Biocatalytic Synthesis of Amino Alcohols I n a u g u r a l d i s s e r t a t i o n zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Ernst-Moritz-Arndt-Universität Greifswald vorgelegt von Hannes Kohls geboren am 16.04.1987 in Hoyerswerda Greifswald, September 2015

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Biocatalytic Synthesis of Amino Alcohols
I n a u g u r a l d i s s e r t a t i o n
zur
Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften (Dr. rer. nat.)
der
Mathematisch-Naturwissenschaftlichen Fakultät
der
Ernst-Moritz-Arndt-Universität Greifswald
vorgelegt von
Hannes Kohls
geboren am 16.04.1987
in Hoyerswerda
Greifswald, September 2015

Dekan: Prof. Dr. Klaus Fesser
1. Gutachter: Jun. Prof. Dr. Matthias Höhne
2. Gutachter: Prof. Dr. Harald Gröger
Tag der Promotion: 09.12.2015

................
III
Table of Contents
Abbreviations ............................................................................................................................... 4
Scope and Outline ........................................................................................................................ 5
Introduction .......................................................................................................................... 7 1
What is Chirality and Why Does it Matter? ............................................................................ 7 1.1
Chiral Amines and Amino Alcohols ......................................................................................... 8 1.2
Routes towards Chiral Amines and Amino Alcohols .......................................................... 10 1.3
1.3.1 Synthesis of Amines and Amino Alcohols Employing Non-Biological Catalysts ..... 10
1.3.2 Biocatalytic Synthesis of Chiral Amines ....................................................................... 11
Can Biocatalysis Compete? - Article I ............................................................................... 14 2
Recently Identified and Newly Developed Biocatalysts ..................................................... 14 2.1
Engineered Enzymes, Smart Substrates and Process Optimisation ................................ 15 2.2
Examples of Industrial Scale Biotransformations ............................................................... 16 2.3
Biocatalytic Syntheses of Amino Alcohols ....................................................................... 18 3
Established Routes towards 1,2- and 1,3-Amino Alcohols ................................................ 18 3.1
3.1.1 Chemoenzymatic Approaches ...................................................................................... 18
3.1.2 Biocatalytic Approaches ................................................................................................. 20
A Novel Route for the Synthesis of an 1,3-Amino Alcohol - Article II .............................. 22 3.2
An Engineered ATA Suitable for the Synthesis of an 1,2-Amino Alcohol - Article III .... 26 3.3
In-Silico Analysis of the Class III TA Family - Article IV .................................................... 30 4
A Family Wide Comparison of Specificity Determining Amino Acid Residues ................ 30 4.1
The Sequence-Function Matrix.............................................................................................. 31 4.2
Challenges of Sequence-Function Prediction ...................................................................... 34 4.3
Conclusion ........................................................................................................................... 36 5
References ........................................................................................................................... 38 6
Author Contribution .................................................................................................................. 44
Articles ......................................................................................................................................... 45
Article I .................................................................................................................................................. 47
Article II ................................................................................................................................................. 63
Article III ................................................................................................................................ 115
Article IV ................................................................................................................................................ 139
Affirmation.................................................................................................................................. 217
Curriculum vitae ......................................................................................................................... 218
Scientific Publications ................................................................................................................ 219
Acknowledgements .................................................................................................................... 220
IV
Abbreviations % (v/v) Volume percent LDH Lactate DH
(R)-ATA (R)-selective ATA Lysε L-Lysine ε-amino group
(S)-ATA (S)-selective ATA MAO Monoamine oxidase
1-PEA 1-Phenylethylamine MAO-N MAO from Aspergillus niger
AcOrn N-2-Acetyl-L-ornithine MS Mass spectrometry
ADH Alcohol DH NAD(P)+ Nicotinamide adenine dinucleotide (phosphate),
oxidised form
ADH-Lbr ADH from Lactobacillus brevis NAD(P)H Nicotinamide adenine dinucleotide (phosphate),
reduced form
ADH-Rsp ADH from Ralstonia species NCS Norcoclaurine synthase
AG Aktiengesellschaft Orn L-Ornithine
AHAS-I-Eco Acetohydroxyacid synthase I from Eschrichia
coli
OrnTL Ornithine transaminase-like
AmDH Amine DH p.a. per anno
APPO 2-Amino-1-phenylpropan-1-one PAC Phenylacetylcarbinol
ATA Amine transaminase PDB Protein data bank
ATA-Ate ATA from Aspergillus terreus PDC Pyruvate decarboxylase
ATA-Cvi ATA from Chromobacterium violaceum PDK-1 3-Phosphoinositide-dependent protein kinase-1
ATA-Vfl ATA from Vibrio fluvialis pH pondus hydrogenii
BASF Badische Anilin- & Soda-Fabrik PLP Pyridoxal 5’-phosphate
BBE Berberine bridge enzyme PPDO 1-Phenylpropane-1,2-dione
Boc tert-Butyloxycarbonyl pyr Pyruvate
C Conversion rpm rounds per minute
CalB Lipase B from Candida antarctica SA Specific activity
COX Cyclooxygenase SAM S-Adenosyl-L-methionine
DB Database SI Supporting information
de Diastereomeric excess sp. species
DH Dehydrogenase STR Strictosidine synthase
DMSO Dimethyl sulfoxide STY Space-time yield
e.g. for example (exempli gratia) SuOrn N-2-Succinyl-L-ornithine
EC Enzyme Commission number TA Transaminase
ee Enantiomeric excess ThDP Thiamine pyrophosphate
equiv Equivalents TK Transketolase
et al. and others (et alii) TOF Turnover frequency
GABA γ-Aminobutyrate TON Turnover number
GC Gas chromatography U Unit [µmol min-1]
GDH Glucose DH WT Wild-type
HEPES 4-(2-Hydroxyethyl)-1-piperazineethane-
sulfonic acid
αAA α-Amino acid
IPA Isopropylamine αKG α-Ketoglutarate
IRED Imine reductase βAla β-Alanine
JAK2 Janus kinase 2 βPhe β-Phenylalanine
KAPA 7-Keto-8-aminopelargonic acid ωAA ω-Amino acid
KRED Keto reductase
Moreover, SI units and the usual one and three letter codes for amino acids were used.

V
Scope and Outline
This thesis deals with the biocatalytic synthesis of amino alcohols. To provide a background,
chapter one briefly introduces the concept of chirality in organic molecules. It provides insight
in the consequences of chirality regarding the synthesis of biologically active compounds. The
thesis continues with an overview about the available strategies, which can be applied for the
synthesis of amines. For this purpose, synthetic routes employing non-biological catalysts are
shortly outlined, followed by an overview about biocatalytic options. The second chapter ad-
dresses the question whether biocatalysis is actually useful for the synthesis of amines - not
only at the lab bench, but also at industrial scale. In light of Article I, newly discovered and engi-
neered enzyme activities are discussed. Thereafter, examples for the application of biocatalytic
amine synthesis at industrial scale are given and compared to traditional synthesis routes. Hav-
ing evaluated applicability and usefulness of biocatalysis, the third chapter of this thesis contin-
ues with a summary of Article II, which reports a novel route for the biocatalytic synthesis of all
four diastereomers of a 1,3-amino alcohol by the combination of different enzymes. Subse-
quently, chapter three also discusses Article III, which deals with protein engineering of an
amine transaminase (ATA) to overcome substrate scope limitations. The engineered enzyme
was successfully employed for the synthesis of an important 1,2-amino alcohol, eventually. Fi-
nally, chapter four discusses the findings described in Article IV: A bioinformatic analysis was
conducted on the ATA containing class III transaminase (TA) family to broaden the understand-
ing of sequence- and structure-function relationships of these enzymes. This approach addi-
tionally aimed to identify new enzymes, thereby expanding the biocatalytic toolbox for chiral
amine synthesis.
Article I Recent Achievements in Developing the Biocatalytic Toolbox for Chiral
Amine Synthesis
Kohls, H; Steffen-Munsberg, F; Höhne, M, Curr Opin Chem Biol 2014, 19, 180-192,
DOI: 10.1016/j.cbpa.2014.02.021
This review discusses the recent developments in the field of chiral amine synthesis using bio-
catalysis over the three preceding years prior to its publication. In this paper we compared bio-
catalytic and traditional organic syntheses routes which utilise non-biological chiral catalysts.
For this purpose, the different chiral amine synthesis methods have been evaluated by means
of turnover number (TON), turnover frequency (TOF) and space-time yield (STY). Furthermore,
newly discovered biocatalysts are presented and the maturation process of already known ones
towards large scale applicability is outlined.
Article II Selective Access to All Four Diastereomers of a 1,3-Amino Alcohol by Com-
bination of a Keto Reductase- and an Amine Transaminase-Catalysed Reac-
tion
Kohls, H; Anderson, M; Dickerhoff, J; Weisz, K; Córdova,A; Berglund, P; Brundiek,
H; Bornscheuer, UT; Höhne, M, Adv Synth Catal 2015, 357, 1808-1814, DOI:
10.1002/adsc.201500214

VI
In this paper we reported a novel and selective route for the biocatalytic synthesis of the 1,3-
amino alcohol 4-amino-1-phenylpentane-2-ol. It employs a keto reductase (KRED) and two en-
antiocomplementary ATAs in stepwise biocatalytic reactions to selectively obtain all four dia-
stereomers of this compound. Starting from a racemic hydroxy ketone, a kinetic resolution pro-
vided optically active (R)-hydroxy ketone (yield 50%, 86% ee) and the corresponding diketone
(yield 27%). Further transamination of the hydroxy ketone with either an (R)- or (S)-selective ATA
yielded the two corresponding 1,3-amino alcohol diastereomers. The two remaining diastere-
omers were accessible in two subsequent asymmetric steps. For this purpose, the diketone was
reduced regio- and enantioselectively by the same KRED, which yielded the (S)-configured hy-
droxy ketone (yield 86%, 71% ee). Eventually, a subsequent transamination with (R)- or (S)-
selective ATA yielded the two remaining diastereomers of the amino alcohol.
Article III Engineering the Active Site of the Amine Transaminase from Vibrio fluvialis
for the Asymmetric Synthesis of Aryl–Alkyl Amines and Amino Alcohols
Nobili, A; Steffen-Munsberg, F; Kohls, H; Trentin, I; Schulzke, C; Höhne, M; Born-
scheuer, UT, ChemCatChem 2015, 7, 757-760, DOI: 10.1002/cctc.201403010
In this work, variants of the title enzyme were generated in order to evolve a catalyst that is
applicable in the asymmetric transamination of ketones with two bulky substituents. For this
purpose, the small binding pocket of the enzyme was enlarged by means of protein engineer-
ing. This was realised by a systematic (partial) saturation of the amino acid residues that set up
the small binding pocket. Eventually, two mutants were obtained which could be successfully
employed in the preparative synthesis of the 1,2-amino alcohol (R)-phenylglycinol and the bulky
amine (S)-1-phenylbutylamine.
Article IV Bioinformatic Analysis of a PLP-Dependent Enzyme Superfamily Suitable
for Biocatalytic Applications
Steffen-Munsberg, F; Vickers, C; Kohls, H; Land, H; Mallin, H; Nobili, A; Skalden, L;
van den Bergh, T; Joosten, H-J; Berglund, P; Höhne, M; Bornscheuer, UT, Biotech-
nol Adv 2015, 33, 566-604, DOI: 10.1016/j.biotechadv.2014.12.012
As comprehensively shown in Articles I-III, ATAs are highly useful biocatalysts for the synthesis
of chiral amines. Putting the focus on the class III TA family (which includes (S)-ATAs) we ana-
lysed sequence- and structure-function relationships within the superfamily of pyridoxal 5’-
phosphate (PLP) dependent enzymes in this research review paper. A bioinformatic analysis of
the structure-based alignment of ~13.000 sequences in combination with structural inspection
and literature research showed that the active site architecture reflects the substrate and reac-
tion specificity within this family. These observations were assembled in a sequence–function
matrix, which can be used for annotation and identification of enzymes by specific active site
amino acids.

Introduction 1
What is Chirality and Why Does it Matter? 1.1
Chirality is a common feature occurring in everyday situations: The left and right hand or the
left and right foot are non-superimposable mirror images of one another, and therefore, they
are chiral. The symmetric difference becomes obvious when one tries to put the left shoe on
the right foot. The very same phenomenon can occur in molecules. In organic molecules it is
frequently found in the form of a carbon atom, which is bound to four different substituents.
This carbon atom constitutes a chiral centre of the molecule. Each chiral centre can occur in two
different symmetrical forms - just as ones hands or feet. These two different forms are called
enantiomers and, in a symmetric context, they behave like the image and the mirror image of a
molecule. The two enantiomers of one compound are composed of the same atoms (four fin-
gers and one thumb on both hands) which are connected towards each other in the same se-
quence (on the thumb follows the index finger, than the middle finger and so on) and despite
these similarities, there are still different kinds of this molecule (left and right hand). This differ-
ent spatial arrangement of the atoms within a molecule is called stereoisomerism. Anyhow,
unlike a hand, more than on stereocentre can occur in a molecule. In that case, non-
superimposable stereoisomers can occur that are not enantiomers (mirror images) of one an-
other. These isomers are called diastereomers (diastereoisomers). In general, for a compound
with n chiral centres a maximum of 2n stereoisomers can occur.
In nature, the principle of homochirality (single-handedness) prevails. Many biochemical
compounds occur in both enantiomeric forms, but nature usually utilises one specific enantio-
mer for a given task within an organism.[1]
For instance, 21 of the 22 proteinogenic amino acids
are chiral but all natural occurring enzymes discovered so far consist solely of the L-
enantiomers of these amino acids.[2, 3]
Again, this principle can be found in everyday situations:
A right-handed person will always use the same enantiomer of her two hands to write a letter.
Since enzymes are built from chiral subunits, they are chiral molecules themselves, which is also
true for other biomolecules such as nucleic acids and carbohydrates. Enzymes are usually high-
ly selective towards the stereo configuration of their substrate. Consequently, as the majority of
biotransformations in a cell is catalysed by enzymes, two enantiomers of a compound must be
regarded as two distinct molecules in a biological context.[4]
With this in mind it becomes obvious why chirality matters in biological active compounds
such as drugs or pesticides. As most of these compounds interact with enzymes or receptors of
an organism the enantiomers of a biologically active compound can differ in their pharmacody-
namic- and pharmacokinetic properties like efficacy, potency, adsorption, distribution within an
organism, the metabolic manipulation they undergo and the elimination route they take.[5, 6]
In this context one frequently mentioned example is the drug thalidomide (Contergan),
which was distributed as a racemate (a 50/50 mixture of both enantiomers). It gained notoriety
in the early sixties when the connection between an increased rate of malformation of infants
and its administration to pregnant women was recognised. Later it was believed[7]
that the (R)-
configured enantiomer shows the desired sedative effect, whereas the (S)-enantiomer exhibits
serious embryotoxic and teratogenic activity, which lead to malformation of the limps (phoco-
melia) in newborns. Anyhow, in that case, the application of the single (R)-enantiomer would not

8 1 Introduction
have prevented this tragedy as both forms are transformed into one another (racemised) within
the body, giving a 50/50 mixture of both enantiomers (racemic mixture) - even if the single en-
antiomer is administered.[8]
This effect of chiral inversion was also shown for ibuprofen, which
is sold as a racemic mixture for over 45 years. In contrast to thalidomide, its chiral inversion is
unidirectional – that is, the active (S)-enantiomers, which is a potent inhibitor of the enzyme
cyclooxygenase (COX), remains untouched.[9]
However, 50-60% of the non-inhibiting (R)-
enantiomer undergo metabolic inversion towards the active (S)-enantiomer. Therefore, and for
other reasons the administration of enantiopure (S)-ibuprofen might be beneficial for patients;
e.g. the rate of chiral inversion of (R)-ibuprofen can greatly differ in individuals, the (S)-isomer is
considered metabolically cleaner and the total dose could be reduced.[10, 11]
In general, one enantiomer can have a beneficial effect while the other can have no effect,
some effect due to chiral inversion occurring in the body, antagonist activity against the active
enantiomer or even completely different activity from the active enantiomer.[12]
This is why the
application of enantiopure drugs is favoured and why methods for obtaining them are highly
desired.
Another important aspect of chirality in drug molecules is marketing and profit.[13]
In a pro-
cess called chiral switch, “new” single enantiomer drugs are derived from their racemate coun-
terparts, which are already successfully claimed, approved and marketed.[14]
Immediately be-
fore the expiration of the patent that covers the racemic product, the single enantiomer is in-
troduced onto the market by the proprietor of the racemic drug. That way, the new product
avoids competition with generic forms of the preceding racemic formulation. This provides a
useful option for owners of racemates to achieve line extensions of their products and prevent
loss of exclusivity - thereby maintaining their market share.[7]
Sadly, in some cases, these “new”
drugs do not necessarily need to be superior to the racemic counterpart in order to gain ap-
proval. Instead, it is sufficient to outperform a placebo.[15-17]
Chiral Amines and Amino Alcohols 1.2
Amines are ubiquitous in nature and occur as macromolecules such as nucleic acids, proteins
and sugars. They also occur as smaller molecules, which are often biologically active com-
pounds. Molecules, which carry at least one amino function adjacent to a chiral carbon are
called -chiral amines and are referred to as chiral amines in the following.
Chiral amines are of great importance in the pharmaceutical and agrochemical industry.
Around 40% of chiral drugs are amines[18]
and around 30% of pesticides are chiral molecules.[19]
These compounds cover a broad range of applications and includes drugs that are used for the
treatment of very diverse conditions such as depression, pain, obesity or malaria.[20]
Also func-
tionalised amines, such as amino alcohols, occur in many biological active compounds.[21]
The
amino alcohol motif is found in many drugs used for treatment of diverse diseases (Figure 1).[22]
Furthermore, amino alcohols such as phenylglycinol, ephedrine or 1-amino-2-indanol are useful
chiral auxiliaries.[23]
For instance, phenygylcinol is used as chiral auxiliary for the synthesis of
drugs such as Saxagliptin[24]
(treatment of type II diabetes) and the antidepressants femoxetine
and paroxetine.[25]
Furthermore, the amino alcohol (R)-phenylglycinol is a building block for a
promising drug being under investigation in cancer therapy (“compound 33”, Figure 1), due to
its function as potent inhibitor of 3-phosphoinositide-dependent protein kinase-1 (PDK1).[26]
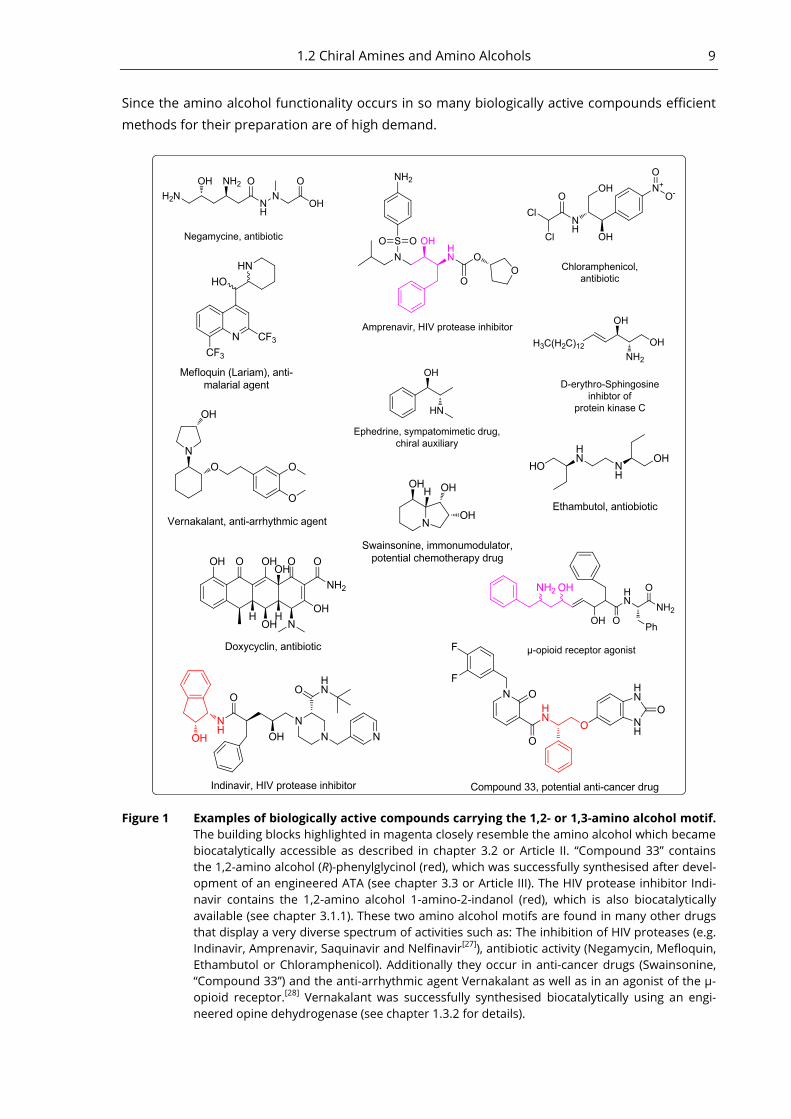
1.2 Chiral Amines and Amino Alcohols 9
Since the amino alcohol functionality occurs in so many biologically active compounds efficient
methods for their preparation are of high demand.
Figure 1 Examples of biologically active compounds carrying the 1,2- or 1,3-amino alcohol motif.
The building blocks highlighted in magenta closely resemble the amino alcohol which became
biocatalytically accessible as described in chapter 3.2 or Article II. “Compound 33” contains
the 1,2-amino alcohol (R)-phenylglycinol (red), which was successfully synthesised after devel-
opment of an engineered ATA (see chapter 3.3 or Article III). The HIV protease inhibitor Indi-
navir contains the 1,2-amino alcohol 1-amino-2-indanol (red), which is also biocatalytically
available (see chapter 3.1.1). These two amino alcohol motifs are found in many other drugs
that display a very diverse spectrum of activities such as: The inhibition of HIV proteases (e.g.
Indinavir, Amprenavir, Saquinavir and Nelfinavir[27]
), antibiotic activity (Negamycin, Mefloquin,
Ethambutol or Chloramphenicol). Additionally they occur in anti-cancer drugs (Swainsonine,
“Compound 33”) and the anti-arrhythmic agent Vernakalant as well as in an agonist of the µ-
opioid receptor.[28]
Vernakalant was successfully synthesised biocatalytically using an engi-
neered opine dehydrogenase (see chapter 1.3.2 for details).

10 1 Introduction
Routes towards Chiral Amines and Amino Alcohols 1.3
The enantioselective synthesis of chiral amines and functionalised amines such as 1,2- or 1,3-
amino alcohols is rather tedious and step intensive. This is illustrated by the proverb that each
nitrogen in a molecule increases a graduate student’s career by one year.[29]
In order to perform
asymmetric syntheses usually transition metal complexes are employed as chiral catalysts. In
the biocatalytic approach, however, chirality is introduced by enzymes. Owing to the often ex-
cellent stereo- and regioselectivity of enzymes, they can provide a useful option in asymmetric
synthesis of amines or kinetic resolution of racemic mixtures. In particular, biocatalytic routes to
compounds with more than one chiral centre have a great potential, as in theory all diastere-
omers can be accessed in a step-efficient manner.[30]
As this thesis deals with the biocatalytic
approach to chiral amines, the traditional approach is outlined only briefly. For further infor-
mation on that topic, the reader is kindly referred to the comprehensive book “Chiral Amine
Synthesis” edited by Thomas C. Nugent.[29]
1.3.1 Synthesis of Amines and Amino Alcohols Employing Non-Biological
Catalysts
Many strategies employing non-biological chiral catalysts have been developed for the synthesis
of chiral amines. Figure 2 provides an overview of the commonly used approaches towards -
chiral primary amines. The three most common methods include the reduction of N-acetyl pro-
tected or unprotected[31]
enamines, reduction of imines[32, 33]
and the reductive
amination.[20, 34, 35]
All methods use transition metal catalysts for the asymmetric addition of
hydrogen or a hydride to a prochiral educt. Furthermore, hydroamination of alkenes provides a
route to chiral amines.[36]
Again, metal complexes are employed to catalyse this reaction, if the
alkene is not activated and electron deficient.[37]
Additionally, the activation of a C-H bond pro-
vides an option to transform it directly to a C-N bond by C-H-Insertion.[38, 39]
For this activation
several transition metals are utilised such as iron, manganese, rhodium, ruthenium, copper or
silver.[40]
Figure 2 Common strategies for the synthesis of -chiral primary amines. Adapted from Nugent et
al.[20]
Nevertheless, the structural scope of amines accessible with high yields and high optical pu-
rities by these methods is limited.[20]
Therefore, additional steps are often required to increase
optical purity, which can be achieved, for instance, by crystallisation of a diastereomeric salt.

1.3 Routes towards Chiral Amines and Amino Alcohols 11
Using this method, the desired enantiomer forms a salt with the anion of an optically active
carboxylic acid. If the carboxylic acid is applied in enantiopure form, only one specific enantio-
mer of the amine forms the salt with the corresponding carboxylic acid anion. At the same time,
the other enantiomer stays in solution, thereby facilitating the separation of the two enantio-
mers.[41]
Synthetic routes towards 1,3-amino alcohols are mostly based upon the diastereoselective
reduction, whereas only a few methods describe the enantioselective preparation of this class
of amines.[21]
An important method is the reduction of enaminones, which is accomplished by
the hydrogenation or a hydride reduction.[42]
Another approach employs proline as organo-
catalyst, which allows for the enantioselective synthesis of N-Boc protected 1,3-amino ketones
in a Mannich reaction.[43]
Stereoselective reduction of the ketone yields the corresponding 1,3-
amino alcohol. This approach was also utilised in combination with a lipase in a dynamic kinetic
asymmetric transformation (see Chapter 3.1.1 and Figure 6 for this example).
One route towards 1,2-amino alcohols is a three-component Mannich reaction as described
by List et al. This asymmetric synthesis employs proline as organocatalyst and allows for the
selective synthesis of amino alcohols.[44]
For an overview of methods for stereoselective synthe-
sis of 1,2-amino alcohols the reader is kindly referred to a review by Sehl et al.[45]
1.3.2 Biocatalytic Synthesis of Chiral Amines
This section is designed to provide the reader with a general overview of the available biocata-
lysts applicable to the synthesis of chiral amines (Figure 3). For most of the enzymes that are
going to be presented in this section, recent achievements and specific examples for their ap-
plication are given in Article I. Therefore, at the same time, the following information provides
the background for the subsequent chapter, which discusses Article I.
Biocatalysis has become a valuable option for the synthesis of chiral amines and many dif-
ferent enzymes classes are available for a biocatalytic approach to this diverse compound class.
The first generally applicable strategy was realised via enantioselective acylation using lipases to
catalyse a kinetic resolution of racemic amines.[46]
Although this reaction mode is limited to 50%
theoretical yield it proofed economically feasible for the synthesis of chiral amines. Consequent-
ly, this method is implemented at industrial scale by BASF since 1999.[47]
This method provides
access to -chiral primary amines and secondary amides.
The limited yield of a kinetic resolution can be circumvented by asymmetric synthesis of the
chiral product. ATAs provide a viable and versatile option to achieve this.[48]
These PLP depend-
ent enzymes catalyse the transamination of an amino group to a prochiral ketone or aldehyde.
Additionally, deracemisation of a racemic mixture with two enantiocomplementary ATAs is ap-
plicable to obtain enantiopure amines in 100% theoretical yield. This approach was also shown
to work in one pot reactions.[49]
The application of ATAs for the asymmetric transamination is
usually limited by the unfavourable equilibrium. Anyhow, several methods to shift the equilibri-
um toward the product are established allowing quantitative conversions.[48]
Furthermore, wild-
type ATAs are usually limited to the transamination of ketones having at least one substituent
not larger than a methyl group. This drawback was successfully addressed by protein engineer-
ing, as shown in chapter 2.2 (Article I) and chapter 3.3 (Article III). ATAs provide access to -chiral
primary amines, though, in some cases, a spontaneous ring closure can also yield-chiral sec-
ondary amines, if suitable substrates are employed (see chapter 2.2 for further details).[50, 51]

12 1 Introduction
Amine Dehydrogenases (AmDH) resemble the overall reaction catalysed by ATAs. However,
AmDH employ ammonia and nicotinamide adenine dinucleotide (NADH) for the reductive ami-
nation of ketones. This kind of biotransformation is highly desirable, since ammonia is a cheap
achiral amino donor that facilitates convenient downstream processing because it is easily sep-
arated from the amine product. No wild-type enzyme displaying this activity is known to date,
but protein engineering gave rise to two (R)-selective AmDH recently[52]
(see chapter 2.1 for fur-
ther details). Additionally, a chimeric AmDH was obtained via domain shuffling of the two
aforementioned enzymes[53]
To date, the compounds accessible with AmDH are limited to a few
-chiral primary amines.[54]
The biocatalytic synthesis of secondary amines can be achieved with imine reductases
(IREDs). These enzymes catalyse the asymmetric reduction of cyclic imines to the corresponding
secondary amines.[55]
Additionally, it was recently demonstrated, that IREDs also catalyse the
reduction of acyclic imines.[56]
Furthermore, two IREDs from Streptomyces were shown to cata-
lyse a direct asymmetric reductive amination of three different aryl-aliphatic ketones, although
with low yields (<9%).[57]
Similarly to AmDH, the selection of IREDs was limited to a few (R)- and
(S)-selective enzymes that were reported to be expressed recombinant,[56-61]
but only recently 32
new enzymes were described.[62, 63]
The current limitations of IREDs are low activities, possibly
because imine reduction is not the main activity of these enzymes (promiscuous activity), or
because the natural substrate is not yet discovered. Furthermore, some enzymes show a nar-
row substrate scope.[59]
Another option for the selective synthesis of optically active secondary and tertiary (as well
as primary) amines arises by the combination of a monoamine oxidase (MAOs) with a non-
selective chemical reducing agent.[64]
MAOs are flavin-dependent enzymes that catalyse the
aerobic oxidation of amines to the corresponding imines. These enzymes can be employed in a
deracemisation strategy. For this purpose, one enantiomer of a racemic amine is oxidised to the
corresponding imine by MAO. A non-selective chemical reduction of the imine yields racemic
amine (e.g. with borane). Subsequent stereoselective oxidation to the imine by MAO leads to
the accumulation of the non-reactive enantiomer to up to 100%, eventually.[48]
Unlike ATAs, the
current drawback of MAOs is not an unfavourable thermodynamic equilibrium but a strong
substrate and product inhibition[48]
, which can be circumvented, for instance, with substrate
feeding and product trapping. For an example of the successful application of an engineered
MAO in the synthesis of a tertiary amine in an upscaled reaction see chapter 2.2.
Additionally, secondary and tertiary chiral amines became available recently by engineered
variants of the opine dehydrogenase (OpineDH) from Arthrobacter sp. 1C.[65]
Naturally, this en-
zyme catalyses the condensation of L-amino acids with either pyruvate, 2-oxobutyrate or glyox-
ylate to yield secondary amines called opines.[66]
The wild-type enzyme lacks the ability to con-
dense non-functionalised ketones without a carboxylic function with non-functionalised amines
instead of amino acids. This drawback was overcome by protein engineering, which evolved
variants applicable in the synthesis several secondary and tertiary amines. One of them carrying
29 mutations (corresponding to an 8% mutation rate) was applied in the synthesis of Ver-
nakalant, a 1,2-amino alcohol used as antiarrhythmic drug (see Figure 1 for the structure of
Vernakalant).[67, 68]

1.3 Routes towards Chiral Amines and Amino Alcohols 13
Figure 3 Overview of the available enzyme activities applicable for the synthesis of -chiral
amines. ATA = amine transaminase, AmDH = amine dehydrogenase, IRED = imine reductase,
MAO = monoamine oxidase, OpineDH = opine dehydrogenase, BBE = berberine bridge en-
zyme
Pictet-spenglerases, such as norcoclaurine synthase (NCS, EC 4.2.1.78) and strictosidine syn-
thase (STR, EC 4.3.3.2) provide access to secondary amines. These plant enzymes catalyse an
asymmetric C-C-bond formation (Pictet-Spengler reaction). The substrates - an aldehyde and an
amine - are condensed to tetrahydroisoquinolines. Thus, chirality is introduced by a C-C-bond
formation, which is in contrast to most other biocatalysts employed for amine synthesis. NCS
shows a rather broad substrate tolerance concerning the aldehyde and it was demonstrated to
be useful for the synthesis of non-natural tetrahydroisoquinolines.[69, 70]
Since asymmetric pic-
tet-spengler reactions are challenging to achieve[71]
, these enzymes represent a potentially val-
uable alternative for the synthesis of tetrahydroisoquinolines and indole alkaloids.[72]
Optically pure tertiary amines are accessible biocatalytically by kinetic resolution via an oxi-
dative enantioselective C-C-bond formation. This reaction is catalysed by the flavoenzyme ber-
berine bridge enzyme (BBE, EC 1.21.3.3) at the expanse of molecular oxygen. Its biocatalytic
potential was demonstrated for the synthesis of berbines using the BBE originating from the
organism Eschscholzia californica. The enzyme accepts modifications at the isoquinoline ring of
the substrate, as well as a methoxy substituent at the phenol moiety. This potential was ex-
ploited in the synthesis of seven different derivatives.[73]
Applicability of the enzyme could be
demonstrated with the successful synthesis of four non-natural derivatives at small preparative
scale.[74]
In conclusion, the options for the enzymatic preparation of chiral amines are manifold. This
field of biocatalysis is an active area of research and recent achievements contributed to the
extension of the enzymatic toolbox of amine synthesis. This recent progress is reviewed in Arti-
cle I and is summarised in the following chapter.
-chiral amine

Can Biocatalysis Compete? - Article I 2Biocatalysis has become an important area in synthetic organic chemistry.
[75-77] Almost 10% of
all organic chemistry papers contain elements of biocatalysis and over 130 processes in indus-
try employ enzymes.[4]
To evaluate the usefulness and applicability of biocatalysis for the syn-
thesis of chiral amines, this chapter compares several enzymatic approaches with traditional
chemical synthesis approaches that apply non-biological chiral catalysts. Therewith it sums up
review Article I, which is focused on the last three years prior to its publication. This chapter, as
well as Article I, is structured in three development phases that are usually passed in order to
evolve a useful biocatalyst. In the first step, new enzymes with new activities or other improved
properties need to be discovered. In the next phase these enzymes are tuned by adjusting
properties such as the catalytic scope or stability under desired conditions. Finally, the biotrans-
formation is further optimised to allow applications at industrial scale.
Recently Identified and Newly Developed Biocatalysts 2.1
Amine dehydrogenases (AmDH) are novel biocatalysts, which were successfully engineered
from amino acid dehydrogenases. At the time of writing this thesis no wild-type enzyme was
known to catalyse the asymmetric reduction of non-functionalised ketones with ammonia. Pro-
tein engineering of L-leucine dehydrogenase and L-phenylalanine dehydrogenase furnished two
(R)-selective variants which catalysed this reaction with good activities (0.69 U/mg for amination
of methyl isobutyl ketone[52]
and 4 U/mg for the amination of para-fluorophenylacetone[78]
) and
with perfect enantioselectivity in both cases (see Article I, Scheme 1a for the latter example). To
achieve this, the carboxylate binding site of the wild-type enzymes was altered by mutagenesis
to accept aliphatic substituents. Small preparative scale synthesis of the two products [(R)-1,3-
dimethylbutylamine and (R)-1-(4-fluorophenyl)-propyl-2-amine] proofed the concept successful-
ly. This reaction is highly desirable as it is very step-efficient compared to chemical hydrogena-
tions using non-biological transition-metal catalysts or chiral auxiliaries. Drawbacks of these
new biocatalysts are the very narrow substrate scope and an unfavourable equilibrium for the
biocatalytic interesting reaction direction.
The hydroamination of alkenes is also a highly demanded reaction, but to date no enzymes
are known to catalyse the addition of ammonia (or another amine) to non-functionalised al-
kenes. In contrast, wild-type enzymes catalyse the addition of ammonia to ,-unsaturated car-
boxylic acids, which yields the corresponding amino acids. Protein engineering was successfully
applied for the generation of variants capable of the synthesis of non-canonical amino acids.
Furthermore, a single mutation of a 3-methylaspartase lyase proofed sufficient to alter the nu-
cleophile specificity from ammonia to amines, however, only with low turnover numbers. An-
other mutation enabled the change of the electrophile specificity towards a broad range of C2-
fumarate derivatives. In another study, the screening of 300.000 mutants revealed a variant of
aspartase capable of the synthesis of the -amino acid 3-amino butanoic acid (Article I, Scheme
1b,1c and 1d, respectively). Nonetheless, the biocatalytic hydroamination of non-functionalised
alkenes stays currently out of reach.
The examples mentioned before aimed to change the substrate specificity of wild-type en-
zymes through protein engineering. Another way to obtain an enzyme catalysing a certain reac-
tion is to alter the reaction specificity. This strategy was successfully applied by McIntosh et al.,

2.2 Engineered Enzymes, Smart Substrates and Process Optimisation 15
who created C-H amination activity in the P450BM3 monooxygenase. The engineered variant
carried 15 mutations and catalysed an intramolecular asymmetric C-H amination, which fur-
nished sultam with 87% ee at 77% conversion and the by-product sulfonamide (Article I, Scheme
1e).[79]
Interestingly, although the reaction is known from synthetic chemistry it is not observed
in nature.
Protein engineering is a powerful tool to generate new enzymes with improved properties.
The field of biocatalysis encountered a stage of development, at which enzymes are designed to
meet the requirements of a certain process, rather than building the process around the needs
of the biocatalyst.[80]
Anyhow, despite these advances, the discovery of new enzyme activities is
still a rewarding endeavour.[81]
Only recently, imine reductases (IRED, EC 1.5.1.X) were recog-
nised for their biocatalytic potential in chiral amine synthesis. (R)- and (S)-selective IREDs from
Streptomyces sp. with unknown metabolic function were discovered by enrichment cultures. The
(S)-selective enzyme proofed useful for the reduction of 1-methyl-3,4-dihydroisoquinoline to the
corresponding amine[55]
(Article I, Scheme 1f). Additionally, IREDs were only recently shown to
catalyse the reduction of at least nine acyclic substrates[56]
and the direct reductive aminationof
three different aryl-aliphatic ketones.[57]
Pictet-Spenglerases catalyse a C-C-bond formation, which has been known for over three
decades. However, the biocatalytic potential of a plant NCS (a Pictet-Spenglerase) was demon-
strated only recently in the synthesis of (S)-norcoclaurine (Article I, Scheme 1g) and three addi-
tional derivatives with good to excellent isolated yields.[70]
Engineered Enzymes, Smart Substrates and Process Op-2.2
timisation
Thanks to available crystal structures, the rational design of enzymes was successfully applied
to obtain biocatalysts for chiral amine synthesis. For instance, an eight-fold mutant of ATA Vibrio
fluvialis (ATA-Vfl) displayed a 60-fold improved activity towards a bulky ketone. The correspond-
ing amine product is a valuable intermediate in the asymmetric synthesis of imagabaline (Article
I, Scheme 1h).[82]
As reported in detail in chapter 3.3 (Article III), we designed other mutants of
ATA-Vfl semi-rationally to achieve an enlarged small binding pocket, which accepted ketones
with two bulky substituents (Table 1). A double mutant was successfully applied in the small-
scale preparative synthesis of the valuable 1,2-amino alcohol (R)-phenylglycinol. As mentioned
earlier, ATAs were also successfully applied in the syntheses of secondary amines. To achieve
this, the substrate must be chosen in a way that allows spontaneous cyclisation of the transam-
ination product. This strategy was successfully applied in the synthesis of a building block of an
Orexin receptor agonist (treatment of insomnia),[51]
a building block of niraparib (anti-cancer
drug) and (+)-dihydropinidine,[50]
a potential antifeedant against the pine weevil (Article I,
Scheme 2a-b). Especially noteworthy is the synthesis of the Niraparib building block as the dy-
namic kinetic resolution employs an ATA which displays high -enantioselectivity, a feature rare-
ly found in ATAs.[83]
Also MAOs were successfully engineered to broaden their applicability. The engineering of a
MAO from Aspergillus niger (MAO-N) yielded a variant capable of deracemisation of an amine
with two bulky substituents, which is an intermediate in the synthesis of the antihistamine
levocetirizine (Article I, Scheme 1i).[84]
Applicability of MAOs was further improved by the devel-
opment of an (R)-selective enzyme, since this enantioselectivity was formerly not available.[85]
An

16 2 Can Biocatalysis Compete? - Article I
example for the synthesis of a tertiary amine is the application of the MAO-N-D9 variant applied
in the synthesis of the alkaloid (R)-harmicine, which displays strong anti-Leishmania activity. In
the first step MAO-N-D9 generates an imium ion, which undergoes symmetric spontaneous
cyclisation yielding rac-harmicine. Subsequent deracemisation by the same enzyme furnishes
the (R)-enantiomer with 83% conversion and perfect enantiomeric excess (>99% ee, Article I,
Scheme 2d).[84]
Examples of Industrial Scale Biotransformations 2.3
The successful upscaling of a biotransformation is the ultimate goal in the maturation process
of a biocatalyst. For instance, this was achieved in a process for the synthesis of a Janus kinase 2
(JAK2) inhibitor. The application of the (S)-selective wild-type ATA-Vfl in a large scale process
allowed for the synthesis of the intermediate (S)-1-(5-fluoropyrimidin-2-yl)-ethanamine, which
was obtained in good yields (66%) and high optical purity (97% ee). The substrate concentrations
which could be applied in this process was ~290 mM, which corresponds to a STY of 51 g d-1
l-1
(Article I: Scheme 3a, Table 1 entry 10).[86]
However, the most important advantages are the mild
conditions under which the reaction occurs. Due to the instability of the pyrimidine ring of the
substrate of this reaction, numerous other strategies failed, as they gave an oily tar. This could
be circumvented by the use of the ATA. Later, the engineered enzyme ATA TA-P1-A06 from Co-
dexis improved the process even further, as higher substrate concentration could be applied
and slightly higher conversion and enantiomeric excess was obtained, resulting in a slightly bet-
ter STY (350 mM, C = 68%, >99% ee, STY = 57 g d-1
l-1
, respectively, Article I, Table 1, entry 11).[87]
A second successful example is the chemoenzymatic asymmetric synthesis of a key building
block of boceprivir, a new drug for the treatment of hepatitis C. The biocatalytic step is an en-
zymatic oxidative desymmetrisation of the prochiral amine substrate (Article I: Scheme 3b). It is
catalysed by an engineered MAO with 8-fold higher activity, improved enzyme solubility and
thermal stability, when compared to the wild-type. Anyhow, the major drawback of substrate
and product inhibition of the enzyme still remained. On the one hand, this was solved by care-
ful substrate feeding, and on the other, by the trapping of the imine product during the reac-
tion. This route is superior in terms of efficiency and is considered more environmentally be-
nign compared to the preceding manufacturing process.[88]
The most famous example for the successful implementation of an engineered ATA in a
large scale process is the manufacturing of sitagliptin (Januvia).[89]
This blockbuster drug (= rev-
enue > $1 billion p.a.) was developed by Merck & Co for the treatment of diabetes type II. Its
biocatalytic manufacturing process can be considered as one of the benchmark reactions in
asymmetric amine biocatalysis for several reason: (i) extensive protein design yielded an en-
zyme which was stable under process conditions, (ii) the enzyme accepts a substrate not con-
verted by the wild-type (two very bulky substituents) at high concentrations, (iii) it displays per-
fect enantioselectivity (99% ee) and affords high conversion (92%), (iv) a decreased waste pro-
duction and no need for transition-metal catalysts, (v) a high STY is achieved, (vi) the possibility
of easy separation and reusability of the enzyme due to immobilisation is possible and (vi)
thanks to immobilisation, activity and stability of the enzymes is retained in organic solvent,
thereby simplifying workup even further (Article I: Table 1, entry 16, Scheme 3c).[89-91]
As shown in this summary of Article I, biocatalytic amine synthesis is an active field of re-
search. Remarkably, genuine novel enzymes such as AmDH have been developed. New enzyme

2.3 Examples of Industrial Scale Biotransformations 17
activities such as IREDs and pictet-spenglerases have been utilised for biocatalytic amine syn-
thesis recently. Established biocatalysts such as ATA and MAO have been improved further to
meet certain process parameters. The current enzymatic toolbox contains new advanced tools
and is competitive with chemical synthesis employing non-biological catalysts. In light of these
developments, the scope of biocatalysis in chiral amine synthesis can be expected to be broad-
ened even more.
The general need for cleaner and environmentally more benign chemical processes calls for
utmost efficient use of the limited resources.[4]
Although this feature is not a privilege of bio-
catalysis, it has a huge potential for more efficient and greener approaches towards chiral
amines. This becomes obvious in the biocatalytic manufacturing process of sitagliptin employ-
ing an ATA or the synthesis of a key intermediate of boceprivir using a MAO. Exploiting this po-
tential will render a significant advantage for synthetic chemists formerly restricted to non-
biological catalysts only.
We were able to exploit this potential for the successful synthesis of an 1,3- and an 1,2-
amino alcohol as presented in the following chapter (chapter 3.2 and 3.3, respectively).

Biocatalytic Syntheses of Amino Alcohols 3This section deals with biocatalytic routes towards amino alcohols. The first subchapter intends
to give an overview of the approaches already described in literature. With this context in mind,
the second subsection discusses a newly developed route towards 1,3-amino alcohols, realised
by the combination of an ATA and a KRED. This work was published in Article II of this thesis.
Finishing the third chapter, the third subsection discusses Article III, which describes the protein
engineering of ATA-Vfl to allow asymmetric synthesis of bulky amines including the 1,2-amino
alcohol (R)-phenylglycinol.
Established Routes towards 1,2- and 1,3-Amino Alcohols 3.1
This subsection is divided into examples that combine biocatalysis with traditional organic
chemistry (chemoenzymatic approaches) and strategies, which solely rely on biocatalysts (bio-
catalytic approaches).
3.1.1 Chemoenzymatic Approaches
In 1930 a patent was filed by the Knoll AG in which the inventors Hildebrandt and Klavehn de-
scribe the synthesis of ephedrine in two steps using baker’s yeast and a diastereoselective re-
duction step.[92]
In this process, benzaldehyde is fermented with baker’s yeast to afford
(R)-phenylacetylcarbinol [(R)-PAC]. This biotransformation is catalysed by the enzyme pyruvate
decarboxylase (PDC): First, a decarboxylation of pyruvate yields acetaldehyde, which is ligated
with benzaldehyde by PDC to yield (R)-PAC. Finally, this intermediate is reacted with methyla-
mine and the resulting imine is reduced diastereoselectivly to the corresponding amine, yielding
ephedrine (Figure 4). This strategy is still employed today almost unchanged for the production
of > 120 per year ephedrine[4]
and is still a subject of research and further improvement.[93]
Figure 4 Manufacturing process of ephedrine patented 85 years ago, which is still employed
today. Decarboxylation by pyruvate decarboxylase (PDC) yields acetaldehyde which under-
goes carboligation with benzaldehyde yielding (R)-phenylacetylcarbinol ((R)-PAC). The carboli-
gation is also catalysed by PDC. A subsequent diastereoselective reductive amination yields
ephedrine.
Another example for the combination of enzymes with synthetic chemistry is the synthesis
of 1-amino-2-indanol (Figure 5). This compound is an important building block of the HIV prote-
ase inhibitor indinavir (Figure 1). Furthermore it was successfully employed as a basic compo-
nent for chiral auxiliaries used in the asymmetric catalysis of Diels-Alder reactions and it repre-
sents a valuable ligand for the asymmetric transfer hydrogenation of ketones.[94]
After oxidation
of indanone with manganese-(III)-acetate, the corresponding 2-acetoxy indanone was subjected
to enantioselective hydrolysis using a lipase from Pseudomonas sp. The corresponding hydroxy
ketone was then transaminated by the (S)-selective ATA-Vfl to yield the (1R,2R)-isomer (trans-
isomer). Furthermore, the cis-isomer is obtained via the synthesis of the racemic 1,2-amino al-

3.1 Established Routes towards 1,2- and 1,3-Amino Alcohols 19
cohol by asymmetric reductive amination. A subsequent kinetic resolution using the same ATA
yielded the (1S,2R)-isomer (cis-isomer).[95]
Figure 5 The combination of a lipase and an ATA allows for the synthesis of the trans-isomer of
1-amino-2-indanol via asymmetric synthesis (A) and the cis-isomer via reductive amina-
tion (B) followed by a kinetic resolution. Note that the ATA is (S)-selective but according to
priority rule the (R)-enantiomer is obtained.
A drawback of this method is the low theoretical yield, which is limited to 50% for the trans-
isomer since the hydroxy ketone is obtained in a kinetic resolution. The maximum possible yield
of the cis-isomer is even lower (25%) since it is obtained in two consecutive kinetic resolutions.
One method to avoid the drawback of low theoretical yields is applied in the following ex-
ample. The study by Millet et al. demonstrates the synthesis of 1,3-amino alcohols using a com-
bination of organo-, organometallic- and biocatalysis.[21]
In this approach, the first stereocentre
is installed in an asymmetric fashion. Proline is utilised as organocatalyst in an enantioselective
Mannich reaction to yield N-Boc protected 1,3-amino ketones. The second step is a dynamic
kinetic asymmetric transformation. The 1,3-amino ketone is reduced to the corresponding alco-
hol using Shvo’s catalyst, which is also cable of the epimerisation of it (Figure 6). As the enanti-
oselective lipase is also present in the same pot, the (R)-configured alcohol is constantly con-
sumed in a stereoselective transesterification, yielding the corresponding acetate. This combi-
nation of reduction, epimerisation and stereoselective transesterification yields diastereo- and
enantiomerically pure N-Boc protected 1,3-amino acetates. This approach was proven to be
useful for the synthesis of seven compounds (yields between 73-93%, 99% ee in all cases, dia-
stereomeric ratio > 92:8) and represents the only described synthesis of 1,3-amino alcohols
employing an enzyme, besides the new approach described in Article II (chapter 3.2).

20 3 Biocatalytic Syntheses of Amino Alcohols
Figure 6 The combination of organocatalysis (proline), organometallic catalysis (Shvo’s catalyst)
and biocatalysis [lipase B from Candida antarctica (CalB)] allows for the synthesis of 1,3-
amino alcohols in a dynamic kinetic asymmetric transformation. The enantiopure 1,3-
amino ketones are available by organocatalysis using either L- or D-proline in a Mannich reac-
tion. The reduction to the corresponding alcohol and its epimerisation is catalysed by the
Shvo catalyst. Stereoselective transesterification by CalB yields diastereo and enantiomerical-
ly pure N-Boc protected 1,3-amino acetates.
3.1.2 Biocatalytic Approaches
The following study is an example for the setup of two stereocentres using exclusively asym-
metric synthesis steps, thereby circumventing the drawback of low theoretical yield as dis-
cussed for 1-amino-2-indanol (Figure 5). First, an engineered transketolase [(TK (D496T)] from
Escherichia coli is used for the catalysis of a stereo selective carboligation yielding an α-α‘-
dihydroxy ketone (Figure 7). A subsequent transamination using ATA2025 from Chromobacte-
rium violaceum DSM30191 (ATA-Cvi) yields the corresponding aminodiol (2S,3S)-2-aminopentan-
1,3-diol.[96]
This strategy was applicable in preparative scale yielding ~185 mg of the final isolat-
ed product. The drawback of this approach is the relatively low selectivity of the carboligation
reaction. The final diastereomeric excess was 61% de with the impurity being the (2S,3R)-isomer.
This indicates that the ATA shows no selectivity for the C3 position, which is in agreement with a
similar example.[97]
Figure 7 The combination of an engineered transketolase (TK) and an amine transaminase (ATA)
allows for the asymmetric synthesis of the aminodiol (2S,3S)-2-aminopentan-1,3-diol.
Starting from the achiral substrates propanal and hydroxypyruvate a stereoselective enzy-
matic carboligation gives the α-α‘-dihydroxy ketone (S)-1,3-dihydroxypentan-2-one. This is fur-
ther transaminated by the ATA from Chromobacterium violaceum to yield (2S,3S)-2-
aminopentan-1,3-diol. ThDP = Thiamine pyrophosphate
Another example for the combination of two enzymes is the biocatalytic synthesis of
norephedrine. Its synthesis was accomplished by the same strategy discussed above. In a first
step, a stereoselective carboligation is conducted, which is catalysed by the (R)-selective enzyme
acetohydroxyacid synthase I (AHAS-I) from Escherichia coli (AHAS-I-Eco). This reaction furnishes
the 1,2-hydroxy ketone (R)-PAC, which is then subsequently transaminated to yield
nor(pseudo)ephedrine. This strategy is applicable with enantiocomplementary ATAs, thereby
providing access to (1R,2S)-norephedrine employing (S)-selective ATA-Cvi and (1R,2R)-

3.1 Established Routes towards 1,2- and 1,3-Amino Alcohols 21
norpseudoephedrine employing (R)-selective ATA from Aspergillus terreus (ATA-Ate) with high
conversion (>95%) and optical purity of 95% ee and >98% de (Figure 8, Route A)[30]
Since alanine
is used as amino donor, pyruvate builds up during the transamination reaction, which is recy-
cled in the carboligation of the first step, thereby shifting the equilibrium towards the product
(see also Article I, Scheme 2e for recycling mode).
Figure 8 Biocatalytic synthesis of all four stereoisomers of the 1,2-amino alcohols
nor(pseudo)ephedrine. Route A: combines the (R)-selective AHAS-I-Eco catalysing a carboli-
gation, with an ATA suitable for transamination of the 1,2-hydroxy ketone (R)-PAC resulting
from the carboligation step. Since two enantiocomplementary ATAs are available the synthe-
sis of (1R,2R)-norpseudoephedrine and (1R,2S)-norephedrine is possible via this route. Route
B: combines two enantiocomplementary ATAs with two enantiocomplementary ADHs. In the
first step, an asymmetric transamination of the 1,2-diketone PPDO is conducted with either
(S)- or (R)-selective ATAs. The 1,2-amino ketone is then further reduced to the corresponding
1,2-amino alcohols (1S,2S)-norpseudoephedrine or (1S,2R)-norephedrine using the (S)-
selective enzyme ADH-Lbr. Additionally, Route B provides access to (1R,2S)-norephedrine via
reduction of (S)-APPO with (R)-selective ADH-Rsp in lower yields but higher optical purity com-
pared to Route A. Abbreviations: ATA-Cvi = amine transaminase from Chromobacterium vio-
laceum, ATA-Ate = amine transaminase from Aspergillus terreus, ADH-Rsp = alcohol dehydro-
genase from Ralstonia sp., ADH-Lbr = alcohol dehydrogenase from Lactobacillus brevis, AHAS-I-
Eco acetohydroxyacid synthase I from Escherichia coli, PPDO = 1-phenylpropane-1,2-dione,
APPO = 2-amino-1-phenylpropan-1-one, PAC = phenylacetyl-carbinol, C = conversion
Access towards the two remaining stereoisomers of nor(pseudo)ephedrine is not yet rea-
sonable via Route A. The only known (S)-selective carboligase catalysing the described reaction
(mutant E469G of pyruvate decarboxylase from Acetobacter pasteurianus)[98]
displays a low ste-
reoselectivity for the carboligation step (~70% ee for (S)-PAC).[99]
Sehl et al. addressed this lack of
access recently: The combination of an ATA with an ADH allowed for the synthesis of the re-

22 3 Biocatalytic Syntheses of Amino Alcohols
maining (1S,2S)-isomer (cathine) and the (1S,2R)-isomer [Figure 8, Route B].[99]
For the synthesis
of cathine, the 1,2-diketone 1-phenylpropane-1,2-dione (PPDO) was transaminated regio- and
stereoselectivly by the (S)-selective ATA-Cvi. The second stereocentre is set up in a subsequent
step by stereoselective reduction of (S)-APPO catalysed by (S)-selective ADH from Lactobacillus
brevis (ADH-Lbr). The amino ketone (S)-APPO is also a substrate for the (R)-selective ADH from
Ralstonia sp. (ADH-Rsp). Its reduction yields the (1R,2S)-isomer in a way being inferior to Route A
in terms of conversion but superior concerning optical purity of the product. In contrast to the
synthesis of cathine, the synthesis of the (1S,2R)-isomer was investigated with whole cells in-
stead of purified enzymes, which gave quite poor optical purity. The major product with ~80%
de was the (1S,2S)-isomer.
This chapter provided examples for biocatalytic approaches towards amino alcohols, which
are described in literature. To the best of our knowledge, only the study of Millet et al. described
the synthesis of 1,3-amino alcohols, which employs biocatalysis in combination with an organo-
ruthenium catalyst (Shvo catalyst). We were able to develop a selective route towards all four
diastereomers of the 1,3-amino alcohol 4-amino-1-phenylpentane-2-ol by using a combination
of enzymes, thereby avoiding toxic transition metal catalysis. The following chapter reports
about the findings of this study.
Please note that the numbering of chemical compounds in the following chapters 3.2 and
3.3, which discuss Article II and III respectively, is adopted from the original publications to aid
understanding and easier portability.
A Novel Route for the Synthesis of an 1,3-Amino Alcohol - 3.2
Article II
As comprehensively described in chapter 1.2 chiral amines in general and amino alcohols in
particular are very useful as chiral auxiliaries or as synthons of biological active compounds
(Figure 1). The efficient asymmetric synthesis of 1,3-amino alcohols bearing more than one ste-
reocentre remains challenging and efficient new methods for their enantio- and diastereoselec-
tive preparation are highly demanded.[21]
In the project reported in Article II, and summarised in
this chapter, we envisaged a biocatalytic approach towards the 1,3-amino alcohol 4-amino-1-
phenylpentane-2-ol (5), which was chosen as a representative molecule bearing the 1,3-amino
alcohol motif (Figure 9). To the best of our knowledge, a synthesis towards 1,3-amino alcohols,
which solely relied on biocatalysis was not described before. In fact, when the project was initi-
ated, the combination of the enzymes ADH and ATA had not been explored for the synthesis of
amino alcohols. In the meantime, the combination of these two enzyme activities was also
shown to be successful in the synthesis of the 1,2-amino alcohol nor(pseudo)ephedrine,
demonstrating the general applicability of this synthetic strategy (Chapter 3.1.2 and Figure 8).
Our approach employs the combination of a KRED and two enantiocomplementary ATAs to
provide access to all four diastereomers of 4-amino-1-phenylpentane-2-ol (5). Details of this
synthetic route are depicted in Figure 9: Starting from the racemic hydroxy ketone rac-3, a kinet-
ic resolution using an (S)-selective KRED provided optically active hydroxy ketone (R)-3 (86% ee)
and the corresponding diketone. After separation and isolation of the hydroxy ketone from the
diketone, (R)-3 was further transaminated using either an (R)- or (S)-selective ATA. This gave ac-
cess to two of four possible diastereomers of the amino alcohol synthesised in this study (5c
and 5d). The diketone was reduced to the (S)-configured hydroxy ketone (S)-3 in a highly regi-
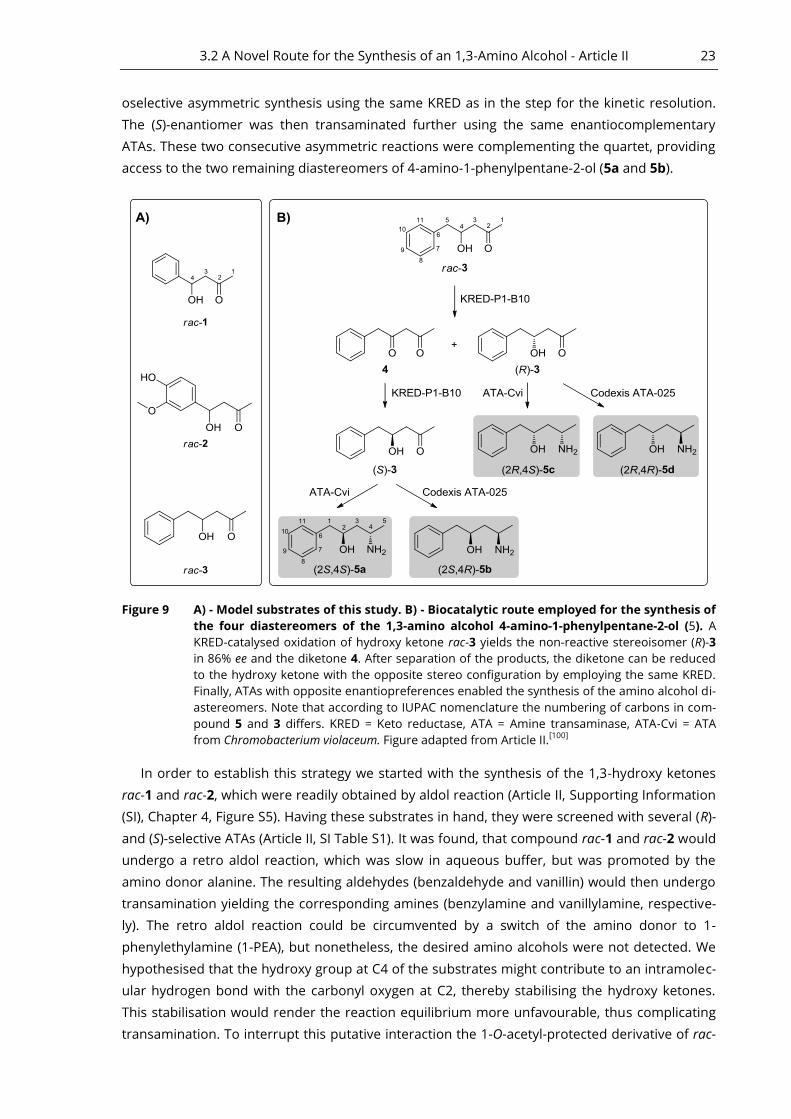
3.2 A Novel Route for the Synthesis of an 1,3-Amino Alcohol - Article II 23
oselective asymmetric synthesis using the same KRED as in the step for the kinetic resolution.
The (S)-enantiomer was then transaminated further using the same enantiocomplementary
ATAs. These two consecutive asymmetric reactions were complementing the quartet, providing
access to the two remaining diastereomers of 4-amino-1-phenylpentane-2-ol (5a and 5b).
Figure 9 A) - Model substrates of this study. B) - Biocatalytic route employed for the synthesis of
the four diastereomers of the 1,3-amino alcohol 4-amino-1-phenylpentane-2-ol (5). A
KRED-catalysed oxidation of hydroxy ketone rac-3 yields the non-reactive stereoisomer (R)-3
in 86% ee and the diketone 4. After separation of the products, the diketone can be reduced
to the hydroxy ketone with the opposite stereo configuration by employing the same KRED.
Finally, ATAs with opposite enantiopreferences enabled the synthesis of the amino alcohol di-
astereomers. Note that according to IUPAC nomenclature the numbering of carbons in com-
pound 5 and 3 differs. KRED = Keto reductase, ATA = Amine transaminase, ATA-Cvi = ATA
from Chromobacterium violaceum. Figure adapted from Article II.[100]
In order to establish this strategy we started with the synthesis of the 1,3-hydroxy ketones
rac-1 and rac-2, which were readily obtained by aldol reaction (Article II, Supporting Information
(SI), Chapter 4, Figure S5). Having these substrates in hand, they were screened with several (R)-
and (S)-selective ATAs (Article II, SI Table S1). It was found, that compound rac-1 and rac-2 would
undergo a retro aldol reaction, which was slow in aqueous buffer, but was promoted by the
amino donor alanine. The resulting aldehydes (benzaldehyde and vanillin) would then undergo
transamination yielding the corresponding amines (benzylamine and vanillylamine, respective-
ly). The retro aldol reaction could be circumvented by a switch of the amino donor to 1-
phenylethylamine (1-PEA), but nonetheless, the desired amino alcohols were not detected. We
hypothesised that the hydroxy group at C4 of the substrates might contribute to an intramolec-
ular hydrogen bond with the carbonyl oxygen at C2, thereby stabilising the hydroxy ketones.
This stabilisation would render the reaction equilibrium more unfavourable, thus complicating
transamination. To interrupt this putative interaction the 1-O-acetyl-protected derivative of rac-
24 3 Biocatalytic Syntheses of Amino Alcohols
1 was synthesised. Unfortunately, after incubation with ATA, the corresponding amino acetate
could not be detected. From these findings we concluded that the hydroxy ketones 1 and 2
were no substrates for the screened ATAs.
We therefore decided to synthesize compound rac-3, assuming that it might be a better sub-
strate since it has a more flexible aromatic substituent compared to 1 and 2. Hydroxy ketone 3
was also accessible via symmetric aldol reaction, however, a two-step strategy implying the
coupling of phenylacetaldehyde with vinylmagnesium bromide and a subsequent Wacker oxida-
tion proved more feasible, as higher yields could be obtained (Article II, SI Scheme S1 for syn-
thesis details).
We found that 3 was stable in buffer and at reaction conditions (including alanine, excluding
ATA). TLC analysis detected the consumption of substrate 3 by several tested ATAs. This could
be confirmed by GC/MS and HPLC after the synthesis of reference compounds by chemical
means (including amino alcohol 5, regioisomer of rac-3 [4-Hydroxy-1-phenylpentan-2-one (6)],
the corresponding diol 1-phenylpentane-2,4-diol (7) and corresponding diketone 1-
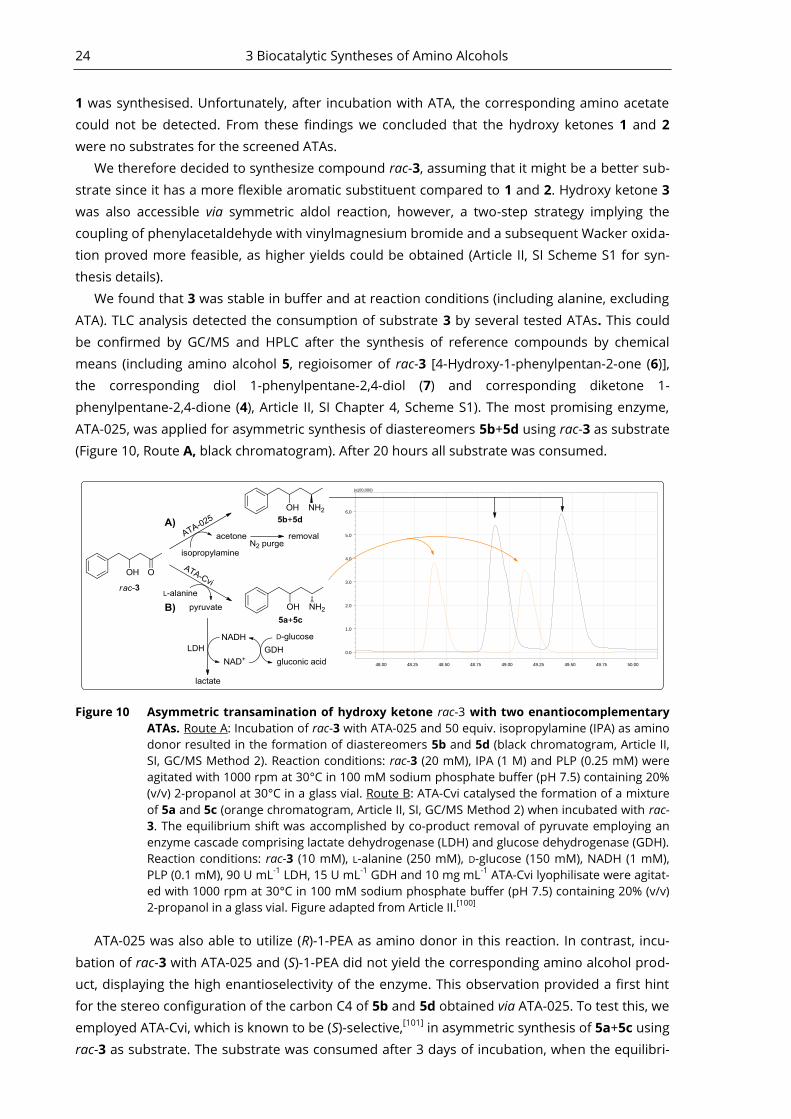
phenylpentane-2,4-dione (4), Article II, SI Chapter 4, Scheme S1). The most promising enzyme,
ATA-025, was applied for asymmetric synthesis of diastereomers 5b+5d using rac-3 as substrate
(Figure 10, Route A, black chromatogram). After 20 hours all substrate was consumed.
48.00 48.25 48.50 48.75 49.00 49.25 49.50 49.75 50.00
0.0
1.0
2.0
3.0
4.0
5.0
6.0
(x100,000)
Figure 10 Asymmetric transamination of hydroxy ketone rac-3 with two enantiocomplementary
ATAs. Route A: Incubation of rac-3 with ATA-025 and 50 equiv. isopropylamine (IPA) as amino
donor resulted in the formation of diastereomers 5b and 5d (black chromatogram, Article II,
SI, GC/MS Method 2). Reaction conditions: rac-3 (20 mM), IPA (1 M) and PLP (0.25 mM) were
agitated with 1000 rpm at 30°C in 100 mM sodium phosphate buffer (pH 7.5) containing 20%
(v/v) 2-propanol at 30°C in a glass vial. Route B: ATA-Cvi catalysed the formation of a mixture
of 5a and 5c (orange chromatogram, Article II, SI, GC/MS Method 2) when incubated with rac-
3. The equilibrium shift was accomplished by co-product removal of pyruvate employing an
enzyme cascade comprising lactate dehydrogenase (LDH) and glucose dehydrogenase (GDH).
Reaction conditions: rac-3 (10 mM), L-alanine (250 mM), D-glucose (150 mM), NADH (1 mM),
PLP (0.1 mM), 90 U mL-1
LDH, 15 U mL-1
GDH and 10 mg mL-1
ATA-Cvi lyophilisate were agitat-
ed with 1000 rpm at 30°C in 100 mM sodium phosphate buffer (pH 7.5) containing 20% (v/v)
2-propanol in a glass vial. Figure adapted from Article II.[100]
ATA-025 was also able to utilize (R)-1-PEA as amino donor in this reaction. In contrast, incu-
bation of rac-3 with ATA-025 and (S)-1-PEA did not yield the corresponding amino alcohol prod-
uct, displaying the high enantioselectivity of the enzyme. This observation provided a first hint
for the stereo configuration of the carbon C4 of 5b and 5d obtained via ATA-025. To test this, we
employed ATA-Cvi, which is known to be (S)-selective,[101]
in asymmetric synthesis of 5a+5c using
rac-3 as substrate. The substrate was consumed after 3 days of incubation, when the equilibri-

3.2 A Novel Route for the Synthesis of an 1,3-Amino Alcohol - Article II 25
um was shifted using the LDH/GDH cascade[102]
(Article II, Experimental Section). Indeed, chiral
analysis of the amino alcohol product gave the two remaining isomers (Figure 10, Route B, or-
ange chromatogram).
Having identified two enantiocomplementary ATAs capable of transaminating rac-3 we
screened for KREDs, which were active on hydroxy ketone 3. To provide (R)-3 a kinetic resolution
of rac-3 was considered (Figure 11). The enzyme KRED-B1-B10 catalysed this reaction. It was
identified within a commercial screening panel of 22 engineered enzymes and catalysed the
reaction with very high regio- and good stereoselectivity (89% ee). Only traces of the regioiso-
mer 4-hydroxy-1-phenylpentan-2-one (6, Article II, SI Chapter 4, Scheme S1) or the correspond-
ing diol 1-phenylpentane-2,4-diol (7) were formed during the reaction, as confirmed via GC/MS
using chemically derived reference compounds (GC/MS Method 3, Article II, SI, Figure S2).
Figure 11 Kinetic resolution of 1,3-hydroxy ketone rac-3. Five of 22 KREDs from a commercial screen-
ing kit were identified to catalyse the desired reaction with the same enantiopreference. As
determined by GC/MS, the reaction was finished after 24 h leaving behind (R)-3, yielding
diketone 4. Reaction conditions: 10 mM rac-3, 1 mM NADP+ , 1 mM MgSO4, 20% (v/v) acetone,
1 mg mL-1
KRED lyophilisate in 100 mM potassium phosphate buffer (pH 7). Agitated with
1200 rpm in a glass vial at 30°C. Figure adapted from Article II.[100]
To provide (S)-3, an asymmetric reduction at C2 of diketone 4 was desired (Figure 12). For
this, a highly regioselective KRED acting exclusively at this position was necessary. The same
KRED employed in the kinetic resolution (KRED-P1-B10) selectively reduced the diketone 4 to
the corresponding hydroxy ketone (S)-3 if the reaction was stopped immediately after the
diketone was consumed. However, if the reaction was continued beyond this time point, the
corresponding diol was formed, leaving behind no hydroxy ketone eventually.
Figure 12 Asymmetric synthesis of hydroxy ketone (S)-3. Reaction conditions: 10 mM rac-3, 1 mM
NADP+, 1 mM MgSO4 , 20% (v/v) 2-propanol, 1 mg mL
-1 KRED-P1-B10 lyophilisate in 100 mM
potassium phosphate buffer (pH 7), agitated at 1200 rpm in a glass vial at 30°C. Figure
adapted from Article II.[100]
Finally, the kinetic resolution of rac-3 (1.8 mmol, 321 mg) was performed in small preparative
scale furnishing the products (R)-3 (160 mg, 50% yield, 86% ee) and 4 (85 mg, 27% yield). Prepar-
ative asymmetric reduction of 4 (0.312 mmol) yielded 48 mg (S)-3 (86% yield; 71% ee).
Using (R)-3 and (S)-3 as substrates for either ATA-025 or ATA-Cvi, all four diastereomers of 5
could be synthesised separately on an analytical scale with high enantioselectivity (>98% ee at

26 3 Biocatalytic Syntheses of Amino Alcohols
carbon C4 carrying the amino function, full consumption of the substrates). Preparative scale
transamination of (R)-3 (0.5 mmol, 90 mg) was conducted with ATA-025, which resulted in 5d
with 73% yield (66 mg).
The amino alcohol 5d was isolated and its absolute stereo configuration was elucidated via
NMR (see Article II, SI, Figure S4). From this data, the absolute configuration of all other prod-
ucts could be derived eventually.
In conclusion, these results proofed that the combination of ATA and KRED enzymes is a val-
uable option for 1,3-amino alcohol synthesis. It allows for a successive introduction of two ste-
reocentres. This approach constitutes a valuable addition to the traditional synthesis strategies
towards this compound class, as it is highly selective, step efficient and avoids protecting groups
as well as transition metal catalysis.
Future research will probably improve the potential of this strategy even further by: (i) the
development of this reaction as a one pot or as a cascade reaction, (ii) increasing substrate con-
centrations and reducing excess of the amino donor alanine to improve scalability, (iii) identifi-
cation of enzymes that facilitate the synthesis of the remaining regioisomer of 5 with the amino
group at the “inner” position (4-amino-5-phenylpentan-2-ol).
The latter remains challenging since wild-type ATAs display a strict selectivity for the reduc-
tive amination of aryl-aliphatic ketones having an aliphatic group not larger than an ethyl group.
This is caused by a “small” and a “large” binding pocket found in all wild-type ATAs described to
date. The “large” binding pocket may accommodate relatively large substituents such as naphtyl
groups. In contrast, the “small” binding pocket is restricted in size and activity drops significantly
for substrates whose small residue exceeds the size of a methyl group.[103, 104]
To address this drawback we conducted protein engineering on the (S)-selective wild-type
ATA from Vibrio fluvialis (ATA-Vfl) which enabled the synthesis of the 1,2-amino alcohol (R)-
phenylglycinol and the amine (S)-1-phenylbutylamine. By the time this project was initiated no
example of an (S)-selective ATA applicable for the synthesis of amines with two bulky substitu-
ents was reported in literature. This work was published in Article III and is summarised in the
following section.
An Engineered ATA Suitable for the Synthesis of an 1,2-3.3
Amino Alcohol - Article III
As discussed in the preceding section, the stereo selective synthesis of amino alcohols bearing
more than one stereocentre remains challenging. Chapter 2 exemplifies how ATAs can be em-
ployed as valuable biocatalysts for enantioselective amine synthesis. Anyhow, one of their big
advantages, the regioselectivity, can become a drawback under certain circumstances. The ap-
plication of wild-type ATAs in the synthesis of amines with two bulky residues stays out of ques-
tion, as the active site is comprised of a “small“ and a “large“ binding pocket. These pockets de-
termine regio- and enantioselectivity by only allowing conversion of substrates bearing a “small“
and a “large“ substituent.[103]
To circumvent this restriction one can use protein engineering,
which provides a powerful option to evolve ATAs accepting substrates with two bulky residues.
As comprehensively reported in Article I, this was shown before by the engineering of an (R)-ATA
applied in the synthesis of sitagliptin[89]
or, only recently, for an (S)-selective ATA employed for
the synthesis of a key intermediate of imagabalin[82]
(Scheme 1h and Scheme 3c; Chapter 2.2
and 2.3 in this thesis).

3.3 An Engineered ATA Suitable for the Synthesis of an 1,2-Amino Alcohol - Article III 27
Since an engineered (S)-selective ATA was lacking by the time this project was initiated, we
were inspired by the design of the (R)-selective ATA applied for the synthesis of sitagliptin and
analysed the amino acids contributing to the small binding pocket of the (S)-selective ATA-Vfl. It
is composed of eight residues: Leu56, Phe85*, Phe19, Val153, Tyr150 and three additional ami-
no acids which form the PLP Binding Cup (Gly320*, Phe321*, and Thr322*; see Figure 13). We
applied a (partial) saturation mutagenesis strategy for the construction of mutants with an in-
creased size of the small binding pocket. Therefor the first five residues were (partly) saturated
with smaller amino acids, whereas the residues belonging to the PLP binding cup were excluded
from mutagenesis, as they are responsible for the coordination of the cofactor.
The mutants were tested with the two bulky ketones 1-phenylbutane-2-one (2a) and 2-
hydroxyacetophenone (3a) as model substrates. Their conversion requires fitting a steric more
challenging substituent in the “small” pocket compared to the benchmark substrate acetophe-
none (1a). Assuming that mutants capable of the kinetic resolution of the corresponding amines
1b-3b would also perform reasonably well in the asymmetric synthesis reaction direction, we
screened for deamination via detection of the ketones. The quantification of the ketones 2a and
3a was accomplished via UV absorption by a customisation of the commonly applied acetophe-
none assay.[105]
Figure 13 The small binding pocket of ATA-Vfl as derived from the crystal structure (PDB 4E3Q).
The cofactor pyridoxal 5’-phosphate (PLP, light grey) is bound to 1-phenylethylamine (1-PEA,
black) forming an aldemine. Going clockwise starting from top: Lys285 is the catalytic residue,
which covalently binds to the cofactor PLP during the catalytic cycle. Black: The large binding
pocket is comprised (among others) of the residues Arp57, Arg415, Ala228, Ile259, Val258.
Bottom: Asp256 is responsible for the protonation of the pyridine nitrogen of PLP. Residues
of the PLP Binding Cup were not considered during mutagenesis as they are responsible for
the binding of PLP. Orange: Small binding pocket comprised of (among others) Tyr150,
Val153, Phe19, Phe85*, Leu56. Some residues were omitted for clarity; see Article III for de-
tails. Figure adapted from Article III.[106]
The single mutation of the residues Phe85*, Tyr150 or Val153 resulted in variants with im-
proved specific activities in kinetic resolution mode for all tested substrates (1b-3b). The variant
Y150M displayed the highest activity in conversion of (R)-phenylglycinol (3b, 40-fold) whereas
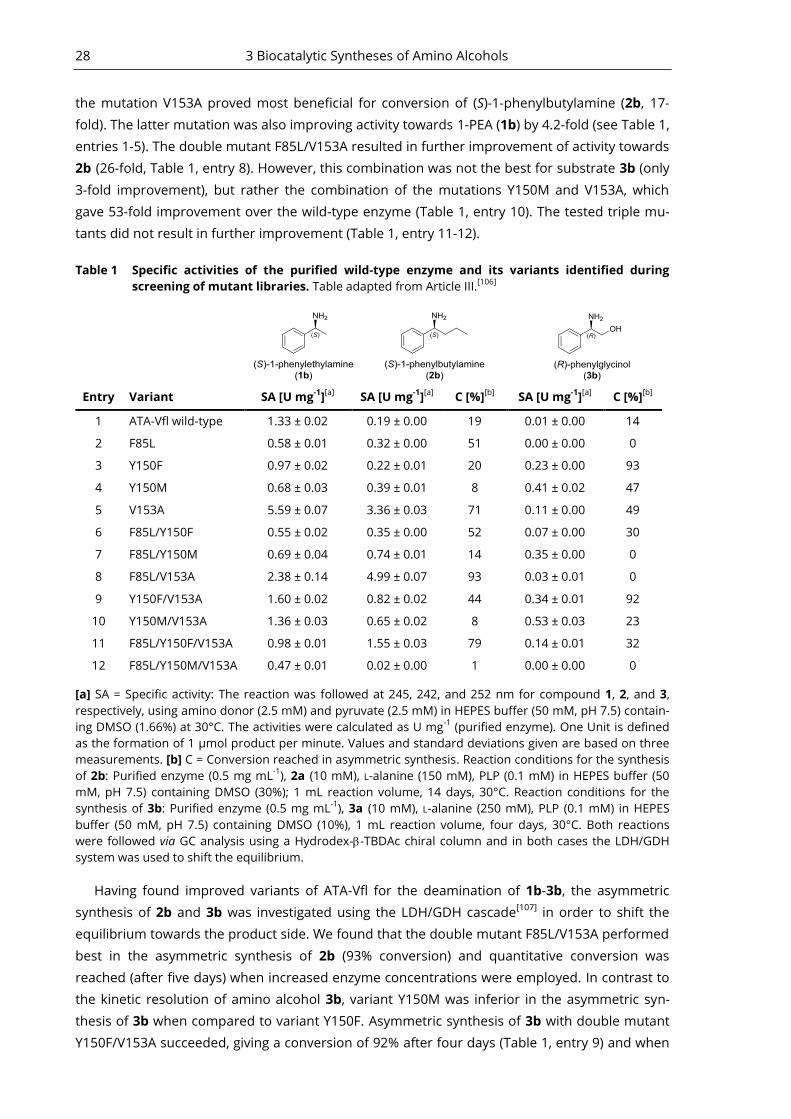
28 3 Biocatalytic Syntheses of Amino Alcohols
the mutation V153A proved most beneficial for conversion of (S)-1-phenylbutylamine (2b, 17-
fold). The latter mutation was also improving activity towards 1-PEA (1b) by 4.2-fold (see Table 1,
entries 1-5). The double mutant F85L/V153A resulted in further improvement of activity towards
2b (26-fold, Table 1, entry 8). However, this combination was not the best for substrate 3b (only
3-fold improvement), but rather the combination of the mutations Y150M and V153A, which
gave 53-fold improvement over the wild-type enzyme (Table 1, entry 10). The tested triple mu-
tants did not result in further improvement (Table 1, entry 11-12).
Table 1 Specific activities of the purified wild-type enzyme and its variants identified during
screening of mutant libraries. Table adapted from Article III.[106]
Entry Variant SA [U mg-1
][a]
SA [U mg-1
][a]
C [%][b]
SA [U mg-1
][a]
C [%][b]
1 ATA-Vfl wild-type 1.33 ± 0.02 0.19 ± 0.00 19 0.01 ± 0.00 14
2 F85L 0.58 ± 0.01 0.32 ± 0.00 51 0.00 ± 0.00 0
3 Y150F 0.97 ± 0.02 0.22 ± 0.01 20 0.23 ± 0.00 93
4 Y150M 0.68 ± 0.03 0.39 ± 0.01 8 0.41 ± 0.02 47
5 V153A 5.59 ± 0.07 3.36 ± 0.03 71 0.11 ± 0.00 49
6 F85L/Y150F 0.55 ± 0.02 0.35 ± 0.00 52 0.07 ± 0.00 30
7 F85L/Y150M 0.69 ± 0.04 0.74 ± 0.01 14 0.35 ± 0.00 0
8 F85L/V153A 2.38 ± 0.14 4.99 ± 0.07 93 0.03 ± 0.01 0
9 Y150F/V153A 1.60 ± 0.02 0.82 ± 0.02 44 0.34 ± 0.01 92
10 Y150M/V153A 1.36 ± 0.03 0.65 ± 0.02 8 0.53 ± 0.03 23
11 F85L/Y150F/V153A 0.98 ± 0.01 1.55 ± 0.03 79 0.14 ± 0.01 32
12 F85L/Y150M/V153A 0.47 ± 0.01 0.02 ± 0.00 1 0.00 ± 0.00 0
[a] SA = Specific activity: The reaction was followed at 245, 242, and 252 nm for compound 1, 2, and 3,
respectively, using amino donor (2.5 mM) and pyruvate (2.5 mM) in HEPES buffer (50 mM, pH 7.5) contain-
ing DMSO (1.66%) at 30°C. The activities were calculated as U mg-1
(purified enzyme). One Unit is defined
as the formation of 1 µmol product per minute. Values and standard deviations given are based on three
measurements. [b] C = Conversion reached in asymmetric synthesis. Reaction conditions for the synthesis
of 2b: Purified enzyme (0.5 mg mL-1
), 2a (10 mM), L-alanine (150 mM), PLP (0.1 mM) in HEPES buffer (50
mM, pH 7.5) containing DMSO (30%); 1 mL reaction volume, 14 days, 30°C. Reaction conditions for the
synthesis of 3b: Purified enzyme (0.5 mg mL-1
), 3a (10 mM), L-alanine (250 mM), PLP (0.1 mM) in HEPES
buffer (50 mM, pH 7.5) containing DMSO (10%), 1 mL reaction volume, four days, 30°C. Both reactions
were followed via GC analysis using a Hydrodex--TBDAc chiral column and in both cases the LDH/GDH
system was used to shift the equilibrium.
Having found improved variants of ATA-Vfl for the deamination of 1b-3b, the asymmetric
synthesis of 2b and 3b was investigated using the LDH/GDH cascade[107]
in order to shift the
equilibrium towards the product side. We found that the double mutant F85L/V153A performed
best in the asymmetric synthesis of 2b (93% conversion) and quantitative conversion was
reached (after five days) when increased enzyme concentrations were employed. In contrast to
the kinetic resolution of amino alcohol 3b, variant Y150M was inferior in the asymmetric syn-
thesis of 3b when compared to variant Y150F. Asymmetric synthesis of 3b with double mutant
Y150F/V153A succeeded, giving a conversion of 92% after four days (Table 1, entry 9) and when

3.3 An Engineered ATA Suitable for the Synthesis of an 1,2-Amino Alcohol - Article III 29
increased enzyme concentrations were applied 88% conversion was reached after 24 hours (see
Table S1 for details). Finally, successful asymmetric syntheses of 2b and 3b in semi-preparative
scale (0.2 mmol) confirmed the results obtained at analytical scale (2b: Conversion > 98%, 98%
ee, 53% yield, 14 days; 3b Conversion > 98%, 98% ee, 60% yield, three days).
In conclusion, the features of wild-type ATA-Vfl were successfully altered by protein engineer-
ing to allow for the synthesis of an amine with two bulky substituents and the 1,2-amino alcohol
(R)-phenylglycinol. An alternative approach towards enzymes with new or altered properties is
the in silico mining of protein sequence databases. It was shown before to provide the commu-
nity with new and very valuable biocatalysts. For instance, the discovery of over a dozen (R)-
selective ATAs, that significantly broadened the selection of enzymes with this enantioselectivi-
ty, were identified in-silico.[81]
We investigated sequence–function relationships of the class III TA family – to which the bio-
catalytically valuable (S)-selective ATAs belong. For this purpose, we conducted a bioinformatic
analysis of ~13.000 protein sequences and derived amino acid fingerprints allowing the predic-
tion of the main reaction and substrate specificity of an enzyme from its sequence. This is ex-
plained in detail in Article IV and summarised in the following chapter.

In-Silico Analysis of the Class III TA 4
Family - Article IV A huge amount of potentially useful enzyme sequences is deposited in public databases. For
instance, the protein data bank (PDB) contains a huge amount of precise information about the
spatial arrangement of a protein. Anyhow, there is no additional information concerning the
enzymes’ reaction and substrate specificities because the function of a certain protein is hard to
predict. Probably, there are sequences of potentially valuable biocatalysts deposited in public
databases, which are buried under an enormous amount of data. The challenge of closing the
information gap between sequence and function of a protein has been the subject of extensive
research.[108-110]
Even though computational protein function prediction is steadily advancing
with algorithms, including literature mining and machine learning, there is still need for im-
provements.[108]
Despite these difficulties, the in-silico discovery of new enzymes (e.g. IREDs[56]
)
or altered stereoselectivity (e.g. (R)-selective ATAs[81]
) is contributing to significant advances in
the field of biocatalysis. Therefore, the ability to efficiently extract information about the func-
tion of proteins from databases is of high demand.
A Family Wide Comparison of Specificity Determining 4.1
Amino Acid Residues
The biocatalytically valuable (S)-selective ATAs, such as ATA-Cvi and ATA-Vfl, belong to the class
III TA family (also referred to as ornithine aminotransferase-like family) - a subfamily of the PLP-
fold type I superfamily. Despite the structural conservation within the class III TA family it con-
tains quite diverse enzyme activities, for instance decarboxylases, racemases and phospholyas-
es. However, the majority of enzymes within this family display TA activity with various sub-
strate scopes. Given the high structural conservation within this family we assumed that it
might be possible to discriminate the different substrate and reaction specificities by certain
amino acid constellations within the active site. For instance, if a given substrate recognition is
realised by a specific spatial arrangement of amino acid residues in or near the active site, this
information may be used to discriminate enzyme specificities within the class III TA family. As-
suming that such a constellation is conserved in enzymes with similar specificities, this ap-
proach would allow prediction of the reaction and/or substrate specificity of an uncharacterised
enzyme with known sequence or identification of new enzyme activities within this class. The
identification of specificity determining residues would require a family wide comparison of
enzyme sequences on a structural basis. To achieve this, we conducted a structure-based se-
quence alignment of the class III TA family using the commercial software 3DM.[111]
With the
help of this software we created alignments that exclusively included enzyme sequences that
shared enough similarity to an enzyme with a solved crystal structure, namely the ornithine:-
ketoglutarate transaminase (Orn:aKG TA, PDB code 2OAT). Sequences matching this criterion
were grouped in subfamilies based on their degree of similarity towards additional “parental”
crystal structures. These parental structures served as subfamily templates and represent en-
zymes with experimentally confirmed reaction and substrate specificity (see Article IV, SI Table
S2 for a list of these specificities). In this way we obtained a database (referred to as ornithine

4.2 The Sequence-Function Matrix 31
TA–like database (OrnTL DB) in the following) that contained about 13.000 protein sequences
that were divided in 21 subfamilies. The structure based alignment of enzymes with known
crystal structure resulted in specific parts of the amino acid sequence that were termed either
“core” or “variable” regions. The core contained residues, which were spatially properly aligned,
and variable regions that did not align because, for instance, they represented spatially flexible
regions of the sequence or a loop region with amino acid insertions. Amino acids belonging to
the core regions were assigned to 3D numbers allowing for comparison of structurally equiva-
lent amino acid residues within the OrnTL DB. More detailed information about the generation
of a 3DM databank can be found elsewhere.[112]
With the focus on the active site and the help of
the OrnTL DB we compared the residues that determine the enzymes’ specificity of all class III
TA enzymes on a structural basis and correlated them with reaction and substrate specificity.
This information provided insight in the different ways of substrate recognition and reaction
specificity and was compiled in a sequence-function matrix.
The Sequence-Function Matrix 4.2
Using literature research and the statistical tools provided by the 3DM software suite we identi-
fied the essential residues that are critical for a certain specificity to occur. These amino acid
residues were summarised in the sequence-function matrix and are referred to as “active site
fingerprints”. This matrix can help to predict the main specificity of an experimentally uncharac-
terised enzyme. If a sequence of interest matches an active site fingerprint of the matrix, the
corresponding enzyme will probably display the same main specificity. If none of the finger-
prints matches, the enzyme may display a yet unknown function or utilises a new method for an
already known specificity. In either case, the detailed investigation would contribute to a better
understanding of the modes of substrate recognition within the class III TA family.
The structural details, which govern substrate and reaction specificities are discussed at a
molecular level for each subfamily in the chapters 3.1-3.8 of Article IV. To aid a quick grasp of
the most important principles for a certain subfamily, each subsection of chapter 3.1 provides a
quick summary containing the fingerprint containing only the amino acids of high relevance for
substrate specificity and catalysis.
Further research is needed to investigate to which extent the sequence–function relation-
ships can be generalised. The following questions might be answered by the information pro-
vided by the sequence-function matrix:
Q: What are the main substrates of a given protein belonging to this superfamily?
A: In an alignment, compare the active site residues of the query sequence to the fingerprints
of the matrix. In case of a match, it is likely that also the activities are matching. For conven-
ience, a multiple sequence alignment of all subfamilies' parental structures of the OrnTL
DB is available (Article IV, Supplementary data Figure S1). It helps to match the 3DM num-
bers of this review with the original numbering of the crystal structures and, therefore, also
to the query sequence.

32 4 In-Silico Analysis of the Class III TA Family - Article IV
Q: How can I identify novel enzymes having the desired activity present in the superfamily?
A: Usually, a BLAST with a query sequence is conducted to identify enzymes with similar/equal
function. We recommend aligning available sequences and check, whether they carry the
specificity determining residues highlighted in the matrix. This helps especially to evaluate
distantly related sequences which otherwise would often not be chosen as candidate be-
cause the outcome would be too uncertain.
Q: Does the superfamily contain novel enzyme activities, which are yet unknown?
A: Enzymes whose active site residues do not fit the pattern of the matrix have either an un-
known activity, or they might contain an alternative active site design to confer a known
specificity.

4.2 The Sequence-Function Matrix 33
Table 2 Truncated sequence–function matrix. Key residues identified in this project are in bold and
represent the fingerprint residues that predict the reaction and substrate specificity. If no res-
idue is bold structural information was lacking. Physicochemical properties of the residues
are indicated by the colour code. The three most frequent amino acids per position are given
in descending order of conservation. The degree of conservation is indicated the following
way: capital letters: >70% conservation (considered conserved residues); lower case letters:
residues with conservation between 30% and 70%. Lower case italic letters indicate that none
of the amino acids at a given position occurs with more than 30% conservation. A minus (–)
indicates that there is no residue that can be aligned to this position. A special character is in-
troduced for sequences that do not belong to the OrnTL DB and were aligned manually to
their closest homologues within the database. At positions, where these manual alignments
were ambiguous, a question mark is shown. Table adapted from Article IV.[113]
See Article IV
for a full version of the matrix including all activities investigated.
Activitya PDB 16 45 46 47 132 145 185 216 267 269 346 348 353
Orn:αKG TA 2OAT Y A Y S R a E Q e G K T R
AcOrn/SuOrn:αKG TA 2ORD Y G I as R y E Q std G l at R
Lysε:αKG TA 2JJG - F F A R K E Q R N L c R
GABA:αKG TA (narrow
substrate scope) 1OHV - Q I sa R H E Q R F G C R
GABA:αKG TA (broad
substrate scope) 3Q8N v G I A R Y E Q g G L c R
Putrescine:αKG TAb 4UOX - g f g k f e q l t n a r
βAla:pyr TA 3A8U f G L W V N S I f G R t a
(S)-ATA (low activity) 3GJU S G L Y S F T V G G R M G
(S)-ATA (high activity) 3HMU f G L W s m A I n G R v i
Taurine:pyr TAc - L G V W T R G V R I G T C
Ala:glyoxylate TA 2d - v G I V C Y V Q lq F G G R
βPhe:αKG/pyr TA 4AO9 - E yf t - - A M a s a R f
D-Phenylglycine:αKG TAe 2CY8 - G H G ? ? H I L Q G R S
SAM:KAPA TA 1MLZ yc S W W de -yf A A ml gs R fl Y
Lys:KAPA TA 3DU4 F S li W D F a- A F G R L v
Activitya PDB 16 45 46 47 132 145 185 216 267 269 346 348 353
a Abbreviations: Orn = ornithine, KG = -ketoglutarate, AcOrn = N-acetylornithine, SuOrn = N-
succinylornithine, Lys lysine -amino group, GABA = -aminobutyrate, Ala = -alanine, pyr = py-
ruvate, (S)-ATA = (S)-selective amine transaminase, AA = -amino acid, Phe = -phenylalanine, SAM
= S-adenosylmethionine, KAPA = 7-keto-8-aminopelargonic acid
b Only based on one sequence [the E. coli YgjG enzyme (UniProt ID: P42588; PDB ID: 4UOX)]
c Only based on four characterised enzymes (see Supplementary data Table S5 entries 137–140, Uni-
Prot ID: Q6JE91 is different at several matrix positions)
d Only based on five characterised mammalian enzymes (see Article IV, Supplementary data, Table S5,
entries 162-166)
e Only based on two sequences (the Pseudomonas stutzeri (UniProt ID: Q6VY99) and the P. putida en-
zyme (GenBank ID: AX467211)), the structure of the P. stutzeri enzyme (PDB ID: 2CY8) is an un-
published apo structure

34 4 In-Silico Analysis of the Class III TA Family - Article IV
Challenges of Sequence-Function Prediction 4.3
There are a few limitations a researcher has to keep in mind when trying to predict the function
of an enzyme from its amino acid sequence. Some of them, concerning structure-based se-
quence alignments and predictions based on short sequence fingerprints, are discussed briefly
here; for a more in-depth discussion please see chapter 4 of Article IV.
The structure based alignment strategy described here enabled us, to some extent, to cir-
cumvent a critical drawback of alignments based on sequence only. In a protein sequence the
amino acid residues of two different C-carbons can occupy the same space within the active
site. This fact cannot be addressed in a sequence alignment and is therefore omitted. For that
reason, among others, alignments of protein structures will always allow for more accurate
conclusions. Anyhow, also structure based alignments suffer from certain drawbacks:
First, a general limitation for the identification of novel activities with the active site finger-
print function prediction is the fact, that the same substrate specificity or enzyme activity can be
realised in different ways. Thus, for instance, a different active site set up does not necessarily
imply another reaction or substrate specificity, but may also constitute a different approach to
achieve the same activity.
Second, some interactions between substrate and active site may not allow for the identifica-
tion of a clear pattern. Complementary electrostatic interactions or hydrogen bonds are easier
to interpret than, for instance, hydrophobic interactions that may be realised by a variety of
different amino acid combinations. Additionally, it can get tricky if water molecules act as medi-
ators for certain interactions. Furthermore, partially (chemically) equivalent residues might re-
sult in comparable interaction patterns (see also Article IV, Figure 6). Therefore, one has to keep
in mind to apply the presented fingerprints with caution, because similar amino acids at the
suggested positions might be found in enzymes with comparable specificities.
Third, enzymes in general and especially the active sites often have various degrees of flexi-
bility, which cannot be predicted by a sequence alignment. Anyhow, to some extent, insight can
be gained by comparing structures of the apo, holo and substrate, intermediate or product
bound enzyme.
Fourth, for the application of the rationale presented here, there is the necessity of a spatial-
ly conserved backbone of the superfamily to be analysed. For instance, if the active site consists
of variable loops, a proper alignment is impossible.
Fifth, creating a superfamily wide structure-based sequence alignment is always a trade-off
between the diversity of specificities it contains and the positions which can be aligned proper-
ly. For example the PLP fold type I database contained too diverse structures and therefore, it
contained too few alignable core regions in the active site for a proper comparision. In this pro-
ject, the compilation of the smaller OrnTL DB solved this issue. It contained structurally more
related enzymes, which increased the alignable core but, at the same time, decreased the diver-
sity in the database.
Sixth, in the pursuit of finding enzymes with new functions one is limited to those, which
switched their specificities by only a few mutations. This is obvious because the structure-based
sequence alignment database only contains sequences that share a certain identity to a known
structure and crystal structures are usually solved for enzymes with already known activities.
Therefore, complex adaptations, which usually go along with many mutations, are probably not

4.3 Challenges of Sequence-Function Prediction 35
easy to detect in such databases. For that matter, structural genomics projects for increasing
the amount of structural information about low identity sequences is important.
Nevertheless, in Article IV, we show that for the class III TA family the combination of struc-
tural information and analysis of multiple sequence alignments allowed to extract active site
amino acid fingerprints for the different reaction and substrate specificities. Using these finger-
prints will allow researchers to connect enzyme activities to sequences and to discover enzymes
with unknown activities or new mechanisms for known properties. This approach should not be
regarded a dogmatic way for qualitative prediction of a substrate scope because, besides the
amino acid distribution in the active site, other factors also effect the catalytic properties of en-
zymes to various degrees. Anyhow, we encourage to apply these patterns in annotation strate-
gies and to apply this methodology also for other superfamilies. It will broaden the scope of
biocatalysis by increasing the number of useful enzymes and help to further understand the
sequence and structure-function relationships of enzymes.

