"Determining the Regulatory Pathway to Market" Classification Heather S. Rosecrans Director, 510(k) Staff Office of Device Evaluation Center for Devices & Radiological Health U.S. Food & Drug Administration (240) 276-4040 [email protected]

"Determining the Regulatory Pathway to Market" Classification Heather S. Rosecrans Director, 510(k) Staff Office of Device Evaluation Center for Devices.
Dec 14, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

"Determining the Regulatory Pathway to Market"
Classification
Heather S. RosecransDirector, 510(k) Staff
Office of Device EvaluationCenter for Devices & Radiological Health
U.S. Food & Drug Administration(240) 276-4040

2
What is a 510(k)
Premarket Notification Section 510(k) of FF,D,&C Act 21 CFR 807 Subpart E Submission for Device Premarket Review Allows FDA to Make a Determination Regarding
Substantial Equivalence (SE) “The” classification process for a device

3
What a 510(k) Is Not
A Form Establishment Registration (FDA-2891) Device Listing (FDA-2892) Premarket Approval (PMA) Product Development Protocol (PDP) Evaluation of Automatic Class III Designation
(De Novo)

4
Medical Device Amendments of 1976 to the FF,D,&C Act
May 28, 1976 Defined a device (201(h) of the Act) Required risk based classification of device
types legally on the market Led to classification of approximately 1,700 different
generic types of devices and grouped into 19 medical specialties
Required premarket review of devices

5
Safe Medical Devices Act (SMDA)
1990 – 513(i)
• Substantial Equivalence Defined
– 513(a)(1)(B)• Special Controls

6
Food & DrugModernization Act (FDAMA)
1997 – Redefined 510(k) Exemption Criteria for Class I – Added Class II Exemption Criteria– De Novo– SE w/Limitations– Class II Petitions for Exemption

7
510(k) & Classification
A 510(k) is the classification process for individual post-amendment devices by:
– Finding the device substantially equivalent (SE) or
– Finding the device not substantially equivalent (NSE)

8
510(k) & Classification Determination regarding substantial equivalence:
– NSE (For reasons other than lack of performance data) PMA, PDP, or De Novo
– NSE for lack of performance data a new 510(k) may be submitted for review
– SE To Market

9
Substantially Equivalent (SE)
If:– In Comparison to a legally marketed device (that
does not require PMA), it:• Has the same intended use, and• Has the same technological characteristics as the
predicate device,
Or . . . . . . . . . . . . .

10
Has the same intended use, and Has different technological characteristics and the
information in the 510(k):– Does not raise a new question(s) of safety and
effectiveness; and– Demonstrates it is at least as safe and effective as
the predicate.
Substantially Equivalent (SE)
11
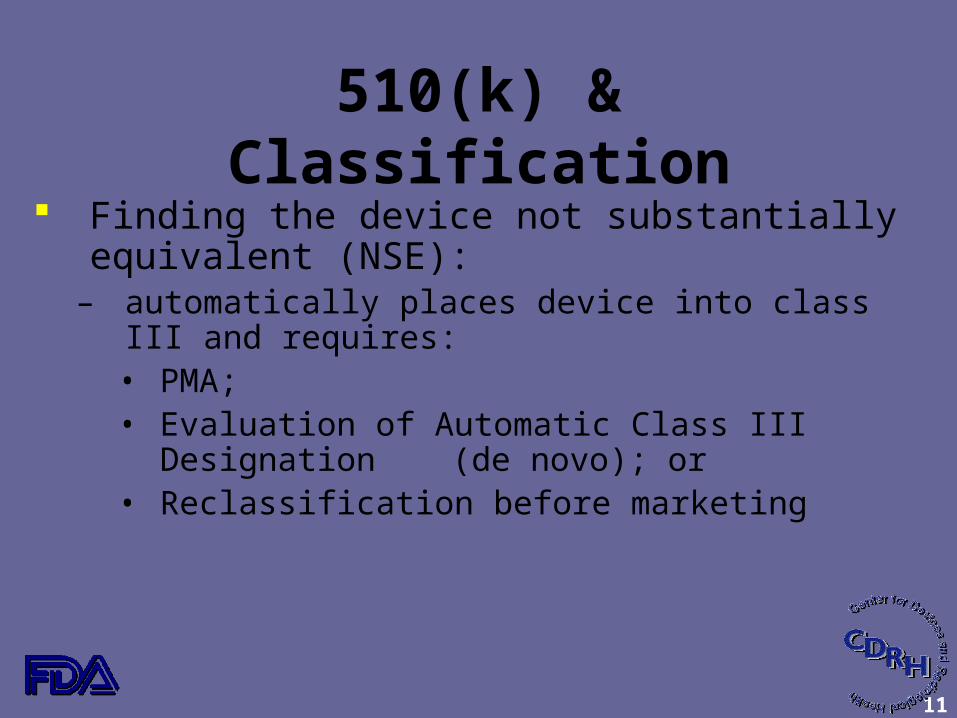
510(k) & Classification Finding the device not substantially equivalent
(NSE):– automatically places device into class III and requires:
• PMA;• Evaluation of Automatic Class III Designation
(de novo); or• Reclassification before marketing

