Dědičná mitochondriální onemocnění a poruchy mitochondriální beta oxidace mastných kyselin prim.MUDr. RNDr. Pavel Ješina, Ph.D. Ústav dědičných metabolických poruch 1.LF UK, VFN Metabolická jednotka Klinika dětského a dorostového lékařství VFN 1.LF UK

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Dědičná mitochondriální onemocnění a poruchy mitochondriální beta oxidace mastných kyselin
prim.MUDr. RNDr. Pavel Ješina, Ph.D.
Ústav dědičných metabolických poruch 1.LF UK, VFN Metabolická jednotka Klinika dětského a dorostového lékařství VFN 1.LF UK

Mitochondrie - struktura

Struktura mitochondrií
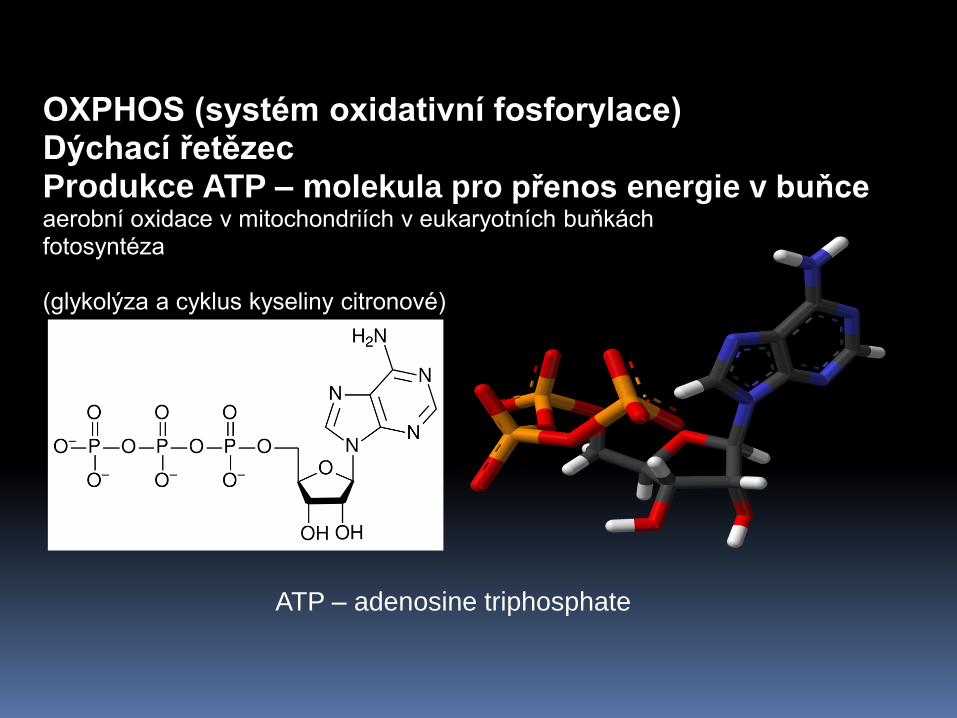
OXPHOS (systém oxidativní fosforylace) Dýchací řetězec Produkce ATP – molekula pro přenos energie v buňce aerobní oxidace v mitochondriích v eukaryotních buňkách fotosyntéza (glykolýza a cyklus kyseliny citronové)
ATP – adenosine triphosphate

Metabolické dráhy v mitochondriích
Krebsův cyklus
oxidace
Ketogeneze
Močovinový cyklus
Syntéza hemu a porfyrinů
…

cykly pro přenos transport NADH.H+
malát-aspartátový cyklus glycerolfosfátový cyklus
přenašeče pro substráty a produkty metab. drah probíhajících v mitochondriích
ADP-ATP transokátor
import proteinů
Mitochondriální transportní systém

Oxidativní fosforylace
43 4 11 13 16
podjednotek

Superkomplexy
Tkáňová specifita (játra, srdce, hnědý tuk ...) Ontogenetické změny (perinatální vývoj, ...)


