This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse
Verna Louhivuori a, Annalisa Vicario b, Marko Uutela a, Tomi Rantamäki c, Lauri M. Louhivuori a,Eero Castrén c, Enrico Tongiorgi b, Karl E. Åkerman a, Maija L. Castrén a,d,⁎a Department of Biomedicine/Physiology, University of Helsinki, PO Box 63, FIN-00014 Helsinki, Finlandb BRAIN Centre for Neuroscience, Department of Life Sciences, University of Trieste, Via Giorgieri, 10, 34127 Trieste, Italyc Neuroscience Center, University of Helsinki, PO Box 56, FIN-00014 Helsinki, Finlandd Rinnekoti Foundation, Rinnekodintie 10, FIN-02980 Espoo, Finland
a b s t r a c ta r t i c l e i n f o
Article history:Received 2 May 2010Revised 14 October 2010Accepted 27 October 2010Available online 1 November 2010
Keywords:BDNFFragile X syndromeTrkBNeuronal progenitor cellsSynapseCalcium imaging
Fragile X syndrome (FXS) is a common cause of inherited mental retardation and the best characterized formof autistic spectrum disorders. FXS is caused by the loss of functional fragile X mental retardation protein(FMRP), which leads to abnormalities in the differentiation of neural progenitor cells (NPCs) and in thedevelopment of dendritic spines and neuronal circuits. Brain-derived neurotrophic factor (BDNF) and its TrkBreceptors play a central role in neuronal maturation and plasticity. We studied BDNF/TrkB actions in theabsence of FMRP and show that an increase in catalytic TrkB expression in undifferentiated NPCs of Fmr1-knockout (KO) mice, a mouse model for FXS, is associated with changes in the differentiation andmigration ofneurons expressing TrkB in neurosphere cultures and in the developing cortex. Aberrant intracellular calciumresponses to BDNF and ATP in subpopulations of differentiating NPCs combined with changes in theexpression of BDNF and TrkB suggest cell subtype-specific alterations during early neuronal maturation in theabsence of FMRP. Furthermore, we show that dendritic targeting of Bdnf mRNA was increased under basalconditions and further enhanced in cortical layer V and hippocampal CA1 neurons of Fmr1-KO mice bypilocarpine-induced neuronal activity represented by convulsive seizures, suggesting that BDNF/TrkB-mediated feedback mechanisms for strengthening the synapses were compromised in the absence of FMRP.Pilocarpine-induced seizures caused an accumulation of Bdnf mRNA transcripts in the most proximalsegments of dendrites in cortical but not in hippocampal neurons of Fmr1-KO mice. In addition, BDNF proteinlevels were increased in the hippocampus but reduced in the cortex of Fmr1-KO mice in line with regionaldifferences of synaptic plasticity in the brain of Fmr1-KO mice. Altogether, the present data suggest thatalterations in the BDNF/TrkB signaling modulate brain development and impair synaptic plasticity in FXS.
© 2010 Elsevier Inc. All rights reserved.
Introduction
The functional loss of fragile X mental retardation protein (FMRP)results in fragile X syndrome (FXS), the most common cause ofinherited mental retardation (Garber et al., 2008). A distinctneurobehavioral phenotype of FXS includes hyperactivity, attentiondeficit, hyper-responsiveness to sensory stimuli, social anxiety, and
autistic behaviors. About 20% of FXS males exhibit epileptic seizuresand polymorphisms in the BDNF gene may modulate the epilepsy inFXS (Louhivuori et al., 2009). The absence of FMRP results inabnormalities of dendritic spines in multiple regions of human FXSbrain and in the brain of Fmr1-knockout mice (Fmr1-KO), a mousemodel for FXS (Rudelli et al., 1985; Comery et al., 1997; Irwin et al.,2001; Galvez and Greenough, 2005). FMRP is an RNA-binding proteinthat is a constituent of messenger ribonucleoprotein complexes,which are transported to dendrites in a microtubule-dependentmanner and through association with polyribosomes, FMRP acts as atranslational regulator of specific dendritic mRNAs (Bassell andWarren, 2008). Its absence causes defects of protein synthesis-dependent plasticity seen as an impairment of long-term potentiation(LTP) in the cortex (Cx) and hippocampus (HC) (Li et al., 2002; Zhaoet al., 2005; Desai et al., 2006; Lauterborn et al., 2007; Meredith et al.,2007) and as an augmentation of long-term depression (LTD) in theHC and cerebellum of Fmr1-KO mice (Huber et al., 2002; Koekkoeket al., 2005; Volk et al., 2006, 2007). We have previously reportedalterations in the differentiation and migration of FMRP-deficient
Neurobiology of Disease 41 (2011) 469–480
Abbreviations: BDNF, brain-derived neurotrophic factor; [Ca2+]i, intracellular Ca2+;Cx, cortex; CxP, cortical plate; E14, embryonic day 14; FMR1 gene, fragile X mentalretardation 1 gene; Fmr1-KO mouse, Fmr1-knockout mouse; FMRP, fragile X mentalretardation protein; FXS, fragile X syndrome; GP, globus pallidus; HC, hippocampus;IHC, immunohistochemistry; INPs, intermediate neuronal progenitors; IZ, intermediatezone; ldt/lpt, lateral dorsal/posterior thalamic nucleus; LTD, long-term depression; LTP,long-term potentiation; mBDNF, mature BDNF; NPCs, neural progenitor cells; proBDNF,precursor BDNF; SVZ, subventricular zone; TrkB, tyrosine receptor kinase B; Trkb.FL,full-length tyrosine kinase receptor B; WT, wild type.⁎ Corresponding author. Rinnekoti Foundation, Rinnekodintie 10, FIN-02980 ESPOO,
Finland.E-mail address: [email protected] (M.L. Castrén).Available online on ScienceDirect (www.sciencedirect.com).
0969-9961/$ – see front matter © 2010 Elsevier Inc. All rights reserved.doi:10.1016/j.nbd.2010.10.018
Contents lists available at ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r.com/ locate /ynbd i

Author's personal copy
neuronal progenitor cells (NPCs), which may contribute to theabnormal development of neuronal circuits in FXS (Castren et al.,2005; Selby et al., 2007; Bureau et al., 2008; Tervonen et al., 2009;Harlow et al., 2010). However, it remains unclear how FMRP deficit inNPCs translates into an abnormal dendritic architecture and function.
An essential role of the brain-derived neurotrophic factor (BDNF)in neuronal maturation and plasticity (Huang and Reichardt, 2001;Mattson, 2008) led us to examine the involvement of BDNF and itstropomyosin-related kinase B (TrkB) receptors in the abnormalitiesseen in the brain development and plasticity of Fmr1-KO mice. NPCsexpress catalytically competent full-length TrkB (TrkB.FL) (Tervonenet al. 2006), which is involved in the regulation of the proliferation,survival, and neuronal differentiation of progenitor cells (Takahashiet al., 1999; Benoit et al., 2001; Barnabe-Heider and Miller, 2003). Ourprevious studies suggested a role for FMRP in BDNF-induced plasticity(Castren et al., 2002), and abnormally high TrkB immunoreactivityhas been found in cortical parvalbumin expressing cells in the brainsof adult Fmr1-KO mice (Selby et al., 2007). Here, we studiedcontribution of BDNF/TrkB signaling to the abnormal maturation ofFXS brain and show enhanced TrkB.FL expression and BDNF-inducedintracellular calcium responses in cortical NPCs lacking FMRP as wellas changes in the expression of TrkB.FL in the embryonic Fmr1-KO Cx.We found that dendritic targeting of Bdnf mRNA was increased inFMRP-deficient neurons in vitro and in vivo under basal conditions.Local increase in dendritic Bdnf mRNA was further enhanced bypilocarpine-induced neuronal activity represented by seizures indi-cating alterations in synapse function. Differences observed betweenthe cortical and hippocampal expression of BDNF likely contribute tothe spatial differences of defects shown in the neuronal plasticity inthe brain of Fmr1-KO mice.
Materials and methods
Mice
Male Fmr1-KO mice and their male wild-type (WT) littermateswere used in all experiments. The inbred FVB Fmr1-KO mice and theirgenotyping by tail-PCR have been previously described (Fragile XConsortium, 1994). All animals were housed in standard laboratoryconditions in qualified animal facility in accordance with the NationalInstitutes of Health guidelines. All animal experiments wereperformed according to the guidelines of the Society for Neuroscienceand European Economic Community Council Directive. The studieswere approved by the Experimental Animal Ethics Committee of theNational Laboratory Animal Center, Finland.
For the induction of sustained seizures by pilocarpine, mice wereprepared according to an established protocol (Turski, et al., 1983).Accordingly, mice were injected intraperitoneally with pilocarpinehydrochloride (300 mg/kg; Sigma) or controls with 1 X phosphatesaline buffer (PBS; 3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl,135 mM NaCl, pH 7.3). Methylscopolamine (Sigma) was injected(1 mg/kg, s.c.) 30 min before pilocarpine to reduce peripheralcholinergic effects. All pilocarpine-treated mice developed withinthe first hour after injection discontinuous seizures, includinginvoluntary oro-facial movements, salivation, eye-blinking, fasttwitching of whiskers, yawning, rearing, and slow tail movements.The mice were sacrificed 3 h after pilocarpine injection, in thisparadigm at the time of highest pan-Bdnf mRNA accumulation inneurons (Tongiorgi et al., 2004).
Brain tissue
The pregnant Fmr1+/− female mice were anesthetized with CO2
and sacrificed by cervical dislocation. The pups were quickly removedand sacrificed by decapitation at embryonic day 14 (E14) or 17 (E17).For paraffin sectioning and Nissl staining, the E17 brains were fixed in
4% paraformaldehyde (PFA) overnight (o/n) and washed twice in PBSafter fixation. Then the brains were dehydrated through increasingalcohol series: 70% ethanol for 2 h, 96% ethanol for 2 h, and 100%ethanol o/n at 4 °C. On the following day, the brains were treatedtwice with xylene (Riedel-de Haën) for 2 h before immersion inparaffin o/n at 60 °C. Next day, the brains were molded into paraffinand cut to 20-μm coronal sections on object glasses with a rotatorymicrotome MICROM HM 355 (MICROM).
For Bdnf mRNA studies, brain sections of 2-month-old adult maleFmr1-KO mice and their male WT littermates were used. The micewere transcardially perfusedwith 4% PFA under ketamine (100 mg/kgi.p.) anesthesia, and the brains were removed and kept in 4% PFA with20% sucrose at 4 °C for at least 5 days before sectioning to 40-μm-thickfree-floating coronal sections for nonradioactive in situ hybridization(Tongiorgi et al., 2004). For BDNF ELISA, brain tissue samples ofprefrontal andmotor corteces and hippocampi were collected from 2-to 4-month-old male Fmr1-KO mice and their WT littermates andsacrificed by cervical dislocation followed by anesthesia with CO2.Samples were frozen on dry ice, and stored at −70 °C until use.
Cell cultures
TrkB and BDNF expression, intracellular calcium responses, andneuronal differentiation were investigated in NPC cultures, whichwere propagated from the wall of lateral ventricles of male Fmr1-KOpups and their WT littermates at E14 as previously described (Castrenet al., 2005). The dissociated cells were plated in Dulbecco's modifiedEagle's medium Nutrient Mix F-12 (DMEM-F12; Life Technologies)culture medium supplemented with 2 mM L-glutamine, 15 mMHEPES, 100 U/ml penicillin, 100 mg/ml streptomycin, B27 supple-ment, 20 ng/ml epidermal growth factor (EGF, Life Technologies), and10 ng/ml fibroblast growth factor (FGF-2, PeproTech EC Ltd., London,UK) and grown in the 5% CO2-humidified incubator. Mitogens (20 ng/ml EGF and 10 ng/ml FGF-2) were added to the medium every thirdday, and the medium was refreshed every 3–4 days. The neurospherecultures were passaged every 7–9 days with papain (0.5 mg/ml,Sigma) and regrown from single cells at cell density 100,000 cells/mlto neurospheres. For immunocytochemistry or Fura2-AM-based Ca2+-imaging, 20 to 30 middle-sized neurospheres (diameter around225 μm) were collected from the free-floating neurosphere culturesand placed on coverslips coated with 10 μg/ml poly-DL-ornithine(Sigma) and differentiated without themitogens for 5, 7, or 14 days asindicated. One-third of the medium was changed every third dayduring the differentiation of neurospheres.
