The FASEB Journal • Research Communication Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators Aksam J. Merched,* ,1 Kerry Ko,* Katherine H. Gotlinger, ‡ Charles N. Serhan, ‡ and Lawrence Chan* ,† *Department of Molecular and Cellular Biology and † Department of Medicine, Division of Diabetes, Endocrinology, and Metabolism, Baylor College of Medicine, and St. Luke’s Episcopal Hospital, Houston, Texas, USA; and ‡ Center for Experimental Therapeutics and Reperfusion Injury, Department of Anesthesiology, Perioperative and Pain Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA ABSTRACT Atherosclerosis is now recognized as an inflammatory disease involving the vascular wall. Recent results indicate that acute inflammation does not simply passively resolve as previously assumed but is actively terminated by a homeostatic process that is governed by specific lipid-derived mediators initiated by lipoxygen- ases. Experiments with animals and humans support a proinflammatory role for the 5-lipoxygenase system. In contrast, results from animal experiments show a range of responses with the 12/15-lipoxygenase pathways in ath- erosclerosis. To date, the only two clinical epidemiology human studies both support an antiatherogenic role for 12/15-lipoxygenase downstream actions. We tested the hypothesis that atherosclerosis results from a failure in the resolution of local inflammation by analyzing apoli- poprotein E-deficient mice with 1) global leukocyte 12/ 15-lipoxygenase deficiency, 2) normal enzyme expression, or 3) macrophage-specific 12/15-lipoxygenase overex- pression. Results from these indicate that 12/15-lipoxy- genase expression protects mice against atherosclerosis via its role in the local biosynthesis of lipid mediators, including lipoxin A 4 , resolvin D1, and protectin D1. These mediators exert potent agonist actions on macrophages and vascular endothelial cells that can control the magni- tude of the local inflammatory response. Taken together, these findings suggest that a failure of local endogenous resolution mechanisms may underlie the unremitting in- flammation that fuels atherosclerosis.—Merched, A. J., Ko, K., Gotlinger, K. H., Serhan, C. N. Chan, L. Athero- sclerosis: evidence for impairment of resolution of vascu- lar inflammation governed by specific lipid mediators. FASEB J. 22, 3595–3606 (2008) Key Words: lipoxygenase innate immunity Atherosclerosis is now widely appreciated as an inflammatory disease involving the vascular wall (1, 2). Advanced complex atheromata that set the stage for overt clinical events in atherosclerosis are preceded by less complex lesions. The earliest lesions are designated type I (an increase in intimal macrophages and pres- ence of foam cells) and type II lesions (grossly visible fatty streaks), which are common in infancy and child- hood (3, 4) that may progress to advanced atheromata or disappear. The factors that enable some lesions to progress while others regress remain unclear. It is clear, however, that lack of regression is associated with persistent inflammation in the vascular wall (reviewed in refs. 1, 2). In this context, it is noteworthy that inflammation does not simply “burn out” on its own as once thought, as specific tissue-level resolution pro- grams are initiated for inflammation that actively gov- ern the process via the biosynthesis of novel proresolv- ing chemical mediators (5). Resolution is programmed within the normal inflam- matory response itself that enables the body to contain inflammation to minimize tissue and organ damage (6). It involves limiting cellular trafficking as well as nonphlo- gistic phagocytic removal of apoptotic cells, key parts of the integrated programs that are orchestrated by special- ized lipid-derived mediators (6). The biosynthesis of these local acting mediators is regulated by availability of fatty acid precursors such as -3 polyunsaturated fatty acids (PUFAs) and the spatial and temporal control of specific lipoxygenase (LO) pathways (5). 12/15-LO (type 1 in humans and its ortholog in mice) and 5-LO (7) are key LO systems in leukocytes and other neighboring cells that can biosynthesize local products that can steer tissues toward chronic inflam- mation or complete resolution. One class of LO prod- ucts, the lipoxins (LX, an acronym for lipoxygenase interaction products), was identified as “braking sig- nals” in acute inflammation that can activate the reso- lution phase of an inflammatory response. Although neutrophil 5-LO initially generates the proinflamma- tory chemoattractants, such as leukotriene B 4 , from 1 Correspondence: Department of Molecular and Cellular Biology, Baylor College of Medicine, 1 Baylor Plaza, N520.11, Houston, TX 77030, USA. E-mail: [email protected] doi: 10.1096/fj.08-112201 3595 0892-6638/08/0022-3595 © FASEB

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The FASEB Journal • Research Communication
Atherosclerosis: evidence for impairment of resolutionof vascular inflammation governed by specific lipidmediators
Aksam J. Merched,*,1 Kerry Ko,* Katherine H. Gotlinger,‡ Charles N. Serhan,‡ andLawrence Chan*,†
*Department of Molecular and Cellular Biology and †Department of Medicine, Division of Diabetes,Endocrinology, and Metabolism, Baylor College of Medicine, and St. Luke’s Episcopal Hospital,Houston, Texas, USA; and ‡Center for Experimental Therapeutics and Reperfusion Injury,Department of Anesthesiology, Perioperative and Pain Medicine, Brigham and Women’s Hospital,Harvard Medical School, Boston, Massachusetts, USA
ABSTRACT Atherosclerosis is now recognized as aninflammatory disease involving the vascular wall. Recentresults indicate that acute inflammation does not simplypassively resolve as previously assumed but is activelyterminated by a homeostatic process that is governed byspecific lipid-derived mediators initiated by lipoxygen-ases. Experiments with animals and humans support aproinflammatory role for the 5-lipoxygenase system. Incontrast, results from animal experiments show a range ofresponses with the 12/15-lipoxygenase pathways in ath-erosclerosis. To date, the only two clinical epidemiologyhuman studies both support an antiatherogenic role for12/15-lipoxygenase downstream actions. We tested thehypothesis that atherosclerosis results from a failure inthe resolution of local inflammation by analyzing apoli-poprotein E-deficient mice with 1) global leukocyte 12/15-lipoxygenase deficiency, 2) normal enzyme expression,or 3) macrophage-specific 12/15-lipoxygenase overex-pression. Results from these indicate that 12/15-lipoxy-genase expression protects mice against atherosclerosisvia its role in the local biosynthesis of lipid mediators,including lipoxin A4, resolvin D1, and protectin D1. Thesemediators exert potent agonist actions on macrophagesand vascular endothelial cells that can control the magni-tude of the local inflammatory response. Taken together,these findings suggest that a failure of local endogenousresolution mechanisms may underlie the unremitting in-flammation that fuels atherosclerosis.—Merched, A. J.,Ko, K., Gotlinger, K. H., Serhan, C. N. Chan, L. Athero-sclerosis: evidence for impairment of resolution of vascu-lar inflammation governed by specific lipid mediators.FASEB J. 22, 3595–3606 (2008)
Key Words: lipoxygenase � innate immunity
Atherosclerosis is now widely appreciated as aninflammatory disease involving the vascular wall (1, 2).Advanced complex atheromata that set the stage forovert clinical events in atherosclerosis are preceded byless complex lesions. The earliest lesions are designated
type I (an increase in intimal macrophages and pres-ence of foam cells) and type II lesions (grossly visiblefatty streaks), which are common in infancy and child-hood (3, 4) that may progress to advanced atheromataor disappear. The factors that enable some lesions toprogress while others regress remain unclear. It is clear,however, that lack of regression is associated withpersistent inflammation in the vascular wall (reviewedin refs. 1, 2). In this context, it is noteworthy thatinflammation does not simply “burn out” on its own asonce thought, as specific tissue-level resolution pro-grams are initiated for inflammation that actively gov-ern the process via the biosynthesis of novel proresolv-ing chemical mediators (5).
Resolution is programmed within the normal inflam-matory response itself that enables the body to containinflammation to minimize tissue and organ damage (6).It involves limiting cellular trafficking as well as nonphlo-gistic phagocytic removal of apoptotic cells, key parts ofthe integrated programs that are orchestrated by special-ized lipid-derived mediators (6). The biosynthesis of theselocal acting mediators is regulated by availability of fattyacid precursors such as �-3 polyunsaturated fatty acids(PUFAs) and the spatial and temporal control of specificlipoxygenase (LO) pathways (5).
12/15-LO (type 1 in humans and its ortholog inmice) and 5-LO (7) are key LO systems in leukocytesand other neighboring cells that can biosynthesize localproducts that can steer tissues toward chronic inflam-mation or complete resolution. One class of LO prod-ucts, the lipoxins (LX, an acronym for lipoxygenaseinteraction products), was identified as “braking sig-nals” in acute inflammation that can activate the reso-lution phase of an inflammatory response. Althoughneutrophil 5-LO initially generates the proinflamma-tory chemoattractants, such as leukotriene B4, from
1 Correspondence: Department of Molecular and CellularBiology, Baylor College of Medicine, 1 Baylor Plaza, N520.11,Houston, TX 77030, USA. E-mail: [email protected]
doi: 10.1096/fj.08-112201
35950892-6638/08/0022-3595 © FASEB

arachidonate (7), 12/15-LO products interact with5-LO in a temporally distinct fashion to generate theanti-inflammatory LXs that are involved in resolution.Along with LXs, specialized lipid mediators known asresolvins and protectins were identified (5), which aregenerated from �-3 essential PUFAs downstream of the12/15-LO in human cells. They possess potent dualanti-inflammatory and proresolving actions that medi-ate resolution of inflammation.
Recent results (8, 9) indicated that leukotrienes andspecifically the 5-LO system play critical roles in athe-romas and cardiovascular diseases in humans. Thepossible proresolving actions of 12/15-LO downstreammolecules in the inflammation associated with athero-sclerosis in humans has been addressed in two recentreports. Wittwer et al. (10) first reported in a case-control study involving 498 Caucasians that heterozy-gotes for a �292C�T variant in the promoter of the12/15-LO gene (which was associated with higherenzyme expression in vitro) showed a tendency towardprotection against atherosclerosis. In another study,Assimes et al. (11) described a coding SNP (T560M)variant in the 12/15-LO gene that is associated with a20-fold reduction in enzyme activity. Genotyping ofatherosclerotic disease, vascular function, and geneticepidemiology (ADVANCE) and atherosclerosis risk incommunities (ARIC) study cohorts (involving 3543individuals) showed that heterozygote carriers of thisnear-null 560 M allele had an increased risk of clinicalcoronary artery disease (adjusted odds ratio, 1.62; P �0.02). Thus, the only two available case-control studiesto date support a protective role of 12/15-LO expres-sion against coronary disease in humans.
