Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease Ira L. Goldknopf a, * , Essam A. Sheta a , Jennifer Bryson a , Brian Folsom a , Chris Wilson a , Jeff Duty a , Albert A. Yen b , Stanley H. Appel b a Power3 Medical Products, Inc., The Woodlands, TX, USA b Methodist Neurological Institute, Houston, TX, USA Received 3 February 2006 Available online 20 February 2006 Abstract We have used quantitative 2D gel electrophoresis to analyze serum proteins from 422 patients with neurodegenerative diseases and normal individuals in an unbiased approach to identify biomarkers. Differences in abnormal serum levels were found between amyotro- phic lateral sclerosis (ALS), Parkinson’s disease (PD), and related disorders for 34 protein biomarker spots, nine of which were related to the complement system. Of these nine, four spots originated from the Complement C3b-a-chain (C3c 1 , C3c 2a , C3c 2b , and C3dg). The C3c spots (C3c 1 , C3c 2a , and C3c 2b ) had the same amino acid sequence and glycosylation, though only C3c 1 was phosphorylated. In addition, Complement Factors H, Bb, and Pre-Serum amyloid protein displayed different serum concentrations in ALS, PD, and normal sera, whereas Complement C4b c-chain and Complement Factor I did not. The differential expression of the complement proteins provides potentially useful biomarkers as well as evidence for the involvement of inflammatory processes in the pathogenesis of ALS and PD. Ó 2006 Elsevier Inc. All rights reserved. Keywords: Amytrophic lateral sclerosis; Parkinson’s; Neurodegenerative; Proteomic; LC–MS/MS; Two-dimensional electrophoresis; Immune; Inflammation; Complement; Phosphorylation Two-dimensional (2D) gel electrophoresis has been used in research laboratories for biomarker discovery since the 1970’s [1–8]. Recently, faster identification of proteins by in-gel digestion and mass spectrometry has expanded and enhanced the application of these techniques [9,10], while the advent of bioinformatics is progressing proteomics towards diagnostics [11–13]. Comprehensive analysis of disease mechanisms and disease markers is now feasible through clinical proteomics [14], which could be of clinical value in amyotrophic lateral sclerosis (ALS) and Parkin- son’s disease (PD), as well as other devastating neurode- generative disorders. These central nervous system disorders are characterized by the progressive impairment and loss of specific neuronal populations. PD is characterized by tremor, bradykinesia, rigidity, and abnormal gait and posture, resulting from dysfunction and cell loss of substantia nigral dopaminergic neurons [15]. Alterations in a-synuclein have been postulated to underlie many of the pathological processes [15,16]. Over 95% of cases are sporadic while a smaller percentage are familial and due to specific mutations, several of which are related to proteosomal function [16]. ALS is characterized by degeneration of both upper and lower motor neurons, manifested by weakness of upper and lower extremities, speech, and swallowing, and ulti- mately resulting in respiratory muscle failure. Over 90% of ALS patients are sporadic while 10% of cases are famil- ial. Mutations in superoxide dismutase are responsible for 20% of the familial cases [17–19]. In both PD and ALS, there is currently no blood test to confirm the diagnosis or monitor the burden of disease. In the present study, we used 2D gel electrophoresis of serum proteins to define possible biomarkers for PD and 0006-291X/$ - see front matter Ó 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.bbrc.2006.02.051 * Corresponding author. Fax: +1 281 466 1481. E-mail address: [email protected] (I.L. Goldknopf ). www.elsevier.com/locate/ybbrc Biochemical and Biophysical Research Communications 342 (2006) 1034–1039 BBRC

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

www.elsevier.com/locate/ybbrc
Biochemical and Biophysical Research Communications 342 (2006) 1034–1039
BBRC
Complement C3c and related protein biomarkers in amyotrophiclateral sclerosis and Parkinson’s disease
Ira L. Goldknopf a,*, Essam A. Sheta a, Jennifer Bryson a, Brian Folsom a, Chris Wilson a,Jeff Duty a, Albert A. Yen b, Stanley H. Appel b
a Power3 Medical Products, Inc., The Woodlands, TX, USAb Methodist Neurological Institute, Houston, TX, USA
Received 3 February 2006Available online 20 February 2006
Abstract
We have used quantitative 2D gel electrophoresis to analyze serum proteins from 422 patients with neurodegenerative diseases andnormal individuals in an unbiased approach to identify biomarkers. Differences in abnormal serum levels were found between amyotro-phic lateral sclerosis (ALS), Parkinson’s disease (PD), and related disorders for 34 protein biomarker spots, nine of which were related tothe complement system. Of these nine, four spots originated from the Complement C3b-a-chain (C3c1, C3c2a, C3c2b, and C3dg). The C3cspots (C3c1, C3c2a, and C3c2b) had the same amino acid sequence and glycosylation, though only C3c1 was phosphorylated. In addition,Complement Factors H, Bb, and Pre-Serum amyloid protein displayed different serum concentrations in ALS, PD, and normal sera,whereas Complement C4b c-chain and Complement Factor I did not. The differential expression of the complement proteins providespotentially useful biomarkers as well as evidence for the involvement of inflammatory processes in the pathogenesis of ALS and PD.� 2006 Elsevier Inc. All rights reserved.
