The EMBO Journal vol. 15 no.7 pp. 1 596-1606, 1996 Cell type-specific inhibition of p53-mediated apoptosis by mdm2 Ygal Haupt, Yaacov Barak1 and Moshe Oren2 Department of Molecular Cell Biology, The Weizmann Institute of Science, Rehovot 76100, Israel 'Present address: The Salk Institute, POB 85800, San Diego, CA 92186, USA 2Corresponding author The effect of excess mdm2 on p53-mediated apoptosis was investigated in two human-derived cell lines, H1299 and HeLa. In H1299 cells, overexpression of mdm2 resulted in effective protection from apoptosis. This protective effect was seen only under conditions allowing the formation of p53-Mdm2 complexes. In contrast, excess mdm2 failed to abolish p53-mediated apoptosis in HeLa cells, despite a complete abrogation of p53-dependent sequence-specific transcriptional activation (SST). These data strongly support the contention that SST is dispensable for at least some types of p53-mediated apoptosis. Further, they suggest that one of the roles of mdm2 may be to modulate the apoptotic activity of p53, in a manner which is dictated by the pathway through which p53 induces apoptosis in a given cell type. Keywords: apoptosis/mdm2/p53/transactivation Introduction The p53 tumor suppressor plays an important role in the regulation of cell growth. In response to signals such as DNA damage and the illegitimate activation of certain viral and cellular oncogenes, accumulated p53 can induce cell growth arrest and/or apoptosis (Levine, 1993; Oren, 1994; Haffner and Oren, 1995). p53 induces growth arrest primarily at the G1 phase of the cell cycle, and to some extent also at the G2 phase. This presumably allows the damaged cell to carry out effective DNA repair, and thereby genomic integrity is maintained (Lane, 1992). In other cases, either when DNA repair cannot be completed successfully or when the cell is not programmed to respond to p53 by a viable growth arrest, p53 activation may destine the cell for apoptosis. p53-dependent apoptosis has been demonstrated in a variety of cell types (reviewed in Oren, 1994). In some cases, p53-mediated apoptosis can be inhibited by the presence of survival factors, including various cytokines (Yonish-Rouach et al., 1991; Johnson et al., 1993; Canman et al., 1995). Similarly, withdrawal of survival factors from factor-dependent cells results in apoptotic death which is at least partially dependent on p53 (Lotem and Sachs, 1993; Gottlieb et al., 1994; Zhu et al., 1994; Canman et al., 1995). The particular cellular outcome in response to activated p53 depends on cell type, cellular context and extracellular signals (Haffner and Oren, 1995). p53 is a bona fide transcription factor (Vogelstein and Kinzler, 1992; Prives and Manfredi, 1993). Sequence- specific transcriptional activation (SST) by p53 correlates well with the ability of p53 to suppress cell growth (Reed et al., 1993; Shaulian et al., 1993; Crook et al., 1994; Ory et al., 1994; Pietenpol et al., 1994). This is believed to be achieved primarily through the induction of p21 wafipl, a broad-spectrum inhibitor of cyclin-dependent kinases (El-Deiry et al., 1993; Harper et al., 1993; Xiong et al., 1993; Dulic et al., 1994). Overexpression of p21 alone, and especially of truncated forms which possess excess kinase inhibitory activity, is on its own sufficient to suppress cell growth (Li et al., 1994; Waga et al., 1994; Zakut and Givol, 1995). SST also appears to be important for the apoptotic activity of p53 (El-Deiry et al., 1994; Sabbatini et al., 1995b). To date at least two targets of p53, bax and Fas/Apo- 1, have been implicated in apoptosis (Miyashita et al., 1994; Selvakumaran et al., 1994; Miyashita and Reed, 1995; Owen-Schaub et al., 1995). While SST is likely to contribute to p53-dependent apoptosis, it may not be essential for every case. Indeed, p53-dependent apoptosis can occur in the presence of RNA and protein synthesis inhibitors (Caelles et al., 1994). More directly, we have recently shown that a deletion mutant of p53, which lacks detectable SST ability, is a potent inducer of apoptosis in HeLa cells (Haupt et al., 1995b). A similar suggestion was recently made by Ishioka et al. (1995). Thus, p53 can potentially induce apoptosis by at least two distinct pathways: one involving SST and another which is independent of SST. Growth suppression by p53, through the induction of either growth arrest or apoptosis, can be modulated by viral and cellular proteins. For a successful viral life cycle, DNA tumor viruses evolved proteins that abrogate p53 functions (Fanning and Knippers, 1992; Ludlow, 1993; Moran, 1993; Vousden, 1993). The SV40 large T and the adenovirus E1B 55 kDa proteins inhibit p53 SST (Farmer et al., 1992; Mietz et al., 1992; Yew et al., 1994). An additional mechanism is used by the human papilloma viral protein, E6, which also targets p53 for degradation through the ubiquitin pathway (Scheffner et al., 1990). Among the cellular proteins that modulate p53 function, the best characterized is Mdm2. The mdm2 gene was identified as an oncogene amplified in a mouse tumor cell line (Cahilly-Snyder et al., 1987; Fakharzadeh et al., 199 1). mdm2 was later found to be amplified in a significant proportion of human sarcomas (Oliner et al., 1992; Leach et al., 1993; Cordon-Cardo et al., 1994), as well as in a variety of other tumors (Ladanyi et al., 1993; Reifenberger et al., 1993; Corvi et al., 1995; McCann et al., 1995). Mdm2 binds p53 both in vivo and in vitro (Hinds et al., 1990; Barak and Oren, 1992; Momand et al., 1992; Oliner 1 6 Oxford University Press 1596

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The EMBO Journal vol. 15 no.7 pp. 1 596-1606, 1996
Cell type-specific inhibition of p53-mediatedapoptosis by mdm2
Ygal Haupt, Yaacov Barak1 andMoshe Oren2Department of Molecular Cell Biology, The Weizmann Institute ofScience, Rehovot 76100, Israel
'Present address: The Salk Institute, POB 85800, San Diego,CA 92186, USA
2Corresponding author
The effect of excess mdm2 on p53-mediated apoptosiswas investigated in two human-derived cell lines, H1299and HeLa. In H1299 cells, overexpression of mdm2resulted in effective protection from apoptosis. Thisprotective effect was seen only under conditionsallowing the formation of p53-Mdm2 complexes. Incontrast, excess mdm2 failed to abolish p53-mediatedapoptosis in HeLa cells, despite a complete abrogationof p53-dependent sequence-specific transcriptionalactivation (SST). These data strongly support thecontention that SST is dispensable for at least sometypes of p53-mediated apoptosis. Further, they suggestthat one of the roles of mdm2 may be to modulate theapoptotic activity of p53, in a manner which is dictatedby the pathway through which p53 induces apoptosisin a given cell type.Keywords: apoptosis/mdm2/p53/transactivation
IntroductionThe p53 tumor suppressor plays an important role in theregulation of cell growth. In response to signals such asDNA damage and the illegitimate activation of certainviral and cellular oncogenes, accumulated p53 can inducecell growth arrest and/or apoptosis (Levine, 1993; Oren,1994; Haffner and Oren, 1995). p53 induces growth arrestprimarily at the G1 phase of the cell cycle, and to someextent also at the G2 phase. This presumably allows thedamaged cell to carry out effective DNA repair, andthereby genomic integrity is maintained (Lane, 1992). Inother cases, either when DNA repair cannot be completedsuccessfully or when the cell is not programmed to respondto p53 by a viable growth arrest, p53 activation maydestine the cell for apoptosis. p53-dependent apoptosishas been demonstrated in a variety of cell types (reviewedin Oren, 1994). In some cases, p53-mediated apoptosiscan be inhibited by the presence of survival factors,including various cytokines (Yonish-Rouach et al., 1991;Johnson et al., 1993; Canman et al., 1995). Similarly,withdrawal of survival factors from factor-dependent cellsresults in apoptotic death which is at least partiallydependent on p53 (Lotem and Sachs, 1993; Gottlieb et al.,1994; Zhu et al., 1994; Canman et al., 1995). The particularcellular outcome in response to activated p53 depends on
cell type, cellular context and extracellular signals (Haffnerand Oren, 1995).
p53 is a bona fide transcription factor (Vogelstein andKinzler, 1992; Prives and Manfredi, 1993). Sequence-specific transcriptional activation (SST) by p53 correlateswell with the ability of p53 to suppress cell growth (Reedet al., 1993; Shaulian et al., 1993; Crook et al., 1994;Ory et al., 1994; Pietenpol et al., 1994). This is believedto be achieved primarily through the induction ofp21wafipl, a broad-spectrum inhibitor of cyclin-dependentkinases (El-Deiry et al., 1993; Harper et al., 1993; Xionget al., 1993; Dulic et al., 1994). Overexpression of p21alone, and especially of truncated forms which possessexcess kinase inhibitory activity, is on its own sufficientto suppress cell growth (Li et al., 1994; Waga et al., 1994;Zakut and Givol, 1995). SST also appears to be importantfor the apoptotic activity of p53 (El-Deiry et al., 1994;Sabbatini et al., 1995b). To date at least two targets ofp53, bax and Fas/Apo- 1, have been implicated in apoptosis(Miyashita et al., 1994; Selvakumaran et al., 1994;Miyashita and Reed, 1995; Owen-Schaub et al., 1995).While SST is likely to contribute to p53-dependentapoptosis, it may not be essential for every case. Indeed,p53-dependent apoptosis can occur in the presence ofRNA and protein synthesis inhibitors (Caelles et al., 1994).More directly, we have recently shown that a deletionmutant of p53, which lacks detectable SST ability, is apotent inducer of apoptosis in HeLa cells (Haupt et al.,1995b). A similar suggestion was recently made by Ishiokaet al. (1995). Thus, p53 can potentially induce apoptosisby at least two distinct pathways: one involving SST andanother which is independent of SST.
