The FASEB Journal express article 10.1096/fj.00-0843fje. Published online June 27, 2001. Aspirin inhibits NF-κB and protects from angiotensin II- induced organ damage Dominik N. Muller,* ,†,‡ Vigo Heissmeyer,* ,†,‡ Ralf Dechend,* ,† Franziska Hampich,* Joon-Keun Park,* ,§ Anette Fiebeler,* ,§ Erdenechimeg Shagdarsuren,* Jürgen Theuer,* Marlies Elger, § Bernhard Pilz,* Volker Breu, ¶ Karsten Schroer,** Detlev Ganten, †,†† Rainer Dietz,* Hermann Haller, § Claus Scheidereit, † Friedrich C. Luft* *Franz Volhard Clinic, Medical Faculty of the Charite´, Humboldt University of Berlin, Germany; † Max Delbrück Center for Molecular Medicine, Berlin, Germany; § Medizinische Hochschule Hannover, Hannover, Germany; ¶ Hoffmann-La Roche, Basel, Switzerland; †† Free University of Berlin, Berlin, Germany; **Heinrich Heine University Düsseldorf, Germany ‡ These authors contributed equally to this work Corresponding author: Friedrich C. Luft, Franz Volhard Clinic, Wiltberg Strasse 50, 13125 Berlin, Germany. E-mail: [email protected] ABSTRACT Angiotensin (Ang)-II induces vascular wall inflammation by activating NF-κB. Aspirin inhibits IKKβ in vitro; however, the in vivo relevance of the phenomenon is unclear. We tested the hypothesis that aspirin protects from Ang II-induced endorgan damage by inhibiting NF-κB activation in vivo. Rats harboring human renin and angiotensinogen genes received high- (600 mg/kg/day) or low- (25 mg/kg/day) dose aspirin. High-dose aspirin reduced mortality, cardiac hypertrophy, fibrosis, and albuminuria independent of blood pressure, whereas both doses reduced cyclooxygenase activity. High-dose aspirin inhibited NF-κB and AP-1 activation and inflammation in heart and kidney. These in vivo results serve to explain the clinical utility of high-dose aspirin in inflammatory disorders and suggest additional therapeutic avenues that may be relevant to cardiovascular disease. Key words: angiotensin II • aspirin, NF-κB • end-organ damage • inflammation ngiotensin (Ang)-II not only causes vascular constriction, aldosterone release, and sodium reabsorption, but it also induces cell growth, proliferation, and inflammatory responses important to hypertension, cardiac, and renal disease (1–3). Various studies have investigated the link between Ang II and nuclear factor κB (NF-κB) (4–6). NF-κB regulates genes involved in the control of the immune and inflammatory responses (7, 8). NF- κB signaling has been shown in cardiovascular diseases featuring renin-angiotensin system activation. (6, 9–13). Intracellular NF-κB resides inactive and bound to the inhibitory protein IκB in the cytoplasm of T lymphocytes, monocytes, macrophages, endothelial cells, and vascular smooth muscle cells. The complex is activated by TNF-α, IL-1, reactive oxygen species, and A

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The FASEB Journal express article 10.1096/fj.00-0843fje. Published online June 27, 2001.