Conclusion 5In this thesis the biocatalytic synthesis of amines and amino alcohols has been investigated. As
a first step, the applicability and economic feasibility of biocatalysis for chiral amine synthesis
have been reviewed. The findings were compared to established chemical processes using rele-
vant process parameters such as TON, TOF and STY. Limited substrate scope and concentra-
tion, the stability of enzymes as well as product and substrate inhibition are issues that usually
hamper the application of biocatalysis, but process and reaction design are effective measures
to address these shortcomings. Furthermore, protein engineering has become a powerful tool
in order to generate novel enzyme activities or to tailor enzymes that fit process requirements.
The comparison of biocatalytic synthesis routes being applied at large scale with established
chemical routes revealed that biocatalytic amine synthesis has matured to a level of competi-
tiveness. New emerging strategies that deploy recently discovered or developed biocatalysts,
enriched the biocatalytic toolbox for asymmetric amine synthesis. This review clearly showcases
the potential of biocatalysis for the synthesis of chiral amines and provides a valuable guide for
synthetic chemists who want to benefit from these new opportunities.
Consequently, we applied biocatalysis for the synthesis of amino alcohols with two stereo-
centres, since conventional synthetic routes towards this compound class are tedious and step
intensive. A novel route for the synthesis of all four stereoisomers of the 1,3-amino alcohol 4-
amino-1-phenylpentane-2-ol has been established. Enzymes were applied to install both stere-
ocentres successively, which allowed the selective synthesis with high yields and optical purities.
A small scale preparative asymmetric transamination yielded one amino alcohol stereoisomer
selectively with a STY of 2.85 g L-1
d-1
. This is comparable with the first generation process for
the biocatalytic synthesis of the amino alcohol (1R,2R)-norpseudoephedrine (STY ~ 2 g L-1
d-1
). As
outlined in Article I, a biocatalyst usually needs to pass certain stages of development during
the maturation process towards application in an economically feasible process. In agreement
with this concept the biocatalytic synthesis of (1R,2R)-norpseudoephedrine was developed fur-
ther by reaction design, which improved the STY by 10-fold.[99]
This shows the potential of the
new approach for the synthesis of the 1,3-amino alcohol presented here, since the optimisation
of reaction parameters is not explored yet. Asymmetric synthesis of functionalised amines with
more than one stereocentre has gained attention lately since they are important building blocks
for many drugs. The novel approach presented in this thesis provides a valuable option for the
synthesis of this compound class as it is highly selective, step efficient and circumvents the need
for protecting groups as well as transition-metal catalysis.
We aimed to broaden the substrate scope of (S)-selective ATAs in order to expand the ap-
plicability further. As the activity of wild-type ATAs is usually limited to -methyl ketones, protein
engineering was conducted to overcome this limitation. In contrast to (S)-selective ATAs, the
engineering of (R)-selective ATAs was shown earlier to be very successful. As these enzymes
belong to different fold types of the PLP enzyme family (fold type I and fold type IV, respectively)
it was not possible to simply transfer the information gained by the manipulation of (R)-ATAs.
Addressing this knowledge gap, we enlarged the small binding pocket of the (S)-selective ATA-
Vfl. Small scale preparative synthesis of the 1,2-amino alcohol (R)-phenylglycinol exemplified the
applicability of the evolved variants for the asymmetric synthesis of this compound. At the mo-
ment, the STY for this reaction is ten times lower compared to the synthesis of the 1,3-amino

5 Conclusion 37
alcohol. With the maturation process of a biocatalyst in mind, the obtained variants can be con-
sidered at the beginning and further optimisation is required to allow viable applications. How-
ever, the information gained during this protein engineering project is an important advance-
ment, since only a few examples of engineered (S)-ATAs with an enlarged small binding pocket
are reported in literature. Furthermore, the designed variants expand the collection of ATAs
that are suitable for the synthesis of amino alcohols with bulkier substituents.
To deepen the understanding of ATAs further we performed for the first time a class III TA
family wide analysis (which includes (S)-selective ATAs) that was based on a structure-based
sequence alignment of ~13.000 sequences. After comparing the active site architectures and
performing literature research we identified amino acids that correlate with the reaction- and
substrate specificity of the enzymes within this family. This information was compiled in a se-
quence-function matrix, which allows the prediction of the main activity of biochemically un-
characterised enzymes from their sequence. These insights provided a better understanding of
the activity determining residues in (S)-ATAs and class III TAs in general.

References 6[1] Voet, D, Voet, J. G.; Biochemie. 1st ed.; Wiley VCH Weinheim, 1994
[2] Milton, R; Milton, S; Kent, S; Total Chemical Synthesis of a D-Enzyme: The Enantiomers
of HIV-1 Protease Show Reciprocal Chiral Substrate Specificity [corrected]. Science 1992,
256, 1445-1448
[3] Weinstock, MT; Jacobsen, MT; Kay, MS; Synthesis and Folding of a Mirror-Image Enzyme
Reveals Ambidextrous Chaperone Activity. PNAS 2014, 111, 11679-11684
[4] Faber, K; Biotransformations in Organic Chemistry. 6th ed.; Springer Berlin Heidelberg,
2011 [5] Natarajan, R; Basak, SC; Numerical Descriptors for the Characterization of Chiral
Compounds and their Applications in Modeling Biological and Toxicological Activities.
Curr Top Med Chem 2011, 11, 771-787
[6] Islam, MR; Mahdi, JG; Bowen, ID; Pharmacological Importance of Stereochemical
Resolution of Enantiomeric Drugs. Drug Saf 1997, 17, 149-65
[7] Agranat, I; Caner, H; Caldwell, J; Putting Chirality to Work: The Strategy of Chiral
Switches. Nat Rev Drug Discov 2002, 1, 753-768
[8] Reist, M; Carrupt, P-A; Francotte, E; Testa, B; Chiral Inversion and Hydrolysis of
Thalidomide: Mechanisms and Catalysis by Bases and Serum Albumin, and Chiral Stability
of Teratogenic Metabolites. Chem Res Toxicol 1998, 11, 1521-1528
[9] Evans, AM; Comparative Pharmacology of S(+)-Ibuprofen and (RS)-Ibuprofen. Clin
Rheumatol 2001, 20 Suppl 1, S9-14
[10] Bonabello, A; Galmozzi, MR; Canaparo, R; Isaia, GC; Serpe, L; Muntoni, E; Zara, GP;
Dexibuprofen (S(+)-Isomer Ibuprofen) Reduces Gastric Damage and Improves Analgesic
and Antiinflammatory Effects in Rodents. Anesth Analg 2003, 97, 402-408
[11] Zamani, O; Böttcher, E; Rieger, JD; Mitterhuber, J; Hawel, R; Stallinger, S; Eller, N;
Comparison of Safety, Efficacy and Tolerability of Dexibuprofen and Ibuprofen in the
Treatment of Osteoarthritis of the Hip or Knee. Wien Klin Wochenschr 2014, 126, 368-375
[12] Khan, AY; Preskorn, SH; Wimalasena, K; Single Enantiomer Drugs: Should They be
Developed? Essent Psychopharmacol 2006, 7, 15-23
[13] Blake, K; Raissy, H; Chiral Switch Drugs for Asthma and Allergies: True Benefit or
Marketing Hype. Pediatr Allergy Immunol Pulmonol 2013, 26, 157-160
[14] Tucker, G; Chiral Switches. The Lancet 2000, 355, 1085-1087
[15] Avorn, J; FDA Standards - Good Enough for Government Work? N Engl J Med 2005, 353,
969-972
[16] Stafford, RS; Wagner, TH; Lavori, PW; New, but Not Improved? Incorporating
Comparative-Effectiveness Information into FDA Labeling. N Engl J Med 2009, 361, 1230-
1233
[17] Mansfield, P; Henry, D; Tonkin, A; Single-Enantiomer Drugs. Clin Pharmacokinet 2004,
43, 287-290
[18] Blaser, HU; Spindler, F; Studer, M; Enantioselective Catalysis in Fine Chemicals
Production. Appl Catal, A 2001, 221, 119-143
[19] Sekhon, BS; Chiral Pesticides. J Pestic Sci 2009, 34, 1-12
[20] Nugent, TC; El-Shazly, M; Chiral Amine Synthesis - Recent Developments and Trends for
Enamide Reduction, Reductive Amination, and Imine Reduction. Adv Synth Catal 2010,
352, 753-819
[21] Millet, R; Traff, AM; Petrus, ML; Backvall, J-E; Enantioselective Synthesis of syn- and anti-
1,3-Aminoalcohols via β-Aminoketones and Subsequent Reduction/Dynamic Kinetic
Asymmetric Transformation. J Am Chem Soc 2010, 132, 15182-15184
[22] Benedetti, F; Norbedo, S; Epoxyalcohol Route to Hydroxyethylene Dipeptide Isosteres: A
New Synthesis of the Diaminoalcohol Core of HIV-Protease Inhibitor ABT-538 (Ritonavir).
Chem Commun 2001, 203-204
[23] Lait, SM; Rankic, DA; Keay, BA; 1,3-Aminoalcohols and Their Derivatives in Asymmetric
Organic Synthesis. Chem Rev 2007, 107, 767-796
[24] Augeri, DJ; Robl, JA; Betebenner, DA, et al.; Discovery and Preclinical Profile of
Saxagliptin (BMS-477118): A Highly Potent, Long-Acting, Orally Active Dipeptidyl

6 References 39
Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J Med Chem 2005, 48, 5025-
5037
[25] Amat, M; Bosch, J; Hidalgo, J, et al.; Synthesis of Enantiopure trans-3,4-Disubstituted
Piperidines. An Enantiodivergent Synthesis of (+)- and (−)-Paroxetine. J Org Chem 2000,
65, 3074-3084
[26] Erlanson, DA; Arndt, JW; Cancilla, MT, et al.; Discovery of a Potent and Highly Selective
PDK1 Inhibitor via Fragment-Based Drug Discovery. Bioorg Med Chem Lett 2011, 21,
3078-3083
[27] Raghavan, S; Krishnaiah, V; Sridhar, B; Asymmetric Synthesis of the Potent HIV-Protease
Inhibitor Nelfinavir. J Org Chem 2009, 75, 498-501
[28] Shi, Z; Harrison, BA; Verdine, GL; Unpredictable Stereochemical Preferences for µ-Opioid
Receptor Activity in an Exhaustively Stereodiversified Library of 1,4-Enediols. Org Lett
2003, 5, 633-636
[29] Nugent, TC; Chiral Amine Synthesis - Methods, Developments and Applications. 1st ed.;
Wiley VCH Weinheim, 2010
[30] Sehl, T; Hailes, HC; Ward, JM; Wardenga, R; von Lieres, E; Offermann, H; Westphal, R;
Pohl, M; Rother, D; Two Steps in One Pot: Enzyme Cascade for the Synthesis of
Nor(pseudo)ephedrine from Inexpensive Starting Materials. Angew Chem Int Ed 2013, 52,
6772-6775
[31] Xie, J-H; Zhu, S-F; Zhou, Q-L; Recent Advances in Transition Metal-Catalyzed
Enantioselective Hydrogenation of Unprotected Enamines. Chem Soc Rev 2012, 41, 4126-
4139
[32] Fleury-Brégeot, N; de la Fuente, V; Castillón, S; Claver, C; Highlights of Transition Metal-
Catalyzed Asymmetric Hydrogenation of Imines. ChemCatChem 2010, 2, 1346-1371
[33] Xie, J-H; Zhu, S-F; Zhou, Q-L; Transition Metal-Catalyzed Enantioselective Hydrogenation
of Enamines and Imines. Chem Rev 2011, 111, 1713-1760
[34] Wang, C; Xiao, J; Asymmetric Reductive Amination. In Stereoselective Formation of
Amines, Li, W; Zhang, X, Eds. Springer Berlin Heidelberg, 2014, Vol. 343, 261-282
[35] Nugent, TC; Marinova, SM; Step-Efficient Access to Chiral Primary Amines. Synthesis
2013, 45, 153-166
[36] Hesp, KD; Stradiotto, M; Rhodium- and Iridium-Catalyzed Hydroamination of Alkenes.
ChemCatChem 2010, 2, 1192-1207
[37] Ephedrin. In RÖMPP Enzyklopädie Online, Thieme Verlag: www.roempp.com,
[38] Collet, F; Dodd, RH; Dauban, P; Catalytic C-H Amination: Recent Progress and Future
Directions. Chem Commun 2009, 5061-5074
[39] Cho, SH; Kim, JY; Kwak, J; Chang, S; Recent Advances in the Transition Metal-Catalyzed
Twofold Oxidative C-H Bond Activation Strategy for C-C and C-N Bond Formation. Chem
Soc Rev 2011, 40, 5068-83
[40] Li, Z; Capretto, DA; Rahaman, R; He, C; Silver-Catalyzed Intermolecular Amination of C-H
Groups. Angew Chem Int Ed 2007, 46, 5184-5186
[41] Breuer, M; Ditrich, K; Habicher, T; Hauer, B; Keßeler, M; Stürmer, R; Zelinski, T;
Industrial Methods for the Production of Optically Active Intermediates. Angew Chem Int Ed
2004, 43, 788-824
[42] Haight, AR; Stuk, TL; Allen, MS, et al.; Reduction of an Enaminone: Synthesis of the
Diamino Alcohol Core of Ritonavir. Org Process Res Dev 1999, 3, 94-100
[43] Yang, JW; Stadler, M; List, B; Proline-Catalyzed Mannich Reaction of Aldehydes with N-
Boc-Imines. Angew Chem Int Ed 2007, 46, 609-611
[44] List, B; Pojarliev, P; Biller, WT; Martin, HJ; The Proline-Catalyzed Direct Asymmetric
Three-Component Mannich Reaction: Scope, Optimization, and Application to the Highly
Enantioselective Synthesis of 1,2-Amino Alcohols. J Am Chem Soc 2002, 124, 827-833
[45] Sehl, T; Maugeri, Z; Rother, D; Multi-Step Synthesis Strategies Towards 1,2-Amino
Alcohols with Special Emphasis on Phenylpropanolamines. J Mol Catal B: Enzym 2015,
114, 65-71
[46] van Rantwijk, F; Sheldon, RA; Enantioselective Acylation of Chiral Amines Catalysed by
Serine Hydrolases. Tetrahedron 2004, 60, 501-519

40 6 References
[47] Balkenhohl, F, Hauer, Bernhard, Ladner, Wolfgang , Pressler, Uwe , Nübling, Christoph;
Racemate Cleavage of Primary and Secondary Amines by Enzyme-Catalysed Acylation.
Patent Application WO1994EP03102 19940916, 1999
[48] Höhne, M; Bornscheuer, UT; Biocatalytic Routes to Optically Active Amines.
ChemCatChem 2009, 1, 42-51
[49] Koszelewski, D; Clay, D; Rozzell, D; Kroutil, W; Deracemisation of α-Chiral Primary
Amines by a One-Pot, Two-Step Cascade Reaction Catalysed by ω-Transaminases. Eur J
Org Chem 2009, 2009, 2289-2292
[50] Simon, RC; Zepeck, F; Kroutil, W; Chemoenzymatic Synthesis of All Four Diastereomers of
2,6-Disubstituted Piperidines through Stereoselective Monoamination of 1,5-Diketones.
Chem Eur J 2013, 19, 2859-2865
[51] Girardin, M; Ouellet, SG; Gauvreau, D; Moore, JC; Hughes, G; Devine, PN; O’Shea, PD;
Campeau, L-C; Convergent Kilogram-Scale Synthesis of Dual Orexin Receptor Antagonist.
Org Process Res Dev 2013, 17, 61-68
[52] Abrahamson, MJ; Vázquez-Figueroa, E; Woodall, NB; Moore, JC; Bommarius, AS;
Development of an Amine Dehydrogenase for Synthesis of Chiral Amines. Angew Chem Int
Ed 2012, 51, 3969-3972
[53] Bommarius, BR; Schurmann, M; Bommarius, AS; A Novel Chimeric Amine Dehydrogenase
Shows Altered Substrate Specificity Compared to its Parent Enzymes. Chem Commun 2014,
50, 14953-14955
[54] Ye, LJ; Toh, HH; Yang, Y; Adams, JP; Snajdrova, R; Li, Z; Engineering of Amine
Dehydrogenase for Asymmetric Reductive Amination of Ketone by Evolving Rhodococcus
Phenylalanine Dehydrogenase. ACS Catalysis 2015, 5, 1119-1122
[55] Leipold, F; Hussain, S; Ghislieri, D; Turner, NJ; Asymmetric Reduction of Cyclic Imines
Catalyzed by a Whole-Cell Biocatalyst Containing an (S)-Imine Reductase. ChemCatChem
2013, 5, 3505-3508
[56] Gand, M; Müller, H; Wardenga, R; Höhne, M; Characterization of Three Novel Enzymes
with Imine Reductase Activity. J Mol Catal B: Enzym 2014, 110, 126-132
[57] Huber, T; Schneider, L; Präg, A; Gerhardt, S; Einsle, O; Müller, M; Direct Reductive
Amination of Ketones: Structure and Activity of S-Selective Imine Reductases from
Streptomyces. ChemCatChem 2014, 6, 2248-2252
[58] Mitsukura, K; Kuramoto, T; Yoshida, T; Kimoto, N; Yamamoto, H; Nagasawa, T; A
NADPH-Dependent (S)-Imine Reductase (SIR) from Streptomyces sp. GF3546 for
Asymmetric Synthesis of Optically Active Amines: Purification, Characterization, Gene
Cloning, and Expression. Appl Microbiol Biotechnol 2013, 97, 8079-8086
[59] Mitsukura, K; Suzuki, M; Shinoda, S; Kuramoto, T; Yoshida, T; Nagasawa, T; Purification
and Characterization of a Novel (R)-Imine Reductase from Streptomyces sp. GF3587. Biosci
Biotechnol Biochem 2011, 75, 1778-1782
[60] Scheller, PN; Fademrecht, S; Hofelzer, S; Pleiss, J; Leipold, F; Turner, NJ; Nestl, BM;
Hauer, B; Enzyme Toolbox: Novel Enantiocomplementary Imine Reductases.
ChemBioChem 2014, 15, 2201-2204
[61] Man, H; Wells, E; Hussain, S; Leipold, F; Hart, S; Turkenburg, JP; Turner, NJ; Grogan, G;
Structure, Activity and Stereoselectivity of NADPH-Dependent Oxidoreductases Catalysing
the S-Selective Reduction of the Imine Substrate 2-Methylpyrroline. ChemBioChem 2015,
16, 1052-1059
[62] Wetzl, D; Berrera, M; Sandon, N; Fishlock, D; Ebeling, M; Müller, M; Hanlon, S; Wirz, B;
Iding, H; Expanding the Imine Reductase Toolbox by Exploring the Bacterial Protein-
Sequence Space. ChemBioChem 2015, 16, 1749-1756
[63] Li, H; Luan, Z-J; Zheng, G-W; Xu, J-H; Efficient Synthesis of Chiral Indolines using an
Imine Reductase from Paenibacillus lactis. Adv Synth Catal 2015, 357, 1692-1696
[64] Turner, NJ; Deracemisation Methods. Curr Opin Chem Biol 2010, 14, 115-121
[65] Dairi, T; Asano, Y; Cloning, Nucleotide Sequencing, and Expression of an Opine
Dehydrogenase Gene from Arthrobacter sp. Strain 1C. Appl Environ Microbiol 1995, 61,
3169-3171
[66] Kato, Y; Yamada, H; Asano, Y; Stereoselective Synthesis of Opine-Type Secondary Amine
Carboxylic Acids by a New Enzyme Opine Dehydrogenase Use of Recombinant Enzymes. J
Mol Catal B: Enzym 1996, 1, 151-160

6 References 41
[67] Haibin, C; Collier, SJ; Nazor, J, et al.; Engineered Imine Reductases and Methods for the
Reductive Animation of Ketone and Amine Compounds. Patent Application
WO2013170050 A1, 2013
[68] Schrittwieser, JH; Velikogne, S; Kroutil, W; Biocatalytic Imine Reduction and Reductive
Amination of Ketones. Adv Synth Catal 2015, 357, 1655-1685
[69] Ruff, BM; Brase, S; O’Connor, SE; Biocatalytic Production of Tetrahydroisoquinolines.
Tetrahedron Lett 2012, 53, 1071-1074
[70] Pesnot, T; Gershater, MC; Ward, JM; Hailes, HC; The Catalytic Potential of Coptis japonica
NCS2 Revealed – Development and Utilisation of a Fluorescamine-Based Assay. Adv Synth
Catal 2012, 354, 2997-3008
[71] Stöckigt, J; Antonchick, AP; Wu, F; Waldmann, H; The Pictet–Spengler Reaction in Nature
and in Organic Chemistry. Angew Chem Int Ed 2011, 50, 8538-8564
[72] Schrittwieser, JH; Resch, V; The Role of Biocatalysis in the Asymmetric Synthesis of
Alkaloids. RSC Adv 2013, 3, 17602-17632
[73] Schrittwieser, JH; Resch, V; Wallner, S; Lienhart, W-D; Sattler, JH; Resch, J; Macheroux, P;
Kroutil, W; Biocatalytic Organic Synthesis of Optically Pure (S)-Scoulerine and Berbine and
Benzylisoquinoline Alkaloids. J Org Chem 2011, 76, 6703-6714
[74] Schrittwieser, JH; Resch, V; Sattler, JH; Lienhart, W-D; Durchschein, K; Winkler, A;
Gruber, K; Macheroux, P; Kroutil, W; Biocatalytic Enantioselective Oxidative C-C
Coupling by Aerobic C-H Activation. Angew Chem Int Ed 2011, 50, 1068-1071
[75] Schmid, A; Dordick, JS; Hauer, B; Kiener, A; Wubbolts, M; Witholt, B; Industrial
Biocatalysis Today and Tomorrow. Nature 2001, 409, 258-268
[76] Koeller, KM; Wong, C-H; Enzymes for Chemical Synthesis. Nature 2001, 409, 232-240
[77] Reetz, MT; Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future.
J Am Chem Soc 2013, 135, 12480-12496
[78] Abrahamson, MJ; Wong, JW; Bommarius, AS; The Evolution of an Amine Dehydrogenase
Biocatalyst for the Asymmetric Production of Chiral Amines. Adv Synth Catal 2013, 355,
1780-1786
[79] McIntosh, JA; Coelho, PS; Farwell, CC; Wang, ZJ; Lewis, JC; Brown, TR; Arnold, FH;
Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P450
Enzymes In Vitro and In Vivo. Angew Chem Int Ed 2013, 52, 9309-9312
[80] Bornscheuer, UT; Huisman, GW; Kazlauskas, RJ; Lutz, S; Moore, JC; Robins, K;
Engineering the Third Wave of Biocatalysis. Nature 2012, 485, 185-194
[81] Höhne, M; Schätzle, S; Jochens, H; Robins, K; Bornscheuer, UT; Rational Assignment of
Key Motifs for Function Guides in silico Enzyme Identification. Nat Chem Biol 2010, 6,
807-813
[82] Midelfort, KS; Kumar, R; Han, S, et al.; Redesigning and Characterizing the Substrate
Specificity and Activity of Vibrio fluvialis Aminotransferase for the Synthesis of Imagabalin.
Protein Eng Des Sel 2012, 26, 25-33
[83] Chung, CK; Bulger, PG; Kosjek, B, et al.; Process Development of C–N Cross-Coupling and
Enantioselective Biocatalytic Reactions for the Asymmetric Synthesis of Niraparib. Org
Process Res Dev 2013,
[84] Ghislieri, D; Green, AP; Pontini, M; Willies, SC; Rowles, I; Frank, A; Grogan, G; Turner,
NJ; Engineering an Enantioselective Amine Oxidase for the Synthesis of Pharmaceutical
Building Blocks and Alkaloid Natural Products. J Am Chem Soc 2013, 135, 10863-10869
[85] Heath, RS; Pontini, M; Bechi, B; Turner, NJ; Development of an R-Selective Amine
Oxidase with Broad Substrate Specificity and High Enantioselectivity. ChemCatChem 2014,
6, 996-1002
[86] Meadows, RE; Mulholland, KR; Schürmann, M, et al.; Efficient Synthesis of (S)-1-(5-
Fluoropyrimidin-2-yl)ethylamine Using an ω-Transaminase Biocatalyst in a Two-Phase
System. Org Process Res Dev 2013, 17, 1117-1122
[87] Frodsham, L; Golden, M; Hard, S, et al.; Use of ω-Transaminase Enzyme Chemistry in the
Synthesis of a JAK2 Kinase Inhibitor. Org Process Res Dev 2013, 17, 1123-1130
[88] Li, T; Liang, J; Ambrogelly, A, et al.; Efficient, Chemoenzymatic Process for Manufacture
of the Boceprevir Bicyclic [3.1.0] Proline Intermediate Based on Amine Oxidase-Catalyzed
Desymmetrization. J Am Chem Soc 2012, 134, 6467-6472

42 6 References
[89] Savile, CK; Janey, JM; Mundorff, EC, et al.; Biocatalytic Asymmetric Synthesis of Chiral
Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305-309
[90] Desai, AA; Sitagliptin Manufacture: A Compelling Tale of Green Chemistry, Process
Intensification, and Industrial Asymmetric Catalysis. Angew Chem Int Ed 2011, 50, 1974-
1976
[91] Truppo, MD; Strotman, H; Hughes, G; Development of an Immobilized Transaminase
Capable of Operating in Organic Solvent. ChemCatChem 2012, 4, 1071-1074
[92] Knoll AG, HG, Klavehn W; Verfahren zur Herstellung von 1-1-Phenyl-2-
methylaminopropan-1-ol. Patent Application DE1930548459D, 1930
[93] Pohl, M; Protein Design on Pyruvate Decarboxylase (PDC) by Site-Directed Mutagenesis. In
New Enzymes for Organic Synthesis, Springer Berlin Heidelberg, 1997, Vol. 58, 15-43
[94] Palmer, M; Walsgrove, T; Wills, M; (1R,2S)-(+)-cis-1-Amino-2-indanol: An Effective
Ligand for Asymmetric Catalysis of Transfer Hydrogenations of Ketones. J Org Chem 1997,
62, 5226-5228
[95] Yun, H; Kim, J; Kinnera, K; Kim, B-G; Synthesis of Enantiomerically Pure trans-(1R,2R)-
and cis-(1S,2R)-1-Amino-2-indanol by Lipase and ω-Transaminase. Biotechnol Bioeng
2006, 93, 391-395
[96] Smith, MEB; Chen, BH; Hibbert, EG, et al.; A Multidisciplinary Approach Toward the
Rapid and Preparative-Scale Biocatalytic Synthesis of Chiral Amino Alcohols: A Concise
Transketolase-ω-Transaminase-Mediated Synthesis of (2S,3S)-2-Aminopentane-1,3-diol.
Org Process Res Dev 2010, 14, 99-107
[97] Smithies, K; Smith, MEB; Kaulmann, U; Galman, JL; Ward, JM; Hailes, HC;
Stereoselectivity of an ω-Transaminase-Mediated Amination of 1,3-Dihydroxy-1-
phenylpropane-2-one. Tetrahedron: Asymmetry 2009, 20, 570-574
[98] Rother, D; Kolter, G; Gerhards, T, et al.; S-Selective Mixed Carboligation by Structure-
Based Design of the Pyruvate Decarboxylase from Acetobacter pasteurianus.
ChemCatChem 2011, 3, 1587-1596
[99] Sehl, T; Hailes, HC; Ward, JM; Menyes, U; Pohl, M; Rother, D; Efficient 2-Step
Biocatalytic Strategies for the Synthesis of All Nor(pseudo)ephedrine Isomers. Green Chem
2014, 16, 3341-3348
[100] Kohls, H; Anderson, M; Dickerhoff, J; Weisz, K; Córdova, A; Berglund, P; Brundiek, H;
Bornscheuer, UT; Höhne, M; Selective Access to All Four Diastereomers of a 1,3-Amino
Alcohol by Combination of a Keto Reductase- and an Amine Transaminase-Catalysed
Reaction. Adv Synth Catal 2015, 357, 1808-1814
[101] Kaulmann, U; Smithies, K; Smith, MEB; Hailes, HC; Ward, JM; Substrate Spectrum of ω-
Transaminase from Chromobacterium violaceum DSM30191 and its Potential for
Biocatalysis. Enzyme Microb Technol 2007, 41, 628-637
[102] Shin, J-S; Kim, B-G; Asymmetric Synthesis of Chiral Amines with ω-Transaminase.
Biotechnol Bioeng 1999, 65, 206-211
[103] Shin, J-S; Kim, B-G; Exploring the Active Site of Amine:Pyruvate Aminotransferase on the
Basis of the Substrate Structure−Reactivity Relationship: How the Enzyme Controls
Substrate Specificity and Stereoselectivity. J Org Chem 2002, 67, 2848-2853
[104] Mutti, FG; Kroutil, W; Asymmetric Bio-Amination of Ketones in Organic Solvents. Adv
Synth Catal 2012, 354, 3409-3413
[105] Schätzle, S; Höhne, M; Redestad, E; Robins, K; Bornscheuer, UT; Rapid and Sensitive
Kinetic Assay for Characterization of omega-Transaminases. Anal Chem 2009, 81, 8244-
8248
[106] Nobili, A; Steffen-Munsberg, F; Kohls, H; Trentin, I; Schulzke, C; Höhne, M; Bornscheuer,
UT; Engineering the Active Site of the Amine Transaminase from Vibrio fluvialis for the
Asymmetric Synthesis of Aryl–Alkyl Amines and Amino Alcohols. ChemCatChem 2015, 7,
757-760
[107] Koszelewski, D; Lavandera, I; Clay, D; Rozzell, D; Kroutil, W; Asymmetric Synthesis of
Optically Pure Pharmacologically Relevant Amines Employing ω-Transaminases. Adv Synth
Catal 2008, 350, 2761-2766
[108] Radivojac, P; Clark, WT; Oron, TR, et al.; A Large-Scale Evaluation of Computational
Protein Function Prediction. Nat Meth 2013, 10, 221-227

6 References 43
[109] Zhao, S; Kumar, R; Sakai, A, et al.; Discovery of New Enzymes and Metabolic Pathways by
Using Structure and Genome Context. Nature 2013, 502, 698-702
[110] Gerlt, JA; Allen, KN; Almo, SC, et al.; The Enzyme Function Initiative. Biochemistry 2011,
50, 9950-9962
[111] Kuipers, RK; Joosten, H-J; van Berkel, WJH, et al.; 3DM: Systematic Analysis of
Heterogeneous Superfamily Data to Discover Protein Functionalities. Proteins Struct Funct
Bioinf 2010, 78, 2101-2113
[112] Kourist, R; Jochens, H; Bartsch, S; Kuipers, R; Padhi, SK; Gall, M; Böttcher, D; Joosten, H-
J; Bornscheuer, UT; The α/β-Hydrolase Fold 3DM Database (ABHDB) as a Tool for Protein
Engineering. ChemBioChem 2010, 11, 1635-1643
[113] Steffen-Munsberg, F; Vickers, C; Kohls, H, et al.; Bioinformatic Analysis of a PLP-
Dependent Enzyme Superfamily Suitable for Biocatalytic Applications. Biotechnol Adv
2015, 33, 566-604

Author Contribution Article I Recent Achievements in Developing the Biocatalytic Toolbox for Chiral
Amine Synthesis
Kohls, H; Steffen-Munsberg, F; Höhne, M, Curr Opin Chem Biol 2014, 19, 180-192,
DOI: 10.1016/j.cbpa.2014.02.021
MH initiated the review and with the help of HK and FS performed the literature search. HK and
FS extracted the data necessary for the calculation of process parameters from literature and
processed them for the calculation of TON, TOF and STY. All authors read and edited the manu-
script
Article II Selective Access to All Four Diastereomers of a 1,3-Amino Alcohol by Com-
bination of a Keto Reductase- and an Amine Transaminase-Catalysed Reac-
tion
Kohls, H; Anderson, M; Dickerhoff, J; Weisz, K; Córdova, A; Berglund, P; Brundiek, H; Bornscheu-
er, UT; Höhne, M, Adv Synth Catal 2015, 357, 1808-1814, DOI: 10.1002/adsc.201500214
MH and UTB initiated the project. HK and MA designed the experiments with the help of MH
UTB and PB. HK, MA and JD performed the experiments. HK and MA wrote the manuscript with
the help of MH and UTB. All authors read and edited the manuscript.
Article III Engineering the Active Site of the Amine Transaminase from Vibrio fluvialis
for the Asymmetric Synthesis of Aryl–Alkyl Amines and Amino Alcohols
Nobili, A; Steffen-Munsberg, F; Kohls, H; Trentin, I; Schulzke, C; Höhne, M; Born-
scheuer, UT, ChemCatChem 2015, 7, 757-760, DOI: 10.1002/cctc.201403010
UTB initiated the project. AN with the help of FS and MH designed and with the help of FS, HK
and IT performed the experiments. AN, CS, MH and UTB wrote and all authors read and edited
the manuscript.
Article IV Bioinformatic Analysis of a PLP-Dependent Enzyme Superfamily Suitable
for Biocatalytic Applications
Steffen-Munsberg, F; Vickers, C; Kohls, H; Land, H; Mallin, H; Nobili, A; Skalden, L;
van den Bergh, T; Joosten, H-J; Berglund, P; Höhne, M; Bornscheuer, UT, Biotech-
nol Adv 2015, 33, 566-604, DOI: 10.1016/j.biotechadv.2014.12.012
MH and UTB initiated the review, FS and MH devised the conceptual design, FS and CV coordi-
nated data and literature analysis of the subsections and together with MH performed the in-
depth analysis of the superfamily. FS, CV, HK, HL, HM, AN and LS analysed literature and 3DM
alignments of subgroups, created fingerprints and were involved in writing subchapters. TvdB
and H-JJ established the 3DM database and wrote the corresponding chapter. MH created the
reaction mechanism video. With the help of CV and UTB, FS and MH did the main editing. FS, CV,
PB, MH and UTB finalised the review.
Jun. Prof. Dr. Matthias Höhne Hannes Kohls

Articles


Articles 47
ARTICLE I


Recent achievements in developing the biocatalytic toolbox forchiral amine synthesisHannes Kohls1, Fabian Steffen-Munsberg1,2 and Matthias Hohne1
Novel enzyme activities and chemoenzymatic reaction
concepts have considerably expanded the biocatalytic toolbox
for chiral amine synthesis. Creating new activities or extending
the scope of existing enzymes by protein engineering is a
common trend in biocatalysis and in chiral amine synthesis
specifically. For instance, an amine dehydrogenase that allows
for the direct asymmetric amination of ketones with ammonia
was created by mutagenesis of an L-amino acid
dehydrogenase. Another trend in chiral amine chemistry is the
development of strategies allowing for the synthesis of
secondary amines. For example the smart choice of substrates
for amine transaminases provided access to secondary amines
by chemoenzymatic reactions. Furthermore novel biocatalysts
for the synthesis of secondary amines such as imine
reductases and Pictet-Spenglerases have been identified and
applied. Recent examples showed that the biocatalytic amine
synthesis is emerging from simple model reactions towards
industrial scale preparation of pharmaceutical relevant
substances, for instance, as shown in the synthesis of a Janus
kinase 2 inhibitor using an amine transaminase. A comparison
of important process parameters such as turnover number
and space–time yield demonstrates that biocatalytic
strategies for asymmetric reductive amination are maturing
and can already compete with established chemical
methods.
Addresses1 Greifswald University, Institute of Biochemistry, Felix-Hausdorff-Str. 4,
17487 Greifswald, Germany2 KTH Royal Institute of Technology, AlbaNova University Center,
Division of Industrial Biotechnology, 10691 Stockholm, Sweden
Corresponding author: Hohne, Matthias
Current Opinion in Chemical Biology 2014, 19:180–192
This review comes from a themed issue on Biocatalysis and
biotransformation
Edited by Jeffrey C Moore and Uwe T Bornscheuer
For a complete overview see the Issue and the Editorial
1367-5931/$ – see front matter, # 2014 Elsevier Ltd. All rights
reserved.
http://dx.doi.org/10.1016/j.cbpa.2014.02.021
IntroductionChiral prim-amines, sec-amines, and tert-amines are
important building blocks for the pharmaceutical indus-
try. As the structural scope of amines is very large
(especially for secondary and tertiary amines), a diverse
spectrum of enzymes has been developed during the past
decade to address specific synthetic problems.
The identified enzymes often feature high regioselectivity
and stereoselectivity and therefore have great potential to
serve as a ‘green alternative’ to common metal catalyzed
chemical reductive aminations [1]. Although the high
optical purities and conversions are often impressive, these
are not always sufficient for industrial applications. There-
fore both, the biocatalyst and the reaction conditions often
need optimization prior to their (industrial) application
[2,3]. These endeavors are often as challenging as the
discovery of the enzymes itself. As an economically feas-
ible industrial process requires a high space–time yield
(STY), the catalyst needs to meet certain criteria in view of
turnover number (TON) and turnover frequency (TOF)
values. Furthermore, a high substrate loading is desired,
which is significantly different from the natural conditions
under which most enzymes have evolved. For a compari-
son of chemical catalysts with established, optimized or
newly developed enzymes, Table 1 summarizes important
reaction characteristics of chosen recent examples.
The current biocatalytic toolbox for amine synthesis
represents a diverse collection of enzymes with respect
to their catalytic scope. Several of the established tools,
such as transaminases (TA) [4–6], monoamine oxidases
(MAO) [7] and lyases [8] have been reviewed in detail
recently. In this review, we structure the major achieve-
ments in the field according to three development phases
of a biocatalytic process: discovery of novel enzyme
activities, advancements by broadening the catalytic
scope and finally the maturation towards industrial scale
applications. In the first two sections we focus on the
discovery phase and highlight proof of concept examples
of novel enzyme activities obtained either by protein
engineering or wild-type enzyme identification. The
second section will summarize how basic research com-
bined with protein and reaction engineering can widen
the scope and improve applicability. In the third and final
section, the last maturation phase in process development
is addressed, which often requires the extensive use of
(bio) reaction engineering methods to facilitate the inte-
gration of a biocatalyst into a chemical manufacturing
process. As stated in a recent review, we are currently
experiencing the third wave of biocatalysis [9��], where
enzymes can be tailored to fit the needs of the process
conditions rather than building the chemical process
around the needs of the biocatalysts. From this process
application perspective we present a comparison of
Available online at www.sciencedirect.com
ScienceDirect
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com
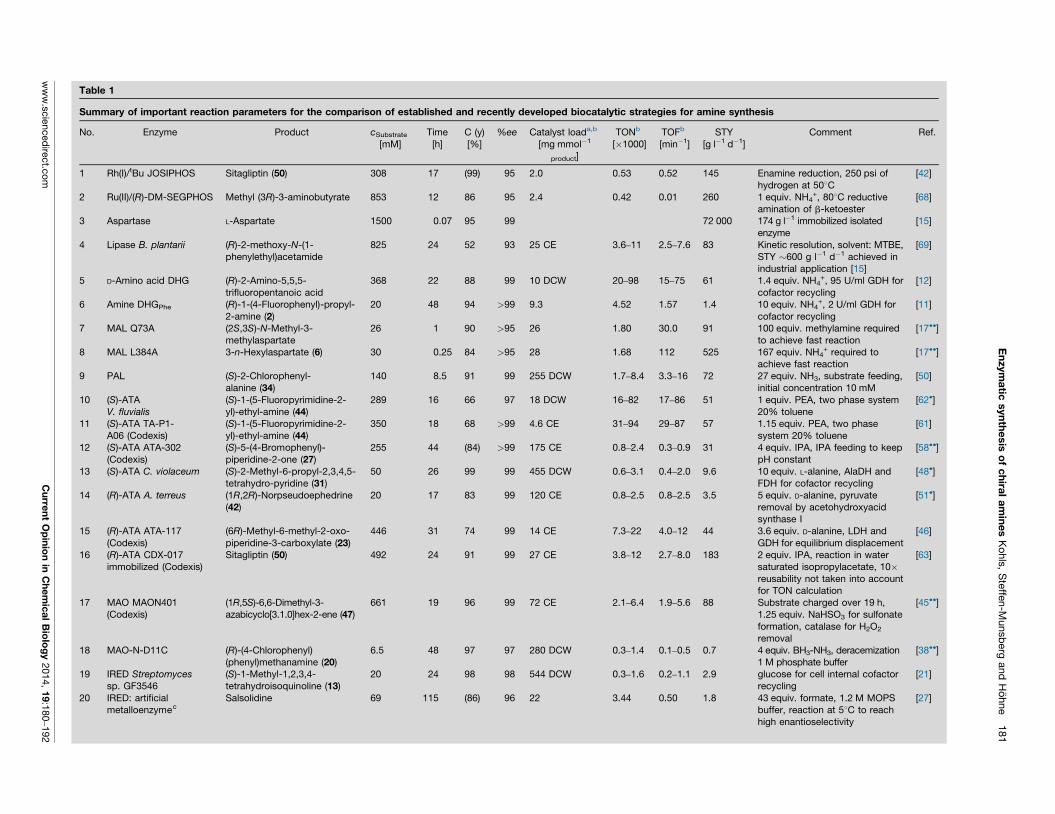
En
zym
atic
syn
the
sis
of
ch
iral
am
ine
s K
ohls
, S
teffe
n-M
unsb
erg
and
Ho
hne
181
Table 1
Summary of important reaction parameters for the comparison of established and recently developed biocatalytic strategies for amine synthesis
No. Enzyme Product cSubstrate
[mM]Time[h]
C (y)[%]
%ee Catalyst loada,b
[mg mmol�1
product]
TONb
[�1000]TOFb
[min�1]STY
[g l�1 d�1]Comment Ref.
1 Rh(I)/tBu JOSIPHOS Sitagliptin (50) 308 17 (99) 95 2.0 0.53 0.52 145 Enamine reduction, 250 psi ofhydrogen at 508C
[42]
2 Ru(II)/(R)-DM-SEGPHOS Methyl (3R)-3-aminobutyrate 853 12 86 95 2.4 0.42 0.01 260 1 equiv. NH4+, 808C reductive
amination of b-ketoester[68]
3 Aspartase L-Aspartate 1500 0.07 95 99 72 000 174 g l�1 immobilized isolatedenzyme
[15]
4 Lipase B. plantarii (R)-2-methoxy-N-(1-phenylethyl)acetamide
825 24 52 93 25 CE 3.6–11 2.5–7.6 83 Kinetic resolution, solvent: MTBE,STY �600 g l�1 d�1 achieved inindustrial application [15]
[69]
5 D-Amino acid DHG (R)-2-Amino-5,5,5-trifluoropentanoic acid
368 22 88 99 10 DCW 20–98 15–75 61 1.4 equiv. NH4+, 95 U/ml GDH for
cofactor recycling[12]
6 Amine DHGPhe (R)-1-(4-Fluorophenyl)-propyl-2-amine (2)
20 48 94 >99 9.3 4.52 1.57 1.4 10 equiv. NH4+, 2 U/ml GDH for
cofactor recycling[11]
7 MAL Q73A (2S,3S)-N-Methyl-3-methylaspartate
26 1 90 >95 26 1.80 30.0 91 100 equiv. methylamine requiredto achieve fast reaction
[17��]
8 MAL L384A 3-n-Hexylaspartate (6) 30 0.25 84 >95 28 1.68 112 525 167 equiv. NH4+ required to
achieve fast reaction[17��]
9 PAL (S)-2-Chlorophenyl-alanine (34)
140 8.5 91 99 255 DCW 1.7–8.4 3.3–16 72 27 equiv. NH3, substrate feeding,initial concentration 10 mM
[50]
10 (S)-ATAV. fluvialis
(S)-1-(5-Fluoropyrimidine-2-yl)-ethyl-amine (44)
289 16 66 97 18 DCW 16–82 17–86 51 1 equiv. PEA, two phase system20% toluene
[62�]
11 (S)-ATA TA-P1-A06 (Codexis)
(S)-1-(5-Fluoropyrimidine-2-yl)-ethyl-amine (44)
350 18 68 >99 4.6 CE 31–94 29–87 57 1.15 equiv. PEA, two phasesystem 20% toluene
[61]
12 (S)-ATA ATA-302(Codexis)
(S)-5-(4-Bromophenyl)-piperidine-2-one (27)
255 44 (84) >99 175 CE 0.8–2.4 0.3–0.9 31 4 equiv. IPA, IPA feeding to keeppH constant
[58��]
13 (S)-ATA C. violaceum (S)-2-Methyl-6-propyl-2,3,4,5-tetrahydro-pyridine (31)
50 26 99 99 455 DCW 0.6–3.1 0.4–2.0 9.6 10 equiv. L-alanine, AlaDH andFDH for cofactor recycling
[48�]
14 (R)-ATA A. terreus (1R,2R)-Norpseudoephedrine(42)
20 17 83 99 120 CE 0.8–2.5 0.8–2.5 3.5 5 equiv. D-alanine, pyruvateremoval by acetohydroxyacidsynthase I
[51�]
15 (R)-ATA ATA-117(Codexis)
(6R)-Methyl-6-methyl-2-oxo-piperidine-3-carboxylate (23)
446 31 74 99 14 CE 7.3–22 4.0–12 44 3.6 equiv. D-alanine, LDH andGDH for equilibrium displacement
[46]
16 (R)-ATA CDX-017immobilized (Codexis)
Sitagliptin (50) 492 24 91 99 27 CE 3.8–12 2.7–8.0 183 2 equiv. IPA, reaction in watersaturated isopropylacetate, 10�reusability not taken into accountfor TON calculation
[63]
17 MAO MAON401(Codexis)
(1R,5S)-6,6-Dimethyl-3-azabicyclo[3.1.0]hex-2-ene (47)
661 19 96 99 72 CE 2.1–6.4 1.9–5.6 88 Substrate charged over 19 h,1.25 equiv. NaHSO3 for sulfonateformation, catalase for H2O2
removal
[45��]
18 MAO-N-D11C (R)-(4-Chlorophenyl)(phenyl)methanamine (20)
6.5 48 97 97 280 DCW 0.3–1.4 0.1–0.5 0.7 4 equiv. BH3-NH3, deracemization1 M phosphate buffer
[38��]
19 IRED Streptomycessp. GF3546
(S)-1-Methyl-1,2,3,4-tetrahydroisoquinoline (13)
20 24 98 98 544 DCW 0.3–1.6 0.2–1.1 2.9 glucose for cell internal cofactorrecycling
[21]
20 IRED: artificialmetalloenzymec
Salsolidine 69 115 (86) 96 22 3.44 0.50 1.8 43 equiv. formate, 1.2 M MOPSbuffer, reaction at 58C to reachhigh enantioselectivity
[27]
ww
w.s
cie
nced
irect.c
om
C
urre
nt
Op
inio
n in
Ch
em
ica
l B
iolo
gy
2014,
19:1
80
–192

biocatalytic routes to developed chemical methods for
asymmetric reductive amination to guide the evaluation
of current biocatalytic processes (Table 1).
Discovery: novel catalytic activities by proteinengineeringIn recent years, novel biocatalytic routes were developed
by altering the substrate specificity or reaction specificity
of known enzymes, or by isolating and exploring the
potential of novel wild-type enzymes.
For the preparation of primary amines, the direct asym-
metric amination of ketones with ammonia is a highly
desirable reaction. Until now, no wild-type enzymes with
this activity are known, but the group of Bommarius
successfully engineered two (S)-selective amine dehydro-
genases, AmDHLeu and AmDHPhe, starting from L-leucine
[10��] or L-phenylalanine dehydrogenase (LeuDH and
PheDH respectively) [11] (Scheme 1a). The carboxylate
binding site of the wild-type enzymes could be modified by
focused mutagenesis of residues in the active site. For
LeuDH, eleven libraries were screened targeting 14 resi-
dues, resulting in a quadruple mutant with activity towards
methyl isobutyl ketone (0.69 U/mg). Interestingly, only
two amino acid residues in PheDH had to be mutated to
obtain an enzyme (AmDHPhe) with activity comparable to
a wild-type amine transaminase (ATA) (4 U/mg for the
amination of p-fluoroacetophenone). As a proof of concept,
in small preparative scale reactions, (R)-1,3-dimethylbu-
tylamine and (R)-1-(4-fluorophenyl)-propyl-2-amine could
be synthesized by AmDHLeu and AmDHPhe, respectively.
Important issues towards broader applicability of these
enzymes are twofold. The first is improving the substrate
scope, as the enzymes show a distinct preference for their
model substrates while other investigated ketones were
converted with only �1–10% relative activity. The
AmDHPhe prefers ketones having a methyl substituent,
similar to wild-type amine transaminases [6]. The second
issue concerns the reaction equilibrium. In the proof of
concept reactions good conversions (85% and 94% maxi-
mum conversion) were achieved if a 10–50 fold excess of
ammonia was employed and glucose dehydrogenase was
used to shift the equilibrium. The non-quantitative con-
versions of the model substrates suggest that the amination
in thermodynamically more unfavored reactions might
suffer from low conversion, for example when acetophe-
none derivatives are used as amino acceptor.
Overall, the optimization of amine dehydrogenases for
synthetic purposes similar to amino acid dehydrogenases
[12,13] (see Table 1, entries 5 and 6, Scheme 3d) would
be clearly advantageous in terms of step efficiency com-
pared to chemical hydrogenations of ketimines or enam-
ides using chiral metal complexes or auxiliaries [14�].Beneficially, ammonia is a cheap amino donor and its
use further simplifies the separation of the obtained
amine product from the reaction mixture.
182 Biocatalysis and biotransformation
Ta
ble
1(C
ontinued
)
No
.E
nzy
me
Pro
duct
cS
ub
str
ate
[mM
]T
ime
[h]
C(y
)[%
]%
ee
Cata
lyst
load
a,b
[mg
mm
ol�
1
pro
duct]
TO
Nb
[�1000]
TO
Fb
[min�
1]
ST
Y[g
l�1
d�
1]
Co
mm
ent
Ref.
21
IRE
D:
art
ificia
lm
eta
lloenzy
me
dS
als
olid
ine
444
82
70
3204
0.0
09
0.0
04
0.4
750
eq
uiv
.fo
rmate
,re
actio
nat
48C
toin
cre
ase
enantio
sele
ctivity,
at
408C�
4�
faste
rw
ith
half
%ee
[28]
22
CjN
CS
2P
icte
t-S
peng
lera
se
(S)-
No
rco
cla
urine
(16)
53
(99)
95
58
0.4
02.2
311
1.5
eq
uiv
.ald
ehyd
e[2
3� ]
23
P411
BM
3-C
IST
438S
(S)-
5,7
-Die
thyl-
3-m
eth
yl-
2,3
-d
ihyd
rob
enzo
[d]is
oth
iazo
le1,1
-dio
xid
e(1
0)
224
77
87
2742
DC
W0.1
–0.5
0.1
–0.4
0.4
Anaero
bic
co
nd
itio
ns,
glu
co
se
for
cell
inte
rnal
co
facto
rre
cyclin
g[1
9� ]
Ab
bre
viatio
ns:
C,
co
nvers
ion;
y,
yie
ld;
ee,
enantio
meric
excess;
TO
N,
turn
over
num
ber;
TO
F,
turn
over
freq
uency;
ST
Y,
sp
ace
–tim
eyie
ld;
CE
,cru
de
extr
act;
DH
G,
dehyd
rog
enase;
DC
W,
dry
cell
weig
ht;
GD
H,g
luco
se
dehyd
rog
enase;M
AL,m
eth
yla
sp
art
ate
am
mo
nia
lyase;P
AL,p
henyla
lanin
eam
mo
nia
lyase;A
TA
,am
ine
transam
inase;P
EA
,1-p
henyl
eth
anam
ine;IP
A,i
so
pro
pyla
min
e;A
laD
H,
ala
nin
ed
ehyd
rog
enase;
FD
H,
form
ate
dehyd
rog
enase;
LD
H,
lacta
ted
ehyd
rog
enase;
MA
O,
mo
no
am
ine
oxid
ase;
IRE
D,
imin
ere
ducta
se.
aC
ata
lyst
load
calc
ula
ted
for
pure
enzy
me,
cell
dry
weig
ht
(CD
W)
or
freeze
dried
cell
extr
act
(CE
),d
ep
end
ing
on
the
availa
ble
info
rmatio
n.
Ad
ditio
nally
ad
ded
enzy
mes
(e.g
.fo
req
uili
brium
dis
pla
cem
ent)
are
no
tta
ken
into
acco
unt.
bA
pp
roxim
atio
ns
for
calc
ula
tio
n:
1O
D600
i1
mg
wet
cells
ml�
1culture
i1
ml w
et
cells
ml�
1culture
i0.2
5m
gd
rycells
ml�
1culture
i0.0
75
–0.1
25
mg
freeze
dry
ed
cru
de
extr
act
ml�
1culture
i0.0
4–0.1
8m
gta
rget
enzym
em
l�1culture
.If
giv
en
as
rang
e,th
ere
sultin
gvalu
es
are
roug
hestim
ate
sd
ue
tom
issin
gin
form
atio
nin
the
co
rresp
ond
ing
pub
licatio
n.T
urn
over
num
ber
(TO
N)and
turn
over
freq
uency
(TO
F)have
been
calc
ula
ted
as
am
ean
valu
eo
ver
the
who
lere
actio
ntim
e.
c[C
p*I
r(B
iot-
p-L
)Cl]�
Str
ep
tavid
ine
S112A
muta
nt.
d[(h
5�
Cp
*)Ir
(pic
o4)C
l]�
hum
an
carb
onic
anhyd
rase
IIW
T.
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com

A similar highly desired reaction would be the hydro-
amination of alkenes, but for this reaction only biocata-
lysts for the synthesis of amino acids are available, as
demonstrated in the astonishingly efficient aspartate pro-
cess using aspartate ammonia lyase (aspartase) [EC
4.3.1.1] [15] (Table 1, entry 3). Additionally, protein
engineering of a 3-methylaspartate lyase (MAL) [EC
4.3.1.2] afforded variants applicable for the synthesis of
non-canonical amino acids. Initially, the nucleophile
specificity was extended from ammonia to amines by a
single mutation located in the ammonia binding region of
the active site [16,17��]; whereas methylamine is not
accepted as a substrate by MAL wild-type at a detectable
level, the Q73A variant utilized methylamine with a kcat
of 0.8 s�1 and a KM of �0.5 M (Scheme 1b). However, the
turnover number is relatively low compared to the ammo-
nia addition by the WT enzyme (kcat 50 s�1). More bulky
amines such as cyclobutylamine or 3-hydroxypropyl-
amine were also accepted and allowed the synthesis
of secondary amines containing the aspartate moiety
(Table 1, entry 7). A second mutation (L384A) allowed
the utilization of a broad range of C2-fumarate deriva-
tives, such as 2-n-hexylfumarate that is converted
with high efficiency (kcat = 36 s�1 and KM = 4.6 mM,
Scheme 1c, Table 1, entry 8). As the L384A/Q73A
double mutant was almost inactive, further research
aims to create double mutants by focused mutagenesis
of both positions simultaneously to yield more versatile
catalysts with both a relaxed electrophile and nucleo-
phile specificity. Additional research also aims to gen-
erate mutants that can be useful for b-amino acid
synthesis, as the a-carboxylate of aspartate seems to
be dispensable for the reaction mechanism, in contrast
to the C4-carboxylate [8]. Researchers at c-LEcta were
able to demonstrate a first proof of concept using aspar-
tase: a mutant converting crotonic acid yielding 3-amino
butanoic acid was identified after screening 300 000
mutant clones (Scheme 1d). Although details of the
protein engineering were not disclosed, the activity
was reported to be 6 U/g DCW (compared to
21 000 U/g DCW for the natural reaction) [18]. Further
evolution of aspartase or MAL could furnish enzymes
suitable for b-amino acid synthesis. Alternative
approaches of b-amino acid synthesis include the appli-
cation of b-aminotransferases or 4-methylideneimida-
zole-5-one (MIO) dependent ammonia lyases or
mutases. However, both strategies are hampered by
certain drawbacks. The substrates for transamination,
b-keto acids, undergo spontaneous decarboxylation.
Using mutases or ammonia lyases, b-amino acids are
accessible from a-amino acids or a,b-unsaturated car-
boxylic acids, respectively. Unfortunately in both cases, a
mixture of a-amino acids and b-amino acids is obtained, as
an enzyme featuring an exclusive b-selectivity is not yet
known. An ammonia lyase for the preparation of amines
from alkenes lacking a carboxylate, however, still seems
out of reach.
In the above-mentioned examples, the substrate speci-
ficity was altered. McIntosh et al. showed that it is also
possible to change the reaction specificity: an engineered
P450BM3 monooxygenase was able to catalyze an intra-
molecular asymmetric C-H amination with reasonable
TON (Table 1, entry 23), which is not observed in nature
but is known from organic synthesis [19�]. As a proof of
principle, resting cells containing a 15-tuple mutant of
P450BM3 were used as a catalyst under anaerobic con-
ditions to afford 39 mg sultam 10 with 87%ee at 77%
conversion, together with sulfonamide 11, which was
generally observed as a by-product (Scheme 1e). There-
fore, exploring enzymatic C-H amination would be very
interesting from a synthetic point of view, especially if the
substrate scope is widened or an intermolecular C-H
amination could be realized. Details for the protein
engineering of P450 enzymes can be found elsewhere
in this issue [20].
Discovery: identification of novel wild-typeenzymesBesides optimization of known enzymes through protein
or reaction engineering, the discovery of novel wild-type
enzymes is still an important strategy to access new
biocatalysts. Two examples of recently recognized
enzymes are imine reductases (IREDs) [EC 1.5.1.X]
[21,22] and ‘Pictet-Spenglerases’ [23�,24,25], which are
useful for the preparation of secondary amines. Strictly
NADPH-dependent (S)-selective and (R)-selective
IREDs from Streptomyces sp. were identified by classical
enrichment cultures, similar to the identification strategy
employed in the discovery of some of the early amine
transaminases. (S)-IREDs prefer six-membered cyclic
imines, such as dihydroisoquinolines and dihydro-b-car-
bolines over 5-membered methylpyrrolines; the larger
cyclic imines being converted with an activity up to
1 U/mg [21] (Scheme 1f). The conversion of an iminium
ion indicated that these enzymes might also be useful for
the preparation of tertiary amines. Whole cell reactions
facilitating cofactor recycling demonstrate the biocataly-
tic potential of (S)-IREDs (Table 1, entry 19). In contrast,
a (R)-IRED showed a very narrow substrate specificity
and activity of only 10 mU/mg for methylpyrroline, the
best model substrate reported [26]. The crystal structure
of the (R)-IRED with bound NADPH revealed a very
large active site [26]. As the metabolic function of these
enzymes has not yet been discovered, the relatively low
activity might indicate that the natural substrates differ
substantially from the compounds investigated so far. In
addition to the naturally occurring imine reductases,
artificial metalloenzymes bearing iridium complexes in
a protein scaffold have been investigated [27,28].
Remarkably, formate acts as reducing agent, and reason-
able turnovers and enantioselectivities have been
achieved with a streptavidin scaffold [27]. Recent results
with iridium complexes in human carbonic anhydrase II
[28] by contrast gave only low turnover numbers with
Enzymatic synthesis of chiral amines Kohls, Steffen-Munsberg and Hohne 183
www.sciencedirect.com Current Opinion in Chemical Biology 2014, 19:180–192
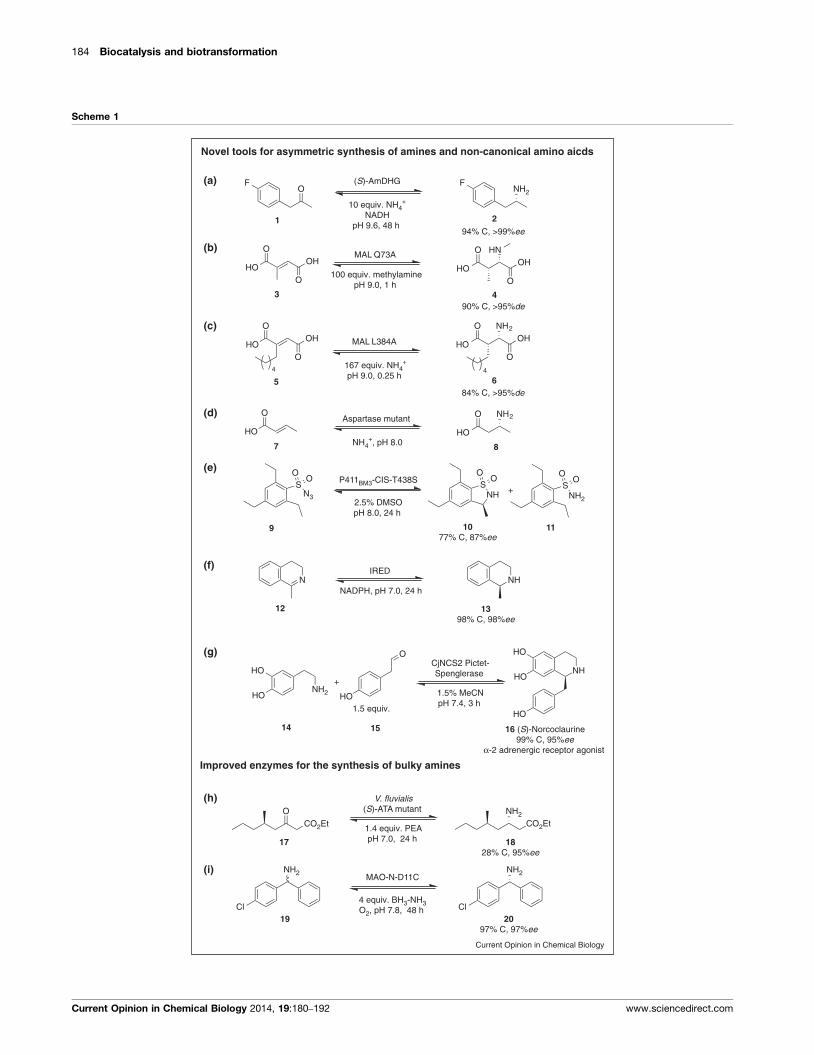
184 Biocatalysis and biotransformation
Scheme 1
NH2F
OF
HNO
HOO
OH
O
HOO
OHMAL Q73A
100 equiv. methylaminepH 9.0, 1 h
NHS
OO
SO
O
N3
NHN
NH
HO
HO
HO
NH2HO
HO
HO
O
+
CO2EtNH2
CO2EtO
NH2O
HOO
OHO
HOO
OH
(S)-AmDHG
10 equiv. NH4+
NADHpH 9.6, 48 h
94% C, >99%ee
90% C, >95%de
MAL L384A
167 equiv. NH4+
pH 9.0, 0.25 h
84% C, >95%de
P411BM3-CIS-T438S
2.5% DMSOpH 8.0, 24 h
IRED
NADPH, pH 7.0, 24 h
1.5 equiv.
CjNCS2 Pictet-Spenglerase
1.5% MeCNpH 7.4, 3 h
V. fluvialis(S)-AT A mutant
1.4 equiv. PEApH 7.0, 24 h
NH2
Cl
NH 2
Cl
MAO-N-D11C
4 equiv. BH3-NH3O2, pH 7.8, 48 h
(a)
(b)
(c)
(d)
Novel tools for asymmetric synthesis of amines and non-canonical amino aicds
NH2O
HO
O
HO
Aspartase mutant
NH4+, pH 8.0
(e)
(f)
(g)
NH2
SO
O
1 2
+
Improved enzymes for the synthesis of bulky amines
(h)
(i)
65
3 4
111077% C, 87%ee
9
87
1828% C, 95%ee
17
16 (S)-Norcoclaurine99% C, 95%ee
α-2 adrenergic receptor agonist
14
1398% C, 98%ee
12
2097% C, 97%ee
19
15
4 4
Current Opinion in Chemical Biology
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com

reasonable %ee, which suggests that the protein scaffold
mainly determined the catalyst’s performance (compare
entries 19–21 in Table 1).
Although norcoclaurine synthase (NCS) [EC 4.2.1.78]
and strictosidine synthases [EC 4.3.3.2] have been known
for over three decades [29], the biocatalytic potential of a
plant NCS was investigated only recently [23�,25].
During the reaction, a C–C bond is formed via an elec-
trophilic addition reaction, which is in stark contrast to the
other available biocatalysts for amine synthesis, where a
C–H or a C–N bond is formed as the key asymmetric step.
Owing to the requirements of the reaction mechanism,
the substrate scope is restricted to 2-phenylethanamines
with a free hydroxyl group in the ortho position. In a proof
of principle reaction, several structurally different alde-
hydes could be used (Scheme 1g) and the final products
were isolated using preparative HPLC, as they racemize
during silica gel chromatography. Strictosidine synthases
seem to have a narrow substrate specificity leading
to products with a common tryptoline skeleton,
although recently products based on the rare piperazino
[1,2-a]indole scaffold could be synthesized [24].
These examples demonstrate that careful analysis of the
secondary metabolism can be an interesting resource for
enzyme mining to find new biocatalysts useful for amine
synthesis.
Advancements by broadening the catalyticscopeUnderstanding the reaction mechanism and the avail-
ability of crystal structures significantly aided in the
identification of novel enzymes and guided the engin-
eering of the substrate scope of known enzymes to
broaden the amine synthesis toolbox. During the last
few years, several structures from (S)-ATAs [30�,31–33], b-amino acid TAs [34,35] and very recently also from
(R)-ATAs [36,37] were solved, along with structures from
MAOs [38��,39] and IREDs [26–28]. Interestingly, since
2009 crystal structures of four uncharacterized proteins
were deposited in the PDB that could only recently be
characterized as (S)-selective transaminases with varying
activities towards amines [40�]. The structures facilitated
an understanding of the mechanism of dual substrate
recognition and of factors influencing the level of ATA
activity [41].
The strategic use of structure guided protein engineering,
the smart application of known enzymes, process optim-
ization and the combination thereof, led to four major
advances that significantly expanded the scope of bioca-
talytic amine synthesis:
(i) Protein engineering allowed for broadening the
substrate scope of several enzymes. Enlarging the
small binding pocket of (S)-selective ATAs enabled
the conversion of substrates with two bulky
substituents. An eight-fold mutant of ATA from V.fluvialis showed a 60-fold improved activity towards
the bulky 1,3-ketoester 17 (kcat of 0.02 s�1), which is
a first step towards an asymmetric synthesis of
imagabaline [30�] (Scheme 1h). Regarding this low
activity, it seems to be more difficult to engineer the
active site in (S)-ATAs compared to the success story
reported for (R)-ATA-117 by Codexis and
Merck&Co [42,43]. Furthermore, engineering
MAO-N yielded an enzyme variant able to
deracemize amines with two bulky substituents,
such as 19, an important intermediate for Levoce-
tirizine [38��] (Scheme 1i). A recent protein
engineering approach to broaden the substrate
scope of a 6-hydroxy-D-nicotine oxidase filled a gap
in the biocatalytic amine toolbox by providing the
missing enantiocomplementary (R)-MAO [44]. In
contrast to transaminases it is striking that MAOs
do not suffer from an unfavourable equilibrium,
however, only low substrate concentrations can be
applied owing to pronounced substrate and product
inhibition [45��].(ii) Secondary and tertiary amines became accessible
from primary amines when the substrate was
properly chosen to enable cyclization after the
enzymatic reaction (Scheme 2a–d). Cyclization has
been demonstrated for a 1,5-amino ester 22 [46],
aminoketones 30 [47,48�,49], and o-halogenated L-
phenylalanine 34 [50], as well as imine 38, which
underwent a Pictet-Spengler reaction to the tertiary
amine harmicine 40 [38��]. The cyclized products
were then further processed by a MAO catalyzed
reaction [49] or by reduction. In the latter case, a
careful choice of reagents and reaction conditions
allowed a diastereoselective reduction and thus
access to the syn-piperidine and anti-piperidine 32.
All four diastereomers were accessible via modular
combination of either a (R)-selective or a (S)-
selective ATA followed by a syn or anit-selective
chemical reduction [48�].(iii) Challenging substrates: the diastereomers of 1,2-
aminoalcohols were synthesized by two consecutive
asymmetric steps combined in a one-pot cascade
reaction (Table 1, entry 14, Scheme 3e). For the
Enzymatic synthesis of chiral amines Kohls, Steffen-Munsberg and Hohne 185
( Scheme 1 Legend ) Reaction schemes of newly developed or further explored enzymes for the synthesis of amines and non-canonical amino acids.
(a) Glucose dehydrogenase (GDH) was applied to accomplish a displacement of equilibrium. (e,f) Whole cells where applied. GDH was used to
facilitate the cofactor recycling. (S)-AmDHG, (S)-selective amine dehydrogenase; MAL, 3-methylaspartate lyase; P411BM3, mutant of P450BM3: the
Soret peak of the ferrous CO-bound enzyme is shifted to 411 nm compared to 450 nm for the wild-type; IRED, imine reductase; (S)-ATA, (S)-selective
amine transaminase; MAO, monoamine oxidase.
www.sciencedirect.com Current Opinion in Chemical Biology 2014, 19:180–192
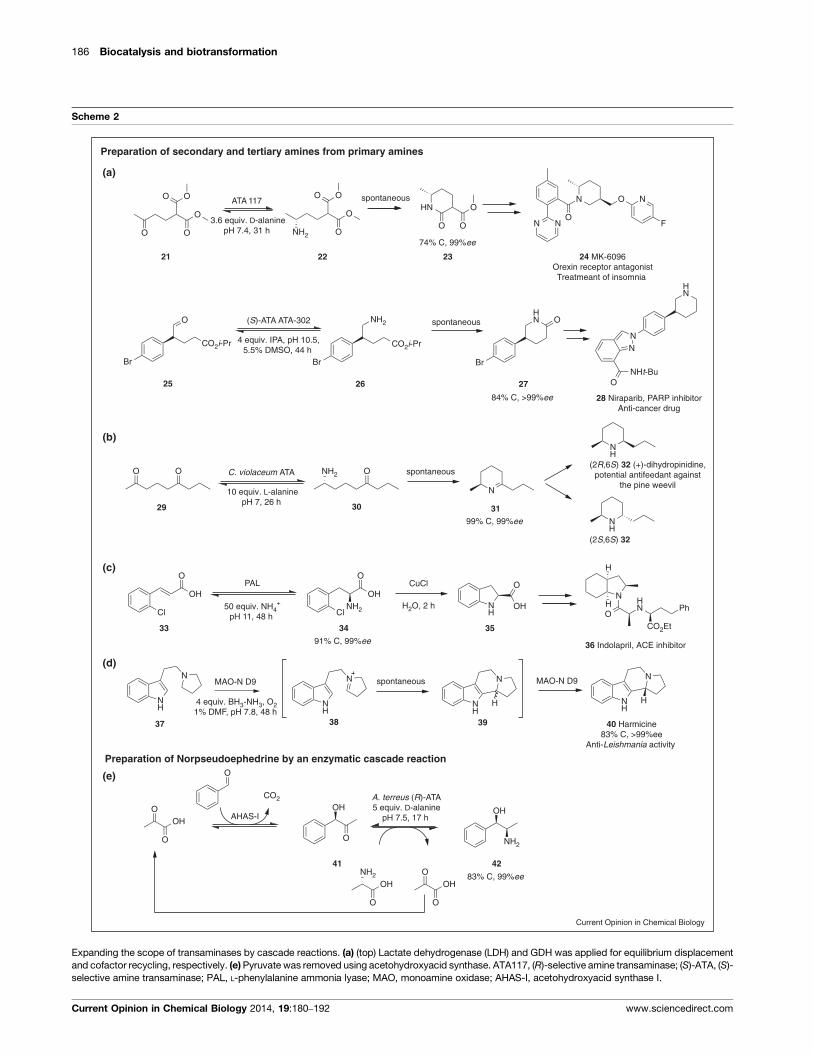
186 Biocatalysis and biotransformation
Scheme 2
OH
O
O
O
CO2
OH
O
OH
O
OOH
O
NH2
OH
NH2
A. terreus ( R)-AT A5 equiv. D-alanine
pH 7.5, 17 h
NH
N
NH
N
HNH
N
NH
N
H
MAO-N D9
4 equiv. BH3-NH3, O21% DMF, pH 7.8, 48 h
NH2
O
OH
Cl
O
OH
ClNH
OH
OCuCl
H2O, 2 h
PAL
50 equiv. NH4+
pH 11, 48 h
N
HN
OO
O O
OO
O
NH2 O
OO
O
O
O O ONH2C. violaceum AT A
10 equiv. L-alaninepH 7, 26 h
AHAS-I
spontaneous
99% C, 99%ee
ATA 117
3.6 equiv. D-alaninepH 7.4, 31 h
74% C, 99%ee
spontaneous
83% C, 99%ee
MAO-N D9
(a)
(b)
(c)
(d)
22
30
34
38
Preparation of Norpseudoephedrine by an enzymatic cascade reaction
(e)
42
Br
NH2
CO2i-Pr
Br
O
CO2i-Pr
spontaneous
Br
HN O(S)-AT A AT A-302
4 equiv. IPA, pH 10.5,5.5% DMSO, 44 h
84% C, >99%ee
21 23
26
29
(2R,6 S) 32 (+)-dihydropinidine,potential antifeedant against
the pine weevil
31
33 35
37 40 Harmicine83% C, >99%ee
Anti-Leishmania activity
39
41
24 MK-6096Orexin receptor antagonistTreatmeant of insomnia
NH
NH
N NO
N O N
F
Preparation of secondary and tertiary amines from primary amines
2725
(2S,6 S) 32
N
HN
N
NHt-BuO
28 Niraparib, PAR P inhibitorAnti-cancer drug
36 Indolapril, ACE inhibitor
N
O
HN
CO2Et
Ph
H
H
91% C, 99%ee
spontaneous
Current Opinion in Chemical Biology
Expanding the scope of transaminases by cascade reactions. (a) (top) Lactate dehydrogenase (LDH) and GDH was applied for equilibrium displacement
and cofactor recycling, respectively. (e) Pyruvate was removed using acetohydroxyacid synthase. ATA117, (R)-selective amine transaminase; (S)-ATA, (S)-
selective amine transaminase; PAL, L-phenylalanine ammonia lyase; MAO, monoamine oxidase; AHAS-I, acetohydroxyacid synthase I.
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com

synthesis of norephedrine and norpseudoephe-
drine, the hydroxyketone generated by a carboliga-
tion was aminated with a (R)-ATA or (S)-ATA.
Surprisingly, the (R)-ATA showed a strict pre-
ference for 41 over benzaldehyde [51�]. This was
unexpected, as aldehydes are considered the much
more reactive substrates. Hence, the cascade could
operate in a simultaneous mode with all enzymes
present in one pot during the whole reaction. The
combination of ketoreductases and transaminases
was also investigated [52] in order to access the
remaining diastereomers of 42. Many different
cascade reactions were conceptualized to enable
deracemization [53], or the conversion of alcohols to
amines [54] and were reviewed in detail recently
[55–57]. Upscaling and optimization of these
cascade reactions to afford higher STY will be an
important issue in the future.
(iv) A dynamic kinetic resolution was developed for the
preparation of b-chiral-1,5-amino ester 26, which
spontaneously cyclized to 3-phenylpiperidone 27.
Luckily, a b-enantioselective TA could be ident-
ified, as most screened ATAs showed low b-
enantioselectivity [58��]. After reduction this com-
pound can be used for the synthesis of Niraparib, a
potential drug for cancer treatment.
Enzymatic synthesis of chiral amines Kohls, Steffen-Munsberg and Hohne 187
Scheme 3
V. fluvialis(S)-AT A
phosphate buffe r, pH 7.520% toluene, 16 h
NH
N
MAON401ox MAON401red
O2H2O2
H2O + 1/2 O2
Catalase
1.25 equiv. NaHSO3pH 7.4, 19 h
N
OO
FF
FNN
N
F3C
N
NH2O
FF
FNN
N
F3C
(R)-AT A CDX017
2.67 equiv. IPA50% DMSO, pH 8.5, 15 h
66% C, 97%ee
Examples for upscaled reactions
96% C, 99%ee
50 Sitagliptin95% C, 99%ee
Treatment for diabetes mellitus type 2
49
4746
4443
(b)
(a)
(c)
HN
HN
OO
HHHN
O
O
NH2O
48 BoceprevirTreatment for Hepatitis C
45 AZD 1480JAK2 kinase inhibitor
N
N
HN
N
NNH
N
F
Cl
N
N
O
F
N
N
NH2
F
OOH
O
NH2
OH
OLeu-DHG
2 equiv. NH4+
NAD+, pH 8, 17 h 50 51
99% C, >99%ee
N
N
N
O
NH
N
O
F
FO
52Corticotropin-releasing factor-1(CRF-1)
receptor antagonist
(d)
NH2 O
1 equiv.
Current Opinion in Chemical Biology
Reactions optimized for larger scale synthesis. (a) The biphasic system phosphate buffer/toluene was used as solvent. The by-product acetophenone
was removed by extraction from the equilibrium. (S)-ATA, (S)-selective amine transaminase; MAON401, monoamine oxidase mutant; IPA,
isopropylamine; Leu-DHG, L-leucine dehydrogenase.
www.sciencedirect.com Current Opinion in Chemical Biology 2014, 19:180–192

Maturation: optimization for industrial scaleapplication
Optimizing a biocatalytic reaction for industrial appli-
cations is a challenging task as many parameters need
to be optimized, which can often only be achieved by a
combination of extensive reaction and protein engineer-
ing. Although biocatalysis often claims to be environmen-
tally friendly because aqueous solvents are employed
(which are usually not considered in the calculation of
the E-factor for a process), water miscible or immiscible
cosolvents are often required to allow high substrate
loadings [59] or to decrease product inhibition. As organic
solvent contaminated water causes high costs for disposal,
increasing the product concentration to minimize reaction
volume is of high importance, especially as many of the
proof of concept reactions employ very low concen-
trations. Secondly, a high activity is crucial to ensure
low enzyme concentration in the reaction, as this is
considered a significant cost factor [2,60]. Additionally,
high protein concentrations often cause problems in
downstream processing.
The successful upscaling of several reactions using amine
transaminases [46,58��,61,62�,63], PAL [50], MAO [45��]and amino acid dehydrogenase [13] were reported
recently. For the synthesis of a Janus kinase 2 (JAK2)
inhibitor, the aryl-alkyl amine 44 bearing a p-fluoropyr-
imidine ring was required as an intermediate (Scheme
3a). The mild reaction conditions were the decisive factor
for developing a transamination reaction, as with numer-
ous other strategies oily tars were formed due to the
instability of the pyrimidine ring. The ‘strong’ amino
donor 1-phenylethanamine (PEA) enabled a near quan-
titative conversion at already 1–1.15 equivalents and a
substrate load of �300 mM was facilitated by using
toluene as a cosolvent to circumvent product inhibition
from acetophenone. The STY of 50 g l�1 d�1 using a
wild-type transaminase is a remarkable success, but
switching to the engineered Codexis enzyme eventually
enabled an as-yet unmatched low catalyst load (Table 1,
entries 10, 11) [62�]. Although asymmetric syntheses like
this reaction featuring near 100% conversion are con-
sidered favorably compared to classical kinetic resolution
with lipases (Table 1, entry 4) because of its higher atom
economy, it is worth mentioning that the utilized chiral
amino donor (S)-PEA is presumably also prepared by
biocatalysis from a classical kinetic resolution with a
lipase.
A second very successful example is the development of
an efficient oxidative desymmetrization of a bicyclic pro-
line intermediate for the boceprevir manufacture
(Scheme 3b) using a MAO [45��]. Protein engineering
afforded a suitable catalyst, which displays 8-fold higher
activity, improved enzyme solubility and thermal stability
at 508C compared to the wild-type. In spite of these
achievements, the main challenge was the still remaining
susceptibility of the MAO for substrate and (irreversible)
product inhibition: already 0.8 mM imine 47 decreased
activity by 40% over time. By careful fine-tuning of the
substrate feeding and the addition of bisulfite to achieve a
trapping of the imine product, these inhibition issues
were overcome. Another successfully applied solution
for inhibition issues is the application of a two phase
system to avoid high substrate and product concentrations
in the aqueous phase [62�].
The sitagliptin process [42,43] published in 2010
(Scheme 3c) can be seen as an ultimate benchmark
reaction and as a true maturation of a transaminase
process in terms of space–time yield, which could not
be outperformed over the last three years. Furthermore,
the engineered (R)-ATA of Codexis was reported to be
active in an immobilized form in water saturated isopro-
pyl acetate [63]. This afforded sitagliptin with a similar
STY to the established process, simplified the workup,
and enabled reusability of the catalyst (Table 1, entry 16).
Recent results suggest that freeze dried crude extracts
containing wild-type ATA can also be employed as
enzyme formulation for performing transamination in
organic solvents [64]. In our opinion, further research is
important to increase the robustness of this method.
ConclusionAs discussed in this review most enzymes have to go
through a certain maturation process to be eventually
applied as a biocatalytic tool on an industrial scale.
Furthermore, important challenges and achievements
at the different stages within this progression were sum-
marized. For example protein engineering of dehydro-
genases [10��] and ammonia lyases [17��] acting on a-
amino acids allowed the creation of enzymes for reductive
amination of ketones and the preparation of b-amino
acids and N-alkyl substituted aspartate derivatives. Also
a P450 monooxygenase could be redesigned to catalyze
intramolecular C-H aminations [19�]. It is remarkable that
these engineering efforts created genuine novel activities,
as these could not be detected in any of the wild-type
enzymes. The biocatalytic potential of NCS [23�] and
reductases [21] acting on imines demonstrate that the
secondary metabolism is an interesting resource for
enzyme discovery, and we expect that future research
will reveal other useful enzymes allowing the synthesis of
secondary and tertiary amines.
In the proof of principle reactions, reasonable TON and
excellent %ee were achieved, but enzyme activity, sub-
strate scope and concentration were limited. We know
these issues are solvable, as this has already been demon-
strated in other cases: reaction engineering and structure
inspired protein design were undertaken to facilitate the
biocatalytic conversion of bulkier substrates [30�,38��].
188 Biocatalysis and biotransformation
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com

Unfortunately, the obstacle of product inhibition and
substrate inhibition could not yet be overcome by rational
design approaches. Having the high substrate load of the
sitagliptin process [43] in mind, directed evolution is a
more promising strategy to address this challenge. A more
tempting approach would be the application of enzymes
that do not suffer from inhibition. One ATA was recently
characterized which has this feature and might be a
promising candidate for further studies [65]. This finding
stresses the importance of having a large collection of
enzymes in the toolbox. The importance of high diversity
is furthermore demonstrated in another case, where only
one ATA screened from a large collection showed high
enantioselectivity in the amination of the chiral aldehyde
25 [58��].
Through these explorations, reactions with reasonable
STY were realized by the application of a high catalyst
load. In favor of simpler downstream processing, enzyme
loadings need to be lowered for the maturation towards
industrial applications. This issue is often addressed by
immobilization [63] or protein engineering [43] to obtain
catalysts with enhanced activity and stability. Special
tricks in process design, such as trapping of reactive
products can further improve economic efficiency
[45��]. The examples of developed processes on indus-
trial scale demonstrated that there is no generally
applicable solution to improve STY. Instead, each bio-
catalytic reaction required an individual optimization to
address important issues such as equilibrium, enzyme
stability, and inhibition.
Lipases are the most mature enzyme class in the biocata-
lysis field today as they have been used synthetically for the
longest time. Industrial processes utilizing lipases have
STY in the 200–600 g l�1 d�1 range (Table 1, entry 4), and
this provides an aspirational target for these relatively new
asymmetric reactions reviewed here. Only a few of the
mature asymmetric chemistries presented have attained
this level of productivity (aspartate ammonia lyase or
aspartase, Table 1, entry 3, methylaspartate ammonia
lyase, Table 1 entry 8 and the sitagliptin (R)-transaminase,
Table 1, entry 16). However even at lower STY, the
asymmetric nature of these reactions has a dramatic
positive impact on the productivity and efficiency of the
overall chemical syntheses by substantially improving step
and atom efficiency and providing enhanced flexibility in
reaction design. These same high STY processes also
compare favorably to the chemocatalytic processes (Table
1, entries 1 and 2) with STYs of 145 and 260 g l�1 d�1
respectively. In addition, research efforts have expanded
the scope of asymmetric amine synthesis. Until recently
the use of lipases constituted the only biocatalytic method
to access b-chiral amines or those with two similar sub-
stituents [1,66,67]. Due to persistent research efforts, these
amines are now accessible asymmetrically via transamin-
ases and by monoamine oxidases. As such, the biocatalytic
toolbox has been substantially extended in terms of
additional possibilities to access such compounds by asym-
metric synthesis. In conclusion, the current biocatalytic
toolbox contains new advanced tools and has matured to a
level competitive with asymmetric chemical synthesis.
Conflicts of interestThe authors are aware of no conflicts of interest regarding
the preparation and submission of this manuscript.
AcknowledgementsWe thank Dr Henrike Brundiek from Enzymicals AG and Dr Clare Vickersand Martin Gand, both from the Institute of Biochemistry, Greifswald, forfruitful discussions.
References and recommended readingPapers of particular interest, published within the period of review,have been highlighted as:
� of special interest�� of outstanding interest
1. Dunn PJ: The importance of green chemistry in processresearch and development. Chem Soc Rev 2012, 41:1452-1461.
2. Tufvesson P, Lima-Ramos J, Jensen JS, Al-Haque N, Neto W,Woodley JM: Process considerations for the asymmetricsynthesis of chiral amines using transaminases. BiotechnolBioeng 2011, 108:1479-1493.
3. Woodley JM: Protein engineering of enzymes for processapplications. Curr Opin Chem Biol 2013, 17:310-316.
4. Kroutil W, Fischereder E-M, Fuchs CS, Lechner H, Mutti FG,Pressnitz D, Rajagopalan A, Sattler JH, Simon RC, Siirola E:Asymmetric preparation of prim-, sec-, and tert-aminesemploying selected biocatalysts. Org Process Res Dev 2013,17:751-759.
5. Rudat J, Brucher BR, Syldatk C: Transaminases for thesynthesis of enantiopure b-amino acids. AMB Express 2012,2:11.
6. Hohne M, Bornscheuer UT: Application of transaminases inorganic synthesis. In Enzymes in Organic Synthesis. Edited byMay O, Groger H, Drauz K. Wiley-VCH; 2012:779-820.
7. Ghislieri D, Turner N: Biocatalytic approaches to the synthesisof enantiomerically pure chiral amines. Top Catal 2013, 57:284-300.
8. Heberling MM, Wu B, Bartsch S, Janssen DB: Priming ammonialyases and aminomutases for industrial and therapeuticapplications. Curr Opin Chem Biol 2013, 17:250-260.
9.��
Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC,Robins K: Engineering the third wave of biocatalysis. Nature2012, 485:185-194.
This review describes the main developments in the field of biocatalysis.Important breakthroughs and persisting limitations are addressed todepict the state of the art in enzyme engineering.
10.��
Abrahamson MJ, Vazquez-Figueroa E, Woodall NB, Moore JC,Bommarius AS: Development of an amine dehydrogenase forsynthesis of chiral amines. Angew Chem Int Ed 2012, 51:3969-3972.
A leucine dehydrogenase has been subjected to eleven rounds of proteinengineering to yield an amine dehydrogenase that converts a leucineanalogue without a carboxyl group. Enzymes of the first library had to bepurified prior to the activity measurements to distinguish the very lowinitial activities from background signal. Interestingly, the ratio of reduc-tive amination over deamination was pronounced differently in distinctmutants. The reiterated site saturation of the most promising positionsfrom the earlier rounds led to an enzyme with reasonable amine dehy-drogenase activity.
11. Abrahamson MJ, Wong JW, Bommarius AS: The evolution of anamine dehydrogenase biocatalyst for the asymmetric
Enzymatic synthesis of chiral amines Kohls, Steffen-Munsberg and Hohne 189
www.sciencedirect.com Current Opinion in Chemical Biology 2014, 19:180–192

production of chiral amines. Adv Synth Catal 2013,355:1780-1786.
12. Hanson RL, Johnston RM, Goldberg SL, Parker WL, Goswami A:Enzymatic preparation of an R-amino acid intermediate for ag-secretase inhibitor. Org Process Res Dev 2013, 17:693-700.
13. Parker WL, Hanson RL, Goldberg SL, Tully TP, Goswami A:Preparation of (S)-1-cyclopropyl-2-methoxyethanamine by achemoenzymatic route using leucine dehydrogenase. OrgProcess Res Dev 2012, 16:464-469.
14.�
Nugent TC, Marinova SM: Step-efficient access to chiralprimary amines. Synthesis 2013, 45:153-166.
A useful minireview as guide for the chemical preparation of opticallyactive primary amines using 1-phenylethanamine as chiral auxiliar. Ithighlights the developments of a one-pot protocol and shows importanttricks to avoid product loss during the workup.
15. Liese A, Seelbach K, Wandrey C: Industrial Biotransformations.edn 2. Weinheim: Wiley-VCH; 2006, .
16. Puthan Veetil V, Raj H, de Villiers M, Tepper PG, Dekker FJ,Quax WJ, Poelarends GJ: Enantioselective synthesis of N-substituted aspartic acids using an engineered variant ofmethylaspartate ammonia lyase. ChemCatChem 2013, 5:1325-1327.
17.��
Raj H, Szymanski W, de Villiers J, Rozeboom HJ, Puthan Veetil V,Reis CR, de Villiers M, Dekker FJ, de Wildeman S, Quax WJ et al.:Engineering methylaspartate ammonia lyase for theasymmetric synthesis of unnatural amino acids. Nat Chem2012, 4:478-484.
The first example for a substantially broadened nucleophile and electro-phile substrate specificity of MAL by focused protein engineering. Inter-estingly, the mutants produced the threo-(2S,3S) diasteromer with >95%de in contrast to the wild-type (5% de), and thus two stereocenters couldbe obtained in a single operation.
18. Casablancas A, Cardenas-Fernandez M, Alvaro G, Benaiges MD,Carminal G, De Mas C, Gonzalez G, Lopez C, Lopez-Santın J: Newammonia lyases and amine transaminases: standardization ofproduction process and preparation of immobilizedbiocatalysts. Electron J Biotechnol 2013, 16 http://dx.doi.org/10.2225/vol2216-issue2223-fulltext-4.
19.�
McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC,Brown TR, Arnold FH: Enantioselective intramolecular C–Hamination catalyzed by engineered cytochrome P450enzymes in vitro and in vivo. Angew Chem Int Ed 2013, 52:9309-9312.
Protein engineering changed the reaction specificity to facilitate C–Hamination, which is not known in nature. The two key mutations, C400Sand T268A, are conserved residues that play an essential role in thereaction mechanism of P450 WT enzymes: C400 ligates the hemecofactor, and T286 protonates the iron-peroxo intermediate.
20. McIntosh JA, Farwell CC, Arnold FH: Expanding P450 catalyticreaction space through evolution and engineering. Curr OpinChem Biol 2014, 19:126-134.
21. Leipold F, Hussain S, Ghislieri D, Turner NJ: Asymmetricreduction of cyclic imines catalyzed by a whole-cellbiocatalyst containing an (S)-imine reductase. ChemCatChem2013, 5:3505-3508.
22. Mitsukura K, Suzuki M, Shinoda S, Kuramoto T, Yoshida T,Nagasawa T: Purification and characterization of a novel (R)-imine reductase from Streptomyces sp., GF3587. BiosciBiotechnol Biochem 2011, 75:1778-1782.
23.�
Pesnot T, Gershater MC, Ward JM, Hailes HC: The catalyticpotential of Coptis japonica NCS2 revealed — developmentand utilisation of a fluorescamine-based assay. Adv SynthCatal 2012, 354:2997-3008.
The developed photometric assay enabled a fast and in depth character-ization of the substrate specificity of NCS. The results obtained by theassay could be confirmed by small scale preparation of these com-pounds. Based on these results and molecular docking studies, analternative binding mode of the substrate is proposed in contrast to apreviously reported reaction mechanism.
24. Wu F, Zhu H, Sun L, Rajendran C, Wang M, Ren X, Panjikar S,Cherkasov A, Zou H, Stockigt J: Scaffold tailoring by a newlydetected Pictet-piperazino-indole framework activity of
strictosidine synthase (STR1): From the common tryptolineskeleton to the rare piperazino-indole framework. J Am ChemSoc 2012, 134:1498-1500.
25. Ruff BM, Brase S, O’Connor SE: Biocatalytic production oftetrahydroisoquinolines. Tetrahedron Lett 2012, 53:1071-1074.
26. Rodrıguez-Mata M, Frank A, Wells E, Leipold F, Turner NJ, Hart S,Turkenburg JP, Grogan G: Structure and activity of NADPHdependent reductase Q1EQE0 from Streptomyceskanamyceticus, which catalyses the R-selective reduction ofan imine substrate. ChemBioChem 2013, 14:1372-1379.
27. Durrenberger M, Heinisch T, Wilson YM, Rossel T, Nogueira E,Knorr L, Mutschler A, Kersten K, Zimbron MJ, Pierron J et al.:Artificial transfer hydrogenases for the enantioselectivereduction of cyclic imines. Angew Chem Int Ed 2011, 50:3026-3029.
28. Monnard FW, Nogueira ES, Heinisch T, Schirmer T, Ward TR:Human carbonic anhydrase II as host protein for the creationof artificial metalloenzymes: the asymmetric transferhydrogenation of imines. Chem Sci 2013, 4:3269.
29. Stockigt J, Antonchick AP, Wu F, Waldmann H: The Pictet-Spengler reaction in nature and in organic chemistry. AngewChem Int Ed 2011, 50:8538-8564.
30.�
Midelfort KS, Kumar R, Han S, Karmilowicz MJ, McConnell K,Gehlhaar DK, Mistry A, Chang JS, Anderson M, Villalobos A et al.:Redesigning and characterizing the substrate specificity andactivity of Vibrio fluvialis aminotransferase for the synthesis ofimagabalin. Protein Eng Des Sel 2012, 26:25-33.
The first initial success in engineering an (S)-selective amine transami-nase towards bulky substrates. With three different strategies of librarydesign, unique beneficial mutations could be obtained which were com-bined in the final mutant. The effect of the mutations could be rationalizedfrom a solved crystal structure of the final mutant.
31. Rausch C, Lerchner A, Schiefner A, Skerra A: Crystal structure ofthe v-aminotransferase from Paracoccus denitrificans and itsphylogenetic relationship with other class IIIaminotransferases that have biotechnological potential.Proteins 2013, 81:774-787.
32. Sayer C, Isupov MN, Westlake A, Littlechild JA: Structural studiesof Pseudomonas and Chromobacterium-aminotransferasesprovide insights into their differing substrate specificity. ActaCrystallogr D 2013, 69:564-576.
33. Svedendahl Humble M, Engelmark Cassimjee K, Hakansson M,Kimbung YR, Walse B, Abedi V, Federsel HJ, Berglund P,Logan DT: Crystal structures of the Chromobacteriumviolaceum v-transaminase reveal major structuralrearrangements upon binding of coenzyme PLP. FEBS J 2012,279:779-792.
34. Crismaru CG, Wybenga GG, Szymanski W, Wijma HJ, Wu B,Bartsch S, de Wildeman S, Poelarends GJ, Feringa BL,Dijkstra BW et al.: Biochemical properties and crystal structureof a b-phenylalanine aminotransferase from Variovoraxparadoxus. Appl Environ Microbiol 2013, 79:185-195.
35. Wybenga GG, Crismaru CG, Janssen DB, Dijkstra BW: Structuraldeterminants of the b-selectivity of a bacterialaminotransferase. J Biol Chem 2012, 287:28495-28502.
36. Thomsen M, Skalden L, Palm GJ, Hohne M, Bornscheuer UT,Hinrichs W: Crystallographic characterisation of the (R)-selective amine transaminase from Aspergillus fumigatus.Acta Crystallogr D 2014, 70:1086-1093 http://dx.doi.org/10.1107/S1399004714001084.
37. Łyskowski A, Gruber C, Steinkellner G, Schurmann M, Schwab H,Gruber K, Steiner K: Crystal structure of an (R)-selective v-transaminase from Aspergillus terreus. PLoS ONE 2014, 9:350http://dx.doi.org/10.1371/journal.pone.0087350.
38.��
Ghislieri D, Green AP, Pontini M, Willies SC, Rowles I, Frank A,Grogan G, Turner NJ: Engineering an enantioselective amineoxidase for the synthesis of pharmaceutical building blocksand alkaloid natural products. J Am Chem Soc 2013, 135:10863-10869.
By protein engineering, the active site volume of MAO-N9 was furtherincreased. This is the first example of a biocatalytic preparation ofenantiopure (S)-amines containing two bulky aryl substituents. Solifena-
190 Biocatalysis and biotransformation
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com

cin, Levocetirizine, (R)-eleagnine, (R)-leptaflorine and (R)-harmicine wereprepared by the evolved MAO.
39. Mirza IA, Burk DL, Xiong B, Iwaki H, Hasegawa Y, Grosse S,Lau PCK, Berghuis AM: Structural analysis of a novelcyclohexylamine oxidase from Brevibacterium oxydans IH-35A. PLOS ONE 2013, 8:072 http://dx.doi.org/10.1371/journal.pone.0060.
40.�
Steffen-Munsberg F, Vickers C, Thontowi A, Schatzle S,Tumlirsch T, Svedendahl Humble M, Land H, Berglund P,Bornscheuer UT, Hohne M: Connecting unexplored proteincrystal structures to enzymatic function. ChemCatChem 2013,5:150-153.
Structural information of four transaminases converting (S)-amines wasalready available (but not recognized) in the PDB three years before acrystal structure of the established C. violaceum ATA was solved. In a onepot screening reaction, various amino acceptors and amino donors werecombined to rapidly elucidate the function of these enzymes. Interest-ingly, the level of activity towards amines is pronounced very different.These findings and mutagenesis studies in the active site might guideidentification of ATAs in the future.
41. Steffen-Munsberg F, Vickers C, Thontowi A, Schatzle S,Meinhardt T, Svedendahl Humble M, Land H, Berglund P,Bornscheuer UT, Hohne M: Revealing the structural basis ofpromiscuous amine transaminase activity. ChemCatChem2013, 5:154-157.
42. Desai AA: Sitagliptin manufacture: a compelling tale of greenchemistry, process intensification, and industrial asymmetriccatalysis. Angew Chem Int 2011, 50:1974-1976.
43. Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR,Colbeck JC, Krebber A, Fleitz FJ, Brands J et al.: Biocatalyticasymmetric synthesis of chiral amines from ketonesapplied to Sitagliptin manufacture. Science 2010,329:305-309.
44. Heath RS, Pontini M, Bechi B, Turner NJ: Development of an R-selective amine oxidase with broad substrate specificity andhigh enantioselectivity. ChemCatChem 2014 http://dx.doi.org/10.1002/cctc.201301008.
45.��
Li T, Liang J, Ambrogelly A, Brennan T, Gloor G, Huisman G,Lalonde J, Lekhal A, Mijts B, Muley S et al.: Efficient,chemoenzymatic process for manufacture of the boceprevirbicyclic [3.1.0]proline intermediate based on amine oxidase-catalyzed desymmetrization. J Am Chem Soc 2012, 134:6467-6472.
First application of a MAO in an industrial process. The net reaction is anoxidative Strecker reaction. For the key step, the oxidative desymme-trization of the prochiral amine substrate, a MAO variant with high activityhad to be engineered. A perfect fine tuning of various parameters, forexample substrate and bisulfite feeding was necessary to circumventsubstrate and especially product inhibition.
46. Girardin M, Ouellet SG, Gauvreau D, Moore JC, Hughes G,Devine PN, O’Shea PD, Campeau L-C: Convergent kilogram-scale synthesis of dual orexin receptor antagonist. OrgProcess Res Dev 2013, 17:61-68.
47. Simon RC, Grischek B, Zepeck F, Steinreiber A, Belaj F, Kroutil W:Regio- and stereoselective monoamination of diketoneswithout protecting groups. Angew Chem Int 2012, 51:6713-6716.
48.�
Simon RC, Zepeck F, Kroutil W: Chemoenzymatic synthesis ofall four diastereomers of 2,6-disubstituted piperidines throughstereoselective monoamination of 1,5-diketones. Chem Eur J2013, 19:2859-2865.
This contribution demonstrates the elegance of combining enantiocom-plementary enzymes and cis/trans diastereoselective reduction reactionsin a modular way to access all possible diastereomers. Reagents andconditions had to be chosen very carefully to optimize the trans-selectivereduction step.
49. O’Reilly E, Iglesias C, Ghislieri D, Hopwood J, Galman JL,Lloyd RC, Turner NJ: A regio- and stereoselective v-transaminase/monoamine oxidase cascade for the synthesisof chiral 2,5-disubstituted pyrrolidines. Angew Chem Int Ed2014, 53:2447-2450.
50. de Lange B, Hyett DJ, Maas PJD, Mink D, van Assema FBJ,Sereinig N, de Vries AHM, de Vries JG: Asymmetric synthesis of
(S)-2-indolinecarboxylic acid by combining biocatalysis andhomogeneous catalysis. ChemCatChem 2011, 3:289-292.
51.�
Sehl T, Hailes HC, Ward JM, Wardenga R, von Lieres E,Offermann H, Westphal R, Pohl M, Rother D: Two steps in onepot: Enzyme cascade for the synthesis ofnor(pseudo)ephedrine from inexpensive starting materials.Angew Chem Int Ed 2013, 52:6772-6775.
An extensive optimization of reaction parameters allowed to install twostereocenters by the simultaneous action of a C-C lyase and an aminetransaminase. Interestingly, the byproduct generated in the transamina-tion (second reaction) was consumed as substrate in the carboligation(first reaction).
52. Sehl T, Pohl M, Rother D: Verfahren zur Herstellung von Cathinedurch Kopplung einer (S)-selektiven Transaminase und einer(S)-selektiven Alkoholdehydrogenase. Ger Patent 2013,German patent application: DE102013 009 145.3.
53. Shin G, Mathew S, Shon M, Kim B-G, Yun H: One-pot one-stepderacemization of amines using v-transaminases. ChemCommun 2013, 49:8629-8631.
54. Tauber K, Fuchs M, Sattler JH, Pitzer J, Pressnitz D,Koszelewski D, Faber K, Pfeffer J, Haas T, Kroutil W: Artificialmulti-enzyme networks for the asymmetric amination of sec-alcohols. Chem Eur J 2013, 19:4030-4035.
55. Simon RC, Richter N, Busto E, Kroutil W: Recent developmentsof cascade reactions involving v-transaminases. ACS Catal2014, 4:129-143.
56. Oroz-Guinea I, Garcia-Junceda E: Enzyme catalysed tandemreactions. Curr Opin Chem Biol 2013, 17:236-249.
57. Ricca E, Brucher B, Schrittwieser JH: Multi-enzymatic cascadereactions: overview and perspectives. Adv Synth Catal 2011,353:2239-2262.
58.��
Chung CK, Bulger PG, Kosjek B, Belyk KM, Rivera N, Scott ME,Humphrey GR, Limanto J, Bachert DC, Emerson KM: Processdevelopment of C–N cross-coupling and enantioselectivebiocatalytic reactions for the asymmetric synthesis ofniraparib. Org Process Res Dev 2014, 18:215-227.
This is the first example where a highly enantioselective ATA was used inan efficient dynamic kinetic resolution to prepare an amine with the chiralcenter in beta position. The prepared aldehyde precursor was isolated asbisulfite adduct in the previous reaction step and could be used directlyas starting material for the transamination reaction, thereby circumvent-ing problems with stability and isolation of the aldehyde. Microscale high-throughput techniques were employed for optimization of single reactionsteps. Thus, a high number of reaction parameters (e.g. solvent, reagents)could be screened to rapidly identify the optimal reaction conditions.
59. Simon RC, Fuchs CS, Lechner H, Zepeck F, Kroutil W: Concisechemoenzymatic three-step total synthesis of isosolenopsinthrough medium engineering. Eur J Org Chem 2013:3397-3402.
60. Tufvesson Pr, Lima-Ramos J, Nordblad M, Woodley JM:Guidelines and cost analysis for catalyst production inbiocatalytic processes. Org Process Res Dev 2010, 15:266-274.
61. Frodsham L, Golden M, Hard S, Kenworthy MN, Klauber DJ,Leslie K, Macleod C, Meadows RE, Mulholland KR, Reilly J et al.:Use of v-transaminase enzyme chemistry in the synthesisof a JAK2 kinase inhibitor. Org Process Res Dev 2013,17:1123-1130.
62.�
Meadows RE, Mulholland KR, Schurmann M, Golden M,Kierkels H, Meulenbroeks E, Mink D, May O, Squire C,Straatman H et al.: Efficient synthesis of (S)-1-(5-fluoropyrimidin-2-yl)ethylamine using an v-transaminasebiocatalyst in a two-phase system. Org Process Res Dev 2013,17:1117-1122.
The first report showing that a process based on a wild-type transami-nase being optimized to achieve a sufficient product concentration. Byapplying a biphasic system, product inhibition of the co-product acet-ophenone could be circumvented.
63. Truppo MD, Strotman H, Hughes G: Development of animmobilized transaminase capable of operating in organicsolvent. ChemCatChem 2012, 4:1071-1074.
64. Mutti FG, Kroutil W: Asymmetric bio-amination of ketones inorganic solvents. Adv Synth Catal 2012, 354:3409-3413.
Enzymatic synthesis of chiral amines Kohls, Steffen-Munsberg and Hohne 191
www.sciencedirect.com Current Opinion in Chemical Biology 2014, 19:180–192

