Correspondence: V. S. Parmar, Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi–110 007, India. Tel/Fax: 91-11-27667206. E-mail: [email protected] (Received 28 May 2009; revised 21 December 2009; accepted 26 February 2010) ISSN 1024-2422 print/ISSN 1029-2446 online © 2010 Informa UK Ltd. (Informa Healthcare, Taylor & Francis AS) DOI: 10.3109/10242421003734704 ORIGINAL ARTICLE Enantioselective biocatalytic reactions on ( )-aryl alkyl ketones with native and modified porcine pancreatic lipase MOFAZZAL HUSAIN 1,2,3 , VINEET KUMAR 1,4 , RAJESH KUMAR 1 , NAJAM A. SHAKIL 1,8 , SUNIL K. SHARMA 1 , ASHOK K. PRASAD 1 , CARL E. OLSEN 5 , RAJENDRA K. GUPTA 2 , SANJAY V. MALHOTRA 4 , ERIK VAN DER EYCKEN 3 , ANTHONY L. DEPASS 6 , KALLE LEVON 7 & VIRINDER S. PARMAR 1 1 Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi, India, 2 School of Biotechnology, GGSIP University, Delhi, India, 3 Laboratory for Organic & Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, Katholieke Universiteit Leuven, Leuven, Belgium, 4 Laboratory of Synthetic Chemistry, Development Therapeutics Program Support, National Cancer Institute–Frederick, Frederick, Maryland, USA, 5 Department of Natural Sciences, Faculty of Life Sciences, University of Copenhagen, Frederiksberg, Denmark, 6 Department of Biology, Long Island University, Brooklyn, New York, USA, 7 Department of Chemistry, Polytechnic University, Brooklyn, New York, USA and 8 Division of Agricultural Chemicals, Indian Agricultural Research Institute, Pusa, New Delhi, India Abstract Porcine pancreatic lipase (PPL), pre-incubated with acetophenone in tetrahydrofuran, fails to recognize ortho- and para- acyloxy functions with respect to the nuclear carbonyl group in ( )-2,4-diacyloxyphenyl alkyl ketones and produces novel aryl alkyl ketones in moderate-to-highly optically active forms; this result supports the hypothesis on the mechanism of action of PPL in deacylation reactions on peracylated polyphenolics. Keywords: Lipase, aryl akyl ketones, enzymatic deacylation, Schiff's base complex Biocatalysis and Biotransformation, May–June 2010; 28(3): 172–184 Introduction Polyphenolics are secondary metabolites of plants and possess an array of biological activities (Amakura et al. 2008; Bracke et al. 2008; Naithani et al. 2008; Ghosh & Scheepens 2009). The synthe- sis of a variety of natural polyphenolic compounds such as coumarins (Ghate et al. 2005), chalcones (Narender et al. 2007; Sashidhara et al. 2009), fla- vones (Tang et al. 2005), flavanones (Wang & Cheng 2006; Pathak et al. 2008) and chromanones (Cham- bers & Marfat 1994) requires polyhydroxyaryl alkyl ketones as substrates. Synthesis of enantiomerically pure heterocyclic polyphenolic compounds can be achieved either by the use of enantiomerically pure substrates or by the resolution of final racemic prod- ucts. The use of enzymes in a synthetic sequence provides unique advantages of efficiency, stereospeci- ficity and environmental friendliness (Walsh 2001; Drauz & Waldmann 2002; Bommarius & Riebel 2004; Faber 2004; Jestin & Kaminski 2004; Valetti & Gilardi 2004; Chica et al. 2005; Khersonsky et al. 2006; Rubin-Pitel & Zhao 2006). We have pre- viously demonstrated the capabilities of lipases from porcine pancreas (PPL), Candida rugosa (CRL), Candida antarctica (CAL) and Pseudomonas spp. for carrying out the selective protection/deprotection of hydroxyl groups in different classes of compounds, such as aryl alkyl ketones (Kumar et al. 1998; Prasad et al. 1999; Kumar et al. 2001), hydroxymethylated phenolic compounds (Parmar et al. 1999), triazolyl sugars (Prasad et al. 2002; Bhattacharya et al. 2003), benzoxazines (Shakil et al. 2003), dihydrocoumarins (Singh et al. 2003), chromanones (Poonam et al. 2001) and modified sugar molecules (Prasad et al. 2007; Maity et al. 2008). It has been observed by us that PPL in tetrahydrofuran (THF) mediates the deacety- lation of all other acetoxy groups except the one at the ortho or peri position(s) to the nuclear carbonyl

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Correspondence: V. S. Parmar, Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi–110 007, India. Tel/Fax: � 91-11-27667206. E-mail: [email protected]
(Received 28 May 2009; revised 21 December 2009; accepted 26 February 2010)
ISSN 1024-2422 print/ISSN 1029-2446 online © 2010 Informa UK Ltd. (Informa Healthcare, Taylor & Francis AS)DOI: 10.3109/10242421003734704
ORIGINAL ARTICLE
Enantioselective biocatalytic reactions on ( � )-aryl alkyl ketones with native and modifi ed porcine pancreatic lipase
MOFAZZAL HUSAIN 1,2,3 , VINEET KUMAR 1,4 , RAJESH KUMAR 1 , NAJAM A. SHAKIL 1,8 , SUNIL K. SHARMA 1 , ASHOK K. PRASAD 1 , CARL E. OLSEN 5 , RAJENDRA K. GUPTA 2 , SANJAY V. MALHOTRA 4 , ERIK VAN DER EYCKEN 3 , ANTHONY L. DEPASS 6 , KALLE LEVON 7 & VIRINDER S. PARMAR 1
1 Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi, India, 2 School of Biotechnology, GGSIP University, Delhi, India, 3 Laboratory for Organic & Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, Katholieke Universiteit Leuven, Leuven, Belgium, 4 Laboratory of Synthetic Chemistry, Development Therapeutics Program Support, National Cancer Institute – Frederick, Frederick, Maryland, USA, 5 Department of Natural Sciences, Faculty of Life Sciences, University of Copenhagen, Frederiksberg, Denmark, 6 Department of Biology, Long Island University, Brooklyn, New York, USA, 7 Department of Chemistry, Polytechnic University, Brooklyn, New York, USA and 8Division of Agricultural Chemicals, Indian Agricultural Research Institute, Pusa, New Delhi, India
Abstract Porcine pancreatic lipase (PPL), pre-incubated with acetophenone in tetrahydrofuran, fails to recognize ortho - and para -acyloxy functions with respect to the nuclear carbonyl group in ( � )-2,4-diacyloxyphenyl alkyl ketones and produces novel aryl alkyl ketones in moderate-to-highly optically active forms; this result supports the hypothesis on the mechanism of action of PPL in deacylation reactions on peracylated polyphenolics.
Keywords: Lipase , aryl akyl ketones , enzymatic deacylation , Schiff's base complex
Biocatalysis and Biotransformation, May–June 2010; 28(3): 172–184
Introduction
Polyphenolics are secondary metabolites of plants and possess an array of biological activities (Amakura et al. 2008; Bracke et al. 2008; Naithani et al. 2008; Ghosh & Scheepens 2009). The synthe-sis of a variety of natural polyphenolic compounds such as coumarins (Ghate et al. 2005), chalcones (Narender et al. 2007; Sashidhara et al. 2009), fl a-vones (Tang et al. 2005), fl avanones (Wang & Cheng 2006; Pathak et al. 2008) and chromanones (Cham-bers & Marfat 1994) requires polyhydroxyaryl alkyl ketones as substrates. Synthesis of enantiomerically pure heterocyclic polyphenolic compounds can be achieved either by the use of enantiomerically pure substrates or by the resolution of fi nal racemic prod-ucts. The use of enzymes in a synthetic sequence provides unique advantages of effi ciency, stereospeci-fi city and environmental friendliness (Walsh 2001; Drauz & Waldmann 2002; Bommarius & Riebel
2004; Faber 2004; Jestin & Kaminski 2004; Valetti & Gilardi 2004; Chica et al. 2005; Khersonsky et al. 2006; Rubin-Pitel & Zhao 2006). We have pre-viously demonstrated the capabilities of lipases from porcine pancreas (PPL), Candida rugosa (CRL), Candida antarctica (CAL) and Pseudomonas spp. for carrying out the selective protection/deprotection of hydroxyl groups in different classes of compounds, such as aryl alkyl ketones (Kumar et al. 1998; Prasad et al. 1999; Kumar et al. 2001), hydroxymethylated phenolic compounds (Parmar et al. 1999), triazolyl sugars (Prasad et al. 2002; Bhattacharya et al. 2003), benzoxazines (Shakil et al. 2003), dihydrocoumarins (Singh et al. 2003), chromanones (Poonam et al. 2001) and modifi ed sugar molecules (Prasad et al. 2007; Maity et al. 2008). It has been observed by us that PPL in tetrahydrofuran (THF) mediates the deacety-lation of all other acetoxy groups except the one at the ortho or peri position(s) to the nuclear carbonyl

Enantioselective biocatalytic reactions with native and modifi ed PPL 173
group in chalcones (Parmar et al. 1992a), acetophe-nones (Parmar et al. 1992b), desoxybenzoins (Parmar et al. 1998b) and fl avones (Parmar et al. 1993). To explain the mechanism of selective deacetylation catalyzed by PPL, we postulated that the nuclear carbonyl group present in the substrate is engaged in the formation of a transient (dynamic) Schiff's base-type complex with the ξ -amino function of the lysine residue present in the active site of PPL (by analogy to the human pancreatic lipase). The for-mation of this complex causes the ortho / peri -acetoxy function to be embedded under the hydrophobic bulk of the active site of the enzyme and the serine-OH of the lipase takes part in deacetylation of other more suitably placed acetoxy function(s) in the same mol-ecule (Bisht et al. 1996). However, no direct proof for this mechanism could be given as the structure of the active site of PPL is not known and also because the Schiff's base formation, being a transient (dynamic) process, could not be verifi ed. However, a similar formation of Schiff's base at the ξ -amino function of the lysine residue present in the active site of CRL was later reported by Weber et al. (1997). Esters and amides of aromatic hydroxy acids do not form Schiff's bases easily; consequently the incubation of esters and amides of polyacetoxy aromatic acids with PPL leads to the formation of ortho hydroxy compounds together with other possible products (Parmar et al. 1997, 1998a). These fi ndings substantiate our hypothesis on the presence of lysine in the active site of the PPL and the formation of the Schiff's base-type complexes during deacetylation of peracetates of polyphenolic compounds bearing a nuclear carbonyl group.
