1 College of Pharmacy 4 th Stage// Lecture 6 Medicinal Chemistry Dr.Narmin H.Amin Central Nervous System Stimulants CNS stimulants speed up mental and physical processes in the body. They increase energy, attention, and alertness, and elevate blood pressure, heart rate and respiratory rate. CNS stimulants are still used to treat attention-deficit hyperactivity disorder (ADHD), narcolepsy, and weight loss. Analeptics -Respiratory stimulants & Convulsants. Psychomotor stimulants- Excitement & Euphoria – Decrease feeling of fatigue & increase motor activity. Psychotomimetic (Hallucinogenic)- Changes in thought patterns & mood Analeptics Drugs: The traditional analeptics are a group of potent and relatively nonselective CNS stimulants. The convulsive dose lies near their analeptic dose. Picrotoxin: Picrotoxinin, the active ingredient of picrotoxin, the hydroxylactonyl moiety is mandatory for activity, with the 2-propenyl group assisting. Picrotoxinin exerts its effects by interfering with the inhibitory effects of γ- aminobutyric acid (GABA) at the level of the GABA A receptor’s chloride channel (Block action of GABA …. Blocks Cl- Conductance). It has been useful in determining mechanisms of action of sedative– hypnotics and anticonvulsants. Butyrolactones bind to the picrotoxinin site. Pentylenetetrazole(Metrazol): Pentylenetetrazole has been used in conjunction with the electroencephalograph to help locate epileptic foci. It is used as a laboratory tool in determining potencies of potential anticonvulsant drugs in experimental animals (In Epilepsy Research Work). The drug acts as a convulsant by interfering with chloride conductance.It binds to an allosteric site on the GABAA receptor and acts as a negative modulator.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
College of Pharmacy 4th
Stage// Lecture 6 Medicinal Chemistry Dr.Narmin H.Amin
Central Nervous System Stimulants
CNS stimulants speed up mental and physical processes in the body. They increase
energy, attention, and alertness, and elevate blood pressure, heart rate and respiratory
rate.
CNS stimulants are still used to treat attention-deficit hyperactivity disorder (ADHD),
narcolepsy, and weight loss.
Analeptics -Respiratory stimulants & Convulsants.
Psychomotor stimulants- Excitement & Euphoria – Decrease feeling of fatigue &
increase motor activity.
Psychotomimetic (Hallucinogenic)- Changes in thought patterns & mood
Analeptics Drugs: The traditional analeptics are a group of potent and relatively nonselective CNS
stimulants. The convulsive dose lies near their analeptic dose.
Picrotoxin: Picrotoxinin, the active ingredient of picrotoxin, the hydroxylactonyl moiety is
mandatory for activity, with the 2-propenyl group assisting.
Picrotoxinin exerts its effects by interfering with the inhibitory effects of γ-
aminobutyric acid (GABA) at the level of the GABAA receptor’s chloride channel
(Block action of GABA …. Blocks Cl- Conductance).
It has been useful in determining mechanisms of action of sedative– hypnotics and
anticonvulsants. Butyrolactones bind to the picrotoxinin site.
Pentylenetetrazole(Metrazol): Pentylenetetrazole has been used in conjunction with the electroencephalograph
to help locate epileptic foci.
It is used as a laboratory tool in determining potencies of potential anticonvulsant
drugs in experimental animals (In Epilepsy Research Work).
The drug acts as a convulsant by interfering with chloride conductance.It binds to
an allosteric site on the GABAA receptor and acts as a negative modulator.

2
Doxapram Hydrochloride ( Dopram): It stimulates respiration by action on peripheral carotid chemoreceptors. It has use as a
respiratory stimulant postanesthetically, after CNS depressant drug overdose, in chronic
obstructive pulmonary diseases, and in the apneas (Occasionally used in pt. with acute
resp. failure).
Dopram is administered exclusively by intravenous injection. Because of the benzyl
alcohol content of the injectable formulation, Dopram must never be given to neonates.
Strychnine (spinal cord Stimulant): Strychnine is an indole alkaloid obtained from the seeds of the Indian tree Strychnos nux-
vomica.
