Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis Nigar Fatma, 1 Prerna Singh, 1 Bhavana Chhunchha, 1 Eri Kubo, 2 T. Shinohara, 1 Biju Bhargavan, 1 and Dhirendra P. Singh 1 1 Department of Ophthalmology and Visual Sciences, University of Nebraska Medical Center, Omaha, Nebraska; and 2 Department of Ophthalmology, Kanazawa Medical University, Ishikawa, Japan Submitted 4 March 2011; accepted in final form 15 June 2011 Fatma N, Singh P, Chhunchha B, Kubo E, Shinohara T, Bhargavan B, Singh DP. Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apop- tosis. Am J Physiol Cell Physiol 301: C954 –C967, 2011. First pub- lished June 15, 2011; doi:10.1152/ajpcell.00061.2011.—The multi- functional cytoprotective protein peroxiredoxin 6 (Prdx6) maintains cellular homeostasis and membrane integrity by regulating expression of intracellular reactive oxygen species (ROS) and phospholipid turnover. Using cells derived from targeted inactivation of Prdx6 gene or its depletion by RNA interference or aging, we showed that Prdx6 deficiency in cells evoked unfolded protein response (UPR), evi- denced by increased expression or activation of proapoptotic factors, CHOP, ATF4, PERK, IRE- and eIF2- and by increased caspases 3 and 12 processing. Those cells displayed enhanced and sustained expression of endoplasmic reticulum (ER) stress-related chaperon proteins, Bip/glucose-regulated protein 78, calnexin, and calreticulin. Under cellular stress induced by hypoxia (1% O 2 or CoCl 2 treatment) or tunicamycin, Prdx6-deficient cells exhibited aberrant activation of ER stress-responsive genes/protein with higher expression of ROS, and died with apoptosis. Wild-type cells exposed to tunicamycin or hypoxia remained relatively insensitive with lower expression of ROS and ER-responsive genes than did Prdx6-deficient cells, but upregu- lation of ER stress responsive proteins or chaperones mimicked the UPR response of Prdx6-deficient or aging cells. Expression of Prdx6 blocked ER stress-induced deleterious signaling by optimizing phys- iologically aberrant expression of ER stress responsive genes/proteins in Prdx6-deficient cells or cells facing stressors, and rescued the cells from apoptosis. These findings demonstrate that impaired ho- meostasis and progression of pathogenesis in Prdx6-deficient lens epithelial cells or in aging cells should be blocked by a supply of Prdx6. The results provide a new molecular basis for understanding the etiology of several age-associated degenerative disorders, and potentially for developing antioxidant Prdx6-based therapeutics. peroxiredoxin 6; antioxidant; unfolded protein response; reactive oxygen species; endoplasmic reticulum OXIDATIVE STRESS HAS AN IMPACT on biological processes in- volved in cell survival and aging as well as on pathogenesis of diseases ranging from cataractogenesis to age-related neurode- generative conditions (3, 9, 14, 19, 39, 75). To counteract oxidative stress, hosts have evolved antioxidant defense sys- tems that include superoxide dismutases (SODs), catalase (Cat), glutathione peroxidase (Gpx), and antioxidant peroxire- doxins (Prdxs). The physiological role of antioxidants depends on their levels of expression and activity in cells. A decrease in expression of antioxidants leads to uncontrolled overproduc- tion of intracellular reactive oxygen species (ROS), resulting in dysfunction of cellular organelles including those in the endo- plasmic reticulum (ER) (63, 85). The normally functioning ER provides a unique and balanced oxidizing microenvironment for protein folding (28, 50, 70). Newly synthesized proteins gain their native conformation in the ER, where an efficient quality control system allows only correctly folded molecules to exit for their allotted destinations (23, 30). Thus, the un- folded protein response (UPR) is an adaptive signaling path- way evolved to prevent the accumulation of misfolded protein in the ER lumen. When cellular homeostasis fails in the increased redox environment generated by reduced expression of antioxidants, spontaneous accumulation of proteins eventu- ally leads to ER stress-induced cell death (70). Furthermore, diverse cellular stresses, as may be caused by chemotherapeu- tic drugs, radiation, hypoxia, or cellular stress, can initiate or accelerate both oxidative and ER stresses, causing greater damage in cells (53, 62, 78). Prdx6 acts to alter gene expres- sion by optimizing ROS expression, which culminates in either apoptosis or cell recovery (9, 14, 18, 38, 75). Thus mainte- nance of nearly constant levels of cellular Prdx6 expression is crucial to maintenance of the physiological levels of ROS necessary to regulate cell survival signaling. Cells deficient in the Prdx6 gene are known to display higher expression of ROS with impaired homeostasis and spontaneous apoptosis (14, 75), but the intracellular mechanism(s) by which Prdx6 deficiency relays adverse signaling and causes cells to be more sensitive to oxidative stress (14) is not known. Prdx6 is a member of the selenium-independent peroxidase family that has GSH peroxidase as well as acidic Ca 2 - independent phospholipase A2 (PLA 2 ) activities. Prdx6 has been found essential for maintaining cellular homeostasis (14, 16, 52, 72). We reported earlier that an extrinsic supply of Prdx6 provides cytoprotection against various stressors and delays the progression of cataractogenesis (14, 16, 35, 56). Although Prdx6 is classified as a peroxiredoxin based on homology of structure, its properties differ from those of other mammalian peroxidase family members, and the sequence associated with the protective activity of Prdx6 is not present in other peroxiredoxins (52). All six mammalian isoforms of Prdxs are relatively expressed at high levels and are differen- tially localized in cytoplasm, mitochondria, ER, nucleus, and peroxisomes (12, 33, 79), protecting them from various stres- sors. On the basis of the number of conserved catalytic cys- teines (peroxidatic cysteine), they are generally divided into two groups, 1-Cys and 2-Cys Prdxs. Prdx6 is the only member of the family that has non-selenium peroxidase and Ca 2 - independent PLA 2 activities (8, 36, 52, 54). In cells, Prdx6 participates in oxidative defense by suppressing intracellular Address for reprint requests and other correspondence: D. P. Singh, Dept. of Ophthalmology and Visual Sciences, Univ. of Nebraska Medical Center, Omaha NE 68198-5840 (e-mail: [email protected]). Am J Physiol Cell Physiol 301: C954–C967, 2011. First published June 15, 2011; doi:10.1152/ajpcell.00061.2011. 0363-6143/11 Copyright © 2011 the American Physiological Society http://www.ajpcell.org C954 on November 7, 2011 ajpcell.physiology.org Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Deficiency of Prdx6 in lens epithelial cells induces ER stressresponse-mediated impaired homeostasis and apoptosis
Nigar Fatma,1 Prerna Singh,1 Bhavana Chhunchha,1 Eri Kubo,2 T. Shinohara,1 Biju Bhargavan,1
and Dhirendra P. Singh1
1Department of Ophthalmology and Visual Sciences, University of Nebraska Medical Center, Omaha, Nebraska;and 2Department of Ophthalmology, Kanazawa Medical University, Ishikawa, Japan
Submitted 4 March 2011; accepted in final form 15 June 2011
Fatma N, Singh P, Chhunchha B, Kubo E, Shinohara T,Bhargavan B, Singh DP. Deficiency of Prdx6 in lens epithelial cellsinduces ER stress response-mediated impaired homeostasis and apop-tosis. Am J Physiol Cell Physiol 301: C954–C967, 2011. First pub-lished June 15, 2011; doi:10.1152/ajpcell.00061.2011.—The multi-functional cytoprotective protein peroxiredoxin 6 (Prdx6) maintainscellular homeostasis and membrane integrity by regulating expressionof intracellular reactive oxygen species (ROS) and phospholipidturnover. Using cells derived from targeted inactivation of Prdx6 geneor its depletion by RNA interference or aging, we showed that Prdx6deficiency in cells evoked unfolded protein response (UPR), evi-denced by increased expression or activation of proapoptotic factors,CHOP, ATF4, PERK, IRE-� and eIF2-� and by increased caspases 3and 12 processing. Those cells displayed enhanced and sustainedexpression of endoplasmic reticulum (ER) stress-related chaperonproteins, Bip/glucose-regulated protein 78, calnexin, and calreticulin.Under cellular stress induced by hypoxia (1% O2 or CoCl2 treatment)or tunicamycin, Prdx6-deficient cells exhibited aberrant activation ofER stress-responsive genes/protein with higher expression of ROS,and died with apoptosis. Wild-type cells exposed to tunicamycin orhypoxia remained relatively insensitive with lower expression of ROSand ER-responsive genes than did Prdx6-deficient cells, but upregu-lation of ER stress responsive proteins or chaperones mimicked theUPR response of Prdx6-deficient or aging cells. Expression of Prdx6blocked ER stress-induced deleterious signaling by optimizing phys-iologically aberrant expression of ER stress responsive genes/proteinsin Prdx6-deficient cells or cells facing stressors, and rescued thecells from apoptosis. These findings demonstrate that impaired ho-meostasis and progression of pathogenesis in Prdx6-deficient lensepithelial cells or in aging cells should be blocked by a supply ofPrdx6. The results provide a new molecular basis for understandingthe etiology of several age-associated degenerative disorders, andpotentially for developing antioxidant Prdx6-based therapeutics.
peroxiredoxin 6; antioxidant; unfolded protein response; reactiveoxygen species; endoplasmic reticulum
OXIDATIVE STRESS HAS AN IMPACT on biological processes in-volved in cell survival and aging as well as on pathogenesis ofdiseases ranging from cataractogenesis to age-related neurode-generative conditions (3, 9, 14, 19, 39, 75). To counteractoxidative stress, hosts have evolved antioxidant defense sys-tems that include superoxide dismutases (SODs), catalase(Cat), glutathione peroxidase (Gpx), and antioxidant peroxire-doxins (Prdxs). The physiological role of antioxidants dependson their levels of expression and activity in cells. A decrease inexpression of antioxidants leads to uncontrolled overproduc-tion of intracellular reactive oxygen species (ROS), resulting in
dysfunction of cellular organelles including those in the endo-plasmic reticulum (ER) (63, 85). The normally functioning ERprovides a unique and balanced oxidizing microenvironmentfor protein folding (28, 50, 70). Newly synthesized proteinsgain their native conformation in the ER, where an efficientquality control system allows only correctly folded moleculesto exit for their allotted destinations (23, 30). Thus, the un-folded protein response (UPR) is an adaptive signaling path-way evolved to prevent the accumulation of misfolded proteinin the ER lumen. When cellular homeostasis fails in theincreased redox environment generated by reduced expressionof antioxidants, spontaneous accumulation of proteins eventu-ally leads to ER stress-induced cell death (70). Furthermore,diverse cellular stresses, as may be caused by chemotherapeu-tic drugs, radiation, hypoxia, or cellular stress, can initiate oraccelerate both oxidative and ER stresses, causing greaterdamage in cells (53, 62, 78). Prdx6 acts to alter gene expres-sion by optimizing ROS expression, which culminates in eitherapoptosis or cell recovery (9, 14, 18, 38, 75). Thus mainte-nance of nearly constant levels of cellular Prdx6 expression iscrucial to maintenance of the physiological levels of ROSnecessary to regulate cell survival signaling. Cells deficient inthe Prdx6 gene are known to display higher expression of ROSwith impaired homeostasis and spontaneous apoptosis (14, 75),but the intracellular mechanism(s) by which Prdx6 deficiencyrelays adverse signaling and causes cells to be more sensitiveto oxidative stress (14) is not known.
