The FASEB Journal • Research Communication Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion Oriol Nicolas,* ,† Rosalina Gavı ´n,* ,† Nathalie Braun, ‡ Jesu ´ s Mariano Uren ˜a, † Xavier Fontana,* Eduardo Soriano, † Adriano Aguzzi, ‡ and Jose ´ Antonio del Rı ´o* ,†,1 *Cellular and Molecular Basis of Neurodegeneration and Neurorepair, Department of Cell Biology, University of Barcelona, Barcelona, Spain; † Developmental Neurobiology and Regeneration, Institute for Research in Biomedicine (IRB Barcelona), Department of Cell Biology, University of Barcelona, Barcelona, Spain; and ‡ Institute of Neuropathology, University Hospital of Zu ¨ rich, Zurich, Switzerland ABSTRACT Prnp knockout mice that overexpress an amino-truncated form of PrPc (PrP) are ataxic and display cerebellar cell loss and premature death. Stud- ies on the molecular and intracellular events that trig- ger cell death in these mutants may contribute to elucidate the functions of PrPc and to the design of treatments for prion disease. Here we examined the effects of Bcl-2 overexpression in neurons on the development of the neurological syndrome and cere- bellar pathology of PrP. We show that PrP overex- pression activates the stress-associated kinases ERK1–2 in reactive astroglia, p38 and the phosphorylation of p53, which leads to the death of cerebellar neurons in mutant mice. We found that the expression of PrP in cell lines expressing very low levels of PrPc strongly induces the activation of apoptotic pathways, thereby leading to caspase-3 activation and cell death, which can be prevented by coexpressing Bcl-2. Finally, we corrob- orate in vivo that neuronal-directed Bcl-2 overexpres- sion in PrP mice (PrP Bcl-2) markedly reduces caspase-3 activation, glial activation, and neuronal cell death in cerebellum by improving locomotor deficits and life expectancy.—Nicolas, O., Gavı ´n, R., Braun, N., Uren ˜a, J. M., Fontana, X., Soriano, E., Aguzzi, A., del Rı ´o,. J. A. Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion- deficient mice that overexpress amino-terminally trun- cated prion. FASEB J. 21, 3107–3117 (2007) Key Words: cerebellar syndrome granule cells oxidative damage prion protein PrPc is a glycosylphosphatidylinositol (GPI)-an- chored protein expressed by neurons and glial cells. It contains an N-terminal octarepeat region (OR), a cen- tral domain (CD) with a charge cluster (CC) and a hydrophobic domain (HR), two glycans, a disulfide bond, and a C-terminal GPI-linked region (1). Al- though many studies have examined the putative roles of PrPc in the CNS, a full repertory of PrPc functions remains to be determined. PrPc is involved in copper metabolism, and cell homeostasis and is considered a neuroprotective molecule (2). Particular attention has been devoted to determine the functions of the OR and HR domains of PrPc. For example, the OR participates in copper binding (3) and its modification increases susceptibility to oxidative attack (e.g., 4). The study of numerous PrPc transgenic mice has provided a relevant advance toward the identification of PrPc functions (5). Several Prnp o/o mouse lines exhibit ataxia and Purkinje cell (PC) degeneration as a result of the overexpression of PrP-like Doppel (Dpl; see ref 5 for review). In addition, the transgenic expression of amino truncated PrPc (e.g., F35), lacking most of the OR and HR in Prnp o/o mice (termed PrP in the present study), in- duces severe ataxia because of extensive cerebellar granule cell (GC) death (6 – 8). In both PrP- and Dpl-transgenic mice, neurodegenerative effects are largely counteracted by Prnp expression. This observa- tion indicates that PrP and Dpl interfere with normal PrPc functions (9); however, they also suggest that the neuroprotective properties of PrPc may reside in the N-terminal region. Recent studies in Prn o/o cells dem- onstrate that Dpl overexpression becomes neurotoxic by increasing oxidative damage (e.g., 10) and that the overexpression of a C-terminal fragment of the PrPc (C1 fragment) sensitizes the cell to caspase-3 activation (11). However, no data are available on PrP-mediated neurotoxicity. The Bcl-2 protein is crucial for the regulation of cell death since it interacts with the proapoptotic protein Bax to prevent mitochondrial permeability and release of proapoptotic molecules (12). Bax-mediated apopto- 1 Correspondence: Department of Cell Biology, Institute for Research in Biomedicine, University of Barcelona, Josep Samitier 1–5, E-08028 Barcelona, Spain. E-mail: [email protected], [email protected] doi: 10.1096/fj.06-7827com 3107 0892-6638/07/0021-3107 © FASEB

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The FASEB Journal • Research Communication
Bcl-2 overexpression delays caspase-3 activation andrescues cerebellar degeneration in prion-deficientmice that overexpress amino-terminallytruncated prion
Oriol Nicolas,*,† Rosalina Gavın,*,† Nathalie Braun,‡ Jesus Mariano Urena,†
Xavier Fontana,* Eduardo Soriano,† Adriano Aguzzi,‡ and Jose Antonio del Rıo*,†,1
*Cellular and Molecular Basis of Neurodegeneration and Neurorepair, Department of Cell Biology,University of Barcelona, Barcelona, Spain; †Developmental Neurobiology and Regeneration, Institutefor Research in Biomedicine (IRB Barcelona), Department of Cell Biology, University of Barcelona,Barcelona, Spain; and ‡Institute of Neuropathology, University Hospital of Zurich, Zurich, Switzerland
ABSTRACT Prnp knockout mice that overexpress anamino-truncated form of PrPc (�PrP) are ataxic anddisplay cerebellar cell loss and premature death. Stud-ies on the molecular and intracellular events that trig-ger cell death in these mutants may contribute toelucidate the functions of PrPc and to the design oftreatments for prion disease. Here we examined theeffects of Bcl-2 overexpression in neurons on thedevelopment of the neurological syndrome and cere-bellar pathology of �PrP. We show that �PrP overex-pression activates the stress-associated kinases ERK1–2in reactive astroglia, p38 and the phosphorylation ofp53, which leads to the death of cerebellar neurons inmutant mice. We found that the expression of �PrP incell lines expressing very low levels of PrPc stronglyinduces the activation of apoptotic pathways, therebyleading to caspase-3 activation and cell death, which canbe prevented by coexpressing Bcl-2. Finally, we corrob-orate in vivo that neuronal-directed Bcl-2 overexpres-sion in �PrP mice (�PrP Bcl-2) markedly reducescaspase-3 activation, glial activation, and neuronal celldeath in cerebellum by improving locomotor deficitsand life expectancy.—Nicolas, O., Gavın, R., Braun,N., Urena, J. M., Fontana, X., Soriano, E., Aguzzi, A.,del Rıo,. J. A. Bcl-2 overexpression delays caspase-3activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally trun-cated prion. FASEB J. 21, 3107–3117 (2007)
Key Words: cerebellar syndrome � granule cells � oxidativedamage � prion protein
PrPc is a glycosylphosphatidylinositol (GPI)-an-chored protein expressed by neurons and glial cells. Itcontains an N-terminal octarepeat region (OR), a cen-tral domain (CD) with a charge cluster (CC) and ahydrophobic domain (HR), two glycans, a disulfidebond, and a C-terminal GPI-linked region (1). Al-though many studies have examined the putative roles
of PrPc in the CNS, a full repertory of PrPc functionsremains to be determined. PrPc is involved in coppermetabolism, and cell homeostasis and is considered aneuroprotective molecule (2). Particular attention hasbeen devoted to determine the functions of the OR andHR domains of PrPc. For example, the OR participatesin copper binding (3) and its modification increasessusceptibility to oxidative attack (e.g., 4). The study ofnumerous PrPc transgenic mice has provided a relevantadvance toward the identification of PrPc functions (5).Several Prnpo/o mouse lines exhibit ataxia and Purkinjecell (PC) degeneration as a result of the overexpressionof PrP-like Doppel (Dpl; see ref 5 for review). Inaddition, the transgenic expression of amino truncatedPrPc (e.g., �F35), lacking most of the OR and HR inPrnpo/o mice (termed �PrP in the present study), in-duces severe ataxia because of extensive cerebellargranule cell (GC) death (6–8). In both �PrP- andDpl-transgenic mice, neurodegenerative effects arelargely counteracted by Prnp expression. This observa-tion indicates that �PrP and Dpl interfere with normalPrPc functions (9); however, they also suggest that theneuroprotective properties of PrPc may reside in theN-terminal region. Recent studies in Prno/o cells dem-onstrate that Dpl overexpression becomes neurotoxicby increasing oxidative damage (e.g., 10) and that theoverexpression of a C-terminal fragment of the PrPc(C1 fragment) sensitizes the cell to caspase-3 activation(11). However, no data are available on �PrP-mediatedneurotoxicity.