Growth suppression by p53, through the induction ofeither growth arrest or apoptosis, can be modulated byviral and cellular proteins. For a successful viral life cycle,DNA tumor viruses evolved proteins that abrogate p53functions (Fanning and Knippers, 1992; Ludlow, 1993;Moran, 1993; Vousden, 1993). The SV40 large T and theadenovirus E1B 55 kDa proteins inhibit p53 SST (Farmeret al., 1992; Mietz et al., 1992; Yew et al., 1994). Anadditional mechanism is used by the human papillomaviral protein, E6, which also targets p53 for degradationthrough the ubiquitin pathway (Scheffner et al., 1990).Among the cellular proteins that modulate p53 function,the best characterized is Mdm2. The mdm2 gene wasidentified as an oncogene amplified in a mouse tumor cellline (Cahilly-Snyder et al., 1987; Fakharzadeh et al.,1991). mdm2 was later found to be amplified in a significantproportion of human sarcomas (Oliner et al., 1992; Leachet al., 1993; Cordon-Cardo et al., 1994), as well as in avariety of other tumors (Ladanyi et al., 1993; Reifenbergeret al., 1993; Corvi et al., 1995; McCann et al., 1995).Mdm2 binds p53 both in vivo and in vitro (Hinds et al.,1990; Barak and Oren, 1992; Momand et al., 1992; Oliner
16 Oxford University Press1596

Modulation of p53-mediated apoptosis by mdm2
et al., 1992; Chen et al., 1993). This binding inhibits p-53SST, as well as the ability of p53 to repress the activityof at least some promoters (Momand et al., 1992; Brownet al., 1993; Oliner et al., 1993; Haines et al., 1994; Chenet al., 1995). The binding of Mdm2 to the transactivationdomain of p53 (Chen et al., 1993; Picksley et al., 1994)has been proposed to conceal this domain from interactionwith the transcriptional machinery (Oliner et al., 1993).The observation that the TATA binding-protein-associated-factors (TAFs) and Mdm2 bind overlapping sites in thep53 protein provides further support for this notion (Linet al., 1994; Lu and Levine, 1995; Thut et al., 1995). Thebinding of Mdm2 to p53 has also been shown to resultin vivo in complexes lacking the ability to bind to specificDNA sites (Zauberman et al., 1993).The physical association between the p53 and Mdm2
polypeptides is part of a more complex picture. Activationof p53 was shown to induce mdm2 expression (Baraket al., 1993; Otto and Deppert, 1993; Wu et al., 1993;Price and Park, 1994). In fact, p53 binding sites wereidentified within the murine mdm2 gene (Juven et al.,1993; Wu et al., 1993) as well as in the human mdm2gene (Zauberman et al., 1995b). These sites constitutepart of a distinct internal p53-dependent promoter, whichbecomes functional only in the presence of activated p53and gives rise to novel, efficiently translatable transcripts(Juven et al., 1993; Barak et al., 1994; Zauberman et al.,1995b). These findings have suggested an autoregulatoryfeedback loop between p53 and mdm2 (Barak et al., 1993;Picksley and Lane, 1993; Wu et al., 1993). As predictedfrom the above observations, excess expression of mdm2can prevent p53-mediated growth arrest (Chen et al.,1994). Yet, the endogenous mdm2 gene is activated byp53 under a variety of conditions where the cells areeffectively growth arrested (Barak et al., 1993, 1994; Ottoand Deppert, 1993). Work by Perry et al. (1993) hasrevealed that in cells exposed to extensive DNA damage,the appearance of Mdm2 proteins is severely delayed.This raises the interesting possibility that the functionalinterplay between p53 and mdm2 involves yet another levelof regulation, namely translation of the mdm2 message.The interaction between the p53 and Mdm2 proteins
has important biological consequences. The oncogeniceffect of mdm2 was directly shown by cooperation withras in the transformation of primary rat embryo fibroblasts(REFs), and further, mdm2 overcomes the ability of p53to inhibit transformation (Finlay, 1993). Consistent withthe latter role of mdm2 is the paucity of p53 mutations inhuman sarcomas containing amplified mdm2 (Oliner et al.,1992). Moreover, tumor cells overexpressing mdm2 cantolerate high levels of wild-type (wt) p53 (Finlay, 1993;Otto and Deppert, 1993).The inhibition of p53 SST, and consequently p53-
dependent growth arrest by mdm2, prompted us to evaluatewhether mdm2 can also modulate p53-mediated apoptosis.In particular, in view of the multiple mechanisms bywhich p53 was proposed to induce apoptosis (Haupt et al.,1995b), it was of interest to find out whether the effectof excess mdm2 on p53-mediated apoptosis will varyamong different cell types. For this purpose, transienttransfection assays in H1299 and HeLa cells were per-formed. We report here that overexpression of mdm2inhibits efficiently p53-mediated apoptosis in H1299 cells.
This inhibitory effect correlates with an ability of theexcess mdm2 to block p53 SST, and is dependent on theability of the transfected Mdm2 protein to form complexeswith p53. In contrast, excess mdm2 failed to block p53-mediated apoptosis in HeLa cells, even when it practicallyabolished p53-specific SST. These findings suggest thatone normal function of the mdm2 gene is to modulate theapoptotic activity of p53, and that the net effect will bedictated by the particular pathways through which p53exerts its apoptotic action in a given cell.
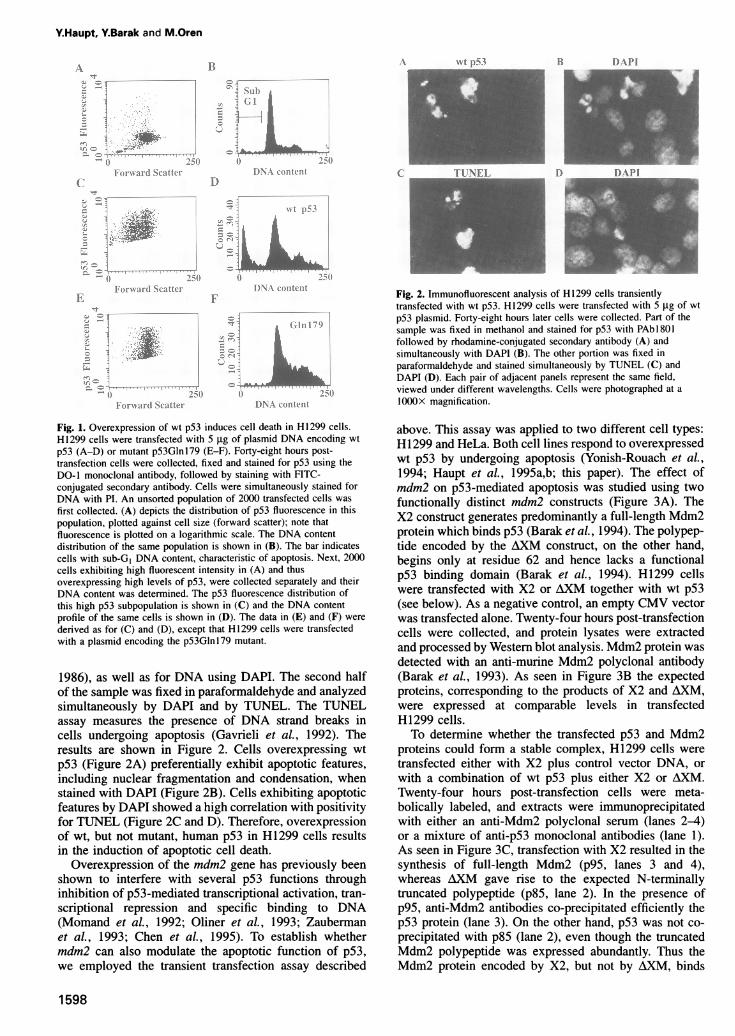
ResultsOverexpression ofmdm2 inhibits p53-mediatedapoptosis in H1299 cellsIn order to identify cellular factors that modulate p53-mediated apoptosis we employed cell lines that respondto activation of p53 by undergoing apoptosis. One suchcell line is the p53-null large cell lung carcinoma, H1299(Mitsudomi et al., 1992). The biological effect of p53 wasstudied in H1299 cells by a transient transfection assayusing flow cytometric analysis as previously described(Haupt et al., 1995a,b). Wild type (wt) human p53 cDNA,driven by the cytomegalovirus promoter/enhancer element(CMV), was transfected into H1299 cells using the calciumphosphate method. Forty-eight hours post-transfectioncells were collected, fixed and stained for p53 by incuba-tion with a specific anti-p53 monoclonal antibody, DO-1(Vojtesek et al., 1992), followed by incubation with afluorescein isothiocyanate (FHTC)-conjugated secondaryantibody. Cells expressing elevated levels of exogenousp53 were detected by virtue of their high fluorescentintensity (Figure lA, note that fluorescent intensity ismeasured on a logarithmic scale). This intensity was wellabove the background fluorescence of the bulk, presumablynon-transfected population, which was ~0.5-1 X 10' inFigure lA. The effect of p53 overexpression on theviability and cell cycle distribution of H1299 cells wasdetermined by measuring the DNA content of the trans-fected cells, through staining with propidium iodide. Tocompare the successfully transfected cells with the non-transfected bulk population, each population was collectedand analyzed separately. Initially, 2000 cells from the totalpopulation were collected. Then, a gate defining cells withhigh fluorescent intensity was set, and 2000 cells fallinginto this gate were collected separately (Figure IC). Thelatter population consists of successfully transfected cellsonly. The cell cycle distributions of the bulk (Figure iB)and of the transfected population (Figure ID) were nextdetermined. A significant fraction of the transfected cellsshowed a DNA content of <2N, and thus fell into thesub-GI fraction (Figure 1D). The sub-GI fraction definesdead cells, and is characteristic of apoptosis (e.g. Hauptet al., 1995a,b). Overexpression of tumor-derived mutantp53, p53Glnl79, had no apoptotic activity in H1299 cells(Figure IE and F). Thus, induction of H1299 cell deathrequires the wt function of p53.To verify that overexpression of wt p53 in H1299
cells induces death by apoptosis, transfected cells wereexamined microscopically for the appearance of apoptoticfeatures 48 h post-transfection. To that end, half of thesample was fixed in methanol and stained for p53 usingthe anti-p53 monoclonal antibody PAb 1801 (Banks et al.,
1597
Y.Haupt, Y.Barak and M.Oren
-r-
-r
-
- Ii 250]Iorir=-t4b'
I)
-_' ffi~~
IN I"LI. 1) 1)AP11
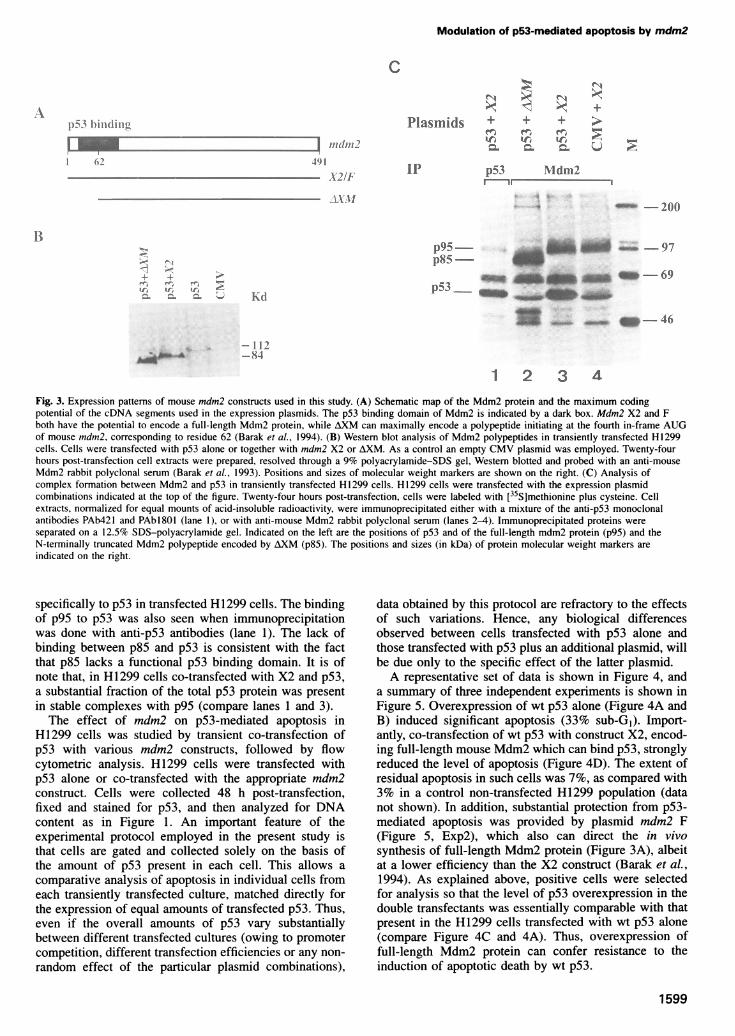
Fig. 2. Immunofluorescent analysis of H 1299 cells transientlytransfected with wt p53. H1299 cells were transfected with 5 ig of wtp53 plasmid. Forty-eight hours later cells were collected. Part of thesample was fixed in methanol and stained for p53 with PAbl 801followed by rhodamine-conjugated secondary antibody (A) andsimultaneously with DAPI (B). The other portion was fixed inparaformaldehyde and stained simultaneously by TUNEL (C) andDAPI (D). Each pair of adjacent panels represent the same field,viewed under different wavelengths. Cells were photographed at a
IOOOX magnification.