Prdx6 is a member of the selenium-independent peroxidasefamily that has GSH peroxidase as well as acidic Ca2�-independent phospholipase A2 (PLA2) activities. Prdx6 hasbeen found essential for maintaining cellular homeostasis (14,16, 52, 72). We reported earlier that an extrinsic supply ofPrdx6 provides cytoprotection against various stressors anddelays the progression of cataractogenesis (14, 16, 35, 56).Although Prdx6 is classified as a peroxiredoxin based onhomology of structure, its properties differ from those of othermammalian peroxidase family members, and the sequenceassociated with the protective activity of Prdx6 is not present inother peroxiredoxins (52). All six mammalian isoforms ofPrdxs are relatively expressed at high levels and are differen-tially localized in cytoplasm, mitochondria, ER, nucleus, andperoxisomes (12, 33, 79), protecting them from various stres-sors. On the basis of the number of conserved catalytic cys-teines (peroxidatic cysteine), they are generally divided intotwo groups, 1-Cys and 2-Cys Prdxs. Prdx6 is the only memberof the family that has non-selenium peroxidase and Ca2�-independent PLA2 activities (8, 36, 52, 54). In cells, Prdx6participates in oxidative defense by suppressing intracellular
Address for reprint requests and other correspondence: D. P. Singh, Dept. ofOphthalmology and Visual Sciences, Univ. of Nebraska Medical Center,Omaha NE 68198-5840 (e-mail: [email protected]).
Am J Physiol Cell Physiol 301: C954–C967, 2011.First published June 15, 2011; doi:10.1152/ajpcell.00061.2011.
0363-6143/11 Copyright © 2011 the American Physiological Society http://www.ajpcell.orgC954
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

enzyme inactivation and membrane phospholipid peroxidation,and by eliminating excess ROS (13, 20, 38, 75).
UPR is primarily an adaptive response aiming to restore ERhomeostasis and protect cells from stress. With prolongedstress, the ER stress receptors can initiate proapoptotic path-ways, leading to cell death (69). CHOP/Gadd153 was the firstmolecule found to mediate ER stress-induced apoptosis (61).Empirically, different cell types have been observed to responddifferently to oxidative stress inducers such as ischemia (47).Because Prdx6-deficient cells are vulnerable to oxidativestress, and hypoxia activates UPR (32, 46, 85), the subsequentabnormal signaling can undermine cellular survival whenthreshold levels of cellular defense are inadequate. In thecurrent study, we found that lens epithelial cells (LECs) iso-lated from Prdx6-deficient mice (14) displayed increased ex-pression of CHOP and Bip, leading to the prediction that Prdx6depletion may be associated with ER stress and that deficiencyof Prdx6 is the initiator of UPR/ER stress in these cells. Inaddition, our investigations revealed the induction of all threearms of UPR—PERK, ATF6, and IRE1-�— and their down-stream targets in these cells, in which aberrant expressionlevels were further modulated by hypoxia (1% O2 or CoCl2, a
hypoxia mimic) (1). These data underscore the role of Prdx6 inmaintaining ER stress homeostasis by regulating cellular ROSat normal physiological levels. However, perturbation of ERhomeostasis may occur under various conditions. Given therole of Prdx6 in maintaining signaling pathways, its deficiencymay influence cell signaling, including both the UPR-mediatedsurvival response and the apoptotic cell death response to ERstress.
Using Prdx6-deficient cells coupled with Prdx6 overexpres-sion and aging cells, we found that loss of Prdx6 led toinitiation of UPR/ER stress signaling as evidenced by selectiveupregulation of UPR/ER stress-associated proteins such as Bip,CHOP, ATF4, and so on. These proteins were further increasedin Prdx6�/� cells exposed to oxidative/ER stressors, leading toapoptosis. In contrast, delivery of Prdx6 prevented overmodu-lation of the proteins and cell death. We also found thatPrdx6-deficient cells had enhanced sensitivity to oxidativestress from ROS accumulation, and these cells displayed al-tered expression of antioxidants and chaperon proteins andmRNA. The data presented here provide evidence that Prdx6deficiency induces/initiates UPR/ER stress-induced abnormal-ity and cell death, at least in lens cells. We propose that Prdx6
Fig. 1. A: photomicrograph of Prdx6�/� cells cultured in vitro showing phenotypic abnormalities. Lens epithelial cells (LECs) were isolated from peroxiredoxin6 (Prdx6)-targeted mutants (Prdx6�/�) and wild-type (Prdx6�/�) mice. Cells were cultured in complete DMEM medium (GIBCO) overnight. The medium wasreplaced with DMEM containing 0.1% BSA. Significant morphological alterations were observed in Prdx6�/� cells; they became elongated and fiberlike, formedcellular aggregates, packed irregularly, and showed apoptosis. Arrows indicate dead cells. B: involvement of oxidative stress in Prdx6�/� cells, showing elevatedlevels of reactive oxygen species (ROS). ROS-responsive fluorescence probe 2=,7=-dichlorofluorescein (H2-DCF-DA) assay was conducted to monitor theintracellular ROS level. Prdx6�/� and Prdx6�/� cells were cultured in 96-well plates in DMEM � 10% FBS. The next day, the medium was replaced with HBSScontaining H2-DCF-DA dye and fluorescence intensity was measured. Histogram values are means � SD of three independent experiments. OD, optical density.**Statistically significant difference (P � 0.001 vs. control). C: Western analysis of Prdx6�/� and Prdx6�/� cells showing expression or activation of Bip,CHOP, calnexin, peIF2-�, pPERK, pIRE-�, ATF4, and ATF6-� (#, nuclear extract) in Prdx6�/� and Prdx6�/� cells. Cells were cultured in 60-mm plates for48 h, cell lysates were prepared, proteins were resolved on 10% SDS-PAGE, and Western analysis was done. Membranes were stripped/restripped andimmunostained with different antibodies. In each experiment, �-actin was used as an internal marker. Protein bands were quantified using a densitometer, andlevels were normalized to corresponding �-actin levels; histograms are shown below the protein bands. Data represent means � SD of three independentexperiments. **P � 0.001 vs. control.
C955Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

is necessary for maintaining a homeostatic regulatory systemand allowing cells to maintain an intracellular microenviron-ment favorable for cellular function.
MATERIALS AND METHODS
Generation and validation of LECs isolated from lenses ofPrdx6�/� and Prdx6�/� mice. All animal experiments followed therecommendations set forth in the “Statement for the Use of Animalsin Ophthalmic and Visual Research” by the Association for Researchin Vision and Ophthalmology. Animal studies were approved by theUniversity of Nebraska Medical Center (UNMC). LECs isolated fromPrdx6-targeted mutants (Prdx6�/�) and wild-type (Prdx6�/�) micewere generated and maintained in Dulbecco’s modified Eagle’s me-dium (DMEM) with 10% fetal bovine serum (FBS), as describedearlier (14). Prdx6�/� 129/Sv mice were generated at Harvard Med-ical School (Boston, MA) under the supervision of Dr. David R.Beier. For the present study, we used Prdx6�/� mutant mice of pure129 background, and, as controls, wild-type 129/Sv inbred mice of thesame sex and age (Prdx6�/�). All animals were maintained under
specific pathogen-free conditions in an animal facility. LECs wereisolated from mice of identical age, and Western analysis was carriedout to confirm the presence of �A-crystallin, a specific marker ofLECs. Cells from 3–5 passages were used for the experiments.
Isolation of LECs from human subjects. Eye lenses isolated fromhuman eyes aged 18, 23, 24, 63, 64, and 74 yr were obtained from theLions Eye Bank, UNMC. LECs were generated as described earlierwith some modification (59). Briefly, clear lenses were washed withDMEM medium containing penicillin-streptomycin (100 �g/ml) andamphotericin B (25 �g/ml). Capsules were spread by forceps with celllayers upwards on the surface of plastic culture petri dishes. CompleteDMEM containing 15% FBS serum was added. The growth ofexplants culture was monitored routinely. For subcultivation, mono-layer of culture was incubated with trypsin (GIBCO), and the disso-ciated cells were split as described earlier (59, 68). LECs obtainedfrom 2 to 3 passages were used for the experiments.
Cell culture and generation of hypoxic stress. LECs were culturedin 96-well plates or 100-mm petri dishes according to the require-ments of the experiment. For each assay, cells were first cultured in
Fig. 2. A: quantitative real-time PCR showing differential expression of Prdx1–6 mRNA in Prdx6�/� and Prdx6�/� LECs. Total RNA was isolated andtranscribed into cDNA. Real-time PCR was performed using specific primers as described in MATERIALS AND METHODS. mRNA expression of each Prdx wasadjusted to the mRNA copies of �-actin. B: Western analysis showing expression of Prdxs 1–6 protein in Prdx6�/� and Prdx6�/� cells. �-Actin bands showequal loading. C: quantitative real-time PCR showing reduced levels of LEDGF and �B-crystallin in Prdx6�/� cells in comparison to Prdx6�/� LECs.D: real-time PCR showing the reduced levels of catalase and glutathione peroxidase in Prdx6�/� cells. E: Western blot showing reduced expression of LEDGFand �B-crystallin (B-crys) protein in Prdx6�/� cells. �-Actin was used to analyze equal loading. *P � 0.05, **P � 0.001, statistically significant difference.