Primary cultures of hippocampal neurons provide an establishedin vitromodel to study neuronal effects and were used to verify the invivo studies of subcellular localization of BdnfmRNA. Rat hippocampalneuron cultures were prepared at P0-1 as described by Tongiorgi et al.(1997). Briefly, cells were plated on 2% Matrigel (BD-Biosciences)coated coverslips in 24-well plates at a density of 4×105/ml cells perwell and cultured for 8 days in a 5% CO2-humidified incubator, inminimum essential medium with Earle's salts (MEM) with GlutaMAX(Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco),35 nM D-glucose (Lancaster), 14 mM HEPES (Sigma), 1 mM vitaminB12 (Sigma), 0.36 mM D-biotin (Sigma), 30 μg/ml insulin (Sigma),100 μg/ml bovine transferring (Sigma), and antibiotics (Euroclone).The culture medium was changed every 2 days from the second dayonwards.
RNAi interference
We used RNAi interference to reduce the FMRP expression incultured hippocampal neurons. Fmr1 siRNAs (sequence #1GGUUUAUUCCAGAGCAAAUtt corresponding nt 460–478 in exon 4and #2 GCAUGUGAUGCUACGUAUAtt nt 554–572 in exon 5; GenBankNM_008031.2) and negative control siRNA were purchased from
470 V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480

Author's personal copy
Ambion. Transfection of cultured hippocampal neurons was per-formed after 11 days in vitro using 30 nM of Fmr1 or control siRNAmixture with 1 μl of siPORT NeoFX (Ambion) per well, following themanufacturer's instructions.
To test the efficiency of siRNA duplexes to reduce FMRP ex-pression, rat pheochromocytoma cells (PC12) were grown for 24 hand then transfected with siRNAs using Lipofectamine 2000(Invitrogen). At 48 h post-transfection, cells were harvested andimmunoblotted with an antibody anti-FMRP (1:1000, Abcam) inWestern analysis. The detection was performed by chemilumines-cence using the ECL++kit (Amersham) (see section Western blotanalysis for detailed procedure).
Nonradioactive in situ hybridization
Nonradioactive in situ hybridization was used to examine theexpression and subcellular localization of Bdnf mRNA in culturedhippocampal neurons and in both hippocampal and cortical neuronsin vivo. Hippocampal neurons were analyzed by in situ hybridizationafter 48 h of silencing, either in normal medium or after depolariza-tion for 3 h with 10 mM KCl as previously described (Tongiorgi et al.,1997). Cells were fixed for 15 min at RT in 4% PFA in PBS, washed inPBS with 0.1% Tween 20 (PBST), and permeabilized in absoluteethanol for 15 min at−20 °C. After rehydration, cells were hybridizedwith antisense or sense probes for the Bdnf coding region. Digox-igenin-labeled probes were synthesizedwith the DIG-RNA labeling kit(La Roche Diagnostics) using linearized plasmids as templates,according to the manufacturer's instructions. Hybridization wasfollowed by high stringency washes with 0.01 X sodium saline citratebuffer containing 0.1% Tween 20 (SSCT) at 60 °C. Thereafter, sampleswere incubated with anti-digoxigenin alkaline phosphatase-coupledantibody (1:1000) for 3 h and developed with NBT (75 mg/ml) andBCIP (50 mg/ml) for 40 min at RT.
In situ hybridization on free-floating, 40-μm coronal sections cut atthe level of dorsal hippocampus was performed as previouslydescribed (Tongiorgi et al., 2004). All in situ hybridizations onpilocarpine-treated brain sections were carried out sequentiallywith brain sections of control animals. In brief, after permeabilizationwith 2.3% NaMetaperiodate, 1% NaBH, slices were treated withproteinase K (Roche) and pre-hybridized for 2 h at 55 °C inhybridization solution. Hybridization was performed o/n at 55 °Cwith 100 ng/ml of DIG-labeled Bdnf antisense probe and followed byhigh stringency washes with SSCT at 60 °C. The immunodetection wasprocessed with anti-digoxigenin alkaline phosphatase-coupled anti-body (1:1000). To obtain reproducible and comparable results and toavoid saturation of the reaction, alkaline phosphatase developmentwas always performed for 5 h at RT.
Calcium imaging
Intracellular calcium imaging allows analysis of functionalresponses in neuronal cells. We used calcium imaging to studyBDNF responses during neuronal differentiation. For Ca2+-imagingexperiments, between 20 and 30 neurospheres (sized on average225 μm) were placed on poly-DL-ornithine-coated coverslips anddifferentiated for 5 days in a culture medium without mitogens. Theintracellular calcium recordings were made as previously described(Kärkkäinen et al., 2009). Briefly, the neurosphere-derived cells wereincubated at 37 °C for 20 minwith 4 μM fura-2 acetoxymethyl-ester ina Na+-Ringer solution consisting of 137 mM NaCl, 5 mM KCl, 1 mMCaCl2, 1 mM MgCl2, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 10 mMglucose, and 1 mM probenecid (pH adjusted to 7.4). Followingincubation, the coverslips were placed on the bottom of a thermo-stated perfusion chamber and perfused at a rate of 2 ml/min (37 °C).Between 35 to 50 single cells near the edge of the neurospheres weresimultaneously recorded using 340- and 380-nm light excitation
achieved through a filter changer under the control of an InCytIM-2system (Intracellular Imaging, Cincinnati, OH) and dichroic mirror(DM430, Nikon). The emission was measured through a 510-nmbarrier filter with an integrating charge-coupled device camera(COHU, San Diego, CA). The cells were excited by alternatingwavelengths of 340 and 380 nm and an image of 340 nm/380 nmratio was taken every second. The differentiating cells werechallenged when specified with 20 ng/ml BDNF (Tocris) or 100 μMATP (Sigma) diluted in Na+-Ringer solution or with 50 μM NMDA(Sigma) without MgCl2 in the Na+-Ringer solution.
Western blot analysis
Protein expression was studied by Western analysis. The sampleswere homogenized and processed in lysis buffer as previouslydescribed (Castren et al., 2002). The protein concentration of thesupernatant samples was determined using Biorad DC protein assay.The protein extracts (60 μg) were electrophoresed on 7.5% SDS-polyacrylamide minigels and transferred to 0.2 mm nitrocellulosemembranes (Schleicher & Schuell) for 1 h at 400 mA. The membraneswere washed 10 min in TBS, pH 7.4 (0.1 M Tris, 0.15 M NaCl) andblocked in 5% nonfat dry milk, in TBS with 0.1% Tween 20 (TBST) for1.5 h. The incubation with the primary mouse TrkB (1:2000, BDTransduction Laboratories™) at +4 °C o/n was followed by washes inTBST and incubation with the secondary goat anti-mouse HRPantibody (1:5000) for 1.5 h at RT. Detection was performed usingthe ECL++ kit (Amersham). Membranes were exposed to X-ray film(Fuji), scannedwith Epson Perfection V750 Pro (Epson, America, Inc.),and analyzed with Image J or imaged with luminescent imageanalyzer Las-3000 (Fuji FILM, Japan) and analyzed with ImageJ. Thespecificity of the Western blot results was confirmed by negativecontrols with lysis buffer and PBS in 2× Laemli buffer without theprotein extract. Ponceu S staining was used to control the laneloading.
BDNF ELISA
The BDNF expression was determined in proliferative corticalneurospheres and brain tissue samples using BDNF ELISA (Quantikinehuman BDNF kit, R&D Systems) following the manufacturer'sinstructions. The samples were homogenized in lysis buffer consistingof 137 mM NaCl, 20 mM Tris (pH 8.0), 1% (vol/vol) NP-40, 10% (vol/vol) glycerol, 50 mM sodium fluoride, 2× Complete Mini (RocheDiagnostics) and 2 mM sodium vanadate. After homogenization andincubation at +4 °C for 20 min, the total protein lysate wascentrifuged at 13,000 rpm for 15 min at +4 °C. The total proteinconcentration was determined using Biorad DC protein assay, and thevalues were used to normalize the BDNF protein concentration. Todetermine the BDNF concentration, the absorbance values of sampleduplicates at 450 nm were averaged and the background value wasreduced (absorbance measured at 540 nm) along with reduction ofzero value. The BDNF content was calculated as pg/ml according to thestandard curve. Used dilution factor (K-diluent) for the brain tissuelysates was 2.5 and no dilution was used for cell culture samples. Thespecificity of the ELISA results was confirmed by using lysis buffer andcell medium without protein extract as negative controls. StandardBDNF sample duplicates were included to every ELISA plate to controland standardize the measured values.
Immunostaining
The cellular TrkB expression was studied by immunostaining ofhistological brain sections, and immunocytochemistry was used toexamine cell populations in differentiated NPCs. Nissl-staining andsample preparation for paraffin-based immunohistochemistry (IHC)was performed as previously described (Tervonen et al., 2009). For
471V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480

Author's personal copy
immunocytochemistry of differentiated NPCs, cells were fixed in 4%PFA followed by washes with PBS three times at RT. Backgroundblocking and permeabilization of the samples were performed with0.5% Triton X-100 in 20% normal goat, horse, or rabbit serum in PBS for1 h at RT before incubation with the primary antibody o/n at +4 °C.After washing in PBS, cells were incubated with the secondaryantibody diluted in PBS for 45 min or 1 h at RT andwashed three timeswith PBS. Primary antibodies used in the study were polyclonal goator rabbit full-length specific anti-TrkB (1:250 and 1:150, respectively;Santa Cruz), polyclonal rabbit anti-BDNF (1:500, Chemicon), rabbitanti-MAP-2 (1:500, Chemicon), and guinea pig anti-GLAST (1:400,Chemicon). Secondary antibodies were Alexa Fluor 488, Alexa Fluor546 or Alexa Fluor 568 anti-rabbit, anti-guinea pig, or anti-goat IgG(all 1:1000 for brain sections and 1:500 for cells, Molecular Probes).The cell nuclei were counterstained with 4′,6-diaminodino-2-pheny-lindole (DAPI, 0.1 mg/ml, Boehringer Mannheim Biochemica,Germany). The sections were mounted with Gel Mount media(Sigma) and the cells with Dapi-Vectashield mounting media(Vector). The samples were visualized and imaged with a LSM 5Pascal system (Zeiss). The specificity of the antibodies was controlledby immunostaining control samples with omitting either the primaryor secondary antibody.