Keywords: Amytrophic lateral sclerosis; Parkinson’s; Neurodegenerative; Proteomic; LC–MS/MS; Two-dimensional electrophoresis; Immune;Inflammation; Complement; Phosphorylation
Two-dimensional (2D) gel electrophoresis has been usedin research laboratories for biomarker discovery since the1970’s [1–8]. Recently, faster identification of proteins byin-gel digestion and mass spectrometry has expanded andenhanced the application of these techniques [9,10], whilethe advent of bioinformatics is progressing proteomicstowards diagnostics [11–13]. Comprehensive analysis ofdisease mechanisms and disease markers is now feasiblethrough clinical proteomics [14], which could be of clinicalvalue in amyotrophic lateral sclerosis (ALS) and Parkin-son’s disease (PD), as well as other devastating neurode-generative disorders.
These central nervous system disorders are characterizedby the progressive impairment and loss of specific neuronalpopulations.
0006-291X/$ - see front matter � 2006 Elsevier Inc. All rights reserved.
doi:10.1016/j.bbrc.2006.02.051
* Corresponding author. Fax: +1 281 466 1481.E-mail address: [email protected] (I.L. Goldknopf ).
PD is characterized by tremor, bradykinesia, rigidity,and abnormal gait and posture, resulting from dysfunctionand cell loss of substantia nigral dopaminergic neurons[15]. Alterations in a-synuclein have been postulated tounderlie many of the pathological processes [15,16]. Over95% of cases are sporadic while a smaller percentage arefamilial and due to specific mutations, several of whichare related to proteosomal function [16].
ALS is characterized by degeneration of both upper andlower motor neurons, manifested by weakness of upperand lower extremities, speech, and swallowing, and ulti-mately resulting in respiratory muscle failure. Over 90%of ALS patients are sporadic while 10% of cases are famil-ial. Mutations in superoxide dismutase are responsible for20% of the familial cases [17–19]. In both PD and ALS,there is currently no blood test to confirm the diagnosisor monitor the burden of disease.
In the present study, we used 2D gel electrophoresis ofserum proteins to define possible biomarkers for PD and

I.L. Goldknopf et al. / Biochemical and Biophysical Research Communications 342 (2006) 1034–1039 1035
ALS. In this unbiased approach, differences in the abnor-mal expression of 7 of 9 complement-related proteins werefound in sera from 422 patients with ALS, PD, related neu-rological disorders and normal individuals, providing inde-pendent evidence for the role of immune-inflammatoryprocesses in these diseases.
Materials and methods
Samples. Serum samples were obtained with informed consent frompatients followed in the Department of Neurology, Baylor College ofMedicine and the Methodist Hospital. Samples were stored at �70 to�80 �C until analysis. Patients with clinical symptoms similar to PD butnot diagnosed as PD (PD-like) included: Lewy Body Dementia; MultipleSystem Atrophy; Vascular Parkinsonism; Cerebellar Ataxia; and Corti-cobasal Ganglionic Degeneration (CBGD). Patients with clinical symp-toms similar to ALS but not diagnosed as ALS (ALS-like) included: spinalmuscular atrophy, primary lateral sclerosis, chronic inflammatory demy-linating polyneuropathy, multifocal motor neuropathy, inclusion bodymyositis, myasthenia gravis, and cervical myelopathy.
Reagents. Protease inhibitors were from Roche Diagnostics (India-napolis, IN). Protein assay and purification reagents were from Bio-RadLaboratories (Hercules, CA). Immobilon-P membranes were from Milli-pore (Billerica, MAL). All other chemicals were from Sigma Chemical (St.Louis, MO). Sheep anti-C3c polyclonal antibody and HP-linked Rabbitanti-sheep IgG were from Abcam (Cambridge, UK).
2D gel electrophoresis and gel image analysis. Serum protein sampleswere suspended in 5 M urea, 2 M thiourea, 1% CHAPS, 2% ASB-14,0.25% Tween 20, 100 mM DTT, 1% ampholytes, pH 3–10, 5% glycerol,and 1 · EDTA-free protease inhibitors. One hundred micrograms ofprotein was run on 11 cm IEF strip, pH 5–8, on IEF cells. Strips were thenequilibrated and run in 375 mM Tris buffer, pH 8.8, containing 6 M urea,20% glycerol, and 2% SDS, 30 mg/ml DTT, treated with 40 mg/ml iodo-acetamide, and loaded onto the second dimension Criterion gradient gels(Bio-Rad, Hercules, CA), acrylamide gradient 8-16%. After seconddimension electrophoresis, gels were then stained using SyproRuby� andscanned (Molecular Imager� FX Pro, Bio-Rad). Digital fluorescent imageanalysis compared diseased sera and normal samples using PDQuest�(Bio-Rad).