porucha mitochondriální respirační funkce jaderné geny geny kodované mitochondriální DNA mtDNA Deficity enzymů respiračního řetězce Deficity enzymů cyklu kyseliny citronové a pyruvátového metabolismu Poruchy mitochondriální fůze a dělení Konzervativní odhad incidence : 12/100 000 1/4 000
Mitochondriální onemocnění

Mitochondriální DNA
2-20 / mito
10-10 000 / bb
16 569 párů bazí
13 mRNA, 2 rRNA; 22 tRNA

Mitochondriální choroby
Frekvence 1:3500 – 1:4000
Vysoce energeticky závislé orgány
Mutace v ncDNA i v mtDNA
13 mRNA, 2 rRNA 22 tRNA - >100 mutací
Leu; Lys; Ser
MELAS; MERRF
Schon et al, 2001

1962 Luft R, et al Luftova nemoc 1963 Nass S, Nass MHK DNA v mitochondriích 1970 Spiro AJ, et al. defekt resp. řetězce 1974 Berk AS, Clayton DA replikace mtDNA 1979 Barrell BG, et al genetický kód mtDNA 1981 Andersson S, et al. sekvence mtDNA 1988 Holt IJ, et al. delece mtDNA
Wallace DC, et al bodová mutace mtDNA 2000 www.gen.emory.edu/mitomap.html > 130 bodových mutací > 70 typů delecí/duplikací ……… další a další objevy
Mitochondriální choroby

mRNA
prekursor
NUCLEUS
CYTOSOL
MATRIX
OXPHOS komplex
nc podjednotka
ncDNA
mRNA
mtDNA
mt podjednotka > 70 nc OXPHOS podjednotek 13 mt OXPHOS podjednotek 26 asemblačních proteinů
~ 150 „biogenesis“ proteinů
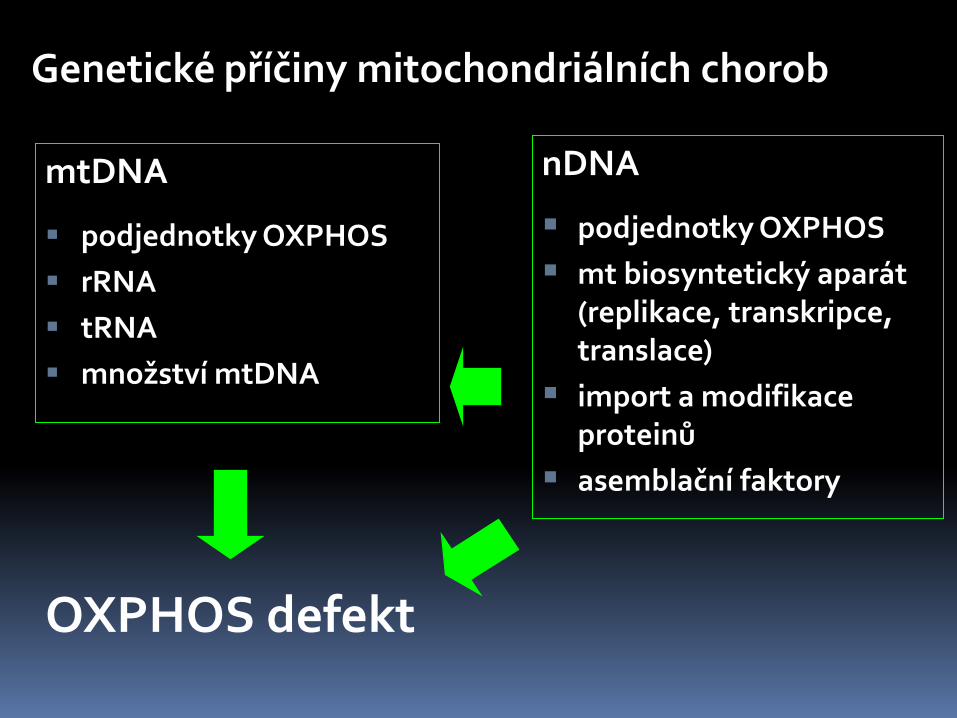
Genetické příčiny mitochondriálních chorob
mtDNA
podjednotky OXPHOS
rRNA
tRNA
množství mtDNA
nDNA
podjednotky OXPHOS
mt biosyntetický aparát (replikace, transkripce, translace)
import a modifikace proteinů
asemblační faktory
OXPHOS defekt

