Emergence of a novel lineage genetically divergent from the predominant Ind2001 lineage of serotype O foot-and-mouth disease virus in India Saravanan Subramaniam, Aniket Sanyal, Jajati K. Mohapatra, Gaurav K. Sharma, Jitendra K. Biswal, Rajeev Ranjan, Manoranjan Rout, Biswajit Das, Punam Bisht, Basavaraj S. Mathapati, Bana B. Dash, Bramhadev Pattnaik ⇑ Project Directorate on Foot-and-Mouth Disease, Mukteswar-Kumaon, Nainital 263138, Uttarakhand, India article info Article history: Received 14 August 2012 Received in revised form 19 April 2013 Accepted 22 April 2013 Available online 3 May 2013 Keywords: FMDV serotype O VP1 coding region Lineage Ind2001 Ind2011 abstract In India, emergence of Ind2001 lineage of foot-and-mouth disease virus (FMDV) serotype O was recorded in the year 2001. After causing sporadic incidences, the Ind2001 lineage that re-surged in 2008 out-com- peted PanAsia from the field during 2009 and continued its dominance during 2010 and 2011 as well. The lineage has diversified in due course of time, leading to two sub-lineages (Ind2001a and Ind2001b). The sub-lineage Ind2001a include isolates collected during 2001–2002 and sub-lineage Ind2001b is consti- tuted largely by isolates collected during 2008–2012. The nucleotide substitution rate of sub-lineage Ind2001b was estimated at 6.58 10 3 substitutions/site/year. The most stable PanAsia lineage is restricted only to few outbreaks. During 2011, emergence of a new genetic group with >9% nucleotide divergence from rest of the lineages circulating in the country was detected and named as lineage Ind2011. Two specific amino acid substitutions at positions VP1–36 (F) and VP2–133 (T) were observed in the Ind2011 lineage. The new lineage at present is restricted only to southern states of the country. It is uncertain whether the emergence was triggered by immune pressure or due to a bottleneck in transmis- sion or selected for higher fitness value. Six sites (4, 68, 83, 135, 138 and 209) in VP1 protein were iden- tified to undergo episodic diversifying selection in serotype O field isolates. Both emerging and re- emerging lineages had appropriate antigenic match with currently used vaccine strain, INDR2/1975. Irre- spective of genetic variability, the field isolates showed remarkable conservation at antigenically critical residues that might contribute to the observed antigenic stability. With the emergence of a new genetic group after a span of 10 years, the overall epidemiological scenario in the region is expected to change in the coming years. Ó 2013 Elsevier B.V. All rights reserved. 1. Introduction Foot-and-mouth disease (FMD) is a highly infectious and conta- gious disease of transboundary importance affecting cloven hoofed animals. India with its huge FMD susceptible livestock population (528 million, Department of Animal Husbandry, Dairying and Fish- eries, Government of India, 2007) suffers heavy economic losses due to endemicity of this dreaded disease. The causative agent, FMD Virus (FMDV) is a single stranded positive sense RNA virus and its 8.5 kb length genome is enclosed in a highly structured icosahedral capsid. FMDV is classified in the genus Aphthovirus of the family Picornaviridae (Racaniello et al., 2001). Currently six ser- otypes viz; O, A, Asia1 and Southern African Territories (SAT 1–3) are prevalent in the world. The last serotype C outbreak was in the Amazon region of Brazil in the year 2004 (WRL-FMD, 2011). Three serotypes including O, A and Asia1 are prevalent in South Asia; approximately 80% of the FMD outbreaks in India are attrib- uted to serotype O (Subramaniam et al., 2012). Comparison of the nucleotide sequence of VP1 coding region forms the basis for genetic classification of FMDV as topotype, line- age and sub-lineage within the serotype (Samuel and Knowles, 2001). Globally, in serotype O, 11 geographically restricted topo- types namely Europe-South America, Middle East-South Asia, South East Asia, Cathay (China and East Tartary), Indonesia (ISA)- 1, ISA-2, East Africa (EA)-1, EA-2, EA-3, EA-4, and West Africa have been identified (Samuel and Knowles, 2001; Ayelet et al., 2009). The serotype O isolates collected in India were found to be of var- ious lineages within the ME-SA topotype (Hemadri et al., 2002). The first appearance of Ind2001 lineage was reported in the year 2001 (Hemadri et al., 2002). The Ind2001 lineage after causing sporadic cases during 2003–2005, re-surged in 2008 and out- competed the then dominant PanAsia lineage in 2009 (Das et al., 2012). The pandemic PanAsia strain was detected as early as 1567-1348/$ - see front matter Ó 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.meegid.2013.04.027 ⇑ Corresponding author. Tel.: +91 5942 286004; fax: +91 5942 286307. E-mail address: [email protected] (B. Pattnaik). Infection, Genetics and Evolution 18 (2013) 1–7 Contents lists available at SciVerse ScienceDirect Infection, Genetics and Evolution journal homepage: www.elsevier.com/locate/meegid

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Infection, Genetics and Evolution 18 (2013) 1–7
Contents lists available at SciVerse ScienceDirect
Infection, Genetics and Evolution
journal homepage: www.elsevier .com/locate /meegid
Emergence of a novel lineage genetically divergent from the predominantInd2001 lineage of serotype O foot-and-mouth disease virus in India
Saravanan Subramaniam, Aniket Sanyal, Jajati K. Mohapatra, Gaurav K. Sharma, Jitendra K. Biswal,Rajeev Ranjan, Manoranjan Rout, Biswajit Das, Punam Bisht, Basavaraj S. Mathapati, Bana B. Dash,Bramhadev Pattnaik ⇑Project Directorate on Foot-and-Mouth Disease, Mukteswar-Kumaon, Nainital 263138, Uttarakhand, India
a r t i c l e i n f o a b s t r a c t
Article history:Received 14 August 2012Received in revised form 19 April 2013Accepted 22 April 2013Available online 3 May 2013
Keywords:FMDV serotype OVP1 coding regionLineageInd2001Ind2011
1567-1348/$ - see front matter � 2013 Elsevier B.V. Ahttp://dx.doi.org/10.1016/j.meegid.2013.04.027
⇑ Corresponding author. Tel.: +91 5942 286004; faxE-mail address: [email protected] (B. Pattnaik