12
Not Substantially Equivalent (NSE)
There is no predicate device; or The device has a new intended use; or The device has different technological
characteristics compared to the predicate device and raises a new type question(s) of safety and effectiveness
*All of the above require no review of data and will
require PMA or De Novo.

13
Not Substantially Equivalent (NSE)
The data provided do not demonstrate that the device is at least as safe and effective as the predicate.
*FDA usually asks for additional information at least once prior to determining the device is NSE for lack of data
*The above NSE reason requires review of the data and would require submission of a new 510(k) with new data to demonstrate SE.

14
Approximately 80% are found SE
2 – 3% are found NSE
Remaining 510(k)s are usually withdrawn
510(k) & Classification

04/18/23
Evaluation of Automatic Class III Designation
“de novo” Classification Section 513(f)(2) De novo received within 30 days of receipt of NSE letter
(NSE reason for other than lack of performance data) No predicate/“Low risk” devices 60-day review period Extensions for submission of additional information
granted
04/18/23
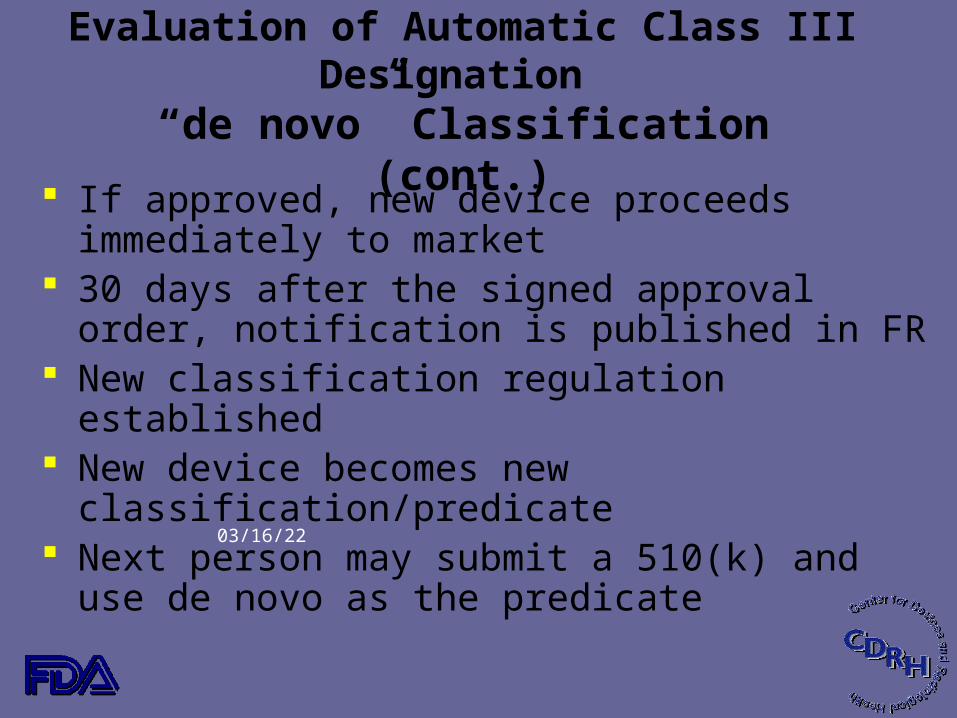
Evaluation of Automatic Class III Designation
“de novo” Classification (cont.) If approved, new device proceeds immediately to market 30 days after the signed approval order, notification is
published in FR New classification regulation established New device becomes new classification/predicate Next person may submit a 510(k) and use de novo as the
predicate

17
Related Documents