Dědičnost mtDNA mutací
Sporadické mtDNA mutace
jednoduché delece/duplikace
Maternálně děděné mtDNA mutace bodové mutace jednoduché delece/duplikace
Autosomálně děděné mtDNA mutace mnohočetné delece deplece

Bodové mutace mtDNA
Homoplasmické – 1forma mtDNA
OXPHOS proteiny (LHON)
Heteroplasmické – 2 formy mtDNA
tRNA (MERRF, MELAS)
rRNA (Kardiomyopatie)
OXPHOS proteiny
Leigh, NARP
LHON
N P
RFLP
N P

Symptomy nemocí způsobených mutacemi v mtDNA se často progresivně zhoršují s věkem. K mitochondriální dysfunkci dochází když je překročen bioenergetický práh . Některé orgány jsou zvláště závislé na funkci mitochondrií: Mozek, kosterní sval, srdeční sval a endokrinní žlázy Buňky ztrácí respirační funkci až při vysokém podílu patogeních mtDNA, obvykle 60-90% pro jednotlivé mtDNA mutace Některé mutantní mtDNA mají replikační výhodu (v porovnaní s nemutovanou mtDNA).
Heteroplasmie, mtDNA a nemoci

Inter- a intramitochondriální heteroplazmie

Segregace mtDNA

MOLECULAR MEDICINE TODAY, NOVEMBER 2000 (VOL. 6)
Mitochondriální genetické „zúžení“ (Mitochondrial genetic bottleneck)

Segregace a distribuce heteroplasmických mtDNA mutací
Germinativní bb Somatické bb („bottleneck“ – nízký (velký počet dělení) počet mtDNA)
rychlá segregace pomalá segregace
(trend k homoplasmii)
rozdílný přenos a meziorgánové i
postižení potomků mezibuněčné
rozdíly

7512T>C mutace v mtDNA
Sval: 95% Krev: 91% Fibroblasty: 92%
Krev: 68% Bukální slizn.: 59% Vlas.folikuly: 73%
Sval: 95% Krev: 93% Fibroblasty: 94% Buk.slznice: 92% Vlas.folikuly: 95%
Krev: 71% Buk.sliznice: 74% Vlas.folikuly: 87%
Krev: 92% Bukál.sliznice: 86% Fibroblasty: 94%

Segregace mtDNA v rodinách s mutacemi NARP (8993), MELAS (3243) a MERRF (8344).
NARP MELAS MELAS
MERRF MERRF MERRF

Klinické projevy
Biochemický defekt
Mutace
OXPHOS
isoenzymy Limitující krok v OXPHOS
počet podjednotek počet komplexů
heteroplasmie
segregace
Heterogenita fenotypu
Heterogenita genotypu

Prahový efekt u OXPHOS defektů
0-100% podíl mutované mtDNA
Nelineární vztah mezi heteroplazmií a poruchou funkce – „recesivní mutace“ tolerance mutací do 80-90%
S věkem klesající aktivita OXPHOS
Tkáňová/bb specifita –energetické nároky


Patogenní mechanismy
mutace mtDNA
porucha OXPHOS
snížená tvorba ATP
porucha bb funkcí
tvorba kyslíkových radikálů
apoptóza nekróza buněk
mutace ncDNA

Defektu v mitochondriálním respiračním
řetězci by se měl zvažovat u pacientů, kteří
mají jakoukoli nevysvětlenou kombinaci
neuromuskulárních a/nebo jiných příznaků,
s progredujícím průběhem a postihující
zdánlivě nesouvisející orgány nebo tkáně. Munnich et al , OMMBD, Chapter 99