The Bcl-2 protein is crucial for the regulation of celldeath since it interacts with the proapoptotic proteinBax to prevent mitochondrial permeability and releaseof proapoptotic molecules (12). Bax-mediated apopto-
1 Correspondence: Department of Cell Biology, Institutefor Research in Biomedicine, University of Barcelona,Josep Samitier 1–5, E-08028 Barcelona, Spain. E-mail:[email protected], [email protected]
doi: 10.1096/fj.06-7827com
31070892-6638/07/0021-3107 © FASEB

sis can be triggered by oxidative stress, while Baxsynthesis is activated by the tumor suppressor proteinp53 (13). The hypothesis of the present study is thattight regulation of Bcl-2/Bax functions suppresses neu-rodegeneration induced by �F35 overexpression invitro and in vivo. We tested this hypothesis by analyzingthe effects of Bcl-2 overexpression on �F35-transfectedcells and on the onset of the neurological syndromeand cerebellar pathology in �PrP mice by generating atransgenic mouse that also overexpresses Bcl-2 underthe neuronal specific enolase (NSE)-promoter (�PrPBcl-2). In contrast to �PrP animals, in most �PrP Bcl-2mice, neuronal-directed Bcl-2 overexpression reducedp53 activation, caspase-3 activation, and cerebellar de-generation. These alterations correlated with clear im-provements in gait and behavior deficits in these mu-tant mice.
MATERIALS AND METHODS
Antibodies
The Bcl-2 antibody was obtained from Santa Cruz Biotechnol-ogy (Santa Cruz, CA, USA). p53 Phospho-serine-15, ERKphospho-threonine 202/phospho-tyrosine 204 and p38 phos-pho-threonine-180/phospho-tyrosine-182 were from Cell Sig-naling Technology (Beverly, MA, USA). Total ERK and p38antibodies were from Transduction Laboratories (Lexington,KY, USA). Total p53 was from Novocastra (Newcastle, UK),and monoclonal antibodies against actin and GFAP werefrom Chemicon (Temecula, CA, USA). The Calbindin anti-body was purchased from Swant antibodies (Bellinzona, Swit-zerland). The antibody against PrPc (6H4) was from Prionics(Schlieren, Switzerland). The antibody dilutions used inWestern blotting (WB) and immunocytochemistry (ICC)procedures were as follows: Bcl-2 (1:500 for WB), p53 phos-pho-serine-15 (1:2000 for WB), p53 (1:1000 for WB) ERKphospho-threonine 202/phospho-tyrosine 204 (1:1000 forWB, 1:150 for ICC), ERK and p38 (1:2500 for WB), Calbindin(1:2000 for ICC), p38 phospho-threonine-180/phospho-ty-rosine-182 (1:1000 for WB, 1:150 for ICC), actin (1:10000 forWB), GFAP (1:100 for ICC), and 6H4 (1:5000 for WB and1:100 for ICC).
Animals
Prnpo/o (Zurich I) and Tg20/Tga20 mice were purchasedfrom EMMA (Monterotondo, Italy). Prnpo/o mice overexpress-ing N-terminal truncated (lacking aa32–134) PrPc protein(called �PrP mice in our study) were generated previously(6). NSEa-Bcl-2 transgenic mice were a gift from M. Barbacid(CNIO, Madrid, Spain) and I. Silos-Santiago (The ScrippsResearch Institute, La Jolla, CA, USA). All experimentalprocedures were performed in accordance with the guide-lines of the Spanish Ministry of Science and Technology,following European Standards.
Breeding and PCR genotyping of mice
Wild-type and neomycin-targeted Prnp loci and the �F35transgene were detected as described previously (6, 14). Bcl-2transgene detection under Neuronal Specific Enolase (NSE)promoter was performed as described (15). NSEa-Bcl-2 micepresent many Bcl-2 copies in several genome alleles (13).
Prnpo/o �F35�/o mice overexpressing Bcl-2 (called �PrPBcl-2,see below) were generated by mating Prnpo/o with NSEa-Bcl-2mice to obtain Prnp�/0 Bcl-2 F1 offspring. Finally, these micewere crossed with Prnp�/0 �F35�/o to obtain Prnpo/o �F35�/o
Bcl-2, termed �PrP Bcl-2 in this study. To ensure that Bcl-2overexpression was maintained during breeding, we per-formed RT-PCR and WB techniques, which revealed that Bcl-2expression and protein levels in �PrP Bcl-2 mice were higherthan in wild-type, Prnpo/o and �PrP mice (not shown, see Fig.4G for WB). A total of 136 animals were used to produce thetransgenic mice. �PrP and �PrP Bcl-2 mice were examinedevery 2 to 5 days for gait deficit.
Histological methods
Mice were perfused with 0.1 M PBS buffered paraformalde-hyde pH 7.3 and cryoprotected in 30% sucrose. Coronalsections (30 �m thick) were obtained in a freezing mi-crotome. Sections from individual animals were processed inparallel. Free-floating sections were rinsed in 0.1 M PBS, andendogenous peroxidases were blocked in 3% H2O2 and 10%methanol dissolved in 0.1 M PBS. After being rinsed, sectionswere incubated in 0.1 M PBS containing 0.2% gelatin, 10%normal serum, 0.2% glycine, and 0.5% Triton-X 100 for 1 h atroom temperature. Sections were then incubated overnight at4°C with the different primary antibodies. Tissue-boundprimary antibody was detected by the ABC method, followingthe manufacturer’s instructions (Vector Labs, Burlingame,CA, USA), and revealed with 0.03% diaminobenzidine and0.003% H2O2. Revealed sections were mounted onto gelati-nized slides and coverslipped with Eukitt (Merck, Darmstadt,Germany). Selected sections were processed for double im-munofluorescence (e.g., phospho-ERK1–2 � GFAP) usingAlexa-Fluor 488- and Alexa-Fluor 568-tagged secondary anti-bodies (Molecular Probes, Eugene, OR, USA). Sections weremounted on Fluoromount (Vector Labs) and analyzed on anOlympus Fluoview SV 500 confocal microscope. All imageswere obtained in sequential scanning laser mode to avoidfluorochrome cross-excitation.
Cell culture transfection and propidium iodide labeling
EBNA-293T cells were cultured on 12 mm Ø coverslips in a6-well plate (Nunc, Denmark) to obtain 50–60% confluencebefore transfection using Lipofectamine Plus (Invitrogen,Carlsbad, CA, USA). Transfection was performed with �F35-,PrPc-, and Bcl-2-pcDNA3-encoding plasmids following themanufacturer’s instructions. Two days after transfection, cellswere incubated with propidium iodide (PI, 1 �g/ml, Sigma-Aldrich, Andover, UK) or collected for caspase-3 assay (seebelow) or WB techniques. PI-treated cultures were rinsed inPBS 0.1 M, fixed with 2% buffered paraformaldehyde, coun-terstained with Bisbenzimide, mounted on Fluoromount, andanalyzed on a Nikon Eclipse Te200 inverted microscope. Forquantification, the percentage of PI-labeled cells with respectto bisbenzimide labeled cells was determined using theQuantity one Image Software Analysis (Bio-Rad, Hercules,CA, USA).
WB techniques and measurement of caspase-3 activity
Tissue samples from cerebella were homogenized (10% w/v)in RIPA buffer containing protease and phosphatase inhibi-tors, using a motor-driven glass-Teflon homogenizer in ice.Transfected EBNA-293T cells were collected using 150 �l ofRIPA buffer per well. After protein quantification, tissue andcell extracts (20 �g) were boiled in Laemmli sample buffer at100°C for 10 min, followed by 12% SDS-PAGE electrophore-
3108 Vol. 21 October 2007 NICOLAS ET AL.The FASEB Journal

sis. They were then electrotransferred to nitrocellulose mem-branes and processed for immunoblotting using primaryantibodies and the ECL-plus kit (Amersham-Pharmacia Bio-tech, Buckinghamshire, UK). In our experiments, each nitro-cellulose membrane was used for detecting both phosphory-lated and total kinase or protein levels. For quantification,revealed films were scanned at 1200 � 1200 dpi (HP 5530photo scanner) and the densitometric analysis was performedusing the Quantity one Image Software Analysis (Bio-Rad).The caspase-3 activity assay was performed using Ac-DEVD-AFC (Sigma-Aldrich) as substrate (see (16) for details).