Failure in mounting endogenous resolution mecha-nisms is increasingly being recognized as an importantfeature in diverse inflammatory disorders such as glomer-ulonephritis (12, 13), bronchial asthma (14), and inflam-matory bowel disease (15). Hence, we hypothesized thatatherosclerosis may result, in part, from local nonresolv-ing forms of vascular inflammation. Here, we report that12/15-LO is pivotal in protecting from atherosclerosis andthat several of its products, namely, LXA4, resolvin D1(RvD1), and protectin D1 (PD1), are potent local-actingproresolving mediators that exhibit robust proresolutionactions regulating multiple proinflammatory cytokinesproduced by macrophages. These 12/15-LO-derived me-diators also exert proresolution actions on vascular endo-thelial cells, which together suggest that a failure toeffectively resolve local inflammatory insults initiated inthe vessel wall may result in persistent inflammation andatherosclerosis progression.
MATERIALS AND METHODS
Mice
12/15-LO�/� and apoE�/� mice in C57BL/6J backgroundwere purchased from Jackson Laboratories (Bar Harbor, ME,USA). 12/15-LO�/� mice were backcrossed onto the C57BL/6J
background for �11 generations. All mice were maintainedunder normal chow diet. We measured total cholesterol andtriglyceride concentrations in plasma at the end of diet feedingusing enzymatic procedures (Sigma, St. Louis, MO, USA). Fastprotein liquid chromatography (FPLC) separation of lipopro-tein particles was achieved as described previously (16). Thecholesterol content of the FPLC fractions was measured by usingan enzymatic kit (Sigma).
For the creation of the mouse transgenic line targetinggene expression to macrophages, we used a gene expressionsystem containing a scavenger receptor promoter that hadshown highly specific expression approach (17). We sub-cloned the human 15-LO cDNA into the EcoRV site of thescavenger receptor promoter expression cassette. Male pro-nuclei of fertilized ova from C57BL/6J mice were microin-jected with this construct, and transgenic offspring andsubsequent progeny were genotyped by polymerase chainreaction (PCR) of tail DNA. 12/15-LO specific mRNA expres-sion in macrophages was characterized by RNase protectionanalyses performed on RNA by using the 510 bp fragment of15-LO as a probe. For bone marrow transplantation, 15-wk-old female apoE�/�/12/15-LO�/� mice were subjected to10 Gy total body irradiation to eliminate endogenous bonemarrow stem cells and most of the bone marrow-derived cells.Bone marrow cells for repopulation were prepared fromapoE�/�/12/15-LO�/� or apoE�/� mice, and transplanta-tion was performed as described previously (18). After bonemarrow transplantation, mice were kept on a regular chowdiet for 15 wk. All procedures were approved by the animalprotocol review committee of our institution.
Quantitative morphometry and immunohistochemistry
We performed cross-section analysis of the aortic sinus ofmice at 22 wk. En face study of atherosclerotic lesion area wasperformed on all the other experiments, using completeaortas spanning from the root of the aorta to the iliacbifurcation. We prepared aortas for analysis as describedpreviously (18). Cryostat sections of the aortic root and aortawere fixed in acetone for 10 min and air dried for at least 30min. After blocking the endogenous peroxidase activity andwashing in PBS (pH 7.4), we incubated the sections for 30min with different monoclonal antibodies individually andexposed the sections to peroxidase or alkaline phosphatase-conjugated secondary antibody for 30 min. Primary antibod-ies used included rat anti-mouse macrophages Mac3 (SantaCruz Biotechnology, Santa Cruz, CA, USA), rat anti-mouseVCAM-1 (Santa Cruz), and rat anti-mouse CD18 (Pharmin-gen, San Diego, CA, USA). Collagen content of lesions wasanalyzed using trichrome staining.
Gene expression and macrophage functional studies
Macrophages were collected 3 days after activation withintraperitoneal injection of 1 ml of 3% aged Brewer’s thio-glycolate. Cells were cultured and RNA was extracted asdescribed in our previous study (19). The expression ofspecific genes was studied at the mRNA level by quantitativeRT-PCR using a multiplex quantitative PCR (qPCR) system(Stratagene, La Jolla, CA, USA; ref. 19) with the followingmodifications. For normalization of gene-expression analysis,we used 8 mouse housekeeping genes as endogenous con-trols: cyclophilin A gene (PPIA), B2M (�-microglobulin)GAPDH, 5-aminolevulinate synthase (ALAS1), hydroxymethyl-bilane synthase (HMBS), 18S rRNA (RN18S), eukaryoticelongation factor 1g (EeE1g), and �-actin (ACT�). Moreover,using GeNorm (20), we selected PPIA and EeF1g as the twomost stable housekeeping genes within our experimental
3596 Vol. 22 October 2008 MERCHED ET AL.The FASEB Journal

conditions and used the geometric standardization as de-scribed previously (20). Macrophage uptake of apoptoticthymocytes was performed as described previously (19).
Human aortic endothelial cell (HAEC) experiment
HAECs were plated at 2.0 � 105 cells/well in 12-well tissueculture plates coated with 0.1% gelatin and grown in EGM-2culture medium (Lonza, Walkersville, MD, USA) at 37°C in5% CO2 for 20–24 h before the experiments were started. Atthe time of the experiment, the HAEC culture medium wasremoved and replaced by medium containing 5 U/ml tumornecrosis factor (TNF) -�, with either 100 nM of LXA4, PD1,RvD1, or ethanol (used as vehicle at 0.04%). The plate wasincubated at 37°C in 5% CO2 for 2 h. Media were collectedand frozen at 80°C until use for bioplex assay, as describedbelow. For RNA extraction, the same cells were washed twicewith PBS and lyzed for RNA preparation as indicated previ-ously (19). Data are means � sd obtained from threedifferent cultures (n�3). We analyzed gene expression byreal-time qPCR using the same approach as with macro-phages. We identified the most stable housekeeping genesin our experiments e.g., �-actin and GAPDH, which we haveused as internal controls for normalization to quantify theexpression of different adhesion molecules and chemo-kines. Primers for human genes were designed as follows:�-actin: (5-GCCATGTACGTTGCTATCCA-3 and 5-CCTC-GTAGATGGGCACAGT-3); GAPDH: (5-TGGTATCGTGG-AAGGACTCA-3 and 5-CCAGTAGAGGCAGGGATGAT-3);ICAM-1: (5-GGGAGAAGGAGCTGAAACG-3 and 5-CACG-AGAAATTGGCTCCAT-3), VCAM-1: (5-TGTGAATCCATC-CACAAAGC-3 and 5-GGTGAGAGTTGCATTTCCAG-3);P-selectin: 5-CTTCCTCAATGCCAGTCAGA-3 and 5- GCC-GTTCAGTAGCAAGGAA-3); MCP-1: (5-GAATCACCAGCA-GCAAGTGT-3 and 5-GTCTTCGGAGTTTGGGTTTG-3).
Cytokine immunoassays
We used the Bioplex Protein Array system (Bio-Rad, Her-cules, CA, USA) to measure a panel of 18 mouse and 27human cytokines. The mouse panel includes CCL5 (regu-lated on activation, normal T cell expressed and secreted),colony stimulating factor (CSF), granulocyte-macrophage col-ony-stimulating factor (GM-CSF), interferon-gamma (IFN),interleukin (IL) -1�, IL-1�, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10,IL-12P40, IL-12P70, IL-17, keratinocyte-derived chemokine(KC), macrophage inflammatory protein (MIP-1�), andTNF-�. The human panel includes CCL5, eotaxin, FGF basic,G-CSF, GM-CSF, IFN, IL-1�, IL-1ra, IL-2, IL-4, IL-5, IL-6,IL-7, IL-8, IL-9, IL10, IL-12p70, IL-13, IL-15, IL-17, IP10,MCP-1, MIP-1�, MIP-1b, PDGF-bb, TNF-�, and VEGF. Theseare novel multiplexed, particle-based, flow cytometric assaythat uses anti-cytokine monoclonal antibodies linked to mi-crospheres incorporating distinct proportions of two fluores-cent dyes. The minimum detectable dose was �10 pg/ml.Four- or 5-parameter logistic regression algorithms were usedto quantify the standards. Serum samples were stored at�80�C before the assays to avoid protein degradation.
Mediator lipidomics: lipoxygenase pathway markers
Resident peritoneal macrophages were collected, and 12/15-LO-derived mediators were extracted before and after activa-tion with 2 M of the divalent cation ionophore A23187 for20 min at 37°C. We determined plasma Lipoxin A4 andLeukotriene B4 in Sep-Pack-extracted samples (Waters, Mil-ford, MA, USA) using specific enzyme-linked immunosorbentassay as described by the manufacturer (Neogen, Lexington,
KY, USA). At least four animals were included in each group.To analyze other mediators by lipidomics, samples wereextracted using solid-phased extraction C-18 SPE 500 mgcolumns (Alltech, Deerfield, IL, USA) as in Lu et al. (21).Criteria for identification of a specific mediator included aminimum of 4 to 6 diagnostic ions and matching retentiontime with compounds prepared by total organic synthesis (21,22). Aorta and macrophage samples were analyzed usingliquid chromatography-tandem mass spectrometry (LC/MS/MS), which was performed with a LCQ (ThermoFinnigan,San Jose, CA, USA) quadrupole ion trap spectrometer systemequipped with an electrospray ionization probe. Sampleswere suspended in mobile phase immediately before injec-tion into the HPLC, which consisted of a SpectraSYSTEMSP4000 (ThermoFinnigan) quaternary gradient pump, with aThermo Electron BDS Hypersil C18 (100�2 mm, 5 m)column (ThermoFisher Scientific, Waltham, MA, USA).The column was eluted at a flow rate of 0.2 ml/min withmethanol/water/acetic acid (65:35:0.01, v/v/v) from 0 to 8min and then a gradient increasing to 100% methanolfrom 8.01 to 30 min (21). Lipid mediators were quantifiedusing the area beneath the peak obtained for syntheticstandards and linear calibration as in Pouliot et al. (23) andSerhan (24).