15
25
100
250
5kD
4411
3209
1416
1511
15
25
15
25
37
50
4411
3209
1416
1511
Fig. 1. The 2D PAGE pattern of human serum proteins. The position of the(7310 = C3c1, 9311 = C3c2a, and 9312 = C3c2b), C4b (7304), Complement FactAmyloid Protein (3209) are indicated.
Tryptic digestion, MALDI/MS, and LC–MS/MS. Protein spots wereexcised from gels (ProteomeWorks� robotic spot cutter, Bio-Rad) andthen subjected to in-gel trypsin digestion (ProGest�, Genomic Solutions,Ann Arbor, MI). The resulting digest supernatants were concentrated anddesalted using l-C18 ZipTips� (Millipore). Peptides were roboticallyspotted using ProMS� (Genomic Solutions, Ann Arbor, MI).
MALDI/MS data were acquired from the in-gel digests (AppliedBiosystems Voyager DE-STR�) and the spectra were analyzed withProFound� (Proteometrics) for peptide mass fingerprint identification(NCBInr database).
For nano-LC–MS/MS (Micromass Q-TOF 2), LC of digests wasconducted using C18 column (75 mm). MS/MS data were analyzed usingMASCOT�.
Phosphoprotein and glycoprotein staining of 2D gels. Gels were fixed(50% methanol and 10% acetic acid), washed (ultrapure water), andstained (Pro-Q Diamond� dye phosphoprotein stain, or ProQ Emerald�488 glycoprotein stain).
Western immunoassay. Proteins resolved by 2D gel electrophoresiswere electroblotted to Immobilon-P membranes and immunoblotted withsheep anti-human C3c polyclonal antibody. A secondary antibody ofrabbit anti-sheep IgG-peroxidase conjugate was then applied followed byvisualization of spots using Super Signal� West Femto detection system(Pierce).
Results
Serum proteins were separated by 2D gel electrophoresisas indicated under Materials and methods and a represen-tative 2D-gel is shown in Fig. 1. Quantitative digital imageanalysis revealed differences in serum levels of 34 spots ingels of serum from patients with ALS, PD, related disor-ders, and normal controls. The identification of these spotswas accomplished by Maldi-TOF peptide fingerprint anal-ysis and independently by LC–MS/MS peptide fragmenta-tion analysis. Nine of these spots were identified as proteinsinvolved in the complement cascade (Numbered spots,Fig. 1; ID data, Table 1). As shown in Fig. 2, using percentof normal PPM serum levels, seven of the nine complement
8
77616
9311
93127310
7304
8
7
8
7616
9311
93127310
7304
spots corresponding to the Complement C3 biomarkers: C3d (1511), C3cor H, truncated form (4411), Factor I (1416), Factor Bb (7616), and Serum
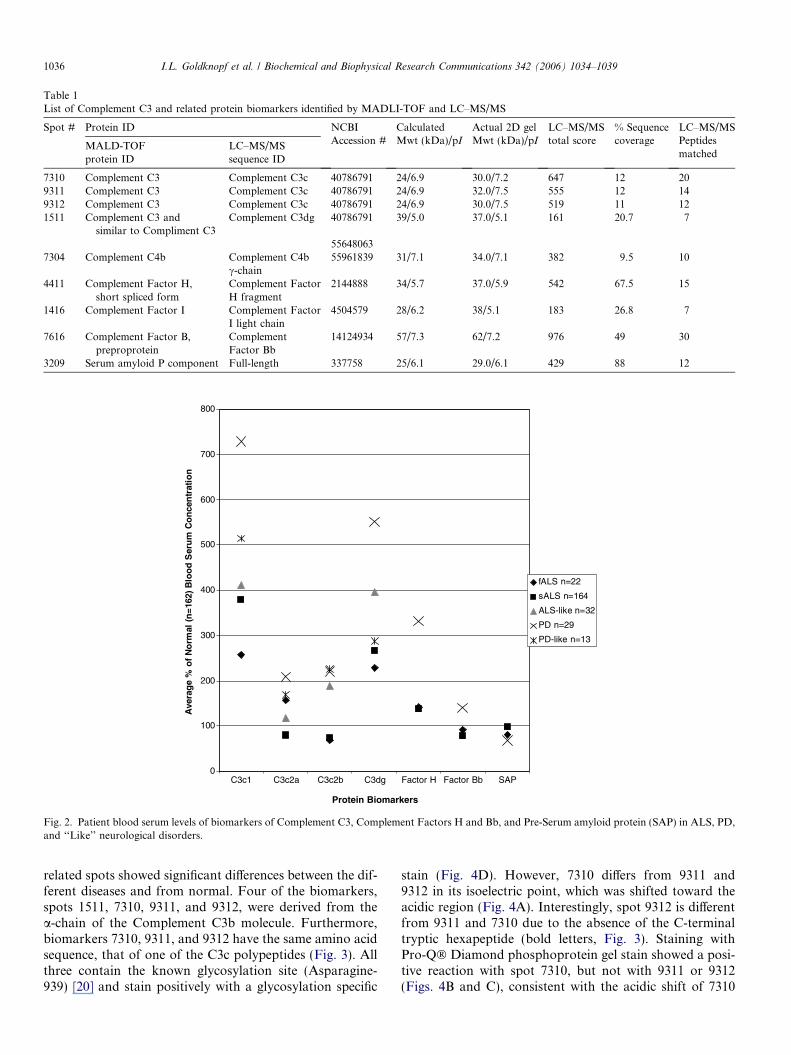
Table 1List of Complement C3 and related protein biomarkers identified by MADLI-TOF and LC–MS/MS
Spot # Protein ID NCBIAccession #
CalculatedMwt (kDa)/pI
Actual 2D gelMwt (kDa)/pI
LC–MS/MStotal score
% Sequencecoverage
LC–MS/MSPeptidesmatched
MALD-TOFprotein ID
LC–MS/MSsequence ID
7310 Complement C3 Complement C3c 40786791 24/6.9 30.0/7.2 647 12 209311 Complement C3 Complement C3c 40786791 24/6.9 32.0/7.5 555 12 149312 Complement C3 Complement C3c 40786791 24/6.9 30.0/7.5 519 11 121511 Complement C3 and
similar to Compliment C3Complement C3dg 40786791 39/5.0 37.0/5.1 161 20.7 7
556480637304 Complement C4b Complement C4b
c-chain55961839 31/7.1 34.0/7.1 382 9.5 10
