carcinogenesis [136]3 Whey-acidic protein (WAP) None Mammary hyperplasia and cancer [137]
Mouse mammary tumour virus (MMTV) None Mammary hyperplasia and cancer [138]7 MMTV None Mammary hyperplasia [99]
MMTV-HER2/neu Increased mammary carcinogenesis [99]14 MMTV None Mammary hyperplasia and cancer [139]
MMP deletion2 RIP-Tag Reduced pancreatic carcinogenesis [102]7 ApcMin Reduced Intestinal adenoma
formation[140]
9 K14-HPV16 Reduced skin carcinogenesis [106]RIP-Tag Reduced pancreatic carcinogenesis [102]
11 DMBA Reduced mammary carcinogenesis [100]
ones requires at the very least the creation of a pathalong which endothelial cells can migrate and formnew tubular structures. Intuitively, ECM degradationshould therefore provide one mechanism wherebyMMPs facilitate angiogenesis. This notion is supportedby the observation that degradation of collagen typeI is necessary for endothelial cell invasion of theECM and vascular development [105]. However, thereappear to be multiple ways in which MMPs regulateangiogenesis. Convincing evidence that MMPs playa role in angiogenesis has been provided by theMMP-9 knockout mouse, which displays a delay inendochondral bone formation that has been attributedto delayed neovascularization [106]. Subsequently,pancreatic islet tumours that develop in the RIP-Tagmodel were shown to lack the angiogenic switch,characteristic of tumour progression, in the absenceof MMP-9 [102]. Similar behaviour was observed inthe K14-HPV16 skin cancer model in the absenceof MMP-9 [106]. In addition to helping degradecollagen IV and other ECM components, MMP-9 hasbeen proposed to augment VEGF bioavailability. Inparallel studies, MMP-9 activation of latent TGF-β hasbeen shown to contribute to capillary tube formationin vitro [42]. MMP-2 is also implicated in tumourangiogenesis. A reduction in MMP-2 expression intumour cells decreases angiogenesis in the chickenchorioallantoic membrane model [107] and tumourangiogenesis and growth were reduced in MMP-2deficient mice compared to normal counterparts (108).In contrast to MMP-9, MMP-2 does not appear tobe required for the angiogenic switch in the RIP-Tagmodel [102].
MMP-14 also appears to play a role in angiogenesisas MMP-14-deficient mice display defective angio-genesis during development [109]. MMP-14 degradesthe fibrin matrix that surrounds newly formed bloodvessels [110], potentially facilitating endothelial cellpenetration of tumour tissue. In addition, inhibitionof MMP-14 function blocks endothelial cell migrationand capillary tube formation in vitro [111].
Although there is strong support for MMP par-ticipation in the promotion of angiogenesis, there isalso evidence that MMPs may regulate angiogene-sis by generating angiogenic inhibitors from substratecleavage products. Angiostatin, a cleavage product ofplasminogen, is generated by MMP-2, -3, -9, and -12[112,113], and endostatin, a cleavage product of base-ment membrane collagen type XVIII, can be producedby the proteolytic activity of MMP-3, -9, -12, and -13[114]. Angiostatin and endostatin can inhibit endothe-lial cell proliferation and endostatin has been proposedto block MMP-14 activity [115]. However, the contri-bution of MMP-mediated generation of these cleavageproducts to the control of physiological and tumourangiogenesis remains to be defined.
MMPs in invasion and metastasis
Metastasis is the most devastating event associatedwith cancer because it heralds an irreversible stageof progression that responds poorly if at all tocurrent therapeutic regimens. Cancer metastasis isa complex, multistage process, which includes celldetachment from the primary tumour mass, migra-tion through the ECM, degradation of the vascularendothelial basement membrane and penetration intothe vascular lumen — a process known as intravasa-tion — survival within the circulation, which reflectsresistance to both shear stress and immune surveil-lance, adhesion to and proliferation on distal vas-cular endothelia, and finally penetration into a newhost tissue microenvironment and establishment of arelationship with the local stroma that is conducive tonew tumour colony outgrowth. Several if not all ofthese steps depend at least in part on MMP activity.
Detachment of cells from the primary tumour massrequires the downregulation of cell–cell adhesionmechanisms. Loss of E-cadherin expression in variouscancers has been associated with increased metastaticproclivity [116], and recent evidence suggests that one
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

Extracellular matrix remodelling 459
mechanism whereby cell surface E-cadherin is lostin tumour cells is proteolytic cleavage by MMP-3and -7 [56]. Moreover, E-cadherin proteolytic cleav-age fragments generated by MMPs appear to promotetumour cell migration [56], although the precise mech-anism remains to be determined. E-cadherin sheddingis associated with epithelial to mesenchymal transi-tion, which often correlates with cancer aggressiveness[116].
Migration of tumour cells through the host tissuestroma requires partial degradation of the ECM andcoordinated sequential attachment to and detachmentfrom the ECM scaffold. Recent work using two-photon microscopy has provided spectacular real-time evidence that MMP proteolytic activity causescontrolled degradation of collagen fibrils that arein contact with the invading tumour cell surface,leaving trail of released cell surface molecules inthe cell’s wake [117,118]. Interestingly, inhibitionof MMPs does not result in abrogation of tumourcell migration through the collagen gel but rathertransforms the crawling movement associated withcollagen fibril cleavage into amoeboid movement thatleaves the collagen lattice intact [117,118]. Accordingto this model, MMP expression does not influencetumour cell migration rate through collagen but playsa defining role in the type of migratory activity thattumour cells display, and, by extension, in the typesof signals they receive from the host tissue stroma.Moreover, this model strongly supports the notionthat the MMP activity relevant to ECM degradationis associated with the tumour cell surface.