Quantitative image analysis and statistics
The fluorescent-stained brain sections and differentiated NPCs wereviewed with AxioPLAN 2 imaging microscope (Zeiss), and the confocalmicroscopic images were obtained with a LSM 5-Pa system (Zeiss) andanalyzed with the image analysis program, Image-ProPlus version 6.1(Media Cybernetics, Inc., Silver Spring, MD, USA). The cell countinganalysis of the immunostained brain sections (10× magnification) anddifferentiated NPCs (25× magnification) was performed semiquantita-tively frommaximumprojection confocal images. Image stack sizeswere750 μm height×750 μm width×23.7 μm in z-dimension for corticalanalysis; 900 μm height×900 μm width×23.7 μm in z-dimension forhippocampal and thalamic analysis; 1272.8 μm height×1272.8 μmwidth×23.7 μm in z-dimension for the analysis of globus pallidus (GP);509.1 μm height×509.1 μm width×13.2 μm in z-dimension for differ-entiating NPCs. Nissl and DAPI staining was used for the layerdetermination in the developing Cx (Fig. 3B). The number of cellsexpressing the fluorescent stain was analyzed from the intermediatezone (IZ) and cortical plate (CxP) of the coronal somatosensoryneocortex at E17 approximately 860 μm and 1.3 mm posterior to rostralborder of the olfactory bulbs (Jacobowitz and Abbott, 1998). The numberof TrkB.FL positive cells was counted fromdefined counting frames (CxP:100×200 μm, IZ: 175×500 μm, HC: 100×200 μm, globus pallidus (GP):220×220 μm, lateral dorsal/posterior thalamic nucleus (ldt/lpt):150×150 μm) in 3–6 sequential coronal sections from each 3–6 malemice per group, then averaged and pooled together. Brightness andcontrast of inverted grayscale maximum projection pictures wereoptimized, and the cell counting analysis was performed with “autocount” function with defined cell size limits and threshold values.Independent two population Student's t-test was used in the statisticalanalysis; statistical significance was set at level 0.05. All the confocalimaging data are represented as means±SEM.
Quantification of in situ hybridization data were performed aspreviously described (Tongiorgi et al., 2004). Stained neurons wereanalyzed by viewing stained cultures (60× objective) and brainsections (20× objective) under bright-field illumination with a NikonE800 microscope with a differential interference contrast-equippedlens (60× magnification). Images of the stained neurons wereacquired with a charge-coupled device (CCD) camera (Nikon ADX-1200) mounted on the microscope and analyzed with the imageanalysis program Image-ProPlus (Media Cybernetics, Inc., SilverSpring, MD, USA). The function “trace” was used to measure, startingfrom the base of the dendrites, the maximal distance of dendritic
labeling (MDDL) as previously described (Tongiorgi et al., 1997).Dendrites were traced, in a conservative manner, up to the point atwhich the in situ labeling was no longer clearly distinguishable fromthe background. The background level obtained in sister cell cultureshybridized with the sense probes was used as a reference to set thethreshold for specific labeling obtained with the antisense probes.Between 100 and 150 dendrites were measured for each condition(three independent experiments). Individual preparations werecoded and analyzed in a blind manner. Statistical data analysis wasperformed with Sigma Stat 3.2 software (Systat Software, Inc.) andgraph elaborations with Sigma Plot 11 software (Systat Software,Inc.). Statistical significance among groups was evaluated performingone-way analysis of variance (ANOVA) and Dunn's post hoccomparison following ANOVA.
Fura2-AM calcium-imaging data and BDNF ELISA were analyzedwith Excel and Microcal Software Origin 6.1 (Northampton, MA).Independent two population Student's t-test was used in thestatistical analysis, statistical significance was set at level *p≤0.05.Values are means±SEM.
Results
TrkB expression in NPCs lacking FMRP
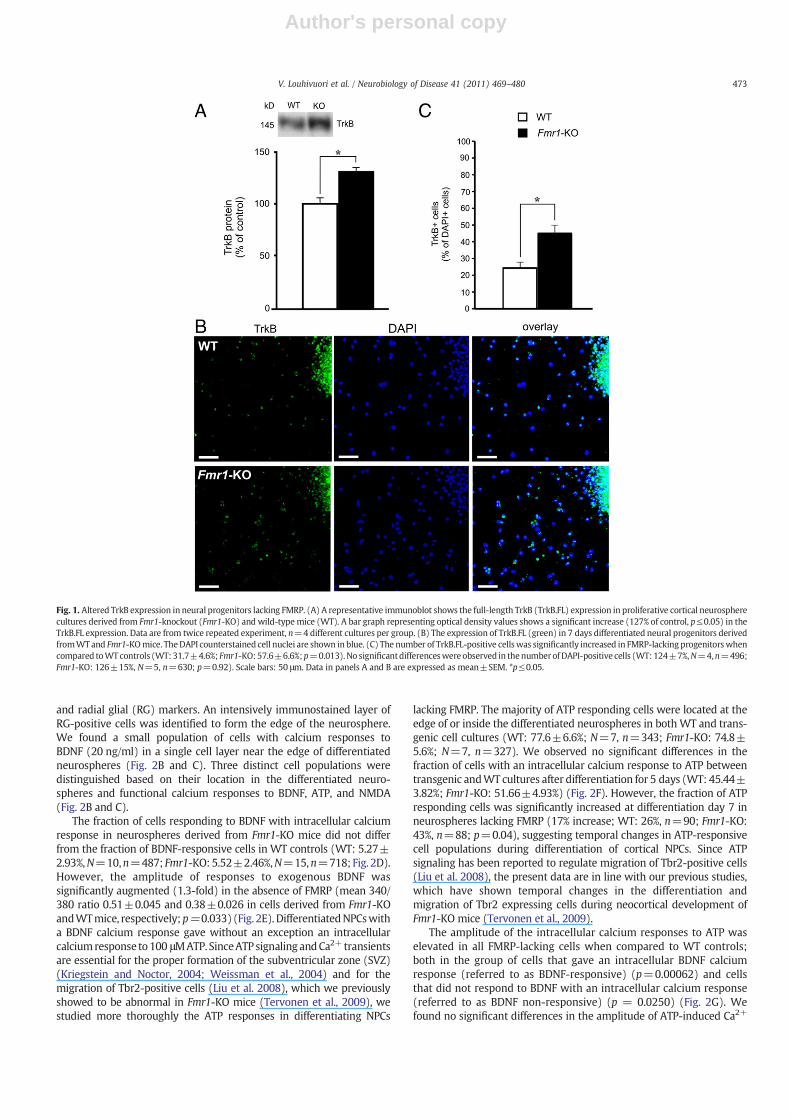
Both BDNF and the lack of FMRP have been shown to promoteneuronal differentiation of NPCs, suggesting that BDNF and FMRPmayplay a role in the control of common intracellular pathways thatregulate early neuronal fate determination (Takahashi et al., 1999;Benoit et al., 2001; Barnabe-Heider and Miller, 2003; Castren et al.,2005). To examine the commonmechanisms involved in the action ofBDNF and FMRP, we investigated TrkB protein expression inproliferative cortical neurosphere cultures generated from Fmr1-KOand WT mouse embryos. As shown in Fig. 1A, the catalytic TrkBexpression was significantly increased (127% of control, p≤0.05) inNPCs lacking FMRP when compared to WT controls in Westernanalysis. We studied TrkB expression also during the differentiation ofNPCs with immunostaining of cells migrated out from the neuro-spheres using a catalytic TrkB-specific antibody (Fig. 1B). The fractionof TrkB expressing cells was significantly increased in 7 daysdifferentiated NPCs lacking FMRP when compared to WT controls(WT: 31.7±4.6%; Fmr1-KO: 57.6±6.6%; p=0.013; Fig. 1C). Since thenumber of DAPI immunostained cells was similar in WT and FMRPlacking NPCs (WT: 124±7%, N=4, n=496; Fmr1-KO: 126±15%,N=5, n=630; p=0.92), the data suggested differences specifically inthe fate determination of differentiating NPCs. Primary corticalprogenitors derived from WT and Fmr1-KO mouse embryos formedneurospheres in an identical manner in the clonal analysis. Theproliferation and survival of transgenic primary progenitors did notsignificantly differ from WT controls (data not shown), and thefindings were consistent with our previous studies, which haveshown a defect in the differentiation but not in the proliferation ofembryonic NPCs in the absence of FMRP (Castren et al., 2005).
Enhanced calcium responses to BDNF and ATP in NPCs lacking FMRP
Changes in intracellular free Ca2+ play an important role in thedifferentiation of NPCs (Ciccolini et al. 2003, Kärkkäinen et al., 2009).BDNF elicits Ca2+ increases in neurons (Berninger and Poo, 1996; Zhangand Poo, 2002) and sustained intracellular calcium elevations inducedby BDNF application may trigger cytoskeletal rearrangements essentialfor dendritic spine formation and remodeling. Therefore, we nextexamined the effects of brief BDNF exposure on the intracellular calciummobilization in differentiated WT and FMRP-deficient cortical NPCs.Fura2-AM-based calcium imagingwasperformedon thefifthdayofNPCdifferentiation in neurosphere cultures propagated at E14. As shown inFig. 2A, the differentiated NPCs exhibited immunopositivity to neuronal
472 V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Author's personal copy
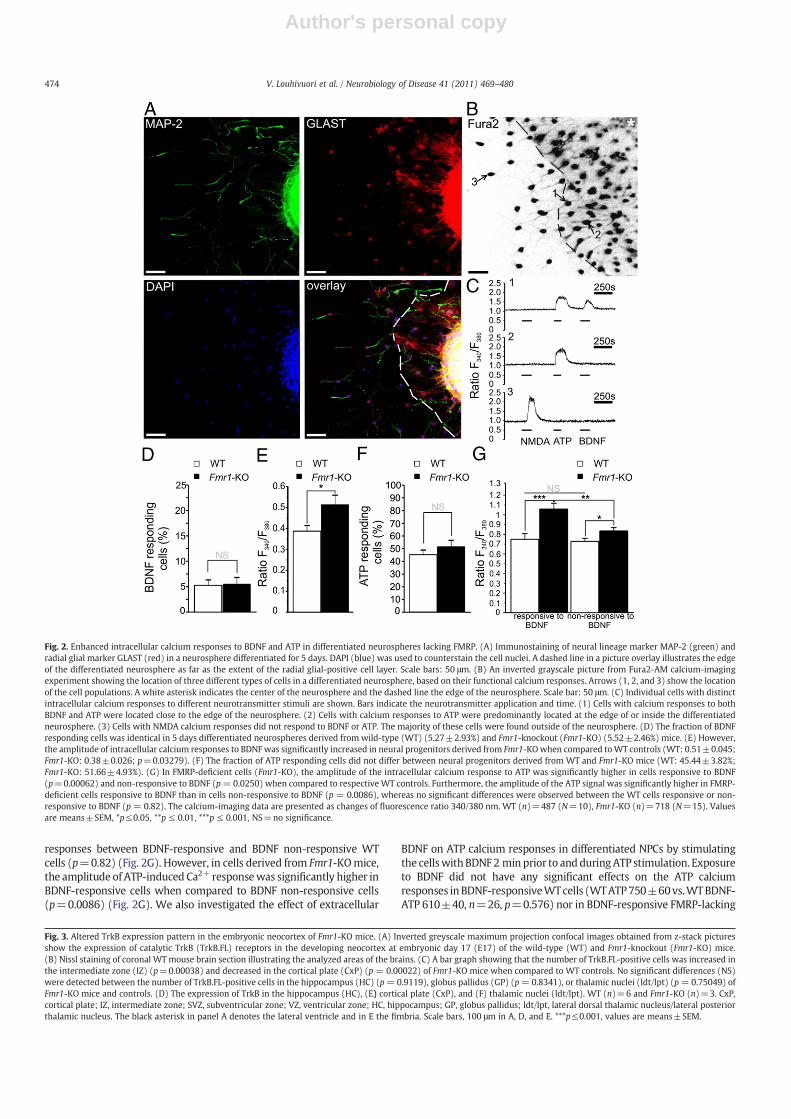
and radial glial (RG) markers. An intensively immunostained layer ofRG-positive cells was identified to form the edge of the neurosphere.We found a small population of cells with calcium responses toBDNF (20 ng/ml) in a single cell layer near the edge of differentiatedneurospheres (Fig. 2B and C). Three distinct cell populations weredistinguished based on their location in the differentiated neuro-spheres and functional calcium responses to BDNF, ATP, and NMDA(Fig. 2B and C).