4411 Complement Factor H,short spliced form
Complement FactorH fragment
2144888 34/5.7 37.0/5.9 542 67.5 15
1416 Complement Factor I Complement FactorI light chain
4504579 28/6.2 38/5.1 183 26.8 7
7616 Complement Factor B,preproprotein
ComplementFactor Bb
14124934 57/7.3 62/7.2 976 49 30
3209 Serum amyloid P component Full-length 337758 25/6.1 29.0/6.1 429 88 12
0
100
200
300
400
500
600
700
800
C3c1 C3c2a C3c2b C3dg Factor H Factor Bb SAP
Protein Biomarkers
Ave
rag
e %
of
No
rmal
(n
=16
2) B
loo
d S
eru
m C
on
cen
trat
ion
fALS n=22
sALS n=164
ALS-like n=32
PD n=29
PD-like n=13
Fig. 2. Patient blood serum levels of biomarkers of Complement C3, Complement Factors H and Bb, and Pre-Serum amyloid protein (SAP) in ALS, PD,and ‘‘Like’’ neurological disorders.
1036 I.L. Goldknopf et al. / Biochemical and Biophysical Research Communications 342 (2006) 1034–1039
related spots showed significant differences between the dif-ferent diseases and from normal. Four of the biomarkers,spots 1511, 7310, 9311, and 9312, were derived from thea-chain of the Complement C3b molecule. Furthermore,biomarkers 7310, 9311, and 9312 have the same amino acidsequence, that of one of the C3c polypeptides (Fig. 3). Allthree contain the known glycosylation site (Asparagine-939) [20] and stain positively with a glycosylation specific
stain (Fig. 4D). However, 7310 differs from 9311 and9312 in its isoelectric point, which was shifted toward theacidic region (Fig. 4A). Interestingly, spot 9312 is differentfrom 9311 and 7310 due to the absence of the C-terminaltryptic hexapeptide (bold letters, Fig. 3). Staining withPro-Q� Diamond phosphoprotein gel stain showed a posi-tive reaction with spot 7310, but not with 9311 or 9312(Figs. 4B and C), consistent with the acidic shift of 7310

721 KKVFLDCCNY ITELRRQHAR ASHLGLARSN LDEDIIAEEN IVSRSEFPES WLWNVEDLKE781 PPKNGISTKL MNIFLKDSIT TWEILAVSMS DKKGICVADP FEVTVMQDFF IDLRLPYSVV841 RNEQVEIRAV LYNYRQNQEL KVRVELLHNP AFCSLATTKR RHQQTVTIPP KSSLSVPYVI901 VPLKTGLQEV EVKAAVYHHF ISDGVRKSLK VVPEGIRMNK TVAVRTLDPE RLGREGVQKE
73109311
*
9312
AR LDEDIIAEEN IVSRSEFPES WLWNVEDLKE
L TKR VPYVIHF NK T TLDPE R
Missing in 9312
*= C3c1
= C3c2a
= C3c2b
Fig. 3. Amino acid sequence spanned by peptides identified by LC–MS/MS of spots 7310, 9311, and 9312 (underlined). The C3c biomarkers 7310 and9311 have the same amino acid sequence derived from the C3c domain of C3 parent. C3 biomarker 9312 is identical to 9311 in sequence and very close inpI, but is missing a short peptide at the C-terminal (bold letters), consistent with its slightly lower molecular weight (Fig. 1). All three sequences span theglycosylation site, asparagine-939 (bold N*).
9311
9312
73047310
73047310
73047310
9311
9312
7304
7310
9311
9312
7304
7310
9311
9312
7304
7310
9311
9312
7304
7310
7310 9311
9312
A
B
C
D
E
Fig. 4. Enlarged section of 2D gel enclosed spots 7304, 7310, 9311, and9312. Gels are stained with (A) SyproRuby� protein stain, (B) Pro-QDiamond Phosphoprotein stain, (C) Pro-Q Diamond� Phosphoproteinstain followed by SyproRuby� protein stain, and (D) Pro-Q Emerald�-488 glycoprotein stain, and (E) Western immunoassay with sheep anti-C3cpolyclonal antibody (Abcam).