Earlier we reported the deacetylation of 2,4-diacetoxyphenyl alkyl ketone catalyzed by modifi ed PPL (pre-incubated with acetophenone for 1 h) in con-fi rmation of our hypothesis on dynamic Schiff's base complex formation (Shakil et al. 2001). We report herein the expanded study to prove our dynamic Schiff's base complex formation hypothesis by studying the Raman spectra of native and modifi ed PPL. Further, deacylation reactions on a series of novel ( � )-2,4-diacyloxyphenyl alkyl ketones by modifi ed PPL have been carried out, providing access to a large number of enantiomerically enriched, useful novel synthons.
Experimental
Melting points were determined either on a Mettler FP62 (Columbus, OH, USA) instrument or in a sul-furic acid bath and are uncorrected. The IR spectra were recorded on a Perkin–Elmer (Waltham, MA, USA) model 2000 FT-IR spectrophotometer. The 1 H NMR and 13 C NMR spectra were recorded on a Bruker Avance 300 spectrometer (Billerica, MA,
USA) at 300 and 75.5 MHz, respectively, using TMS as an internal standard. The chemical shift values are on the δ scale and the coupling constants ( J ) are in Hz. Optical rotations were measured with a Bellingham–Stanley (Kent, UK) AD 220 pola-rimeter. The accurate masses were determined by measuring FAB-HRMS spectra on a JEOL (Tokyo, Japan) JMS-AX505W high-resolution mass spec-trometer in positive mode using the matrix HEDS (bishydroxydiethylsulfi de) doped with sodium acetate or on a Kratos (Manchester, UK) MS50TC instru-ment in the EI mode at a resolution of 10 000. The Raman spectra of different samples were recorded on a Thermo Electron Corporation (Waltham, MA, USA), data handling NXR FT-Raman module cou-pled to Nicolet (Waltham, MA, USA) 5700 FT-IR Omnic version 7.3 software using laser power of 1.0 W. The enzyme, PPL (porcine pancreatic lipase, Type-II), was purchased from Sigma Chemical Co. (St Louis, MO, USA) and used after storing in vacuo over P 2 O 5 for 12 h. THF was redistilled and dried over molecular sieves (4 Å). TLC was carried out on commercially available Merck (Darmstadt, Germany) silica gel 60F 254 plates.
General procedure for preparation of ( � )-2,4-dihydroxyphenyl alkyl ketones, 2a–2d (Scheme 1)
The heterogeneous mixture of fused ZnCl 2 (2 g, 15 mmol) and the racemic acid ( 1a – 1d , 10 mmol) was heated slowly with stirring to make the mixture homogeneous. Resorcinol (1.1 g, 10 mmol) was added and the reaction mixture was kept for about 2 h at 150°C, poured onto crushed ice containing hydrochloric acid (1:1) and extracted with dichlo-romethane (2 � 50 mL). The composition is v/v. The organic layer was washed with sodium bicarbonate solution (5%), the % composition is w/v, dried over anhydrous Na 2 SO 4 and the solvent removed under reduced pressure. The residue thus obtained was purifi ed by column chromatography using petro-leum ether–ethyl acetate as eluting solvent to afford racemic 2,4-dihydroxyphenyl alkyl ketones 2a – 2d as viscous oils in 50–55% yields (Shakil et al. 2001). The composition of the petroleum ether-ethyl ace-tate mixture is: ‘90/10 (v/v) throughout the text.
General procedure for acetylation of ( � )-2,4-dihydroxyphenyl alkyl ketones, 2a–2d: preparation of ( � )-diacetates, 3a–3d (Scheme 1)
( � )-2,4-Dihydroxyphenyl alkyl ketone ( 2a – 2d , 3 mmol) was stirred with acetic anhydride and a cata-lytic amount of N , N -dimethylaminopyridine (DMAP) at 22–25°C. The reaction was monitored periodically
174 M. Husain et al .
by TLC and, on completion, poured into ice-cold water (50 mL). The compound was extracted with dichloromethane (2 � 25 mL), the combined organic layer dried over anhydrous Na 2 SO 4 and the solvent removed under reduced pressure. The crude product thus obtained was purifi ed by column chromatogra-phy using petroleum ether–ethyl acetate as eluent to afford ( � )-2,4-diacetoxyphenyl alkyl ketones 3a – 3d as viscous oils in 90–95% yields (Shakil et al. 2001).
General procedure for deacetylation of ( � )-2,4-diacetoxyphenyl alkyl ketones (3a–3d) mediated by modifi ed porcine pancreatic lipase
To a solution of acetophenone (0.25 g, 2 mmol) in dry THF (30 mL), PPL (250 mg) was added and the suspension was stirred at 40–42°C for 1 h in an incubator shaker. A solution of ( � )-2,4-diacetoxyphenyl alkyl ketone ( 3a – 3d , 2 mmol) in THF (10 mL) was added to the incubated suspen-sion, followed by the addition of n-butanol (5 molar equivalents) and stirring continued at the same tem-perature. The reaction was stopped at three different stages (Scheme 2).
(1) When the dihydroxy compounds just started to appear; reaction was quenched by fi ltering off the enzyme, solvent was removed under reduced pressure and the crude product was purifi ed by column chromatography over silica gel using a gradient solvent system of chloroform–acetone (The composition of the chloroform-acetone mixture is ‘85/15 (v/v) throughout the text) to afford the opti-cally enriched, recovered ( � )-2,4-diacetoxyphenyl alkyl ketones 3a – 3d and the mono-deacetylated (–)-2-acetoxy-4-hydrox-yphenyl alkyl ketones 4a – 4d ; yields and
optical rotation values are given in Table I. All of the monoacetates 4a – 4d were charac-terized on the basis of their spectral data and also by comparing their spectral data with those given in the literature. The spectral data of ( � )-2,4-diacetoxyphenyl alkyl ketones 3a – 3d were found to be identical to the cor-responding data of ( � )-2,4-diacetoxyphenyl alkyl ketones 3a – 3d synthesized by acetyla-tion of ( � )-2,4-dihydroxyphenyl alkyl ketones 2a – 2d and also found to be identical to spectral data given in the literature (Shakil et al. 2001).
(2) After 50% conversion of the starting materi-als 3a – 3d into the products, the reaction was quenched by fi ltering off the enzyme, the sol-vent was removed under reduced pressure and the crude products were purifi ed by col-umn chromatography over silica gel using a gradient solvent system of chloroform–acetone to afford the optically enriched, ( � )-2,4-diacetoxyphenyl alkyl ketones 3a – 3d , mono-deacetylated (–)-2-acetoxy-4-hydrox-yphenyl alkyl ketones 4a – 4d and the ( � )-2,4-dihydroxyphenyl alkyl ketones 2a – 2d ; yields and optical rotation values are given in Table II. All of the monoacetates 4a – 4d , dia-cetates 3a – 3d and dihydroxy ketones 2a – 2d were characterized on the basis of their spec-tral data and also by comparison with spec-tral data given in the literature (Shakil et al. 2001).
(3) After total conversion of starting materials 3a – 3d into the products, the reaction mix-ture was quenched by fi ltering off the enzyme and the solvent removed under reduced pres-sure; the crude products were purifi ed by
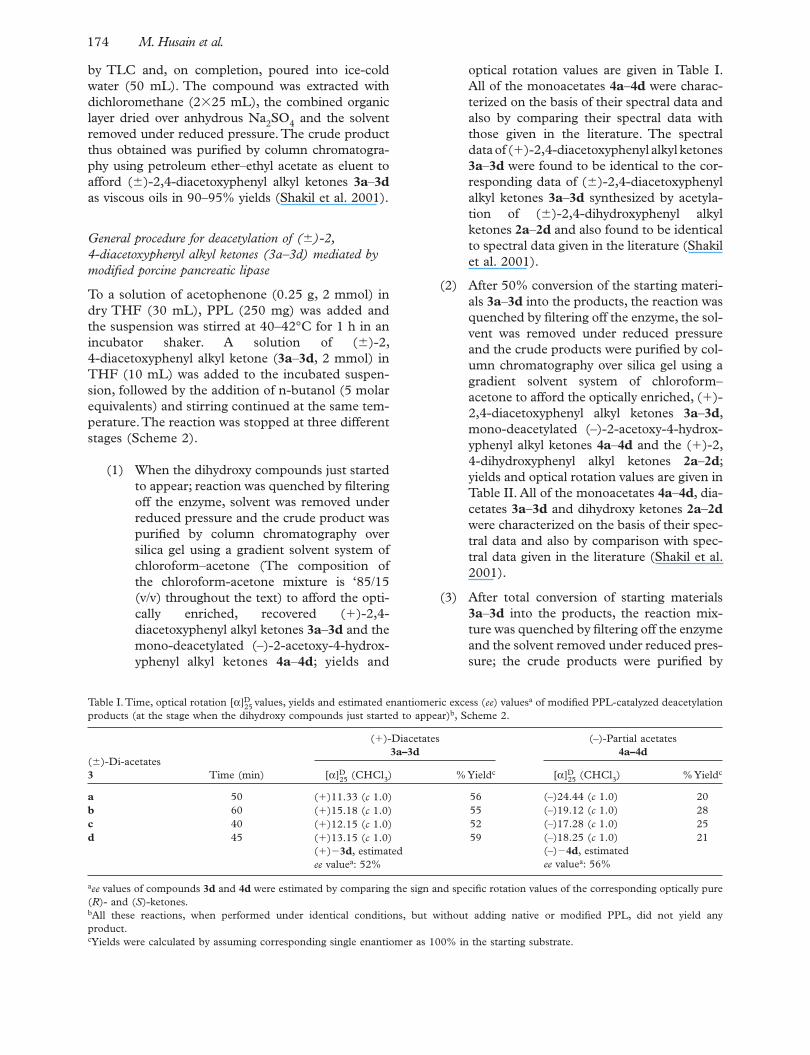
Table I. Time, optical rotation [α]D25 values, yields and estimated enantiomeric excess ( ee ) values a of modifi ed PPL-catalyzed deacetylation
products (at the stage when the dihydroxy compounds just started to appear) b , Scheme 2.
( � )-Di-acetates 3 Time (min)
( � )-Diacetates 3a – 3d
(–)-Partial acetates 4a – 4d
[α]D25 (CHCl 3 ) % Yield c [α]D
25 (CHCl 3 ) % Yield c
a 50 ( � )11.33 ( c 1.0) 56 (–)24.44 ( c 1.0) 20 b 60 ( � )15.18 ( c 1.0) 55 (–)19.12 ( c 1.0) 28 c 40 ( � )12.15 ( c 1.0) 52 (–)17.28 ( c 1.0) 25 d 45 ( � )13.15 ( c 1.0)
( � )� 3d , estimated ee value a : 52%
59 (–)18.25 ( c 1.0)(–)� 4d , estimated ee value a : 56%
21
a ee values of compounds 3d and 4d were estimated by comparing the sign and specifi c rotation values of the corresponding optically pure ( R )- and ( S )-ketones. b All these reactions, when performed under identical conditions, but without adding native or modifi ed PPL, did not yield any product. c Yields were calculated by assuming corresponding single enantiomer as 100% in the starting substrate.