Fig. 1. Overexpression of wt p53 induces cell death in H1299 cells.H1299 cells were transfected with 5 gg of plasmid DNA encoding wtp53 (A-D) or mutant p53Glnl79 (E-F). Forty-eight hours post-transfection cells were collected, fixed and stained for p53 using theDO-1 monoclonal antibody, followed by staining with FITC-conjugated secondary antibody. Cells were simultaneously stained forDNA with PI. An unsorted population of 2000 transfected cells was
first collected. (A) depicts the distribution of p53 fluorescence in thispopulation, plotted against cell size (forward scatter); note thatfluorescence is plotted on a logarithmic scale. The DNA contentdistribution of the same population is shown in (B). The bar indicatescells with sub-GI DNA content, characteristic of apoptosis. Next, 2000cells exhibiting high fluorescent intensity in (A) and thusoverexpressing high levels of p53, were collected separately and theirDNA content was determined. The p53 fluorescence distribution ofthis high p53 subpopulation is shown in (C) and the DNA contentprofile of the same cells is shown in (D). The data in (E) and (F) were
derived as for (C) and (D), except that H1299 cells were transfectedwith a plasmid encoding the p53Glnl79 mutant.
1986), as well as for DNA using DAPI. The second halfof the sample was fixed in paraformaldehyde and analyzedsimultaneously by DAPI and by TUNEL. The TUNELassay measures the presence of DNA strand breaks incells undergoing apoptosis (Gavrieli et al., 1992). Theresults are shown in Figure 2. Cells overexpressing wtp53 (Figure 2A) preferentially exhibit apoptotic features,including nuclear fragmentation and condensation, whenstained with DAPI (Figure 2B). Cells exhibiting apoptoticfeatures by DAPI showed a high correlation with positivityfor TUNEL (Figure 2C and D). Therefore, overexpressionof wt, but not mutant, human p53 in H1299 cells resultsin the induction of apoptotic cell death.
Overexpression of the mdm2 gene has previously beenshown to interfere with several p53 functions throughinhibition of p53-mediated transcriptional activation, tran-scriptional repression and specific binding to DNA(Momand et al., 1992; Oliner et al., 1993; Zaubermanet al., 1993; Chen et al., 1995). To establish whethermdm2 can also modulate the apoptotic function of p53,we employed the transient transfection assay described
above. This assay was applied to two different cell types:H11299 and HeLa. Both cell lines respond to overexpressedwt p53 by undergoing apoptosis (Yonish-Rouach et al.,1994; Haupt et al., 1995a,b; this paper). The effect ofmdm2 on p53-mediated apoptosis was studied using twofunctionally distinct mdm2 constructs (Figure 3A). TheX2 construct generates predominantly a full-length Mdm2protein which binds p53 (Barak et al., 1994). The polypep-tide encoded by the AXM construct, on the other hand,begins only at residue 62 and hence lacks a functionalp53 binding domain (Barak et al., 1994). H1299 cellswere transfected with X2 or AXM together with wt p53(see below). As a negative control, an empty CMV vectorwas transfected alone. Twenty-four hours post-transfectioncells were collected, and protein lysates were extractedand processed by Western blot analysis. Mdm2 protein wasdetected with an anti-murine Mdm2 polyclonal antibody(Barak et al., 1993). As seen in Figure 3B the expectedproteins, corresponding to the products of X2 and AXM,were expressed at comparable levels in transfectedH11299 cells.To determine whether the transfected p53 and Mdm2
proteins could form a stable complex, H1299 cells were
transfected either with X2 plus control vector DNA, or
with a combination of wt p53 plus either X2 or AXM.Twenty-four hours post-transfection cells were meta-bolically labeled, and extracts were immunoprecipitatedwith either an anti-Mdm2 polyclonal serum (lanes 2-4)or a mixture of anti-p53 monoclonal antibodies (lane 1).As seen in Figure 3C, transfection with X2 resulted in thesynthesis of full-length Mdm2 (p95, lanes 3 and 4),whereas AXM gave rise to the expected N-terminallytruncated polypeptide (p85, lane 2). In the presence ofp95, anti-Mdm2 antibodies co-precipitated efficiently thep53 protein (lane 3). On the other hand, p53 was not co-
precipitated with p85 (lane 2), even though the truncatedMdm2 polypeptide was expressed abundantly. Thus theMdm2 protein encoded by X2, but not by AXM, binds
1598
Bt A N-t ps153 IDI)API
1)
Modulation of p53-mediated apoptosis by mdm2
C
Plasmids + +M, et,t.i ,rQ Cinmd2
p53 bAlid({i "1-
m Dl(}n 4'} l~~~~~~~~
X21F IP p53I 11
.\Avi
1Bp95 -p85
+
Ir,Z.
4...
MI
-112-X4
- -
__.
-~ mw -m__ __ __ _
-X Mot .*4M40
69a~~~~ -46
1 2 3 4
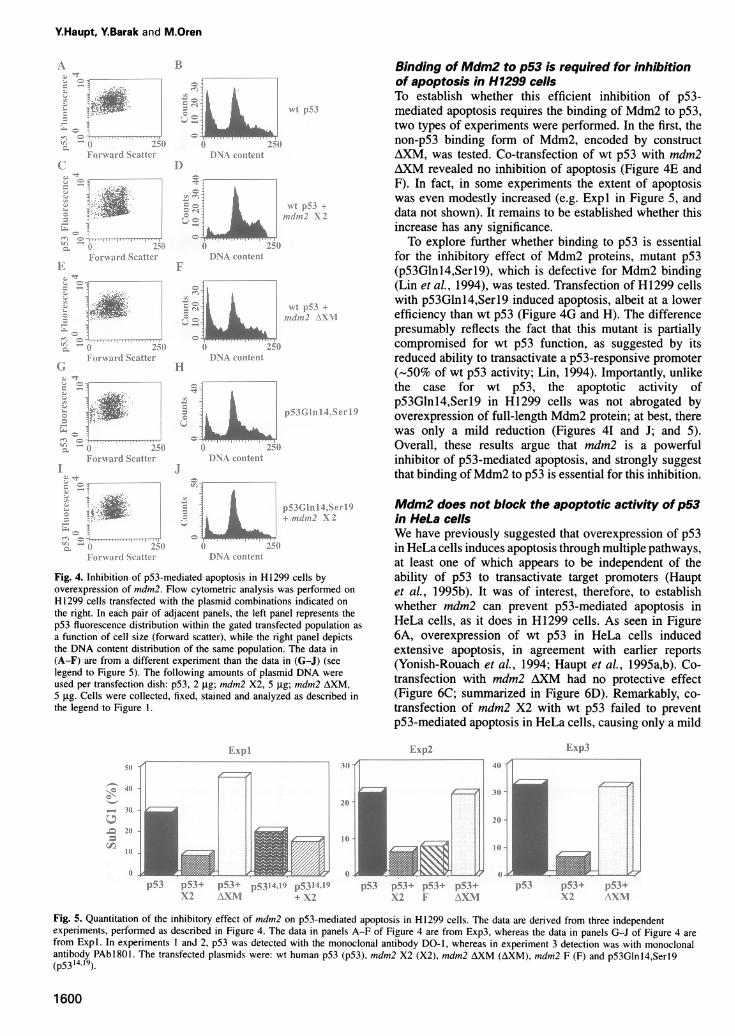
Fig. 3. Expression patterns of mouse mdm2 constructs used in this study. (A) Schematic map of the Mdm2 protein and the maximum codingpotential of the cDNA segments used in the expression plasmids. The p53 binding domain of Mdm2 is indicated by a dark box. Mdm2 X2 and Fboth have the potential to encode a full-length Mdm2 protein, while AXM can maximally encode a polypeptide initiating at the fourth in-frame AUGof mouse mdmn2, corresponding to residue 62 (Barak et al., 1994). (B) Western blot analysis of Mdm2 polypeptides in transiently transfected H1299cells. Cells were transfected with p53 alone or together with mdm2 X2 or AXM. As a control an empty CMV plasmid was employed. Twenty-fourhours post-transfection cell extracts were prepared, resolved through a 9% polyacrylamide-SDS gel, Western blotted and probed with an anti-mouseMdm2 rabbit polyclonal serum (Barak et al., 1993). Positions and sizes of molecular weight markers are shown on the right. (C) Analysis ofcomplex formation between Mdm2 and p53 in transiently transfected H1299 cells. H1299 cells were transfected with the expression plasmidcombinations indicated at the top of the figure. Twenty-four hours post-transfection, cells were labeled with [35S]methionine plus cysteine. Cellextracts, normalized for equal mounts of acid-insoluble radioactivity, were immunoprecipitated either with a mixture of the anti-p53 monoclonalantibodies PAb421 and PAb1801 (lane 1), or with anti-mouse Mdm2 rabbit polyclonal serum (lanes 2-4). Immunoprecipitated proteins were
separated on a 12.5% SDS-polyacrylamide gel. Indicated on the left are the positions of p53 and of the full-length mdm2 protein (p95) and theN-terminally truncated Mdm2 polypeptide encoded by AXM (p85). The positions and sizes (in kDa) of protein molecular weight markers are
indicated on the right.
specifically to p53 in transfected H1299 cells. The bindingof p95 to p53 was also seen when immunoprecipitationwas done with anti-p53 antibodies (lane 1). The lack ofbinding between p85 and p53 is consistent with the factthat p85 lacks a functional p53 binding domain. It is ofnote that, in H1299 cells co-transfected with X2 and p53,a substantial fraction of the total p53 protein was presentin stable complexes with p95 (compare lanes 1 and 3).The effect of mdm2 on p53-mediated apoptosis in
H1299 cells was studied by transient co-transfection ofp53 with various mdm2 constructs, followed by flowcytometric analysis. H1299 cells were transfected withp53 alone or co-transfected with the appropriate mdm2construct. Cells were collected 48 h post-transfection,fixed and stained for p53, and then analyzed for DNAcontent as in Figure 1. An important feature of theexperimental protocol employed in the present study isthat cells are gated and collected solely on the basis ofthe amount of p53 present in each cell. This allows a
comparative analysis of apoptosis in individual cells fromeach transiently transfected culture, matched directly forthe expression of equal amounts of transfected p53. Thus,even if the overall amounts of p53 vary substantiallybetween different transfected cultures (owing to promotercompetition, different transfection efficiencies or any non-
random effect of the particular plasmid combinations),
data obtained by this protocol are refractory to the effectsof such variations. Hence, any biological differencesobserved between cells transfected with p53 alone andthose transfected with p53 plus an additional plasmid, willbe due only to the specific effect of the latter plasmid.A representative set of data is shown in Figure 4, and
a summary of three independent experiments is shown inFigure 5. Overexpression of wt p53 alone (Figure 4A andB) induced significant apoptosis (33% sub-GI). Import-antly, co-transfection of wt p53 with construct X2, encod-ing full-length mouse Mdm2 which can bind p53, stronglyreduced the level of apoptosis (Figure 4D). The extent ofresidual apoptosis in such cells was 7%, as compared with3% in a control non-transfected H1299 population (datanot shown). In addition, substantial protection from p53-mediated apoptosis was provided by plasmid mdm2 F(Figure 5, Exp2), which also can direct the in vivosynthesis of full-length Mdm2 protein (Figure 3A), albeitat a lower efficiency than the X2 construct (Barak et al.,1994). As explained above, positive cells were selectedfor analysis so that the level of p53 overexpression in thedouble transfectants was essentially comparable with thatpresent in the H1299 cells transfected with wt p53 alone(compare Figure 4C and 4A). Thus, overexpression offull-length Mdm2 protein can confer resistance to theinduction of apoptotic death by wt p53.