Různorodé orgánové postižení u deficitů respiračního řetězce Munnich et al, OMMBID, Ch 99
“jakýkoli příznak, v jakomkoli orgánu, v jakémkoli věku a s jakoukoli dědičností“

Defekty mtDNA
LHON – Leberova hereditární optická neuropatie
- 11778 G>A; 3460G>A; 14484 T>C – podjednotky komplexu I - akutní/subakutní ztráta zraku v dopělosti - 4x u mužů
NARP/Leigh syndrom – neurogenní svalová slabost, ataxie, retinitis
pigmentosa - 8993 T>G a další - psychomotorická retardace, laktátová acidóza, nekróza bazálních ganglií
MERRF – myoclonic epilepsy, ragged red fibres
- 8344 G>A (tRNA pro Lys) a další - hluchota senzorineurální
MELAS – mit. encephalomyopathy, laktátová acidóza, stroke-like episodes
- 3243 A>G (tRNA pro Leu) a další - diaetes mellitus …….

Neurodegenerativní onemocnění se začátkem obvykle před 1 rokem věku vedoucí k úmrtí během několika měsíců nebo let. „subakutní sklerozující encefalomyelopatie“ Degenerace bazálních ganglií, progresivní průběh se zhoršováním motoriky a vývoje, nepravidelné dýchání, ataxie, hyperlaktacidemie, svalová slabost, křeče. Přechodné fenotypy
Deficity : komplex pyruvát dehydrogenázy (PDH)(E1α gene), Cytochrom c oxidáza (complex IV) – často deficit SURF-1, asemblačního proteinu komplexu IV NADH-ubichinon oxidoreduktáza (komplex I) – jak jaderně tak mitochondriálně kodované geny (jiné komplexy respiračního řetězce)
Leighův syndrom

Mutant mtDNA level
Threshold
Defekt
mut mtDNA 31 % 82 % 93 % 95 %
nástup - dospělost dětství novorozenci
symptomy zdraví ataxia NARP Leigh
retinopatie syndrom syndrom
Threshold efekt

CPEO – Chronická progresivní externí oftalmoplegie
Bodové mutace v mitochondriální DNA

http://www.snof.org/maladies/kearnsSayre.html
Syndrom Kearns-Sayre
Oftalmoplegie, ptóza a mitochondriální myopatie před 20 lety věku Další příznaky : pigmentová retinitida a nejméně jeden z následujících příznaků: převodní srdeční porucha, mozečková ataxie, zvýšená koncentrace bílkoviny v likvoru (více než 100 mg/dl) Obvykle způsobeno delecemi či duplikacemi v mtDNA

Leberova hereditární optická neuropatie-LHON
Theodore Leber
LHON je maternálně dědičná akutní optická atrofie s pozdním začátkem. V některých rodinách se vyskytuje i neuritida optiku. Neúplná penetrance (U 40% mužů a 10% žen se vyvinou příznaky) LHON je způsoben homoplasmickými mutacemi mitochondriální DNA Více než 90% evropských a asijských případů LHON je způsobeno třemi bodovými mutacemi mtDNA Mutace G na A v genu MTND4 v posici 11778 (MTND4*LHON11778A) způsobuje asi 50% evropských a asi 90% asijských případů LHON Dále MTND1*LHON3460A (ND1 Ala52Thr) a MTND6*LHON14484C (ND6 Met64Val) . Řada vzácnějších mutací mtDNA zřejmě také způsobuje LHON
http://www.snof.org/maladies/leber.html

Rodokmen rodiny s LHON – maternální
dědičnost

Defekty mtDNA – MERRF Svalová biopsie
Ragged red fibres

Defekty mtDNA Svalová biopsie
COX negativní vlákna
Subsarkelemální reaktivita SDH