Enantioselective biocatalytic reactions with native and modifi ed PPL 175
column chromatography over silica gel using a gradient solvent system of chloroform–acetone to afford the optically enriched ( � )-2,4-dihydroxyphenyl alkyl ketones 2a – 2d and the mono-deacetylated (–)-2-acetoxy-4-hydroxyphenyl alkyl ketones 4a – 4d ; yields and optical rotation values are given in Table III. All of the monoacetates 4a – 4d and the dihydroxy ketones 2a – 2d were characterized on the basis of their spectral data and also by comparison with spectral data given in the literature (Shakil et al. 2001).
General procedure for preparation of ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones, 6a–6c (Scheme 3)
( � )-2,4-Dihydroxyphenyl (1-phenyl)propyl ketone ( 2d , 3 mmol) was stirred with the correspond-ing acid anhydride 5a – 5c (6.5 mmol) in pyridine (10 mL) at 40–45°C. The reaction was monitored
periodically by TLC and, on completion, poured into ice-cold water (50 mL). The compound thus obtained was extracted with dichloromethane (2 � 25 mL), the combined organic layer dried over anhydrous Na 2 SO 4 and the solvent removed under reduced pressure. The products were puri-fi ed by column chromatography using petroleum ether–ethyl acetate as eluting solvent to afford the ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a – 6c .
( � )-2,4-Dibutanoyloxyphenyl (1-phenyl)propyl ketone ( 6a ) was obtained as a viscous oil in 92% yield. IR (Nujol, cm– 1 ): 1768 (O–C � O); 1703 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.87 (3H, t, J � 7.3 Hz, C-3′H); 0.9–1.06 (6H, m, C-4′′′H); 1.68–1.78 (5H, m, C-2′H a and C-3′′′H); 2.09–2.16 (1H, m, C-2′H b ); 2.40–2.50 (4H, m, C-2′′′H); 4.20 (1H, t, J � 7.3 Hz, C-1′H); 6.88 (1H, d, J � 2.2 Hz, C-3H); 6.96 (1H, dd, J � 6.3 and 2.2 Hz, C-5H); 7.20–7.30 (5H, m, aromatic protons); 7.60 (1H, d, J � 8.6 Hz, C-6H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm):
Table III. Time, optical rotation [α]D25 values, yields and estimated enantiomeric excess ( ee ) values a of modifi ed PPL-catalyzed deacetylation
products (at the stage of total conversion of substrate) b , Scheme 2.
( � )-Diacetates 3 Time (h)
( � )-Dihydroxy compounds 2a – 2d
(–)-Partial acetates 4a – 4d
[α]D25 (CHCl 3 ) % Yield c [α]D
25 (CHCl 3 ) % Yield c
3a 15 ( � )24.81 ( c 0.8) 59 (–)40.00 ( c 0.8) 7 3b 16 ( � )20.68 ( c 0.8) 60 (–)25.16 ( c 0.8) 8 3c 14 ( � )23.20 ( c 0.8) 61 (–)28.30 ( c 0.8) 8 3d 13 ( � )28.18 ( c 0.8)
( � )- 2d , estimated ee value a : 80%
58 (–)32.66 ( c 0.8)(–)- 4d , estimated ee value a : 80%
8
a ee values of compounds 2d and 4d were estimated by comparing the sign and specifi c rotation values of the corresponding optically pure ( R )- and ( S )-ketones. b All these reactions, when performed under identical conditions, but without adding native or modifi ed PPL, did not yield any product. c Yields were calculated by assuming corresponding single enantiomer as 100% in the starting substrate.
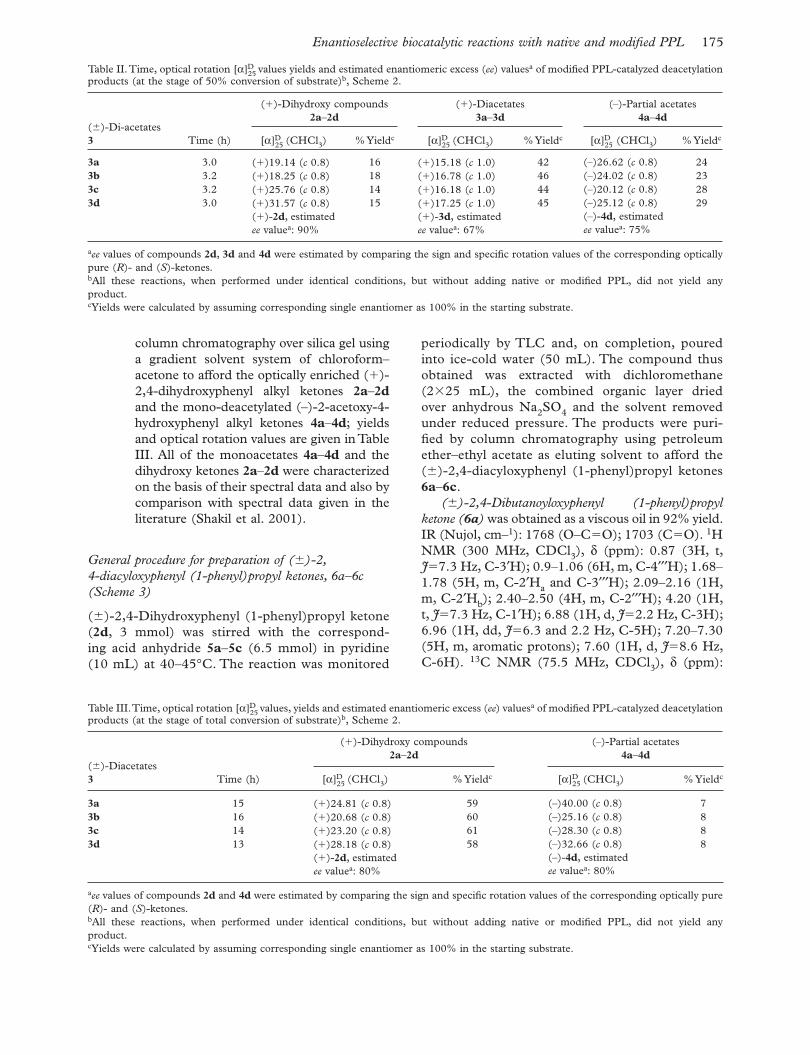
Table II. Time, optical rotation [α]D25 values yields and estimated enantiomeric excess ( ee ) values a of modifi ed PPL-catalyzed deacetylation
products (at the stage of 50% conversion of substrate) b , Scheme 2.
( � )-Di-acetates 3 Time (h)
( � )-Dihydroxy compounds 2a – 2d
( � )-Diacetates 3a – 3d
(–)-Partial acetates 4a – 4d
[α]D25 (CHCl 3 ) % Yield c [α]D
25 (CHCl 3 ) % Yield c [α]D25 (CHCl 3 ) % Yield c
3a 3.0 ( � )19.14 ( c 0.8) 16 ( � )15.18 ( c 1.0) 42 (–)26.62 ( c 0.8) 24 3b 3.2 ( � )18.25 ( c 0.8) 18 ( � )16.78 ( c 1.0) 46 (–)24.02 ( c 0.8) 23 3c 3.2 ( � )25.76 ( c 0.8) 14 ( � )16.18 ( c 1.0) 44 (–)20.12 ( c 0.8) 28 3d 3.0 ( � )31.57 ( c 0.8)
( � )- 2d , estimated ee value a : 90%
15 ( � )17.25 ( c 1.0)( � )- 3d , estimated ee value a : 67%
45 (–)25.12 ( c 0.8)(–)- 4d , estimated ee value a : 75%
29
a ee values of compounds 2d , 3d and 4d were estimated by comparing the sign and specifi c rotation values of the corresponding optically pure ( R )- and ( S )-ketones. b All these reactions, when performed under identical conditions, but without adding native or modifi ed PPL, did not yield any product. c Yields were calculated by assuming corresponding single enantiomer as 100% in the starting substrate.

176 M. Husain et al .
12.21 (C-3′); 13.52 (2 � C-4′′′); 18.08 (2 � C-3′′′); 26.96 (C-2′); 36.93 (2 � C-2′′′); 58.38 (C-1′); 115.94 (C-1); 117.27 (C-3); 118.64 (C-5); 127.05, 128.48 and 129.11 (C-2″, C-3″, C-4″, C-5″ and C-6″); 130.39 (C-6); 138.84 (C-1″); 149.74 and 153.23 (C-2 and C-4); 170.98 and 171.53 (2 � OCO); 199.45 (C � O). FAB-HRMS, m / z : 419.1839 ([M � Na] � ). Calc. for C 24 H 28 O 5 � Na: 419.1834.
( � )-2,4-Diheptanoyloxyphenyl (1-phenyl)propyl ketone ( 6b ) was obtained as a viscous oil in 92% yield. IR (Nujol, cm– 1 ): 1768 (O–C � O); 1706 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.84–0.91 (9H, m, C-3′H and C-7′′′H); 1.25–1.35 (12H, m, C-3′′′H, C-4′′′H and C-5′′′H); 1.65–1.81 (5H, m, C-2′H a and C-6′′′H); 2.11–2.16 (1H, m, C-2′H b ); 2.48–2.58 (4H, m, C-2′′′H); 4.20 (1H, t, J � 7.3 Hz, C-1′H); 6.87 (1H, d, J � 2.0 Hz, C-3H); 6.95 (1H, dd, J � 6.4 and 2.0 Hz, C-5H); 7.20–7.28 (5H, m, aromatic protons); 7.59 (1H, d, J � 8.5 Hz, C-6H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.56 (C-3′); 14.40 (2 � C-7′′′); 22.85 (2 � C-6′′′); 24.98 (2 � C-3′′′); 27.41 (C-2′); 29.17 (2 � C-4′′′); 31.81 (2 � C-5′′′); 34.65 (2 � C-2′′′); 58.90 (C-1′); 115.50 (C-1); 117.70 (C-3); 119.07 (C-5); 127.51, 128.52 and 129.33 (C-2″, C-3″, C-4″, C-5″ and C-6″); 130.83 (C-6); 139.30 (C-1″), 150.22 and 153.72 (C-2 and C-4); 171.62 and 172.15 (2 � OCO); 199.95(C � O). FAB-HRMS, m / z : 503.2771 ([M � Na] � ). Calc. for C 30 H 40 O 5 � Na: 503.2773.