The fraction of cells responding to BDNF with intracellular calciumresponse in neurospheres derived from Fmr1-KO mice did not differfrom the fraction of BDNF-responsive cells in WT controls (WT: 5.27±2.93%,N=10,n=487; Fmr1-KO: 5.52±2.46%,N=15,n=718; Fig. 2D).However, the amplitude of responses to exogenous BDNF wassignificantly augmented (1.3-fold) in the absence of FMRP (mean 340/380 ratio 0.51±0.045 and 0.38±0.026 in cells derived from Fmr1-KOandWTmice, respectively;p=0.033) (Fig. 2E). DifferentiatedNPCswitha BDNF calcium response gave without an exception an intracellularcalciumresponse to100 μMATP. SinceATP signaling andCa2+ transientsare essential for the proper formation of the subventricular zone (SVZ)(Kriegstein and Noctor, 2004; Weissman et al., 2004) and for themigration of Tbr2-positive cells (Liu et al. 2008), which we previouslyshowed to be abnormal in Fmr1-KO mice (Tervonen et al., 2009), westudied more thoroughly the ATP responses in differentiating NPCs
lacking FMRP. The majority of ATP responding cells were located at theedge of or inside the differentiated neurospheres in bothWT and trans-genic cell cultures (WT: 77.6±6.6%; N=7, n=343; Fmr1-KO: 74.8±5.6%; N=7, n=327). We observed no significant differences in thefraction of cells with an intracellular calcium response to ATP betweentransgenic andWT cultures after differentiation for 5 days (WT: 45.44±3.82%; Fmr1-KO: 51.66±4.93%) (Fig. 2F). However, the fraction of ATPresponding cells was significantly increased at differentiation day 7 inneurospheres lacking FMRP (17% increase; WT: 26%, n=90; Fmr1-KO:43%, n=88; p=0.04), suggesting temporal changes in ATP-responsivecell populations during differentiation of cortical NPCs. Since ATPsignaling has been reported to regulate migration of Tbr2-positive cells(Liu et al. 2008), the present data are in line with our previous studies,which have shown temporal changes in the differentiation andmigration of Tbr2 expressing cells during neocortical development ofFmr1-KO mice (Tervonen et al., 2009).
The amplitude of the intracellular calcium responses to ATP waselevated in all FMRP-lacking cells when compared to WT controls;both in the group of cells that gave an intracellular BDNF calciumresponse (referred to as BDNF-responsive) (p=0.00062) and cellsthat did not respond to BDNF with an intracellular calcium response(referred to as BDNF non-responsive) (p = 0.0250) (Fig. 2G). Wefound no significant differences in the amplitude of ATP-induced Ca2+
Fig. 1. Altered TrkB expression in neural progenitors lacking FMRP. (A) A representative immunoblot shows the full-length TrkB (TrkB.FL) expression in proliferative cortical neurospherecultures derived from Fmr1-knockout (Fmr1-KO) and wild-typemice (WT). A bar graph representing optical density values shows a significant increase (127% of control, p≤0.05) in theTrkB.FL expression. Data are from twice repeated experiment, n=4 different cultures per group. (B) The expression of TrkB.FL (green) in 7 days differentiated neural progenitors derivedfromWTand Fmr1-KOmice. TheDAPI counterstained cell nuclei are shown in blue. (C) The number of TrkB.FL-positive cellswas significantly increased in FMRP-lacking progenitorswhencompared toWTcontrols (WT:31.7±4.6%; Fmr1-KO:57.6±6.6%;p=0.013).No significantdifferenceswere observed in thenumberofDAPI-positive cells (WT:124±7%,N=4,n=496;Fmr1-KO: 126±15%, N=5, n=630; p=0.92). Scale bars: 50 μm. Data in panels A and B are expressed as mean±SEM. *p≤0.05.
473V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Author's personal copy
responses between BDNF-responsive and BDNF non-responsive WTcells (p=0.82) (Fig. 2G). However, in cells derived from Fmr1-KOmice,the amplitude of ATP-induced Ca2+ responsewas significantly higher inBDNF-responsive cells when compared to BDNF non-responsive cells(p=0.0086) (Fig. 2G). We also investigated the effect of extracellular
BDNF on ATP calcium responses in differentiated NPCs by stimulatingthe cellswithBDNF 2 min prior to and during ATP stimulation. Exposureto BDNF did not have any significant effects on the ATP calciumresponses in BDNF-responsiveWTcells (WTATP750±60vs.WTBDNF-ATP 610±40, n=26, p=0.576) nor in BDNF-responsive FMRP-lacking
Fig. 2. Enhanced intracellular calcium responses to BDNF and ATP in differentiated neurospheres lacking FMRP. (A) Immunostaining of neural lineage marker MAP-2 (green) andradial glial marker GLAST (red) in a neurosphere differentiated for 5 days. DAPI (blue) was used to counterstain the cell nuclei. A dashed line in a picture overlay illustrates the edgeof the differentiated neurosphere as far as the extent of the radial glial-positive cell layer. Scale bars: 50 μm. (B) An inverted grayscale picture from Fura2-AM calcium-imagingexperiment showing the location of three different types of cells in a differentiated neurosphere, based on their functional calcium responses. Arrows (1, 2, and 3) show the locationof the cell populations. A white asterisk indicates the center of the neurosphere and the dashed line the edge of the neurosphere. Scale bar: 50 μm. (C) Individual cells with distinctintracellular calcium responses to different neurotransmitter stimuli are shown. Bars indicate the neurotransmitter application and time. (1) Cells with calcium responses to bothBDNF and ATP were located close to the edge of the neurosphere. (2) Cells with calcium responses to ATP were predominantly located at the edge of or inside the differentiatedneurosphere. (3) Cells with NMDA calcium responses did not respond to BDNF or ATP. The majority of these cells were found outside of the neurosphere. (D) The fraction of BDNFresponding cells was identical in 5 days differentiated neurospheres derived from wild-type (WT) (5.27±2.93%) and Fmr1-knockout (Fmr1-KO) (5.52±2.46%) mice. (E) However,the amplitude of intracellular calcium responses to BDNF was significantly increased in neural progenitors derived from Fmr1-KOwhen compared toWT controls (WT: 0.51±0.045;Fmr1-KO: 0.38±0.026; p=0.03279). (F) The fraction of ATP responding cells did not differ between neural progenitors derived from WT and Fmr1-KO mice (WT: 45.44±3.82%;Fmr1-KO: 51.66±4.93%). (G) In FMRP-deficient cells (Fmr1-KO), the amplitude of the intracellular calcium response to ATP was significantly higher in cells responsive to BDNF(p=0.00062) and non-responsive to BDNF (p=0.0250) when compared to respective WT controls. Furthermore, the amplitude of the ATP signal was significantly higher in FMRP-deficient cells responsive to BDNF than in cells non-responsive to BDNF (p = 0.0086), whereas no significant differences were observed between the WT cells responsive or non-responsive to BDNF (p = 0.82). The calcium-imaging data are presented as changes of fluorescence ratio 340/380 nm. WT (n)=487 (N=10), Fmr1-KO (n)=718 (N=15). Valuesare means±SEM, *p≤0.05, **p ≤ 0.01, ***p ≤ 0.001, NS=no significance.
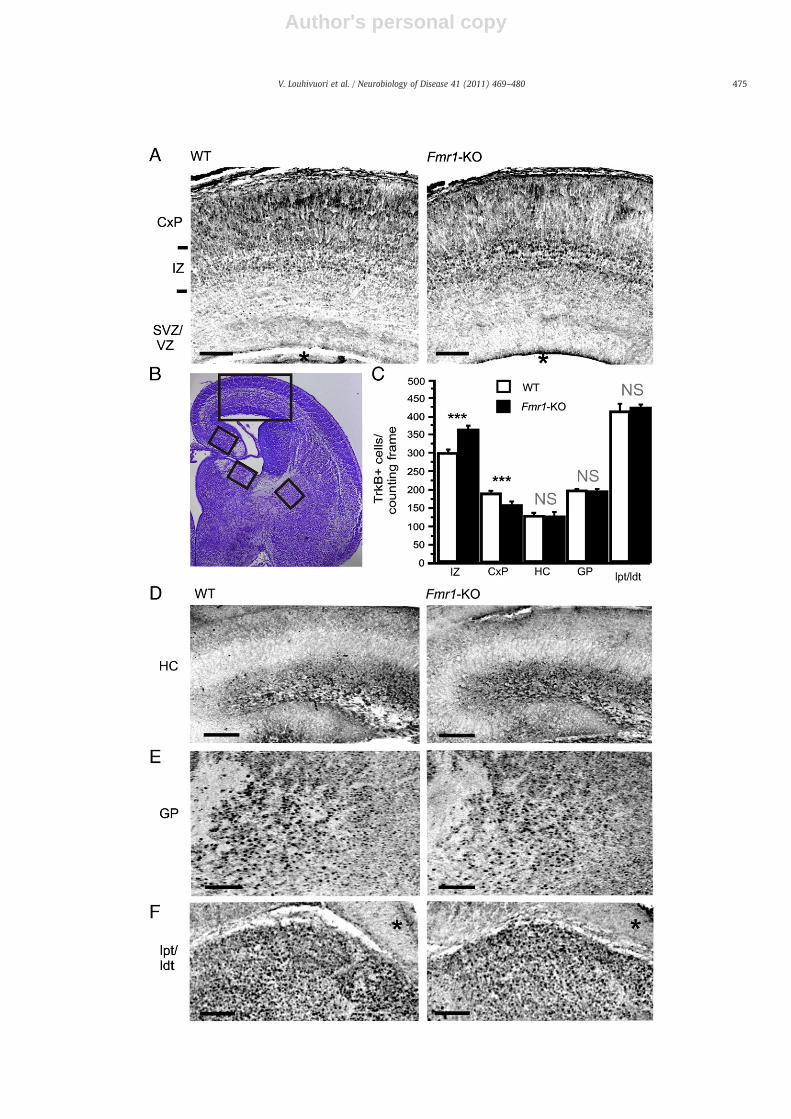
Fig. 3. Altered TrkB expression pattern in the embryonic neocortex of Fmr1-KO mice. (A) Inverted greyscale maximum projection confocal images obtained from z-stack picturesshow the expression of catalytic TrkB (TrkB.FL) receptors in the developing neocortex at embryonic day 17 (E17) of the wild-type (WT) and Fmr1-knockout (Fmr1-KO) mice.(B) Nissl staining of coronal WTmouse brain section illustrating the analyzed areas of the brains. (C) A bar graph showing that the number of TrkB.FL-positive cells was increased inthe intermediate zone (IZ) (p=0.00038) and decreased in the cortical plate (CxP) (p = 0.00022) of Fmr1-KO mice when compared to WT controls. No significant differences (NS)were detected between the number of TrkB.FL-positive cells in the hippocampus (HC) (p=0.9119), globus pallidus (GP) (p=0.8341), or thalamic nuclei (ldt/lpt) (p=0.75049) ofFmr1-KO mice and controls. (D) The expression of TrkB in the hippocampus (HC), (E) cortical plate (CxP), and (F) thalamic nuclei (ldt/lpt). WT (n)=6 and Fmr1-KO (n)=3. CxP,cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone; HC, hippocampus; GP, globus pallidus; ldt/lpt, lateral dorsal thalamic nucleus/lateral posteriorthalamic nucleus. The black asterisk in panel A denotes the lateral ventricle and in E the fimbria. Scale bars, 100 μm in A, D, and E. ***p≤0.001, values are means±SEM.