Aspirin inhibits NF-κB and protects from angiotensin II-induced organ damage Dominik N. Muller,*,†,‡ Vigo Heissmeyer,*,†,‡ Ralf Dechend,*,† Franziska Hampich,* Joon-Keun Park,*,§ Anette Fiebeler,*,§ Erdenechimeg Shagdarsuren,* Jürgen Theuer,* Marlies Elger,§ Bernhard Pilz,* Volker Breu,¶ Karsten Schroer,** Detlev Ganten,†,†† Rainer Dietz,* Hermann Haller,§ Claus Scheidereit,† Friedrich C. Luft*
*Franz Volhard Clinic, Medical Faculty of the Charite´, Humboldt University of Berlin, Germany; † Max Delbrück Center for Molecular Medicine, Berlin, Germany; §Medizinische Hochschule Hannover, Hannover, Germany; ¶Hoffmann-La Roche, Basel, Switzerland; ††Free University of Berlin, Berlin, Germany; **Heinrich Heine University Düsseldorf, Germany ‡These authors contributed equally to this work Corresponding author: Friedrich C. Luft, Franz Volhard Clinic, Wiltberg Strasse 50, 13125 Berlin, Germany. E-mail: [email protected] ABSTRACT Angiotensin (Ang)-II induces vascular wall inflammation by activating NF-κB. Aspirin inhibits IKKβ in vitro; however, the in vivo relevance of the phenomenon is unclear. We tested the hypothesis that aspirin protects from Ang II-induced endorgan damage by inhibiting NF-κB activation in vivo. Rats harboring human renin and angiotensinogen genes received high- (600 mg/kg/day) or low- (25 mg/kg/day) dose aspirin. High-dose aspirin reduced mortality, cardiac hypertrophy, fibrosis, and albuminuria independent of blood pressure, whereas both doses reduced cyclooxygenase activity. High-dose aspirin inhibited NF-κB and AP-1 activation and inflammation in heart and kidney. These in vivo results serve to explain the clinical utility of high-dose aspirin in inflammatory disorders and suggest additional therapeutic avenues that may be relevant to cardiovascular disease. Key words: angiotensin II • aspirin, NF-κB • end-organ damage • inflammation
ngiotensin (Ang)-II not only causes vascular constriction, aldosterone release, and sodium reabsorption, but it also induces cell growth, proliferation, and inflammatory responses important to hypertension, cardiac, and renal disease (1–3). Various studies
have investigated the link between Ang II and nuclear factor κB (NF-κB) (4–6). NF-κB regulates genes involved in the control of the immune and inflammatory responses (7, 8). NF-κB signaling has been shown in cardiovascular diseases featuring renin-angiotensin system activation. (6, 9–13). Intracellular NF-κB resides inactive and bound to the inhibitory protein IκB in the cytoplasm of T lymphocytes, monocytes, macrophages, endothelial cells, and vascular smooth muscle cells. The complex is activated by TNF-α, IL-1, reactive oxygen species, and
A

numerous other stimuli. The activation is mediated by increased activity of an IκB kinase (IKK) complex. Two kinases termed IKK-α and IKK-β phosphorylate IκBβ, leading to ubiquitination and degradation by the 26S proteasome. The liberated heterodimer p50-p65 translocates into the nucleus, where it activates the genes for IL-1, IL-6, TNF-α, intracellular adhesion molecule-1, vascular cell adhesion molecule (VCAM)-1, and others participating in inflammation (8, 14). Aspirin acetylates cyclooxygenase leading to the irreversible inhibition of prostaglandin synthesis (15). In the intact organism, aspirin is rapidly deacetylated to salicylate. Low-dose aspirin inhibits platelet aggregation and has utility in preventing myocardial infarction and stroke. At higher doses, aspirin and salicylates promote anti-inflammatory/antiproliferative effects independent of cyclooxygenase-1 and 2 activity (16–18). Aspirin and sodium salicylate both inhibit NF-κB in vitro (19, 20). Yin et al. (21) showed in cell culture that high-dose aspirin inhibited IKKβ activity by competing with ATP for the ATP binding site. This observation may explain the use of high-dose aspirin for inflammatory disorders such as rheumatoid arthritis. IKK inhibition by aspirin has not been shown in vivo. Furthermore, whether or not aspirin might inhibit other transcription factors is uncertain (22–24). Frantz et al. showed that sodium salicylate also inhibites the activator protein (AP)-1 promotor activity after phorbol ester treatment (24). Ang II is a potent activator of AP-1 regulating vascular