( � )-2,4-Dibenzoyloxyphenyl (1-phenyl)propyl ketone ( 6c ) was obtained as a white solid in 94% yield; mp 87–89°C. IR (KBr, cm– 1 ): 1745 (O–C � O); 1677 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.87 (3H, t, J � 7.0 Hz, C-3′H); 1.80–1.87 (1H, m, C-2′H a ); 2.14–2.23 (1H, m, C-2′H b ); 4.31 (1H, t, J � 6.9 Hz, C-1′H); 6.87 (1H, d, J � 2.0 Hz, C-3H); 6.95 (1H, dd, J � 6.4 and 2.0 Hz, C-5H); 7.20–7.75 and 8.22 (16H, 2m, 12H and 4H each, aromatic protons and C-6H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.50 (C-3′); 27.16 (C-2′); 59.26 (C-1′); 115.45 (C-1); 117.82 (C-3); 119.23 (C-5); 127.51, 128.78, 129.04, 129.19, 130.42 and 130.62 (C-2″, C-3″, C-4″, C-5″, C-6″, 2 � C-2′′′, 2 � C-3′′′, 2 � C-4′′′, 2 � C-5′′′ and 2 � C-6′′′); 130.84 (C-6); 134.17 and 134.33 (2 � C-1′′′); 139.00 (C-1″); 150.13 and 153.82 (C-2 and C-4); 161.12 and 161.48 (2 � OCO); 200.15 (C � O). FAB-HRMS, m / z : 487.1560 ([M � Na] � ). Calc. for C 30 H 24 O 5 � Na: 487.1521.
( � )-2,4-Dicyclohexanoyloxyphenyl (1-phenyl)pro-pyl ketone ( 6d ) . ( � )-2,4-Dihydroxyphenyl (1-phe-nyl)propyl ketone ( 2d , 3 mmol) was stirred with cyclohexanoyl chloride 5d (6.5 mmol) and trieth-ylamine (Et 3 N) (6.5 mmol) in dichloromethane
(10 mL) at 30–35°C. The reaction was monitored periodically by TLC and, on completion, poured into ice-cold water (50 mL). The compound thus obtained was extracted with dichloromethane (2 � 25 mL), the combined organic layer dried over anhy-drous Na 2 SO 4 and the solvent removed under reduced pressure; the product was purifi ed by column chromatography using petroleum ether–ethyl acetate as eluting solvent to afford ( � )-2,4-dicyclohexanoylxyphenyl (1-phenyl)propyl ketone ( 6d ) as a viscous oil in 96% yield. IR (Nujol, cm– 1 ): 1760 (O–C � O); 1688 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.86 (3H, t, J � 7.35 Hz, C-3′H); 1.22–2.21 (22H, m, cyclohexyl rings protons and C-2′H); 2.50–2.54 (2H, m, C-1′′′H); 4.19 (1H, t, J � 7.21 Hz, C-1′H); 6.84 (1H, d, J � 2.11 Hz, C-3H); 6.92 (1H, dd, J � 6.43 and 2.10 Hz, C-5H); 7.20–7.30 (5H, m, aromatic protons); 7.55 (1H, d, J � 8.55 Hz, C-6H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.08 (C-3′); 25.18 and 25.65 (2 � C-3′′′, 2 � C-4′′′ and 2 � C-5′′′); 26.80 (C-2′); 28.73 (2 � C-2′′′ and 2 � C-6′′′); 43.08 (2 � C-1′′′); 58.51 (C-1′); 115.32 (C-1); 117.13 (C-3); 118.51 (C-5); 127.03, 128.72 and 129.45 (C-2″, C-3″, C-4″, C-5″ and C-6″); 130.17 (C-6); 138.76 (C-1″); 149.70 and 153.30 (C-2 and C-4); 173.41 and 173.79 (2 � OCO); 199.57 (C � O). FAB-HRMS: 476.2570 found. Calc. for C 30 H 36 O 5 : 476.2563.
( � )-2,4-Di- α -naphthoyloxyphenyl (1-phenyl)propyl ketone ( 6e ) was prepared as 6d (except that the acid chloride 5e was used). It was obtained as a viscous oil in 93% yield. IR (Nujol, cm– 1 ): 1738 (O–C � O); 1687 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.79 (3H, t, J � 7.32 Hz, C-3′H); 1.71–1.79 (1H, m, C-2′H a ); 2.12–2.14 (1H, m, C-2′H b ); 4.32 (1H, t, J � 7.23 Hz, C-1′H); 7.17–7.30 (7H, m, C-3H, C-5H, C-2″H, C-3″H, C-4″H, C-5″H and C-6″H); 7.53–7.80 (6H, m, C-3′′′H, C-6′′′H and C-7′′′H); 7.90–7.94 (2H, m, C-5′′′H); 8.09–8.19 (3H, m, C-6H and C-4′′′H); 8.48 (2H, dd, J � 9.90 and 7.34 Hz, C-2′′′H); 9.01 (2H, d, J � 8.52 Hz, C-8′′′H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.03 (C-3′); 26.80 (C-2′); 58.70 (C-1′); 115.50 (C-1); 117.95 (C-3); 119.52 (C-5); 124.52, 125.73, 126.31, 127.03, 127.51, 128.12, 128.78, 129.04, 129.19, 130.42, 130.57, 130.62 and 131.61 (C-2″, C-3″, C-4″, C-5″, C-6″, 2 � C-1′′′, 2 � C-2′′′, 2 � C-3′′′, 2 � C-4′′′, 2 � C-5′′′, 2 � C-6′′′, 2 � C-7′′′ and 2 � C-8′′′); 131.84 and 133.86 (2 � C-10′′′ and C-6); 134.17 (2 � C-9′′′); 138.62 (C-1″); 150.33 and 153.57 (C-2 and C-4); 161.12 and 161.48 (2 � OCO); 200.15 (C � O). FAB-HRMS: 564.1930 found. Calc. for C 38 H 28 O 5 : 564.1937.

Enantioselective biocatalytic reactions with native and modifi ed PPL 177
General procedure of enzymatic deacylation of ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a–6e by native porcine pancreatic lipase (Scheme 4)
To a solution of the ( � )-2,4-diacyloxyphenyl (1-phe-nyl)propyl ketones ( 6a – 6e , 2 mmol) in anhydrous THF (20–25 mL), n-butanol (5 equivalents) was added, followed by the addition of native PPL (300 mg). The suspension was incubated in a shaker at 40–42°C and the progress of the reaction was mon-itored periodically by HPLC and/or TLC examina-tion. After about 50% conversion of the starting material into the product, the reaction mixture was quenched by fi ltering off the enzyme and the solvent removed under reduced pressure to afford a thick oil that was purifi ed by column chromatography over silica gel using a gradient solvent system of chloro-form–acetone to afford optically enriched, ( S )-( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a – 6d and the mono-deacylated ( R )-(–)-2-acyloxy-4-hy-droxyphenyl (1-phenyl)propyl ketones 7a – 7d in 65–70% and 60–68% yields, respectively (Table V). The spectral data of ( S )-( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a – 6d were found to be identical with the data of ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a – 6d given above.
( R )-( – )-2-Butanoylxy-4-hydroxyphenyl (1-phenyl)propyl ketone ( 7a ) was obtained as a viscous oil in 60% yield. [α]D
25 : –31.42 ( c 0.4, CHCl 3 ). IR (Nujol, cm– 1 ): 3305 (OH); 1731 (O-C � O); 1670 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.84 (3H, t, J � 7.3 Hz, C-3′H); 1.01 (3H, t, J � 7.3 Hz, C-4′′′H); 1.72–1.79 (3H, m, C-2′H a and C-3′′′H); 2.07–2.09 (1H, m, C-2′H b ); 2.58 (2H, t, J � 7.4 Hz, C-2′′′H); 4.20 (1H, t, J � 7.2 Hz, C-1′H); 6.88 (1H, d, J � 2.2 Hz, C-3H); 6.99 (1H, dd, J � 6.4 and 2.14 Hz, C-5H); 7.18–7.30 (5H, m, aromatic protons); 7.42 (1H, d, J � 9.0 Hz, C-6H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.45 (C-3′); 14.01 (C-4′′′); 18.43 (C-3′′′); 27.46 (C-2′); 36.50 (C-2′′′); 57.81 (C-1′); 111.7 (C-3); 113.77 (C-5); 123.42 (C-1); 127.36, 128.57 and 129.17 (C-2″, C-3″, C-4″, C-5″ and C-6″); 132.04 (C-6); 139.94 (C-1″); 151.45 and 160.89 (C-4 and C-2); 173.70 (OCO); 199.57(C � O). FAB-HRMS, m / z : 349.1439 ([M � Na] � ). Calc. for C 20 H 22 O 4 � Na: 349.1416.
( R )-( – )-2-Heptanoyloxy-4-hydroxyphenyl (1-phenyl)propyl ketone ( 7b ) was obtained as a viscous oil in 62% yield. [α]D
25: –26.32 ( c 0.4, CHCl 3 ). IR (Nujol, cm– 1 ): 3367 (OH); 1734 (O–C � O); 1675 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.83–0.93 (6H, m, C-3′H and C-7′′′H); 1.25–1.43 (6H, m, C-3′′′H, C-4′′′H and C-5′′′H); 1.69–1.78 (3H, m, C-2′H a and C-6′′′H); 2.06–2.13 (1H, m, C-2′H b ); 2.60 (2H, t, J � 7.5 Hz, C-2′′′H); 4.20 (1H, t, J � 7.2 Hz, C-1′H); 6.37–6.40 (2H, m, C-3H and C-5H); 7.19–7.29 (5H, m, aromatic protons); 7.49 (1H, d, J � 8.3 Hz, C-6H).
13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.58 (C-3′); 14.39 (C-7′′′); 22.88 (C-6′′′); 24.90 (C-3′′′); 27.47 (C-2′); 29.17 (C-4′′′); 31.86 (C-5′′′); 34.71 (C-2′′′); 57.87 (C-1′); 111.67 (C-3); 113.28 (C-5); 123.77 (C-1); 127.33, 128.59 and 129.14 (C-2″, C-3″, C-4″, C-5″ and C-6″); 132.35 (C-6); 140.01 (C-1″); 151.49 and 160.47 (C-4 and C-2); 173.53 (OCO); 199.19 (C � O). FAB-HRMS, m / z : 391.1893 ([M � Na] � ). Calc. for C 23 H 28 O 4 � Na: 391.1885.