C956 Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

DMEM containing 10% FBS for 24 h. Then the medium was replacedwith DMEM containing either 0.1% bovine serum albumin (BSA) or1% FBS. For hypoxic treatment, cells were either exposed to 1%oxygen (O2) in a hypoxic chamber (24) or treated with differentconcentrations of cobalt chloride (CoCl2) (37, 84). Tunicamycin, aknown ER stress inducer (27, 86), was used as a positive control.
Construction of Prdx6 antisense. Human LEC cDNA library wasused to isolate Prdx6 cDNA having a full-length open reading frame.A full-length Prdx6 antisense construct was made by subcloningPrdx6 cDNA into a pcDNA3.1/NT-GFP-TOPO vector in reverseorientation. Plasmid was amplified following TOP 10 bacterial celltransformation as described earlier (17).
Western blot analysis and antibodies. Cytoplasmic and nuclearextracts or total cell lysates were prepared in ice-cold radio immuno-precipitation assay (RIPA) lysis buffer, as described previously (15,17). Equal amounts of protein samples were loaded onto a 10% SDSgel, blotted onto polyvinylidene fluoride membrane (PerkinElmer,Waltham, MA), and immunostained with primary antibodies at theappropriate dilutions. The antibodies were Prdx6 monoclonal anti-body (Lab Frontier, Seoul, Korea), pIRE1-� (sc-20790, Santa CruzBiotechnology), ATF4 (ab50546, Abcam, Cambridge, MA), ATF6-�(sc-22799, Santa Cruz Biotechnology), tropomyocin (Tmp 1� and 1�,sc-28543, Santa Cruz Biotechnology), Bip (no. 3183, Cell SignalingTechnology), calnexin (SPA-860, Stressgen), CHOP (sc-7351, SantaCruz Biotechnology), peIF2-� (sc-11386, Santa Cruz Biotechnology),pPERK (sc-32577, Santa Cruz Biotechnology), caspase 3 (no. 9665,Cell Signaling), and caspase 12 (no. 2202, Cell Signaling, ab8117,Abcam; sc-5627, Santa Cruz Biotechnology). Membranes were incu-bated with horseradish peroxidase-conjugated secondary antibodies.Specific protein bands were visualized by incubating the membranewith luminal reagent (Santa Cruz Biotechnology), and the imageswere recorded with FUJIFILM-LAS-4000 luminescent image ana-
lyzer (FUJIFILM Medical Systems). To ascertain comparative expres-sion and equal loading of the protein samples, the membrane stainedearlier was stripped and reprobed with �-actin antibody (Sigma).
Expression and purification of TAT-HA-Prdx6 fusion protein. Con-struction and purification of Prdx6 linked to TAT were performed asdescribed previously (38). Briefly, a full-length cDNA of Prdx6 wasisolated from human LEC cDNA library and cloned into TAT-HA-Prdx6 prokaryotic expression vector. Recombinant protein was puri-fied using Ni2�-nitrilotriacetic acid Sepharose column. Escherichiacoli BL21 (DE3) was transformed with pTAT-HA-Prdx6, and thecells were harvested in binding buffer and sonicated. Immediatelyafter centrifugation, supernatant containing TAT-HA-Prdx6 wasloaded onto a 2.5-ml column. The fusion protein was washed, elutedwith an elution buffer, and dialyzed. The purified protein was eitherused directly for protein transduction or aliquoted and stored frozen in10% glycerol at �80°C for later use. A batch of recombinant proteinTAT-HA-Prdx6 was passed through a Detoxi-Gel endotoxin-remov-ing gel column (no. 20344; Pierce, Rockford, IL) to exclude endotoxincontamination, if any. In a parallel experiment, this preparation wasused to compare protective efficacy of Prdx6-linked to TAT-HA thatwas not purified through the column.
Quantitative real-time PCR. Quantitative real-time PCR was per-formed using LightCycler 480II as described earlier (38, 72). Prdxs and�-actin primers were purchased from Roche Applied Sciences. Gpx1 andcatalase-specific primers were used as described earlier (72). The com-parative Cp method was used to calculate relative fold expression levelsusing LightCycler 480 software (release 1.5.0 SP3). The Cps of targetgenes were normalized to the levels of �-actin as an endogenous controlin each group. Primers were as follows: �-actin forward: 5=-CTAAGGCCAACCGTGAAAAG-3= and reverse: 5=-ACCAGAG-GCATACAGGGACA-3=; Prdx1 forward: 5=-GTGAGACCTGTG-GCTCGAC-3= and reverse: 5=-TGTCCATCTGGCATAACAGC-3=;
Fig. 3. A: photomicrograph of Prdx6�/� cellsfollowing transfection with either green fluores-cent protein (GFP)-vector (a) or Prdx6-anti-sense (Prdx6-AS) (b). Prdx6�/� cells exhibitmorphological changes and cell death (whiterounded cells, indicated by arrows) similarto Prdx6�/� cells, showing the importanceof Prdx6 in maintaining cellular homeosta-sis. B: intracellular ROS level increased inPrdx6�/� cells transfected with Prdx6-AS.Cells were transfected with either Prdx6-AS orGFP-vector, and ROS levels were measuredusing H2-DCF-DA dye. *P � 0.05. C: annexinV-FITC/propidium iodide (PI) binding assayshowing apoptotic cell death in cells trans-fected with Prdx6-AS. Cells transfected witheither GFP-vector or Prdx6-AS were sub-jected to apoptotic cell death assay usingannexin V-FITC/PI staining followed by flowcytometric analysis. Representative histo-gram shows % annexin V-FITC/PI staining ofcells (gray and black bars). **P � 0.001.D: Western analysis showing reduced level ofPrdx6 and increased expression of endoplas-mic reticulum (ER) stress-response proteinsin Prdx6�/� cells following transfection ofPrdx6-AS. This result shows that Prdx6downregulation or depletion initiates ERstress. Histograms represent means � SD ofthree independent Western blot experiments.
C957Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

Prdx2 forward: 5=-GACGAGCATGGGGAAGTCT-3= and reverse: 5=-TCCTTGCTGTCATCCACATT-3=; Prdx3 forward: 5=-GTGCCTCTT-GCGTGCTCT-3= and reverse: 5=-ACTTGCATGACGAGCAACC-3=;Prdx4 forward: 5=-TGACAAGCATGGAGAAGTCTG-3= and reverse:5=-CAGCTGGATCTGGGATTATTG-3=; Prdx5 forward: 5=-GATT-GAAGAGTGGGGTCGAG-3= and reverse: 5=-TCTGTCGCCTTC-CCAAAG-3=; Prdx6 forward: 5=-TTTCAATAGACAGTGTTGAG-GATCA-3= and reverse: 5=- CGTGGGTGTTTCACCATTG-3=.
Assay of intracellular ROS level. Intracellular ROS level wasmeasured by use of fluorescent dye dichlorofluorescin diacetate (H2-DCF-DA), a nonpolar compound that is converted into a polarderivative (dichlorofluorescein) by cellular esterase after incorpora-tion into cells (14, 38). Cells were cultured in 96-well plates for 24 hwith DMEM having 10% FBS. Cells were then either exposed to 1%O2 or treated with CoCl2 and tunicamycin at various doses fordifferent time intervals. The medium was replaced with Hanks’solution containing 10 �M H2-DCF-DA dye, and cells were incu-bated. Following 30 min of incubation at room temperature, intracel-lular fluorescence was detected with excitation at 485 nm and emis-sion at 530 nm as measured by a Spectra Max Gemini EM (MolecularDevices, Sunnyvale, CA).
Cell viability assay. A colorimetric MTS assay (Promega, Madison,MI) was performed as described earlier (14, 38). This assay of cellularproliferation/viability uses 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2 to 4-(sulfophenyl)2H-tetrazolium salt. Whenadded to medium containing viable cells, MTS is reduced to awater-soluble formazan salt. The A490 nm value was measured after 4h with an ELISA reader. Results were normalized with absorbance ofthe untreated control(s).
Cell apoptosis assay. Apoptosis was measured by flow cytometryto quantify the levels of oxidative stress-induced apoptotic cells. Theannexin V binding assay was performed with the annexin V-FITC/PI
Apoptosis Detection Kit I (BD Biosciences), following the company’sprotocol. Briefly, Prdx6�/� and Prdx6�/� cells were seeded in100-mm plates and were subjected to stressors or chemical chaperone[4-phenylbutyrate (4-PBA)] for variable periods. After an incubationperiod, cells were washed twice with ice-cold phosphate-bufferedsaline solution (PBS). Finally, 105 cells were resuspended in 100 �l ofbinding buffer. The cell suspension (1 � 105/100 �l) was thenincubated with 5 �l of annexin V-FITC and 5 �l of propidium iodide(PI) for 15 min at room temperature in the dark. A volume of 500 �lwas maintained by adding 400 �l of binding buffer to each mixture,and samples were analyzed by flow cytometry within 1 h (UNMCcore facilities). All experiments were carried out in triplicate.
Statistical methods. Data are means � SD of the indicated numberof experiments. Data were analyzed by Dunnett’s multiple conversiontests or Student’s t-test when appropriate. P � 0.05 was defined asindicating a statistically significant difference.
RESULTS
Cells deficient in Prdx6 had enhanced expression of ERstress response genes, were sensitive to oxidative stress, andshowed abnormal phenotypes with spontaneous apoptosis.ER stress and oxidative stress are closely linked, and oxidativestress is known to disrupt protein folding (50). Prdx6 plays avital role in maintaining cellular survival signaling by optimiz-ing ROS expression at physiological levels, as shown by thefinding that LECs and lenses isolated from mice in whichPrdx6 has been genetically deleted (Prdx6�/�) are susceptibleto oxidative stress-induced deleterious signaling (14, 38–40,72–74, 76). To examine whether Prdx6 deficiency in cellsevokes ER stress, we used LECs isolated from Prdx6-mutant
Fig. 4. A: enhanced expression of ER stress-responsive proteins in primary LECs obtainedfrom aging human subjects. Western analysisis a representative of 24- and 64-yr-old sub-jects. Cell lysate was prepared from LECsisolated from lenses of young (18 or 24 yr old)and aging (63, 64, or 74 yr old) human subjectsand analyzed by Western analysis. Proteinblots were quantified using a densitometer, andlevels were normalized to corresponding �-ac-tin levels; histograms are shown beside theprotein bands. B: ROS expression in aging andyounger cells. Data represent means � SD ofthree independent experiments. hLECs, humanLECs. C: increased expression of Bip in cellsafter H2O2 exposure. Cells were treated with50 �M H2O2 for 24 h, and cell lysate wasprepared. A marked increase in the expressionof Bip was observed in aged cells after H2O2
exposure (top). Results are derived from threeindependent experiments. *P �0.05, **P �0.001.