As already discussed, cleavage of ECM componentsby MMPs generates proteolytic fragments that enhancetumour cell migration. Thus, cleavage of laminin-5 byMMP-2 and -14 results in laminin fragments that trig-ger migration signals in cells [45,119], and cleavage ofcollagen IV discloses cryptic sites that are recognizedby integrins and contribute to migration stimuli [46].MMPs also cleave adhesion receptors responsible forcell–matrix interaction, thereby presumably participat-ing in the detachment of cells from the ECM. The cellsurface hyaluronan receptor and facultative proteogly-can CD44 is cleaved by MMP-14, and its cleavagepromotes migration. Expression of CD44 containing amutation of the proteolytic cleavage site abrogates cellmigration on ECM [57].
Intravasation, the process whereby tumour cellspenetrate the vascular endothelial wall, has beenproposed to be a rate-limiting event in metastasis[120]. Although it is likely that a variety of MMPsmay be involved in the degradation of the vascularendothelial basement membrane, MMP-9 has thusfar been shown to play a potentially leading role[120,121].
Survival in the face of the immune response is keyfor the ability of tumour cells with metastatic poten-tial to establish new colonies. Among the wide rangeof mechanisms that have been proposed to explaintumour cell evasion of immune surveillance, several
are MMP-dependent. Tumour cells typically inter-act with neutrophils, macrophages, cytotoxic T cells(CTLs), and natural killer (NK) cells. T cell prolifer-ation is controlled in large part by the engagement ofthe interleukin-2 receptor (IL-2R) by its natural ligandIL-2. MMPs, including MMP-9, have been shown tocleave the α-chain of IL-2R [65], resulting in the inhi-bition of T cell proliferation. MMP-9-mediated activa-tion of latent TGF-β may also contribute to immunesuppression, since TGF-β is a potent inhibitor of T cellfunction [122]. Cleavage by MMPs of FasL mayconfer partial protection from CTL-dependent Fas-mediated cell death [64] and recent evidence indicatesthat MMP-9-mediated shedding of cell surface ICAM-1 may block the ability of CTLs and NK cells to inter-act with target cells, thereby reducing the effectivenessof their cytotoxicity [39]. In MMP-11-deficient mice,tumours display a significant increase in the numberof infiltrating neutrophils and macrophages, consis-tent with the possibility that MMP-11 may cleaveand inactivate a chemokine [123]. Interestingly, anMMP-11 cleavage product of α1-proteinase inhibitorreduces the sensitivity of tumour cells to NK-mediatedkilling [124].
Recent evidence indicates that MMPs cleave a vari-ety of chemokines in ways that can either enhanceor block their function. MMP-2 cleaves and inac-tivates MCP-3 (CXCL7). Interestingly, the cleavedfragment becomes an antagonist to the correspondingreceptors [65]. MMP-9 augments IL-8 activity, whichshould have a potent chemotactic effect on neutrophils,but proteolytically blocks the activity of several otherchemokines that may have an important effect on leu-cocyte recruitment and survival [66]. Thus, tumourcell expression of MMP-9 or its induction in stro-mal cells may help contain the immune response byseveral mechanisms. SDF-1/CXCL12, which is inacti-vated by several MMPs [67] (Table 1) is a ligand forthe CXC chemokine receptor 4 (CXCR4) on leuco-cytes and breast carcinoma cells [125]. Inhibition ofCXCR4 engagement by its ligand using monoclonalantibodies reduces metastasis to lung and lymph nodesin vivo [125]. In an elegant recent study, MMP-9 hasbeen shown to cleave cell surface KitL in bone mar-row cells [68]. Shedding of KitL plays an importantrole in the transfer of endothelial and haematopoieticstem cells (HSC) from a quiescent to a proliferativeniche. In MMP-9 null mice, KitL shedding is impaired,resulting in reduced HSC mobility and a correspond-ingly reduced haematopoietic recovery following bonemarrow ablation. Thus, MMP-9-mediated KitL releaseallows bone marrow repopulating cells to home to anmicroenvironment that facilitates their differentiationand promotes the reconstitution of the progenitor/stemcell pool [68].
Extravasation was believed to be a key step in can-cer metastasis. However, increasing evidence indicatesthat extravasation is not rate limiting (reviewed in[10,126]), and that it occurs following the proliferationof immobilized tumour cells on vascular endothelium.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

460 I Stamenkovic
This process does not appear to require the proteolyticaction of MMPs but results from the mechanical dis-ruption of blood vessels by locally growing tumourcells.
The final step in metastasis is the establishment oftumour colonies at sites distant to that of the origin,which relies on interactions between the tumour cellsand host tissue stroma. It is likely that most if not all ofthe MMP functions discussed thus far are harnessed bythe tumour cells into helping establish a relationshipwith the stroma that is favourable to tumour devel-opment (Figure 3). Invading tumour cells may havetheir own repertoire of MMPs, but it is becomingincreasingly clear that they direct, either by physi-cal contact or in paracrine manner, MMP expressionand secretion by stromal cells, including fibroblasts,endothelial cells, and leucocytes (Figure 3). MMPsproduced by the stroma are likely to play an impor-tant role in tumour-directed tissue remodelling. Notonly can they accelerate the remodelling process itself,but their action can augment the release of ECM-sequestered growth factors, which may help enhancetumour survival, promote angiogenesis, and contributeto further tumour dissemination. A key question iswhether reliance on MMP activity lasts throughoutmetastatic tumour growth or whether MMP-dependentevents serve to initiate colony development, whichmay then proceed in the absence of further MMP-mediated proteolysis. This is an important consider-ation for therapeutic strategies targeted toward con-trolling MMP activity and one that remains to beadequately addressed.
MMPs as targets for cancer therapy
The perceived role of MMPs in cancer developmentand progression, based principally on mouse mod-els, places them in the front line for cancer ther-apy targets. Accordingly, numerous MMP-inhibitingstrategies have been tried both in experimental mod-els of cancer and in clinical trials. Conceptually, oneof the simplest approaches consists of blocking MMPactivity.