I.L. Goldknopf et al. / Biochemical and Biophysical Research Communications 342 (2006) 1034–1039 1037
as a result of phosphorylation. In addition, all three C3cspots gave a positive Western reaction with anti-C3c poly-clonal antibody (Fig. 4E). To differentiate the three C3c,component spots 7310, 9311, and 9312, we named themC3c1, C3c2a, and C3c2b, respectively.
Mass spectral analysis of tryptic peptides of biomarker1511 identified a protein ‘‘Similar to Complement C3’’(Table 1). However, a databank sequence comparisonwas employed with Complement C3 and ‘‘Similar to C3’’,using the SIM-alignment tool for protein sequences (datanot shown), combined with an alignment of tryptic pep-tides identified by Maldi-TOF–MS and LC–MS/MS. Anal-
ysis indicated that the sequence of 1511 is identical toComplement C3dg.
Levels of biomarker 4411, which corresponds to theshort spliced form of Complement Factor H (52.7 kDa),were also elevated in ALS and PD (Fig. 2). The 2D-gelposition of this biomarker, with an estimated molecularmass of �37 kDa, indicates that biomarker 4411 is a trun-cated version or a cleavage product of the Factor H shortsplice form. Biomarker 7304, identified by LC–MS/MS asComplement C4, and the matched peptides spanned thesequence of C4b c-chain. Biomarker 1416 was identifiedas a cleavage product of Complement Factor I. Peptidesmatched from this protein contain the amino acid sequencethat matches the C-terminal trypsin-like domain. NeitherComplement C4bc nor Factor I was significantly increasedin either ALS or PD. Biomarker 7616, identified as FactorBb, showed significant differences in serum levels in ALSvs. PD. It was increased significantly in PD, whereas itwas decreased significantly in serum levels of sALS butnot in fALS. Finally, biomarker 3209, identified as Pre-Serum amyloid protein (SAP), displayed a significantreduction in PD, while no difference was found in sALS.
Discussion
There is now compelling evidence that inflammatoryprocesses involving complement activation are associatedwith a broad spectrum of neurodegenerative diseasesincluding ALS, Alzheimer’s and Parkinson’s diseases(PD), the Parkinson-dementia complex of Guam, all ofthe tauopathies, and age-related macular degeneration[21,22]. Examples of localized inflammatory processesrelated to the present study include elevated tissue levelsof C3c and C3d cleavage products in immunohistochemicalstudies of ALS and PD [23–27] as well as reduced levels offull-length Factors H and B in CSF of PD and MS patients[28]. Corresponding examples of systemic immune/inflam-matory processes include evidence of MCP-1 and the pres-ence of dendritic cells and activated microglia in ALS[29,30] as well as evidence of neuronal injury resulting fromactivated microglial release of free radicals in ALS and PD[31,32]. In ALS, such oxidative stress is likely reflectedlocally and systemically by increased levels of peroxidizedlipids such as hydroxynonenal in CSF and serum of ALSpatients [33].

1038 I.L. Goldknopf et al. / Biochemical and Biophysical Research Communications 342 (2006) 1034–1039
In the present study, both ALS and PD patient popula-tions showed marked increases of Complement C3 compo-nents C3c and C3dg, and Complement Factor H. In PDpatient serum, C3dg and C3c1 levels were 6- and 7- foldthat of normal controls and twice that of ALS patients.All three identified C3c generated spots (C3c1, 2a, and 2b)were consistently increased in PD, while only the phos-phorylated C3c spot (C3c1) was selectively increased inALS. Phosphorylation of Complement C3 parent proteinhas been shown to block cleavage by trypsin [34] or by Fac-tor I with Factor H as cofactor [35]. However, phosphory-lation has also been shown to selectively activate thecleavage of C3 by membrane proteases and leukocyte elas-tases [36]. Furthermore, inasmuch as binding of Fc recep-tor ligands also stimulates C3 phosphorylation andprocessing [36], it is likely that serum elevation of the phos-phorylated form, C3c1, found in ALS and PD in the pres-ent study, constitutes an independent proteomicmeasurement of immune/inflammatory aspects of ALSand Parkinson’s disease, probably mediated by microgliastimulation of Fc receptors [32,37].