Strychnine inhibits competitively and reversibly the inhibitory neurotransmitter glycine at
postsynaptic neuronal sites in the spinal cord and medulla. This results in unchecked
reflex stimulation of motor neurons affecting all the striated muscles.
Because the extensor muscles are relatively more powerful than the flexor muscles, they
predominate to produce generalized rigidity and tonic-clonic seizures. Death results from
anoxia and exhaustion.

3
Psychomotor stimulants: SAR activity of psychomotor agents: Structural features for many of the agents can be visualized easily by considering that
within their structure, they contain a β-phenethylamine moiety, and this grouping can give
some selectivity for presynaptic or postsynaptic noradrenergic systems.
β-Phenethylamine, given peripherally, lacks central activity. Facile metabolic inactivation
by monoamine oxidases (MAOs) is held responsible.
Branching with lower alkyl groups on the carbon atom adjacent (α) to the amino nitrogen
increases CNS rather than peripheral activity (e.g., amphetamine, presumably by retarding
metabolism).
The dextro(S)-isomer of amphetamine is up to 10 times as potent as the levo(R)-isomer for
alerting activity and about twice as active as a psychotomimetic agent.
Hydroxylation of the ring or hydroxylation on the β-carbon (to the nitrogen) decreases
activity, largely by decreasing the ability to cross the blood brain barrier.
Halogenation (F, Cl, and Br) of the aromatic ring decreases sympathomimetic activity.
Other activities may increase. P-Chloroamphetamine has strong central serotoninergic
activity.
Methoxyl or methylenedioxy substitution on the ring tends to produce psychotomimetic
agents, suggesting tropism for dopaminergic (D2) receptors.
N-methylation increases activity (e.g,compare methamphetamine with
dextroamphetamine). Di-N-methylation decreases activity. Mono-N substituents larger than
methyl decrease excitatory properties.
Amphetamine Sulfate: Amphetamine, (±)-1-phenyl-2-aminopropane (Benzedrine), as the racemic mixture has a
higher proportion of cardiovascular effects than the dextro isomer. For most medical
uses, the dextrorotatory isomer is preferred.
Amphetamines include methamphetamine (meth) and phentermine.
Amphetamine is a commonly used street drug. It makes users feel very alert and have lots
of energy. Stimulants like amphetamine and methamphetamine can also make the user
feel very happy.

4
Dextroamphetamine Sulfate and Dextroamphetamine Phosphate: Dextroamphetamine, (+)-(S)-methylphenethylamine, forms salts with sulfuric acid
(Dexedrine) and with phosphoric acids. The phosphate is the more water-soluble salt and
is preferred if parenteral administration is required.
The dextrorotatory isomer has the (S) configuration and fewer cardiovascular effects than
the levorotatory (R)-isomer. Additionally, it may be up to 10 times as potent as the (R)-
isomer as an alerting agent and about twice as potent a psychotomimetic agent. Although
it is a more potent psychotomimetic agent than the (R)-isomer, it has a better ratio of
alerting to psychotomimetic effects.
The psychotomimetic effects are linked to release of DA and activation of postsynaptic
receptors. D2 and mesolimbic D3 receptors would be involved.
Dextroamphetamine is a strongly basic amine, with values from 9.77 to 9.94 reported.
Absorption from the gastrointestinal tract occurs as the lipid-soluble amine. The drug is
not extensively protein bound.
Varying amounts of the drug are excreted intact under ordinary conditions. The amount is
insignificant under conditions of alkaline urine. Under conditions producing systemic
acidosis, 60% to 70% of the drug can be excreted unchanged. This fact can be used to
advantage in treating drug overdose.
Under most conditions, the bulk of a dose of dextroamphetamine is metabolized by N-
dealkylation to phenylacetone and ammonia. Phenylacetone is degraded further to
benzoic acid.
In experimental animals, about 5% of a dose accumulates in the brain, especially the
cerebral cortex, the thalamus, and the corpus callosum. It is first p-hydroxylated and then
β-hydroxylated to produce p-hydroxynorephedrine, which has been reported to be the
major active metabolite involved in NE and DA release.