RESULTS
Sequential GC degeneration, reactive gliosis, and PCdeath in the cerebellum of �PrP mice
�PrP mice have a life expectancy around of 90–95 days(6, 7). The first signs of locomotor deficits, ascertainedas reduced exploratory activities and slight staggeringgait (score �), were observed around 35–40 days of age(Table 1). In the following days, these mice displayedemerging ataxia with increased trembling gait andprogressive paralysis in hind legs (scores �� and���, Table 1 and see Supplemental Video 2 for gaitsymptoms of score ��). At three months of age, thedeteriorated condition of the mice precluded furthermaintenance and survival since they were unable tostand alone and displayed profound paralysis in hindlegs (score ����, Table 1 and Supplemental Video3). Correlation of gait deficits with the histologicalanalysis of the cerebellum showed that the first signs ofGC pyknosis was also detected in the anterior cerebellarfolia (vermis region) of affected mice around 35–40days (not shown). Degeneration progressed with
emerging disorganization of cerebellar lamination asthe result of a reduction in GC cells, numerous macro-phage inclusions, and relevant vacuolization. Theseeffects extended in a rostro-caudal gradient, first affect-ing rostral then posterior cerebellar folia (Fig. 1).Staining using Calbindin or GFAP antibodies showedrelevant PC disorganization and death, accompaniedby general gliosis (Fig. 1G-I). At �90 days of age,cerebellar cytoarchitecture was lost in �PrP mice andthe shrinkage of the cerebellar layers was striking (Fig.1C-F).
Activation of stress-associated kinases (ERK1–2 andp38), p53 phosphorylation, and caspase-3 activity in�PrP cerebellum
In postmitotic neurons, p53 may mediate cell death bycaspase activation under conditions of oxidative stressor DNA damage (13). p53 Activity can be induced,among other factors, by p38 through serine phosphor-ylation. p38 Activity also down-regulates murine doubleminute-2 (MDM2) protein, which has key roles inphospho-p53 degradation (17). Thus, we examined theactivation of ERK1–2 and p38 and levels of phospho-p53 in �PrP cerebellum at several stages of neurode-generation (Fig. 2; see also Fig. 4H-I). First, our resultscorroborated previous studies illustrating endogenousactivation of ERK1–2 in Prnp0/0 mice compared tocontrols (18) and decreased levels of total p53 in theformer compared to the latter (18, 19; Fig. 2A). More-over, WB analysis of cerebellar protein extracts andfurther quantification of revealed films showed a strongincrease in activated ERK1–2, p38, and phospho-p53 in�PrP compared to Prnp�/� and Prnp0/0 mice (Fig.
TABLE 1. Description of phenotypic alterations in �PrP and �PrPBcl-2 mice
Mouse code GenotypeOnset of Locomotor
SymptomsLocomotor Symptoms
at Death
14 Prnpo/o �PrP 36 days 99 days (����)10nn Prnpo/o �PrP 34 days 76 days (���)15 Prnpo/o �PrP 34 days 76 days (���)321r Prnpo/o �PrP 32 days 46 (��)33nn Prnpo/o �PrP 34 days 46 (��)231nn Prnpo/o �PrP 33 days 34 (�)36nn Prnpo/o �PrP 33 days 34 (�)166 Prnpo/o �PrP Bcl-2 55 days 108 (��)169 Prnpo/o �PrP Bcl-2 69 days 111 ( �)154 Prnpo/o �PrP Bcl-2 75 days 131 (�)133 Prnpo/o �PrP Bcl-2 69 days 76 (�)217 Prnpo/o �PrP Bcl-2 80 days 85 (�)128 Prnpo/o �PrP Bcl-2 70 days 78 (�)109 Prnpo/o �PrP Bcl-2 64 days 76 (�)117 Prnpo/o �PrP Bcl-2 none 50 (�)233 Prnpo/o �PrP Bcl-2 none 55 (�)152 Prnpo/o �PrP Bcl-2 none 96 (�)186 Prnpo/o �PrP Bcl-2 none 96 (�)176 Prnpo/o �PrP Bcl-2 none 111 (�)172 Prnpo/o �PrP Bcl-2 none 111 (�)225 Prnpo/o �PrP Bcl-2 none 214 (�)
Onset of gait alterations and behavioural stages and time of death are shown in each case.
3109ANTIAPOPTOTIC PROTEINS IN �PrP MICE
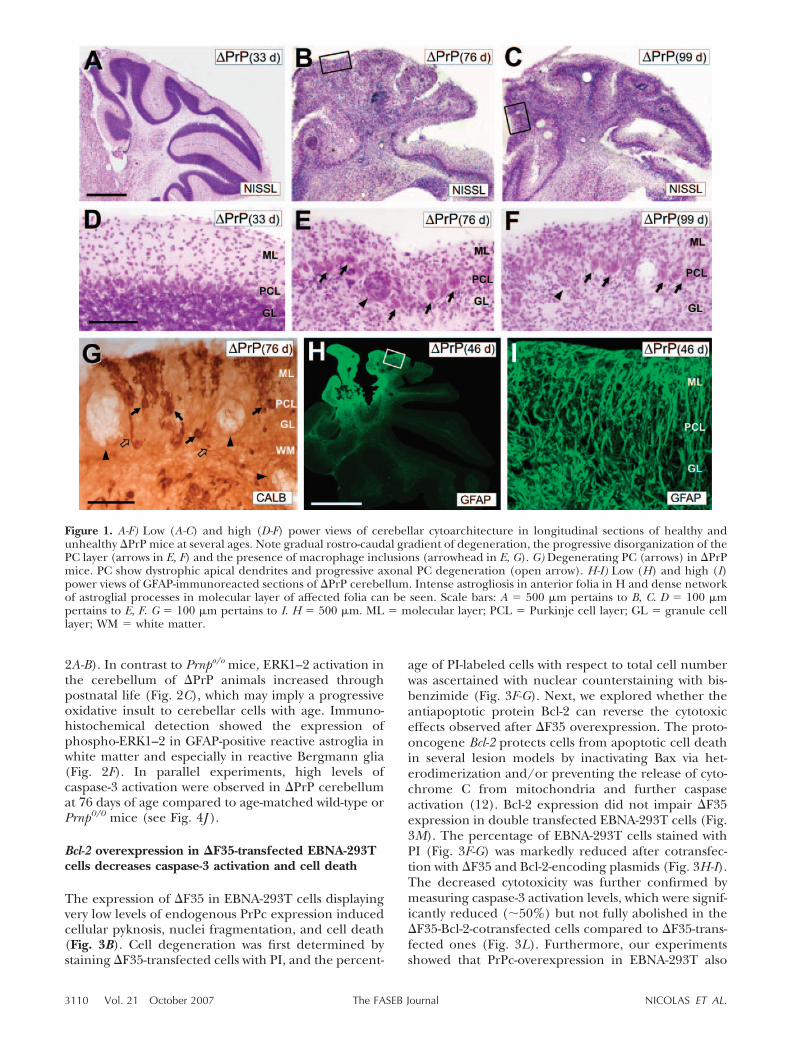
2A-B). In contrast to Prnpo/o mice, ERK1–2 activation inthe cerebellum of �PrP animals increased throughpostnatal life (Fig. 2C), which may imply a progressiveoxidative insult to cerebellar cells with age. Immuno-histochemical detection showed the expression ofphospho-ERK1–2 in GFAP-positive reactive astroglia inwhite matter and especially in reactive Bergmann glia(Fig. 2F). In parallel experiments, high levels ofcaspase-3 activation were observed in �PrP cerebellumat 76 days of age compared to age-matched wild-type orPrnp0/0 mice (see Fig. 4J ).
Bcl-2 overexpression in �F35-transfected EBNA-293Tcells decreases caspase-3 activation and cell death
The expression of �F35 in EBNA-293T cells displayingvery low levels of endogenous PrPc expression inducedcellular pyknosis, nuclei fragmentation, and cell death(Fig. 3B). Cell degeneration was first determined bystaining �F35-transfected cells with PI, and the percent-
age of PI-labeled cells with respect to total cell numberwas ascertained with nuclear counterstaining with bis-benzimide (Fig. 3F-G). Next, we explored whether theantiapoptotic protein Bcl-2 can reverse the cytotoxiceffects observed after �F35 overexpression. The proto-oncogene Bcl-2 protects cells from apoptotic cell deathin several lesion models by inactivating Bax via het-erodimerization and/or preventing the release of cyto-chrome C from mitochondria and further caspaseactivation (12). Bcl-2 expression did not impair �F35expression in double transfected EBNA-293T cells (Fig.3M). The percentage of EBNA-293T cells stained withPI (Fig. 3F-G) was markedly reduced after cotransfec-tion with �F35 and Bcl-2-encoding plasmids (Fig. 3H-I).The decreased cytotoxicity was further confirmed bymeasuring caspase-3 activation levels, which were signif-icantly reduced (�50%) but not fully abolished in the�F35-Bcl-2-cotransfected cells compared to �F35-trans-fected ones (Fig. 3L). Furthermore, our experimentsshowed that PrPc-overexpression in EBNA-293T also
Figure 1. A-F) Low (A-C) and high (D-F) power views of cerebellar cytoarchitecture in longitudinal sections of healthy andunhealthy �PrP mice at several ages. Note gradual rostro-caudal gradient of degeneration, the progressive disorganization of thePC layer (arrows in E, F) and the presence of macrophage inclusions (arrowhead in E, G). G) Degenerating PC (arrows) in �PrPmice. PC show dystrophic apical dendrites and progressive axonal PC degeneration (open arrow). H-I) Low (H) and high (I)power views of GFAP-immunoreacted sections of �PrP cerebellum. Intense astrogliosis in anterior folia in H and dense networkof astroglial processes in molecular layer of affected folia can be seen. Scale bars: A � 500 �m pertains to B, C. D � 100 �mpertains to E, F. G � 100 �m pertains to I. H � 500 �m. ML � molecular layer; PCL � Purkinje cell layer; GL � granule celllayer; WM � white matter.