Several synthetic MMP inhibitors have been devel-oped to that end and shown to be effective in blockingtumour growth in experimental animals, validating theconcept of MMPs as therapeutic targets for cancer.These observations led to high expectations of syn-thetic MMP blocking compounds in human clinicaltrials. Unfortunately, synthetic MMP inhibitors havethus far failed to live up to their expectations andin fact some cancer patients had poorer survival asa result of their administration (reviewed in [127]).
Two main categories of synthetic MMP inhibitorcompounds have been developed: the collagen pep-tidomimetics and the collagen non-peptidomimetics.The peptidomimetics batimastat and marimastat mimicthe cleavage sites of MMP substrates. Batimastat can-not be administered orally and has been removed from
testing for human cancer treatment. Marimastat hasundergone numerous Phase III clinical trials, but hasfailed to prolong survival in patients with advanced,metastatic cancer [127], although at maximal dosesits effectiveness equalled that of conventional ther-apy [128]. However, in groups of patients withoutmetastases at the time of diagnosis, survival was sig-nificantly higher among those treated with marimastatthan among placebo-treated patients.
The non-peptidomimetic MMP inhibitors, also kn-own as ‘deep pocket’ MMP inhibitors, were syn-thesized on the basis of the conformation of theMMP active site. These inhibitors include prinoma-stat and tanomastat and several related compounds.However, studies with most of these compounds inadvanced cancer were terminated in Phase III trialseither because the patient survival was worse thanthat of the placebo group or because the compoundsin combination with conventional therapy providedno advantage over conventional therapy alone [127].Some of these compounds are still undergoing evalu-ation in patients with less advanced cancer.
Several other less conventional drugs are underdevelopment or in early stages of clinical trials. Exam-ples include tetracycline derivatives that block boththe synthesis and activity of MMPs [129]. A compo-nent of green tea, currently in Phase III clinical trials,blocks MMP-2 and MMP-9 activity in vitro [130],while acetylsalicylic acid, a potent anti-inflammatorydrug known to reduce the risk of colon cancer, directlyblocks MMP-2 activity and inhibits tumour cell inva-siveness in vitro [131]. Finally, small inhibitor pep-tides selected for high specificity for individual MMPshave shown some promise in experimental animalmodels [132].
Other experimental approaches have been triedwith potentially interesting results. MMP synthesisinhibition by targeting mRNA with ribozymes [121]or transfection of antisense constructs [133,134] canprovide a successful means of reducing tumour growthand dissemination. However, the applicability of suchapproaches in the clinic is still remote.
A more attractive approach consists of preventingMMP localization to the cell surface by disruptingtheir interaction with cell surface docking recep-tors. Recently, a compound has been discovered thatblocks MMP-2 interaction with αvβ3-integrin [135].Although the effect of this compound still awaits vali-dation in clinical trials, it has shown promising resultsin blocking tumour angiogenesis and growth in exper-imental animal models.
The failure of synthetic MMP inhibitors in clinicaltrials in patients with advanced cancer raises ques-tions as to the suitability of MMPs as therapeutictargets. However, in view of our current understand-ing of how MMPs function, a new assessment ofanti-MMP strategies is warranted. Part of the expla-nation for the failure of synthetic MMP inhibitorsmay lie in the fact that none of those used thusfar have been shown to be highly selective for any
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

Extracellular matrix remodelling 461
individual MMP. Furthermore, since the compoundswere typically administered at the maximal tolerateddose, whatever selectivity they may display at lowerdoses was most likely overridden. Non-selective inhi-bition of MMP activity may result in the abrogationof potentially beneficial effects of MMPs, with anadverse net effect on patient survival. Finally, theseinhibitors were administered to patients with advancedmetastatic disease and it remains an open question towhat extent tumours rely on MMP activity at suchlate stages. A more selective approach that takes intoaccount the identity of MMPs that any given tumourrelies on will be more desirable, and one strategy thatappears highly attractive is to block MMP interac-tion with cell surface docking molecules. Because itappears increasingly clear that location in general andcell surface localization in particular are important forMMP-mediated tumour invasion and metastasis, dis-ruption of the mechanisms that help retain secretedMMPs on the cell surface may provide an effectivemeans to inhibit the function of individual MMPswithout impairing MMP activity in general.
Perspectives
Recent experimental data provide strong evidence thatrather than being mere processors of ECM proteins,MMPs are major regulators of normal and malignanttissue physiology. While the discovery that MMPshave a broad range of substrates other than ECMproteins provides new insight into the complexity oftheir role in tissue remodelling, it also raises numerousquestions as to the mechanisms whereby the net effectof their activity is determined. The various cleavageproducts of an individual MMP can both promoteand inhibit a defined process such as angiogenesis.What determines whether the process proceeds or isinhibited? Could it be conceivable to design therapeu-tic strategies that might shift the equilibrium towardpromoting the beneficial effects of substrate cleavagewhile inhibiting the detrimental ones? Why are themajority of known MMPs secreted when it appearsthat membrane localization promotes the effectivenessof their proteolytic activity? What other functionalsignificance does cell surface localization of secretedMMPs have, ie does it play a role in physiologicalmechanisms of MMP activation in general? Does ithelp maintain proteolytic activity and, if so, how? Withrespect to cancer, it will be important to determinewhich MMPs different types of tumour cells depend onfor growth and dissemination and the duration of MMPdependence of each stage of progression. Finally, theold questions regarding MMPs remain: why are thereso many MMPs with overlapping substrate specificityand why is it necessary to have two such closelyrelated families as MMPs and ADAMs that appear tobe capable of many of the same functions? The MMPfield has reached a new level of complexity and will
provide a fertile soil for cancer researchers in the nextfew years.
Acknowledgements
This work was supported by a grant from the Fonds Nationalpour la Recherche Scientifique and a Swiss Institute forExperimental Cancer Research (ISREC) Molecular OncologyNCCR grant.