Defekty jaderné DNA
jaderná DNA - replikace, transkripce, translace, oprava … asemblace, strukturální podjednotky… defekty v genech pro strukturální podjednotky komplex I – Leigh syndrom, kardiomyopatie, encefalomyopatie, myoklonická epilepsie komplex II – ataxie, atrofie optiku - hereditaární paragangliomy defekty ve assemblačních genech COX – SURF-1, SCO2, SCO1, COX10, COX15, … ATPáza – ATP12

Leigh syndrom
defekt SURF1 těžké neurodegenerativní onemocnění dětská populace nekróza basálních ganglií a mozkového kmene fatální průběh

Nekompletní formy COX Chybění regulačních nc-kódovaných podjednotek Labilita subkomplexů porucha H+transportu
670
440
230
140
70
kDa
Model poruchy COX na podkladě chybění Surf1 proteinu
C P P

Defekty ATPázy jaderného původu TMEM70
Nástup v novoroz.věku 14/14
Úmrtí 7/14 (4; 3)
Žijící (≥3;≥5;≥10 let) 7/14 (3;2;3)
Kardiomyopatie 13/14
Hypotonie 12/13
Psychomotor.retardace 10/10
Hepatomegalie 6/14
Dysmorfie obličeje 5/14
Hyperlaktacidémie 14/14
3-methylglutakon.acidurie 12/12
ATP hydrolýza <30% 13/13
Snížený obsah ATPázy 13/13 Sperl, Ješina et al, Neuromuscul.Disord., 2006 Study of mitochondrial proteins was based on electrophoretic andimmunochemical analysis. Blue-Native PAGE showed a decreasedcontent of the whole ATPase complex which retained normal sizeof approximately 640 kDa. It was detected in fibroblasts, skeletal
and heart muscle tissue, liver and in one case in brain.
ATP produkce <30% 4/4

Defekty ATPázy
„kvalitativní“ „kvantitativní“
mutace v mtDNA v jaderné DNA
strukturálně změněný ATPázový komplex s poškozenou funkcí
výrazně snížený obsah ATPázy
maternální dědičnost
Mendelovská dědičnost
neurologická symptomatologie
kardiomyopatie
X
X
X

Poruchy mitochondriálního meta-bolismu pyruvátu a cyklu kyseliny citronové
Deficit pyruvát karboxylázy Deficit fosfoenolkarboxykinázy Deficit pyruvát dehydrogenázy (PDH) Deficit dihydrolipoamid dehydrogenázy, E3 podjednotky PDH, mnohočetný deficit dehydogenáz 2-oxokyselin: deficit PDH, deficit dehydogenázy 2-oxoglutarátu, deficit dehydrogenáz větvených 2-oxo kyselin Deficit fumarázy Deficit sukcinát dehydrogenázy Deficit transportétu pro pyruvát Laktátová acidosa, často neurologické příznaky, progresivní průběh, příznaky postižení kosterního svalu Autosomálně recesivní onemocnění, deficit podjednotky α PDH E1 je dědičný gonosomálně recesivně

Komplex pyruvát dehydogenázy
Pyruvát → acetyl-CoA Dehydrogenázová komponenta:E1, podjednotka α je X vázaná Deficit PDHE1α : psychomotorická retardace, ataxie, křeče Charakteristické fenotypy: neonatální laktátová acidóza, Leighova encefalopatie: abnormální dýchání, apnoe, ataxie, svalová slabost, vývojové opoždění, degenerace bazálních ganglií U žen: křeče, subkortikální a kortikální atrofie Deficity ostatních podjednotek jsou vzácné laktátová acidóza, zvýšení koncentrace laktátu po jídle, snížení během hladovění Léčba: ketogení dieta, thiamin Nepříznivá prognóza

Defekty jaderné DNA
jaderná DNA - replikace, transkripce, translace, oprava … asemblace, strukturální podjednotky… Integrita a replikace mtDNA – thymidin fosforyláza ANT1, twinkle, polymeráza (POLG) Transport proteinů Fission / fuse mitochondrií – OPA1 Stabilita mitochondriální membrány – Bartův syndrom