growth and fibrosis (25, 26). We tested the utility of aspirin in ameliorating Ang II-induced vascular injury in a model of human disease, specifically focusing on NF-κB and AP-1 activation. We studied rats harboring both the human renin and angiotensinogen genes (dTGR). These animals generate large amounts of Ang II in their tissues, develop hypertension, and die of cardiac and renal damage at about 7 weeks of age. In earlier studies, we documented an inflammatory cascade, NF-κB, and AP-1 activation (27, 28). We treated the animals with aspirin intraperitoneally (600 mg/kg/day or 25 mg/kg/day) from weeks 4 through 7. The low-dose resembles that given to patients at risk for cardiovascular disease and the high dose on a body surface area basis is similar to doses used years ago in the treatment of acute flares in rheumatoid arthritis. Chronic treatment with 600 mg/kg/day seems excessive. However, the aspirin clearance is threefold higher in rats than in humans (29, 30). For example the ED50 ranges from 150 to 350 mg/kg for aspirin in various analgesic test models. Therefore, dose comparison with the human situation on the basis of body weight is not useful. In addition, the intraperitoneal route of administration obviated gastrointestinal complications in our rats. MATERIALS AND METHODS Animals Four-week-old male age- and body weight-matched dTGR and Sprague-Dawley (SD) rats were used as described elsewhere (28). All procedures were done according to guidelines from the American Physiological Society. We compared untreated dTGR (n=30), high-dose aspirin treated (600 mg/kg/d DL lysin mono-acetylsalicylicate i.p.; n=27), low-dose aspirin treated dTGR (25 mg/kg/d; n=27), and SD control rats (n=15) receiving vehicle (0.9% saline i.p.). Systolic blood pressure was measured at week 5, 6, and 7 by the tail-cuff method under light ether anesthesia. Echocardiography (M-mode tracings and short axis; n=5 per group) was performed using a commercially available system equipped with a 7-MHz phased-array

transducer under light thiopental anesthesia. Three measurements per heart were determined, averaged, and statistically analyzed. Urine was collected over a 24-h period. Albumin was determined by ELISA (CellTrend, Luckenwalde, Germany). Rats were killed at age 7 weeks. The kidneys and hearts were washed with ice-cold saline, blotted dry, and weighed. For Western blot and NF-κB analysis, the tissues were snap-frozen in liquid nitrogen, and for immunohistochemistry in isopentane (–35°C), and stored at –80°C. Electrophoretic mobility shift assay (EMSA) Tissue preparation for EMSA was performed as described earlier (28). Nuclear extracts (5 µg) were incubated in binding reaction medium with 0.5 ng of 32P-dATP end-labeled oligonucleotide, containing the NF-κB binding site from the MHC-enhancer (H2K, 5'-gatcCAGGGCTGGGGATTCCCCATCTCCACAGG). For AP-1 double-stranded oligonucleotides containing the consensus sequence for AP-1 (Santa Cruz Biotechnology, Santa Cruz, CA; 5'-CGCTTGATGACTCAGCCGGAA-3') were radiolabeled with γ-32P with the use of T4 polynucleotide kinase by standard methods and purified over a column. The DNA-protein complexes were analyzed on a 5% polyacrylamide gel, dried, and autoradiographed. In competition assays, 50 ng of unlabeled H2K or AP-1 oligonucleotides were used. Homogenates (30 µg) were used for Western blot and stained with monoclonal VCAM-1 antibody (PharMingen, Heidelberg, Germany). In vitro kinase assay and immunoprecipitation Rat kidneys were homogenized in liquid nitrogen and resuspended in lysis buffer as described previously (31). In brief, lysates were shaken for 15 min at 4°C and centrifuged at 120,000 g for 30 min. Supernatants were precleared with Protein A Sepharose for 1 h at 4°C, and 1.8 mg of tissue extract was immunoprecipitated with anti-IKKγ polyclonal antisera (32) for 3 h. Immunoprecipitations were washed two times in lysis buffer and once in kinase buffer as described previously (31). Kinase reactions were performed with 1 µg IκBα- or 1 µg IκBαS32/36A-peptides comprising amino acids 1–53 of IκBα or the respective pointmutant. Kinase reactions were separated on 15% SDS-PAGES and visualized by 15 min autoradiography. IKKγ-immunoprecipitates were analyzed with