References
1. Rohrbach DH, Timpl R. Molecular and cellular aspects ofbasement membranes. In Cell Biology: A Series of Monographs.Academic Press: New York, 1993.
2. Iozzo RV. Matrix proteoglycans: from molecular design tocellular function. Annu Rev Biochem 1998; 76: 609–652.
3. Laurent TC, Fraser JRE. Hyaluronan. FASEB J 1992; 6:2397–2404.
4. Rawlings ND, Barrett AJ. Evolutionary families of metallopepti-dases. Methods Enzymol 1995; 248: 183–228.
5. Stocker W, Grams F, Baumann U, et al. The metzincins:topological and sequential relations between the astracins,adamalysins, serralysins and matrixins (collagenases) define asuperfamily of zinc peptidases. Protein Sci 1995; 4: 823–840.
6. Nagase H, Woesner JF. Matrix metalloproteinases. J Biol Chem1999; 274: 21 491–21 494.
7. Gross J, Lapierre CM. Collagenolytic activity in amphibiantissues: a tissue culture assay. Proc Natl Acad Sci USA 1962;48: 1014–1022.
8. McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’renot just for matrix anymore! Curr Opin Cell Biol 2001; 13:534–540.
9. Egeblad M, Werb Z. New functions for the matrix metallopro-teinases in cancer progression. Nature Rev 2002; 2: 161–174.
10. Stamenkovic I. Matrix metalloproteinases in tumor invasion andmetastasis. Cancer Biol 2000; 10: 415–433.
11. Nagase H. Activation mechanisms of matrix metalloproteinases.Biol Chem Hoppe Seyler 1997; 378: 151–160.
12. Sternlicht MD, Werb Z. How matrix metalloproteinases regulatecell behavior. Annu Rev Cell Dev Biol 2001; 17: 463–516.
13. Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA,Goldberg GI. Mechanism of cell surface activation of 72-kDatype IV collagenase. Isolation of the activated form of themembrane metalloprotease. J Biol Chem 1995; 270: 5331–5338.
14. Sato H, Takino T, Okada Y, et al. A matrix metalloproteinaseexpressed on the surface of invasive tumor cells. Nature 1994;370: 61–65.
15. Deryugina EI, Ratnikov B, Monosov E, et al. MT1-MMPinitiates activation of pro-MMP-2 and integrin αvβ3 promotesmaturation of MMP-2 in breast carcinoma cells. Exp Cell Res2001; 263: 209–223.
16. Sottrup-Jensen L, Birkedal-Hansen H. Human fibroblast collage-nase–α-macroglobulin interactions. Localization of cleavage sitesin the bait regions of five mammalian α-macroglobulins. J BiolChem 1989; 264: 393–401.
17. Yang Z, Strickland DK, Bornstein P. Extracellular matrixmetalloproteinase 2 levels are regulated by the low densitylipoprotein-related scavenger receptor and thrombospondin 2. JBiol Chem 2001; 276: 8403–8408.
18. Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissueinhibitors of metalloproteinases: structure, regulation andbiological functions. Eur J Cell Biol 1997; 74: 111–122.
19. Goldberg GI, Strongin A, Collier IE, Genrich LT, Marmer BL.Interaction of 92-kDa type IV collagenase with the tissue inhibitorof metalloproteinases prevents dimerization, complex formationwith interstitial collagenase, and activation of the proenzyme withstromelysin. J Biol Chem 1992; 267: 4583–4591.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

462 I Stamenkovic
20. Kolkenbrock H, Orgel D, Hecker-Kia A, Zimmermann J,Ulbrich N. Generation and activity of ternary gelatinase B/TIMP-1/LMW-stomelysin-1 complex. Biol Chem Hoppe Seyler 1995;376: 495–500.
21. Amour A, Slocombe PM, Webster A, et al. TNF-α convertingenzyme (TACE) is inhibited by TIMP-3. FEBBS Lett 1998; 435:39–44.
22. Liu YE, Wang M, Greene J, et al. Preparation and character-ization of recombinant tissue inhibitor of metalloproteinase 4(TIMP-4). J Biol Chem 1997; 272: 20 479–20 483.
23. Wang Z, Juttermann R, Soloway PD. TIMP-2 is required forefficient activation of proMMP-2 in vivo. J Biol Chem 2000;275: 26 411–26 415.
24. Bein K, Simons M. Thrombospondin type 1 repeats interactwith matrix metalloproteinase 2: regulation of metalloproteinaseactivity. J Biol Chem 2000; 275: 32 167–32 173.
25. Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO,Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppressesspontaneous tumor growth and inhibits activation of matrixmetalloproteinase-9 and mobilization of vascular endothelialgrowth factor. Proc Natl Acad Sci USA 2001; 98: 12 485–12 490.
26. Taraboletti G, Morbidelli L, Donnini S, et al. The heparinbinding 25kDa fragment of thrombospondin-1 promotesangiogenesis a modulates gelatinase and TIMP-2 production inendothelial cells. FASEB J 2000; 14: 1674–1676.
27. Oh J, Takahashi R, Kondo S, et al. The membrane-anchoredMMP inhibitor RECK is a key regulator of extracellular matrixintegrity and angiogenesis. Cell 2001; 107: 789–800.
28. Werb Z. ECM and cell surface proteolysis regulating cellularecology. Cell 1997; 91: 439–442.
29. Nakahara H, Howard L, Thompson EW, et al. Transmem-brane/cytoplasmic domain-mediated membrane type 1-matrixmetalloprotease docking to invadopodia is required for cell inva-sion. Proc Natl Acad Sci USA 1997; 94: 7959–7964.
30. Chen W-T. Proteases associated with invadopodia, and their rolein degradation of extracellular matrix. Enzyme Protein 1996; 49:59–71.
31. Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. Regulationof cell invasion and morphogenesis in a three dimensional typecollagen I matrix by membrane type matrix metalloproteinases 1,2 and 3. J Cell Biol 2000; 149: 1309–1323.