65. Park E-S, Shin J-S: v-Transaminase from Ochrobactrumanthropi is devoid of substrate and product inhibitions. ApplEnviron Microbiol 2013, 79:4141-4144.
66. Koszelewski D, Cwiklak M, Ostaszewski R: A newchemoenzymatic approach to the synthesis of chiral 4-aryl-1,4-dihydro-2H-isoquinolines via the enzymatic resolution of2-acetyl-4-phenyl-1,4-dihydro-2H-isoquinolin-3-one.Tetrahedron Asymmetry 2012, 23:1256-1261.
67. Busto E, Martınez-Montero L, Gotor V, Gotor-Fernandez V:Chemoenzymatic asymmetric synthesis of serotonin
receptor agonist (R)-Frovatriptan. Eur J Org Chem 2013:4057-4064.
68. Matsumura K, Zhang X, Hori K, Murayama T, Ohmiya T,Shimizu H, Saito T, Sayo N: Practical, catalyticenantioselective hydrogenation to synthesizeN-unprotected b-amino esters. Org Process Res Dev 2011,15:1130-1137.
69. Balkenhohl F, Ditrich K, Hauer B, Ladner W: Optically activeamines via lipase-catalyzed methoxyacetylation. J Prakt Chem1997, 339:381-384.
192 Biocatalysis and biotransformation
Current Opinion in Chemical Biology 2014, 19:180–192 www.sciencedirect.com


Articles 63
ARTICLE II


DOI: 10.1002/adsc.201500214
Selective Access to All Four Diastereomers of a 1,3-AminoAlcohol by Combination of a Keto Reductase- and an AmineTransaminase-Catalysed Reaction
Hannes Kohls,a Mattias Anderson,b Jonathan Dickerhoff,a Klaus Weisz,a
Armando Cýrdova,c Per Berglund,b Henrike Brundiek,d Uwe T. Bornscheuer,a,*and Matthias Hçhnea,*a Institute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germany
Fax: (++49)-3834-86-794367; phone: (++ 49)-3834-86-4367 (UTB), (++49)-3834-86-22832 (MH);e-mail: [email protected] or [email protected]
b KTH Royal Institute of Technology, Division of Industrial Biotechnology, School of Biotechnology, AlbaNova UniversityCenter, SE-106 91 Stockholm, Sweden
c Department of Natural Sciences, Engineering and Mathematics, Mid Sweden University, SE-851 70 Sundsvall, Swedend Enzymicals AG, Walther-Rathenau-Straße 49a, 17489 Greifswald, Germany
Received: March 2, 2015; Revised: April 20, 2015; Published online: May 18, 2015
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201500214.
Abstract: The biocatalytic synthesis of chiral amineshas become a valuable addition to the chemistsÏ tool-box. However, the efficient asymmetric synthesis offunctionalised amines bearing more than one stereo-centre, such as 1,3-amino alcohols, remains challeng-ing. By employing a keto reductase (KRED) andtwo enantiocomplementary amine transaminases(ATA), we developed a biocatalytic route towardsall four diastereomers of 4-amino-1-phenylpentane-2-ol as a representative molecule bearing the 1,3-amino alcohol functionality. Starting from a racemichydroxy ketone, a kinetic resolution using an (S)-se-lective KRED provided optically active hydroxyketone (86% ee) and the corresponding diketone.
Further transamination of the hydroxy ketone wasperformed by either an (R)- or an (S)-selective ATA,yielding the (2R,4R)- and (2R,4S)-1,3-amino alcoholdiastereomers. The remaining two diastereomerswere accessible in two subsequent asymmetric steps:the diketone was reduced regio- and enantioselec-tively by the same KRED, which yielded the (S)-con-figured hydroxy ketone. Eventually, the subsequenttransamination of the crude product with (R)- and(S)-selective ATAs yielded the remaining (2S,4R)-and (2S,4S)-diastereomers, respectively.
Keywords: amine transaminase; amino alcohols;enzyme catalysis; keto reductase
Introduction
Chiral 1,3-amino alcohols (g-amino alcohols) arefound as a structural motif in many natural productsand biologically active compounds and are thereforepromising synthons for the pharmaceutical industry.[1]
Furthermore, they are applied as chiral auxiliaries inorganic synthesis.[2] However, the synthesis of the 1,3-amino alcohol motif is tedious and step-intensive, asexpressed in a proverb that each nitrogen in a mole-cule increases a graduate studentÏs career by at leastone year.[3] Consequently, efficient new methods forthe enantio- and diastereoselective preparation of thiscompound class are in high demand.[4]
The chemical synthesis of 1,3-amino alcohols canbe accomplished by asymmetric assembly of b-aminoketones[4,5] or b-hydroxy imines[6] with subsequent ste-reoselective reduction of either the ketone or theimine, respectively. Further strategies imply transitionmetal-catalysed C¢H amination starting from allylic[7]
or aliphatic[8] educts. The incubation of tosylated aldi-mines with a palladium catalyst yields 1,3-amino alco-hol derivatives with low-to-moderate selectivity asshown recently by Menche et al.[9] Furthermore, thering opening of 3-trifluoromethyl-2-isoxazolines yields1,3-amino alcohols, but unfortunately with low diaste-reomeric excess.[10]
Due to the constant innovation in protein engineer-ing, the application of biocatalysts represents a valua-
1808 Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2015, 357, 1808 – 1814
FULL PAPERS

ble addition to conventional chemistry.[11] In particu-lar, biocatalytic routes to compounds with more thanone chiral centre have a great potential, as in theoryall diastereomers can be accessed in a step-efficientmanner[12] with high optical purity owing to the oftenexcellent stereo- and regioselectivity of enzymes.[13] Ifthe reaction parameters are suitable, both enzyme re-actions can be conducted in one pot or even in a cas-cade reaction. Furthermore, the modularity of this ap-proach allows the combination of enzymes with oppo-site regio- or enantioselectivity yielding products withdifferent diastereomeric configuration.
For example, the use of an engineered ATA in thesynthesis of a 1,2-amino alcohol was recentlyshown.[14] Combining the (R)-selective thiamine di-phosphate-dependent (ThDP) acetohydroxyacid syn-thase I (AHAS-I) and either an (S)- or (R)-selectiveamine transaminase (ATA) in a stepwise fashion al-lowed the synthesis of either norephedrine (NE) ornorpseudoephedrine (NPE), respectively.[12] Theisomer (1S,2S)-NPE, also known as cathine, was ac-cessible by combination of an (S)-selective ATA withan (S)-selective alcohol dehydrogenase in high opticalpurity.[15] Another cascade was established by thecombination of a transketolase and an ATA yielding(2S,3S)-aminopentane-1,3-diol in two sequentialsteps.[16]
The use of enzymes in the synthesis of 1,3-amino al-cohols was explored recently. The combination of or-ganocatalysis, organometallic catalysis and biocataly-sis using a lipase was successfully applied for the syn-thesis of enantio- and diastereomerically pure N-Bocprotected 1,3-amino acetates.[4] This route providedaccess to two of four diastereomers. To the best ofour knowledge the synthesis of 1,3-amino alcohols in-cluding enzymes is restricted to this example.
We envisioned the biocatalytic synthesis of the 1,3-amino alcohol motif by the combination of a keto re-ductase (KRED)- and an ATA-catalysed reaction.One option for the assembly of the 1,3-amino alcoholwould be the asymmetric synthesis in two consecutivesteps starting from a prochiral 1,3-diketone:a KRED-catalysed reduction would yield the enantio-pure 1,3-hydroxy ketone (b-hydroxy ketone), which issubsequently aminated by an ATA. Alternatively, rac-emic 1,3-hydroxy ketones such as 1 and 3(Scheme 1A) could be used as starting material, asthese compounds are easily available by aldol reac-tions. Both hydroxy ketone enantiomers could be pre-pared by kinetic resolution or deracemisation employ-ing KREDs as reported for secondary alcohols.[17] Asubsequent transamination would yield the desired1,3-amino alcohol. The advantage of this approach isits modularity: depending on the enantiopreference ofthe employed KRED and ATA, the desired stereoiso-mers can be assembled.
In this work we report the synthesis of all four dia-stereomers of 4-amino-1-phenylpentane-2-ol (5,Scheme 1B) using a KRED and two enantiocomple-mentary ATAs by stepwise biocatalytic reactions fol-lowing the strategy discussed above (see Scheme 1B).The racemic 1,3-hydroxy ketone rac-3 was applied ina kinetic resolution catalysed by the (S)-selectiveKRED-P1-B10, providing enantioenriched (R)-3 andthe corresponding diketone 4. Transamination of (R)-3 with enantiocomplementary ATAs provided accessto either 5c or 5d. Diketone 4 was converted in step-wise biocatalytic reactions by KRED-P1-B10 to (S)-3,which was then further transaminated using ATA-Cvior ATA-025 to yield 5a or 5b, respectively.
Scheme 1. (A) Model substrates of this study. (B) Biocata-lytic route for the synthesis of the four diastereomers of 4-amino-1-phenylpentane-2-ol (5): A KRED-catalysed oxida-tion of the hydroxy ketone rac-3 yields the non-reactive ste-reoisomer (R)-3 in 86% ee and the diketone 4. After separa-tion of the products, the diketone can be reduced to the hy-droxy ketone with the opposite stereo configuration by em-ploying the same KRED. Finally, ATAs with opposite enan-tiopreferences facilitate synthesis of the amino alcoholdiastereomers. Note that the numbering of carbons in 5 and3 differs, according to IUPAC nomenclature.
Adv. Synth. Catal. 2015, 357, 1808 – 1814 Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 1809
FULL PAPERS Selective Access to All Four Diastereomers of a 1,3-Amino Alcohol

Results and Discussion
We started by screening several ATAs for acceptanceof the 1,3-hydroxy ketones 1 and 2 (Scheme 1A) assubstrates, which were prepared by an aldol reaction(see Supporting Information, Figure S5). Several at-tempts to convert 4-hydroxy-4-phenylbutan-2-one(rac-1) to the corresponding amino alcohol were un-successful. Neither the (S)-selective ATA from Chro-mabacterium violaceum (ATA-Cvi) and its mutantW60C,[18] nor six (R)-selective ATAs led to a conver-sion when combined with a large alanine excess (80–200 equiv., see Supporting Information, Table S1 fortested enzymes). Also, when combined with anenzyme cascade (l-alanine dehydrogenase and glu-cose dehydrogenase[19]) to shift the equilibrium, ATA-Cvi did not yield the desired amino alcohol. However,benzylamine was formed as a side-product with con-versions up to 52% (HPLC Method 1, see SupportingInformation for all analytics). Similarly, a reactionwith hydroxy ketone rac-2 and the same enzyme cas-cade gave vanillylamine as a side product with conver-sion of 40% after 90 h. Further experiments revealedthat the hydroxy ketone rac-1 degraded to benzalde-hyde by a retro aldol reaction under the conditionsused.[19] Although the retro aldol reaction was slowwhen compound rac-1 was dissolved in buffer, it wasfound to be promoted by alanine. Once formed, ben-zaldehyde was converted to benzylamine by the trans-aminase.
As an alternative to alanine, 1-phenylethylamine(1-PEA) was applied as amino donor (see SupportingInformation, Table S2 for tested conditions), as itdoes not promote the retro aldol reaction and onlytrace amounts of benzylamine were formed duringthe reaction. Nonetheless, the desired amino alcoholwas not detected. We hypothesised that the hydroxygroup of 1 or 2 is involved in an intramolecular hy-drogen bond with the carbonyl oxygen. This interac-tion might stabilise the hydroxy ketones and conse-quently render the reaction equilibrium more unfav-ourable, thereby complicating transamination. An at-tempt to convert the 1-O-acetyl-protected derivativeof rac-1, which lacks this putative stabilisation, wasunsuccessful. Finally, to investigate the reaction in thethermodynamically favoured direction, the corre-sponding amino alcohol was synthesised (SupportingInformation) and employed as substrate with pyru-vate as amino acceptor. As no conversion was ob-served (HPLC Method 1), we concluded that the in-vestigated ATAs were not able to act on this sub-strate.
We therefore synthesised the 1,3-hydroxy ketone 4-hydroxy-5-phenylpentane-2-one (rac-3, Scheme 1A,see Supporting Information, Scheme S1 for synthesisdetails), assuming that it might be accepted moreeasily as substrate since, in contrast to the phenyl sub-
stituent of compounds 1 and 2, the benzyl substituentof 3 generates a more flexible substrate. Furthermore,it prevents the degradation by retro aldol reaction: 3was stable when dissolved in aqueous phosphatebuffer together with alanine or isopropylamine (IPA),PLP and up to 20% (v/v) 2-propanol as co-solvent. Incorrelation with our assumptions, no degradationproducts could be observed using thin layer chroma-tography (TLC) and GC/MS analysis (GC/MSMethod 1) after three days of incubation. This is inline with our failed attempts to prepare 3 by a pro-line-catalysed asymmetric aldol reaction.
We were pleased to find that several ATAs actedon substrate rac-3 as detected by TLC (Supporting In-formation, Table S3). Conversion could be confirmedby HPLC (HPLC Method 2), after standards for ana-lytics [3, its regioisomer 4-hydroxy-1-phenylpentan-2-one (rac-6), the diol 1-phenylpentane-2,4-diol (7) andrac-5] were synthesised (Supporting Information,Scheme S1). Furthermore, a chiral GC method wasdeveloped facilitating the separation of all four diaste-reomers after derivatisation using MBTFA (GC/MSMethod 2, see Supporting Information, Figure S1 forchromatograms).
The most promising transaminase – ATA-025 – wasapplied for the asymmetric synthesis of amino alcohol5b and 5d using 50 equiv. isopropylamine (IPA) asamino donor. The substrate was consumed after 20 hand two of the four amino alcohol isomers where de-tected using GC/MS (Scheme 2A).
ATA-025 was able to utilise (R)-1-PEA as aminodonor, and no product was detected when (S)-1-PEAwas used instead. This indicates (R)-selectivity ofATA-025 towards substrate rac-3. To test this assump-tion, we applied the (S)-selective ATA-Cvi for theasymmetric synthesis of 5a and 5c (Scheme 2B). Toour delight, we found that the substrate was con-sumed after three days and the two remaining isomers5a and 5c were detected (GC/MS Method 2).
Having found two enantiocomplementary ATAsacting on rac-3, we aimed to find a KRED acting ex-clusively on the ÐinnerÏ C-2 carbon (hydroxy groupand carbonyl carbon for 3 and 4, respectively), as thiswould provide access to (R)-3 via kinetic resolutionand asymmetric synthesis of (S)-3, respectively. Byscreening the panel of engineered KREDs from Co-dexisÔ, we found five out of 22 enzymes were able tooxidise the carbonyl group at C-2 of rac-3 (GCMethod 3). The most active enzyme was KRED-P1-B10. All enzymes displayed the same enantioprefer-ence, oxidising (S)-3 to yield the corresponding dike-tone 4, thereby leaving behind (R)-3 (89% ee at 50%conversion, Scheme 3). The corresponding regioiso-mer 6 was detected only in trace amounts.
To obtain the 1,3-hydroxy ketone with the oppositeabsolute configuration – (S)-3 – it was necessary tofind an enzyme able to selectively reduce the C-2 car-
1810 asc.wiley-vch.de Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2015, 357, 1808 – 1814
FULL PAPERSHannes Kohls et al.

bonyl atom of diketone 4. The screening of the avail-able enzymes revealed that by the use of the sameKRED as in the previous step – KRED-P1-B10 – itwas possible to install the stereocentre at C-2 with
high regio- and enantioselectivity (86% ee, Scheme 4).Importantly, the corresponding hydroxy ketone regio-isomer 6 was detected in trace amounts only (1%,GC/MS Method 1), displaying the high regioselectivi-ty of the enzyme. Since the reaction was very fast, itwas crucial to stop it immediately after all the sub-strate was consumed. If the reaction was run for ex-tended time, (S)-3 was reduced further and eventuallyconverted to the corresponding diol quantitatively, asobserved via GC/MS (GC/MS Method 1). We foundthat the enzyme was able to convert up to 9 mmol(100 mM) of 4 quantitatively in less than 24 h. It tookless than 60 min to convert 1.8 mmol (20 mM) of 4and after two hours all substrate was converted to thecorresponding diol.
Having found a way to obtain the two optically en-riched enantiomers of 3, we set to synthesise them ona preparative scale, applying the conditions describedabove: 321 mg (1.8 mmol) of rac-3 were resolved to(R)-3 (160 mg, 50%, 86% ee) with the concomitantisolation of 4 (85 mg, 27%). The low yield of the dike-tone is probably due to its instability during columnchromatography.[20] Then, the isolated diketone 4(0.312 mmol) was converted to (S)-3 (48 mg, 86%,71% ee). Using the substrates (R)-3 and (S)-3, thefour diastereomers 5a–5d were synthesised on an ana-lytical scale with high enantioselectivity (>98% ee atcarbon C-4 carrying the amino group, full consump-tion of the substrates). Finally, the transamination of(R)-3 on a preparative scale (90 mg, 0.5 mmol) wasconducted with ATA-025, which yielded 5d (66 mg,73%).
The isolated product 5d was used to elucidate theabsolute stereo configuration at C-2 of the amino al-cohol. To this end, the absolute configuration of C-4of 5d was anticipated to be (R) for the following rea-sons: (i) enzyme ATA-025 was reported earlier to be(R)-selective for a range of substrates,[21] (ii) amino al-cohol 5 was only detected when the (R)-enantiomerof PEA was used as the amino donor, (iii), incubationof rac-3 with (S)-selective ATA-Cvi provided the tworemaining isomers as detected by GC/MS Method 2(Supporting Information). Consequently, as the bioca-talysis product 5d was identified as the anti-isomer viaNMR coupling constants, the absolute configuration
Scheme 2. Asymmetric synthesis of amino alcohol 5 usingenantiocomplementary ATAs. A) Incubation of rac-3 withATA-025 and 50 equiv. isopropylamine (IPA) as aminodonor resulted in the formation of diastereomers 5b and 5d(GC/MS Method 2). The co-product acetone was removedby purging with N2. Reaction conditions: 20 mM rac-3, 1 MIPA and 0.25 mM PLP were agitated in 100 mM sodiumphosphate buffer (pH 7.5) containing 20% (v/v) 2-propanolat 30 88C in a glass vial. B) ATA-Cvi facilitated formation ofa mixture of 5a and 5c (GC/MS Method 2). The equilibriumshift was accomplished by co-product removal of pyruvateby lactate dehydrogenase (LDH). The consumed NADHwas regenerated by glucose dehydrogenase (GDH). Reac-tion conditions: 10 mM rac-3, l-alanine (250 mM), d-glucose(150 mM), NADH (1 mM), PLP (0.1 mM), 90 U mL¢1 LDH,15 UmL¢1 GDH and 10 mg mL¢1 ATA-Cvi lyophilisatewere agitated with 1000 rpm at 30 88C in 100 mM sodiumphosphate buffer (pH 7.5) containing 20% (v/v) 2-propanolin a glass vial.
Scheme 3. Kinetic resolution of 1,3-hydroxy ketone rac-3.Five KREDs from the CodexisÔ Screening Kit were identi-fied to catalyse the reaction with the same enantioprefer-ence. As determined by GC/MS, the reaction was finishedafter 24 h leaving behind (R)-3, yielding diketone 4. Reac-tion conditions: 10 mM rac-3, 1 mM NADP++, 1 mM MgSO4,20% (v/v) acetone, 1 mg mL¢1 KRED lyophilisate in100 mM potassium phosphate buffer (pH 7). Agitated at1200 rpm in a glass vial at 30 88C.
Scheme 4. Asymmetric synthesis of 1,3-hydroxy ketone (S)-3. Reaction conditions: 10 mM rac-3, 1 mM NADP++, 1 mMMgSO4, 20% (v/v) 2-propanol, 1 mgmL¢1 KRED lyophili-sate in 100 mM potassium phosphate buffer (pH 7), agitatedat 1200 rpm in a glass vial at 30 88C.
Adv. Synth. Catal. 2015, 357, 1808 – 1814 Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 1811
FULL PAPERS Selective Access to All Four Diastereomers of a 1,3-Amino Alcohol

at C-2 could be assigned to be (R) (see the Support-ing Information for details).
This study demonstrates the proof of principle fora new synthesis strategy towards 1,3-amino alcohols.Compared to classical reductive amination reactionsconducted for the preparation of amino alcohols, de-protection steps are not necessary. At the moment,the kinetic resolution step limits the possible yield to50% for a desired diastereomer. Therefore, an (R)-se-lective KRED is in high demand, since it would facili-tate the enzymatic deracemisation of 3, thereby maxi-mising the theoretical yield to 100%.[17] Alternatively,if the diketone is prepared by other means,[20,22] alldiastereomers could be accessed via two consecutiveasymmetric steps. Furthermore, we aim to improvethis approach by (i) performing this reaction as a onepot or as a cascade reaction and (ii) increasing sub-strate concentrations and reducing excess of theamino donor alanine to improve scalability and (iii)identifying enzymes that facilitate the synthesis of theremaining regioisomer of 5 (4-amino-5-phenylpentan-2-ol), having the amino group at the ÐinnerÏ position.
Conclusions
The asymmetric synthesis of functionalised amineswith more than one stereocentre has gained attentionlately. Our results prove the applicability of a new ap-proach towards the 1,3-amino alcohol motif by thecombination of the enzymes ATA and KRED, allow-ing for a successive introduction of two stereocentres.We consider this approach a valuable addition to thetraditional strategies towards this compound class, asit is highly selective, step efficient, and avoids transi-tion metal catalysis.
Experimental Section
Chemicals, Enzymes and Strains
All chemicals were purchased from Fluka (Buchs, Switzer-land), Sigma (Steinheim, Germany), Merck (Darmstadt,Germany), VWR (Hannover, Germany), or Carl Roth(Karlsruhe, Germany) and were used without further purifi-cation unless otherwise specified. Purification of productswas accomplished by flash column chromatography silica gel(Fluka 60, particle size 0.069–0.2 mm). Aluminium foil silicagel plates (Fluka 60 F254) were used for thin-layer chroma-tography (TLC). The synthesis of analytical standards is de-scribed in the Supporting Information.
Lactate dehydrogenase (LDH, order number L2500-5KU)and glucose dehydrogenase (GDH, order number 19359-10MG-F) were bought from Sigma–Aldrich. ATAs werebought as “CodexÔ TA Screening Kit” as lyophilised en-zymes. KREDs were bought as “CodexÔ KRED ScreeningKit” from CodexisÔ as lyophilised enzymes. The (R)-selec-tive wild-type amine transaminases AspFum, NeoFis,
GibZea, AspTer and AspOry were either supplied by En-zymicals AG or expressed as described later.
E. coli BL21 (DE3) {fhuA2 [lon] ompT gal (l DE3) [dcm]DhsdS} was purchased from New England Biolabs (Beverly,MA, USA). The plasmid pET24b bearing the gene encodingthe amine transaminase from Vibrio fluvialis (UniProt:F2XBU9) was kindly provided by Prof. Byung-Gee Kim(Seoul National University, Seoul, South Korea).
Cultivation and Expression Conditions
Expression and purification of the Chromobacterium viola-ceum wild-type amine transaminase (ATA-Cvi wt) was car-ried out as described previously by Anderson et al.[19] Thesame procedure was used for expression and purification ofthe W60C mutant of the enzyme (ATA-Cv W60C). Site-di-rected mutagenesis to obtain the mutant was performed pre-viously by Humble et al.[18] The concentration of ATA-Cviand ATA-Cvi W60C was measured as described by Cassim-jee et al.[23]
The codon-optimised gene for the transaminase ATA-117was inserted into the pET-22b plasmid between the restric-tion sites NdeI and BamHI. Cloning and expression of theamine transaminases from Aspergillus terreus (AspTer), As-pergillus oryzae (AspOry), Aspergillus fumigatus (AspFum),Neosartorya fischeri (NeoFis) and Gibberella zeae (GibZea)was carried out as described previously by Hçhne et al. ,[24]
and the same procedure was used for expression of theamine transaminase ATA-117. The cultivation volume usedfor protein expression was 100 mL. The cell pellets were re-suspended in 5 mL HEPES buffer (50 mM, pH 8.2 at 37 88C)containing 0.1 mM pyridoxal-5’-phosphate (PLP) and dis-rupted with sonication. Ammonium sulfate precipitation at60% saturation was then performed as described previouslyby Sch�tzle et al.[25] After the precipitation, the lysates werecentrifuged, the supernatants were discarded and each pelletwas resuspended in 5 mL HEPES buffer (50 mM, pH 8.2 at37 88C) containing 0.1 mM PLP and stored in 4 88C. The aceto-phenone assay[26] confirmed that all these transaminaseswere active.
Biocatalysis on the Analytical Scale
All biocatalytic reactions on an analytical scale were per-formed with a final volume of 1 mL with either 2-propanol(in transamination or reduction reactions) or with acetone(in oxidation reactions) as co-solvent in glass vials at 30 88Cwith shaking at 1000 rpm. Details for all analytics are givenin the Supporting Information.
Transaminations employing the LDH/GDH cascade: Bio-catalysis employing wild-type ATAs were coupled with lac-tate dehydrogenase (LDH) and glucose dehydrogenase(GDH) in 100 mM sodium phosphate buffer (pH 7.5). De-pending on the enantiopreference of the ATA, d- or l-ala-nine (250 mM) was used, and the reactions further con-tained d-glucose (150 mM), NADH (1 mM), PLP (0.1 mM),90 U mL¢1 LDH, 15 U mL¢1 GDH, 5–40 mgmL¢1 ATAcrude cell lyophilisate, 10–20% (v/v) co-solvent (2-propanol)and 10–50 mM amino acceptor. The reaction solutions weremixed thoroughly and were shaken in glass vials at 30 88Cand 1200 rpm.
Transaminations employing Codexis ATAs: Reactionswith ATAs from CodexisÔ were performed at 30 88C in
1812 asc.wiley-vch.de Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2015, 357, 1808 – 1814
FULL PAPERSHannes Kohls et al.

sodium phosphate buffer (100 mM, pH 7.5) containing0.25 mM PLP and 1 M isopropylamine, which was preparedfreshly prior to reaction. After addition of lyophilisedenzyme (5 mgmL¢1) a stock solution of the amino acceptorwas added to adjust the final concentration to 20 mM ofsubstrate and 10% (v/v) 2-propanol. After 17 to 18 h of re-action, the glass vial was shaken without cap for one to twohours until all substrate was consumed.
Redox reactions employing KRED from CodexisÔ : Reac-tions were performed at 30 88C in potassium phosphatebuffer (125 mM, pH 7) containing 1.25 mM MgSO4 and1 mM NADP++, which was prepared freshly prior to the reac-tion. After addition of lyophilised enzyme (1–2.5 mg mL¢1),a stock solution of the substrate in either acetone (for oxida-tion reactions) or 2-propanol (for reduction reactions) wasused to adjust the final concentration to 10–30 mM substrateand 10–50% (v/v) co-solvent.
Biocatalysis on the Preparative Scale
(R)-3: Preparative kinetic resolution of rac-3 was performedon a 1.8-mmol scale (320.9 mg, 25 mM) in a two-neckedflask equipped with a thermometer and a magnetic stirringbar. 180 mg of KRED-P1-B10 lyophilisate (2.5 mg mL¢1)were dissolved in 57.6 mL freshly prepared potassium phos-phate buffer (125 mM, pH 7) containing 1.25 mM MgSO4
and 1 mM NADP++. The reaction mixture was stirred at30 88C and while stirring rac-3 (14.4 mL; 125 mM in acetone)was added. After one day the reaction stalled at 75% ee for3 days. The addition of acetone [15% (v/v), 10.8 mL] pushedthe reaction towards the product side (85% ee, after 4 days).Further addition of acetone the fifth and sixth days [5%(v/v) per day] gave an enantiomeric excess of 89% ee after 7days reaction time. The reaction mixture was extracted fivetimes with ethyl acetate. Column chromatography (petrole-um ether/ethyl acetate 5:1) afforded (R)-3 (yield: 160 mg,50%; 86% ee) and diketone 4 (yield: 85 mg, 26.6%).
(S)-3: Preparative asymmetric reduction of diketone 4was performed on a 0.312-mmol scale (55 mg, 30 mM) ina 10-mL glass vial equipped with a magnetic stirring bar. Tothis end, 10.4 mg KRED-P1-B10 lyophilisate (1 mgmL¢1)were dissolved in 8.216 mL freshly prepared potassiumphosphate buffer (125 mM, pH 7) containing 1.25 mMMgSO4 and 1 mM NADP++. The reaction mixture was stirredat 30 88C and diketone 4 (2.08 mL; 150 mM in 2-propanol)was added while stirring. After 70 min the reaction mixturewas extracted five times with ethyl acetate. The crude prod-uct (S)-3 (yield: 48 mg, 86%; 71% ee) was applied to transa-mination without further purification.
5d: Preparative asymmetric transamination of (R)-3 wasperformed on a 0.5-mmol scale (90 mg, 20 mM) in a 50-mLflask equipped with a magnetic stirring bar. 105 mg of ATA-025 lyophilisate (5 mgmL¢1) were dissolved in 24 mL freshlyprepared sodium phosphate buffer (100 mM, pH 7.5) con-taining 0.25 mM PLP and 1 M isopropylamine (50 equiv.).The reaction mixture was stirred at 30 88C while (R)-3(1.262 mL; 400 mM in 2-propanol) was added. After 22 hthe reaction was stopped by addition of 10 M aqueousNaOH solution until a pH >10 was reached. This mixturewas extracted five times with ethyl acetate. Silica filtration[ethyl acetate and MeOH++ 2% (v/v) trimethylamine] af-forded 5d ; yield: 66 mg (73%).
Acknowledgements
We thank the “Deutsche Bundesstiftung Umwelt” (grant No.AZ29937) for financial support and Prof. Byung Gee Kim(Seoul National University, Seoul, South Korea) for provid-ing the gene for the Vibrio fluvialis ATA. Furthermore wewould like to thank Dr. Samson Afewerki and Dr. Guangn-ing Ma, Mid Sweden University, for synthesis of some com-pounds. KTH Royal Institute of Technology is acknowledgedfor an excellence PhD student position to Mattias Anderson.His contribution is a result of the COST Action CM1303“Systems Biocatalysis”. Moreover, HK wants to express hisdeep gratitude to Philine Pia.
References
[1] F. Benedetti, S. Norbedo, Chem. Commun. 2001, 203–204.
[2] S. M. Lait, D. A. Rankic, B. A. Keay, Chem. Rev. 2007,107, 767–796.
[3] T. C. Nugent, Chiral Amine Synthesis Methods, Devel-opments and Applications, Wiley, Hoboken, 2010.
[4] R. Millet, A. M. Tr�ff, M. L. Petrus, J.-E. B�ckvall, J.Am. Chem. Soc. 2010, 132, 15182–15184.
[5] a) F. Huguenot, T. Brigaud, J. Org. Chem. 2006, 71,2159–2162; b) V. A. Sukach, N. M. Golovach, V. V. Pir-ozhenko, E. B. Rusanov, M. V. Vovk, Tetrahedron:Asymmetry 2008, 19, 761–764.
[6] T. Kochi, T. P. Tang, J. A. Ellman, J. Am. Chem. Soc.2002, 124, 6518–6519.
[7] G. T. Rice, M. C. White, J. Am. Chem. Soc. 2009, 131,11707–11711.
[8] C. G. Espino, P. M. Wehn, J. Chow, J. D. Bois, J. Am.Chem. Soc. 2001, 123, 6935–6936.
[9] B. Tang, L. Wang, D. Menche, Synlett 2013, 24, 625–629.
[10] R. S. B. GonÅalves, M. Dos Santos, G. Bernadat, D.Bonnet-Delpon, B. Crousse, Beilstein J. Org. Chem.2013, 9, 2387–2394.
[11] H. Kohls, F. Steffen-Munsberg, M. Hçhne, Curr. Opin.Chem. Biol. 2014, 19, 180–192.
[12] T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. vonLieres, H. Offermann, R. Westphal, M. Pohl, D.Rother, Angew. Chem. 2013, 125, 6904–6908; Angew.Chem. Int. Ed. 2013, 52, 6772–6775.
[13] R. C. Simon, N. Richter, E. Busto, W. Kroutil, ACSCatal. 2013, 4, 129–143.
[14] A. Nobili, F. Steffen-Munsberg, H. Kohls, I. Trentin, C.Schulzke, M. Hçhne, U. T. Bornscheuer, ChemCatCh-em 2015, 7, 757–760.
[15] a) T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M.Pohl, D. Rother, Green Chem. 2014, 16, 3341–3348;b) T. Sehl, Z. Maugeri, D. Rother, J. Mol. Catal. B:Enzym. 2015, 114, 65–71.
[16] M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaul-mann, K. Smithies, J. L. Galman, F. Baganz, P. A.Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Wood-ley, M. Micheletti, Org. Process Res. Dev. 2010, 14, 99–107.
Adv. Synth. Catal. 2015, 357, 1808 – 1814 Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 1813
FULL PAPERS Selective Access to All Four Diastereomers of a 1,3-Amino Alcohol

[17] C. V. Voss, C. C. Gruber, W. Kroutil, Angew. Chem.2008, 120, 753–757; Angew. Chem. Int. Ed. 2008, 47,741–745.
[18] M. S. Humble, K. E. Cassimjee, V. Abedi, H.-J. Feder-sel, P. Berglund, ChemCatChem 2012, 4, 1167–1172.
[19] M. Anderson, S. Afewerki, P. Berglund, A. Cýrdova,Adv. Synth. Catal. 2014, 356, 2113–2118.
[20] S. L. Bartlett, C. M. Beaudry, J. Org. Chem. 2011, 76,9852–9855.
[21] C. E. Paul, M. Rodr�guez-Mata, E. Busto, I. Lavandera,V. Gotor-Fern�ndez, V. Gotor, S. Garc�a-Cerrada, J.Mendiola, . de Frutos, I. Collado, Org. Process Res.Dev. 2013, 18, 788–792.
[22] R. Pellicciari, R. Fringuelli, E. Sisani, M. Curini, J.Chem. Soc. Perkin Trans. 1 1981, 2566–2569.
[23] a) K. E. Cassimjee, M. S. Humble, V. Miceli, C. G. Co-lomina, P. Berglund, ACS Catal. 2011, 1, 1051–1055;b) K. E. Cassimjee, M. S. Humble, H. Land, V. Abedi,P. Berglund, Org. Biomol. Chem. 2012, 10, 5466–5470.
[24] M. Hçhne, S. Sch�tzle, H. Jochens, K. Robins, U. T.Bornscheuer, Nat. Chem. Biol. 2010, 6, 807–813.
[25] S. Sch�tzle, F. Steffen-Munsberg, A. Thontowi, M.Hçhne, K. Robins, U. T. Bornscheuer, Adv. Synth.Catal. 2011, 353, 2439–2445.
[26] S. Sch�tzle, M. Hçhne, E. Redestad, K. Robins, U. T.Bornscheuer, Anal. Chem. 2009, 81, 8244–8248.
1814 asc.wiley-vch.de Õ 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2015, 357, 1808 – 1814
FULL PAPERSHannes Kohls et al.


Supporting Information

Selective access to all four diastereomers of a 1,3-amino alcohol by
combination of a keto reductase- and an amine transaminase-
catalysed reaction Hannes Kohlsa, Mattias Andersonb, Jonathan Dickerhoffa, Klaus Weisza, Armando Córdovac,
Per Berglundb, Henrike Brundiekd, Uwe T. Bornscheuera* and Matthias Höhnea*
a Institute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17487 Greifswald,
Germany
Fax: (+49)-3834-86-794367; phone: (+49)-3834-86-22832; e-mail: matthias.hoehne@uni-
greifswald.de
Fax: (+49)-3834-86-794367; phone: (+49)-3834-86-4367; e-mail: uwe.bornscheuer@uni-
greifswald.de b KTH Royal Institute of Technology, Division of Industrial Biotechnology, School of
Biotechnology, AlbaNova University Center, SE-106 91 Stockholm, Sweden c Department of Natural Sciences, Mid Sweden University, Holmgatan 10, Sundsvall, Sweden d Enzymicals AG, Walther-Rathenau-Straße 49a, 17489 Greifswald, Germany
* To whom correspondence should be addressed.

S2
TABLE OF CONTENTS
1 ANALYTICAL METHODS 3
1.1 GC/MS analysis 3 1.2 GC analysis 4 1.3 HPLC analysis 5 1.4 Thin layer chromatography (TLC) 6
2 SCREENED ENZYMES 7
2.1 Amine transaminases (ATA) 7 2.2 Keto reductases (KRED) 10
3 NMR ANALYSES 10
4 CHEMICAL SYNTHESIS OF REFERENCE COMPOUNDS 12
5 SPECTRA 17
5.1 Reference Compounds and Substrates 17 5.1.1 3-Amino-1-phenylbutan-1-ol (rac-8) 17 5.1.2 4-Hydroxy-4-(4-hydroxy-3methoxyphenyl)butan-2-one (rac-2) 19 5.1.3 3-Oxo-1-phenylbutyl acetate (rac-9) 21 5.1.4 4-Hydroxy-5-phenylpentan-2-one (rac-3) 22 5.1.5 1-Phenylpentane-2,4-diol (7) 25 5.1.6 1-Phenylpentane-2,4-dione (4) 27 5.1.7 4-Hydroxy-1-phenylpentan-2-one (rac-6) 29 5.1.8 4-Amino-1-phenylpentan-2-ol (5) 32
5.2 Products of preparative biocatalysis 34 5.2.1 4-Hydroxy-5-phenylpentan-2-one ((R)-3 or (S)-3) 34 5.2.2 1-Phenylpentane-2,4-dione (4) 36 5.2.3 (2R,4R)-4-Amino-1-phenylpentan-2-ol (5d) 38

S3
1 Analytical Methods
1.1 GC/MS analysis
GC/MS analysis was conducted with a Shimadzu GC2010 GC coupled to a QP2010 MS device with helium as carrier gas using a HYDRODEX β-TBDAc column (25 m, 0.25 mm, 0.25 µm) heptakis-(2,3-di-O-acetyl-6-O-t-butyldimethylsilyl)-β-cyclodextrin). GC/MS Method 1 was applied for determination of conversion and enantiomeric excess of hydroxy ketone 3 (note: for numbering of compounds see main paper, Scheme 1 and also Figure S5 and Scheme S1) during transamination reactions. This method also allowed for detection of the corresponding regioisomer rac-6, diketone 4 and diol 7. Aqueous samples (150 µl) were withdrawn, extracted with ethyl acetate (150 µl) and centrifuged to separate the organic phase. The organic phase was dried over anhydrous Na2SO4 and subjected to analysis. Temperature profile: 150°C/hold 45 min, heating to 200°C (rate 20°C/min), hold 200°C for 4.5 min; GC program parameters: injector 220°C, pressure 62.7 kPa, column flow 0.86 ml/min; MS parameters: ion source temperature 220°C, interface temperature 220°C. GC/MS Method 2 was applied for detection of the diastereomeric composition of amino alcohols 5. Aqueous samples (150 µl) were withdrawn and 10 % (v/v) NaOH (10 M in water) was added followed by extraction with ethyl acetate (150 µl). The organic phase was dried over anhydrous Na2SO4, centrifuged and subjected to derivatization in glass vials. For this, 30 % (v/v) of MBTFA (N-Methyl-bis(trifluoroacetamide)) was added and the sample was agitated for 45 min at 60°C prior to injection. Temperature profile: 150°C/hold 45 min, heating to 200°C (rate 20°C/min), hold 200°C for 4.5 min; GC program parameters: injector 220°C, pressure 62.7 kPa, column flow 0.86 ml/min; MS parameters: ion source temperature 220°C, interface temperature 220°C.

S4
Figure S1. Separation of derivatized diastereomers of 5. The top left chromatogram depicts four peaks, which are detected after injection of the chemically synthesized amino alcohol 5 after derivatization with MBTFA. The first peak (tR = 47.154 min) is an unidentified impurity. The second represents the (2R,4S)-diastereomer (5c). The third peak is concealing another smaller peak and is actually a mixture of 5a and 5b (the sum of the peak areas of the second and fourth peak is 98% of the area of the third peak). The fourth peak represents the (2R,4R)-diastereomer (5d). Two of the four amino alcohol peaks are smaller, since the applied synthetic route prefers the formation of the syn-isomers.[1] The second, third and fourth peak do all have an identical fragmentation pattern, according to mass spectrometry (top right). The molecule peak is m/z 257, which is explained by the elimination of water occuring during ionization. The base peak is m/z 144. The two peaks in black (middle left) are shown with the chemical synthesized reference (orange) and were detected, when rac-3 was incubated with ATA-025. The two orange peaks depicted in the chromatogram in the middle right were detected after incubation of rac-3 with ATA-Cvi and are shown together with the black peaks, which were detected when ATA-025 was applied. The blue peak was detected after incubation of (R)-3 with ATA-025. Finally, derivatized samples of the reaction of rac-3 with either ATA-025 or ATA-Cvi were co-injected, as depicted in the chromatogram on the bottom left.
1.2 GC analysis
GC analysis was conducted on an Shimadzu GC2010 Plus GC device equipped with an HYDRODEX γ-TBDAc octakis-(2,3-di-O-acetyl-6-O-t-butyldimethylsilyl)-γ-cyclodextrin (25 m, 0.25 mm, 0.25 µm) column. Hydrogen was applied as carrier gas. GC Method 3 was applied for screening of the “KRED Screening Kit”. For this, aqueous samples were extracted with ethyl acetate containing 2 mM 1-phenylbutan-2-one as standard. The organic phase was dried over anhydrous Na2SO4, centrifuged and injected into
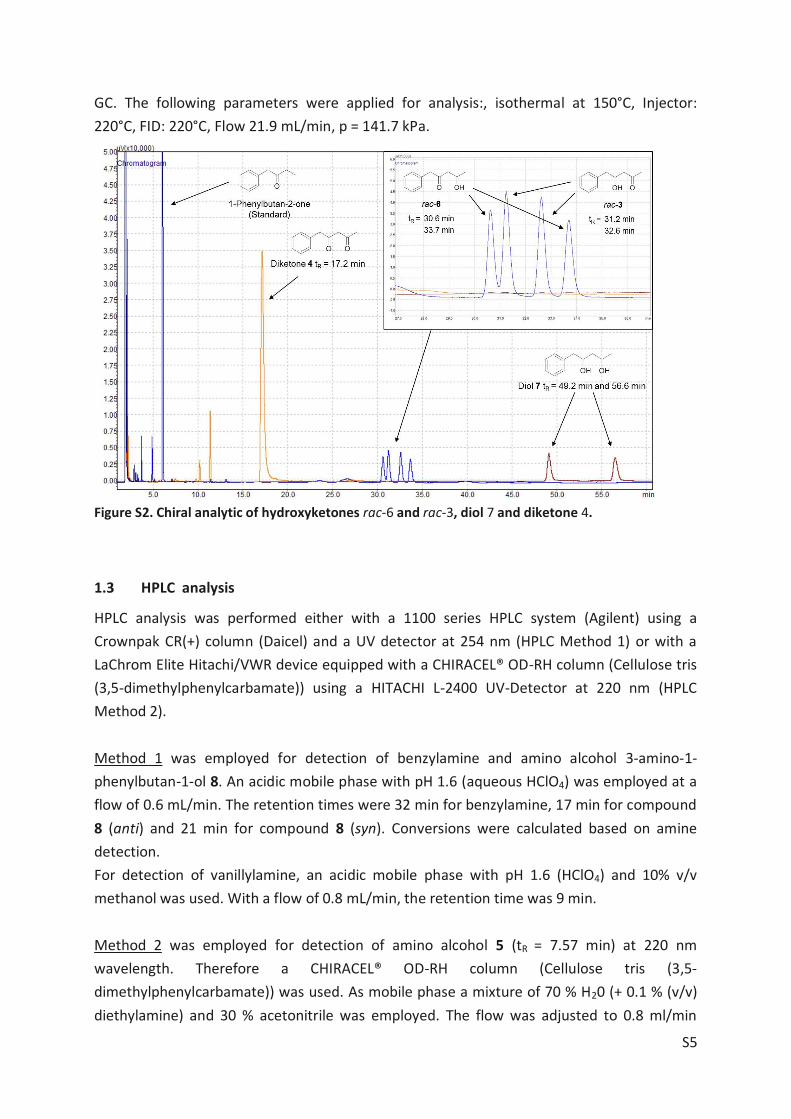
S5
GC. The following parameters were applied for analysis:, isothermal at 150°C, Injector: 220°C, FID: 220°C, Flow 21.9 mL/min, p = 141.7 kPa.
Figure S2. Chiral analytic of hydroxyketones rac-6 and rac-3, diol 7 and diketone 4.
1.3 HPLC analysis
HPLC analysis was performed either with a 1100 series HPLC system (Agilent) using a Crownpak CR(+) column (Daicel) and a UV detector at 254 nm (HPLC Method 1) or with a LaChrom Elite Hitachi/VWR device equipped with a CHIRACEL® OD-RH column (Cellulose tris (3,5-dimethylphenylcarbamate)) using a HITACHI L-2400 UV-Detector at 220 nm (HPLC Method 2). Method 1 was employed for detection of benzylamine and amino alcohol 3-amino-1-phenylbutan-1-ol 8. An acidic mobile phase with pH 1.6 (aqueous HClO4) was employed at a flow of 0.6 mL/min. The retention times were 32 min for benzylamine, 17 min for compound 8 (anti) and 21 min for compound 8 (syn). Conversions were calculated based on amine detection. For detection of vanillylamine, an acidic mobile phase with pH 1.6 (HClO4) and 10% v/v methanol was used. With a flow of 0.8 mL/min, the retention time was 9 min. Method 2 was employed for detection of amino alcohol 5 (tR = 7.57 min) at 220 nm wavelength. Therefore a CHIRACEL® OD-RH column (Cellulose tris (3,5-dimethylphenylcarbamate)) was used. As mobile phase a mixture of 70 % H20 (+ 0.1 % (v/v) diethylamine) and 30 % acetonitrile was employed. The flow was adjusted to 0.8 ml/min
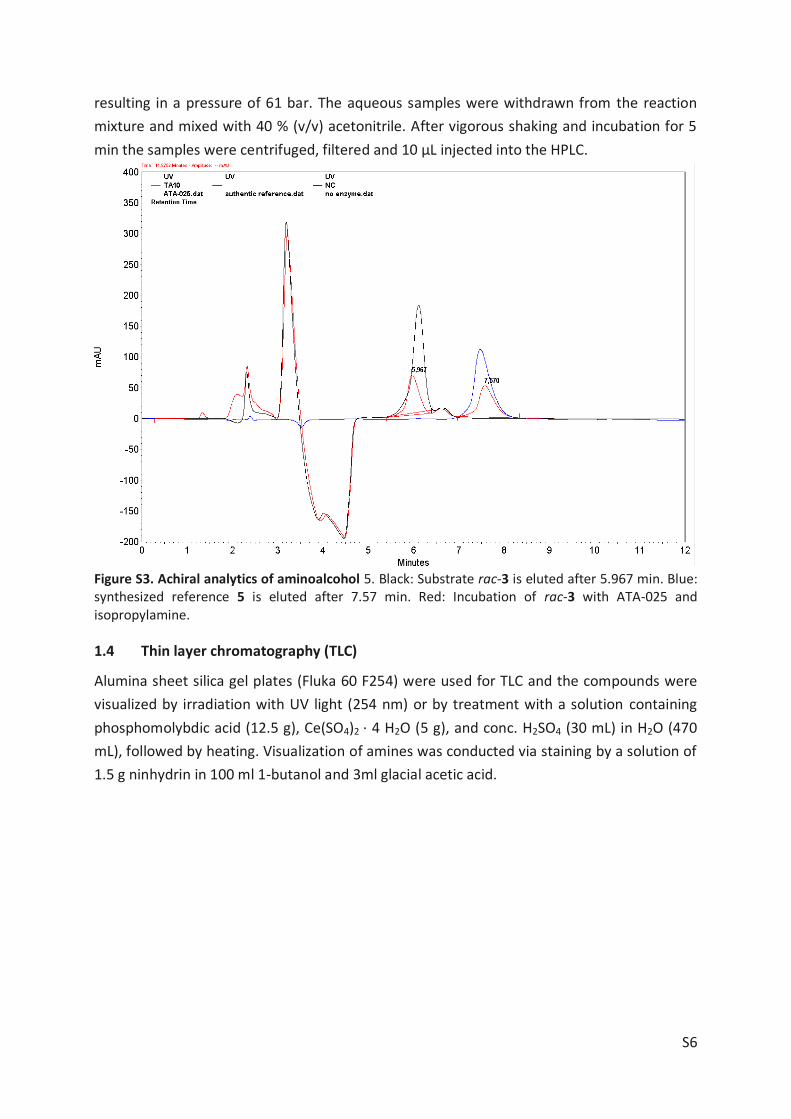
S6
resulting in a pressure of 61 bar. The aqueous samples were withdrawn from the reaction mixture and mixed with 40 % (v/v) acetonitrile. After vigorous shaking and incubation for 5 min the samples were centrifuged, filtered and 10 µL injected into the HPLC.
Figure S3. Achiral analytics of aminoalcohol 5. Black: Substrate rac-3 is eluted after 5.967 min. Blue: synthesized reference 5 is eluted after 7.57 min. Red: Incubation of rac-3 with ATA-025 and isopropylamine.
1.4 Thin layer chromatography (TLC)
Alumina sheet silica gel plates (Fluka 60 F254) were used for TLC and the compounds were visualized by irradiation with UV light (254 nm) or by treatment with a solution containing phosphomolybdic acid (12.5 g), Ce(SO4)2 · 4 H2O (5 g), and conc. H2SO4 (30 mL) in H2O (470 mL), followed by heating. Visualization of amines was conducted via staining by a solution of 1.5 g ninhydrin in 100 ml 1-butanol and 3ml glacial acetic acid.

S7
2 Screened Enzymes
2.1 Amine transaminases (ATA)
Each reaction had a total volume of 1 mL in a 1.5 mL Eppendorf tube. The reactions were run overnight in 37° C and darkness with no stirring, and analyzed with HPLC Method 1. Attempts with substrate rac-1 and alanine The reactions were run in 50 mM HEPES buffer, pH 8.2 at 37°C for all enzymes except ATA-Cvi W60C, where the pH was 7. The reactions with ATA-Cvi W60C also contained an additional 0.2 mM PLP. Table S1. Transaminases investigated for the synthesis of amino alcohol rac-8.
Entry rac-1 (mM) Alanine a) (mM) Enzyme Conc. 1 2.5 200 ATA-Cvi wt 0.5 mg/mL 2 2.5 300 ATA-Cvi wt 0.5 mg/mL 3 2.5 500 ATA-Cvi wt 0.5 mg/mL 4 3.2 500 ATA- AspTer 200 µLb) 5 3.2 500 ATA- AspFum 200 µL b) 6 3.2 500 ATA- AspOry 200 µL b) 7 3.2 500 ATA- NeoFis 200 µL b) 8 3.2 500 ATA- GibZea 200 µL b) 9 3.2 500 ATA117 200 µL b) 10 2.0 300 ATA-Cvi W60C 0.5 mg/mL 11 2.0 400 ATA-Cvi W60C 0.25 mg/mL a) L-Alanine was used for the ATA-Cvi enzymes and D-alanine was used for the others. b) Lysate solution, see Cultivation and expression conditions in the experimental section.
Experimental details on the retro aldol reactions of the hydroxyketones 1 and 2, as well as the use of an enzyme cascade system with these compounds, can be found elsewhere.[2] Attempts with substrate rac-1 and 1-phenylethylamine (1-PEA) The reactions were run in 50 mM HEPES buffer, pH 8.2 at 37°C for ATA-Cvi wt and pH 7 at 37°C for ATA-Cvi W60C. The reactions with ATA-Cvi W60C also contained an additional 0.2 mM PLP.
S8
Table S2. Investigated conditions for transamination applying (S)-1-PEA as amino donor.
Entry rac-1 (mM) (S)-1-PEA (mM) Enzyme Conc. 1 0.25 3 ATA-Cvi wt 0.1 mg/mL 2 0.5 3 ATA-Cvi wt 0.1 mg/mL 3 1.5 3 ATA-Cvi wt 0.1 mg/mL 4 2.5 3 ATA-Cvi wt 0.1 mg/mL 5 2.0 4 ATA-Cvi W60C 0.5 mg/mL 6 2.0 8 ATA-Cvi W60C 0.25 mg/mL
Attempts with rac-9 (O-Acetyl protected derivative of rac-1) The reaction composition was 2.5 mM of compound rac-9, 5 mM (S)-1-PEA and 1 mg/mL ATA-Cvi wt in HEPES buffer (45 mM, pH 8.2 at 37°C) with 10 % (v/v) DMSO as co-solvent in a total volume of 1 mL. Compound 3 was dissolved in DMSO before mixing it with the other components. Attempts with substrate rac-8 The reaction composition was <2.5 mM of amino alcohol rac-8 (crude), 100 mM pyruvate and 1 mg/mL ATA-Cvi wt in HEPES buffer (45 mM, pH 8.2 at 37°C) with 10 % (v/v) DMSO as co-solvent in a total volume of 1 mL. The amino alcohol rac-8 was dissolved in DMSO before mixing it with the other components. HPLC analysis showed no formation of the hydroxyketone rac-1 and no consumption of the starting material rac-8. Conversion of substrate rac-3 All reactions were performed in 1 ml scale in glass vials at 30° C with shaking at 1000 rpm. If not otherwise stated all reactions were performed using the LDH/GDH cascade, as described in the experimental section.
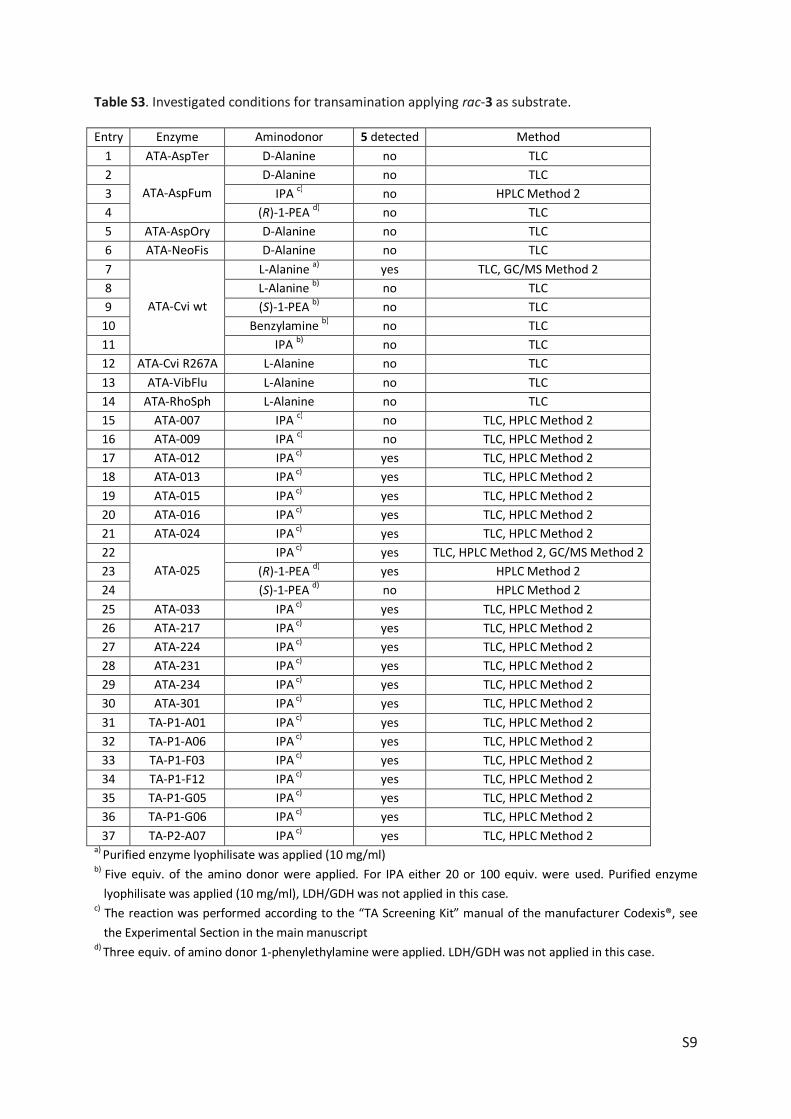
S9
Table S3. Investigated conditions for transamination applying rac-3 as substrate.
Entry Enzyme Aminodonor 5 detected Method 1 ATA-AspTer D-Alanine no TLC 2
ATA-AspFum D-Alanine no TLC
3 IPA c) no HPLC Method 2 4 (R)-1-PEA d) no TLC 5 ATA-AspOry D-Alanine no TLC 6 ATA-NeoFis D-Alanine no TLC 7
ATA-Cvi wt
L-Alanine a) yes TLC, GC/MS Method 2 8 L-Alanine b) no TLC 9 (S)-1-PEA b) no TLC
10 Benzylamine b) no TLC 11 IPA b) no TLC 12 ATA-Cvi R267A L-Alanine no TLC 13 ATA-VibFlu L-Alanine no TLC 14 ATA-RhoSph L-Alanine no TLC 15 ATA-007 IPA c) no TLC, HPLC Method 2 16 ATA-009 IPA c) no TLC, HPLC Method 2 17 ATA-012 IPA c) yes TLC, HPLC Method 2 18 ATA-013 IPA c) yes TLC, HPLC Method 2 19 ATA-015 IPA c) yes TLC, HPLC Method 2 20 ATA-016 IPA c) yes TLC, HPLC Method 2 21 ATA-024 IPA c) yes TLC, HPLC Method 2 22
ATA-025 IPA c) yes TLC, HPLC Method 2, GC/MS Method 2
23 (R)-1-PEA d) yes HPLC Method 2 24 (S)-1-PEA d) no HPLC Method 2 25 ATA-033 IPA c) yes TLC, HPLC Method 2 26 ATA-217 IPA c) yes TLC, HPLC Method 2 27 ATA-224 IPA c) yes TLC, HPLC Method 2 28 ATA-231 IPA c) yes TLC, HPLC Method 2 29 ATA-234 IPA c) yes TLC, HPLC Method 2 30 ATA-301 IPA c) yes TLC, HPLC Method 2 31 TA-P1-A01 IPA c) yes TLC, HPLC Method 2 32 TA-P1-A06 IPA c) yes TLC, HPLC Method 2 33 TA-P1-F03 IPA c) yes TLC, HPLC Method 2 34 TA-P1-F12 IPA c) yes TLC, HPLC Method 2 35 TA-P1-G05 IPA c) yes TLC, HPLC Method 2 36 TA-P1-G06 IPA c) yes TLC, HPLC Method 2 37 TA-P2-A07 IPA c) yes TLC, HPLC Method 2
a) Purified enzyme lyophilisate was applied (10 mg/ml) b) Five equiv. of the amino donor were applied. For IPA either 20 or 100 equiv. were used. Purified enzyme
lyophilisate was applied (10 mg/ml), LDH/GDH was not applied in this case. c) The reaction was performed according to the “TA Screening Kit” manual of the manufacturer Codexis®, see
the Experimental Section in the main manuscript d) Three equiv. of amino donor 1-phenylethylamine were applied. LDH/GDH was not applied in this case.

S10
2.2 Keto reductases (KRED)
All KREDs were screened according to the “KRED Screening Kit” manual (see also Experimental Section) provided by the manufacturer Codexis®. Evaluation of the experiments was done by TLC or GC Method 3. This analysis identified the enzymes KRED-P1-B02, KRED-P1-B10, KRED-P1-B12, KRED-P2-B02 and KRED-P1-C02 to act on rac-3 to yield diketone 4 and the disfavored enantiomer (R)-3. The reaction with KRED-P1-B10 was the fastest and this enzyme was therefore used for further experiments. For the reduction of diketone 4 to yield (S)-3 only KRED-P1-B10 was found to be active.
3 NMR analyses
NMR spectra were recorded from samples dissolved in CDCl3 containing TMS as internal standard. All chemical shifts are reported in ppm. 1H-NMR measurements were performed at 298 K with a Bruker Avance 600 MHz spectrometer equipped with an inverse 1H/13C/15N/31P quadruple resonance cryoprobe head and z-field gradients. 200 MHz NMR-spectra were recorded on a Bruker AC 200, 300 MHz spectra on a Bruker ARX300 and 400 MHz spectra on a Bruker Advance UltraShield 400 spectrometer. Discrimination of syn- and anti-isomers Scalar coupling constants of the diastereotopic H3 protons were evaluated to discriminate between the syn-(5b, 5c) and anti-isomers (5a, 5d). These are correlated with the dihedral angle between the coupled protons and generally average to a value of 6-7 Hz in case of free rotation about the single bonds in alkyl chains. However, the rotation appears to be hindered and observed coupling constants significantly deviating from expected values indicate a strong preference for a single conformer. The characteristic coupling patterns that include a geminal coupling constant of about 14 Hz are shown in Figure S4 and exhibit a three-fold doublet splitting for the biocatalysis product 5d (Figure S4 A) and doublets further split into triplets of very different coupling constants for the chemically synthesized amino alcohol (Figure S4 B). The syn-isomer is proposed for the latter and confirmed by the vicinal coupling constants of 2.0 Hz and 10.7 Hz as determined for the resonance at 1.5 ppm and 1.21 ppm, respectively. Based on these couplings, the two protons H2 and H4 must both be oriented gauche or trans with respect to the H3 protons in line with the syn-isomer. In contrast, the unequal vicinal coupling constants with H2 or H4 of 5d derive from the anti-isomer. Due to the different orientation of the hydroxyl- and amino-substituent, the H3 protons will necessarily have a different dihedral angle with both neighbors, also excluding two trans orientations.
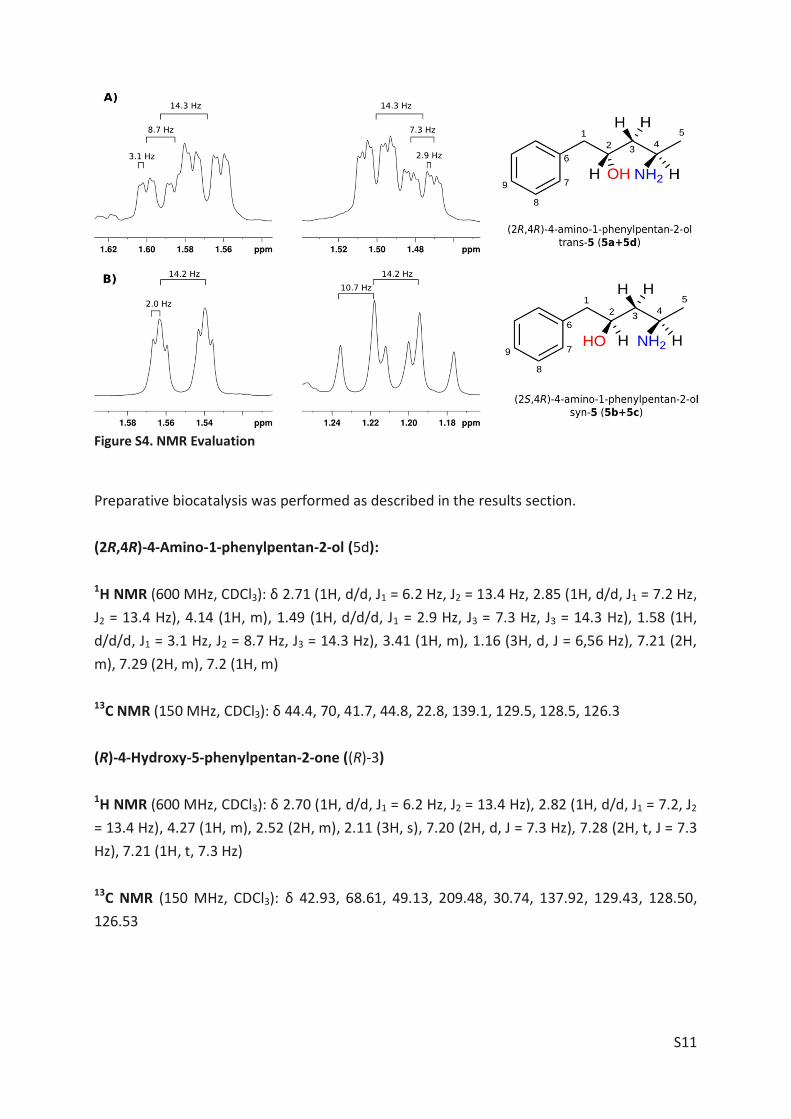
S11
Figure S4. NMR Evaluation
Preparative biocatalysis was performed as described in the results section. (2R,4R)-4-Amino-1-phenylpentan-2-ol (5d): 1H NMR (600 MHz, CDCl3): δ 2.71 (1H, d/d, J1 = 6.2 Hz, J2 = 13.4 Hz, 2.85 (1H, d/d, J1 = 7.2 Hz, J2 = 13.4 Hz), 4.14 (1H, m), 1.49 (1H, d/d/d, J1 = 2.9 Hz, J3 = 7.3 Hz, J3 = 14.3 Hz), 1.58 (1H, d/d/d, J1 = 3.1 Hz, J2 = 8.7 Hz, J3 = 14.3 Hz), 3.41 (1H, m), 1.16 (3H, d, J = 6,56 Hz), 7.21 (2H, m), 7.29 (2H, m), 7.2 (1H, m) 13C NMR (150 MHz, CDCl3): δ 44.4, 70, 41.7, 44.8, 22.8, 139.1, 129.5, 128.5, 126.3 (R)-4-Hydroxy-5-phenylpentan-2-one ((R)-3) 1H NMR (600 MHz, CDCl3): δ 2.70 (1H, d/d, J1 = 6.2 Hz, J2 = 13.4 Hz), 2.82 (1H, d/d, J1 = 7.2, J2 = 13.4 Hz), 4.27 (1H, m), 2.52 (2H, m), 2.11 (3H, s), 7.20 (2H, d, J = 7.3 Hz), 7.28 (2H, t, J = 7.3 Hz), 7.21 (1H, t, 7.3 Hz) 13C NMR (150 MHz, CDCl3): δ 42.93, 68.61, 49.13, 209.48, 30.74, 137.92, 129.43, 128.50, 126.53

S12
4 Chemical synthesis of reference compounds
Figure S5. Synthesized substrates.
The 1,3-hydroxy ketones rac-1 and rac-2 were synthesized as described previously.[2] 4-Hydroxy-4-phenylbutan-2-one (rac-1): The determined NMR data was in accordance to the values given in the literature. 4-Hydroxy-4-(4-hydroxy-3-methoxyphenyl)butan-2-one (rac-2): 74 % yield. 1H NMR (500 MHz, CDCl3): δ 6.93 (d, J = 1.9 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.80 (dd, J = 8.1, 1.9 Hz, 1H), 5.60 (br s, 1H), 5.09 (dt, J = 9.2, 3.0, Hz, 1H), 3.90 (s, 3H), 3.23 (d, J = 3.0 Hz, 1H), 2.88 (dd, J = 17.6, 9.3 Hz, 1H), 2.80 (dd, J = 17.6, 3.3 Hz, 1H), 2.20 (s, 3H). 3-Oxo-1-phenylbutyl acetate (rac-9): The hydroxy ketone rac-1 was acetylated at 0° C using standard acetylation conditions (Ac2O/pyridine/DMAP) in CH2Cl2 to give compound rac-9. 77 % yield. 1H NMR (400 MHz, CDCl3): δ 7.37–7.28 (m, 5H), 6.18 (dd, J = 8.7, 4.9 Hz, 1H), 3.12 (dd, J = 16.6, 8.7 Hz, 1H), 2.82 (dd, J = 16.7, 4.9 Hz, 1H), 2.15 (s, 3H), 2.04 (s, 3H) 13C NMR (100 MHz, CDCl3): δ = 204.7, 169.8, 139.6, 128.6, 128.2, 126.4, 71.6, 49.8, 30.4, 21.0. 3-Amino-1-phenylbutan-1-ol (rac-8): The amino alcohol rac-8 was synthesized according to Narasaka[3] by reducing the O-benzyl oxime derived from the hydroxy ketone rac-1 with LiAlH4 (see also Scheme S2). The corresponding 1,3-amino alcohol was obtained in a 5:1 syn:anti ratio according to 1H NMR analysis. 1H NMR data of the 1,3-amino alcohol was in accordance to the literature.[4]

S13
Scheme S1. Synthesis of diketone 4, hydroxy ketone rac-3, its regioisomer rac-6 and diol 7. Left: Wacker oxidation of homoallyl alcohol 10 yielded the hydroxy ketone rac-3. Swern and PCC oxidation of 10 gave the desired product as a mixture with the rearranged α,β-unsaturated ketone and was therefore omitted. An oxidation using Dess-Martin periodinane (DMP) yielded the desired product 11 almost quantitatively, but the subsequent Wacker oxidation did not yield the desired diketone 4. The α,β-unsaturated ketone was isolated instead. Eventually, the synthesis of 4 was accomplished by the oxidation with DMP. Reduction of rac-3 with LiAlH4 yielded diol 7. Right: The hydroxy ketone rac-6 was synthesized via Weinreb ketone synthesis.
1-Phenylpent-4-en-2-ol (10): Freshly distilled phenylacetaldehyde (4.5 g, 37.45 mmol) was stirred in 180 ml dry diethylether at –10° C under an argon atmosphere. Allylmagnesiumbromide (1.2 equiv., 44.94 ml, 1 M in Et2O) was added carefully, avoiding an increase of the temperature above 0°C. After 90 min, 150 ml of saturated aqueous NH4Cl solution were added drop wise. The aqueous phase was extracted five times with CH2Cl2. The combined organic phases were washed with brine and dried over Na2SO4. The organic solvent was filtered and removed which yielded 6.015 g (99%) of 1-phenylpent-4-en-2-ol (10, pale yellow oil). 1H NMR (200 MHz, CDCl3): δ 1.71 (brs, 1H), 2.14-2.41 (m, 2H), 2.66-2.88 (dd, 1H, J1=13.6 Hz, J2=7.7 Hz), 2.76 (dd, 1H, J1=13.6 Hz, J2=5.1 Hz), 3.82-3.95 (m, 1H), 5.12-5.20 (m, 2H), 5.76-5.97 (m, 1H), 7.20-7.36 (m, 5H) 4-Hydroxy-5-phenylpentan-2-one (rac-3) via Wacker Oxidation of 10: CuCl (5 equiv., 1.525 g) and PdCl2 (0.25 equiv.; 136.6 mg) were suspended in 8 ml of a mixture of DMF and water (4:1). The apparatus was flushed with O2 and the mixture was stirred at room temperature for 3 h. Then 10 (500 mg; 3.082 mmol) was added via syringe and the mixture was stirred under O2 atmosphere for 19 h. Next, the reaction mixture was diluted with 15 ml water and then extracted four times with ethyl acetate. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and the solvent removed in vacuo. Silica gel column chromatography (petrolether/ethyl acetate 3:1 + 2 % (v/v) triethylamine) yielded

S14
325 mg (61% yield) of 4-hydroxy-5-phenylpentan-2-one (rac-3, yellow liquid) after removal of eluent in vacuo. 4-Hydroxy-5-phenylpentan-2-one (rac-3) via aldol reaction: To a solution of KOH (1ml, 5 M in methanol) and acetone (50 % (v/v)) 500 mg of freshly distilled phenylacetaldehyde was added drop wise at 0°C while stirring. After 2 hours the reaction mixture was neutralized 1H NMR (300 MHz, CDCl3): δ 2.15 (s, 3H), 2.55-2.61 (m, 2H), 2.72 (dd, 1H, J1= 6.3 Hz, J2= 13.5 Hz), 2.85 (dd, 1H, J1= 7.2 Hz, J2= 13.5 Hz), 2.96 (d, 1H), 4.23-4.36 (m, 1H), 7.17-7.37 (m, 5H)
13C NMR (125MHz, CDCl3): δ 30.71, 42.79, 48.97, 68.54, 126.51, 128.48, 129.34, 137.78, 209.39 Spectral data were found to be in accordance to those reported in literature.[5] 1-Phenylpentane-2,4-diol (7): LiAlH4 (5.3 mg, 0.5 equiv.) was stirred in 1 ml dry diethylether at 0°C. A solution of hydroxy ketone rac-3 (0.281 mmol, 50 mg) in CH2Cl2 was added dropwise. Then, the mixture was slowly warmed to room temperature and quenched with saturated aqueous NH4Cl solution followed by extraction with CH2Cl2. The organic phase was dried over anhydrous Na2SO4 and the solvent was evaporated. Silica column chromatography (petrolether/ethyl acetate 2:1 + 1 % (v/v) triethylamine) yielded 8 mg (18 %) of 1-phenylpentane-2,4-diol (6, pale yellow oil). 1H NMR (600 MHz, CDCl3): δ 1.23 (3H, d, J = 6.3 Hz), 1.67 (2H, t, J = 5.7 Hz), 2.79 (2H, m), 4.18 (1H, m), 4.2 (1H, m), 7.22 (2H, d, J = 7.4 Hz), 7.32 (2H, t, J = 7,4 Hz), 7.24 (1H, t, J = 7.5 Hz)
13C NMR (125MHz, CDCl3): δ 23.71, 43.72, 44.12, 65.6, 70.26, 126.67, 128.76, 129.5, 138.49 1-Phenylpentane-2,4-dione (4): To a slurried solution of 0.606 mmol (1.2 equiv., 257 mg) Dess-Martin periodinane in 6 ml CH2Cl2, 0.505 mmol (90 mg), hydroxy ketone rac-3 was added. After 3 h another equivalent (214 mg) of DMP was added followed by two more equivalents (428 mg) after 18 h. After 8 days the substrate was consumed as indicated by TLC. The reaction mixture was washed twice with 40 ml of a 1:1 mixture of a saturated aqueous solution of anhydrous Na2SO3 and a saturated aqueous solution of NaHCO3. The aqueous phase was extracted five times with CH2Cl2 (15 ml each). The combined organic phases were washed with brine and dried over anhydrous Na2SO4 and the solvent was evaporated under vacuum. The crude product was subjected to silica column chromatography (petrolether/ethyl acetate 20:1) which gave 22.3 mg of 1-phenylpentane-2,4-dione (4, pale yellow oil).

S15
1H NMR (300 MHz, CDCl3): δ 2.01 (s, 3H), 3.58 (s, 2H), 5.43 (s, 1H), 7.17-7.38 (m), 15.39 (s, 1H) 13C NMR (125MHz, CDCl3): δ 24.82, 45.18, 99.94, 127.1, 128.73, 129.4, 135.14, 191.32, 192.47 4-Hydroxy-1-phenylpentan-2-one (rac-6): The synthesis started from ethyl 3-hydroxybutyrate, which was converted to the corresponding Weinreb amide 12 as described elsewhere.[6] A solution of 12 (1.678 mmol, 247 mg) in 15 ml dry THF was stirred at 0°C in a three necked flask equipped with a thermometer and a magnetic stirring bar under an argon atmosphere. To this solution 5.9 ml benzylmagnesium chloride (7 equiv., 2 M in THF) was added dropwise via an addition funnel. The mixture was aged at 0°C for 30 minutes and then warmed to room temperature and aged for 1 hour or until TLC indicated full consumption of amide 12. Eventually, the reaction mixture was cooled to 0°C and quenched with 25 ml of an ice cold aqueous solution of NH4Cl. The aqueous phase was extracted four times with CH2Cl2, the combined organic phases were dried over anhydrous Na2SO4 and the solvent was evaporated in vacuo. Silica column chromatography (petrol ether/ethyl acetate 3:1) furnished 253 mg (85%) of 4-hydroxy-1-phenylpentan-2-one (rac-6, yellow oil). 1H NMR (300 MHz, CDCl3): δ 1.14 (d, 3H, J = 6.4 Hz), 2.54 (dd, 1H, J1= 8.5 Hz, J2= 17.8 Hz), 2.64 (dd, 1H, J1= 3.4 Hz, J2= 17.8 Hz), 3.03 (d, 1H, J = 3,3 Hz), 3.7 (s, 2H), 4.13-4.25 (m, 1H), 7.18-7.37 (m, 5H) 13C NMR (125MHz, CDCl3): δ 22.22, 49.71, 50.57, 63.71, 127.07, 128.67, 129.31, 133.51, 209.27
Scheme S2. Synthesis of 1,3-amino alcohol 5. 4-Hydroxy-5-phenylpentan-2-one O-benzyl oxime (13): A solution of rac-3 (2.665 mmol, 475 mg) and O-benzylhydroxylamine hydrochloride (2 , 850.8 mg) in pyridine (1.2 ml) and methanol (18.8 ml) was agitated under refluxing for 22 hours. Then, most of the methanol was removed in vacuo and a yellow solid remained, to which 10 ml distilled water was added. This mixture was extracted four times with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4 and the solvent was evaporated. Purification by silica column chromatography (petrolether/ethyl acetate 6:1) yielded O-benzyl oxime 13 (634 mg, 84%) as a pale yellow liquid.

S16
4-Amino-1-phenylpentan-2-ol hydrochloride (5•HCl): In a three necked flask flushed with argon and equipped with a thermometer, 100 ml THF were stirred at 0°C employing a magnetic stirring bar. To this, 33.6 mmol LiAlH4 (15 equiv., 33.6 ml, 1 M in THF) was added. While stirring 2.237 mmol of 13 (634 mg in 16.4 ml THF) was added via an addition funnel and stirring was continued for 23 hours. The reaction was cooled to 0°C and quenched with a saturated aqueous solution of Na2SO4 and the resulting precipitate was filtered off. As known from former experiments, the isolation of 5 proved difficult. Therefore, the condensed filtrate was divided for several individual workup procedures. The following attempt yielded the purest compound: 92.4 mg of the crude product were dissolved in ethyl acetate (500 µL) and 200 µL of a 10 % solution of HCl in methanol was added. After evaporation of solvent the resin-like crude product was dissolved in acetone, which was slowly evaporated. After three days solid particles were observed. Washing with CH2Cl2 and diethylether yielded 9.2 mg (12%) of 4-amino-1-phenylpentan-2-ol hydrochloride (5•HCl, white powder). This was dissolved in water. Approximately two parts of that mixture were separated and extracted. For this, the pH was adjusted to >12 and the aqueous phase was extracted three times with ethyl acetate. The solvent was removed in vacuo which yielded 2.4 mg of a transparent liquid. 1H NMR (600 MHz, CDCl3): δ 2.64 (1H, d/d, J1 = 13.4 Hz, J2 = 6.8 Hz), 2.86 (1H, d/d, J1 = 13.4 Hz, J2 = 6.2 Hz), 4.04 (1H, m), 1.21 (1H, d/t, J1 = 14.2 Hz, J2 = 10.7 Hz), 1.55 (1H, d/t, J1 = 14.2 Hz, J2 = 2.0 Hz), 2.98 (1H, m), 1.11 (3H, d, J = 6.3 Hz), 7.22 (2H, m), 7.29 (2H, m), 7.21 (1H, m) 13C NMR (125 MHz, CDCl3): δ 44.72, 74.08, 43.06, 48.69, 27.62, 139.11, 129.54, 128.31, 126.12
S17
5 Spectra
5.1 Reference Compounds and Substrates
5.1.1 3-Amino-1-phenylbutan-1-ol (rac-8)

S18

S19
5.1.2 4-Hydroxy-4-(4-hydroxy-3methoxyphenyl)butan-2-one (rac-2)

S20
S21
5.1.3 3-Oxo-1-phenylbutyl acetate (rac-9)

S22
5.1.4 4-Hydroxy-5-phenylpentan-2-one (rac-3)

S23
S24

S25
5.1.5 1-Phenylpentane-2,4-diol (7)
S26
S27
5.1.6 1-Phenylpentane-2,4-dione (4)
S28
S29
5.1.7 4-Hydroxy-1-phenylpentan-2-one (rac-6)

S30
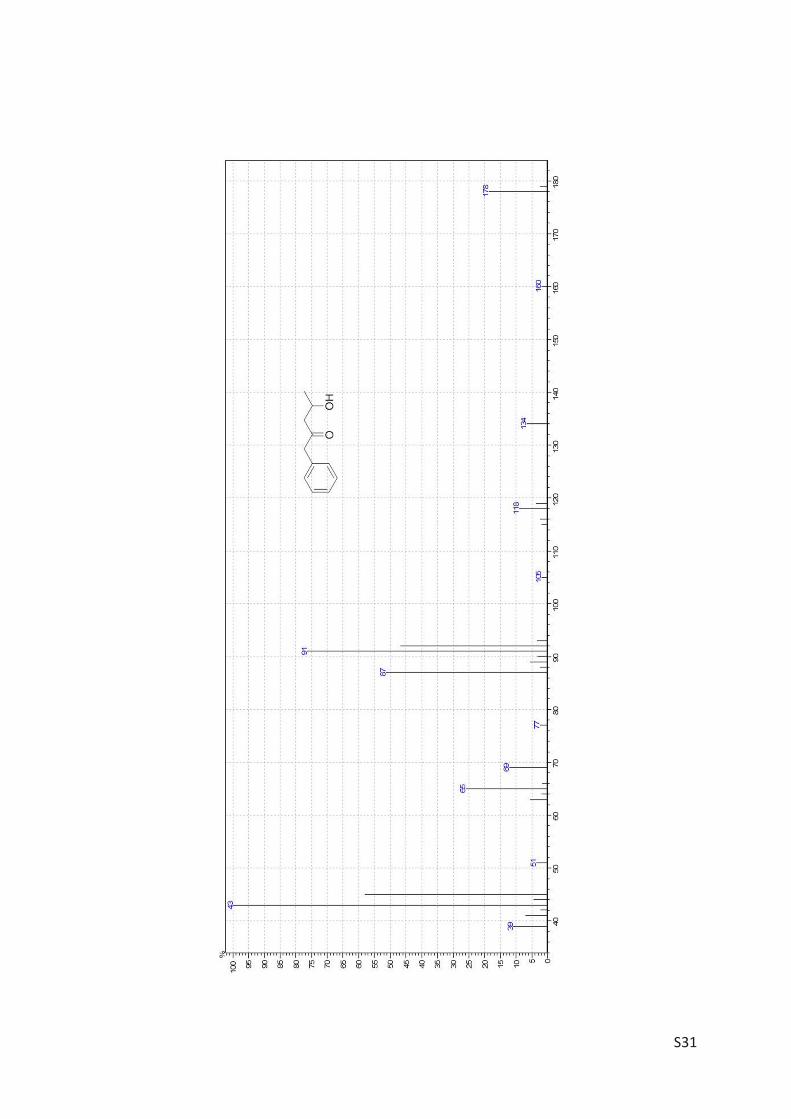
S31
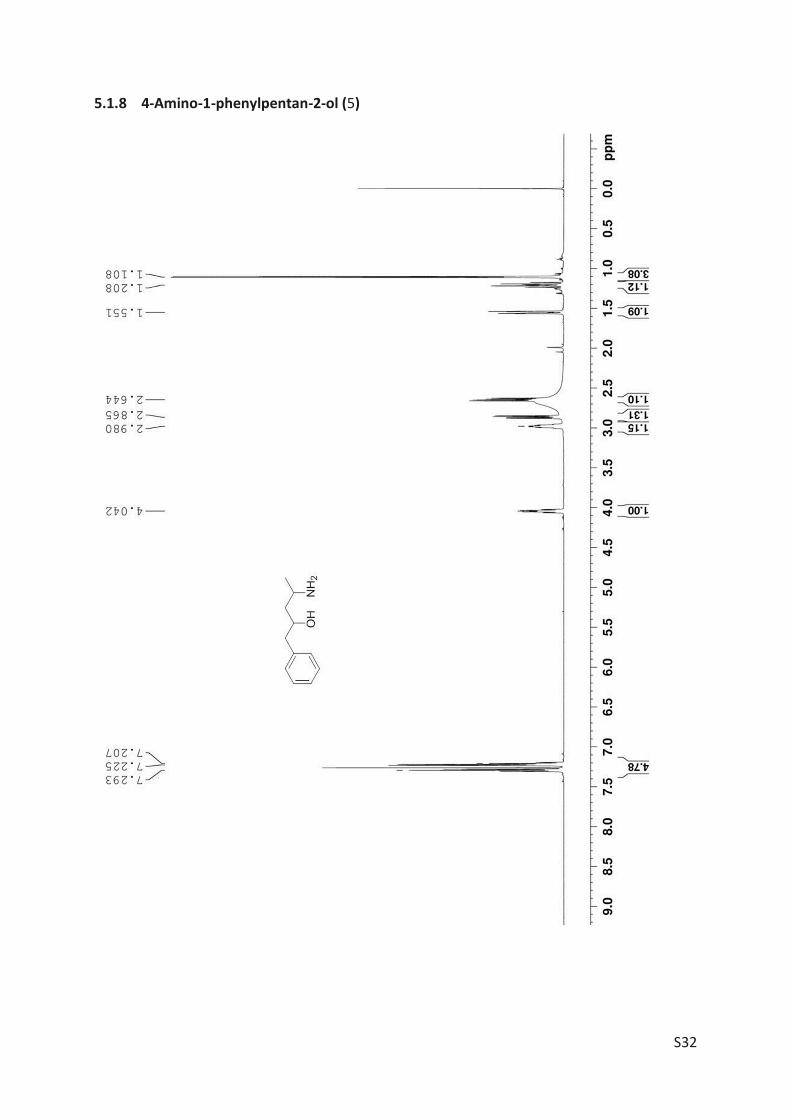
S32
5.1.8 4-Amino-1-phenylpentan-2-ol (5)
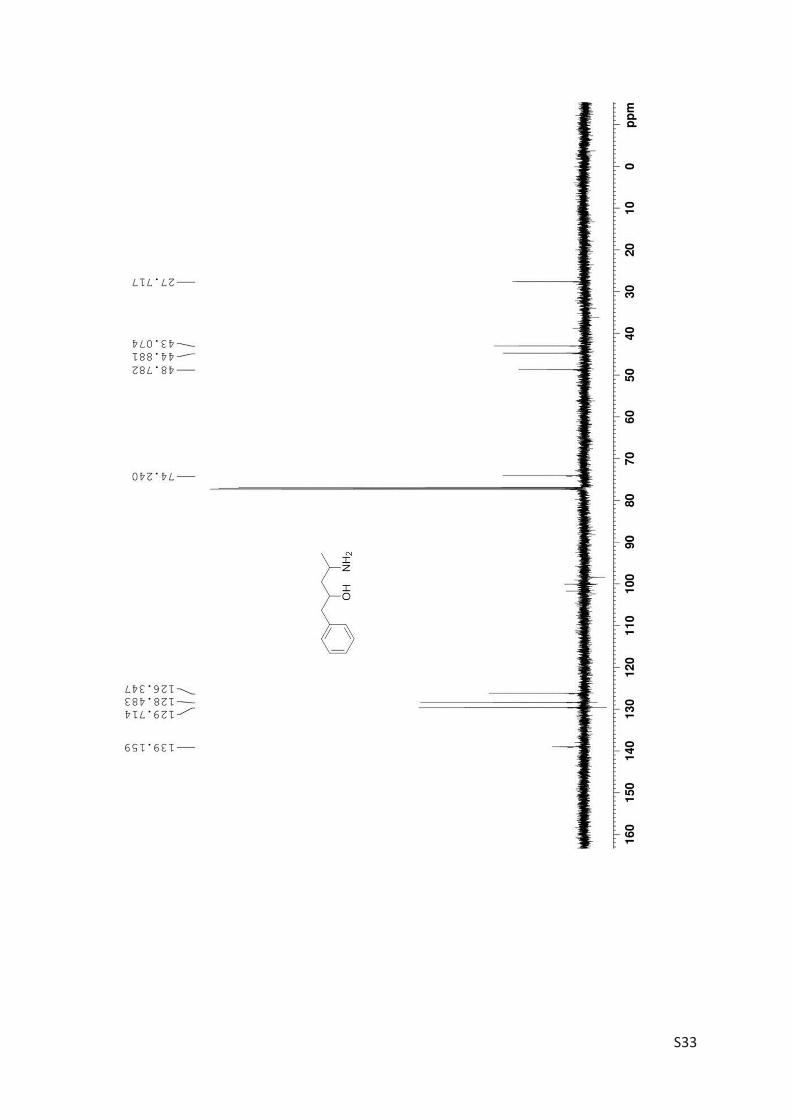
S33
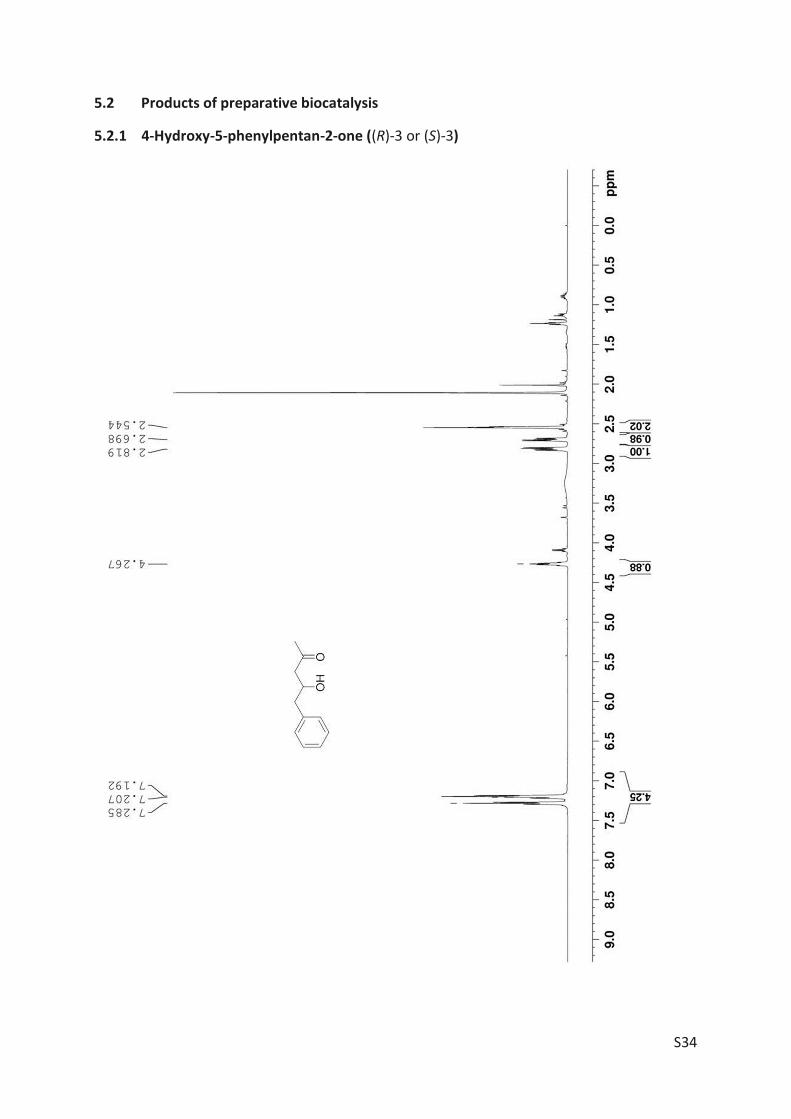
S34
5.2 Products of preparative biocatalysis
5.2.1 4-Hydroxy-5-phenylpentan-2-one ((R)-3 or (S)-3)
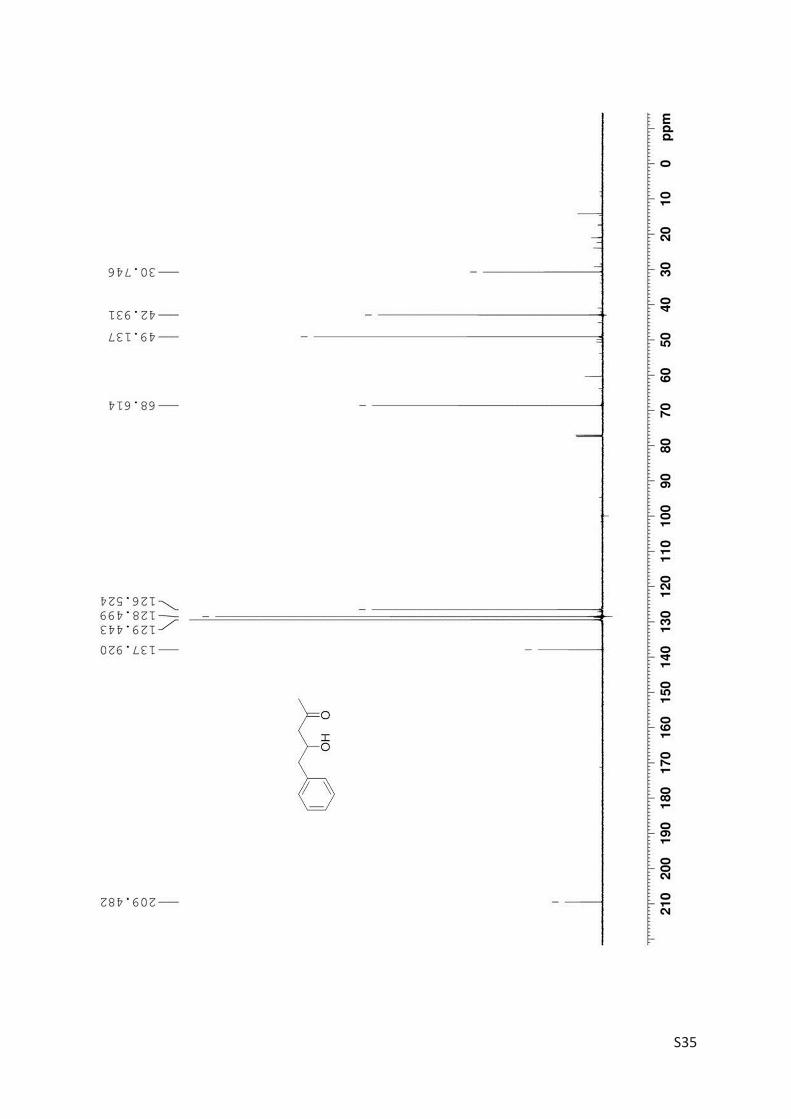
S35
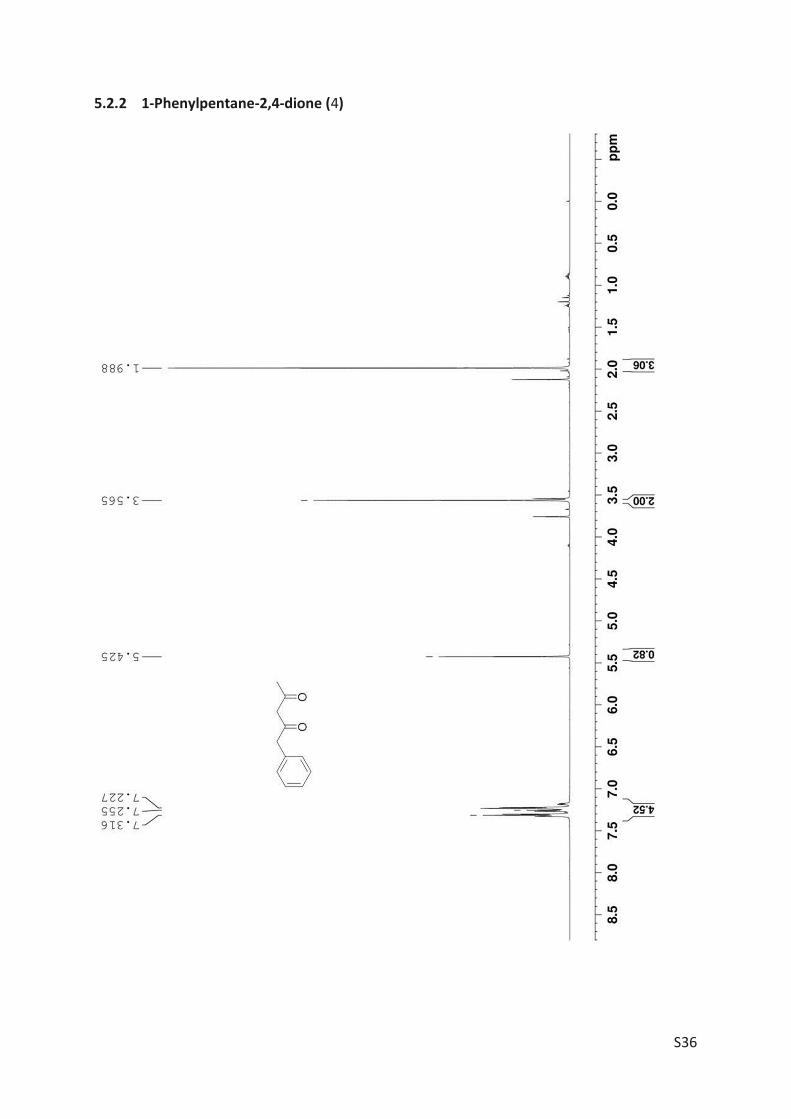
S36
5.2.2 1-Phenylpentane-2,4-dione (4)
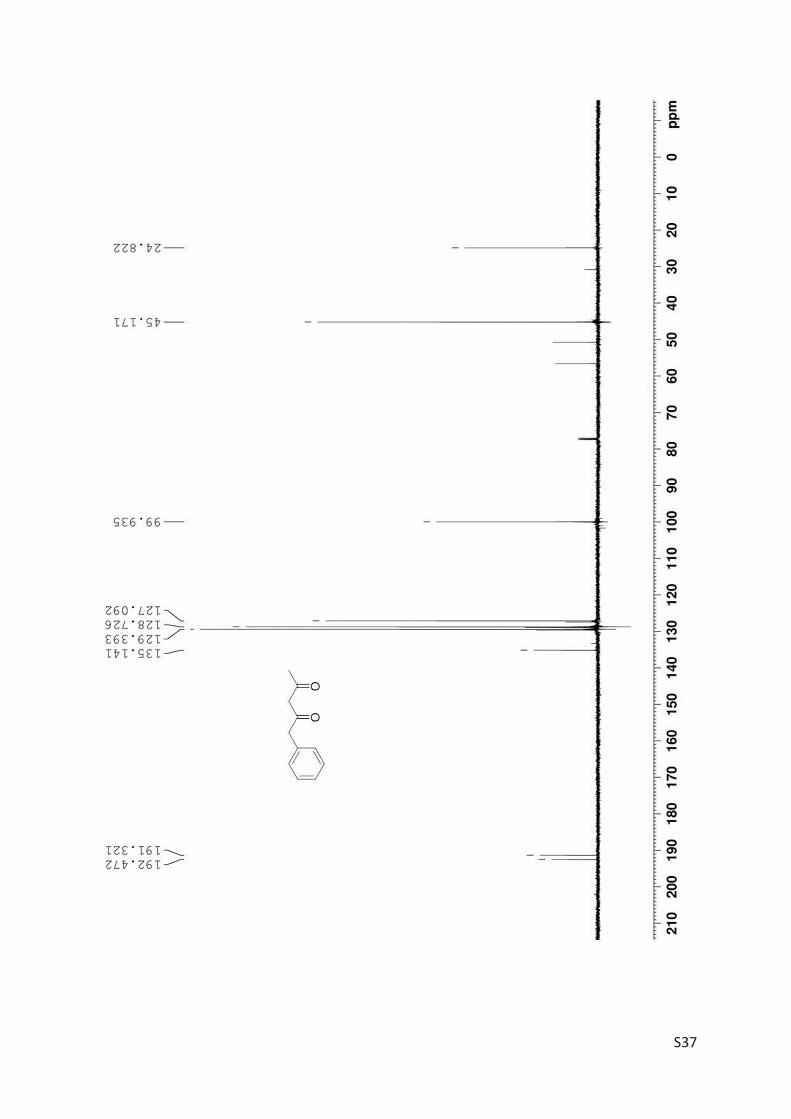
S37
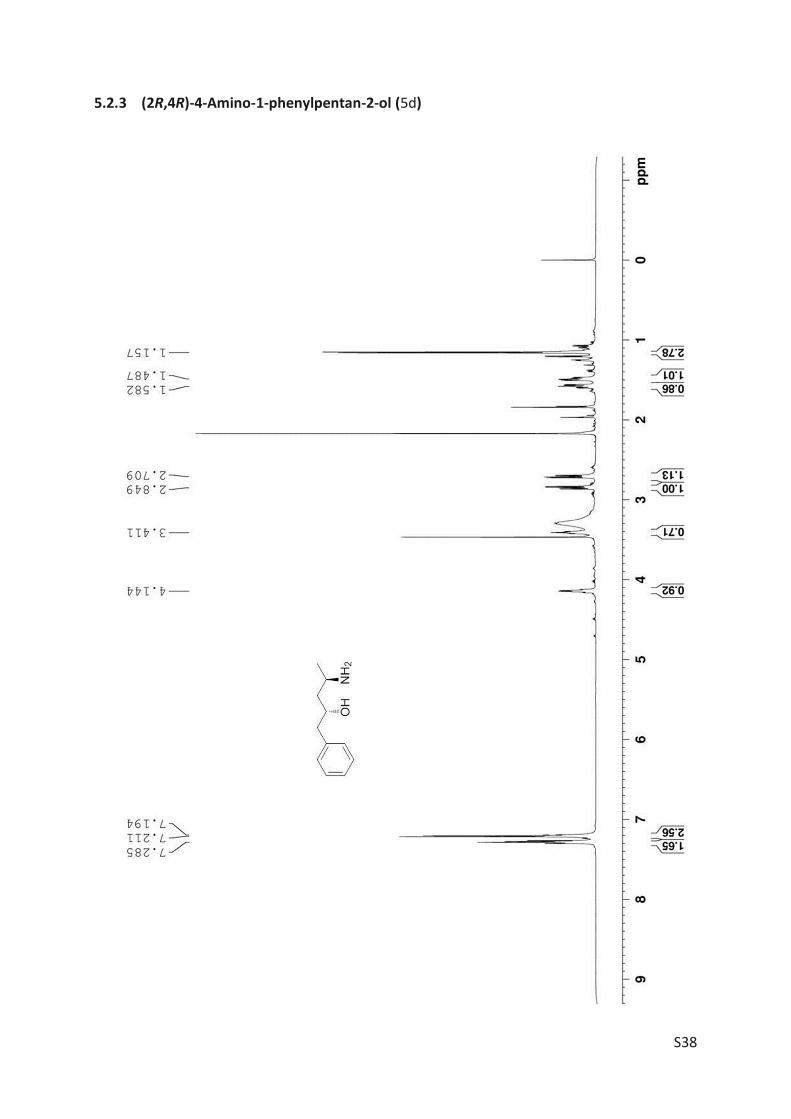
S38
5.2.3 (2R,4R)-4-Amino-1-phenylpentan-2-ol (5d)

S39

S40
[1] K. Narasaka, Y. Ukaji, S. Yamazaki, Bull. Chem. Soc. Jpn. 1986, 59, 525-533. [2] M. Anderson, S. Afewerki, P. Berglund, A. Córdova, Adv. Synth. Catal. 2014, 356,
2113-2118. [3] K. Narasaka, in Pure Appl. Chem., Vol. 57, 1985, p. 1883. [4] G. Bartoli, G. Cupone, R. Dalpozzo, A. De Nino, L. Maiuolo, A. Procopio, A. Tagarelli,
Tetrahedron Lett. 2002, 43, 7441-7444. [5] S. Joly, M. S. Nair, J. Mol. Catal. B: Enzym. 2003, 22, 151-160. [6] J. M. Williams, R. B. Jobson, N. Yasuda, G. Marchesini, U.-H. Dolling, E. J. J. Grabowski,
Tetrahedron Lett. 1995, 36, 5461-5464.