5
Methamphetamine Hydrochloride: Methamphetamine,(+)-1-phenyl-2-methylaminopropanehydrochloridedesoxyephedrine
hydrochloride (Desoxyn), is the N-methyl analog of dextroamphetamine.
It has more marked central and less peripheral action than dextroamphetamine.
It has a very high abuse potential, and by the intravenous route, its salts are known as
“speed.”
Medicinally acceptable uses of methamphetamine are analogous to those of
dextroamphetamine.
Methylphenidate Hydrochloride (Ritalin): Methylphenidate is a potent CNS stimulant. Indications include narcolepsy and
attention-deficit disorder.
Methylphenidate is the most commonly used medication for ADHD, which work by
increasing activity in the brain, particularly in areas that play a part in controlling
attention and behaviour.
Because methylphenidate has two asymmetric centers, there are four possible
isomers. Methylphenidate, probably largely via its p-hydroxy metabolite, blocks NE
reuptake, acts as a postsynaptic agonist, depletes the same NE pools as reserpine, and
has effects on dopaminergic systems, such as blocking DA reuptake.
Methylphenidate is an ester drug with interesting pharmacokinetic properties arising
from its structure. The pKa values are 8.5 and 8.8. The protonated form in the
stomach reportedly resists ester hydrolysis.
After absorption from the gastrointestinaltract, however, 80% to 90% of the drug is
hydrolyzed rapidly to inactive ritalinic acid. Another 2% to 5% of the racemate is
oxidized by liver microsomes to the inactive cyclic amide.
6
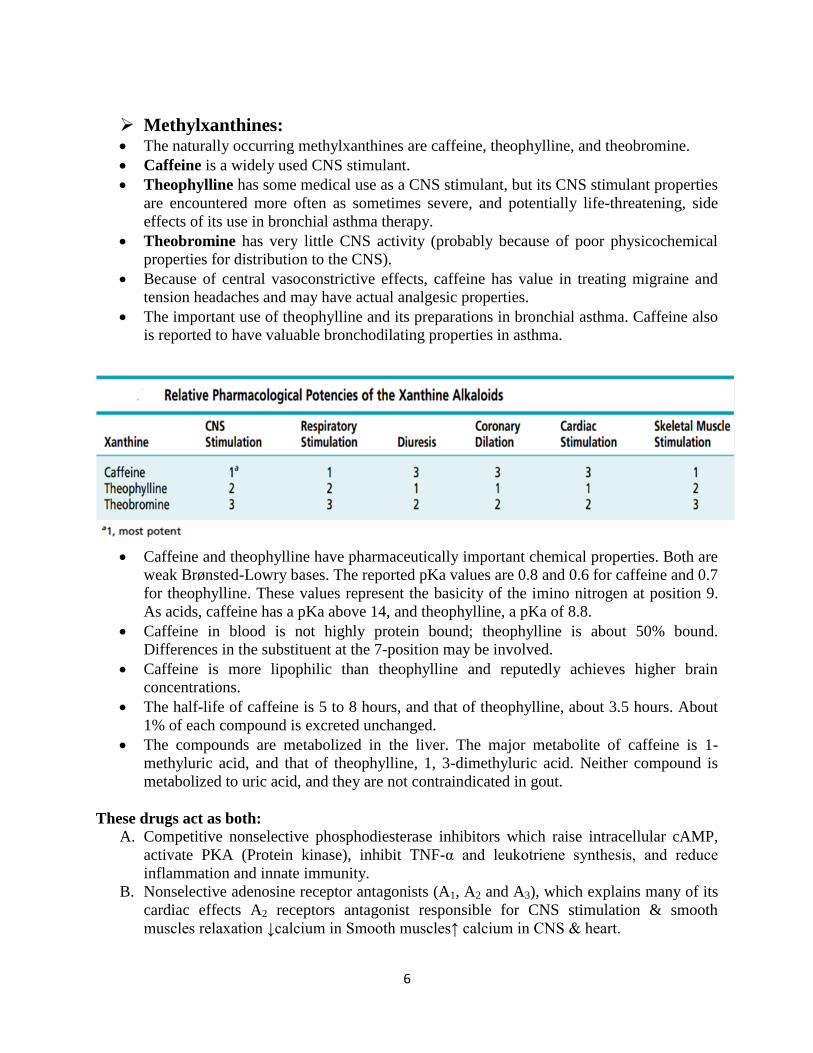
Methylxanthines: The naturally occurring methylxanthines are caffeine, theophylline, and theobromine.