3110 Vol. 21 October 2007 NICOLAS ET AL.The FASEB Journal
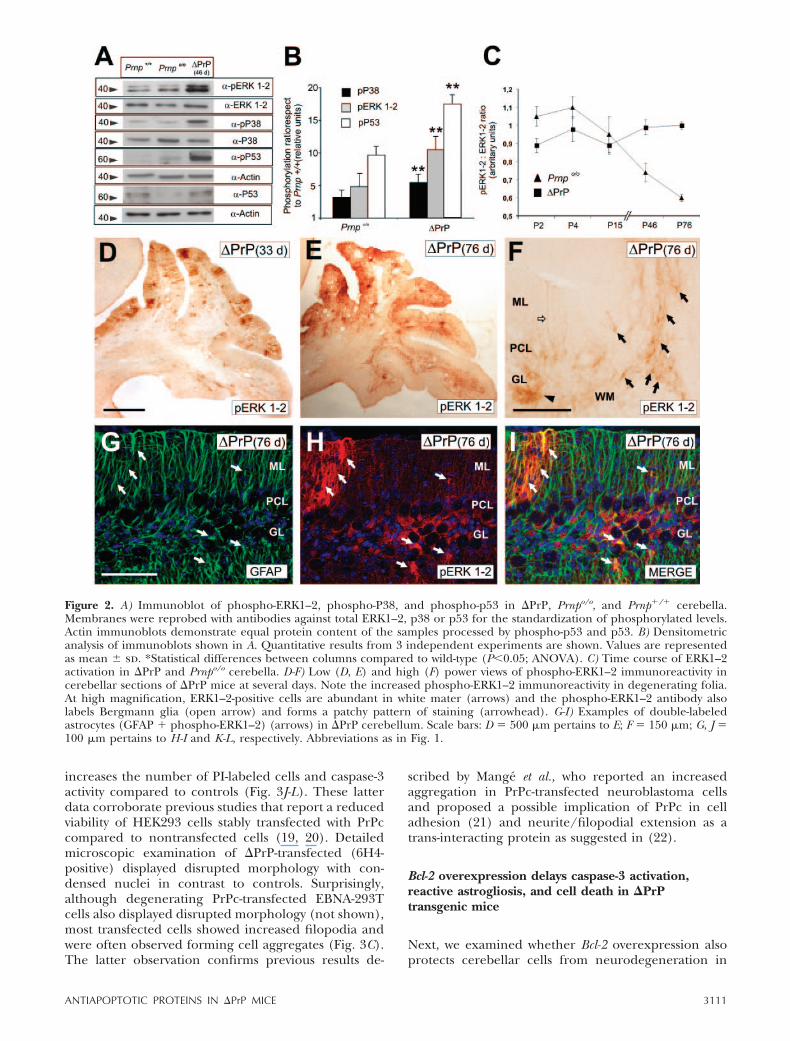
increases the number of PI-labeled cells and caspase-3activity compared to controls (Fig. 3J-L). These latterdata corroborate previous studies that report a reducedviability of HEK293 cells stably transfected with PrPccompared to nontransfected cells (19, 20). Detailedmicroscopic examination of �PrP-transfected (6H4-positive) displayed disrupted morphology with con-densed nuclei in contrast to controls. Surprisingly,although degenerating PrPc-transfected EBNA-293Tcells also displayed disrupted morphology (not shown),most transfected cells showed increased filopodia andwere often observed forming cell aggregates (Fig. 3C).The latter observation confirms previous results de-
scribed by Mange et al., who reported an increasedaggregation in PrPc-transfected neuroblastoma cellsand proposed a possible implication of PrPc in celladhesion (21) and neurite/filopodial extension as atrans-interacting protein as suggested in (22).
Bcl-2 overexpression delays caspase-3 activation,reactive astrogliosis, and cell death in �PrPtransgenic mice
Next, we examined whether Bcl-2 overexpression alsoprotects cerebellar cells from neurodegeneration in
Figure 2. A) Immunoblot of phospho-ERK1–2, phospho-P38, and phospho-p53 in �PrP, Prnpo/o, and Prnp�/� cerebella.Membranes were reprobed with antibodies against total ERK1–2, p38 or p53 for the standardization of phosphorylated levels.Actin immunoblots demonstrate equal protein content of the samples processed by phospho-p53 and p53. B) Densitometricanalysis of immunoblots shown in A. Quantitative results from 3 independent experiments are shown. Values are representedas mean � sd. *Statistical differences between columns compared to wild-type (P0.05; ANOVA). C) Time course of ERK1–2activation in �PrP and Prnpo/o cerebella. D-F) Low (D, E) and high (F) power views of phospho-ERK1–2 immunoreactivity incerebellar sections of �PrP mice at several days. Note the increased phospho-ERK1–2 immunoreactivity in degenerating folia.At high magnification, ERK1–2-positive cells are abundant in white mater (arrows) and the phospho-ERK1–2 antibody alsolabels Bergmann glia (open arrow) and forms a patchy pattern of staining (arrowhead). G-I) Examples of double-labeledastrocytes (GFAP � phospho-ERK1–2) (arrows) in �PrP cerebellum. Scale bars: D � 500 �m pertains to E; F � 150 �m; G, J �100 �m pertains to H-I and K-L, respectively. Abbreviations as in Fig. 1.
3111ANTIAPOPTOTIC PROTEINS IN �PrP MICE

Figure 3. A-C) Immunocytochemical detection of PrPc using the 6H4 antibody in nontransfected (A), �F35 (B), andPrPc-transfected (C) EBNA-293T cell cultures. Note the disrupted cell morphology of some EBNA-293T cells after �F35transfection (arrow) and the increase in filopodial-like membrane extension in PrPc transfected cells. D-K) PI-labeling andparallel Bisbenzimide staining of nontransfected (D, E), �F35 (F, G), �F35-Bcl-2 (H, I), and PrPc transfected (J, K) cell cultures.Note the significant increase in PI-labeled cells in �F35-transfected cells compared to nontransfected or pcDNA3-transfectedcells. Moreover, cultures transfected with PrPc also displayed numerous PI-labeled nuclei. Cotransfection with Bcl-2 decreasesnumber of PI-labeled cells (J, K). L) Quantification of PI-labeled cells (open bars) as percentage respect to Bisbenzimide labeledcells and fluorimetry analysis of caspase-3 activation (filled bars) in EBNA-293T transfected cells (see Materials and Methods fordetails). Both PI-quantification and caspase-3 activation are expressed as mean � sd of duplicates from 3 independentexperiments. A relevant decrease of caspase-3 activation and PI-labeled cells in �F35-Bcl-2 cotransfected cultures can be seen incontrast to �F35 transfected EBNA-293T cells. Asterisk indicates statistical differences between treatments and controls columns(P0.05; ANOVA). M) Western blot illustrating the presence of short form of PrPc in �F35-transfected cells and doubletransfected (�F35-Bcl-2) EBNA-293T cells. Membranes were reprobed with antibodies against Actin for protein standardization.Note the relevant band (arrow) of around 19 kDa in both �F35 and �F35-Bcl-2 transfected EBNA-293T cells. Scale bars: A �25 �m pertains to B, C; D � 100 �m pertains to E-K.