For macrophage incubations with DHA, an Applied Biosys-tems (Foster City, CA, USA) 3200 Q-trap LC/MS/MS systemequipped with a TurboV ionization source with a turbo ionspray probe was used. After extraction, samples were sus-pended in mobile phase and injected into the HPLC compo-nent, which consisted of an Agilent 1100 series binary gradi-ent pump, with an Aglient Eclipse plus C18 (50�4.6 mm, 1.8 m) column (Agilent Technologies, Santa Clara, CA, USA).The column was eluted at a flow rate of 0.4 ml/min withmethanol/water/acetic acid (60:40:0.01, v/v/v) from 0 to 5min and then a gradient increasing to 100% methanol from5.01 to 13 min. Information-dependent acquisition (IDA)used multiple reaction monitoring (MRM) with a dwell timeof 25 ms for each lipid mediator of interest, with sourceparameters set as follows: ion spray voltage, �4200 V; curtaingas, 20 U; ion source gas flow rates 1 and 2 at 50 U each; andtemperature at 400°C. IDA criteria were as follows: the mostabundant compound was chosen to fragment with no exclu-sion of former target and above the threshold of 200 countsper second (cps). For enhanced product ion (EPI) collision,energy was set at �25 V, with a spread of �5 V and �5 V,using dynamic fill-time. The mass range was 100–400 m/z,with a scan rate of 4000 atomic mass units (amu)/s and acomplete cycle (MRM, IDA, and EPI) of �1 s. Lipid media-tors prepared by total organic synthesis were used to obtaincalibration curves for quantitation as in Hong et al. (22).Quantification and identification of products from macro-phage incubations with 17HDHA was carried out postextrac-tion with an Applied Biosystems Q-Star LC/MS/MS systemequipped with a TurboIonSpray ionization source as inSchwab et al. (25).
Statistics
For two-group comparison, we used the t test routinely exceptwhen value distribution failed the normality test, in whichcase we used the Mann-Whitney rank sum test as specified(SigmaStat; Jandel Scientific, Corte Madera, CA, USA). One-way ANOVA was used to compare three groups. Values areexpressed as means � se. Values of P � 0.05 were consideredto be significant.
3597ATHEROSCLEROSIS: A CASE OF UNRESOLVED INFLAMMATION
RESULTS
Transgenic overexpression of 12/15-LO isatheroprotective
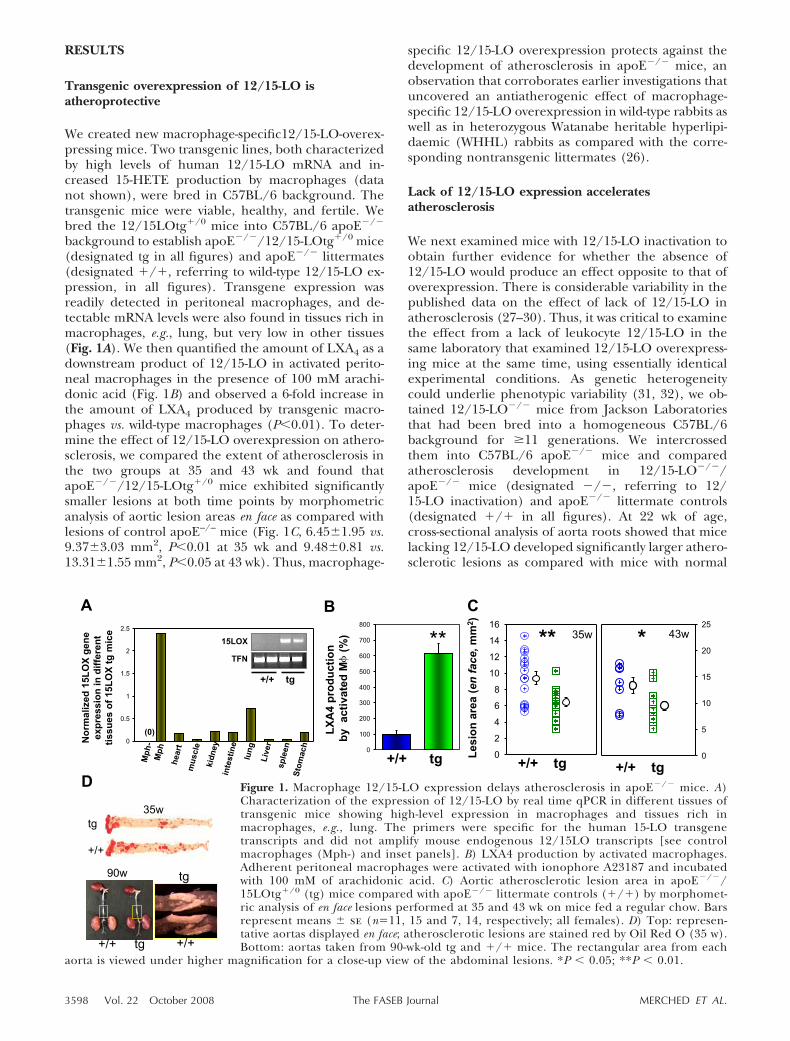
We created new macrophage-specific12/15-LO-overex-pressing mice. Two transgenic lines, both characterizedby high levels of human 12/15-LO mRNA and in-creased 15-HETE production by macrophages (datanot shown), were bred in C57BL/6 background. Thetransgenic mice were viable, healthy, and fertile. Webred the 12/15LOtg�/0 mice into C57BL/6 apoE�/�
background to establish apoE�/�/12/15-LOtg�/0 mice(designated tg in all figures) and apoE�/� littermates(designated �/�, referring to wild-type 12/15-LO ex-pression, in all figures). Transgene expression wasreadily detected in peritoneal macrophages, and de-tectable mRNA levels were also found in tissues rich inmacrophages, e.g., lung, but very low in other tissues(Fig. 1A). We then quantified the amount of LXA4 as adownstream product of 12/15-LO in activated perito-neal macrophages in the presence of 100 mM arachi-donic acid (Fig. 1B) and observed a 6-fold increase inthe amount of LXA4 produced by transgenic macro-phages vs. wild-type macrophages (P�0.01). To deter-mine the effect of 12/15-LO overexpression on athero-sclerosis, we compared the extent of atherosclerosis inthe two groups at 35 and 43 wk and found thatapoE�/�/12/15-LOtg�/0 mice exhibited significantlysmaller lesions at both time points by morphometricanalysis of aortic lesion areas en face as compared withlesions of control apoE–/– mice (Fig. 1C, 6.45�1.95 vs.9.37�3.03 mm2, P�0.01 at 35 wk and 9.48�0.81 vs.13.31�1.55 mm2, P�0.05 at 43 wk). Thus, macrophage-
specific 12/15-LO overexpression protects against thedevelopment of atherosclerosis in apoE�/� mice, anobservation that corroborates earlier investigations thatuncovered an antiatherogenic effect of macrophage-specific 12/15-LO overexpression in wild-type rabbits aswell as in heterozygous Watanabe heritable hyperlipi-daemic (WHHL) rabbits as compared with the corre-sponding nontransgenic littermates (26).
Lack of 12/15-LO expression acceleratesatherosclerosis
We next examined mice with 12/15-LO inactivation toobtain further evidence for whether the absence of12/15-LO would produce an effect opposite to that ofoverexpression. There is considerable variability in thepublished data on the effect of lack of 12/15-LO inatherosclerosis (27–30). Thus, it was critical to examinethe effect from a lack of leukocyte 12/15-LO in thesame laboratory that examined 12/15-LO overexpress-ing mice at the same time, using essentially identicalexperimental conditions. As genetic heterogeneitycould underlie phenotypic variability (31, 32), we ob-tained 12/15-LO�/� mice from Jackson Laboratoriesthat had been bred into a homogeneous C57BL/6background for �11 generations. We intercrossedthem into C57BL/6 apoE�/� mice and comparedatherosclerosis development in 12/15-LO�/�/apoE�/� mice (designated �/�, referring to 12/15-LO inactivation) and apoE�/� littermate controls(designated �/� in all figures). At 22 wk of age,cross-sectional analysis of aorta roots showed that micelacking 12/15-LO developed significantly larger athero-sclerotic lesions as compared with mice with normal
Figure 1. Macrophage 12/15-LO expression delays atherosclerosis in apoE�/� mice. A)Characterization of the expression of 12/15-LO by real time qPCR in different tissues oftransgenic mice showing high-level expression in macrophages and tissues rich inmacrophages, e.g., lung. The primers were specific for the human 15-LO transgenetranscripts and did not amplify mouse endogenous 12/15LO transcripts [see controlmacrophages (Mph-) and inset panels]. B) LXA4 production by activated macrophages.Adherent peritoneal macrophages were activated with ionophore A23187 and incubatedwith 100 mM of arachidonic acid. C) Aortic atherosclerotic lesion area in apoE�/�/15LOtg�/0 (tg) mice compared with apoE�/� littermate controls (�/�) by morphomet-ric analysis of en face lesions performed at 35 and 43 wk on mice fed a regular chow. Barsrepresent means � se (n�11, 15 and 7, 14, respectively; all females). D) Top: represen-tative aortas displayed en face; atherosclerotic lesions are stained red by Oil Red O (35 w).Bottom: aortas taken from 90-wk-old tg and �/� mice. The rectangular area from each
aorta is viewed under higher magnification for a close-up view of the abdominal lesions. *P � 0.05; **P � 0.01.