C958 Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

(Prdx6�/�) and wild-type (Prdx6�/�) mice and cultured thecells in DMEM containing 10% FBS. Cells were analyzed atdifferent time intervals and photomicrographed. As reportedearlier, 48 h after culture, Prdx6�/� cells displayed significantabnormal morphological changes with elevated expression ofROS as determined by the ROS-responsive fluorescence probe2=,7=-dichlorofluorescein (Fig. 1B). The cells became elon-gated and fiberlike, were packed irregularly, formed cellularaggregates, and detached more frequently. They also under-went spontaneous cell death (Fig. 1A). These morphologicalchanges in Prdx6�/� LECs were similar to those observed intransdifferentiation in anterior subcapsular cataracts and pos-terior capsule opacifications (45) after cataract surgery.
Oxidative stress delays recovery of unfolded proteins andinitiates the ER stress response (UPR) (48, 49). ER stresspathways involve three major distinct stress sensor proteins,PERK, pIRE1-�, and ATF6; Bip is the master regulator ofthose sensor proteins. Therefore, we first monitored the expres-sion levels of Bip in Prdx6�/� cells and found that the cells hadincreased Bip expression. Dissociation of Bip from PERKleads to the autophosphorylation of PERK, which in turnphosphorylates eIF2-� and activates ATF4 translation. ATF4increases the expression of proapoptotic factor CHOP (43, 61,66, 67, 87). Investigating the expression levels of ER stress-related proteins in Prdx6�/� cells, we found that, indeed,Prdx6-depleted cells carried increased expression of phosphor-ylated PERK and eIF2-�, and that CHOP and ATF4 proteinlevels were also higher (Fig. 1C), indicating the prevalence of
UPR/ER stress response in Prdx6-deficient cells. Furthermore,the dissociation of Bip from IRE1-� permits the activation ofthat protein, and the activated IRE1-� is a proapoptotic factor.On the other side, the dissociation of Bip from ATF6 permitsthe translocation of ATF6 into the Golgi compartment ofintramembrane proteolysis. To test whether these two factorsare also elevated in Prdx6�/� cells, we extracted cytosolic andnuclear extracts from Prdx6-depleted cells. Western analysisdemonstrated increased levels of pIRE1-� and ATF6 expres-sion in cytosolic and nuclear fraction of Prdx6�/� cells, re-spectively. Notably, ATF6-� was cleaved, giving rise to 37-kDa protein band (Fig. 1C; ATF6-� #) in Prdx6�/� cells, butnot in Prdx6�/� cells. Taken together, Western analysis andthe relative densitometry of protein bands as shown in Fig. 1Crevealed that Prdx6 depletion activated ER stress signaling inLECs.
Prdx6 deficiency influenced the expression of other membersof the Prdx family (Prdxs 1–5) and antioxidant/chaperoneprotein. Our previous report and present data indicate thatPrdx6�/� cells are under oxidative pressure (redox state), thatsuch cells are unable to maintain homeostasis, and that theyundergo apoptosis (14, 16, 38, 39, 52, 54, 72). In the presentstudy, Prdx6-deficient cells harbored activated ER stress sig-naling with phosphorylation of eIF2-�, a global translationalinhibition (Fig. 1C). Notably, the cells were obviously dam-aged during oxidative stress, behaving like aging cells with lowantioxidant levels. Other enzymes appeared unable to maintainLEC homeostasis. We hypothesized that levels of other en-
Fig. 5. A: Prdx6�/� and Prdx6�/� cells with orwithout hypoxia stress showing morphologicalchanges (photomicrograph) and cell death(rounded white cells, indicated by arrows). Cellswere cultured in 60-mm plates and exposed to1% O2 with a hypoxic chamber. Prdx6�/� cellswere highly sensitive to hypoxic stress (d). Incontrast, Prdx6�/� cells were resistant to hyp-oxia stress (b), demonstrating the protective roleof Prdx6. B: measurement of intracellular redoxstate of Prdx6�/� and Prdx6�/� cells after hyp-oxia. Cells were exposed to 1% oxygen, andROS level was measured with H2-DCF-DA dye.ROS were further elevated in Prdx6�/� cellsafter hypoxia exposure (B, gray bar vs. blackbar). C: effect of hypoxia on viability ofPrdx6�/� and Prdx6�/� cells. Cells were cul-tured in 48-well plates containing DMEM sup-plemented with 10% FBS; 24 h later, cells wereexposed to 1% O2 for 48 h and cell viability wasestimated using colorimetric MTS assay. Grayand black bars denote relative cell viability ofPrdx6�/� and Prdx6�/� cells with or withouthypoxia following normalization with absor-bance. Data represent means � SD of threeindependent experiments. **P � 0.001, statisti-cally significant difference.
C959Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

zymes may be altered in Prdx6�/� cells. By using real-timePCR or Western analysis, we monitored the levels of mRNAand protein expression of known major cytoprotective mole-cules: Prdxs 1–5, Cat, Gpx1, chaperone protein �B-crystallin,and survival transcriptional protein LEDGF, using probes spe-cific to corresponding genes/proteins (14, 16, 38–40, 72). Wefound that the mRNA and protein expression levels of Prdx 1and 5 were decreased. In contrast, Prdx 2 was significantlyincreased, and expression of Prdx 3 and 4 was also increased(Fig. 2, A and B). Furthermore, these cells displayed a reduc-tion in expression of Cat and Gpx-1 accompanied by dimin-ished expression of �B-crystallin and LEDGF (Fig. 2, C–E).The increased expression of Prdx 2, 3, and 4 (Fig. 2, A and B)was not able to attenuate the adverse reaction in Prdx6�/�
cells. We propose that Prdx6�/� cells represent a model thatcan be used to elucidate the signaling pathways involved inaging cells, because aging is related to a decline in expressionof protective proteins.
Prdx6 knockdown initiated ER response similar to that inPrdx6-deficient cells. Prdx6�/� cells are under chronic oxida-tive stress that may not provide the underlying injurious sig-naling that occurs during acute oxidative stress. Therefore, wedepleted Prdx6 by transfecting cells with antisense specific toPrdx6 (17). The transfection caused elevation of ROS expres-sion and significant cell death (Fig. 3, A and B). Next, toexamine the type of cell death triggered by knockdown of Prdx6,we analyzed the transfected cells for annexin V-FITC/PI assay(Fig. 3C, black bar). Results indicated that reduced expression ofPrdx6-induced apoptosis in LECs. In parallel experiments, cellextracts were prepared and subjected to Western analysis toexamine whether ER stress genes were upregulated. Of thesegenes, CHOP and Bip/glucose-regulated protein 78 (GRP/78)were often used as UPR/ER stress markers. As expected, Prdx6
knockdown strikingly induced the expression of CHOP and Bip,and the cells showed activated expression of caspase 12 (Fig. 3D,right lane and histogram).
Aging cells displayed UPR. The ability to engender resis-tance to stress under adverse conditions is a fundamentalcellular defense. The resistance capability of cells is evidentlydecreased during aging due to reduced expression of defensemolecules (65). Earlier research revealed a decline of Prdx6mRNA and protein in aging mouse LECs. In the present studywe sought to extend that finding by determining whetherreduced expression of Prdx6 in aging cells contributes tobaseline UPR/ER stress, making these cells susceptible tooxidative stress. We cultured LECs isolated from human sub-jects aged 24 and 64 yr. Using Western analysis, we examinedexpression of Prdx6, Bip, and ATF4 and found that, indeed, ERstress-associated genes or their products were activated (butnot dramatically) in the aged cells than in the younger ones(Fig. 4A). The aged cells also had elevated ROS expression(Fig. 4B). In addition, among cells facing oxidative stress fromH2O2, the aged cells were more sensitive to ER stress and hadincreased expression of the UPR marker Bip, a key indicator ofER stress (Fig. 4C). Collectively, the results revealed that lossof Prdx6 was a major event in initiation/activation of ER stresssignaling as observed in Prdx6-deficient cells. Since such cellscharacteristically behave similarly to aged cells, in later exper-iments, Prdx6�/� cells were utilized as a model system foraging cells.
Prdx6�/� cells were sensitive to hypoxia-induced cytotoxic-ity and displayed increased ROS expression. Prdx6 is a mul-tifunctional protein that protects cells against oxidative stressby optimizing ROS (14, 38, 54, 72). Hypoxia increases intra-cellular ROS production in a variety of cells (7, 26, 46, 55, 69,77). Mitochondria have been proposed as a primary source of
Fig. 6. A: photomicrograph of Prdx6�/� andPrdx6�/� cells with or without CoCl2 treat-ment. Cells were treated with different concen-trations of CoCl2 (50, 100, 150 �M) for 48 h.No significant cell death occurred in Prdx6�/�
cells (top: b, c, d). In contrast, significant celldeath was observed in Prdx6�/� cells (bottom:f, g, h). Arrows indicate dead cells (roundedwhite cells). B: H2-DCF-DA assay showingincreased ROS in Prdx6�/� cells after CoCl2treatment. Dye was added to the cells with orwithout CoCl2 treatment, and fluorescence in-tensity was measured. C: MTS assay showinghigher susceptibility of Prdx6�/� cells toCoCl2 treatment. Prdx6�/� or Prdx6�/� cellswere exposed to various concentrations ofCoCl2, and MTS assay was conducted after 48h. A significant decrease in survival ofPrdx6�/� cells was observed compared withPrdx6�/� cells, suggesting that Prdx6 is essen-tial to protect cells from hypoxia-induced dam-age. Results were normalized according to theabsorbance of the untreated control at termi-nation of experiments. Data represent means �SD of three independent experiments. **P �0.001, statistically significant difference.