Caffeine is a widely used CNS stimulant.
Theophylline has some medical use as a CNS stimulant, but its CNS stimulant properties
are encountered more often as sometimes severe, and potentially life-threatening, side
effects of its use in bronchial asthma therapy.
Theobromine has very little CNS activity (probably because of poor physicochemical
properties for distribution to the CNS).
Because of central vasoconstrictive effects, caffeine has value in treating migraine and
tension headaches and may have actual analgesic properties.
The important use of theophylline and its preparations in bronchial asthma. Caffeine also
is reported to have valuable bronchodilating properties in asthma.
Caffeine and theophylline have pharmaceutically important chemical properties. Both are
weak Brønsted-Lowry bases. The reported pKa values are 0.8 and 0.6 for caffeine and 0.7
for theophylline. These values represent the basicity of the imino nitrogen at position 9.
As acids, caffeine has a pKa above 14, and theophylline, a pKa of 8.8.
Caffeine in blood is not highly protein bound; theophylline is about 50% bound.
Differences in the substituent at the 7-position may be involved.
Caffeine is more lipophilic than theophylline and reputedly achieves higher brain
concentrations.
The half-life of caffeine is 5 to 8 hours, and that of theophylline, about 3.5 hours. About
1% of each compound is excreted unchanged.
The compounds are metabolized in the liver. The major metabolite of caffeine is 1-
methyluric acid, and that of theophylline, 1, 3-dimethyluric acid. Neither compound is
metabolized to uric acid, and they are not contraindicated in gout.
These drugs act as both:
A. Competitive nonselective phosphodiesterase inhibitors which raise intracellular cAMP,
activate PKA (Protein kinase), inhibit TNF-α and leukotriene synthesis, and reduce
inflammation and innate immunity.
B. Nonselective adenosine receptor antagonists (A1, A2 and A3), which explains many of its
cardiac effects A2 receptors antagonist responsible for CNS stimulation & smooth
muscles relaxation ↓calcium in Smooth muscles↑ calcium in CNS & heart.

7
Cocaine: Cocaine as a euphoriant–stimulant and drug of abuse could as well be discussed with
amphetamine and methamphetamine, with which it shares many biological properties. At
low doses, it produces feelings of well-being, decreased fatigue, and increased alertness.
Cocaine is blockade of reuptake of the monoamines (NE, serotonin and dopamine).
A phenethylamine moiety with added steric bulk may suffice for this action. An
interaction between a hydrogen atom on the nitrogen of the protonated form of cocaine
and oxygen of the benzoyl ester group, or alternatively, an interaction between the
unshared electron pair of the freebase nitrogen and the carbonyl of the benzoyl ester
group, could approximate this moiety.

8
Antidepressants: Antidepressant therapy usually implies therapy directed against major depressive
disorders of the unipolar type and is centered on three groups of chemical agents: the
MAOIs, the monoamine reuptake inhibitors, and autoreceptor desensitizers and
antagonists.
A-Monoamine Oxidase Inhibitors: MAOIs act by inhibiting the activity of monoamine oxidase, thus preventing the
breakdown of monoamine neurotransmitters and thereby increasing their availability.
There are two isoforms of monoamine oxidase, MAO-A and MAO-B. MAO-A
preferentially deaminates serotonin, melatonin, epinephrine, and norepinephrine. MAO-
B preferentially deaminates phenethylamine and certain other trace amines; in contrast,
MAO-A preferentially deaminates other trace amines, like tyramine, whereas dopamine
is equally deaminated by both types.
Patients taking MAOIs generally need to change their diets to limit or avoid foods and
beverages containing tyramine. Examples of foods and beverages with potentially high
levels of tyramine include liver and fermented substances, such as alcoholic beverages
and aged cheeses.