32. Dumin JA, Dickerson SK, Stricker TO, et al. Pro-collagenase-1(matrix metalloproteinase-1) binds the alpha(2)beta(1) integrinupon release from keratinocytes migrating on type I collagen. JBiol Chem. 2001; 276: 29 368–29 374.
33. Guo H, Li R, Zucker S, Toole BP. EMMPRIN (CD147) aninducer of matrix metalloproteinase synthesis also bindsinterstitial collagenase to the tumor cell surface. Cancer Res 2000;60: 888–891.
34. Guo H, Zucker S, Gordon MK, Toole BP, Biswas C. Stimulationof matrix metalloproteinase production by recombinant extracel-lular matrix metalloproteinase inducer from transfected Chinesehamster ovary cells. J Biol Chem 1997; 272: 24–27.
35. Brooks PC, Stromblad S, Sanders LC, et al. Localization ofmatrix metalloproteinase MMP-2 to the surface of invasive cellsby interaction with integrin αvβ3. Cell 1996; 85: 683–693.
36. Yu WH, Woessner JF Jr, McNeish JD, Stamenkovic I. CD44anchors the assembly of matrilysin/MMP-7 with heparin-bindingepidermal growth factor precursor and ErbB4 and regulatesfemale reproductive organ remodeling. Genes Dev 2002; 16:307–323.
37. Yu Q, Stamenkovic I. Localization of matrix metalloproteinase9 to the cell surface provides a mechanism for CD44-mediatedtumor invasion. Genes Dev 1999; 13: 35–48.
38. Bourguignon LY, Gunja-Smith Z, Iida N, et al. CD44v (3,8–10)is involved in cytoskeleton-mediated tumor cell migration andmatrix metalloproteinase (MMP-9) association in metastaticbreast cancer cells. J Cell Physiol 1998; 176: 206–215.
39. Fiore E, Fusco C, Romero P, Stamenkovic I. Matrix metallopro-teinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene 2002; 21: 5213–5223.
40. Olson MW, Toth M, Gervasi DC, Sado Y, Ninomiya Y, Frid-man R. High affinity binding of latent matrix metalloproteinase-9to the α2 (IV) chain of collagen IV. J Biol Chem 1998; 273:10 672–10 681.
41. Mauch S, Kolb C, Kolb B, Sadowski T, Sedlacek R. Matrixmetalloproteinase-19 is expressed in myeloid cells in an adhesion-dependent manner and associates with the cell surface. J Immunol.2002; 168: 1244–1251.
42. Yu Q, Stamenkovic I. Cell surface-localized matrix metallo-proteinase-9 proteolytically activates TGF-β and promotes tumorinvasion and angiogenesis. Genes Dev 2000; 14: 163–176.
43. Mueller SC, Ghersi G, Akiyama SK, et al. A novel protease-docking function of integrin at invadopodia. J Biol Chem 1999;247: 24 947–24 952.
44. Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh YY,Chen WT. Activation of β1 integrin signaling stimulatestyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J Biol Chem 1998; 273:9–12.
45. Gianelli G, Falk-Marzillier J, Schiraldi O, Stetler-StevensonWG, Quaranta V. Induction of cell migration by matrixmetalloprotease-2 cleavage of laminin-5. Science 1997; 277:225–228.
46. Xu J, Rodriguez D, Petitclerc E, et al. Proteolytic exposure of acryptic site within collagen type IV is required for angiogenesisand tumor growth in vivo. J Cell Biol 2001; 154: 1069–1080.
47. Manes S, Mira E, Barbacid MM, et al. Identification of insulin-like growth factor-binding protein-1 as a potential physiologicsubstrate for human stromelysin-3. J Biol Chem 1997; 272:25 706–25 712.
48. Manes S, Llorente M, Lacalle RA, et al. The matrix metallo-proteinase-9 regulates the insulin-like growth factor-triggeredautocrine response in DU-145 carcinoma cells. J Biol Chem 1999;274: 6935–6945.
49. Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. Thedegradation of human endothelial cell-derived perlecan andrelease of bound basic fibroblast growth factor by stromelysin,collagenase, plasmin, and heparanases. J Biol Chem 1996; 271:10 079–10 086.
50. Mu D, Cambier S, Fjellbirkeland L, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002; 157:493–507.
51. Suzuki M, Raab G, Moses MA, Fernandez CA, Klagsbrun M.Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site.J Biol Chem 1997; 272: 31 730–31 737.
52. Codony-Servat J, Albanell J, Lopez-Talavera JC, Arribas J,Baselga J. Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor ofmetalloproteases-1 in breast cancer cells. Cancer Res 1999; 59:1196–1201.
53. Vecchi M, Rudolph-Owen LA, Brown CL, Dempsey PJ, Carpen-ter G. Tyrosine phosphorylation and proteolysis: pervanadate-induced, metalloprotease-dependent cleavage of the ErbB-4 receptor and amphiregulin. J Biol Chem 1998; 273:20 589–20 595.
54. Nath D, Williamson NJ, Jarvis R, Murphy G. Shedding of c-Metis regulated by crosstalk between a G-protein coupled receptorand EGF receptor and is mediated by a TIMP-3 sensitivemetalloproteinase. J Cell Sci 2001; 114: 1213–1220.
55. Levi E, Fridman R, Miao HQ, Ma YS, Yayon A, Vlodavsky I.Matrix metalloproteinase 2 releases active soluble ectodomain offibroblast growth factor receptor 1. Proc Natl Acad Sci USA 1996;93: 7069–7074.
56. Noe V, Fingleton B, Jacobs K, et al. Release of an invasionpromoter E-cadherin fragment by matrilysin and stromelysin-1. JCell Sci 2001; 114: 111–118.