3598 Vol. 22 October 2008 MERCHED ET AL.The FASEB Journal
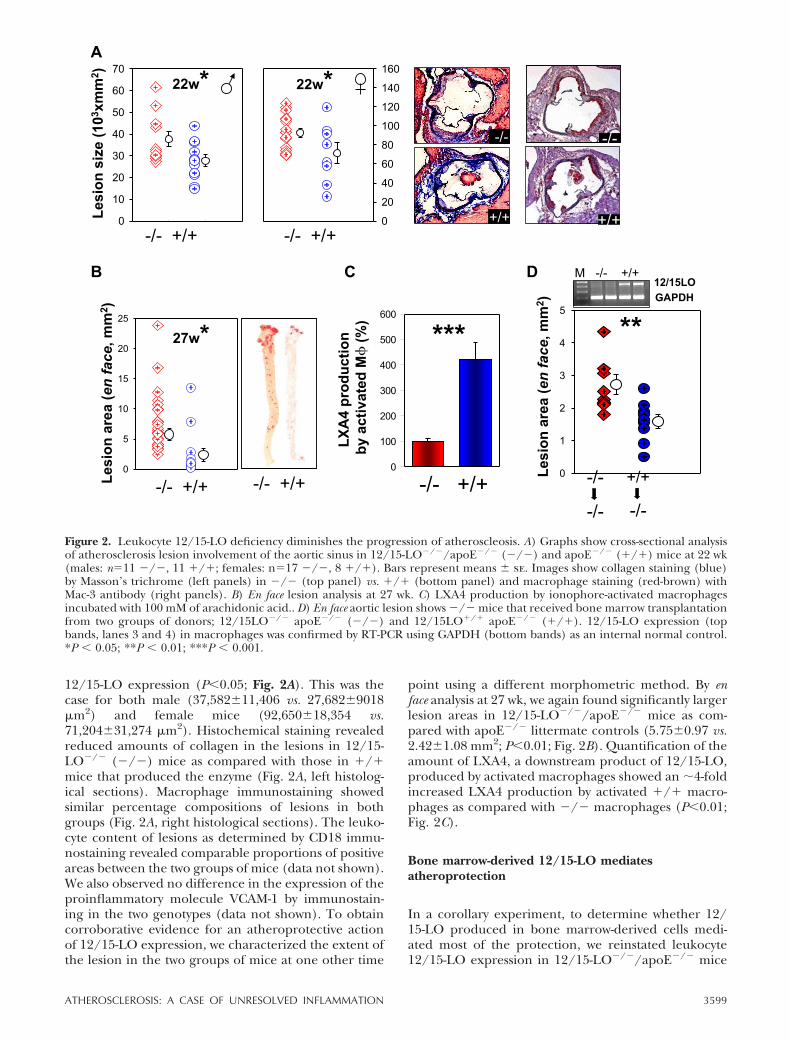
12/15-LO expression (P�0.05; Fig. 2A). This was thecase for both male (37,582�11,406 vs. 27,682�9018 m2) and female mice (92,650�18,354 vs.71,204�31,274 m2). Histochemical staining revealedreduced amounts of collagen in the lesions in 12/15-LO�/� (�/�) mice as compared with those in �/�mice that produced the enzyme (Fig. 2A, left histolog-ical sections). Macrophage immunostaining showedsimilar percentage compositions of lesions in bothgroups (Fig. 2A, right histological sections). The leuko-cyte content of lesions as determined by CD18 immu-nostaining revealed comparable proportions of positiveareas between the two groups of mice (data not shown).We also observed no difference in the expression of theproinflammatory molecule VCAM-1 by immunostain-ing in the two genotypes (data not shown). To obtaincorroborative evidence for an atheroprotective actionof 12/15-LO expression, we characterized the extent ofthe lesion in the two groups of mice at one other time
point using a different morphometric method. By enface analysis at 27 wk, we again found significantly largerlesion areas in 12/15-LO�/�/apoE�/� mice as com-pared with apoE�/� littermate controls (5.75�0.97 vs.2.42�1.08 mm2; P�0.01; Fig. 2B). Quantification of theamount of LXA4, a downstream product of 12/15-LO,produced by activated macrophages showed an �4-foldincreased LXA4 production by activated �/� macro-phages as compared with �/� macrophages (P�0.01;Fig. 2C).
Bone marrow-derived 12/15-LO mediatesatheroprotection
In a corollary experiment, to determine whether 12/15-LO produced in bone marrow-derived cells medi-ated most of the protection, we reinstated leukocyte12/15-LO expression in 12/15-LO�/�/apoE�/� mice
Figure 2. Leukocyte 12/15-LO deficiency diminishes the progression of atheroscleosis. A) Graphs show cross-sectional analysisof atherosclerosis lesion involvement of the aortic sinus in 12/15-LO�/�/apoE�/� (�/�) and apoE�/� (�/�) mice at 22 wk(males: n�11 �/�, 11 �/�; females: n�17 �/�, 8 �/�). Bars represent means � se. Images show collagen staining (blue)by Masson’s trichrome (left panels) in �/� (top panel) vs. �/� (bottom panel) and macrophage staining (red-brown) withMac-3 antibody (right panels). B) En face lesion analysis at 27 wk. C) LXA4 production by ionophore-activated macrophagesincubated with 100 mM of arachidonic acid.. D) En face aortic lesion shows �/� mice that received bone marrow transplantationfrom two groups of donors; 12/15LO�/� apoE�/� (�/�) and 12/15LO�/� apoE�/� (�/�). 12/15-LO expression (topbands, lanes 3 and 4) in macrophages was confirmed by RT-PCR using GAPDH (bottom bands) as an internal normal control.*P � 0.05; **P � 0.01; ***P � 0.001.
3599ATHEROSCLEROSIS: A CASE OF UNRESOLVED INFLAMMATION
by transplanting bone marrow cells from 12/15-LO�/�/apoE�/� and 12/15-LO�/�/apoE�/� donors to 12/15-LO�/�/apoE�/� mice (Fig. 2D). RT-PCR analysisconfirmed that macrophages of recipient mice ac-quired 12/15-LO expression only after they had re-ceived bone marrow cells from 12/15-LO�/�/apoE�/�
mice but not from 12/15-LO�/�/apoE�/� donors(Fig. 2D). Morphometric analysis of aortic atheroscle-rotic lesions in the recipients 15 wk after transplanta-tion revealed that mice receiving 12/15-LO�/�/apoE�/� bone marrow displayed 41% smaller lesionareas (P�0.01) as compared with mice receiving 12/15-LO�/�/apoE�/� bone marrow (1.60�0.62 vs.2.72�0.95 mm2; Fig. 2D). Thus, in support of conclu-sions from an overexpression model, use of a geneticknockout demonstrates a protective role of leukocyte12/15-LO expression against atherosclerosis develop-ment in apoE�/� mice. Moreover, we found that most,if not all, of the atheroprotective effects of 12/15-LOexpression were mediated by bone marrow-derivedcells.
Anti-inflammatory actions of 12/15-LO
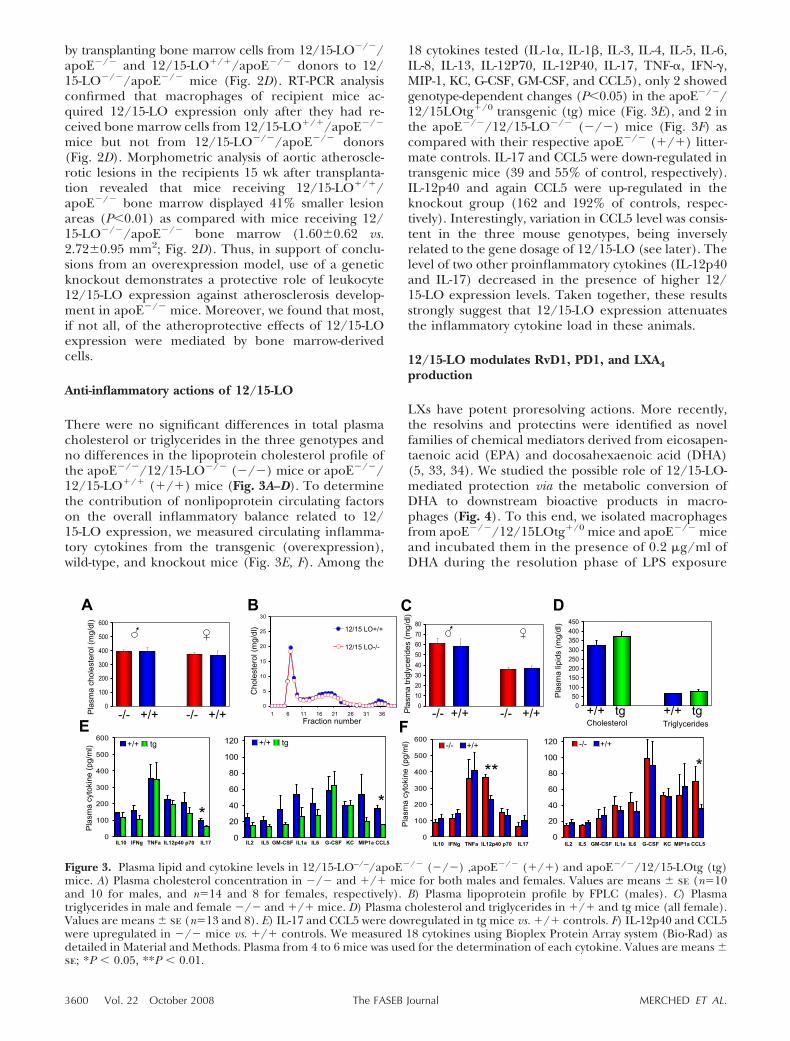
There were no significant differences in total plasmacholesterol or triglycerides in the three genotypes andno differences in the lipoprotein cholesterol profile ofthe apoE�/�/12/15-LO�/� (�/�) mice or apoE�/�/12/15-LO�/� (�/�) mice (Fig. 3A–D). To determinethe contribution of nonlipoprotein circulating factorson the overall inflammatory balance related to 12/15-LO expression, we measured circulating inflamma-tory cytokines from the transgenic (overexpression),wild-type, and knockout mice (Fig. 3E, F). Among the
18 cytokines tested (IL-1�, IL-1�, IL-3, IL-4, IL-5, IL-6,IL-8, IL-13, IL-12P70, IL-12P40, IL-17, TNF-�, IFN-,MIP-1, KC, G-CSF, GM-CSF, and CCL5), only 2 showedgenotype-dependent changes (P�0.05) in the apoE�/�/12/15LOtg�/0 transgenic (tg) mice (Fig. 3E), and 2 inthe apoE�/�/12/15-LO�/� (�/�) mice (Fig. 3F) ascompared with their respective apoE�/� (�/�) litter-mate controls. IL-17 and CCL5 were down-regulated intransgenic mice (39 and 55% of control, respectively).IL-12p40 and again CCL5 were up-regulated in theknockout group (162 and 192% of controls, respec-tively). Interestingly, variation in CCL5 level was consis-tent in the three mouse genotypes, being inverselyrelated to the gene dosage of 12/15-LO (see later). Thelevel of two other proinflammatory cytokines (IL-12p40and IL-17) decreased in the presence of higher 12/15-LO expression levels. Taken together, these resultsstrongly suggest that 12/15-LO expression attenuatesthe inflammatory cytokine load in these animals.
12/15-LO modulates RvD1, PD1, and LXA4production
LXs have potent proresolving actions. More recently,the resolvins and protectins were identified as novelfamilies of chemical mediators derived from eicosapen-taenoic acid (EPA) and docosahexaenoic acid (DHA)(5, 33, 34). We studied the possible role of 12/15-LO-mediated protection via the metabolic conversion ofDHA to downstream bioactive products in macro-phages (Fig. 4). To this end, we isolated macrophagesfrom apoE�/�/12/15LOtg�/0 mice and apoE�/� miceand incubated them in the presence of 0.2 g/ml ofDHA during the resolution phase of LPS exposure
Figure 3. Plasma lipid and cytokine levels in 12/15-LO–/–/apoE�/� (�/�) ,apoE�/� (�/�) and apoE�/�/12/15-LOtg (tg)mice. A) Plasma cholesterol concentration in �/� and �/� mice for both males and females. Values are means � se (n�10and 10 for males, and n�14 and 8 for females, respectively). B) Plasma lipoprotein profile by FPLC (males). C) Plasmatriglycerides in male and female �/� and �/� mice. D) Plasma cholesterol and triglycerides in �/� and tg mice (all female).Values are means � se (n�13 and 8). E) IL-17 and CCL5 were dowregulated in tg mice vs. �/� controls. F) IL-12p40 and CCL5were upregulated in �/� mice vs. �/� controls. We measured 18 cytokines using Bioplex Protein Array system (Bio-Rad) asdetailed in Material and Methods. Plasma from 4 to 6 mice was used for the determination of each cytokine. Values are means �se; *P � 0.05, **P � 0.01.