Tyramine leads to hypertensive crisis by increasing the release of norepinephrine (NE),
which causes blood vessels to constrict, When MAO-A is inhibited, though, NE levels
get too high, leading to dangerous increases in blood pressure.
Tranylcypromine Sulfate: Tranylcypromine sulfate, (±)-trans-2-phenylcyclopropylamine sulfate (Parnate), was
synthesized to be an amphetamine analog (visualize the -methyl of amphetamine
condensed onto the β-carbon atom). It does have some amphetamine-like properties,
which may be why it has more immediate CNS-stimulant effects than agents that act by
MAO inhibition alone.
Tranylcypromine is a mechanism-based inactivator. It is metabolized by MAO, with one
electron of the nitrogen pair lost to flavin. This, in turn, produces hemolytic fission of a
carbon–carbon bond of cyclopropane, with one electron from the fission pairing with the
remaining lone nitrogen electron to generate an imine (protonated) and with the other
residing on a methylene carbon.
Thus, a free radical is formed that reacts to form a covalent bond with the enzyme or with
reduced flavin to inactivate the enzyme.

9
B- Monoamine Reuptake Inhibitors: Is a drug that acts as a reuptake inhibitor of one or more of the three major monoamine
neurotransmitters serotonin, norepinephrine, and dopamine by blocking the action of one
or more of the respective monoamine transporters (MATs), which include the serotonin
transporter (SERT), norepinephrine transporter (NET), and dopamine transporter (DAT).
The net effect of the drug is to increase the level of the monoamine in the synapse.
Sustained high synaptic levels of 5-HT, NE, or both appear to be the basis for the
antidepressant effect of these agents.
Tricyclic Antidepressants:
The TCAs are extremely lipophilic and, accordingly, very highly tissue bound outside the
CNS. Because they have anticholinergic and noradrenergic effects, both central and
peripheral side effects are often unpleasant and sometimes dangerous. In overdose, the
combination of effects, as well as a quinidine-like cardiac depressant effect, can be lethal.
Overdose is complicated because the agents are so highly protein bound that dialysis is
ineffective.
Imipramine Hydrochloride: It is also a close relative of the antipsychotic phenothiazines (replace the 10–11 bridge
with sulfur, and the compound is the antipsychotic agent promazine). It has weaker D2
postsynaptic blocking activity than promazine and mainly affects amines (5-HT, NE, and
DA) via the transporters. As is typical of dimethylamino compounds, anticholinergic and
sedative (central H1 block) effects tend to be marked.
Metabolic inactivation proceeds mainly by oxidative hydroxylation in the 2-position,
followed by conjugation with glucuronic acid of the conjugate. Urinary excretion
predominates (about 75%), but some biliary excretion (up to 25%) can occur, probably
because of the large nonpolar grouping. Oxidative hydroxylation is not as rapid or
complete as that of the more nucleophilic ring phenothiazine antipsychotics;
consequently, appreciable N-demethylation occurs, with a buildup of norimipramine (or
desimipramine).
The demethylated metabolite is less anticholinergic, less sedative, and more stimulatory
and is a SNERI.Consequently, a patient treated with imipramine has two compounds that
contribute to activity. Overall, the effect is nonselective 5-HT versus NE reuptake. The
activity of desimipramine or norimipramine is terminated by 2-hydroxylation, followed
by conjugation and excretion. A second N-demethylation can occur, which in turn is
followed by 2-hydroxylation, conjugation, and excretion

10
Clomipramine Hydrochloride: Clomipramine (Anafranil) is up to 50 times as potent as imipramine in some bioassays.
The chloro replacing the H-substituent could increase potency by increasing distribution
to the CNS.
It might be conjectured that an H-bond between the protonated amino group (as in vivo)
and the unshared electrons of the chloro substituent might stabilize a β-arylamine–like
shape and give more efficient competition for the transporter. The drug is an
antidepressant. It is used in obsessive compulsive disorder, an anxiety disorder that may
have an element of depression.