Koordinovaná fůze jak vnější, tak vnitří mitochondriální membrány Mitofusiny jsou GTPasy lokalizované do zevní mitochondriální membrány Mfn1 a Mfn2 Mutace mitofusinu 2 způsobují neuropatii typu Charcot-Marie-Tooth neuropathy 2A(Hereditární motorická a senzorická neuropatie) GTPasa OPA1 z rodiny dynaminu: mutatovaná u autosomálně dominantní optické atrofie, gen je na chromozomu 3q28. Kjerova choroba – ztráta zrakové ostrosti, ztráta retinálních gangliových buněk
Mitochondriální fůze a štěpení

Mitochondriální fůze a štěpení

Struktura mitochondrií

Poruchy mitondriální beta-oxidace mastných kyselin
Karnitinový cyklus Beta oxidace Transfer elektronů na komplex II (deficit vede ke glutarové acidurii II typu) Syntéza ketolátek, ketolýza Deficity beta oxidace: Příznaky se často objevují po hladovění (obvykle 12-16h) Hypoglykémie Nízké koncentrace ketolátek v krvi a moči (U některých onemocnění svalová slabost, rhabdomyolýza, kardiomyopatie)

Porucha transportu MK
Porucha b-oxidace MK
Kardiomyopatie
Hepatopatie
Neketot.hypoglykémie
Myopatie
U-org.kyseliny
Profil karnitinů
Porucha b-oxidace MK
Metabolické myopatie
www.annualreviews.org (Bennett MJ, Fatty acid oxidation disorders, 2002)

Karnitinový cyklus Volné mastné kyseliny s dlouhým řetězcem jsou aktivovány na acyl-CoA estery v cytosolu a jsou importovány do mitochondrie pomocí karnitinového cyklu Mastné kyseliny o středně dlouhém a krátkém řetězci vstupují do mitochondrie přímo a jsou aktivovány na acyl-CoA estery v mitochondriální matrix CPT1 – karnitin palmitoyl transferasa I CPT2 - karnitin palmitoyl transferasa II CACT – karnitine /acylkarnitine translokase
www.bioscience.org

CPT I a CACT – kardiomyopatie, srdeční selhání,
arytmie, jaterní selhání
CPT II – časná forma – podobná CPT I
- late onset – sval.slabost, rhabdomyolýza,
netolerance dlouhého cvičení, není „sec.wind“
- dg – profil karnitinů, U-org.kyselin – bpn
EMG, CK mezi atakami v pořádku
- th – režimová opatření, strava
Porucha beta-oxidace MK

VLCAD – infantilní forma – SIDS
- late onset – rhabdomyolýza, intol.cvičení
LCHAD a MTP (trifunkční protein) – kardiomyopatie,
intol.cvičení, hepatopatie, retinopatie,
neuropatie
gravidní matky - HELLP
- dg – OH-acylkarnitiny, U-OK
- th – dieta, antihypoglyk.režim
Porucha beta-oxidace MK

MCAD – nejčastější; prevalentní mutace
- není primární myopatie
SCAD – široké klinické spektrum
myopatie (těžké,hepat.encefalopatie)…asympt.
Glutarová acidurie II typu – (ETF)
těžká forma (Reye sy, hypoglykémie,
progr.encefalopatie, (kardio)myopatie)
lehčí myopatická forma
Porucha beta-oxidace MK

Hlavní symptomy - sval.slabost, hypotonie, intolerance
cvičení, rhabdomyolýzy, laboratorní (z ataky)
Intolerance krátkého cvičení – porucha
glykogenu/glykolýzy
Intolerance vytrvalostního cvičení – utilizace MK,
mitopatie
Hladovění, chlad, infekce, léky –zhoršují myopatie
Široké spektrum – akutní x chronické
- infantilní x late onset
- izolované x multisystémové
Souhrn klinických projevů
Poruch beta-oxidace MK

Děkuji za pozornost
Related Documents