3600 Vol. 22 October 2008 MERCHED ET AL.The FASEB Journal
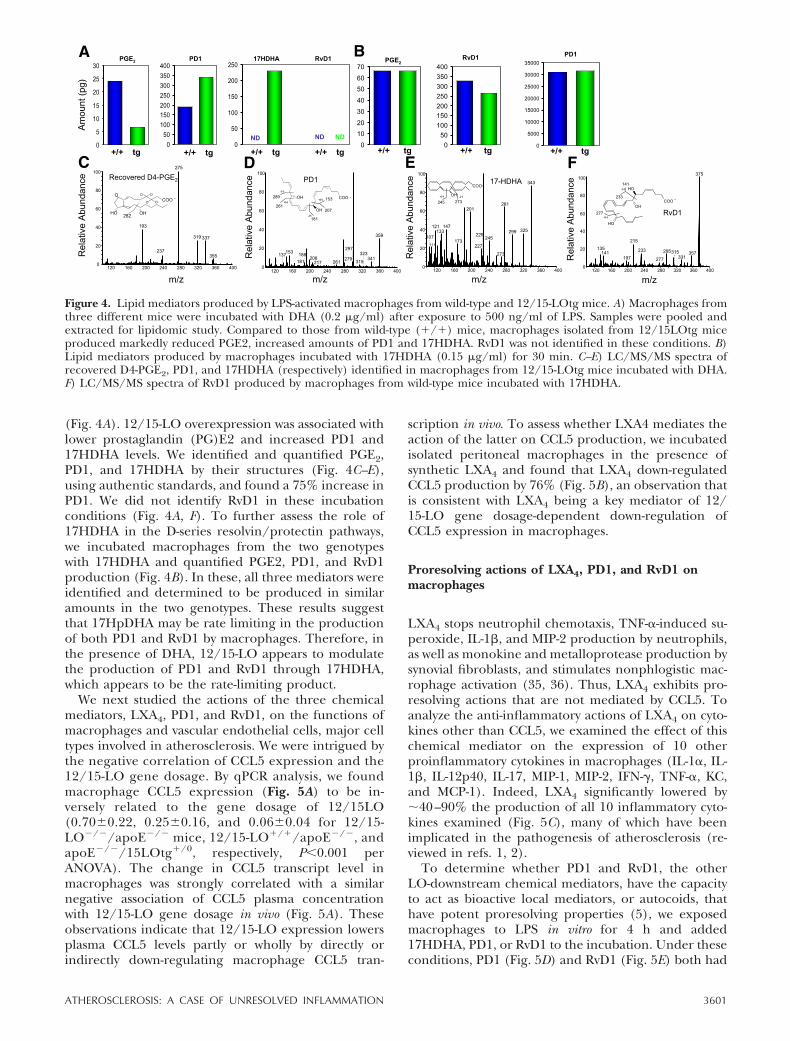
(Fig. 4A). 12/15-LO overexpression was associated withlower prostaglandin (PG)E2 and increased PD1 and17HDHA levels. We identified and quantified PGE2,PD1, and 17HDHA by their structures (Fig. 4C–E),using authentic standards, and found a 75% increase inPD1. We did not identify RvD1 in these incubationconditions (Fig. 4A, F). To further assess the role of17HDHA in the D-series resolvin/protectin pathways,we incubated macrophages from the two genotypeswith 17HDHA and quantified PGE2, PD1, and RvD1production (Fig. 4B). In these, all three mediators wereidentified and determined to be produced in similaramounts in the two genotypes. These results suggestthat 17HpDHA may be rate limiting in the productionof both PD1 and RvD1 by macrophages. Therefore, inthe presence of DHA, 12/15-LO appears to modulatethe production of PD1 and RvD1 through 17HDHA,which appears to be the rate-limiting product.
We next studied the actions of the three chemicalmediators, LXA4, PD1, and RvD1, on the functions ofmacrophages and vascular endothelial cells, major celltypes involved in atherosclerosis. We were intrigued bythe negative correlation of CCL5 expression and the12/15-LO gene dosage. By qPCR analysis, we foundmacrophage CCL5 expression (Fig. 5A) to be in-versely related to the gene dosage of 12/15LO(0.70�0.22, 0.25�0.16, and 0.06�0.04 for 12/15-LO�/�/apoE�/� mice, 12/15-LO�/�/apoE�/�, andapoE�/�/15LOtg�/0, respectively, P�0.001 perANOVA). The change in CCL5 transcript level inmacrophages was strongly correlated with a similarnegative association of CCL5 plasma concentrationwith 12/15-LO gene dosage in vivo (Fig. 5A). Theseobservations indicate that 12/15-LO expression lowersplasma CCL5 levels partly or wholly by directly orindirectly down-regulating macrophage CCL5 tran-
scription in vivo. To assess whether LXA4 mediates theaction of the latter on CCL5 production, we incubatedisolated peritoneal macrophages in the presence ofsynthetic LXA4 and found that LXA4 down-regulatedCCL5 production by 76% (Fig. 5B), an observation thatis consistent with LXA4 being a key mediator of 12/15-LO gene dosage-dependent down-regulation ofCCL5 expression in macrophages.
Proresolving actions of LXA4, PD1, and RvD1 onmacrophages
LXA4 stops neutrophil chemotaxis, TNF-�-induced su-peroxide, IL-1�, and MIP-2 production by neutrophils,as well as monokine and metalloprotease production bysynovial fibroblasts, and stimulates nonphlogistic mac-rophage activation (35, 36). Thus, LXA4 exhibits pro-resolving actions that are not mediated by CCL5. Toanalyze the anti-inflammatory actions of LXA4 on cyto-kines other than CCL5, we examined the effect of thischemical mediator on the expression of 10 otherproinflammatory cytokines in macrophages (IL-1�, IL-1�, IL-12p40, IL-17, MIP-1, MIP-2, IFN-, TNF-�, KC,and MCP-1). Indeed, LXA4 significantly lowered by�40–90% the production of all 10 inflammatory cyto-kines examined (Fig. 5C), many of which have beenimplicated in the pathogenesis of atherosclerosis (re-viewed in refs. 1, 2).
To determine whether PD1 and RvD1, the otherLO-downstream chemical mediators, have the capacityto act as bioactive local mediators, or autocoids, thathave potent proresolving properties (5), we exposedmacrophages to LPS in vitro for 4 h and added17HDHA, PD1, or RvD1 to the incubation. Under theseconditions, PD1 (Fig. 5D) and RvD1 (Fig. 5E) both had
Figure 4. Lipid mediators produced by LPS-activated macrophages from wild-type and 12/15-LOtg mice. A) Macrophages fromthree different mice were incubated with DHA (0.2 g/ml) after exposure to 500 ng/ml of LPS. Samples were pooled andextracted for lipidomic study. Compared to those from wild-type (�/�) mice, macrophages isolated from 12/15LOtg miceproduced markedly reduced PGE2, increased amounts of PD1 and 17HDHA. RvD1 was not identified in these conditions. B)Lipid mediators produced by macrophages incubated with 17HDHA (0.15 g/ml) for 30 min. C–E) LC/MS/MS spectra ofrecovered D4-PGE2, PD1, and 17HDHA (respectively) identified in macrophages from 12/15-LOtg mice incubated with DHA.F) LC/MS/MS spectra of RvD1 produced by macrophages from wild-type mice incubated with 17HDHA.
3601ATHEROSCLEROSIS: A CASE OF UNRESOLVED INFLAMMATION
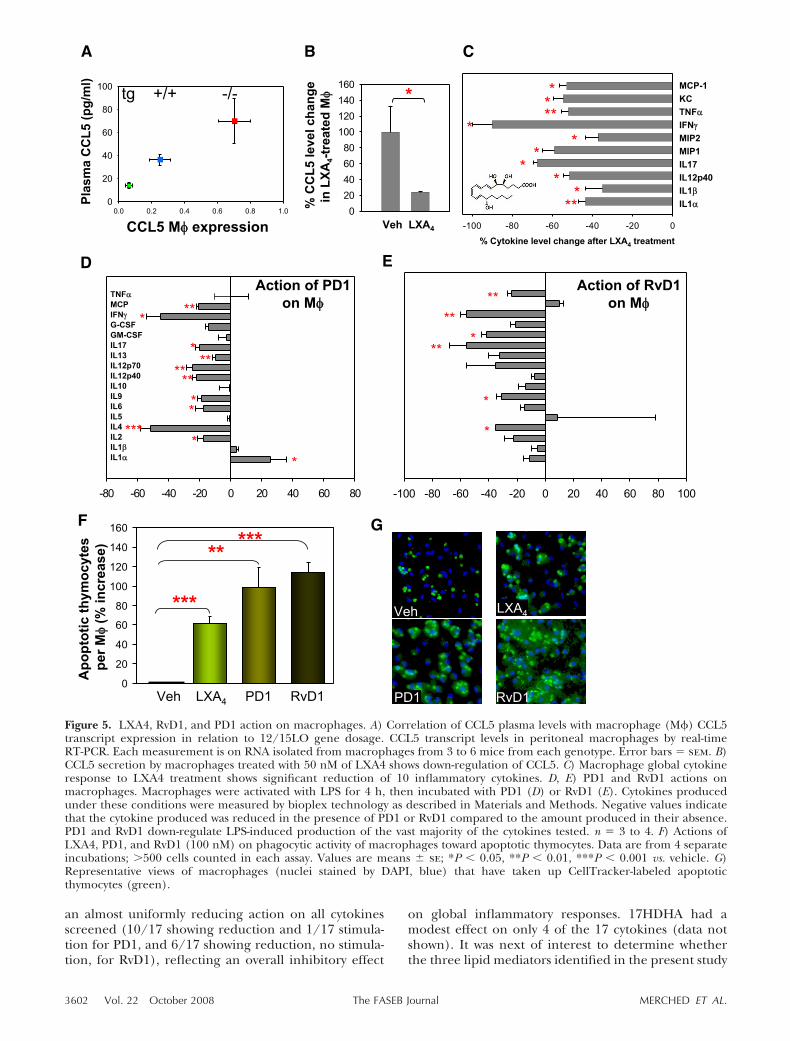
an almost uniformly reducing action on all cytokinesscreened (10/17 showing reduction and 1/17 stimula-tion for PD1, and 6/17 showing reduction, no stimula-tion, for RvD1), reflecting an overall inhibitory effect
on global inflammatory responses. 17HDHA had amodest effect on only 4 of the 17 cytokines (data notshown). It was next of interest to determine whetherthe three lipid mediators identified in the present study
Figure 5. LXA4, RvD1, and PD1 action on macrophages. A) Correlation of CCL5 plasma levels with macrophage (M�) CCL5transcript expression in relation to 12/15LO gene dosage. CCL5 transcript levels in peritoneal macrophages by real-timeRT-PCR. Each measurement is on RNA isolated from macrophages from 3 to 6 mice from each genotype. Error bars � sem. B)CCL5 secretion by macrophages treated with 50 nM of LXA4 shows down-regulation of CCL5. C) Macrophage global cytokineresponse to LXA4 treatment shows significant reduction of 10 inflammatory cytokines. D, E) PD1 and RvD1 actions onmacrophages. Macrophages were activated with LPS for 4 h, then incubated with PD1 (D) or RvD1 (E). Cytokines producedunder these conditions were measured by bioplex technology as described in Materials and Methods. Negative values indicatethat the cytokine produced was reduced in the presence of PD1 or RvD1 compared to the amount produced in their absence.PD1 and RvD1 down-regulate LPS-induced production of the vast majority of the cytokines tested. n � 3 to 4. F) Actions ofLXA4, PD1, and RvD1 (100 nM) on phagocytic activity of macrophages toward apoptotic thymocytes. Data are from 4 separateincubations; �500 cells counted in each assay. Values are means � se; *P � 0.05, **P � 0.01, ***P � 0.001 vs. vehicle. G)Representative views of macrophages (nuclei stained by DAPI, blue) that have taken up CellTracker-labeled apoptoticthymocytes (green).