Amitriptyline Hydrochloride: Amitriptyline is one of the most anticholinergic and sedative of the TCAs. Because it
lacks the ring-electron–enriching nitrogen atom of imipramine, metabolic inactivation
mainly proceeds not at the analogous 2-position but at the benzylic 10-position (i.e.,
toluene-like metabolism predominates).
Because of the 5- exocyclic double bond, E- and Z-hydroxy isomers are produced by
oxidation metabolism. Conjugation produces excretable metabolites. As is typical of the
dimethyl compounds, N-demethylation occurs, and nortriptyline is produced, which has a
less anticholinergic, less sedative, and more stimulant action than amitriptyline.
Nortriptyline is a SNERI; the composite action of drug and metabolite is nonselective.
Nortriptyline metabolic inactivation and elimination are like those of amitriptyline.
Amitriptyline Nortriptyline

11
C- Selective Serotonin Reuptake Inhibitors: Structurally, the SSRIs differ from the tricyclics, in that the tricyclic system has been
taken apart in the center.
Many of the dimethylamino tricyclics are, in fact, SSRIs. Because they are extensively N-
demethylated in vivo to norcompounds, which are usually SNERIs, however, the overall
effect is not selective. Breaking up the tricyclic system breaks up an anticholinergic
pharmacophoric group and gives compounds with diminished anticholinergic effects.
Overall, this diminishes unpleasant CNS effects ad increases cardiovascular safety.
Instead, side effects related to serotonin predominate.
Fluoxetine (Prozac): In fluoxetine, protonated in vivo, the protonated amino group can H-bond to the ether
oxygen electrons, which can generate the β-arylamino–like group, with the other aryl
serving as the characteristic “extra” aryl. The S-isomer is much more selective for SERT
than for NET. The major metabolite is the N-demethyl compound, which is as potent as
the parent and more selective (SERT versus NET).
Therapy for 2 or more weeks is required for the antidepressant effect. Somatodendritic 5-
HT1A autoreceptor desensitization with chronic exposure to high levels of 5-HT is the
accepted explanation for the delayed effect for this and other serotonin reuptake
inhibitors.
Paroxetine (Paxil): In the structure of paroxetine (Paxil), an amino group, protonated in vivo could H-bond
with the –CH2–O– unshared electrons. A β-arylamine–like structure with an extra aryl
group results. The compound is a very highly selective SERT. As expected, it is an
effective antidepressant and anxiolytic

12
Sertraline (Zoloft): Inspection of sertraline reveals the pharmacophore for SERT inhibition. The Cl
substituents also predict tropism for a 5-HT system. The depicted stereochemistry is
important for activity.
Citalopram (Celexa): Citalopram is a racemic mixture and is very SERT selective. The N-monodemethylated
compound is slightly less potent but is as selective. The aryl substituents are important
for activity. The ether function is important and probably interacts with the protonated
amino group to give a suitable shape for SERT binding.