In India, emergence of Ind2001 lineage of foot-and-mouth disease virus (FMDV) serotype O was recordedin the year 2001. After causing sporadic incidences, the Ind2001 lineage that re-surged in 2008 out-com-peted PanAsia from the field during 2009 and continued its dominance during 2010 and 2011 as well. Thelineage has diversified in due course of time, leading to two sub-lineages (Ind2001a and Ind2001b). Thesub-lineage Ind2001a include isolates collected during 2001–2002 and sub-lineage Ind2001b is consti-tuted largely by isolates collected during 2008–2012. The nucleotide substitution rate of sub-lineageInd2001b was estimated at 6.58 � 10�3 substitutions/site/year. The most stable PanAsia lineage isrestricted only to few outbreaks. During 2011, emergence of a new genetic group with >9% nucleotidedivergence from rest of the lineages circulating in the country was detected and named as lineageInd2011. Two specific amino acid substitutions at positions VP1–36 (F) and VP2–133 (T) were observedin the Ind2011 lineage. The new lineage at present is restricted only to southern states of the country. It isuncertain whether the emergence was triggered by immune pressure or due to a bottleneck in transmis-sion or selected for higher fitness value. Six sites (4, 68, 83, 135, 138 and 209) in VP1 protein were iden-tified to undergo episodic diversifying selection in serotype O field isolates. Both emerging and re-emerging lineages had appropriate antigenic match with currently used vaccine strain, INDR2/1975. Irre-spective of genetic variability, the field isolates showed remarkable conservation at antigenically criticalresidues that might contribute to the observed antigenic stability. With the emergence of a new geneticgroup after a span of 10 years, the overall epidemiological scenario in the region is expected to change inthe coming years.
� 2013 Elsevier B.V. All rights reserved.
1. Introduction
Foot-and-mouth disease (FMD) is a highly infectious and conta-gious disease of transboundary importance affecting cloven hoofedanimals. India with its huge FMD susceptible livestock population(528 million, Department of Animal Husbandry, Dairying and Fish-eries, Government of India, 2007) suffers heavy economic lossesdue to endemicity of this dreaded disease. The causative agent,FMD Virus (FMDV) is a single stranded positive sense RNA virusand its �8.5 kb length genome is enclosed in a highly structuredicosahedral capsid. FMDV is classified in the genus Aphthovirus ofthe family Picornaviridae (Racaniello et al., 2001). Currently six ser-otypes viz; O, A, Asia1 and Southern African Territories (SAT 1–3)are prevalent in the world. The last serotype C outbreak was inthe Amazon region of Brazil in the year 2004 (WRL-FMD, 2011).
ll rights reserved.
: +91 5942 286307.).
Three serotypes including O, A and Asia1 are prevalent in SouthAsia; approximately 80% of the FMD outbreaks in India are attrib-uted to serotype O (Subramaniam et al., 2012).
Comparison of the nucleotide sequence of VP1 coding regionforms the basis for genetic classification of FMDV as topotype, line-age and sub-lineage within the serotype (Samuel and Knowles,2001). Globally, in serotype O, 11 geographically restricted topo-types namely Europe-South America, Middle East-South Asia,South East Asia, Cathay (China and East Tartary), Indonesia (ISA)-1, ISA-2, East Africa (EA)-1, EA-2, EA-3, EA-4, and West Africa havebeen identified (Samuel and Knowles, 2001; Ayelet et al., 2009).The serotype O isolates collected in India were found to be of var-ious lineages within the ME-SA topotype (Hemadri et al., 2002).The first appearance of Ind2001 lineage was reported in the year2001 (Hemadri et al., 2002). The Ind2001 lineage after causingsporadic cases during 2003–2005, re-surged in 2008 and out-competed the then dominant PanAsia lineage in 2009 (Das et al.,2012). The pandemic PanAsia strain was detected as early as

2 S. Subramaniam et al. / Infection, Genetics and Evolution 18 (2013) 1–7
1982 in India and was responsible for most of the outbreaks be-tween 1996 and 2008 (Hemadri et al., 2002; Subramaniam et al.,2012). The PanAsia lineage has also established itself in South Asiaas the most dominant lineage within ME-SA topotype (Knowleset al., 2005). Vaccination based control programme is being con-stantly challenged by the emergence of new strains as sometimesemerging genotypes/lineages may be antigenically divergent(Mohapatra et al., 2008). In this study, a total of 141 serotype Ofield outbreak viruses collected during 2009–2012 were character-ized genetically at VP1 coding region and 35 isolates were evalu-ated antigenically using Bovine Vaccinate Serum (BVS) againstthe currently used serotype O Indian vaccine strain INDR2/1975.The study revealed emergence of a new genetic group henceforthnamed as Ind2011 in the year 2011 after a gap of 10 years sinceInd2001 lineage was identified. To get more insight, four isolatesof Ind2011 lineage were also sequenced at complete capsid codingregion.
2. Materials and methods
2.1. Sample processing and sequencing
A total of 141 serotype O FMD field outbreak viruses were usedin this study (Supplementary Table). The samples were processed(chloroform extracted 10% PBS suspension) and serotype con-firmed using sandwich ELISA and multiplex PCR. Virus isolationwas attempted in BHK-21 cell monolayer. Total RNA was extractedeither directly from the clinical materials or infected BHK-21 cellculture supernatants (2–3 passages) using RNeasy Mini Kit (QIA-GEN). The genomic RNA was reverse transcribed using M-MLV Re-verse transcriptase (Promega) and oligo d(T)15 primer. PCRamplification of VP1 and P1 coding regions were carried out usingPfu DNA polymerase (Fermentas) essentially as described earlier(Mittal et al., 2005; Hemadri et al., 2002) and the PCR productswere purified using QIA quick Gel Extraction Kit (QIAGEN). Cyclesequencing reactions were carried out on PCR products using Big-dyeV3.1 terminator kit and majority population sequence was re-solved on ABI 3130 genetic analyzer (Applied Biosystems).