3112 Vol. 21 October 2007 NICOLAS ET AL.The FASEB Journal
�PrP mice. We obtained transgenic mice overexpress-ing Bcl-2 under the NSE promoter carrying the �PrPtransgene in a Prnpo/o background (see Materials andMethods; Fig. 4). None of the these mice showed anygait deficits before 50 days of age and most survivedmore than 90 days without apparent deficits untilsacrifice (Table 1, see below). At the time of death(from 50 to 214 days), 13 out of the 14 PrP Bcl-2transgenic mice did not show altered gait and displayednormal behavior (Suppl. video 1) or very few gaitsymptoms (score �). When minor gait alterations werepresent, a delay of around 33–35 days in the onset ofthe first gait symptoms was observed between �PrP Bcl-2
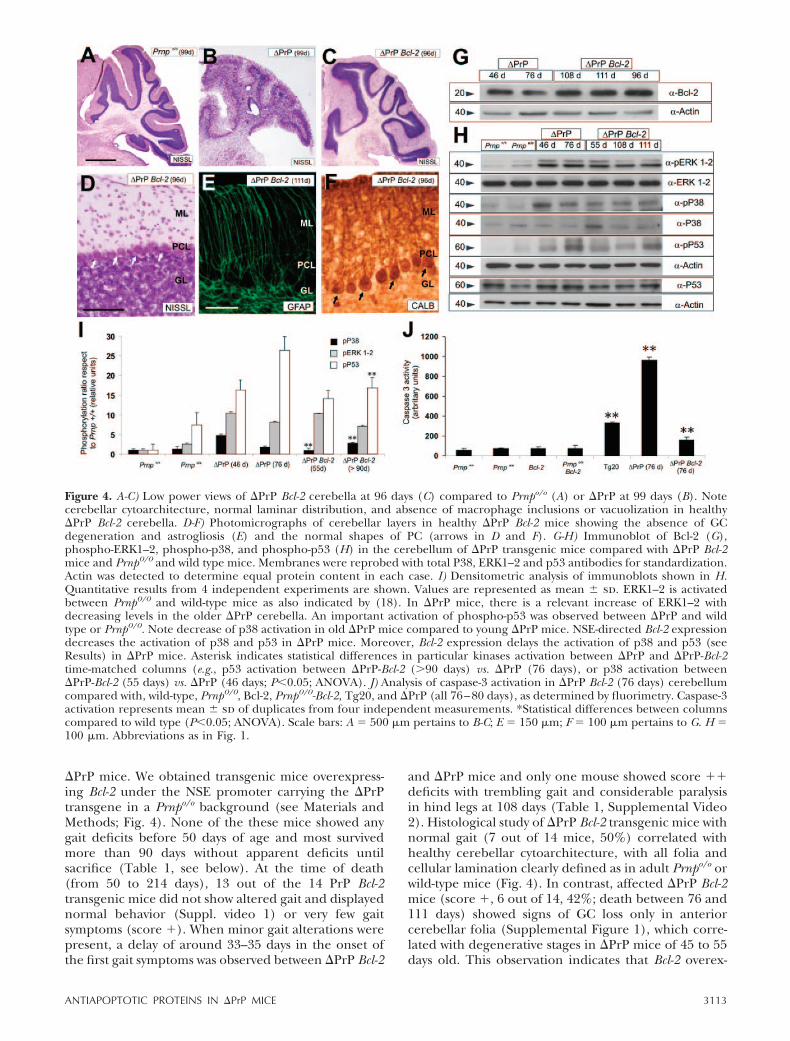
and �PrP mice and only one mouse showed score ��deficits with trembling gait and considerable paralysisin hind legs at 108 days (Table 1, Supplemental Video2). Histological study of �PrP Bcl-2 transgenic mice withnormal gait (7 out of 14 mice, 50%) correlated withhealthy cerebellar cytoarchitecture, with all folia andcellular lamination clearly defined as in adult Prnpo/o orwild-type mice (Fig. 4). In contrast, affected �PrP Bcl-2mice (score �, 6 out of 14, 42%; death between 76 and111 days) showed signs of GC loss only in anteriorcerebellar folia (Supplemental Figure 1), which corre-lated with degenerative stages in �PrP mice of 45 to 55days old. This observation indicates that Bcl-2 overex-
Figure 4. A-C) Low power views of �PrP Bcl-2 cerebella at 96 days (C) compared to Prnpo/o (A) or �PrP at 99 days (B). Notecerebellar cytoarchitecture, normal laminar distribution, and absence of macrophage inclusions or vacuolization in healthy�PrP Bcl-2 cerebella. D-F) Photomicrographs of cerebellar layers in healthy �PrP Bcl-2 mice showing the absence of GCdegeneration and astrogliosis (E) and the normal shapes of PC (arrows in D and F). G-H) Immunoblot of Bcl-2 (G),phospho-ERK1–2, phospho-p38, and phospho-p53 (H) in the cerebellum of �PrP transgenic mice compared with �PrP Bcl-2mice and Prnp0/0 and wild type mice. Membranes were reprobed with total P38, ERK1–2 and p53 antibodies for standardization.Actin was detected to determine equal protein content in each case. I) Densitometric analysis of immunoblots shown in H.Quantitative results from 4 independent experiments are shown. Values are represented as mean � sd. ERK1–2 is activatedbetween Prnp0/0 and wild-type mice as also indicated by (18). In �PrP mice, there is a relevant increase of ERK1–2 withdecreasing levels in the older �PrP cerebella. An important activation of phospho-p53 was observed between �PrP and wildtype or Prnp0/0. Note decrease of p38 activation in old �PrP mice compared to young �PrP mice. NSE-directed Bcl-2 expressiondecreases the activation of p38 and p53 in �PrP mice. Moreover, Bcl-2 expression delays the activation of p38 and p53 (seeResults) in �PrP mice. Asterisk indicates statistical differences in particular kinases activation between �PrP and �PrP-Bcl-2time-matched columns (e.g., p53 activation between �PrP-Bcl-2 (90 days) vs. �PrP (76 days), or p38 activation between�PrP-Bcl-2 (55 days) vs. �PrP (46 days; P0.05; ANOVA). J) Analysis of caspase-3 activation in �PrP Bcl-2 (76 days) cerebellumcompared with, wild-type, Prnp0/0, Bcl-2, Prnp0/0-Bcl-2, Tg20, and �PrP (all 76–80 days), as determined by fluorimetry. Caspase-3activation represents mean � sd of duplicates from four independent measurements. *Statistical differences between columnscompared to wild type (P0.05; ANOVA). Scale bars: A � 500 �m pertains to B-C; E � 150 �m; F � 100 �m pertains to G. H �100 �m. Abbreviations as in Fig. 1.
3113ANTIAPOPTOTIC PROTEINS IN �PrP MICE

pression delays the onset of clinical illness and in-creases the life expectancy of �PrP mice. Histologicalanalysis of healthy �PrP Bcl-2 mice revealed normalBergmann glia and PC distribution with dendritesexpanding the molecular layer, as ascertained withGFAP and Calbindin antibodies (Fig. 4E-F).
Next, we explored kinase activation in �PrP and�PrP Bcl-2 mice over time to ascertain whether Bcl-2overexpression alters this activation (Fig. 4). WB anal-ysis of cerebellar extracts revealed that phospho-ERK1–2 remained constant in �PrP cerebella duringneurodegeneration. In contrast, phospho-p38 levelsdecreased at advanced stages of cerebellar neurodegen-eration in �PrP mice (Fig. 4H). This decrease may beassociated with the increased neuronal damage (GCand PC) observed in older �PrP mice. Densitometricanalysis of revealed films demonstrated that phospho-ERK1–2 activation was similar between �PrP Bcl-2 and�PrP, and phospho-p38 and phospho-p53 followed asimilar evolution in �PrP Bcl-2, being reduced in �PrPBcl-2 mice aged 55 days but progressively increased inolder �PrP Bcl-2 (90 days; Fig. 4H-I). However, thelevels of p38 and p53 activation reached in �PrP Bcl-2were lower (�25–50% reduction, depending on theage) than in �PrP age-matched animals. Moreover,levels of p38 and p53 activation were similar in �PrPmice of 46 days and �PrP Bcl-2 of 90 days, therebyindicating protracted kinase activation (Fig. 4H-I).However, the level of caspase-3 activity was stronglyreduced in �PrP Bcl-2 mice (�80% reduction) com-pared to age-matched �PrP animals (Fig. 4J). Thesedata indicate that Bcl-2 overexpression largely delaysphospho-p38 and phospho-p53 activation in �PrP micebut strongly reduces caspase-3 activation. On the otherhand, Tg20 transgenic PrPc-overexpressing mice (23)showed increased caspase-3 activity compared to wild-type, Prnp0/0, Bcl-2, and Prnp0/0-Bcl-2 mice but lowerthan �PrP. These data are similar to those observedpreviously in EBNA-293T cells after PrPc overexpres-sion (Fig. 3). In parallel experiments, we observed thatoverexpression of the Bcl-2 transgene did not preclude�F35 expression (not shown). Taken together, ourresults indicate that although �F35-expressing cere-bella showed relevant cellular stress (as ascertained bythe phospho-ERK1–2, phospho-p38, and phospho-p53levels), the neuronal-directed overexpression of Bcl-2largely reduced caspase-3 activation and rescued cere-bellar degeneration and gait deficits in most �PrP Bcl-2mice.