immunoblots probed with IKKα (H-744, Santa Cruz Biotechnology, Heidelberg, Germany) and IKKβ (Biosource, Camarillo, CA). IKKγ-immunoprecipitates were prepared identically to the IP procedure used for in vitro kinase reactions. Precipitates were washed four times in lysis buffer, boiled in twofold SDS-loading buffer, separated on 9% SDS-PAGE, and stained with anti-IKKα (H-744, Santa Cruz) antisera or anti-IKKβ antibodies (Biosource) with chemiluminescence system. For bacterial protein expression and purification, IκBα or IκBα-S32/36A peptides were expressed as C-terminal fusions of glutathion S-transferase in pGEX-expression vectors (Pharmacia, Freiburg, Germany). Fusion protein expression was induced by 3 h of IPTG-stimulation of BL-21pLys S bacteria, transformed with IκBα-expression vectors. Cells were lyzed by sonication, induced proteins were bound to glutathion Sepharose (Pharmacia), and the GST-fusionpart was cleaved off with Precission protease (Pharmacia), according to the manufacturer’s protocol. Purity and the amount of peptides were controlled on 15% SDS-PAGE. Immunofluorescence staining

Ice-cold acetone-fixed cryosections (6 µm) were air dried and immersed in TBS (0.05 M Tris buffer and 0.15 M NaCl, pH 7.6). All incubations were performed in a humid chamber at room temperature. At first, the sections were incubated in 10% normal donkey serum (Dianova, HamburGermany) for 30 min to block any nonspecific binding. The sections were incubated for 60 min with the following monoclonal antibodies: anti-VCAM-1, anti-VLA-4, anti-CD4 (all PharMingen), anti-ED-1, anti-CD8 (both Serotec, Eching, Germany), polyclonal antibodies anti-laminin (Sigma Chemie, Deisenhofen, Germany), and anti-fibronectin (Paessel, Frankfurt, Germany). After washing with TBS, the sections were incubated with Cy3-conjugated secondary antibodies (donkey anti-mouse IgG-Cy3, donkey anti-rabbit IgG-Cy3, or donkey anti-goat IgG-Cy3; Dianova) for 60 min. After a final washing with TBS, slides were mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA). In controls, where a primary antibody was substituted by isotype control antibody CBL 600 mouse IgG1-ve control (Cymbus Biotechnology, Hampshire, UK) at the same final concentration, no specific immunolabeling was observed. The nonspecific binding of secondary antibodies was excluded by omitting the primary antibody. Preparations were analyzed under a Zeiss Axioplan-2 microscope (Carl Zeiss, Jena, Germany) and were digitally photographed using an AxioVision 2 multichannel image processing system (Carl Zeiss). Electron microscopy was performed as described earlier (33). Statistics Data are presented as means ± SE. Statistically significant differences in mean values were tested by ANOVA, blood pressure by repeated measures ANOVA, and the Scheffé test as indicated. A value of P < 0.05 was considered statistically significant. The data were analyzed using Statview statistical software. RESULTS Renal damage in dTGR Light and electron microscope investigations of untreated dTGR kidneys revealed glomerular and renal vascular damage (Fig. 1A, upper panel). Glomeruli showed all stages, from apparently normal structure to advanced sclerosis. Glomeruli with moderate changes exhibited mesangial proliferation and numerous electron dense inclusions in podocytes. In sclerotic glomeruli, severe foot process effacement of podocytes resulted in flat cell portions without interdigitation. These glomeruli were further characterized by a reduction in endothelial fenestration, marked matrix accumulation, and capillary obliteration. Small renal arteries (mainly cortical radial arteries) and arterioles showed frequently increased intimal thickness and marked thickening of the media due to proliferation of smooth muscle cells, and deposition of matrix and lipid were accompanied by reduction of the vessel lumen. In contrast, nontransgenic SD rats showed no vascular or glomerular damage (Fig. 1A, lower panel). High-dose aspirin reduces mortality and organ damage

