Born-Oppenheimer ab initio QM/MM Molecular Dynamics Simulations of Enzyme Reactions Yingkai Zhang Department of Chemistry New York University Dr. Po Hu Dr. Shenglong Wang $$$ NIH $$$ NSF Dr. Yihan Shao (Q-Chem Inc.)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Born-Oppenheimer ab initio QM/MM
Molecular Dynamics Simulations of
Enzyme Reactions
Yingkai Zhang
Department of Chemistry
New York University
Dr. Po Hu
Dr. Shenglong Wang$$$ NIH
$$$ NSF
Dr. Yihan Shao (Q-Chem Inc.)

Two Key Requirements for Reliably
Simulating Enzyme Reactions
• 1. A reasonably accurate potential energy surface
A. Describing bond forming/breaking
B. Modeling heterogeneous
enzyme environment.

Combined Ab initio Quantum Mechanical and Molecular
Mechanical (QM/MM) Approach
MM
QM
Interaction
Enzyme reactive site: small The rest: large
Allows for modeling the chemistry at the enzyme active site
with the state-of-the-art quantum mechanical methods
Properly including the heterogeneous enzyme environment
bondedMM
MMQM
vdw
MMQM
ele
MMQMMMQM
MMQMMMQMtotal
EEEE
EEEE
////
/
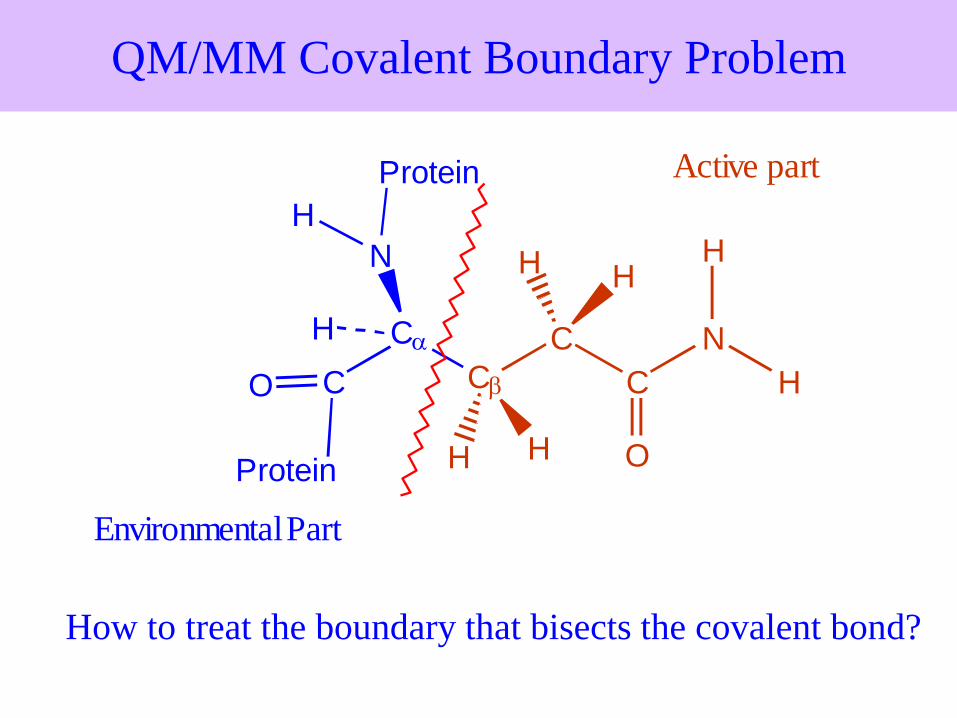
QM/MM Covalent Boundary Problem
How to treat the boundary that bisects the covalent bond?
C
C
C
C
C
N
H
H
O
HH
H H
N
H
Protein
O
Protein
H
Active part
Environmental Part