Articles 115
ARTICLE III


Engineering the Active Site of the Amine Transaminasefrom Vibrio fluvialis for the Asymmetric Synthesis of Aryl–Alkyl Amines and Amino AlcoholsAlberto Nobili,[a] Fabian Steffen-Munsberg,[a, b] Hannes Kohls,[a] Ivan Trentin,[a]
Carola Schulzke,[a] Matthias Hçhne,[a] and Uwe T. Bornscheuer*[a]
Although the amine transaminase from Vibrio fluvialis hasoften been applied as a catalyst for the biocatalytic prepara-tion of various chiral primary amines, it is not suitable for thetransamination of a-hydroxy ketones and aryl-alkyl ketonesbearing an alkyl substituent larger than a methyl group. Weaddressed this problem through a systematic mutagenesisstudy of active site residues to expand its substrate scope to-wards two bulky ketones. We identified two mutants (F85L/V153A and Y150F/V153A) showing 30-fold increased activity inthe conversion of (S)-phenylbutylamine and (R)-phenylglycinol,respectively. Notably, they facilitated asymmetric synthesis ofthese amines with excellent enantiomeric purities of 98 % ee.
Enantiomerically pure amines and amino alcohols play a funda-mental role in the pharmaceutical industry. One in four of the200 top-sold drugs contains a chiral amine moiety and thesedrugs had a total market value of more than 88 billion USD in2013 according to Weber and Sedelmeier.[1] When it comes tothe choice of the synthetic strategy for the preparation of theamine building blocks, amine transaminases (ATAs) are increas-ingly recognized as an attractive option as they facilitatea one-step asymmetric synthesis starting from the correspond-ing prochiral ketone.[2] A very impressive example is the appli-cation of an engineered (R)-selective ATA from Arthrobacter sp.(ATA117-mut), which is currently being used for the productionof sitagliptin, the active ingredient of the drugs Januvia and Ja-numet.[3] This example demonstrates the importance of proteinengineering of wild-type amine transaminases to expand theirlimited substrate scope. Known wild-type ATAs are not able toconvert bulky compounds demanded by the pharmaceuticalindustry. Compared to the success story of engineered (R)-se-lective transaminases with relaxed substrate specificity, (S)-se-lective ATAs that convert a range of bulky ketones with similarefficiency as the engineered ATA117-mut are still not available,
despite the progress of first engineering studies.[4] The crystalstructures of several (S)-selective ATAs were solved recently, en-abling a detailed understanding of the mechanism of substratebinding.[5]
Both (R)- and (S)-selective ATAs that were found in naturepossess a large and a small pocket in their active sites (Fig-ure 1 a).[5a, 6] Although the large pocket can accommodate sub-stituents with a rather broad size distribution, such as smallalkyl to naphthyl groups, the small pocket creates a strict stericconstraint: if the size of the small substituent exceeds that ofa methyl group, activity drops significantly.[6] For instance, ke-tones with a hydroxymethyl group as small substituent arehardly accepted.[7] This active site architecture limits the sub-strate scope, but at the same time contributes to the usuallyhigh enantioselectivity of these ATAs.
Midelfort et al.[4a] and Park et al.[4b] recently reported the firstattempts of rational engineering: they identified key residuesvia bioinformatic methods or structural inspection and investi-gated up to two substitutions per position by site-directedmutagenesis to achieve the transamination of their bulkytarget ketones. By combining eight mutations in Vibrio fluvialisATA, a b-keto ester bearing a long (6 carbon) alkyl chain couldbe converted employing 1-phenylethylamine 1 b as aminodonor, affording the amine imagabalin at 28 % yield via asym-metric synthesis. A single mutant in Paracoccus denitrificansATA[4b] showed increased activity in the deamination of 1-alkylsubstituted benzyl amines and the amination of 2-oxo-octa-noate. Interestingly, this study showed that larger n-alkyl sub-stituents are accepted in the small binding pocket if the sub-strate bears an a-carboxylate functional group instead ofa large hydrophobic substituent such as a phenyl group.
Despite these first successes, further efforts are needed tocreate an (S)-selective ATA that is useful for asymmetric synthe-sis of bulky amines. In the present study, we systematically ad-dress this problem by a (partial) saturation mutagenesis of allamino acids that form the small binding pocket of the ATA ofVibrio fluvialis.
We employed 1-phenylbutane-1-one 2 a and the hydroxyketone 2-hydroxyacetophenone 3 a as model substrates(Table 1). The amine product (R)-phenylglycinol 3 b is a buildingblock for many important pharmaceuticals, such as an inhibitorof the 3-phosphoinositide-dependent protein kinase-1 (PDK1),which was identified as a target enzyme for cancer therapy.[8]
Additionally, 3 b is applied as a chiral auxiliary in the synthesisof some of the top selling drugs, saxagliptin[9] (treatment oftype 2 diabetes), femoxetine and paroxetine[10] (antidepres-
[a] A. Nobili, F. Steffen-Munsberg, H. Kohls, I. Trentin, Prof. Dr. C. Schulzke,Prof. Dr. M. Hçhne, Prof. Dr. U. T. BornscheuerInstitute of Biochemistry, University of GreifswaldFelix-Hausdorff Str. 4, 17487 Greifswald (Germany)Fax: (+ 49)3834-86-794367E-mail : [email protected]
[b] F. Steffen-MunsbergKTH Royal Institute of Technology, School of BiotechnologyDivision of Industrial BiotechnologyAlbaNova University Center, SE-106 91 Stockholm (Sweden)
Supporting information for this article is available on the WWW underhttp://dx.doi.org/10.1002/cctc.201403010.
ChemCatChem 2015, 7, 757 – 760 � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim757
CommunicationsDOI: 10.1002/cctc.201403010

sants). These substrates exert an increased steric demand onthe small binding pocket compared to the well-known aceto-phenone 1 a, which is often employed as a benchmark sub-strate. Furthermore, (S)-phenylbutylamine 2 b and (R)-phenyl-glycinol 3 b facilitate an easy activity screening owing to theincrease in UV-absorption upon deamination to their corre-sponding ketones 2 a and 3 a. Hence, the acetophenoneassay[11] could be used with slight modifications to screen foractive variants with high throughput and sensitivity (see Fig-ure S1 in the Supporting Information). We assumed that muta-tions which would facilitate asymmetric synthesis of 2 b and
3 b would also lead to higher activity in the deamination reac-tion, and thus be detected during screening.
When pyruvate is used as the amino acceptor, the reactionis virtually irreversible, reducing the screening time and reac-tion complexity compared to asymmetric synthesis. As thisassay works with both enantiomers, we were also able to de-termine if a mutation affected the enzyme’s enantioselectivity.Based on the crystal structure of the transaminase of Vibrio flu-vialis (PDB-code: 4E3Q), the small binding pocket, located atthe interface of the homodimer, is composed of eight residuesthat completely surround the cofactor, from Tyr 150 at theactive site entrance (re-face of PLP[12]) to the catalytic Lys 285on the other (si-) side (Figure 1 B, the residues that belong tothe second monomer are indicated with an asterisk).[4a] Resi-dues Tyr 165, Tyr 150, Phe 19, Phe 85*, and Phe 86* create a con-tinuous p–p stacked shell and thus form a relatively hydropho-bic environment. Three of the eight residues, Gly 320*,Phe 321*, and Thr 322*, were excluded in our mutagenesisstrategy, as they are known as the “phosphate-binding cup”[13]
and are responsible for the coordination of the PLP phosphategroup. All the other positions (Phe 19, Leu 56, Phe 85*, Tyr 150and Val 153) were sifted through for improved properties inthe transamination between the enantiopure amines and pyru-vate. In a first step, we generated libraries with a (partial) satu-ration at each position. The allowed residues in the partially sa-turated libraries included those amino acids that aim to createmore space in the pocket, while still maintaining structural sol-idity (Figure 1 B). The quality of each library was checked toensure 99 % library coverage during the screening,[14] theneach library was screened using the modified acetophenoneassay.[11] Variants with improved activities were purified and fur-ther investigated to confirm their improved properties. Finally,the best mutants were combined to elucidate positive additiveeffects. When positions 19 and 56 were randomized, no im-proved variants were found for the substrates tested (data notshown).
Single mutations at positions 85, 150, and 153 led to up to40-fold improvements. For the conversion of 3 b, the resultssuggest that the positioning of its hydroxyl group in the smallbinding pocket is hindered by the presence of the Tyr 150 inthe wild-type scaffold (Figure 1), whose hydroxyl group occu-pies the required space. The simple substitution Y150F im-proves the template’s performance 23-fold, whereas the bestmutant discovered in this project is the double mutantY150M/V153A for a final improvement of the reaction velocityof 53-fold. By combining the mutations F85L and V153A it waspossible to achieve a 26-fold improvement towards 2 b. Wethen tested the ability of the best mutants to catalyze theasymmetric synthesis of our target amines, using the LDH/GDHsystem to shift the equilibrium.[15]
Regrettably, the mutants carrying the mutation Y150M hada decreased activity towards 2 a. The Y150M-containing mu-tants were not among the best mutants for the transaminationof 3 a, either. Consequently, we investigated with increasedenzyme concentrations all the different improved variantsidentified by the screening in the asymmetric synthesis reac-tion and we found that variant Y150F/V153A formed 3 b with
Figure 1. Active site architecture of Vibrio fluvialis ATA (PDB-code: 4E3Q).A) Schematic drawing of residues that form the large and small bindingpocket around the external aldimine intermediate (PLP-Schiff’ base with (S)-1-phenylethylamine). B) View of the small binding pocket. The side-chains offive residues form a hydrophobic shell by p-p stacking interactions. Y165and F86* are omitted for clarity. The catalytic lysine and PMP are shown asgray sticks; the targeted residues for mutagenesis and their surface areshown in orange and are labeled by one-letter abbreviation and number.The libraries were constructed by choosing those residues that were poten-tially able to decrease the steric hindrance in the pocket, that is, smaller andaliphatic residues. Residues with different chemical properties derive fromthe selection of the randomized codons (see the Supporting Information).The phosphate-binding cup is shown in blue.
ChemCatChem 2015, 7, 757 – 760 www.chemcatchem.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim758
Communications

88 % conversion after 24 h. In agreement with the previous ob-servations, the asymmetric synthesis of 3 b is possible whenthe active site is freed from the Tyr 150’s hydroxyl group andY150F is now the key mutation to accomplish this transamina-tion. Variant F85L/V153A afforded 2 b with quantitative conver-sion after 5 days (Table S1).
The asymmetric synthesis of 2 b and 3 b was then confirmedon a semi-preparative scale (Table 2) employing 0.2 mmolketone and 0.2 mol % catalyst. We also observed a beneficialeffect of the above-identified mutations on the activity of theATA towards branched-chain a-keto acids, where the activity
was increased by up to 6-fold inthe synthesis of l-Leu, l-Ile, andl-Val (Table S2).
Our results demonstrate thedifficulty of broadening the sub-strate specificity of (S)-selectiveATAs. All amino acids that con-tribute to the small bindingpocket are located at the dimerinterface of the enzymes (exceptY150) and they are placed ondifferent loops. Although theplacement of smaller residues inpositions 19, 56, and 85 wouldtheoretically excavate the smallbinding pocket and providemore space for bulky substrates,mutations at these sites seem toaffect other properties of theenzyme (such as flexibility) im-portant for folding, catalysis orstability. Other than the effect ofF85L on 2 b, all mutations inthese positions were detrimen-tal. This supports previous find-ings by Park et al. and Humbleet al. where mutations at thestructurally equivalent positions
led to a drastic drop in activity in the ATAs from Paracoccus de-nitrificans and Chromobacterium violaceum.[4b, 16]
Most of the variants screened from those libraries showedlittle-to-no residual activity in our initial screening assays com-pared to the wild-type. In position 150, a Phe or Tyr is found invirtually all ATA-sequences, as this position is important for thepositioning and p-stacking with PLP. The presence of the Tyrhydroxyl group, however, determines the acceptance of thehydroxylated substrates 3 a and 3 b in the forward or backwardreaction, respectively. This result is in agreement with thosepresented in an independent study performed on the ATA
from Chromobacterium violaceum, and published byDeszcz et al. contemporaneously to ours.[17]
The different activities of Y150F- and Y150M-con-taining variants in the kinetic resolution or asymmet-ric synthesis underline the importance of screeningvariants under conditions as close to the desired syn-thetic application as possible. V153A showed thelargest contribution towards a relaxed active site ableto accept bulkier substrates, which was also observedby Park and coworkers.[4b] This second shell residue isnot in contact with the bound substrate, but its mu-tation to alanine might increase the active site’s flexi-bility.
Finally, we conclude that 1) there is no easy solu-tion to generally expand the substrate scope ofVibrio fluvialis ATA by modifying single residues ofthe small binding pocket, and 2) creating space isnot sufficient to yield an efficient amine transaminase
Table 1. Specific activities of the purified wild-type and variants identified during the screening.
1 b 2 b 3 bVariant SA [U mg�1][a] SA [U mg�1][a] Conv [%][b] SA [U mg�1][a] Conv [%][b]
VF-wt 1.33�0.02 0.19�0.00 19 0.01�0.00 14F85L 0.58�0.01 0.32�0.00 51 0.00�0.00 0Y150F 0.97�0.02 0.22�0.01 20 0.23�0.00 93Y150M 0.68�0.03 0.39�0.01 8 0.41�0.02 47V153A 5.59�0.07 3.36�0.03 71 0.11�0.00 49F85L/Y150F 0.55�0.02 0.35�0.00 52 0.07�0.00 30F85L/Y150M 0.69�0.04 0.74�0.01 14 0.35�0.00 0F85L/V153A 2.38�0.14 4.99�0.07 93 0.03�0.01 0Y150F/V153A 1.60�0.02 0.82�0.02 44 0.34�0.01 92Y150M/V153A 1.36�0.03 0.65�0.02 8 0.53�0.03 23F85L/Y150F/V153A 0.98�0.01 1.55�0.03 79 0.14�0.01 32F85L/Y150M/V153A 0.47�0.01 0.02�0.00 1 0.00�0.00 0
[a] SA = Specific activity : The reaction was followed at 245, 242, and 252 nm for the detection of 1 a, 2 a, and3 a, respectively, using 2.5 mm of the amino donor and 2.5 mm of pyruvate in 50 mm HEPES buffer pH 7.5 con-taining 1.66 % DMSO at 30 8C. The activities were calculated as U mg�1 (purified enzyme). One Unit is definedas the conversion of 1 mmol of product per minute. Values and standard deviations given are based on threemeasurements. [b] Conversion reached in asymmetric synthesis: Reaction conditions for the synthesis of 2 b :0.5 mg mL�1 purified enzyme, 1 mL reaction volume, 10 mm 2 a, 150 mm L-alanine, 30 % DMSO, 14 days, 30 8C,50 mm HEPES buffer pH 7.5, 0.1 mm PLP. Reaction conditions for the synthesis of 3 b : 0.5 mg mL�1 purifiedenzyme, 1 mL reaction volume, 10 mm 3 a, 250 mm L-alanine, 10 % DMSO, 4 days, 30 8C, 50 mm HEPES bufferpH 7.5, 0.1 mm PLP. Both reactions were followed via GC analysis using a Hydrodex-b-TBDAC chiral column andin both cases the LDH/GDH system was used to shift the equilibrium.[15]
Table 2. Asymmetric synthesis results with the best (purified) variants.
Mutant Product Conv. ee Yield[%] [%] [%]
F85L/V153A 2 b >98[a] 98 53
Y150F/V153A 3 b >98[b] 98 60
The reaction progress was followed via TLC analysis (0.2 mm detection limit of theketone). To shift the equilibrium, the LDH/GDH system was applied.[15] [a] Reactionconditions: 1 mg mL�1 purified enzyme, 20 mL reaction volume, 10 mm 2 a, 150 mm L-alanine, 30 % DMSO, 14 days, 30 8C, 50 mm HEPES buffer pH 7.5, 0.1 mm PLP. [b] Reac-tion conditions: 1 mg mL�1 purified enzyme 20 mL reaction volume, 10 mm 3 a,250 mm L-alanine, 10 % DMSO, 3 days, 30 8C, 50 mm HEPES buffer pH 7.5, 0.1 mm PLP.
ChemCatChem 2015, 7, 757 – 760 www.chemcatchem.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim759
Communications

activity. Instead, second shell residues should be included infurther mutagenesis studies. The prediction of further usefulmutations is not possible at the moment, because the underly-ing factors governing catalytic efficiency are not yet under-stood and hence, depending on the chemical structure of thesubstrate, different mutations are needed to enhance activity.
Experimental Section
All experimental details are presented in the SupportingInformation.
Acknowledgements
FS thanks the “Fonds der Chemischen Industrie” for financial sup-port. We especially thank the European Union (KBBE-2011-5,grant No. 289350) for financial support within the EuropeanUnion Seventh Framework Programme. We also thank Dr. Ioan-nis V. Pavlidis and Anders M. Knight for their useful feedbackduring the preparation of the manuscript.
Keywords: amine transaminase · biocatalysis · proteinengineering · substrate scope
[1] F. Weber, G. Sedelmeier, Nachr. Chem. 2013, 61, 528 – 529.[2] a) H. Kohls, F. Steffen-Munsberg, M. Hçhne, Curr. Opin. Chem. Biol. 2014,
19, 180 – 192; b) W. Kroutil, E.-M. Fischereder, C. S. Fuchs, H. Lechner,F. G. Mutti, D. Pressnitz, A. Rajagopalan, J. H. Sattler, R. C. Simon, E. Siiro-la, Org. Process Res. Dev. 2013, 17, 751 – 759; c) M. Hçhne, U. T. Born-scheuer, in Enzymes in Organic Synthesis (Eds. : O. May, H. Grçger, W.Drauz), Wiley-VCH, Weinheim, 2012, pp. 779 – 820; d) D. Ghislieri, N.Turner, Top. Catal. 2014, 57, 284 – 300; e) D. Koszelewski, K. Tauber, K.Faber, W. Kroutil, Trends Biotechnol. 2010, 28, 324 – 332.
[3] C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis,J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huis-man, G. J. Hughes, Science 2010, 329, 305 – 309.
[4] a) K. S. Midelfort, R. Kumar, S. Han, M. J. Karmilowicz, K. McConnell, D. K.Gehlhaar, A. Mistry, J. S. Chang, M. Anderson, A. Villalobos, J. Minshull,S. Govindarajan, J. W. Wong, Protein Eng. Des. Sel. 2013, 26, 25 – 33;b) E.-S. Park, S.-R. Park, S.-W. Han, J.-Y. Dong, J.-S. Shin, Adv. Synth. Catal.2014, 356, 212 – 220.
[5] a) F. Steffen-Munsberg, C. Vickers, A. Thontowi, S. Sch�tzle, T. Meinhardt,M. S. Humble, H. Land, P. Berglund, U. T. Bornscheuer, M. Hçhne, Chem-CatChem 2013, 5, 154 – 157; b) F. Steffen-Munsberg, C. Vickers, A. Thon-towi, S. Sch�tzle, T. Tumlirsch, M. S. Humble, H. Land, P. Berglund, U. T.Bornscheuer, M. Hçhne, ChemCatChem 2013, 5, 150 – 153.
[6] J.-S. Shin, B.-G. Kim, J. Org. Chem. 2002, 67, 2848 – 2853.[7] F. G. Mutti, W. Kroutil, Adv. Synth. Catal. 2012, 354, 3409 – 3413.[8] D. A. Erlanson, J. W. Arndt, M. T. Cancilla, K. Cao, R. A. Elling, N. English,
J. Friedman, S. K. Hansen, C. Hession, I. Joseph, G. Kumaravel, W.-C. Lee,K. E. Lind, R. S. McDowell, K. Miatkowski, C. Nguyen, T. B. Nguyen, S.Park, N. Pathan, D. M. Penny, M. J. Romanowski, D. Scott, L. Silvian, R. L.Simmons, B. T. Tangonan, W. Yang, L. Sun, Bioorg. Med. Chem. Lett. 2011,21, 3078 – 3083.
[9] D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G.Robertson, A. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S. P. Han, B.Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J.Marcinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker, L. G.Hamann, J. Med. Chem. 2005, 48, 5025 – 5037.
[10] M. Amat, J. Bosch, J. Hidalgo, M. Canto, M. Perez, N. Llor, E. Molins, C.Miravitlles, M. Orozco, J. Luque, J. Org. Chem. 2000, 65, 3074 – 3084.
[11] S. Sch�tzle, M. Hçhne, E. Redestad, K. Robins, U. T. Bornscheuer, Anal.Chem. 2009, 81, 8244 – 8248.
[12] K. Soda, T. Yoshimura, N. Esaki, Chem. Rec. 2001, 1, 373 – 384.[13] M. S. Humble, K. E. Cassimjee, M. Hakansson, Y. R. Kimbung, B. Walse, V.
Abedi, H. J. Federsel, P. Berglund, D. T. Logan, FEBS J. 2012, 279, 779 –792.
[14] S. Kille, C. G. Acevedo-Rocha, L. P. Parra, Z.-G. Zhang, D. J. Opperman,M. T. Reetz, J. P. Acevedo, ACS Synth. Biol. 2013, 2, 83 – 92.
[15] D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell, W. Kroutil, Adv. Synth.Catal. 2008, 350, 2761 – 2766.
[16] M. S. Humble, K. E. Cassimjee, V. Abedi, H.-J. Federsel, P. Berglund,ChemCatChem 2012, 4, 1167 – 1172.
[17] D. Deszcz, P. Affaticati, A. Gegel, J. M. Ward, H. C. Hailes, P. A. Dalby,FEBS J. in press.
Received: December 11, 2014Published online on February 2, 2015
ChemCatChem 2015, 7, 757 – 760 www.chemcatchem.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim760
Communications

Supporting Information� Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2015
Engineering the Active Site of the Amine Transaminasefrom Vibrio fluvialis for the Asymmetric Synthesis of Aryl–Alkyl Amines and Amino AlcoholsAlberto Nobili,[a] Fabian Steffen-Munsberg,[a, b] Hannes Kohls,[a] Ivan Trentin,[a]
Carola Schulzke,[a] Matthias Hçhne,[a] and Uwe T. Bornscheuer*[a]
cctc_201403010_sm_miscellaneous_information.pdf

1""
Supporting*Information!"
Experimental!Section"• Chemicals"
• Library"cultivation"
• Primers"
• Parameters"for"creation"of"the"mutants"via"QuikChange®"
• Acetophenone"assay"conditions"
• Enzyme"purification"
• Single"variants"cultivation"for"purification"
• Asymmetric"synthesis"conditions"
• Analytics"for"asymmetric"synthesis"experiments"
Supporting!Tables!and!Figures"• Acetophenone"assay"
• Asymmetric"synthesis"experiment"with"increased"enzyme"concentrations"
• Activities"on"branchedCchain"amino"acids"
References!"
" "

2""
Experimental!Section"
Chemicals!
The"following"chemicals"were"purchased"from"Sigma"Aldrich"Co.:"Pyruvic"acid"(127C17C3),"LCalanine"(56C41C7),"(S)CαCmethylbenzylamine"(2627C86C3),"(R)C1Cphenylbutylamine"(6150C01C2),"butyrophenone"(495C40C9)," (S)C1Cphenylbutylamine" (3789C60C4),"2Chydroxyacetophenone" (582C24C1)," (R)C(−)C2Cphenylglycinol"(56613C80C0)." The" following" enzymes"were" purchased" from" Sigma"Aldrich" Co.:" LClactic" dehydrogenase"from" bovine" heart" (9001C60C9)," glucose" dehydrogenase" from" Pseudomonas, sp." (9028C53C9)." (S)C(+)C2Cphenylglycinol" (20989C17C7)"was"purchased"from"AlfaCAesar"Co,"DC(+)Cglucose"monohydrate"(14431C43C7)" was" from" AppliChem" GmbH," NADHCNa2" (606C68C8)" was" obtained" from" Roth" GmbH." Unless" stated"otherwise,"all"other"chemicals"were"from"Sigma"Aldrich"Co.""Library!cultivation!
The"pool"of"circularized"plasmids"was"transformed"in"E.,coli,BL21"(DE3)"cells."Single"colonies"were"picked"using" sterile" toothpicks" and" inoculated" into" sterile" polystyrene" 96Cwells" test" plates" (Sarstedt,"82.1581.001)"containing"LB"media"(Fluka,"L3152)"with"50"μg/mL"Kanamycin.""The"seed"cultivation"was"carried"on"for"16"hours"at"37°C"under"sufficient"agitation"(Incubator"TH30"and"shaker"TiMix""are"from"Edmund"Bühler"GmbH)."Then,"a"glycerol"stock"was"created"by"mixing"one"part"of"the"culture"with"one"part"of"a"50%"sterile"glycerol"solution."The"remaining"amount"of"seed"cultivation"was" reCinoculated" in" sterile" deepCwellCblocks" (Greiner" BioCOne," 780271)" containing" 1" mL/well" of" TB"media" (TB"media"containing"50.75"g"TB"–"Roth,"X972.3,"and"4"g"glycerol" in"one" liter"deCionized"water)"kept" at" 37°C" until" OD600" reached" ca." 0.7." Induction" of" the" gene" encoding" the" transaminase"was" then"performed"with" IPTG" (0.1"mM"final"concentration)" for"16"hours"at"23°C"at"450"rpm."During" the"entire"period,"the"plates"were"covered"with"sterile"breathable"membranes"(Aeraseal"BS25,"Excel"Scientific)."The"cultures"were"then"centrifuged"for"15"min"at"4000"g"at"4°C"(Multifuge"3SCR,"Heraeus)."Immediately"after"centrifugation,"the"supernatant"was"removed"by"tilting"the"block"and"thus"discarding"the"media."While"facing"down,"the"edge"of"the"wells"were"dried"with"paper"to"prevent"crossCcontaminations.",Cells,disruption,
The"pellets"were"mixed"thoroughly"with"the"pipette"and"then"the"disruption"was"carried"on" for"3"h"at"30°C" in" the" following" lysis" solution:"PLP" (Final" concentration"0.1"mM)," lysozyme" (final" concentration"1"mg/mL)"and"DNAse"I"(final"concentration"1"μg/mL),"HEPES"buffer"50"mM,"pH"7.5.""! !

3""
Primers!
The"gene"encoding" the" transaminase" from"Vibrio, fluvialis, (1362"pb)"was"cloned" into"a"pET"24b"vector"(6635"bpClong"construct)."The"primers"used"for"the"construction"of"libraries"or"of"the"mutants"were"the"following:"Randomization"at"F85."Residues"allowed:"GAVLISTCPQD"
• 01_FW1:"CGTTTTCCCGGTTATCACGCCRNCTTCGGCCGCATGT"• 02_FW2:"CGTTTTCCCGGTTATCACGCCCHSTTCGGCCGCATGT"• 03_FW3:"CGTTTTCCCGGTTATCACGCCKGCTTCGGCCGCATGT"• 04_RV1:"CTGATCGGACATGCGGCCGAAGNYGGCGTGATAACC"• 05_RV2:"CTGATCGGACATGCGGCCGAASDGGGCGTGATAACC"• 06_RV3:"CTGATCGGACATGCGGCCGAAGCMGGCGTGATAACC"
Randomization"at"F19."Residues"allowed:"ILMVPGRA"• 07_FW1:"CGCTCTATGGTVTSACCGACATGC"• 08_FW2:"CGCTCTATGGTSSGACCGACATGC"• 09_RV1:"GCATGTCGGTSABACCATAGAGCG"• 10_RV2:"GCATGTCGGTCSSACCATAGAGCG"
Randomization"at"Y150:"full"saturation"• 11_FW1:"GGAACGCCNDTCACGGCGTGACCGCCGTTTCG"• 12_FW2:"GGAACGCCVHGCACGGCGTGACCGCCGTTTCG"• 13_FW3:"GGAACGCCTGGCACGGCGTGACCGCCGTTTCG"• 14_RV1:"GGTCACGCCGTGAHNGGCGTTCCAGCGGGTCAGG"• 15_RV2:"GGTCACGCCGTGCDBGGCGTTCCAGCGGGTCAGG"• 16_RV3:"GGTCACGCCGTGCCAGGCGTTCCAGCGGGTCAGG"
Randomization"at"L56:"full"saturation"• 17_FW1:"CTCGGGCNDTTGGAACATGGTCGCGGGC"• 18_FW2:"CTCGGGCVHGTGGAACATGGTCGCGGGC"• 19_FW3:"CTCGGGCTGGTGGAACATGGTCGCGGGC"• 20_RV1:"CCATGTTCCAAHNGCCCGAGTTGGCGTCC"• 21_RV2:"CCATGTTCCACDBGCCCGAGTTGGCGTCC"• 22_RV3:"CCATGTTCCACCAGCCCGAGTTGGCGTCC"
Mutant"F85A"• 23_FW:"GGTTATCACGCCGCGTTCGGCCGCATGTCC"• 24_RV:"GGACATGCGGCCGAACGCGGCGTGATAACC"
Mutant"F85L"• 25_FW:"GGTTATCACGCCCTCTTCGGCCGCATGTCC"• 26_RV:"GGACATGCGGCCGAAGAGGGCGTGATAACC"
Mutant"V153A"• 27_FW:"CTATCACGGCGCGACCGCCGTTTC"• 28_RV:"GAAACGGCGGTCGCGCCGTGATAG"
Mutant"Y150M/V153A"• 29_FW:"CATGCACGGCGCGACCGCCGTTTC"• 30_RV:"GAAACGGCGGTCGCGCCGTGCATG"
Mutant"Y150F/V153A"• 31_FW:"CTTTCACGGCGCGACCGCCGTTTC"• 32_RV:"GAAACGGCGGTCGCGCCGTGAAAG"
"! !

4""
Parameters!for!creation!of!the!mutants!via!QuikChange®"
The"master"mix"necessary" for" a"QuikChange®"was" composed"of" sterilized"deionized"H2O" (41"μL)," Pfu+"buffer" (10x,"5"μL,"EURx),"dNTPs" (1"μL,"10"mM"each),"plasmid"template" (1"μL,"10"ng/μL)"containing"the"gene"encoding"for"the"transaminase"of"Vibrio,fluvialis,"a"1:1"mixture"of"forward"and"reverse"primer"(1"μL,"0.5"μM"for"each"primer),"PfuPlus!"DNA"Polymerase"(0.2"μL,"EURx).""The"mixture"was"divided"in"two"different"PCR"tubes"and"used"then"at"the"following"conditions:"1)"95°C,"300"s;"2)"30"cycles:"95°C,"45"s;"one"aliquot"at"50°C"and"one"aliquot"at"65°C,"45"s;"72°C"420"s;"3)"72°C,"900"s."Afterwards,"the"PCR"products"were"digested"by"DpnI"(0.5"μL,"NEB)"for"2"hours"at"37°C"and"then"80°C"for" 20" minutes." ChemoCcompetent" E., coli" cells" (Top10)" were" transformed" with" the" PCR" product" for"plasmid" amplification." Once" the" correct" cloning" was" confirmed" by" sequencing" (Eurofins" Genomics,"Germany)," the"corresponding"plasmid"was" transformed" into"chemoCcompetent"E., coli" cells" (BL21"DE3)"and"plated"onto"LBKANCplates."Acetophenone!assay!conditions"
The" quick" and" sensitive" 'acetophenone'" photometric" assay[1]" was" used" to" determine" the" enzyme"activities." The" best" conditions" for" asymmetric" synthesis" with" the" LDH/GDH" system"were" observed" to"require"a"neutral"pH"and"30°C" (data"not" shown)."This" supported" the" findings"described" in" literature.[2]"Thus,"the"assay"was"performed"at"the"following"conditions:"HEPES"buffer"50"mM"pH"7.5,"30"°C,"DMSO"1.66%,"2.5"mM"amino"donor"and"2.5"mM"amino"acceptor,"200"μL" final" reaction"volume," (UV"96Cwells"plate,"655801,"Greiner"BioCone)."The"production"of"acetophenone"was"followed"at"245"nm,"as"described"by"Schätzle"et,al.,[1]"while"in"present"work"work,"we"followed"2a"at"242"nm"and"3a"at"252"nm"(see"Figure"S1"in"the"dedicated"chapter"of"the"Supporting"Information)."The"detection"limit"was"3"mU/mL."
Enzyme!purification!
The" enzymes" were" purified" according" to" the" instructions" provided" in" the" supporting" information" by"SteffenCMunsberg" et, al.[3]:" The" cell" pellet" was" washed" with" HEPES" buffer" (50"mM," pH" 7.5)" and" then"resuspended"in"50"mM"HEPES"buffer"pH"7.5,"containing"0.3"M"NaCl,"and"0.1"mM"PLP."After"disruption"by"French"press"(Thermo"Fischer"Scientific,"MA,"USA)"at"0°C,"the"suspension"was"centrifuged"(10,000"x"g,"45"min)" and" the" supernatant" was" passed" through" a" 0.2" µm" filter" prior" to" chromatography." Affinity"chromatography"was"performed"using"an"Äkta"Purifier"(GE"Healthcare,"Chalfont"St"Giles,"GB)"and"a"5"mL"IMAC"Sepharose"6"Fast"Flow"column"(GE"Healthcare)."After"washing"the"column"with"HEPES"buffer"(50"ml,"50"mM,"pH"7.5;"containing"0.3"M"NaCl,"0.1"mM"PLP"and"0.03"M"imidazole;"5"mL"minC1"flow"rate),"the"crude"extract"was" injected."The"enzymes"were"eluted"by"0.3"M" imidazole"containing"HEPES"buffer" (50"mM,"pH"7.5,"0.3"M"NaCl"and"0.1"mM"PLP;"5"mL"minC1"flow"rate)"and"the"fractions"containing"the"desired"protein" were" pooled." For" desalting" size" exclusion" chromatography" using" 50"mM"HEPES," 0.1"mM" PLP"buffer"pH"7.5"was"performed.""The" enzyme" solutions"were" aliquoted" in" different" 1.5"mL" stored" at" C80°C" in" 20%" glycerol" stocks." The"protein"concentration"was"calculated"via"the"Bradford"assay.""Single!variants!cultivation!for!purification!
The"sequenced"single"E.,coli,BL21"(DE3)"colonies"were"inoculated"for"16"h"in"LB"(seed"cultivation,"5"mL)"and" then" reCinoculated" in" fresh" TB"media" (4"mL" seed" in" 400"mL" TB)." The" cultures"were" kept" at" 37°C"under"moderate" shaking" until" the" culture" reached" OD600" reached" 0.7;" then" the" gene" expression" was"induced"with"IPTG"(0.1"mM"final"concentration)"and"the"culture"was"shaken"for"16"h"at"20°C."The"culture"was"then"harvested"(15"min,"4000"g"and"4°C)"and"the"pellet"was"washed"in"HEPES"buffer"(pH"7.5,"50"mM,"

5""
300"mM"NaCl"and"0.1"mM"PLP)."The"suspension"was"mixed"with"DNAse"I"(final"concentration"1"μg/mL)"and"then"homogenized"(French"pressure"cell"press,"Thermo"Spectronic,"1800"psi)."The"resulting"solution"was"centrifuged"(1"h"at"10,000"g)"and"the"supernatant"was"filtered"(Filtropur"Syringe"Filter,"0.20"μm"pore"size,"sterile)."The"thus"prepared"solution"was"then"ready"to"be"loaded"onto"the"purification"column.""
Asymmetric!synthesis!conditions:!
All"reactions"were"performed"in"1"mL"volume"in"1.5"mL"glass"vials"containing"0.5"mg"or"2.5"mg"purified"ATA"enzyme," 90"U"mLC1" lactate"dehydrogenase," 15"U"mLC1" glucose"dehydrogenase," 150"or" 250"mM"LCalanine" (for" the" synthesis" of" (S)Cphenylbutyl" amine" or" " (R)Cphenylglycinol," respectively)," 150" mM" DCglucose," 1" mM" NADH," 0.1" mM" PLP" and" 10" mM" keto" substrate" in" 50" mM" HEPES" buffer" pH" 7.5." The"solutions"were"sealed"and"shaken"at"30°C"and"600"rpm."Then"100"µL"samples"were"taken"for"chiral"gas"chromatography"(GC)"or"thinClayerCchromatography"(TLC)"quantification.[3]"During"the"preparation"of"the"reaction"mixture,"to"obtain"complete"solution"of"the"ketones"it"was"necessary"to"dissolve"them"in"DMSO"before" that" in" the"HEPES"buffer."High" initial" concentrations" of"2a," in" fact,"were" seen" to" be" extremely"harmful"to"the"enzymes."3a"could"only"be"dissolved"after"being"solvatated"by"DMSO."
Analytics!for!asymmetric!synthesis!experiments!
GC,Analysis,–,Column,
The"evaluation"of"the"most"promising"mutants"in"the"asymmetric"synthesis"reaction"for"both"compounds"was" performed" via" GC" analysis" (chiral" column"HydrodexCβCTBDAc" from"MachereyCNagel," 25"m"× 0.25"mm"× 0.25"μm).""
Quantification,of,conversion,
Extraction"procedure"for"GC"analysis:"For"the"analysis"of"the"reaction,"a"sample"of"the"reaction"mixture"was"taken"(0.1"mL)"and"treated"with"NaOH" (10"M,"0.01"mL)."The"basified" solution"was" saturated"with"NaCl," shortly"mixed" (5" sec)"and" then"centrifuged"(5"sec)."The"solution"was"considered"saturated"with"NaCl" if"after"mixing"and"centrifuging"a"NaCl"pellet"was"formed"at"the"bottom"of"the"1.5"mL"tube.""The"extraction"of"both"substrate"and"product"was"performed"with"ethylacetate"(0.3"mL)"after"vigorous"mixing"(30"sec)."The"separation"of"the"two"phases"was"obtained"via"centrifugation"(180"sec)."The"organic"phase"was" transferred" into"a" clean"container"and"dried"with"NaS04."The"solution"was"again"mixed" (30"sec)"and"centrifuged"(180"sec)"before"being"filtered"and"analyzed"on"TLC"or"GC"(for"compounds"2)."For"the"analysis"on"GC"of"compound"3,"the"reaction"was"at"this"moment"treated"with"the"derivatizing"agent,"MBTFA." The" sample" was" injected" after" thorough" mixing" of" MBTFA" with" the" organic" fraction" and"incubation"at"room"temperature.""Comments"to"the"derivatization"via"MBTFA:"A"5"μL"aliquot"of"the"MBTFA"commercial"solution"(98%"pure),"previously"stored"at"4°C,"was"mixed"with"200" μL" reaction" solution," extracted" as" described" above." At" this" stage," the" solutions" were" thoroughly"mixed"by"pipetting"upCandCdown"100"μL"and"incubation"was"performed"at"room"temperature.""According"to"our"results,"it"seems"that"the"derivatization"at"the"hydroxyl"group"takes"place"immediately"after"mixing"and"slowly"over"time"the"second"derivatization"at"the"amine"group"takes"place."This"results"in"two"pairs"of"enantiomeric"products"that"shift"from"49.8"and"50.6"min"(derivatized"alcohol"enantiomers"of"3b!and"3c)"to"45.7"and"46.0"min"(both"functional"groups"derivatized)."It"is"important"to"mention"that"after" the"derivatization," the"signal" corresponding" to" the"substrate"3a"decreased"with"a" rate"of"2%"per"hour"after"derivatization"probably"due"to" instability"of" the"derivatized"product."Therefore,"samples" for"
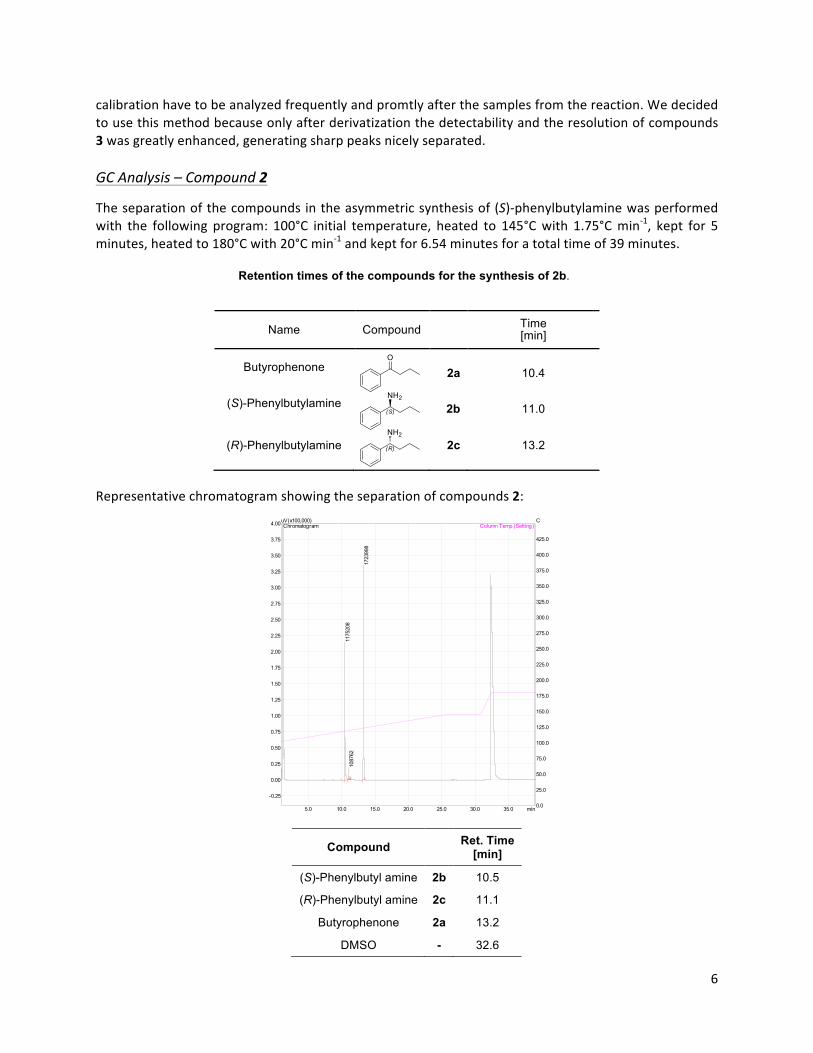
6""
calibration"have"to"be"analyzed"frequently"and"promtly"after"the"samples"from"the"reaction."We"decided"to"use"this"method"because"only"after"derivatization"the"detectability"and"the"resolution"of"compounds"3"was"greatly"enhanced,"generating"sharp"peaks"nicely"separated."""GC,Analysis,–,Compound,2,
The"separation"of"the"compounds"in"the"asymmetric"synthesis"of"(S)Cphenylbutylamine"was"performed"with" the" following" program:" 100°C" initial" temperature," heated" to" 145°C"with" 1.75°C"minC1," kept" for" 5"minutes,"heated"to"180°C"with"20°C"minC1"and"kept"for"6.54"minutes"for"a"total"time"of"39"minutes.""
Retention times of the compounds for the synthesis of 2b.
Name Compound Time [min]
Butyrophenone 2a
10.4
(S)-Phenylbutylamine 2b
11.0
(R)-Phenylbutylamine
2c 13.2
Representative"chromatogram"showing"the"separation"of"compounds"2:"
Compound Ret. Time [min]
(S)-Phenylbutyl amine 2b 10.5
(R)-Phenylbutyl amine 2c 11.1
Butyrophenone 2a 13.2
DMSO - 32.6
!
5.0 10.0 15.0 20.0 25.0 30.0 35.0 min
-0.25
0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
2.75
3.00
3.25
3.50
3.75
4.00uV(x100,000)
0.0
25.0
50.0
75.0
100.0
125.0
150.0
175.0
200.0
225.0
250.0
275.0
300.0
325.0
350.0
375.0
400.0
425.0
CColumn Temp.(Setting) Chromatogram
1175
208
1087
62
1723
998

7""
GC,Analysis,–,Compound,3,
The" quantification" of" compounds"3"was" performed"with"MBTFA" (NCMethylCbis(trifluoroacetamide))" at"least"one"hour"prior"to"the"analysis.""The"separation"of"the"compounds"in"the"asymmetric"synthesis"of"(R)Cphenylglycinol"was"obtained"with"the" following" program:" 65°C" initial" temperature," heated" to" 100°C"with" 1.00°C"minC1," heated" to" 180°C"with"20°C"minC1"and"kept"for"15"minutes"for"a"total"time"of"54.""
Retention times of the compounds for the synthesis of 3b.
Name Compound Time [min]
Hydroxyacetophenone 3a
39.9
(R)-Phenylglycinol 3b
45.7/49.8
(S)-Phenylglycinol 3c 46.0/50.6
Representative"chromatogram"showing"the"separation"of"compounds"3:"
!!
Chromatogram showing the trace of pure MBTFA in EtOAc: four additional peaks were detected (see table below). !
Compound Ret. Time [min]
MBTFA 1 4.4 MBTFA 2 23.4 MBTFA 3 31.2 MBTFA 4 38.4
DMSO 41.7
5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 min-0.50
-0.25
0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
2.75
3.00
3.25
3.50
3.75
4.00
4.25
4.50
4.75
5.00uV(x10,000)
0.0
25.0
50.0
75.0
100.0
125.0
150.0
175.0
200.0
225.0
250.0
275.0
300.0
325.0
350.0
375.0
400.0
425.0
CColumn Temp.(Setting) Chromatogram
6498
90
4477
310
2102
93
2196
750
2716
80
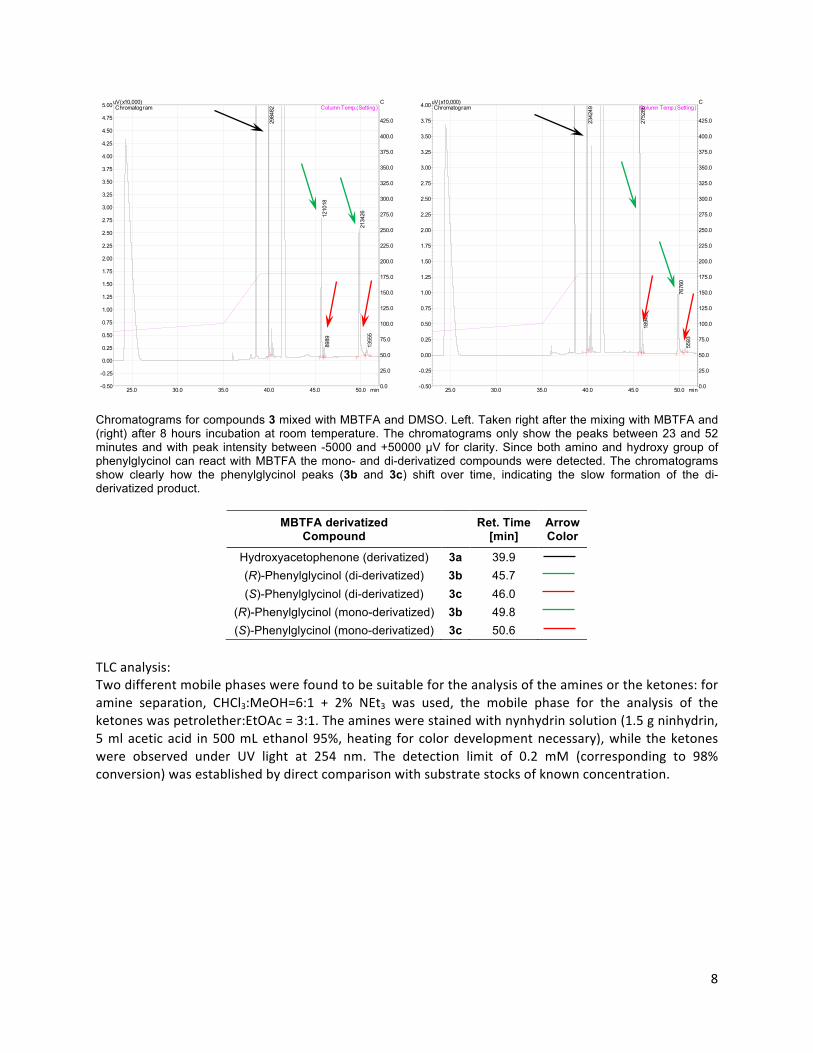
8""
"TLC"analysis:"Two"different"mobile"phases"were"found"to"be"suitable"for"the"analysis"of"the"amines"or"the"ketones:"for"amine" separation," CHCl3:MeOH=6:1" +" 2%" NEt3" was" used," the" mobile" phase" for" the" analysis" of" the"ketones"was"petrolether:EtOAc"="3:1."The"amines"were"stained"with"nynhydrin"solution"(1.5"g"ninhydrin,"5"ml"acetic"acid" in"500"mL"ethanol"95%,"heating" for" color"development"necessary),"while" the"ketones"were" observed" under" UV" light" at" 254" nm." The" detection" limit" of" 0.2" mM" (corresponding" to" 98%"conversion)"was"established"by"direct"comparison"with"substrate"stocks"of"known"concentration.""
Chromatograms for compounds 3 mixed with MBTFA and DMSO. Left. Taken right after the mixing with MBTFA and (right) after 8 hours incubation at room temperature. The chromatograms only show the peaks between 23 and 52 minutes and with peak intensity between -5000 and +50000 µV for clarity. Since both amino and hydroxy group of phenylglycinol can react with MBTFA the mono- and di-derivatized compounds were detected. The chromatograms show clearly how the phenylglycinol peaks (3b and 3c) shift over time, indicating the slow formation of the di-derivatized product.
MBTFA derivatized
Compound Ret. Time [min]
Arrow Color
Hydroxyacetophenone (derivatized) 3a 39.9
(R)-Phenylglycinol (di-derivatized) 3b 45.7
(S)-Phenylglycinol (di-derivatized) 3c 46.0
(R)-Phenylglycinol (mono-derivatized) 3b 49.8
(S)-Phenylglycinol (mono-derivatized) 3c 50.6
25.0 30.0 35.0 40.0 45.0 50.0 min-0.50
-0.25
0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
2.75
3.00
3.25
3.50
3.75
4.00
4.25
4.50
4.75
5.00uV(x10,000)
0.0
25.0
50.0
75.0
100.0
125.0
150.0
175.0
200.0
225.0
250.0
275.0
300.0
325.0
350.0
375.0
400.0
425.0
CColumn Temp.(Setting) Chromatogram
2984
82
1210
1889
89
2134
2613
555
25.0 30.0 35.0 40.0 45.0 50.0 min-0.50
-0.25
0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
2.75
3.00
3.25
3.50
3.75
4.00uV(x10,000)
0.0
25.0
50.0
75.0
100.0
125.0
150.0
175.0
200.0
225.0
250.0
275.0
300.0
325.0
350.0
375.0
400.0
425.0
CColumn Temp.(Setting) Chromatogram
2342
49
2752
8618
945
7676
055
93

9""
" " " "
Depletion of 2a
Production of 2b
Depletion of 3a
Production of 3b
"
Exemplary TLC plates showing the two different detection methods. At each time point, the analysis of the best variant (second lane) was combined with the analysis of the wild-type sample (first lane), a negative control containing all the components of the asymmetric synthesis except the transaminase (third lane) and on the TLC a final spot containing just the pure standard reference. The plates show the reactions at the end of the asymmetric synthesis.
Extraction"of"(S)Cphenylbutyl"amine"after"semiCpreparative"biocatalysis:"The"pH"of"the"20"mL"reaction"mixture"was"shifted"to"pH"2"by"the"addition"of"HCl"(12"M)."The"solution"was"extracted"once"with"20"mL"Cl2CH2" (DCM)"and" successively"with"30"mL"DCM" for"another" two" times."At"every"washing" step," the" solution"was" centrifuged" for" the" removal" of" the" precipitated" enzymes" at" the"interphase"between" the"organic"and" the"aqueous"solution."The"pH"was" then"shifted" to"a"value"higher"than"12"by"the"addition"of"NaOH"(10"M),"for"the"extraction"of"the"amine"with"50"mL"nChexane"for"three"times."We"chose"to"extract"with"nChexane"because"in"this"solvent"DMSO"is"almost"not"soluble."The" solution"was" dried"with"MgSO4" before" being" concentrated" at" 335"mbar" and"40°C" in" a" RotaVapor"station."The"final"volume"of"around"1"mL"was"dried"under"nitrogen"flux"in"a"GC"vial,"which"was"then"used"to"calculate"the"final"extracted"yield."When"this"procedure"was"applied"for"extracting"a"reference"sample"simulating" 91%" conversion," the" extracted" yield" was" equal" to" 66%" (data" not" shown)." The" reference"solution"contained"all"the"components"except"the"transaminase.""Extraction"of"(R)Cphenylglycinol"after"semiCpreparative"biocatalysis:"The" procedure" for" the" extraction" of" (R)Cphenylglycinol" had" only" slight"modifications" compared" to" the"one" performed" for" (S)Cphenylbutyl" amine." To" increase" the" extraction" yield," after" basification" of" the"reaction"mixture" to"a"pH"higher" than"12," the"aqueous"solution"was"saturated"with"NaCl."Due"to"a" low"solubility"of"the"aminoCalcohol"in"nChexane"the"extraction"was"performed"with"DCM"(3x50"mL)."Once"the"organic" solution" was" concentrated" to" a" volume" of" approximately" 2" mL," the" product" was" dried" from"DMSO"at"0.4"mbar"and"25"°C"under"agitation"by"a"magnetic"stirrer."In"a"preliminary"test"simulating"99%"conversion," the" extracted" yield" was" equal" to" 68%" (data" not" shown)." The" solution" contained" all" the"components"except"the"transaminase."! !
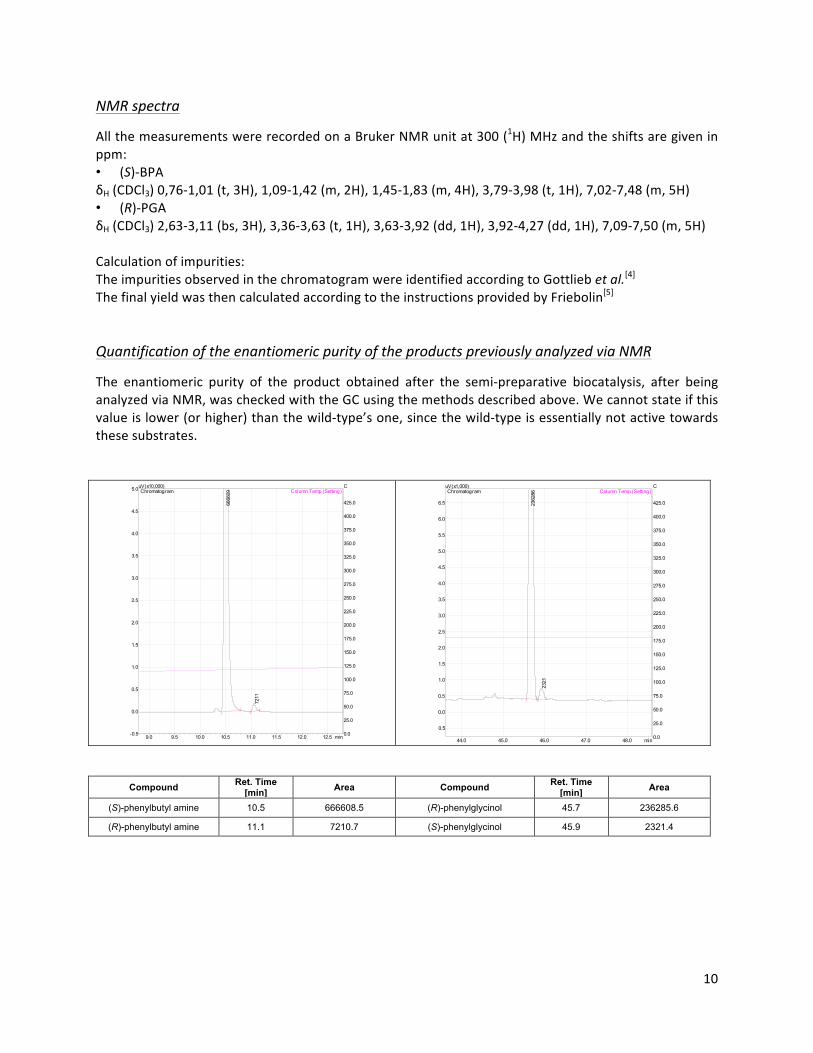
10""
NMR,spectra,
All"the"measurements"were"recorded"on"a"Bruker"NMR"unit"at"300"(1H)"MHz"and"the"shifts"are"given"in"ppm:"• (S)CBPA"δH"(CDCl3)"0,76C1,01"(t,"3H),"1,09C1,42"(m,"2H),"1,45C1,83"(m,"4H),"3,79C3,98"(t,"1H),"7,02C7,48"(m,"5H)"• (R)CPGA"δH"(CDCl3)"2,63C3,11"(bs,"3H),"3,36C3,63"(t,"1H),"3,63C3,92"(dd,"1H),"3,92C4,27"(dd,"1H),"7,09C7,50"(m,"5H)""Calculation"of"impurities:"The"impurities"observed"in"the"chromatogram"were"identified"according"to"Gottlieb"et,al.[4]"The"final"yield"was"then"calculated"according"to"the"instructions"provided"by"Friebolin[5]"!
Quantification,of,the,enantiomeric,purity,of,the,products,previously,analyzed,via,NMR,
The" enantiomeric" purity" of" the" product" obtained" after" the" semiCpreparative" biocatalysis," after" being"analyzed"via"NMR,"was"checked"with"the"GC"using"the"methods"described"above."We"cannot"state"if"this"value"is"lower"(or"higher)"than"the"wildCtype’s"one,"since"the"wildCtype"is"essentially"not"active"towards"these"substrates.""""
" ""
Compound Ret. Time [min] Area Compound Ret. Time
[min] Area
(S)-phenylbutyl amine 10.5 666608.5 (R)-phenylglycinol 45.7 236285.6
(R)-phenylbutyl amine 11.1 7210.7 (S)-phenylglycinol 45.9 2321.4
9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 min-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0uV(x10,000)
0.0
25.0
50.0
75.0
100.0
125.0
150.0
175.0
200.0
225.0
250.0
275.0
300.0
325.0
350.0
375.0
400.0
425.0
CColumn Temp.(Setting) Chromatogram
6666
09
7211
44.0 45.0 46.0 47.0 48.0 min
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
uV(x1,000)
0.0
25.0
50.0
75.0
100.0
125.0
150.0
175.0
200.0
225.0
250.0
275.0
300.0
325.0
350.0
375.0
400.0
425.0
CColumn Temp.(Setting) Chromatogram
2362
86
2321
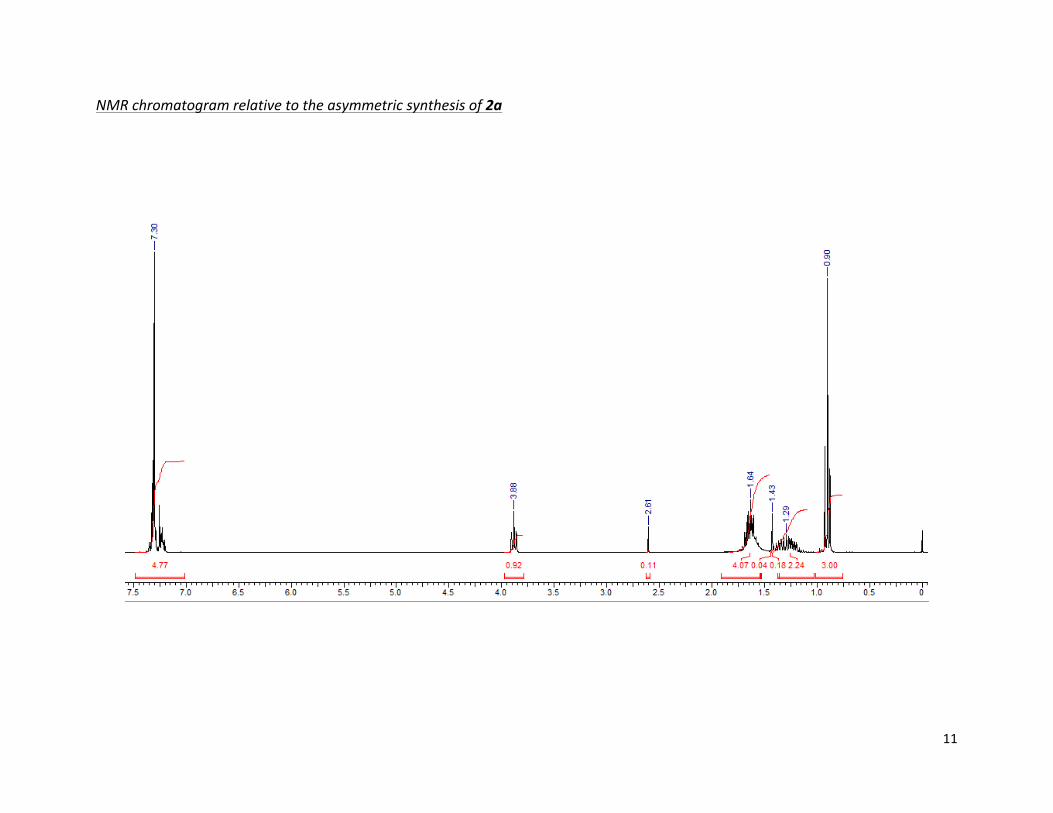
11""
NMR$chromatogram$relative$to$the$asymmetric$synthesis$of$2a#
#
#
$
"
!
"
"

12""
NMR$chromatogram$relative$to$the$asymmetric$synthesis$of$3a#
"
"
"
"20140926B_ALN_BO_PGA-FA1.001.001.1R.ESP
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Nor
mal
ized
Inte
nsity
0.172.831.000.830.880.554.88
0.00
2.60
2.81
3.58
3.744.07
5.29
7.26

13#
#
Supplementary,Tables,and,Figures,
Acetophenone,assay,
Absorbance#spectra#for#the#compounds#2#and#3#
#
Standard#curve#of#2a#and#3a,#
, Figure S2. Standard curves for the compounds 2a (R² = 0.9909) and 3a (R² = 0.9996). ,Molar#exctintion#coefficients#for#the#studied#compounds#
In#the#acetophenone#assay#the#ketone#production#is#followed#at#the#following#wavelengths:#
• Production#of#acetophenone#(ketoBderivative#of#1)#measured#at#245#nm#(ε=3.66#AU#mMB1),[6]#
• Production#of#butyrophenone#2a#measured#at#242#nm#(ε=3.91#AU#mMB1),#
• Production#of#hydroxyacetophenone#3a#measured#at#252#nm#(ε=4.25#AU#mMB1).#
#
, , Figure S1. Comparison between the absorbance spectra between amino-donor (2b and 3b, 2.5 mM) and their corresponding ketones formed after transamination (2a and 3a, 0.125 mM).
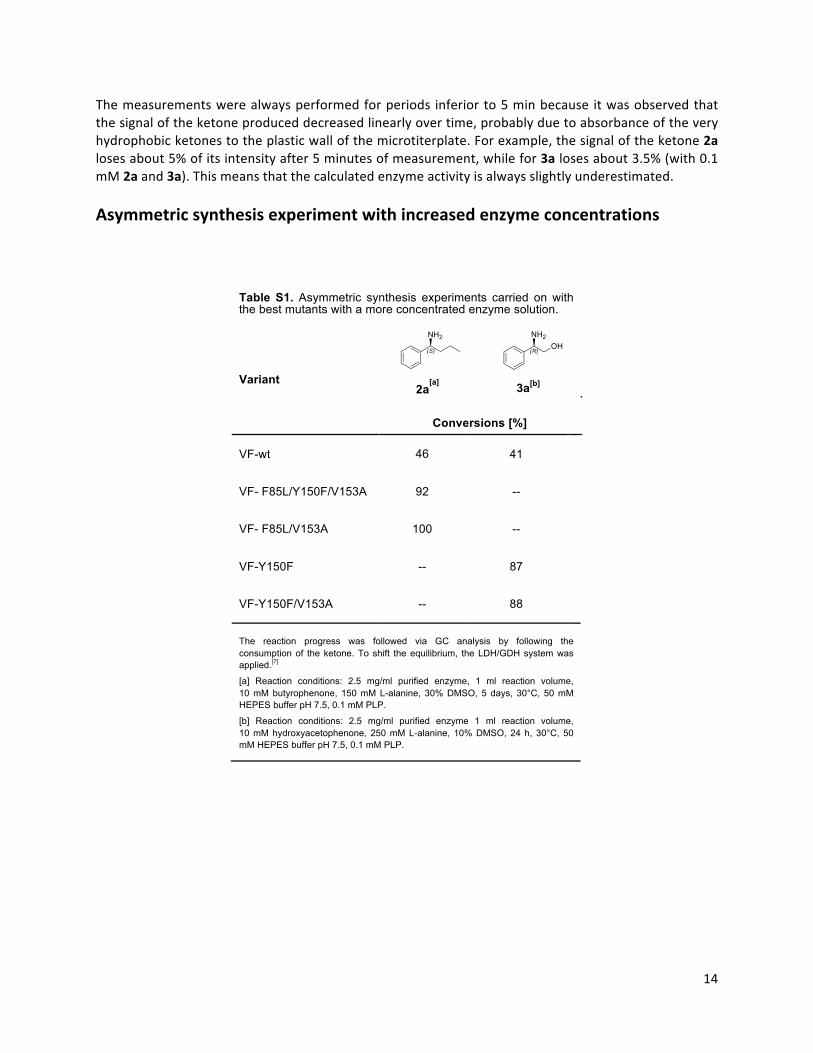
14#
#
The#measurements#were#always#performed#for#periods# inferior#to#5#min#because# it#was#observed#that#
the#signal#of#the#ketone#produced#decreased#linearly#over#time,#probably#due#to#absorbance#of#the#very#
hydrophobic#ketones#to#the#plastic#wall#of#the#microtiterplate.#For#example,#the#signal#of#the#ketone#2a#loses#about#5%#of#its#intensity#after#5#minutes#of#measurement,#while#for#3a#loses#about#3.5%#(with#0.1#mM#2a#and#3a).#This#means#that#the#calculated#enzyme#activity#is#always#slightly#underestimated.#
#
Asymmetric,synthesis,experiment,with,increased,enzyme,concentrations,
Table S1. Asymmetric synthesis experiments carried on with the best mutants with a more concentrated enzyme solution.
Variant
2a[a]
3a[b]
Conversions [%]
VF-wt 46 41
VF- F85L/Y150F/V153A 92 --
VF- F85L/V153A 100 --
VF-Y150F -- 87
VF-Y150F/V153A -- 88
The reaction progress was followed via GC analysis by following the consumption of the ketone. To shift the equilibrium, the LDH/GDH system was applied.[7]
[a] Reaction conditions: 2.5 mg/ml purified enzyme, 1 ml reaction volume, 10 mM butyrophenone, 150 mM L-alanine, 30% DMSO, 5 days, 30°C, 50 mM HEPES buffer pH 7.5, 0.1 mM PLP.
[b] Reaction conditions: 2.5 mg/ml purified enzyme 1 ml reaction volume, 10 mM hydroxyacetophenone, 250 mM L-alanine, 10% DMSO, 24 h, 30°C, 50 mM HEPES buffer pH 7.5, 0.1 mM PLP.
,
, ,
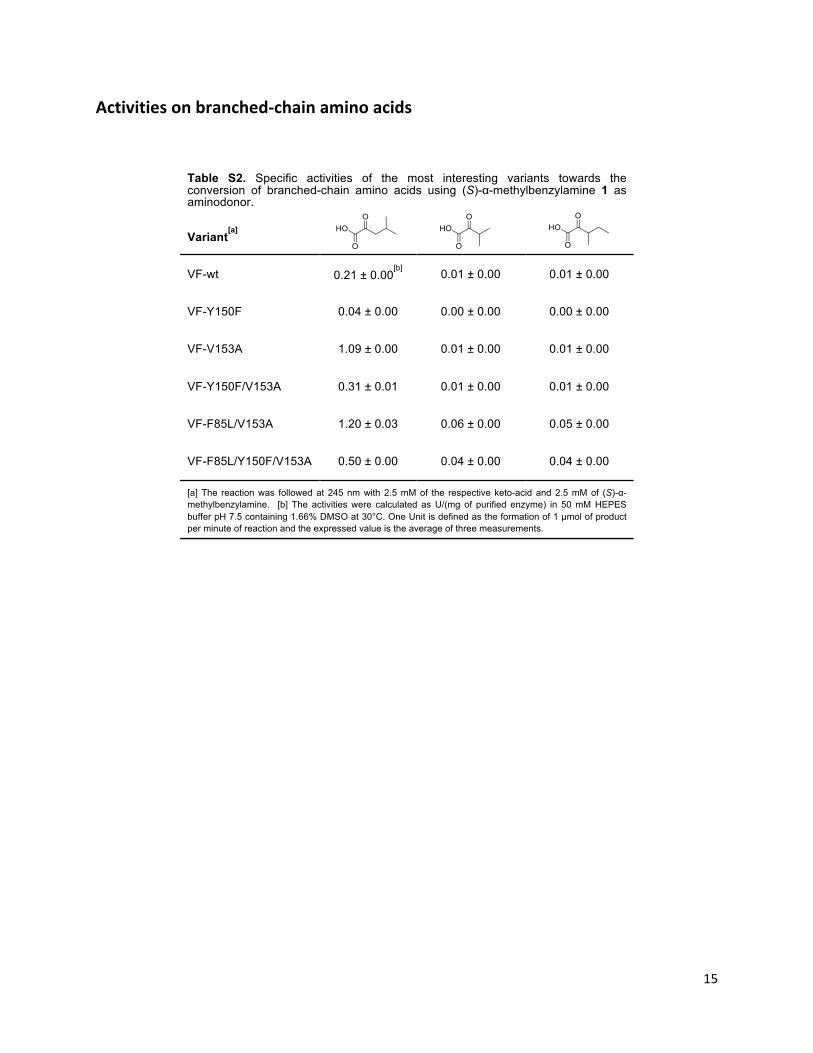
15#
#
Activities,on,branched?chain,amino,acids,
Table S2. Specific activities of the most interesting variants towards the conversion of branched-chain amino acids using (S)-α-methylbenzylamine 1 as aminodonor.
Variant[a]
VF-wt 0.21 ± 0.00[b]
0.01 ± 0.00 0.01 ± 0.00
VF-Y150F 0.04 ± 0.00 0.00 ± 0.00 0.00 ± 0.00
VF-V153A 1.09 ± 0.00 0.01 ± 0.00 0.01 ± 0.00
VF-Y150F/V153A 0.31 ± 0.01 0.01 ± 0.00 0.01 ± 0.00
VF-F85L/V153A 1.20 ± 0.03 0.06 ± 0.00 0.05 ± 0.00
VF-F85L/Y150F/V153A 0.50 ± 0.00 0.04 ± 0.00 0.04 ± 0.00
[a] The reaction was followed at 245 nm with 2.5 mM of the respective keto-acid and 2.5 mM of (S)-α-methylbenzylamine. [b] The activities were calculated as U/(mg of purified enzyme) in 50 mM HEPES buffer pH 7.5 containing 1.66% DMSO at 30°C. One Unit is defined as the formation of 1 µmol of product per minute of reaction and the expressed value is the average of three measurements.
#
# #

16#
#
References,#
[1]# S.#Schätzle,#M.#Höhne,#E.#Redestad,#K.#Robins,#U.#T.#Bornscheuer,#Anal.&Chem.&2009,#81,#8244B8248.#
[2]# a)#J.#S.#Shin,#B.#G.#Kim,#Biotechnol.&Bioeng.&1999,#65,#206B211;#b)#M.#S.#Humble,#K.#E.#Cassimjee,#
M.#Hakansson,#Y.#R.#Kimbung,#B.#Walse,#V.#Abedi,#H.#J.#Federsel,#P.#Berglund,#D.#T.#Logan,#FEBS&J.&2012,#279,#779B792.#
[3]# F.#SteffenBMunsberg,#C.#Vickers,#A.#Thontowi,#S.#Schätzle,#T.#Tumlirsch,#M.#Svedendahl#Humble,#
H.#Land,#P.#Berglund,#U.#T.#Bornscheuer,#M.#Höhne,#ChemCatChem&2013,#5,#150B153.#[4]# H.#E.#Gottlieb,#V.#Kotlyar,#A.#Nudelman,#J.&Org.&Chem.&1997,#62,#7512B7515.#[5]# H.#Friebolin,#Ein>&und&zweidimensionale&NMR>Spektroskopie,#Wiley,#2006.#[6]# S.# Schätzle,# F.# SteffenBMunsberg,# A.# Thontowi,#M.# Höhne,# K.# Robins,# U.# T.# Bornscheuer,# Adv.&
Synth.&Catal.&2011,#353,#2439B2445.#[7]# D.#Koszelewski,#I.#Lavandera,#D.#Clay,#D.#Rozzell,#W.#Kroutil,#Adv.&Synth.&Catal.&2008,#350,#2761B
2766.#
#
#


Articles 139
ARTICLE IV


Research review paper
Bioinformatic analysis of a PLP-dependent enzyme superfamily suitablefor biocatalytic applications
Fabian Steffen-Munsberg a,c, Clare Vickers a, Hannes Kohls a,b, Henrik Land c, Hendrik Mallin a, Alberto Nobili a,Lilly Skalden a, Tom van den Bergh d, Henk-Jan Joosten d, Per Berglund c,Matthias Höhne b,⁎, Uwe T. Bornscheuer a,⁎⁎a Dept. of Biotechnology & Enzyme Catalysis, Institute of Biochemistry, Greifswald University, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germanyb Protein Biochemistry, Institute of Biochemistry, Greifswald University, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germanyc KTH Royal Institute of Technology, School of Biotechnology, Division of Industrial Biotechnology, AlbaNova University Center, SE-106 91 Stockholm, Swedend Bio-Prodict, Nieuwe Marktstraat 54E, 6511 AA Nijmegen, The Netherlands
a b s t r a c ta r t i c l e i n f o
Available online 7 January 2015
Keywords:Protein functionAnnotationPLP-dependent enzymesBioinformaticsBiocatalysisEnzyme discoveryTransaminase
In this review we analyse structure/sequence–function relationships for the superfamily of PLP-dependentenzymes with special emphasis on class III transaminases. Amine transaminases are highly important for appli-cations in biocatalysis in the synthesis of chiral amines. In addition, other enzyme activities such as racemases ordecarboxylases are also discussed. The substrate scope and the ability to accept chemically different types ofsubstrates are shown to be reflected in conserved patterns of amino acids around the active site. These findingsare condensed in a sequence–function matrix, which facilitates annotation and identification of biocatalyticallyrelevant enzymes and protein engineering thereof.
© 2015 Elsevier Inc. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5671.1. Motivation and learning objectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5671.2. How the review is structured and where do I find what? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5671.3. The protein environment of PLP-dependent enzymes diversifies reaction specificity . . . . . . . . . . . . . . . . . . . . . . . . . . 5681.4. PLP-dependent biocatalysts as a short cut for multistep chemical syntheses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5701.5. The ‘predicting function from sequence’-problem: how analysis of sequence fingerprints of active site residues can provide functional insights 572
2. Analysing sequence–function relationships of PLP-dependent enzymes using 3DM — high quality alignments meet powerful analysis tools . . . . . 5742.1. The PLP fold type I and ornithine transaminase-like (OrnTL) 3DM databases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5742.2. Special features of the ornithine TA-like family exemplify the structural flexibility of PLP-fold type I . . . . . . . . . . . . . . . . . . . 5752.3. Reaction and substrate specificity determining residues revealed by correlated mutations analysis (CMA) . . . . . . . . . . . . . . . . 5762.4. The sequence–function matrix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 576
3. Activities represented in the ornithine TA-like database . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5773.1. ω-Amino acid:α-ketoglutarate transaminases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 578
3.1.1. Dual substrate recognition: the glutamate switch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 581
Biotechnology Advances 33 (2015) 566–604
Abbreviations:AA, amino acid; AAA, amino acid amide; 3AcOc, 3-acetyloctanal; AcOrn,N-acetylornithine; DAIB, D-aminoisobutyrate; ATA, amine transaminase; CoAβAA, coenzymeAβ-amino acid thioester; DABA,α,γ-diaminobutyrate; DAPA, 7,8-diaminopelargonic acid; DGD, 2,2-dialkylglycine decarboxylase; DTS, dethiobiotin synthase; fumB1, fumonisin B1; GABA,γ-aminobutyrate; glyox, glyoxylate; GSAM, glutamate-1-semialdehyde aminomutase; Lysε, lysine ε-amino group; HfumB1, hydrolysed fumonisin B1; Orn, ornithine; KAPA, 7-keto-8-aminopelargonic acid;αKG,α-ketoglutarate; OrnTL DB, ornithine transaminase-like database;βAla,β-alanine;βPhe,β-phenylalanine; PLP, pyridoxal 5'-phosphate; PUT, putrescine; pyr,pyruvate; SAM, S-adenosylmethionine; SuOrn,N-succinylornithine; TA, transaminase; tau, taurine⁎ Corresponding author. Tel: +49 3834 8622832; fax: +49 3834 86 794391.⁎⁎ Corresponding author. Tel.: +49 3834 864367; fax: +49 3834 86 794367.
E-mail addresses: [email protected] (M. Höhne), [email protected] (U.T. Bornscheuer).
http://dx.doi.org/10.1016/j.biotechadv.2014.12.0120734-9750/© 2015 Elsevier Inc. All rights reserved.
Contents lists available at ScienceDirect
Biotechnology Advances
j ourna l homepage: www.e lsev ie r .com/ locate /b iotechadv

3.1.2. Ornithine, acetylornithine and succinylornithine:α-ketoglutarate TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5833.1.3. Lysine-ε:α-ketoglutarate TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5833.1.4. γ-Aminobutyrate:α-ketoglutarate TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5843.1.5. Putrescine and cadaverine:α-ketoglutarate TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5863.1.6. 3-Acetyloctanal transaminase (PigE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5863.1.7. 2-amino-4-oxobutyrate transaminases (diaminobutyrate TAs) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 587
3.2. ω-Amino acid:pyruvate transaminases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5873.2.1. Dual substrate recognition: the flipping arginine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5883.2.2. Natural function of amine transaminases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5883.2.3. Discriminating high and low activity amine transaminases and βAla:pyr TAs . . . . . . . . . . . . . . . . . . . . . . . . . . 5883.2.4. Cadaverine/putrescine:pyruvate TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5903.2.5. Taurine:pyruvate TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 590
3.3. ω-Transaminases with unusual acceptor spectrum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5903.3.1. Dual substrate recognition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5913.3.2. β-Phenylalanine aminotransferases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5913.3.3. Acyl-CoA-β-TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5923.3.4. D-p-hydroxyphenylglycine:αKG TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5923.3.5. Diamino pelargonic acid transaminases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5923.3.6. Alanine:glyoxylate transaminase 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 593
3.4. Glutamate-1-semialdehyde transaminases (2,1-amino mutases) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5943.5. Decarboxylation dependent TAs: the 2,2-dialkylglycine decarboxylases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5953.6. α-H-amino acid amide/α-amino-ε-caprolactam racemases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5963.7. Isoleucine 2-epimerase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5973.8. Enzymes with unclear substrate recognition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 597
3.8.1. Neamine TAs, 2′-deamino-2′-hydroxyneamine and neomycin C TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5973.8.2. (Hydrolysed) fumonisin B1 TAs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5983.8.3. Phospholyases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5983.8.4. Multi-domain or non-enzymes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 598
4. Challenges for fingerprint-based sequence–function predictions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5994.1. Limitations of the active site amino acid fingerprint-based approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5994.2. 3DM database related issues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5994.3. The literature mining problem . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5994.4. The challenge to identify unknown specificities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 600
5. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 600Author contributions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 600Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 600Appendix A. Supplementary data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 601References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 601
1. Introduction
1.1. Motivation and learning objectives
What's the function of a certain gene or protein? Answering this ques-tion precisely is still a challenging, but very important task. An over-whelming number of potentially interesting enzymes for biocatalysis areavailable in public protein databases. However, this resource is only par-tially useful, because often the function and properties of enzymes cannotbe predicted reliably. This review exemplifies how structural knowledgeof enzymes and bioinformatics tools can be integrated to increase the pre-cision of function prediction. As an example, we analysed enzymes of thePLP fold type I superfamily with special focus on class III transaminases.
With the help of the review, the reader should be able to:
• understand the fascinating mechanisms and features that govern re-action and substrate specificity of PLP-fold type I enzymes,
• understand howbioinformatics tools and structural knowledge can becombined to study structure–function relationships,
• understand how the enzymes' activities are reflected in small aminoacid sequence fingerprints,
• take a class III transaminase amino acid sequence and easily assign themost probable function (out of 28 different known functions),
• apply this knowledge to guide experiments for the discovery of novelenzymes,
• apply the guidelines and tools covered in this review to analyse otherenzyme superfamilies
1.2. How the review is structured and where do I find what?
Some basic introduction about the diversity of PLP chemistry, PLP-dependent enzyme classification and the biotechnological relevance oftransaminases is given in the introductory sections 1.3 and 1.4. The sec-tion 1.5 introduces the active site fingerprint concept, which forms thebasis of our structure–function relationship analysis. The most impor-tant terms and concepts of sections 1.3, 1.4 and 1.5, which are usedthroughout the review, are summarised in Boxes 1 and 2. Section 2 con-denses all information from literature and our bioinformatic analysis:first, in section 2.1 we provide a brief description of the algorithms be-hind 3DM, the bioinformatics platform used for our analyses. Generalstructure and sequence features of the class III transaminase familyand specificity determining residues are analysed in sections 2.2 and2.3. Section 2.4 presents the sequence–activity matrix, the central partof our analysis. It shows a correlation of the function of different pro-teinswith amino acid patterns of a few active site residues (fingerprint).The most important structural details behind these analyses are pre-sented in section 3. In this section we aim to illustrate the artful mech-anisms and active site adaptations that facilitated the development of28 different enzyme activities. On the one hand, specificity is createdby providing a binding pocket that is complementary to the substratein shape and polarity and provides electrostatic interactions. On theother hand, different mechanisms render the active site very flexibleand allow two or more chemically different substrates to bind in thesame pocket (so called dual substrate recognition). An overview of sec-tion 3 is given by Table 3, which contains structures of substrates and
567F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604
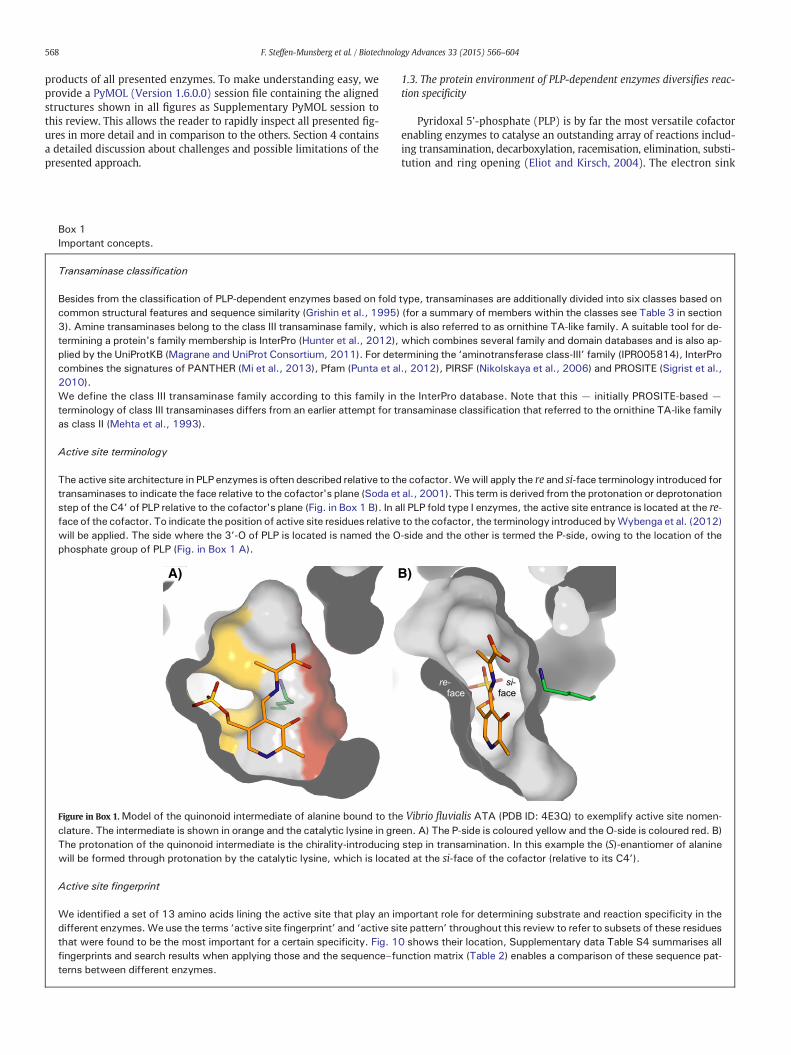
products of all presented enzymes. To make understanding easy, weprovide a PyMOL (Version 1.6.0.0) session file containing the alignedstructures shown in all figures as Supplementary PyMOL session tothis review. This allows the reader to rapidly inspect all presented fig-ures in more detail and in comparison to the others. Section 4 containsa detailed discussion about challenges and possible limitations of thepresented approach.
1.3. The protein environment of PLP-dependent enzymes diversifies reac-tion specificity
Pyridoxal 5'-phosphate (PLP) is by far the most versatile cofactorenabling enzymes to catalyse an outstanding array of reactions includ-ing transamination, decarboxylation, racemisation, elimination, substi-tution and ring opening (Eliot and Kirsch, 2004). The electron sink
Box 1Important concepts.
Transaminase classification
Besides from the classification of PLP-dependent enzymes based on fold type, transaminases are additionally divided into six classes based oncommon structural features and sequence similarity (Grishin et al., 1995) (for a summary of members within the classes see Table 3 in section3). Amine transaminases belong to the class III transaminase family, which is also referred to as ornithine TA-like family. A suitable tool for de-termining a protein's family membership is InterPro (Hunter et al., 2012), which combines several family and domain databases and is also ap-plied by the UniProtKB (Magrane and UniProt Consortium, 2011). For determining the ‘aminotransferase class-III’ family (IPR005814), InterProcombines the signatures of PANTHER (Mi et al., 2013), Pfam (Punta et al., 2012), PIRSF (Nikolskaya et al., 2006) and PROSITE (Sigrist et al.,2010).We define the class III transaminase family according to this family in the InterPro database. Note that this — initially PROSITE-based —
terminology of class III transaminases differs from an earlier attempt for transaminase classification that referred to the ornithine TA-like familyas class II (Mehta et al., 1993).
Active site terminology
The active site architecture in PLP enzymes is often described relative to the cofactor.Wewill apply the re and si-face terminology introduced fortransaminases to indicate the face relative to the cofactor's plane (Soda et al., 2001). This term is derived from the protonation or deprotonationstep of the C4’ of PLP relative to the cofactor's plane (Fig. in Box 1 B). In all PLP fold type I enzymes, the active site entrance is located at the re-face of the cofactor. To indicate the position of active site residues relative to the cofactor, the terminology introduced byWybenga et al. (2012)will be applied. The side where the 3’-O of PLP is located is named the O-side and the other is termed the P-side, owing to the location of thephosphate group of PLP (Fig. in Box 1 A).
A) B)
Figure in Box 1.Model of the quinonoid intermediate of alanine bound to the Vibrio fluvialis ATA (PDB ID: 4E3Q) to exemplify active site nomen-clature. The intermediate is shown in orange and the catalytic lysine in green. A) The P-side is coloured yellow and the O-side is coloured red. B)The protonation of the quinonoid intermediate is the chirality-introducing step in transamination. In this example the (S)-enantiomer of alaninewill be formed through protonation by the catalytic lysine, which is located at the si-face of the cofactor (relative to its C4’).
Active site fingerprint
We identified a set of 13 amino acids lining the active site that play an important role for determining substrate and reaction specificity in thedifferent enzymes.Weuse the terms ‘active site fingerprint’ and ‘active site pattern’ throughout this review to refer to subsets of these residuesthat were found to be the most important for a certain specificity. Fig. 10 shows their location, Supplementary data Table S4 summarises allfingerprints and search results when applying those and the sequence–function matrix (Table 2) enables a comparison of these sequence pat-terns between different enzymes.
568 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

nature of the PLP cofactor allows for this vast variety of chemistry. It isthe enzyme scaffold, however, which elegantly determines whichreaction pathway is followed. Toney (2011) recently reviewed the sub-ject of reaction specificity and summarised three main themes.1) Stereoelectronic control: the enzyme forces the substrate/aldimineintermediate to adopt a certain conformation. The bond to be cleavedhas to be aligned parallel to the π-orbitals (perpendicular to the PLPplane) for a π–σ-orbital overlap, facilitating bond cleavage and reso-nance stabilisation of the developing charge by the conjugatedπ-electron system of PLP. This principle that explains the preferencefor cleavage of one of the Cα substituents over another was initiallypublished by Dunathan (1966) and is therefore known as Dunathanprinciple (see Fig. 1). 2) The electrophilic strength of the Schiff base—pyridine ring π-electron system: this property governs the capabilityof negative charge delocalisation. Different reactions, i.e. racemisationor transamination, require a lower or higher degree of negative chargestabilisation, respectively. Thus, different enzymes affect the electro-philic strength of the aldimine intermediate by controlling the proton-ation state of the pyridoxyl N atom. Its protonation increases thecapability of resonance stabilisation and a quinonoid intermediate canbe formed. 3) Catalytic side chain placements: to control the outcome
of the reaction, the enzyme provides catalytic functional groupspossessing a certain positional flexibility, which promotes the desiredbond formation(s). Enzymes achieve the above-mentioned tasks by anartful design of their active sites. For a deeper understanding of thesefascinating details we highly recommend to read the review by Toney(2011).
Besides controlling reaction specificity, a second importantissue is controlling substrate specificity and enantioselectivity.For many PLP-dependent enzymes, this is a complex task, aschemically different substrates have to be accepted (e.g. acidicand aromatic amino acids). This phenomenon of multiple sub-strate recognition is discussed in detail in sections 3.1.1, 3.2.1and 3.3.1.
Altogether, the majority of PLP-dependent enzymes catalysing thisplethora of reactions have been evolved in a very small number of dif-ferent tertiary structures: only seven different fold types of PLP-dependent enzymes were discovered until now (Supplementary dataTable S1) (Percudani and Peracchi, 2009).
The fold type I, also referred to as the ‘aspartate aminotransferasesuperfamily’, combines the highest quantity and diversity of mem-bers, compared to the other fold types (Schneider et al., 2000). It
Box 2Definitions used in the 3DM-based creation and analysis of the superfamily database.
3DM-database3DM databases comprise sequence alignments based on a superfamily wide structure alignment and thus offer a way of reliably comparing allsequences independent from low sequence similarity. For this review, a large PLP-fold type I database and a small ornithine TA-like database(OrnTL DB) were built. The latter offers a larger core (for details see Table 1, and section 2.1).
SubfamilyBy the alignment of all available sequences to the structures of the initial structural alignment, so-called subfamilies are formed as the smallestbuilding blocks of the superfamily alignment. Each subfamily is formed using one structure of the structural alignment as ‘template structure’and all sequences that could be aligned to it within a certain cut-off ( section 2.1, steps 4–7). For the OrnTL DB the template structure's PDBcode was applied to name each subfamily. Note: one subfamily can contain several proteins for which a structure is available, but these are se-quentially so similar (see cut-off step 4, section 2.1) that they are not used as a template for a new subfamily; it is also possible that a subfamilycomprises enzymes with different activities. As long as there is a structure in the initial structure alignment with sufficient similarity to a se-quence of interest, this sequence can be compared to the whole database.
Core and variable regionsTo allow for facilitated comparisons and statistics, 3DM introduces the concepts of core and variable regions. The initial structure alignment isevaluated to find the common structural ‘core’ that is conserved in all structures available in the database. Positions in these regions are calledcore positionswhile all structurally non-aligned areas are referred to as variable regions or positions. The larger the database (i.e. themore struc-tural variation), the smaller the core, as local differences in the structures caused by mutations, insertions or deletions prevent a meaningfulalignment.
3D numberA unified numbering scheme, called 3D numbers, is created for all sequences and structures in the 3DM database by renumbering them accord-ing to the core positions: all structural equivalent residues get the same number. This enables easy comparison of the amino acid distribution ofall proteins in the database. All residue numbers used in this review are the 3D numbers of the OrnTL DB if not stated otherwise. Residue num-bers within variable regions are given in italics in combination with the accession number of the enzyme to which the numbers refer.
Correlated mutation analysis (CMA)Structural or functionally important amino acid positions are often not conserved, but mutations at these positions are made possible by addi-tional mutation(s) within the protein. Therefore several positions, often randomly scattered over a protein's sequence, are commonly mutatedtogether during evolution. Compared to conserved residues, these networks of correlated mutations are much more difficult to detect in a mul-tiple sequence alignment by visual inspection, but they can be identified and visualised by CMA.Hence, theCMAnetwork analysis tool integrat-ed in the 3DM suite, called CorNet (publication in progress, free web based version available from www.3dm.bio-prodict.nl/Comulator), is apowerful tool to reveal structural or functional important residues. The CMA algorithm, called Comulator, behind the CorNet tool was publishedpreviously (Kuipers et al., 2009).
569F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604
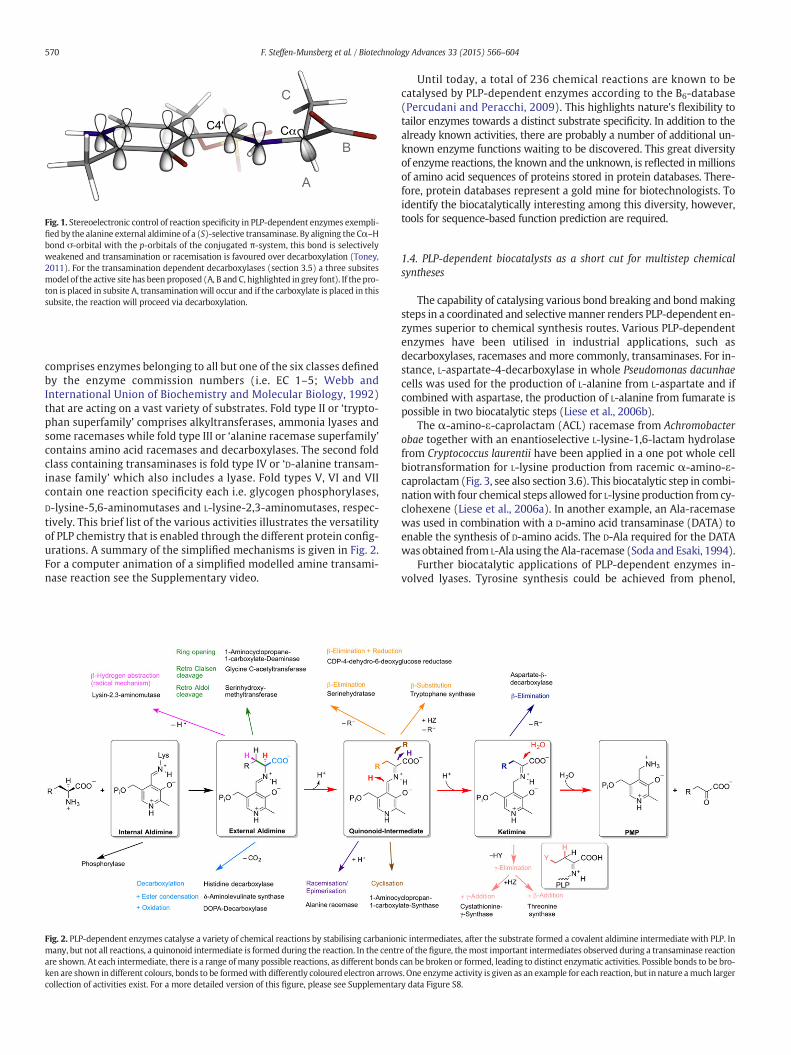
comprises enzymes belonging to all but one of the six classes definedby the enzyme commission numbers (i.e. EC 1–5; Webb andInternational Union of Biochemistry and Molecular Biology, 1992)that are acting on a vast variety of substrates. Fold type II or ‘trypto-phan superfamily’ comprises alkyltransferases, ammonia lyases andsome racemases while fold type III or ‘alanine racemase superfamily’contains amino acid racemases and decarboxylases. The second foldclass containing transaminases is fold type IV or ‘D-alanine transam-inase family’ which also includes a lyase. Fold types V, VI and VIIcontain one reaction specificity each i.e. glycogen phosphorylases,D-lysine-5,6-aminomutases and L-lysine-2,3-aminomutases, respec-tively. This brief list of the various activities illustrates the versatilityof PLP chemistry that is enabled through the different protein config-urations. A summary of the simplified mechanisms is given in Fig. 2.For a computer animation of a simplified modelled amine transami-nase reaction see the Supplementary video.
Until today, a total of 236 chemical reactions are known to becatalysed by PLP-dependent enzymes according to the B6-database(Percudani and Peracchi, 2009). This highlights nature's flexibility totailor enzymes towards a distinct substrate specificity. In addition to thealready known activities, there are probably a number of additional un-known enzyme functions waiting to be discovered. This great diversityof enzyme reactions, the known and the unknown, is reflected inmillionsof amino acid sequences of proteins stored in protein databases. There-fore, protein databases represent a gold mine for biotechnologists. Toidentify the biocatalytically interesting among this diversity, however,tools for sequence-based function prediction are required.
1.4. PLP-dependent biocatalysts as a short cut for multistep chemicalsyntheses
The capability of catalysing various bond breaking and bondmakingsteps in a coordinated and selectivemanner renders PLP-dependent en-zymes superior to chemical synthesis routes. Various PLP-dependentenzymes have been utilised in industrial applications, such asdecarboxylases, racemases and more commonly, transaminases. For in-stance, L-aspartate-4-decarboxylase in whole Pseudomonas dacunhaecells was used for the production of L-alanine from L-aspartate and ifcombined with aspartase, the production of L-alanine from fumarate ispossible in two biocatalytic steps (Liese et al., 2006b).
The α-amino-ε-caprolactam (ACL) racemase from Achromobacterobae together with an enantioselective L-lysine-1,6-lactam hydrolasefrom Cryptococcus laurentii have been applied in a one pot whole cellbiotransformation for L-lysine production from racemic α-amino-ε-caprolactam (Fig. 3, see also section 3.6). This biocatalytic step in combi-nationwith four chemical steps allowed for L-lysine production from cy-clohexene (Liese et al., 2006a). In another example, an Ala-racemasewas used in combination with a D-amino acid transaminase (DATA) toenable the synthesis of D-amino acids. The D-Ala required for the DATAwas obtained from L-Ala using the Ala-racemase (Soda and Esaki, 1994).
Further biocatalytic applications of PLP-dependent enzymes in-volved lyases. Tyrosine synthesis could be achieved from phenol,
Fig. 1. Stereoelectronic control of reaction specificity in PLP-dependent enzymes exempli-fied by the alanine external aldimine of a (S)-selective transaminase. By aligning the Cα–Hbond σ-orbital with the p-orbitals of the conjugated π-system, this bond is selectivelyweakened and transamination or racemisation is favoured over decarboxylation (Toney,2011). For the transamination dependent decarboxylases (section 3.5) a three subsitesmodel of the active site has been proposed (A, B and C, highlighted in grey font). If the pro-ton is placed in subsite A, transamination will occur and if the carboxylate is placed in thissubsite, the reaction will proceed via decarboxylation.
Fig. 2. PLP-dependent enzymes catalyse a variety of chemical reactions by stabilising carbanionic intermediates, after the substrate formed a covalent aldimine intermediate with PLP. Inmany, but not all reactions, a quinonoid intermediate is formed during the reaction. In the centre of the figure, themost important intermediates observed during a transaminase reactionare shown. At each intermediate, there is a range ofmany possible reactions, as different bonds can be broken or formed, leading to distinct enzymatic activities. Possible bonds to be bro-ken are shown in different colours, bonds to be formedwith differently coloured electron arrows. One enzyme activity is given as an example for each reaction, but in nature amuch largercollection of activities exist. For a more detailed version of this figure, please see Supplementary data Figure S8.
570 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

pyruvate and ammoniawith tyrosine phenol lyases from various organ-isms (Lütke-Eversloh et al., 2007). If a tyrosine phenol lyase is combinedwith the PLP-dependent tyrosine decarboxylase in a second step, dopa-mine could be synthesised from catechol, pyruvate and ammonia (Leeet al., 1999). Furthermore, threonine aldolases have been applied inasymmetric aldol reactions to form α,α-dialkyl-α-amino acids fromAla, Cys and Ser and various acceptor aldehydes (Fesko et al., 2010).
Currently, the most useful and widely applied PLP-dependent en-zymes are transaminases (TA) as they can be used in biocatalytic asym-metric synthesis of amino acids and amines. Compared to organo- ormetallo-catalysis, transaminases are superior in step efficiency as theycatalyse several steps in a one-pot reaction: 1) reaction of the ketonewith a N-source (pyridoxamine 5'-phosphate, PMP) to form the imine,2) reduction to yield a protected amine, and 3) liberation of the freeamine by cleavage of theN-protecting group (PLP) (see Fig. 4 for a com-parison to the chemical routes).1 The enzymatic reaction substantiallyhelps to increase process efficiency as intermediate work-up proce-dures as well as toxic heavy metals can be avoided. This has been nicely
demonstrated for the large scale synthesis of the antidiabetic drugSitagliptin, whereMerck & Co (USA) initially used an asymmetric trans-fer hydrogenation process catalysed by a rhodium-complex, which waslater replaced by a transaminase-catalysed route, in which simplyisopropylamine could be used as amino donor. The highly active andstable (R)-selective amine transaminasewas developed in collaborationbetween Merck & Co with the company Codexis (USA) as published bySavile et al. (2010). This improved process substantially reduced thewaste and E-factor of the process as summarised in a highlight article(Desai, 2011). Both companies were awarded the ‘Presidential GreenChemistry Award USA’ for this green chemistry route. For detailsabout the application of transaminases in biocatalysis, readers are re-ferred to recent reviews (Berglund et al., 2012; Höhne andBornscheuer, 2009, 2012; Kohls et al., 2014; Kroutil et al., 2013; Rudatet al., 2012).
In nature, two types of PLP-dependent transaminases have beendiscovered, according to the type of substrate that is converted:α-transaminases (α-TAs) and ω-transaminases (ω-TAs). Whereasα-TAs (the majority of TAs) exclusively convert α-amino and α-ketoacids, ω-TA also accept substrates having a distal carboxylic acid groupinstead of a carboxylate function in α-position. The term ω-TA is usedto summarise a very heterogeneous group of activities (see sections3.1, 3.2 and 3.3). Two subgroups of ω-TAs studied for biocatalytic
Fig. 4. Transaminases are useful for the manufacture of amines. A) TA allow for amine synthesis in a single step, compared to traditional metal-catalysed chemical procedures likeB) enamide reduction or C) imine reduction (Nugent and El-Shazly, 2010).
1 Interestingly, the intermediates and steps in the enzymatic reaction resemble those ofthe chemical synthesis, but most transaminase research overlooks these details and sim-ply considers this as amino group transfer — matching the classification in the enzymecommission class (EC) 2, transferases.
Fig. 3. Biocatalytic process for L-lysine production from racemicα-amino-ε-caprolactam. As both enzymes have a comparable pH optimum, itwas possible to run thewhole cell process inone reactor.
571F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