Hallucinogens (psychotomimetic):
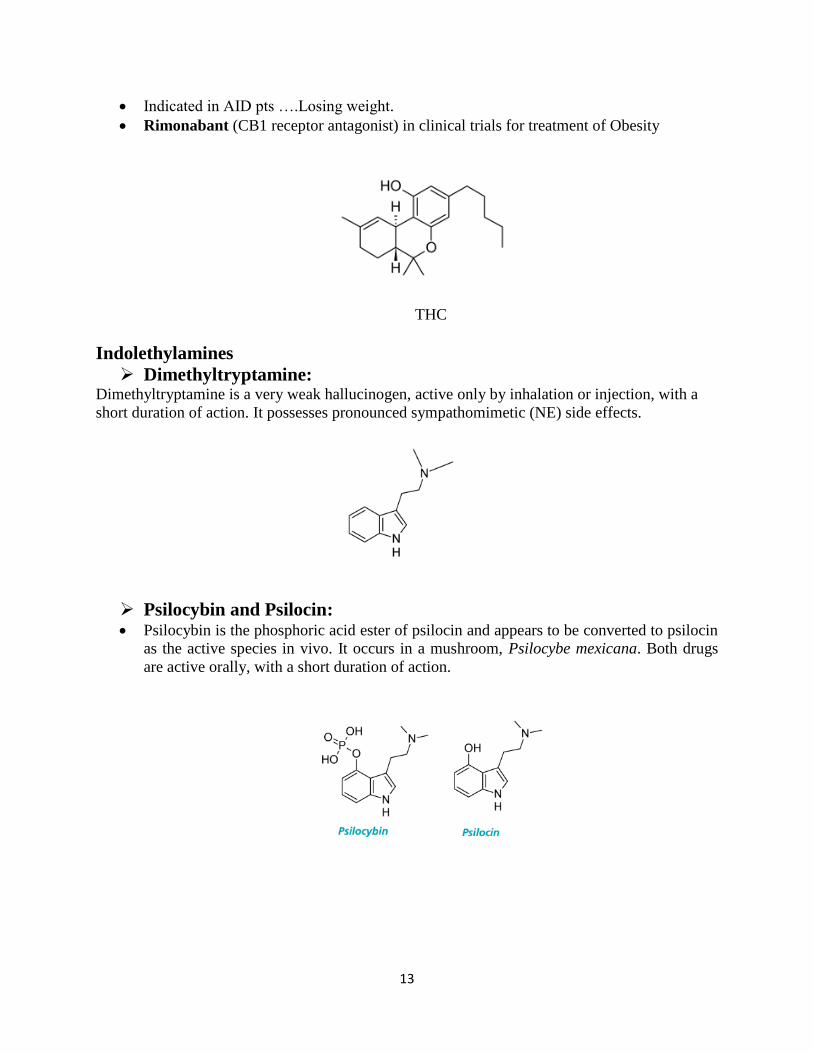
Tetrahydrocannabinol (THC) Tetrahydrocannabinoid is a depressant with apparent stimulant sensations arising from
depression of higher centers. The main psychoactive alkaloid contained in marijuana is
tetrahydrocannabinol (THC).
Many effects, reputedly subjectively construed as pleasant, are evident at low doses. At
higher doses, psychotomimetic actions, including dysphoria, hallucinations, and paranoia,
can be marked.
Structural features associated with activity among cannabis-derived compounds, the
phenolic OH is required for activity.
Two receptors for THC have been discovered. The relevant receptor for CNS actions is
CB1. CB2 occurs in immune tissues. The first natural ligand found for the receptor is the
amide derivative of arachidonic acid, anandamide. Other natural cannabinoids are
arachidonic acid 2-glycerol ester and 2-arachidonyl glycerol ether.
The endogenous cannabinoid system appears to function as a retrograde messenger
system at both stimulatory synapses and depressant synapses.
13
Indicated in AID pts ….Losing weight.
Rimonabant (CB1 receptor antagonist) in clinical trials for treatment of Obesity
THC
Indolethylamines
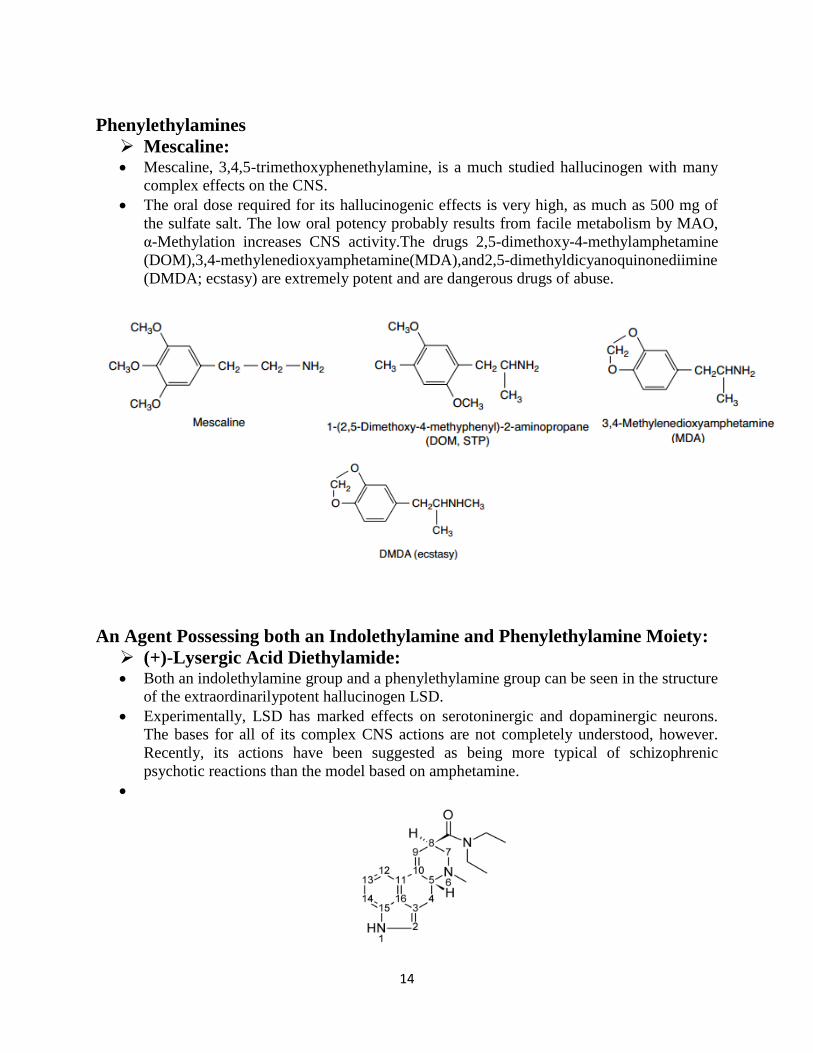
Dimethyltryptamine: Dimethyltryptamine is a very weak hallucinogen, active only by inhalation or injection, with a
short duration of action. It possesses pronounced sympathomimetic (NE) side effects.
Psilocybin and Psilocin: Psilocybin is the phosphoric acid ester of psilocin and appears to be converted to psilocin
as the active species in vivo. It occurs in a mushroom, Psilocybe mexicana. Both drugs
are active orally, with a short duration of action.
14
Phenylethylamines
Mescaline: Mescaline, 3,4,5-trimethoxyphenethylamine, is a much studied hallucinogen with many