1599
X ++ ;>
Mdm2
-- -200
Y.Haupt, Y.Barak and M.Oren
l .id 2531x
i ? ,f311U 251
:,C;l'&
_ \t j
250 11251w1 _rdScatter D\A co 1l11
253 11 (1
.1
221 1)'>sN%Z t/ 1) ^;-I l
p1 3G(1 14.St r1-19
pS.3tll 14.S r 2)-~,mnm2 X 2'r,
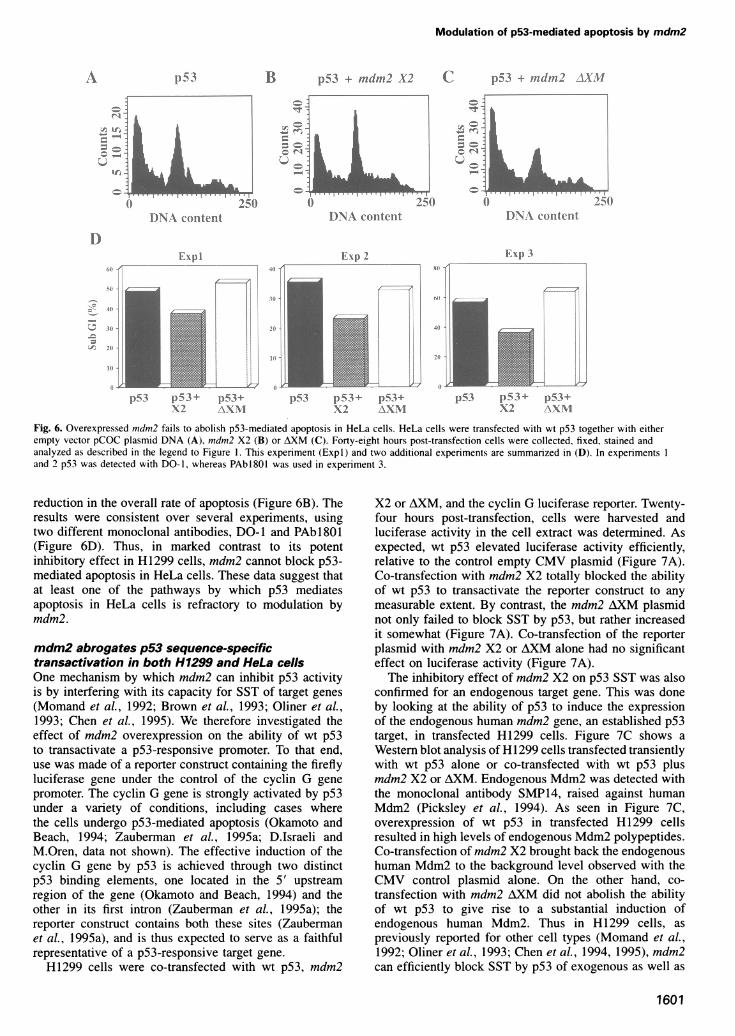
Fig. 4. Inhibition of p53-mediated apoptosis in H1299 cells byoverexpression of mdm2. Flow cytometric analysis was performed on
H1299 cells transfected with the plasmid combinations indicated on
the right. In each pair of adjacent panels, the left panel represents thep53 fluorescence distribution within the gated transfected population as
a function of cell size (forward scatter), while the right panel depictsthe DNA content distribution of the same population. The data in(A-F) are from a different experiment than the data in (G-J) (seelegend to Figure 5). The following amounts of plasmid DNA were
used per transfection dish: pS3, 2 ,ug; mdm2 X2, 5 rg; mdm2 AXM,5 gg. Cells were collected, fixed, stained and analyzed as described inthe legend to Figure 1.
F jIx I
411
21 -
11
Binding of Mdm2 to p53 is required for inhibitionof apoptosis in H1299 cellsTo establish whether this efficient inhibition of p53-mediated apoptosis requires the binding of Mdm2 to p53,two types of experiments were performed. In the first, thenon-p53 binding form of Mdm2, encoded by constructAXM, was tested. Co-transfection of wt p53 with mdm2AXM revealed no inhibition of apoptosis (Figure 4E andF). In fact, in some experiments the extent of apoptosiswas even modestly increased (e.g. Expl in Figure 5, anddata not shown). It remains to be established whether thisincrease has any significance.To explore further whether binding to p53 is essential
for the inhibitory effect of Mdm2 proteins, mutant p53(pS3Glnl4,Serl9), which is defective for Mdm2 binding(Lin et al., 1994), was tested. Transfection of H11299 cellswith p53Glnl4,Serl9 induced apoptosis, albeit at a lowerefficiency than wt p53 (Figure 4G and H). The differencepresumably reflects the fact that this mutant is partiallycompromised for wt p53 function, as suggested by itsreduced ability to transactivate a p53-responsive promoter(~50% of wt p53 activity; Lin, 1994). Importantly, unlikethe case for wt p53, the apoptotic activity ofpS3Glnl4,Serl9 in H1299 cells was not abrogated byoverexpression of full-length Mdm2 protein; at best, therewas only a mild reduction (Figures 41 and J; and 5).Overall, these results argue that mdm2 is a powerfulinhibitor of p53-mediated apoptosis, and strongly suggestthat binding ofMdm2 to p53 is essential for this inhibition.
Mdm2 does not block the apoptotic activity ofp53in HeLa cellsWe have previously suggested that overexpression of p53in HeLa cells induces apoptosis through multiple pathways,at least one of which appears to be independent of theability of p53 to transactivate target promoters (Hauptet al., 1995b). It was of interest, therefore, to establishwhether mdm2 can prevent p53-mediated apoptosis inHeLa cells, as it does in H1299 cells. As seen in Figure6A, overexpression of wt p53 in HeLa cells inducedextensive apoptosis, in agreement with earlier reports(Yonish-Rouach et al., 1994; Haupt et al., 1995a,b). Co-transfection with mdm2 AXM had no protective effect(Figure 6C; summarized in Figure 6D). Remarkably, co-transfection of mdm2 X2 with wt p53 failed to preventp53-mediated apoptosis in HeLa cells, causing only a mild
Fxl')2 F ^,p
p53 p53+ p)-534 1 5314,11) pSSl,314-19\2 \XNl + X?
p53 p53+ )53k+ Fp53+X2 F A\XI
l53 p53k+ p)3+
xi \XNl
Fig. 5. Quantitation of the inhibitory effect of mdm2 on p53-mediated apoptosis in H1299 cells. The data are derived from three independentexperiments, performed as described in Figure 4. The data in panels A-F of Figure 4 are from Exp3, whereas the data in panels G-J of Figure 4 are
from Expl. In experiments 1 and 2, p53 was detected with the monoclonal antibody DO-1, whereas in experiment 3 detection was with monoclonalantibody PAbI801. The transfected plasmids were: wt human p53 (p53), mdm2 X2 (X2), mdm2 AXM (AXM), mdm2 F (F) and p53Glnl4,Serl9(p5314'19).
1600
rd cIt..
25t i._ ..I . 11........1...
{l V 11( Scilr
1K -t; ] > 1
I
h
Modulation of p53-mediated apoptosis by mdm2
1)53
D)NA\ content254
F I) I
B p5-3 + indmn2 A'-"
) f r25()D)NA Coniteilt
EIxi) 2
C p53 + mndn2 A.X11
D)NA Colntentt
Exp 3
E ....
21i -
III
1)5 3 Z)5 3 + p53+X2 ANNI
p5-s}S3+ p5>3+\;2 VY)
Fig. 6. Overexpressed mndmn2 fails to abolish p53-mediated apoptosis in HeLa cells. HeLa cells were transfected with wt p53 together with eitherempty vector pCOC plasmid DNA (A), mndmn2 X2 (B) or AXM (C). Forty-eight hours post-transfection cells were collected, fixed, stained andanalyzed as described in the legend to Figure 1. This experiment (Expl) and two additional experiments are summarized in (D). In experiments Iand 2 p53 was detected with DO-1, whereas PAbl8O0 was used in experiment 3.
reduction in the overall rate of apoptosis (Figure 6B). Theresults were consistent over several experiments, usingtwo different monoclonal antibodies, DO-1 and PAbl801(Figure 6D). Thus, in marked contrast to its potentinhibitory effect in H1299 cells, mdm2 cannot block p53-mediated apoptosis in HeLa cells. These data suggest thatat least one of the pathways by which p53 mediatesapoptosis in HeLa cells is refractory to modulation bymdm2.