approaches are β-TAs (Rudat et al., 2012) and amine TAs (ATAs). Thelatter became particularly popular during the last decade (see Fig. 5and legend for details).
1.5. The ‘predicting function from sequence’-problem: how analysis of se-quence fingerprints of active site residues can provide functional insights
The ability to predict an enzyme's function based on its amino acidsequence is central for a variety of scientific disciplines:
• Physiology and system biology aim to understand an organism's(or habitat's) metabolic capability based on (meta)genomic data.With this capability they could predict ecophysiological functions ordiscover novel metabolic pathways.
• For biotechnological applications, researchers aim to identify and usespecific enzymes to catalyse a given reaction. Besides the targetedidentification of enzymes, discovering novel enzymes is important toexpand the enzymatic toolbox for biocatalysis.
• Protein engineers wish to apply their understanding of structure–function relationships to the reverse direction:which (also bymodifi-cation of an) amino acid sequence will generate the desired activity?
An important aspect complicating sequence–function prediction ispromiscuity, which is a consequence of evolution. In the case of PLP-dependent enzymes, Christen and Mehta (2001) proposed that PLPbinding developed first, while evolution of reaction specificity precededsubstrate specificity. There are often less genes encoding PLP-dependent enzymes than PLP-catalysed reactions needed in anorganism's metabolism (Percudani and Peracchi, 2003). Therefore, it isnot surprising that both reaction and especially substrate promiscuity(Bornscheuer and Kazlauskas, 2004; Hult and Berglund, 2007; Humbleand Berglund, 2011) occur across all fold types of PLP-dependent en-zymes. The fact that enzymes of very low sequence similarity can havesimilar specificities, while closely related enzymes will not accept thesame substrates, makes reliable prediction of function with respect touncharacterised enzymes challenging (Percudani and Peracchi, 2003).
These difficulties result in error prone functional annotations of se-quences of PLP-dependent enzymes. While reaction specificity predic-tion is achieved with a relatively high success rate, the prediction ofthe detailed function at the substrate specificity level is complicated.For example, ATAs belong to a subfamily of PLP fold type I that isreferred to as class III transaminase family (Rausch et al., 2013) (seeBox 1). This enzyme class harbours approximately 28 different enzymeactivities (see Table 3). If one was to BLASTP (Altschul et al., 1997) thesequence of the ATA from Ruegeria pomeroyi (PDB ID: 3HMU)(Steffen-Munsberg et al., 2013b) against the non-redundant protein se-quences database with pre-set parameters, one will obtain results thatdo not allow any conclusion on its substrate specificity. Within thefirst 100 results (all have 61–100% sequence identity to 3HMU) foursequences are annotated as ‘class III aminotransferases’ (as is 3HMU it-self), three are termed ‘adenosylmethionine-8-amino-7-oxononanoateaminotransferase’ and the remaining 93 are referred to as ‘aminotrans-ferase’, from which it is impossible to predict 3HMU's substrate scopefor small amino acids, such as pyruvate andγ-aminobutyrate or amines.This example highlights that sequence similarity might not be sufficientfor a protein's function prediction.
The challenge of closing the gap between sequence and function in-formation has been subject to extensive research over recent years, andis addressed by bioinformatic annotation solutions (Radivojac et al.,2013), solving protein structures (Jaskolski et al., 2014) and biochemicalcharacterisation of single enzymes and combinations thereof. For in-stance, the Enzyme Function Initiative characterises proteins of un-known function structurally and functionally to systematically closeknowledge gaps to enable further predictions (Gerlt et al., 2011), whileother groups predicted function by docking of metabolites to homologymodels and the evaluation of genetic contexts (Zhao et al., 2013). The
additional sequence–function connections that are gained by these stud-ies can then be applied for further predictions and annotations.
Even though computational protein function prediction is steadilyadvancing with algorithms, including literature mining and machinelearning, there is still a need for improvements: in the Critical Assess-ment of Functional Annotation (CAFA) experiment, several methodshave recently been evaluated (Radivojac et al., 2013). Unfortunately,many of thesemethods are not yet available for standard large-scale an-notation projects. Radivojac et al. (2013) conclude that there is a needfor improving the availability of stand-alone tools to allow the predic-tion of an enzyme's function independently from the slow updatingrate of sequence databases according to the recent advances in annota-tion technology.
In our past research, we have focussed on investigating active sitedesign/amino acid composition for analysis and prediction of enzymefunction (Gand et al., 2014; Höhne et al., 2010; Steffen-Munsberget al., 2013b). The advantage we see in this approach is that predictionsare guided from hypotheses; it can be easily performed by all re-searchers and it deepens the understanding of structure–function rela-tionships of a given superfamily of proteins. If structural information forthe enzymes of interest is available, the structural alignment of only ac-tive site residues provides a powerful tool for sequence independentfunction prediction in evolutionary distant enzymes. For instance ene-reductases that have completely different sequences and structuralfolds, but similar active site geometry, were recently shown to possess
Fig. 5. Common nomenclature of transaminases based on the distance of the transferredamino group from the carboxylic function. A) α-Transaminases (α-TA) catalyse theconversion of α-amino acids to the corresponding α-keto acid and vice versa. Note that(S)- and (R)-selectiveα-TA occur in nature: typical examples are aspartate transaminases(Asp:α-ketoglutarate TA) and D-amino acid transaminases (DATA). B) ω-Transaminases(ω-TA) transfer amino groups that are more distant from a carboxylic group (e.g. in γ, δor ε position). Note that β-TAs are a subgroup of ω-TAs: these enzymes transaminateβ-amino groups with respect to the acid function (n = 0). A typical example for β-TAsare β-phenylalanine TAs. Enzymes converting the ω-amino group of ω-amino acids(e.g. γ-aminobutyrate (GABA), n = 1, R = H) and α,ω-diamino acids (e.g. Lys, n = 3,R = NH2) are both referred to as ω-TAs. A subgroup of ω-TAs, the amine transaminases(ATAs), also allow for the conversion of chiral amines independently from the presenceof carboxylic groups in the substrate as exemplified in C) these ATAs are very useful forbiocatalysis as they can be applied for asymmetric chiral amine synthesis from the corre-sponding prochiral ketones if applied in reverse direction. Owing to their ability to convertω-amino acids aswell, the term ‘ω-TA’ has been used equally to the term ‘ATA’ in biocatal-ysis focussing publications. This terminology is misleading because there are severalω-TAs known that do not convert any amine substrate. As the activity towards amines isthe biocatalytically most relevant one, we prefer to term these enzymes ATAs to empha-sise their independency of carboxylic groups in the substrate and not to confuse withother ωAA converting enzymes, throughout this review. Both, (R)- and (S)-selective ATAhave been found in nature.
572 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

comparable activity (Steinkellner et al., 2014). Sequence independentactive site alignments can also be applied for functional comparisonsof structurally unrelated superfamilies and thereby reveal reactionspecificity determining features as recently demonstrated for the PLP
fold types I–IV (Catazaro et al., 2014). However, these approaches re-quire detailed structural data for the target enzymes, which is oftennot available.
Another, inmany casesmore applicable, strategy is to investigate se-quence–function relationships employing structural based sequencealignments. This approach was recently applied to identify selectivitydetermining positions in P450 monooxygenases and thiamine diphos-phate dependent decarboxylases, thereby enabling targeted mutagene-sis to improve the enzymes' selectivities (Pleiss, 2014). Furthermore,discrimination between dehydrogenase and oxidase reaction specificitywas shown to bedetermined by the presence of Ala or Gly in a single po-sition (Leferink et al., 2009) and oxaloacetate hydrolyase activity withinthe lyase/PEP mutase enzyme superfamily can be predicted based onthe existence of a single Ser in the active site (Joosten et al., 2008).
The importance of tools to predict an enzyme's function if only thesequences have been deposited in public databases but no biochemicalcharacterisation or evidence of their function is available can be exem-plified by an example from our research: until 2010 (R)-selective ATAactivity was only found in two wild type strains, but the sequences ofthe responsible enzymes had been unknown. By analysing the determi-nants for substrate and reaction specificity in PLP fold type IV enzymes,we identified sequence motifs that allowed for the rapid annotationof all known enzymes of this fold type (i.e. branched chain amino acidtransaminases (BCAT), D-amino acid transaminases (DATA) and4-amino-4-deoxychorismate lyases). Then, we predicted keymutationsthat should facilitate amine conversion in the scaffold of a BCAT toachieve patterns for (R)-ATA prediction (Höhne et al., 2010). Throughthese fingerprints we identified 17 (R)-ATAs that had been depositedin the sequence databases, butwithmisleading annotations. Further ex-periments revealed that these new (R)-ATAs have a great potential inasymmetric amine synthesis (Schätzle et al., 2011). This example indi-cates how understanding of substrate and reaction specificity-determining residues can result in the identification of new versatilebiocatalysts.
Whereas information about (R)-ATA (found in PLP fold type IV) wasscarce until our study, (S)-selective ATAs (belonging to fold type I) havebeen explored for biocatalytic applications for more than 15 years (Shinand Kim, 1997), the first sequence being described in 2003 (Shin et al.,2003). However, the methods for (S)-selective amine transaminasesdiscovery had been restricted to enrichment cultures (Shin et al.,2003) and sequence homology searches (E.S Park et al., 2010) until2011 when Park et al. proposed substrate specificity determining resi-dues based on homologymodels (E.S. Park et al., 2011). In the followingyears, solved crystal structures (Humble et al., 2012; Midelfort et al.,2013; Rausch et al., 2013; Sayer et al., 2013) displayed the spatial ar-rangement of these residues in the active site. Interestingly, four crystalstructures of (S)-ATAs were deposited in the database since 2009, butnot recognised as ATAs because of the lacking experimental data forthese enzymes. Their detailed characterisation combined with a muta-genesis study (Steffen-Munsberg et al., 2013a) unravelled the detailedmechanism of dual substrate specificity and factors affecting catalyticefficiency towards amines. All these investigations strongly focused on(S)-ATAswithout discussing the important residues in related enzymes,
A) Substrate 1 bound by X100, Y200, Z300
B) Substrate 1 bound by X150, Y250, Z300
C) Substrate 2 bound by X100, Y250, Z300
D) Sequence alignment guides annotation
Specificity determining Catalytic
amino acids residue
Probable
Substrate
A100
Z400
C300
B200
A150
Z400
C300
B250
D100
Z400
C300
E250
Fig. 6. Active site patterns can be used to predict enzyme function. Substrate and reactionspecificity of enzymes are governed by the presence of key residues in the active site,which can be detected in a multiple sequence alignment. A) & B) The same substrate 1can be converted in enzymes by placing similar residues, but at different positions in theamino acid sequence. Because of the flexibility of the amino acid side chains, importantfunctional groups might be in a similar geometric position. Therefore, important residuesare not conserved in all cases. C) Chemically different residues realise conversion of a dif-ferent substrate 2. D) In amultiple sequence alignment, sequencesmatching thepattern ofEnzyme A) or B) can be identified. Sequences 6–8 have different pattern, and thus mighthave different substrate specificities. Sequence 9 differs in the catalytic residue. Thereforeit is either not catalytically competent, or catalyses a different reaction. Conserved aminoacids are shown in different colours.
573F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

which would be required to broaden the insight into mechanisms ofsubstrate recognition and catalysis within the whole PLP fold type I.
From these resultswe hypothesise that reaction specificity aswell assubstrate specificity is mainly reflected by the presence of certain activesite residues, which interact with the substrate or the cofactor duringthe reaction. This pattern of active site residues also referred to as ‘activesite fingerprint’ can be used to assign a function from a simple align-ment, if the sequence matches the known pattern (Fig. 6). It is impor-tant to keep in mind that the situation in nature is more complex:there might exist more than one solution to realise the same substratespecificity (as shown in Fig. 6A, B and D, sequences 1–4), and especiallyPLP-dependent enzymes bind more than one substrate in the same ac-tive site (for dual substrate recognition see sections 3.1.1, 3.2.1 and3.3.1).
In the following, we analyse important residues of well-describedenzymes of the class III transaminases of PLP-fold type I. Together
with an analysis of respective multiple sequence alignments, we showthat activities can indeed correlate with a few active site residues,which enables a more detailed annotation compared to standard toolswhich are available online. Furthermore, we elucidate, how conservedthese sequence motifs are, and which fraction of sequences cannot beannotated by this approach at themoment and hencemight be interest-ing for further research.
2. Analysing sequence–function relationships of PLP-dependentenzymes using 3DM — high quality alignments meet powerfulanalysis tools
2.1. The PLP fold type I and ornithine transaminase-like (OrnTL) 3DMdatabases
To identify and compare residues that govern substrate and reactionspecificity in an enzyme superfamily, multiple structure and sequencealignments have to be computed to generate an overall alignment. Asthey are the basis of all further analysis steps, their quality is of extraor-dinary importance.
The commercial software 3DM, a protein superfamily analysis suite,employs highly sophisticated algorithms to ensure that the alignment isreliable, and at the same time integrates tools that allow one to generatehypothesis of relevant structure/sequence–function relationships.Many different data types are collected for all the proteins of a super-family in a 3DM system by extensive data collection tools (i.e. all
AcOrn:αKG TAs
DGD
DAPA TAs
ATAs
βAla:pyr TAs
GABA:pyr TAs
Tau:pyr TAs
Ala:glyox TAs 2
GSAM
racemases
Orn:αKG TAs
Lysε:αKG TAs
GABA:αKG TAs
Not characterised
Phe:αβ KG/pyr TAs
AAAα
1.0
Fig. 7. Phylogenetic tree comprising all 12,956 sequences of the OrnTL DB. Colouring highlights the characterised enzymes. For each group of substrate/reaction specificity the whole in-duced network is highlighted. Colouring: grey: not characterised; yellow: GABA:αKG TAs; red: Lysε:αKG TAs; light orange: Orn:αKG TAs; dark orange: AcOrn/SuOrn:αKG TAs; darkgreen: DGD; light blue: DAPA TAs; pink: ATAs, βAla:pyr TAs and GABA:pyr TAs; black: Tau:pyr TAs; brown: Ala:glyox TAs 2; dark blue: αAAA racemases; light green: GSAM; violet:βPhe: αKG/pyr TAs. Abbreviations are explained in Table 3. The unrooted tree was calculated based on the core alignment of the OrnTL DB using FastTree 2.1.3 (Price et al., 2010) andformatted using Dendroscope (Version 3.2.10) (Huson and Scornavacca, 2012).
Table 1Comparison of the size of the PLP fold type I database and the OrnTL database.
Amount of PLP fold type I OrnTL
Crystal structures found initially 717 170Crystal structures in final alignment 406 170Subfamilies 94 21Sequences found initially 120,870 31,000Sequences aligned 42,080 12,956Core residues 290 379
574 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

available structures are examined and full-text articles are text-minedto extract mutation studies from the literature; Kuipers et al., 2010a).Besides the description of the 3DM database on PLP-dependent en-zymes and the sub-database on ornithine TA like (OrnTL) enzymes inthis section, the most important concepts of 3DM relevant for this re-view are briefly summarised in Box 2.
The PLP fold type I 3DM database was generated with the humanornithine:α-ketoglutarate transaminase (Orn:αKGTA) crystal structureas the starting template for the structural alignment. This enzyme waschosen as the template because the biocatalytically most relevant en-zymes of this fold type ((S)-ATAs) belong to the class III transaminasefamily, which are also referred to as ornithine transaminase-like family(see Box 1). The method of the generation of 3DM databases is, in eightsteps, briefly described below. Further details were published else-where (Kourist et al., 2010; Kuipers et al., 2010b).
1) 3DM collects and superimposes all structures that share a commonstructural fold with the starting template structure (717 structuresin the PLP fold type I superfamily). Structures that are difficult to su-perimpose on the template — therefore resulting in only a smallnumber of superimposed residues — are discarded. The defaultcut-off is at least 60 residues that are within a sphere of 2.5 Å fromthe equivalent template residues leaving 406 structures in the PLPfold type I database.
2) A structural alignment is generated for each combination of twostructures in an all-to-all comparison. The resulting structure align-ments are merged into one large alignment that represents thestructurally conserved core of the superfamily.
3) The positions in this core alignment are numbered (called 3Dnumbers) such that all structural equivalent residues in the 3DMsystem have the same number.
4) 3DMgenerates subfamilies by grouping all structures that arewithina defined sequence identity cut-off (in the PLP fold type I databasethis cut-off was set to 50% resulting in 94 distinct subfamilies).
5) From each subfamily the structure that structurally matches bestwith the starting template, thereby maximising the number of coreresidues in each subfamily, is chosen as the representative subfamilytemplate (see Supplementary data Table S2 for a list of enzyme ac-tivities represented by the subfamilies' template structures).
6) The subfamily templates are used in a UniProt BLAST search to col-lect protein sequences for which no structures are available(120,870 proteins were collected for the PLP fold type I database).
7) For each subfamily template a profile based iterative alignment isperformed (this is beyond the scope of this article and publishedelsewhere; Kuipers et al., 2010a; Kuipers et al., 2010b). The inclusioncut-off for aligned sequences is 30% sequence identity compared tothe last profile used in this alignment procedure, which is approxi-mately 20% identical to the starting subfamily template. Aligning se-quences of this low sequence similarity with high quality is difficult,but a high quality alignment is established by a quality controlmechanismdescribed by Kuipers et al. (2010b). To ensure high qual-ity alignments, the sequences for which no evident solution can bedetermined are simply deleted from the alignment. Althoughmany aligned sequences are removed this way, this method still al-lows for the generation of very large superfamily alignments. In thecase of the PLP fold type I database, the final alignment generatedfrom the 120,870 sequences collected in step 6 contains 42,080 se-quences.
8) The 3Dnumbering scheme is applied to all aligned sequences,whichconnects all sequences, all structures, and all alignments from thedifferent subfamilies to each other.
The size of the conserved core is determined by the structural diver-sity in the superfamily: the more slightly different enzymes are includ-ed, the smaller are the structurally conserved regions that make up thecore. Not all active site residues are covered in the conserved core of the
PLP fold type I database due to structural diversity in the superfamily.This protein fold was extremely adaptive during evolution, thusallowing a large quantity of reactions to be catalysed. This database cov-ering a large fraction of PLP fold type I enzymes might be employed forfold type overarching questions, but more detailed investigations likesubstrate specificity require a smaller ‘sub-database’ comprisingstructurally more related enzymes. We therefore created a smaller3DM database, containing only sequences that belong to the class IIItransaminases, also referred to as the ‘ornithine transaminase-like’ fam-ily (see Box 1, compare Table 3). This yielded the ornithine TA-like data-base (OrnTL DB), comprising 21 subfamilies with a much larger corecompared to the large PLP fold type I database (see Table 1 for a compar-ison of the two databases, see Fig. 7 for a phylogenetic tree of the wholeOrnTL DB). Almost all active site residues of class III TAs were covered inthe core and 81% of all residues in the (S)-ATA from Chromobacteriumviolaceum (Cvi-ATA) belong to core regions in the OrnTL DB comparedto 62% in the larger PLP fold type I database (see Supplementary dataFigure S2 and S3 for more details). Besides the subfamily selection,this database was generated with the same settings as the full PLP foldtype I database.
3DM was able to generate a high quality alignment that consists of12,956 protein sequences from the 31,000 sequences collected by theinitial BLAST searches. The discrepancy between the amount of collect-ed class III TA sequences and the sequences present in the OrnTL DBreflects the portion of sequence space with insufficient structuralinformation. This demonstrates the need of further research to enhancethe structural coverage of the class III TA family.
Using this 3DM OrnTL DB we aim to extend recent studies, whicheither used sequence similarity to classify ATA related enzymes andonly investigated ATAs' functional residues in detail (Rausch et al.,2013) or focused only on a limited number of residues in the firstshell of the active site and was therefore limited to enzymes with struc-tural information (Catazaro et al., 2014). In this review we summariserecent literature and our findings concerning substrate and reactionspecificity determinants within PLP fold type I enzymes with specialfocus on the class III transaminase family.
2.2. Special features of the ornithine TA-like family exemplify the structuralflexibility of PLP-fold type I
The PLP-fold type I is an interesting example how very distantlyrelated sequences still form similar tertiary structures. The diversity ofenzyme activities within PLP fold type I is reflected in extensive adapta-tions at the amino acid sequence level: for example, aromatic aminoacid transaminase (PDB ID: 1AY4) and ornithine aminotransferase(PDB ID: 2OAT) share only 7% sequence identity. Within the OrnTL da-tabase low sequence identities down to 13%2 are observed. Interesting-ly, the backbone arrangement of the majority of the active site residuesis still very similar. From an inspection of the structure alignments, weidentified several features that are conserved on the structure or se-quence level in the OrnTL family, but differ in other fold type I enzymes.Four regions are especially conserved among class III transaminases(see Fig. 8 for secondary structure elements numbering):
1) A small antiparallel β-sheet (residues 23–36, comprising strands β1,β2 and β3) close to the N-terminus ‘on top’ of the small domain isconserved in most structures of the OrnTL family. This region is lo-cated at the domain interface and we speculate that this sheet con-tributes to the suppressed domain movements during substratebinding, which is, in contrast to other fold type I transaminases(McPhalen et al., 1992), commonly observed in this family (Cha
2 Pairwise identity within core regions of the sequences with UniProt IDs G9N4G9 andB8MF32 belonging to subfamilies 4A0G and 4AO9, respectively.
575F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

et al., 2014; Käck et al., 1999; Liu et al., 2004;Wybenga et al., 2012).2) Another region that might prevent OrnTL enzymes' domain closure
is located ‘below’ the small domain at the domain interface (residues218–234, including the helix α10).
3) All active sites (of available holoWT structures) comprise one turn ofa left-handed helix (α2) at the si-face of PLP (residues 44–47), as waspreviously described for Orn:αKG TAs, γ-aminobutyrate:αKG TAs(GABA:αKG TAs), and S-adenosylmethionine:7-keto-8-amino-pelargonic acid TAs (SAM:KAPA TAs) (Novotny and Kleywegt,2005). The influence of this exceptional secondary structure elementfor substrate recognition was shown for the Escherichia coli SAM:KAPA TA. The R221A mutation inverted this region to a right handedα-helix which strongly declined the catalytic efficiency for SAM(Sandmark et al., 2004). An arginine coordinating the left-handedhelix at the O-side is conserved in 80.1% of the sequences in theOrnTL family (R221 might be substituted by a lysine or R220 e.g. in2GSA or K243 e.g. in 1OHV). The helix is additionally stabilised by ahighly conserved D41 (97.9% of all sequences in the OrnTL DB, seeSupplementary data Table S3), which caps the N-terminus of thehelix. These two conserved residues and the left-handed helix to-gether are specific for the OrnTL family and are not found in otherfold type I enzymes.
4) A second OrnTL specific region in the active site is located at theO-side (residues 267–272). This loop contains the conserved T/S271that is involved in PLP binding (Humble et al., 2012) andmight addi-tionally be involved in catalysis by hydrogen bonding the catalyticK242 as it was shown for a conserved cysteine in a comparable posi-tion in ornithine decarboxylases (Oliveira et al., 2011). This regionsubstantially differs in the whole fold type I database and thereforebelongs to the variable region there, whereas it could be properlyaligned in the proteins of the OrnTL DB.
Several amino acid positions were identified as conserved andunique within proteins of the OrnTL family (see supplementary data
Table S3). Of particular interest is that the catalytic lysine is present inthis list: in themajority of sequences, which are not in the OrnTL family,the catalytic lysine is found two positions later at position 244 (for a dis-cussion see section 4.1). Most other positions in this list are not directlyexplained by their functional role, which is not surprising, as the OrnTLDB comprises a variety of substrate and reaction specificities that de-mand different recognition mechanisms. The only amino acid that isconserved over the whole fold type I database is D213, which is the res-idue responsible for protonating the pyridine nitrogen of the cofactor.
2.3. Reaction and substrate specificity determining residues revealed bycorrelated mutations analysis (CMA)
Evaluating sequence conservation is a fast way to identify positionsthat are probably relevant for catalysis or protein architecture. However,this strategy will only display a fraction of important residues. Often,more than one solution (amino acid composition) exists to realise afunction in a protein. For example, in the case of a salt bridge at a do-main interface that is required for protein stabilisation, it is probablynot important on which of the two domains the acidic or basic residueis localised. Thus, these two positions might not be conserved but mu-tated simultaneously. The degree to which mutations occur in a corre-lated fashion depends on the strength of the selection pressure, whichis related to the functional relevance of these positions. Halabi et al.(2009) demonstrated the power of such analyses using proteases:clusters of amino acids (called protein sectors) relevant for substratespecificity, thermostability and catalysis could be discovered from cor-relatedmutationswithout looking at crystal structures. Thus, correlatedmutations analysis (CMA) is a powerful statistical evaluation tool(Kuipers et al., 2009; Kuipers et al., 2010b). The informative value of aCMA depends on the size, composition and quality of the multiple se-quence alignment. The OrnTL DB mainly contains amino acidsequences of transaminases with different substrate specificities. As ex-pected, the network that resulted from a CMA on the OrnTL DB (Fig. 9)includes 10 active site positions that are important for substrate recog-nition (we found 13 key amino acids during our literature research andstructure inspection, see section 3; for a picture of the human Orn:αKGTA highlighting the active site residues, see Fig. 10).
Residues 132, 185, 216, and 353 and residues 47 and 346 are part ofthe CMA network; they are key positions determining amino acceptorspecificity in the transaminases (α-ketoglutarate versus pyruvate).The majority of the sequences of the OrnTL DB are transaminases withat least 28 significantly different substrate specificities. As more thanone amino acid residue is necessary to create a distinct substrate speci-ficity, it makes sense that these residues mutated simultaneously. Thisclearly demonstrates the potential of CMA. The CMA also detected resi-dues involved in cofactor binding, at the dimer interface, but also resi-dues whose relevance is not yet known.
2.4. The sequence–function matrix
From the 3DMdatabase statistics, structure inspection and literatureresearch concerning each substrate and reaction specificity, we con-structed a sequence–function matrix. This is the centrepiece of this re-view: the matrix summarises 13 active site residues within the OrnTLfamily that determine reaction or substrate specificity (Table 2). As cer-tain patterns are unique for each enzyme activity, we suggest that theseactive site fingerprints can be used to predict themain activity of a givensequence belonging to the OrnTL family. Not all 13 positions are equallyrelevant for each enzyme. The most important residues for eachspecificity are shown in bold. In the specificity-dedicated subsections,we describe the details of these sequence–function relationships at amolecular level.
We constructed the matrix in the following way: for each activity,we built small alignments containing only enzymes with experimental-ly confirmed activities. From crystal structures and literature, we
Fig. 8. Topology plot of the class III transaminases exemplified on chain A of the humanOrn:αKG TA structure (PDB ID: 2OAT). The regions with specifically conserved secondarystructure in class III TAs are highlighted red (see main text for more information). Con-served residues among the whole family are highlighted in grey: the aspartate ‘on top’of the left-handed helix α2 (D41), the aspartate ‘below’ PLP, coordinating its pyridine ni-trogen (D213) and the catalytic lysine (K242).
576 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

extracted the relevant residues and then identified all sequences of theOrnTL DB carrying these minimal sequence patterns. Finally, we usedthe larger alignment derived from the sequence pattern search to ex-tract the most frequently observed amino acids at the other matrix po-sitions for each specificity.
Owing to the lack of crystal structures with sufficient sequence sim-ilarity, several class III transaminases have not been included in theOrnTL DB (see Table 3 entries with a minus (−) sign in the ‘subfamily’column). Most of these enzymes could be aligned to their closest struc-ture and sequence in the 3DM database manually and thereby also becompared to the other class III TAs. The results from thesemanual align-ments are discussed in the corresponding sections and are alsodisplayed in the sequence–function matrix (see Table 2).
Further research is needed to investigate to which extent the se-quence–function relationships can be generalised. We suggest using
the information stored in the matrix to address the followingquestions:
Question Solution
1. What are the mainsubstrates of a givenprotein belonging tothis superfamily?
In an alignment, compare the active site residues of thequery sequence to the fingerprints of the matrix. In caseof a match, it is likely that also the activities are matching.For convenience, a multiple sequence alignment of allsubfamilies' parental structures of the OrnTL DB isavailable (Supplementary data Figure S1) that helps tomatch the 3DM numbers of this review with the originalnumbering of the crystal structures and therefore also tothe query sequence.
2. How can I identifynovel enzymeshaving the desiredactivity present inthe superfamily?
Usually, a BLAST with a query sequence is conducted toidentify enzymes with similar/equal function. Werecommend to align available sequences and check,whether they carry the specificity determining residueshighlighted in the matrix. This helps especially toevaluate distantly related sequences which otherwisewould often not be chosen as candidate because theoutcome would be too uncertain.
3. Does the superfamilycontain novel enzymeactivities, which arenot yet known?
Enzymes whose active site residues do not fit the patternof the matrix have either an unknown activity, or theymight contain an alternative active site design to confera known specificity.
3. Activities represented in the ornithine TA-like database
The class III transaminase family represents only a small fraction ofknown sequences within the PLP fold class I. Nevertheless, it containsat least 28 distinct enzymatic activities (not all have a separate EC num-ber assigned, yet). This section aims to summarise structural details thatgovern these substrate and reaction specificities.
A table of contents of this section's subsections is given in Table 3,which provides an overview of enzyme activities by providing struc-tures of their substrates and products, as well as a list of abbreviations.We will discuss these enzymes in the order mentioned in Table 3. Theentries in the sequence–function matrix and the scenes in the Supple-mentary PyMOL session are in the same order to facilitate a convenientcomparison of active site fingerprints and their underlying structuralfeatures. Enzymes displaying transaminase activities will be describedfirst, followed by a smaller group of enzymes with other reaction spec-ificities, namely decarboxylation dependent transaminases, 1,2-aminomutases and α-amino acid amide (αAAA) racemases. Besides thesewell-investigated enzymes, we mention fumonisin B1 TAs, aminosugarTAs, phospholyases and multi-domain enzymes grouped together atthe end: due to a lack of structural information, the substrate or reaction
c
c
c
c c
c c
c
c
M
M
M
M
M
M M
M
PLP
stacking
dimer
interface
Fig. 9. CMA of the OrnTL database detects a network of key residues responsible for substrate specificity. A 0.8 cut-off for the correlation scorewas applied (see Kuipers et al. (2009) for anexplanation of this score). The strength of the correlation of two positions is indicated by the colour of the connecting lines (red: 1, yellow: 0.8). Residues, which we collected fromliterature as key residues for determining substrate and reaction specificity are marked with anM (as they are part of the sequence–function matrix Table 2). To indicate that correlatingresidues are in contact, the lines are labelled with ‘c’.
45
46
47
16
353
352
351
347
349
185
216
215
129
132
213
268
271270
73
348
242
41
346
Orn: KG TA, 2OAT
Fig. 10.Active site residues and 3DMnumbering of theOrn TA-like database highlighted inthe human Orn:αKG TA (PDB ID: 2OAT). Core regions are shown as grey, variable regionsas yellow cartoon. The PLP bound ornithine mimicking analogue is shown in orange. Thisand all other crystal structure figures were created using PyMOL (Version 1.6.0.0).
577F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

specificity determining residues of these enzymes cannot be predictedfrom the sequence alignments at the moment. A list of sequences,which have been experimentally characterised, is given in Supplemen-tary data Table S5.
The majority of characterised enzymes found in the OrnTL databasecan be classified as ω-transaminases. Whereas α-transaminases transferthe amino group at the Cα position with respect to the carboxylategroup, ω-transaminases can accept amino acids where the position ofthe amino group with respect to the carboxylate group varies (seeFig. 5). These enzymes can further be clustered into three groupsbased on the preferred amino acceptor. The first group is accepting α-ketoglutarate (αKG) (section 3.1), the second pyruvate (pyr) (section
3.2) and the third is converting either both αKG and pyr or other aminoacceptors (section 3.3). As all ω-TAs convert structurally and chemicallydifferent amino donors and acceptors in the same active site, they allrequire a mechanism for dual substrate recognition. Within the OrnTLdatabase three different solutions for this task have been found,which are each discussed in the beginning of the sections dedicatedto the three amino acceptors (sections 3.1.1, 3.2.1 and 3.3.1). Thealanine:glyoxylate transaminases 2 (Ala:glyox TA 2) are the onlycharacterised transaminases within the OrnTL DB that do not requirea dual substrate recognition because only α- and βAA are converted(section 3.3.6). The second known enzyme that only converts αAA,D-p-hydroxyphenylglycine:αKG TA, however, requires dual sub-strate recognition because its substrate and product have inversedabsolute configuration (D-p-hydroxyphenylglycine and L-Glu, sec-tion 3.3.4).
Among theω-transaminases, a few have been found to additionallyconvert amines independently from the presence of carboxylic groupsin the substrate. Those enzymes, referred to as amine transaminases(ATAs), are the biocatalytically most interesting enzymes within thisfamily (Rausch et al., 2013), and are described in sections 3.2.2 and 3.2.3.
To facilitate the understanding of the most important features of agiven subfamily discussed below and to provide a quick summary, wefirst present the active site fingerprint containing only amino acids,which are of high relevance for substrate recognition or catalysis for thisenzyme followed by a brief summary of the essentials of each subsection.
3.1. ω-Amino acid:α-ketoglutarate transaminases
Fingerprint ωAA:αKG TAs: R132, D/E185, Q216, R353.Summary: the specificity forω-amino acid:α-ketoglutarate (ωAA:αKG)
transamination can be reliably predicted because the four fingerprintresidues, involved in dual substrate recognition, are found in allcharacterised enzymes. However, amino donor specificity prediction ismore delicate as most ωAA:αKG TAs show a relatively broad substratescope. Since broad substrate spectra are achieved by unspecific binding ofdifferent substrates, predictions by use of active site fingerprints are oftennot reliable. Nevertheless, the ωAA:αKG TAs with narrow substrate scopes
Table 2Sequence–function matrix: overview of the function determining positions in correlationto reaction and substrate specificities. Residues in bold are thefingerprint residues that de-termine reaction and substrate specificity (as retrieved from mutagenesis or crystallo-graphic studies). If not sufficient information was available to assign a fingerprint for acertain specificity, no residue is bold. The colour code indicates the physicochemical prop-erties of the residues. The degree of conservation is indicated by the following notations:capital letters— conserved residues (more than 70% of the subset); lowercase — residueswith a conservation between 30% and 70%, up to 3 amino acids are listed per positionwithdescending conservation; lowercase italic letters indicate that none of the amino acids at agiven position occurs with more than 30%, the threemost frequent amino acids are given.Aminus (−) indicates that there is no residue that can be aligned to this position. A specialcharacter is introduced for sequences that donot belong to theOrnTL DB andwere alignedmanually to their closest homologs within the database. At positions where thesemanualalignments were ambiguous a questionmark is shown. For details on substrates, productsand abbreviations of the enzymes corresponding to this matrix, see Table 3.
Notes to Table 2:aOnly based on one sequence (Flavobacterium lutescens Lysε:αKG TA (UniProt ID: Q9EVJ7)that was aligned to its closest homolog A9MMF7 in the OrnTL DB to determine the 3Dnumbers.bOnly based on one sequence (the E. coli YgjG enzyme (UniProt ID: P42588; PDB ID:4UOX))cOnly based on one sequence (the Serratia sp. 3-acetyloctanal TA (UniProt ID: Q5W267),crystal structure (PDB ID: 4PPM))dOnly based on nine characterised enzymes (see Supplementary data Table S5 entries76–85)eOnly based on two sequences (the Pseudomonas aeruginosa spuC (UniProt ID: Q9I6J2)and its homolog from P. putida (UniProt ID: Q88CJ8))fOnly based on four characterised enzymes (see Supplementary data Table S5 entries137–140) (UniProt ID: Q6JE91 is different at several matrix positions)gOnly based on one characterised enzyme (from Candidatus cloacamonas acidaminovorans(UniProt ID: B0VH76))hOnly based on two sequences (the Pseudomonas stutzeri (UniProt ID: Q6VY99) and theP. putida enzyme (GenBank ID: AX467211)) the structure of the P. stutzeri enzyme (PDBID: 2CY8) is an unpublished apo structureiOnly based onfive characterisedmammalian enzymes (see Supplementary data, Table S5,entries 162–166)jOnly based on one sequence (the Lactobacillus buchneri Ile-2-epimerase (UniProt ID:F4FWH4))kOnly based on two characterised sequences (the human O-phosphoethanolaminephospholyase (UniProt ID: Q8TBG4) and 5-phosphohydroxy-L-lysine phospholyase(UniProt ID: Q8IUZ5)); the only difference among these residues is 185 where Cys is re-placed by a Val in the latter enzymelOnly based on two characterised sequences (the enzymes from Sphingopyxismacrogoltabida (UniProt ID: D2D3B2) and bacterium ATCC 55552 (UniProt ID: E2E0Q4))mOnly based on one characterised sequence (Seq. ID 56 in patent WO2004085624)nOnly based on one characterised sequence (the Mycosubtilin synthase subunit A fromBacillus subtilis (UniProt ID: Q9R9J1))
578 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604
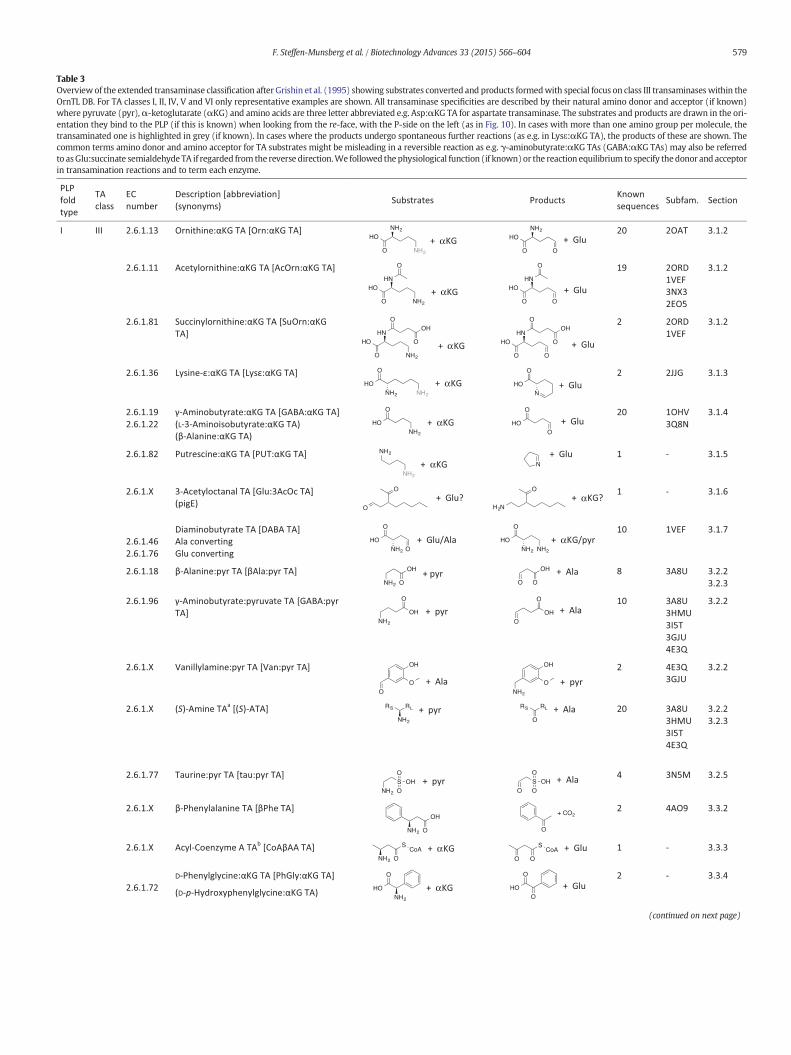
Table 3Overviewof the extended transaminase classification after Grishin et al. (1995) showing substrates converted and products formedwith special focus on class III transaminaseswithin theOrnTL DB. For TA classes I, II, IV, V and VI only representative examples are shown. All transaminase specificities are described by their natural amino donor and acceptor (if known)where pyruvate (pyr),α-ketoglutarate (αKG) and amino acids are three letter abbreviated e.g. Asp:αKG TA for aspartate transaminase. The substrates and products are drawn in the ori-entation they bind to the PLP (if this is known) when looking from the re-face, with the P-side on the left (as in Fig. 10). In cases with more than one amino group per molecule, thetransaminated one is highlighted in grey (if known). In cases where the products undergo spontaneous further reactions (as e.g. in Lysε:αKG TA), the products of these are shown. Thecommon terms amino donor and amino acceptor for TA substrates might be misleading in a reversible reaction as e.g. γ-aminobutyrate:αKG TAs (GABA:αKG TAs) may also be referredto asGlu:succinate semialdehyde TA if regarded from the reverse direction.We followed the physiological function (if known)or the reaction equilibrium to specify thedonor and acceptorin transamination reactions and to term each enzyme.
α
α
α
α
α
α
α
α
α
α
(continued on next page)
579F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

α
α
Table 3 (continued)
580 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

developed mechanisms to distinguish the different ωAAs, thereby enablingplausible predictions of their specificity.
ω-Transaminases, which are selective for αKG as the amino accep-tor, but differ in their amino donor specificity, include 72 characterisedenzymes of the 12,956 sequences in the OrnTL DB. The above shownfingerprint residues are characteristic for these αKG specific enzymes.Already small substitutions of additional active site residues vary the ac-ceptance of amino donors (mainly ω-amino acids of different lengthand substitution pattern). It is of special interest to understand howthese different enzymes learned to favour one of the very similar sub-strates over another and to distinguish between GABA, Orn, AcOrnand Lys. The majority of these enzymes, however, are not absolutelyspecific and convert more than one of those substrates with reasonableactivity. Enzyme redundancy and substrate promiscuity seems to be acommon feature of many class III transaminases (Lal et al., 2014;Schneider and Reitzer, 2012). Unfortunately, the substrate scope ofseveral enzymes has not been investigated systematically and oftenonly the substrate pair suggested from sequence identity to other
known enzymes was tested (Seong Gyu et al., 2001; Tripathi andRamachandran, 2006). Therefore it is often not possible to unambigu-ously classify certain ωAA:αKG TAs and we focused on enzymes thatwere tested for more than one substrate to compare active site featuresthat are important for amino donor discrimination.
3.1.1. Dual substrate recognition: the glutamate switchThe dual substrate recognition mechanism is conserved among the
ωAA:αKG TAs as all substrate pairs demand for the same requirements.Given the fact that the L-enantiomer of glutamate is formed in eachreaction, the O-side (for active site nomenclature, see Box 1) has to ac-commodateαKG's 1-carboxylate. This is achieved by a highly conservedarginine (R353, see Fig. 11B) (Hirotsu et al., 2005).
In theω-amino acid converting half reaction the O-side only accom-modates a proton (instead of a carboxyl group) from the terminal car-bon of the substrate and therefore the pocket needs to be otherwise‘filled’. For this purpose, the side chain of E185 switches into the activesite and forms a salt bridge with R353 to neutralise its positive charge.
α
α
α
α
aRS: small residue, RL: large residue.bThe full coenzyme A structure is shown in Supplementary data Figure S5 A.cFor six of these the amino donor is unknown, the sequences, however, suggest SAM:KAPA TA.dAdditional substrates are shown in Supplementary data Figure S4 A.eSubstrates and products are shown in Supplementary data Figure S4 B–E.fSubstrate structures are shown in Supplementary data Figure S5 B and C.gTransaminase class VI is equal to InterPro's DegT/DnrJ/EryC1 family.
Table 3 (continued)
581F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

Due to this movement, the dual substrate recognition in these enzymesis often referred to as ‘glutamate switch’ (Hirotsu et al., 2005). The‘switched-in’ position of the side chain of E185 is further stabilised bya hydrogen bond from ‘below’ by Q216 (see Fig. 11A).
On the P-side another highly conserved arginine residue (R132)forms a salt bridge with the 1-carboxylate of the ωAA substrate in thefirst half reaction (Fig. 11A) and in most ωAA:αKG TAs also with the5-carboxylate of αKG in the second half reaction (Fig. 12B) (Newmanet al., 2013). One exception is the AcOrn:αKG TA from Thermusthermophilus, (Fig. 11B) where R132 is only involved in weak electronicinteractions to αKG (Hirotsu et al., 2005).
R132 also determines the specificity towards transamination of theω-amino group in α,ω-diamino acids: R132 specifically interacts withthe substrate's 1-carboxylate, thereby keeping the substrate in theright orientation for ω-transamination. Additionally, R132 would repelthe ω-amino group when the di-amino acid would be oriented in thealterative binding mode at the P-side; thereby the conversion of the
α-amino group is prevented (Markova et al., 2005). The importance ofR132 for substrate recognition, which is probably the reason for its con-servation in ωAA:αKG TAs, becomes evident in the human inbornGABA:αKG TA deficiency, which may result from a R132K mutation inthese enzymes that reduces the vmax by 25% (Medina-Kauwe et al.,1999). A second confirmation of its influence on αKG recognitionwas provided in a mutagenesis study on a dialkylglycine decarbo-xylase (see section 3.5) where the M132R mutation conferred theability to convert L-glutamate (Fogle and Toney, 2010).
The functional importance of the interplay of the three residuesE185 Q216 and R353 is also reflected by the CMA: these positionsare highly correlated in the OrnTL DB and mainly occur together (seeCMA network in Fig. 9). Therefore the presence of these three residuestogether might be regarded as a strong indication forωAA:αKG activityof an uncharacterised enzyme.
The conserved R353 is additionally found in other enzymes thataccept α-amino acids such as the 2,2-dialkylglycine decarboxylase
82113
15
16
132
129
46
353
185
216
242
271
215
AcOrn:αKG TA, 1WKG AcOrn:αKG TA, 1WKH
Orn:αKG TA, 2OATAcOrn:αKG TA, 1WKG
A) B)
C) D)
Fig. 11. The dual substrate recognition in AcOrn:αKG TAs is achieved by the E185 ‘switch’ (A & B) and Y16 side chain conformation determines AcOrn:αKG TAs fromOrn:αKG TAs (C &D).The substrate (analogue) PLP adducts are shown in orange, variable region residues are shown in yellow. A) E185 neutralises R353 when AcOrn is bound (PDB ID: 1WKG). B) E185‘switched’ out of the active site to allow for the coordination of αKG's 1-carboxylic group by R353 (PDB ID: 1WKH) C) In AcOrn:αKG TAs Y16 points out of the active site to allow forAcOrn binding (Q82 is 1WKG numbering, between core positions 73 & 74) R132 is omitted for clarity reasons. D) In Orn:αKG TAs Y16 points towards the active site to coordinateOrn's α-amino group. This side chain conformation is caused by a second water molecule (compared to one in 1WKG), which is held in place by a hydrogen-bonding network involvingthe other water, N15 and R113 (2OAT numbering, between core positions 73 & 74). R132 is omitted for clarity reasons.
582 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

subfamily 1D7V and the α-TAs found in PLP fold type I, where it is alsocoordinating the substrate's carboxylate (Sun et al., 1998).
3.1.2. Ornithine, acetylornithine and succinylornithine:α-ketoglutarate TAsFingerprint Orn:αKG TA: Y16, Y46, R132, E185, Q216, R353, R113
(2OAT numbering, between core positions 73 & 74)Fingerprint Orn/AcOrn/SuOrn:αKG TA: Y16, R132, E185, Q216, R353Summary: AcOrn/SuOrn:αKG TAs have a broad substrate specificity and
convert both AcOrn, SuOrn, and at higher pH values also Orn. The lack ofspecific interactions allows for this broad substrate spectrum. On the con-trary, Orn:αKG TAs are highly specific because the free α-amino group isplaced between the two conserved Y16 and Y46. The presence of thesetwo tyrosines, however, is not sufficient for creating the high specificity.R113, an arginine residue of the variable region between position 73 and74 is important for the correct positioning of the side chain of Y46.
Ornithine aminotransferases (EC 2.6.1.13, Orn:αKG TA, found in sub-family 2OAT), acetylornithine aminotransferases (EC2.6.1.11, AcOrn:αKGTA, subfam. 2ORD, 1VEF, 3NX3 and 2EO5) and succinylornithine amino-transferases (EC 2.6.1.81, SuOrn:αKG TA, subfam 2ORD) are found inArchaea, Bacteria and Eukaryota, where they are involved in ornithinehomeostasis and proline biosynthesis (Jortzik et al., 2010) or the arginineand lysine biosynthetic pathways (Xu et al., 2007).
Due to the narrow substrate scopes of Orn:αKG TAs, which arelimited to biotechnologically rather uninteresting compounds, thereare not many biocatalytic or biotechnological applications described.However, they were utilised for equilibrium displacement in otherαKG forming transaminations as the formed aldehyde product(glutamate-5-semialdehyde) is instantly removed from the equilibriumby spontaneous cyclisation (Tufvesson et al., 2011).
In general AcOrn:αKG, SuOrn:αKG and often also N-succinyl-L,L-diaminopimelate:αKG activity is found in the same, broad substratescope, enzymes. These AcOrn:αKG TAs have a preference for the acylat-ed ornithine species over ornithine itself at neutral pH (Heimberg et al.,1990; Ledwidge and Blanchard, 1999; Newman et al., 2013), whereasthe Orn:αKG TAs do not convert AcOrn (Heimberg et al., 1990) andGABA is only converted with very low activity (Markova et al., 2005).
The substrate preference of Orn:αKGTAs can be explained by specif-ic interactions of Y16 with the free (non-converted) α-amino group(Markova et al., 2005). The Y16A/G mutations drastically reducedOrn:αKG activity in the human enzyme (PDB ID: 2OAT). Additionally,the role of the highly conserved Y46 in determining Orn:αKG TA speci-ficity was shown by the Y46I mutation in the human enzyme, whichswitched the substrate specificity towards GABA:αKG transamination.Y16 and Y46 might therefore, together with the dual substrate re-cognition residues (i.e. R132, E185, Q216, R353, see section 3.1.1) beemployed to distinguish Orn:αKG TAs from other class III transami-nases. Nevertheless, these features alone are not sufficient to discrimi-nate Orn:αKG TAs from all AcOrn:αKG TAs, as the AcOrn and AcLysconverting enzyme from T. thermophilus also has these two tyrosines(PDB ID: 1WKG, see Fig. 11) (Miyazaki et al., 2001; Rajaram et al.,2008). Rajaram et al. (2008) proposed that the difference in substratespecificity between these enzymes arises from side chain conformation-al changes of Y16, rather than from differing active site residues. InOrn:αKG TAs the side chain is pointing towards the active side, therebypreventing the productive binding of AcOrn, whereas this tyrosinepoints out of the active site in AcOrn:αKG TAs (see Fig. 11C & D) to cre-ate additional space resulting in a more relaxed substrate scope. Thecomparison of the 2OAT and 1WKG structures implies that this subtledifference is mediated by the presence of an additional water moleculebehind Y16 in 2OAT.We propose that N15 and R113 (2OAT numbering,between core positions 73 and 74), that are highly conserved inOrn:αKG TAs, but not in AcOrn:αKG TAs are responsible for the correctpositioning of the two water molecules (Fig. 11C & D). R113 (2OATnumbering) in Orn:αKG TAs corresponds to a conserved Q82 (1WKGnumbering) or N79 (2PB0 numbering) in AcOrn:αKG TAs, while posi-tion 15 is not conserved in these enzymes. The influence of the R113
(2OAT numbering) on AcOrn/Orn activity has probably already been in-vestigated as the structure of the N79R mutant of the Salmonellatyphimurium enzyme has been deposited in the PDB recently (PDB IDs:4JF0 & 4JEZ) but unfortunately the mutant is misfolded at the P-side inboth structures (Bisht et al., 2014; unpublished crystal structures).
Searching for fingerprint Y16, Y46, R132, E185, Q216, R353 in theOrnTL DB resulted in 816 sequences (97% of which are found in the2OAT subfamily). 786 of those sequences have R113 (2OAT numbering,between core positions 73 & 74) that are therefore predicted to encodefor enzymes highly specific for Orn:αKG transamination.
The fact that AcOrn:αKG and SuOrn:αKG TAs prefer the acylated or-nithine over the free di-amino acid at neutral pH values cannot be ex-plained by any specific interaction with the substrate. This preference,which is not found at higher pH values (Heimberg et al., 1990), is prob-ably established by steric and desolvation effects (Newman et al., 2013).Newman et al. (2013) concluded that these enzymes possess a broadersubstrate scope or some degree of substrate promiscuity compared toOrn:αKG TAs due to non-specific interactions which enables the bind-ing of different substrates in a similar orientation. Specific interactions(e.g. hydrogen bonds) to the substratewould not allow for a broad spec-trum because substrates not satisfying these interactions would sufferenergetic penalties.
Unspecific binding of AcOrn is probably also responsible forAcOrn:αKG TA activity that was detected in ‘broad spectrum’ GABA:αKGTAs (Lal et al., 2014; Voellym and Leisinger, 1976) which are discussed insection 3.1.4. Additionally, two enzymes from thermophiles that had,based on sequence similarity, initially been annotated as GABA:αKG TAsturned out to be AcOrn:αKG TAs (Koma et al., 2006). These are alsodiscussed in the GABA:αKG TA dedicated section 3.1.4.
The inhomogeneity of enzymes possessingAcOrn TA activity and theunspecific binding of the substratemakes thefingerprint based discrim-ination of these enzymes from otherωAA:αKG TAs impossible. To someextent, however, it is possible to distinguish the AcOrn:αKG TAs thatshare sequence similarity to Orn:αKG TAs from the other class III trans-aminases. Most Orn:αKG TA similar AcOrn:αKG and SuOrn:αKG haveY16 on the P-side but not Y46 like the Orn:αKG TAs (1WKG is an excep-tion). Even though Y16 does not coordinate AcOrn or SuOrn, its conser-vation implies that it is structurally or functionally important for thetransaminases converting both free and acylated ornithine (seesequence–function matrix Table 2).
3.1.3. Lysine-ε:α-ketoglutarate TAsFingerprint Lysε:αKG TAs similar to 2JJG: R132, E185, Q216, N/S269,
R353Summary: three different kinds of Lysε:αKG TAs are known: 1) enzymes
similar to 2JJG with a broad substrate spectrum that match the fingerprint;2) enzymes similar to the Flavobacterium lutescens enzyme, thatmatch thefingerprint of broad spectrum GABA:αKG TAs, but do not convert GABA;3) enzymes which are highly specific for Lys and do not convert Orn (se-quence unknown).
L-Lysine-ε-transaminases (Lysε:αKG TA), which are found in the2JJG subfamily, catalyse thefirst step in a bacterial biosynthetic pathwaytowards β-lactam, the building block of several antibiotic families suchas penicillins and cephalosporins (Tobin et al., 1991). This enzyme hasbeen used to convert Nα-protected-L-lysine into precursors of ACE(Angiotensin Converting Enzyme) inhibitors, which are used as antihy-pertensive drugs (Patel et al., 1999). Other examples include the use ofLysε:αKG TA from Sphingomonas paucimobilis to synthesise a precursorof the vasopeptidase inhibitor Omapatrilat (Patel et al., 2000) as well as5-hydroxy-L-proline and some protected variants thereof (Hanson et al.,2011). The application of these enzymes (similar to that of Orn:αKGTAs, described in section 3.1.2) to shift the equilibrium of other αKGforming reactions is possible because the aldehyde product isinstable (Tufvesson et al., 2011): Lysε:αKG transamination formsα-aminoadipate-δ-semialdehyde and Glu, while α-aminoadipate-δ-
583F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

semialdehyde undergoes spontaneous intramolecular imine formationto form 1-piperideine-6-carboxylic acid (Soda et al., 1968) (see Table 3).
Unfortunately the only two characterised enzymes in the OrnTL DBhave been tested for very few substrates (Romero et al., 1997; Tripathiand Ramachandran, 2006), which makes general conclusions about se-quence–function relations in Lysε:αKG TA difficult. We suggest thatthere are at least three types of Lysε:αKG TAs: the first one is found inthe 2JJG subfamily and accepts lysine, ornithine,αKG and to a lower ex-tent also pyr and oxaloacetate as found for the Streptomyces clavuligerusenzyme (UniProt ID: Q01767) (Romero et al., 1997). The second type ofLysε:αKG TAswith known sequence and substrate scope is representedby the enzyme from F. lutescens (UniProt D: Q9EVJ7) (Fujii et al., 2000;Yagi et al., 1991), which is, however, not aligned to the OrnTL DB as itshares too little sequence identitywith the 2JJG enzymeor anyother en-zyme with known structure. This enzyme is also found to convert orni-thine in addition to the favoured lysine (Yagi et al., 1991). The third typeof Lysε:αKG TAs, which is in contrast to the two other mentioned typesby not converting ornithine at all, was found in Candida utilis (Hammerand Bode, 1992) but its sequence is unknown. Even though it would beinteresting to investigate how this third type achieves the lysine–ornithine discrimination, sequence–function relationships can only beinvestigated for the first two types.
The dual substrate recognition in the enzymeswith known sequenceis performed as described for all ωAA:αKG TAs in section 3.1.1 andR132, E185, Q216 and R353 are therefore conserved (Tripathi andRamachandran, 2006).
The creation of a sequence fingerprint for this substrate specificityhas proven to be challenging owing to its similarity with the otherωAA:αKG TAs, which all convert very similar substrates (e.g. ornithine,acetylornithine and γ-aminobutyrate). A closer structural comparisonof the 2JJG enzymewith the GABA:αKG TAs andOrn/AcOrn/SuOrn:αKGTAs showed very similar active sites and it is therefore likely that GABAand AcOrn is converted by these enzymes as well. There is only oneposition (N/S269) in the active site which is fairly unique for the twocharacterised Lysε:αKG TAs in the OrnTL DB (Supplementary dataTable S5 entry 42 & 43) and the whole subfamily 2JJG. This residue,however, is not involved in lysine or αKG coordination (see Fig. 12)and therefore the function of this additional hydrogen-bonding donorat the P-side cannot be rationalised, yet. Nevertheless, we suggestincluding it in a sequence fingerprint to identify enzymes with compa-rable substrate specificities but its function should be investigated bymutagenesis studies. The fingerprint search for R132, E185, Q216,
N/S269, R353 resulted in 112 sequences of which 88% belong to the2JJG subfamily.
The Lysε:αKG TAs of the second type are not found in the OrnTL DB,but the enzyme from F. lutescens was aligned to the closest homolog inthe database to compare the active site residues (see sequence–functionmatrix Table 2). It was found that it also possesses the common dual sub-strate recognition residues but a G269 and an I46 instead of N/S269 andV/F46 in the 2JJG subfamily. Interestingly, this enzyme accepts lysineand ornithine, but not GABA (Yagi et al., 1991) even though it sharesthe common active site residues with the GABA:αKG TAs as describedin thenext section. Regions that couldnot be alignedproperlymust there-fore additionally determine substrate specificity in this enzyme.
3.1.4. γ-Aminobutyrate:α-ketoglutarate TAsFingerprint ‘narrow spectrum’ GABA:αKG TAs: I46, R132, E185,
F269, Q216 and R353Summary: I46 is important for GABA:αKG TA activity but no unique fea-
ture, as I46 is also found in some other enzymes within the class III TAs. Theactive site of eukaryotic GABA:αKG TAs is narrowed by F269 and there-fore only GABA, LAIB and βAla are accepted as amino donors. Subtleamino acid exchanges not covered by the sequence–function matrixcan substantially shift the preference from GABA towards βAla.
Fingerprint ‘broad spectrum’ GABA:αKG TAs: (NOT Y16), I46, R132,E185, G269, Q216 and R353
Summary: bacterial GABA:αKG TAs have, due to G269, more space inthe active site and therefore have a pH dependent relaxed substratescope. AcOrn is also converted at neutral pH. At higher pH values substrateswith additional free amino groups like Orn, Lys and putrescine (PUT) areaccepted as well. Unidentified differences to these enzymes (as found inthermophiles) result in transaminases prefering AcOrn over GABA.
γ-Aminobutyrate (GABA) is a neurotransmitter and catabolicGABA:αKG TAs (EC 2.6.1.19) are involved in neurological disordersand therefore are targets for the treatment of e.g. epilepsy. For instancethe suicide inhibitor Vigabatrin is active for epilepsy treatment (Grantand Heel, 1991), as it irreversibly inhibits mammalian GABA:αKGTAs by covalently linking PLP and the catalytic lysine and therebypreventing further catalysis. Interestingly, Orn:αKGTAs are onlyweaklyand reversibly inhibited by this highly selective inhibitor (Lee et al.,2014). The differences of mammalian Orn:αKG and GABA:αKG TAsfrom a pharmaceutical point of view have been recently reviewed byLee et al. (2014).
129
185
353
216
132
269
46
47
242
215
Lysε:αKG TA, 2CJHLysε:αKG TA, 2CJDA) B)
Fig. 12.Dual substrate recognition in Lysε:αKGTAs exemplified for the enzyme fromMycobacterium tuberculosis. A) Lysine's carboxylate is coordinated byR132 and E185 is in contactwithQ216 and R353 (PDB ID: 2CJD) B) α-Ketoglutarate is coordinated by R132 on the P-side and R353 and Q216 at the O-side, while E185 ‘switched’ out of the active site (PDB ID: 2CJH).
584 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

From sequence similarity and substrate scope we suggest todistinguish the GABA:αKG TAs in two groups: eukaryotic enzymesthat possess a very narrow substrate scope and are only acceptingGABA and β-alanine (βAla), and the bacterial enzymes which have abroader substrate spectrum.
Due to the very narrow substrate scope the options for biotechno-logical applications of eukaryotic enzymes is limited, but also for thosewith bacterial origin only few biocatalytic applications have been de-scribed. Bacterial GABA:αKG TAs can be applied as effective biocatalystsfor the production of GABA or Glu similar substances as demonstratedfor the herbicide L-phosphinotricin by the E. coli gabT enzyme (PDBID: 1SFF) (Schulz et al., 1990).
All described GABA:αKG TAs share the common residues for dualsubstrate recognition with the other ωAA:αKG TAs (i.e. R132, E185,Q216, R353, see section 3.1.1 and Fig. 13A & B) and I46, which hasbeen found to enhance GABA:αKG TA activity in Orn:αKG TAs (seesection 3.1.2) (Markova et al., 2005). However, residue I46 is notspecific for GABA:αKG TAs among the class III transaminases, as sev-eral AcOrn or SuOrn:αKG TAs also have a I46. The discrimination ofenzymes with preference for GABA from the AcOrn/SuOrn:αKG TAsthat are similar to the Orn:αKG TAs, however, is possible if se-quences with Y16 are excluded. This residue is highly conserved inthe AcOrn/SuOrn:αKG TAs (see section 3.1.2), but not in the knownGABA:αKG TAs.
Structural features limiting the substrate scope of mammalianGABA:αKG TAs (e.g. from pig (UniProt ID: P80147) and mouse (UniProtID: P61922) in subfamily 1OHV) to GABA, L-3-aminoisobutyrate (LAIB)and βAla (Buzenet et al., 1978; Schousboe et al., 1973; Tamaki et al.,2000) have been investigated in detail (Markova et al., 2005; Storiciet al., 1999). The F269, which is highly conserved in the eukaryotic en-zymes, narrows the active site at the ‘top’ of PLP, thereby preventingthe binding of larger substrates. We propose that most of the 233 se-quences in the OrnTL DB that match the fingerprint I46, R132, E185,Q216, F269, R353 encode GABA:αKG TAs with a narrow substrate scope.
A unique structural feature is found in the pig enzyme, where posi-tions C98 and C101 of both subunits form a [2Fe–2S]-cluster at thedimer interface, but its function is unknown (Storici et al., 2004; Sungand Kim, 2000) (see Fig. 13B). It is suggested to be involved in an activa-tion mechanism, especially in higher organisms, because this [2Fe–2S]-cluster was not found in bacteria or yeast except for the basidiomycete
Ustilago maydis. This explains why both, C98 and C101, are onlyconserved in the mammal enzymes within the 1OHV subfamily.
In bacterial GABA:αKG TAs (found in subfamily 3Q8N) F269 is inmost cases replaced by a glycine (found in 91% of sequences in subfam-ily 3Q8N), thereby creating space and allowing for a more relaxed sub-strate spectrum (compare Fig. 13A & B). E. coli, for instance, has twosuch ‘broad spectrum’GABA:αKGTAs: gabT (PDB ID: 1SFF) is constantlyexpressed and puuE (UniProt ID: P50457) is induced by putrescine.These two enzymes have recently been shown to convert AcOrnin vitro and in vivo (Lal et al., 2014). Lal et al. (2014) for the first time in-vestigated the function of all E. coli class III transaminases systematicallyand demonstrated that four of its transaminases possess partly redun-dant substrate spectra. The gabT enzyme that was further characterised,was additionally shown to convert L-aspartate (Liu et al., 2005) butinterestingly, in contrast to the eukaryotic enzymes, not βAla (Parket al., 1993). The GABA:αKG TA from Pseudomonas aeruginosa (UniProtID: Q9I6M4) also efficiently converts AcOrn at physiological pH and ad-ditionally Orn, putrescine (PUT) and Lys with a higher pH optimumcompared to the GABA:αKG reaction (Voellym and Leisinger, 1976).
We suggest that other class III transaminases, also matching theactive site fingerprint (NOT Y16), I46, R132, E185, Q216, G269 andR353 could also be able to convert AcOrn at physiological and freeα,ω-diamino acids like Orn and Lys at higher pH values.
The prediction of a GABA:αKG TAs substrate scope based on the twoproposed active site fingerprints is unfortunately not possible in allcases. There are three examples described where very subtle aminoacid substitutions in the active sites or even in their entrances changedthe substrate spectra.
The first example is from the yeast Lachancea kluyveri, which hastwo ‘narrow spectrum’ GABA:αKG TAs (UniProt IDs: A5H0J5 andA5H0J6, 57% sequence identity), the first one favours βAla three foldover GABA and the second is selective for only GABA (Andersen et al.,2007). Homologymodelling of these two enzymeswith the pig enzymestructure (PDB ID: 1OHV) as template revealed that the only active sitedifferences are found relatively far away from the cofactor at the P-sideof the active site entrance (substitutions from the βAla converting en-zyme to the GABAαKG TA: P266A, F349Y (A5H0J5 numbering, betweencore positions 266 and 267) and C108D (A5H0J5 numbering, betweencore positions 73 and 74)) (data not shown). How these mutationsare able to effect substrate recognition in such a drastic mannerwithout
16
271 46
353
216
185
215
242
132
129
GABA:αKG TA, 4ATQA)
101
98 269
101
98
46
185
129
215
242
271
216
353
132
GABA:αKG TA, 1OHYB)
Fig. 13. Coordination of GABA (or γ-ethynyl-GABA) by GABA:αKG TA from A) Arthrobacter aurescens (PDB ID: 4ATQ) (Bruce et al., 2012) and B) from pig (PDB ID: 1OHY) (Storici et al.,2004). The substrate/inhibitor-PLP adducts are shown in orange, which are coordinated by R132. R353, responsible for αKG's 1-carboxylate recognition in the other half reaction, isneutralised by E185 at the O-side. F269 in the pig enzyme narrows the P-side and forces R132 and the GABA analogue in a ‘lower’ position, thereby causing the narrow substrate scopeof this enzyme. In theA. aurescens enzyme, due toG269, the P-side providesmore space,which allowsmore freedom for R132 and therefore longer chainωAA likeOrn and Lys are acceptedas well. The iron in the [2Fe–2S] cluster of the pig enzyme is coloured brown.
585F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

reaching the substrate–cofactor complexes remains puzzling withoutstructural information.
The second example of a few active site substitutions influencingGABA:αKG TA relevance in vivo was discovered in Pseudomonassyringae, which has three gabT variants (gabT1, UniProt ID: Q48QA9;gabT2, UniProt ID: Q88AT5 and gabT3, UniProt ID: Q885E5), which pre-sumably have different functions (D.H. Park et al., 2010). Knockout of allthree and separate complementation through recombinant expressionindicated that only gabT2 is able to restore the ability to grow onGABA as the sole carbon and nitrogen source. The in vivo function ofgabT1 and gabT3 remains unclear. Homology modelling with theE. coli enzyme structure (PDB ID: 1SFF) as the template revealed thatthe only active site differences that might have a direct influence onGABA:αKG TA activity due to involvement in coordination from gabT2to gabT1 are K142M and E185D, and from gabT2 to gabT3 only isQ80N (Q88AT5 numbering, between core positions 73 & 74) (data notshown). Even though the exchange of glutamate to aspartate at position185 seems very subtle at a first glance, especially as it is not directly in-volved in substrate binding, a different substrate or reaction specificityof gabT1 seems reasonable with the knowledge that it might preventdual substrate recognition and that an aspartate at position 185was (to our knowledge) only found in the racemases among theclass III transaminase family (see sections 3.6 and 3.7). The case ofgabT3, however, is not explicable without further experimental data. Ifthis enzyme lacks GABA:αKG TA activity, Q80 (Q88AT5 numbering,equal to Q79 in the E. coli gabT (PDB ID: 1SFF))might have a greater im-pact on substrate recognition in these enzymes than anticipated. In thePutrescine:αKG TA from E. coli this residue was found to coordinateputrescine's non-reacting amino group (section 3.1.5).
The third example is a report of two enzymes from thermophilesthat match the ‘broad spectrum’ GABA:αKG TA fingerprint, but preferAcOrn over GABA (Koma et al., 2006) (the exact values, however, re-main unfortunately unknown). A comparison of all hypothetical activesite residues of these enzymes to those of characterised GABA:αKGTAs revealed either a S269 or S267 at the P-side as the only differencethat can be seen from the alignment. However, the N-terminus maynot be aligned properly and its contribution to the active site remainsunclear. These two enzymes' preference for AcOrn over GABA can there-fore not be rationalised without additional structural information.
3.1.5. Putrescine and cadaverine:α-ketoglutarate TAsFingerprint PUT:αKG TAs: F46, K132, E185, Q216, R353, Q119
(P42588 numbering, between OrnTL DB core positions 73 and 74)Summary: the dual substrate recognition is probably achieved as in
ωAA:αKG TAs with the exception that R132 is replaced by a lysine.Putrescine's second amino group is coordinated by Q119 (P42588 number-ing), which is located at the O-side in the variable region between corepositions 73 & 74. The substrate scope is additionally determined bybulky hydrophobic residues narrowing the active site. An additionalN-terminal helix provides increased stability by interactions with theother subunit.
Putrescine (PUT) and cadaverine are biogenic diamines that arefound in almost all living organisms and are known for modulatingtranslation and transcription (Schneider and Wendisch, 2011). Owingto their drastic influences on metabolism, their cellular levels need tobe carefully tuned. Three different pathways for PUT degradation havebeen proposed (Kurihara et al., 2005; Lu et al., 2002; Shaibe et al.,1985). These catabolic routes proceed via acetylation, glutamylation ordirect oxidation and all yield GABA. The oxidation from amine to alde-hyde is realised by amine oxidases or transaminases, the second oxida-tion to GABA is achieved by dehydrogenases. Organisms that applytransamination for the first oxidation step have been shown to eitheruse αKG (Kim, 1964) or pyr (Yorifuji et al., 1997) as amino acceptor.For the discussion of PUT:pyr TAs see section 3.2.4. An additional optionfor PUT transamination can be ‘broad substrate spectrum’ GABA:αKGTAs (Voellym and Leisinger, 1976) or ω-amino acid:pyr TAs (Yonaha
et al., 1977) that accept PUT as the amino donor. Transamination ofnon-acylated PUT leads to free γ-aminobutyraldehyde, which sponta-neously forms the cyclic imine 1-pyrolline (Shaibe et al., 1985).
To date only three cadaverine or PUT:αKG TAs (EC 2.6.1.82) havebeen described: the YgjG enzyme from E. coli (UniProt ID: P42588),which has been extensively studied (Schneider and Reitzer, 2012), acadaverine:αKG-converting enzyme involved in the lysine catabolismin Streptomyces ambofaciens (Untrau et al., 1992) and a partially investi-gated enzyme found in a methanogenic coculture (Roeder and Schink,2009). Unfortunately, only the sequence of the E. coli enzyme isknown, which was found to accept PUT and cadaverine as good aminodonors while GABA and Orn showed only low activity and Lys was notconverted at all (Kim, 1964; Samsonova et al., 2003). Both αKG andpyr were converted, while αKG was a ten times better amino acceptor.The structure of the crystallised E. coli enzyme (PDB ID: 4UOX)was pub-lished recently by Cha et al. (2014) after the OrnTL DB was created andis therefore not included in the database. Even though its sequence isnot found in the OrnTL DB, a protein with 97% identity (UniProt ID:A8APX8) is found in the 1VEF subfamily, which, together with thestructure, allowed for aligning the characterised PUT:αKG TA to theother sequences in the database.
A special feature of this enzyme compared to other class III TAs wasrevealed by its structure: the extended N-terminus folds to an addition-al helix (residues 9–23 in P42588 numbering), that interacts with theother subunit. By this additional interaction, the dimer and thereforetemperature stability is increased (Cha et al., 2014).
Even though there is no structure with bound αKG available, itsbinding at the O-side is clear: E185, Q216 and R353 are present toachieve the dual substrate recognition like in the ωAA:αKG TAs (seesection 3.1.1) (Cha et al., 2014). The αKG coordination at the P-side isprobably achieved by K132 (instead of R132 in the other αKGconverting enzymes) and maybe additionally by T269 (see Fig. 14).
Cha et al. (2014) state that specific PUT recognition is realised byQ119 (P42588 numbering, between OrnTL DB core positions 73 and74), because it is found to hydrogen bond PUT's non-reacting aminogroup in chain B of the structure. However, in the two other PUT con-taining active sites (with K242 from chain A and C) PUT adopts orienta-tions that do not allow this H-bond (Fig. 14 shows the active site ofchain B, a scene highlighting PUT's orientation in chain A is providedin the Supplementary PyMOL session). A mutagenesis study wouldtherefore be required to investigate Q119's (P42588 numbering) rolein PUT binding in more detail. This residue is also commonly foundamong the ‘broad spectrum’ GABA:αKG TAs (section 3.1.4), whichmight explain their ability to convert PUT as well. The reduced activityof the PUT:αKG TA towards GABA and AcOrn may be explained by thenarrow, hydrophobic active site of the enzyme. Residues F46, F145,F327 (P42588 numbering between core positions 266 and 267) andL419 (P42588 numbering between core positions 348 and 350), proba-bly hamper their binding. The insertion of amino acids between posi-tions 348 and 350 and their orientation (protruding into the activesite; L419 in particular) is characteristic for this enzyme and not foundin other class III TA structures (Cha et al., 2014). Additionally, K132 isnot as efficient for the binding of ωAA's carboxylate as found for thehuman Orn:αKG TA (see section 3.1.1).
A fingerprint identifying PUT:αKG TAs should therefore contain F46,K132, E185, Q216, R353 and Q119 (P42588 numbering, between OrnTLDB core positions 73 and 74), which matches only 15 sequences in theOrnTL DB. However, as most active site residues (except the dual sub-strate recognition residues and Q119) determine the substrate specific-ity by unspecific hydrophobic interactions or by narrowing the activesite, several other combinations of hydrophobic active site residuesmay probably achieve a comparable substrate scope.
3.1.6. 3-Acetyloctanal transaminase (PigE)Summary: only one enzyme with this activity is known. Structural in-
formation (PDB ID: 4PPM) became available after the revision of this
586 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

article. The substrate scope is unknown; we propose that it prefers Glu asdonor and has a broad spectrum for aldehydes as acceptor.
The biosynthesis of the red tripyrrole pigment prodigiosin in Serratiasp. was found to involve the class III transaminase pigE (UniProt ID:Q5W267) for aminating the aldehyde of the precursor 3-acetyloctanal(3AcOc) (Williamson et al., 2005). This enzyme has unfortunately onlybeen investigated for 3AcOc conversion and amino donor and acceptorspectra remain unknown. The crystal structure of this enzyme has beensolved recently (Lou et al., 2014), but its structure (PDB ID: 4PPM) wasnot available in the PDB until the revision of this article was completed.The sequence alignment to its closest homolog in the OrnTL DB, howev-er, due to the standard dual substrate recognition residues at the O-side(E185, Q216, R353, see section 3.1.1) and K132 at the P-side, suggeststhat Glu is the preferred donor. 3AcOc recognition cannot be predictedwithout structural information, but is probably achieved by unspecificinteractions. This enzymemust, however, be able to somehow discrim-inate the aldehyde from the keto function in 3AcOc. We therefore pro-pose that pigE might possess a broad substrate scope for aldehydes. Incase this enzyme is enantioselective at C3, it might be a valuable toolfor biocatalytic kinetic resolutions of aldehydes.
3.1.7. 2-amino-4-oxobutyrate transaminases (diaminobutyrate TAs)Fingerprint for DABA TAs unknown — P-side substrate recognition
remains unclearSummary: substrate coordination at the O-side is achieved as in
ωAA:αKG TAs (E185 & Q216), only R353 is replaced by a lysine. Substratebinding at the P-side and the molecular basis for α/γ-amino group discrim-ination in DABA cannot be predicted.
Diaminobutyrate transaminases (DABA TAs) catalyse thefirst step inthe biosynthesis of the compatible solute ectoine and are thereforemainly found in halophile or halotolerant species (Schwibbert et al.,2011). DABA:αKG transamination in some species does not need an
additional enzyme and can be achieved by ‘broad spectrum’ GABA:αKGTAs, as found in Pseudomonas species (Brohn and Tchen, 1971), butmany halotolerant organisms were shown to have a separate enzymefor DABA synthesis. The amino donor in the reaction from L-2-amino-4-oxo-butyrate to L-2,4-diaminobutyrate might be either glutamatein Glu:2-amino-4-oxobutyrate TAs (Vandenende et al., 2004) (EC2.6.1.76) or alanine in Ala:2-amino-4-oxobutyrate TAs (Rao et al.,1969) (EC 2.6.1.46), but for most characterised DABA TAs the aminodonor specificity was not investigated, hence we refer to all of them asDABA TAs.
Unfortunatelymost DABA TAs are not similar enough to any enzymewith solved crystal structure and are therefore not aligned to the OrnTLDB. The only aligned sequence is ectB from Virgibacillus pantothenticus(UniProt ID: Q6PR32), which belongs to subfamily 1VEF. To be able toinvestigate sequence conservation among these enzymes, a manualalignment of 20 characterised and predicted (Reshetnikov et al., 2006)sequences has been created (for details see Supplementary dataTable S5 entries 76–96). From this alignment in comparison to theOrnTL DB, the probable substrate coordination enabling residues couldbe derived.
The substrate coordination in the DABA TAs is comparable to that inthe otherωAA:αKG TAs only at the O-side. The commonαKG dual sub-strate recognition residues E185 and Q216 are found there (see section3.1.1). Only R353 is replaced by K353, which is probably able to substi-tute R353's role in the substrate's α-carboxyl coordination. The R353Kreplacement in the E. coli Asp:αKG TA, for instance, was shown to retainthe enzyme's activity (Cánovas et al., 1998). K353might additionally becoordinated from the ‘top’ by E346, which is also conserved in this smallalignment but is found relatively seldom in the OrnTL DB (only 134sequences).
The main difference to other ωAA:αKG TAs is located at the P-side,where position 132 is not conserved and contains mainly non-polar amino acids instead of an arginine. The coordination of theα-carboxylic group of DABA and the 5-carboxylate of αKG need there-fore to be achieved in a different way. When comparing the conserva-tion of all residues at the P-side to other families in the OrnTL DB,mainly three residues attracted attention: Y16, H72 and R273. Thesethree amino acids might be involved in the substrates' carboxylatecoordination. Furthermore, position 269 contains a suitable hydrogenbond donor (N/T/S269) in all DABA TA sequences (except A269 in theP. aeruginosa enzyme, UniProt ID: A3KUH7) and might also be involvedin substrate recognition. However, how these enzymes achieve thediscrimination of DABA's α- and γ-amino group remains unclear. Theelucidation of this interesting feature and the amino donor coordinationat the P-side will require a solved crystal structure.
3.2. ω-Amino acid:pyruvate transaminases
Fingerprint ωAA:pyr TAs: R346 and (NOT D/E132)Summary: amine transaminases (ATAs) are valuable catalysts for
asymmetric amine synthesis, but not all ωAA:pyr TAs possess high ATA ac-tivity. Based on their biocatalytic usefulness,ωAA:pyr TAs can be grouped in‘high activity’ ATAs and ‘low activity’ ATAs. Note that stereoselectivity isusually excellentwithin this enzyme class and thus activity— and substratescope— is the main property of interest when interrogating the protein se-quence for novel useful enzymes.
In addition to theω-TAs, which accept α-ketoglutarate as an aminoacceptor, the class III transaminases of PLP fold type I also includeω-TAswhich accept pyruvate as an amino acceptor (ωAA:pyr TAs). This groupof transaminases comprises GABA:pyr TAs, Taurine:pyr TAs, βAla:pyrTAs, vanillylamine:pyr TAs and also includes examples, which catalysethe transamination of substrates lacking carboxylic acid moieties(amine transaminases, amine:pyr TAs, ATAs, see Fig. 5). ATAs are ofgreat biotechnological interest as they can be utilised for asymmetricamine synthesis. These enzymes proved to be able to compete withestablished chemical methods for industrial amine production (Kohls
215185
216
242
353
119
46
PUT:αKG TA, 4UOX
129
145
132
327269
271
419
Fig. 14. Substrate recognition in the PUT:αKG TA from E. coli (PDB ID: 4UOX). The sub-strate-PLP adduct is shown in orange, which is coordinated by Q119 (P42588 numberingbetween core positions 73 and 74) in chain B of the structure (see scene ‘Fig14_chainA’in the Supplementary PyMOL session for the PUT orientation in chain A). Residues of thevariable regions are shown in yellow (Q119, F327 and L416 (P42588 numbering)). The hy-drophobic residues F46, F145, F327 and L416 form a narrow and hydrophobic active site(entrance), which is supposed to prevent binding of bulkier and more polar substrates.The ‘glutamate switch’ (E185, Q216, R353) ismost probably responsible for dual substraterecognition and togetherwith K132 bindsαKG. The loop including L416narrowing the ac-tive site entrance is a major difference of the PUT:αKG TA compared to other enzymes inthe OrnTL DB and therefore belongs to the variable regions.
587F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

et al., 2014). The broad applicability of these enzymes in biocatalysishave been reviewed recently (Berglund et al., 2012; Höhne andBornscheuer, 2009, 2012; Kroutil et al., 2013; Rudat et al., 2012).
The different amino donor specificities within this group are oftennot clearly distinguishable, as most of the characterised enzymes havea rather relaxed substrate specificity, but we attempted to group themby their usefulness for biocatalytic amine synthesis. Originally, ωAA:pyr TAs were described to be identical to βAla:pyr TAs (EC 2.6.1.18)(Yun et al., 2004), but it turned out that not all βAla:pyr TAs couldalso convert a variety of different ω-amino acids and amines and thatothers convert several amines and ω-amino acids, but not β-alanine(Sayer et al., 2013). Among the ωAA:pyr TAs, it is not possible toconclude an enzyme's ability to convert amines from only regardingsequence similarity to known ATAs as the group of ωAA:pyr TAs is rel-atively diverse and are found in six different subfamilies of the OrnTLDB (4E3Q, 3HMU, 3I5T, 3GJU, 3A8U and 3N5M). In each subfamilyamine converting enzymes have been described. By experimentallycharacterising four TAs belonging to subfamilies 3HMU, 3I5T and 3GJU(PDB IDs: 3HMU, 3I5T, 3FCR and 3GJU), we found that only twoof them (3HMU and 3I5T) possessed relatively high activity towardsstandard ATA substrates such as 1-phenylethylamine (around 0.5 U/mg or higher). The ‘low activity’ ATAs showed at least 20 times less ac-tivity towards those amines. Interestingly, the GABA:pyr activity (mea-sured as reverse reaction) was comparable in all of them (Steffen-Munsberg et al., 2013b). We therefore hypothesised that the naturalfunction of these enzymes is GABA:pyr transamination, while amineconversion is based on substrate promiscuity, which is pronounced dif-ferently among the four enzymes. By comparing their active sites, it wasfound that the mechanism for dual substrate recognition was the samein all four (see section 3.2.1), while there were main differences at theO-side between the ‘high activity’ and the ‘low activity’ ATAs (see sec-tion 3.2.3).
3.2.1. Dual substrate recognition: the flipping arginineAs most knownωAA:pyr TAs transaminate a variety of amino donors
with pyruvate, their substrate recognition has necessarily to be quite flex-ible. While the P-side only needs to accommodate small alkyl groups(ethyl or methyl) or only a proton, the O-side must on the one hand beable to coordinate the carboxylate of pyruvate and ω-amino acids, buton the other hand also accommodate the bulky hydrophobic substituentof the amine substrates. The dual substrate recognition that enables car-boxylate and hydrophobic group binding is achieved by a flexible, so-called ‘flipping’ arginine R346 (Steffen-Munsberg et al., 2013a). This argi-nine is highly conserved in the ωAA:pyr TA containing subfamilies (ex-cept 3N5M) and it is most probable that the same dual substraterecognition mechanism is utilised in all these enzymes: the flexible argi-nine might form a salt bridge with the 1-carboxylate of the amino accep-tor (e.g. pyruvate)when it is in its ‘flipped in’ conformation (see Fig. 15A).By ‘flipping upwards’ and thereby out of the O-side, it allows for the ac-commodation of large hydrophobic groups of the amine (e.g. the phenylgroup of (S)-1-phenylethylamine, see Fig. 15B). Furthermore, the coordi-nation of ω-amino acids' 1-carboxyl group is also achieved by R346 (foran animation of the dual substrate recognition involving R346 in ATAs,see Supplementary video). Its side chain is able to adopt sufficient confor-mations that are required to bind ω-amino acids of different length (seeFig. 15C & D and section 3.2.3 for a more detailed discussion of the sub-strate recognition). The central role of this arginine for the ‘dual’ substraterecognition was proven by the R346A mutation, which drastically de-creased the activity towards keto acids (pyruvate and succinic semialde-hyde), whereas amine transamination was hardly effected (Steffen-Munsberg et al., 2013a). We therefore suggest the fingerprint R346 andNOT D/E132 (to discriminate from the DAPA TAs, which also have R346for DAPA coordination; see section 3.3.5) to identify enzymes withωAA:pyr TA activity among the class III transaminase family. There are,however, also enzymes with a substrate specificity for pyr or Ala that donot match this fingerprint, such as Ala:glyoxylate TAs 2 (see section
3.3.6). These enzymes are, however, not comparable as they do notneed a dual substrate recognition at the O-side as only α- and β-aminoacids are converted. A notable difference is found in the vanillylamine:pyr TAs from chilli pepper and a GABA:pyr TA from tomato (see Sup-plementary data Table S5 entries 125, 126 and 131) that have a S346.In this case pyruvate is probably only coordinated by W47, which isalso involved in the ‘high activity’ ATAs (see Fig. 15A), as there isno other residue at the O-side of this enzyme to replace R346.Whether an asparagine at position 353, which is present in thesethree enzymes, is additionally involved in this coordination remainsunclear.
3.2.2. Natural function of amine transaminasesThe natural function of enzymes with ATA activity is not known in
most cases. There are βAla:pyr TAs (Yonaha et al., 1977), GABA:pyr TAs(Steffen-Munsberg et al., 2013a) and vanillylamine:pyr TAs (Weberet al., 2014) described to accept amines, but several enzymes have onlybeen investigated for amine productionwithout testing their in vivo func-tion. We hypothesised that the conversion of amines in many cases is a‘substrate promiscuous’ activity and their natural function might be theconversion of small ω-amino acids (e.g. βAla or GABA) with pyruvate orglyoxylate as the acceptor (Steffen-Munsberg et al., 2013a). Rausch et al.(2013) together with our study further strengthened this hypothesis, asthe known ATAs share high sequence identity with characterised βAla:pyr TAs (Supplementary data Table S5 entries 109–116), GABA:pyr TAs(Supplementary data Table S5 entries 127–136) and vanillylamine:pyrTAs (Supplementary data Table S5 entries 125 & 126). Another enzymewith high similarity to known ATAs is spuC from P. aeruginosa that wasdescribed as putrescine:pyr (PUT:pyr) TA, some ATAs, however, werefound to not accept PUT as substrate (section 3.2.4). All these enzymesare found in the same OrnTL DB subfamilies and in most cases havethe same active site residues. Their substrate spectra are relativelyrelaxed and it is therefore likely that many of them possess morethan one natural function (i.e. are involved in more than onemetabolic pathway). However, the conversion of amines is in mostcases (except for vanillylamine:pyr or PUT:pyr TAs) not likely to betheir in vivo purpose. As it is impossible to clearly define an ωAA:pyr TA's function, we focused on their biocatalytic usefulness andonly attempted to distinguish between enzymes with high and lowATA activity and enzymes that show a preference for small β-aminoacids (βAla:pyr TAs, for enzymes converting bulkier β-amino acids seesection 3.3.2 and section 3.3.3 for enzymes converting thioesters of β-amino acids).
3.2.3. Discriminating high and low activity amine transaminases and βAla:pyr TAs
Fingerprint high activity ATAs: W47, A185, R346, NOT D/E132Fingerprint low activity ATAs: Y47, S/T185, R346, NOT D/E132Fingerprint βAla:pyr TAs: W47, S185, R346, NOT D/E132Summary: subtle amino acid exchanges at position 47 and 185 deter-
mine ωAA:pyr TAs' ability to convert amines and β-alanine.Comparisons of the ‘high activity’ATAs to other class III transaminases
revealed that only thosewithW47 and A185 turned out to possess a high‘substrate promiscuous’ ATA activity (Rausch et al., 2013; Steffen-Munsberg et al., 2013a). Enzymes with hydrogen bond donors at thesepositions (e.g. Y47 and T/S185) showed less pronounced activity foramines (Steffen-Munsberg et al., 2013a). Site directed mutagenesisproved that the residuesW47 and A185, among all active site differencesto ‘low activity’ ATAs, are the most important ones for high ATA activity.The ‘low activity’ ATAs from Reugeria sp. (PDB ID: 3FCR) andMesorhizobium loti (PDB ID: 3GJU) showed substantially increased ATAactivity when the Y47W or the T185A single mutations had beenintroduced.
We therefore suggest using W47, A185, R346 and NOT D/E132(to discriminate from the DAPA TAs (see section 3.3.5) to identifyATAs with high activity within the class III transaminase family.
588 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