474 V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Author's personal copy
475V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Author's personal copy
transgenic NPCs (FX ATP 1060±60 vs. FX BDNF-ATP 910±95, n=27,p=0.205) and the ATP responses were still elevated in BDNF-treatedtransgenic cells when compared toWT (FX BDNF-ATP 910±95, n=27vs. WT ATP 750±60, n=26, p=0.0057) (Supplemental Fig. 1A).Whereas in cells that were non-responsive to BDNF, the amplitude ofATP responseswas significantly reduced in both transgenic andWT cells(FX ATP 850±30 vs. FX BDNF-ATP 695±30, n=139, p=0.0014, andWT ATP 735±30 vs. WT BDNF-ATP 625±40, n=79, p=0.035)(Supplemental Fig. 1B). In BDNF non-responsive transgenic cells,BDNF treatment prior and during ATP stimulation decreased theamplitude of the intracellular calcium response to ATP to WT levels(FX BDNF-ATP 695±30, n=139 vs. WT ATP 735±30, n=79,p=0.2005) (Supplemental Fig. 1B) implicating that BDNF-mediatedcellular mechanisms were involved in aberrant ATP responses ofprecursors lacking FMRP.
Themajority of cells responding toBDNF and/or ATPdid not respondto 50 μM NMDA (Fig. 2B and C). Most of the NMDA-responsive cells
were migrated out from the neurosphere (WT: 90.3±3.6%, N=7,n=343; Fmr1-KO: 83.7±7.3%, N=7, n=327), and only a smallproportion of cells with NMDA response was found at the edge of theneurosphere (Fig. 2B and C) (WT: 10.4±3.99%, N=7, n=343; Fmr1-KO: 16.3±7.3%,N=7, n=327). The number of NMDA-responsive cellsdid not significantly differ between transgenic and WT cultures (WT:8.25±2.16%; Fmr1-KO: 7.13±1.37%).
TrkB expression during early development of Fmr1-KO mouse brain
To examine whether changes in catalytic TrkB expression observedin FMRP-deficient NPCs could be seen in vivo, we investigated TrkBexpression in the developing brain of Fmr1-KO and WT mice with IHC.In the E17 neocortex, when only neurons of the deep cortical layershave completed their migration, the TrkB.FL-specific antibody recog-nized catalytic TrkB expression primarily in the IZ and CxP (Fig. 3A).Only faint cellular staining was observed in the SVZ. Semiquantitative
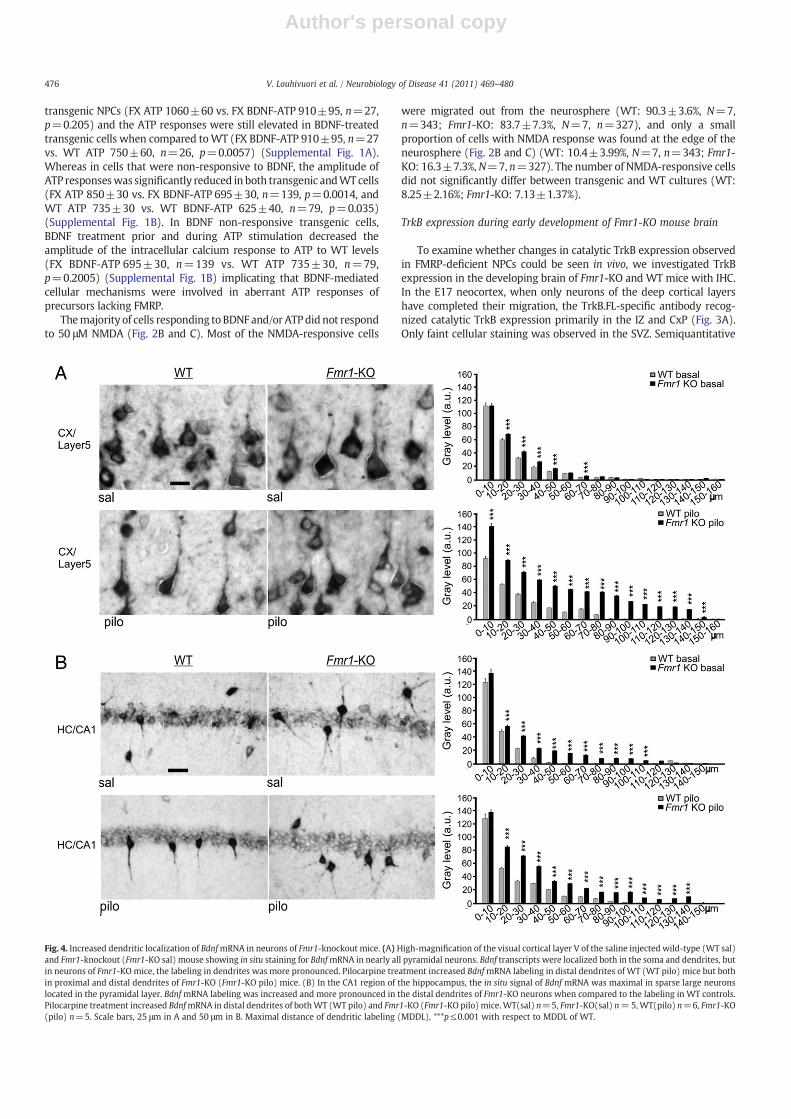
Fig. 4. Increased dendritic localization of BdnfmRNA in neurons of Fmr1-knockout mice. (A)High-magnification of the visual cortical layer V of the saline injected wild-type (WT sal)and Fmr1-knockout (Fmr1-KO sal) mouse showing in situ staining for BdnfmRNA in nearly all pyramidal neurons. Bdnf transcripts were localized both in the soma and dendrites, butin neurons of Fmr1-KO mice, the labeling in dendrites was more pronounced. Pilocarpine treatment increased BdnfmRNA labeling in distal dendrites of WT (WT pilo) mice but bothin proximal and distal dendrites of Fmr1-KO (Fmr1-KO pilo) mice. (B) In the CA1 region of the hippocampus, the in situ signal of Bdnf mRNA was maximal in sparse large neuronslocated in the pyramidal layer. BdnfmRNA labeling was increased and more pronounced in the distal dendrites of Fmr1-KO neurons when compared to the labeling in WT controls.Pilocarpine treatment increased BdnfmRNA in distal dendrites of bothWT (WT pilo) and Fmr1-KO (Fmr1-KO pilo) mice.WT(sal) n=5, Fmr1-KO(sal) n=5,WT(pilo) n=6, Fmr1-KO(pilo) n=5. Scale bars, 25 μm in A and 50 μm in B. Maximal distance of dendritic labeling (MDDL), ***p≤0.001 with respect to MDDL of WT.
476 V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Author's personal copy
immunofluorescent-based analysis of maximum projection confocalimages revealed that the number of TrkB.FL-positive cells wassignificantly increased (121±3.3% of control, p=0.00038) in the IZ(Fig. 3C)but the TrkB.FL expressionwas significantly reduced (82±3.5%of control, p=0.00022) in the CxP of the embryonic neocortex in Fmr1-KO mice (Fig. 3C). These data are consistent with our previous studies,which revealed abnormalities in the differentiation and migration ofnewborn cells with impaired FMRP function, particularly in the IZ ofembryonic Fmr1-KO brain (Tervonen et al., 2009) and suggested thatchanges of TrkB expression in NPCs lacking FMRP might contribute tothe aberrances of neocortex formation in Fmr1-KO mice. No significantchanges in the TrkB.FL expression were found in the HC (p = 0.9119),globus pallidus GP (p=0.8341), nor in the in the ldt/lpt (p=0.75049)of Fmr1-KO mice when compared to WT controls (Fig. 3C, D, E, and F)indicating region-specific spatial differences in the alterations of TrkBexpression.
The effects of FMRP deficits on Bdnf mRNA expression in Fmr1-KO mousebrain and in cultured neurons
Since there is evidence that TrkB contributes to the signals thatcontrol the dendritic targeting of Bdnf mRNA (Righi et al., 2000), weexamined whether alterations of the TrkB expression in neuronslacking FMRP were associated with abnormalities of the subcellularexpression of Bdnf mRNA. To this end, subcellular Bdnf mRNAexpression was analyzed in neurons of the cortical layer V and CA1region of the hippocampus of Fmr1-KO and WT mice by in situhybridization. Under basal conditions, Bdnf mRNA was observed innearly all pyramidal neurons of layer V in the visual Cx (Fig. 4A). In thehippocampal CA1 region, the in situ signal was maximal in sparselarge neurons located in the pyramidal layer (Fig. 4B). We observedthat the basal levels of dendritic Bdnf mRNA were significantlyincreased in neurons of the cortical layer V and hippocampal CA1region of Fmr1-KO mice when compared to the levels in neurons ofWT mice (ANOVA, pb0.001; Fig. 4A and B).
Bdnf mRNA is known to be targeted to dendrites in an activity-dependentmanner, both in vitro and in vivo (Tongiorgi, 2008; Tongiorgiet al., 1997). We studied the effects of neuronal activity on thesubcellular Bdnf mRNA expression in neurons lacking FMRP afterneuronal activation through seizures initiated by a muscarine acetyl-choline receptor agonist, pilocarpine. Pilocarpine-induced seizures havepreviously been shown to increase dendritic targeting of Bdnf mRNA(Tongiorgi et al., 2004). In saline-injected control condition, the BdnfmRNA was mainly localized in the soma and proximal dendrites, butafter pilocarpine treatment, the in situ signal was found also in moredistal dendrites (Fig. 4A and B). Pilocarpine treatment enhancedthe expression of Bdnf mRNA in more distal dendrites of both WT andFmr1-KO neurons and augmented the difference between the dendriticBdnfmRNA expression of Fmr1-KO andWT neurons (ANOVA, pb0.001;Fig. 4A and B). The Bdnf mRNA expression was increased in the mostproximal dendrites in cortical neurons but not hippocampal neurons ofpilocarpine-treated Fmr1-KO mice (Fig. 4A and 4B) suggesting regionaldifferences in the regulation of subcellular Bdnf mRNA expression.
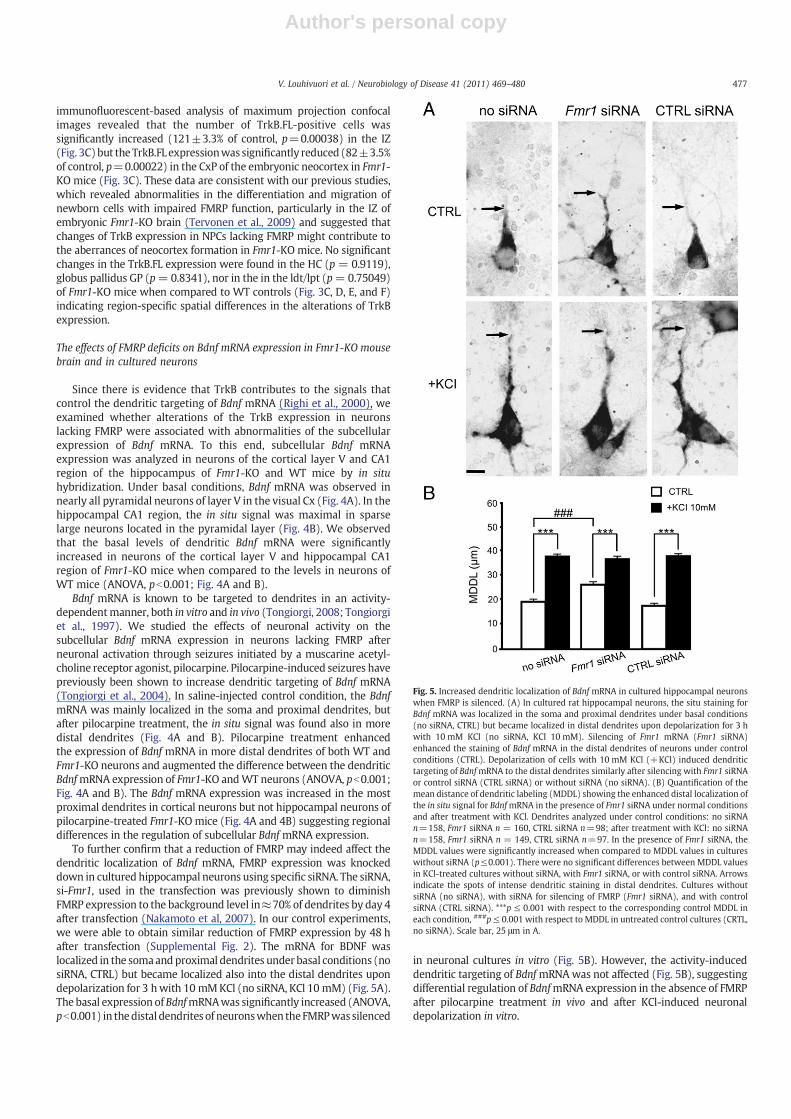
To further confirm that a reduction of FMRP may indeed affect thedendritic localization of Bdnf mRNA, FMRP expression was knockeddown in cultured hippocampal neurons using specific siRNA. The siRNA,si-Fmr1, used in the transfection was previously shown to diminishFMRP expression to the background level in≈70% of dendrites by day 4after transfection (Nakamoto et al, 2007). In our control experiments,we were able to obtain similar reduction of FMRP expression by 48 hafter transfection (Supplemental Fig. 2). The mRNA for BDNF waslocalized in the somaandproximal dendrites under basal conditions (nosiRNA, CTRL) but became localized also into the distal dendrites upondepolarization for 3 hwith 10 mMKCl (no siRNA, KCl 10 mM) (Fig. 5A).The basal expression ofBdnfmRNAwas significantly increased (ANOVA,pb0.001) in thedistal dendrites of neuronswhen theFMRPwassilenced
in neuronal cultures in vitro (Fig. 5B). However, the activity-induceddendritic targeting of BdnfmRNAwas not affected (Fig. 5B), suggestingdifferential regulation of BdnfmRNA expression in the absence of FMRPafter pilocarpine treatment in vivo and after KCl-induced neuronaldepolarization in vitro.