High-dose aspirin reduced mortality by 85%. In contrast, mortality in low-dose aspirin was not different from no treatment (Fig. 1B). However, neither dose lowered blood pressure. Only nontransgenic SD rats had low blood pressure levels (Fig. 1C). dTGR had a 24-h albumin excretion 150-fold greater than SD rats. High-dose aspirin reduced albuminuria by 86% (P<0.0001), whereas low-dose aspirin did not (P=0.68; Fig. 1D). We then investigated whether low-dose aspirin was insufficient to affect cyclooxygenase activity. Plasma thromboxane (TXB)2 (Fig. 1E) was significantly increased in dTGR compared with all other groups (P<0.05). Low-dose and high-dose aspirin both reduced TXB2 to SD levels. High-dose aspirin reduced cardiac fibronectin (Fig. 2A) and collagen I production (data not shown), whereas low-dose aspirin did not. Left ventricular hypertrophy and diminished left ventricular cavity dimensions were observed in untreated and low-dose treated dTGR by M-mode echocardiography. High-dose aspirin treated rats had markedly improved left ventricles similar to normal rats (Fig. 2B). Cavity diameters of untreated dTGR (3.1±0.09 mm) and low-dose aspirin (4.4±0.06 mm) were significantly lower compared with high-dose aspirin (5.2±0.2 mm) and nontransgenic SD (5.9±0.1 mm) (P<0.05). Cardiac hypertrophy index, expressed as the ratio of heart weight to body weight, confirmed the data on matrix formation and echocardiography. Body weights between untreated and aspirin-treated dTGR were not different. However, SD were heavier than dTGR. High-dose aspirin also reduced renal interstitial fibronectin and laminin, perivascular collagen I, and glomerular collagen IV expression (data not shown), whereas low-dose aspirin had no effect. High-dose aspirin inhibits renal and cardiac NF-κB and AP-1 We used EMSAs for the detection of NF-κB and AP-1 DNA binding activity. Nuclear extracts of untreated dTGR show increased NF-κB DNA binding activity in kidney (Fig. 3A) and heart (Fig. 3C) compared with SD controls. High-dose aspirin reduced NF-κB binding, whereas low-dose aspirin had no effect. Binding specificity was demonstrated by competition of excess unlabeled oligonucleotides containing the κB site from the MHC-enhancer (H2K). Nuclear extracts were supershifted with antibodies against the NF-κB subunits p50 and p65. High-dose aspirin also inhibited AP-1 DNA binding activity in kidney (Fig. 3B) and heart (Fig. 3D). The effects of low-dose aspirin could not be distinguished from no treatment. Complexes were supershifted with antibodies against c-fos and c-jun. AP-1 binding was prevented by competition with excess of unlabeled oligonucleotides containing the AP-1 site. Each lane represents a separate animal. Semiquantification for NF-κB and AP-1 is shown in Figures 3E and 3F. The densitometric signal is expressed as a percentage of untreated dTGR. Hearts and kidney nuclear extracts were prepared from three representative animals per group. Each extract was analyzed by EMSA in three separate runs. The shifts were quantitated with the NIH image program, and the data were analyzed by ANOVA followed by Scheffé post hoc test. Aspirin inhibits IKK activity To test whether the increased NF-κB activity in dTGR kidneys was caused by chronically activated IκB kinases, we set up an in vitro kinase assay with extracts prepared from kidney tissue. Immunoprecipitations against IKKγ were performed to coimmunoprecipitate the signal-dependently regulated IKK complex. We tested the ability of the precipitates to phosphorylate

IκBα at serines 32 and 36. A specific IKK activity in precipitates of dTGR kidneys was observed that was clearly markedly increased compared with activity from SD controls (Fig. 4A). Specific phosphorylation of serines 32 and 36 of IκBα from IKK complexes from high-dose and low-dose aspirin groups was indistinguishable. The kinase activity in both groups was reproducibly intermediate, compared with untreated dTGR and SD rats. Thus, aspirin at either dose inhibited IκB kinase activity. IKKγ immunoprecipitation and subsequent IKKβ Western blotting consistently revealed more IKKγ from dTGR than from SD kidneys (Fig. 4B). Control immunoprecipitations from dTGR kidneys with anti-CDK4 antibody showed no increased CDK4 protein, compared with kidneys from SD rats (not shown). We also ruled out any Ang II-induced stimulation of IKKβ protein expression in human coronary artery vascular smooth muscle cells (data not shown). The detection of IKKγ-IKKβ, but not