2.2. Sequence analysis
Nucleotide sequences were processed using ABI sequence anal-ysis v5.3.1 software, and consensus was assembled from sequencesof both the strands using EditSeq programme of Lasergene coresuite 10 (DNASTAR, Inc., USA). Sequences were aligned using clus-tal W algorithm (Thompson et al., 1994). Phylogenetic analysis wasconducted using MEGA 5.05 software (Tamura et al., 2011)employing the best fit nucleotide substitution model, TN93 + G + I.The gamma shape parameter was estimated to be 1.83. Phyloge-netic tree was reconstructed using maximum likelihood (ML)method employing above mentioned parameters. Percent nucleo-tide and amino acid divergence was calculated using Megalignmodule of Lasergene core suite 10 (DNASTAR, Inc., USA). Selectionpressure analysis was performed on 115 sequences (26 similar se-quences were removed) using datamonkey web interface (Delportet al., 2010) which uses HyPhy (Hyphothesis testing using Phylog-eny) package (Pond et al., 2005) as its computational engine. Fourcomplementary methods; Single likelihood ancestor counting(SLAC), Fixed effects likelihood (FEL), Internal branch FEL (IFEL)and Mixed effects model of evolution (MEME) were used. Analysiswas done using TrN93 substitution model with a cut-off p value of<0.1 on neighbour-joining phylogenetic tree. The rate of evolution(substitution rate) was calculated by linear regression. For this,total nucleotide substitutions were estimated using Kimura2-parameter model (Kimura, 1980) implemented in MEGA 5.05.
The genetic distance of each isolates from the oldest one in thecluster and the time of isolation (year) were plotted along the Yand X axis, respectively. The nucleotide sequences of FMDV isolatesgenerated in the study are available at GenBank under accessionnumbers KC506424–KC506564. The other FMDV serotype O se-quences used in phylogenetic comparison and evolutionary ratecalculation were obtained from GenBank and Genetic database ofProject Directorate on FMD, Mukteswar.
2.3. Antigenic analysis
Two dimensional micro-neutralization test (2D-MNT) was per-formed as described earlier (Rweyemamu et al., 1978) using BVSagainst the currently used vaccine strain (INDR2/1975). BHK 21cells were used as indicator system in the neutralization test. Theend point titre of the serum was calculated as the reciprocal ofthe last dilution of serum that neutralizes 100TCID50 in 50% ofthe wells. One way antigenic relationship (r1-value) was calculatedas the ratio between heterologous and homologous serum titre.The test was repeated three times and the titre was averaged forcalculation of r1-value.
3. Results and discussion
In India, FMD Control Programme (FMDCP) was launched in theyear 2003–04 in 54 districts and later in 2010–11, expanded to 221districts with the aim of creating Disease Free Zones (DFZs)(Pattnaik et al., 2012). Inactivated trivalent oil adjuvanted vaccineformulation containing strains of serotypes O, A and Asia1 has beenused in the region for prophylactic six monthly vaccinations.Efforts to create DFZs are often threatened by unrestricted animalmovement across the interstate borders. Serotype O isolatescollected to date in the country belong to ME-SA topotype. Therehas been circulation of two prominent lineages during the last10–12 years; PanAsia and Ind2001. The VP1 region sequence basedmolecular epidemiology provides important insights into virusevolution and virus movements. Antigenic profiling of the fieldviruses in relation to vaccine strain provides a measure to assessits suitability in an ever changing epidemiological scenario. Inthe present analysis, we have sequenced the VP1 coding regionof recent FMDV serotype O isolates collected from 141 outbreaksand also carried out antigenic matching exercise for 35 fieldisolates with vaccine strain, INDR2/1975.