Link Atom Approach
• Additional atoms to cap the free valence
• Introducing additional degree of freedoms
C
C
C
C
C
N
H
H
O
HH
H H
N
H
Protein
O
Protein
H
Active part
Environmental Part
C
C
C
C
C
N
H
H
O
HH
H H
N
H
Protein
O
Protein
H
QM
MM
HL

The Pseudobond Approach
• Cps: a seven-valence-electron atom with an effective core potential
• Cps-C: a pseudobond mimics the original C-C bond with the similar bond length and strength, and the similar effects on the rest of the system
• No additional atoms or degrees of freedom
• Offers smooth connection between the QM and MM regions
Y. Zhang, T. S. Lee, W. Yang, J. Chem. Phys, 1999,110,46-54
C
C
C
C
C
N
H
H
O
HH
H H
N
H
Protein
O
Protein
H
C
Cps
C
C
C
N
H
H
O
HH
H H
N
H
Protein
O
Protein
H
QM
MM
HL
QM
MM
Link atom Pseudobond

• Significantly improved C(sp3)-C(sp3) pseudobonds for cutting of protein side chains for 6-31G* basis set
• Developed C(sp3)-C(sp2) and C(sp3)-N(sp3) pseudobonds for the cutting of protein backbones, DNAs and RNAs for the first time
• Independent of force field
• DFT (B3LYP, BLYP, PW91), HF , MP2
HH
CH2
OH
H H
O
N
NN
N
NH2
5'
3'
C
C
Side-chain
HN
C
O
HN
O
C
H
C
C
C
C
C
C
N
H
H
O
HH
H H
N
H
Protein
O
Protein
H
Active part
Environmental PartY. Zhang, J. Chem. Phys., 122, 024114 (2005)
New Development: the Cps with its own basis set

Two Key Requirements for Reliably
Simulating Enzyme Reactions
• 1. A reasonably accurate potential energy surface
Describing bond forming/breaking
Modeling heterogeneous enzyme environment
• 2. Extensive sampling due to the complexity of
energy landscape
Free energy change instead of potential energy change
Molecular dynamics instead of minimization

Allows for modeling the chemistry at the enzyme active site with the state-of-the-art quantum mechanical methods, while properly including the heterogeneous enzyme environment
• It takes account of dynamics of the reaction active site and its environment on an equal footing.
Born-Oppenheimer ab initio QM/MM MD simulation
At each time step, the atomic forces as well as the total
energy of the whole QM/MM system are calculated with
the ab initio QM/MM method on the fly, and Newton
equations of motion are integrated..