mdm2 abrogates p53 sequence-specifictransactivation in both H1299 and HeLa cellsOne mechanism by which mdm2 can inhibit p53 activityis by interfering with its capacity for SST of target genes
(Momand et al., 1992; Brown et al., 1993; Oliner et al.,1993; Chen et al., 1995). We therefore investigated theeffect of mdm2 overexpression on the ability of wt p53to transactivate a p53-responsive promoter. To that end,use was made of a reporter construct containing the fireflyluciferase gene under the control of the cyclin G gene
promoter. The cyclin G gene is strongly activated by p53under a variety of conditions, including cases wherethe cells undergo p53-mediated apoptosis (Okamoto andBeach, 1994; Zauberman et al., 1995a; D.Israeli andM.Oren, data not shown). The effective induction of thecyclin G gene by p53 is achieved through two distinctp53 binding elements, one located in the 5' upstreamregion of the gene (Okamoto and Beach, 1994) and theother in its first intron (Zauberman et al., 1995a); thereporter construct contains both these sites (Zaubermanet al., 1995a), and is thus expected to serve as a faithfulrepresentative of a p53-responsive target gene.H1299 cells were co-transfected with wt p53, mdm2
X2 or AXM, and the cyclin G luciferase reporter. Twenty-four hours post-transfection, cells were harvested andluciferase activity in the cell extract was determined. Asexpected, wt p53 elevated luciferase activity efficiently,relative to the control empty CMV plasmid (Figure 7A).Co-transfection with mdm2 X2 totally blocked the abilityof wt p53 to transactivate the reporter construct to any
measurable extent. By contrast, the mdm2 AXM plasmidnot only failed to block SST by p53, but rather increasedit somewhat (Figure 7A). Co-transfection of the reporterplasmid with mdm2 X2 or AXM alone had no significanteffect on luciferase activity (Figure 7A).The inhibitory effect of mdm2 X2 on p53 SST was also
confirmed for an endogenous target gene. This was doneby looking at the ability of p53 to induce the expressionof the endogenous human mdm2 gene, an established p53target, in transfected H1299 cells. Figure 7C shows a
Western blot analysis of HI 299 cells transfected transientlywith wt p53 alone or co-transfected with wt p53 plusmdm2 X2 or AXM. Endogenous Mdm2 was detected withthe monoclonal antibody SMP14, raised against humanMdm2 (Picksley et al., 1994). As seen in Figure 7C,overexpression of wt p53 in transfected H1299 cellsresulted in high levels of endogenous Mdm2 polypeptides.Co-transfection of mdm2 X2 brought back the endogenoushuman Mdm2 to the background level observed with theCMV control plasmid alone. On the other hand, co-
transfection with mdm2 AXM did not abolish the abilityof wt p53 to give rise to a substantial induction ofendogenous human Mdm2. Thus in H1299 cells, as
previously reported for other cell types (Momand et al.,1992; Oliner et al., 1993; Chen et al., 1994, 1995), mdm2can efficiently block SST by p53 of exogenous as well as
1601
iA
I()
D
.4
-!I
I-
iri
j- Jr, :
I
c =
if-,-ALL
= T
4._/
.2.%. I,.;.:
p-5%3 1) 5 3 + 1)53+X2 \XNI
Y.Haupt, Y.Barak and M.Oren
-A
:-
z
;-
:_dZ;
11199
|~~~~~~
[).; X1).;+xl W-, I \x\1 \1 %AN1
ful})-,
;01 -
-41
I11 -
(i1\
lII La
f1! ~:i(.' 1X' pt+xrp4 .\ 1 \n \x\
N1 r
+ +C1 I
lb
Kd
-112-84
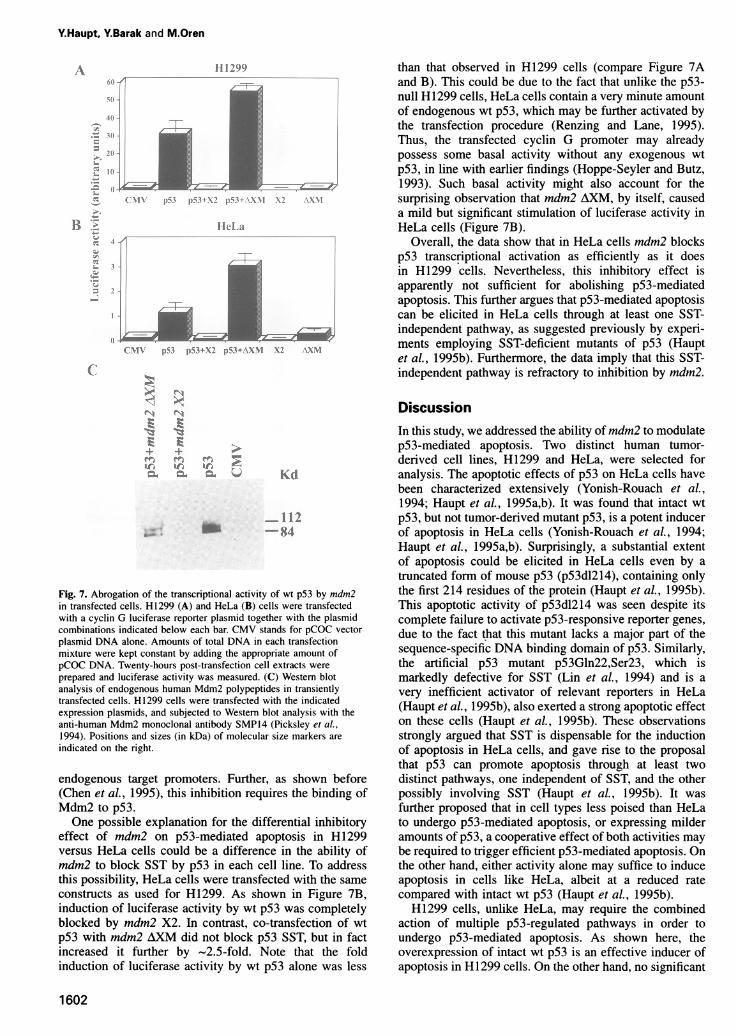
Fig. 7. Abrogation of the transcriptional activity of wt p53 by mdm2in transfected cells. H1299 (A) and HeLa (B) cells were transfectedwith a cyclin G luciferase reporter plasmid together with the plasmidcombinations indicated below each bar. CMV stands for pCOC vectorplasmid DNA alone. Amounts of total DNA in each transfectionmixture were kept constant by adding the appropriate amount ofpCOC DNA. Twenty-hours post-transfection cell extracts wereprepared and luciferase activity was measured. (C) Western blotanalysis of endogenous human Mdm2 polypeptides in transientlytransfected cells. H1299 cells were transfected with the indicatedexpression plasmids, and subjected to Western blot analysis with theanti-human Mdm2 monoclonal antibody SMP14 (Picksley et al.,1994). Positions and sizes (in kDa) of molecular size markers areindicated on the right.
endogenous target promoters. Further, as shown before(Chen et al., 1995), this inhibition requires the binding ofMdm2 to p53.One possible explanation for the differential inhibitory
effect of mdm2 on p53-mediated apoptosis in H1299versus HeLa cells could be a difference in the ability ofmdm2 to block SST by p53 in each cell line. To addressthis possibility, HeLa cells were transfected with the sameconstructs as used for H1299. As shown in Figure 7B,induction of luciferase activity by wt p53 was completelyblocked by mdm2 X2. In contrast, co-transfection of wtp53 with mdm2 AXM did not block p53 SST, but in factincreased it further by ~2.5-fold. Note that the foldinduction of luciferase activity by wt p53 alone was less
than that observed in HI 299 cells (compare Figure 7Aand B). This could be due to the fact that unlike the p53-null H1299 cells, HeLa cells contain a very minute amountof endogenous wt p53, which may be further activated bythe transfection procedure (Renzing and Lane, 1995).Thus, the transfected cyclin G promoter may alreadypossess some basal activity without any exogenous wtp53, in line with earlier findings (Hoppe-Seyler and Butz,1993). Such basal activity might also account for thesurprising observation that mdm2 AXM, by itself, causeda mild but significant stimulation of luciferase activity inHeLa cells (Figure 7B).
Overall, the data show that in HeLa cells mdm2 blocksp53 transcriptional activation as efficiently as it doesin H1299 cells. Nevertheless, this inhibitory effect isapparently not sufficient for abolishing p53-mediatedapoptosis. This further argues that p53-mediated apoptosiscan be elicited in HeLa cells through at least one SST-independent pathway, as suggested previously by experi-ments employing SST-deficient mutants of p53 (Hauptet al., 1995b). Furthermore, the data imply that this SST-independent pathway is refractory to inhibition by mdm2.
DiscussionIn this study, we addressed the ability of mdm2 to modulatep53-mediated apoptosis. Two distinct human tumor-derived cell lines, H1299 and HeLa, were selected foranalysis. The apoptotic effects of p53 on HeLa cells havebeen characterized extensively (Yonish-Rouach et al.,1994; Haupt et al., 1995a,b). It was found that intact wtp53, but not tumor-derived mutant p53, is a potent inducerof apoptosis in HeLa cells (Yonish-Rouach et al., 1994;Haupt et al., 1995a,b). Surprisingly, a substantial extentof apoptosis could be elicited in HeLa cells even by atruncated form of mouse p53 (p53dl214), containing onlythe first 214 residues of the protein (Haupt et al., 1995b).This apoptotic activity of p53dl214 was seen despite itscomplete failure to activate p53-responsive reporter genes,due to the fact that this mutant lacks a major part of thesequence-specific DNA binding domain of p53. Similarly,the artificial p53 mutant p53Gln22,Ser23, which ismarkedly defective for SST (Lin et al., 1994) and is avery inefficient activator of relevant reporters in HeLa(Haupt et al., 1995b), also exerted a strong apoptotic effecton these cells (Haupt et al., 1995b). These observationsstrongly argued that SST is dispensable for the inductionof apoptosis in HeLa cells, and gave rise to the proposalthat p53 can promote apoptosis through at least twodistinct pathways, one independent of SST, and the otherpossibly involving SST (Haupt et al., 1995b). It wasfurther proposed that in cell types less poised than HeLato undergo p53-mediated apoptosis, or expressing milderamounts of p53, a cooperative effect of both activities maybe required to trigger efficient p53-mediated apoptosis. Onthe other hand, either activity alone may suffice to induceapoptosis in cells like HeLa, albeit at a reduced ratecompared with intact wt p53 (Haupt et al., 1995b).H1299 cells, unlike HeLa, may require the combined
action of multiple p53-regulated pathways in order toundergo p53-mediated apoptosis. As shown here, theoverexpression of intact wt p53 is an effective inducer ofapoptosis in H1299 cells. On the other hand, no significant
1602

Modulation of p53-mediated apoptosis by mdm2
extent of apoptotic death could be elicited in H 1299 cellsby p53dl214 (Y.Haupt and M.Oren, data not shown).Furthermore, a tumor-derived mutant of human p53,p53Prol75, also failed to trigger apoptosis in H1299 cells,despite its ability to exhibit a pronounced degree of SST(Rowan et al., 1996). Taken together, these data suggestthat induction of apoptosis in H 1299 cells can only beachieved by a p53 protein which retains the ability toturn on simultaneously both SST-dependent and SST-independent downstream pathways. This situation mayresemble that observed in baby rat kidney cells, wherethe SST-deficient p53Gln22,Ser23 mutant is unable totrigger cell death (Sabbatini et al., 1995b).
In the present study, it was found that co-expressionof mdm2 with wt p53 efficiently inhibits p53-mediatedapoptosis in H1299 cells. This inhibition persisted for atleast 72 h (data not shown), indicating that mdm2 blocksrather than delays the apoptotic activity of wt p53. Underphysiological conditions, the activation of endogenousmdm2 may serve to limit p53-mediated apoptosis underconditions where the cell can cope adequately with thedamage which triggered the p53 response. A lag in theactual accumulation of Mdm2 protein (Perry et al., 1993)might allow some of the cells, presumably those leastcapable of dealing with the damage, to become irreversiblycommitted to the apoptotic pathway before this process isblocked by build-up of sufficient amounts of Mdm2. Onthe other hand the simultaneous co-expression of mdm2with p53, as accomplished in this study, will not allowsuch a lag. Consequently, the inhibitory effect of mdm2on p53-mediated apoptosis will be far more complete.The inhibitory activity of mdm2 in H1299 cells appears
to require a physical interaction between the Mdm2 proteinand p53. Thus, the mutant p53Glnl4,Serl9 which cannotbind Mdm2 but can induce apoptosis in H1299 cells, wasnot blocked by overexpression of mdm2. Furthermore, theMdm2 protein encoded by AXM, which is incapable ofassociating stably with p53, also failed to block p53-dependent apoptosis in H1299 cells.The inhibitory effect of mdm2 on p53-mediated
apoptosis in H1299 cells correlates very well with itsability to abrogate p53 SST. This strict correlation isconsistent with the notion that SST is essential for p53-mediated apoptosis in H1299 cells. To date, the mostlikely target genes of p53 in the apoptotic pathway arebax and Fas/Apo- 1 (Miyashita et al., 1994; Selvakumaranet al., 1994; Miyashita and Reed, 1995; Owen-Schaubet al., 1995). In line with this suggestion, wt p53 stronglyactivates a reporter gene driven by the human bax promoterin H1299 cells, and this activation is efficiently inhibitedby mdm2 (Y.Haupt, data not shown). Other target genes,yet unidentified, may also contribute to the induction ofapoptosis by p53. The inhibitory effect of mdm2 on p53SST may involve competition between mdm2 and certaintranscription factors on binding to p53. This notion isbased primarily on the recent findings that TAFs andMdm2 bind the same site in p53 (Lin et al., 1994; Lu andLevine, 1995; Thut et al., 1995).