57. Kajita M, Itoh Y, Chiba T, et al. Membrane-type 1 matrixmetalloproteinase cleaves CD44 and promotes cell migration. JCell Biol 2001; 153: 893–904.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

Extracellular matrix remodelling 463
58. Deryugina EI, Ratnikov BI, Postnova TI, Rozanov DV, Stron-gin AY. Processing of integrin αV subunit by MT1-MMP stim-ulates migration of breast carcinoma cells on vitronectin andenhances tyrosine phosphorylation of FAK. J Biol Chem 2002;277: 9749–9756.
59. Von Bredow DC, Nagle RB, Bowden GT, Cress AE. Cleavageof b4 integrin by matrilysin. Exp Cell Res 1997; 236: 341–345.
60. Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding ofsyndecan-1 regulates chemokine mobilization and transepithelialefflux of neutrophils in acute lung injury. Cell 2002; 111:635–646.
61. Haro H, Crawford HC, Fingleton B, Shinomiya K, Spen-gler DM, Matrisian LM. Matrix metalloproteinase-7-dependentrelease of tumor necrosis factor α in a model of herniated discresorption. J Clin Invest 105: 143–150.
62. Powell WC, Fingleton B, Wilson CL, Boothby M, Matrisian LM.The metalloproteinase matrilysin proteolytically generates activesoluble Fas ligand and potentiates epithelial cell apoptosis. CurrBiol 1999; 9: 1441–1447.
63. Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I.Matrix metalloproteinase-7-mediated cleavage of Fas ligandprotects tumor cells from chemotherapeutic drug cytotoxicity.Cancer Res 2001; 61: 577–581.
64. Sheu BC, Hsu SM, Ho HN, Lien HC, Huang SC, Lin RH. Anovel role of metalloproteinase in cancer-mediated immunosup-pression. Cancer Res 61: 237–242.
65. McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase Acleavage of monocyte chemoattractant protein-3. Science 2000;289: 1202–1206.
66. Opdenakker G, Van des Steen PE, Van Damme J, Gelatinase. B:a tuner and amplifier of immune functions. Trends Immunol 2001;22: 571–579.
67. McQuibban GA, Butler GS, Gong JH, et al. Matrix metallopro-teinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem 2001; 276: 43 503–43 508.
68. Heissig B, Hattori K, Dias S, et al. Recruitment of stem andprogenitor cells from the bone marrow niche requires MMP-9mediated release of kit ligand. Cell 2002; 109: 625–637.
69. Brown LM, Fox HL, Hazen SA, LaNoue KF, Rannels SR,Lynch CJ. Role of the matrixin MMP-2 in multicellularorganization of adipocytes cultured in basement membranecomponents. Am J Physiol 1997; 272: C937–C949.
70. Miralles F, Battelino T, Czernichow P, Scharfmann R. TFG-beta plays a key role in morphogenesis of the pancreaticislets of Langerhans by controlling the activity of the matrixmetalloproteinase MMP-2. J Cell Biol 1998; 143: 827–836.
71. Lelongt B, Trugnan G, Murphy G, Ronco PM. Matrix metallo-proteinases MMP2 and MMP9 are produced in early stages ofkidney morphogenesis but only MMP9 is required for renalorganogenesis in vitro. J Cell Biol 1997; 136: 1363–1373.
72. Kanwar YS, Ota K, Yang Q, et al. Role of membrane-type matrixmetalloproteinase 1 (MT-1-MMP), MMP-2, and its inhibitor innephrogenesis. Am J Physiol 1999; 277: F934–F947.
73. Fukuda Y, Masuda Y, Kishi J, et al. The role of interstitialcollagens in cleft formation of mouse embryonic submandibulargland during initial branching. Development 1988; 103: 259–267.
74. Sympson CJ, Talhouk RS, Alexander CM, et al. Targetedexpression of stromelysin-1 in mammary gland provides evidencefor a role of proteinases in branching morphogenesis and therequirement for an intact basement membrane for tissue-specificgene expression. J Cell Biol 1994; 125: 681–693.
75. Witty JP, Wright JH, Matrisian LM. Matrix metalloproteinasesare expressed during ductal and alveolar mammary morpho-genesis, and misregulation of stromelysin-1 in transgenic miceinduces unscheduled alveolar development. Mol Biol Cell 1995;6: 1287–1303.
76. Alexander CM, Howard EW, Bissel MJ, Werb Z. Rescue ofmammary epithelial cell apoptosis and entactin degradation bya tissue inhibitor of metelloproteinases-1 transgene. J Cell Biol1996; 135: 1669–1677.
77. Woessner JF. Regulation of matrilysin in the rat uterus. BiochemJ 1996; 74: 777–784.
78. Karsenty G. The genetic transformation of bone biology. GenesDev 1999; 13: 3037–3051.
79. Vu Th, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is akey regulator of growth plate angiogenesis and apoptosis ofhypertrophic chondrocytes. Cell 1998; 93: 411–422.
80. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N.VEGF couples hypertrophic cartilage remodeling, ossificationand angiogenesis during endochondral bone formation. Nat Med1999; 5: 623–628.
81. Holmbeck K, Bianco P, Caterina J, et al. MT1-MMP-deficientmice develop dwarfism, osteopenia, arthritis, and connectivetissue disease due to inadequate collagen turnover. Cell 1999;99: 81–92.
82. Liu X, Wu H, Byrne M, Jeffrey J, Krane S, Jaenisch R. Atargeted mutation at the known collagenase cleavage site in mousetype I collagen impairs tissue remodeling. J Cell Biol 1995; 130:227–237.
83. Alexander CM, Hansell EJ, Behrendtsen O, et al. Expressionand function of matrix metalloproteinases and their inhibitorsat the maternal–embryonic boundary during mouse embryoimplantation. Development 1996; 122: 1723–1736.
84. Behrendtsen O, Werb Z. Metalloproteinases regulate parietalendoderm differentiation and migrating in cultured mouseembryos. Develop Dyn 1997; 208: 255–265.