3602 Vol. 22 October 2008 MERCHED ET AL.The FASEB Journal
modulate the phagocytic activity of macrophages to-ward apoptotic cells, because removal of apoptotic cellsin atherosclerosis is an important proresolving functionof macrophages (37). To this end, we exposed isolatedperitoneal macrophages to LXA4, PD1, or RvD1 andfound that they each increased macrophage uptake ofapoptotic thymocytes by 60, 100, and 115%, respec-tively, compared to vehicle-treated cells (Fig. 5F, G).These findings corroborate and extend our recentdemonstration that PD1 and another lipid mediator,RvE1, increase macrophage ingestion of apoptotic poly-morphonuclear leukocytes in an acute inflammationmodel (25). Interestingly, 12/15-LO has been shown tomodulate actin polymerization and enhance phagocy-tosis of apoptotic cells (38, 39). Herein we have iden-tified specific downstream products that mediate thisaction of 12/15-LO. Thus, the protective effect ofmacrophage expression of 12/15-LO is mediated byfacilitated production of PD1 and RvD1 (the latterthrough up-regulation of the rate-limiting 17HpDHA),which orchestrate proresolving actions via multiplemechanisms. These DHA-dependent lipid mediatorscomplement the action of LXA4, which is also turnedon by the transcellular action of 12/15-LO and 5-LO.
Proresolving actions of LXA4, PD1, and RvD1 onvascular endothelial cells
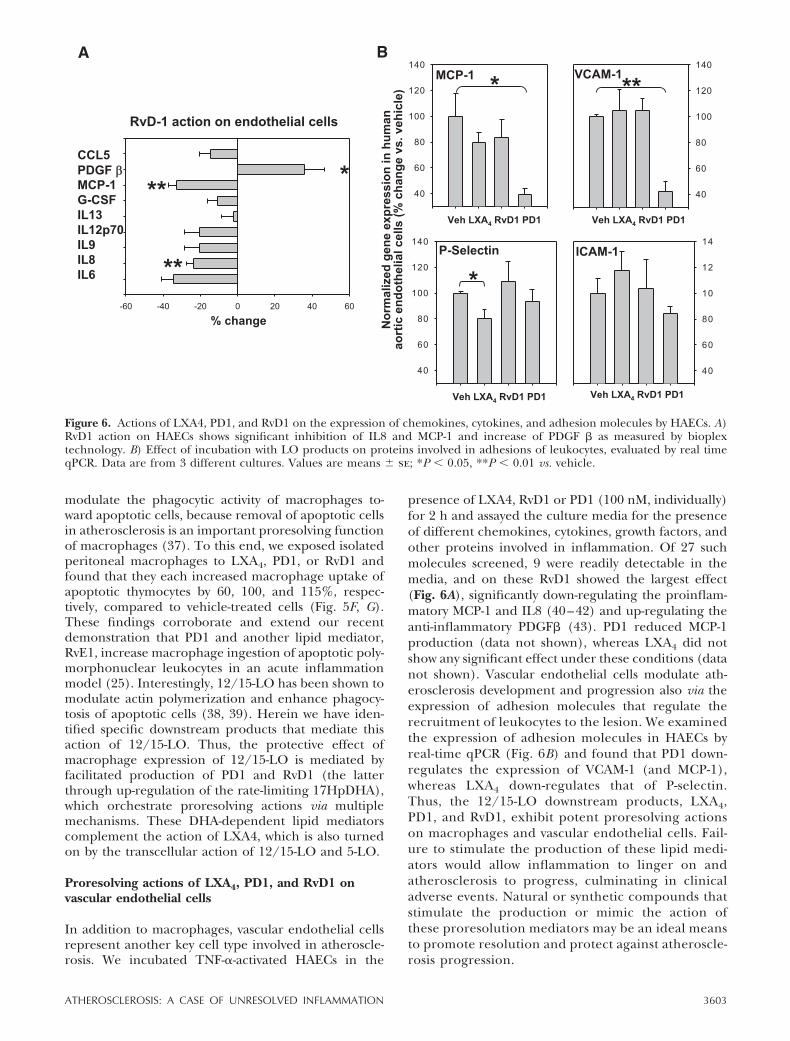
In addition to macrophages, vascular endothelial cellsrepresent another key cell type involved in atheroscle-rosis. We incubated TNF-�-activated HAECs in the
presence of LXA4, RvD1 or PD1 (100 nM, individually)for 2 h and assayed the culture media for the presenceof different chemokines, cytokines, growth factors, andother proteins involved in inflammation. Of 27 suchmolecules screened, 9 were readily detectable in themedia, and on these RvD1 showed the largest effect(Fig. 6A), significantly down-regulating the proinflam-matory MCP-1 and IL8 (40–42) and up-regulating theanti-inflammatory PDGF� (43). PD1 reduced MCP-1production (data not shown), whereas LXA4 did notshow any significant effect under these conditions (datanot shown). Vascular endothelial cells modulate ath-erosclerosis development and progression also via theexpression of adhesion molecules that regulate therecruitment of leukocytes to the lesion. We examinedthe expression of adhesion molecules in HAECs byreal-time qPCR (Fig. 6B) and found that PD1 down-regulates the expression of VCAM-1 (and MCP-1),whereas LXA4 down-regulates that of P-selectin.Thus, the 12/15-LO downstream products, LXA4,PD1, and RvD1, exhibit potent proresolving actionson macrophages and vascular endothelial cells. Fail-ure to stimulate the production of these lipid medi-ators would allow inflammation to linger on andatherosclerosis to progress, culminating in clinicaladverse events. Natural or synthetic compounds thatstimulate the production or mimic the action ofthese proresolution mediators may be an ideal meansto promote resolution and protect against atheroscle-rosis progression.
Figure 6. Actions of LXA4, PD1, and RvD1 on the expression of chemokines, cytokines, and adhesion molecules by HAECs. A)RvD1 action on HAECs shows significant inhibition of IL8 and MCP-1 and increase of PDGF � as measured by bioplextechnology. B) Effect of incubation with LO products on proteins involved in adhesions of leukocytes, evaluated by real timeqPCR. Data are from 3 different cultures. Values are means � se; *P � 0.05, **P � 0.01 vs. vehicle.
3603ATHEROSCLEROSIS: A CASE OF UNRESOLVED INFLAMMATION

DISCUSSION
Increasing evidence and awareness point to aberrantinflammation as a principal factor in the progression ofatherosclerosis in humans (1, 2). The appreciation thatthe natural course of acute inflammation is resolution (5,6), together with the present results showing that LXA4,RvD1, and PD1 mediate the antiatherosclerosis actionswith macrophage-specific 12/15-LO overexpression,strongly supports the conclusion that atherosclerosis is anonresolving form of vascular inflammation. The disrup-tion of atherosclerotic plaques in humans via percutane-ous transluminal coronary angioplasty leads to rapidappearance (within 10 s) of leukotrienes and lipoxins inthe lumen of the vessel (44). The ratio of leukotrienes tolipoxins favors leukotriene formation in the lumen andlesion, which suggests that the absence and/or deficiencyof intraluminal LXA4 generation in humans may lead toan inability to counterregulate local vascular inflamma-tion and hence the progression of local insult from acuteto chronic, resulting in the disease phenotype, i.e., athero-sclerosis.
Recent results (45, 46) also showed that 5-LO and the5-LO-activating protein (FLAP) is associated with in-creased incidence of cardiovascular disease. LeukotrieneB4 production is upregulated during atherosclerosis inhumans, and 5-LO is found associated with atheroscle-rotic lesions (8, 47, 48). The enhanced production ofproinflammatory mediators, such as leukotrienes, under-scores the balance toward a proinflammatory milieuaround atherosclerotic lesions, as well as the genetictendency toward acquiring this disease in select humanpopulations. Thus, diminished capacity to generate localLXA4 at the sites of vascular inflammation or local vascu-lar insults may contribute to the nonresolving vascularinflammation and its progression to atherosclerosis. Fur-thermore, as discussed in the introduction, only two casecontrol studies (10, 11) on human genetic variants of12/15-LO, a key enzyme involved in LXA4 biosynthesis,
have been published, and both supported a protectiverole for 12/15-LO expression against coronary arterydisease.