Whereas significant reduction in the full-length Factor Bin the CSF of PD patients was recently reported [28], thepresent work indicates elevated levels of fragment Bb ofFactor B were detected in the serum of PD but not ALSpatients. This elevation of fragment Bb in PD most proba-bly arose, at least in part, from the elevated levels of FactorH, which has been found to induce dissociation of C3 con-vertase (C3bBb(Mg2+))[38], liberating Factor Bb and inter-rupting the Alternative Pathway of the complementactivation cascade. Factor H acts as a cofactor for the ser-ine protease Factor I, which cleaves the a-chain of C3b atthree sites to form iC3b, similarly interrupting the ClassicalPathway of the complement activation cascade [39–42]. Wehave identified a cleavage product (Mwt �37 kDa, pI 5.9)of the short spliced form of Factor H (Mwt 57.2 kDa) asa serum biomarker that is significantly higher in PD andALS when compared to normal serum. Generally, the2-fold increased level of Complement C3dg, C3c, FactorsH short splice form, and Bb, in PD over that of ALSmay indicate a broader and higher level of abnormalComplement C3b processing and differential effects onthe complement cascade in PD than in ALS, possiblyinvolving additional complement pathways in PD. In addi-tion, the present results of reduced serum levels of SAP andincreased serum levels of C3c2a, and/or C3c2b betweensALS, fALS, and PD, may reflect increased incorporationof SAP into neurofibrillary tangles and Lewy Bodies inPD [43–46], accompanied by increased Complement FactorI mediated processing of Complement C3 [47–50]. Similarchanges in complement components may also be presentin CSF although they were not noted in recently identifiedbiomarkers in ALS CSF [51].
In summary, the present results are an independentquantitative proteomic measure in patient sera of localizedand systemic inflammatory and immune/inflammatoryprocesses in ALS and PD. To our knowledge, this is the
first proteomic study of patient sera to show differencesin the pathways of complement, including C3 phosphoryla-tion differences, between ALS and PD. These differences inserum concentrations of Complement C3 and related pro-teins in patients with the two diseases may potentially pro-vide useful biomarkers, not only for differential diagnosis,but also for measuring burden of disease and monitoringtherapeutic interventions [52].
Acknowledgments
The authors gratefully acknowledge Baylor College ofMedicine, the Methodist Hospital, and Dr. Thomas Wattsfor providing samples under informed consent. The cost ofanalysis and labor of this work was fully supported byPower3 Medical. Discovery of these biomarkers is theexclusive right of Power3 Medical, through research andlicense agreements.
References
[1] L. Anderson, N.G. Anderson, High resolution two-dimensionalelectrophoresis of human plasma proteins, Proc. Natl. Acad. Sci.USA 74 (1977) 5421–5425.
[2] I.L. Goldknopf, M.F. French, R. Musso, H. Busch, Presence ofprotein A24 in rat liver nucleosomes, Proc. Natl. Acad. Sci. USA 74(1977) 5492–5495.
[3] I.L. Goldknopf, C.W. Taylor, R.M. Baum, L.C. Yeoman, M.O.Olson, A.W. Prestayko, H. Busch, Isolation and characterization ofprotein A24, a ‘‘histone-like’’ non-histone chromosomal protein, J.Biol. Chem. 250 (1975) 7182–7187.
[4] J. Klose, Protein mapping by combined isoelectric focusing andelectrophoresis of mouse tissues. A novel approach to testing forinduced point mutations in mammals, Human Genet. 26 (1975)231–243.
[5] J. Margolis, K.G. Kenrick, 2-Dimensional resolution of plasmaproteins by combination of polyacrylamide disc and gradient gelelectrophoresis, Nature 221 (1969) 1056–1057.
[6] P.Z. O’Farrell, H.M. Goodman, P.H. O’Farrell, High resolution two-dimensional electrophoresis of basic as well as acidic proteins, Cell 12(1977) 1133–1141.
[7] L.R. Orrick, M.O. Olson, H. Busch, Comparison of nucleolarproteins of normal rat liver and Novikoff hepatoma ascites cells bytwo-dimensional polyacrylamide gel electrophoresis, Proc. Natl.Acad. Sci. USA 70 (1973) 1316–1320.
[8] S. Raymond, Protein purification by elution convection electropho-resis, Science 146 (1964) 406–407.
[9] R. Aebersold, M. Mann, Mass spectrometry-based proteomics,Nature 422 (2003) 198–207.
[10] H. Kuruma, S. Egawa, M. Oh-Ishi, Y. Kodera, T. Maeda, Proteomeanalysis of prostate cancer, Prostate Cancer Prostatic Dis. 8 (2005)14–21.
[11] N.L. Anderson, N.G. Anderson, The human plasma proteome:history, character, and diagnostic prospects, Mol. Cell. Proteomics 1(2002) 845–867.
[12] V. Rachakonda, T.H. Pan, W.D. Le, Biomarkers of neurodegen-erative disorders: how good are they? Cell Res. 14 (2004)347–358.
[13] C.N. White, D.W. Chan, Z. Zhang, Bioinformatics strategies forproteomic profiling, Clin. Biochem. 37 (2004) 636–641.
[14] S. Hanash, HUPO initiatives relevant to clinical proteomics, Mol.Cell. Proteomics 3 (2004) 298–301.
[15] T.M. Dawson, V.L. Dawson, Molecular pathways of neurodegener-ation in Parkinson’s disease, Science 302 (2003) 819–822.

I.L. Goldknopf et al. / Biochemical and Biophysical Research Communications 342 (2006) 1034–1039 1039
[16] W.D. Lee, S.H. Appel, Mutant genes responsible for Parkinson’sdisease, Curr. Opin. Pharmacol. 4 (2004) 79–84.