In addition to SST, transcriptional repression by p53has also been proposed to contribute to apoptosis (Shenand Shenk, 1994; Sabbatini et al., 1995a). Recently, it hasbeen demonstrated that Mdm2 can alleviate the ability ofp53 to repress various promoters lacking p53 binding
sites (Chen et al., 1995). Thus, it remains possible thattransrepression may represent a crucial mdm2 target inthe inhibition of apoptosis.The effect of mdm2 overexpression on apoptosis is
strikingly different in HeLa cells. Unlike the completeblock seen in H1299, HeLa cells transfected with full-length mdm2 continue to exhibit a substantial extent ofp53-mediated cell death. Most remarkably, this apoptosistakes place under conditions where all detectable SST isabrogated by mdm2. These findings confirm and reinforceour earlier suggestion that SST is dispensable for p53-mediated apoptosis in HeLa cells (Haupt et al., 1995b).Moreover, the new data allow us to rule out the concern thatthe overexpressed SST-deficient p53 proteins, employed inthe previous study (Haupt et al., 1995b), promotedapoptosis by somehow inducing the SST function of thelow amount of endogenous HeLa wt p53. Since apoptosiswas also maintained under conditions where there wasenough Mdm2 to silence completely the SST activity ofa much larger amount of transfected wt p53, we canformally conclude that p53 can induce apoptosis in HeLain an SST-independent manner.The nature of the SST-independent activity responsible
for apoptosis in HeLa remains unclear. The fact thatexcess mdm2 blocks not only SST but also transcriptionalrepression by p53 (Chen et al., 1995), may argue that thelatter activity of wt p53 is equally unlikely to account forapoptosis in HeLa cells. However, it should be kept inmind that there may be multiple ways through whichexcess wt p53 could repress transcription, and only partof those may be inhibited by mdm2. For instance, differentgenes may be repressed through interaction of p53 withdifferent components of the cellular transcriptionmachinery. Some critical transcription factors may bebound by p53 through sites which are not blocked byMdm2. Hence, if certain genes are repressed through thelatter type of interaction, it still remains possible thatsuch repression could explain the mdm2-insensitive, SST-independent apoptosis seen in transfected HeLa cells.Other, yet undefined mechanisms are equally likely.
In conclusion, the data presented here suggest thatmdm2 may act as a physiological modulator of p53-mediated apoptosis. This capacity of mdm2, along with itsability to relieve p53-mediated growth inhibition (Finlay,1993; Otto and Deppert, 1993; Chen et al., 1994), mayconstitute the basis for its oncogenic effects, in particularsince the apoptotic activity of p53 has been suggested tomediate its ability to suppress tumorigenesis (Howes et al.,1994; Morgenbesser et al., 1994; Pan and Griep, 1994;Symonds et al., 1994). Inhibition of p53-mediatedapoptosis by mdm2 is consistent with the clinical observa-tion that, in many of the tumors containing amplifiedmdm2, p53 is not mutated (Oliner et al., 1992; Leachet al., 1993; Reifenberger et al., 1993; Cordon-Cardoet al., 1994). In line with this suggested anti-apoptoticrole of mdm2, Kondo et al. (1995) have reported thatoverexpression of mdm2 protects U87-MG glioblastomacells from cisplatin-induced apoptosis; the involvement ofp53 in this response still needs to be determined. Moreover,in view of the more aggressive phenotype of tumorscontaining mutant p53 plus amplified mdm2, relative tothose containing mutant p53 only (Cordon-Cardo et al.,
1603

Y.Haupt, Y.Barak and M.Oren
1994), one may wish to consider the possibility that excessmdm2 may inhibit apoptosis even in cells lacking wt p53.
The recent description of mdm2-null ('knock-out') miceargues strongly in favor of a tight physiological relation-ship between p53 and mdm2. The complete abrogation ofmdm2 expression results in early embryonic lethality(Jones et al., 1995; Montes de Oca Luna et al., 1995).However, this lethality is efficiently rescued when suchmice are crossed with p53-null mice (Jones et al., 1995;Montes de Oca Luna et al., 1995). These findings indicatethat mdm2 plays a crucial role in restraining a physiologicalactivity of p53 during normal development, and that thisactivity is extremely deleterious to animal survival in theabsence of mdm2 function. Our data, which demonstratethe ability of Mdm2 protein to act as an effective inhibitorof p53-mediated apoptosis, thus provide a possiblemechanistic explanation for these in vivo observations.Our findings imply that the anti-apoptotic effect of
mdm2 will depend on the cellular context. Thus, amplifiedmdm2 may block apoptosis in some cells, but not in others.In particular, cells undergoing p53-mediated apoptosisthrough an SST-independent pathway (Caelles et al., 1994;Wagner et al., 1994; Haupt et al., 1995b) may be lesssusceptible to protection by excess mdm2. Further workshould reveal whether these predictions are indeed borneout in actual tumors.
Materials and methodsCells and transfectionsHeLa cells and H1299 cells were maintained in DMEM and RPMI,respectively, supplemented with 10% fetal calf serum (FCS), at 37.50C.Cells were seeded 24 h before transfection at 1.2X 106 cells per 10 cm dish(for flow cytometric analysis and immunostaining) or at 6x 105 cells per6 cm dish (for luciferase assays). Cells of both lines were transfectedby the calcium phosphate method in DMEM. The DNA precipitate wasleft on the cells for 6 h (luciferase assays) or overnight (flow cytometricand immunostaining analysis). Cells were subsequently shocked for1 min with medium containing 10% glycerol. Wherever appropriate, thepCOC vector was used in order to maintain a constant amount of DNAin each transfection mixture.
Luciferase assays and Western blot analysis were carried out essentiallyas previously described (Haupt et al., 1995a). The following antibodieswere used: anti-human p53 monoclonal antibodies (mAb) PAbI 801(Banks et al., 1986), PAb42l (Harlow et al., 1981) and DO-1 (Vojteseket al., 1992), anti-human Mdm2 mAb SMP14 (Picksley et al., 1994)and an anti-mouse Mdm2 rabbit polyclonal serum (Barak et al., 1993).
ImmunoprecipitationH1299 cells were transfected as described above. Twenty hours post-transfection cells were washed in PBS and starved for 30 min inmethionine and cysteine-free DMEM, supplemented with 2% FCS. Cellswere then labeled for 2 h with [35S]methionine plus cysteine (NewEngland Nuclear) in starvation medium. Cell extracts were prepared inextraction buffer (50 mM Tris-HCI, pH 8.0, 5 mm EDTA, 150 mMNaCl and 0.5% NP-40, 1% aprotinin and 30 gg/ml PMSF). Extractscontaining equal amounts of trichloroacetic acid (TCA)-insoluble radio-activity were precleared with preimmune rabbit serum or with controlhybridoma culture medium for precipitation with anti-Mdm2 polyclonalserum and anti-p53 monoclonal antibodies respectively. Followingincubation for 30 min at 4°C, 35 g1 of formalin-fixed Staphylococcusaureus (Staph A) bacteria [prewashed three times in buffer A (50 mMTris-HCl pH 7.4, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40 and30 ,ug/ml PMSF)] were added for another 10 min. Bacteria were pelletedout, and the supernatant was taken for immunoprecipitation with either2 g1 of anti-Mdm2 serum or with a combination of anti-p53 monoclonalantibodies PAb42l plus PAbl8O1 (25 g1 of each culture supernatant) for2 h at 4°C. Immune complexes were pelleted with bacteria as before,and the pellets were washed three times in buffer B (5% sucrose, 50 mMTris-HCI pH 7.4, 500 mM NaCI, 5 mM EDTA and 0.5% NP-40),
followed by one wash with buffer C (50 mM Tris-HCI pH 7.4, 150 mMNaCl and 5 mM EDTA). The precipitated proteins were resuspended inprotein sample buffer and separated on a 12.5% SDS-polyacrylamidegel. The gel was then fixed in 10% acetic acid for 1 h, fluorographed in1 M sodium salicylate for 30 min, dried and exposed to X-ray film.
PlasmidsExpression plasmids for human wt p53 (pCMV-Neo-Bam-p53; Bakeret al., 1990) and mutant human p53 (pRCp53Glnl4Serl9 and pCMV-Neo-Bam-p53Gln 179; Lin et al., 1994; Unger et al., 1992, respectively)were all under the CMV immediate early enhancer/promoter. Murinemdm2 cDNA was inserted into the pCOC vector, which is a pBluescript-based plasmid containing (in that order) the CMV enhancer/promoter, amultiple cloning site, the SV40 small t splice and the SV40 earlypolyadenylation site (Y.Barak, unpublished). The mdm2 constructs were:pCOC-mdm2 F, pCOC-mdm2 X2 and pCOC-mdm2 AXM. The formertwo plasmids encode full-length Mdm2 protein plus varying amounts ofN-terminally truncated polypeptides (Barak et al., 1994), whereas thelatter contains an internal deletion and consequently encodes a polypep-tide initiating at the AUG at amino acid position 62 (Barak et al., 1994).The cyclin G-luc reporter plasmid contains the promoter, exon I and
part of intron I of the rat cyclin G gene, including two p53 binding sites(Zauberman et al., 1995a), linked to the firefly luciferase gene (kindlyprovided by A.Zauberman).
Indirect immunofluorescenceH 1299 cells were seeded and transfected as described above. Forty-eighthours post-transfection cells were collected, fixed and stained essentiallyas previously described (Haupt et al., 1995b). Human p53 protein wasdetected with monoclonal antibody PAbl 801. Fluorescently labeled cellswere examined under the fluorescent microscope (Zeiss Axioskop) forred fluorescence (rhodamine) at 570 nm and for DAPI at 420 nm. TheTUNEL assay was carried out as described (Haupt et al., 1995b).
Flow cytometryAt the indicated times post-transfection, adherent and floating cells werecombined and washed in cold PBS. Cells were fixed in methanol for atleast 20 min at -20°C, rehydrated in PBS for a minimum of 30 min at4°C, and then reacted with the primary antibody (DO-1 or PAbl801 forp53) for 30 min at room temperature. Cells were washed twice in PBSand incubated with a goat anti-mouse FITC-conjugated secondaryantibody (Jackson Labs) for 30 min at room temperature. Followingincubation, cells were washed in PBS and treated with RNase A(50 ,ug/ml) for 20 min. To stain DNA, propidium iodide (PI; 25 gg/mI,Sigma) was added to the cells. Samples were then analyzed in a cellsorter (FACSORT, Becton Dickinson). Relative levels of p53 per cellwere measured by FITC fluorescence intensity. A region defining highFITC fluorescence was determined, and cells falling into this region('gate') were collected separately. Equal numbers of events from thetotal population and from the gated sub-population were recordedseparately. The PI staining was recorded simultaneously in the redchannel.