( R )-( – )-2-Benzoyloxy-4-hydroxyphenyl (1-phenyl)propyl ketone ( 7c ) was obtained as a viscous oil in 68% yield. [α]D
25: –30.25 ( c 0.4, CHCl 3 ). IR (Nujol, cm– 1 ): 3345 (OH); 1750 (O–C � O); 1670 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.78 (3H, t, J � 7.0 Hz, C-3′H); 1.65–1.75 (1H, m, C-2′H a ); 2.01–2.11 (1H, m, C-2′H b ); 4.21 (1H, t, J � 6.8 Hz, C-1′H); 6.41–6.48 (2H, m, C-3H and C-5H); 7.18–7.50 (8H, m, aromatic protons); 7.60 (1H, d, J � 6.9 Hz, C-6H); 8.13–8.18 (2H, m, aromatic protons). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.54 (C-3′); 27.31 (C-2′); 58.17 (C-1′); 111.74 (C-3); 113.64 (C-5); 124.09 (C-1); 127.35, 128.75, 129.02, 129.61, 130.57 and 130.84 (C-2″, C-3″, C-4″, C-5″, C-6″, C-2′′′, C-3′′′, C-4′′′, C-5′′′ and C-6′′′); 132.27 (C-6); 134.21 (C-1′′′;, 139.78 (C-1′); 151.28 and 160.67 (C-4 and C-2); 166.31 (OCO); 199.59 (C � O). FAB-HRMS, m / z : 383.1245 ([M � Na] � ). Calc. for C 23 H 20 O 4 � Na: 383.1259.
( R )-( – )-2-Cyclohexanoylxy-4-hydroxyphenyl (1-phenyl)propyl ketone ( 7d ) was obtained as a vis-cous oil in 63% yield. [α]D
25: –25.78 ( c 0.4, CHCl 3 ). IR (Nujol, cm– 1 ): 3350 (OH); 1761 (O–C � O); 1700 (C � O). 1 H NMR (300 MHz, CDCl 3 ), δ (ppm): 0.86 (3H, t, J � 7.31 Hz, C-3′H); 1.25–2.11 (12H, m, cyclohexyl ring protons and C-2′H); 2.50–2.54 (1H, m, C-1′′′H); 4.19 (1H, t, J � 7.2 Hz, C-1′H); 6.34–6.38 (2H, m, C-3H and C-5H); 7.21–7.30 (5H, m, aromatic protons); 7.45 (1H, d, J � 8.4 Hz, C-6H). 13 C NMR (75.5 MHz, CDCl 3 ), δ (ppm): 12.13 (C-3′); 25.18 and 25.56 (C-3′′′, C-4′′′ and C-5′′′); 26.76 (C-2′); 28.74 (C-2′′′ and C-6′′′); 43.09 (C-1′′′); 54.86 (C-1′); 111.16 (C-3); 112.69 (C-5); 116. 97 (C-1); 127.23, 128.02 and 128.92 (C-2″, C-3″, C-4″, C-5″ and C-6″); 131.17 (C-6); 139.12 (C-1″); 156.70 and 164.67 (C-4 and C-2); 173.44 (OCO); 205.21 (C � O). FAB-HRMS: 366.1845 found. Calc. for C 23 H 26 O 4 : 366.1831.
General procedure for chemical acylation of (R)-(–)-2-acyloxy-4-hydroxyphenyl (1-phenyl)propyl ketones, 7a–7d
Compounds (–)- 6a – 6d were prepared by acylation of the corresponding monohydroxy compounds (–)- 7a – 7d as discussed above in 98–99% yields. The spectral data of the ( R )-(–)-diacylates 6a – 6d were identical to the data of the ( � )-diacylates 6a – 6d reported above.
178 M. Husain et al .
General procedure for deacylation of ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones, 6a–6e, mediated by modifi ed porcine pancreatic lipase (Scheme 3)
To a solution of acetophenone (0.25 g, 2 mmol) in dry THF (30 mL), PPL (250 mg) was added and the suspension was stirred at 40–42°C for 1 h in an incu-bator shaker. A solution of ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a – 6e (2 mmol) in THF (10 mL) was added into the incubated suspension, followed by the addition of n-butanol (5 molar equiv-alents) and stirring continued at the same tempera-ture. The reaction was stopped after 50% conversion of substrate into products by fi ltering off the enzyme and the solvent was removed under reduced pressure. The crude product was purifi ed by column chroma-tography over silica gel using a gradient solvent sys-tem of chloroform–acetone to afford optically enriched, ( S )-( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones 6a – 6d , mono-deacylated ( R )-(–)-2-acyloxy-4-hydroxyphenyl (1-phenyl)propyl ketones
7a – 7d and ( S )-( � )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone 2d ; the yields, optical rotation values and enantiomeric excess values of all the products are given in Table IV.
Results and discussion
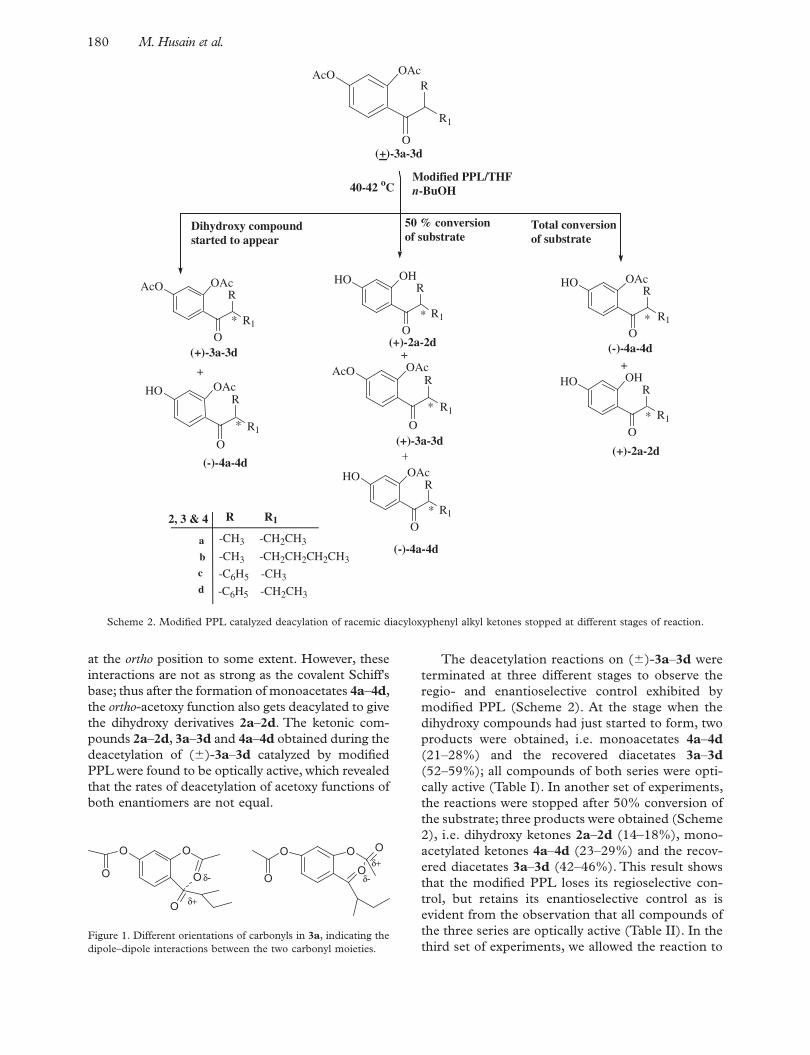
The racemic aryl alkyl ketones 2a – 2d were prepared by the Nencki reaction (Nencki & Sieber 1881) of resorcinol with the corresponding racemic aliphatic acids 1a – 1d in the presence of fused ZnCl 2 at 150°C in 50–55% yields; the diacetates 3a – 3d were pre-pared by acetylation of 2a – 2d using the acetic anhydride/DMAP method in quantitative yields (Scheme 1). All of the ( � )-dihydroxyphenyl alkyl ketones 2a – 2d and the ( � )-diacetoxyphenyl alkyl ketones 3a – 3d were characterized by comparing their spectral data with those given in the literature (Shakil et al. 2001). We have earlier reported the lack of selectivity of modifi ed PPL during the deacetylation of 2,4-diacetoxyphenyl methyl ketone
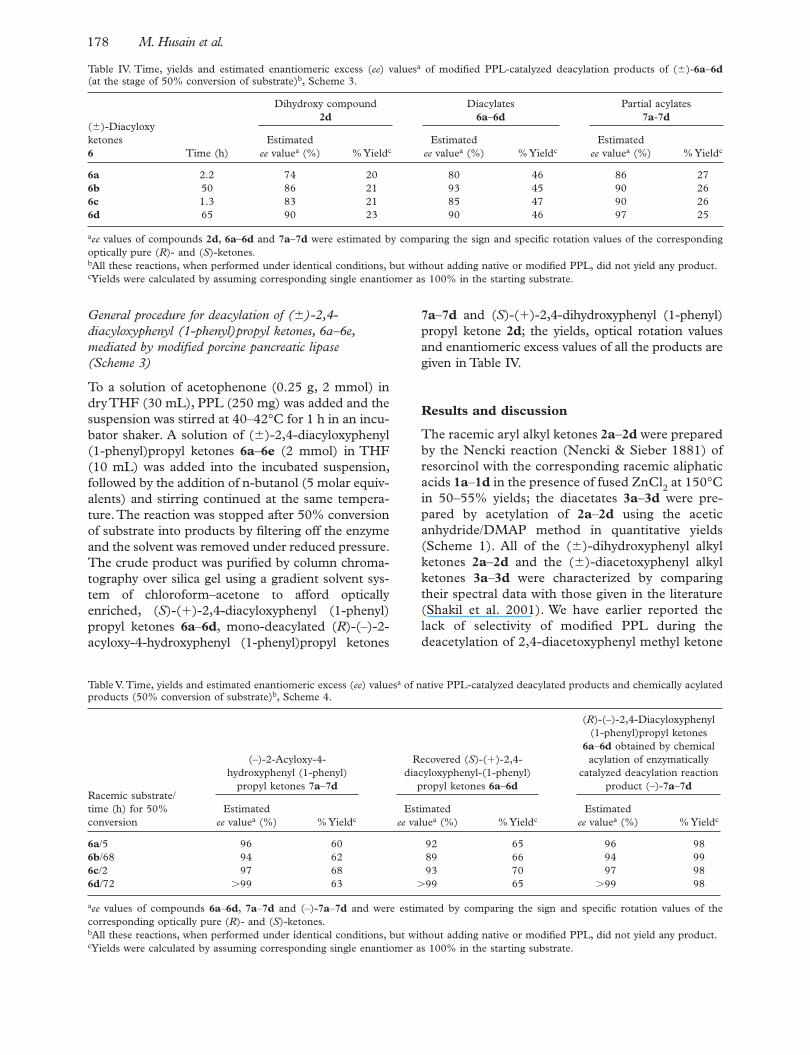
Table IV. Time, yields and estimated enantiomeric excess ( ee ) values a of modifi ed PPL-catalyzed deacylation products of ( � )- 6a – 6d (at the stage of 50% conversion of substrate) b , Scheme 3.