C960 Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

ROS production during hypoxic stress. Prdx6 has been foundto be translocated into mitochondria during ischemia, suggest-ing that Prdx6 may eliminate hypoxia-induced ROS-mediatedcell injury (12, 14, 38, 72) and thereby maintain survivalsignaling (14, 38). We tested the ability of Prdx6 to mountcytoprotection against hypoxic stress. Exposure of cells tohypoxia (1% O2 for 48 h) or to variable concentrations of thehypoxia mimic CoCl2 significantly increased cell injury inPrdx6-deficient cells, as shown in photomicrographs (Fig. 5Advs. Ab and Fig. 6Ab vs. Af, Ac vs. Ag, and Ad vs. Ah). Thesecells displayed an abundance of ROS expression when mea-sured by H2-DCF-DA fluorescence dye (Fig. 5B, black bar,and Fig. 6B, black bar), in contrast to Prdx6�/� cells. How-ever, photomicrographs showed cell death in Prdx6�/� cellswithout hypoxia, because these cells are always in redox state(14, 38, 72). In contrast, Prdx6�/� cells showed some growthinhibition.
Next we evaluated the protective efficacy of Prdx6.Prdx6�/� and Prdx6�/� cells were exposed to hypoxia (1%O2) or treated with increasing concentrations of CoCl2 andwere analyzed for cell viability by MTS assay and apoptosisassay (annexin V-FITC/PI binding assay). The Prdx6�/� cellswere significantly viable and survived well even with exposure tohypoxic stressors (1% O2: Fig. 5C, gray bar, or hypoxia mimic,
CoCl2; Fig. 6C, gray bars), demonstrating the ability of Prdx6 toprotect cells against hypoxia-induced apoptosis (Fig. 7A, B, and C,gray bars).
Hypoxia-exposed Prdx6�/� cells showed enhanced ERstress. ER stress is triggered by a variety of stressors includinghypoxia (81). Prdx6�/� cells display ER stress signaling andare vulnerable to stress-induced cell death. To examinewhether hypoxia would stimulate ER stress signaling in Prdx6-deficient cells, we first exposed the cells to variable concen-trations of CoCl2, a hypoxia mimic (10), and measured theexpression and activation of caspase 12 (no. 2202, Cell Sig-naling) and the induction of CHOP, known to be involved inER stress-induced apoptosis. Compared with Prdx6�/� cells,Prdx6�/� cells exposed to hypoxia were found to be morevulnerable to cell death and underwent apoptosis (Figs. 5 and6) and showed increased expression of CHOP and caspase 12(and cleaved form of caspase 12) (Fig. 8, right, and black bars).However, Prdx6-deficient cells harbored relatively higheramounts and activated caspase 12. Densitometry analysis ofprotein bands also showed elevation of Bip and calnexin (Fig.8), but these chaperone proteins were not able to suppress celldeath, suggesting that ER stress occurs downstream of oxida-tive stress.
Fig. 7. Prdx6�/� cells are more susceptible to CoCl2 (hypoxia mimic)-induced apoptosis. Cells were treated with different concentrations of CoCl2 (a, untreated;b, 50 �M; c, 100 �M; d, 150 �M). After 48 h of treatment, apoptosis was evaluated using annexin V-FITC/PI staining followed by flow cytometry. A andB: representative plots showing annexin V-FITC/PI staining of Prdx6�/� and Prdx6�/� cells. The proportion of late or apoptotic cells is shown. C: histogramshowing results of three independent experiments. A significant increase of apoptotic cells is observed (compare gray vs. black bars). Data represent means �SD of three independent experiments.
C961Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from
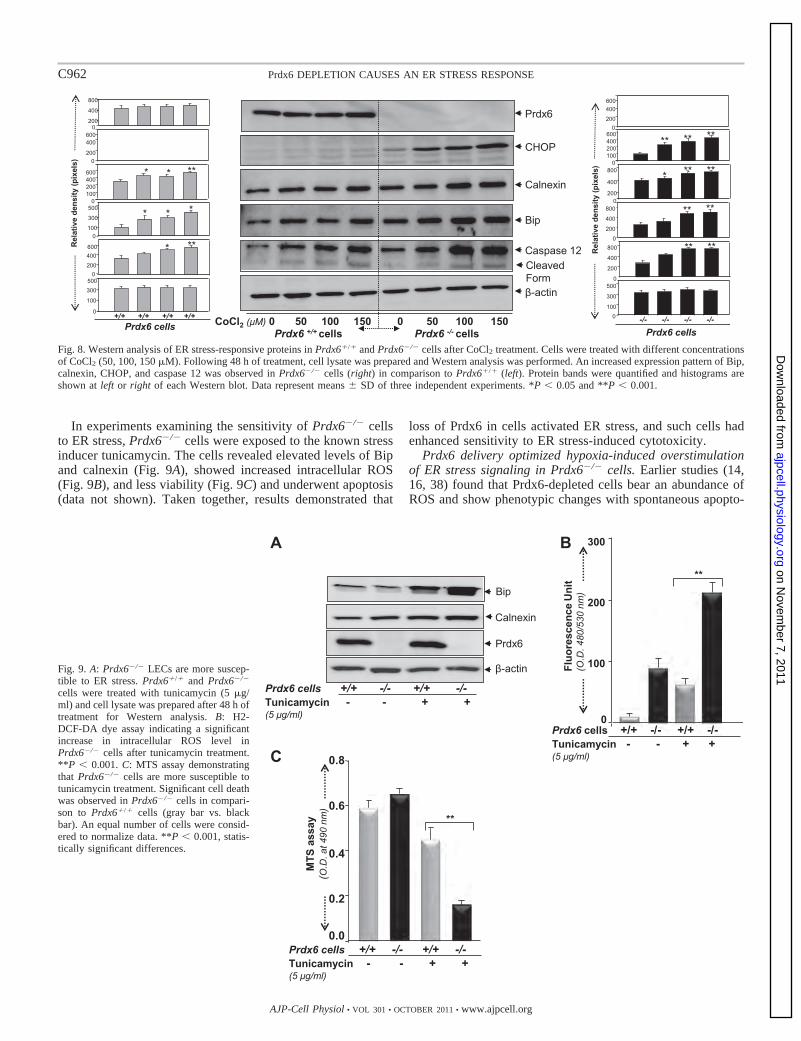
In experiments examining the sensitivity of Prdx6�/� cellsto ER stress, Prdx6�/� cells were exposed to the known stressinducer tunicamycin. The cells revealed elevated levels of Bipand calnexin (Fig. 9A), showed increased intracellular ROS(Fig. 9B), and less viability (Fig. 9C) and underwent apoptosis(data not shown). Taken together, results demonstrated that
loss of Prdx6 in cells activated ER stress, and such cells hadenhanced sensitivity to ER stress-induced cytotoxicity.
Prdx6 delivery optimized hypoxia-induced overstimulationof ER stress signaling in Prdx6�/� cells. Earlier studies (14,16, 38) found that Prdx6-depleted cells bear an abundance ofROS and show phenotypic changes with spontaneous apopto-
Fig. 8. Western analysis of ER stress-responsive proteins in Prdx6�/� and Prdx6�/� cells after CoCl2 treatment. Cells were treated with different concentrationsof CoCl2 (50, 100, 150 �M). Following 48 h of treatment, cell lysate was prepared and Western analysis was performed. An increased expression pattern of Bip,calnexin, CHOP, and caspase 12 was observed in Prdx6�/� cells (right) in comparison to Prdx6�/� (left). Protein bands were quantified and histograms areshown at left or right of each Western blot. Data represent means � SD of three independent experiments. *P � 0.05 and **P � 0.001.
Fig. 9. A: Prdx6�/� LECs are more suscep-tible to ER stress. Prdx6�/� and Prdx6�/�
cells were treated with tunicamycin (5 �g/ml) and cell lysate was prepared after 48 h oftreatment for Western analysis. B: H2-DCF-DA dye assay indicating a significantincrease in intracellular ROS level inPrdx6�/� cells after tunicamycin treatment.**P � 0.001. C: MTS assay demonstratingthat Prdx6�/� cells are more susceptible totunicamycin treatment. Significant cell deathwas observed in Prdx6�/� cells in compari-son to Prdx6�/� cells (gray bar vs. blackbar). An equal number of cells were consid-ered to normalize data. **P � 0.001, statis-tically significant differences.
C962 Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

sis. Treatment with MnSOD mimetic or Prdx6 supply attenu-ates these adverse cellular events (14, 16, 38). But it wasunknown whether a supply of Prdx6 would attenuate the basalprevalence of ER stress in Prdx6�/� cells or the stimulation ofER stress in these cells during stress. Hypoxic stress is asso-ciated with increased production of free radicals. Also, accu-mulation of unfolded proteins triggers ER stress and is consid-ered a part of the cellular response to hypoxia (32). We usedTAT-linked Prdx6 to supply Prdx6 to deficient cells facinghypoxic stress (38) and measured expression levels of ERstress-associated genes or gene products as well as intracellularlevel of TAT-linked Prdx6 (data not shown). Western analysisshowed that, in Prdx6�/� cells exposed to 1% O2, the level ofER stress-related genes/products PERK, ATF4, eIF2-�, Bip,and CHOP along with caspase 12 (sc-5627, Santa Cruz Bio-technology) and 3 (no. 9665, Cell Signaling) were overstimu-lated significantly (Fig. 10, A and B) compared with the levelsin Prdx6�/� cells. We also examined levels of tropomyosinprotein (Tmp 1� and 2�) expression, suggested to be unregu-lated during adverse signaling. Indeed, expression of theseproteins was increased. The cells that received an extrinsicsupply of Prdx6 showed inhibition of activated ER stress-related genes under hypoxic conditions, as evidenced by re-duced expression of their protein level (Fig. 10, A and B, lane3 vs. lane 5 and gray bars vs. black bars). Interestingly, theexpression of cleaved caspases (activated forms) was reduced,suggesting that Prdx6 attenuates the processing of caspases(Fig. 10, caspase 12 cleaved form or caspase 3 cleaved form,and histogram). No effect was observed on �-actin used as
control, indicating the role of Prdx6 deficiency-mediated acti-vation of ER stress.