For ωAA:pyr TAs with low ATA activity we suggest the fingerprintY47, S/T185, R346 and NOT D/E132. Additionally, L46 might havean important role for ωAA:pyr TA activity as it is highly conserved(91% of all sequences that match the fingerprint R346, NOT D/E132). Its function, however, is not yet known.
In addition to thewell described enzymes above, there are some high-ly active amine converting enzymes known that do not match the sug-gested patterns, e.g., the two very similar enzymes from Arthrobacter
citreus (Garcia et al., 2006) and Bacillus megaterium (Hanson et al.,2008) (98% sequence identity), both of which are not included in any se-quence database (see Supplementary data section 5 for their sequences).Furthermore, low ATA activity was found in additional class III transami-nases that are comparably evolutionary distant fromωAA:pyr TAs. Two ofthese enzymes were found in M. loti (UniProt IDs: Q98AI1 and Q98NJ9)that both converted several amines with pyr (Seo et al., 2012), whilenot matching the ωAA:pyr TA fingerprint. These two enzymes, however,
271
46
346
129
185
216
215
A)
47
242
“high activity” ATA, 4E3Q
332
B) “high activity” ATA, 4E3Q
“low activity” ATA, 3GJUC) Ala:pyr TA, 3A8UβD)
Fig. 15. Substrate recognition of amines and α-, β- and γ-amino acids in ωAA:pyr TAs. The modelled quinonoid intermediates of A) alanine in 4E3Q, B) 1-phenylethylamine in 4E3Q,C) γ-aminobutyrate in 3GJU and D) β-alanine in 3A8U are shown in orange. R346 is sufficiently flexible to coordinate the carboxylate of α-, β- and γ-amino acids, but can also ‘flipout’ of the active site to create space for e.g. the phenyl ring of PEA (B). In particular positions 47 and 185 are different in enzymes with high ATA activity (A & B) compared to thosewith low ATA activity (C) and those with preference for β-amino acids (D) as described in Discriminating high and low activity amine transaminases and βAla:pyr TAs section. The inter-mediates were modelled with YASARA Structure (Version 13.6.16) as described elsewhere (Steffen-Munsberg et al., 2013a).
589F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

only possess very low ATA activity (80 times lower activity towardsbenzylamine compared to the Vfl-ATA) (Kwon et al., 2010) and are there-fore not discussed here in detail. Two enzymes with high ATA activitywere found among the βPhe TAs, which are discussed with their relatedenzymes in section 3.3.2. These findings indicate that there might bemany more enzymes having a promiscuous ATA activity, but most en-zymes are not characterised systematically at the moment.
Still puzzling are the structural features that allow for β-alaninetransamination as some amine accepting enzymes convert β-alanine while others do not (Sayer et al., 2013). The first describedamine-converting transaminase (from Pseudomonas putida, PDBID: 3A8U (Watanabe et al., 1989)) for instance prefers βAla overcarboxylate free substrates (Yonaha et al., 1977), while severallater described ‘high activity’ ATAs do not convert βAla (e.g. the en-zymes from Vibrio fluvialis (Vfl-ATA, PDB ID: 4E3Q) (Shin et al.,2003) and C. violaceum (Cvi-ATA, PDB ID: 4A6T) (Kaulmann et al.,2007)). Sayer et al. (2013) reasoned that a difference in activesite flexibility is important for amine conversion, but prevents theconversion of βAla in the broad spectrum ATAs: the left-handedhelix (α2, see section 2.2) in Cvi-ATA was inverted in thegabaculine (a common GABA homolog TA inhibitor) bound crystalstructure (PDB ID: 4BA5), compared to the gabaculine structure ofthe βAla:pyr TA from P. aeruginosa (PDB ID: 4B98), where the left-handed helix stayed intact. The observed inversion of the left-handed helix in Cvi-TA might, however, not be necessary foramine binding, but instead be forced by the inhibitor's rigidity:the inhibitor would clash with L46 and W47 if the helix α2 wasnot inverted. Normal ATA substrates are less rigid and thereforeshould be able to bind without α2 inversion.
However, when comparing the active site residues of ATAs withpreference for βAla (see Supplementary data Table S5 entries109–116 and sequence–function matrix Table 2) with those ofbroader spectrum ATAs or GABA:pyr TAs that do not accept βAla(see Supplementary data Table S5 entries 99–105), the previouslymentioned positions 47 and 185 are found to be specifically con-served. The occurrence of W47 and S185 among the βAla:pyr TAsmight be necessary to fix βAla's carboxylate in the proper positionfor catalysis, as found when modelling the quinonoid intermediateof βAla in the structure 3A8U (see Fig. 15D). Other ATAs have A185or Y47 instead of S185 orW47 (Fig. 15A–C), indicating that they arenot able to fix the carboxylate in the right position for βAA conver-sion, whereas αAA and γAA are bound in a productive way.3 Fur-ther experiments are needed to clarify, whether these are themain factors that facilitate βAla conversion.
However, class III transaminase enzymes developed severalways for βAla binding, which is found in other pyr and βAla convertingenzymes like tau:pyr TAs (section 3.2.5), Ala:glyox TAs 2 (section 3.3.6)and βPhe-TAs (section 3.3.2). Owing to the different requirements forthe binding of their natural substrates these enzymes are equipped withdifferent solutions for substrate recognition, but all allow for βAlacoordination.
3.2.4. Cadaverine/putrescine:pyruvate TAsSummary: the only two known sequences have not been tested for PUT:
pyr activity in purified form and share exactly the same active site residueslike the ‘high activity’ ATA 3HMU. 3HMU, however, does not convert freePUT but its acylated derivatives.
For an introduction to putrescine and cadaverine, see section3.1.5 about PUT:αKG TAs.
The knowledge of PUT:pyr TAs ismore limited compared to theαKGaccepting enzymes. This activity has been found in crude extracts of
Nocardioides simplex (Kaneoke et al., 1994) and was purified fromArthrobacter sp. TMP-1 (Yorifuji et al., 1997), both without sequenceinformation of the responsible enzymes. In P. aeruginosa the enzymespuC (UniProt ID: Q9I6J2) has been suggested to catalyse the PUT:pyrtransamination due to its PUT dependent up regulation, the disruptionof the organism's ability to utilise PUT as sole N- or C-source by knock-out mutations and PUT:pyr activity measurements in crude extracts(Chou et al., 2008; Lu et al., 2002). An analogue enzyme (UniProt ID:Q88CJ8) was induced by PUT in P. putida as well (Bandounas et al.,2011). Furthermore, spuC homologs have also been induced by PUT incoastal bacterioplankton as shown by a metatranscriptomic analysis(Mou et al., 2011).
The spuC enzyme from P. aeruginosa and its closest homolog inP. putida, that are found in the 3HMU subfamily, share 59 and 58% iden-titywith the amine transaminase 3HMU, respectively and all three haveidentical active site residues (see sequence–function matrix Table 2).However, when 3HMU was tested with different PUT derivatives, itturned out to convert glutamyl-PUT and acetyl-PUT but not PUT (LeaKennel, unpublished results). The connection of spuC and its homologsto the putrescine catabolism will therefore need to be investigated inmore detail, as free putrescine conversion by spuC seems to be unlikely.Unfortunately, P. aeruginosa was only tested for PUT:pyr TA activity incrude extracts and spuC was never purified. The involvement of addi-tional enzymes in the detected activity can therefore not be ruled out.
3.2.5. Taurine:pyruvate TAsSummary: sulfonate/carboxylate coordination is different from all other
ωAA:pyr TAs. The left-handed helix α2 is probably also oriented differentlybecause tau:pyr TAs have a glycine inserted before position 46.
As taurine (tau) is one of themost abundant small organic solutes inseveral animals, many bacteria have developed ways to utilise it as a S-,C- or N-source, where tau:pyr transamination (EC 2.6.1.77) is involvedinmost cases (Laue and Cook, 2000). Unfortunately only four sequencesof transaminases with proven tau:pyr activity are known and no struc-tural information is available (three in 3N5Mand one in 3HMU subfam-ily, see Supplementary data Table S5 entries 137–140). The enzymefrom Bilophila wadsworthia (UniProt ID: Q9APM5) is the only examplethat has been characterised for its substrate scope (Laue and Cook,2000): small α- and β-amino acids are accepted. Hypotaurine, taurineand β-alanine are the best amino donors (in that order), while pyruvateand 2-ketobutyrate can be employed as the amino acceptor.
The three enzymes in the 3N5M subfamily share the majority of ac-tive site residues but the enzyme from Rhodococcus opacus (UniProt ID:Q6JE91) (Denger et al., 2004) is completely different. All these enzymesemploy a different mechanism for substrate recognition compared toother ωAA:pyr TAs, as they do not have R346 or other basic residuesat the O-side (see sequence–function matrix Table 2). Within thethree enzymes in 3N5M subfamily the sulfonate coordination mightbe realisedwith R145 pointing towards the active site from the entrance‘bottom’ and R414 (Q9APM5 numbering, first of seven amino acids be-tween core positions 349 and 350), which might also point towardsthe active site if this loop is folded like in the 3N5M structure. W47,which is found in these three enzymes, might additionally be involvedin carboxylate coordination, but the region of the left-handed helix α2has a glycine insertion (before core position 46) that might completelychange the tryptophan's orientation compared to ATAs. It is not possibleto elucidate these enzymes' substrate recognition without structural in-formation and we are therefore not able to suggest a sequence finger-print for this specificity.
3.3. ω-Transaminases with unusual acceptor spectrum
Even though most characterised enzymes within the group of classIII transaminases are specific for transaminations with αKG or pyr asacceptor, there are several exceptions known as well. In this sectionwe summarise enzymes with an ‘unusual’ substrate scope that either
3 Exception: the enzyme from Rhodobacter sphaeroides (PDB ID: 3I5T) has W47 andS185. A possible explanation for its lack of βAla:pyr TA activity (Steffen-Munsberg et al.,2013b) might be its R142 that is pointing in the active site from the ‘entrance bottom’
and thus probably disrupts the proper coordination (Supplementary data Figure S5).
590 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604
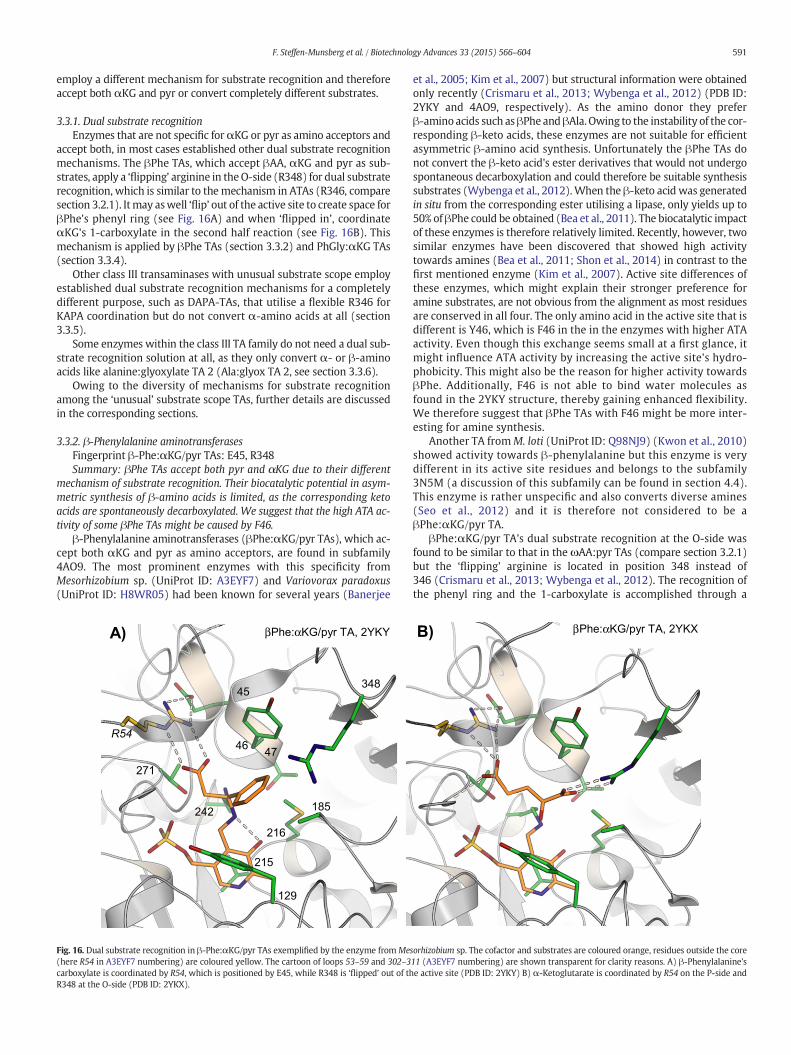
employ a different mechanism for substrate recognition and thereforeaccept both αKG and pyr or convert completely different substrates.
3.3.1. Dual substrate recognitionEnzymes that are not specific forαKG or pyr as amino acceptors and
accept both, in most cases established other dual substrate recognitionmechanisms. The βPhe TAs, which accept βAA, αKG and pyr as sub-strates, apply a ‘flipping’ arginine in theO-side (R348) for dual substraterecognition, which is similar to themechanism in ATAs (R346, comparesection 3.2.1). Itmay aswell ‘flip’ out of the active site to create space forβPhe's phenyl ring (see Fig. 16A) and when ‘flipped in’, coordinateαKG's 1-carboxylate in the second half reaction (see Fig. 16B). Thismechanism is applied by βPhe TAs (section 3.3.2) and PhGly:αKG TAs(section 3.3.4).
Other class III transaminases with unusual substrate scope employestablished dual substrate recognition mechanisms for a completelydifferent purpose, such as DAPA-TAs, that utilise a flexible R346 forKAPA coordination but do not convert α-amino acids at all (section3.3.5).
Some enzymes within the class III TA family do not need a dual sub-strate recognition solution at all, as they only convert α- or β-aminoacids like alanine:glyoxylate TA 2 (Ala:glyox TA 2, see section 3.3.6).
Owing to the diversity of mechanisms for substrate recognitionamong the ‘unusual’ substrate scope TAs, further details are discussedin the corresponding sections.
3.3.2. β-Phenylalanine aminotransferasesFingerprint β-Phe:αKG/pyr TAs: E45, R348Summary: βPhe TAs accept both pyr and αKG due to their different
mechanism of substrate recognition. Their biocatalytic potential in asym-metric synthesis of β-amino acids is limited, as the corresponding ketoacids are spontaneously decarboxylated. We suggest that the high ATA ac-tivity of some βPhe TAs might be caused by F46.
β-Phenylalanine aminotransferases (βPhe:αKG/pyr TAs), which ac-cept both αKG and pyr as amino acceptors, are found in subfamily4AO9. The most prominent enzymes with this specificity fromMesorhizobium sp. (UniProt ID: A3EYF7) and Variovorax paradoxus(UniProt ID: H8WR05) had been known for several years (Banerjee
et al., 2005; Kim et al., 2007) but structural information were obtainedonly recently (Crismaru et al., 2013; Wybenga et al., 2012) (PDB ID:2YKY and 4AO9, respectively). As the amino donor they preferβ-amino acids such asβPhe andβAla. Owing to the instability of the cor-responding β-keto acids, these enzymes are not suitable for efficientasymmetric β-amino acid synthesis. Unfortunately the βPhe TAs donot convert the β-keto acid's ester derivatives that would not undergospontaneous decarboxylation and could therefore be suitable synthesissubstrates (Wybenga et al., 2012).When the β-keto acid was generatedin situ from the corresponding ester utilising a lipase, only yields up to50% of βPhe could be obtained (Bea et al., 2011). The biocatalytic impactof these enzymes is therefore relatively limited. Recently, however, twosimilar enzymes have been discovered that showed high activitytowards amines (Bea et al., 2011; Shon et al., 2014) in contrast to thefirst mentioned enzyme (Kim et al., 2007). Active site differences ofthese enzymes, which might explain their stronger preference foramine substrates, are not obvious from the alignment as most residuesare conserved in all four. The only amino acid in the active site that isdifferent is Y46, which is F46 in the in the enzymes with higher ATAactivity. Even though this exchange seems small at a first glance, itmight influence ATA activity by increasing the active site's hydro-phobicity. This might also be the reason for higher activity towardsβPhe. Additionally, F46 is not able to bind water molecules asfound in the 2YKY structure, thereby gaining enhanced flexibility.We therefore suggest that βPhe TAs with F46 might be more inter-esting for amine synthesis.
Another TA fromM. loti (UniProt ID: Q98NJ9) (Kwon et al., 2010)showed activity towards β-phenylalanine but this enzyme is verydifferent in its active site residues and belongs to the subfamily3N5M (a discussion of this subfamily can be found in section 4.4).This enzyme is rather unspecific and also converts diverse amines(Seo et al., 2012) and it is therefore not considered to be aβPhe:αKG/pyr TA.
βPhe:αKG/pyr TA's dual substrate recognition at the O-side wasfound to be similar to that in the ωAA:pyr TAs (compare section 3.2.1)but the ‘flipping’ arginine is located in position 348 instead of346 (Crismaru et al., 2013; Wybenga et al., 2012). The recognition ofthe phenyl ring and the 1-carboxylate is accomplished through a
46
348
216
185
129
271
242
47
215
R54
45
Phe:αβ KG/pyr TA, 2YKYA)Phe:αβ KG/pyr TA, 2YKXB)
Fig. 16. Dual substrate recognition in β-Phe:αKG/pyr TAs exemplified by the enzyme fromMesorhizobium sp. The cofactor and substrates are coloured orange, residues outside the core(here R54 in A3EYF7 numbering) are coloured yellow. The cartoon of loops 53–59 and 302–311 (A3EYF7 numbering) are shown transparent for clarity reasons. A) β-Phenylalanine'scarboxylate is coordinated by R54, which is positioned by E45, while R348 is ‘flipped’ out of the active site (PDB ID: 2YKY) B) α-Ketoglutarate is coordinated by R54 on the P-side andR348 at the O-side (PDB ID: 2YKX).
591F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

movement of the flexible R348 (compare Fig. 16A and B). On the P-side,coordination of the 1-carboxylate of (S)-β-phenylalanine and the 5-carboxylate of α-ketoglutarate is realised via salt-bridge formationwith R54 (A3EYF7 numbering, between core positions 17 and 18, seeFig. 16). An E45 controls the position of R54's side chain (A3EYF7 num-bering) and allows for the conversion of pyruvate as it permanentlyneutralises the arginine via salt-bridge formation.
As these residues are important for βPhe:αKG/pyr TA activity and allfour characterised enzymes have them, we suggest the fingerprint E45and R348 for the identification of βPhe:αKG/pyr TAs among class IIItransaminases. The combination of E45 and R348 is unique for theseenzymes and the suggested pattern separates subfamily 4AO9 from allother in the OrnTL DB.
3.3.3. Acyl-CoA-β-TAsSummary: this enzyme might be interesting for asymmetric β-amino
acid production, if, in addition to the tested N-acetylcysteamine thioesters,simple esters are accepted as well. The carboxylate coordination is(in contrast to themajority of all class III transaminases) probably achievedby R216.
Recently, a metagenomic approach within a wastewater treatmentplant has discovered a new substrate specificity within the class IIItransaminases (Perret et al., 2011). The enzyme with UniProt IDB0VH76, whose closest homolog in the OrnTL DB is that with UniProtID D8F1V2 (66% identity) in the 2GSA subfamily, was shown totransaminate coenzyme A (CoA) thioesters of β-amino acids with pref-erably αKG but also pyr and it was therefore termed Acyl-CoA β TA(CoAβAA TA). Its physiological role is supposed to be the conversionof β-aminobutyryl-CoA:αKG in an alternative Lys catabolic pathway inthe anaerobic lysine digester Candidatus cloacamonas acidaminovorans.Substrate scope investigations showed that not the whole CoA moietyis needed for substrate recognition, but N-acetylcysteamine thioestersof β-aminobutyryl, β-homoleucine and β-phenylalanine were also ac-cepted. Unfortunately no other esters have been investigated so far,but if the enzyme would also convert e.g. ethyl esters efficiently, itcould be valuable for asymmetric β-amino acid synthesis. Owing tothe instability of the free β-keto acids and the low activity of knownβ-Phe:αKG TAs towards the corresponding esters (Wybenga et al.,2012) (see section 3.3.2), this acyl-CoA-β-TA could be a beneficial alter-native for biocatalysis.
By aligning the sequence (UniProt ID: B0VH76) to its closest homo-log in the OrnTL DB some of its active site residues could be predicted(see sequence–function matrix Table 2) but the substrate recognitionin this enzyme cannot be elucidated without structural information asit is too different from other class III TAs. The only part that we dare tohypothesise is the carboxylate coordination at the O-side by R216,which is the only basic residue there.
3.3.4. D-p-hydroxyphenylglycine:αKG TAsSummary: PhGly:αKG TAs are the only class III transaminases accepting
solely α-amino acids. A dual substrate recognition mechanism is, however,required as both substrates are bound in inverted orientation. The therebygained enantioselectivity for one (S)- and one (R)-amino acid is virtuallyunique among transaminases. Substrate recognition at the O-side isachieved like in β-Phe TAs.
D-Phenylglycine (D-PhGly) and its p-hydroxy derivative are buildingblocks for the commonly used β-lactam antibiotics ampicillin andamoxicillin, respectively. Biocatalytic approaches for their productionusually start from the carbamoyl, involving D-selective carbamoylasesand hydantoinases (Wegman et al., 2001). A second option is the appli-cation of D-p-hydroxyphenylglycine:αKG TAs (PhGly:αKG TA, EC2.1.6.72) in asymmetric syntheses from the corresponding keto acids(Müller et al., 2006). These class III transaminases differ from mostother TAs by converting substrates with inversed enantio preference.Recently it was proven that these enzymes bind D-PhGly in an inverted
fashion compared to other L-α-transaminases (Jomrit et al., 2011). Thephenyl ring is accommodated at the O-side, while the carboxylic func-tion is bound at the P-side. Until now only the two Pseudomonas speciesstutzeri (Wiyakrutta and Meevootisom, 1997) and putida (Müller et al.,2006; Townsend et al., 2002) have been described to possess enzymeswith this activity. Unfortunately the attempt to solve the crystal struc-ture (Kongsaeree et al., 2003) of the P. stutzeri enzymewasonly partiallysuccessful with important parts missing at the P-side in the final (apo)structure (PDB ID: 2CY8, Fig. 17), which still remains unpublished.Owing to the missing regions in this structure it was not included inthe OrnTL DB, but it and the other sequence (see Supplementary dataTable S5, entries 146 and 147) have beenmanually aligned to the closesthomolog structures 2GSA and 4AO9 to compare it to the OrnTL DB.
The O-side of the P. stutzeri enzyme apo structure (Fig. 17) is mainlyfolded as in the other class III TAs and is therefore assumed to be in theactive conformation. From this structure in combination with theknowledge of substrate coordination in βPhe:αKG/pyr TAs (see section3.3.2), it can be concluded that the recognition of the 1-carboxylic groupof Glu is achieved by R348. This arginine may ‘switch’ out of the activesite (as found in the apo structure) to leave a hydrophobic pocket com-prising several Phe and His residues. Unfortunately the P-side is notpresent in the crystal structure and the dual substrate recognition ofGlu's 5-carboxylate and D-PhGly's 1-carboxylate therefore remain un-clear. An E45 in combination with an arginine, which was found in β-Phe:αKG/pyr TAs is missing in these enzymes but the uncommonQ269 (only 6 sequences in the OrnTL DB) might be involved in the rec-ognition here. The fact that the P. putida enzyme accepts pyruvate(Townsend et al., 2002) as acceptor whereas the P. stutzeri enzymedoes not (Wiyakrutta and Meevootisom, 1997) cannot be explainedfrom the sequence alignment. Structural information preferably withbound inhibitors is highly desirable to finally understand the coordina-tion at the P-side.
3.3.5. Diamino pelargonic acid transaminasesFingerprint SAM:KAPA TAs: Y129, D/E132, R346, Y353Fingerprint Lys:KAPA TAs: Y129, D/E132, R346, not Y353Summary: DAPA TAs are highly specific due to fine tuned substrate rec-
ognition. The discrimination between 7- and 8-amino group of DAPA andbetween (8R)- and (8S)-DAPA is achieved by a hydrogen bondingnetwork of Y129, D/E132 and in some cases Y16. Y353 is proposed to deter-mine SAM coordination and is therefore suggested to discriminate Lys:KAPA TAs from SAM:KAPA TAs.
Diamino pelargonic acid transaminases (DAPA TAs) convert 7-keto-8-aminoperlargonic acid (KAPA) to 7,8-diaminopelargonic acid (DAPA)employing either S-adenosyl-L-methionine (SAM) or L-lysine (for sub-strate and product structures, see Table 3) as the amino donor andthereby play an important role in the biotin biosynthesis (Mann andPloux, 2011). Owing to the limitation of biotin anabolic pathways toonly plants and bacteria, DAPA-TAs are potential antibiotic targets.Therefore several studies focusing on selective inhibition of theseenzymes have been conducted (Mann et al., 2009).
Most known DAPA-TAs convert SAM and KAPA to S-adenosyl-2-oxo-4-methylthiobutyric acid (which undergoes β-elimination to resultin 5-methylthioadenosine and 2-oxo-3,4-butenoic acid) and DAPA(Stoner and Eisenberg, 1975). These SAM:KAPA-TAs (EC 2.6.1.62)have been discovered in several bacteria and plants (Supplementarydata Table S5, entries 148–158, 160 and 161). The only examplewhere a DAPA transaminase did not utilise SAM as amino donor wasfound in Bacillus subtilis (PDB ID: 3DU4) (Van Arsdell et al., 2005).This enzyme is closely related to SAM:KAPA-TAs but employs L-lysineas the amino donor and will therefore be referred to as Lys:KAPA-TA(EC 2.6.1.105). Unfortunately, it remains unclear whether this enzymetransaminates the α- or ε-amino group of lysine. The transfer of theε-amino function, however, seems more likely owing to the resultingirreversibility of the overall reaction. As described for Lysε:αKG TAs,the formed product, Δ1 piperideine-6-carboxylic acid, would undergo
592 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

internal Schiff-base formation and therefore virtually disable the backreaction (Soda et al., 1968; Van Arsdell et al., 2005).
All characterised DAPA-TAs have a very narrow substrate scope andonly convert substrate analogues that are very similar to the naturalones (Cobessi et al., 2012; Izumi et al., 1975; Izumi et al., 1981; Mannand Ploux, 2006; Stoner and Eisenberg, 1975; Van Arsdell et al., 2005).Owing to the narrow substrate scope and to their low activity (e.g. asteady-state kcat of 0.013 s−1 for the E. coli enzyme (PDB ID: 1MLZ)(Eliot et al., 2002)) these enzymes are of low value for biocatalyticapplications. But this perfect selectivity combined with the fact ofbeing absent in humans makes them valuable targets for potentialantibiotic substances.
Compared to other class III transaminases, DAPA-TAs selectively con-vert extraordinary substrates and therefore need characteristic patternsto bind these. In particular, the recognition of KAPA needs to be finelytuned to allow the discrimination between the 7-keto and 8-aminogroup. By selectively coordinating the 8-amino group and keeping itaway from the C4′ atom of PLP, the enzymes are able to prevent sidereactions at this position. The positioning is achieved by Y129 and addi-tionally either Y16 or the carbonyl oxygen of residue 269 that bind the8-amino group (see Fig. 18). A highly conserved aspartate at position132, coordinates the tyrosine(s) and thereby keeps them in the rightplace (Käck et al., 1999). This finely tuned recognition even allows theenzyme to discriminate between the (R)- and (S)-configuration at C8of KAPA (Mann et al., 2009). On the O-side of the active site, the carboxylgroup of KAPA forms a salt bridge to R346 (same as the ‘flipping’ arginineinωAA:pyr TAs, section 3.2.1),which is also highly conserved (Eliot et al.,2002; Wybenga et al., 2012). Since these features are unique among theclass III transaminases, KAPA converting enzymes can be selectivelyidentified by the presence of Y129, D132 and R346.
Even though SAMbinding has been studied extensively, its exact ori-entation in the active site still remains unclear. Dey et al. (2010) havebeen able to solve a structure of theMycobacterium tuberculosis enzymecontaining the SAM analogue sinefungin. Unfortunately this structure(PDB ID: 3LV2) was not sufficient to unravel the binding of SAM. Theorientation of sinefungin in this structure must represent an unproduc-tive binding mode for the following three reasons. The first and mostimportant reason is motivated by the enantioselectivity of the reaction.The carboxylic group of sinefungin is located at the P-side, which, owing
to (S)-configuration, results in the amino function pointing away fromPLP. Even though unlikely, the formation of the external aldiminemight still be possible in this orientation but the deprotonation of thesubstrate by the catalytic K242 to form the quinonoid intermediate(see Fig. 2) is definitely not. Since there is no base available on the re-side of the cofactor that could do the deprotonation instead, the carbox-ylic group of SAM and sinefunginwould have to be located at the O-sideto enable transamination. The second reason to doubt SAM bindingbeing represented in the 3LV2 structure is an unexplained mutation atthe active site entrance (H268R), which substantially reshapes theP-side, while thirdly, the substrate analogue differs from SAM in an im-portant feature. An amino group replaces themethyl group at the sulfo-nium ion in SAM and the sulphur itself is replaced by a carbon, whichmodifies the electronic properties of the analogue and therefore mostprobably also its binding. The importance of the sulfonium ion forsubstrate recognition was proven by the absence of activity forS-adenosylhomocysteine, which is a SAM analogue lacking the methylgroup at the sulphur (Dey et al., 2010). Owing to the high conservationof Y353 in all SAM:KAPA-TAs, it was hypothesised that the sulfoniumion is coordinated by the hydroxyl group of this tyrosine (Dey et al.,2010). This proposal can be strengthened by the fact that the onlycharacterised DAPA-TA lacking the tyrosine in that position is the Lys:KAPA-TA from B. subtilis. One could, therefore, hypothesise that thepresence of Y353 might be employed to distinguish the SAM and Lysconverting DAPA-TAs.
3.3.6. Alanine:glyoxylate transaminase 2Fingerprint Ala:glyox TAs2: Y/W145, F269, R353, NOT D/E185Summary: Ala:glyox TA 2, in contrast to most other class III transami-
nases, does not require a dual substrate recognition mechanism at the O-side because only αAA and βAA are converted. The broad substrate scopeincludes small αAA such as Ala, βAA, such as βAla and 3-aminoisobutyrate (AIB) but also bulky αAA such as NG,NG-dimethylarginine. For the conversion of βAA a (R)-enantioselectivity atthe α-position was found (selectivity for DAIB).
The mammalian mitochondrial Ala:glyox TA 2 (AGXT2, EC 2.6.1.44)is one of the class III transaminases with the broadest substrate scopebut is limited to α- and β-amino acids and therefore requires no dualsubstrate recognition at the O-side. Owing to the specificity for severalkey metabolites, its annotation proved to be difficult: the rat Ala:glyoxTA 2 (UniProt ID: Q64565) had previously also been described asD-3-aminoisobutyrate:pyr TA (EC 2.6.1.40), Ala:4,5-dioxopentanoateTA (EC 2.6.1.43), β-Ala:pyr TA 2 (EC 2.6.1.18), 2-aminobutyrate:pyru-vate TA and NG,NG-dimethylarginine:pyr TA (Kontani et al., 1993;Tamaki et al., 2000). This enzyme is supposed to be the mitochondrialcounterpart of the peroxisomal Ala:glyox TA 1 (AGXT1, EC 2.6.1.44)which keeps physiological glyoxylate levels low and is involved in thehyperoxaluria disease (Baker et al., 2004). Although both enzymesshare the ability to catalyse the Ala:glyox conversion, their sequencesand substrate spectra differ substantially. AGXT1, also referred to asSer:pyr TA (EC 2.6.1.51), which only shares 12% identity with AGXT2(UniProt IDs: P21549 and Q9BYV1 in humans, respectively), belongsto a different class of transaminases (class V) and its substrate scope fa-vours small α-amino or keto acids (i.e. Ser, pyr, glyox), but Phe Arg andGlu are also converted by the human Ser:pyr TA (Cellini et al., 2007).These enzymes are of special interest regarding the connection betweensequence and function as Ala:glyox TAs 2 and Ser:pyr TAs both convertAla:glyox transamination despite their low sequence similarity, where-as a close homolog to Ala:glyox TA 2 in Arabidopsis thaliana differs in itssubstrate spectrum and additionally catalyses the Glu:glyox conversion(Liepman and Olsen, 2003).
The Ala:glyox TAs 2, which are found in the 3N5M subfamily, alsoconvert β-amino acids like βAla and D-3-aminoisobutyrate (DAIB)where they showed strict (R)-selectivity for the α-methyl group.Its DAIB:pyr TA activity together with the enantiocomplemen-tary LAIB:αKG TA (EC 2.6.1.22, which is identical to the above described
215
185
325
186
216
353
348
43
46
D-PhGly:αKG TA, 2CY8
Fig. 17. O-side of the D-PhGly:αKG TA from Pseudomonas stutzeri apo structure (PDB ID:2CY8) . The PLP, taken from the aligned β-phenylalanine:αKG TA structure (PDB ID:4AO9), is shown transparently in orange for orientation reasons.
593F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

GABA:αKG TA EC 2.6.1.19, see section 3.1.4), interconvert both enantio-mers of AIB in mammals (Tamaki et al., 2000). In contrast to theGABA:αKG TAs, the Ala:glyox TAs 2 do not convert amino groupsmore distant from the carboxylic function than in β-position (Tamakiet al., 2000).
The conversion of these various substrates raises the question howsubstrate recognition is realised in these enzymes. There is unfortunate-ly no structural information for Ala:glyox TAs 2 available and thereforeonly hypotheses based on conserved positions within these enzymescan be developed. The coordination of the carboxylic group of α-amino acids is probably undertaken by the conserved R353 like in theω-amino acid:αKG TAs (see section 3.1.1), but in contrast to those the‘switching’ glutamate in position 185 is replaced by Val or Ile. Thismight also contribute to the fact that the common dual substrate recog-nition is not possible and no ω-amino acids (except for β-amino acids)are converted. The unique feature of these enzymes within the class IIITA family, namely the conversion of huge α-amino acids likedimethylarginine and the enantioselectivity at the α-position whenconverting the β-amino acid DAIB, might be explained by the uniquecombination of residues Y145 and F269 (only 21 sequences in theOrnTL DB). The aromatic ring of F269 at the P-side might be involvedin cation–π interactions with the guanidinium group ofdimethylarginine, whereas Y145 that points from the entrance towardsthe active site and might be responsible for the recognition of the 4-carboxylic group of aspartate. This hypothesis is strengthened by thefact that glutamate, due to its additional carbon, is not accepted as sub-strate butwhen Y145 is exchanged to a tryptophan like in two Ala:glyoxTA 2 homologous enzymes from Arabidopsis, glutamate is converted aswell (Liepman and Olsen, 2003; Tamaki et al., 2000).
3.4. Glutamate-1-semialdehyde transaminases (2,1-amino mutases)
Fingerprint GSAM: N185, Y267Summary: GSAM transaminates glutamate-1-semialdehyde intramo-
lecularly to form 5-aminolevulinate. A gating loop closes the active site en-trance after substrate binding to prevent the intermediate diamine'srelease. After product formation the entrance is opened again. GSAM isthe only known class III TA that uses PMP in its resting state in contrast toPLP (internal aldimine) in other enzymes.
Glutamate-1-semialdehyde 2,1-aminomutase (GSAM, EC 5.4.3.8) isa unique enzyme within the class III transaminase family, owing to thefact that it does not catalyse the interconversion between an amino-and a keto function of two molecules. GSAM instead catalyses theintraconversion between an amino- and a keto function within thesame molecule i.e. the conversion of glutamate-1-semialdehyde to5-aminolevulinate (Hoober et al., 1988). Another unique feature ofGSAM is that the resting state of this enzyme is in the PMP-form whilethe resting state of the majority of the class III transaminases is in thePLP-form (Smith et al., 1991). This feature might be induced by thehighly conserved N185 (see Fig. 19), which is supposed to modify thecofactor's electron sink properties by coordinating its 3′-hydroxylgroup (Orriss et al., 2010). To which extent the coordination by N185is different from that of Q216, as present in the ωAA:αKG TAswhere the internal aldimine is the resting state (see section 3.1.1), can-not be predicted without mutagenesis studies.
GSAM belongs to the subfamily 2GSA, which consists of 2048 se-quences. Out of these entries, 16 enzymes were experimentallycharacterised and several crystal structures have been solved (Supple-mentary data Table S5 entries 170–185) (Ge et al., 2010; Hennig et al.,1997; Schulze et al., 2006; Stetefeld et al., 2006). Most of these struc-tures showed a third unique feature of GSAM when compared to theother class III TAs, an active site gating loop (residues between corepositions 131 & 155, which cannot be aligned to the OrnTL DB) whichopens and closes the entrance to the active site during catalysis(Contestabile et al., 2000; Stetefeld et al., 2006). The movement of thegating loop is controlled by allosteric communication between thetwo active sites of the homodimeric enzyme. The ability to close the ac-tive site gives GSAM the ability to control substrate entry and productrelease. The intermediate 4,5-diaminovalerate is kept in the active siteto avoid its release as unwanted product. The general opinion aboutthe gating loop is that it is controlled through negative cooperativity:when the substrate binds to one subunit the affinity for the substratedecreases in the other subunit (Hennig et al., 1997; Stetefeld et al.,2006). This leads to the enzyme converting the substrate one subunitat the time. This negative cooperativity has been shown to be controlledby the communication of a network of the amino acids S98, E101, R108,H153, D155 (Q31QJ2 (from Synechococcus elongatus) numberingbetween core positions 131 and 155) and R111 that connects the activesites (Stetefeld et al., 2006). This theory is, however, challenged by
B) Lys:KAPA TA, 3DU4
16
346
353
46
47
132
129
242
271
A) SAM:KAPA TA, 1QJ3
185
216
Fig. 18. Coordination of KAPA in A) SAM:KAPA TA (enzyme from E. coli PDB ID: 1QJ3) and B) Lys:KAPA TA (Enzyme from B. subtilis, PDB ID: 3DU4). PLP and KAPA are shown in orange.
594 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

a published structure where both monomers are in the PMP-form(Ge et al., 2010).
GSAM is, like many other PLP-dependent enzymes, inhibited bygabaculine (Grimm et al., 1991). This inhibition has, however, beenshown to decrease significantly when an active site methionine is mu-tated to an isoleucine (M216I) (Ferradini et al., 2011; Grimm et al.,1991; Orriss et al., 2010). Since GSAM is not present in animals it is apromising target for herbicides and antibiotics and this mutation alsomakes selective growth via the addition of gabaculine possible(Hennig et al., 1997).
Asmany residues of GSAM that are involved in substrate binding be-long to the variable regions, establishment of an active site fingerprintonly based on core positions was not trivial. We suggest applying thetwo amino acids N185 and Y267 to identify enzymes performing the in-tramolecular transamination of glutamate-1-semialdehyde to form5-aminolevulinate. N185 modulates the cofactors electronic properties(Orriss et al., 2010) and Y267 is involved in a hydrogen bondingnetwork with the substrate and with S163 (Q31QJ2 numbering, in thegating loop) (see Fig. 19) (Stetefeld et al., 2006). Additional hints forGSAM activity could be R108, which is highly conserved in the subunitcommunication network (Stetefeld et al., 2006) involved in inter-subunit signalling and the presence of a gating loop between core posi-tions 131–155 that cannot be aligned to the other known class III TAs.
3.5. Decarboxylation dependent TAs: the 2,2-dialkylglycine decarboxylases
Fingerprint DGD: Q46, (W129), R353Summary: DGD is the only known PLP fold type I enzyme that catalyses
both, transamination and oxidative decarboxylation to a comparable ex-tent. The reaction specificity is determined by the orientation of thesubstrate's substituents relatively to the cofactor's plane: if the Cα-H ispointing towards the si-face, transamination is preferred, if Cα–CO2 ispointing towards the si-face, decarboxylation is favoured.
In addition to the above-mentioned transaminases, the family ofclass III TAs also comprises the decarboxylation dependent transami-nase, 2,2-dialkylglycine decarboxylase (DGD, EC 4.1.1.64). This enzyme
catalyses the decarboxylation and transamination of dialkylglycine sub-strates with an α-keto acid (e.g. pyr) as the amino acceptor (Sun et al.,1998). This enzyme is uniquewithin PLP fold type I because it can cleaveboth the substrate's Cα–CO2 bond and the Cα–H bond in the sameactive site. Furthermore, it catalyses an oxidative decarboxylation(forming α-keto product), which is in contrast to most other aminoacid decarboxylases where non-oxidative decarboxylation is favoured(forming an amino product) (Li et al., 2012). By protonating the C4′ ofthe quinonoid intermediate after decarboxylation instead of the Cα asin non-oxidative decarboxylases, the ketimine is formed, which leadsto transamination and release of the keto product from PMP. PMP'samino group is then transferred to an amino acceptor in the second,transamination half reaction restoring the internal aldimine (see Fig. 1).
The selectivity over the site of protonation (Cα over C4′ or viceversa) has been investigated in both non-oxidative and oxidativedecarboxylases (Bertoldi et al., 2008; Jackson et al., 2000; Sun et al.,1998). Sun et al. (1998) proposed an active site model for the DGDfrom Pseudomonas cepacia, whereby the site of protonation by thecatalytic lysine is controlled by the tilt of the cofactor, which in turn, isinfluenced by the identity of the substrate. Themodel consists accordingto the Dunathan principle (see section 1.3) of three binding subsites:(A) the activated position for bond cleavage (may bind Cα–H or Cα–
CO2), which is perpendicular to the plane of the cofactor and locatedat the si-face; (B) a second carboxylate binding subsite at the O-side ofthe cofactor and (C) a hydrophobic subsite at the P-side of the cofactor(see Figs. 1 and 20). For α-H-α-amino acids, for which both reactionsare possible, the accommodation of the substrate's carboxylate in theA subsite would lead to decarboxylation, whereas binding in the B sub-site would place the Cα–H in the A pocket and would lead to deproton-ation and therefore to transamination. However, dialkylglycinesubstrates cannot be transaminated due to their lack of a Cα–H. There-fore binding of their carboxylates in the A subsite leads to decarboxyl-ation, while binding in the B subsite would represent an unreactivebindingmode. Structural investigationswith phosphonate substrate an-alogues, however, revealed that the position of the scissile carboxylate isprobably not perfectly perpendicular, as the orbital overlap with the
185
E400
A)
46
R26
267
271
S23
242
S157
215
129
GSAM, 2HP1
216
B) GSAM, 2HP2
Fig. 19. Substrate and product binding in GSAM. The substrate-cofactor intermediates are shown in orange and residues that belong to the variable regions (S29, R32, S163 and E406(Q31QJ2 numbering)) are shown in yellow. The cartoon is shown transparent for clarity reasons. A) After binding glutamate-1-semialdehyde, the gating loop is closed to prevent the re-lease of the intermediate 4,5-diaminovalerate (PDB ID: 2HP1). Y267 indirectly coordinates the substrate's carboxylate and the gating loop's S163 (Q31QJ2 numbering) B) The gating loopopenswhen 4,5-diaminovalerate is bound at its 4-amino group, opening up the active site entrance. Thereafter the product 5-aminolevulinate is formed throughwater attack andmay bereleased from the active site (PDB ID: 2HP2).
595F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604
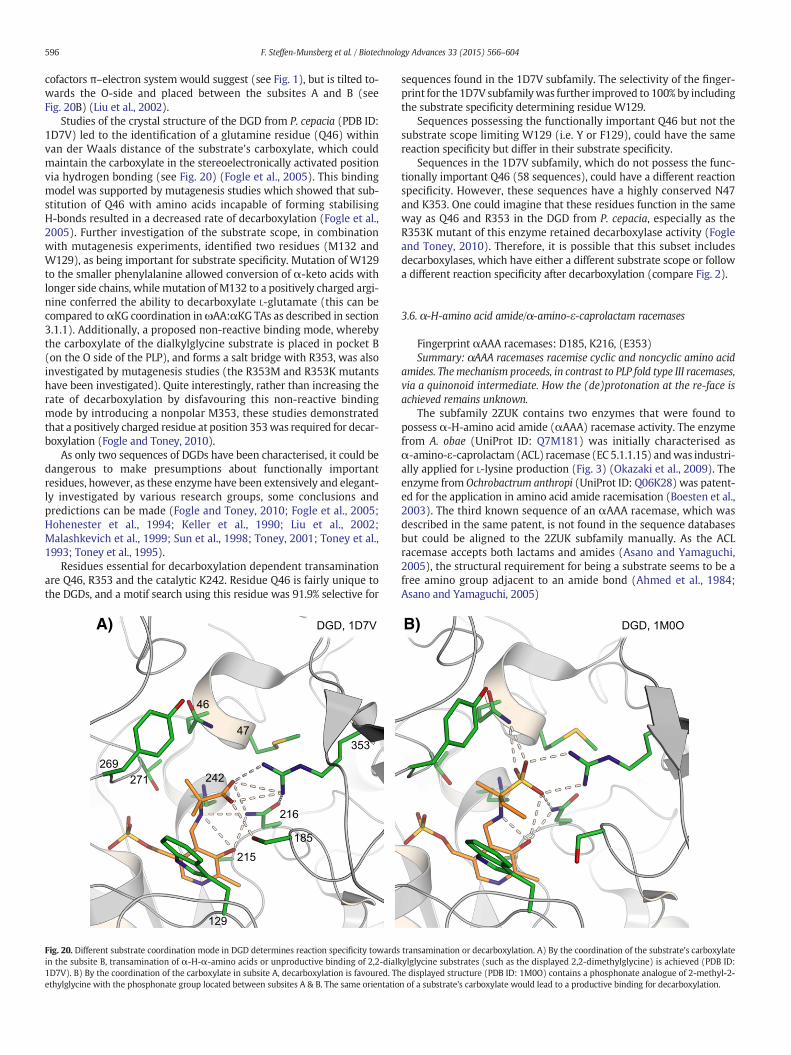
cofactors π–electron system would suggest (see Fig. 1), but is tilted to-wards the O-side and placed between the subsites A and B (seeFig. 20B) (Liu et al., 2002).
Studies of the crystal structure of the DGD from P. cepacia (PDB ID:1D7V) led to the identification of a glutamine residue (Q46) withinvan der Waals distance of the substrate's carboxylate, which couldmaintain the carboxylate in the stereoelectronically activated positionvia hydrogen bonding (see Fig. 20) (Fogle et al., 2005). This bindingmodel was supported by mutagenesis studies which showed that sub-stitution of Q46 with amino acids incapable of forming stabilisingH-bonds resulted in a decreased rate of decarboxylation (Fogle et al.,2005). Further investigation of the substrate scope, in combinationwith mutagenesis experiments, identified two residues (M132 andW129), as being important for substrate specificity. Mutation of W129to the smaller phenylalanine allowed conversion of α-keto acids withlonger side chains, whilemutation of M132 to a positively charged argi-nine conferred the ability to decarboxylate L-glutamate (this can becompared toαKG coordination inωAA:αKG TAs as described in section3.1.1). Additionally, a proposed non-reactive binding mode, wherebythe carboxylate of the dialkylglycine substrate is placed in pocket B(on the O side of the PLP), and forms a salt bridge with R353, was alsoinvestigated by mutagenesis studies (the R353M and R353K mutantshave been investigated). Quite interestingly, rather than increasing therate of decarboxylation by disfavouring this non-reactive bindingmode by introducing a nonpolar M353, these studies demonstratedthat a positively charged residue at position 353was required for decar-boxylation (Fogle and Toney, 2010).
As only two sequences of DGDs have been characterised, it could bedangerous to make presumptions about functionally importantresidues, however, as these enzyme have been extensively and elegant-ly investigated by various research groups, some conclusions andpredictions can be made (Fogle and Toney, 2010; Fogle et al., 2005;Hohenester et al., 1994; Keller et al., 1990; Liu et al., 2002;Malashkevich et al., 1999; Sun et al., 1998; Toney, 2001; Toney et al.,1993; Toney et al., 1995).
Residues essential for decarboxylation dependent transaminationare Q46, R353 and the catalytic K242. Residue Q46 is fairly unique tothe DGDs, and a motif search using this residue was 91.9% selective for
sequences found in the 1D7V subfamily. The selectivity of the finger-print for the 1D7V subfamilywas further improved to 100% by includingthe substrate specificity determining residue W129.
Sequences possessing the functionally important Q46 but not thesubstrate scope limiting W129 (i.e. Y or F129), could have the samereaction specificity but differ in their substrate specificity.
Sequences in the 1D7V subfamily, which do not possess the func-tionally important Q46 (58 sequences), could have a different reactionspecificity. However, these sequences have a highly conserved N47and K353. One could imagine that these residues function in the sameway as Q46 and R353 in the DGD from P. cepacia, especially as theR353K mutant of this enzyme retained decarboxylase activity (Fogleand Toney, 2010). Therefore, it is possible that this subset includesdecarboxylases, which have either a different substrate scope or followa different reaction specificity after decarboxylation (compare Fig. 2).
3.6. α-H-amino acid amide/α-amino-ε-caprolactam racemases
Fingerprint αAAA racemases: D185, K216, (E353)Summary: αAAA racemases racemise cyclic and noncyclic amino acid
amides. Themechanism proceeds, in contrast to PLP fold type III racemases,via a quinonoid intermediate. How the (de)protonation at the re-face isachieved remains unknown.
The subfamily 2ZUK contains two enzymes that were found topossess α-H-amino acid amide (αAAA) racemase activity. The enzymefrom A. obae (UniProt ID: Q7M181) was initially characterised asα-amino-ε-caprolactam (ACL) racemase (EC 5.1.1.15) andwas industri-ally applied for L-lysine production (Fig. 3) (Okazaki et al., 2009). Theenzyme from Ochrobactrum anthropi (UniProt ID: Q06K28) was patent-ed for the application in amino acid amide racemisation (Boesten et al.,2003). The third known sequence of an αAAA racemase, which wasdescribed in the same patent, is not found in the sequence databasesbut could be aligned to the 2ZUK subfamily manually. As the ACLracemase accepts both lactams and amides (Asano and Yamaguchi,2005), the structural requirement for being a substrate seems to be afree amino group adjacent to an amide bond (Ahmed et al., 1984;Asano and Yamaguchi, 2005)
242
269
271
46
47
353
216
185
129
215
A) DGD, 1D7V B) DGD, 1M0O
Fig. 20. Different substrate coordination mode in DGD determines reaction specificity towards transamination or decarboxylation. A) By the coordination of the substrate's carboxylatein the subsite B, transamination of α-H-α-amino acids or unproductive binding of 2,2-dialkylglycine substrates (such as the displayed 2,2-dimethylglycine) is achieved (PDB ID:1D7V). B) By the coordination of the carboxylate in subsite A, decarboxylation is favoured. The displayed structure (PDB ID: 1M0O) contains a phosphonate analogue of 2-methyl-2-ethylglycine with the phosphonate group located between subsites A & B. The same orientation of a substrate's carboxylate would lead to a productive binding for decarboxylation.
596 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

Racemases are unique among the other types of PLP-dependent en-zymes, as they are capable of deprotonating and protonating on boththe si- and the re-face. For the ACL racemase two mechanisms havebeen proposed, a two base mechanism (where two acid–base groupsare situated on either side of the substrate–PLP complex), and a singlebase mechanism (with a single base capable of accessing both faces).Ahmed et al. (1986) found evidence for a single base mechanism,which is at odds with the two base mechanism described by Okazakiet al. (2009) which was proposed based on structural information. Byanalogy to alanine racemases in PLP fold type III, Okazaki et al. (2009)proposed Y129 at the re-face and K242 at the si-face to enableracemisation in the ACL racemase (see Fig. 21). The mechanism of ACLracemases, in contrast to alanine racemases, is believed to proceed viaa quinonoid intermediate because the cofactor's pyridine nitrogen iskept protonated by the D213 ‘below’ it (Okazaki et al., 2009), whereasit is deprotonated by an arginine residue in PLP fold type III racemases.Fold type III racemases do not require the quinonoid formation forstabilising the carbanionic intermediate: the negative charge is mainlystabilised at the protonated Schiff base (Griswold and Toney, 2011).
The above-mentioned role of Y129 at the re-face for protonation inthe racemisation mechanism has not been confirmed by mutagenesisstudies and the fact that Y129 is found in 58% of the sequences in theOrnTL family (e.g. in ATAs, DAPA TAs and βPhe TAs), places doubt onits suitability as a candidate as a 2nd base (if indeed a two base mecha-nism is followed by these enzymes). Since the mechanism in the αAAAracemases is not yet fully elucidated, no residues determining the reac-tion specificity may be included in the fingerprint and we thereforefocus on the substrate specificity determining residues.
Okazaki et al. (2009) proposed that amide nitrogen recognition inthis enzyme is achieved by D185 (see section 3.7 for a discussion ofthis residue) and that the carbonyl O is coordinated by K216. In the ε-caprolactam bound internal aldimine structure (PDB ID: 2ZUK), K216is kept in place by the coordination of E353 (see Fig. 21). The two resi-dues D185 and K216 that are believed to be essential for substrate rec-ognition in the characterised enzymes (and optionally E353), aretherefore suggested for the active site pattern identifying αAAAracemases. As all 18 sequences in the OrnTL DB with K216 and D185also have E353, including this glutamate in the fingerprint is notnecessary.
3.7. Isoleucine 2-epimerase
Summary: Ile-2-racemisation probably, like in the αAAA racemases,also proceeds via a quinonoid intermediate. The racemisation mechanismis also unknown but we suggest that D185 is important for racemaseactivity in the class III transaminase family because it was found in all sofar characterised enzymes.
The recent discovery of an isoleucine-2-epimerase (Ile-2-epimerase,from Lactobacillus buchneri (UniProt ID: F4FWH4)) which racemises theC2 (=Cα) in aliphatic α-amino acids, further provided insight to theversatility of the family of class III transaminases (Mutaguchi et al.,2013). This enzyme is especially interesting because it shares relativelyhigh sequence similarity with GABA:αKG TAs (e.g. 41% sequence iden-tity to B. subtilis GABA:αKG TA (UniProt ID: P94427)) and is found inthe 3Q8N subfamily but most of the active site residues that are impor-tant for substrate recognition differ from the GABA:αKG TAs (see se-quence–function matrix Table 2) and therefore GABA and αKG are nottransaminated by this enzyme. Owing to the lack of structural informa-tion the mechanism of racemisation cannot be elucidated and from thealignment it is not obvious how the protonation/deprotonation of thesubstrate at the re-face is achieved (the possible involvement of Y129therein is discussed for theαAAA racemases in section 3.6). The cataly-sis of this enzyme, however, seems to be comparable to the αAAAracemases because it also has D213, which presumably leads to PLP'spyridine nitrogen protonation and therefore a mechanism that pro-ceeds via a quinonoid intermediate. The substrate's 1-carboxylate
recognition is most probably achieved, as in GABA:αKG TAs, withR353. The extent to which the relatively uncommon D185 and N216(only present in 30 sequences in the whole OrnTL DB) or A46 and S47(found in only 109 sequences in the whole OrnTL DB) are involved can-not be predicted without further experimental data. Notably, position185 seems to be important for racemase activity within the class IIItransaminase family because the αAAA racemases and the isoleucine2-epimerase share the uncommon D185 (present in 234 of the 12,956sequences in the OrnTL DB). Additionally, position 216 is harbouring avery uncommon K216 or N216 in the two racemases (only 28 se-quences have K216 and only 30 have N216 in the OrnTL DB).
3.8. Enzymes with unclear substrate recognition
In this sectionwe grouped recently discovered class III transaminaseenzymes, for which not sufficient structural or functional data isavailable to suggest specificity determining residues. In particular,the aminosugar TAs (section 3.8.1) and the multi-domain enzymes(section) share too low sequence similarity with any known structureto reliably align their sequences to the OrnTL DB. These examples,above all, highlight the lack of structural information within the classIII transaminase family.
3.8.1. Neamine TAs, 2′-deamino-2′-hydroxyneamine and neomycin C TAsSummary: Glu:6′-oxoglucos(amin)yl TAs are the only known
aminosugar converting TAs among the class III transaminases. They are in-volved in aminoglycoside biosynthesis, by selectively aminating 6′-oxoglucos(amin)yl moieties.
Aminosugar converting transaminases are commonly found to be-long to the class VI transaminases (degT/dnrJ/eryC1 family in InterPro),which were recently reviewed by Romo and Liu (2011). The biosynthe-sis of several aminoglycoside antibiotics, such as neomycin, butirosinand kanamycin however, was found to involve class III transaminasesto aminate the C6 atoms of the glucos(amin)yl substituents withglutamate as amino donor. The three different substrate specificitiesGlu:6′-dehydroparomamine TA (EC 2.6.1.93, forming neamine),Glu:2′-Deamino-2′-hydroxy-6′dehydroparomamine TA (EC 2.6.1.94,forming 2′-deamino-2′-hydroxyneamine) and Glu:6‴-deamino-6‴-oxoneomycin C TA (EC 2.6.1.95, forming neomycin C), that may besummarised as Glu:6′-oxoglucos(amin)yl TAs, have been found in
46353
185
216
213
129
215
271
242
ACL racemase, 2ZUK
Fig. 21. Substrate recognition in ACL racemase exemplified by the ε-caprolactam boundinternal aldimine structure (PDB ID: 2ZUK). The cofactor and ε-caprolactam are show inorange.
597F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

different aminoglycoside producing organisms (see Supplementarydata Figure S4 for substrate and product structures). As the EC 2.6.1.93and EC 2.6.1.95 reactions are catalysed by the same enzymes (e.g.NeoB, BtrB and LivB, see Supplementary data Table S5 entries192–194; Clausnitzer et al., 2011; Huang et al., 2007), and the EC2.9.1.93 also occurred in the kacL enzyme that was originallycharacterised for EC 2.6.1.94 specificity (see Supplementary dataTable S5 entry 19; J.W. Park et al., 2011), we suggest that all threeGlu:6′-oxoglucos(amin)yl transaminations (EC 2.6.1.93–95) arecatalysed by one class III transaminase enzyme. Interestingly, the C6′is deaminated with αKG as the acceptor in the pseudodisaccharideneamine and pseudotrisaccharide kanamycin A (J.W. Park et al.,2011), but not when incorporated in neomycin C, where the C6‴ is ex-clusively deaminated (compare Supplementary data Figure S4)(Huang et al., 2007).
Unfortunately these enzymes share low sequence identity with theenzymes in the OrnTL DB and therefore the alignments to compare ac-tive site residues for substrate recognition hypothesis gave unsatisfacto-ry results. Structural information of Glu:6′-oxoglucos(amin)yl TAs ishighly desired to explain the uncommon substrate scope that allowsfor the discrimination of C6′ and C6‴ in neomycin C.
3.8.2. (Hydrolysed) fumonisin B1 TAsSummary: (H)fum B1 TAs were applied to detoxify the poly-hydroxy
amine fumonisin B1. Further substrate scope investigationsmight beworth-while, as these enzymes could have potential for amino-alcohol synthesis.
The carcinogenic mycotoxin fumonisin B1 (Supplementary dataFigure S6 B) is a common contaminant of maize produced in warmclimate areas (Hartinger et al., 2011). The studies of its degradation inbacteria has led to the discovery of class III transaminases responsiblefor the deamination of fumonisin B1 (fumB1) (Leslie et al., 2004), orafter hydrolysis, to 2-amino-12,16-dimethylicosane-3,5,10,14,15-pentol (hydrolysed fumonisin B1, HfumB1, Supplementary dataFigure S6 C) (Hartinger et al., 2011; Heinl et al., 2011) with αKG orpyr as the acceptor, respectively. These enzymes have been investigatedfor the application as food additives to detoxify fumB1 biocatalytically(Leslie et al., 2004; Moll et al., 2010). Unfortunately, the scope ofamino donors has not been examined and it is unknown, whether sub-strates other than (hydrolysed) fumonisins are converted. A further in-vestigation of their substrate specificity might be beneficial as theycould probably be applied for asymmetric amino alcohol synthesis.Even though they share high sequence similarities, the substrate scopeof the fumB1:αKG TA andHfumB1:pyr TAs differ substantially as the lat-ter only applies pyr as the acceptor and does not accept the non-hydrolysed fumB1 (Hartinger et al., 2011), whereas the fumB1:αKG TAconverted fumB1 with αKG (Leslie et al., 2004). These differences can-not be explained based on the active site residues identified by thealignment to the OrnTL DB, as they are almost all identical among theenzymes with these two specificities (see sequence–function matrixTable 2). (H)fumB1 conversion seem to demand a very specialised activesite compared to the other enzymes in the OrnTL DB, as no sequence inthis database has the combination of E185 and R216 found in all(H)fumB1 TAs.
3.8.3. PhospholyasesSummary: the two characterised phospholyases among the class III
transaminase family were found to lack transaminase activity. Factors de-termining this switch in reaction specificity are not known as the catalyticmachinery is the same as in the related transaminases. A substantial differ-ence, however, is the deletion of two amino acids in the left-handed helixα2, which could reshape the active site and thereby might shift the reactionspecificity.
By discovering twophospholyases, Veiga-da-Cunhaet al. (2012) fur-ther broadened the spectrum of known reaction specificities among theclass III transaminases. They found that the two human Ala:glyox TA 2homologs AGXT2L1 (UniProt ID: Q8TBG4) and AGXT2L2 (UniProt ID:
Q8IUZ5) possess no TA activity but are a O-phosphoethanolaminephospholyase (EC 4.2.3.2) and a 5-phosphohydroxy-L-lysine phos-pholyase (EC 4.2.3.134), respectively. Both enzymes do not belong tothe OrnTL DB, but enzymes with high sequence identity (N68%) arefound in the 2ZUK subfamily, which allowed for the elucidation of sev-eral of their active site residues by aligning them to the 3DM database(see sequence–function matrix Table 2). The active site of these en-zymes must substantially differ from the other class III transaminasesbecause it lacks two amino acids in the common left-handed helix α2(positions 45 and 46). At least one of the polar side chains of N/S44and N47 (found in both enzymes) are presumably pointing towardsthe active site. Whether this deletion and the polar amino acids at posi-tions 44 and 47 are required for phospholyase activity remains unclearwithout structural information. The PLP coordination and the catalyticmachinery is the same as in the transaminases of the OrnTL DB, proba-bly because the β-elimination follows the same mechanism until thequinonoid intermediate (see Fig. 2) and therefore has comparable re-quirements. Owing to the high similarity between these two reactionspecificities and (given a good leaving group) the facility of this reaction,β-elimination is commonly found among transaminase families (Eliotand Kirsch, 2004). β-Elimination was also found to occur in class IIItransaminases when certain inhibitors, especially those with good leav-ing groups were applied as substrates (e.g. Ala:glyox TA 2 catalysesβ-elimination of β-chloro-β-alanine and halogenated alkene–cysteineconjugates; Cooper et al., 2003). Several known transaminase inhibitorsinactivate the enzymes by forming reactive products throughβ-elimination that can covalently bind to the cofactor or active site res-idues (Eliot and Kirsch, 2004).
Given the fact that phosphate is an excellent leaving group, the oc-currence of β-elimination with β-phosphate substrates in a class III TAis not surprising. The selectivity for β-elimination of these enzymes,caused by the complete lack of TA activity, however, is of specialinterest. As the whole catalytic TA machinery is available, other factorsin these enzymes must prevent transamination. Since these twophospholyases are the only characterised enzymes of the class III trans-aminase family that lack the common left-handed helix at positions44–47, it might be hypothesised that it is essential for TA activityamong this group of enzymes. The characterisation of additional en-zymes with deletions in this region, however, is required to strengthenthis hypothesis.
A varied carboxylate recognition is probably not the reason for thediffering reaction specificity because the phospholyases have K353and Q216 that should be able to position αAA properly in the O-sideas achieved by R353 and Q216 in Ala:glyox TA 2 (section 3.3.6). Thephosphate recognition in the lyases probably also involves K355 andS346, the presence of E267 and D348, however, is puzzling becausethese residues should repel the phosphate and the carboxylic acid moi-ety on both sides of the active site entrance. Sequence fingerprints thatstrictly determine the different substrate specificity of these two en-zymes cannot be foundwithout structural information or the character-isation of similar enzymes. The common active site residues present inthe sequence–function matrix (Table 2) did not show obvious differ-ences compared to the transaminases except for the deletion at posi-tions 45 and 46 and the polar residues N/S44 and N47.
3.8.4. Multi-domain or non-enzymesSummary: PLP fold type I and in particular, the class III transaminase
family's common fold are versatile building blocks for multi-domain en-zymes or transcriptional factors.
The versatility of the commonPLP fold type I is expressly highlightedby recent studies that discovered non-enzymatic or multi-domain ex-amples of this fold. The MocR-like transcriptional factors belong to theclass I transaminase family (also belonging to PLP fold type I, seeTable 3) attached to a helix–turn–helix domain for DNA binding(Bramucci et al., 2011). Recent structural investigations of gabR, thetranscriptional activator of the GABA:αKG TA in B. subtilis (PDB ID:
598 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

4N0B), revealed that PLP binding is achieved as in other fold type I en-zymes, GABA, however, is bound differently and not transaminated bythis transcription factor (Edayathumangalam et al., 2013).
Another notable example is the presence of PLP fold type I and espe-cially class III transaminase domains in multimodular nonribosomalpeptide synthetase or polyketide synthase assembly lines (Milanoet al., 2013). The class III domains required some structural variations(e.g. insertions of ~12 amino acids between core positions 266 and267), compared to the stand-alone enzymes to enable integration inthe multi-domain complexes. However most active site residues andtherefore probably also substrate scopes of these enzymes differsubstantially from those in all other characterised enzymes and predic-tions are therefore not possible. One characterised example illustratesthese differences: the class III TA domain from MycA (UniProt ID:Q9R9J1, from B. subtilis, involved in cyclic lipopeptidemycosubtilin syn-thesis), which has been investigated for substrate scope independentlyof the whole multi-domain complex (Aron et al., 2005). This domainprefers glutamine as the amino donor and converts acyl carrier protein(ACP) thioesters of β-ketobutyrate. Active site residues, identified byaligning this sequence to its closest homolog in the OrnTL DB, are notsufficient to suggest substrate recognition as they differ substantiallyfrom those in all other characterised class III transaminases (see se-quence–function matrix Table 2).
4. Challenges for fingerprint-based sequence–function predictions
During the process of discovering and evaluating sequence–functionrelationships within the OrnTL DB, we encountered some challengesregarding this approach. Some resulted from intrinsic limitations ofstructure-based sequence alignments; others are challenges for theapproach of predicting function based on short sequence fingerprints.In this section we attempt to highlight the current challenges or limita-tions for a general evaluation of this method thereby deepening itsunderstanding and to motivate future approaches to solve currentproblems.
4.1. Limitations of the active site amino acid fingerprint-based approach
Enzyme function prediction by a few residues with known impor-tance for catalysis or substrate recognition, as introduced in section1.5 and summarised in Fig. 6, is a powerful approach as highlighted bythis account. It is, however, important to keep inmind that the situationin nature is often more complex than this.
1) There often exists more than one solution to realise the same sub-strate specificity, which leads to a general limitation of sequencealignments: amino acid side chains of different Cα (and alignment)positions might fill the same space in the active site (exemplified inFig. 6A, B and D, sequences 1–4). PLP fold type I provides a perfectexample for this case: the catalytic K242 is fully conserved in theOrnTL DB, whereas only 20% of the full PLP fold type I databasehave a lysine there. Interestingly the catalytic lysine is located twopositions later (K244) in many of the other sequences of that data-base (40% of the whole database have K244, the additional 40%could not be aligned properly at this position). The structural super-position of the human Orn:αKG TA (PDB ID: 1OAT, has K242) andthe 2-aminoethylphosphonate:pyr TA from Salmonella enterica(PDB ID: 1M32, has K244) revealed that both lysine ε-amino groupsare located close to each other, while their Cα positions differ(Supplementary data, Figure S4). Structural alignments therefore al-ways allow further and more accurate conclusions than sequencealignments, but for many activities, crystal structures are not yetavailable. This lack may lead to erroneous sequence alignments, forinstance if the catalytic lysine is aligned at the same commonposition (242) before the crystal structure reveals that it actuallyoccupies another position (244).
2) Some interactions of amino acids to the substrate might not be con-served. Complementary electrostatic interactions or hydrogen bond-ing are easily analysed, but if the contacts are mainly realised byhydrophobic interactions or if water molecules mediate them, it ismore difficult to formulate a clear pattern. Additionally, chemicallypartially equivalent residues might result in comparable substratescopes. The suggested fingerprints in this review therefore need tobe applied with caution, always considering that similar aminoacids at the suggested positions might be found in enzymes withrelated specificities. Furthermore, not all sequences thatmatch a cer-tainfingerprint need to possess a related specificity as demonstratedfor a GABA:αKGTA that has been transformed into aDGD. In thefirstattempt Liu et al. (2005) exchanged all active site residues of theE. coli GABA:αKG TA (PDB ID: 1SFF) that were different to thosefound in DGD. The I46Q, E185S, V215A, and G269Y quadruple mu-tant showed, however, no increased decarboxylase activity, whichcould be explained by slightly different spatial positions of the Cαatoms of these residues in the GABA:αKG TA compared to the DGD(Liu et al., 2005). A final mutant, that had switched reaction specific-ity, only contained one of the residues (S185) that were predicted bythose found in DGD.
3) Proteins including the active sites have various degrees of flexibility,which cannot be seen from sequence patterns. Further informationof potential flexibility is only accessible by substrate or productbound crystal structures.
4) The superfamily to be analysed needs a spatially conserved back-bone to allow for a proper alignment. If the active site ismainly com-posed of variable loops, the position of relevant amino acids mightdiffer significantly, although they are the same in the sequencealignment. This fact highlights the class III transaminase fold's versa-tility as all active site structural elements (except the N-terminusand the loop between core positions 73 and 74, see Fig. 10) are con-served and allow for such a variety of catalysed reactions and accept-ed substrates.
4.2. 3DM database related issues
Structural based sequence alignments and their organisation in crys-tal structure derived subfamilies also have a few issues to be aware of.
Superfamily diversity is a major criterion deciding the database'scontent of information. For instance the database created for the PLPfold type I contained too diverse crystal structures, which resulted in arelatively small structural core. Even though subgroups within thisalignment (e.g. the class III transaminases) share more structural fea-tures, these regions cannot be analysed in the large database becausethey belong to the variable positions. Creating the OrnTL DB, whichonly contains structurally more related enzymes — thereby increasingthe core, but also reducing the reaction diversity in the database —
overcame this problem. The database size will in most cases be atrade-off between contained reaction or substrate specificities andcovered positions.
4.3. The literature mining problem
A general difficulty for connecting sequence and function is gatheringall available literature that describes enzymes' sequence or specificitycharacterisations. Attempts are made to connect publications regardingfunction and sequence in databases like BRENDA (Schomburg et al.,2013). These are constantly growing, but unfortunately still far fromcomplete. Additional hints for sequences with available experimentaldata can be retrieved from the sequences' ‘evidence on protein existence’annotations in the UniProtKB (Magrane and UniProt Consortium, 2011).
3DM integrates the literature mining software Mutator (Kuiperset al., 2010a) and the PDF reader Utopia Documents (Attwood et al.,2010) to provide a link between 3DMpositions in the alignment and ar-ticles where these positions were mentioned in a certain context, such
599F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

as mutagenesis studies to increase specificity or stability. This newfeature supports literature research regarding mutagenesis studies.However, the current versions of Mutator and Utopia are made for ex-traction ofmutation data from literature and they do not include proteincharacterisation studies making manual literature studies still indis-pensable. Although these tools are optimised for finding mutationdata from literature, only approximately one third of the characterisedenzyme list (Supplementary data, Table S5) were not found by the au-tomated methods in combination with characterisation data retrievedfrom BRENDA and UniProt. The development of such tools is a step inthe right direction as the gap between sequence information and relat-ed literature data is a main obstacle and requires substantial reduction.
4.4. The challenge to identify unknown specificities
The attempts to identify proteins with unknown function, asdescribed in section 2.4, are limited to enzymes that switched reactionor substrate specificity by only mutating a few residues compared tothe known enzymes. As the structure-based sequence alignment data-bases only contain sequences that share a certain identity to a knownstructure, and crystal structures are usually solved for already function-ally characterised enzymes, the identification of specificities that re-quired several mutations to develop are probably not easy torecognise in such databases. To allow for a wider identification of bio-technologically interesting enzymes within the databases increasingthe amount of structural information for low identity sequences(which otherwise cannot be included in such a database) is of para-mount importance. Therefore, projects like the Protein Structure Initia-tive (Berman et al., 2009) or the Enzyme Function Initiative (Gerlt et al.,2011) are of substantial value for the biotechnological community.
Most notably, the recently discovered new substrate and reactionspecificities (see section 3.8) showed that the potential of the familyof class III transaminases had been underestimated for several years.This common structural fold allows for a variety of reaction and sub-strate specificities that have not yet been explored or exploited suffi-ciently. We therefore attempted to exemplify how the knowledgegained by this work can be applied to discover new enzyme activitiesor new active site designs for achieving already known specificities (asdiscussed in section 2.4). The subfamilies of a 3DM database make thistask relatively easy because these groups can be searched as an evolu-tionary related set to identify sequences that differ from known activi-ties by not matching the common fingerprints found in eachsubfamily. The investigation of such a group separately from thewhole superfamily provides a faster overview of present activities andpotentially new ones because some of the active site residues withineach group are still conserved and differences can be more easily com-pared and evaluated when not all positions are varied at the same time.
For instance the 2ZUK subfamily contains 18 (putative) αAAAracemases (2 characterised and 16 additional sequences match the fin-gerprint D185/K216) and 190 sequences that do not match any of theactive site patterns for known activities (summarised in Supplementarydata, Table S4). Further analysis showed that many of those 190 se-quences have R/K353, but no E185. Therefore, they probably convertαAA but not ωAA because the usual mechanism for dual substraterecognition for this specificity is not present (see section 3.1.1). Further-more, these might be clustered by certain patterns foundwithin the se-quence–functionmatrix (Table 2) positions. A few of these, for instance,contain D185 and N216 (like the Ile-2-epimerase, section 3.7) andmight therefore also be able to catalyse amino acid racemisation. Alarger fraction contains a modified left-handed helix (N44, N47and a deletion at positions 45 and 46) as found in characterisedphospholyases (section 3.8.3). It is likely, however, that many of theseenzymes have a different substrate scope than the two knownphospholyases because they do not have E267 and E348 and further-more, several have N185 instead of the unpolar residues found in theknown enzymes. It would be interesting to determine if these enzymes
show phospholyase and/or transaminase activity to investigate themechanisms responsible for the different specificities.
For the discovery of unknown functions or other mechanisms of al-ready known ones, the 3N5M subfamily is of special interest. Not only isthe template structure's function unknown (PDB ID: 3N5M), but alsomost of the entries within this subfamily cannot be assigned with a pu-tative specificity by the active site fingerprints. This subfamily that onlycontains six characterised enzymes (tau:pyr TAs, Ala:glyox TAs 2 and aβAla:pyr TA that is also able to convert amines, Supplementary data,Table 5) is very heterogeneous regarding the sequence–functionmatrixpositions (Table 2). Several sequences have R346, while several haveR353 instead, and others have neither. The positions 185 and 269, how-ever, are relatively conserved and also correlated, as revealed by CMA(35% sequences of the subfamily have A185 and G269, while 41% haveG185 and I/V269). Regardless of whether these enzymes possess sofar unknown specificities or established other ways to achieve knownactivities, the characterisation of such enzymes with unknown functionwill be worthwhile as either new biocatalysts will be obtained or theknowledge of functional determinants within the class III transaminasefamily will be extended.
5. Conclusion
In this review we show that the combination of structural informa-tion and analysis of multiple sequence alignments as exemplified forthe class III transaminase family allowed to extract active site aminoacid fingerprints that correlate with the different enzymatic activities.The different active site designs identified allowed covering 28 knownreaction and substrate specificities of enzymes within the ornithinetransaminase like family. This analysis should be regarded as a hint fora qualitative prediction of the substrate scope rather than a fixed rulebecause, besides the amino acid distribution of the active site, additionalfactors also affect the catalytic properties to a smaller or greater extend.Nevertheless, we encourage to apply these patterns in annotation strat-egies and to apply this methodology also for other superfamilies, whichare amenable to such analyses. A critical mass of crystal structures,however, is necessary to build up high quality sequence alignments,which include a possibly large fraction of the available sequences ofthe superfamily. The fingerprint approach allows not only to connectknown enzyme activities to given sequences, but also to discover en-zymes with yet unknown specificities and to suggest key mutations toverify hypotheses derived from the bioinformatics guided in-depthanalysis of an enzyme superfamily. These kinds of systematic investiga-tions will expand the number of useful enzymes and thereby providethe community with potentially interesting biocatalysts as well as itsubstantially helps to improve our understanding of sequence– andstructure–function relationships of enzymes.
Author contributions
MH and UTB initiated the review, FS andMH devised the conceptualdesign, FS and CV coordinated data and literature analysis of the subsec-tions and together with MH performed the in-depth analysis of thesuperfamily. FS, CV, HK, HL, HM, AN and LS analysed literature and3DM alignments of subgroups, created fingerprints and were involvedin writing subchapters. TvdB and H-JJ established the 3DM databaseand wrote the corresponding chapter. MH created the reaction mecha-nism video. With the help of CV and UTB, FS and MH did the mainediting. FS, CV, PB, MH and UTB finalised the review.
Acknowledgements
We thank Maika Genz for kindly providing Fig. 8, SebastianWenskefor his support in the creation of the reference list and Lea Kennel forproviding activity data of the ATA 3HMU towards acylated and non-acylated putrescine. FS thanks the Fonds der Chemischen Industrie
600 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

(Chemiefonds-Stipendium) and HK thanks the Deutsche BundesstiftungUmwelt (GrantNo. AZ29937) forfinancial support. PB andUB are gratefulfor support by the COST Action (CM1303 Systems Biocatalysis). We espe-cially thank the European Union (KBBE-2011-5, grant No. 289350) for fi-nancial support within the European Union Seventh FrameworkProgramme.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.biotechadv.2014.12.012.
References
Ahmed SA, Esaki N, Tanaka H, Soda K. L-α-Amino-β-thio-ε-caprolactam, a new sulfur-containing substrate forα-amino-ε-caprolactam racemase. FEBS Lett 1984;174:76–9.
Ahmed SA, Esaki N, Tanaka H, Soda K. Mechanism of α-amino-ε-caprolactam racemasereaction. Biochemistry 1986;25:385–8.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST andPSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res1997;25:3389–402.
Andersen G, Andersen B, Dobritzsch D, Schnackerz KD, Piškur J. A gene duplication led tospecialized γ-aminobutyrate and β-alanine aminotransferase in yeast. FEBS J 2007;274:1804–17.
Aron ZD, Dorrestein PC, Blackhall JR, Kelleher NL, Walsh CT. Characterization of a newtailoring domain in polyketide biogenesis: the amine transferase domain of MycAin the mycosubtilin gene cluster. J Am Chem Soc 2005;127:14986–7.
Asano Y, Yamaguchi S. Discovery of amino acid amides as new substrates for α-amino-ɛ-caprolactam racemase from Achromobacter obae. J Mol Catal B: Enzym 2005;36:22–9.
Attwood TK, Kell DB, McDermott P, Marsh J, Pettifer SR, Thorne D. Utopia documents:linking scholarly literature with research data. Bioinformatics 2010;26:i568–74.
Baker PRS, Cramer SD, Kennedy M, Assimos DG, Holmes RP. Glycolate and glyoxylatemetabolism in HepG2 cells. Am J Physiol Cell Physiol 2004;287:C1359–65.
Bandounas L, Ballerstedt H, de Winde JH, Ruijssenaars HJ. Redundancy in putrescinecatabolism in solvent tolerant Pseudomonas putida S12. J Biotechnol 2011;154:1–10.
Banerjee A, Chase M, Clayton RA, Landis B. Methods for the stereoselective synthesis andenantiomeric enrichment of β-amino acids. WO International Patent ApplicationWO2005005633, 2005.
Bea H-S, Park H-J, Lee S-H, Yun H. Kinetic resolution of aromatic β-amino acids byω-transaminase. Chem Commun 2011;47:5894–6.
Berglund P, Humble MS, Branneby C. C–X Bond formation: transaminases as chiralcatalysts: mechanism, engineering, and applications. In: Carreira EM, Yamamoto H,editors. Comprehensive chirality. Amsterdam: Elsevier; 2012. p. 390–401.
Berman HM, Westbrook JD, Gabanyi MJ, Tao W, Shah R, Kouranov A, et al. The proteinstructure initiative structural genomics knowledgebase. Nucleic Acids Res 2009;37:D365–8.
Bertoldi M, Cellini B, Montioli R, Borri Voltattorni C. Insights into themechanism of oxida-tive deamination catalyzed by DOPA decarboxylase. Biochemistry 2008;47:7187–95.
Bisht S, Bharath SR, Murthy MRN. Conformational transitions, ligand specificity and catal-ysis in N-acetylornithine aminotransferase: implications on drug designing and ratio-nal enzyme engineering in omega aminotransferases. Unpublished crystal structuresreleased in the Protein Data Bank; 2014.
Boesten WHJ, Raemakers-Franken PC, Sonke T, Eu-Verink GJW, Grijpstra P. Polypeptideshaving α-H-α-amino acid amide racemase activity and nucleic acids encoding thesame. WO International Patent Application WO2003106691, 2003.
Bornscheuer UT, Kazlauskas RJ. Catalytic promiscuity in biocatalysis: using old enzymes toform new bonds and follow new pathways. Angew Chem Int Ed 2004;43:6032–40.
Bramucci E, Milano T, Pascarella S. Genomic distribution and heterogeneity of MocR-liketranscriptional factors containing a domain belonging to the superfamily of thepyridoxal-5′-phosphate dependent enzymes of fold type I. Biochem Biophys ResCommun 2011;415:88–93.
Brohn F, Tchen TT. A single transaminase for 1,4-diaminobutane and 4-aminobutyrate ina Pseudomonas species. Biochem Biophys Res Commun 1971;45:578–82.
Bruce H, Tuan AN, Sanchez JM, Leese C, Hopwood J, Hyde R, et al. Structures of aγ-aminobutyrate (GABA) transaminase from the s-triazine-degrading organismArthrobacter aurescens TC1 in complex with PLP and with its external aldiminePLP-GABA adduct. Acta Crystallogr F Struct Biol Commun 2012;68:1175–80.
Buzenet AM, Fages C, Bloch-Tardy M, Gonnard P. Purification and properties of4-aminobutyrate 2-ketoglutarate aminotransferase from pig liver. Biochim BiophysActa 1978;522:400–11.
Cánovas D, Vargas C, Calderón MI, Ventosa A, Nieto JJ. Characterization of the genes forthe biosynthesis of the compatible solute ectoine in the moderately halophilic bacte-rium Halomonas elongata DSM 3043. Syst Appl Microbiol 1998;21:487–97.
Catazaro J, Caprez A, Guru A, Swanson D, Powers R. Functional evolution ofPLP-dependent enzymes based on active-site structural similarities. Proteins 2014;82:2597–608.
Cellini B, Bertoldi M, Montioli R, Paiardini A, Borri Voltattorni C. Human wild-typealanine:glyoxylate aminotransferase and its naturally occurring G82E variant:functional properties and physiological implications. Biochem J 2007;408:39–50.
Cha HJ, Jeong J-H, Rojviriya C, Kim Y-G. Structure of putrescine aminotransferase fromEscherichia coli provides insights into the substrate specificity among class III amino-transferases. PLoS One 2014;9:e113212.
Chou HT, Kwon DH, Hegazy M, Lu CD. Transcriptome analysis of agmatine and putrescinecatabolism in Pseudomonas aeruginosa PAO1. J Bacteriol 2008;190:1966–75.
Christen P, Mehta PK. From cofactor to enzymes. Themolecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Chem Rev 2001;1:436–47.
Clausnitzer D, Piepersberg W, Wehmeier UF. The oxidoreductases LivQ and NeoQ areresponsible for the different 6′-modifications in the aminoglycosides lividomycinand neomycin. J Appl Microbiol 2011;111:642–51.
Cobessi D, Dumas R, Pautre V, Meinguet C, Ferrer JL, Alban C. Biochemical and structuralcharacterization of the Arabidopsis bifunctional enzyme dethiobiotin synthetase–diaminopelargonic acid aminotransferase: evidence for substrate channeling in biotinsynthesis. Plant Cell 2012;24:1608–25.
Contestabile R, Angelaccio S, Maytum R, Bossa F, John RA. The contribution of aconformationally mobile, active site loop to the reaction catalyzed by glutamatesemialdehyde aminomutase. J Biol Chem 2000;275:3879–86.
Cooper AJL, Krasnikov BF, Okuno E, Jeitner TM. L-Alanine-glyoxylate aminotransferase II ofrat kidney and liver mitochondria possesses cysteine S-conjugate β-lyase activity: acontributing factor to the nephrotoxicity/hepatotoxicity of halogenated alkenes?Biochem J 2003;376:169–78.
Crismaru CG, Wybenga GG, Szymanski W, Wijma HJ, Wu B, Bartsch S, et al. Biochemicalproperties and crystal structure of a β-phenylalanine aminotransferase fromVariovorax paradoxus. Appl Environ Microbiol 2013;79:185–95.
Denger K, Ruff J, Schleheck D, Cook AM. Rhodococcus opacus expresses the xsc gene toutilize taurine as a carbon source or as a nitrogen source but not as a sulfur source.Microbiology 2004;150:1859–67.
Desai AA. Sitagliptin manufacture: a compelling tale of green chemistry, process intensi-fication, and industrial asymmetric catalysis. Angew Chem Int Ed 2011;50:1974–6.
Dey S, Lane JM, Lee RE, Rubin EJ, Sacchettini JC. Structural characterization of theMycobacterium tuberculosis biotin biosynthesis enzymes 7,8-diaminopelargonic acidsynthase and dethiobiotin synthetase. Biochemistry 2010;49:6746–60.
Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes.Proc Natl Acad Sci U S A 1966;55:712–6.
Edayathumangalam R, Wu R, Garcia R, Wang Y, Wang W, Kreinbring CA, et al. Crystalstructure of Bacillus subtilis GabR, an autorepressor and transcriptional activator ofgabT. Proc Natl Acad Sci U S A 2013;110:17820–5.
Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolution-ary considerations. Annu Rev Biochem 2004;73:383–415.
Eliot AC, Sandmark J, Schneider G, Kirsch JF. The dual-specific active site of 7,8-diaminopelargonic acid synthase and the effect of the R391A mutation. Biochemistry2002;41:12582–9.
Ferradini N, Nicolia A, Capomaccio S, Veronesi F, Rosellini D. A point mutation in theMedicago sativa GSA gene provides a novel, efficient, selectable marker for plantgenetic engineering. J Biotechnol 2011;156:147–52.
Fesko K, Uhl M, Steinreiber J, Gruber K, Griengl H. Biocatalytic access to α, α-dialkyl-α-amino acids by a mechanism-based approach. Angew Chem Int Ed 2010;49:121–4.
Fogle EJ, Toney MD. Mutational analysis of substrate interactions with the active site ofdialkylglycine decarboxylase. Biochemistry 2010;49:6485–93.
Fogle EJ, Liu W, Woon ST, Keller JW, Toney MD. Role of Q52 in catalysis of decarboxylationand transamination in dialkylglycine decarboxylase. Biochemistry 2005;44:16392–404.
Fujii T, Narita T, Agematu H, Agata N, Isshiki K. Characterization of L-lysine 6-aminotransferase and its structural gene from Flavobacterium lutescens IFO3084. JBiochem 2000;128:391–7.
Gand M, Müller H, Wardenga R, Höhne M. Characterization of three novel enzymes withimine reductase activity. J Mol Catal B: Enzym 2014;110:126–32.
Garcia ARM, Kamat SV, Pannuri S. Thermostable ω-transaminases. WO InternationalPatent Application WO2006063336, 2006.
Ge H, Lv X, Fan J, Gao Y, Teng M, Niu L. Crystal structure of glutamate1-semialdehydeaminotransferase from Bacillus subtilis with bound pyridoxamine-5′-phosphate.Biochem Biophys Res Commun 2010;402:356–60.
Gerlt JA, Allen KN, Almo SC, Armstrong RN, Babbitt PC, Cronan JE, et al. The enzymefunction initiative. Biochemistry 2011;50:9950–62.
Grant SM, Heel RC. Vigabatrin. Drugs 1991;41:889–926.Grimm B, Smith AJ, Kannangara CG, Smith M. Gabaculine-resistant glutamate 1-
semialdehyde aminotransferase of Synechococcus. Deletion of a tripeptide close to theNH2 terminus and internal amino acid substitution. J Biol Chem 1991;266:12495–501.
Grishin NV, Phillips MA, Goldsmith EJ. Modeling of the spatial structure of eukaryoticornithine decarboxylases. Protein Sci 1995;4:1291–304.
Griswold WR, Toney MD. Role of the pyridine nitrogen in pyridoxal 5′-phosphatecatalysis: activity of three classes of PLP enzymes reconstituted with deazapyridoxal5′-phosphate. J Am Chem Soc 2011;133:14823–30.
Halabi N, Rivoire O, Leibler S, Ranganathan R. Protein sectors: evolutionary units ofthree-dimensional structure. Cell 2009;138:774–86.
Hammer T, Bode R. Purification and characterization of an inducible L-lysine: 2-oxoglutarate 6-aminotransferase from Candida utilis. J Basic Microbiol 1992;32:21–7.
Hanson RL, Davis BL, Chen Y, Goldberg SL, Parker WL, Tully TP, et al. Preparation of(R)-amines from racemic amines with an (S)-amine transaminase from Bacillusmegaterium. Adv Synth Catal 2008;350:1367–75.
Hanson RL, Johnston RM, Goldberg SL, Parker WL, Patel RN. Enzymatic preparation of5-hydroxy-L-proline, N-Cbz-5-hydroxy-L-proline, and N-boc-5-hydroxy-L-prolinefrom (α-N-protected)-L-ornithine using a transaminase or an amine oxidase. EnzymeMicrob Technol 2011;48:445–53.
Hartinger D, Schwartz H, Hametner C, Schatzmayr G, Haltrich D, Moll WD. Enzymecharacteristics of aminotransferase FumI of Sphingopyxis sp. MTA144 for deaminationof hydrolyzed fumonisin B1. Appl Microbiol Biotechnol 2011;91:757–68.
Heimberg H, Boyen A, Crabeel M, Glansdorff N. Escherichia coli and Saccharomycescerevisiae acetylornithine aminotransferase: evolutionary relationship with ornithineaminotransferase. Gene 1990;90:69–78.
601F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

Heinl S, Hartinger D, ThamheslM, Schatzmayr G,MollWD, Grabherr R. An aminotransfer-ase from bacterium ATCC 55552 deaminates hydrolyzed fumonisin B1. Biodegrada-tion 2011;22:25–30.
Hennig M, Grimm B, Contestabile R, John RA, Jansonius JN. Crystal structure of glutamate-1-semialdehyde aminomutase: An α2-dimeric vitamin B6-dependent enzyme withasymmetry in structure and active site reactivity. Proc Natl Acad Sci U S A 1997;94:4866–71.
Hirotsu K, GotoM, Okamoto A,Miyahara I. Dual substrate recognition of aminotransferases.Chem Rec 2005;5:160–72.
Hohenester E, Keller JW, Jansonius JN. An alkali metal ion size-dependent switch in theactive site structure of dialkylglycine decarboxylase. Biochemistry 1994;33:13561–5570.
Höhne M, Bornscheuer UT. Biocatalytic routes to optically active amines. ChemCatChem2009;1:42–51.
Höhne M, Bornscheuer UT. Application of transaminases in organic synthesis. In: May O,Gröger H, Drauz W, editors. Enzymes in organic synthesis. Weinheim: Wiley-VCH;2012. p. 779–820.
Höhne M, Schätzle S, Jochens H, Robins K, Bornscheuer UT. Rational assignment of keymotifs for function guides in silico enzyme identification. Nat Chem Biol 2010;6:807–13.
Hoober JK, Kahn A, Ash DE, Gough S, Kannangara CG. Biosynthesis of Δ-aminolevulinatein greening barley leaves. IX. structure of the substrate, mode of gabaculine inhibi-tion, and the catalytic mechanism of glutamate 1-semialdehyde aminotransferase.Carlsberg Res Commun 1988;53:11–25.
Huang F, Spiteller D, Koorbanally NA, Li Y, Llewellyn NM, Spencer JB. Elaboration ofneosamine rings in the biosynthesis of neomycin and butirosin. ChemBioChem2007;8:283–8.
Hult K, Berglund P. Enzyme promiscuity: mechanism and applications. Trends Biotechnol2007;25:231–8.
Humble MS, Berglund P. Biocatalytic promiscuity. Eur J Org Chem 2011;2011:3391–401.Humble MS, Cassimjee KE, Hakansson M, Kimbung YR, Walse B, Abedi V, et al. Crystal
structures of the Chromobacterium violaceum ω-transaminase reveal major structuralrearrangements upon binding of coenzyme PLP. FEBS J 2012;279:779–92.
Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011:new developments in the family and domain prediction database. Nucleic Acids Res2012;40:D306–12.
Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetictrees and networks. Syst Biol 2012;61:1061–7.
Izumi Y, Sato K, Tani Y, Ogata K. 7,8-Diaminopelargonic acid aminotransferase, anenzyme involved in biotin biosynthesis by microorganisms. Agric Biol Chem 1975;39:175–81.
Izumi Y, Kano Y, Inagaki K, Kawase N, Tani Y, Yamada H. Characterization of biotin biosyn-thetic enzymes of Bacillus sphaericus: a dethiobiotin producing bacterium. Agric BiolChem 1981;45:1983–9.
Jackson LK, Brooks HB, Osterman AL, Goldsmith EJ, Phillips MA. Altering the reactionspecificity of eukaryotic ornithine decarboxylase. Biochemistry 2000;39:11247–57.
Jaskolski M, Dauter Z, Wlodawer A. A brief history of macromolecular crystallography,illustrated by a family tree and its Nobel fruits. FEBS J 2014;281:3985–4009.
Jomrit J, Summpunn P, Meevootisom V, Wiyakrutta S. Sensitive non-radioactive determi-nation of aminotransferase stereospecificity for C-4′ hydrogen transfer on the coen-zyme. Biochem Biophys Res Commun 2011;405:626–31.
Joosten HJ, Han Y, Niu W, Vervoort J, Dunaway-Mariano D, Schaap PJ. Identification offungal oxaloacetate hydrolyase within the isocitrate lyase/PEP mutase enzymesuperfamily using a sequence marker-based method. Proteins 2008;70:157–66.
Jortzik E, Fritz-Wolf K, Sturm N, Hipp M, Rahlfs S, Becker K. Redox regulation ofPlasmodium falciparum ornithine δ-aminotransferase. J Mol Biol 2010;402:445–59.
Käck H, Sandmark J, Gibson K, Schneider G, Lindqvist Y. Crystal structure ofdiaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymes. J Mol Biol 1999;291:857–76.
Kaneoke M, Shimizu E, Yorifuji T. Metabolism of L-arginine, agmatine, and relatedcompounds in Nocardioides simplex. Biosci Biotechnol Biochem 1994;58:244–9.
Kaulmann U, Smithies K, Smith MEB, Hailes HC, Ward JM. Substrate spectrum ofω-transaminase from Chromobacterium violaceum DSM30191 and its potential forbiocatalysis. Enzyme Microb Technol 2007;41:628–37.
Keller JW, Baurick KB, Rutt GC, O'MalleyMV, Sonafrank NL, Reynolds RA, et al. Pseudomonascepacia 2,2-dialkylglycine decarboxylase. sequence and expression in Escherichia coli ofstructural and repressor genes. J Biol Chem 1990;265:5531–9.
Kim KH. Purification and properties of a mine α-ketoglutarate transaminase fromEscherichia coli. J Biol Chem 1964;239:783–6.
Kim J, Kyung D, Yun H, Cho BK, Seo JH, Cha M, et al. Cloning and characterization of a novelβ-transaminase fromMesorhizobium sp. strain LUK: a new biocatalyst for the synthesisof enantiomerically pure β-amino acids. Appl Environ Microbiol 2007;73:1772–82.
Kohls H, Steffen-Munsberg F, Höhne M. Recent achievements in developing the biocata-lytic toolbox for chiral amine synthesis. Curr Opin Chem Biol 2014;19:180–92.
Koma D, Sawai T, Harayama S, Kino K. Overexpression of the genes from thermophiles inEscherichia coli by high-temperature cultivation. Appl Microbiol Biotechnol 2006;73:172–80.
Kongsaeree P, Samanchart C, Laowanapiban P,Wiyakrutta S, Meevootisom V. Crystallizationand preliminary X-ray crystallographic analysis of D-phenylglycine aminotransferasefrom Pseudomonas stutzeri ST201. Acta Crystallogr D Biol Crystallogr 2003;59:953–4.
Kontani Y, Kaneko M, Kikugawa M, Fujimoto S, Tamaki N. Identity of D-3-amino-isobutyrate-pyruvate aminotransferase with alanine-glyoxylate aminotransferase 2.Biochim Biophys Acta 1993;1156:161–6.
Kourist R, Jochens H, Bartsch S, Kuipers RKP, Padhi SK, Gall M, et al. The α/β-hydrolase fold3DM database (ABHDB) as a tool for protein engineering. ChemBioChem 2010;11:1635–43.
Kroutil W, Fischereder EM, Fuchs CS, Lechner H, Mutti FG, Pressnitz D, et al. Asymmetricpreparation of prim-, sec-, and tert-amines employing selected biocatalysts. OrgProcess Res Dev 2013;17:751–9.
Kuipers RKP, Joosten HJ, Verwiel E, Paans S, Akerboom J, van der Oost J, et al. Correlatedmutation analyses on super-family alignments reveal functionally importantresidues. Proteins 2009;76:608–16.
Kuipers RKP, van den Bergh T, Joosten H-J, Lekanne dit Deprez RH, Mannens MMAM,Schaap PJ. Novel tools for extraction and validation of disease-related mutations ap-plied to fabry disease. Hum Mutat 2010a;31:1026–32.
Kuipers RKP, Joosten HJ, van Berkel WJH, Leferink NGH, Rooijen E, Ittmann E, et al. 3DM:systematic analysis of heterogeneous superfamily data to discover protein function-alities. Proteins 2010b;78:2101–13.
Kurihara S, Oda S, Kato K, Kim HG, Koyanagi T, Kumagai H, et al. A novel putrescineutilization pathway involves γ-glutamylated intermediates of Escherichia coli K-12. JBiol Chem 2005;280:4602–8.
Kwon YC, Lee KH, Kim HC, Han K, Seo JH, Kim B-G, et al. Cloning-independent expressionand analysis of ω-transaminases by use of a cell-free protein synthesis system. ApplEnviron Microbiol 2010;76:6295–8.
Lal PB, Schneider BL, Vu K, Reitzer L. The redundant aminotransferases in lysine andarginine synthesis and the extent of aminotransferase redundancy in Escherichiacoli. Mol Microbiol 2014;94:843–56.
Laue H, Cook AM. Biochemical andmolecular characterization of taurine:pyruvate amino-transferase from the anaerobe Bilophila wadsworthia. Eur J Biochem 2000;267:6841–8.
Ledwidge R, Blanchard JS. The dual biosynthetic capability of N-acetylornithine amino-transferase in arginine and lysine biosynthesis. Biochemistry 1999;38:3019–24.
Lee S-G, Hong S-P, Sung M-H. Development of an enzymatic system for the production ofdopamine from catechol, pyruvate, and ammonia. Enzyme Microb Technol 1999;25:298–302.
Lee H, Juncosa JI, Silverman RB. Ornithine aminotransferase versus GABA aminotransfer-ase: implications for the design of new anticancer drugs. Med Res Rev 2014;35:286–305.
Leferink NGH, FraaijeMW, Joosten HJ, Schaap PJ, Mattevi A, van BerkelWJH. Identificationof a gatekeeper residue that prevents dehydrogenases from acting as oxidases. J BiolChem 2009;284:4392–7.
Leslie H, Paul WD, Lishan Z. Transaminases, deaminases and aminomutases and compo-sitions and methods for enzymatic detoxification. WO International Patent Applica-tion WO2004085624, 2004.
Li T, Huo L, Pulley C, Liu A. Decarboxylation mechanisms in biological system. BioorgChem 2012;43:2–14.
Liepman AH, Olsen LJ. Alanine aminotransferase homologs catalyze the glutamate:glyoxylate aminotransferase reaction in peroxisomes of arabidopsis. Plant Physiol2003;131:215–27.
Liese A, Seelbach K, Buchholz A, Haberland J. Processes: hydrolases EC 3 — EC 3.4.21.4 toEC 3.8.X.X. In: Liese A, Seelbach K, Wandrey C, editors. Industrial biotransformations.Wiley-VCH; 2006a. p. 350–446.
Liese A, Seelbach K, Buchholz A, Haberland J. Processes: lyases EC 4. In: Liese A, SeelbachK, Wandrey C, editors. Industrial biotransformations. Wiley-VCH; 2006b. p. 447–503.
LiuW, Rogers CJ, Fisher AJ, ToneyMD. Aminophosphonate inhibitors of dialkylglycine decar-boxylase: structural basis for slow binding inhibition. Biochemistry 2002;41:12320–8.
LiuW, Peterson PE, Carter RJ, Zhou X, Langston JA, Fisher AJ, et al. Crystal structures of un-bound and aminooxyacetate-bound Escherichia coli γ-aminobutyrate aminotransfer-ase. Biochemistry 2004;43:10896–905.
LiuW, Peterson PE, Langston JA, Jin X, Zhou X, Fisher AJ, et al. Kinetic and crystallographicanalysis of active site mutants of Escherichia coli γ-aminobutyrate aminotransferase.Biochemistry 2005;44:2982–92.
Lou X, Ran T, Han N, Gao Y, He J, Tang L, et al. Crystal structure of the catalytic domainof PigE: a transaminase involved in the biosynthesis of 2-methyl-3-n-amyl-pyr-role (MAP) from Serratia sp. FS14. Biochem Biophys Res Commun 2014;447:178–83.
Lu CD, Itoh Y, Nakada Y, Jiang Y. Functional analysis and regulation of the divergentspuABCDEFGH-spuI operons for polyamine uptake and utilization in Pseudomonasaeruginosa PAO1. J Bacteriol 2002;184:3765–73.
Lütke-Eversloh T, Santos C, Stephanopoulos G. Perspectives of biotechnological produc-tion of L-tyrosine and its applications. Appl Microbiol Biotechnol 2007;77:751–62.
Magrane M, UniProt Consortium. UniProt knowledgebase: a hub of integrated proteindata. Database 2011;2011.
Malashkevich VN, Strop P, Keller JW, Jansonius JN, Toney MD. Crystal structures ofdialkylglycine decarboxylase inhibitor complexes. J Mol Biol 1999;294:193–200.
Mann S, Ploux O. Pyridoxal-5′-phosphate-dependent enzymes involved in biotin biosyn-thesis: structure, reaction mechanism and inhibition. Biochim Biophys Acta 2011;1814:1459–66.
Mann S, Ploux O. 7,8-Diaminoperlargonic acid aminotransferase from Mycobacteriumtuberculosis, a potential therapeutic target. FEBS J 2006;273:4778–89.
Mann S, Colliandre L, Labesse G, Ploux O. Inhibition of 7,8-diaminopelargonic acidaminotransferase from Mycobacterium tuberculosis by chiral and achiral anologs ofits substrate: biological implications. Biochimie 2009;91:826–34.
Markova M, Peneff C, Hewlins MJ, Schirmer T, John RA. Determinants of substratespecificity in ω-aminotransferases. J Biol Chem 2005;280:36409–16.
McPhalen CA, Vincent MG, Picot D, Jansonius JN, Lesk AM, Chothia C. Domain closure inmitochondrial aspartate aminotransferase. J Mol Biol 1992;227:197–213.
Medina-Kauwe LK, Tobin AJ, De Meirleir L, Jaeken J, Jakobs C, Nyhan WL, et al.4-Aminobutyrate aminotransferase (GABA-transaminase) deficiency. J InheritMetab Dis 1999;22:414–27.
Mehta PK, Hale TI, Christen P. Aminotransferases: demonstration of homology and divi-sion into evolutionary subgroups. Eur J Biochem 1993;214:549–61.
602 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of genefunction, and other gene attributes, in the context of phylogenetic trees. NucleicAcids Res 2013;41:D377–86.
Midelfort KS, Kumar R, Han S, Karmilowicz MJ, McConnell K, Gehlhaar DK, et al.Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialisaminotransferase for the synthesis of imagabalin. Protein Eng Des Sel 2013;26:25–33.
Milano T, Paiardini A, Grgurina I, Pascarella S. Type I pyridoxal 5′-phosphate dependentenzymatic domains embedded within multimodular nonribosomal peptide synthe-tase and polyketide synthase assembly lines. BMC Struct Biol 2013;13:26.
Miyazaki J, Kobashi N, Nishiyama M, Yamane H. Functional and evolutionary relationshipbetween arginine biosynthesis and prokaryotic lysine biosynthesis throughα-aminoadipate. J Bacteriol 2001;183:5067–73.
Moll WD, Hartinger D, Grieβler K, Binder EM, Schatzmayr G. Method for the productionof an additive for the enzymatic decomposition of mycotoxins, additive, and usethereof. WO International Patent Application WO2010031101, 2010.
Mou X, Vila-Costa M, Sun S, Zhao W, Sharma S, Moran MA. Metatranscriptomic signatureof exogenous polyamine utilization by coastal bacterioplankton. Environ MicrobiolRep 2011;3:798–806.
Müller U, van Assema F, Gunsior M, Orf S, Kremer S, Schipper D, et al. Metabolic engineer-ing of the E. coli L-phenylalanine pathway for the production of D-phenylglycine(D-Phg). Metab Eng 2006;8:196–208.
Mutaguchi Y, Ohmori T, Wakamatsu T, Doi K, Ohshima T. Identification, purification, andcharacterization of a novel amino acid racemase, isoleucine 2-epimerase, from Lacto-bacillus species. J Bacteriol 2013;195:5207–15.
Newman J, Seabrook S, Surjadi R, Williams CC, Lucent D, Wilding M, et al. Determinationof the structure of the catabolic N-succinylornithine transaminase (AstC) fromEscherichia coli. PLoS One 2013;8:e58298.
Nikolskaya AN, Arighi CN, Huang H, BarkerWC,Wu CH. PIRSF family classification systemfor protein functional and evolutionary analysis. Evol Bioinform Online 2006;2:197–209.
Novotny M, Kleywegt GJ. A survey of left-handed helices in protein structures. J Mol Biol2005;347:231–41.
Nugent TC, El-Shazly M. Chiral amine synthesis — recent developments and trends forenamide reduction, reductive amination, and imine reduction. Adv Synth Catal2010;352:753–819.
Okazaki S, Suzuki A, Mizushima T, Kawano T, Komeda H, Asano Y, et al. The novel struc-ture of a pyridoxal 5′-phosphate-dependent fold-type I racemase, α-amino-ε-caprolactam racemase from Achromobacter obae. Biochemistry 2009;48:941–50.
Oliveira EF, Cerqueira NM, Fernandes PA, RamosMJ. Mechanism of formation of the inter-nal aldimine in pyridoxal 5′-phosphate-dependent enzymes. J Am Chem Soc 2011;133:15496–505.
Orriss GL, Patel TR, Sorensen J, Stetefeld J. Absence of a catalytic water confers resistanceto the neurotoxin gabaculine. FASEB J 2010;24:404–14.
Park J, Osei YD, Churchich JE. Isolation and characterization of recombinantmitochondrial4-aminobutyrate aminotransferase. J Biol Chem 1993;268:7636–9.
Park DH, Mirabella R, Bronstein PA, Preston GM, Haring MA, Lim CK, et al. Mutations inγ-aminobutyric acid (GABA) transaminase genes in plants or Pseudomonas syringaereduce bacterial virulence. Plant J 2010a;64:318–30.
Park ES, Kim M, Shin J-S. One-pot conversion of L-threonine into L-homoalanine: biocata-lytic production of an unnatural amino acid from a natural one. Adv Synth Catal2010b;352:3391–8.
Park ES, Kim M, Shin J-S. Molecular determinants for substrate selectivity ofω-transaminases. Appl Microbiol Biotechnol 2011a;93:2425–35.
Park JW, Park SR, Nepal KK, Han AR, Ban YH, Yoo YJ, et al. Discovery of parallel pathwaysof kanamycin biosynthesis allows antibiotic manipulation. Nat Chem Biol 2011b;7:843–52.
Patel RN, Banerjee A, Hanson RL, Brzozowski DB, Parker LW, Szarka LJ. Oxidation ofNα-protected-L-lysine by Rhodotorula graminis to produce novel chiral compounds.Tetrahedron Asymmetry 1999;10:31–6.
Patel RN, Banerjee A, Nanduri VB, Goldberg SL, Johnston RM, Hanson RL, et al. Biocatalyticpreparation of a chiral synthon for a vasopeptidase inhibitor: enzymatic conversionof N2-[N-Phenylmethoxy)carbonyl] L-homocysteinyl]-L-lysine (1 → 1′)-disulfideto [4S-(4I,7I,10aJ)] 1-octahydro-5-oxo-4-[phenylmethoxy)carbonyl]amino]-7H-pyrido-[2,1-b] [1,3]thiazepine-7-carboxylic acid methyl ester by a novel L-lysineε-aminotransferase. Enzyme Microb Technol 2000;27:376–89.
Percudani R, Peracchi A. A genomic overview of pyridoxal-phosphate-dependentenzymes. EMBO Rep 2003;4:850–4.
Percudani R, Peracchi A. The B6 database: a tool for the description and classification ofvitamin B6-dependent enzymatic activities and of the corresponding protein families.BMC Bioinform 2009;10:273.
Perret A, Lechaplais C, Tricot S, Perchat N, Vergne C, Pelle C, et al. A novel acyl-CoA beta-transaminase characterized from a metagenome. PLoS One 2011;6:e22918.
Pleiss J. Systematic analysis of large enzyme families: identification of specificity- andselectivity-determining hotspots. ChemCatChem 2014;6:944–50.
Price MN, Dehal PS, Arkin AP. FastTree 2 — approximately maximum-likelihood trees forlarge alignments. PLoS One 2010;5:e9490.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam proteinfamilies database. Nucleic Acids Res 2012;40:D290–301.
Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, et al. A large-scaleevaluation of computational protein function prediction. Nat Methods 2013;10:221–7.
Rajaram V, Prasuna PR, Savithri HS, Murthy MRN. Structure of biosyntheticN-acetylornithine aminotransferase from Salmonella typhimurium: studies onsubstrate specificity and inhibitor binding. Proteins 2008;70:429–41.
Rao DR, Hariharan K, Vijayalakshmi KR. A study of the metabolism of L-αγ-diaminobutyric acid in a Xanthomonas species. Biochem J 1969;114:107–15.
Rausch C, Lerchner A, Schiefner A, Skerra A. Crystal structure of the ω-aminotransferase from Paracoccus denitrificans and its phylogenetic relationshipwith other class III amino-transferases that have biotechnological potential. Pro-teins 2013;81:774–87.
Reshetnikov AS, Khmelenina VN, Trotsenko YA. Characterization of the ectoine biosynthe-sis genes of haloalkalotolerant obligate methanotroph “Methylomicrobiumalcaliphilum 20Z”. Arch Microbiol 2006;184:286–97.
Roeder J, Schink B. Syntrophic degradation of cadaverine by a defined methanogeniccoculture. Appl Environ Microbiol 2009;75:4821–8.
Romero J, Martin JF, Liras P, Demain AL, Rius N. Partial purification, characterization andnitrogen regulation of the lysine ε-aminotransferase of Streptomyces clavuligerus. JInd Microbiol Biotechnol 1997;18:241–6.
Romo AJ, Liu HW. Mechanisms and structures of vitamin B6-dependent en-zymes involved in deoxy sugar biosynthesis. Biochim Biophys Acta 2011;1814:1534–47.
Rudat J, Brucher BR, Syldatk C. Transaminases for the synthesis of enantiopure beta-amino acids. AMB Express 2012;2:11.
Samsonova NN, Smirnov SV, Altman IB, Ptitsyn LR. Molecular cloning and characterizationof Escherichia coli K12 ygjG gene. BMC Microbiol 2003;3:2.
Sandmark J, Eliot AC, Famm K, Schneider G, Kirsch JF. Conserved and nonconservedresidues in the substrate binding site of 7,8-diaminopelargonic acid synthase fromEscherichia coli are essential for catalysis. Biochemistry 2004;43:1213–22.
Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, JarvisWR, et al. Biocatalytic asymmet-ric synthesis of chiral amines from ketones applied to sitagliptin manufacture.Science 2010;329:305–9.
Sayer C, Isupov MN, Westlake A, Littlechild JA. Structural studies of Pseudomonas andChromobacterium ω-aminotransferases provide insights into their differing substratespecificity. Acta Crystallogr D Biol Crystallogr 2013;69:564–76.
Schätzle S, Steffen-Munsberg F, Thontowi A, Höhne M, Robins K, Bornscheuer UT.Enzymatic asymmetric synthesis of enantiomerically pure aliphatic, aromatic andarylaliphatic amines with (R)-selective amine transaminases. Adv Synth Catal 2011;353:2439–45.
Schneider BL, Reitzer L. Pathway and enzyme redundancy in putrescine catabolism inEscherichia coli. J Bacteriol 2012;194:4080–8.
Schneider J, Wendisch VF. Biotechnological production of polyamines by bacteria: recentachievements and future perspectives. Appl Environ Microbiol 2011;91:17–30.
Schneider G, Käck H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes.Structure 2000;8:R1–6.
Schomburg I, Chang A, Placzek S, Söhngen C, Rother Ml, Lang M, et al. BRENDA in2013: integrated reactions, kinetic data, enzyme function data, improved diseaseclassification: new options and contents in BRENDA. Nucleic Acids Res 2013;41:D764–72.
Schousboe A, Wu JY, Roberts E. Purification and characterization of the 4-aminobutyrate-2-ketoglutarate transaminase from mouse brain. Biochemistry 1973;12:2868–73.
Schulz A, Taggeselle P, Tripier D, Bartsch K. Stereospecific production of the herbicidephosphinothricin (glufosinate) by transamination: isolation and characterization ofa phosphinothricin-specific transaminase from Escherichia coli. Appl EnvironMicrobiol 1990;56:1–6.
Schulze JO, Schubert WD, Moser J, Jahn D, Heinz DW. Evolutionary relationship betweeninitial enzymes of tetrapyrrole biosynthesis. J Mol Biol 2006;358:1212–20.
Schwibbert K, Marin-Sanguino A, Bagyan I, Heidrich G, Lentzen G, Seitz H, et al. A blue-print of ectoine metabolism from the genome of the industrial producer Halomonaselongata DSM 2581T. Environ Microbiol 2011;13:1973–94.
Seo J-H, Hwang J-Y, Seo S-H, Kang H, Hwang B-Y, Kim B-G. Computational selection,identification and structural analysis of ω-aminotransferases with various substratespecificities from the genome sequence of Mesorhizobium loti MAFF303099. BiosciBiotechnol Biochem 2012;76:1308–14.
Seong Gyu J, Jae Hoon B, Joong Sik J, Sang Ho J, Byung Ryong L, Kil Soo L, et al. Molecularcloning and functional expression of bovine brain GABA transaminase. Mol Cells2001;12:91–6.
Shaibe E, Metzer E, Halpern YS. Metabolic pathway for the utilization of L-arginine, L-or-nithine, agmatine, and putrescine as nitrogen sources in Escherichia coli K-12. JBacteriol 1985;163:933–7.
Shin J-S, Kim B-G. Kinetic resolution of α-methylbenzylamine with ω-transaminasescreened from soil microorganisms: application of a biphasic system to overcomeproduct inhibition. Biotechnol Bioeng 1997;55:348–58.
Shin J-S, Yun H, Jang JW, Park I, Kim B-G. Purification, characterization, and molecularcloning of a novel amine:pyruvate transaminase from Vibrio fluvialis JS17. ApplMicrobiol Biotechnol 2003;61:463–71.
Shon M, Shanmugavel R, Shin G, Mathew S, Lee SH, Yun H. Enzymatic synthesis of chiralγ-amino acids using ω-transaminase. Chem Commun 2014;50:12680–3.
Sigrist CJA, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, et al.PROSITE, a protein domain database for functional characterization and annotation.Nucleic Acids Res 2010;38:D161–6.
Smith MA, Kannangara CG, Grimm B, von Wettstein D. Characterization of glutamate-1-semialdehyde aminotransferase of Synechococcus. Steady-state kinetic analysis. EurJ Biochem 1991;202:749–57.
Soda K, Esaki N. Pyridoxal enzymes acting on D-amino acids. Pure Appl Chem 1994;66:709–14.
Soda K, Misono H, Yamamoto T. L-Lysine-α-ketoglutarate aminotransferase. I. identifica-tion of a product, Δ1-piperideine-6-carboxylic acid. Biochemistry 1968;7:4102–9.
Soda K, Yoshimura T, Esaki N. Stereospecificity for the hydrogen transfer of pyridoxalenzyme reactions. Chem Rec 2001;1:373–84.
Steffen-Munsberg F, Vickers C, Thontowi A, Schätzle S, Meinhardt T, Svedendahl HumbleM, et al. Revealing the structural basis of promiscuous amine transaminase activity.ChemCatChem 2013a;5:154–7.
603F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604