Fig. 5. Increased dendritic localization of Bdnf mRNA in cultured hippocampal neuronswhen FMRP is silenced. (A) In cultured rat hippocampal neurons, the situ staining forBdnf mRNA was localized in the soma and proximal dendrites under basal conditions(no siRNA, CTRL) but became localized in distal dendrites upon depolarization for 3 hwith 10 mM KCl (no siRNA, KCl 10 mM). Silencing of Fmr1 mRNA (Fmr1 siRNA)enhanced the staining of Bdnf mRNA in the distal dendrites of neurons under controlconditions (CTRL). Depolarization of cells with 10 mM KCl (+KCl) induced dendritictargeting of BdnfmRNA to the distal dendrites similarly after silencing with Fmr1 siRNAor control siRNA (CTRL siRNA) or without siRNA (no siRNA). (B) Quantification of themean distance of dendritic labeling (MDDL) showing the enhanced distal localization ofthe in situ signal for BdnfmRNA in the presence of Fmr1 siRNA under normal conditionsand after treatment with KCl. Dendrites analyzed under control conditions: no siRNAn=158, Fmr1 siRNA n = 160, CTRL siRNA n=98; after treatment with KCl: no siRNAn=158, Fmr1 siRNA n = 149, CTRL siRNA n=97. In the presence of Fmr1 siRNA, theMDDL values were significantly increased when compared to MDDL values in cultureswithout siRNA (p≤0.001). There were no significant differences between MDDL valuesin KCl-treated cultures without siRNA, with Fmr1 siRNA, or with control siRNA. Arrowsindicate the spots of intense dendritic staining in distal dendrites. Cultures withoutsiRNA (no siRNA), with siRNA for silencing of FMRP (Fmr1 siRNA), and with controlsiRNA (CTRL siRNA). ***p ≤ 0.001 with respect to the corresponding control MDDL ineach condition, ###p≤ 0.001 with respect to MDDL in untreated control cultures (CRTL,no siRNA). Scale bar, 25 μm in A.
477V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Author's personal copy
BDNF expression in NPCs and in the brain of Fmr1-KO mice
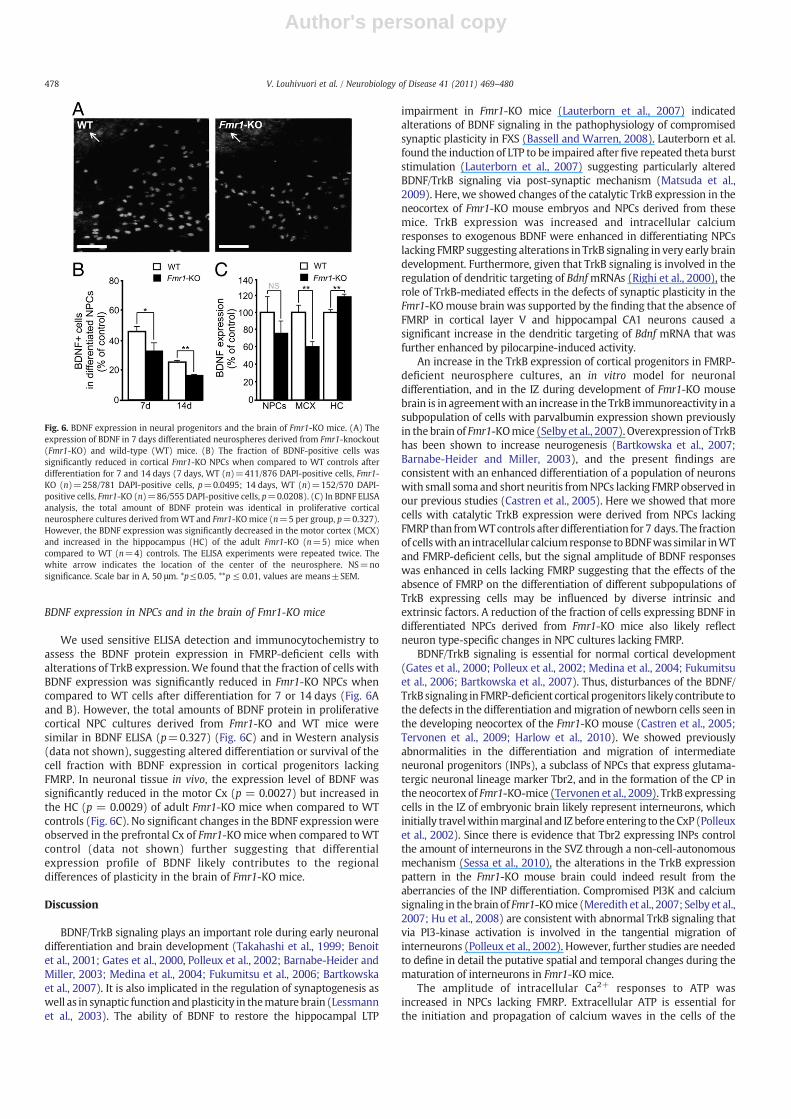
We used sensitive ELISA detection and immunocytochemistry toassess the BDNF protein expression in FMRP-deficient cells withalterations of TrkB expression.We found that the fraction of cells withBDNF expression was significantly reduced in Fmr1-KO NPCs whencompared to WT cells after differentiation for 7 or 14 days (Fig. 6Aand B). However, the total amounts of BDNF protein in proliferativecortical NPC cultures derived from Fmr1-KO and WT mice weresimilar in BDNF ELISA (p=0.327) (Fig. 6C) and in Western analysis(data not shown), suggesting altered differentiation or survival of thecell fraction with BDNF expression in cortical progenitors lackingFMRP. In neuronal tissue in vivo, the expression level of BDNF wassignificantly reduced in the motor Cx (p = 0.0027) but increased inthe HC (p = 0.0029) of adult Fmr1-KO mice when compared to WTcontrols (Fig. 6C). No significant changes in the BDNF expressionwereobserved in the prefrontal Cx of Fmr1-KOmice when compared toWTcontrol (data not shown) further suggesting that differentialexpression profile of BDNF likely contributes to the regionaldifferences of plasticity in the brain of Fmr1-KO mice.
Discussion
BDNF/TrkB signaling plays an important role during early neuronaldifferentiation and brain development (Takahashi et al., 1999; Benoitet al., 2001; Gates et al., 2000, Polleux et al., 2002; Barnabe-Heider andMiller, 2003; Medina et al., 2004; Fukumitsu et al., 2006; Bartkowskaet al., 2007). It is also implicated in the regulation of synaptogenesis aswell as in synaptic function andplasticity in themature brain (Lessmannet al., 2003). The ability of BDNF to restore the hippocampal LTP
impairment in Fmr1-KO mice (Lauterborn et al., 2007) indicatedalterations of BDNF signaling in the pathophysiology of compromisedsynaptic plasticity in FXS (Bassell and Warren, 2008). Lauterborn et al.found the induction of LTP to be impaired afterfive repeated theta burststimulation (Lauterborn et al., 2007) suggesting particularly alteredBDNF/TrkB signaling via post-synaptic mechanism (Matsuda et al.,2009). Here, we showed changes of the catalytic TrkB expression in theneocortex of Fmr1-KO mouse embryos and NPCs derived from thesemice. TrkB expression was increased and intracellular calciumresponses to exogenous BDNF were enhanced in differentiating NPCslacking FMRP suggesting alterations in TrkB signaling in very early braindevelopment. Furthermore, given that TrkB signaling is involved in theregulation of dendritic targeting of BdnfmRNAs (Righi et al., 2000), therole of TrkB-mediated effects in the defects of synaptic plasticity in theFmr1-KOmouse brain was supported by the finding that the absence ofFMRP in cortical layer V and hippocampal CA1 neurons caused asignificant increase in the dendritic targeting of Bdnf mRNA that wasfurther enhanced by pilocarpine-induced activity.
An increase in the TrkB expression of cortical progenitors in FMRP-deficient neurosphere cultures, an in vitro model for neuronaldifferentiation, and in the IZ during development of Fmr1-KO mousebrain is in agreementwith an increase in the TrkB immunoreactivity in asubpopulation of cells with parvalbumin expression shown previouslyin the brain of Fmr1-KOmice (Selby et al., 2007). Overexpressionof TrkBhas been shown to increase neurogenesis (Bartkowska et al., 2007;Barnabe-Heider and Miller, 2003), and the present findings areconsistent with an enhanced differentiation of a population of neuronswith small soma and short neuritis fromNPCs lacking FMRPobserved inour previous studies (Castren et al., 2005). Here we showed that morecells with catalytic TrkB expression were derived from NPCs lackingFMRP than fromWTcontrols after differentiation for 7 days. The fractionof cellswith an intracellular calciumresponse toBDNFwas similar inWTand FMRP-deficient cells, but the signal amplitude of BDNF responseswas enhanced in cells lacking FMRP suggesting that the effects of theabsence of FMRP on the differentiation of different subpopulations ofTrkB expressing cells may be influenced by diverse intrinsic andextrinsic factors. A reduction of the fraction of cells expressing BDNF indifferentiated NPCs derived from Fmr1-KO mice also likely reflectneuron type-specific changes in NPC cultures lacking FMRP.
BDNF/TrkB signaling is essential for normal cortical development(Gates et al., 2000; Polleux et al., 2002; Medina et al., 2004; Fukumitsuet al., 2006; Bartkowska et al., 2007). Thus, disturbances of the BDNF/TrkB signaling in FMRP-deficient cortical progenitors likely contribute tothe defects in the differentiation andmigration of newborn cells seen inthe developing neocortex of the Fmr1-KO mouse (Castren et al., 2005;Tervonen et al., 2009; Harlow et al., 2010). We showed previouslyabnormalities in the differentiation and migration of intermediateneuronal progenitors (INPs), a subclass of NPCs that express glutama-tergic neuronal lineage marker Tbr2, and in the formation of the CP inthe neocortex of Fmr1-KO-mice (Tervonen et al., 2009). TrkB expressingcells in the IZ of embryonic brain likely represent interneurons, whichinitially travelwithinmarginal and IZ before entering to the CxP (Polleuxet al., 2002). Since there is evidence that Tbr2 expressing INPs controlthe amount of interneurons in the SVZ through a non-cell-autonomousmechanism (Sessa et al., 2010), the alterations in the TrkB expressionpattern in the Fmr1-KO mouse brain could indeed result from theaberrancies of the INP differentiation. Compromised PI3K and calciumsignaling in thebrain of Fmr1-KOmice (Meredith et al., 2007; Selby et al.,2007; Hu et al., 2008) are consistent with abnormal TrkB signaling thatvia PI3-kinase activation is involved in the tangential migration ofinterneurons (Polleux et al., 2002). However, further studies are neededto define in detail the putative spatial and temporal changes during thematuration of interneurons in Fmr1-KO mice.