3.1. Phylogenetic relationships
A maximum of 15.6% nucleotide divergence was observedamong Indian serotype O field isolates in contrast to an earlier re-port, where a maximum divergence of 11% was noted (Hemadriet al., 2002). The ML tree depicting phylogenetic relationships ofserotypes O isolates is indicated in the Figs. 1 and 2. Distributionof serotype O lineages in different states of the country is depictedin the Fig. 3. Earlier we reported the dominance of Ind2001 lineageover PanAsia during 2009 (Das et al., 2012). In this study, maxi-mum number of isolates (n = 95) clustered within the Ind2001lineage indicating its continued dominance in the field during2010 and 2011 as well. This lineage has been in circulation in thecountry since 2001; with time it has diversified and formed twosub-lineages. One that largely includes isolates collected during2001–2002 is named as Ind2001a, and another cluster that con-tains mainly isolates collected after 2008 designated as Ind2001b.Overall, a maximum of 10.6% and 11.4% divergence at nucleotideand amino acid level, respectively was observed within theInd2001 lineage. The lineage is distributed widely in as many as20 states. Within Ind2001 lineage, very high genetic diversity
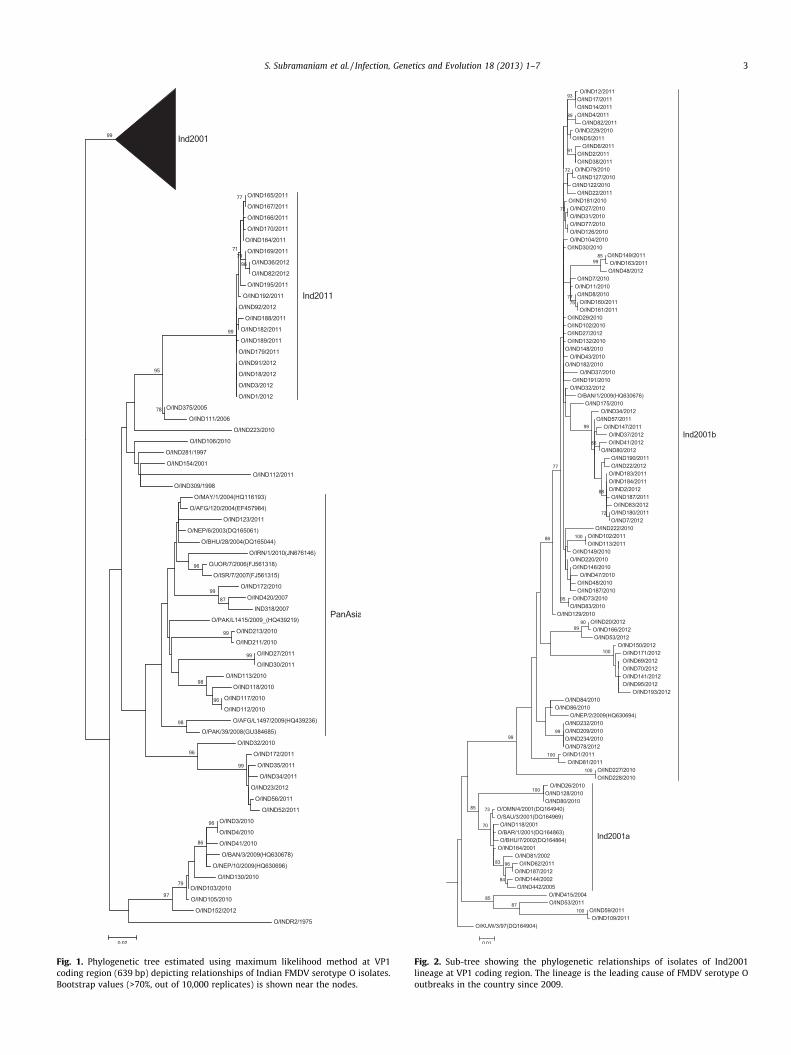
Fig. 1. Phylogenetic tree estimated using maximum likelihood method at VP1coding region (639 bp) depicting relationships of Indian FMDV serotype O isolates.Bootstrap values (>70%, out of 10,000 replicates) is shown near the nodes.
Fig. 2. Sub-tree showing the phylogenetic relationships of isolates of Ind2001lineage at VP1 coding region. The lineage is the leading cause of FMDV serotype Ooutbreaks in the country since 2009.
S. Subramaniam et al. / Infection, Genetics and Evolution 18 (2013) 1–7 3
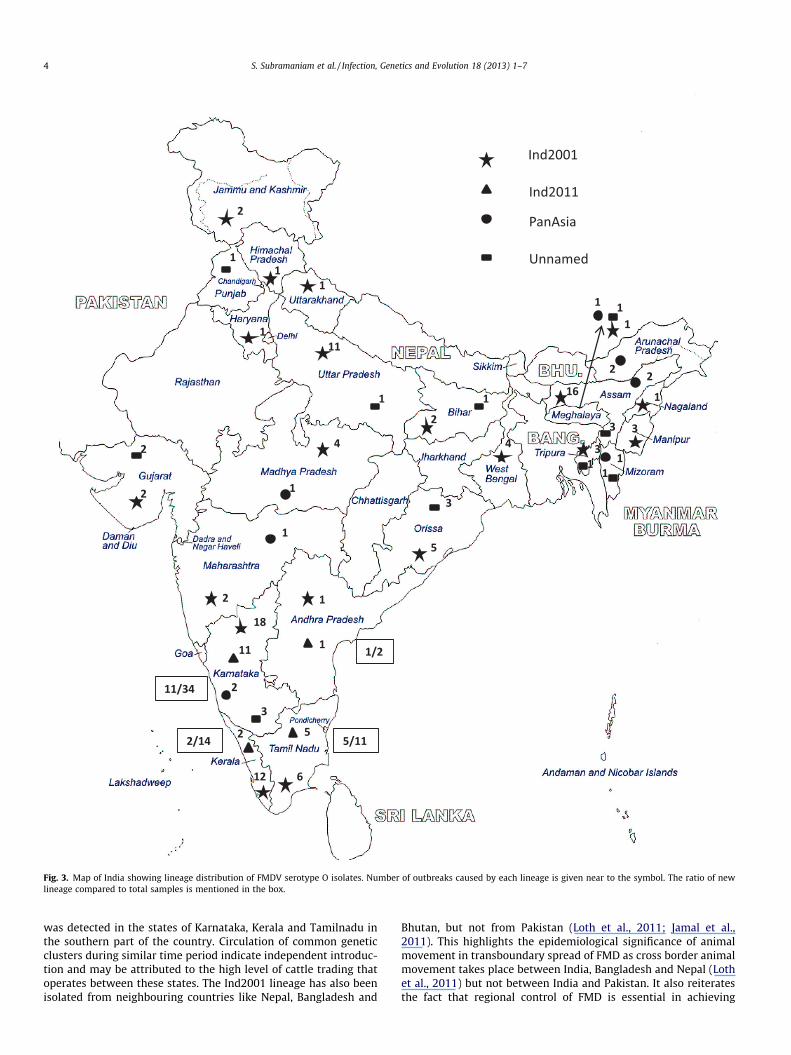
Fig. 3. Map of India showing lineage distribution of FMDV serotype O isolates. Number of outbreaks caused by each lineage is given near to the symbol. The ratio of newlineage compared to total samples is mentioned in the box.
4 S. Subramaniam et al. / Infection, Genetics and Evolution 18 (2013) 1–7
was detected in the states of Karnataka, Kerala and Tamilnadu inthe southern part of the country. Circulation of common geneticclusters during similar time period indicate independent introduc-tion and may be attributed to the high level of cattle trading thatoperates between these states. The Ind2001 lineage has also beenisolated from neighbouring countries like Nepal, Bangladesh and
Bhutan, but not from Pakistan (Loth et al., 2011; Jamal et al.,2011). This highlights the epidemiological significance of animalmovement in transboundary spread of FMD as cross border animalmovement takes place between India, Bangladesh and Nepal (Lothet al., 2011) but not between India and Pakistan. It also reiteratesthe fact that regional control of FMD is essential in achieving

Table 1One way antigenic relationships (r1 value) of FMDV serotype O field isolates withvaccine strain INDR2/1975.