Steffen-Munsberg F, Vickers C, Thontowi A, Schätzle S, Tumlirsch T, Svedendahl HumbleM, et al. Connecting unexplored protein crystal structures to enzymatic function.ChemCatChem 2013b;5:150–3.
Steinkellner G, Gruber CC, Pavkov-Keller T, Binter A, Steiner K, Winkler C, et al. Identificationof promiscuous ene-reductase activity by mining structural databases using active siteconstellations. Nat Commun 2014;5.
Stetefeld J, Jenny M, Burkhard P. Intersubunit signaling in glutamate-1-semialdehyde-aminomutase. Proc Natl Acad Sci U S A 2006;103:13688–93.
Stoner GL, Eisenberg MA. Purification and properties of 7,8-diaminopelargonic acidaminotransferase. J Biol Chem 1975;250:4029–36.
Storici P, Capitani G, De Biase D, Moser M, John RA, Jansonius JN, et al. Crystal structure ofGABA-aminotransferase, a target for antiepileptic drug therapy. Biochemistry 1999;38:8628–34.
Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, et al. Structures ofγ-aminobutyric acid (GABA) aminotransferase, a pyridoxal 5′-phosphate, and[2Fe–2S] cluster-containing enzyme, complexed with γ-ethynyl-GABA and with theantiepilepsy drug Vigabatrin. J Biol Chem 2004;279:363–73.
Sun S, Zabinski RF, Toney MD. Reactions of alternate substrates demonstratestereoelectronic control of reactivity in dialkylglycine decarboxylase. Biochemistry1998;37:3865–75.
Sung BK, Kim YT. Structural arrangement for functional requirements of brain recombi-nant 4-aminobutyrate aminotransferase. J Biochem Mol Biol 2000;33:43–8.
Tamaki N, Sakata SF, Matsuda K. [35] Purification, properties, and sequencing ofaminoisobutyrate aminotransferases from rat liver. In: Sokatch JR, Harris RA, editors.Methods in enzymology. San Diego, CA: Academic Press; 2000. p. 376–89.
The PyMOL Molecular Graphics System, Version 1.6.0.0 Schrödinger, LCC, 2013.Tobin MB, Kovacevic S, Madduri K, Hoskins JA, Skatrud PL, Vining LC, et al. Localization of
the lysine ε-aminotransferase (lat) and δ-(L-α-aminoadipyl)-L-cysteinyl-D-valinesynthetase (pcbAB) genes from Streptomyces clavuligerus and production of lysineε-aminotransferase activity in Escherichia coli. J Bacteriol 1991;173:6223–9.
Toney MD. Computational studies on nonenzymatic and enzymatic pyridoxal phosphatecatalyzed decarboxylations of 2-aminoisobutyrate. Biochemistry 2001;40:1378–84.
Toney MD. Controlling reaction specificity in pyridoxal phosphate enzymes. BiochimBiophys Acta 2011;1814:1407–18.
Toney MD, Hohenester E, Cowan SW, Jansonius JN. Dialkylglycine decarboxylase struc-ture: bifunctional active site and alkali metal sites. Science 1993;261:756–9.
Toney MD, Hohenester E, Keller JW, Jansonius JN. Structural and mechanistic analysis oftwo refined crystal structures of the pyridoxal phosphate-dependent enzymedialkylglycine decarboxylase. J Mol Biol 1995;245:151–79.
Townsend C, Gunsior M, Muller U, Van FA, Sonke T. Fermentative production ofD-hydroxyphenylglycine and D-phenylglycine. WO International Patent ApplicationWO2002034921, 2002.
Tripathi SM, Ramachandran R. Direct evidence for a glutamate switch necessary forsubstrate recognition: crystal structures of lysine ε-aminotransferase (Rv3290c)from Mycobacterium tuberculosis H37Rv. J Mol Biol 2006;362:877–86.
Tufvesson P, Lima-Ramos J, Jensen JS, Al-Haque N, Neto W, Woodley JM. Process consid-erations for the asymmetric synthesis of chiral amines using transaminases.Biotechnol Bioeng 2011;108:1479–93.
Untrau S, Lebrihi A, Germain P, Lefebvre G. Lysine catabolism in Streptomyces ambofaciensproducer of macrolide antibiotic, spiramycin. Curr Microbiol 1992;25:313–8.
Van Arsdell SW, Perkins JB, Yocum RR, Luan L, Howitt CL, Prasad Chatterjee N, et al.Removing a bottleneck in the Bacillus subtilis biotin pathway: BioA utilizes lysine
rather than S-adenosylmethionine as the amino donor in the KAPA-to-DAPA reaction.Biotechnol Bioeng 2005;91:75–83.
Vandenende CS, Vlasschaert M, Seah SYK. Functional characterization of an aminotrans-ferase required for pyoverdine siderophore biosynthesis in Pseudomonas aeruginosaPAO1. J Bacteriol 2004;186:5596–602.
Veiga-da-Cunha M, Hadi F, Balligand T, Stroobant V, Van Schaftingen E. Molecularidentification of hydroxylysine kinase and of ammoniophospholyases acting on5-phosphohydroxy-L-lysine and phosphoethanolamine. J Biol Chem 2012;287:7246–55.
Voellym R, Leisinger T. Role of 4-aminobutyrate aminotransferase in the arginine metab-olism of Pseudomonas aeruginosa. J Bacteriol 1976;128:722–9.
Watanabe N, Sakabe K, Sakabe N, Higashi T, Sasaki K, Aibara S, et al. Crystal structure anal-ysis ofω-amino acid:pyruvate aminotransferase with a newly developedWeissenbergcamera and an imaging plate using synchrotron radiation. J Biochem 1989;105:1–3.
Webb EC, International Union of Biochemistry andMolecular Biology. Enzyme nomencla-ture 1992. Recommendations of the Nomenclature Committee of the InternationalUnion of Biochemistry and Molecular Biology on the Nomenclature and Classificationof Enzymes. San Diego, California: Academic Press; 1992.
Weber N, Ismail A, Gorwa-Grauslund M, Carlquist M. Biocatalytic potential of vanillinaminotransferase from Capsicum chinense. BMC Biotechnol 2014;14:25.
Wegman MA, Janssen MHA, van Rantwijk F, Sheldon RA. Towards biocatalytic synthesisof β-lactam antibiotics. Adv Synth Catal 2001;343:559–76.
Williamson NR, Simonsen HT, Ahmed RAA, Goldet G, Slater H, Woodley L, et al. Biosyn-thesis of the red antibiotic, prodigiosin, in Serratia: identification of a novel2-methyl-3-n-amyl-pyrrole (MAP) assembly pathway, definition of the terminalcondensing enzyme, and implications for undecylprodigiosin biosynthesis inStreptomyces. Mol Microbiol 2005;56:971–89.
Wiyakrutta S, Meevootisom V. A stereo-inverting D-phenylglycine aminotransferase fromPseudomonas stutzeri ST-201: purification, characterization and application forD-phenylglycine synthesis. J Biotechnol 1997;55:193–203.
Wybenga GG, Crismaru CG, Janssen DB, Dijkstra BW. Structural determinants of theβ-selectivity of a bacterial aminotransferase. J Biol Chem 2012;287:28495–502.
Xu Y, Labedan B, Glansdorff N. Surprising arginine biosynthesis: a reappraisal of theenzymology and evolution of the pathway in microorganisms. Microbiol Mol BiolRev 2007;71:36–47.
Yagi T, Misono H, Tanizawa K, Yoshimura T, Soda K. Characterization of the half andoverall reactions catalyzed by L-lysine:2-oxoglutarate 6-aminotransferase. J Biochem1991;109:61–5.
YASARA Structure, Version 13.6.16 YASARA Biosciences, 2013.Yonaha K, Toyama S, Yasuda M, Soda K. Properties of crystalline ω-amino acid: pyruvate
aminotransferase of Pseudomonas sp. F-126. Agric Biol Chem 1977;41:1701–6.Yorifuji T, Ishihara T, Naka T, Kondo S, Shimizu E. Purification and characterization of
polyamine aminotransferase of Arthrobacter sp. TMP-1. J Biochem 1997;122:537–43.Yun H, Lim S, Cho BK, Kim B-G. ω-Amino acid:pyruvate transaminase from Alcaligenes
denitrificans Y2k-2: a new catalyst for kinetic resolution of β-amino acids and amines.Appl Environ Microbiol 2004;70:2529–34.
Zhao S, Kumar R, Sakai A, Vetting MW, Wood BM, Brown S, et al. Discovery of newenzymes and metabolic pathways by using structure and genome context. Nature2013;502:698–702.
604 F. Steffen-Munsberg et al. / Biotechnology Advances 33 (2015) 566–604


1
Supplementary data
1 Remarks to the provided PyMOL session The session was created using PyMOL Version 1.6.0.0[1]. This software can be obtained from Schrödinger Inc. (for free for students) or compiled from its open source code. An introduction to its usage can be found in the PyMOL Wiki (http://www.pymolwiki.org/). Chain A of the structures have been aligned with the cealign command. Because all structures are aligned to the human Orn:αKG TA structure 2OAT, they might easily be compared. The first object ‘2oat_3DM_num’ contains 3D numbers derived from the 3D numbering scheme of the OrnTL DB, applied to the structure of the human Orn:αKG TA structure 2OAT, while the object ‘2oat_label’ contains this numbers as labels for the Cαs of each amino acid. All other objects contain the original numbering from the crystal structures. On the left of the PyMOL window, the reader will find scene buttons that recall the figures of the main text when clicked. If performance is an issue, reducing the ‘display quality’ will help (in the menu Display/Quality).
2 Remarks to the provided dual substrate recognition mechanism movie The movie visualises the dual substrate recognition of amine transaminases and presents a simplified mechanism of the transamination reaction. In the first half reaction, the internal aldimine of the amine transaminase (PDB ID: 3HMU) is converted to the PMP form with the concomitant conversion of L-‐alanine to pyruvate. R346 points towards the cofactor to coordinate alanine’s carboxylate. The movement of R346’s side chain out of the active site creates space to facilitate acetophenone binding and conversion to (S)-‐1-‐phenylethylamine in the second half reaction. The movie was prepared using the YASARA[2] and PyMOL[1] programs. 3HMU was used as the starting structure. The reaction intermediates were modelled using YASARA applying docking and molecular dynamics simulation methods. The purpose of the movie is mainly to visualize dual substrate recognition and the main steps during the first half reaction. Therefore, the intermediates and especially the transition states do not represent structures with the lowest possible energies as one could obtain by using QM simulations. Some (de)protonation steps are omitted, especially in the late stages oft the first half reaction (carbinol amine formation and hydrolysis), as the details are not yet known. Based on the intermediate structures generated with YASARA, the animations between the reaction intermediates were created by using PyMOL’s morphing routine. The second half reaction was modelled only as a shortened version.
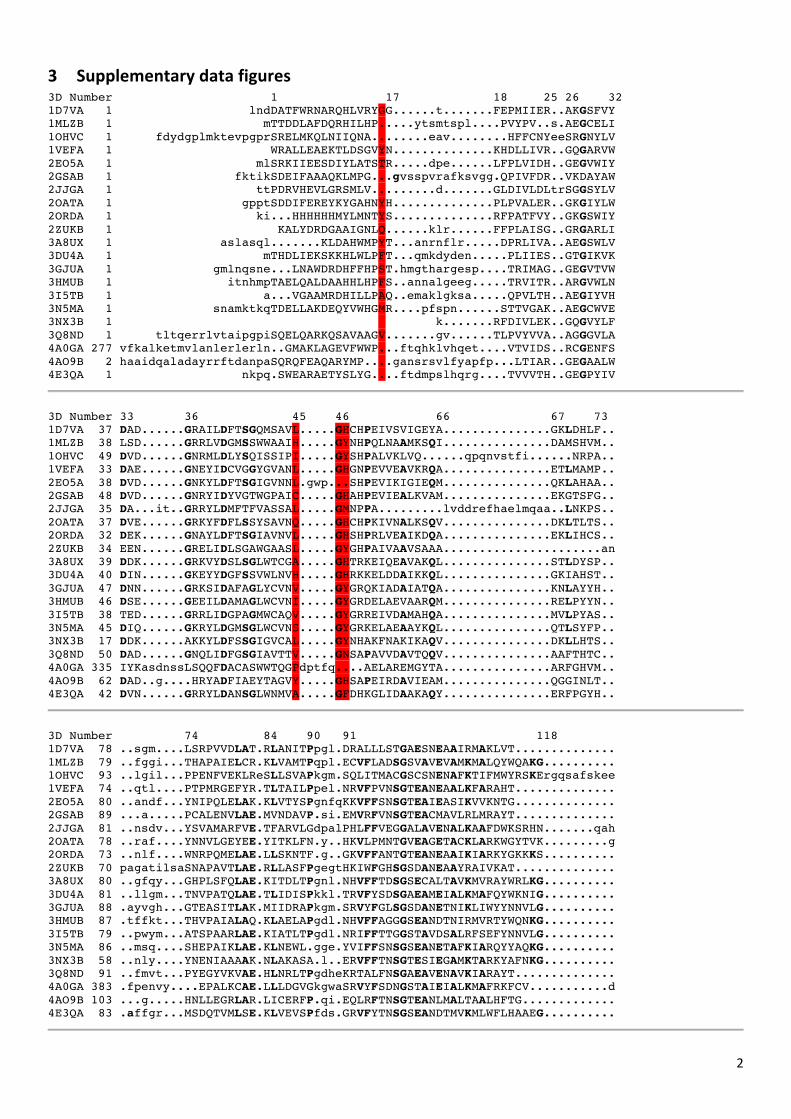
2
3 Supplementary data figures 3D Number 1 17 18 25 26 32 1D7VA 1 lndDATFWRNARQHLVRYGG......t.......FEPMIIER..AKGSFVY 1MLZB 1 mTTDDLAFDQRHILHP.....ytsmtspl....PVYPV..s.AEGCELI 1OHVC 1 fdydgplmktevpgprSRELMKQLNIIQNA........eav........HFFCNYeeSRGNYLV 1VEFA 1 WRALLEAEKTLDSGVYN..............KHDLLIVR..GQGARVW 2EO5A 1 mlSRKIIEESDIYLATSTR.....dpe......LFPLVIDH..GEGVWIY 2GSAB 1 fktikSDEIFAAAQKLMPG...gvsspvrafksvgg.QPIVFDR..VKDAYAW 2JJGA 1 ttPDRVHEVLGRSMLV.........d.......GLDIVLDLtrSGGSYLV 2OATA 1 gpptSDDIFEREYKYGAHNYH..............PLPVALER..GKGIYLW 2ORDA 1 ki...HHHHHHMYLMNTYS..............RFPATFVY..GKGSWIY 2ZUKB 1 KALYDRDGAAIGNLQ......klr......FFPLAISG..GRGARLI 3A8UX 1 aslasql.......KLDAHWMPYT...anrnflr.....DPRLIVA..AEGSWLV 3DU4A 1 mTHDLIEKSKKHLWLPFT...qmkdyden.....PLIIES..GTGIKVK 3GJUA 1 gmlnqsne...LNAWDRDHFFHPST.hmgthargesp....TRIMAG..GEGVTVW 3HMUB 1 itnhmpTAELQALDAAHHLHPFS..annalgeeg.....TRVITR..ARGVWLN 3I5TB 1 a...VGAAMRDHILLPAQ..emaklgksa.....QPVLTH..AEGIYVH 3N5MA 1 snamktkqTDELLAKDEQYVWHGMR....pfspn......STTVGAK..AEGCWVE 3NX3B 1 k.......RFDIVLEK..GQGVYLF 3Q8ND 1 tltqerrlvtaipgpiSQELQARKQSAVAAGV.......gv......TLPVYVVA..AGGGVLA 4A0GA 277 vfkalketmvlanlerlerln..GMAKLAGEVFWWP...ftqhklvhqet....VTVIDS..RCGENFS 4AO9B 2 haaidqaladayrrftdanpaSQRQFEAQARYMP....gansrsvlfyapfp...LTIAR..GEGAALW 4E3QA 1 nkpq.SWEARAETYSLYG....ftdmpslhqrg....TVVVTH..GEGPYIV
3D Number 33 36 45 46 66 67 73 1D7VA 37 DAD......GRAILDFTSGQMSAVL.....GHCHPEIVSVIGEYA...............GKLDHLF.. 1MLZB 38 LSD......GRRLVDGMSSWWAAIH.....GYNHPQLNAAMKSQI...............DAMSHVM.. 1OHVC 49 DVD......GNRMLDLYSQISSIPI.....GYSHPALVKLVQ......qpqnvstfi......NRPA.. 1VEFA 33 DAE......GNEYIDCVGGYGVANL.....GHGNPEVVEAVKRQA...............ETLMAMP.. 2EO5A 38 DVD......GNKYLDFTSGIGVNNL.gwp...SHPEVIKIGIEQM...............QKLAHAA.. 2GSAB 48 DVD......GNRYIDYVGTWGPAIC.....GHAHPEVIEALKVAM...............EKGTSFG.. 2JJGA 35 DA...it..GRRYLDMFTFVASSAL.....GMNPPA.........lvddrefhaelmqaa..LNKPS.. 2OATA 37 DVE......GRKYFDFLSSYSAVNQ.....GHCHPKIVNALKSQV...............DKLTLTS.. 2ORDA 32 DEK......GNAYLDFTSGIAVNVL.....GHSHPRLVEAIKDQA...............EKLIHCS.. 2ZUKB 34 EEN......GRELIDLSGAWGAASL.....GYGHPAIVAAVSAAA......................an 3A8UX 39 DDK......GRKVYDSLSGLWTCGA.....GHTRKEIQEAVAKQL...............STLDYSP.. 3DU4A 40 DIN......GKEYYDGFSSVWLNVH.....GHRKKELDDAIKKQL...............GKIAHST.. 3GJUA 47 DNN......GRKSIDAFAGLYCVNV.....GYGRQKIADAIATQA...............KNLAYYH.. 3HMUB 46 DSE......GEEILDAMAGLWCVNI.....GYGRDELAEVAARQM...............RELPYYN.. 3I5TB 38 TED......GRRLIDGPAGMWCAQV.....GYGRREIVDAMAHQA...............MVLPYAS.. 3N5MA 45 DIQ......GKRYLDGMSGLWCVNS.....GYGRKELAEAAYKQL...............QTLSYFP.. 3NX3B 17 DDK......AKKYLDFSSGIGVCAL.....GYNHAKFNAKIKAQV...............DKLLHTS.. 3Q8ND 50 DAD......GNQLIDFGSGIAVTTV.....GNSAPAVVDAVTQQV...............AAFTHTC.. 4A0GA 335 IYKasdnssLSQQFDACASWWTQGPdptfq....AELAREMGYTA...............ARFGHVM.. 4AO9B 62 DAD..g....HRYADFIAEYTAGVY.....GHSAPEIRDAVIEAM...............QGGINLT.. 4E3QA 42 DVN......GRRYLDANSGLWNMVA.....GFDHKGLIDAAKAQY...............ERFPGYH..
3D Number 74 84 90 91 118 1D7VA 78 ..sgm....LSRPVVDLAT.RLANITPpgl.DRALLLSTGAESNEAAIRMAKLVT.............. 1MLZB 79 ..fggi...THAPAIELCR.KLVAMTPqpl.ECVFLADSGSVAVEVAMKMALQYWQAKG.......... 1OHVC 93 ..lgil...PPENFVEKLReSLLSVAPkgm.SQLITMACGSCSNENAFKTIFMWYRSKErgqsafskee 1VEFA 74 ..qtl....PTPMRGEFYR.TLTAILPpel.NRVFPVNSGTEANEAALKFARAHT.............. 2EO5A 80 ..andf...YNIPQLELAK.KLVTYSPgnfqKKVFFSNSGTEAIEASIKVVKNTG.............. 2GSAB 89 ...a.....PCALENVLAE.MVNDAVP.si.EMVRFVNSGTEACMAVLRLMRAYT.............. 2JJGA 81 ..nsdv...YSVAMARFVE.TFARVLGdpalPHLFFVEGGALAVENALKAAFDWKSRHN.......qah 2OATA 78 ..raf....YNNVLGEYEE.YITKLFN.y..HKVLPMNTGVEAGETACKLARKWGYTVK.........g 2ORDA 73 ..nlf....WNRPQMELAE.LLSKNTF.g..GKVFFANTGTEANEAAIKIARKYGKKKS.......... 2ZUKB 70 pagatilsaSNAPAVTLAE.RLLASFPgegtHKIWFGHSGSDANEAAYRAIVKAT.............. 3A8UX 80 ..gfqy...GHPLSFQLAE.KITDLTPgnl.NHVFFTDSGSECALTAVKMVRAYWRLKG.......... 3DU4A 81 ..llgm...TNVPATQLAE.TLIDISPkkl.TRVFYSDSGAEAMEIALKMAFQYWKNIG.......... 3GJUA 88 .ayvgh...GTEASITLAK.MIIDRAPkgm.SRVYFGLSGSDANETNIKLIWYYNNVLG.......... 3HMUB 87 .tffkt...THVPAIALAQ.KLAELAPgdl.NHVFFAGGGSEANDTNIRMVRTYWQNKG.......... 3I5TB 79 ..pwym...ATSPAARLAE.KIATLTPgdl.NRIFFTTGGSTAVDSALRFSEFYNNVLG.......... 3N5MA 86 ..msq....SHEPAIKLAE.KLNEWL.gge.YVIFFSNSGSEANETAFKIARQYYAQKG.......... 3NX3B 58 ..nly....YNENIAAAAK.NLAKASA.l..ERVFFTNSGTESIEGAMKTARKYAFNKG.......... 3Q8ND 91 ..fmvt...PYEGYVKVAE.HLNRLTPgdheKRTALFNSGAEAVENAVKIARAYT.............. 4A0GA 383 .fpenvy....EPALKCAE.LLLDGVGkgwaSRVYFSDNGSTAIEIALKMAFRKFCV...........d 4AO9B 103 ...g.....HNLLEGRLAR.LICERFP.qi.EQLRFTNSGTEANLMALTAALHFTG............. 4E3QA 83 .affgr...MSDQTVMLSE.KLVEVSPfds.GRVFYTNSGSEANDTMVKMLWFLHAAEG..........
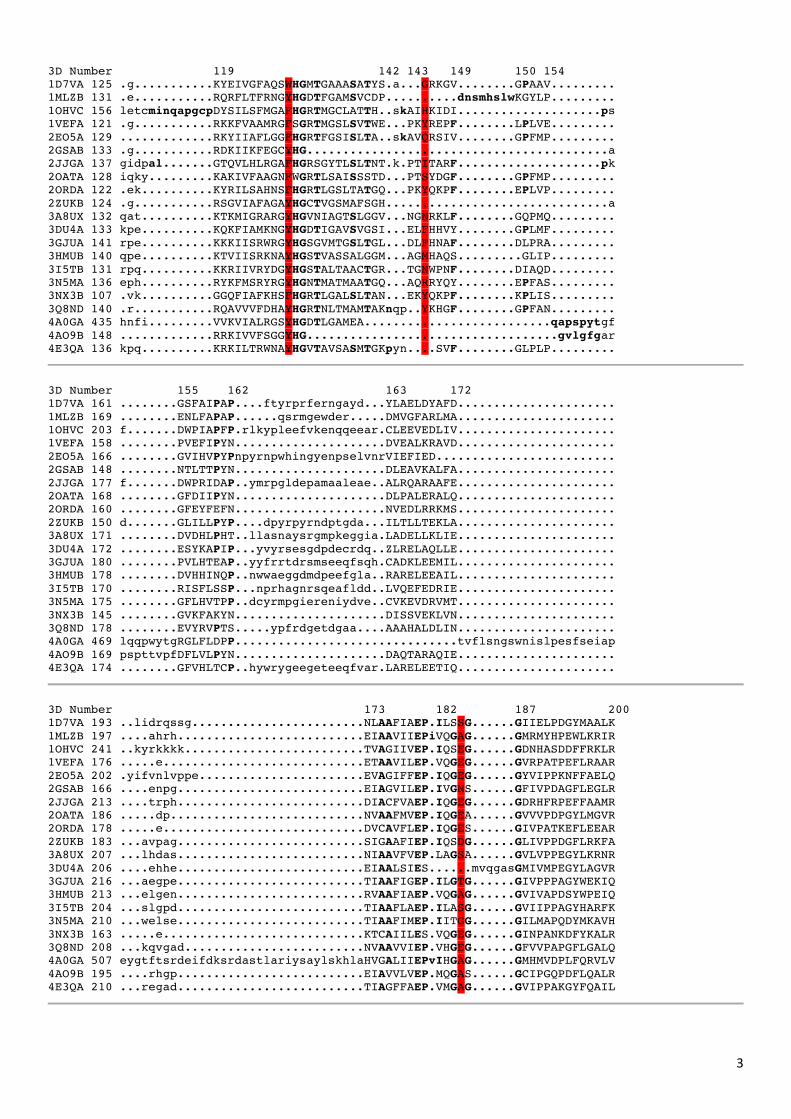
3
3D Number 119 142 143 149 150 154 1D7VA 125 .g...........KYEIVGFAQSWHGMTGAAASATYS.a...GRKGV........GPAAV......... 1MLZB 131 .e...........RQRFLTFRNGYHGDTFGAMSVCDP..........dnsmhslwKGYLP......... 1OHVC 156 letcminqapgcpDYSILSFMGAFHGRTMGCLATTH..skAIHKIDI....................ps 1VEFA 121 .g...........RKKFVAAMRGFSGRTMGSLSVTWE...PKYREPF........LPLVE......... 2EO5A 129 .............RKYIIAFLGGFHGRTFGSISLTA..skAVQRSIV........GPFMP......... 2GSAB 133 .g...........RDKIIKFEGCYHG..........................................a 2JJGA 137 gidpal.......GTQVLHLRGAFHGRSGYTLSLTNT.k.PTITARF....................pk 2OATA 128 iqky.........KAKIVFAAGNFWGRTLSAISSSTD...PTSYDGF........GPFMP......... 2ORDA 122 .ek..........KYRILSAHNSFHGRTLGSLTATGQ...PKYQKPF........EPLVP......... 2ZUKB 124 .g...........RSGVIAFAGAYHGCTVGSMAFSGH...............................a 3A8UX 132 qat..........KTKMIGRARGYHGVNIAGTSLGGV...NGNRKLF........GQPMQ......... 3DU4A 133 kpe..........KQKFIAMKNGYHGDTIGAVSVGSI...ELFHHVY........GPLMF......... 3GJUA 141 rpe..........KKKIISRWRGYHGSGVMTGSLTGL...DLFHNAF........DLPRA......... 3HMUB 140 qpe..........KTVIISRKNAYHGSTVASSALGGM...AGMHAQS.........GLIP......... 3I5TB 131 rpq..........KKRIIVRYDGYHGSTALTAACTGR...TGNWPNF........DIAQD......... 3N5MA 136 eph..........RYKFMSRYRGYHGNTMATMAATGQ...AQRRYQY........EPFAS......... 3NX3B 107 .vk..........GGQFIAFKHSFHGRTLGALSLTAN...EKYQKPF........KPLIS......... 3Q8ND 140 .r...........RQAVVVFDHAYHGRTNLTMAMTAKnqp..YKHGF........GPFAN......... 4A0GA 435 hnfi.........VVKVIALRGSYHGDTLGAMEA..........................qapspytgf 4AO9B 148 .............RRKIVVFSGGYHG...................................gvlgfgar 4E3QA 136 kpq..........KRKILTRWNAYHGVTAVSASMTGKpyn....SVF........GLPLP.........
3D Number 155 162 163 172 1D7VA 161 ........GSFAIPAP....ftyrprferngayd...YLAELDYAFD...................... 1MLZB 169 ........ENLFAPAP......qsrmgewder.....DMVGFARLMA...................... 1OHVC 203 f.......DWPIAPFP.rlkypleefvkenqqeear.CLEEVEDLIV...................... 1VEFA 158 ........PVEFIPYN.....................DVEALKRAVD...................... 2EO5A 166 ........GVIHVPYPnpyrnpwhingyenpselvnrVIEFIED......................... 2GSAB 148 ........NTLTTPYN.....................DLEAVKALFA...................... 2JJGA 177 f.......DWPRIDAP..ymrpgldepamaaleae..ALRQARAAFE...................... 2OATA 168 ........GFDIIPYN.....................DLPALERALQ...................... 2ORDA 160 ........GFEYFEFN.....................NVEDLRRKMS...................... 2ZUKB 150 d.......GLILLPYP....dpyrpyrndptgda...ILTLLTEKLA...................... 3A8UX 171 ........DVDHLPHT..llasnaysrgmpkeggia.LADELLKLIE...................... 3DU4A 172 ........ESYKAPIP...yvyrsesgdpdecrdq..ZLRELAQLLE...................... 3GJUA 180 ........PVLHTEAP..yyfrrtdrsmseeqfsqh.CADKLEEMIL...................... 3HMUB 178 ........DVHHINQP..nwwaeggdmdpeefgla..RARELEEAIL...................... 3I5TB 170 ........RISFLSSP...nprhagnrsqeafldd..LVQEFEDRIE...................... 3N5MA 175 ........GFLHVTPP..dcyrmpgiereniydve..CVKEVDRVMT...................... 3NX3B 145 ........GVKFAKYN.....................DISSVEKLVN...................... 3Q8ND 178 ........EVYRVPTS.....ypfrdgetdgaa....AAAHALDLIN...................... 4A0GA 469 lqqpwytgRGLFLDPP...............................tvflsngswnislpesfseiap 4AO9B 169 pspttvpfDFLVLPYN.....................DAQTARAQIE...................... 4E3QA 174 ........GFVHLTCP..hywrygeegeteeqfvar.LARELEETIQ......................
3D Number 173 182 187 200 1D7VA 193 ..lidrqssg........................NLAAFIAEP.ILSSG......GIIELPDGYMAALK 1MLZB 197 ....ahrh..........................EIAAVIIEPiVQGAG......GMRMYHPEWLKRIR 1OHVC 241 ..kyrkkkk.........................TVAGIIVEP.IQSEG......GDNHASDDFFRKLR 1VEFA 176 .....e............................ETAAVILEP.VQGEG......GVRPATPEFLRAAR 2EO5A 202 .yifvnlvppe.......................EVAGIFFEP.IQGEG......GYVIPPKNFFAELQ 2GSAB 166 ....enpg..........................EIAGVILEP.IVGNS......GFIVPDAGFLEGLR 2JJGA 213 ....trph..........................DIACFVAEP.IQGEG......GDRHFRPEFFAAMR 2OATA 186 .....dp...........................NVAAFMVEP.IQGEA......GVVVPDPGYLMGVR 2ORDA 178 .....e............................DVCAVFLEP.IQGES......GIVPATKEFLEEAR 2ZUKB 183 ...avpag..........................SIGAAFIEP.IQSDG......GLIVPPDGFLRKFA 3A8UX 207 ...lhdas..........................NIAAVFVEP.LAGSA......GVLVPPEGYLKRNR 3DU4A 206 ....ehhe..........................EIAALSIES......mvqgasGMIVMPEGYLAGVR 3GJUA 216 ...aegpe..........................TIAAFIGEP.ILGTG......GIVPPPAGYWEKIQ 3HMUB 213 ...elgen..........................RVAAFIAEP.VQGAG......GVIVAPDSYWPEIQ 3I5TB 204 ...slgpd..........................TIAAFLAEP.ILASG......GVIIPPAGYHARFK 3N5MA 210 ...welse..........................TIAAFIMEP.IITGG......GILMAPQDYMKAVH 3NX3B 163 .....e............................KTCAIILES.VQGEG......GINPANKDFYKALR 3Q8ND 208 ...kqvgad.........................NVAAVVIEP.VHGEG......GFVVPAPGFLGALQ 4A0GA 507 eygtftsrdeifdksrdastlariysaylskhlaHVGALIIEPvIHGAG......GMHMVDPLFQRVLV 4AO9B 195 ....rhgp..........................EIAVVLVEP.MQGAS......GCIPGQPDFLQALR 4E3QA 210 ...regad..........................TIAGFFAEP.VMGAG......GVIPPAKGYFQAIL
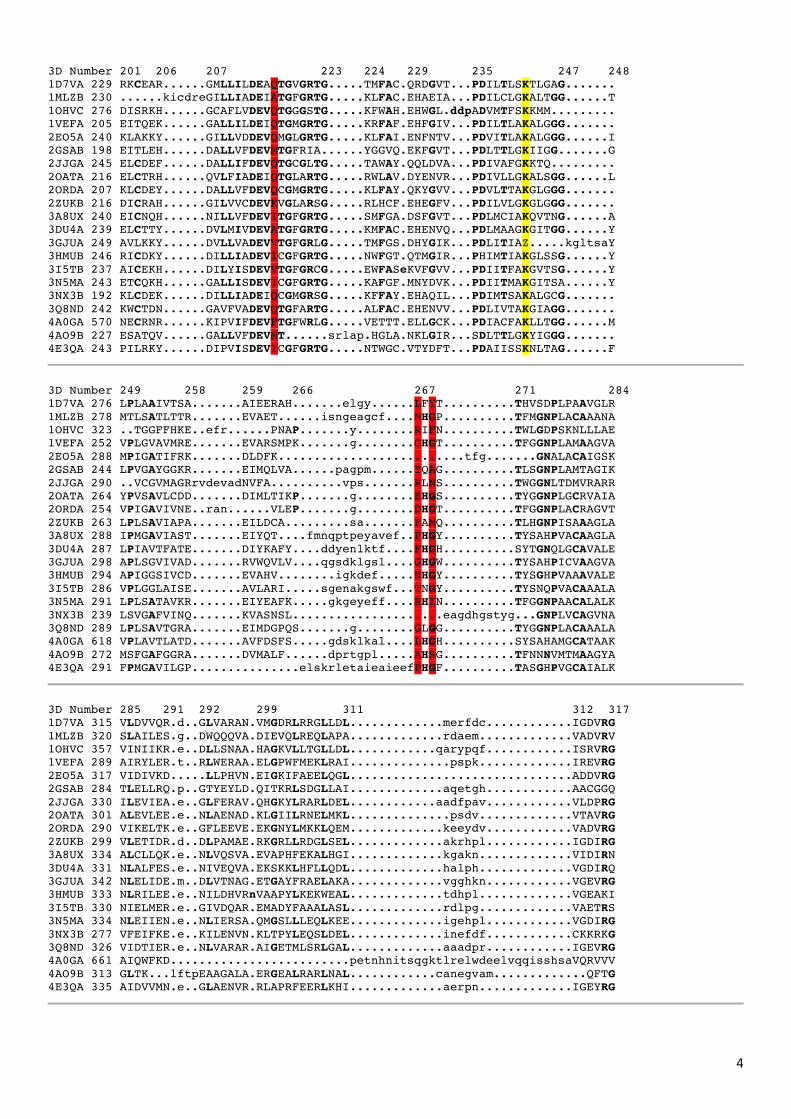
4
3D Number 201 206 207 223 224 229 235 247 248 1D7VA 229 RKCEAR......GMLLILDEAQTGVGRTG.....TMFAC.QRDGVT...PDILTLSKTLGAG....... 1MLZB 230 ......kicdreGILLIADEIATGFGRTG.....KLFAC.EHAEIA...PDILCLGKALTGG......T 1OHVC 276 DISRKH......GCAFLVDEVQTGGGSTG.....KFWAH.EHWGL.ddpADVMTFSKKMM......... 1VEFA 205 EITQEK......GALLILDEIQTGMGRTG.....KRFAF.EHFGIV...PDILTLAKALGGG....... 2EO5A 240 KLAKKY......GILLVDDEVQMGLGRTG.....KLFAI.ENFNTV...PDVITLAKALGGG......I 2GSAB 198 EITLEH......DALLVFDEVMTGFRIA......YGGVQ.EKFGVT...PDLTTLGKIIGG.......G 2JJGA 245 ELCDEF......DALLIFDEVQTGCGLTG.....TAWAY.QQLDVA...PDIVAFGKKTQ......... 2OATA 216 ELCTRH......QVLFIADEIQTGLARTG.....RWLAV.DYENVR...PDIVLLGKALSGG......L 2ORDA 207 KLCDEY......DALLVFDEVQCGMGRTG.....KLFAY.QKYGVV...PDVLTTAKGLGGG....... 2ZUKB 216 DICRAH......GILVVCDEVKVGLARSG.....RLHCF.EHEGFV...PDILVLGKGLGGG....... 3A8UX 240 EICNQH......NILLVFDEVITGFGRTG.....SMFGA.DSFGVT...PDLMCIAKQVTNG......A 3DU4A 239 ELCTTY......DVLMIVDEVATGFGRTG.....KMFAC.EHENVQ...PDLMAAGKGITGG......Y 3GJUA 249 AVLKKY......DVLLVADEVVTGFGRLG.....TMFGS.DHYGIK...PDLITIAZ.....kgltsaY 3HMUB 246 RICDKY......DILLIADEVICGFGRTG.....NWFGT.QTMGIR...PHIMTIAKGLSSG......Y 3I5TB 237 AICEKH......DILYISDEVVTGFGRCG.....EWFASeKVFGVV...PDIITFAKGVTSG......Y 3N5MA 243 ETCQKH......GALLISDEVICGFGRTG.....KAFGF.MNYDVK...PDIITMAKGITSA......Y 3NX3B 192 KLCDEK......DILLIADEIQCGMGRSG.....KFFAY.EHAQIL...PDIMTSAKALGCG....... 3Q8ND 242 KWCTDN......GAVFVADEVQTGFARTG.....ALFAC.EHENVV...PDLIVTAKGIAGG....... 4A0GA 570 NECRNR......KIPVIFDEVFTGFWRLG.....VETTT.ELLGCK...PDIACFAKLLTGG......M 4AO9B 227 ESATQV......GALLVFDEVMT......srlap.HGLA.NKLGIR...SDLTTLGKYIGGG....... 4E3QA 243 PILRKY......DIPVISDEVICGFGRTG.....NTWGC.VTYDFT...PDAIISSKNLTAG......F
3D Number 249 258 259 266 267 271 284 1D7VA 276 LPLAAIVTSA.......AIEERAH.......elgy......LFYT..........THVSDPLPAAVGLR 1MLZB 278 MTLSATLTTR.......EVAET......isngeagcf....MHGP..........TFMGNPLACAAANA 1OHVC 323 ..TGGFFHKE..efr......PNAP.......y........RIFN..........TWLGDPSKNLLLAE 1VEFA 252 VPLGVAVMRE.......EVARSMPK.......g........GHGT..........TFGGNPLAMAAGVA 2EO5A 288 MPIGATIFRK.......DLDFK..........................tfg.......GNALACAIGSK 2GSAB 244 LPVGAYGGKR.......EIMQLVA......pagpm......YQAG..........TLSGNPLAMTAGIK 2JJGA 290 ..VCGVMAGRrvdevadNVFA..........vps.......RLNS..........TWGGNLTDMVRARR 2OATA 264 YPVSAVLCDD.......DIMLTIKP.......g........EHGS..........TYGGNPLGCRVAIA 2ORDA 254 VPIGAVIVNE..ran......VLEP.......g........DHGT..........TFGGNPLACRAGVT 2ZUKB 263 LPLSAVIAPA.......EILDCA.........sa.......FAMQ..........TLHGNPISAAAGLA 3A8UX 288 IPMGAVIAST.......EIYQT....fmnqptpeyavef..PHGY..........TYSAHPVACAAGLA 3DU4A 287 LPIAVTFATE.......DIYKAFY....ddyenlktf....FHGH..........SYTGNQLGCAVALE 3GJUA 298 APLSGVIVAD.......RVWQVLV....qgsdklgsl....GHGW..........TYSAHPICVAAGVA 3HMUB 294 APIGGSIVCD.......EVAHV........igkdef.....NHGY..........TYSGHPVAAAVALE 3I5TB 286 VPLGGLAISE.......AVLARI.....sgenakgswf...TNGY..........TYSNQPVACAAALA 3N5MA 291 LPLSATAVKR.......EIYEAFK.....gkgeyeff....RHIN..........TFGGNPAACALALK 3NX3B 239 LSVGAFVINQ.......KVASNSL.....................eagdhgstyg...GNPLVCAGVNA 3Q8ND 289 LPLSAVTGRA.......EIMDGPQS.......g........GLGG..........TYGGNPLACAAALA 4A0GA 618 VPLAVTLATD.......AVFDSFS.....gdsklkal....LHGH..........SYSAHAMGCATAAK 4AO9B 272 MSFGAFGGRA.......DVMALF......dprtgpl.....AHSG..........TFNNNVMTMAAGYA 4E3QA 291 FPMGAVILGP...............elskrletaieaieefPHGF..........TASGHPVGCAIALK
3D Number 285 291 292 299 311 312 317 1D7VA 315 VLDVVQR.d..GLVARAN.VMGDRLRRGLLDL.............merfdc............IGDVRG 1MLZB 320 SLAILES.g..DWQQQVA.DIEVQLREQLAPA.............rdaem.............VADVRV 1OHVC 357 VINIIKR.e..DLLSNAA.HAGKVLLTGLLDL............qarypqf............ISRVRG 1VEFA 289 AIRYLER.t..RLWERAA.ELGPWFMEKLRAI..............pspk.............IREVRG 2EO5A 317 VIDIVKD.....LLPHVN.EIGKIFAEELQGL...............................ADDVRG 2GSAB 284 TLELLRQ.p..GTYEYLD.QITKRLSDGLLAI.............aqetgh............AACGGQ 2JJGA 330 ILEVIEA.e..GLFERAV.QHGKYLRARLDEL............aadfpav............VLDPRG 2OATA 301 ALEVLEE.e..NLAENAD.KLGIILRNELMKL..............psdv.............VTAVRG 2ORDA 290 VIKELTK.e..GFLEEVE.EKGNYLMKKLQEM.............keeydv............VADVRG 2ZUKB 299 VLETIDR.d..DLPAMAE.RKGRLLRDGLSEL.............akrhpl............IGDIRG 3A8UX 334 ALCLLQK.e..NLVQSVA.EVAPHFEKALHGI.............kgakn.............VIDIRN 3DU4A 331 NLALFES.e..NIVEQVA.EKSKKLHFLLQDL.............halph.............VGDIRQ 3GJUA 342 NLELIDE.m..DLVTNAG.ETGAYFRAELAKA.............vgghkn............VGEVRG 3HMUB 333 NLRILEE.e..NILDHVRnVAAPYLKEKWEAL.............tdhpl.............VGEAKI 3I5TB 330 NIELMER.e..GIVDQAR.EMADYFAAALASL.............rdlpg.............VAETRS 3N5MA 334 NLEIIEN.e..NLIERSA.QMGSLLLEQLKEE.............igehpl............VGDIRG 3NX3B 277 VFEIFKE.e..KILENVN.KLTPYLEQSLDEL.............inefdf............CKKRKG 3Q8ND 326 VIDTIER.e..NLVARAR.AIGETMLSRLGAL.............aaadpr............IGEVRG 4A0GA 661 AIQWFKD.........................petnhnitsqgktlrelwdeelvqqisshsaVQRVVV 4AO9B 313 GLTK...lftpEAAGALA.ERGEALRARLNAL............canegvam.............QFTG 4E3QA 335 AIDVVMN.e..GLAENVR.RLAPRFEERLKHI.............aerpn.............IGEYRG

5
3D Number 318 328 329 341 349 350 361 1D7VA 355 RGLLLGVEIVK....drrtkepad....GLGAKITRECMN.LGLSMNIVQ.lpgmg.GVFRIAPPLTVS 1MLZB 359 LGAIGVVETTH........p........VNMAALQKFFVE.QGVWIRPFG.......KLIYLMPPYIIL 1OHVC 398 RGTFCSFDTPD........e........SIRNKLISIARN.KGVMLGGCG...d...KSIRFRPTLVFR 1VEFA 327 MGLMVGLELKE..................KAAPYIARLEKeHRVLALQAG...p...TVIRFLPPLVIE 2EO5A 349 IGLAWGLEYNE........k........KVRDRIIGESFK.RGLLLLPAG...r...SAIRVIPPLVIS 2GSAB 324 VSGMFGFFFT..egpvhnyedakksdl.QKFSRFHRGMLE.QGIYLAPSQ...fe.....AGFTSLAHT 2JJGA 371 RGLMCAFSLPT........t........ADRDELIRQLWQ.RAVIVLPAG...a...DTVRFRPPLTVS 2OATA 339 KGLLNAIVIKE.......tkd.......WDAWKVCLRLRD.NGLLAKPTH...g...DIIRFAPPLVIK 2ORDA 330 MGLMIGIQFRE........e........VSNREVATKCFE.NKLLVVPAG...n...NTIRFLPPLTVE 2ZUKB 339 RGLACGMELVC....drqsrepar....AETAKLIYRAYQ.LGLVVYYVG..mng..NVLEFTPPLTIT 3A8UX 373 FGLAGAIQIAP......rdgdai.....VRPFEAGMALWK.AGFYVRFGG.......DTLQFGPTFNSK 3DU4A 370 LGFMCGAELVR...sketkepypadr..RIGYKVSLKMRE.LGMLTRPLG.......DVIAFLPPLAST 3GJUA 382 DGMLAAVEFVA...dkddrvffdasq..KIGPQVATALAA.SGVIGRAMP...qg..DILGFAPPLCLT 3HMUB 373 VGMMASIALTP..nkasrakfasepg..TIGYICRERCFA.NNLIMRHVG.......DRMIISPPLVIT 3I5TB 369 VGLVGCVQCL......lgtaedk.....AFTLKIDERCFE.LGLIVRPLG.......DLCVISPPLIIS 3N5MA 374 KGLLVGIELVN....dketkepidn...DKIASVVNACKE.KGLIIGRNGmttagynNILTLAPPLVIS 3NX3B 317 LGFMQGLSLDK........s........VKVAKVIQKCQE.NALLLISCG...e...NDLRFLPPLILQ 3Q8ND 366 RGAMIAVELVK.....pgttepda....DLTKRVAAAAHA.QGLVVLTCG..tyg..NVLRFLPPLSMP 4A0GA 705 IGTLFALELKS........l.........YAKSLLIMLRE.DGIFTRPLG.......NVIYLMCGPCTS 4AO9B 353 IGSLMNAHF..vqgdvrssedlaavdgr.LRQLLFFHLLN.EDIYSSPR..........GFVVLSLPLT 4E3QA 374 IGFMWALEAVK...dkasktpfdgnl..SVSERIANTCTD.LGLICRPLG.......QSVVLCPPFILT
3D Number 362 379 1D7VA 413 EDEIDLGLSLLGQAIERAl 1MLZB 404 PQQLQRLTAAVNRAVQDEtffc 1OHVC 444 DHHAHLFLNIFSDILADF 1VEFA 372 KEDLERVVEAVRAVLA 2EO5A 395 EEEAKQGLDILKKVIKVV 2GSAB 381 EEDIDATLAAARTVMSAL 2JJGA 417 TAEIDAAIAAVRSALPVVt 2OATA 387 EDELRESIEIINKTILSF 2ORDA 376 YGEIDLAVETLKKVLQGI 2ZUKB 395 ETDIHKALDLLDRAFSELsavsneeiaqfagw 3A8UX 423 PQDLDRLFDAVGEVLNKLl 3DU4A 426 AEELSEMVAIMKQAIHEVtsled 3GJUA 440 REQADIVVSKTADAVKSVfa 3HMUB 430 PAEIDEMFVRIRKSLDEAqaeiekqglmkse 3I5TB 419 RAQIDEMVAIMRQAITEVsaahgl 3N5MA 435 SEEIAFVIGTLKTAMERI 3NX3B 363 KEHIDEMSEKLRKALKSF 3Q8ND 421 DHLLDEGLDILAAVFAEVk 4A0GA 749 PEICRRLLTKLYKRLGEFnrt 4AO9B 408 DADIDRYVAAIGSFIGGHgallpran 4E3QA 430 EAQMDEMFDKLEKALDKVfaeva
Figure S1: Structural based sequence alignment of all OrnTL DB subfamily template structures. The 3D numbers are given in the first row, sequence numbers correspond to the residues present in the aligned chain (indicated by the last letter of the enzymes’ names). Core regions are shown in capital letters and residues in variable regions are not aligned. The sequence-‐function matrix (Table 2) residues are highlighted red and the catalytic lysine is highlighted yellow.
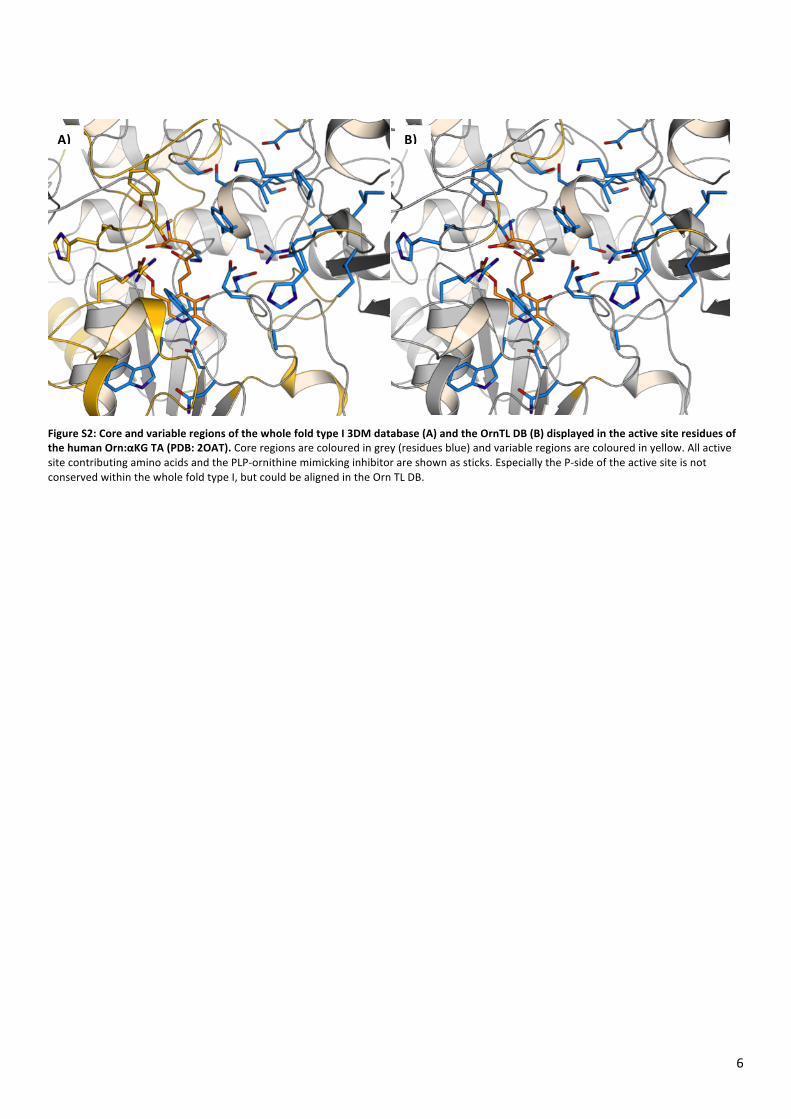
6
Figure S2: Core and variable regions of the whole fold type I 3DM database (A) and the OrnTL DB (B) displayed in the active site residues of the human Orn:αKG TA (PDB: 2OAT). Core regions are coloured in grey (residues blue) and variable regions are coloured in yellow. All active site contributing amino acids and the PLP-‐ornithine mimicking inhibitor are shown as sticks. Especially the P-‐side of the active site is not conserved within the whole fold type I, but could be aligned in the Orn TL DB.
A) B)
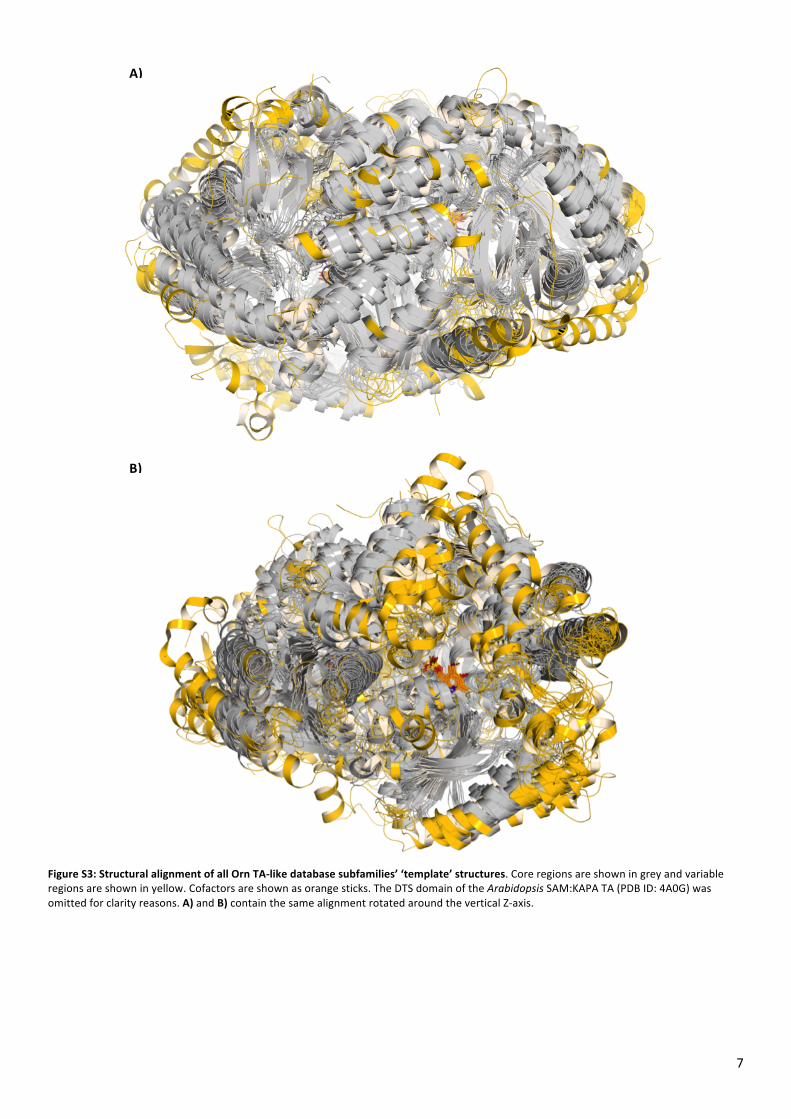
7
Figure S3: Structural alignment of all Orn TA-‐like database subfamilies’ ‘template’ structures. Core regions are shown in grey and variable regions are shown in yellow. Cofactors are shown as orange sticks. The DTS domain of the Arabidopsis SAM:KAPA TA (PDB ID: 4A0G) was omitted for clarity reasons. A) and B) contain the same alignment rotated around the vertical Z-‐axis.
A)
B)
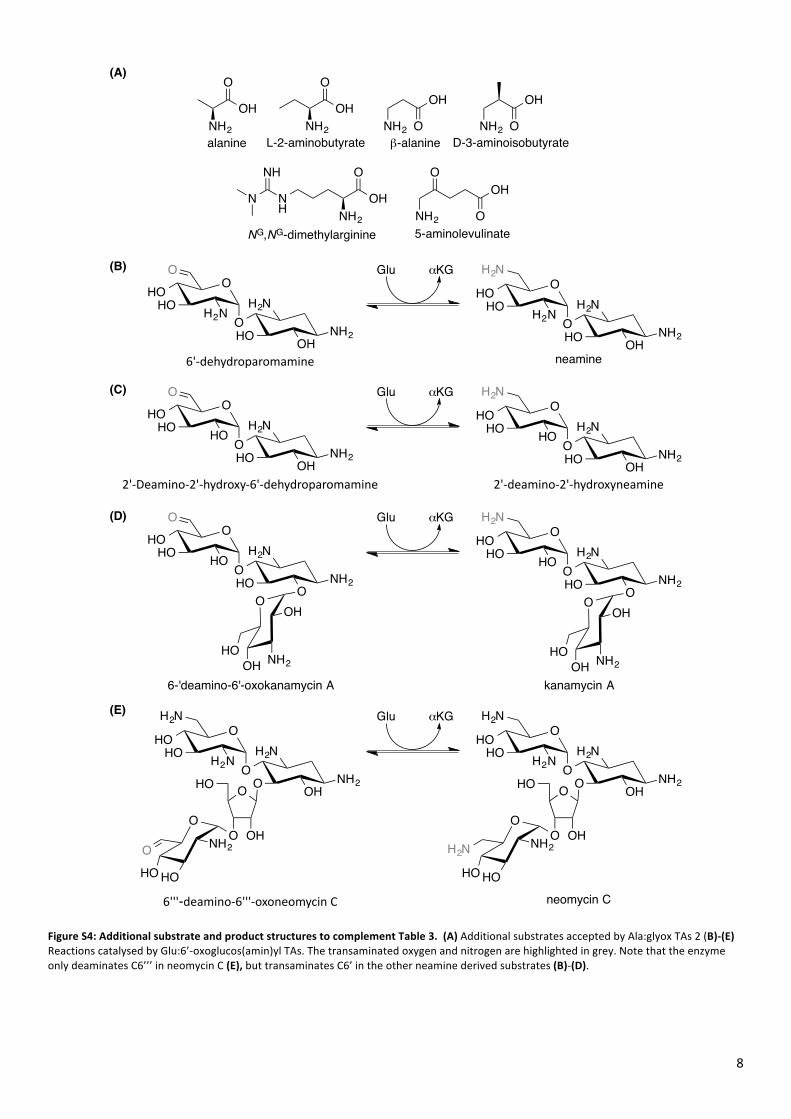
8
Figure S4: Additional substrate and product structures to complement Table 3. (A) Additional substrates accepted by Ala:glyox TAs 2 (B)-‐(E) Reactions catalysed by Glu:6’-‐oxoglucos(amin)yl TAs. The transaminated oxygen and nitrogen are highlighted in grey. Note that the enzyme only deaminates C6’’’ in neomycin C (E), but transaminates C6’ in the other neamine derived substrates (B)-‐(D).
OHO
HOO
O
H2N
HO NH2O
H2NH
OHO
HOO
N
H2N
HO NH2OH
H2NH2
neamine
OHO
HOO
O
H2N
HO NH2OH
H2NH
2'#deamino#2'#hydroxyneamine
O
OH NH2
OH
HO
kanamycin A
OHO
HOO
N
H2N
O NH2OH
H2NH2
OHO
O OHO
HO HO
NH2H2N
neomycin C
OHO
HOO
O
O
HO NH2OH
H2NH
2'#Deamino#2'#hydroxy#6'#dehydroparomamine
OHO
HOO
N
O
HO NH2OH
H2NH2
6'#dehydroparomamine
Glu αKG
Glu αKG
OHO
HOO
O
O
HO NH2O
H2NH
O
OH NH2
OH
HO
6-'deamino-6'-oxokanamycin A
6'''-deamino#6'''#oxoneomycin3C
OHO
HOO
N
H2N
O NH2OH
H2NH2
OHO
O OHO
HO HO
NH2O
Glu αKG
Glu αKG
(B)
(C)
(D)
(E)
NH2
OH
O
NH2
OH
O NH2
OH
O
NH2
OOH
O
NH2
OH
O
NH2
OH
O
NH
N
NH
alanine β-alanine D-3-aminoisobutyrate
5-aminolevulinate
L-2-aminobutyrate
NG,NG-dimethylarginine
(A)
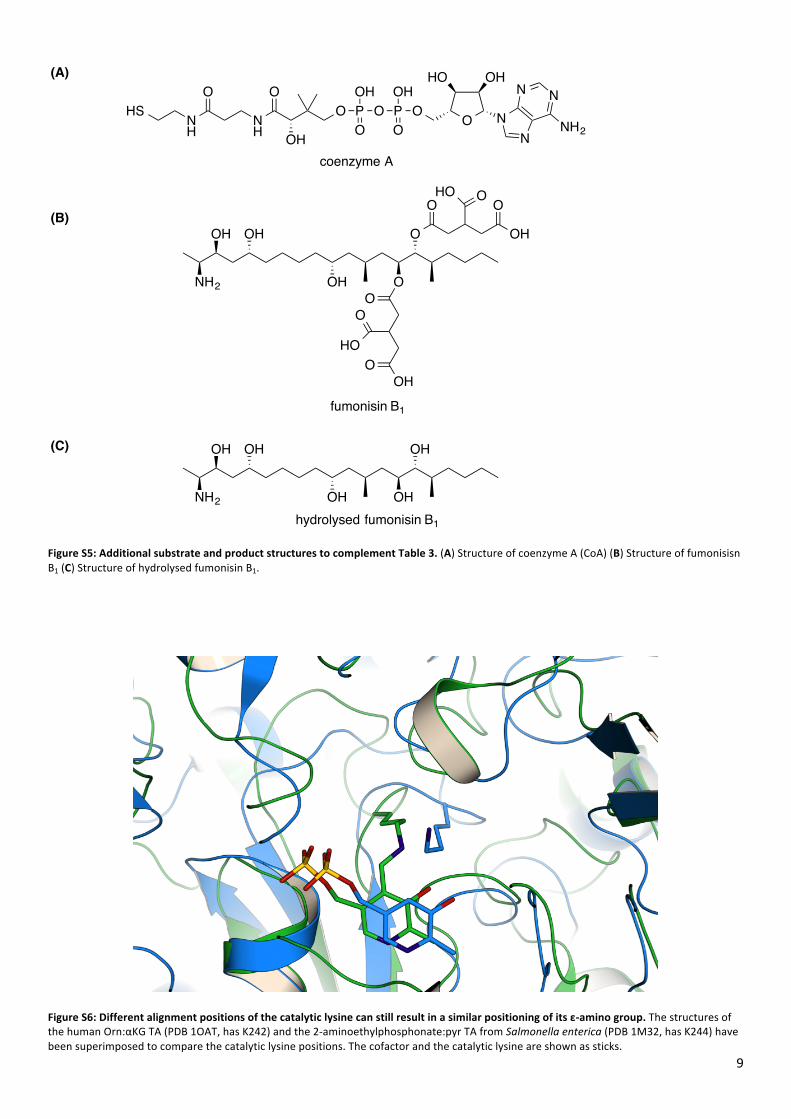
9
Figure S5: Additional substrate and product structures to complement Table 3. (A) Structure of coenzyme A (CoA) (B) Structure of fumonisisn B1 (C) Structure of hydrolysed fumonisin B1.
Figure S6: Different alignment positions of the catalytic lysine can still result in a similar positioning of its ε-‐amino group. The structures of the human Orn:αKG TA (PDB 1OAT, has K242) and the 2-‐aminoethylphosphonate:pyr TA from Salmonella enterica (PDB 1M32, has K244) have been superimposed to compare the catalytic lysine positions. The cofactor and the catalytic lysine are shown as sticks.
O NN
N N
NH2
OHHO
OPOH
OOP
OH
OO
OH
O
NH
O
NH
HS
coenzyme A
(A)
NH2
OH
OHOH
OH OH
NH2
OH
OOH
OH O
O O
OH
HO O
O
OOH
O
HO
fumonisin B1
hydrolysed fumonisin B1
(B)
(C)

10
Figure S7: Active site of the ATA from Rhodobacter sphaeroides (PDB ID: 3I5T). R142 is probably disrupting βAla binding between W47, S185 and R353 and therefore 3I5T possesses only low βAla:pyr activity. The side chains of residues S185 and R142 seem to be relatively flexible because of the electron density of the crystal structure and therefore also the model deposited in the PDB indicates more then one orientation. Residues are shown in green and PLP in orange sticks.
46
47
346
142
185
242
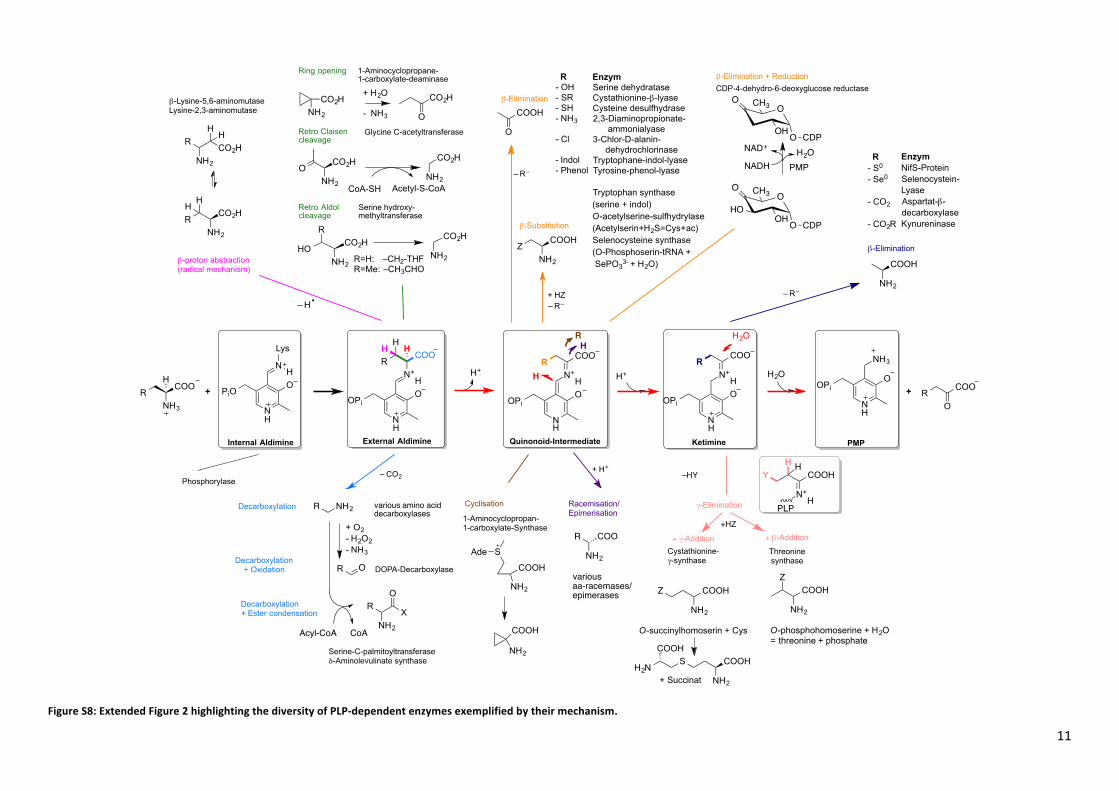
11
Figure S8: Extended Figure 2 highlighting the diversity of PLP-‐dependent enzymes exemplified by their mechanism.
N
OOPi
NH3
N
OPiO
N
Lys
N
OOPi
N+
COOH
R
NH
OOPi
N+
COOR
H
HH
H
H H
N
OOPi
N+
COOR
H
H
Ring opening
Decarboxylation
Internal Aldimine External Aldimine Quinonoid-Intermediate Ketimine
Retro Aldolcleavage
Retro Claisencleavage
Decarboxylation+ Oxidation
β-Elimination
β-Substitution
γ-Elimination
Cystathionine-γ-synthase
Threonine synthase
Phosphorylase
PMP
CDP-4-dehydro-6-deoxyglucose reductase
1-Aminocyclopropane-1-carboxylate-deaminase
H+ H+ H2O
– CO2
Racemisation/Epimerisation
+ H+
Decarboxylation+ Ester condensation
DOPA-Decarboxylase
+ HZ– R–
Glycine C-acetyltransferase
Serine hydroxy-methyltransferase
β-Elimination + Reduction
RNH3
COOH
+ RO
COO+
N+
COOHH
Y
Cyclisation
1-Aminocyclopropan-1-carboxylate-Synthase
H
H
– R–
R
H
H2O
β-proton abstraction(radical mechanism)
– H
various amino acid decarboxylases
β-Elimination
– R–
–HY
+ γ-Addition + β-Addition+HZ
PLP
R
HCO2H
NH2
CO2HR
NH2
H
β-Lysine-5,6-aminomutaseLysine-2,3-aminomutase
CO2HNH2
CO2H
O
+ H2O
- NH3
O CO2H
NH2NH2
CO2H
CoA-SH Acetyl-S-CoA
HOCO2H
NH2
CO2H
NH2R=H: –CH2-THFR=Me: –CH3CHO
R
COOH
O
ZCOOH
NH2
R- OH - SR- SH - NH3
- Cl
- Indol - Phenol
EnzymSerine dehydrataseCystathionine-β-lyaseCysteine desulfhydrase 2,3-Diaminopropionate- ammonialyase3-Chlor-D-alanin- dehydrochlorinaseTryptophane-indol-lyaseTyrosine-phenol-lyase
Tryptophan synthase (serine + indol)O-acetylserine-sulfhydrylase (Acetylserin+H2S=Cys+ac)Selenocysteine synthase(O-Phosphoserin-tRNA + SePO3
3- + H2O)
NADH
OO
HOOOH
CH3
CDP
NAD+H2O
PMP
OO
OOH
CH3
CDP
COOH
SAde
NH2
COOH
NH2
COOH
NH2
R- S0
- Se0
- CO2
- CO2R
EnzymNifS-ProteinSelenocystein-Lyase Aspartat-β-decarboxylase Kynureninase
R
NH2
X
O
Serine-C-palmitoyltransferaseδ-Aminolevulinate synthase
R NH2
R O
+ O2- H2O2- NH3
Acyl-CoA
variousaa-racemases/epimerases
NH2
COOHZ
NH2
COOHZ
O-phosphohomoserine + H2O= threonine + phosphate
COOH
NH2
SH2N
COOH
+ Succinat
O-succinylhomoserin + Cys
R
NH2
COO
H
H
H+
H
H
CoA

12
4 Supplementary data tables Table S1: Overview of the reaction specificities found in the different PLP fold types.
Fold type Representative name EC number Description
I Aspartate aminotransferase superfamily
1.17.1 (CH/CH2) Oxidoreductase
1.4.4 (CHNH2) Oxidoreductase 2.1.2 (1C) Transferase
2.3.1 Acyl transferase 2.5.1 Alkyl/aryl transferase
2.6.1 Amino transferase
2.8.1 Sulfur transferase 2.9.1 Seleno transferase
3.7.1 Hydrolase (C-‐C) 4.1.1 Carboxyl transferase
4.1.2 Aldehyde lyase 4.1.99 C-‐C Lyase
4.2.1 Hydro lyase
4.3.1 Ammonia lyase 4.4.1 C-‐S Lyase
5.1.1 Racemase 5.4.3 Isomerase
II Tryptophan synthase superfamily
2.5.1 Alkyl/aryl transferase
3.5.99 C-‐N Hydro lyase 4.2.1 C-‐O Hydro lyase
4.2.3 C-‐O Lyase (phosphates) 4.3.1 Ammonia lyase
4.4.1 C-‐S Lyase 5.1.1 Amino acid racemase
III Alanine racemase superfamily
4.1.1 Carboxy lyase
4.1.2 Aldehyde lyase 4.3.1 Ammonia lyase
5.1.1 Amino acid racemase
IV D-‐alanine aminotransferase superfamily
2.6.1 Amino transferase
4.1.3 Oxo acid lyase
V Glycogen phosphorylase superfamily
2.4.1 Glycosyltransferases
VI D-‐Lysine-‐5,6-‐aminomutase superfamily 5.4.3 Isomerase
VII Lysine-‐2,3-‐aminomutase superfamily 5.4.3 Isomerase
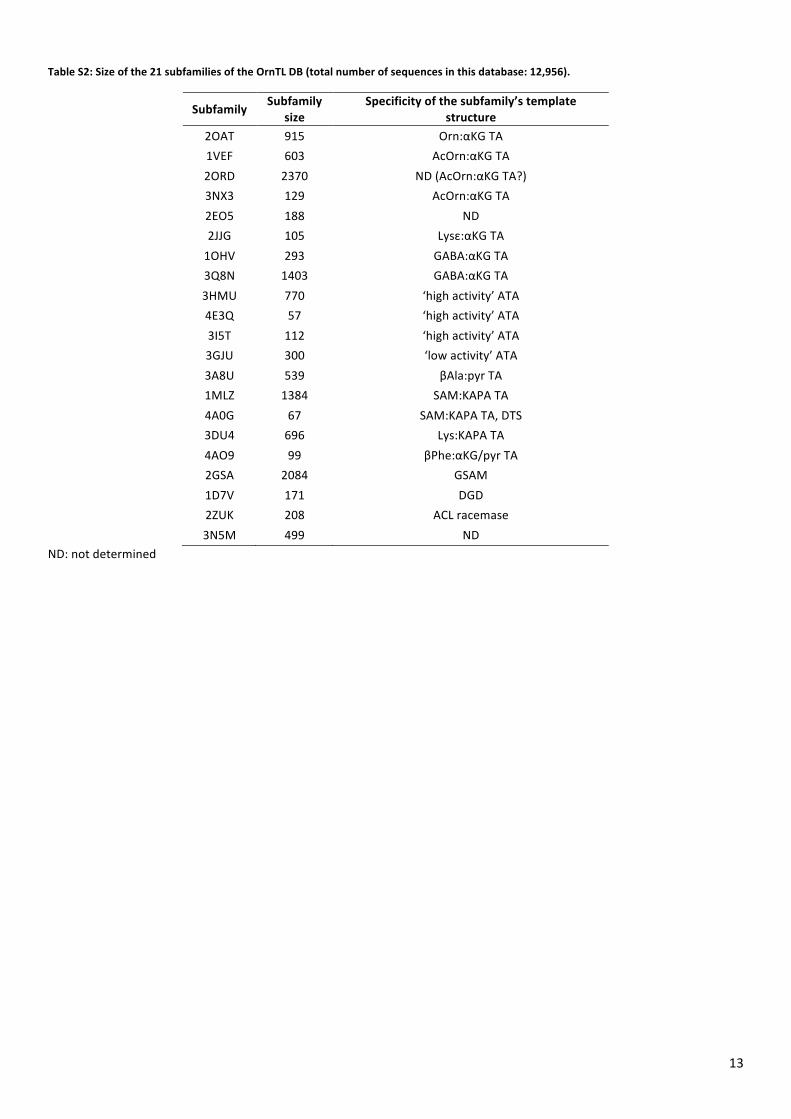
13
Table S2: Size of the 21 subfamilies of the OrnTL DB (total number of sequences in this database: 12,956).
Subfamily Subfamily
size Specificity of the subfamily’s template
structure 2OAT 915 Orn:αKG TA 1VEF 603 AcOrn:αKG TA 2ORD 2370 ND (AcOrn:αKG TA?) 3NX3 129 AcOrn:αKG TA 2EO5 188 ND 2JJG 105 Lysε:αKG TA 1OHV 293 GABA:αKG TA 3Q8N 1403 GABA:αKG TA 3HMU 770 ‘high activity’ ATA 4E3Q 57 ‘high activity’ ATA 3I5T 112 ‘high activity’ ATA 3GJU 300 ‘low activity’ ATA 3A8U 539 βAla:pyr TA 1MLZ 1384 SAM:KAPA TA 4A0G 67 SAM:KAPA TA, DTS 3DU4 696 Lys:KAPA TA 4AO9 99 βPhe:αKG/pyr TA 2GSA 2084 GSAM 1D7V 171 DGD 2ZUK 208 ACL racemase 3N5M 499 ND
ND: not determined
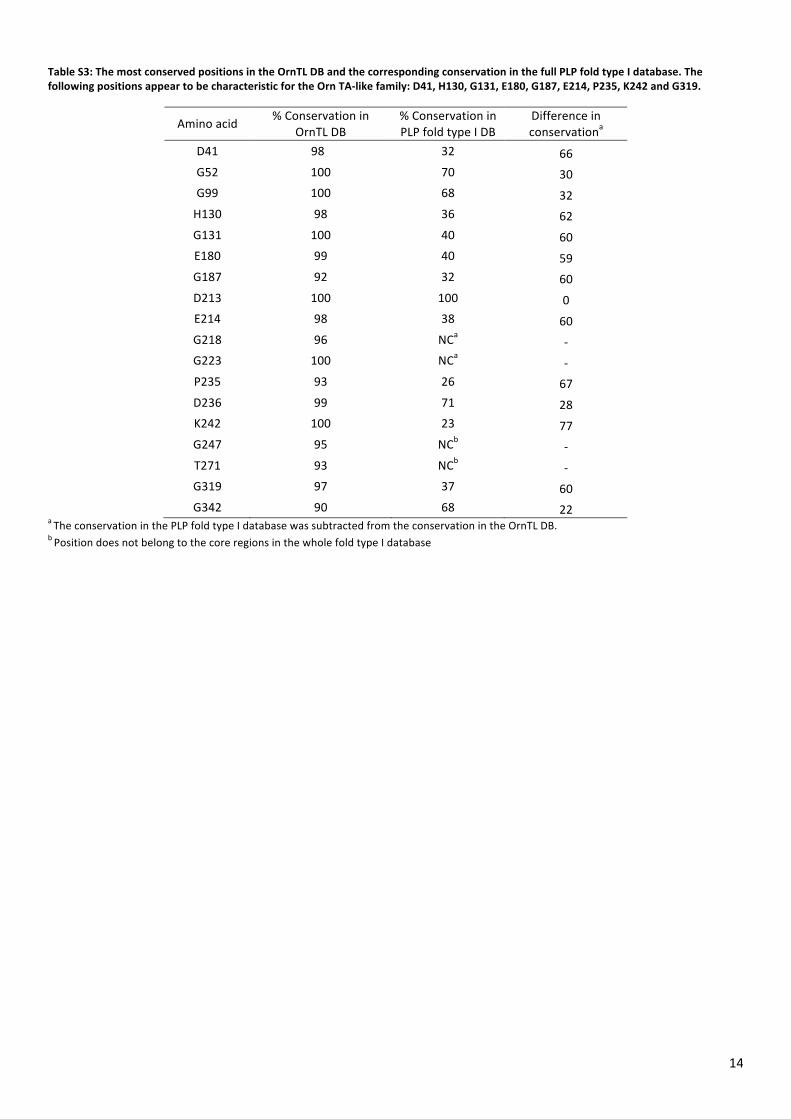
14
Table S3: The most conserved positions in the OrnTL DB and the corresponding conservation in the full PLP fold type I database. The following positions appear to be characteristic for the Orn TA-‐like family: D41, H130, G131, E180, G187, E214, P235, K242 and G319.
Amino acid % Conservation in OrnTL DB
% Conservation in PLP fold type I DB
Difference in conservationa
D41 98 32 66 G52 100 70 30 G99 100 68 32 H130 98 36 62 G131 100 40 60 E180 99 40 59 G187 92 32 60 D213 100 100 0 E214 98 38 60 G218 96 NCa -‐ G223 100 NCa -‐ P235 93 26 67 D236 99 71 28 K242 100 23 77 G247 95 NCb -‐ T271 93 NCb -‐ G319 97 37 60 G342 90 68 22
a The conservation in the PLP fold type I database was subtracted from the conservation in the OrnTL DB. b Position does not belong to the core regions in the whole fold type I database
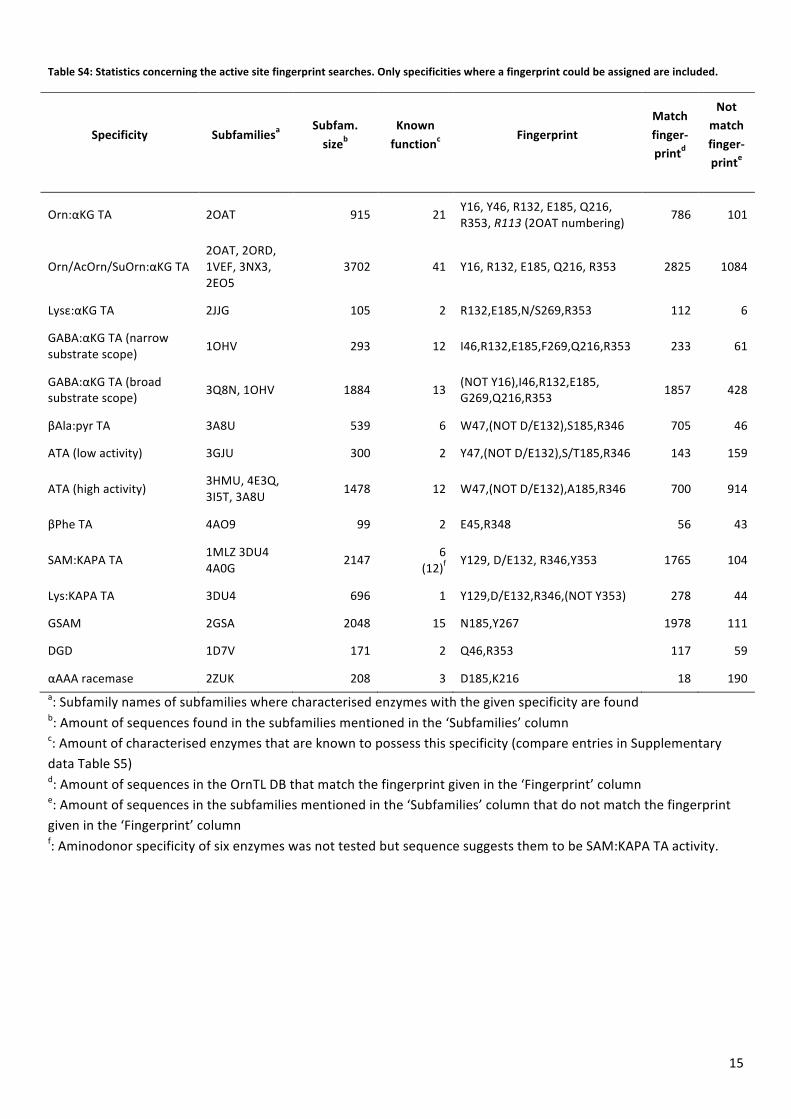
15
Table S4: Statistics concerning the active site fingerprint searches. Only specificities where a fingerprint could be assigned are included.
Specificity Subfamiliesa Subfam. sizeb
Known functionc
Fingerprint Match finger-‐printd
Not match finger-‐printe
Orn:αKG TA 2OAT 915 21 Y16, Y46, R132, E185, Q216, R353, R113 (2OAT numbering)
786 101
Orn/AcOrn/SuOrn:αKG TA 2OAT, 2ORD, 1VEF, 3NX3, 2EO5
3702 41 Y16, R132, E185, Q216, R353 2825 1084
Lysε:αKG TA 2JJG 105 2 R132,E185,N/S269,R353 112 6
GABA:αKG TA (narrow substrate scope) 1OHV 293 12 I46,R132,E185,F269,Q216,R353 233 61
GABA:αKG TA (broad substrate scope) 3Q8N, 1OHV 1884 13 (NOT Y16),I46,R132,E185,
G269,Q216,R353 1857 428
βAla:pyr TA 3A8U 539 6 W47,(NOT D/E132),S185,R346 705 46
ATA (low activity) 3GJU 300 2 Y47,(NOT D/E132),S/T185,R346 143 159
ATA (high activity) 3HMU, 4E3Q, 3I5T, 3A8U 1478 12 W47,(NOT D/E132),A185,R346 700 914
βPhe TA 4AO9 99 2 E45,R348 56 43
SAM:KAPA TA 1MLZ 3DU4 4A0G
2147 6 (12)f
Y129, D/E132, R346,Y353 1765 104
Lys:KAPA TA 3DU4 696 1 Y129,D/E132,R346,(NOT Y353) 278 44
GSAM 2GSA 2048 15 N185,Y267 1978 111
DGD 1D7V 171 2 Q46,R353 117 59
αAAA racemase 2ZUK 208 3 D185,K216 18 190 a: Subfamily names of subfamilies where characterised enzymes with the given specificity are found b: Amount of sequences found in the subfamilies mentioned in the ‘Subfamilies’ column c: Amount of characterised enzymes that are known to possess this specificity (compare entries in Supplementary data Table S5) d: Amount of sequences in the OrnTL DB that match the fingerprint given in the ‘Fingerprint’ column e: Amount of sequences in the subfamilies mentioned in the ‘Subfamilies’ column that do not match the fingerprint given in the ‘Fingerprint’ column f: Aminodonor specificity of six enzymes was not tested but sequence suggests them to be SAM:KAPA TA activity.
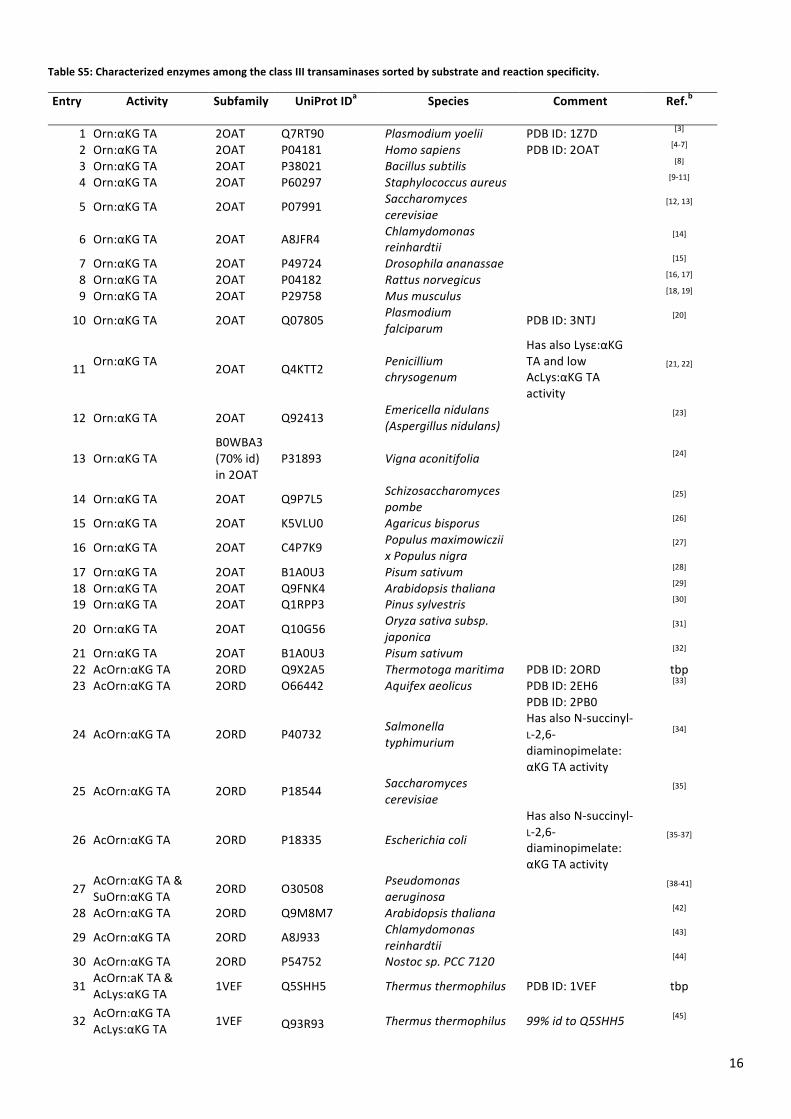
16
Table S5: Characterized enzymes among the class III transaminases sorted by substrate and reaction specificity.
Entry Activity Subfamily UniProt IDa Species Comment Ref.b
1 Orn:αKG TA 2OAT Q7RT90 Plasmodium yoelii PDB ID: 1Z7D [3] 2 Orn:αKG TA 2OAT P04181 Homo sapiens PDB ID: 2OAT [4-‐7] 3 Orn:αKG TA 2OAT P38021 Bacillus subtilis [8] 4 Orn:αKG TA 2OAT P60297 Staphylococcus aureus [9-‐11]
5 Orn:αKG TA 2OAT P07991 Saccharomyces cerevisiae
[12, 13]
6 Orn:αKG TA 2OAT A8JFR4 Chlamydomonas reinhardtii [14]
7 Orn:αKG TA 2OAT P49724 Drosophila ananassae [15] 8 Orn:αKG TA 2OAT P04182 Rattus norvegicus [16, 17] 9 Orn:αKG TA 2OAT P29758 Mus musculus [18, 19]
10 Orn:αKG TA 2OAT Q07805 Plasmodium falciparum
PDB ID: 3NTJ [20]
11 Orn:αKG TA 2OAT Q4KTT2 Penicillium chrysogenum
Has also Lysε:αKG TA and low AcLys:αKG TA activity
[21, 22]
12 Orn:αKG TA 2OAT Q92413 Emericella nidulans (Aspergillus nidulans) [23]
13 Orn:αKG TA B0WBA3 (70% id) in 2OAT
P31893 Vigna aconitifolia [24]
14 Orn:αKG TA 2OAT Q9P7L5 Schizosaccharomyces pombe [25]
15 Orn:αKG TA 2OAT K5VLU0 Agaricus bisporus [26]
16 Orn:αKG TA 2OAT C4P7K9 Populus maximowiczii x Populus nigra
[27]
17 Orn:αKG TA 2OAT B1A0U3 Pisum sativum [28] 18 Orn:αKG TA 2OAT Q9FNK4 Arabidopsis thaliana [29] 19 Orn:αKG TA 2OAT Q1RPP3 Pinus sylvestris [30]
20 Orn:αKG TA 2OAT Q10G56 Oryza sativa subsp. japonica [31]
21 Orn:αKG TA 2OAT B1A0U3 Pisum sativum [32] 22 AcOrn:αKG TA 2ORD Q9X2A5 Thermotoga maritima PDB ID: 2ORD tbp 23 AcOrn:αKG TA 2ORD O66442 Aquifex aeolicus PDB ID: 2EH6 [33]
24 AcOrn:αKG TA 2ORD P40732 Salmonella typhimurium
PDB ID: 2PB0 Has also N-‐succinyl-‐L-‐2,6-‐diaminopimelate: αKG TA activity
[34]
25 AcOrn:αKG TA 2ORD P18544 Saccharomyces cerevisiae
[35]
26 AcOrn:αKG TA 2ORD P18335 Escherichia coli
Has also N-‐succinyl-‐L-‐2,6-‐diaminopimelate: αKG TA activity
[35-‐37]
27 AcOrn:αKG TA & SuOrn:αKG TA 2ORD O30508
Pseudomonas aeruginosa [38-‐41]
28 AcOrn:αKG TA 2ORD Q9M8M7 Arabidopsis thaliana [42]
29 AcOrn:αKG TA 2ORD A8J933 Chlamydomonas reinhardtii
[43]
30 AcOrn:αKG TA 2ORD P54752 Nostoc sp. PCC 7120 [44]
31 AcOrn:aK TA & AcLys:αKG TA 1VEF Q5SHH5 Thermus thermophilus PDB ID: 1VEF tbp
32 AcOrn:αKG TA AcLys:αKG TA
1VEF Q93R93 Thermus thermophilus 99% id to Q5SHH5 [45]
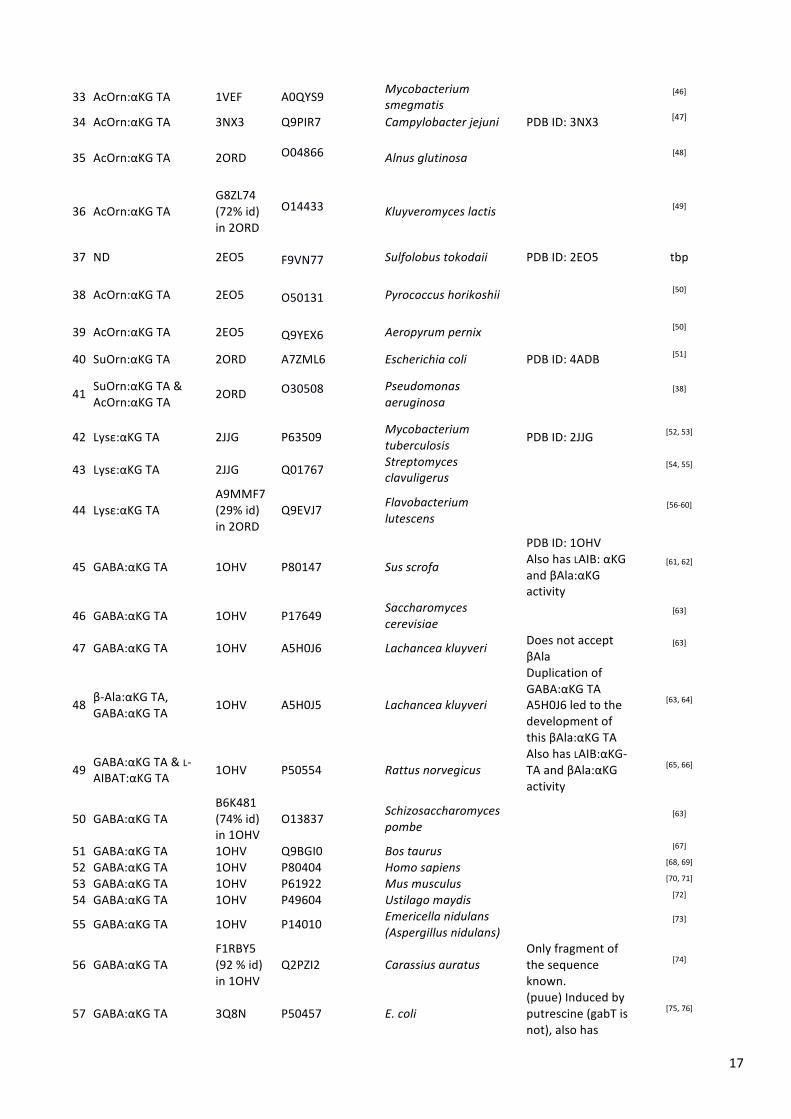
17
33 AcOrn:αKG TA 1VEF A0QYS9 Mycobacterium smegmatis [46]
34 AcOrn:αKG TA 3NX3 Q9PIR7 Campylobacter jejuni PDB ID: 3NX3 [47]
35 AcOrn:αKG TA 2ORD O04866
Alnus glutinosa [48]
36 AcOrn:αKG TA G8ZL74 (72% id) in 2ORD
O14433
Kluyveromyces lactis [49]
37 ND 2EO5 F9VN77 Sulfolobus tokodaii PDB ID: 2EO5 tbp
38 AcOrn:αKG TA 2EO5 O50131 Pyrococcus horikoshii [50]
39 AcOrn:αKG TA 2EO5 Q9YEX6 Aeropyrum pernix [50]
40 SuOrn:αKG TA 2ORD A7ZML6 Escherichia coli PDB ID: 4ADB [51]
41 SuOrn:αKG TA & AcOrn:αKG TA
2ORD O30508
Pseudomonas aeruginosa
[38]
42 Lysε:αKG TA 2JJG P63509 Mycobacterium tuberculosis
PDB ID: 2JJG [52, 53]
43 Lysε:αKG TA 2JJG Q01767 Streptomyces clavuligerus [54, 55]
44 Lysε:αKG TA A9MMF7 (29% id) in 2ORD
Q9EVJ7 Flavobacterium lutescens
[56-‐60]
45 GABA:αKG TA 1OHV P80147 Sus scrofa
PDB ID: 1OHV Also has LAIB: αKG and βAla:αKG activity
[61, 62]
46 GABA:αKG TA 1OHV P17649 Saccharomyces cerevisiae [63]
47 GABA:αKG TA 1OHV A5H0J6 Lachancea kluyveri Does not accept βAla
[63]
48 β-‐Ala:αKG TA, GABA:αKG TA
1OHV A5H0J5 Lachancea kluyveri
Duplication of GABA:αKG TA A5H0J6 led to the development of this βAla:αKG TA
[63, 64]
49 GABA:αKG TA & L-‐AIBAT:αKG TA 1OHV P50554 Rattus norvegicus Also has LAIB:αKG-‐TA and βAla:αKG activity
[65, 66]
50 GABA:αKG TA B6K481 (74% id) in 1OHV
O13837 Schizosaccharomyces pombe [63]
51 GABA:αKG TA 1OHV Q9BGI0 Bos taurus [67] 52 GABA:αKG TA 1OHV P80404 Homo sapiens [68, 69] 53 GABA:αKG TA 1OHV P61922 Mus musculus [70, 71] 54 GABA:αKG TA 1OHV P49604 Ustilago maydis [72]
55 GABA:αKG TA 1OHV P14010 Emericella nidulans (Aspergillus nidulans)
[73]
56 GABA:αKG TA F1RBY5 (92 % id) in 1OHV
Q2PZI2 Carassius auratus Only fragment of the sequence known.
[74]
57 GABA:αKG TA 3Q8N P50457 E. coli (puue) Induced by putrescine (gabT is not), also has
[75, 76]
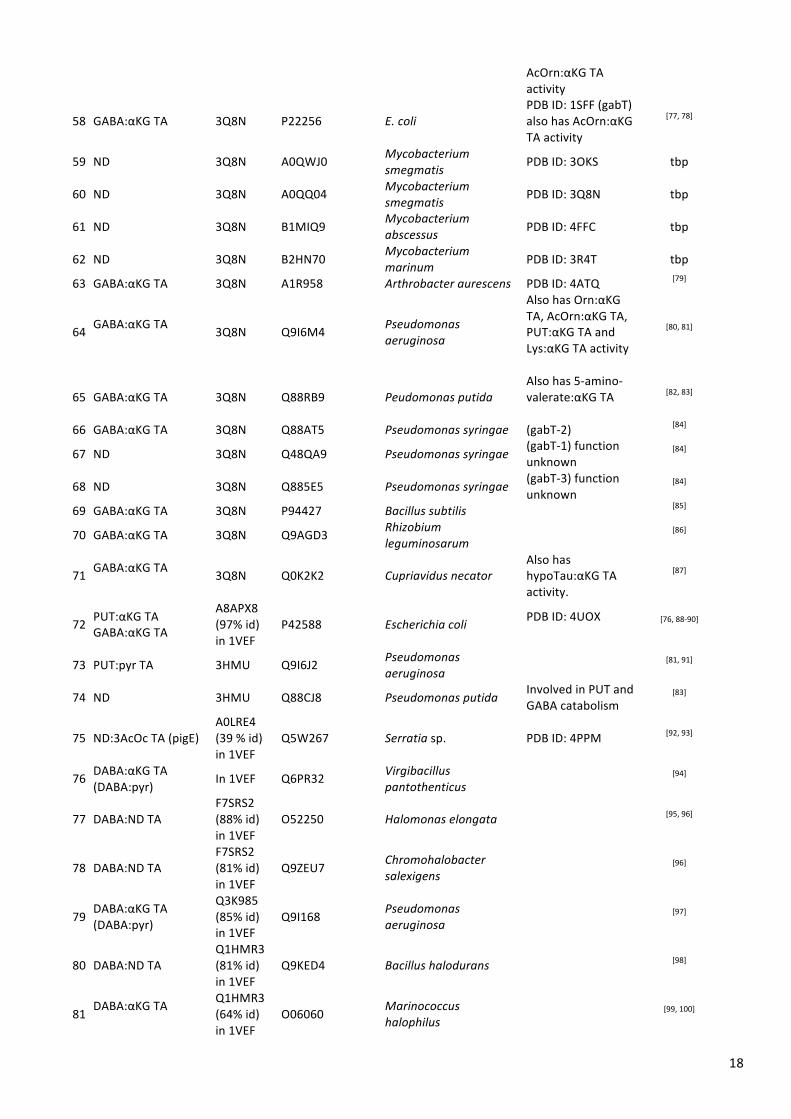
18
AcOrn:αKG TA activity
58 GABA:αKG TA 3Q8N P22256 E. coli PDB ID: 1SFF (gabT) also has AcOrn:αKG TA activity
[77, 78]
59 ND 3Q8N A0QWJ0 Mycobacterium smegmatis
PDB ID: 3OKS tbp
60 ND 3Q8N A0QQ04 Mycobacterium smegmatis
PDB ID: 3Q8N tbp
61 ND 3Q8N B1MIQ9 Mycobacterium abscessus PDB ID: 4FFC tbp
62 ND 3Q8N B2HN70 Mycobacterium marinum PDB ID: 3R4T tbp
63 GABA:αKG TA 3Q8N A1R958 Arthrobacter aurescens PDB ID: 4ATQ [79]
64 GABA:αKG TA
3Q8N Q9I6M4 Pseudomonas aeruginosa
Also has Orn:αKG TA, AcOrn:αKG TA, PUT:αKG TA and Lys:αKG TA activity
[80, 81]
65 GABA:αKG TA 3Q8N Q88RB9 Peudomonas putida Also has 5-‐amino-‐valerate:αKG TA
[82, 83]
66 GABA:αKG TA 3Q8N Q88AT5 Pseudomonas syringae (gabT-‐2) [84]
67 ND 3Q8N Q48QA9 Pseudomonas syringae (gabT-‐1) function unknown
[84]
68 ND 3Q8N Q885E5 Pseudomonas syringae (gabT-‐3) function unknown
[84]
69 GABA:αKG TA 3Q8N P94427 Bacillus subtilis [85]
70 GABA:αKG TA 3Q8N Q9AGD3 Rhizobium leguminosarum
[86]
71 GABA:αKG TA
3Q8N Q0K2K2 Cupriavidus necator Also has hypoTau:αKG TA activity.
[87]
72 PUT:αKG TA GABA:αKG TA
A8APX8 (97% id) in 1VEF
P42588 Escherichia coli PDB ID: 4UOX
[76, 88-‐90]
73 PUT:pyr TA 3HMU Q9I6J2 Pseudomonas aeruginosa
[81, 91]
74 ND 3HMU Q88CJ8 Pseudomonas putida Involved in PUT and GABA catabolism
[83]
75 ND:3AcOc TA (pigE) A0LRE4 (39 % id) in 1VEF
Q5W267 Serratia sp. PDB ID: 4PPM [92, 93]
76 DABA:αKG TA (DABA:pyr)
In 1VEF Q6PR32 Virgibacillus pantothenticus
[94]
77 DABA:ND TA F7SRS2 (88% id) in 1VEF
O52250 Halomonas elongata [95, 96]
78 DABA:ND TA F7SRS2 (81% id) in 1VEF
Q9ZEU7 Chromohalobacter salexigens [96]
79 DABA:αKG TA (DABA:pyr)
Q3K985 (85% id) in 1VEF
Q9I168 Pseudomonas aeruginosa [97]
80 DABA:ND TA Q1HMR3 (81% id) in 1VEF
Q9KED4 Bacillus halodurans [98]
81 DABA:αKG TA
Q1HMR3 (64% id) in 1VEF
O06060 Marinococcus halophilus [99, 100]