DISCUSSION
Cytotoxic effects of the overexpression of N-terminally truncated variants of PrPc in cellslacking PrPc
Mice lacking Prnp present intrinsic resistance to prioninfection but mild phenotypic alterations (5). However,mouse lines with extensive deletions in Prnp show
severe ataxia and PC death caused by Dpl overexpres-sion (5). The overall structure of Dpl is similar to the�F35 protein, both lack the OR, the CC, and the HRbut preserve the C-terminal domain of PrPc (9). Theoverexpression of these two PrPc variants in Prnpo/o
mice induces cell death in several cerebellar cell types(GC or PC) as the result of their cell-specific expression(23, 24). However, it is plausible that Dpl and �F35induce cell death by the same or similar mechanismssince the targeted expression of �F35 or Dpl in PCcauses similar degenerative phenotypes (25, 26). Dpl-mediated neurotoxicity is produced by increasing oxi-dative stress (10). Our results demonstrate that �F35overexpression is toxic in non-neuronal cells with lowPrPc levels and induces relevant activation of the stress-inducible kinases ERK1–2 and p38 in �PrP cerebellum,both indicating an oxidative insult. Truncated forms ofPrPc lacking the N-terminal polybasic regions and theOR interfere with PrPc endocytosis via clathrin-coatedvesicles and beta-cleavage of PrPc, respectively, therebyimpairing the antioxidative functions of PrPc (27–29).Moreover, the HR binds to the stress-inducible protein1 (STI1), which plays a crucial role in preventingoxidative cell damage (30). Thus, the absence of theOR and HR regions in �PrP may induce the loss of thenatural protective functions of PrPc, thereby leading toincreased intracellular oxidative damage and celldeath. The fusion of the OR and the most N-terminalportion of the HR region to Dpl overcomes cell deathwhen serum is removed (31). However, �F35 mayalternatively arrest a survival ligand, which transducesantiapoptotic signals through PrPc signaling (6, 32,33). In this regard, a 37KD/67/KD laminin receptor orthe adhesion molecule NCAM bind PrPc can triggercell survival signals (34, 35).
During recent months, several studies have focusedon delimiting the region of the PrPc implicated inpremature neuronal death in mice overexpressing N-truncated variants of PrPc (11, 32, 33). For example,Sunyach and coworkers (11) demonstrated that theC-terminal product derived from the endoproteoliticprocessing of PrPc (C1 fragment), which lacks all theN-terminal region of the PrPc until the 110 residue,positively controls p53 transcription and activity. More-over, this C1 portion potentiates staurosporine-inducedcaspase-3 activation through a p53-mechanism inHEK293 cells stably overexpressing C1. In contrast,these effects are not elicited by a C2 fragment thatcontains the amino acids 90–110, which are missing inC1 (11). In a broad sense, the C1 fragment and theproduct of �F35 construct are similar, both lack themost N-terminal portion of the PrPc, including the CCflanked by the OR region and the HR respectively. Ourpresent findings demonstrate that p53 and caspase-3are activated in the degenerating cerebella of �PrPmice and that �F35 overexpression in EBNA-293T cellsincreases cell death (accessed by PI-uptake), whichcorrelates with enhanced caspase-3 activity. Taken to-gether, these results open up the interesting questionof whether the neurotoxic mechanisms observed in
3114 Vol. 21 October 2007 NICOLAS ET AL.The FASEB Journal

�PrP mice or �F35-transfected cells are similar to thosederived from C1 overproduction, as proposed by Sun-yach and coworkers. In this regard, a recent study byBaumann et al. (33) showed that mice overexpressing aPrPc variant lacking 94–134 amino acids (PrP�CD),comprising the CC and the HR of the CD, displayed asevere and fast neurodegeneration. Surprisingly, trans-genic mice lacking only 114–121 residues (PrP�pHC)corresponding to the HR but preserving the CC did notelicit any pathological phenotype in Prnpo/o mice (33).Moreover, the neurodegenerative phenotype of thePrP�CD mice was completely counteracted by coexpress-ing high levels of full-length PrPc and partially over-come by coexpressing a PrPc variant lacking all octare-peats (33). A parallel study by Li et al. (32) illustratedthat mice lacking the 105–125 amino acids of the CD[Tg(�PrP105–125)] in a Prnp0/0 background showedpremature death (few days after birth). Taken together,the findings of these studies suggest that the CC of thePrPc molecule is involved in both neuroprotective andneurodegenerative function. However, additional ex-periments including those performed with Prnp0/0 de-ficient mice overexpressing the C1 fragment, togetherwith additional biochemical experiments in vitro incellular models overexpressing PrP�CD and PrP�pHCand �PrP105–125 constructs, are required to ascertainthis hypothesis.
The cellular prion protein: a protective orproapoptotic molecule?
We observed that PrPc overexpression in EBNA-293Tcells increased cell death and caspase-3 activity andcaused relevant changes in cell morphology. Enhancedcaspase-3 activity was also observed in Tg20 transgenicmice overexpressing PrPc. Our observations confirmprevious studies by Paitel and coworkers, which dem-onstrated that the stable overexpression of PrPc sensi-tizes HEK293 cells to a proapoptotic phenotype de-scribed by Paitel et al. (19, 36) and by Westaway andcoworkers (37), which reported that older mice harbor-ing high copy numbers of wild-type PrPc showed,among other symptoms, cell degeneration in the ner-vous system. However, Tg20 mice are described withoutclinical illness (23), and the 4-fold expression of PrPc intransgenic mice (Tg(WT-E1) never causes the develop-ment of clinical symptoms (38).
Although considerable effort has been devoted todetermining the function of PrPc, the full repertory ofphysiological functions in healthy neurons is still un-known. PrPc has been implicated in copper homeosta-sis (39, 40), neuronal plasticity (41), cell proliferation(42), and neurite outgrowth (22, 35). In addition,several studies also point to a neuroprotective role ofPrPc (22, 43). Indeed, PrPc is overexpressed in isch-emic brain and correlates with neuroprotective effects(44, 45). PrPc also protects human primary neurons inculture against bax-mediated apoptosis (46). Moreover,although the first descriptions of Prnp0/0 mice indicatenormal behavior and few deficits (14), several studies in
recent years have described several phenotypic alter-ations in Prnp mutant mice (reviewed in ref 5).
Taken together, our results indicate that PrPc isinvolved directly or indirectly in numerous cellularfunctions and that its absence or overexpression sensi-tizes neurons to cell death. Thus, it is tempting tospeculate that basal PrPc expression may play a crucialrole in controlling neural homeostatic equilibrium byacting through several processes and that these pro-found changes in basal PrPc expression (e.g., completeabsence or high overexpression) may predispose neu-rons to cell death under certain physiological situa-tions.
Delayed caspase-3 activation by Bcl-2, a putative clueto reversing cerebellar degeneration in �PrP mice
Our findings also demonstrate that the overexpressionof Bcl-2 under the NSE promoter blocked or greatlydelayed caspase-3 activation and rescued most (�50%)�PrP mice from global GC loss and cerebellar degen-eration. In vitro experiments also corroborated thatcaspase-3 activation and cell death induced by �F35overexpression were blocked by Bcl-2 in a similar man-ner. Moreover, in �PrP Bcl-2 mice, activated p53 andp38 were decreased; however, phosphorylated levels ofERK1–2 were similar to �PrP mice. These results indi-cate that oxidative insults occur in �PrP Bcl-2 cerebellarneurons, although GC and PC cell death is largelyblocked by Bcl-2 overexpression in most of these mice.
Taken together, our findings also suggest that �F35overexpression in the absence of PrPc triggers celldeath pathways that differ to those associated with Baxactivation, since the overexpression of Bcl-2 in vitro andin vivo is unable to completely overcome cell death in�F35-expressing cells. This hypothesis has also beenrecently illustrated by Li and coworkers (8) using Bax0/0
�PrP transgenic mice. In these mice, Bax deletiondelays the development of clinical illness and slowsapoptosis of cerebellar GC but has no effect on whitematter degeneration, which also suggests that �PrP-induced neurodegeneration involves Bax-independentpathways. In a previous study by Radovanovic et al. (7),NSE-Prnp expression rescued GC degeneration in �PrPmice but these animals showed myelin defects as aresult of the absence of PrPc in oligodendrocytes.Although healthy �PrP Bcl-2 mice displayed normal gaitand no relevant changes were observed at the stagesanalyzed, we cannot exclude a delayed myelin alter-ation in older mice. However, neither macrophageinfiltration nor apparent leucoencephalopathy was de-tected in healthy �PrP Bcl-2 animals.