Isolate ID Lineage r1 Value
O/IND48/2010 Ind2001 1.00O/IND77/2010 Ind2001 0.76O/IND79/2010 Ind2001 0.69O/IND80/2010 Ind2001 0.60O/IND84/2010 Ind2001 1.00O/IND104/2010 Ind2001 0.91O/IND122/2010 Ind2001 1.00O/IND126/2010 Ind2001 1.00O/IND127/2010 Ind2001 1.00O/IND12/2011 Ind2001 1.00O/IND22/2011 Ind2001 0.43O/IND53/2011 Ind2001 1.00O/IND57/2011 Ind2001 0.50O/IND62/2011 Ind2001 1.00O/IND102/2011 Ind2001 0.52O/IND147/2011 Ind2001 0.73O/IND160/2011 Ind2001 0.47O/IND161/2011 Ind2001 0.91O/IND22/2012 Ind2001 0.31O/IND37/2012 Ind2001 0.59O/IND69/2012 Ind2001 0.48O/IND80/2012 Ind2001 0.57O/IND141/2012 Ind2001 0.44O/IND165/2011 Ind2011 0.63O/IND170/2011 Ind2011 0.79O/IND182/2011 Ind2011 0.49O/IND188/2011 Ind2011 0.86O/IND192/2011 Ind2011 0.61O/IND195/2011 Ind2011 0.60O/IND213/2010 PanAsia 1.00O/IND34/2011 Unnamed 0.58O/IND52/2011 Unnamed 0.59O/IND4/2010 Unnamed 1.00O/IND56/2011 Unnamed 0.62O/IND23/2012 Unnamed 0.52
S. Subramaniam et al. / Infection, Genetics and Evolution 18 (2013) 1–7 5
successful national control. Though the PanAsia lineage is reportedto be the dominant lineage in South Asia, this lineage hasbeen slowly replaced from the field by the Ind2001 lineage(Subramaniam et al., 2012) in India. In the present analysis also,decline in circulation of PanAsia lineage was noticed as only 10outbreak strains clustered within this lineage.
A new genetic group in serotype O appeared in the year 2011with 9.8–14.8% and 9.7–12.8% nucleotide divergence from contem-porary viruses of Ind2001 and PanAsia lineages circulating in India,respectively. The genetic cluster is named as Ind2011 lineage andhad 0.5–1.4% divergence at the nucleotide level among the constit-uent isolates. Divergence at the amino acid level for this group ran-ged from 0.9% to 1.9%. The Ind2011 had minimum of 7% nucleotidedivergence from global isolates and appeared to share ancestrywith PanAsia lineage rather than with Ind2001. Geographically,the Ind2011 lineage is so far restricted to southern region in thestates of Karnataka, Tamilnadu, Andhra Pradesh and Kerala. Earli-est outbreak as per sample collection date was in September2011 in Andhra Pradesh. Until January 2012, this lineage hascaused 19 outbreaks involving mostly unvaccinated animals. Thevirus possibly appeared first (reported late) in the state of Tamiln-adu (Erode district) where large cattle markets happen in themonth of August every year when movement of thousands of ani-mals occur between the southern states. Extensive spread of thevirus was observed in the state of Karnataka where 11 outbreaksdue to this lineage were recorded within a short span of time.The lineage has accumulated 12 synonymous and 5 non-synony-mous mutations in a span of 5 months in the VP1 region. It corrob-orate with the established fact that higher mutation rate in earlierphase of emergence is essential for rapid adaptability in viruses. Itis uncertain whether the emergence of new genetic lineage wastriggered by immune pressure or selected for fitness value or dueto a bottleneck in transmission. The level of amino acid divergenceis more than nucleotide divergence in both Ind2001 and Ind2011lineages indicating faster rate of non-synonymous replacements.
Correlation of time, space and species with genetic variationsobserved in the virus isolates could not be established in some in-stances in this analysis. For example, two isolates (IND209/2010and IND78/2012) collected in a time span of 16 months were foundto be same and maintenance of such a genetic conservation underextreme selective pressure in the field due to vaccination/infectionis surprising. Incidentally both the strains were sampled from thestate of Madhya Pradesh. In another instance, the strain (IND102/2011) isolated from a cattle in January 2011 and strain (IND113/2011) collected from a sheep in April 2011 in a spatial distanceof 500 km from the state of Karnataka were found to be same.Again, viruses (IND69/2012, IND70/2012, IND95/2012, IND141/2012, IND150/2012, IND171/2012 and IND193/2012) isolated fromdifferent states during similar period of time were genetically verymuch similar. These observations indicate existence of extremeepidemiological complexity in the field.
3.2. Antigenic relationships
The antigenic relationships of serotype O field isolates to thevaccine strain are given in Table 1. The test results were inter-preted as per criteria set by Rweyemamu (1984). From the resultit is evident that none of the isolates had r1-value of <0.3 indicatingsuitable antigenic coverage. For Ind2001 lineage, r1-value variedfrom 0.31 to 1.00. Isolates of Ind2011 lineage also had a close anti-genic match and r1-value for this group ranged from 0.49 to 0.86.Currently used serotype O vaccine strain, INDR2/1975 has beenin use for decades in the country. The vaccine strain belongs tolineage Branch B that was in circulation until 2003. Recent fieldisolates are divergent from the vaccine strain by 10.9% to 15.6%at nucleotide level and 3.8% to 6.9% at amino acid level in the
VP1 coding region. Despite considerable genetic variability, thevaccine strain is expected to cover all the field isolates antigeni-cally and there is no necessity for change of vaccine strain as ofnow.