( � )-Diacyloxy ketones 6 Time (h)
Dihydroxy compound 2d
Diacylates 6a – 6d
Partial acylates 7a - 7d
Estimated ee value a (%) % Yield c
Estimated ee value a (%) % Yield c
Estimated ee value a (%) % Yield c
6a 2.2 74 20 80 46 86 27 6b 50 86 21 93 45 90 26 6c 1.3 83 21 85 47 90 26 6d 65 90 23 90 46 97 25
a ee values of compounds 2d , 6a – 6d and 7a – 7d were estimated by comparing the sign and specifi c rotation values of the corresponding optically pure ( R )- and ( S )-ketones. b All these reactions, when performed under identical conditions, but without adding native or modifi ed PPL, did not yield any product. c Yields were calculated by assuming corresponding single enantiomer as 100% in the starting substrate.
Table V. Time, yields and estimated enantiomeric excess ( ee ) values a of native PPL-catalyzed deacylated products and chemically acylated products (50% conversion of substrate) b , Scheme 4.
Racemic substrate/time (h) for 50% conversion
(–)-2-Acyloxy-4-hydroxyphenyl (1-phenyl)
propyl ketones 7a – 7d
Recovered ( S )-( � )-2,4-diacyloxyphenyl-(1-phenyl)
propyl ketones 6a – 6d
( R )-(–)-2,4-Diacyloxyphenyl (1-phenyl)propyl ketones
6a – 6d obtained by chemical acylation of enzymatically
catalyzed deacylation reaction product (–)- 7a – 7d
Estimated ee value a (%) % Yield c
Estimated ee value a (%) % Yield c
Estimated ee value a (%) % Yield c
6a /5 96 60 92 65 96 98 6b /68 94 62 89 66 94 99 6c /2 97 68 93 70 97 98 6d /72 � 99 63 � 99 65 � 99 98
a ee values of compounds 6a – 6d , 7a – 7d and (–)- 7a – 7d and were estimated by comparing the sign and specifi c rotation values of the corresponding optically pure ( R )- and ( S )-ketones. b All these reactions, when performed under identical conditions, but without adding native or modifi ed PPL, did not yield any product. c Yields were calculated by assuming corresponding single enantiomer as 100% in the starting substrate.

Enantioselective biocatalytic reactions with native and modifi ed PPL 179
(Shakil et al. 2001). Herein, PPL was incubated with acetophenone in THF at 40–42°C in an incu-bator shaker for 1 h and this modifi ed PPL was used to catalyze the deacetylation of ( � )-2,4-diacetoxyphenyl alkyl ketones 3a – 3d , leading to the formation of completely deacetylated compounds 2a – 2d , mono-deacetylated compounds 4a – 4d together with the remaining diacetoxy compounds 3a – 3d at the stage of 50% conversion of the reac-tants (Scheme 2).
The formation of dihydroxy ketones 2a – 2d indicates that PPL, which in its native form exclu-sively mediates the deacetylation of the para - acetoxy function of 2,4-diacetoxyphenyl alkyl ketones 3a – 3d over the ortho -acetoxy group with respect to the car-bonyl group (Parmar et al. 1992a,b, 1993), does not show any preference for the ortho - or para -acetoxy functions when it is pre-incubated with acetophe-none. This observation supports our proposed mech-anism of action of PPL in THF involving formation of a dynamic Schiff ’s base complex between the ξ -amino group of the lysine residue in the active site of PPL and the keto group directly attached to the benzenoid ring (Bisht et al. 1996). In the modifi ed PPL (pre-incubated with acetophenone), the ξ -amino group of its lysine residue gets engaged in Schiff ’s base formation with the carbonyl group of acetophe-none and therefore the PPL–acetophenone system fails to differentiate between the ortho - and para -ace-toxy functions of 2,4-diacetoxyphenyl alkyl ketones 3a – 3d as the active-site lysine residue(s) of PPL may no longer be available for Schiff's base formation with the carbonyl group(s) of the substrates 3a – 3d .
In order to study the changes occurring at the active site of PPL on incubation with acetophenone, we have recorded the Raman spectra of native PPL and the modifi ed PPL (i.e. PPL pre-treated with acetophenone). A band at 3347.9 cm– 1 (presumably for the NH 2 group of the lysine residue of the enzyme) was observed in the Raman spectrum of native PPL, which disappeared in the Raman spectrum of modi-fi ed PPL and a new band at 1599.7 cm– 1 was observed due to the C � N bond of the Schiff's base complex (Dollish et al. 1974). Also a band at 1665.1 cm– 1 in the case of native PPL shifted to 1682.2 cm– 1 in the Raman spectrum of modifi ed PPL. These observa-tions indicate modifi cations at the active site of PPL by involvement of acetophenone and form ation of a Schiff's base complex between the nuclear carbonyl group of the aryl alkyl ketone and the lysine residue of the enzyme. As the structure of the active site of PPL is not known, it is diffi cult to delineate which lysine residue in PPL may be involved in Schiff's base formation. However, the structure of human pancre-atic lipase is known and contains an active-site lysine residue which would be capable of forming a tran-sient Schiff's bases with ketonic compounds as previ-ously postulated (Stryer 1981; Bisht et al. 1996).
Interestingly, in the case of deacetylation reac-tions on 3a – 3d with modifi ed PPL, there was no indication of the formation of ortho -hydroxy- para -acetoxy derivatives throughout the reaction. This may be attributed to the dipole–dipole interactions between the carbonyl groups of the ortho -acetoxy moiety and that of the keto group attached to the benzenoid ring (Figure 1), which resists deacylation
OHHOOHHO
O
R1
R
OAc
O
R1
R
+
(+)-2a-2d
1, 2 & 3
R1 CH-COOHR
150 0C
AcO
R1R
Ac2ODMAP
(+)-3a-3d
abcd
-CH3 -CH2CH3
-CH3 -CH2CH2CH2CH3
-C6H5 -CH3
-C6H5 -CH2CH3
Fused ZnCl2
(+)-1a-1d
Scheme 1. Synthesis of racemic diacyloxyphenyl alkyl ketones.
180 M. Husain et al .
at the ortho position to some extent. However, these interactions are not as strong as the covalent Schiff's base; thus after the formation of monoacetates 4a – 4d , the ortho -acetoxy function also gets deacylated to give the dihydroxy derivatives 2a – 2d . The ketonic com-pounds 2a – 2d , 3a – 3d and 4a – 4d obtained during the deacetylation of ( � )- 3a – 3d catalyzed by modifi ed PPL were found to be optically active, which revealed that the rates of deacetylation of acetoxy functions of both enantiomers are not equal.
The deacetylation reactions on ( � )- 3a – 3d were terminated at three different stages to observe the regio- and enantioselective control exhibited by modifi ed PPL (Scheme 2). At the stage when the dihydroxy compounds had just started to form, two products were obtained, i.e. monoacetates 4a – 4d (21–28%) and the recovered diacetates 3a – 3d (52–59%); all compounds of both series were opti-cally active (Table I). In another set of experiments, the reactions were stopped after 50% conversion of the substrate; three products were obtained (Scheme 2), i.e. dihydroxy ketones 2a – 2d (14–18%), mono-acetylated ketones 4a – 4d (23–29%) and the recov-ered diacetates 3a – 3d (42–46%). This result shows that the modifi ed PPL loses its regioselective con-trol, but retains its enantioselective control as is evident from the observation that all compounds of the three series are optically active (Table II). In the third set of experiments, we allowed the reaction to
O
O
O
O
O
OO
O
O
Oδ-
δ+
δ+
δ-
Figure 1. Different orientations of carbonyls in 3a , indicating the dipole–dipole interactions between the two carbonyl moieties.
AcO
O
R1
ROAc
*
HO
O
R1
ROAc
*
HO
O
R1
ROH
*
HO
O
R1
ROH
*
HO
O
R1
ROAc
*
2, 3 & 4
a
b
c
d
AcO
O
R1
ROAc
AcO
O
R1
ROAc
HO
O
R1
ROAc
+
++
Modified PPL/THFn-BuOH40-42 oC
*
*
Dihydroxy compoundstarted to appear
50 % conversion of substrate
Total conversionof substrate
(+)-3a-3d
(+)-3a-3d
(-)-4a-4d(+)-2a-2d
(+)-3a-3d
(-)-4a-4d
(-)-4a-4d(+)-2a-2d
R R1
-CH3 -CH2CH3
-CH3 -CH2CH2CH2CH3
-C6H5 -CH2CH3
-C6H5 -CH3
Scheme 2. Modifi ed PPL catalyzed deacylation of racemic diacyloxyphenyl alkyl ketones stopped at different stages of reaction.
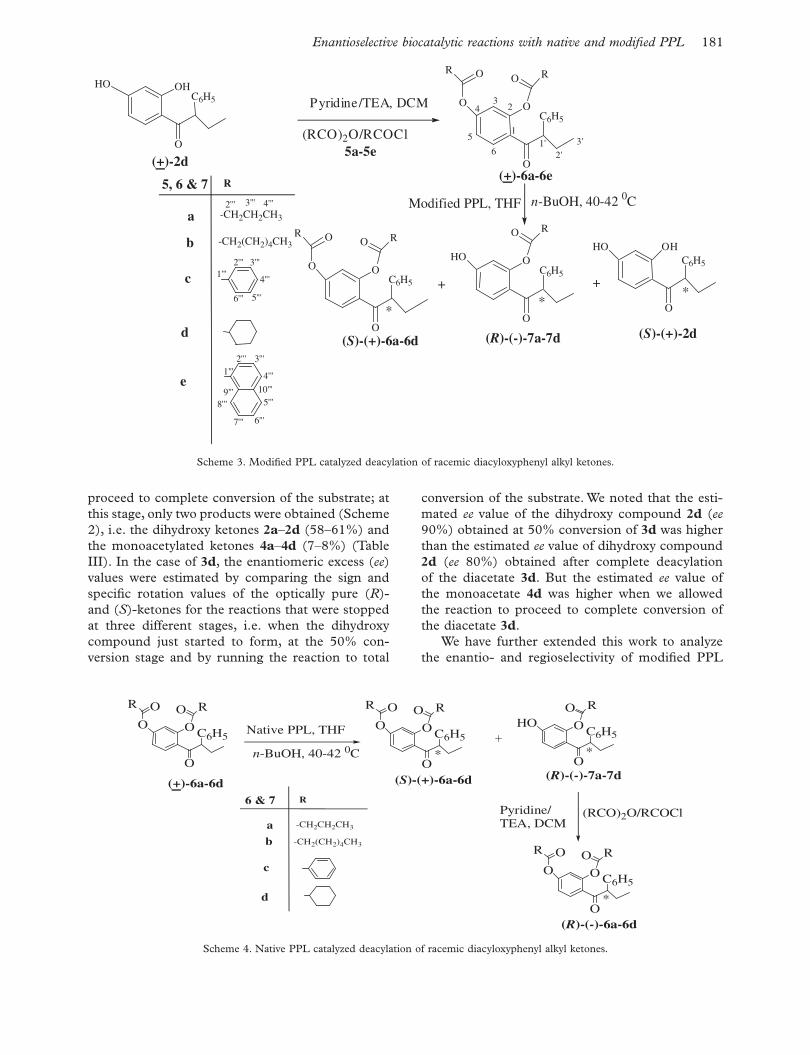
Enantioselective biocatalytic reactions with native and modifi ed PPL 181
proceed to complete conversion of the substrate; at this stage, only two products were obtained (Scheme 2), i.e. the dihydroxy ketones 2a – 2d (58–61%) and the monoacetylated ketones 4a – 4d (7–8%) (Table III). In the case of 3d , the enantiomeric excess ( ee ) values were estimated by comparing the sign and specifi c rotation values of the optically pure ( R )- and ( S )-ketones for the reactions that were stopped at three different stages, i.e. when the dihydroxy compound just started to form, at the 50% con-version stage and by running the reaction to total
conversion of the substrate. We noted that the esti-mated ee value of the dihydroxy compound 2d ( ee 90%) obtained at 50% conversion of 3d was higher than the estimated ee value of dihydroxy compound 2d ( ee 80%) obtained after complete deacylation of the diacetate 3d . But the estimated ee value of the monoacetate 4d was higher when we allowed the reaction to proceed to complete conversion of the diacetate 3d .