complex effects on the CNS.
The oral dose required for its hallucinogenic effects is very high, as much as 500 mg of
the sulfate salt. The low oral potency probably results from facile metabolism by MAO,
α-Methylation increases CNS activity.The drugs 2,5-dimethoxy-4-methylamphetamine
(DOM),3,4-methylenedioxyamphetamine(MDA),and2,5-dimethyldicyanoquinonediimine
(DMDA; ecstasy) are extremely potent and are dangerous drugs of abuse.
An Agent Possessing both an Indolethylamine and Phenylethylamine Moiety:
(+)-Lysergic Acid Diethylamide: Both an indolethylamine group and a phenylethylamine group can be seen in the structure
of the extraordinarilypotent hallucinogen LSD.
Experimentally, LSD has marked effects on serotoninergic and dopaminergic neurons.
The bases for all of its complex CNS actions are not completely understood, however.
Recently, its actions have been suggested as being more typical of schizophrenic
psychotic reactions than the model based on amphetamine.

15
Dissociative Agents:
Phencyclidine: It blocks glutaminergic N-methyl-D-aspartate receptors.
Phencyclidine was introduced as a dissociative anesthetic for animals. Its close
structural relative ketamine is still so used and may be used in humans .In
humans, PCP produces a sense of intoxication, hallucinogenic experiences not
unlike those produced by the anticholinergic hallucinogens, and often, amnesia
The drug affects many systems, including those of NE, DA, and 5-HT. It has been
proposed that PCP (and certain other psychotomimetics) produces a unique
pattern of activation of ventral tegumental area dopaminergic neurons.
The psychotic state produced by this drug is also cited as a better model than
amphetamine psychosis for the psychotic state of schizophrenia.
Newer Drugs:
Modafinil Armodafinil
Questions: Are Stimulants Addictive? How?
Related Documents