3.3. Amino acid variations
Comparison of deduced amino acid sequences of VP1 coding re-gion revealed 56 variable positions and of which 31 positions ac-cepted single amino acid replacement and 25 positionsaccommodated multiple amino acid changes. Earlier, single aminoacid deletion at position 139 in VP1 region was reported in Indianserotype O isolates (Das et al., 2012). None of the field isolates ofthis study had deletion at position 139 in VP1. Though RGD tripep-tide in the bG-bH loop is essential for receptor binding on cells, res-idue L at +1 and +4 positions, and residue A at +2 position to alesser extent were shown to be critical (Mateu et al., 1996). In thissequence set, RGDLXXL motif (145–149) is found conserved fullyexcept two samples which have RGDMXXL (O/IND12/2011) andRGDLXXF (O/IND73/2010). The residue Q at position +2 is fullyconserved in all the isolates.
The Ind2001 lineage that appeared first in the year 2001(Ind2001a) had five consensus variations (A13T, T68A, T96K,S139G and T/S/P140A) compared to other field isolates (Hemadriet al., 2002). The Ind2001 lineage that re-surged in 2008 and circu-lating thereafter (Ind2001b), have four consensus variations (A13T,T/K96A, P/A/G140D and E 198Q). Except position 13, substitutionsfound in other three positions (96, 140 and 198) are unique forInd2001b sub-lineage. The site 13 has evolved as potentialInd2001 lineage inclusive signature. Two specific changes observed

6 S. Subramaniam et al. / Infection, Genetics and Evolution 18 (2013) 1–7
earlier for Ind2001a sub-lineage at positions 68 and 139 have re-verted back to consensus (68T and 139S) in Ind2001b sub-lineage.These observations highlight the differential evolution pattern oflineages in an endemic setting.
The complete capsid coding sequences of 4 isolates (IND165/2011, IND182/2011, IND188/2011 and IND192/2011) of Ind2011lineage were compared with the sequences of vaccine strain and60 other field isolates available in our database. Compared to thevaccine strain, the new genetic cluster has variations at 33 posi-tions (VP4-T60N; VP2-D41G, S74P, E82A, C130Y, L132I, Q133T,L137Q, Y171H, T191N; VP3-F30Y, T99A, E195D, A218T, R219T,E220Q; VP1-P4T, L36F, I48T, A96T, R108K, N133S, K135R, G137S,D138E, G139S, S140P, V141A, T142A, I144V, R172Q, N197S) inone or more field isolates. Position 133 of VP2 and 36 of VP1 hasevolved as potential Ind2011 specific signature. Three substitu-tions (N133S, T142A and K135R) in VP1 fixed in all the Ind2011isolates are also observed in few of the isolates of other lineages.
3.4. Conservation at antigenically critical positions
Five neutralizing antigenic sites have been reported for serotypeO (Xie et al., 1987; Kitson et al., 1990; Crowther et al., 1993;McCahon et al., 1989) and of which sites 1, 3 and 5 are found inVP1, and sites 2 and 4 are in VP2 and VP3, respectively. The aminoacid positions that are critical for antigenic sites were comparedbetween field isolates and the vaccine strain. At antigenic site 1(critical residues 144, 148, 154 and 208), I ? V replacement wasobserved at position 144 in the field isolates and similar observa-tion was also made earlier (Pattnaik et al., 1998; Hemadri et al.,2002). Interestingly none of the field isolates had any substitutionother than V at this position. At position 148, L ? M substitutionwas observed in one isolate (O/IND12/2011) which had close anti-genic match with vaccine strain (r1 = 1). It was earlier observedthat L ? R replacement at position 148 in mutants conferred com-plete resistance to neutralization (Xie et al., 1987). The amino acidchange noticed in the field isolate is of conservative nature com-pared to modification observed in the mutants. It is likely that con-servative replacements can be accommodated without change inantigenecity. The isolates did not reveal any substitutions at othertwo critical positions (154 and 208). At antigenic site 3 (criticalresidues 43 and 44), T was replaced by A at position 43 in twoisolates (O/IND41/2012 and O/IND80/2012) and position 44 wasfully conserved. Position 149 critical for antigenic site 5 was100% conserved in the field isolates. In the four isolates wherethe entire capsid coding region was sequenced, positions criticalfor site 2 in VP2 (70–73, 75, 77 and 131) and site 4 in VP3 (58) werefound preserved fully. Though the field isolates are geneticallydivergent from vaccine strain, such a remarkable conservationat antigenically critical positions might contribute to observedantigenic stability in Indian serotype O field isolates.
3.5. Evolutionary rate in Ind2001 lineage
Nucleotide substitution rates provide measure of virus evolu-tion. The rate of evolution of FMDV over a period of six decades(1.4 � 10�3 substitutions/nt/year, Martínez et al., 1992) was re-ported to be lower than the rate obtained for FMDV during persis-tent infection (0.9–7.4 � 10�2 substitutions/site/year, Gebaueret al., 1988) and during the defined epizootic outbreak(6.5 � 10�3 substitutions/site/year, Villaverde et al., 1991). In thisanalysis evolutionary rate of Ind2001b sub-lineage was estimatedat 6.58 � 10�3 substitutions/site/year (R2 = 0.603; p = 0.001). Therate is very much similar to the one observed for defined epizooticoutbreaks. Earlier similar rates for PanAsia II and III sub-lineages ofserotype O have been reported (Jamal et al., 2011). The rate esti-mated here is significantly higher than the rates reported earlier
for serotype O virus circulating in India from 1989 to 2001(2.0 � 10�3 substitutions/site/year, Hemadri et al., 2002), and EastAfrica from 1964 to 2008 (2.7 � 10�3 substitutions/site/year, Balin-da et al., 2010). It is possible that since isolate collection periodspanned between 12 and 44 years, long term evolution might haveresulted in evolutionary stasis (Martínez et al., 1992). High evolu-tionary rate observed in the current analysis may be due to shorterisolation period (2008–2012) and correlated well with the high ge-netic diversity existing within the Ind2001 lineage.