[17] R.W. Orrell, J.J. Habgood, I. Gardiner, A.W. King, F.A. Bowe, R.A.Hallewell, S.L. Marklund, J. Greenwood, R.J. Lane, J. deBelleroche,Clinical and functional investigation of 10 missense mutations and anovel frameshift insertion mutation of the gene for copper–zincsuperoxide dismutase in UK families with amyotrophic lateralsclerosis, Neurology 48 (1997) 746–751.
[18] C.E. Shaw, Z.E. Enayat, B.A. Chioza, A. Al-Chalabi, A. Radunovic,J.F. Powell, P.N. Leigh, Mutations in all five exons of SOD-1 maycause ALS, Ann. Neurol. 43 (1998) 390–394.
[19] T. Siddique, D.A. Figlewicz, M.A. Pericak-Vance, J.L. Haines, G.Rouleau, A.J. Jeffers, P. Sapp, W.Y. Hung, J. Bebout, D. McKenna-Yasek, et al., Linkage of a gene causing familial amyotrophic lateralsclerosis to chromosome 21 and evidence of genetic-locus heteroge-neity, N. Engl. J. Med. 324 (1991) 1381–1384.
[20] M.D. Crispin, G.E. Ritchie, A.J. Critchley, B.P. Morgan, I.A. Wilson,R.A. Dwek, R.B. Sim, P.M. Rudd, Monoglucosylated glycans in thesecreted human complement component C3: implications for proteinbiosynthesis and structure, FEBS Lett. 566 (2004) 270–274.
[21] P.L. McGeer, E.G. McGeer, Inflammation and neurodegeneration inParkinson’s disease, Parkinsonism Relat. Disord. 10 (Suppl. 1) (2004)S3–S7.
[22] H. Rus, F. Niculescu, The complement system in central nervoussystem diseases, Immunol. Res. 24 (2001) 79–86.
[23] P. Annunziata, N. Volpi, High levels of C3c in the cerebrospinal fluidfrom amyotrophic lateral sclerosis patients, Acta Neurol. Scand. 72(1985) 61–64.
[24] T. Kawamata, H. Akiyama, T. Yamada, P.L. McGeer, Immunologicreactions in amyotrophic lateral sclerosis brain and spinal cord tissue,Am. J. Pathol. 140 (1992) 691–707.
[25] C. Schwab, J.C. Steele, P.L. McGeer, Neurofibrillary tangles ofGuam Parkinson-dementia are associated with reactive microglia andcomplement proteins, Brain Res. 707 (1996) 196–205.
[26] T. Yamada, P.L. McGeer, E.G. McGeer, Lewy bodies in Parkinson’sdisease are recognized by antibodies to complement proteins, ActaNeuropathol. (Berlin) 84 (1992) 100–104.
[27] P.L. McGeer, H. Akiyama, S. Itagaki, E.G. McGeer, Activation ofthe classical complement pathway in brain tissue of Alzheimerpatients, Neurosci. Lett. 107 (1989) 341–346.
[28] E.J. Finehout, Z. Franck, K.H. Lee, Complement protein isoforms inCSF as possible biomarkers for neurodegenerative disease, Dis.Markers 21 (2005) 93–101.
[29] J.S. Henkel, J.I. Engelhardt, L. Siklos, E.P. Simpson, S.H. Kim, T.Pan, J.C. Goodman, T. Siddique, D.R. Beers, S.H. Appel, Presence ofdendritic cells, MCP-1, and activated microglia/macrophages inamyotrophic lateral sclerosis spinal cord tissue, Ann. Neurol. 55 (2)(2004) 221–235.
[30] J.S. Henkel, D.R. Beers, L. Siklos, S.H. Appel, The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in themSOD1 mouse model of ALS, Mol. Cell Neurosci. 2005, In press.
[31] W. Le, D. Rowe, W. Xie, I. Ortiz, Y. He, S.H. Appel, Microglialactivation and dopaminergic cell injury: and in vitro model relevant toParkinson’s disease, J. Neurosci. 21 (2001) 8447–8455.
[32] Y. He, W.D. Le, S.H. Appel, Role of Fcgamma receptors in nigralcell injury induced by Parkinson’s disease immunoglobulin injectioninto mouse substantia nigra, Exp. Neurol. 176 (2002) 322–327.
[33] E.P. Simpson, Y.K. Henry, J.S. Henkel, R.G. Smith, S.H. Appel,Increased lipid peroxidation in sera of ALS patients: a potentialbiomarker of disease burden, Neurology 62 (2004) 1758–1765.
[34] P.O. Forsberg, S.C. Martin, B. Nilsson, P. Ekman, U.R. Nilsson, L.Engstrom, In vitro phosphorylation of human complement factor C3by protein kinase A and protein kinase C. Effects on the classical andalternative pathways, J. Biol. Chem. 265 (1990) 2941–2946.
[35] K.N. Ekdahl, B. Nilsson, Phosphorylation of complement componentC3 and C3 fragments by a human platelet protein kinase. Inhibition
of factor I-mediated cleavage of C3b, J. Immunol. 154 (1995) 6502–6510.