On-the-fly Born-Oppenheimer ab initio QM/MM MD
simulations are becoming increasingly feasible !
One pico-second MD simulation ( 1000 force and energy evaluations)
[intel 64 linux cluster (2.33Ghz, quad core) ], [Q-Chem3.0/Tinker4.2]
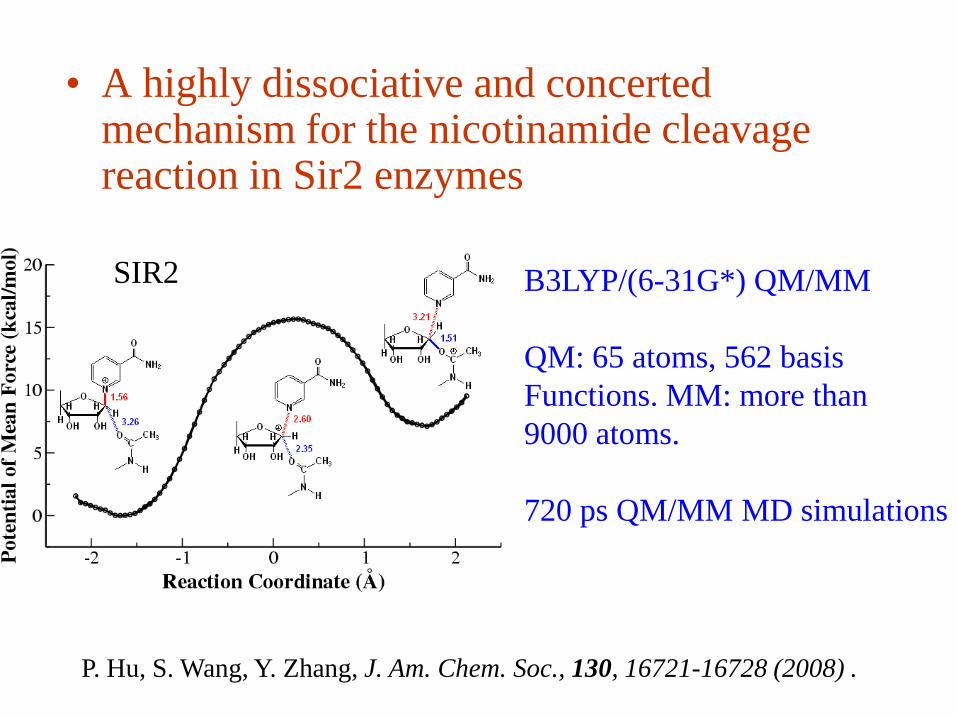
• A highly dissociative and concerted mechanism for the nicotinamide cleavage reaction in Sir2 enzymes
B3LYP/(6-31G*) QM/MM
QM: 65 atoms, 562 basis
Functions. MM: more than
9000 atoms.
720 ps QM/MM MD simulations
SIR2
P. Hu, S. Wang, Y. Zhang, J. Am. Chem. Soc., 130, 16721-16728 (2008) .


What can we really learn from such
advanced simulations ?
On-the-fly Born-Oppenheimer ab initio QM/MM MD
simulations are becoming increasingly feasible !
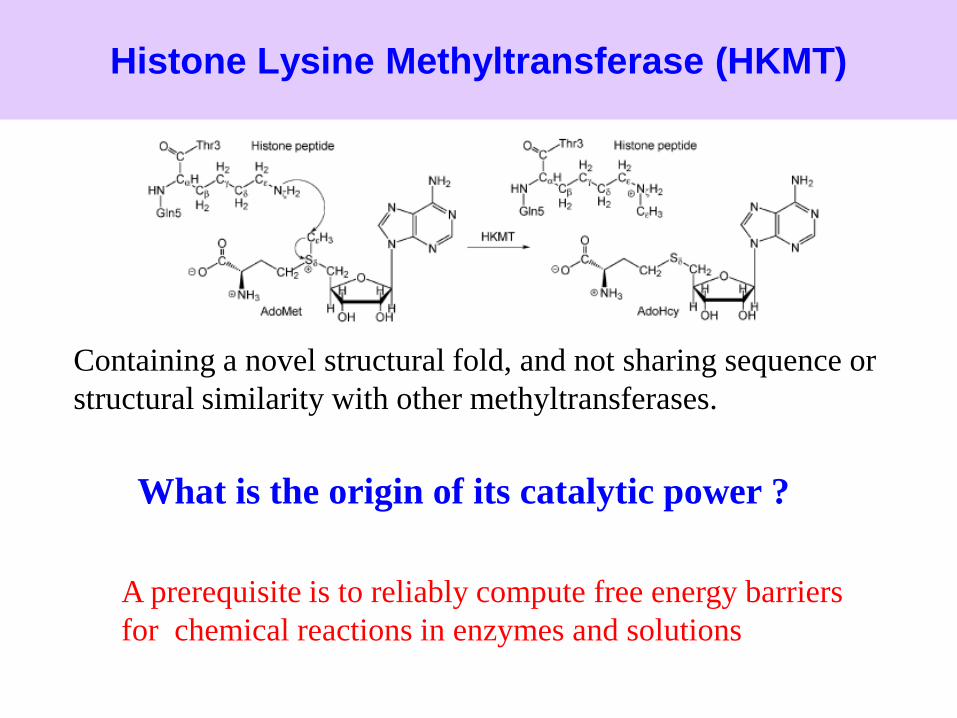
Histone Lysine Methyltransferase (HKMT)
Containing a novel structural fold, and not sharing sequence or
structural similarity with other methyltransferases.
What is the origin of its catalytic power ?
A prerequisite is to reliably compute free energy barriers
for chemical reactions in enzymes and solutions