Indeed, we found that the addition of LXA4 to isolatedmacrophages leads to a down-regulation of macrophagegenes, such as CCL5 (Fig. 5), that are involved in control-ling the local balance of inflammatory mediators in vivo.Of interest, proresolving lipid mediators up-regulateCCR5 on the surface of human leukocytes that are directlyinvolved with the binding of local chemokines and cyto-kines and their accelerated clearance during apoptosis viamacrophages (49). In fact, CCL5 is but one of manyproinflammatory cytokines normally produced by macro-phages that are globally suppressed by exposure to LXA4,consistent with the lipid mediator producing a proresolu-tion milieu locally, putting a brake on atherosclerosisdevelopment.
In addition to the lipoxins, resolvins, and protectins aretwo novel families of locally generated lipid mediatorsderived from �-3 fatty acids (EPA and DHA) that displaypotent anti-inflammatory and proresolving actions in vivo.In murine systems, the 12/15-LO plays a critical role inthe biosynthesis of protectins and resolvins of the D series(34, 50). Recent results (51) demonstrate that 12/15-LO-deficient mice have an impaired ability to generateD-series resolvins and protectins and thus are unable toefficiently repair wounds of the corneal epithelia. Alzhei-mer’s plaque lesions in humans and mouse models alsoappear to have defective 12/15-LO expression and areunable to generate the DHA-derived protectins (52). Inthis study, we found that 12/15-LO expression stimulatesthe production of protectin (PD)1 from 17-HDHA inmacrophages during the resolution phase of inflamma-tion. Furthermore, these cells also have the capacity toproduce resolvin (Rv)D1. Both PD1 and RvD1 are potentproresolution lipid mediators derived from �-3 fatty acidsdownstream of 12/15-LO. Individually, they suppress alarge proportion of the proinflammatory cytokines pro-
Figure 7. Model of atherosclerosis as a nonre-solving form of vascular inflammation. Theessential �-6 PUFA arachidonic acid (AA) isreleased from phospholipids in cells by theaction of cytosolic phospholipase A2. After spe-cific enzymatic steps, AA is converted into dif-ferent families of mediators: prostaglandins(PGs) and leukotrienes (LTs), which are mostlyproinflammatory molecules, and lipoxins suchas LXA4, a stop-inflammation mediator. Theessential �-3 PUFA DHA is converted to twonovel mediators, RvD1 and PD1, that promoteresolution. We propose a model in which ab-sence of macrophage 12/15LO leads to a defi-ciency in proresolving end products, RvD1 andPD1, as well as LXA4, locally at the site of theongoing inflammation, crippling multiple pro-resolving functions, leaving the proinflamma-tory milieu unabated, and fueling atherosclero-sis progression. LOX, lipoxygenase.
3604 Vol. 22 October 2008 MERCHED ET AL.The FASEB Journal

duced by macrophages (Fig. 5D, E). Like LXA4, PD1 andRvD1 also stimulate the phagocytic activity of macro-phages toward apoptotic cells (Fig. 5F, E), an anti-inflam-matory and proresolution function that is thought to beimportant in both acute inflammation (25) and in ath-erosclerosis (37). Complementing their action on macro-phages, the three lipid mediators downstream of 12/15-LO also exhibit concerted inhibitory actions onadhesion molecule and chemokine expression by vascularendothelial cells, putting a brake on the recruitment ofinflammatory cells, to allow the resolution phase to set inand give the vascular wall a chance to return to normality.Hence, it appears that deficiencies in 12/15-LO, whichplays a critical role in initiating the biosynthesis of anti-inflammatory and proresolving lipid mediators includinglipoxins, resolvins, and protectins during the course oflocal acute inflammation, can lead to an inability toefficiently resolve recurring bouts of inflammation andhence chronic inflammatory states that can give rise toprogressive atherosclerosis.
In summation (Fig. 7), the present results demonstratethat regulated expression of the 12/15-LO and some of itspathway products derived from arachidonic acid, specifi-cally LXA4, and from �-3 fatty acids, RvD1 and PD1, caneach directly regulate macrophage functions and geneexpression of interest in the progression of atherosclero-sis. Adding back these specific proresolving lipid media-tors down-regulates gene expression held to play criticalroles in controlling local inflammation and the develop-ment of atherosclerosis. Hence, the present results sug-gest that appropriate targeted regulation of 12/15-LOand/or the use of stable mimetics of resolvins, protectins,and/or lipoxin may be a new approach in reducing theprogression of atherosclerosis. These results also point tothe possibility that global deficiencies in 12/15-LO, my-eloid lineage and/or specific macrophages in humanscan lead to aberrant local vascular inflammation and theatherosclerotic phenotype.
We thank Dr. Colin Funk (University of Pennsylvania, Phila-delphia, PA, USA) for sending 12/15-LO�/� mice that we usedfor pilot experiments before purchasing them from JacksonLaboratory for the definitive experiments. This work was sup-ported by American Heart Association grants to A.M. (0465093Yand 0730172N) and grants from the National Institutes ofHealth (P50 DE016191 to C.N.S., and HL-51586 to L.C.).
REFERENCES
1. Hansson, G. K. (2005) Mechanisms of disease-Inflammation,atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352,1685–1695
2. Libby, P. (2002) Inflammation in atherosclerosis. Nature 420,868–874
3. Stary, H. C., Chandler, A. B., Glagov, S., Guyton, J. R., Insull, W.,Rosenfeld, M. E., Schaffer, S. A., Schwartz, C. J., Wagner, W. D.,and Wissler, R. W. (1994) A definition of initial, fatty streak, andintermediate lesions of atherosclerosis—a report from the com-mittee on vascular-lesions of the Council on Arteriosclerosis,American-Heart-Association. Circulation 89, 2462–2478
4. Stary, H. C. (1989) Evolution and progression of atheroscleroticlesions in coronary-arteries of children and young-adults. Arte-riosclerosis 9, I19–I32
5. Serhan, C. N. (2007) Resolution phase of inflammation: novelendogenous anti-inflammatory and proresolving lipid mediatorsand pathways. Ann. Rev. Immunol. 25, 101–137
6. Serhan, C. N., Savill, J. (2005) Resolution of inflammation: thebeginning programs the end. Nat. Immunol. 6, 1191–1197
7. Samuelsson, B. (1983) Leukotrienes: mediators of immediatehypersensitivity reactions and inflammation. Science 220, 568–575
8. Qiu, H., Gabrielsen, A., Agardh, H. E., Wan, M., Wetterholm, A.,Wong, C. H., Hedin, U., Swedenborg, J., Hansson, G. K.,Samuelsson, B., Paulsson-Berne, G., and Haeggstrom, J. Z.(2006) Expression of 5-lipoxygenase and leukotriene A4 hydro-lase in human atherosclerotic lesions correlates with symptomsof plaque instability. Proc. Natl. Acad. Sci. U. S. A. 103, 8161–8166
9. Spanbroek, R., Grabner, R., Lotzer, K., Hildner, M., Urbach, A.,Ruhling, K., Moos, M. P., Kaiser, B., Cohnert, T. U., Wahlers, T.,Zieske, A., Plenz, G., Robenek, H., Salbach, P., Kuhn, H.,Radmark, O., Samuelsson, B., and Habenicht, A. J. (2003)Expanding expression of the 5-lipoxygenase pathway within thearterial wall during human atherogenesis. Proc. Natl. Acad. Sci.U. S. A. 100, 1238–1243
10. Wittwer, J., Bayer, M., Mosandl, A., Muntwyler, J., and Hers-berger, M. (2007) The c.-292C�T promoter polymorphismincreases reticulocyte-type 15-lipoxygenase-1 activity and couldbe atheroprotective. Clin. Chem. Lab. Med. 45, 487–492
11. Assimes, T. L., Knowles, J. W., Priest, J. R., Basu, A., Borchert, A.,Volcik, K. A., Grove, M. L., Tabor, H. K., Southwick, A.,Tabibiazar, R., Sidney, S., Boerwinkle, E., Go, A. S., Iribarren,C., Hlatky, M. A., Fortmann, S. P., Myers, R. M., Kuhn, H., Risch,N., and Quertermous, T. (2007) A near null variant of 12/15-LOX encoded by a novel SNP in ALOX15 and the risk ofcoronary artery disease. Atherosclerosis 198, 136–144
12. Duffield, J. S., Hong, S., Vaidya, V. S., Lu, Y., Fredman, G.,Serhan, C. N., and Bonventre, J. V. (2006) Resolvin D Series andprotectin D1 mitigate acute kidney injury. J. Immunol. 177,5902–5911
13. Munger, K. A., Montero, A., Fukunaga, M., Uda, S., Yura, T., Imai,E., Kaneda, Y., Valdivielso, J. M., Badr, K. F. (1999) Transfection ofrat kidney with human 15-lipoxygenase suppresses inflammationand preserves function in experimental glomerulonephritis. Proc.Natl. Acad. Sci. U. S. A. 96, 13375–13380
14. Levy, B. D., DeSanctis, G. T., Devchand, P. R., Kim, E., Acker-man, K., Schmidt, B. A., Szczeklik, W., Drazen, J. M., andSerhan, C. N. (2002) Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat. Med. 8,1018–1023
15. Goh, J., Godson, C., Brady, H. R., and MacMathuna, P. (2003)Lipoxins: pro-resolution lipid mediators in intestinal inflamma-tion. Gastroenterology 124, 1043–1054
16. Oka, K., Pastore, L., Kim, I. H., Merched, A., Nomura, S., Lee,H. J., Merched-Sauvage, M., Arden-Riley, C., Lee, B., Finegold,M., Beaudet, A., and Chan, L. (2001) Long-term stable correc-tion of low-density lipoprotein receptor-deficient mice with ahelper-dependent adenoviral vector expressing the very low-density lipoprotein receptor. Circulation 103, 1274–1281
17. Horvai, A., Palinski, W., Wu, H., Moulton, K. S., Kalla, K., andGlass, C. K. (1995) Scavenger receptor A gene regulatoryelements target gene expression to macrophages and to foamcells of atherosclerotic lesions. Proc. Natl. Acad. Sci. U. S. A. 92,5391–595