The amplitude of intracellular Ca2+ responses to ATP wasincreased in NPCs lacking FMRP. Extracellular ATP is essential forthe initiation and propagation of calcium waves in the cells of the
Fig. 6. BDNF expression in neural progenitors and the brain of Fmr1-KO mice. (A) Theexpression of BDNF in 7 days differentiated neurospheres derived from Fmr1-knockout(Fmr1-KO) and wild-type (WT) mice. (B) The fraction of BDNF-positive cells wassignificantly reduced in cortical Fmr1-KO NPCs when compared to WT controls afterdifferentiation for 7 and 14 days (7 days, WT (n)=411/876 DAPI-positive cells, Fmr1-KO (n)=258/781 DAPI-positive cells, p=0.0495; 14 days, WT (n)=152/570 DAPI-positive cells, Fmr1-KO (n)=86/555 DAPI-positive cells, p=0.0208). (C) In BDNF ELISAanalysis, the total amount of BDNF protein was identical in proliferative corticalneurosphere cultures derived fromWT and Fmr1-KOmice (n=5 per group, p=0.327).However, the BDNF expression was significantly decreased in the motor cortex (MCX)and increased in the hippocampus (HC) of the adult Fmr1-KO (n=5) mice whencompared to WT (n=4) controls. The ELISA experiments were repeated twice. Thewhite arrow indicates the location of the center of the neurosphere. NS=nosignificance. Scale bar in A, 50 μm. *p≤0.05, **p ≤ 0.01, values are means±SEM.
478 V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480

Author's personal copy
proliferative ventricular zone (Kriegstein and Noctor, 2004; Weiss-man et al., 2004) and for the migration of INPs (Liu et al. 2008), whichdo not lose their ATP responses as postmitotic neurons do. Thus, anincrease in the amplitude of intracellular Ca2+ responses to ATP and inthe fraction of ATP-responsive cells after differentiation for 7 days inNPCs lacking FMRP is in line with a migration defect of an increasednumber of INPs shown previously in the neocortex of Fmr1-KO mice(Tervonen et al., 2009). Moreover, BDNF exposure restored theenhanced amplitude of ATP responses toWT levels in a subpopulationof differentiating NPCs lacking FMRP suggesting that perturbations ofBDNF/TrkB signaling were involved in the enhanced ATP responsesand thereby in the impairedmigration of INPs in the absence of FMRP.
BDNF exerts a positive feedback to synapse function by a processthat requires local protein synthesis in the postsynaptic dendrites(Kang and Schuman, 1996). FMRP is an RNA-binding protein that isimplicated in the localization, translation, and stability of mRNAs inneurons (Bagni and Greenough 2005; Zalfa et al., 2007; Zhang et al.,2007; Bassell andWarren, 2008). Recent studies have demonstrated arole for FMRP in the stimulus-induced transport of mRNAs (Dicten-berg et al., 2008) and the overall motility of mRNA granules (Esteset al., 2008). FMRP has been shown to bind and regulate thetranslation of a subset of dendritic mRNAs (Zalfa et al., 2003, 2007,Antar et al., 2005; Muddashetty et al., 2007; Schutt et al., 2009). BdnfmRNA is targeted to dendrites in an activity-dependent manner(Tongiorgi et al., 2006), and a pro-convulsant agent pilocarpinepromotes a subset of Bdnf transcripts to localize into distal dendrites(Tongiorgi et al., 2006). We showed that dendritic targeting of BdnfmRNA was increased under basal conditions and further enhanced bypilocarpine-induced neuronal activity in cortical layer V and hippo-campal CA1 neurons lacking FMRP. Defects in the regulation of theBDNF expression in the brain of Fmr1-KO mice are in agreement withan impaired LTP in the Cx and HC of Fmr1-KO mice (Li et al., 2002;Zhao et al., 2005; Desai et al., 2006; Meredith et al., 2007). In the HC ofthe Fmr1-KOmice, basal levels of BDNF proteinwere increased as seenafter increased synaptic activity (Lau et al., 2010) and the finding isconsistent with enhanced hippocampal mTOR signaling reported inthe brain of Fmr1-KO mouse (Sharma et al., 2010). However, theincreased Bdnf mRNA targeting to dendrites of the hippocampal CA1neurons was not associated with an accumulation of transcripts in themost proximal segments of the dendrites contrary to the finding incortical layer V neurons, suggesting spatial differences of feedbackmechanisms in the regulation of BDNF expression. The expression ofBDNF protein was also shown to be reduced in the Cx of Fmr1-KOmice. Differential expression of BDNF protein in the HC and Cx likelyindicates regional alterations in BDNF/TrkB feedback signaling forsynaptic strengthening. The findings are in agreement with observeddifferences in spine density and neuronal plasticity between the Cxand HC of Fmr1-KO mice (Braun and Segal, 2000).
The present study demonstrates that alterations of BDNF/TrkBsignaling bring about cell type-specific changes in the differentiationand migration of neuronal cells lacking FMRP and provide molecularmechanism that may account for specific changes in the cellular andsynaptic plasticity resulting in cognitive defects in FXS. We found thatthe absence of FMRP resulted in increased TrkB expression inneuronal progenitors, changes of the BDNF and TrkB expressing cellpopulations during neuronal differentiation, and spatial differences inthe subcellular and regional BDNF expression in the brain of Fmr1-KOmice. The findings suggested aberrant differentiation of GABAergicinhibitory interneurons. Although we found that a fraction of cellswith intracellular calcium responses to NMDA in differentiating cellsderived from Fmr1-KO mice did not differ from WT controls afterdifferentiation for 5 days, our previous studies have identifiedalterations in the differentiation of cells responsive to metabotropicglutamate receptor activation and further studies are needed toexamine the effects of the absence of FMRP on both GABAergic andglutamatergic signaling during neuronal differentiation and synapse
function in different neuronal circuits causing the clinical phenotypeof FXS, including cognitive and sensory defects, behavioral aberrances,as well as susceptibility to epilepsy.
Supplementarymaterials related to this article can be found onlineat doi: 10.1016/j.nbd.2010.10.018.
Acknowledgments
The authors thank Outi Nikkilä, Erja Huttu, and Xuefei Wu forskilful technical assistance. The study was supported by grants fromthe Academy of Finland and University of Helsinki.
Author contributionThe experimental design, implementation of the studies, and
writing are by V.L., M.C, K.Å.; A.V., and E.T. performed BDNF in situ onmice arranged by V.L.; M.U. studied the BDNF expression in NPCs, T.R.and E.C. contributed to Western analyses, and L.L., to calcium studiesand proofreading of the article.
References
Antar, L.N., Dictenberg, J.B., Plociniak, M., Afroz, R., Bassell, G.J., 2005. Localization ofFMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 4, 350–359.
Bagni, C., Greenough, W.T., 2005. From mRNP trafficking to spine dysmorphogenesis:the roots of fragile X syndrome. Nat. Rev. Neurosci. 6, 376–387.
Barnabe-Heider, F., Miller, F.D., 2003. Endogenously produced neurotrophins regulatesurvival and differentiation of cortical progenitors via distinct signaling pathways.J. Neurosci. 23, 5149–5160.
Bartkowska, K., Paquin, A., Gauthier, A.S., Kaplan, D.R., Miller, F.D., 2007. Trk signalingregulates neural precursor cell proliferation and differentiation during corticaldevelopment. Development 134, 4369–4380.
Bassell, G.J., Warren, S.T., 2008. Fragile X syndrome: loss of local mRNA regulation alterssynaptic development and function. Neuron 60, 201–214.
Benoit, B.O., Savarese, T., Joly, M., Engstrom, C.M., Pang, L., Reilly, J., Recht, L.D., Ross, A.H., Quesenberry, P.J., 2001. Neurotrophin channeling of neural progenitor celldifferentiation. J. Neurobiol. 46, 265–280.
Berninger, B., Poo, M., 1996. Fast actions of neurotrophic factors. Curr. Opin. Neurobiol.6, 324–330.
Braun, K., Segal, M., 2000. FMRP involvement in formation of synapses among culturedhippocampal neurons. Cereb. Cortex 10 (10), 1045–1052.
Bureau, I., Shepherd,G.M., Svoboda, K., 2008. Circuit andplasticitydefects in thedevelopingsomatosensory cortex of FMR1 knock-out mice. J. Neurosci. 28, 5178–5188.
Castrén, M., Lampinen, K.E., Miettinen, R., Koponen, E., Sipola, I., Bakker, C.E., Oostra, B.A.,Castrén, E., 2002. BDNF regulates theexpressionof fragile Xmental retardationproteinmRNA in the hippocampus. Neurobiol. Dis. 11, 221–229.
Castrén, M., Tervonen, T., Kärkkäinen, V., Heinonen, S., Castrén, E., Larsson, K., Bakker, C.E.,Oostra, B.A., Åkerman, K., 2005. Altered differentiation of neural stem cells in fragile Xsyndrome. Proc. Natl Acad. Sci. USA 102, 17834–17839.
Ciccolini, F., Collins, T.J., Sudhoelter, J., Lipp, P., Berridge, M.J., Bootman, M.D., 2003. Localand global spontaneous calcium events regulate neurite outgrowth and onset ofGABAergic phenotype during neural precursor differentiation. J. Neurosci. 23,103–111.
Comery, T.A., Harris, J.B., Willems, P.J., Oostra, B.A., Irwin, S.A., Weiler, I.J., Greenough,W.T.,1997. Abnormal dendritic spines in fragile X knockout mice: maturation and pruningdeficits. Proc. Natl Acad. Sci. USA 94, 5401–5404.
Desai, N.S., Casimiro, T.M., Gruber, S.M., Vanderklish, P.W., 2006. Early postnatalplasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745.
Dictenberg, J.B., Swanger, S.A., Antar, L.N., Singer, R.H., Bassell, G.J., 2008. A direct rolefor FMRP in activity-dependent dendritic mRNA transport links filopodial-spinemorphogenesis to fragile X syndrome. Dev. Cell 14, 926–939.
The Dutch-Belgian Fragile X Consortium, 1994. FMR1 knockout mice: a model to studyfragile X mental retardation. Cell 78, 23–33.
Estes, P.S., O'Shea, M., Clasen, S., Zarnescu, D.C., 2008. Fragile X protein controls theefficacy of mRNA transport in Drosophila neurons. Mol. Cell. Neurosci. 39, 170–179.
Fukumitsu, H., Ohtsuka, M., Murai, R., Nakamura, H., Itoh, K., Furukawa, S., 2006. Brain-derived neurotrophic factor participates in determination of neuronal laminar fatein the developing mouse cerebral cortex. J. Neurosci. 26, 13218–13230.
Galvez, R., Greenough, W.T., 2005. Sequence of abnormal dendritic spine developmentin primary somatosensory cortex of a mouse model of the fragile X mentalretardation syndrome. Am. J. Med. Genet. A 135, 155–160.
Garber, K.B., Visootsak, J., Warren, S.T., 2008. Fragile X syndrome. Eur. J. Hum. Genet. 16,666–672.