Most approaches used to overcome neurodegenera-tion in �PrP and Dpl mice have addressed Prnp expres-sion in distinct cerebellar cell types to abrogate neuro-nal degeneration (e.g., 7). Bcl-2 overexpression or Baxsuppression has been used to ameliorate neurodegen-erative processes in transgenic mice overexpressingPrPc with 14 octapeptide repeats (47) or recently in�PrP mice (8). However, our study is the first to report
3115ANTIAPOPTOTIC PROTEINS IN �PrP MICE

the use of neuronal-directed Bcl-2 overexpression toovercome cerebellar degeneration in �PrP mice. How-ever, it has been reported that Bcl-2 may interact withPrPc (aa 115–156) by its C-terminal domain and thatthis interaction can be proapoptotic for several iso-forms of cytosolic PrP (48). Prnpo/o cerebella showedhigher levels of Bcl-2 protein, which may putativelyinteract with �PrP as in wild-type mice (18). Althoughthis putative interaction was not addressed in thepresent study, a recent report indicates that PrPclacking the OR is not endocyted and remains in lipidrafs (28). Thus a putative interaction of �PrP and Bcl-2in these conditions is unlikely, and in �PrP Bcl-2 mice,Bcl-2 acts as an antiapoptotic protein, probably by adirect effect on Bax activation and mitochondrial ho-meostasis.
Bax deletion prevents the granule cell apoptosis lossbut not other neurological symptoms (synaptic loss) inthe cerebella of Tg(PG14) mice (47). In this study theneurological symptoms (synaptic loss) observed in theBax0/0 Tg(PG14) mice are likely to be associated withPrP accumulation (47). Taken together, the resultsobtained in the present study and those reported inothers (8, 47) indicate that the antiapoptotic therapiesalone have the capacity to rescue neuronal degenera-tion but require other therapies such as pharmacolog-ical intervention to completely overcome neurologicalsymptoms in �PrP mice or Tg(PG14) mice. However, itwould be of interest to establish whether the regulationof the antiapoptotic mechanisms exerted by Bcl-2 areuseful to abrogate PC degeneration in Dpl mice.
We thank Drs. M. Barbacid and I. Silos Santiago for theNSEa-Bcl-2 mice; J.X. Comella and D.A. Harris for theencoding plasmids of Bcl-2 and PrPc, respectively; and I.Palmero for the gift of the antip53 antibody. We also thankM. Lopez, E. Pastor, and E. Marquez for technical assis-tance and also thank T. Yates for correcting the Englishmanuscript. This work was supported by grants from theSpanish Ministry of Science and Technology (MCYT),EET2002–05149, BFU2004–0365-E and BFU2006–13651 toJ.A. del Rıo, FIS (PI042280) to J.M. Urena, SAF2005–00171and ISCIII (CIBERNED) to E. Soriano and grants SGR2005–0382 and SGR2005–00830 awarded by the Generalitat ofCatalunya to J.A. del Rıo and E. Soriano, respectively. O.Nicolas was supported by the Ramon Areces Foundation, X.Fontana by MEC, and R. Gavın by the Juan de la CiervaProgramme of the MCYT.
REFERENCES
1. Prusiner, S. B. (1998) Prions. Proc. Natl. Acad. Sci. U. S. A. 95,13363–13383
2. Roucou, X., Gains, M., and LeBlanc, A. C. (2004) Neuroprotec-tive functions of prion protein. J. Neurosci. Res. 75, 153–161
3. Chattopadhyay, M., Walter, E. D., Newell, D. J., Jackson, P. J.,Aronoff-Spencer, E., Peisach, J., Gerfen, G. J., Bennett, B.,Antholine, W. E., and Millhauser, G. L. (2005) The octarepeatdomain of the prion protein binds Cu(II) with three distinctcoordination modes at pH 7.4. J. Am. Chem. Soc. 127, 12647–12656
4. Yin, S., Yu, S., Li, C., Wong, P., Chang, B., Xiao, F., Kang, S. C.,Yan, H., Xiao, G., Grassi, J., Tien, P., and Sy, M. S. (2006) Prionproteins with insertion mutations have altered N-terminal con-
formation and increased ligand binding activity and are moresusceptible to oxidative attack. J. Biol. Chem. 281, 10698–10705
5. Sakudo, A., Onodera, T., Suganuma, Y., Kobayashi, T., Saeki, K.,and Ikuta, K. (2006) Recent advances in clarifying prion proteinfunctions using knockout mice and derived cell lines. Mini Rev.Medicinal Chem. 6, 589–601
6. Shmerling, D., Hegyi, I., Fischer, M., Blattler, T., Brandner, S.,Gotz, J., Rulicke, T., Flechsig, E., Cozzio, A., von Mering, C., et al.(1998) Expression of amino-terminally truncated PrP in themouse leading to ataxia and specific cerebellar lesions. Cell 93,203–214
7. Radovanovic, I., Braun, N., Giger, O. T., Mertz, K., Miele, G.,Prinz, M., Navarro, B., and Aguzzi, A. (2005) Truncated prionprotein and Doppel are myelinotoxic in the absence of oligo-dendrocytic PrPC. J. Neurosci. 25, 4879–4888
8. Li, A., Barmada, S. J., Roth, K. A., and Harris, D. A. (2007)N-terminally deleted forms of the prion protein activate bothBax-dependent and Bax-independent neurotoxic pathways.J. Neurosci. 27, 852–859
9. Behrens, A., and Aguzzi, A. (2002) Small is not beautiful:antagonizing functions for the prion protein PrP(C) and itshomologue Dpl. Trends Neurosci. 25, 150–154
10. Cui, T., Holme, A., Sassoon, J., and Brown, D. R. (2003) Analysisof doppel protein toxicity. Mol. Cell Neurosci. 23, 144–155
11. Sunyach, C., Cisse, M. A., da Costa, C. A., Vincent, B., andChecler, F. (2007) The C-terminal products of cellular prionprotein processing, C1 and C2, exert distinct influence onp53-dependent staurosporine-induced caspase-3 activation.J. Biol. Chem. 282, 1956–1963
12. Er, E., Oliver, L., Cartron, P. F., Juin, P., Manon, S., and Vallette,F. M. (2006) Mitochondria as the target of the pro-apoptoticprotein Bax. Biochim. Biophys. Acta
13. Cregan, S. P., MacLaurin, J. G., Craig, C. G., Robertson, G. S.,Nicholson, D. W., Park, D. S., and Slack, R. S. (1999) Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci. 19, 7860–7869
14. Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P.,DeArmond, S. J., Prusiner, S. B., Aguet, M., and Weissmann, C.(1992) Normal development and behaviour of mice lacking theneuronal cell-surface PrP protein. Nature 356, 577–582
15. Martinou, J. C., Dubois-Dauphin, M., Staple, J. K., Rodriguez, I.,Frankowski, H., Missotten, M., Albertini, P., Talabot, D., Catsi-cas, S., Pietra, C., and et al. (1994) Overexpression of BCL-2 intransgenic mice protects neurons from naturally occurring celldeath and experimental ischemia. Neuron 13, 1017–1030
16. Gavin, R., Braun, N., Nicolas, O., Parra, B., Urena, J. M.,Mingorance, A., Soriano, E., Torres, J. M., Aguzzi, A., and delRio, J. A. (2005) PrP(106–126) activates neuronal intracellularkinases and Egr1 synthesis through activation of NADPH-oxi-dase independently of PrPc. FEBS Lett. 579, 4099–4106
17. Zhu, Y., Mao, X. O., Sun, Y., Xia, Z., and Greenberg, D. A.(2002) p38 Mitogen-activated protein kinase mediates hypoxicregulation of Mdm2 and p53 in neurons. J. Biol. Chem. 277,22909–22914
18. Brown, D. R., Nicholas, R. S., and Canevari, L. (2002) Lack ofprion protein expression results in a neuronal phenotypesensitive to stress. J. Neurosci. Res. 67, 211–224
19. Paitel, E., Sunyach, C., Alves da Costa, C., Bourdon, J. C.,Vincent, B., and Checler, F. (2004) Primary cultured neuronsdevoid of cellular prion display lower responsiveness to stauro-sporine through the control of p53 at both transcriptional andpost-transcriptional levels. J. Biol. Chem. 279, 612–618