85. Sato T, Foged NT, Delaisse JM. The migration of purified osteo-clasts through collagen is inhibited by matrix metalloproteinaseinhibitors. J Bone Mineral Res 1998; 13: 59–66.
86. Chambaut-Guerin AM, Herigault S, Rouet-Benzineb P, RouherC, Lafuma C. Induction of matrix metalloproteinase MMP-9 (92-kDa gelatinase) by retinoic acid in human neuroblastoma SKNBEcells: relevance to neuronal differentiation. J Neurochem 2000;74: 508–517.
87. Oh LY, Larsen PH, Krekoski CA, et al. Matrix metallo-proteinase-9/gelatinase B is required for process outgrowth byoligodendrocytes. J Neurosci 1999; 19: 8464–8475.
88. Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG,Parks WC. The activity of collagenase-1 is required forkeratinocyte migration on a type 1 collagen matrix. J Cell Biol1997; 137: 1445–1457.
89. Lund LR, Romer J, Bugge TH, et al. Functional overlap betweentwo classes of matrix-degrading proteases in wound healing.EMBO J 1999; 18: 4645–4656.
90. Romer J, Bugge TH, Pyke C, et al. Impaired wound healing inmice with a disrupted plasminogen gene. Nat Med 1996; 2:287–292.
91. Bullard KM, Lund L, Mudgett JS, et al. Impaired woundcontraction in stromelysin-1-deficient mice. Ann Sur 1999; 230:260–265.
92. Bullard KM, Mudgett JS, Scheuenstuhl H, Hunt TK, Banda MJ.Stromelysin-1-deficient fibroblasts display impaired contractionin vitro. J Surg Res 1999; 84: 31–34.
93. Coussens LM, Werb Z. Matrix metalloproteinases and thedevelopment of cancer. Chem Biol 1996; 3: 895–904.
94. Yang W, Arii S, Gorin-Rivas MJ, Mori A, Onodera H, Ima-mura M. Human macrophage metalloelastase gene expression incolorectal carcinoma and its clinicopathologic significance. Can-cer 2001; 91: 1277–1283.
95. Takeha S, Fujiyama Y, Bamba T, Sorsa T, Nagura H, Ohtani H.Stromal expression of MMP-9 and urokinase receptor isinversely associated with liver metastasis and with infiltratinggrowth in human colorectal cancer: a novel approach fromimmune/inflammatory aspect. Jpn J Cancer Res 1997; 88: 72–81.
96. Yoshiji H, Harris SR, Raso E, et al. Mammary carcinoma cellsover-expression tissue inhibitor of metalloproteinases-1 showenhanced vascular endothelial growth factor expression. Int JCancer 1998; 75: 81–87.
97. Hayakawa T, Yamashita K, Ohuchi E, Shinagawa A. Cellgrowth-promoting activity of tissue inhibitor of metallo-proteinases-2 (TIMP-2). J Cell Sci 1994; 107: 2373–2379.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.

464 I Stamenkovic
98. Jiang Y, Wang M, Celiker MY, et al. Stimulation of mammarytumorigenesis by systemic tissue inhibitor of matrix metallopro-teinase 4 gene delivery. Cancer Res 2001; 61: 2365–2370.
99. Rudolph-Owen LA, Chan R, Muller WJ, Matrisian LM. Thematrix metalloproteinase matrilysin influences early-stagemammary tumorigenesis. Cancer Res 1998; 58: 5500–5506.
100. Masson R, Lefebvre O, Noel A, et al. In vivo evidence that thestromelysin-3 metalloproteinase contributes in a paracrine mannerto epithelial cell malignancy. J Cell Biol 1998; 140: 1535–1541.
101. Wu E, Mari BP, Wang F, Anderson IC, Sunday ME, Shipp MA.Stromelysin-3 suppresses tumor cell apoptosis in a murine model.J Cell Biochem 2001; 82: 549–555.
102. Bergers G, Brekken R, McMahon G, et al. Matrix metallo-proteinase-9 triggers the angiogenic switch during carcinogenesis.Nature Cell Biol 2000; 2: 737–744.
103. Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumorsuppression and cancer progression. Nature Genet 2001; 29:117–129.
104. Heissig B, Hattori K, Friedrich M, Rafii S, Werb Z. Angiogen-esis: vascular remodeling of the extracellular matrix involvesmetalloproteinases. Curr Opin Hematol 2003; 10: 136–141.
105. Seandel M, Noack-Kunnmann K, Zhu D, Aimes RT, Quigley JP.Growth factor-induced angiogenesis in vivo requires specificcleavage of fibrillar type I collagen. Blood 2001; 97: 2323–2332.
106. Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 suppliedby bone marrow-derived cells contributes to skin carcinogenesis.Cell 2000; 103: 481–490.
107. Fang J, Shing Y, Wiederschain D, et al. Matrix metallo-proteinase-2 is required for the switch to the angiogenicphenotype in a tumor model. Proc Natl Acad Sci USA 2000; 97:3884–3889.
108. Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H,Itohara S. Reduced angiogenesis and tumor progression ingelatinase A-deficient mice. Cancer Res 1998; 58: 1048–1051.
109. Zhou Z, Apte SS, Soininen R, et al. Impaired endochondralossification and angiogenesis in mice deficient in membrane-typematrix metalloproteinase I. Proc Natl Acad Sci USA 2000; 97:4052–4057.
110. Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrixmetalloproteinases regulate neovascularization by acting aspericellular fibrinolysins. Cell 1998; 95: 365–377.
111. Galvez BG, Matias-Roman S, Albar JP, Sanchez-Madrid F,Arroyo AG. Membrane type 1-matrix metalloproteinase isactivated during migration of human endothelial cells andmodulates endothelial motility and matrix remodeling. J BiolChem 2001; 276: 37 491–37 500.
112. Dong Z, Kumar R, Yang X, Fidler IJ. Macrophage-derivedmetalloelastase is responsible for the generation of angiostatinin Lewis lung carcinoma. Cell 1997; 88: 801–810.