We have further extended this work to analyze the enantio- and regioselectivity of modifi ed PPL
HOC6H5
O
OH
*
OHO
RO
C6H5
O
*
-CH2CH2CH3
-CH2(CH2)4CH3
a
5, 6 & 7
b
c
d
e
OO
RO OR
C6H5
O
OO
RO OR
C6H5
O
Modified PPL, THF n-BuOH, 40-42 0C
(S)-(+)-6a-6d (R)-(-)-7a-7d (S)-(+)-2d
+ +
*
(+)-6a-6eR
OHC6H5
O(RCO)2O/RCOCl
5a-5e(+)-2d
HO
2''' 3'''
4'''
2''' 3''' 4'''
1'''
1'''2''' 3'''
4'''
5'''
5'''
6'''
6'''
7'''
8'''9''' 10'''
1
23
4
5
61'
2'
3'
Pyridine/TEA, DCM
Scheme 3. Modifi ed PPL catalyzed deacylation of racemic diacyloxyphenyl alkyl ketones.
(+)-6a-6d
OO
RO OR
C6H5
O
Native PPL, THF
n-BuOH, 40-42 0C
(S)-(+)-6a-6d
*
(RCO)2O/RCOClPyridine/TEA, DCM
(R)-(-)-6a-6d
OO
RO OR
C6H5
O
(R)-(-)-7a-7d
OHO
RO
C6H5
O
*
*
-CH2CH2CH3
-CH2(CH2)4CH3
a
6 & 7
b
c
d
R
OO
RO OR
C6H5
O
Scheme 4. Native PPL catalyzed deacylation of racemic diacyloxyphenyl alkyl ketones.

182 M. Husain et al .
by carrying out reactions on racemic diacyl deriva-tives of 2d , i.e. ( � )-2,4-dibutanoyloxyphenyl (1-phe-nyl)propyl ketone ( 6a ), ( � )-2,4-diheptanoyloxyphenyl (1-phenyl)propyl ketone ( 6b ), ( � )-2,4-dibenzoyloxyphenyl (1-phenyl)propyl ketone ( 6c ), ( � )-2,4-dicyclohexanoyloxyphenyl (1-phenyl)propyl ketone ( 6d ) and ( � )-2,4-dinaphthanoyloxyphenyl (1-phenyl)propyl ketone ( 6e ) (Scheme 3).
These diacyloxy compounds were then subjected to enzymatic deacylation with modifi ed PPL at 40–42°C and the reaction stopped after 50% conversion of the substrates 6a – 6d , affording the corresponding monoacylated, diacylated and dihydroxy ketones (Scheme 3), thus supporting our hypothesis of the formation of a Schiff ’s base complex. In all these cases, we found similar results to those seen in the case of diacetates 3a – 3d (Scheme 2). The ee values were estimated by comparing the sign and specifi c rotation values of the corresponding optically pure ( R )- and ( S )-ketones and were found to be moder-ate to high (Table IV).
The diacyloxy compounds 6a – 6e were then sub-jected to enzymatic deacylation by suspending them in THF (containing n-butanol) with native PPL at 40–42°C, affording the corresponding monoacylated ketones 7a – 7d along with the recovered diacylated ketones 6a – 6d (Scheme 4). The progress of deacylation reaction was monitored by TLC and/or HPLC exami-nation. In the case of ( � )-2,4-dinaphthoyloxyphenyl (1-phenyl)propyl ketone ( 6e ), the reaction did not proceed with either native or modifi ed PPL. All other reactions were worked up at about 50% conversion of the starting compounds into the products by fi lter-ing off the enzyme. The enzymatic reaction products,
i.e. ( R )-(–)-2-butanoyloxy-4-hydroxyphenyl (1-phe-nyl)propyl ketone ( 7a ), ( R )-(–)-2-heptanoyloxy-4-hydroxyphenyl (1-phenyl)propyl ketone ( 7b ), ( R )-(–)-2-benzoyloxy-4-hydroxyphenyl (1- phenyl)pro-pyl ketone ( 7c ) and ( R )-(–)-2-cyclohexanoyloxy-4- hydroxyphenyl (1-phenyl)propyl ketone ( 7d ), and the corresponding recovered diacyloxy compounds 6a – 6d were separated by column chromatography. The monoacyloxy and diacyloxy compounds 7a – 7d and 6a – 6e are new compounds and were fully char-acterized on the basis of their spectral data (IR, 1 H NMR, 13 C NMR and HRMS). All of the bio-catalytic reaction products were found to be opti-cally active (Tables IV and V) as indicated by their observed optical rotations. In order to know their absolute confi guration and the extent of enanti-oselectivity during the enzymatic deacylation reac-tions, we synthesized the optically pure ( R )- and ( S )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone through the nuclear acylation of resorcinol with the ( R )- and ( S )-2-phenylbutanoic acids; both of these were acylated by the corresponding anhydrides or acid chloride/DMAP or Et 3 N methods to afford the diacylated compounds. The ee values of enzy-matically deacylated compounds were estimated by comparing the sign and specifi c rotation values of the optically pure ( R )- and ( S )-ketones and were found to be in the range of 89–99% (Table V).
The acyl derivatives of ( R )-/( S )-2,4-dihydroxy-phenyl (1-phenyl)propyl ketone, i.e. dibutanoyl, diheptanoyl, dibenzoyl and dicyclohexanoyl ( R )-/( S )- 6a – 6d , were prepared by using the corresponding acid anhydride or cyclohexanoyl chloride in dichlo-romethane in the presence of pyridine and Et 3 N,
C6H5
OHHO
O
C6H5
O
O
C6H5
OHHO
O
C6H5
OAcAcO
O
C6H5
OAcAcO
O
C6H5
OHHO
(S)-(+)-2,4-dihydroxyphenyl(1-phenyl)propyl ketone (2d)[α]25D +35.00 (c 0.80, CHCl3)
C6H11COCl-Et3N
(S)-(+)-6d
(S)-(+)-2,4-dihydroxyphenyl(1-phenyl)propyl ketone (2d)
(S)-(+)-2,4-dihydroxyphenyl(1-phenyl)propyl ketone (2d)[α]25D +34.38 (c 0.80, CHCl3)
Ac2O-pyridine
[α]25D +25.00 (c 1.00, CHCl3)
[α]25D +25.51(c 1.00, CHCl3)
Ac2O-AcOH(S)-(+)-3d
(S)-(+)-3d
[α]25D +35.63 (c 0.80, CHCl3)
O
OCOC6H11COC6H11
MeOH-HCl
MeOH-HCl
Scheme 5. Synthesis and chemical deacylation of optically pure diacyloxyphenyl (1-phenyl)propyl ketone.

Enantioselective biocatalytic reactions with native and modifi ed PPL 183
respectively. The acyl derivatives have been used as standards to estimate the ee values of the prod-ucts obtained during lipase-catalyzed deacylation reactions carried out on ( � )-2,4-diacyloxyphenyl (1-phenyl)propyl ketones (Schemes 2, 3 and 4).
A few chemical transformations were carried out in one case to confi rm that there was no racemization during acylation of ( R )-/( S )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone with acid anhydrides or acid chloride in dichloromethane in the presence of bases like pyridine or Et 3 N (Scheme 5). Thus, ( S )-( � )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone ( 2d ) was converted into ( S )-( � )-2,4-diacetoxyphenyl (1-phe-nyl)propyl ketone ( 3d ) by refl uxing with a mixture of acetic anhydride–acetic acid (Ac 2 O–AcOH). The rotation value and the sign of rotation of 2,4-diace-toxyphenyl (1-phenyl)propyl ketone ( 3d ) obtained by the Ac 2 O–AcOH and Ac 2 O–pyridine methods were compared and found to be identical, thereby indicat-ing that there was no racemization during preparation of different acylates of ( R )-/( S )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone under acid anhydride–pyri-dine condition (Scheme 5). Further, the ( S )-( � )-2,4-diacetoxyphenyl (1-phenyl)propyl ketone ( 3d ) prepared from enantiomerically pure ( S )-( � )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone ( 2d ) by the acid anhydride–pyridine method was deacetylated under acidic conditions (MeOH–HCl) and the rota-tion value and the sign of rotation of the compound thus obtained were compared with those of the enantiomerically pure ( S )-( � )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone ( 2d ); the rotations of both these hydroxy compounds were identical, which fur-ther indicated that no racemization occurs during the acid anhydride–pyridine acylation procedure of the enantiomerically pure hydroxy ketone.
It has also been proved that there was no race-mization during conversion of enatiomerically pure ( S )-( � )-2,4-dihydroxyphenyl (1-phenyl)pro-pyl ketone ( 2d ) into 2,4-dicyclohexanoyloxyphenyl (1-phenyl)propyl ketone ( 6d ) by the cyclohexanoyl chloride–Et 3 N method. Thus the rotation value and the sign of rotation of 2,4-dihydroxyphenyl (1-phe-nyl)propyl ketone ( 2d ) prepared by hydrolysis of 2,4-dicyclohexanoyloxyphenyl (1-phenyl)propyl ketone ( 6d ), which was obtained from enantiomeri-cally pure ( S )-( � )-2,4-dihydroxyphenyl (1-phenyl)propyl ketone ( 2d ), were found to be identical, thus indicating that there is no racemization during cyclo-hexanoylation with acid chloride in the presence of Et 3 N (Scheme 5).