20. Paitel, E., Alves da Costa, C., Vilette, D., Grassi, J., and Checler,F. (2002) Overexpression of PrPc triggers caspase 3 activation:potentiation by proteasome inhibitors and blockade by anti-PrPantibodies. J. Neurochem. 83, 1208–1214
21. Mange, A., Milhavet, O., Umlauf, D., Harris, D., and Lehmann,S. (2002) PrP-dependent cell adhesion in N2a neuroblastomacells. FEBS Lett. 514, 159–162
22. Chen, S., Mange, A., Dong, L., Lehmann, S., and Schachner, M.(2003) Prion protein as trans-interacting partner for neurons isinvolved in neurite outgrowth and neuronal survival. Mol. CellNeurosci. 22, 227–233
23. Fischer, M., Rulicke, T., Raeber, A., Sailer, A., Moser, M., Oesch,B., Brandner, S., Aguzzi, A., and Weissmann, C. (1996) Prionprotein (PrP) with amino-proximal deletions restoring suscep-tibility of PrP knockout mice to scrapie. EMBO J. 15, 1255–1264
3116 Vol. 21 October 2007 NICOLAS ET AL.The FASEB Journal

24. Li, A., Sakaguchi, S., Shigematsu, K., Atarashi, R., Roy, B. C.,Nakaoke, R., Arima, K., Okimura, N., Kopacek, J., and Kat-amine, S. (2000) Physiological expression of the gene forPrP-like protein, PrPLP/Dpl, by brain endothelial cells and itsectopic expression in neurons of PrP-deficient mice ataxic dueto Purkinje cell degeneration. Am. J. Pathol. 157, 1447–1452
25. Flechsig, E., Hegyi, I., Leimeroth, R., Zuniga, A., Rossi, D.,Cozzio, A., Schwarz, P., Rulicke, T., Gotz, J., Aguzzi, A., andWeissmann, C. (2003) Expression of truncated PrP targeted toPurkinje cells of PrP knockout mice causes Purkinje cell deathand ataxia. EMBO J. 22, 3095–3101
26. Anderson, L., Rossi, D., Linehan, J., Brandner, S., and Weiss-mann, C. (2004) Transgene-driven expression of the Doppelprotein in Purkinje cells causes Purkinje cell degeneration andmotor impairment. Proc. Natl. Acad. Sci. U. S. A. 101, 3644–3649
27. Sakudo, A., Hamaishi, M., Hosokawa-Kanai, T., Tuchiya, K.,Nishimura, T., Saeki, K., Matsumoto, Y., Ueda, S., and Onodera,T. (2003) Absence of superoxide dismutase activity in a solublecellular isoform of prion protein produced by baculovirusexpression system. Biochem. Biophys. Res. Commun. 307, 678–683
28. Taylor, D. R., Watt, N. T., Perera, W. S., and Hooper, N. M.(2005) Assigning functions to distinct regions of the N-terminusof the prion protein that are involved in its copper-stimulated,clathrin-dependent endocytosis. J. Cell Sci. 118, 5141–5153
29. Watt, N. T., Taylor, D. R., Gillott, A., Thomas, D. A., Perera,W. S., and Hooper, N. M. (2005) Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellularresponse to oxidative stress. J. Biol. Chem. 280, 35914–35921
30. Sakudo, A., Lee, D. C., Li, S., Nakamura, T., Matsumoto, Y.,Saeki, K., Itohara, S., Ikuta, K., and Onodera, T. (2005) PrPcooperates with STI1 to regulate SOD activity in PrP-deficientneuronal cell line. Biochem. Biophys. Res. Commun. 328, 14–19
31. Lee, D. C., Sakudo, A., Kim, C. K., Nishimura, T., Saeki, K.,Matsumoto, Y., Yokoyama, T., Chen, S. G., Itohara, S., andOnodera, T. (2006) Fusion of Doppel to octapeptide repeat andN-terminal half of hydrophobic region of prion protein confersresistance to serum deprivation. Microbiol. Immunol. 50, 203–209
32. Li, A., Christensen, H. M., Stewart, L. R., Roth, K. A., Chiesa, R.,and Harris, D. A. (2007) Neonatal lethality in transgenic miceexpressing prion protein with a deletion of residues 105–125.EMBO J. 26, 548–558
33. Baumann, F., Tolnay, M., Brabeck, C., Pahnke, J., Kloz, U.,Niemann, H. H., Heikenwalder, M., Rulicke, T., Burkle, A., andAguzzi, A. (2007) Lethal recessive myelin toxicity of prionprotein lacking its central domain. EMBO J. 26, 538–547
34. Gauczynski, S., Peyrin, J. M., Haik, S., Leucht, C., Hundt, C.,Rieger, R., Krasemann, S., Deslys, J. P., Dormont, D., Lasmezas,C. I., and Weiss, S. (2001) The 37-kDa/67-kDa laminin receptoracts as the cell-surface receptor for the cellular prion protein.EMBO J. 20, 5863–5875
35. Santuccione, A., Sytnyk, V., Leshchyns’ka, I., and Schachner, M.(2005) Prion protein recruits its neuronal receptor NCAM tolipid rafts to activate p59fyn and to enhance neurite outgrowth.J. Cell Biol. 169, 341–354
36. Paitel, E., Fahraeus, R., and Checler, F. (2003) Cellular prionprotein sensitizes neurons to apoptotic stimuli through Mdm2-regulated and p53-dependent caspase 3-like activation. J. Biol.Chem. 278, 10061–10066
37. Westaway, D., DeArmond, S. J., Cayetano-Canlas, J., Groth, D.,Foster, D., Yang, S. L., Torchia, M., Carlson, G. A., and Prusiner,S. B. (1994) Degeneration of skeletal muscle, peripheral nerves,and the central nervous system in transgenic mice overexpress-ing wild-type prion proteins. Cell 76, 117–129
38. Chiesa, R., Piccardo, P., Ghetti, B., and Harris, D. A. (1998)Neurological illness in transgenic mice expressing a prionprotein with an insertional mutation. Neuron 21, 1339–1351
39. Brown, D. R., Qin, K., Herms, J. W., Madlung, A., Manson, J.,Strome, R., Fraser, P. E., Kruck, T., von Bohlen, A., Schulz-Schaeffer, W., et al. (1997) The cellular prion protein bindscopper in vivo. Nature 390, 684–687
40. Vassallo, N., and Herms, J. (2003) Cellular prion proteinfunction in copper homeostasis and redox signalling at thesynapse. J. Neurochem. 86, 538–544
41. Maglio, L. E., Perez, M. F., Martins, V. R., Brentani, R. R., andRamirez, O. A. (2004) Hippocampal synaptic plasticity in micedevoid of cellular prion protein. Brain Res. Mol. Brain Res. 131,58–64
42. Steele, A. D., Emsley, J. G., Ozdinler, P. H., Lindquist, S., andMacklis, J. D. (2006) Prion protein (PrPc) positively regulatesneural precursor proliferation during developmental and adultmammalian neurogenesis. Proc. Natl. Acad. Sci. U. S. A. 103,3416–3421
43. Chiarini, L. B., Freitas, A. R., Zanata, S. M., Brentani, R. R.,Martins, V. R., and Linden, R. (2002) Cellular prion proteintransduces neuroprotective signals. EMBO J. 21, 3317–3326
44. Weise, J., Crome, O., Sandau, R., Schulz-Schaeffer, W., Bahr, M.,and Zerr, I. (2004) Upregulation of cellular prion protein(PrPc) after focal cerebral ischemia and influence of lesionseverity. Neurosci. Lett. 372, 146–150
45. McLennan, N. F., Brennan, P. M., McNeill, A., Davies, I.,Fotheringham, A., Rennison, K. A., Ritchie, D., Brannan, F.,Head, M. W., Ironside, J. W., Williams, A., and Bell, J. E. (2004)Prion protein accumulation and neuroprotection in hypoxicbrain damage. Am. J. Pathol. 165, 227–235
46. Bounhar, Y., Zhang, Y., Goodyer, C. G., and LeBlanc, A. (2001)Prion protein protects human neurons against Bax-mediatedapoptosis. J. Biol. Chem. 276, 39145–39149
47. Chiesa, R., Piccardo, P., Dossena, S., Nowoslawski, L., Roth,K. A., Ghetti, B., and Harris, D. A. (2005) Bax deletion preventsneuronal loss but not neurological symptoms in a transgenicmodel of inherited prion disease. Proc. Natl. Acad. Sci. U. S. A.102, 238–243
48. Rambold, A. S., Miesbauer, M., Rapaport, D., Bartke, T., Baier,M., inklhofer, K. F., and Tatzelt, J. (2006) Association of Bcl-2with misfolded prion protein is linked to the toxic potential ofcytosolic PrP. Mol. Biol. Cell 17, 3356–3368
Received for publication January 25, 2007.Accepted for publication April 12, 2007.
3117ANTIAPOPTOTIC PROTEINS IN �PrP MICE
Related Documents