113. Cornelius LA, Nehring LC, Harding E, et al. Matrix metallopro-teinases generate angiostatin: effects on neovascularization. Jimmunol 1998; 161: 6845–6852.
114. Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaisse J. Gener-ation and degradation of human endostatin proteins by variousproteinases. FEBS Lett 2000; 486: 247–251.
115. Kim YM, Jang JW, Lee OH, et al. Endostatin inhibits endothelialand tumor cellular invasion by blocking the activation andcatalytic activity of matrix metalloproteinase. Cancer Res 2000;60: 5410–5413.
116. Birchmeier C, Birchmeier W, Brand-Saberi B. Epithelial–mes-enchymal transitions in cancer progression. Acta Anat (Basel)1996; 156: 217–226.
117. Hegerfeldt Y, Tusch M, Brocker EB, Friedl P. Collective cellmovement from primary melanoma explants: plasticity ofcell–cell interaction, β1 integrin function and migrationstrategies. Cancer Res 2002; 62: 2125–2130.
118. Wolf K, Mazo I, Leung H, et al. Compensation mechanism intumor cell migration: mesenchymal–ameboid transition afterblocking of pericellular proteolysis. J. Cell Biol. 160: 267–277.
119. Koshikawa N, Gianelli G, Cirulli V, Miyazaki K, Quaranta V.Role of cell surface metalloprotease MT1-MMP in epithelial cellmigration over laminin-5. J Cell Biol 2000; 148: 615–624.
120. Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specificproteases in cancer cell intravasation as revealed by a novelsemiquantitative PCR-based assay. Cell 1998; 94: 353–362.
121. Hua J, Muschel RJ. Inhibition of matrix metalloproteinase 9expression by a ribozyme blocks metastasis in a rat sarcomamodel system. Cancer Res 1996; 56: 5279–5284.
122. Gorelik L, Flavell RA. Immune-mediated eradication of tumorsthrough the blockade of transforming growth factor-β signalingin T cells. Nature Med 2001; 7: 1118–1122.
123. Boulay A, Masson R, Chenard MP, et al. High cancer cell deathin syngeneic tumors developed in host mice deficient for thestromelysin-3 matrix metalloproteinase. Cancer Res 2001; 61:2189–2193.
124. Kataoka H, Uchimo H, Iwamura T, Seiki M, Nabeshima K,Koono M. Enhanced tumor growth and invasiveness in vivo by acarboxyl-terminal fragment of α1-proteinase inhibitor generatedby matrix metalloproteinases: a possible modulatory role innatural killer cytotoxicity. Am J Pathol 1999; 154: 457–468.
125. Muller A, Homey B, Soto H, et al. Involvement of chemokinereceptors in breast cancer metastasis. Nature 2001; 410: 50–56.
126. Chambers AF, Matrisian LM. Changing views of the role ofmatrix metalloproteinases in metastasis. J Natl Cancer Inst 1997;89: 1260–1270.
127. Coussens LM, Fingleton B, Matrisian LM. Matrix metallopro-teinase inhibitors and cancer: trials and tribulations. Science 2002;295: 2387–2392.
128. Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA.Marimastat as first-line therapy for patients with unresectablepancreatic cancer: a randomized trial. J Clin Oncol 2001; 19:3447–3455.
129. Hidaldo M, Eckhardt SG. Development of matrix metallopro-teinase inhibitors in cancer therapy. J Natl Cancer Inst 2001;93: 178–193.
130. Garbisa S, Biggin S, Cavallarin N, Sartor L, Benelli R, Albini A.Tumor invasion: molecular shears blunted by green tea. NatureMed 1999; 5: 1216.
131. Jiang MC, Liao CF, Lee PH. Aspirin inhibits matrix metallo-proteinase-2 activity, increases E-cadherin production, andinhibits in vitro invasion of tumor cells. Biochem Biophys ResCommun 2001; 282: 671–677.
132. Koivunen E, Arap W, Valtanen H, et al. Tumor targeting witha selective gelatinase inhibitor. Nature Biotechnol 1999; 17:768–774.
133. Yonemura Y, Endo Y, Fujita H, et al. Inhibition of peritonealdissemination in human gastric cancer by MMP-7-specificantisense oligonucleotide. J Exp Clin Cancer Res 2001; 20:205–212.
134. Kondraganti S, Mohanam S, Chintala SK, et al. Selectivesuppression of matrix metalloproteinase-9 in human glioblastomacells by antisense gene transfer impairs glioblastoma cellinvasion. Cancer Res 2000; 60: 6851–6855.
135. Silletti S, Kessler T, Goldberg J, Boger DL, Cheresh DA.Disruption of matrix metalloproteinase 2 binding to integrin αvβ3by an organic molecule inhibits angiogenesis and tumor growthin vivo. Proc Natl Acad Sci USA 2001; 98: 119–124.
136. D’Armiento, DiColandrea T, Dalal SS, et al. Collagenaseexpression in transgenic mouse skin causes hyperkeratosis andacanthosis and increases susceptibility to tumorigenesis. Mol CellBiol 1995; 15: 5732–5739.
137. Sternlicht MD, Lochter A, Sympson CJ, et al. The stromal pro-teinase MMP3/stromelysin-1 promotes mammary carcinogenesis.Cell 1999; 98: 137–146.
138. Matrisian LM. Cancer biology: extracellular proteinases inmalignancy. Curr Biol 1999; 9: R776–R778.
139. Ha HY, Moon HB, Nam MS, et al. Overexpression of mem-brane-type matrix metalloproteinase-1 gene induces mammarygland abnormalities and adenocarcinoma in transgenic mice.Cancer Res 2001; 61: 984–990.
140. Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM.Intestinal tumorigenesis is suppressed in mice lacking the met-alloproteinase matrilysin. Proc Natl Acad Sci USA 1997; 94:1402–1407.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 200: 448–464.
Related Documents