3.6. Evidence of positive selection
The ratio of nonsynonymous to synonymous substitution rates(x = dN/dS) provide a measure of positive selection in virus se-quences. The selection pressure analyses were performed usingDatamonkey web interface employing four different methods.The dN/dS ratio of serotype O isolates was estimated at0.106908. The ratio obtained for Ind2011 lineage (0.188541) washigher than Ind2001 (0.115545) and PanAsia lineage (0.087755).Overall, positively selected sites could be identified in the back-ground of strong purifying selection. For instance out of 213 codonpositions, 52%, 65% and 58% were found to experience purifyingselection using SLAC, FEL and IFEL methods, respectively. Ingeneral, purifying selection is reported to be the most dominantevolutionary force influencing both genotype and phenotype (Le-wis-Rogers et al., 2008), and strong negative selection operatedin the FMDV population subjected to enhanced mutagenesis(Ojosnegros et al., 2008).
Using MEME approach, 6 codon positions [4 (p = 0.08), 68(p = 0.07), 83 (p = 0.01), 135 (p = 0.09), 138 (p = 0.04) and 209(p = 0.01)] were identified to be under episodic diversifying selec-tion. In MEME analysis, x values were allowed to vary alongbranches and this method was reported to be the most appropriateto detect episodic diversifying selection affecting individual codonsites (Murrell et al., 2012). Sites 4 (p = 0.08), 85 (p = 0.04) and 135(p = 0.07) and, site 135 (p = 0.08) were identified using FEL andIFEL, respectively. None of the sites identified here are antigeni-cally crucial for serotype O and exact effect of diversifying selectionat these positions are unclear. However, two sites at 83 and 209were identified as antigenically critical in FMDV serotypes A byanalyses of neutralization escape mutants (Mittal et al., 2005 andreferences therein). Further, the positions 68 and 138 were identi-fied as signature residues for Ind2001a and Ind2011 lineages,respectively. The sites identified to undergo positive selection inthis study are found in loops, a helix and c-terminus of VP1.
4. Conclusions
To sum up, the present phylogenetic study identified theappearance of a new genetic group (Ind2011) with >9% nucleotidedivergence from rest of the lineages currently prevalent in thecountry. Sequence comparison with global serotype O isolates re-veals that the new group is currently contained only in India, par-ticularly in the southern part of the country. Though geneticallydivergent, the emerging lineage showed adequate antigenic matchwith the vaccine strain. There seems to be complex epidemiologi-cal scenario in the southern region of the country with co-circula-tion of various lineages. Though, PanAsia virus was present in Indiasince 1982, its dominance in the field was evident after a gap of14 years in 1996. Similarly, Ind2001 lineage was identified as adivergent strain from PanAsia in the year 2001; its predominancewas noted in 2009. With the emergence of new genetic group,overall epidemiological picture of FMD in the region is expectedto change in coming years and hence is required to be monitoredcontinually. Irrespective of the genetic variability, the antigenic

S. Subramaniam et al. / Infection, Genetics and Evolution 18 (2013) 1–7 7
characteristics of Indian serotype O isolates seems to be verystable.
Acknowledgements
Indian Council of Agricultural Research provided necessaryfacilities to carry out this work. We sincerely thank all the scientificand technical staff associated with network laboratories for provid-ing clinical materials and required data.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.meegid.2013.04.027.
References
Ayelet, G., Mahapatra, M., Gelaye, E., Egziabher, B.G., Rufeal, T., Sahle, M., Ferris, N.P.,Wadsworth, J., Hutchings, G.H., Knowles, N.J., 2009. Genetic characterization offoot-and-mouth disease viruses, Ethiopia, 1981–2007. Emerg. Infect. Dis. 15 (9),1409–1417.
Balinda, S.N., Sangula, A.K., Heller, R., Muwanika, V.B., Belsham, G.J., Masembe, C.,Siegismund, H.R., 2010. Diversity and transboundary mobility of serotype Ofoot-and-mouth disease virus in East Africa: implications for vaccinationpolicies. Infect. Genet. Evol. 10, 1058–1065.
Crowther, J.R., Farias, S., Carpenter, W.C., Samuel, A.R., 1993. Identification of a fifthneutralizable site on type O foot-and-mouth disease virus followingcharacterization of single and quintuple monoclonal antibody escapemutants. J. Gen. Virol. 74, 1547–1553.
Das, B., Sanyal, A., Subramaniam, S., Mohapatra, J.K., Pattnaik, B., 2012. Fieldoutbreak strains of serotype O foot-and-mouth disease virus from India with adeletion in the immunodominant bG-bH loop of the VP1 protein. Arch. Virol.157 (10), 1967–1970.
Delport, W., Poon, A.F., Frost, S.D., Pond, S.L.K., 2010. Datamonkey 2010: a suite ofphylogenetic analysis tools for evolutionary biology. Bioinformatics 26 (19),2455–2457.
Gebauer, F., De La Torre, J.C., Gomes, I., Mateu, M.G., Barahona, H., Tiraboschi, B.,Bergmann, I., de Mello, P.A., Domingo, E., 1988. Rapid selection of genetic andantigenic variants of foot-and-mouth disease virus during persistence in cattle.J. Virol. 62 (6), 2041–2049.
Hemadri, D., Tosh, C., Sanyal, A., Venkataramanana, R., 2002. Emergences of a newstrain of type O foot-and-mouth disease virus: its phylogenetic andevolutionary relationship with PanAsia pandemic strain. Virus Genes 25, 23–34.
Jamal, S.M., Ferrari, G., Ahmed, S., Normann, P., Belsham, G.J., 2011. Genetic diversityof foot-and-mouth disease virus serotype O in Pakistan and Afghanistan, 1997–2009. Infect. Genet. Evol. 11 (6), 1229–1238.
Kimura, M., 1980. A simple method for estimating evolutionary rates of basesubstitutions through comparative studies of nucleotide sequences. J. Mol. Evol.16, 111–120.
Kitson, J.D., McCahon, D., Belsham, G.J., 1990. Sequence analysis of monoclonalantibody resistant mutants of type O foot and mouth disease virus: evidence forthe involvement of the three surface exposed capsid proteins in four antigenicsites. Virology 179, 26–34.
Knowles, N.J., Samuel, A.R., Davies, P.R., Midgley, R.J., Valarcher, J.F., 2005. Pandemicstrain of foot-and-mouth disease virus serotype O. Emerg. Infect. Dis. 11 (12),1887–1893.