18. Merched, A. J., Williams, E., and Chan, L. (2003) Macrophage-specific p53 expression plays a crucial role in atherosclerosisdevelopment and plaque remodeling. Arterioscler. Thromb. Vasc.Biol. 23, 1608–1614
19. Merched, A. J., and Chan, L. (2004) Absence of p21Waf1/Cip1/Sdi1 modulates macrophage differentiation and inflammatoryresponse and protects against atherosclerosis. Circulation 110,3830–3841
20. Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy,N., De Paepe, A., and Speleman, F. (2002) Accurate normaliza-tion of real-time quantitative RT-PCR data by geometric averag-ing of multiple internal control genes. Genome Biol. 3, research0034
21. Lu, Y., Hong, S., Gotlinger, K., and Serhan, C. N. (2006) Lipidmediator informatics and proteomics in inflammation resolu-tion. ScientificWorldJournal 6, 589–614
3605ATHEROSCLEROSIS: A CASE OF UNRESOLVED INFLAMMATION

22. Hong, S., Lu, Y., Yang, R., Gotlinger, K. H., Petasis, N. A., andSerhan, C. N. (2007) Resolvin D1, protectin D1, and relateddocosahexaenoic acid-derived products: Analysis via electros-pray/low energy tandem mass spectrometry based on spectraand fragmentation mechanisms. J. Am. Soc. Mass. Spectrom. 18,128–144
23. Pouliot, M., Clish, C. B., Petasis, N. A., Van Dyke, T. E., andSerhan, C. N. (2000) Lipoxin A(4) analogues inhibit leukocyterecruitment to Porphyromonas gingivalis: a role for cyclooxygen-ase-2 and lipoxins in periodontal disease. Biochemistry 39, 4761–4768
24. Serhan, C. N. (1990) High-performance liquid chromatographyseparation and determination of lipoxins. Methods Enzymol. 187,167–175
25. Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007)Resolvin E1 and protectin D1 activate inflammation-resolutionprogrammes. Nature 447, 869–874
26. Shen, J., Herderick, E., Cornhill, J. F., Zsigmond, E., Kim, H. S.,and Kuhn, H., Guevara, N. V., Chan, L. (1996) Macrophage-mediated 15-lipoxygenase expression protects against athero-sclerosis development. J. Clin. Invest. 98, 2201–2208
27. Cyrus, T., Pratico, D., Zhao, L., Witztum, J. L., Rader, D. J.,Rokach, J., FitzGerald, G. A., and Funk, C. D. (2001) Absence of12/15-lipoxygenase expression decreases lipid peroxidationand atherogenesis in apolipoprotein e-deficient mice. Circula-tion 103, 2277–2282
28. George, J., Afek, A., Shaish, A., Levkovitz, H., Bloom, N., Cyrus,T., Zhao, L., Funk, C. D., Sigal, E., and Harats, D. (2001)12/15-Lipoxygenase gene disruption attenuates atherogenesisin LDL receptor-deficient mice. Circulation 104, 1646–1650
29. Huo, Y., Zhao, L., Hyman, M. C., Shashkin, P., Harry, B. L.,Burcin, T., Forlow, S. B., Stark, M. A., Smith, D. F., Clarke, S.,Srinivasan, S., Hedrick, C. C., Pratico, D., Witztum, J. L., Nadler,J. L., Funk, C. D., and Ley, K. (2004) Critical role of macrophage12/15-lipoxygenase for atherosclerosis in apolipoprotein E-de-ficient mice. Circulation 110, 2024–2031
30. Reilly, K. B., Srinivasan, S., Hatley, M. E., Patricia, M. K.,Lannigan, J., Bolick, D. T., Vandenhoff, G., Pei, H., Natarajan,R., Nadler, J. L., and Hedrick, C. C. (2004) 12/15-Lipoxygenaseactivity mediates inflammatory monocyte/endothelial interac-tions and atherosclerosis in vivo. J. Biol. Chem. 279, 9440–9450
31. Moore, K. J., Kunjathoor, V. V., Koehn, S. L., Manning, J. J.,Tseng, A. A., Silver, J. M., McKee, M., and Freeman, M. W.(2005) Loss of receptor-mediated lipid uptake via scavengerreceptor A or CD36 pathways does not ameliorate atherosclero-sis in hyperlipidemic mice. J. Clin. Invest. 115, 2192–2201
32. Reardon, C. A., Blachowicz, L., Lukens, J., Nissenbaum, M., andGetz, G. S. (2003) Genetic background selectively influencesinnominate artery atherosclerosis: immune system deficiency asa probe. Arterioscler. Thromb. Vasc. Biol. 23, 1449–1454
33. Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang,N., and Gronert, K. (2000) Novel functional sets of lipid-derivedmediators with antiinflammatory actions generated from ome-ga-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflamma-tory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204
34. Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand,P. R., Mirick, G., and Moussignac, R. L. (2002) Resolvins: afamily of bioactive products of omega-3 fatty acid transforma-tion circuits initiated by aspirin treatment that counter proin-flammation signals. J. Exp. Med. 196, 1025–1037
35. Hachicha, M., Pouliot, M., Petasis, N. A., and Serhan, C. N.(1999) Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 in-hibit tumor necrosis factor 1alpha-initiated neutrophil re-sponses and trafficking: regulators of a cytokine-chemokineaxis. J. Exp. Med. 189, 1923–1930
36. Sodin-Semrl, S., Taddeo, B., Tseng, D., Varga, J., and Fiore, S.(2000) Lipoxin A4 inhibits IL-1 beta-induced IL-6, IL-8, andmatrix metalloproteinase-3 production in human synovial fibro-blasts and enhances synthesis of tissue inhibitors of metallopro-teinases. J. Immunol. 164, 2660–2666
37. Fadok, V. A., and Chimini, G. (2001) The phagocytosis ofapoptotic cells. Semin. Immunol. 13, 365–372
38. Miller, Y. I., Chang, M. K., Funk, C. D., Feramisco, J. R., andWitztum, J. L. (2001) 12/15-lipoxygenase translocation en-
hances site-specific actin polymerization in macrophages phago-cytosing apoptotic cells. J. Biol. Chem. 276, 19431–19439
39. Miller, Y. I., Worrall, D. S., Funk, C. D., Feramisco, J. R., andWitztum, J. L. (2003) Actin polymerization in macrophages inresponse to oxidized LDL and apoptotic cells: role of 12/15-lipoxygenase and phosphoinositide 3-kinase. Mol. Biol. Cell 14,4196–4206
40. Boisvert, W. A., Santiago, R., Curtiss, L. K., and Terkeltaub, R. A.(1998) A leukocyte homologue of the IL-8 receptor CXCR-2mediates the accumulation of macrophages in atheroscleroticlesions of LDL receptor-deficient mice. J. Clin. Invest. 101,353–363
41. Boring, L., Gosling, J., Cleary, M., and Charo, I. F. (1998)Decreased lesion formation in CCR2�/� mice reveals a role forchemokines in the initiation of atherosclerosis. Nature 394,894–897
42. Gosling, J., Slaymaker, S., Gu, L., Tseng, S., Zlot, C. H., Young,S. G., Rollins, B. J., and Charo, I. F. (1999) MCP-1 deficiencyreduces susceptibility to atherosclerosis in mice that overexpresshuman apolipoprotein B. J. Clin. Invest. 103, 773–778
43. Tang, J., Kozaki, K., Farr, A. G., Martin, P. J., Lindahl, P.,Betsholtz, C., and Raines, E. W. (2005) The absence of platelet-derived growth factor-B in circulating cells promotes immuneand inflammatory responses in atherosclerosis-prone ApoE�/�
mice. Am. J. Pathol. 167, 901–91244. Brezinski, D. A., Nesto, R. W., and Serhan, C. N. (1992)
Angioplasty triggers intracoronary leukotrienes and lipoxin A4.Impact of aspirin therapy. Circulation 86, 56–63
45. Helgadottir, A., Manolescu, A., Thorleifsson, G., Gretarsdottir,S., Jonsdottir, H., Thorsteinsdottir, U., Samani, N. J., Gud-mundsson, G., Grant, S. F., Thorgeirsson, G., Sveinbjornsdottir,S., Valdimarsson, E. M., Matthiasson, S. E., Johannsson, H.,Gudmundsdottir, O., Gurney, M. E., Sainz, J., Thorhallsdottir,M., Andresdottir, M., Frigge, M. L., Topol, E. J., Kong, A.,Gudnason, V., Hakonarson, H., Gulcher, J. R., and Stefansson,K. (2004) The gene encoding 5-lipoxygenase activating proteinconfers risk of myocardial infarction and stroke. Nat. Genet. 36,233–239
46. Helgadottir, A., Gretarsdottir, S., St Clair, D., Manolescu, A.,Cheung, J., Thorleifsson, G., Pasdar, A., Grant, S. F., Whalley,L. J., Hakonarson, H., Thorsteinsdottir, U., Kong, A., Gulcher,J., Stefansson, K., and MacLeod, M. J. (2005) Associationbetween the gene encoding 5-lipoxygenase-activating proteinand stroke replicated in a Scottish population. Am. J. Hum. Genet76, 505–509
47. Dwyer, J. H., Allayee, H., Dwyer, K. M., Fan, J., Wu, H., Mar, R.,Lusis, A. J., and Mehrabian, M. (2004) Arachidonate 5-lipoxy-genase promoter genotype, dietary arachidonic acid, and ath-erosclerosis. N. Engl. J. Med. 350, 29–37
48. Mehrabian, M., Allayee, H., Wong, J., Shi, W., Wang, X. P.,Shaposhnik, Z., Funk, C. D., Lusis, A. J. (2002) Identification of5-lipoxygenase as a major gene contributing to atherosclerosissusceptibility in mice. Circ. Res. 91, 120–126
49. Ariel, A., Fredman, G., Sun, Y. P., Kantarci, A., Van Dyke, T. E.,Luster, A. D., and Serhan, C. N. (2006) Apoptotic neutrophilsand T cells sequester chemokines during immune responseresolution through modulation of CCR5 expression. Nat. Immu-nol. 7, 1209–1216
50. Serhan, C. N., Gotlinger, K., Hong, S., Lu, Y., Siegelman, J.,Baer, T., Yang, R., Colgan, S. P., and Petasis, N. A. (2006)Anti-inflammatory actions of neuroprotectin D1/protectin D1and its natural stereoisomers: assignments of dihydroxy-contain-ing docosatrienes. J. Immunol. 176, 1848–1859
51. Gronert, K., Maheshwari, N., Khan, N., Hassan, I. R., Dunn, M.,and Laniado, S. M. (2005) A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healingand host defense. J. Biol. Chem. 280, 15267–15278
52. Lukiw, W. J., Cui, J. G., Marcheselli, V. L., Bodker, M., Botkjaer,A., Gotlinger, K., Serhan, C. N., and Bazan, N. G. (2005) A rolefor docosahexaenoic acid-derived neuroprotectin D1 in neuralcell survival and Alzheimer disease. J. Clin. Invest. 115, 2774–2783
Received for publication April 30, 2008.Accepted for publication May 22, 2008.
3606 Vol. 22 October 2008 MERCHED ET AL.The FASEB Journal
Related Documents