[36] K. Nilsson Ekdahl, B. Nilsson, Phosphorylation of complementcomponent C3 after synthesis in U937 cells by a putative proteinkinase, casein kinase 2, which is regulated by CD11b: evidence thatmembrane-bound proteases preferentially cleave phosphorylated C3,Biochem. J. 328 (Pt. 2) (1997) 625–633.
[37] C.F. Orr, D.B. Rowe, Y. Mizuno, H. Mori, G.M. Halliday, Apossible role for humoral immunity in the pathogenesis of Parkin-son’s disease, Brain 128 (2005) 2665–2674.
[38] D.E. Hourcade, L. Mitchell, L.A. Kuttner-Kondo, J.P. Atkinson,M.E. Medof, Decay-accelerating factor (DAF), complement receptor1 (CR1), and factor H dissociate the complement AP C3 convertase(C3bBb) via sites on the type A domain of Bb, J. Biol. Chem. 277(2002) 1107–1112.
[39] D.E. Isenman, E.R. Podack, N.R. Cooper, The interaction of C5 withC3b in free solution: a sufficient condition for cleavage by a fluidphase C3/C5 convertase, J. Immunol. 124 (1980) 326–331.
[40] M.K. Pangburn, H.J. Muller-Eberhard, Complement C3 convertase:cell surface restriction of beta1H control and generation of restrictionon neuraminidase-treated cells, Proc. Natl. Acad. Sci. USA 75 (1978)2416–2420.
[41] M.K. Pangburn, R.D. Schreiber, H.J. Muller-Eberhard, Humancomplement C3b inactivator: isolation, characterization, and demon-stration of an absolute requirement for the serum protein beta1H forcleavage of C3b and C4b in solution, J. Exp. Med. 146 (1977)257–270.
[42] J.M. Weiler, M.R. Daha, K.F. Austen, D.T. Fearon, Control of theamplification convertase of complement by the plasma proteinbeta1H, Proc. Natl. Acad. Sci. USA 73 (1976) 3268–3272.
[43] G.A. Tennent, L.B. Lovat, M.B. Pepys, Serum amyloid P componentprevents peoteolysis of the amyloid fibrils of Alzheimer disease andsystemic amyloidosis, Proc. Natl. Acad. Sci. USA 92 (1995) 4299–4303.
[44] S.M. Lippa, C.F. Lippa, H. Mori, Alpha-synuclein aggregation inpathological aging and Alzheimer’s disease: the impact pf beta-amyloidplaque level, Am. J. Alzheimers Dis. Other Demen. 20 (2005) 315–318.
[45] O. Pletnikova, N. West, M.K. Lee, G.L. Rudow, R.L. Skolasty, T.M.Dawson, L. Marsh, J.C. Troncoso, Abeta deposition is associatedwith enhanced cortical alpha-synuclein lesions in Lewy body diseases,Neurobiol. Aging 26 (2004) 1183–1192.
[46] H. Yagi, E. Kusaka, H. Kunihiro, T. Mizobata, Y. Kawata, Amyloidfibril formation of a-synuclein is accelerated by amyloid seeds of otherproteins, J. Biol. Chem. 280 (2005) 38609–38616.
[47] P.G. de Frutos, Y. Hardig, B. Dahlback, Serum amyloid P componentbinding to C4b-binding protein, J. Biol. Chem. 45 (1995) 26950–26955.
[48] C. Schwab, J.C. Steele, E.G. McGeer, P.L. McGeer, Amyloid Pimmunoreactivity precedes C4d deposition on extracellular neurofi-brillary tangles, Acta Neuropathol. (Berlin) 93 (1997) 87–92.
[49] R. Veerhuis, I. Janssen, C.J. De Groot, F.L. Van Muiswinkel, C.E.Hack, P. Eikelenboom, Cytokines associated with amyloid plaques inAlzheimer’s disease brain stimulate human glial and neuronal cellcultures to secrete early complement proteins, but not C1-inhibitor,Exp. Neurol. 160 (1999) 289–299.
[50] R. Veerhuis, M.J. Van Breemen, J.M. Hoozemans, M. Morbin, J.Ouladhadj, F. Tagliavini, P. Eikelenboom, Amyloid beta plaque-associated proteins C1q and SAP enhance the Abeta1-42 peptide-induced cytokine secretion by adult human microglia in vitro, ActaNeuropathol. (Berlin) 105 (2003) 135–144.
[51] S. Ranganathan, E. Williams, P. Ganchev, V. Gopalakrishnan, D.Lacomis, L. Urbinelli, K. Newhall, M.E. Cudkowicz, R.H. Brown Jr.,R. Bowser, Proteomic profiling of cerebrospinal fluid identifiesbiomarkers for amyotrophic lateral sclerosis, J. Neurochem. 95(2005) 1461–1471.
[52] M.E. Alexianu, M. Kozovska, S.H. Appel, Immune reactivity in amouse model of familial ALS correlates with disease progression,Neurology 57 (2001) 1282–1289.
Related Documents