Ab initio QM/MM Molecular Dynamics Simulation
with Umbrella Sampling
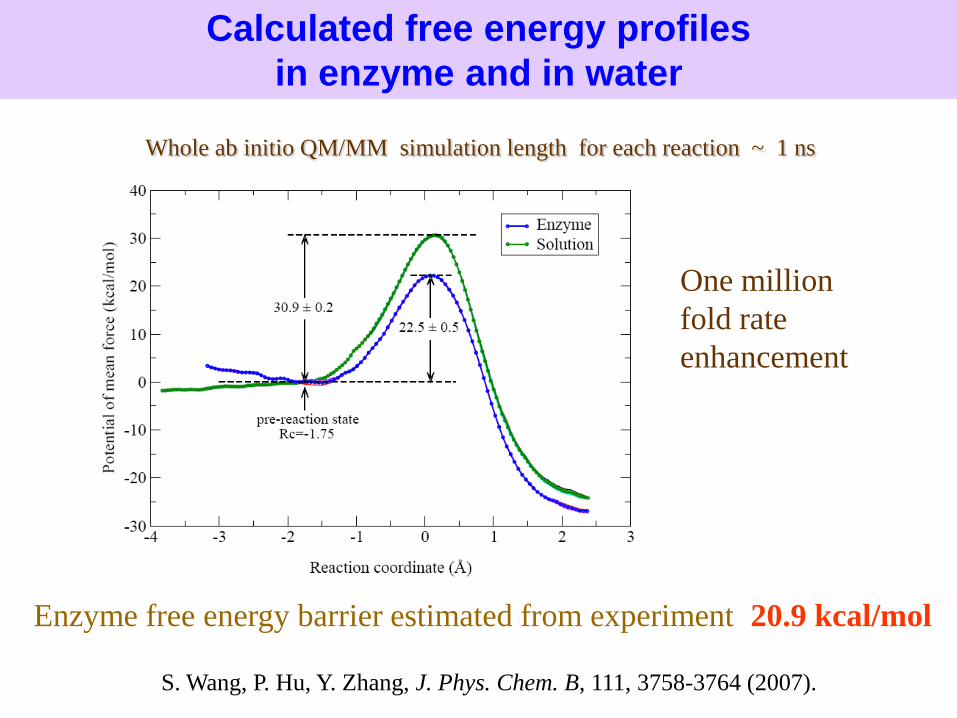
Enzyme free energy barrier estimated from experiment: 20.9 kcal/mol
Calculated free energy profiles
in enzyme and in water
Whole ab initio QM/MM simulation length for each reaction ~ 1 ns
One million
fold rate
enhancement
S. Wang, P. Hu, Y. Zhang, J. Phys. Chem. B, 111, 3758-3764 (2007).