Conclusion
We have successfully explained the mechanism of deacylation of aryl aklyl ketones catalyzed by both
native PPL and modifi ed PPL (PPL pre-treated with acetophenone). The results prove our earlier hypothesis regarding the formation of a transient Schiff's base-type complex between nuclear carbo-nyl groups present in the substrate (aryl alkyl ketone) and the ξ -amino function of the lysine residue pres-ent in the active site of PPL, which governs the regi-oselectivity of these reactions. Furthermore, the novel optically active aryl alkyl ketones obtained in this study may fi nd application in the synthesis of bioactive, chiral, naturally occurring chromanones, fl avanones and dihydrocoumarins, which are cum-bersome to make in optically enriched forms by chemical routes.
Acknowledgements
The authors thank the University of Delhi (Delhi, India), Polytechnic University (New York, USA) and the Department of Biotechnology (DBT, Gov-ernment of India, New Delhi) for fi nancial assis-tance for this work. We also thank Dr Hemant Sharma (APL Research Center, Aurobindo Pharma Ltd., Hyderabad, India) for his help in recording the Raman spectra of several of our samples.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Amakura Y, Tsutsumi T, Sasaki K, Nakamura M, Yoshida T, Maitani T. 2008. Infl uence of food polyphenols on aryl hydrocarbon receptor-signaling pathway estimated by in vitro bioassay. Phytochemistry 69:3117–3130.
Bhattacharya A, Prasad AK, Maity J, Himanshu, Poonam, Olsen CE, Gross RA, Parmar VS. 2003. Highly effi cient and selective biocatalytic acylation studies on triazolylsugars. Tetrahedron 59:10269–10277.
Bisht KS, Kumar A, Kumar N, Parmar VS. 1996. Preparative and mechanistic aspects of interesterifi cation reactions on diols and peracetylated polyphenolic compounds catalyzed by lipases. Pure Appl Chem 68:749–752.
Bommarius AS, Riebel BR. 2004. Biocatalysis. Vol 1. Weinheim: Wiley–VCH.
Bracke ME, Vanhoecke BWA, Derycke L, Bolca S, Possemiers S, Heyerick A, Stevens CV, DeKeukeleire D, Depypere HT, Verstraete W, Williams CA, Mckenna ST, Tomar S, Sharma D, Prasad AK, DePass AL, Parmar VS. 2008. Plant polyphenolics as anti-invasive cancer agents. Anti-Cancer Agents Med Chem 8:171–185.
Chambers RJ, Marfat A. 1994. Development of a stereoselective and chemoselective approach to trans-2,3-disubstituted-4-chromanones. J Heterocyclic Chem 31:1401–1405.
Chica RA, Doucet N, Pelletier JN. 2005. Enantioselective bioca-talysis optimized by directed evolution. Semi-rational approaches to engineering enzyme activity: combining the benefi ts of directed evolution and rational design. Curr Opin Biotechnol 16:378–384.

184 M. Husain et al .
Dollish FR, Fateley WG, Bentley FF. 1974. Characteristic Raman frequencies of organic compounds. New York: Wiley-Interscience. p. 36, p. 136.
Drauz K, Waldmann H. 2002. Enzyme catalysis in organic synthesis. Vol. I, II & III. 2nd ed. Weinheim: Wiley-VCH.
Faber K. 2004. Biotransformations in organic chemistry. 4th ed. Springer.
Ghate M, Kusanur RA, Kulkarni MV. 2005. Synthesis and in vivo analgesic and anti-infl ammatory activity of some bi heterocyclic coumarin derivatives. Eur J Med Chem 40:882–887.
Ghosh D, Scheepens, A. 2009. Vascular action of polyphenols. Mol Nutrition Food Res 53:322–331.
Jestin JL, Kaminski PA. 2004. Directed enzyme evolution and selections for catalysis based on product formation. J Biotechnol 113:85–103.
Khersonsky O, Roodveldt C, Tawfi k DS. 2006. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol 10:498–508.
Kumar A, Poonam, Pati HN, Saxena RK, Davidson S, Gupta R. 1998. Potential use of a novel lipase from Aspergillus carneus in deacetylation reactions. Biochim Biophys Acta 1387:325–330.
Kumar R, Azim A, Kumar V, Sharma SK, Prasad AK, Howarth OW, Olsen CE, Jain SC, Parmar VS. 2001. Lipase-catalyzed chemo- and enantioselective acetylation of 2-alkyl/aryl-3-hydroxypropi-ophenones. Bioorgan Med Chem 9:2643–2652.
Maity J, Shakya G, Singh SK, Ravikumar VT, Parmar VS, Prasad AK. 2008. Effi cient and selective enzymatic acylation reaction: separation of furanosyl and pyranosyl nucleosides. J Org Chem 73:5629–5632.
Naithani R, Huma LC, Holland LE, Shukla D, McCormick DL, Mehta RG, Moriarty RM. 2008. Antiviral activity of phytochemicals: a comprehensive review. Mini-Rev Med Chem 8:1106–1133.
Narender T, Papi Reddy K, Shweta, Srivastava K, Mishra DK, Puri SK. 2007. Total synthesis of munchiwarin, a triprenylated chalcone from Crotalaria medicagenia . Org Lett 9:5369–5372.
Nencki M, Sieber N. 1881. Compounds of mono- and di-basic acids with phenol. J Prak Chem 23:147.
Parmar VS, Khanduri CH, Tyagi OD, Prasad AK, Gupta S, Bisht KS, Pati HN, Sharma NK. 1992a. Regioselective hydrol-ysis of polyacetoxy aromatic ketones with lipases in organic solvents. Indian J Chem B 31:925–929.
Parmar VS, Prasad AK, Sharma NK, Singh SK, Pati HN, Gupta S. 1992b. Regioselective deacylation of polyacetoxyaryl methyl ketones by lipases in organic solvents. Tetrahedron 48:6495–6498.
Parmar VS, Prasad AK, Sharma NK, Vardhan A, Pati HN, Sharma SK, Bisht KS. 1993. Lipase-catalyzed selective deacetylation of peracetylated benzopyranones. J Chem Soc Chem Commun 27–29.
Parmar VS, Kumar A, Bisht KS, Mukherjee S, Prasad AK, Sharma SK, Wengel J, Olsen CE. 1997. Novel chemoselective de-esterifi cation of esters of polyacetoxy aromatic acids by lipases. Tetrahedron 53:2163–2176.
Parmar VS, Kumar A, Prasad AK, Kumar R, Bisht KS, Poonam, Jain SC, Olsen CE. 1998a. Novel enzymic de-esterifi cation studies on substituted polyacetoxybenzamides. J Indian Chem Soc 75:810–822.
Parmar VS, Pati HN, Azim A, Kumar R, Himanshu, Bisht KS, Prasad AK, Errington W. 1998b. Lipase-catalyzed selective deacetylation of phenolic/enolic acetoxy groups in peracetylated benzyl phenyl ketones. Bioorgan Med Chem 6:109–118.
Parmar VS , Prasad AK, Pati HN, Kumar R, Azim A, Roy S, Errington W. 1999. Enzyme-catalyzed chemoselective transes-terifi cation reactions on hydroxymethylated phenolic com-pounds. Bioorg Chem 27:119–134.
Pathak VN, Gupta R, Varshney B. 2008. A ‘one pot’ synthesis of 2-aryl-4 H -1-benzopyran-4-ones under coupled microwave phase transfer catalysis (PTC) and ultrasonic irradiation PTC. J Heterocyclic Chem 45:589–592.
Poonam, Prasad AK, Azim A, Kumar R, Jain SC, Parmar VS, Olsen CE, Errington W. 2001. Synthesis and lipase-mediated stereoselective deacetylation of ( � )-3-acetoxymethyl-3-alkyl-7-methoxychroman-4-ones. Tetrahedron 57:7395–7402.
Prasad AK, Pati HN, Azim A, Trikha S, Poonam. 1999. Lipase-catalysed regio- and enantioselective deacetylation of 2,4-diacetoxyphenyl alkyl ketones. Bioorgan Med Chem 7: 1973–1977.
Prasad AK, Himanshu, Bhattacharya A, Olsen CE, Parmar VS. 2002. Novel lipase-catalyzed highly selective acetylation studies on d-arabino- and d-threo-polyhydroxyalkyltriazoles. Bioorgan Med Chem 10:947–951.
Prasad AK, Kalra N, Yadav Y, Singh SK, Sharma SK, Patkar S, Lange L,Olsen CE, Wengel J, Parmar VS. 2007. Selective bio-catalytic deacylation studies on furanose triesters: a novel and effi cient approach towards bicyclonuclosides. Org Biomol Chem 5:3524–3530.
Rubin-Pitel SB, Zhao H. 2006. Recent advances in biocatalysis by directed enzyme evolution. Comb Chem High Throughput Screen 9:247–257.
Sashidhara KV, Rosaiah JN, Kumar A. 2009. Iodine-catalyzed mild and effi cient method for the synthesis of chalcones. Syn Commun 39:2288–2296.
Shakil NA, Singh A, Prasad AK, Kumar V, Olsen CE, Jain SC, Cholli AL, Watterson AC, Parmar VS. 2001. Enzymatic enanti-oselective deacetylation studies on novel ( � )-2,4-diacetoxyphenyl alkyl ketones. J Macromol Sci Pure A 38:1275–1290.
Shakil NA, Dhawan A, Sharma NK, Kumar V, Kumar S, Bose M, Raj HG, Olsen CE, Cholli AL, Samuelson LA, Kumar J, Watterson AC, Parmar VS, Prasad AK. 2003. Synthetic, biocatalytic acetylation and anti-tuberculosis activity evalua-tion studies on ( � )-4-alkyl-3,4-dihydro-3-hydroxyalkyl-2 H -1,3-benzoxazines. Indian J Chem B 42:1958–1969.
Singh I, Prasad AK, Sharma AK, Saxena RK, Olsen CE, Cholli AL, Samuelson LA, Kumar J, Watterson AC, Parmar VS. 2003. Syn-thetic and novel biocatalytic resolution studies on ( � )-5/6/7-acetoxy-4-aryl-3,4-dihydrocoumarins. Bioorgan Med Chem 11:529–538.
Stryer L. 1981. Biochemistry. 2nd ed. New York: WH Freeman and Company. p. 342.
Tang L, Zhang S, Yang J, Gao W, Cui J, Zhuang T. 2005. Novel and convenient one-pot synthesis of 3-aroyl-7-hydroxy-6-nitrofl avones. Syn Commun 35:315–323.
Valetti F, Gilardi G. 2004. Directed evolution of enzymes for product chemistry. Nat Prod Rep 21:490–511.
Walsh C. 2001. Enabling the chemistry of life. Nature 409:226–231.
Wang X, Cheng S. 2006. Solvent-free synthesis of fl avanones over aminopropyl-functionalized SBA-15. Catal Commun 7:689–695.
Weber HK, Zuegg J, Faber K, Pleiss J. 1997. Molecular reasons for lipase-sensitivity against acetaldehyde. J Mol Catal B: Enzym 3:131–138.
Related Documents