Sodium 4-PBA, a chemical chaperone, blocked apoptosis inPrdx6�/� LECs. Recently, several researchers have reportedthat sodium 4-PBA acts as a chemical chaperone to reverse themislocalization or aggregation of proteins in ER and thereforeto inhibit UPR/ER stress-induced apoptosis (22, 25, 83). Cellsdeficient in Prdx6 are more susceptible to internal and externalstresses leading to apoptosis (38, 72). To determine whetherapoptosis in Prdx6-deficient cells is due to ER stress, we used4-PBA, an inhibitor of ER stress-induced apoptosis. Cells werecultured with increasing amounts of 4-PBA, and after 16 h,cells were analyzed for annexin V-FITC/PI binding. The re-sults showed that 4-PBA delivery attenuated apoptosis inPrdx6�/� cells (Fig. 11, gray vs. black bars), demonstratingprevalence of ER stress-induced apoptotic signaling in thesecells. However, none of the 4-PBA concentrations used in theexperiment provided 100% protection, indicating the possibleinvolvement of some other type of ER-stress-induced apoptoticsignaling that is not attenuated by the addition of 4-PBA.
DISCUSSION
An imbalance between cellular antioxidant defense sys-tem(s) and ROS-driven oxidative stress has been implicated inpathogenesis of cancer and degenerative disorders such asAlzheimer’s and Parkinson’s diseases and cataractogenesis(13, 14, 16, 38, 40, 71). In cells, Prdx6 participates in oxidativedefense by eliminating excess ROS and thereby optimizing
Fig. 10. A and B: Western blot analysis of protein extracts from Prdx6�/� and Prdx6�/� cells showing upregulation of ER-stress related proteins followinghypoxia and its normalization by Prdx6 delivery. Prdx6�/� cells were supplied with TAT-HA-Prdx6 before hypoxia stress (1% O2) to evaluate its ability toprevent hypoxia-induced overexpression of ER stress-related proteins (40). Cell lysates were prepared after 48 h of stress using RIPA buffer, and Western analysiswas performed. Notably, extrinsic supply of Prdx6 to Prdx6�/� cells could restore hypoxia-induced overmodulation of ER stress-related proteins (compare lane3 vs. lane 5 and lane 3 vs. lane 4). Expression levels (protein bands) were quantified and values are presented as histograms. Data represent the mean � S.D.of three independent experiments. **P � 0.001, statistically significant differences vs. control.
C963Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from
them at cellular physiological levels to maintain cell survivalsignaling (13, 14, 16, 38, 40, 72, 74, 76). In our earlier studies,we have shown that those Prdx6-deficient cells undergo spon-taneous apoptosis and are more susceptible to oxidative stress(14, 38). Recent accumulating evidence reveals that oxidativestress is closely linked to UPR and activates UPR/ER stress(32, 46, 50, 82, 85). In the present study, by using Prdx6-deficient cells as a model system, we have unveiled deathsignaling that causes abnormalities and apoptosis (13, 16, 38,40, 72). Our study suggests a model in which deficiency or lossof the antioxidant Prdx6 causes initiation of UPR, and thisprocess becomes overstimulated in response to stressors. Fig-ure 1B shows that Prdx6-deficient cells contain higher thannormal levels of ROS and undergo spontaneous apoptosis.These results were consistent with previous published reports(14, 38). These cells express activated and upregulated ERstress genes/gene products such as Bip/GRP/78 and calnexin,markers of ER stress (Fig. 1C). Bip and calnexin are ER-resident chaperon proteins that play major roles in maintainingER quality control. Under resting or unstressed conditions, Bipbinds to the luminal domain of all three ER stress receptors andinhibits activation of PERK, IRE1-�, and ATF6 (4, 42). During
UPR/ER stress, these molecules dissociate from Bip and be-come activated. Thus UPR activation is correlated with Bipdissociation with these receptors. In our Western analysisexperiment, we found upregulation and activation of ATF6 andpPERK, suggesting that Prdx6�/� cells are under ER stress(Fig. 1C).
Overproduction of ROS induced by various internal orexternal stresses is injurious to cells and tissues. To cope withcellular stressors, cells have evolved defense systems thatinclude chaperones like �B-crystallin survival factor and avariety of antioxidants such as Cat, GPxs, and Prdxs. Byexamining the expression levels of other antioxidants andmolecules involved in protecting cells from stressors inPrdx6�/� cells, we found that the mRNA and protein expres-sion of Prdx2 and Prdx4 were significantly upregulated. Otherantioxidants, including �B-crystallin and the survival factorLEDGF, were downregulated (Fig. 2, C and E). On the basisof that finding and because other Prdxs and protectivemolecules did not counteract the changes in Prdx6�/�
LECs, we think that Prdx6 plays a pivotal role in blockingER stress, at least in LECs. Wang et al. (72) and Manevichand Fisher (52), in work with Prdx6�/� mice, reported thatendogenously produced mouse Prdx6 functions in vivo as anantioxidant enzyme, and its function is not redundant withthat of other Prdxs and antioxidant enzymes. Prdx6 has bothGSH peroxidase and PLA2 activities (52), and recently wefound that Prdx6 maintains Ca2� homeostasis (13, 16).These features make Prdx6 different from other antioxi-dants.
Moreover, antioxidants also play a pivotal in maintaining areductive cytosolic environment through both catalytic andnonenzymatic processes (56). In particular, antioxidants arelikely to regulate intracellular ROS in a localized fashion. Therole of Prdx6 in maintaining cellular survival signaling path-ways and its expression in mitochondria (12) indicate that theexpression level may influence both the UPR-mediated sur-vival and apoptotic cell death responses to ER stress. We positthat deficiency or reduced expression of Prdx6 may initiate ERstress due to cellular redox-environment, and that ER stress canbe overstimulated during hypoxia or oxidative stresses, leadingto apoptotic signaling. Prdx6-antisense experiments, whichrevealed upregulated expression of ER stress-associated pro-teins involved in ER stress signaling, support the notion thatPrdx6 deficiency is one cause of initiation of ER stress (Fig.3C). Oxidative stress causes ER dysfunction (30). Our datarevealed a decline in Prdx6 expression in aging cells (Fig. 4A).These cells showed an increase in Bip with higher expressionof ATF4 and caspases (Fig. 4A), suggesting that aged cells bearactivated ER stress signaling that is associated with reducedexpression of Prdx6. The ER provides a unique and balancedoxidizing microenvironment for protein folding (28, 50, 70).However, after dissociation from luminal surface of ER, Bip/GRP78 activates three ER bound proteins: 1) type 1 ERtransmembrane protein kinase (IRE1) (4, 11); 2) activatingtranscriptional factor 6 (ATF6); and 3) PKR-like ER kinase(PERK) (43, 66, 67). Procaspase 12 is an ER-associatedproximal effector of apoptosis (64). Activated caspase 12further activates caspase 9, which in turn activates caspase 3,leading to apoptosis (51, 80). Free ATF6 translocates to nu-cleus to activate transcription of many unfolded protein-re-sponsive genes including Bip/GRP78 and CHOP. PERK, on
Fig. 11. A supply of sodium phenylbutyrate (4-PBA) inhibits apoptosis inPrdx6�/� cells cultured in vitro. Cells were cultured in DMEM supplementedwith increasing concentrations of 4-PBA (25, 50, and 100 �M); 16 h later, cellswere analyzed for apoptosis using annexin V-FITC/PI staining followingcompany’s protocol (Apoptosis Detection Kit, BD Biosciences). A: represen-tative plots displaying annexin V-FITC/PI staining of Prdx6�/� cells treated(b, 25 �M; c, 50 �M; d, 100 �M) or untreated (a) with 4-PBA. B: resultsshowing inhibition of apoptosis in Prdx6�/� cells in presence of 4-PBA (blackbars). Experiments were done three times and values are represented asmeans � SD. **P � 0.001, statistically significant differences.
C964 Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

the other hand, is a kinase that phosphorylates a subunit of thetranslation initiation factor (eIF2-�). Phosphorylated eIF2-�activates a transcriptional factor, ATF4, and the upregulationof ATF4 activates the transcription of Bip/GRP78 and CHOP.The elevation of CHOP results in a downregulation of Bcl2,activation of caspases and, finally, apoptosis (57). Thus, Bip/GRP78, ATF4, CHOP, and caspase 12 are the key enzymesduring ER stress, and the signaling cascade is activated inPrdx6-deficient cells (Figs. 1, 2, 3, 5, and 6). Recent studieshave indicated a close link between ER stress and caspase 12expression and activation. Indeed, constitutive expression andactivation of this protein was observed in Prdx6�/� cells.Furthermore, the observation that caspase 12 processing occursduring ER stress induced apoptosis has supported the idea thatcaspase 12 could be the initiator caspase in ER stress-mediatedapoptosis. However, the phenotype of mouse embryonic fibro-blasts (MEF) cells isolated from caspase 12-deficient miceconcerning this notion is moderate as the absence of caspase 12in ER stress-induced apoptosis (60), arguing the involvementof other mechanisms. Although our data suggest that Prdx6�/�
cells bear higher expression and activation of caspase 12, itmay be dispensable for the execution of cell death prompted byER stress, and that other molecular mechanisms may be in-volved (34, 41, 44).
Diverse cellular stresses, such as chemotherapeutic drugs,radiation, and hypoxia, can initiate or accelerate both oxidativeand ER stresses, the damaging effects of which are morepronounced in cells with reduced expression of antioxidants.Because hypoxia is considered a triggering stimulus for redoxdisturbances in cellular microenvironment and is known toinduce ER stress (1), we exposed LECs to 1% O2 or CoCl2, ahypoxia-mimicking agent. Our data showed that the hypoxiccondition further activated the ER stress response genes inPrdx6�/� cells (Fig. 8), and the cells displayed a dramaticincrease in ROS level, had reduced viability, and underwentapoptosis (Figs. 6, A–C, and Fig. 7). To validate this finding,we used 1% O2 to evoke hypoxic stress in cultured cellssupplemented with or without Prdx6. We found that a supplyof Prdx6 reversed abnormal ER stress signaling during hyp-oxia, indicating that Prdx6 depletion may be associated withER stress-mediated apoptosis. Furthermore, CHOP/Gadd153was the first molecule observed to mediate ER stress-inducedapoptosis (61), and was found to be accelerated in Prdx6-deficient cells (Figs. 8 and 10).