How does SET7/9 achieve such one million fold
rate enhancement ?
• Is it due to the mechanism difference ?
Compression hypothesis (The TS is more compressed in enzyme than in solution)
Near-attack conformer hypothesis (the reactant conformations in enzyme are more ready for reactions than in solutions)
LYS4
N
H
H
S
C
H
H
H
CH2
CH2
AdoMet
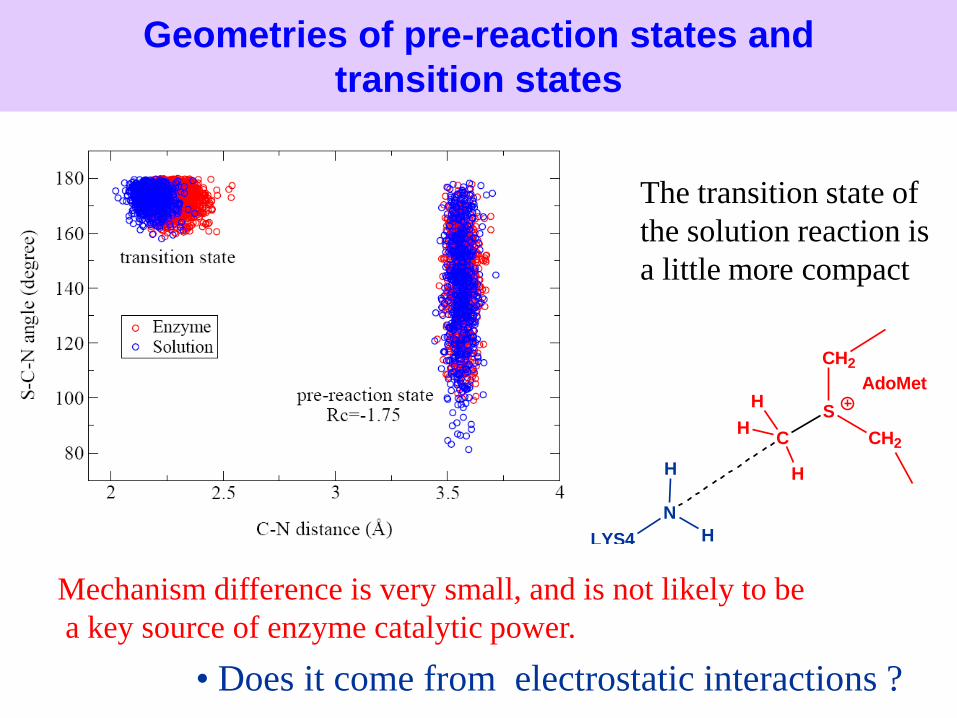
Geometries of pre-reaction states and
transition states
The transition state of
the solution reaction is
a little more compact.
Mechanism difference is very small, and is not likely to be
a key source of enzyme catalytic power.
LYS4
N
H
H
S
C
H
H
H
CH2
CH2
AdoMet
• Does it come from electrostatic interactions ?
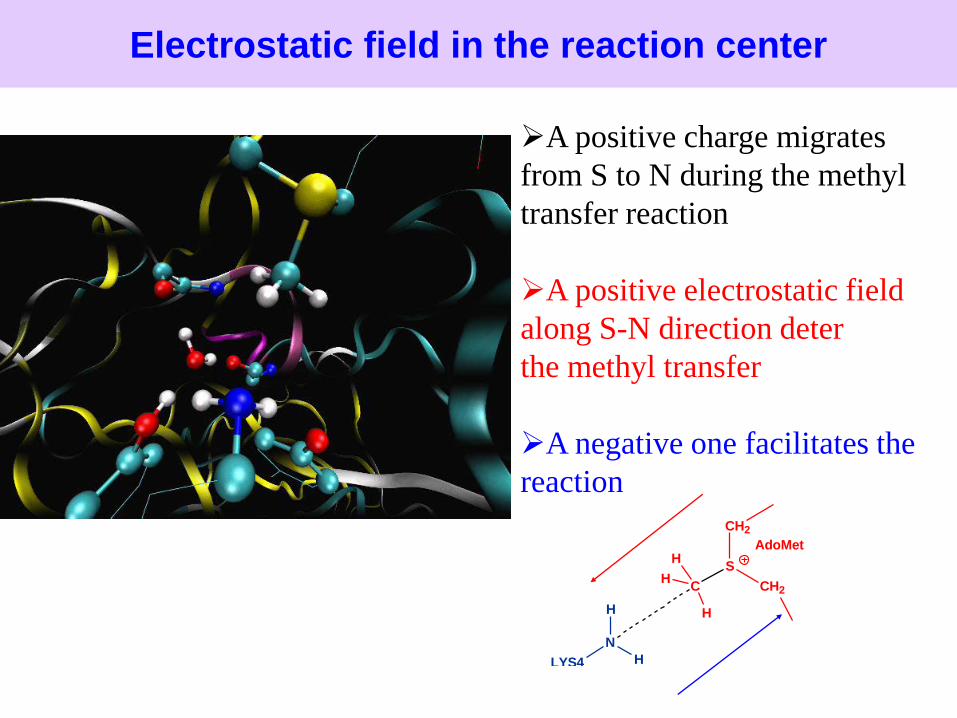
Electrostatic field in the reaction center
LYS4
N
H
H
S
C
H
H
H
CH2
CH2
AdoMet
S
N
A positive charge migrates
from S to N during the methyl
transfer reaction
A positive electrostatic field
along S-N direction deter
the methyl transfer
A negative one facilitates the
reaction