AcknowledgementsWe thank A.Kazaz for outstanding technical assistance, J.Lin, J.Chenand A.J.Levine for the generous gift of the pS3Glnl4,Serl9 mutant,A.Zauberman and A.Lupo for the cyclin-G luciferase plasmid, M.Harigaiand J.Reed for bax promoter DNA, D.Lane and S.Picksley for the giftof DO- 1, PAb 1801 and SMP 14, T.Unger for pCMV-Neo-Bam-p53Gln 179and B.Vogelstein for pCMV-Neo-Bam-p53. This work was supported inpart by The Minerva Foundation (Munich), by PHS grant ROI CA40099 from the National Cancer Institute, and by grants from the RevsonScience Foundation administered by the Israel Academy of Sciencesand Humanities, and the Leo and Julia Forchheimer Center for Molecu-lar Genetics.
ReferencesBaker,S.J., Markowitz,S., Fearson,E.R., Willson,J.K.V. and Vogelstein,
B. (1990) Suppression of human colorectal carcinoma cell growth bywild-type p53. Science, 249, 912-915.
Banks,L., Matlashewski,G. and Crawford,L. (1986) Isolation of human-p53-specific monoclonal antibodies and their use in the studies ofhuman p53 expression. Eur. J. Biochem., 159, 529-534.
1604

Modulation of p53-mediated apoptosis by mdm2
Barak.Y. and Oren.M. (1992) Enhanced binding of a 95-kDa protein top53 in cells undergoing, p53-mediated growth arrest. EMBO J., 11,2115-2121.
Barak.Y.. Juven.T.. Haffner,R. and Oren,M. (1993) indm2 expression isinduced by wild-type p53 activity. EMBO J.. 12. 461-468.
Barak.Y.. Gottlieb.E.. Juven.G.T. and Oren.M. (1994) Regulation ofdmnl2 expression by p53: alternative promoters produce transcriptswith nonidentical translation potential. Genes Dei:. 8, 1739-1749.
Browvn.D.R.. Deb.S., Munoz,R.M., Subler,M.A. and Deb.S.P. (1993) Thetumor suppressor p53 and the oncoprotein simian virus-40 T-Antigenbind to overlapping domains on the Mdm2 protein. Mol. Cell. Biol..13, 6849-6857.
Caelles,C.. Helmberg.A. and Karin.M. (1994) p53-dependent apoptosisin the absence of transcriptional activation of p53-target genes. Natuire.370. 220-223.
Cahilly-Snyder,L., Yang-Feng.T.. Francke.U. and George,D.L. (1987)Molecular analysis and chromosomal mapping of amplified genesisolated from a transformed mouse 3T3 cell line. Soami. Cell Mol.Geniet.. 13, 235-244.
Canman.C.E., Gilmer,T.M., Coutts.S.B. and Kastan,M.B. (1995) Growthfactor modulation of p53-mediated growth arrest versus apoptosis.Genies Dei:. 9. 600-611.
Chen.C.Y.. Oliner,J.D., Zhan.Q.. Fornace.A.J., Vogelstein,B. andKastan,M.B. (1994) Interactions between p53 and Mdm2 in amammalian cell cycle checkpoint pathway. Proc. Natl Acad. Sci. USA,91, 2684-2688.
Chen.J.D., Marechal.V. and Levine,A.J. (1993) Mapping of the p53 and,nldm-2 interaction domains. Mol. Cell. Biol.. 13. 4107-4114.
Chen,J.D., Lin,J.Y. and Levine.A.J. (1995) Regulation of transcriptionfunctions of the p53 tumour suppressor by the ,nd,n-2 oncogene. Mol.Med.. 1. 141-152.
Cordon-Cardo,C. et al. (1994) Molecular abnormalities of indm2 andp53 genes in adult soft tissue sarcomas. Cancer Res.. 54. 794-799.
Corvi.R. et al. (1995) Non-syntenic amplification of MDM2 and MYCNin human neuroblastoma. Oncogene. 10, 1081-1086.
Crook,T., Marston,N.J., Sara,E.A. and Vousden,K.H. (1994)Transcriptional activation by p53 correlates with suppression of growthbut not transformation. Cell. 79. 817-827.
Dulic,V., Kaufmann,W.K.. Wilson,S.J., Tlsty,T.D., Lees,E., Harper,J.W.,Elledge,S.J. and Reed.S.I. (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced GI arrest. Cell. 76. 1013-1023.
El-Deiry,W.S. et al. (1993) WAF1, a potential mediator of p53 tumorsuppression. Cell. 75. 817-825.
El-Deiry,W.S. et al. (1994) WAFl/CIPl is induced in p53-mediated GIarrest and apoptosis. Cancer Res.. 54, 1169-1174.
Fakharzadeh.S.. Trusko.R.S. and George,D.L. (1991) Tumorigenicpotential associated with enhanced expression of a gene that isamplified in a mouse tumour line. EMBO J., 10, 1565-1569.
Fanning.E. and Knippers.R. (1992) Structure and function of simianvirus-40 large tumorantigen. Annuii. Rei: Biochein.. 61, 55-85.
Farmer,G.. Bargonetti.J.. Zhu.H., Friedman,P., Prywes.R. and Prives,C.( 1992) Wild-type p53 activates transcription in vitro. Natuire. 358.83-86.
Finlay.C.A. (1993) The mtidmtz-2 oncogene can overcome wild-type p53suppression of transformed cell growth. Mol. Cell. Biol.. 13. 301-306.
Gavrieli,Y., Sherman.Y. and Ben-Sasson,S.A. (1992) Identification ofprogrammed cell death in situi via specific labeling of nuclear DNAfragmentation. J. cell Biol.. 119, 493-501.
Gottlieb,E., Haffner,R., von Ruden,T., Wagner.E.F. and Oren,M. (1994)Down-regulation of wild-type p53 activity interferes with apoptosisof IL-3-dependent hematopoietic cells following IL-3 withdrawal.EMBO J. 13. 1368-1374.
Haffner,R. and Oren,M. (1995) p53: biochemical properties andbiological effects. Clurr: Oi)n. Geniet. Dei:. 5. 84-90.
Haines,D.S., Landers,J.E., Engle,L.J. and George,D.L. (1994) Physicaland functional interaction between wild-type p53 and Mdm2 proteins.Mol. Cell. Biol.. 14. 1171-1178.
Harlow.E., Crawford,L.V.. Pim,D.C. and Williamson.N.M. (1981)Monoclonal antibodies specific for simian virus 40 tumor antigens.J. Virol.. 34. 752-763.
Harper,J.W.. Adami.G.R.. Wei.N., Keyomarsi.K. and Elledge,S.J. (1993)The p2l CDK-interacting protein cipl is a potent inhibitor of Glcycvlin-dependent kinases. Cell. 75. 805-816.
Haupt.Y.. Rowan.S. and Oren.M. (1995a) p53-mediated apoptosis in
HeLa cells can be overcome by excess pRB. Oncogene. 10. 1563-1571.
Haupt,Y., Rowan,S., Shaulian.E., Vousden,H.K. and Oren,M. (1995b)Induction of apoptosis in HeLa cells by trans-activation-deficient p53.Genes Del., 9, 2170-2183.
Hinds,P.W., Finlay,C.A., Quartin,S.J., Baker,E.R., Fearon.B.,Vogelstein,B. and Levine,A.J. (1990) Mutant p53 DNA clones fromhuman colon carcinomas cooperate with ras in transforming primaryrat cells - a comparison of the hot spot mutant phenotype. Cell Growi thDiffer., 1, 571-580.
Hoppe-Seyler.F. and Butz,K. (1993) Repression of endogenous p53transactivation function in HeLa cervical carcinoma cells by humancolon papillomavirus type-16 E6. human Mdm-2 and mutant p53.J. Virol., 67, 3111-3117.
Howes.K.A.. Ransom,N., Papermaster,D.S.. Lasudry.J.G.H.. Albert,D.M.and Windle,J.J. (1994) Apoptosis or retinoblastoma: alternative fatesof photoreceptors expressing the HPV-16 E7 gene in the presence orabsence or p53. Genes Dei:,. 8. 1300-1310.
Ishioka,C., Englert,C., WingeP., Yan,Y.X.. Engelstein,M. and Friend,S.H.(1995) Mutational analysis of the carboxy-terminal portion of p53using both yeast and mammalian cell assays in vivo. Oncogene. 10.1485-1492.
Johnson,P., Chung,S. and BenchimolS. (1993) Growth suppression ofFriend virus-transformed erythroleukemia cells by p53 protein isaccompanied by hemoglobin production and is sensitive toerythropoietin. Mol. Cell. Biol., 13, 1456-1463.
Jones,S.N.. Roe,A.E., Donehower,L.A. and Bradly,A. (1995) Rescue ofembryonic lethality in Mdm2-deficient mice by absence of p53.Nature, 378, 206-208.
Juven,T., Barak.Y., Zauberman.A.. George.D.L. and Oren,M. (1993)Wild type p53 can mediate sequence-specific transactivation of aninternal promoter within the tndtn2 gene. Oncogene. 8, 3411-3416.
Kondo,S.. Barnett.G.H.. Hara,H., Morimura,T. and Takeuchi,J. (1995)Mdm2 protein confers the resistance of a human glioblastoma cellline to cisplatin-induced apoptosis. Oncogene, 10, 2001-2006.
Ladanyi.M., Cha,C.. Lewis,R., Jhanwar.S.C., Huvos.A.G. andHealey,J.H. (1993) indtn2 gene amplification in metastaticosteosarcoma. Cancer Res.. 53, 16-18.
Lane,D.P. (1992) p53, guardian of the genome. Natuire, 358. 15-16.Leach.F.S. et al. (1993) p53 mutation and mndm2 amplification in human
soft tissue sarcomas. Cancer Res., 53, 2231-2234.Levine.A.J. (1993) The tumor suppressor genes. Annu. Rei: Bioclein..
62, 623-651.Li.R., Waga,S._ Hannon,G.J., Beach,D. and Stillman,B. (1994)
Differential effects by the p21 CDK inhibitor on PCNA-dependentDNA replication and repair. Natutre, 371. 534-537.
Lin,J.Y., Chen,J.D., Elenbaas,B. and Levine,A.J. (1994) Severalhydrophobic amino acids in the p53 amino-terminal domain arerequired for transcriptional activation, binding to Mdm-2 and theadenovirus 5 EIB 55-kD protein. Genes Deil, 8, 1235-1246.
Lotem,J. and Sachs,L. (1993) Hematopoietic cells from mice deficientin wild-type p53 are more resistant to induction of apoptosis by someagents. Blood. 82, 1092-1096.
Lu,H. and Levine.A.J. (1995) Human TAF(II)31 protein is atranscriptional coactivator of the p53 protein. Proc. Natl Acad. Sci.USA, 92, 5154-5158.
Ludlow,J.W. (1993) Interactions between SV40 large-tumor antigen andthe growth suppressor proteins pRB and p53. FASEB J., 7. 866-871.
McCann,A.H., Kirley,A.. Caney,D.N., Corbally.N., Magee,H.M.,Keating,G. and Dervan,P.A. (1995) Amplification of the tndtn2 genein human breast cancer and its association with Mdm2 and p53 proteinstatus. Br. J. Canicer. 71, 981-985.