Our data also showed elevated expression of calnexin inPrdx6�/� cells. Calnexin is an ER-resident molecular chaper-one that plays an essential role in the correct folding ofmembrane proteins. eIF2 is a regulatory protein involved inpolypeptide chain initiation. Kaufman and colleagues (6) re-ported that eIF2-� phosphorylation levels increase in mamma-lian cells that are undergoing apoptosis. We found that cellsdeficient in Prdx6 showed elevated levels of peIF2-�. Becausephosphorylation of eIF2-� is mediated by four distinct proteinkinases—heme-regulated inhibitor kinase (HRI), protein ki-nase RNA (PKR), PKR-like ER kinase (PERK), and generalcontrol non-derepressible-2 (GCN2)—we sought to identifythe kinase responsible for eIF2-� phosphorylation. We foundthat the level of pPERK was high in Prdx6-depleted cells.
Empirically, it has been observed that different cell typesrespond differently to ischemia (47). Particular cell types oraging cells (redox state) with reduced antioxidant may be more
susceptible to hypoxia-induced injury, while cells repeatedlyexposed to hypoxia may adapt and gain resistance againsthypoxic stress (5, 58). Upregulation of hypoxia-inducible fac-tor (HIF)-dependent proteins such as heme oxygenase-1(HO-1) and Glut-1 has been found and has been shown to beprotective. Thus expression level of hypoxia may potentiallymodify cell survival, but the effect may be related to cell type(47). The lens, which is excluded from circulation, is supposedto be in a hypoxic environment (2, 21, 29, 31). However,recently, mild hypoxia was shown to elevate the defensesystem of cells, enabling such cells to adapt to hypoxic stress.We believe that the lens has naturally adapted to minimizedamage caused by the external and internal environments andhas higher expression of several enzymes, including Cat, SOD,Gpx, and Prdx6. We have reported that Prdx6 is relativelyenriched in the lens (39). These vitally encoded proteins maywell form potential targets for treatment of age-associateddegenerative disorders.
In summary, the study presented here describes, for the firsttime, the finding that Prdx6 deficiency activates ER responsesin mammalian cells. Using aging cells and Prdx6-antisenseexperiments, we have demonstrated that loss or lack of Prdx6results in ER stress with increased sensitivity to cell injuryfrom hypoxia or oxidative stress. Raising the expression levelof Prdx6 in cells by extrinsic supply was found to attenuatedeleterious ER stress signaling by normalizing ER stress re-sponses and ROS expression. The results extend our under-standing of a plausible mechanism in cells with Prdx6 defi-ciency and cells facing stress, and add evidence of the impor-tant role of Prdx6 and its potential use in treatment.
GRANTS
Grants provided by the National Eye Institute, National Institutes of Health(EY-13394 and EY-17613; to D. P. Singh) and Research to Prevent Blindnessare gratefully acknowledged. Grant support by American Health AssistanceFoundation (to N. Fatma) is gratefully acknowledged.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
1. Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activationof endoplasmic reticulum stress response during the development ofischemic heart disease. Am J Physiol Heart Circ Physiol 291: H1411–H1420, 2006.
2. Barbazetto IA, Liang J, Chang S, Zheng L, Spector A, Dillon JP.Oxygen tension in the rabbit lens and vitreous before and after vitrectomy.Exp Eye Res 78: 917–924, 2004.
3. Beckman KB, Ames BN. The free radical theory of aging matures.Physiol Rev 78: 547–581, 1998.
4. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamicinteraction of BiP and ER stress transducers in the unfolded-proteinresponse. Nat Cell Biol 2: 326–332, 2000.
5. Bolli R. Cardioprotective function of inducible nitric oxide synthase androle of nitric oxide in myocardial ischemia and preconditioning: anoverview of a decade of research. J Mol Cell Cardiol 33: 1897–1918,2001.
6. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D,Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor ofeIF2alpha dephosphorylation protects cells from ER stress. Science 307:935–939, 2005.
7. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC,Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95: 11715–11720, 1998.
C965Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

8. Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-Cys perox-iredoxin, a bifunctional enzyme with glutathione peroxidase and phospho-lipase A2 activities. J Biol Chem 275: 28421–28427, 2000.
9. Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellulartargets that govern survival during neurodegenerative disease. Prog Neu-robiol 75: 207–246, 2005.
10. Corley KM, Taylor CJ, Lilly B. Hypoxia-inducible factor 1alpha mod-ulates adhesion, migration, and FAK phosphorylation in vascular smoothmuscle cells. J Cell Biochem 96: 971–985, 2005.
11. DeGracia DJ, Kumar R, Owen CR, Krause GS, White BC. Molecularpathways of protein synthesis inhibition during brain reperfusion: impli-cations for neuronal survival or death. J Cereb Blood Flow Metab 22:127–141, 2002.
12. Eismann T, Huber N, Shin T, Kuboki S, Galloway E, Wyder M,Edwards MJ, Greis KD, Shertzer HG, Fisher AB, Lentsch AB.Peroxiredoxin-6 protects against mitochondrial dysfunction and liver in-jury during ischemia-reperfusion in mice. Am J Physiol Gastrointest LiverPhysiol 296: G266–G274, 2009.
13. Fatma N, Kubo E, Sen M, Agarwal N, Thoreson WB, Camras CB,Singh DP. Peroxiredoxin 6 delivery attenuates TNF-alpha-and glutamate-induced retinal ganglion cell death by limiting ROS levels and maintainingCa2� homeostasis. Brain Res 1233: 63–78, 2008.
14. Fatma N, Kubo E, Sharma P, Beier DR, Singh DP. Impaired homeo-stasis and phenotypic abnormalities in Prdx6�/� mice lens epithelial cellsby reactive oxygen species: increased expression and activation of TGF-beta. Cell Death Differ 12: 734–750, 2005.
15. Fatma N, Kubo E, Takamura Y, Ishihara K, Garcia C, Beebe DC,Singh DP. Loss of NF-kappaB control and repression of Prdx6 genetranscription by reactive oxygen species-driven SMAD3-mediated trans-forming growth factor beta signaling. J Biol Chem 284: 22758–22772,2009.
16. Fatma N, Kubo E, Toris CB, Stamer WD, Camras CB, Singh DP.PRDX6 attenuates oxidative stress- and TGFbeta-induced abnormalities ofhuman trabecular meshwork cells. Free Radic Res 43: 783–795, 2009.
17. Fatma N, Singh DP, Shinohara T, Chylack LT Jr. Transcriptionalregulation of the antioxidant protein 2 gene, a thiol-specific antioxidant, bylens epithelium-derived growth factor to protect cells from oxidativestress. J Biol Chem 276: 48899–48907, 2001.
18. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology ofageing. Nature 408: 239–247, 2000.
19. Fisher AB, Beers MF. Letter to the editor: Hyperoxia and acute lunginjury. Am J Physiol Lung Cell Mol Physiol 295: L1066, 2008. Authorreply L1067, 2008.
20. Fisher AB, Dodia C, Feinstein SI, Ho YS. Altered lung phospholipidmetabolism in mice with targeted deletion of lysosomal-type phospho-lipase A2. J Lipid Res 46: 1248–1256, 2005.
21. Fitch CL, Swedberg SH, Livesey JC. Measurement and manipulation ofthe partial pressure of oxygen in the rat anterior chamber. Curr Eye Res20: 121–126, 2000.
22. Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK,Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotectiveeffects of phenylbutyrate in the N171–82Q transgenic mouse model ofHuntington’s disease. J Biol Chem 280: 556–563, 2005.
23. Gething MJ, Sambrook J. Protein folding in the cell. Nature 355: 33–45,1992.
24. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J ClinInvest 115: 500–508, 2005.
25. Gong B, Zhang LY, Lam DS, Pang CP, Yam GH. Sodium 4-phenyl-butyrate ameliorates the effects of cataract-causing mutant gammaD-crystallin in cultured cells. Mol Vis 16: 997–1003, 2010.
26. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, SimonMC, Hammerling U, Schumacker PT. Mitochondrial complex III isrequired for hypoxia-induced ROS production and cellular oxygen sens-ing. Cell Metab 1: 401–408, 2005.
27. Han C, Nam MK, Park HJ, Seong YM, Kang S, Rhim H. Tunicamycin-induced ER stress upregulates the expression of mitochondrial HtrA2 andpromotes apoptosis through the cytosolic release of HtrA2. J MicrobiolBiotechnol 18: 1197–1202, 2008.
28. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N,Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, LeidenJM, Ron D. An integrated stress response regulates amino acid metabo-lism and resistance to oxidative stress. Mol Cell 11: 619–633, 2003.
29. Helbig H, Hinz JP, Kellner U, Foerster MH. Oxygen in the anteriorchamber of the human eye. Ger J Ophthalmol 2: 161–164, 1993.
30. Helenius A, Marquardt T, Braakman I. The endoplasmic reticulum asa protein-folding compartment. Trends Cell Biol 2: 227–231, 1992.
31. Holekamp NM, Shui YB, Beebe DC. Vitrectomy surgery increasesoxygen exposure to the lens: a possible mechanism for nuclear cataractformation. Am J Ophthalmol 139: 302–310, 2005.
32. Hotokezaka Y, van Leyen K, Lo EH, Beatrix B, Katayama I, Jin G,Nakamura T. alphaNAC depletion as an initiator of ER stress-inducedapoptosis in hypoxia. Cell Death Differ 16: 1505–1514, 2009.
33. Immenschuh S, Baumgart-Vogt E. Peroxiredoxins, oxidative stress, andcell proliferation. Antioxid Redox Signal 7: 768–777, 2005.
34. Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, MeeusA, Declercq W, Vandenabeele P. Regulation of the expression andprocessing of caspase-12. J Cell Biol 162: 457–467, 2003.
35. Kang JH, Shin I, Han JS. Changes of phospholipase D activity inTNF-alpha and anti-Fas/Apo1 monoclonal antibody induced apoptosis inHL-60 and A20 cells. Exp Mol Med 30: 21–27, 1998.
36. Kang SW, Baines IC, Rhee SG. Characterization of a mammalianperoxiredoxin that contains one conserved cysteine. J Biol Chem 273:6303–6311, 1998.