In solution reaction, there is a strong shift in electrostatic field from
the pre-reaction state to the transition state
For enzyme reaction, SET7/9 provides a pre-organized electrostatic
environment

Hydrogen network around the reaction center
in solution
It undergoes a significant change from the pre-reaction state
to the transition state

Summary (I)
• A combination of the electrostatic pre-
organization in enzyme and the hydrogen
bond network reorganization in solution is a
key source to the enzymatic power of
HKMT
S. Wang, P. Hu, Y. Zhang, J. Phys. Chem. B, 111, 3758-3764 (2007)
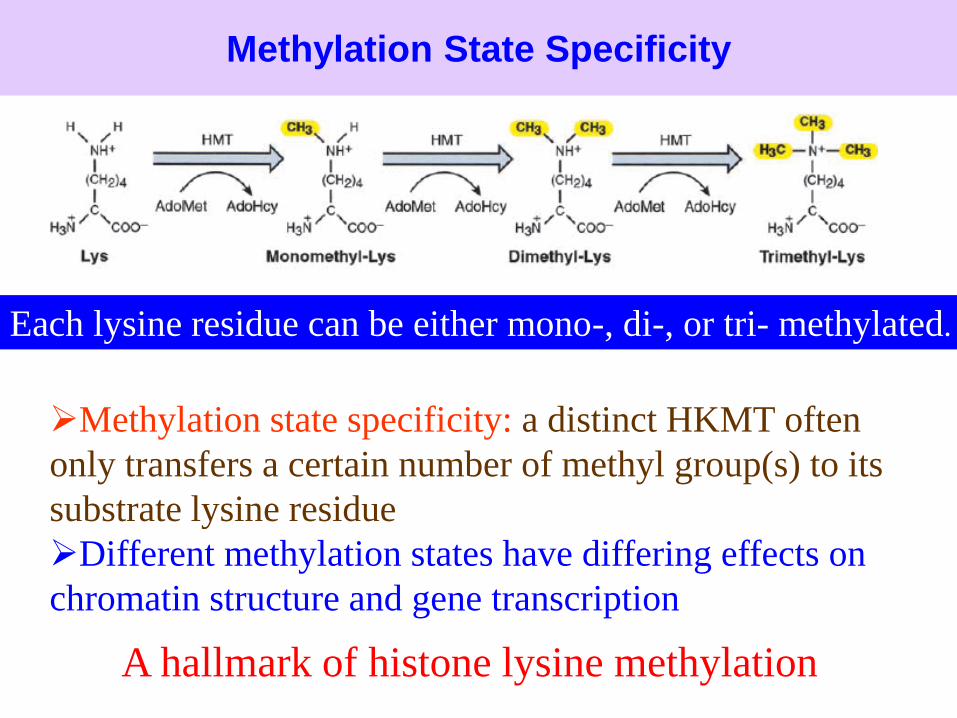
Methylation State Specificity
Methylation state specificity: a distinct HKMT often
only transfers a certain number of methyl group(s) to its
substrate lysine residue.
Different methylation states have differing effects on
chromatin structure and gene transcription
Each lysine residue can be either mono-, di-, or tri- methylated.
A hallmark of histone lysine methylation