19
82 DABA:αKG TA B0VCM6 (97% id) in 3Q8N
P56744 Acinetobacter baumannii [101]
83 DABA:αKG TA GABA:αKG TA
D0BXR7 (71% id) in 3Q8N
P44951 Haemophilus influenzae
[102]
84 DABA:ND TA F9DS30 (76% id) in 3Q8N
Q9AP34 Sporosarcina pasteurii (Bacillus pasteurii) [103]
85 DABA:ND TA B6BTZ4 (70% id) in 2EO5
Q4JQJ4 Methylomicrobium alcaliphilum
[104]
86 ND Q9KLC2 Vibrio cholerae Predicted DABA TA [104]
87 ND Q87NZ7 Vibrio parahaemolyticus
Predicted DABA TA [104]
88 ND Q93RW1 Streptomyces coelicolor Predicted DABA TA [104]
89 ND Q8ESU8 Oceanobacillus iheyensis Predicted DABA TA [104]
90 ND Q7WHI8 Bordetella bronchiseptica
Predicted DABA TA [104]
91 ND Q5YW77 Nocardia farcinica Predicted DABA TA [104] 92 ND Q5DYF3 Vibrio fischeri Predicted DABA TA [104]
93 ND Q829L4 Streptomyces avermitilis Predicted DABA TA [104]
94 ND Q5WL78 Bacillus clausii Predicted DABA TA [104] 95 ND Q6QUY9 Streptomyces anulatus Predicted DABA TA [104] 96 ND Q7M9K2 Wolinella succinogenes Predicted DABA TA [104]
97 ATA high activity A7GNT9 (67% id) in 3GJU
Seq. ID 2 in patent WO2006063336
Arthrobacter citreus [105]
98 ATA high activity A7GNT9 (68% id) in 3GJU
Ref. [106]: Figure 2 Bacillus megaterium
98% pairwise identity with Arthrobacter citreus
[106, 107]
99 ATA high activity 3HMU Q7NWG4 Chromobacterium violaceum
PDB ID: 446T [108]
100 ATA high activity 3HMU Q5LMU1 Silicibacter pomeroyi PDB ID: 3HMU [109]
101 ATA high activity 3GJU A6WVC6 Ochrobactrum anthropi
Neither product nor substrate inhibition
[110]
102 ATA high activity 4E3Q A1B956 Paracoccus denitrificans
PDB ID: 4GRX [111]
103 ATA high activity 4E3Q F2XBU9 Vibrio fluvialis PDB ID: 4E3Q [112]
104 ATA high activity 4E3Q F5M2X9 Rhodobacter sphaeroides [113]
105 ATA high activity 3I5T Q3IWE9 Rhodobacter sphaeroides PDB ID: 3I5T [109]
106 ATA high activity A0B1I8 (54% id) in 3N5M
Seq. ID 6 in patent WO2007139055
Pseudomonas fluorescens [114, 115]
107 ATA high activity 3A8U B7IC89 Acinetobacter baumannii
βAla:pyr TA activity not tested.
[116]
108 ATA high activity 3A8U C7JE89 Acetobacter pasteurianus
βAla:pyr TA activity not tested.
[116]
109 βAla:pyr TA 3A8U Q7WWK8
Alcaligenes denitrificans
Also highly active on amines
[117]
110 βAla:pyr TA
3A8U Q9A3Q9 Caulobacter crescentus Also highly active on amines
[118]
111 βAla:pyr TA 3A8U Q9I700 Pseudomonas PDB ID: 4B9B [119]
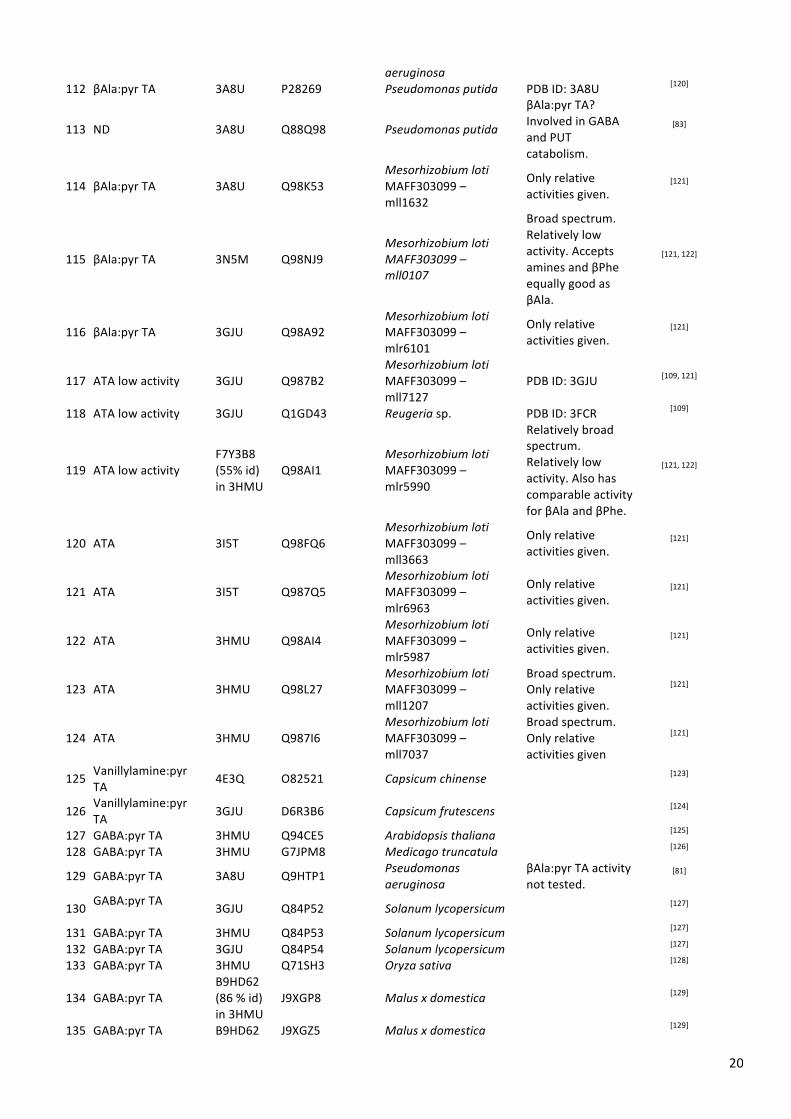
20
aeruginosa 112 βAla:pyr TA 3A8U P28269 Pseudomonas putida PDB ID: 3A8U [120]
113 ND 3A8U Q88Q98 Pseudomonas putida
βAla:pyr TA? Involved in GABA and PUT catabolism.
[83]
114 βAla:pyr TA 3A8U Q98K53 Mesorhizobium loti MAFF303099 – mll1632
Only relative activities given.
[121]
115 βAla:pyr TA 3N5M Q98NJ9 Mesorhizobium loti MAFF303099 – mll0107
Broad spectrum. Relatively low activity. Accepts amines and βPhe equally good as βAla.
[121, 122]
116 βAla:pyr TA 3GJU Q98A92 Mesorhizobium loti MAFF303099 – mlr6101
Only relative activities given.
[121]
117 ATA low activity 3GJU Q987B2 Mesorhizobium loti MAFF303099 – mll7127
PDB ID: 3GJU [109, 121]
118 ATA low activity 3GJU Q1GD43 Reugeria sp. PDB ID: 3FCR [109]
119 ATA low activity F7Y3B8 (55% id) in 3HMU
Q98AI1 Mesorhizobium loti MAFF303099 – mlr5990
Relatively broad spectrum. Relatively low activity. Also has comparable activity for βAla and βPhe.
[121, 122]
120 ATA 3I5T Q98FQ6 Mesorhizobium loti MAFF303099 – mll3663
Only relative activities given.
[121]
121 ATA 3I5T Q987Q5 Mesorhizobium loti MAFF303099 – mlr6963
Only relative activities given.
[121]
122 ATA 3HMU Q98AI4 Mesorhizobium loti MAFF303099 – mlr5987
Only relative activities given.
[121]
123 ATA 3HMU Q98L27 Mesorhizobium loti MAFF303099 – mll1207
Broad spectrum. Only relative activities given.
[121]
124 ATA 3HMU Q987I6 Mesorhizobium loti MAFF303099 – mll7037
Broad spectrum. Only relative activities given
[121]
125 Vanillylamine:pyr TA
4E3Q O82521 Capsicum chinense [123]
126 Vanillylamine:pyr TA 3GJU D6R3B6 Capsicum frutescens [124]
127 GABA:pyr TA 3HMU Q94CE5 Arabidopsis thaliana [125] 128 GABA:pyr TA 3HMU G7JPM8 Medicago truncatula [126]
129 GABA:pyr TA 3A8U Q9HTP1 Pseudomonas aeruginosa
βAla:pyr TA activity not tested.
[81]
130 GABA:pyr TA
3GJU Q84P52 Solanum lycopersicum [127]
131 GABA:pyr TA 3HMU Q84P53 Solanum lycopersicum [127] 132 GABA:pyr TA 3GJU Q84P54 Solanum lycopersicum [127] 133 GABA:pyr TA 3HMU Q71SH3 Oryza sativa [128]
134 GABA:pyr TA B9HD62 (86 % id) in 3HMU
J9XGP8 Malus x domestica [129]
135 GABA:pyr TA B9HD62 J9XGZ5 Malus x domestica [129]

21
(86 % id) in 3HMU
136 GABA:pyr TA 3HMU Q7XN11 Oryza sativa [128] 137 Tau:pyr TA 3N5M Q9APM5 Bilophila wadsworthia [130] 138 Tau:pyr TA 3N5M Q5LVM7 Ruegeria pomeroyi [131] 139 Tau:pyr TA 3HMU Q6JE91 Rhodococcus opacus [132]
140 HypoTau:pyr TA Tau:pyr TA
3N5M A1B9Z3 Paracoccus denitrificans
[133]
141 βPhe:αKG/pyr TA 4AO9 H8WR05 Variovorax paradoxus PDB ID: 4AO9 [134, 135] 142 βPhe:αKG/pyr TA 4AO9 A3EYF7 Mesorhizobium sp. LUK PDB ID: 2YKY [136, 137]
143 βPhe:αKG/pyr TA 4AO9 B1G3Y1 Burkholderia graminis Also active on amines
[138]
144 βAla:pyr TA 4AO9 Q12DH7 Polaromonas sp.
Also highly active on βPhe and amines
[139]
145 CoAβAA TA D8F1V2 (66% id) in 2GSA
B0VH76 Candidatus cloacamonas acidaminovorans
[140]
146 PhGly:αKG TA B4VLD1 (36 % id) in 4AO9
Q6VY99 Pseudomonas stutzeri PDB ID: 2CY8 [141, 142]
147 PhGly:αKG TA B4VLD1 (34 % id) in 4AO9
GenBank AX467211 Pseudomonas putida [143, 144]
148 SAM:KAPA TA 1MLZ P12995 Escherichia coli PDB ID: 1MLZ [145-‐147]
149 SAM:KAPA TA 1MLZ P9WQ80 Mycobacterium tuberculosis PDB ID: 3TFT [148-‐152]
150 SAM:KAPA TA 1MLZ P50277 Saccharomyces cerevisiae
[153]
151 SAM:KAPA TA 1MLZ P36568 Serratia marcescens [154] 152 ND:KAPA TA 1MLZ BAA03167 Brevibacterium flavum [155]
153 ND 1MLZ P46395 Corynebacterium glutamicum
Very similar to B. flavum enzyme (96% identity), not tested for activity
[156]
154 ND:KAPA TA 1MLZ Q9APS9 Uncharacterized bacterium pCosAS1
Only tested in whole biotin biosynthetic pathway
[157]
155 ND:KAPA TA 1MLZ Q9APR4 Uncharacterized bacterium pCosFS1 [157]
156 ND:KAPA TA 1MLZ Q9APP2 Uncharacterized bacterium pCosHE2
[157]
157 ND:KAPA TA D5CGJ4 (87% id) in 1MLZ
Q9APQ7 Uncharacterized bacterium pCosHE1 [157]
158 ND:KAPA TA 1MLZ P53656 Pantoea agglomerans (Erwinia herbicola) [158]
159 Lys:KAPA TA 3DU4 P53555 Bacillus subtilis PDB ID: 3DU4 [149, 159]
160 SAM:KAPA TA 3DU4 P22805 Lysinibacillus sphaericus (Bacillus sphaericus)
[160]
161 SAM:KAPA TA 4A0G B0F481 Arabidopsis thaliana
PDB ID: 4A0G Fusion protein of SAM:KAPA TA and dethiobitin synthase (DTBS)
[161]
162 Ala:glyox TA 2 H2QQR4 (99 % id) in 3N5M
Q9BYV1 Homo sapiens [162]
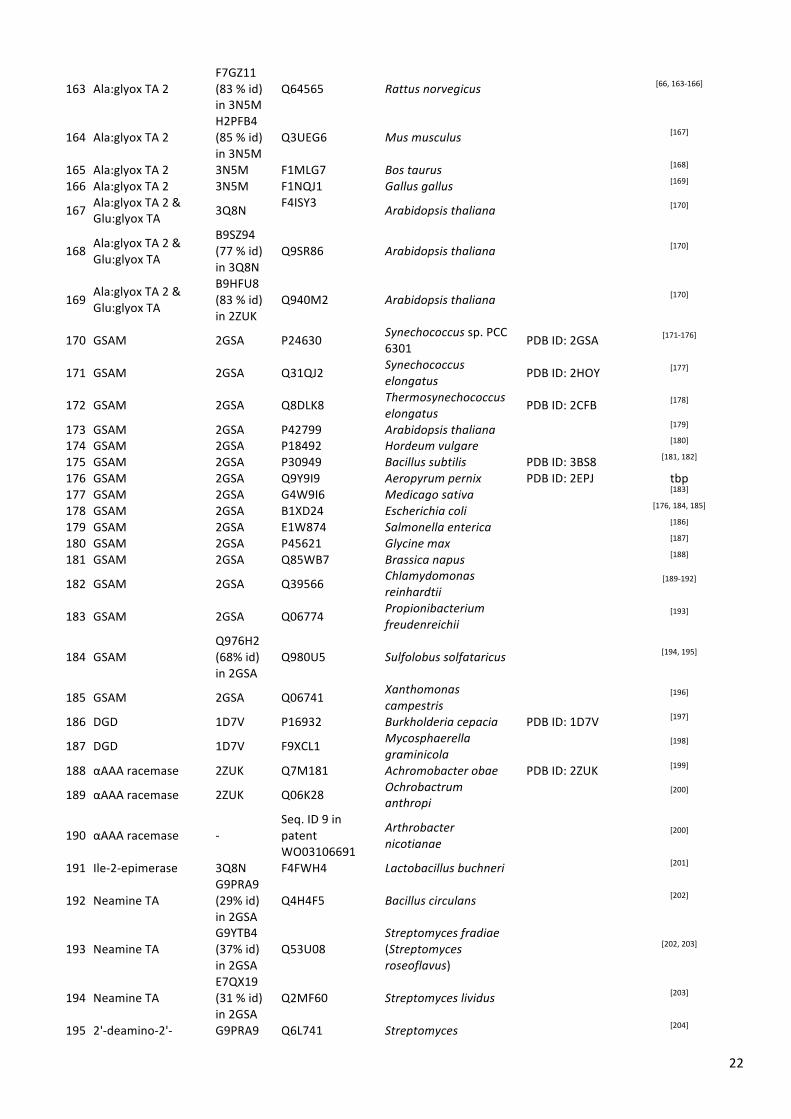
22
163 Ala:glyox TA 2 F7GZ11 (83 % id) in 3N5M
Q64565 Rattus norvegicus [66, 163-‐166]
164 Ala:glyox TA 2 H2PFB4 (85 % id) in 3N5M
Q3UEG6 Mus musculus [167]
165 Ala:glyox TA 2 3N5M F1MLG7 Bos taurus [168] 166 Ala:glyox TA 2 3N5M F1NQJ1 Gallus gallus [169]
167 Ala:glyox TA 2 & Glu:glyox TA 3Q8N F4ISY3 Arabidopsis thaliana [170]
168 Ala:glyox TA 2 & Glu:glyox TA
B9SZ94 (77 % id) in 3Q8N
Q9SR86 Arabidopsis thaliana [170]
169 Ala:glyox TA 2 & Glu:glyox TA
B9HFU8 (83 % id) in 2ZUK
Q940M2 Arabidopsis thaliana [170]
170 GSAM 2GSA P24630 Synechococcus sp. PCC 6301 PDB ID: 2GSA [171-‐176]
171 GSAM 2GSA Q31QJ2 Synechococcus elongatus PDB ID: 2HOY [177]
172 GSAM 2GSA Q8DLK8 Thermosynechococcus elongatus
PDB ID: 2CFB [178]
173 GSAM 2GSA P42799 Arabidopsis thaliana [179] 174 GSAM 2GSA P18492 Hordeum vulgare [180] 175 GSAM 2GSA P30949 Bacillus subtilis PDB ID: 3BS8 [181, 182] 176 GSAM 2GSA Q9Y9I9 Aeropyrum pernix PDB ID: 2EPJ tbp 177 GSAM 2GSA G4W9I6 Medicago sativa [183] 178 GSAM 2GSA B1XD24 Escherichia coli [176, 184, 185] 179 GSAM 2GSA E1W874 Salmonella enterica [186] 180 GSAM 2GSA P45621 Glycine max [187] 181 GSAM 2GSA Q85WB7 Brassica napus [188]
182 GSAM 2GSA Q39566 Chlamydomonas reinhardtii [189-‐192]
183 GSAM 2GSA Q06774 Propionibacterium freudenreichii [193]
184 GSAM Q976H2 (68% id) in 2GSA
Q980U5 Sulfolobus solfataricus [194, 195]
185 GSAM 2GSA Q06741 Xanthomonas campestris [196]
186 DGD 1D7V P16932 Burkholderia cepacia PDB ID: 1D7V [197]
187 DGD 1D7V F9XCL1 Mycosphaerella graminicola
[198]
188 αAAA racemase 2ZUK Q7M181 Achromobacter obae PDB ID: 2ZUK [199]
189 αAAA racemase 2ZUK Q06K28 Ochrobactrum anthropi [200]
190 αAAA racemase -‐ Seq. ID 9 in patent WO03106691
Arthrobacter nicotianae
[200]
191 Ile-‐2-‐epimerase 3Q8N F4FWH4 Lactobacillus buchneri [201]
192 Neamine TA G9PRA9 (29% id) in 2GSA
Q4H4F5 Bacillus circulans [202]
193 Neamine TA G9YTB4 (37% id) in 2GSA
Q53U08 Streptomyces fradiae (Streptomyces roseoflavus)
[202, 203]
194 Neamine TA E7QX19 (31 % id) in 2GSA
Q2MF60 Streptomyces lividus [203]
195 2'-‐deamino-‐2'-‐ G9PRA9 Q6L741 Streptomyces [204]

23
hydroxyneamine TA
(34% id) in 2GSA
kanamyceticus
196 hFumonisin:pyr TA Q0BZI0 (57% id) in 2GSA
D2D3B2
Sphingopyxis macrogoltabida (Sphingomonas macrogoltabidus)
Pyr no αKG [205]
197 hFumonisin:pyr TA Q0BZI0 (61% id) in 2GSA
E2E0Q4 bacterium ATCC 55552 pyr [206]
198 Fumonisin B1: αKG TA
D2ATX4 (47% id) in 2GSA
Seq. ID 56 in patent WO2004085624
unknown [207]
199
O-‐phosphoethanolamine phospholyase (AGTX2L1)
G1L4L7 (88% id) in 2ZUK
Q8TBG4 Homo sapiens [208, 209]
200
5-‐phosphohydroxy-‐L-‐lysine phospholyase (AGTX2L2)
I3K5C7 (68% id) in 2ZUK
Q8IUZ5 Homo sapiens [209]
201 Mycosubtilin synthase subunit A
B8I979 (42 % id) in 4AO9
Q9R9J1 Bacillus subtilis [210]
a All sequences that are not present in the sequence databases and therefore needed to be extracted from the publications are given as FASTA amino acid sequence in section 5 below b tbp: to be published
5 Sequences not included in the sequence databases The sequences that have been extracted from patents or publications are listed below. They might contain errors, therefore please check the original publications. >ATA (high activity)|Arthrobacter citreus|WO2006063336 Seq. ID 2 MGLTVQKINWEQVKEWDRKYLMRTFSTQNEYQPVPIESTEGDYLIMPDGTRLLDFFNQLYCVNLGQKNQKVNAAIKEALD RYGFVWDTYATDYKAKAAKIIIEDILGDEDWPGKVRFVSTGSEAVETALNIARLYTNRPLVVTREHDYHGWTGGAATVTR LRSYRSGLVGENSESFSAQIPGSSYNSAVLMAPSPNMFQDSNGNCLKDENGELLSVKYTRRMIENYGPEQVAAVITEVSQ GAGSAMPPYEYIPQFRKMTKELGVLWINDEVLTGFGRTGKWFGYQHYGVQPDIITMGKGLSSSSLPAGAVVVSKEIAAFM DKHRWESVSTYAGHPVAMAAVCANLEVMMEENLVEQAKNSGEYIRSKLELLQEKHKSIGNFDGYGLLWIVDYVKLDRNFT HGMNPNQIPTQIIMKKALEKGVLIGGVMPNTMRIGASLNVSRGDIDKAMDALDYALDYLESGEWQQS >ATA (high activity)|Bacillus megaterium|Ref.[106], Figure 2 MSLTVQKINWEQVKEWDRKYLMRTFSTQNEYQPVPIESTEGDYLIMPDGTRLLDFFNQLYCVNLGQKNQKVNAAIKEALD RYGFVWDTYATDYKAKAAKIIIEDILGDEDWPGKVRFVSTGSEAVETALNIARLYTNRPLVVTREHDYHGWTGGAATVTR LRSYRSGLVGENSESFSAQIPGSSYNSAVLMAPSPNMFQDSDGNLLKDENGELLSVKYTRRMIENYGPEQVAAVITEVSQ GAGSAMPPYEYIPQIRKMTKELGVLWINDEVLTGFGRTGKWFGYQHYGVQPDIITMGKGLSSSSLPAGAVLVSKEIAAFM DKHRWESVSTYAGHPVAMAAVCANLEVMMEENFVEQAKDSGEYIRSKLELLQEKHKSIGNFDGYGLLWIVDIVNAKTKTP YVKLDRNFTHGMNPNQIPTQIIMKKALEKGVLIGGVMPNTMRIGASLNVSRGDIDKAMDALDYALDYLESGEWQ >ATA (high activity)|Pseudomonas fluorescens|WO2007139055 Seq. ID 6 MNSNNKAWLKEHNTVHMMHPMQDPKALHEQRPLIIQSGKGVHITDVDGRRFIDCQGGLWCVNAGYGRREIIDAVTRQMEE LAYYSLFPGSTNAPAIALSQKLTEVAAEEGMVKASFGLGGSDAVETALKIARQYWKLEGQPDKVKFVSLYNGYHGLNFGG MSACGGNAWKSSYEPLMPGFFQVESPHLYRNPFTNDPEELAEICAQILERQIEMQAPGTVAALIAEPIQGAGGVIVPPAS YWPRLRQICDKYDILLIADEVITGLGRSGSLFGSRGWGVKPDIMCLAKGISSGYVPLSATLVNSRVARAWERDAGFTSVY MHGYTYSGHPVSCAAALAAIDIVLQENLAENARVVGDYFLEKLLILKDKHRAIGDVRGKGLMLAVELVKERATKEPFGPA

24
DAYPLAISEACVNNGVMIRTIVNKLIISPPLTFTTEHVDEVIEVLDRAFVANPW >alphaAAA racemase|Arthrobacter nicotianae|WO03106691 Seq. ID 9 MLEDSLYARDGRVIAGVEKLRFFPLETASGRGSMLVEPGGRELFDFSASWTAAGLGHGNPEITAAIARAAVDSPGASILS ATHSEAVGLAERLLDMVPTRRSGPGGRRVYLGHAGTDSNDVAIRGCRHASGRPGVIAFEGGYHGGLGIAQRISGVHVDSG VPADPHVAFVPYPDLFRPHTGDPETVLPDVLTRVRQNLQRGMTAAVIVEPLLSDGGVIVPPPEFLRGLRELCDAHNAYLI VDEVKVGLGRTGSLHAFEHDGILPDIVTLGKVLGGGLPLSAAIGPSEVLDRPVASALMTTTGNPISCAAGRAAVEIVCRG DVIRNAAERGEQIRDLLAAYARETGRPGAAHVGDVRGRGLSIGIEIVTDRDENVSDPGLTAKAVYRAWELGVVVHPVRGN VLELTPPLTVSADEVQQAMDLLTCALDDAARGLVSDEQIAPYAGW >fumonisin B1:pyr TA||WO2004085624 Seq. ID 56 MRSFTNSEKLFAKAKTIIPGGMPRHLSPASLVPGQSPCFIERAKGCRFWDVDGNEYIDYMCAYGPMILGYNNPKVDAAVD AQKTFGDCLPLPSPILIELAEKMVQLIAAADWATFAKNGSDVCNHAVRIARAHTGRNKVIMARDSYHGIGAWCTPYPTGV TESERIDVLHFPYNDVAALEFLLKKHANEVAAIIVTPFKHETGHDQEMPTDAFIRMLKEQTNVDGPLLIMDDVRCGFRLH MGGSAEYIGLKPHLSCFSKAMGNGYPISALCGVKELMGAANRVFFTGSFFAGAQAIAASIATMDEMVASNALEHAFRMGE MLKAGMLKQASQLGLNVNYTGPVTIPFMSFENDASFDKAKIFCAAAYQEGAFFHPTHNWFVSAAHQEKDIEETLAATEKA FAAVKAKSAS
6 References [1] PyMOL The PyMOL Molecular Graphics System, Version 1.6.0.0; Schrödinger, LCC: [2] YASARA Structure YASARA Structure, Version 13.6.16; YASARA Biosciences: [3] Carlton, J. M.; Angiuoli, S. V.; Suh, B. B.; Kooij, T. W.; Pertea, M.; Silva, J. C.; Ermolaeva, M. D.; Allen, J. E.;
Selengut, J. D.; Koo, H. L.; Peterson, J. D.; Pop, M.; Kosack, D. S.; Shumway, M. F.; Bidwell, S. L.; Shallom, S. J.; van Aken, S. E.; Riedmuller, S. B.; Feldblyum, T. V.; Cho, J. K.; Quackenbush, J.; Sedegah, M.; Shoaibi, A.; Cummings, L. M.; Florens, L.; Yates, J. R.; Raine, J. D.; Sinden, R. E.; Harris, M. A.; Cunningham, D. A.; Preiser, P. R.; Bergman, L. W.; Vaidya, A. B.; van Lin, L. H.; Janse, C. J.; Waters, A. P.; Smith, H. O.; White, O. R.; Salzberg, S. L.; Venter, J. C.; Fraser, C. M.; Hoffman, S. L.; Gardner, M. J.; Carucci, D. J., Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature 2002, 419, 512-‐519.
[4] Markova, M.; Peneff, C.; Hewlins, M. J.; Schirmer, T.; John, R. A., Determinants of substrate specificity in ω-‐aminotransferases. J Biol Chem 2005, 280, 36409-‐36416.
[5] Shah, S. A.; Shen, B. W.; Brunger, A. T., Human ornithine aminotransferase complexed with L-‐canaline and gabaculine: structural basis for substrate recognition. Structure 1997, 5, 1067-‐1075.
[6] Shen, B. W.; Hennig, M.; Hohenester, E.; Jansonius, J. N.; Schirmer, T., Crystal structure of human recombinant ornithine aminotransferase. J Mol Biol 1998, 277, 81-‐102.
[7] Storici, P.; Capitani, G.; Müller, R.; Schirmer, T.; Jansonius, J. N., Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-‐fluoromethylornithine. J Mol Biol 1999, 285, 297-‐309.
[8] Jhee, K. H.; Yoshimura, T.; Esaki, N.; Yonaha, K.; Soda, K., Thermostable ornithine aminotransferase from Bacillus sp. YM-‐2: purification and characterization. J Biochem 1995, 118, 101-‐108.
[9] Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K. I.; Nagai, Y.; Lian, J. Q.; Ito, T.; Kanamori, M.; Matsumaru, H.; Maruyama, A.; Murakami, H.; Hosoyama, A.; Mizutani-‐Ui, Y.; Takahashi, N. K.; Sawano, T.; Inoue, R. I.; Kaito, C.; Sekimizu, K.; Hirakawa, H.; Kuhara, S.; Goto, S.; Yabuzaki, J.; Kanehisa, M.; Yamashita, A.; Oshima, K.; Furuya, K.; Yoshino, C.; Shiba, T.; Hattori, M.; Ogasawara, N.; Hayashi, H.; Hiramatsu, K., Whole genome sequencing of meticillin-‐resistant Staphylococcus aureus. Lancet 2001, 357, 1225-‐1240.
[10] Townsend, D. E.; Kaenjak, A.; Jayaswal, R. K.; Wilkinson, B. J., Proline is biosynthesized from arginine in Staphylococcus aureus. Microbiology 1996, 142, 1491-‐1497.
[11] Li, C.; Sun, F.; Cho, H.; Yelavarthi, V.; Sohn, C.; He, C.; Schneewind, O.; Bae, T., CcpA mediates proline auxotrophy and is required for Staphylococcus aureus pathogenesis. J Bacteriol 2010, 192, 3883-‐3892.

25
[12] Degols, G., Functional analysis of the regulatory region adjacent to the carg B gene of Saccharomyces cerevisiae. Nucleotide sequence, gene fusion experiments and cis-‐dominant regulatory mutation analysis. Eur J Biochem 1987, 169, 193-‐200.
[13] Degols, G.; Jauniaux, J. C.; Wiame, J. M., Molecular characterization of transposable-‐element-‐associated mutations that lead to constitutive L-‐ornithine aminotransferase expression in Saccharomyces cerevisiae. Eur J Biochem 1987, 165, 289-‐296.
[14] Merchant, S. S.; Prochnik, S. E.; Vallon, O.; Harris, E. H.; Karpowicz, S. J.; Witman, G. B.; Terry, A.; Salamov, A.; Fritz-‐Laylin, L. K.; Maréchal-‐Drouard, L.; Marshall, W. F.; Qu, L. H.; Nelson, D. R.; Sanderfoot, A. A.; Spalding, M. H.; Kapitonov, V. V.; Ren, Q.; Ferris, P.; Lindquist, E.; Shapiro, H.; Lucas, S. M.; Grimwood, J.; Schmutz, J.; Cardol, P.; Cerutti, H.; Chanfreau, G.; Chen, C. L.; Cognat, V.; Croft, M. T.; Dent, R.; Dutcher, S.; Fernández, E.; Fukuzawa, H.; González-‐Ballester, D.; González-‐Halphen, D.; Hallmann, A.; Hanikenne, M.; Hippler, M.; Inwood, W.; Jabbari, K.; Kalanon, M.; Kuras, R.; Lefebvre, P. A.; Lemaire, S. D.; Lobanov, A. V.; Lohr, M.; Manuell, A.; Meier, I.; Mets, L.; Mittag, M.; Mittelmeier, T.; Moroney, J. V.; Moseley, J.; Napoli, C.; Nedelcu, A. M.; Niyogi, K.; Novoselov, S. V.; Paulsen, I. T.; Pazour, G.; Purton, S.; Ral, J. P.; Riaño-‐Pachón, D. M.; Riekhof, W.; Rymarquis, L.; Schroda, M.; Stern, D.; Umen, J.; Willows, R.; Wilson, N.; Zimmer, S. L.; Allmer, J.; Balk, J.; Bisova, K.; Chen, C. J.; Elias, M.; Gendler, K.; Hauser, C.; Lamb, M. R.; Ledford, H.; Long, J. C.; Minagawa, J.; Page, M. D.; Pan, J.; Pootakham, W.; Roje, S.; Rose, A.; Stahlberg, E.; Terauchi, A. M.; Yang, P.; Ball, S.; Bowler, C.; Dieckmann, C. L.; Gladyshev, V. N.; Green, P.; Jorgensen, R.; Mayfield, S.; Mueller-‐Roeber, B.; Rajamani, S.; Sayre, R. T.; Brokstein, P.; Dubchak, I.; Goodstein, D.; Hornick, L.; Huang, Y. W.; Jhaveri, J.; Luo, Y.; Martínez, D.; Ngau, W. C. A.; Otillar, B.; Poliakov, A.; Porter, A.; Szajkowski, L.; Werner, G.; Zhou, K.; Grigoriev, I. V.; Rokhsar, D. S.; Grossman, A. R., The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245-‐250.
[15] Yoshida, K. M.; Juni, N.; Hori, S. H., Molecular cloning and characterization of Drosophila ornithine aminotransferase gene. Genes Genet Syst 1997, 72, 9-‐17.
[16] Mueckler, M. M.; Pitot, H. C., Sequence of the precursor to rat ornithine aminotransferase deduced from a cDNA clone. J Biol Chem 1985, 260, 12993-‐12997.
[17] Oyama, R.; Suzuki, M.; Matsuzawa, T.; Titani, K., Complete amino acid sequence of rat kidney ornithine aminotransferase: identity with liver ornithine aminotransferase. J Biochem 1990, 108, 133-‐138.
[18] Daune, G.; Gerhart, F.; Seiler, N., 5-‐Fluoromethylornithine, an irreversible and specific inhibitor of L-‐ornithine :2-‐oxo-‐acid aminotransferase. Biochem J 1988, 253, 481-‐488.
[19] Lim, S. N.; Rho, H. W.; Park, J. W.; Jhee, E. C.; Kim, J. S.; Kim, H. R., A variant of ornithine aminotransferase from mouse small intestine. Exp Mol Med 1998, 30, 131-‐135.
[20] Gafan, C.; Wilson, J.; Berger, L. C.; Berger, B. J., Characterization of the ornithine aminotransferase from Plasmodium falciparum. Mol Biochem Parasitol 2001, 118, 1-‐10.
[21] Naranjo, L.; Lamas-‐Maceiras, M.; Ullán, R. V.; Campoy, S.; Teijeira, F.; Casqueiro, J.; Martín, J. F., Characterization of the oat1 gene of Penicillium chrysogenum encoding an omega-‐aminotransferase: induction by L-‐lysine, L-‐ornithine and L-‐arginine and repression by ammonium. Mol Genet Genomics 2005, 274, 283-‐294.
[22] Valmaseda, E. M. M.; Campoy, S.; Naranjo, L.; Casqueiro, J.; Martín, J. F., Lysine is catabolized to 2-‐aminoadipic acid in Penicillium chrysogenum by an ω-‐aminotransferase and to saccharopine by a lysine 2-‐ketoglutarate reductase. characterization of the ω-‐aminotransferase. Mol Genet Genomics 2005, 274, 272-‐282.
[23] Dzikowska, A.; Swianiewicz, M.; Talarczyk, A.; Wisniewska, M.; Goras, M.; Scazzocchio, C.; Weglenski, P., Cloning, characterisation and regulation of the ornithine transaminase (otaA) gene of Aspergillus nidulans. Curr Genet 1999, 35, 118-‐126.
[24] Delauney, A. J.; Hu, C. A. A.; Kishor, P. B. K.; Verma, D. P. S., Cloning of ornithine δ-‐aminotransferase cDNA from Vigna aconitifolia by trans-‐complementation in Escherichia coli and regulation of proline biosynthesis. J Biol Chem 1993, 268, 18673-‐18678.
[25] Bicho, C. C.; de Lima Alves, F.; Chen, Z. A.; Rappsilber, J.; Sawin, K. E., A genetic engineering solution to the "arginine conversion problem" in stable isotope labeling by amino acids in cell culture (SILAC). Mol Cell Proteomics 2010, 9, 1567-‐77.
[26] Wagemaker, M. J. M.; Eastwood, D. C.; Welagen, J.; van der Drift, C.; Jetten, M. S. M.; Burton, K.; Van Griensven, L. J. L. D.; Op den Camp, H. J. M., The role of ornithine aminotransferase in fruiting body formation of the mushroom Agaricus bisporus. Mycol Res 2007, 111, 909-‐918.

26
[27] Page, A. F.; Minocha, R.; Minocha, S. C., Living with high putrescine: expression of ornithine and arginine biosynthetic pathway genes in high and low putrescine producing poplar cells. Amino acids 2012, 42, 295-‐308.
[28] Stránská, J.; Kopečný, D.; Tylichová, M.; Snégaroff, J.; Šebela, M., Ornithine δ-‐aminotransferase: an enzyme implicated in salt tolerance in higher plants. Plant Signaling Behav 2008, 3, 929-‐935.
[29] Roosens, N. H. C. J.; Thu, T. T.; Iskandar, H. M.; Jacobs, M., Isolation of the ornithine-‐δ-‐aminotransferase cDNA and effect of salt stress on its expression in Arabidopsis thaliana. Plant Physiol 1998, 117, 263-‐271.
[30] Cañas, R. A.; Villalobos, D. P.; Díaz-‐Moreno, S. M.; Cánovas, F. M.; Cantón, F. R., Molecular and functional analyses support a role of ornithine-‐δ-‐aminotransferase in the provision of glutamate for glutamine biosynthesis during pine germination. Plant Physiol 2008, 148, 77-‐88.
[31] You, J.; Hu, H.; Xiong, L., An ornithine δ-‐aminotransferase gene OsOAT confers drought and oxidative stress tolerance in rice. Plant Sci 2012, 197, 59-‐69.
[32] Stránská, J.; Tylichová, M.; Kopečný, D.; Snégaroff, J.; Šebela, M., Biochemical characterization of pea ornithine-‐δ-‐aminotransferase: substrate specificity and inhibition by di-‐ and polyamines. Biochimie 2010, 92, 940-‐948.
[33] Deckert, G.; Warren, P. V.; Gaasterland, T.; Young, W. G.; Lenox, A. L.; Graham, D. E.; Overbeek, R.; Snead, M. A.; Keller, M.; Aujay, M.; Huber, R.; Feldman, R. A.; Short, J. M.; Olsen, G. J.; Swanson, R. V., The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 1998, 392, 353-‐358.
[34] Rajaram, V.; Prasuna, P. R.; Savithri, H. S.; Murthy, M. R. N., Structure of biosynthetic N-‐acetylornithine aminotransferase from Salmonella typhimurium: studies on substrate specificity and inhibitor binding. Proteins 2008, 70, 429-‐441.
[35] Heimberg, H.; Boyen, A.; Crabeel, M.; Glansdorff, N., Escherichia coli and Saccharomyces cerevisiae acetylornithine aminotransferase: evolutionary relationship with ornithine aminotransferase. Gene 1990, 90, 69-‐78.
[36] Ledwidge, R.; Blanchard, J. S., The dual biosynthetic capability of N-‐acetylornithine aminotransferase in arginine and lysine biosynthesis. Biochemistry 1999, 38, 3019-‐3024.
[37] Link, A. J.; Robison, K.; Church, G. M., Comparing the predicted and observed properties of proteins encoded in the genome of Escherichia coli K-‐12. Electrophoresis 1997, 18, 1259-‐1313.
[38] Itoh, Y., Cloning and characterization of the aru genes encoding enzymes of the catabolic arginine succinyltransferase pathway in Pseudomonas aeruginosa. J Bacteriol 1997, 179, 7280-‐7290.
[39] Jann, A.; Stalon, V.; Wauven, C. V.; Leisinger, T.; Haas, D., N2-‐succinylated intermediates in an arginine catabolic pathway of Pseudomonas aeruginosa. Proc Natl Acad Sci USA 1986, 83, 4937-‐4941.
[40] Stalon, V.; Vander Wauven, C.; Momin, P.; Legrain, C., Catabolism of arginine, citrulline and ornithine by Pseudomonas and related bacteria. J Gen Microbiol 1987, 133, 2487-‐95.
[41] Vander Wauven, C.; Jann, A.; Haas, D.; Leisinger, T.; Stalon, V., N2-‐succinylornithine in ornithine catabolism of Pseudomonas aeruginosa. Arch Microbiol 1988, 150, 400-‐404.
[42] Lee, M. W.; Jelenska, J.; Greenberg, J. T., Arabidopsis proteins important for modulating defense responses to Pseudomonas syringae that secrete HopW1-‐1. Plant J 2008, 54, 452-‐465.
[43] Remacle, C.; Cline, S.; Boutaffala, L.; Gabilly, S.; Larosa, V.; Barbieri, M. R.; Coosemans, N.; Hamel, P. P., The ARG9 gene encodes the plastid-‐resident N-‐acetyl ornithine aminotransferase in the green alga Chlamydomonas reinhardtii. Eukaryotic Cell 2009, 8, 1460-‐1463.
[44] Floriano, B.; Herrero, A.; Flores, E., Analysis of expression of the argC and argD genes in the cyanobacterium Anabaena sp. strain PCC 7120. J Bacteriol 1994, 176, 6397-‐6401.
[45] Miyazaki, J.; Kobashi, N.; Nishiyama, M.; Yamane, H., Functional and evolutionary relationship between arginine biosynthesis and prokaryotic lysine biosynthesis through α-‐aminoadipate. J Bacteriol 2001, 183, 5067-‐5073.
[46] Watrous, J.; Burns, K.; Liu, W. T.; Patel, A.; Hook, V.; Bafna, V.; Barry, C. E., 3rd; Bark, S.; Dorrestein, P. C., Expansion of the mycobacterial "PUPylome". Mol Biosyst 2010, 6, 376-‐385.
[47] Hani, E. K.; Ng, D.; Chan, V. L., Arginine biosynthesis in Campylobacter jejuni TGH9011: determination of the argCOBD cluster. Can J Microbiol 1999, 45, 959-‐969.
[48] Guan, C.; Ribeiro, A.; Akkermans, A. D. L.; Jing, Y.; van Kammen, A.; Bisseling, T.; Pawlowski, K., Nitrogen metabolism in actinorhizal nodules of Alnus glutinosa: expression of glutamine synthetase and acetylornithine transaminase. Plant Mol Bio 1996, 32, 1177-‐1184.
[49] Janssen, A.; Chen, X. J., Cloning, sequencing and disruption of the ARG8 gene encoding acetylornithine aminotransferase in the petite-‐negative yeast Kluyveromyces lactis. Yeast 1998, 14, 281-‐285.

27
[50] Koma, D.; Sawai, T.; Harayama, S.; Kino, K., Overexpression of the genes from thermophiles in Escherichia coli by high-‐temperature cultivation. Appl Microbiol Biotechnol 2006, 73, 172-‐180.
[51] Rasko, D. A.; Rosovitz, M. J.; Myers, G. S. A.; Mongodin, E. F.; Fricke, W. F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N. R.; Chaudhuri, R.; Henderson, I. R.; Sperandio, V.; Ravel, J., The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 2008, 190, 6881-‐6893.
[52] Tripathi, S. M.; Ramachandran, R., Direct evidence for a glutamate switch necessary for substrate recognition: crystal structures of lysine ε-‐aminotransferase (Rv3290c) from Mycobacterium tuberculosis H37Rv. J Mol Biol 2006, 362, 877-‐886.
[53] Tripathi, S. M.; Ramachandran, R., Overexpression, purification and crystallization of lysine ε-‐aminotransferase (Rv3290c) from Mycobacterium tuberculosis H37Rv. Acta Crystallogr F Struct Biol Commun 2006, 62, 572-‐575.
[54] Tobin, M. B.; Kovacevic, S.; Madduri, K.; Hoskins, J. A.; Skatrud, P. L.; Vining, L. C.; Stuttard, C.; Miller, J. R., Localization of the lysine ε-‐aminotransferase (lat) and δ-‐(L-‐α-‐aminoadipyl)-‐L-‐cysteinyl-‐D-‐valine synthetase (pcbAB) genes from Streptomyces clavuligerus and production of lysine ε-‐aminotransferase activity in Escherichia coli. J Bacteriol 1991, 173, 6223-‐6229.
[55] Romero, J.; Martin, J. F.; Liras, P.; Demain, A. L.; Rius, N., Partial purification, characterization and nitrogen regulation of the lysine ε-‐aminotransferase of Streptomyces clavuligerus. J Ind Microb Biotechnol 1997, 18, 241-‐246.
[56] Soda, K.; Misono, H., L-‐lysine-‐α-‐ketoglutarate aminotransferase. II. purification, crystallization, and properties. Biochemistry 1968, 7, 4110-‐4119.
[57] Yagi, T.; Misono, H.; Tanizawa, K.; Yoshimura, T.; Soda, K., Characterization of the half and overall reactions catalyzed by L-‐lysine:2-‐oxoglutarate 6-‐aminotransferase. J Biochem 1991, 109, 61-‐65.
[58] Fujii, T.; Narita, T.; Agematu, H.; Agata, N.; Isshiki, K., Characterization of L-‐lysine 6-‐aminotransferase and its structural gene from Flavobacterium lutescens IFO3084. J Biochem 2000, 128, 391-‐397.
[59] Soda, K.; Misono, H.; Yamamoto, T., L-‐lysine-‐α-‐ketoglutarate aminotransferase. I. identification of a product, Δ1-‐piperideine-‐6-‐carboxylic acid. Biochemistry 1968, 7, 4102-‐4109.
[60] Soda, K.; Misono, H.; Yamamoto, T., L-‐lysine-‐α-‐ketoglutaric acid transaminase reaction: identification of the product from L-‐lysine. Agric Biol Chem 1966, 30, 944-‐946.
[61] Storici, P.; De Biase, D.; Bossa, F.; Bruno, S.; Mozzarelli, A.; Peneff, C.; Silverman, R. B.; Schirmer, T., Structures of γ-‐aminobutyric acid (GABA) aminotransferase, a pyridoxal 5ʹ′-‐phosphate, and [2Fe-‐2S] cluster-‐containing enzyme, complexed with γ-‐ethynyl-‐GABA and with the antiepilepsy drug Vigabatrin. J Biol Chem 2004, 279, 363-‐373.
[62] Kakimoto, Y.; Kanazawa, A.; Taniguchi, K.; Sano, I., β-‐aminoisobutyrate-‐α-‐ketoglutarate transaminase in relation to β-‐aminoisobutyric aciduria. Biochim Biophys Acta 1968, 156, 374-‐380.
[63] Andersen, G.; Andersen, B.; Dobritzsch, D.; Schnackerz, K. D.; Piškur, J., A gene duplication led to specialized γ-‐aminobutyrate and β-‐alanine aminotransferase in yeast. FEBS J 2007, 274, 1804-‐1817.
[64] Schnackerz, K. D.; Andersen, G.; Dobritzsch, D.; Piškur, J., Degradation of pyrimidines in Saccharomyces Kluyveri: transamination of β-‐Alanine. Nucleosides Nucleotides Nucleic Acids 2008, 27, 794-‐799.
[65] Medina-‐Kauwe, L. K.; Tillakaratne, N. J. K.; Wu, J. Y.; Tobin, A. J., A rat brain cDNA encodes enzymatically active GABA transaminase and provides a molecular probe for GABA-‐catabolizing cells. J Neurochem 1994, 62, 1267-‐1275.
[66] Tamaki, N.; Sakata, S. F.; Matsuda, K., [35] Purification, properties, and sequencing of aminoisobutyrate aminotransferases from rat liver. In Methods in Enzymology, Sokatch, J. R.; Harris, R. A., Eds. Academic Press: San Diego, CA, 2000; Vol. 324, pp 376-‐389.
[67] Seong Gyu, J.; Jae Hoon, B.; Joong Sik, J.; Sang Ho, J.; Byung Ryong, L.; Kil Soo, L.; Jinseu, P.; Tae-‐Cheon, K.; Moo Ho, W.; Hyong Bai, K.; Oh-‐Shin, K.; Sung-‐Woo, C.; Soo Young, C., Molecular cloning and functional expression of bovine brain GABA transaminase. Mol Cells 2001, 12, 91-‐96.
[68] Osei, Y. D.; Churchich, J. E., Screening and sequence determination of a cDNA encoding the human brain 4-‐aminobutyrate aminotransferase. Gene 1995, 155, 185-‐187.
[69] Yoon, C. S.; Kim, D. W.; Jang, S. H.; Lee, B. R.; Choi, H. S.; Choi, S. H.; Kim, S. Y.; An, J. J.; Kwon, O. S.; Kang, T. C.; Won, M. H.; Cho, S. W.; Lee, K. S.; Park, J. S.; Eum, W. S.; Choi, S. Y., Cysteine-‐321 of human brain GABA transaminase is involved in intersubunit cross-‐linking. Mol Cells 2004, 18, 214-‐219.
[70] Schousboe, A.; Wu, J. Y.; Roberts, E., Purification and characterization of the 4-‐aminobutyrate-‐2-‐ketoglutarate transaminase from mouse brain. Biochemistry 1973, 12, 2868-‐2873.

28
[71] The FANTOM Consortium; Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M. C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; Kodzius, R.; Shimokawa, K.; Bajic, V. B.; Brenner, S. E.; Batalov, S.; Forrest, A. R. R.; Zavolan, M.; Davis, M. J.; Wilming, L. G.; Aidinis, V.; Allen, J. E.; Ambesi-‐Impiombato, A.; Apweiler, R.; Aturaliya, R. N.; Bailey, T. L.; Bansal, M.; Baxter, L.; Beisel, K. W.; Bersano, T.; Bono, H.; Chalk, A. M.; Chiu, K. P.; Choudhary, V.; Christoffels, A.; Clutterbuck, D. R.; Crowe, M. L.; Dalla, E.; Dalrymple, B. P.; de Bono, B.; Gatta, G. D.; di Bernardo, D.; Down, T.; Engstrom, P.; Fagiolini, M.; Faulkner, G.; Fletcher, C. F.; Fukushima, T.; Furuno, M.; Futaki, S.; Gariboldi, M.; Georgii-‐Hemming, P.; Gingeras, T. R.; Gojobori, T.; Green, R. E.; Gustincich, S.; Harbers, M.; Hayashi, Y.; Hensch, T. K.; Hirokawa, N.; Hill, D.; Huminiecki, L.; Iacono, M.; Ikeo, K.; Iwama, A.; Ishikawa, T.; Jakt, M.; Kanapin, A.; Katoh, M.; Kawasawa, Y.; Kelso, J.; Kitamura, H.; Kitano, H.; Kollias, G.; Krishnan, S. P. T.; Kruger, A.; Kummerfeld, S. K.; Kurochkin, I. V.; Lareau, L. F.; Lazarevic, D.; Lipovich, L.; Liu, J.; Liuni, S.; McWilliam, S.; Babu, M. M.; Madera, M.; Marchionni, L.; Matsuda, H.; Matsuzawa, S.; Miki, H.; Mignone, F.; Miyake, S.; Morris, K.; Mottagui-‐Tabar, S.; Mulder, N.; Nakano, N.; Nakauchi, H.; Ng, P.; Nilsson, R.; Nishiguchi, S.; Nishikawa, S.; Nori, F.; Ohara, O.; Okazaki, Y.; Orlando, V.; Pang, K. C.; Pavan, W. J.; Pavesi, G.; Pesole, G.; Petrovsky, N.; Piazza, S.; Reed, J.; Reid, J. F.; Ring, B. Z.; Ringwald, M.; Rost, B.; Ruan, Y.; Salzberg, S. L.; Sandelin, A.; Schneider, C.; Schönbach, C.; Sekiguchi, K.; Semple, C. A. M.; Seno, S.; Sessa, L.; Sheng, Y.; Shibata, Y.; Shimada, H.; Shimada, K.; Silva, D.; Sinclair, B.; Sperling, S.; Stupka, E.; Sugiura, K.; Sultana, R.; Takenaka, Y.; Taki, K.; Tammoja, K.; Tan, S. L.; Tang, S.; Taylor, M. S.; Tegner, J.; Teichmann, S. A.; Ueda, H. R.; van Nimwegen, E.; Verardo, R.; Wei, C. L.; Yagi, K.; Yamanishi, H.; Zabarovsky, E.; Zhu, S.; Zimmer, A.; Hide, W.; Bult, C.; Grimmond, S. M.; Teasdale, R. D.; Liu, E. T.; Brusic, V.; Quackenbush, J.; Wahlestedt, C.; Mattick, J. S.; Hume, D. A.; Group, R. G. E. R.; Genome Science, G.; Kai, C.; Sasaki, D.; Tomaru, Y.; Fukuda, S.; Kanamori-‐Katayama, M.; Suzuki, M.; Aoki, J.; Arakawa, T.; Iida, J.; Imamura, K.; Itoh, M.; Kato, T.; Kawaji, H.; Kawagashira, N.; Kawashima, T.; Kojima, M.; Kondo, S.; Konno, H.; Nakano, K.; Ninomiya, N.; Nishio, T.; Okada, M.; Plessy, C.; Shibata, K.; Shiraki, T.; Suzuki, S.; Tagami, M.; Waki, K.; Watahiki, A.; Okamura-‐Oho, Y.; Suzuki, H.; Kawai, J.; Hayashizaki, Y., The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559-‐1563.
[72] Straffon, M. J.; Hynes, M. J.; Davis, M. A., Characterization of the ugatA gene of Ustilago maydis, isolated by homology to the gatA gene of Aspergillus nidulans. Curr Genet 1996, 29, 360-‐369.
[73] Bailey, C. R.; Arst, H. N.; Penfold, H. A., A third gene affecting GABA transaminase levels in Aspergillans nidulus. Genet Res, Camb 1980, 36, 167-‐180.
[74] Martyniuk, C. J.; Awad, R.; Hurley, R.; Finger, T. E.; Trudeau, V. L., Glutamic acid decarboxylase 65, 67, and GABA-‐transaminase mRNA expression and total enzyme activity in the goldfish (Carassius auratus) brain. Brain Res 2007, 1147, 154-‐166.
[75] Kurihara, S.; Kato, K.; Asada, K.; Kumagai, H.; Suzuki, H., A putrescine-‐inducible pathway comprising PuuE-‐YneI in which γ-‐aminobutyrate is degraded into succinate in Escherichia coli K-‐12. J Bacteriol 2010, 192, 4582-‐4591.
[76] Schneider, B. L.; Reitzer, L., Pathway and enzyme redundancy in putrescine catabolism in Escherichia coli. J Bacteriol 2012, 194, 4080-‐4088.
[77] Liu, W.; Peterson, P. E.; Langston, J. A.; Jin, X.; Zhou, X.; Fisher, A. J.; Toney, M. D., Kinetic and crystallographic analysis of active site mutants of Escherichia coli γ-‐aminobutyrate aminotransferase. Biochemistry 2005, 44, 2982-‐2992.
[78] Lal, P. B.; Schneider, B. L.; Vu, K.; Reitzer, L., The redundant aminotransferases in lysine and arginine synthesis and the extent of aminotransferase redundancy in Escherichia coli. Mol Microbiol 2014, n/a-‐n/a.
[79] Bruce, H.; Tuan, A. N.; Sanchez, J. M.; Leese, C.; Hopwood, J.; Hyde, R.; Hart, S.; Turkenburg, J. P.; Grogan, G., Structures of a γ-‐aminobutyrate (GABA) transaminase from the s-‐triazine-‐degrading organism Arthrobacter aurescens TC1 in complex with PLP and with its external aldimine PLP-‐GABA adduct. Acta Crystallogr F Struct Biol Commun 2012, 68, 1175-‐1180.
[80] Voellym, R.; Leisinger, T., Role of 4-‐aminobutyrate aminotransferase in the arginine metabolism of Pseudomonas aeruginosa. J Bacteriol 1976, 128, 722-‐729.
[81] Chou, H. T.; Kwon, D. H.; Hegazy, M.; Lu, C. D., Transcriptome analysis of agmatine and putrescine catabolism in Pseudomonas aeruginosa PAO1. J Bacteriol 2008, 190, 1966-‐1975.
[82] Espinosa-‐Urgel, M.; Ramos, J. L., Expression of a Pseudomonas putida aminotransferase involved in lysine catabolism is induced in the rhizosphere. Appl Environ Microbiol 2001, 67, 5219-‐5224.
[83] Bandounas, L.; Ballerstedt, H.; de Winde, J. H.; Ruijssenaars, H. J., Redundancy in putrescine catabolism in solvent tolerant Pseudomonas putida S12. J Biotechnol 2011, 154, 1-‐10.

29
[84] Park, D. H.; Mirabella, R.; Bronstein, P. A.; Preston, G. M.; Haring, M. A.; Lim, C. K.; Collmer, A.; Schuurink, R. C., Mutations in γ-‐aminobutyric acid (GABA) transaminase genes in plants or Pseudomonas syringae reduce bacterial virulence. Plant J 2010, 64, 318-‐330.
[85] Belitsky, B. R.; Sonenshein, A. L., GabR, a member of a novel protein family, regulates the utilization of γ-‐aminobutyrate in Bacillus subtilis. Mol Microbiol 2002, 45, 569-‐583.
[86] Prell, J.; Boesten, B.; Poole, P.; Priefer, U. B., The Rhizobium leguminosarum bv. viciae VF39 γ-‐aminobutyrate (GABA) aminotransferase gene (gabT) is induced by GABA and highly expressed in bacteroids. Microbiology 2002, 148, 615-‐623.
[87] Mayer, J.; Cook, A. M., Homotaurine metabolized to 3-‐sulfopropanoate in Cupriavidus necator H16: enzymes and genes in a patchwork pathway. J Bacteriol 2009, 191, 6052-‐6058.
[88] Kim, K. H., Purification and properties of a mine α-‐ketoglutarate transaminase from Escherichia coli. J Biol Chem 1964, 239, 783-‐786.
[89] Yeo, S.-‐J.; Jeong, J.-‐H.; Yu, S.-‐N.; Kim, Y.-‐G., Crystallization and preliminary X-‐ray crystallographic analysis of YgjG from Escherichia coli. Acta Crystallogr F Struct Biol Commun 2012, 68, 1070-‐1072.
[90] Cha, H. J.; Jeong, J.-‐H.; Rojviriya, C.; Kim, Y.-‐G., Structure of putrescine aminotransferase from Escherichia coli provides insights into the substrate specificity among class III aminotransferases. PLoS ONE 2014, 9, e113212.
[91] Lu, C. D.; Itoh, Y.; Nakada, Y.; Jiang, Y., Functional analysis and regulation of the divergent spuABCDEFGH-‐spuI operons for polyamine uptake and utilization in Pseudomonas aeruginosa PAO1. J Bacteriol 2002, 184, 3765-‐3773.
[92] Williamson, N. R.; Simonsen, H. T.; Ahmed, R. A. A.; Goldet, G.; Slater, H.; Woodley, L.; Leeper, F. J.; Salmond, G. P. C., Biosynthesis of the red antibiotic, prodigiosin, in Serratia: identification of a novel 2-‐methyl-‐3-‐n-‐amyl-‐pyrrole (MAP) assembly pathway, definition of the terminal condensing enzyme, and implications for undecylprodigiosin biosynthesis in Streptomyces. Mol Microbiol 2005, 56, 971-‐989.
[93] Lou, X.; Ran, T.; Han, N.; Gao, Y.; He, J.; Tang, L.; Xu, D.; Wang, W., Crystal structure of the catalytic domain of PigE: a transaminase involved in the biosynthesis of 2-‐methyl-‐3-‐n-‐amyl-‐pyrrole (MAP) from Serratia sp. FS14. Biochem Biophys Res Commun 2014, 447, 178-‐183.
[94] Kuhlmann, A. U.; Bursy, J.; Gimpel, S.; Hoffmann, T.; Bremer, E., Synthesis of the compatible solute ectoine in Virgibacillus pantothenticus is triggered by high salinity and low growth temperature. Appl Environ Microbiol 2008, 74, 4560-‐4563.
[95] Ono, H.; Sawada, K.; Khunajakr, N.; Tao, T.; Yamamoto, M.; Hiramoto, M.; Shinmyo, A.; Takano, M.; Murooka, Y., Characterization of biosynthetic enzymes for ectoine as a compatible solute in a moderately halophilic eubacterium, Halomonas elongata. J Bacteriol 1999, 181, 91-‐99.
[96] Cánovas, D.; Vargas, C.; Calderón, M. I.; Ventosa, A.; Nieto, J. J., Characterization of the genes for the biosynthesis of the compatible solute ectoine in the moderately halophilic bacterium Halomonas elongata DSM 3043. Syst Appl Microbiol 1998, 21, 487-‐497.
[97] Vandenende, C. S.; Vlasschaert, M.; Seah, S. Y. K., Functional characterization of an aminotransferase required for pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa PAO1. J Bacteriol 2004, 186, 5596-‐5602.
[98] Rajan, L. A.; Joseph, T. C.; Thampuran, N.; James, R.; Kumar, K. A.; Viswanathan, C.; Bansal, K. C., Cloning and heterologous expression of ectoine biosynthesis genes from Bacillus halodurans in Escherichia coli. Biotechnol Lett 2008, 30, 1403-‐1407.
[99] Louis, P.; Galinski, E. A., Characterization of genes for the biosynthesis of the compatible solute ectoine from Marinococcus halophilus and osmoregulated expression in Escherichia coli. Microbiology 1997, 143, 1141-‐1149.
[100] Göller, K.; Ofer, A.; Galinski, E. A., Construction and characterization of an NaCl-‐sensitive mutant of Halomonas elongata impaired in ectoine biosynthesis. FEMS Microbiol Lett 1998, 161, 293-‐300.
[101] Ikai, H.; Yamamoto, S., Identification and analysis of a gene encoding L-‐2,4-‐diaminobutyrate:2-‐ketoglutarate 4-‐aminotransferase involved in the 1,3-‐diaminopropane production pathway in Acinetobacter baumannii. J Bacteriol 1997, 179, 5118-‐5125.
[102] Ikai, H.; Yamamoto, S., Two genes involved in the 1,3-‐diaminopropane production pathway in Haemophilus influenzae. Biol Pharm Bull 1998, 21, 170-‐173.
[103] Kuhlmann, A. U.; Bremer, E., Osmotically regulated synthesis of the compatible solute ectoine in Bacillus pasteurii and related Bacillus spp. Appl Environ Microbiol 2002, 68, 772-‐783.

30
[104] Reshetnikov, A. S.; Khmelenina, V. N.; Trotsenko, Y. A., Characterization of the ectoine biosynthesis genes of haloalkalotolerant obligate methanotroph “Methylomicrobium alcaliphilum 20Z”. Arch Microbiol 2006, 184, 286-‐297.
[105] Garcia, A. R. M.; Kamat, S. V.; Pannuri, S. Thermostable ω-‐transaminases. 2006. [106] Hanson, R. L.; Davis, B. L.; Chen, Y.; Goldberg, S. L.; Parker, W. L.; Tully, T. P.; Montana, M. A.; Patel, R. N.,
Preparation of (R)-‐amines from racemic amines with an (S)-‐amine transaminase from Bacillus megaterium. Adv Synth Catal 2008, 350, 1367-‐1375.
[107] Fuchs, C. S.; Simon, R. C.; Riethorst, W.; Zepeck, F.; Kroutil, W., Synthesis of (R)-‐ or (S)-‐valinol using ω-‐transaminases in aqueous and organic media. Bioorg Med Chem 2014, 22, 5558-‐5562.
[108] Kaulmann, U.; Smithies, K.; Smith, M. E. B.; Hailes, H. C.; Ward, J. M., Substrate spectrum of ω-‐transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzyme Microb Technol 2007, 41, 628-‐637.
[109] Steffen-‐Munsberg, F.; Vickers, C.; Thontowi, A.; Schätzle, S.; Tumlirsch, T.; Svedendahl Humble, M.; Land, H.; Berglund, P.; Bornscheuer, U. T.; Höhne, M., Connecting unexplored protein crystal structures to enzymatic function. ChemCatChem 2013, 5, 150-‐153.
[110] Park, E.-‐S.; Shin, J.-‐S., ω-‐Transaminase from Ochrobactrum anthropi Is Devoid of Substrate and Product Inhibitions. Appl Environ Microbiol 2013, 79, 4141-‐4144.
[111] Park, E.; Kim, M.; Shin, J. S., One-‐pot conversion of L-‐threonine into L-‐homoalanine: biocatalytic production of an unnatural amino acid from a natural one. Adv Synth Catal 2010, 352, 3391-‐3398.
[112] Shin, J. S.; Yun, H.; Jang, J. W.; Park, I.; Kim, B. G., Purification, characterization, and molecular cloning of a novel amine:pyruvate transaminase from Vibrio fluvialis JS17. Appl Microbiol Biotechnol 2003, 61, 463-‐471.
[113] Schätzle, S.; Höhne, M.; Redestad, E.; Robins, K.; Bornscheuer, U. T., Rapid and sensitive kinetic assay for characterization of ω-‐transaminases. Anal Chem 2009, 81, 8244-‐8248.
[114] Kroutil, W.; Fischereder, E. M.; Fuchs, C. S.; Lechner, H.; Mutti, F. G.; Pressnitz, D.; Rajagopalan, A.; Sattler, J. H.; Simon, R. C.; Siirola, E., Asymmetric preparation of prim-‐, sec-‐, and tert-‐amines employing selected biocatalysts. Org Process Res Dev 2013, 17, 751-‐759.
[115] Ito, N.; Kawano, S.; Yasohara, Y. Method for production of optically active amine compound, recombinant vector, and transformant carrying the vector. WO2007139055, 2007.
[116] Park, E. S.; Kim, M.; Shin, J. S., Molecular determinants for substrate selectivity of ω-‐transaminases. Appl Microbiol Biotechnol 2011, 93, 2425-‐2435.
[117] Yun, H.; Lim, S.; Cho, B. K.; Kim, B. G., ω-‐Amino acid:pyruvate transaminase from Alcaligenes denitrificans Y2k-‐2: a new catalyst for kinetic resolution of β-‐amino acids and amines. Appl Environ Microbiol 2004, 70, 2529-‐2534.
[118] Hwang, B. Y.; Ko, S. H.; Park, H. Y.; Seo, J. H.; Lee, B. S.; Kim, B. G., Identification of ω-‐aminotransferase from Caulobacter crescentus and site-‐directed mutagenesis to broaden substrate specificity. J Microbiol Biotechnol 2008, 18, 48-‐54.
[119] Sayer, C.; Isupov, M. N.; Westlake, A.; Littlechild, J. A., Structural studies of Pseudomonas and Chromobacterium ω-‐aminotransferases provide insights into their differing substrate specificity. Acta Crystallogr D Biol Crystallogr 2013, 69, 564-‐576.
[120] Watanabe, N.; Sakabe, K.; Sakabe, N.; Higashi, T.; Sasaki, K.; Aibara, S.; Morita, Y.; Yonaha, K.; Toyama, S.; Fukutani, H., Crystal structure analysis of ω-‐amino acid:pyruvate aminotransferase with a newly developed Weissenberg camera and an imaging plate using synchrotron radiation. J Biochem 1989, 105, 1-‐3.
[121] Seo, J.-‐H.; Hwang, J.-‐Y.; Seo, S.-‐H.; Kang, H.; Hwang, B.-‐Y.; Kim, B.-‐G., Computational selection, identification and structural analysis of ω-‐aminotransferases with various substrate specificities from the genome sequence of Mesorhizobium loti MAFF303099. Biosci Biotechnol Biochem 2012, 76, 1308-‐1314.
[122] Kwon, Y. C.; Lee, K. H.; Kim, H. C.; Han, K.; Seo, J. H.; Kim, B. G.; Kim, D. M., Cloning-‐independent expression and analysis of ω-‐transaminases by use of a cell-‐free protein synthesis system. Appl Environ Microbiol 2010, 76, 6295-‐6298.
[123] Weber, N.; Ismail, A.; Gorwa-‐Grauslund, M.; Carlquist, M., Biocatalytic potential of vanillin aminotransferase from Capsicum chinense. BMC Biotechnol 2014, 14, 25.
[124] Gururaj, H. B.; Padma, M. N.; Giridhar, P.; Ravishankar, G. A., Functional validation of Capsicum frutescens aminotransferase gene involved in vanillylamine biosynthesis using Agrobacterium mediated genetic transformation studies in Nicotiana tabacum and Capsicum frutescens calli cultures. Plant Sci 2012, 195, 96-‐105.

31
[125] Van Cauwenberghe, O. R.; Makhmoudova, A.; McLean, M. D.; Clark, S. M.; Shelp, B. J., Plant pyruvate-‐dependent gamma-‐aminobutyrate transaminase: identification of an Arabidopsis cDNA and its expression in Escherichia coli. Can J Bot 2002, 80, 933-‐941.
[126] Ricoult, C.; Echeverria, L. O.; Cliquet, J. B.; Limami, A. M., Characterization of alanine aminotransferase (AlaAT) multigene family and hypoxic response in young seedlings of the model legume Medicago truncatula. J Exp Bot 2006, 57, 3079-‐3089.
[127] Clark, S. M.; Di Leo, R.; Van Cauwenberghe, O. R.; Mullen, R. T.; Shelp, B. J., Subcellular localization and expression of multiple tomato γ-‐aminobutyrate transaminases that utilize both pyruvate and glyoxylate. J Exp Bot 2009, 60, 3255-‐3267.
[128] Wu, C.; Zhou, S.; Zhang, Q.; Zhao, W.; Peng, Y., Molecular cloning and differential expression of an γ-‐aminobutyrate transaminase gene, OsGABA-‐T, in rice (Oryza sativa) leaves infected with blast fungus. J Plant Res 2006, 119, 663-‐669.
[129] Trobacher, C. P.; Clark, S. M.; Bozzo, G. G.; Mullen, R. T.; DeEll, J. R.; Shelp, B. J., Catabolism of GABA in apple fruit: subcellular localization and biochemical characterization of two γ-‐aminobutyrate transaminases. Postharvest Biol Technol 2013, 75, 106-‐113.
[130] Laue, H.; Cook, A. M., Biochemical and molecular characterization of taurine:pyruvate aminotransferase from the anaerobe Bilophila wadsworthia. Eur J Biochem 2000, 267, 6841-‐6848.
[131] Gorzynska, A. K.; Denger, K.; Cook, A. M.; Smits, T. H. M., Inducible transcription of genes involved in taurine uptake and dissimilation by Silicibacter pomeroyi DSS-‐3T. Arch Microbiol 2006, 185, 402-‐406.
[132] Denger, K.; Ruff, J.; Schleheck, D.; Cook, A. M., Rhodococcus opacus expresses the xsc gene to utilize taurine as a carbon source or as a nitrogen source but not as a sulfur source. Microbiology 2004, 150, 1859-‐1867.
[133] Felux, A. K.; Denger, K.; Weiss, M.; Cook, A. M.; Schleheck, D., Paracoccus denitrificans PD1222 utilizes hypotaurine via transamination followed by spontaneous desulfination to yield acetaldehyde and, finally, acetate for growth. J Bacteriol 2013, 195, 2921-‐2930.
[134] Banerjee, A.; Chase, M.; Clayton, R. A.; Landis, B. Methods for the stereoselective synthesis and enantiomeric enrichment of β-‐amino acids. 2005.
[135] Crismaru, C. G.; Wybenga, G. G.; Szymanski, W.; Wijma, H. J.; Wu, B.; Bartsch, S.; de Wildeman, S.; Poelarends, G. J.; Feringa, B. L.; Dijkstra, B. W.; Janssen, D. B., Biochemical properties and crystal structure of a β-‐phenylalanine aminotransferase from Variovorax paradoxus. Appl Environ Microbiol 2013, 79, 185-‐195.
[136] Kim, J.; Kyung, D.; Yun, H.; Cho, B. K.; Seo, J. H.; Cha, M.; Kim, B. G., Cloning and characterization of a novel β-‐transaminase from Mesorhizobium sp. strain LUK: a new biocatalyst for the synthesis of enantiomerically pure β-‐amino acids. Appl Environ Microbiol 2007, 73, 1772-‐1782.
[137] Wybenga, G. G.; Crismaru, C. G.; Janssen, D. B.; Dijkstra, B. W., Structural determinants of the β-‐selectivity of a bacterial aminotransferase. J Biol Chem 2012, 287, 28495-‐28502.
[138] Shon, M.; Shanmugavel, R.; Shin, G.; Mathew, S.; Lee, S. H.; Yun, H., Enzymatic synthesis of chiral γ-‐amino acids using ω-‐transaminase. Chem Commun 2014, 50, 12680-‐12683.
[139] Bea, H. S.; Park, H. J.; Lee, S. H.; Yun, H., Kinetic resolution of aromatic β-‐amino acids by ω-‐transaminase. Chem Commun 2011, 47, 5894-‐5896.
[140] Perret, A.; Lechaplais, C.; Tricot, S.; Perchat, N.; Vergne, C.; Pelle, C.; Bastard, K.; Kreimeyer, A.; Vallenet, D.; Zaparucha, A.; Weissenbach, J.; Salanoubat, M., A novel acyl-‐CoA beta-‐transaminase characterized from a metagenome. PLoS ONE 2011, 6, e22918.
[141] Wiyakrutta, S.; Meevootisom, V., A stereo-‐inverting D-‐phenylglycine aminotransferase from Pseudomonas stutzeri ST-‐201: purification, characterization and application for D-‐phenylglycine synthesis. J Biotechnol 1997, 55, 193-‐203.
[142] Kongsaeree, P.; Samanchart, C.; Laowanapiban, P.; Wiyakrutta, S.; Meevootisom, V., Crystallization and preliminary X-‐ray crystallographic analysis of D-‐phenylglycine aminotransferase from Pseudomonas stutzeri ST201. Acta Crystallogr D Biol Crystallogr 2003, 59, 953-‐954.
[143] Townsend, C.; Gunsior, M.; Muller, U.; Van, F. A.; Sonke, T. Fermentative production of D-‐hydroxyphenylglycine and D-‐phenylglycine. WO/2002/034921 A2, 2002.
[144] Müller, U.; van Assema, F.; Gunsior, M.; Orf, S.; Kremer, S.; Schipper, D.; Wagemans, A.; Townsend, C. A.; Sonke, T.; Bovenberg, R.; Wubbolts, M., Metabolic engineering of the E. coli L-‐phenylalanine pathway for the production of D-‐phenylglycine (D-‐Phg). Metab Eng 2006, 8, 196-‐208.
[145] Eliot, A. C.; Sandmark, J.; Schneider, G.; Kirsch, J. F., The dual-‐specific active site of 7,8-‐diaminopelargonic acid synthase and the effect of the R391A mutation. Biochemistry 2002, 41, 12582-‐12589.

32
[146] Käck, H.; Sandmark, J.; Gibson, K.; Schneider, G.; Lindqvist, Y., Crystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-‐5'-‐phosphate-‐dependent enzymes. J Mol Biol 1999, 291, 857-‐876.
[147] Sandmark, J.; Eliot, A. C.; Famm, K.; Schneider, G.; Kirsch, J. F., Conserved and nonconserved residues in the substrate binding site of 7,8-‐diaminopelargonic acid synthase from Escherichia coli are essential for catalysis. Biochemistry 2004, 43, 1213-‐1222.
[148] Bhor, V. M.; Dev, S.; Vasanthakumar, G. R.; Surolia, A., Spectral and kinetic characterization of 7,8-‐diaminopelargonic acid synthase from Mycobacterium tuberculosis. IUBMB Life 2006, 58, 225-‐233.
[149] Dey, S.; Lane, J. M.; Lee, R. E.; Rubin, E. J.; Sacchettini, J. C., Structural characterization of the Mycobacterium tuberculosis biotin biosynthesis enzymes 7,8-‐diaminopelargonic acid synthase and dethiobiotin synthetase. Biochemistry 2010, 49, 6746-‐6760.
[150] Mann, S.; Colliandre, L.; Labesse, G.; Ploux, O., Inhibition of 7,8-‐diaminopelargonic acid aminotransferase from Mycobacterium tuberculosis by chiral and achiral anologs of its substrate: biological implications. Biochimie 2009, 91, 826-‐834.
[151] Mann, S.; Ploux, O., 7,8-‐Diaminoperlargonic acid aminotransferase from Mycobacterium tuberculosis, a potential therapeutic target. FEBS J 2006, 273, 4778-‐4789.
[152] Shi, C.; Geders, T. W.; Park, S. W.; Wilson, D. J.; Boshoff, H. I.; Abayomi, O.; Barry, C. E., 3rd; Schnappinger, D.; Finzel, B. C.; Aldrich, C. C., Mechanism-‐based inactivation by aromatization of the transaminase BioA involved in biotin biosynthesis in Mycobaterium tuberculosis. J Am Chem Soc 2011, 133, 18194-‐18201.
[153] Phalip, V.; Kuhn, I.; Lemoine, Y.; Jeltsch, J. M., Characterization of the biotin biosynthesis pathway in Saccharomyces cerevisiae and evidence for a cluster containing BIO5, a novel gene involved in vitamer uptake. Gene 1999, 232, 43-‐51.
[154] Sakurai, N.; Imai, Y.; Masuda, M.; Komatsubara, S.; Tosa, T., Molecular breeding of a biotin-‐hyperproducing Serratia marcescens strain. Appl Environ Microbiol 1993, 59, 3225-‐3232.
[155] Hatakeyama, K.; Kobayashi, M.; Yukawa, H., [36] Analysis of biotin biosynthesis pathway in coryneform bacteria: Brevibacterium flavum. In Methods in Enzymology, McCormick, D. B.; Suttie, J. W.; Wagner, C., Eds. Academic Press: 1997; Vol. Volume 279, pp 339-‐348.
[156] Marienhagen, J.; Kennerknecht, N.; Sahm, H.; Eggeling, L., Functional analysis of all aminotransferase proteins inferred from the genome sequence of Corynebacterium glutamicum. J Bacteriol 2005, 187, 7639-‐7646.
[157] Entcheva, P.; Liebl, W.; Johann, A.; Hartsch, T.; Streit, W. R., Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl Environ Microbiol 2001, 67, 89-‐99.
[158] Wu, C.-‐H.; Chen, H.-‐Y.; Shiuan, D., Isolation and characterization of the Erwinia herbicola bio operon and the sequences of the bioA and bioB genes. Gene 1996, 174, 251-‐258.
[159] Van Arsdell, S. W.; Perkins, J. B.; Yocum, R. R.; Luan, L.; Howitt, C. L.; Prasad Chatterjee, N.; Pero, J. G., Removing a bottleneck in the Bacillus subtilis biotin pathway: BioA utilizes lysine rather than S-‐adenosylmethionine as the amino donor in the KAPA-‐to-‐DAPA reaction. Biotechnol Bioeng 2005, 91, 75-‐83.
[160] Izumi, Y.; Kano, Y.; Inagaki, K.; Kawase, N.; Tani, Y.; Yamada, H., Characterization of biotin biosynthetic enzymes of Bacillus sphaericus: a dethiobiotin producing bacterium. Agric Biol Chem 1981, 45, 1983-‐1989.
[161] Cobessi, D.; Dumas, R.; Pautre, V.; Meinguet, C.; Ferrer, J. L.; Alban, C., Biochemical and structural characterization of the Arabidopsis bifunctional enzyme dethiobiotin synthetase–diaminopelargonic acid aminotransferase: evidence for substrate channeling in biotin synthesis. Plant Cell 2012, 24, 1608-‐1625.
[162] Baker, P. R. S.; Cramer, S. D.; Kennedy, M.; Assimos, D. G.; Holmes, R. P., Glycolate and glyoxylate metabolism in HepG2 cells. Am J Physiol, Cell Physiol 2004, 287, C1359-‐C1365.
[163] Kaneko, M.; Fujimoto, S.; Kikugawa, M.; Tamaki, N., Irreversible inhibition of D-‐3-‐aminoisobutyrate-‐pyruvate aminotransferase by gabaculine. FEBS Lett 1990, 276, 115-‐118.
[164] Lee, I. S. M.; Muragaki, Y.; Ideguchi, T.; Hase, T.; Tsuji, M.; Ooshima, A.; Okuno, E.; Kido, R., Molecular cloning and sequencing of a cDNA encoding alanine-‐glyoxylate aminotransferase 2 from rat kidney. J Biochem 1995, 117, 856-‐862.
[165] Tamaki, N.; Kaneko, M.; Mizota, C.; Kikugawa, M.; Fujimoto, S., Purification, characterization and inhibition of D-‐3-‐aminoisobutyrate aminotransferase from the rat liver. Eur J Biochem 1990, 189, 39-‐45.
[166] Kontani, Y.; Kaneko, M.; Kikugawa, M.; Fujimoto, S.; Tamaki, N., Identity of D-‐3-‐aminoisobutyrate-‐pyruvate aminotransferase with alanine-‐glyoxylate aminotransferase 2. Biochim Biophys Acta 1993, 1156, 161-‐166.

33
[167] Caplin, B.; Wang, Z.; Slaviero, A.; Tomlinson, J.; Dowsett, L.; Delahaye, M.; Salama, A.; Studies, T. I. C. f. B. P. G.-‐W. A.; Wheeler, D. C.; Leiper, J., Alanine-‐glyoxylate aminotransferase-‐2 metabolizes endogenous methylarginines, regulates NO, and controls blood pressure. Arterioscler Thromb Vasc Biol 2012, 32, 2892-‐2900.
[168] Varticovski, L.; Kushner, J. P.; Burnham, B. F., Biosynthesis of porphyrin precursors. Purification and characterization of mammalian L-‐alanine:γ,δ-‐dioxovaleric acid aminotransferase. J Biol Chem 1980, 255, 3742-‐3747.
[169] Ades, I. Z., Characterization of chicken liver L-‐alanine: 4,5-‐dioxovaleric acid aminotransferase. Int J Biochem 1989, 21, 679-‐687.
[170] Liepman, A. H.; Olsen, L. J., Alanine aminotransferase homologs catalyze the glutamate:glyoxylate aminotransferase reaction in peroxisomes of arabidopsis. Plant Physiol 2003, 131, 215-‐227.
[171] Hennig, M.; Grimm, B.; Contestabile, R.; John, R. A.; Jansonius, J. N., Crystal structure of glutamate-‐1-‐semialdehyde aminomutase: An α2-‐dimeric vitamin B6-‐dependent enzyme with asymmetry in structure and active site reactivity. Proc Natl Acad Sci USA 1997, 94, 4866-‐4871.
[172] Orriss, G. L.; Patel, T. R.; Sorensen, J.; Stetefeld, J., Absence of a catalytic water confers resistance to the neurotoxin gabaculine. FASEB J 2010, 24, 404-‐414.
[173] Grimm, B.; Smith, A. J.; Kannangara, C. G.; Smith, M., Gabaculine-‐resistant glutamate 1-‐semialdehyde aminotransferase of Synechococcus . Deletion of a tripeptide close to the NH2 terminus and internal amino acid substitution. J Biol Chem 1991, 266, 12495-‐12501.
[174] Bishop, K.; Gough, K.; Mahoney, S.; Smith, A.; Rogers, L., Synechococcus mutants resistant to an enamine mechanism inhibitor of glutamate-‐1-‐semialdehyde aminotransferase. FEBS Lett 1999, 450, 57-‐60.
[175] Contestabile, R.; Angelaccio, S.; Maytum, R.; Bossa, F.; John, R. A., The contribution of a conformationally mobile, active site loop to the reaction catalyzed by glutamate semialdehyde aminomutase. J Biol Chem 2000, 275, 3879-‐3886.
[176] Grimm, B.; Bull, A.; Breu, V., Structural genes of glutamate 1-‐semialdehyde aminotransferase for porphyrin synthesis in a cyanobacterium and Escherichia coli. Mol Gen Genet 1991, 225, 1-‐10.
[177] Stetefeld, J.; Jenny, M.; Burkhard, P., Intersubunit signaling in glutamate-‐1-‐semialdehyde-‐aminomutase. Proc Natl Acad Sci USA 2006, 103, 13688-‐13693.
[178] Schulze, J. O.; Schubert, W. D.; Moser, J.; Jahn, D.; Heinz, D. W., Evolutionary relationship between initial enzymes of tetrapyrrole biosynthesis. J Mol Biol 2006, 358, 1212-‐1220.
[179] Ilag, L. L.; Kumar, A. M.; Söll, D., Light regulation of chlorophyll biosynthesis at the level of 5-‐aminolevulinate formation in Arabidopsis. Plant Cell 1994, 6, 265-‐275.
[180] Grimm, B., Primary structure of a key enzyme in plant tetrapyrrole synthesis: glutamate 1-‐semialdehyde aminotransferase. Proc Natl Acad Sci USA 1990, 87, 4169-‐4173.
[181] Ge, H.; Lv, X.; Fan, J.; Gao, Y.; Teng, M.; Niu, L., Crystal structure of glutamate1-‐semialdehyde aminotransferase from Bacillus subtilis with bound pyridoxamine-‐5ʹ′-‐phosphate. Biochem Biophys Res Commun 2010, 402, 356-‐360.
[182] Hansson, M.; Rutberg, L.; Schroder, I.; Hederstedt, L., The Bacillus subtilis hemAXCDBL gene cluster, which encodes enzymes of the biosynthetic pathway from glutamate to uroporphyrinogen III. J Bacteriol 1991, 173, 2590-‐2599.
[183] Ferradini, N.; Nicolia, A.; Capomaccio, S.; Veronesi, F.; Rosellini, D., A point mutation in the Medicago sativa GSA gene provides a novel, efficient, selectable marker for plant genetic engineering. J Biotechnol 2011, 156, 147-‐152.
[184] Ilag, L. L.; Jahn, D.; Eggertsson, G.; Söll, D., The Escherichia coli hemL gene encodes glutamate 1-‐semialdehyde aminotransferase. J Bacteriol 1991, 173, 3408-‐3413.
[185] Ilag, L. L.; Jahn, D., Activity and spectroscopic properties of the Escherichia coli glutamate 1-‐semialdehyde aminotransferase and the putative active site mutant K265R. Biochemistry 1992, 31, 7143-‐7151.
[186] Elliott, T.; Avissar, Y. J.; Rhie, G. E.; Beale, S. I., Cloning and sequence of the Salmonella typhimurium hemL gene and identification of the missing enzyme in hemL mutants as glutamate-‐1-‐semialdehyde aminotransferase. J Bacteriol 1990, 172, 7071-‐7084.
[187] Sangwan, I.; O'Brian, M. R., Expression of the soybean (Glycine max) glutamate 1-‐semialdehyde aminotransferase gene in symbiotic root nodules. Plant Physiol 1993, 102, 829-‐834.
[188] Tsang, E. W. T.; Hu, Z.; Chang, Q.; McGregor, D. I.; Keller, W. A., Expression of a Brassic napus glutamate 1-‐semialdehyde aminotransferase in Escherichia coli and characterization of the recombinant protein. Protein Expr Purif 2003, 29, 193-‐201.

34
[189] Wang, W.-‐Y.; Huang, D.-‐D.; Stachon, D.; Simon, P. G.; Kannangara, C. G., Purification, characterization, and fractionation of the δ-‐aminolevulinic acid synthesizing enzymes from light-‐grown Chlamydomonas reinhardtii cells. Plant Physiol 1984, 74, 569-‐575.
[190] Mau, Y. H. L.; Wang, W. Y., Biosynthesis of δ-‐aminolevulinic acid in Chlamydomonas reinhardtii study of the transamination mechanism using specifically labeled glutamate. Plant Physiol 1988, 86, 793-‐797.
[191] Jahn, D.; Chen, M. W.; Soll, D., Purification and functional characterization of glutamate-‐1-‐semialdehyde aminotransferase from Chlamydomonas reinhardtii. J Biol Chem 1991, 266, 161-‐167.
[192] Matters, G. L.; Beale, S. I., Structure and light-‐regulated expression of the gsa gene encoding the chlorophyll biosynthetic enzyme, glutamate 1-‐semialdehyde aminotransferase, in Chlamydomonas reinhardtii. Plant Mol Biol 1994, 24, 617-‐629.
[193] Murakami, K.; Hashimoto, Y.; Murooka, Y., Cloning and characterization of the gene encoding glutamate 1-‐semialdehyde 2,1-‐aminomutase, which is involved in δ-‐aminolevulinic acid synthesis in Propionibacterium freudenreichii. Appl Environ Microbiol 1993, 59, 347-‐350.
[194] Matters, G. L.; Beale, S. I., Biosynthesis of δ-‐aminolevulinic acid from glutamate by Sulfolobus solfataricus. Arch Microbiol 1994, 161, 272-‐276.
[195] Palmieri, G.; Di Palo, M.; Scaloni, A.; Orru, S.; Marino, G.; Sannia, G., Glutamate-‐1-‐semialdehyde aminotransferase from Sulfolobus solfataricus. Biochem J 1996, 320 ( Pt 2), 541-‐545.
[196] Murakami, K.; Korbsrisate, S.; Asahara, N.; Hashimoto, Y.; Murooka, Y., Cloning and characterization of the glutamate 1-‐semialdehyde aminomutase gene from Xanthomonas campestris pv. phaseoli. Appl Microbiol Biotechnol 1993, 38, 502-‐506.
[197] Sun, S.; Zabinski, R. F.; Toney, M. D., Reactions of alternate substrates demonstrate stereoelectronic control of reactivity in dialkylglycine decarboxylase. Biochemistry 1998, 37, 3865-‐3875.
[198] Adachi, K.; Nelson, G. H.; Peoples, K. A.; DeZwaan, T. M.; Skalchunes, A. R.; Heiniger, R. W.; Shuster, J. R.; Hamer, L.; Tanzer, M. M., Sequence analysis and functional characterization of the dialkylglycine decarboxylase gene DGD1 from Mycosphaerella graminicola. Curr Genet 2003, 43, 358-‐363.
[199] Okazaki, S.; Suzuki, A.; Mizushima, T.; Kawano, T.; Komeda, H.; Asano, Y.; Yamane, T., The novel structure of a pyridoxal 5ʹ′-‐phosphate-‐dependent fold-‐type I racemase, α-‐amino-‐ε-‐caprolactam racemase from Achromobacter obae. Biochemistry 2009, 48, 941-‐950.
[200] Boesten, W. H. J.; Raemakers-‐Franken, P. C.; Sonke, T.; Eu-‐Verink, G. J. W.; Grijpstra, P. Polypeptides having α-‐H-‐α-‐amino acid amide racemase activity and nucleic acids encoding the same. WO03106691, 2003.
[201] Mutaguchi, Y.; Ohmori, T.; Wakamatsu, T.; Doi, K.; Ohshima, T., Identification, purification, and characterization of a novel amino acid racemase, isoleucine 2-‐epimerase, from Lactobacillus species. J Bacteriol 2013, 195, 5207-‐5215.
[202] Huang, F.; Spiteller, D.; Koorbanally, N. A.; Li, Y.; Llewellyn, N. M.; Spencer, J. B., Elaboration of neosamine rings in the biosynthesis of neomycin and butirosin. ChemBioChem 2007, 8, 283-‐288.
[203] Clausnitzer, D.; Piepersberg, W.; Wehmeier, U. F., The oxidoreductases LivQ and NeoQ are responsible for the different 6ʹ′-‐modifications in the aminoglycosides lividomycin and neomycin. J Appl Microbiol 2011, 111, 642-‐651.
[204] Park, J. W.; Park, S. R.; Nepal, K. K.; Han, A. R.; Ban, Y. H.; Yoo, Y. J.; Kim, E. J.; Kim, E. M.; Kim, D.; Sohng, J. K.; Yoon, Y. J., Discovery of parallel pathways of kanamycin biosynthesis allows antibiotic manipulation. Nat Chem Biol 2011, 7, 843-‐852.
[205] Hartinger, D.; Schwartz, H.; Hametner, C.; Schatzmayr, G.; Haltrich, D.; Moll, W. D., Enzyme characteristics of aminotransferase FumI of Sphingopyxis sp. MTA144 for deamination of hydrolyzed fumonisin B1. Appl Microbiol Biotechnol 2011, 91, 757-‐768.
[206] Heinl, S.; Hartinger, D.; Thamhesl, M.; Schatzmayr, G.; Moll, W. D.; Grabherr, R., An aminotransferase from bacterium ATCC 55552 deaminates hydrolyzed fumonisin B1. Biodegradation 2011, 22, 25-‐30.
[207] Leslie, H.; Paul, W. D.; Lishan, Z. Transaminases, deaminases and aminomutases and compositions and methods for enzymatic detoxification. WO2004085624A2, 2004.
[208] Schiroli, D.; Cirrincione, S.; Donini, S.; Peracchi, A., Strict reaction and substrate specificity of AGXT2L1, the human O-‐phosphoethanolamine phospho-‐lyase. IUBMB Life 2013, 65, 645-‐650.
[209] Veiga-‐da-‐Cunha, M.; Hadi, F.; Balligand, T.; Stroobant, V.; Van Schaftingen, E., Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-‐phosphohydroxy-‐L-‐lysine and phosphoethanolamine. J Biol Chem 2012, 287, 7246-‐7255.

35
[210] Aron, Z. D.; Dorrestein, P. C.; Blackhall, J. R.; Kelleher, N. L.; Walsh, C. T., Characterization of a new tailoring domain in polyketide biogenesis: the amine transferase domain of MycA in the mycosubtilin gene cluster. J Am Chem Soc 2005, 127, 14986-‐14987.


Affirmation
Hiermit erkläre ich, dass diese Arbeit bisher von mir weder an der Mathematisch-
Naturwissenschaftlichen Fakultät der Ernst-Moritz-Arndt-Universität Greifswald noch einer an-
deren wissenschaftlichen Einrichtung zum Zwecke der Promotion eingereicht wurde.
Ferner erkläre ich, dass ich diese Arbeit selbstständig verfasst und keine anderen als die darin
angegebenen Hilfsmittel und Hilfen benutzt und keine Textabschnitte eines Dritten ohne Kenn-
zeichnung übernommen habe.
Hannes Kohls

Curriculum vitae
PhD in Biochemistry
Since April 2012 PhD thesis entitled ‘Biocatalytic Synthesis of Amino Alcohols’ in the re-
search groups of Prof. Uwe Bornscheuer, Dept. of Biotechnology & Enzyme
Catalysis and Jun. Prof. Matthias Höhne, Dept. of Protein Biochemistry.
Both at the Institute of Biochemistry, Ernst-Moritz-Arndt-University,
Greifswald, Germany
May 2012 - June
2012
Research internship in the group of Marko Mihovilovic, Institute of Ap-
plied Synthetic Chemistry, TU Wien, Vienna, Austria
Diploma in Biochemistry
May 2011 – Feb-
ruary 2012
Diploma thesis entitled ‘Investigation of (R)-Selective Amine Transaminas-
es’ in the research group of Prof. Dr. Uwe Bornscheuer (Dept. of Biotech-
nology & Enzyme Catalysis) at the Institute of Biochemistry, Ernst-Moritz-
Arndt-University, Greifswald, Germany
March 2010 –
April 2010
Industrial placement in the Research & Development department at TIB
MOLBIOL GmbH, Berlin, Germany
November 2009 –
January 2010
Research assistant for Prof. Uwe Bornscheuer at the Dept. of Biotechnol-
ogy & Enzyme Catalysis at the Institute of Biochemistry, Ernst-Moritz-Arndt-
University, Greifswald, Germany
Stay Abroad
August 2005 –
April 2006
Deepening of communication skills in English language during a stay
abroad in New Zealand
Secondary School
August 2000 –
July 2005
Lessing Gymnasium, Hoyerswerda, Germany

Scientific Publications
Research Papers Journal
2015 Selective Access to All Four Diastereomers of a
1,3-Amino Alcohol by Combination of a Keto Re-
ductase- and an Amine Transaminase-Catalysed
Reaction
Kohls, H; Anderson, M; Dickerhoff, J; Weisz, K;
Córdova, A; Berglund, P; Brundiek, H; Bornscheuer,
UT; Höhne, M
Advanced Synthesis & Catalysis
DOI: 10.1002/adsc.201500214
2015 Engineering the Active Site of the Amine Trans-
aminase from Vibrio fluvialis for the Asymmetric
Synthesis of Aryl-Alkyl Amines and Amino Alco-
hols
Nobili, A; Steffen-Munsberg, F; Kohls, H; Trentin, I;
Schulzke, C; Höhne, M; Bornscheuer, UT
ChemCatChem
DOI: 10.1002/cctc.201403010
Reviews
2015 Bioinformatic Analysis of a PLP-dependent En-
zyme Superfamily Suitable for Biocatalytic Ap-
plications
Steffen-Munsberg, F; Vickers, C; Kohls, H; Land, H;
Mallin, H; Nobili, A; Skalden, L; van den Bergh, T;
Joosten, H-J; Berglund, P; Höhne, M; Bornscheuer,
UT
Biotechnology Advances
DOI:
10.1016/j.biotechadv.2014.12.012
2014 Recent Achievements in Developing the Biocata-
lytic Toolbox for Chiral Amine Synthesis
Kohls, H; Steffen-Munsberg, F; Höhne, M
Current Opinion in Chemical
Biology
DOI: 10.1016/j.cbpa.2014.02.021
Conference Contributions (Posters) Conference
2015 Selective Access to All Four Diastereomers of an
1,3-Amino Alcohol by Combination of a Keto Re-
ductase- and an Amine Transaminase-Catalysed
Reaction
Transam 2.0 - Chiral Amines
Through (Bio)Catalysis,
Greifswald, Germany
2014 Synthesis of Optically Pure Diastereomers with
an 1,3-Amino Alcohol Motif Using an Enzyme
Cascade Reaction
3rd
Multistep Enzyme Catalyzed
Processes (MECP14), Madrid,
Spain

Acknowledgements
Mein erstes und besonderes Dankeschön gilt Uwe und Matthias. Vielen Dank für Euer Vertrau-
en in meine Fähigkeiten und die unzähligen Freiheiten, dir ihr mir gewährt habt. Ich schätze
mich glücklich, von Euch so viel gelernt zu haben. Ich bin sehr dankbar dafür, dass ihr mir den
Auslandsaufenthalt in Wien ermöglicht habt. Dort konnte ich so viele neue Techniken und inte-
ressante Menschen kennen lernen (die Stadt war auch nicht schlecht). An diese Zeit werde ich
mich sicher noch oft mit viel Freude erinnern. Ganz besonders froh war ich, als ihr beide mir
nicht nur eine längere Auszeit, sondern auch Eure Unterstützung zugesichert habt, als es für
mich ganz besonders wichtig war! Sehr schön fand ich auch den gelegentlich Segelschnack und
Segeltörn mit Dir, Uwe. Es war stets ein schönes Erlebnis mit Dir gemeinsam auf dem Wasser zu
sein. Mittwochs gefiel mir Dein Boot besonders gut in meinem Kielwasser :P Dankbar bin ich
dafür, dass ich auch Dich, Matthias, immer besser kennen lernen konnte. Es hat mich sehr ge-
freut, unsere gemeinsamen privaten Interessen zu entdecken. Das hat mich bestärkt und ich
konnte viel davon profitieren.
Weiterhin möchte ich mich bei den Mitarbeitern des Instituts für Angewandte Synthese
Chemie der TU-Wien bedanken. Im Besonderen Marko Mihovilovic, Florian Rudroff und Michael
Fink, welche mich so herzlich in die Arbeitsgruppe eingeführt haben. Sie haben sich nichts aus
dem „Bio“ vor dem Chemiker gemacht und meine ersten Gehversuche im Bereich der organi-
schen Synthese sehr hilfreich und immer vorbehaltslos unterstützt.
Ich möchte mich auch bei Prof. Klaus Weisz und Jonathan, sowie bei Prof. Per Berglund und
Mattias Anderson für die gute Zusammenarbeit bedanken.
Ein großes Dankeschön gilt allen Mitgliedern der Transaminase Crew: Fabian, Alberto, Ioan-
nis, Lilly, Hendrik und Clare – vielen Dank für die großartige Zusammenarbeit. Weiterhin möchte
ich mich natürlich auch bei allen Mitarbeitern des AK Bornscheuer und des AK Höhne bedan-
ken! Besonderer Dank geht an Maria, Daniel, Gandi, Christin und Jan für das konstruktive und
wohlwollende Miteinander sowie dem gelegentlichen gemeinsamen Kaffee. Anita, Ina und An-
gelika danke ich für die Hilfe und Unterstützung, wenn es um Synthesen, Analytik und die Be-
stellungen von Chemikalien ging. Weiterhin möchte ich mich bei Florian, Thomas, Ayad, Anders,
Maika und Lisa bedanken. Schön, dass ihr nach Greifswald gekommen seid. Jedes Käffchen mit
Euch war mir ein wahrer Genuss!
Weiterhin hatte ich die besten Kommilitonen, die man sich nur wünschen kann. Ohne Eure
Unterstützung wäre ich nie an diesem Punkt angelangt. Vielen Dank Sarah, Theresa, Sandra,
Tom und Fabian, dass ich die schöne Zeit des Studentenlebens mit Euch verbringen konnte!
Ganz besonders dankbar bin ich meiner lieben Familie. Mama, Papa, Schwesterlein, alle sol-
len gemeint sein, Oma, Opa vielen Dank; ohne euch wär ich längst blank ;) Vielen, vielen Dank
für Eure jahrelange Unterstützung, Euer Vertrauen in mich und die schöne Zeit, die wir zusam-
men verbracht haben!
Zu guter Letzt möchte ich mich besonders bei Philine bedanken. Sie hat mir in den schwie-
rigsten Zeiten dieser Arbeit zur Seite gestanden und es schien, als wäre es völlig selbstverständ-
lich, mir stets den Rücken frei zu halten. Vielen, vielen Dank, dass du mir immer wieder Mut
gemacht hast! Ohne Dich hätte ich es womöglich nicht geschafft.
Related Documents