37. Kim KS, Sengupta S, Berk M, Kwak YG, Escobar PF, Belinson J,Mok SC, Xu Y. Hypoxia enhances lysophosphatidic acid responsivenessin ovarian cancer cells and lysophosphatidic acid induces ovarian tumormetastasis in vivo. Cancer Res 66: 7983–7990, 2006.
38. Kubo E, Fatma N, Akagi Y, Beier DR, Singh SP, Singh DP. TAT-mediated PRDX6 protein transduction protects against eye lens epithelialcell death and delays lens opacity. Am J Physiol Cell Physiol 294:C842–C855, 2008.
39. Kubo E, Miyazawa T, Fatma N, Akagi Y, Singh DP. Development- andage-associated expression pattern of peroxiredoxin 6, and its regulation inmurine ocular lens. Mech Ageing Dev 127: 249–256, 2006.
40. Kubo E, Singh DP, Fatma N, Akagi Y. TAT-mediated peroxiredoxin 5and 6 protein transduction protects against high-glucose-induced cytotox-icity in retinal pericytes. Life Sci 84: 857–864, 2009.
41. Lamkanfi M, Kalai M, Vandenabeele P. Caspase-12: an overview. CellDeath Differ 11: 365–368, 2004.
42. Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded proteinresponse regulator GRP78/BiP is required for endoplasmic reticulumintegrity and stress-induced autophagy in mammalian cells. Cell DeathDiffer 15: 1460–1471, 2008.
43. Liu CY, Schroder M, Kaufman RJ. Ligand-independent dimerizationactivates the stress response kinases IRE1 and PERK in the lumen of theendoplasmic reticulum. J Biol Chem 275: 24881–24885, 2000.
44. Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol 16:1985–1992, 2005.
45. Liu J, Hales AM, Chamberlain CG, McAvoy JW. Induction of cataract-like changes in rat lens epithelial explants by transforming growth factorbeta. Invest Ophthalmol Vis Sci 35: 388–401, 1994.
46. Liu L, Ning X, Sun L, Zhang H, Shi Y, Guo C, Han S, Liu J, Sun S,Han Z, Wu K, Fan D. Hypoxia-inducible factor-1 alpha contributes tohypoxia-induced chemoresistance in gastric cancer. Cancer Sci 99: 121–128, 2008.
47. Loor G, Schumacker PT. Role of hypoxia-inducible factor in cellsurvival during myocardial ischemia-reperfusion. Cell Death Differ 15:686–690, 2008.
48. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfoldedprotein response. Semin Cell Dev Biol 18: 716–731, 2007.
49. Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidativestress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9:2277–2293, 2007.
50. Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW,Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress andimprove protein secretion. Proc Natl Acad Sci USA 105: 18525–18530,2008.
51. Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin inducesendoplasmic reticulum stress and nucleus-independent apoptotic signal-ing. J Biol Chem 278: 9100–9106, 2003.
52. Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, func-tions in antioxidant defense and lung phospholipid metabolism. FreeRadic Biol Med 38: 1422–1432, 2005.
53. Manevich Y, Shuvaeva T, Dodia C, Kazi A, Feinstein SI, Fisher AB.Binding of peroxiredoxin 6 to substrate determines differential phospho-lipid hydroperoxide peroxidase and phospholipase A(2) activities. ArchBiochem Biophys 485: 139–149, 2009.
C966 Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from

54. Manevich Y, Sweitzer T, Pak JH, Feinstein SI, Muzykantov V, FisherAB. 1-Cys peroxiredoxin overexpression protects cells against phospho-lipid peroxidation-mediated membrane damage. Proc Natl Acad Sci USA99: 11599–11604, 2002.
55. Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, SchumackerPT, Simon MC. Mitochondrial dysfunction resulting from loss of cyto-chrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activa-tion. Cell Metab 1: 393–399, 2005.
56. Mates JM, Perez-Gomez C, Nunez de Castro I. Antioxidant enzymesand human diseases. Clin Biochem 32: 595–603, 1999.
57. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ.Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regu-lating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21:1249–1259, 2001.
58. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: adelay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136, 1986.
59. Nagineni CN, Bhat SP. Alpha B-crystallin is expressed in kidney epi-thelial cell lines and not in fibroblasts. FEBS Lett 249: 89–94, 1989.
60. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J.Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cyto-toxicity by amyloid-beta. Nature 403: 98–103, 2000.
61. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmicreticulum stress. Cell Death Differ 11: 381–389, 2004.
62. Park BJ, Lim YS, Lee HJ, Eum WS, Park J, Han KH, Choi SY, LeeKS. Anti-oxidative effects of Phellinus linteus and red ginseng extracts onoxidative stress-induced DNA damage. BMB Rep 42: 500–505, 2009.
63. Patenaude A, Ven Murthy MR, Mirault ME. Mitochondrial thioredoxinsystem: effects of TrxR2 overexpression on redox balance, cell growth,and apoptosis. J Biol Chem 279: 27302–27314, 2004.
64. Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T,Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE. Couplingendoplasmic reticulum stress to the cell death program: role of the ERchaperone GRP78. FEBS Lett 514: 122–128, 2002.
65. Salminen A, Kaarniranta K. ER stress and hormetic regulation of theaging process. Ageing Res Rev 9: 211–217, 2010.
66. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6localization by dissociation of BiP/GRP78 binding and unmasking ofGolgi localization signals. Dev Cell 3: 99–111, 2002.
67. Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC.Identification and characterization of pancreatic eukaryotic initiation fac-tor 2 alpha-subunit kinase, PEK, involved in translational control. MolCell Biol 18: 7499–7509, 1998.
68. Singh DP, Ohguro N, Chylack LT Jr, Shinohara T. Lens epithelium-derived growth factor: increased resistance to thermal and oxidativestresses. Invest Ophthalmol Vis Sci 40: 1444–1451, 1999.
69. Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediatedapoptosis: the story so far. Ann NY Acad Sci 1010: 186–194, 2003.
70. Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mecha-nisms and consequences. J Cell Biol 164: 341–346, 2004.
71. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Freeradicals and antioxidants in normal physiological functions and humandisease. Int J Biochem Cell Biol 39: 44–84, 2007.
72. Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, BrownA, Lerner CP, Paigen B. Mice with targeted mutation of peroxiredoxin 6
develop normally but are susceptible to oxidative stress. J Biol Chem 278:25179–25190, 2003.
73. Wang X, Phelan SA, Petros C, Taylor EF, Ledinski G, Jurgens G,Forsman-Semb K, Paigen B. Peroxiredoxin 6 deficiency and atheroscle-rosis susceptibility in mice: significance of genetic background for assess-ing atherosclerosis. Atherosclerosis 177: 61–70, 2004.
74. Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. Peroxiredoxin6 gene-targeted mice show increased lung injury with paraquat-inducedoxidative stress. Antioxid Redox Signal 8: 229–237, 2006.
75. Wang Y, Lu Q, Sheldon FS, Ho YS, Phelan SA, Beers MF, Fisher AB.[Antioxidative role of peroxiredoxin 6 in acute lung injury]. Zhonghua ErKe Za Zhi 46: 739–744, 2008.
76. Wang Y, Phelan SA, Manevich Y, Feinstein SI, Fisher AB. Transgenicmice overexpressing peroxiredoxin 6 show increased resistance to lunginjury in hyperoxia. Am J Respir Cell Mol Biol 34: 481–486, 2006.
77. Wu W, Platoshyn O, Firth AL, Yuan JX. Hypoxia divergently regulatesproduction of reactive oxygen species in human pulmonary and coronaryartery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 293:L952–L959, 2007.
78. Wu Y, Feinstein SI, Manevich Y, Chowdhury I, Pak JH, Kazi A,Dodia C, Speicher DW, Fisher AB. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipaseA(2) activity. Biochem J 419: 669–679, 2009.
79. Wu YZ, Manevich Y, Baldwin JL, Dodia C, Yu K, Feinstein SI, FisherAB. Interaction of surfactant protein A with peroxiredoxin 6 regulatesphospholipase A2 activity. J Biol Chem 281: 7515–7525, 2006.
80. Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE,Yoffe B. Effect of tauroursodeoxycholic acid on endoplasmic reticulumstress-induced caspase-12 activation. Hepatology 36: 592–601, 2002.
81. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell lifeand death decisions. J Clin Invest 115: 2656–2664, 2005.
82. Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K,Yagita H, Okumura K, Harding H, Nakano H. Tumor necrosis factoralpha (TNFalpha) induces the unfolded protein response (UPR) in areactive oxygen species (ROS)-dependent fashion, and the UPR counter-acts ROS accumulation by TNFalpha. J Biol Chem 280: 33917–33925,2005.
83. Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. Sodium 4-phenylbu-tyrate acts as a chemical chaperone on misfolded myocilin to rescue cellsfrom endoplasmic reticulum stress and apoptosis. Invest Ophthalmol VisSci 48: 1683–1690, 2007.
84. Yang WW, Shu B, Zhu Y, Yang HT. E2F6 inhibits cobalt chloride-mimetic hypoxia-induced apoptosis through E2F1. Mol Biol Cell 19:3691–3700, 2008.
85. Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A,Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M. Involvementof selective reactive oxygen species upstream of proapoptotic branches ofunfolded protein response. J Biol Chem 283: 4252–4260, 2008.
86. Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1pre-mRNA negatively regulates unfolded protein response activatorpXBP1(S) in mammalian ER stress response. J Cell Biol 172: 565–575,2006.
87. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT,Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed celldeath in response to impaired function of the endoplasmic reticulum.Genes Dev 12: 982–995, 1998.
C967Prdx6 DEPLETION CAUSES AN ER STRESS RESPONSE
AJP-Cell Physiol • VOL 301 • OCTOBER 2011 • www.ajpcell.org
on Novem
ber 7, 2011ajpcell.physiology.org
Dow
nloaded from
Related Documents









![Wnt5a transcription induces epithelial- mesenchymal ... · studies have highlighted a link between canonical Wnt signaling and EMT, particularly in gastric cancer cells [11, 12].](https://static.cupdf.com/doc/110x72/60560c4fbb62fa23cb175b37/wnt5a-transcription-induces-epithelial-mesenchymal-studies-have-highlighted.jpg)