Lewis-Rogers, N., McClellan, D.A., Crandall, K.A., 2008. The evolution of foot-andmouth disease virus: impacts of recombination and selection. Infect. Genet.Evol. 8, 786–798.
Loth, L., Osmani, M.G., Kalam, M.A., Chakraborty, R.K., Wadsworth, J., Knowles, N.J.,Hammond, J.M., Benigno, C., 2011. Molecular characterization of foot-and-mouth disease virus: implications for disease control in Bangladesh.Transbound. Emerg. Dis. 58 (3), 240–246.
Martínez, M.A., Dopazo, J., Hernández, J., Mateu, M.G., Sobrino, F., Domingo, E.,Knowles, N.J., 1992. Evolution of the capsid protein genes of foot-and-mouthdisease virus: antigenic variation without accumulation of amino acidsubstitutions over six decades. J. Virol. 66 (6), 3557–3565.
Mateu, M.G., Valero, M.L., Andreu, D., Domingo, E., 1996. Systematic replacement ofamino acid residues within an Arg-Gly-Asp-containing loop of foot-and-mouthdisease virus and effect on cell recognition. J. Biol. Chem. 271, 12814–12819.
McCahon, D., Crowther, J.R., Belsham, G.J., Kitson, J.D.A., Duchesne, M., Have, P.,Meloen, R.H., Morgan, D.O., DeSimone, F., 1989. Evidence for at least fourantigenic sites on type O foot-and-mouth disease virus involved inneutralization; identification by single and multiple site monoclonalantibody-resistant mutant. J. Gen. Virol. 70, 639–645.
Mittal, M., Tosh, C., Hemadri, D., Sanyal, A., Bandyopadhyay, S.K., 2005. Phylogeny,genome evolution and antigenic variability among endemic foot and mouthdisease virus type A isolates from India. Arch. Virol. 150, 911–928.
Mohapatra, J.K., Hemadri, D., Rao, T.V.S., Sreenivasa, B.P., Subramaniam, S., Sanyal,A., Periyasamy, T.R., Singh, N.K., Pattnaik, B., Venkataramanan, R., 2008.Assessment of suitability of two serotype A candidate vaccine strains forinclusion in FMD vaccine in India. Vet. Microbiol. 131, 65–72.
Murrell, B., Wertheim, J.O., Moola, S., Weighill, T., Scheffler, K., Pond, S.L.K., 2012.Detecting individual sites subject to episodic diversifying selection. PLoS Genet.8 (7), e1002764.
Ojosnegros, S., Agudo, R., Sierra, M., Briones, C., Sierra, S., González-López, C.,Domingo, E., Cristina, J., 2008. Topology of evolving, mutagenized viralpopulations: quasispecies expansion, compression, and operation of negativeselection. BMC Evol. Biol. 17, 8–207.
Pattnaik, B., Venkataramanan, R., Tosh, C., Sanyal, A., Hemadri, D., Samuel, A.R.,Knowles, N.J., Kitching, R.P., 1998. Genetic heterogeneity of Indian field isolatesof foot-and-mouth disease virus serotype O as revealed by partial sequencing of1D gene. Virus Res. 55 (2), 115–127.
Pattnaik, B., Subramaniam, S., Sanyal, A., Mohapatra, J.K., Dash, B.B., Ranjan, R., Rout,M., 2012. Foot-and-mouth disease: global status and future road map forcontrol and prevention in India. Agric. Res. 1 (2), 132–147.
Pond, S.L.K., Frost, S.D.W., Muse, S.V., 2005. HyPhy: hypothesis testing usingphylogenies. Bioinformatics 21 (5), 676–679.
Racaniello, V.R., Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A.,Roizman, B., 2001. Picornaviridae: the viruses and their replication. In: FieldsVirology, . fourth ed., vol. 1 Lippincott Williams & Wilkins, Philadelphia, PA, pp.685–722.
Rweyemamu, M.M., 1984. Antigenic variation in foot-and-mouth disease: studiesbased on the virus neutralization reaction. J. Biol. Stand. 12, 323–337.
Rweyemamu, M.M., Booth, J.C., Head, M., Pay, T.W.F., 1978. Micro-neutralizationtests for serological typing and subtyping of foot-and-mouth disease virusstrains. J. Hyg. Camb. 81, 107–123.
Samuel, A.R., Knowles, N.J., 2001. Foot-and-mouth disease type O viruses exhibitgenetically and geographically distinct evolutionary lineages (topotypes). J.Gen. Virol. 82, 609–621.
Subramaniam, S., Pattnaik, B., Sanyal, A., Mohapatra, J.K., Pawar, S.S., Sharma, G.K.,Das, B., Dash, B.B., 2012. Status of foot-and-mouth disease in India. Transbound.Emerg. Dis. http://dx.doi.org/10.1111/j.1865-1682.2012.01332.x.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S., 2011. MEGA5:molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28(10), 2731–2739.
Thompson, J.D., Higgins, D.G., Gibson, T.J., 1994. CLUSTAL W improving thesensitivity of progressive multiple sequence alignment through sequenceweighting, position-specific gap penalties and weight matrix choice. NucleicAcids Res. 22, 4673–4680.
Villaverde, A., Martinez, M.A., Sobrino, F., Dopazo, J., Moya, A., Domingo, E., 1991.Fixation of mutations at the VP1 gene of foot-and-mouth disease virus, canquasispecies define a transient molecular clock? Genes 103, 147–153.
WRL-FMD, 2011. Annual OIE/FAO FMD Reference Laboratory Network Report.World Reference Laboratory for Foot-and-Mouth Disease, January–December2011, Institute for Animal Health, Pirbright, UK.
Xie, Q.C., McCahon, D., Crowther, J.R., Belsham, G.J., McCullough, K.C., 1987.Neutralization of foot-and-mouth disease virus can be mediated through any ofat least three separate antigenic sites. J. Gen. Virol. 68, 1637–1647.
Related Documents