Steric Hinderance Hypothesis
The active site of mono-HKMT is quite
tight, while that of tri-HKMT LSMT is very
roomy.
• The methylation state specificity is
dependent on the ability of accommodating
the increasing bulk of the lysine group
How can such a steric effect be imposed given
that a protein structure in water is very dynamic
in nature and the methyl group is very small ?
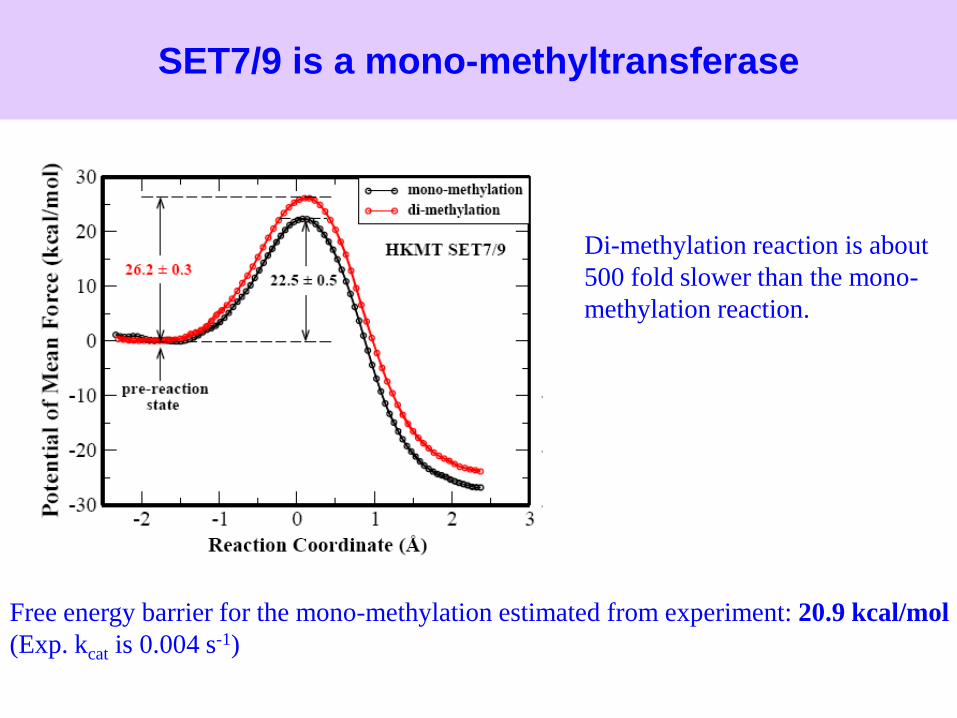
SET7/9 is a mono-methyltransferase
Free energy barrier for the mono-methylation estimated from experiment: 20.9 kcal/mol
(Exp. kcat is 0.004 s-1)
Di-methylation reaction is about
500 fold slower than the mono-
methylation reaction.