Mietz,J.A.. Unger,T., Huibregtse.J.M. and Howley,P.M. (1992) Thetranscriptional transactivation function of wild-type p53 is inhibitedby SV40 large T-antigen and by HPV-16 E6-oncoprotein. EMBO J..11. 5013-5020.
Mitsudomi.T. et al. (1992) p53 gene mutations in non-small-cell lungcancer cell lines and their correlation with the presence of rasmutations and clinical features. Oncogenie, 7, 171-180.
Miyashita,T. and Reed,J.C. (1995) Tumor suppressor p53 is a directtranscriptional activator of the human ba.x gene. Cell, 80. 293-299.
Miyashita,T., Krajewski,S., Krajewska,M., Wang,H.W., Lin,H.K..Liebermann,D.A.. Hoffman.B. and Reed.J.C. (1994) Tumor suppressorp53 is a regulator of bcl-2 and bax gene expression in vitro anditn vli. Onicogene. 9. 1799-1805.
Momand,J.. Zambetti.G.P.. Olson.D.C.. George.D. and Levine.A.J.(1992) The mndmti-2 oncogene product forms a complex with the p53protein and inhibits p53-mediated transactivation. Cell, 69, 1237-1245.
Montes de Oca Luna.R.. Wagner.D.S. and Lozano.G. (1995) Rescue of
1605

Y.Haupt, Y.Barak and M.Oren
early embryonic lethality in mdmn2-deficient mice by deletion of p53.Nature, 378, 203-206.
Moran,E. (1993) Interaction of adenoviral proteins with pRB and p53.FASEB J., 7, 880-885.
Morgenbesser,S.D., Williams,B.O., Jacks,T. and DePinho,R.A. (1994)p53-dependent apoptosis produced by Rb-deficiency in the developingmouse lens. Nature, 371, 72-74.
Okamoto,K. and Beach,D. (1994) Cyclin G is a transcriptional target ofthe p53 tumor suppressor protein. EMBO J., 13, 4816-4822.
Oliner,J.D., Kinzler,K.W., Meltzer,P.S., George,D.L. and Vogelstein,B.(1992) Amplification of a gene encoding a p53-associated protein inhuman sarcomas. Nature, 358, 80-83.
Oliner,J.D., Pietenpol,J.A., Thiagalingam,S., Gvuris,J., Kinzler,K.W. andVogelstein,B. (1993) Oncoprotein Mdm2 conceals the activationdomain of tumour suppressor-p53. Nature, 362, 857-860.
Oren,M. (1994) Relationship of p53 to the control of apoptotic celldeath. Semin. Cancer Biol., 5, 305.1-305.7.
Ory,K., Legros,Y., Auguin,C. and Soussi,T. (1994) Analysis of the mostrepresentative tumor-derived p53 mutants reveals that changes inprotein conformation are not correlated with loss of transactivation orinhibition of cell proliferation. EMBO J., 13, 3496-3504.
Otto,A. and Deppert,W. (1993) Upregulation of mdtn-2 expression inMeth A tumor cells tolerating wild-type p53. Oncogene, 8, 2591-2603.
Owen-Schaub,L.B. et al. (1995) Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO- I expression. Mol. Cell. Biol., 15,3032-3040.
Pan,H. and Griep,A.E. (1994) Altered cell cycle regulation in the lensof HPV-16 E6 or E7 transgeneic mice: implications for tumorsuppressor gene function in development. Genes Dei., 8, 1285-1299.
Perry,M.E., Piette,J., Zawadzki,J.A., Harvey,D. and Levine,A.J. (1993)The mdm-2 gene is induced in response to UV light in a p53-dependentmanner. Proc. Natl Acad. Sci. USA, 90, 11623-11627.
Picksley,S.M. and Lane,D.P. (1993) The p53-mndm2 autoregulatoryfeedback loop: a paradigm for the regulation of growth control byp53? Bioessavs, 15, 689-690.
Picksley,S.M., Vojtesek,B., Sparks,A. and Lane,D.P. (1994)Immunochemical analysis of the interaction of p53 with Mdm2; finemapping of the Mdm2 binding site on p53 using synthetic peptides.Oncogene, 9, 2523-2529.
Pietenpol,J.A., Tokino,T., Thiagalingam,S., Eldeiry,W.S., Kinzler,K.W.and Vogelstein,B. (1994) Sequence-specific transcriptional activationis essential for growth suppression by p53. Proc. Natl Acad. Sci. USA,91, 1998-2002.
Price,B.D. and Park,S.J. (1994) DNA damage increases the levels ofindm2 messenger RNA in wt p53 human cells. Cancer Res., 54,896-899.
Prives,C. and Manfredi,J.J. (1993) The p53 tumor suppressor protein:meeting review. Genes Des., 7, 529-534.
Reed,M., Wang,Y., Mayr,G., Enderson,M.E., Schwedes,J.F. andTegtmeyer,P. (1993) p53 domains: suppression, transformation, andtransactivation. Gene Exp., 3, 95-106.
Reifenberger,G., Liu,L., Ichimura,K., Schmidt,E.E. and Collins,V.P.(1993) Amplification and overexpression of the indin2 gene in a subsetof human malignant gliomas without p53 mutations. Cancer Res., 53,2736-2739.
Renzing,J. and Lane,D.P. (1995) p53-dependent growth arrest followingcalcium phosphate-mediated transfection of murine fibroblasts.Oncogene, 10, 1865-1868.
Rowan,S., Ludwig,R.L., Haupt,Y., Bates,S., Lu,X., Oren,M. andVousden,K.H. (1996) Specific loss of apoptotic but not cell-cyclearrest function in a human tumor derived p53 mutant. EMBO J., 15,827-838.
Sabbatini,P., Chiou,S.K., Rao,L. and White,E. (1995a) Modulation ofp53-mediated transcriptional repression and apoptosis by theadenovirus EIB 19K protein. Mol. Cell. Biol., 15, 1060-1070.
Sabbatini,P., Lin,J., LevineJ.A. and White,E. (1995b) Essential role forp53-mediated transcription in apoptosis but not growth suppression.Genes Dev., 9, 2184-2192.
Scheffner,M., Werness,B.A., Huibregtse,J.M., Levine,A.J. andHowley,P.M. (1990) The E6 oncoprotein encoded by humanpapillomavirus Type- 16 and Type- 18 promotes the degradation ofp53. Cell, 63, 1129-1136.
Selvakumaran,M., Lin,H.K., Miyashita,T., Wang,H.G., Krajewski,S.,Reed,J.C., Hoffman,B. and Liebermann,D. (1994) Immediate earlyup-regulation of bax expression by p53 but not TGF beta 1: a paradigmfor distinct apoptotic pathways. Oncogene, 9, 1791-1798.
Shaulian,E., Zauberman,A., Milner,J., Davies,E.A. and Oren,M. (1993)
Tight DNA binding and oligomerization are dispensable for the abilityof p53 to transactivate target genes and suppress transformation.EMBO J., 12, 2789-2797.
Shen,Y.Q. and Shenk,T. (1994) Relief of p53-mediated transcriptionalrepression by the adenovirus E1B 19-kDa protein or the cellular Bcl-2protein. Proc. Natl Acad. Sci. USA, 91, 8940-8944.
Symonds,H., Krall,L., Remington,L., Saenz-Robles,M., Lowe,S., Jacks,T.and Van Dyke,T. (1994) p53-dependent apoptosis suppresses tumorgrowth and progression in iiWo. Cell, 78, 703-711.
Thut,C.J., Chen,J.L., Klemm,R. and Tjian,R. (1995) p53 transcriptionalactivation mediated by coactivators TAFII40 and TAFII60. Science.267, 100-104.
Unger,T., Nau,M.M., Segal,S. and MinnaJ.D. (1992) p53-Atransdominant regulator of transcription whose function is ablated bymutations occurring in human cancer. EMBO J., 11, 1383-1390.
Vogelstein,B. and Kinzler,K.W. (1992) p53 function and dysfunction.Cell, 70, 523-526.
Vojtesek,B., Bartek,J., Midgley,C.A. and Lane,D.P. (1992) Animmunochemical analysis of the human nuclear phosphoprotein-p53 -new monoclonal antibodies and epitope mapping using recombinant-p53. J. Iminlno. Methods, 151, 237-244.
Vousden,K. (1993) Interactions of human papillomavirus transformingproteins with the products of tumor suppressor genes. FASEB J., 7,872-879.
Waga,S., Hannon,G.J., Beach,D. and Stillman,B. (1994) The p21 inhibitorof cyclin-dependent kinases controls DNA replication by interactionwith PCNA. Nature, 369, 574-578.
Wagner,A.J., Kokontis,J.M. and Hay,N. (1994) invc-mediated apoptosisrequires wild-type p53 in a manner independent of cell cycle arrestand the ability of p53 to induce p21 1(fI//(P. Genes Dev., 8, 2817-2830.
Wu,X.W., Bayle,J.H., Olson,D. and Levine,A.J. (1993) The p53 mdm-2autoregulatory feedback loop. Genes Dev:, 7. 1126-1132.
Xiong,Y., Hannon,G.J., Zhang,H., Casso,D., Kobayashi,R. and Beach,D.(1993) p21 is a universal inhibitor of cyclin kinases. Science, 366,701-704.
Yew,P.R., Liu,X. and Berk,A.J. (1994) Adenovirus EIB oncoproteintethers a transcriptional repression domain to p53. Genes Del:, 8.190-202.
Yonish-Rouach,E., Resnitzky,D., Lotem,J., Sachs,L., Kimchi,A. andOren,M. (1991) Wild-type p53 induces apoptosis of myeloid leukaemiccells that is inhibited by interleukin-6. Nature, 352, 345-347.
Yonish-Rouach,E., Borde,J., Gotteland,M., Mishal,Z., Viron,A. andMay,E. (1994) Induction of apoptosis by transiently transfectedmetabolically stable wt p53 in transformed cell lines. Cell DeatlDiffer., 1, 39-47.
Zakut,R. and Givol,D. (1995) The tumor suppression function of p21l"atis contained in its N-terminal half ('half-WAF'). Oncogene, 11,393-395.
Zauberman,A., Barak,Y., Ragimov,N., Levy,N. and Oren,M. (1993)Sequence-specific DNA binding by p53-identification of targetsites and lack of binding to p53-Mdm2 complexes. EMBO J., 12,2799-2808.
Zauberman,A., Lupo,A. and Oren,M. (1995a) Identification of p53 targetgenes through immune selection of genomic DNA: the cyclin G genecontains two distinct p53 binding sites. Oncogene, 10, 2361-2366.
Zauberman,A., Flusberg,D., Haupt,Y., Barak,Y. and Oren,M. (1995b) Afunctional p53-responsive intronic promoter is contained within thehuman mndmn-2 gene. Nuicleic Acids Res., 23, 2584-2592.
Zhu,Y.M., Bradbury,D.A. and Russell,N.H. (1994) Wild-type p53 isrequired for apoptosis induced by growth factor deprivation in factor-dependent leukaemic cells. Br. J. Cancer, 69, 468-472.
Received oni September 19, 1995; rev,ised oni December 7, 1995
1606
Related Documents