Gates, M.A., Tai, C.C., Macklis, J.D., 2000. Neocortical neurons lacking the protein-tyrosine kinase B receptor display abnormal differentiation and process elongationin vitro and in vivo. Neuroscience 98, 437–447.
Harlow, E.G., Till, S.M., Russell, T.A., Wijetunge, L.S., Kind, P., Contractor, A., 2010. Criticalperiod plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron65, 385–398.
479V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480

Author's personal copy
Hu, H., Qin, Y., Bochorishvili, G., Zhu, Y., van Aelst, L., Zhu, J.J., 2008. Ras signalingmechanisms underlying impaired GluR1-dependent plasticity associated withfragile X syndrome. J. Neurosci. 28, 7847–7862.
Huang, E.J., Reichardt, L.F., 2001. Neurotrophins: roles in neuronal development andfunction. Annu. Rev. Neurosci. 24, 677–736.
Huber, K.M., Gallagher, S.M., Warren, S.T., Bear, M.F., 2002. Altered synaptic plasticity in amousemodel of fragile Xmental retardation. Proc. Natl Acad. Sci. USA 99, 7746–7750.
Irwin, S.A., Patel, B., Idupulapati, M., Harris, J.B., Crisostomo, R.A., Larsen, B.P., Kooy, F.,Willems, P.J., Cras, P., Kozlowski, P.B., et al., 2001. Abnormal dendritic spinecharacteristics in the temporal and visual cortices of patients with fragile-Xsyndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167.
Jacobowitz, D.M., Abbott, L.C., 1998. Chemoarchitectonic Atlas of the Developing MouseBrain. CRC Press LLC, Boca Raton, USA.
Kang, H., Schuman, E.M., 1996. A requirement for local protein synthesis inneurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406.
Kärkkäinen, V., Louhivuori, V., Castrén, M.L., Åkerman, K.E., 2009. Neurotransmitterresponsiveness during early maturation of neural progenitor cells. Differentiation77, 188–198.
Koekkoek, S.K., Yamaguchi, K., Milojkovic, B.A., Dortland, B.R., Ruigrok, T.J., Maex, R., DeGraaf, W., Smit, A.E., VanderWerf, F., Bakker, C.E., et al., 2005. Deletion of FMR1 inPurkinje cells enhances parallel fiber LTD, enlarges spines, and attenuatescerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352.
Kriegstein, A.R., Noctor, S.C., 2004. Patterns of neuronal migration in the embryoniccortex. Trends Neurosci. 27, 392–399.
Lau, A.G., Irier, H.A., Gu, J., Tian, D., Ku, L., Liu, G., Xia, M., Fritsch, B., Zheng, J.Q.,Dingledine, R., Xu, B., Lu, B., Feng, Y., 2010. Distinct 3′UTRs differentially regulateactivity-dependent translation of brain-derived neurotrophic factor (BDNF). Proc.Natl Acad. Sci. USA 107, 15945–15950.
Lauterborn, J.C., Rex, C.S., Kramar, E., Chen, L.Y., Pandyarajan, V., Lynch, G., Gall, C.M.,2007. Brain-derived neurotrophic factor rescues synaptic plasticity in a mousemodel of fragile X syndrome. J. Neurosci. 27, 10685–10694.
Lessmann, V., Gottmann, K., Malcangio, M., 2003. Neurotrophin secretion: current factsand future prospects. Prog. Neurobiol. 69, 341–374.
Li, J., Pelletier, M.R., Perez Velazquez, J.L., Carlen, P.L., 2002. Reduced cortical synapticplasticity and GluR1 expression associated with fragile X mental retardationprotein deficiency. Mol. Cell. Neurosci. 19, 138–151.
Liu, X., Hashimoto-Torii, K., Torii, M., Haydar, T.F., Rakic, P., 2008. The role of ATPsignaling in the migration of intermediate neuronal progenitors to the neocorticalsubventricular zone. Proc. Natl Acad. Sci. USA 105, 11802–11807.
Louhivuori, V., Arvio, M., Soronen, P., Oksanen, V., Paunio, T., Castrén, M.L., 2009. TheVal66Met polymorphism in the BDNF gene is associated with epilepsy in fragile Xsyndrome. Epilepsy Res. 85, 114–117.
Matsuda, N., Lu, H., Fukata, Y., Noritake, J., Gao, H., Mukherjee, S., Nemoto, T., Fukata, M.,Poo, M.M., 2009. Differential activity-dependent secretion of brain-derivedneurotrophic factor from axon and dendrite. J. Neurosci. 29, 14185–14198.
Mattson, M.P., 2008. Glutamate and neurotrophic factors in neuronal plasticity anddisease. Ann. NY Acad. Sci. 1144, 97–112.
Medina, D.L., Sciarretta, C., Calella, A.M., Von Bohlen Und Halbach, O., Unsicker, K.,Minichiello, L., 2004. TrkB regulates neocortex formation through the Shc/PLCgamma-mediated control of neuronal migration. EMBO J. 23, 3803–3814.
Meredith, R.M., Holmgren, C.D., Weidum, M., Burnashev, N., Mansvelder, H.D., 2007.Increased threshold for spike-timing-dependent plasticity is caused by unreliablecalcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–638.
Muddashetty, R.S., Kelic, S., Gross, C., Xu, M., Bassell, G.J., 2007. Dysregulatedmetabotropic glutamate receptor-dependent translation of AMPA receptor andpostsynaptic density-95 mRNAs at synapses in a mouse model of fragile Xsyndrome. J. Neurosci. 27, 5338–5348.
Nakamoto, M., Nalavadi, V., Epstein, M.P., Narayanan, U., Bassell, G.J., Warren, S.T., 2007.Fragile Xmental retardation protein deficiency leads to excessivemGluR5-dependentinternalization of AMPA receptors. Proc. Natl Acad. Sci. USA 104, 15537–15542.
Polleux, F., Whitford, K.L., Dijkhuizen, P.A., Vitalis, T., Ghosh, A., 2002. Control of corticalinterneuron migration by neurotrophins and PI3-kinase signaling. Development129, 3147–3160.
Rudelli, R.D., Brown, W.T., Wisniewski, K., Jenkins, E.C., Laure-Kamionowska, M.,Connell, F., Wisniewski, H.M., 1985. Adult fragile X syndrome. Clinico-neuropath-ologic findings. Acta Neuropathol. 67, 289–295.
Righi, M., Tongiorgi, E., Cattaneo, A., 2000. Brain-derived neurotrophic factor (BDNF)induces dendritic targeting of BDNF and tyrosine kinase B mRNAs in hippocampalneurons through a phosphatidylinositol-3 kinase-dependent pathway. J. Neurosci.20, 3165–3174.
Schutt, J., Falley, K., Richter, D., Kreienkamp, H.J., Kindler, S., 2009. Fragile X mentalretardation protein regulates the levels of scaffold proteins and glutamatereceptors in postsynaptic densities. J. Biol. Chem. 284, 25479–25487.
Selby, L., Zhang, C., Sun, Q.Q., 2007.Major defects inneocortical GABAergic inhibitory circuitsin mice lacking the fragile X mental retardation protein. Neurosci. Lett. 412, 227–232.
Sessa, A., Mao, C.A., Colasante, G., Nini, A., Klein, W.H., Broccoli, V., 2010. Tbr2-positiveintermediate (basal) neuronal progenitors safeguard cerebral cortex expansion bycontrolling amplification of pallial glutamatergic neurons and attraction ofsubpallial GABAergic interneurons. Genes Dev. 24, 1816–1826.
Sharma, A., Hoeffer, C.A., Takayasu, Y., Miyawaki, T., McBride, S.M., Klann, E., Zukin, R.S.,2010.Dysregulation ofmTOR signaling in fragile X syndrome. J.Neurosci. 30, 694–702.
Takahashi, J., Palmer, T.D., Gage, F.H., 1999. Retinoic acid and neurotrophins collaborate toregulateneurogenesis inadult-derivedneural stemcell cultures. J. Neurobiol. 38, 65–81.
Tervonen, T.A., Ajamian, F., De Wit, J., Verhaagen, J., Castrén, E., Castrén, M., 2006.Overexpression of a truncated TrkB isoform increases the proliferation of neuralprogenitors. Eur. J. Neurosci. 24, 1277–1285.
Tervonen, T.A., Louhivuori, V., Sun, X., Hokkanen, M.E., Kratochwil, C.F., Zebryk, P.,Castrén, E., Castrén, M.L., 2009. Aberrant differentiation of glutamatergic cells inneocortex of mouse model for fragile X syndrome. Neurobiol. Dis. 33, 250–259.
Tongiorgi, E., 2008. Activity-dependent expression of brain-derived neurotrophic factorin dendrites: facts and open questions. Neurosci. Res. 61, 335–346.
Tongiorgi, E., Armellin, M., Giulianini, P.G., Bregola, G., Zucchini, S., Paradiso, B., Steward,O., Cattaneo, A., Simonato, M., 2004. Brain-derived neurotrophic factor mRNA andprotein are targeted to discrete dendritic laminas by events that triggerepileptogenesis. J. Neurosci. 24, 6842–6852.
Tongiorgi, E., Domenici, L., Simonato, M., 2006. What is the biological significance ofBDNF mRNA targeting in the dendrites? Clues from epilepsy and corticaldevelopment. Mol. Neurobiol. 33, 17–32.
Tongiorgi, E., Righi, M., Cattaneo, A., 1997. Activity-dependent dendritic targeting ofBDNF and TrkB mRNAs in hippocampal neurons. J. Neurosci. 17, 9492–9505.
Turski, W.A., Cavalheiro, E.A., Schwarz, M., Czuczwar, S.J., Kleinrok, Z., Turski, L., 1983.Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalo-graphic and neuropathological study. Behav. Brain Res. 3, 315–335.
Weissman, T.A., Riquelme, P.A., Ivic, L., Flint, A.C., Kriegstein, A.R., 2004. Calcium wavespropagate through radial glial cells and modulate proliferation in the developingneocortex. Neuron 43, 647–661.
Volk, L.J., Daly, C.A., Huber, K.M., 2006. Differential roles for group 1 mGluR subtypes ininduction and expression of chemically induced hippocampal long-term depres-sion. J. Neurophysiol. 95, 2427–2438.
Volk, L.J., Pfeiffer, B.E., Gibson, J.R., Huber, K.M., 2007. Multiple Gq-coupled receptorsconverge on a common protein synthesis-dependent long-term depression that isaffected in fragile X syndrome mental retardation. J. Neurosci. 27, 11624–11634.
Zalfa, F., Eleuteri, B., Dickson, K.S., Mercaldo, V., De Rubeis, S., di Penta, A., Tabolacci, E.,Chiurazzi, P., Neri, G., Grant, S.G., et al., 2007. A new function for the fragile X mentalretardation protein in regulation of PSD-95mRNA stability. Nat. Neurosci. 10, 578–587.
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., Oostra, B., Bagni, C.,2003. The fragile X syndrome protein FMRP associates with BC1 RNA and regulatesthe translation of specific mRNAs at synapses. Cell 112, 317–327.
Zhang, M., Wang, Q., Huang, Y., 2007. Fragile X mental retardation protein FMRP andthe RNA export factor NXF2 associate with and destabilize Nxf1 mRNA in neuronalcells. Proc. Natl Acad. Sci. USA 104, 10057–10062.
Zhang, X., Poo, M.M., 2002. Localized synaptic potentiation by BDNF requires localprotein synthesis in the developing axon. Neuron 36, 675–688.
Zhao, M.G., Toyoda, H., Ko, S.W., Ding, H.K., Wu, L.J., Zhuo, M., 2005. Deficits in trace fearmemory and long-term potentiation in a mouse model for fragile X syndrome.J. Neurosci. 25, 7385–7392.
480 V. Louhivuori et al. / Neurobiology of Disease 41 (2011) 469–480
Related Documents