What is the key difference between mono- and di-methylation
in SET7/9 ?
The AdoMet binding
channel.
The access of solvent
water molecules
to the active site

What is the consequence for the access of solvent water
molecules? A water chain
The lone pair of N atom of the lysine substrate forms a direct
N…H-O hydrogen bond with a water molecule. The break of
this HB requires extra energy.
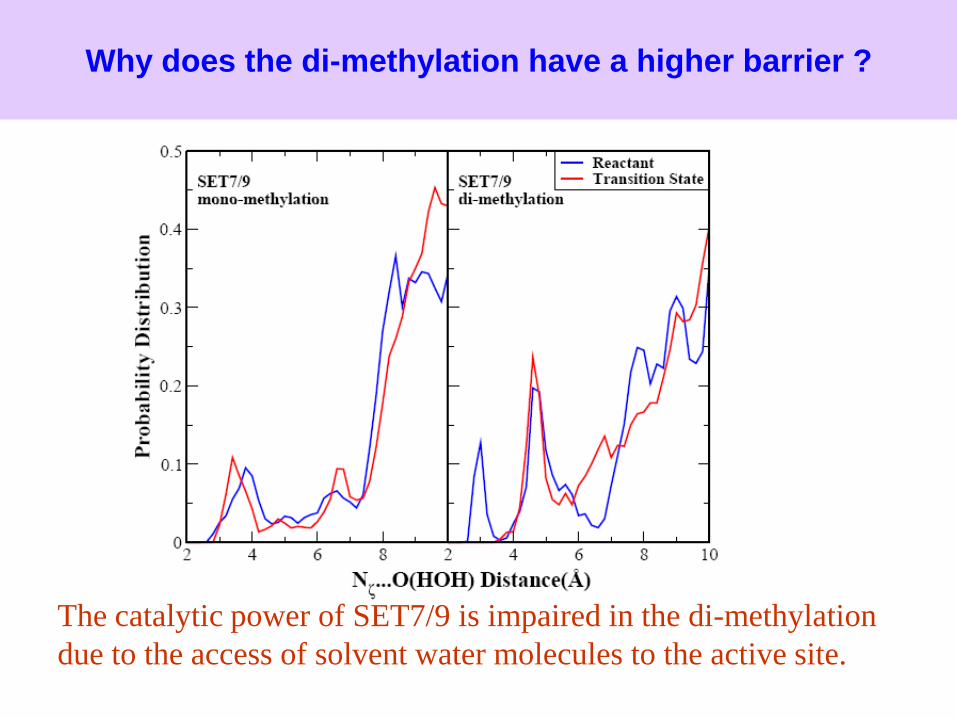
Why does the di-methylation have a higher barrier ?
The catalytic power of SET7/9 is impaired in the di-methylation
due to the access of solvent water molecules to the active site.

LSMT: a SET-domain tri-methyltransferase
(more spacious active site than SET7/9)
The estimated experimental barrier for mono- and di-methylation are
23.3 and 22.5 kcal/mol respectively.

In LSMT, mono- and di- methylation are very similar
The active site of LSMT is larger, so that the methylated lysine can
be accommodated without interfering with its catalytic power.

Summary (II)
• A dynamic mechanism is emerged regarding how the methylation state specificity is achieved.
• The binding of Me-lys in SET7/9 opens up the binding channel so that solvent water molecules get access to the active site, which leads to a higher reaction barrier.
• The active site of LSMT is more roomy so that it can accommodate Me-Lys substrate without disrupting its catalytic power.
• These detailed insights take account of protein dynamics, consistent with available experimental results as well as the theoretical findings regarding the catalytic power.
P. Hu, S. Wang, Y. Zhang, J. Am. Chem. Soc., 130, 3806-3813 (2008). .

Conclusion
• We have advanced the frontier of Born-
Oppenheimer ab initio QM/MM molecular
dynamics simulations.
• Novel insights have been provided into
important biological problems.

Acknowledgement
Dr. Shenglong Wang
(currently at NYU-
ITS)
Dr. Po Hu ( currently
at UC Berkley)
$$$ NIH (R01-GM079223)
$$$ NSF (CAREER Award, MRI)
$$$ NYSTAR (James D. Watson Young Investigator Award)
$$$ NYU (Whitehead Fellowship, start-up fund)
Computing NSF-MRI, NYU-ITS, NCSA
Dr. Yihan Shao (Q-Chem Inc.)
Related Documents