IKKα containing IKK-complexes, has recently been reported for CD4+-T cells (34) and may reflect increased IKKχ, but not IKKα mRNA transcription, in spleen as seen in the Northern blot analysis of multiple tissues (35). We have observed that significantly more IKKβ than IKKα is associated with IKKγ in extracts from murine spleen compared with lung and kidney (data not shown). Elevated IKKβ levels in dTGR kidneys thus may be in part related to infiltrating lymphocytes. These findings prompted us to investigate the infiltration of lymphocytes into kidney and heart tissue in our hypertensive animal model. Inflammatory responses in heart and kidney Untreated dTGR showed leukocyte attachment to the renal endothelium (Fig. 5A) leading to cell infiltration and dramatic inflammatory injury. The endothelium of nontransgenic SD showed no attached leukocytes (data not shown). Monocytes/macrophages (Mo/Ma; ED-1+) were found infiltrating predominantly around the damaged vessels, whereas T helper cells (CD4+) showed perivascular and interstitial and cytotoxic T cells (CD8+) interstitial, periglomerular, and glomerular locations (data not shown). Semiquantification revealed that high-dose and low-dose aspirin both reduced CD4+ (Fig. 5C) as well as CD8+ cell infiltration (Fig. 5D) in heart and kidney. However, the extent of the reduction was significantly different between the aspirin groups. Although low-dose aspirin treatment led to more pronounced CD4+ cell reduction, there was only a slight effect on CD8+ cells and no reduction of Mo/Ma (Fig. 5B). In contrast, high-dose aspirin reduced all infiltrated cell types. Inflammatory responses in the heart were less pronounced than in the kidney. Vascular adhesion molecule VCAM-1 and very late antigen (VLA)-4 expression We investigated the effect of aspirin treatment on NF-κB/AP-1 regulated adhesion molecules. Figure 6A shows immunofluorescence staining of VCAM-1 (red) on the endothelium of a renal vessel. The sections were counterstained with an anti-laminin antibody to visualize the internal elastic lamina (green) in the vessel wall. The activated endothelium of untreated and low-dose aspirin treated dTGR showed marked expression of VCAM-1 and intercellular adhesion molecule-1 (data not shown). In contrast, high-dose aspirin reduced the immunofluorescent staining towards non-transgenic control level. Western blotting (Fig. 6B) of kidney and heart extracts confirmed the VCAM-1 and intercellular adhesion molecule-1 expression data (data not shown). We also stained for the VCAM-1 ligand VLA-4 on infiltrated leukocytes in kidney and

heart. Semiquantitative analysis (Fig. 6C) showed a reduction of VLA-4-positive cells by high-dose aspirin, compared with low-dose aspirin and no treatment. DISCUSSION We present the first in vivo data on IKK/NF-κB inhibition by aspirin in Ang II-induced organ damage. Not only were the effects of high-dose aspirin limited to the IKK/NF-κB pathway, but they also involved AP-1 inhibition. Because NF-κB also functions in concert with AP-1 (7), the finding that high-dose aspirin inhibits both transcription factors may have therapeutic importance. Our transgenic rat model showed severe inflammatory organ damage with increased NF-κB and AP-1 DNA binding activity (27, 28, 36). We treated the rats with both high- and low-dose aspirin to distinguish between direct effects on the NF-κB-inflammatory system and possible organ protection due to cyclooxygenase inhibition. We found that only high-dose aspirin reduced mortality, cardiac hypertrophy, fibrosis, and albuminuria independent of blood pressure. In contrast, both doses blocked cyclooxygenase activity. Ang II has been investigated for decades in terms of causing peripheral vascular constriction, aldosterone release, and renal sodium reabsorption. More recently, the fact that Ang II causes cell growth and proliferation by stimulating tyrosine phosphorylation has become appreciated. That Ang II stimulates an inflammatory cascade, involving NF-κB activation, is more recent still. Increased production of superoxide anion in hypertension was shown a decade ago (37). More recently, the fact that Ang II stimulates the generation of reactive oxygen species by activating NADH/NADPH oxidases has been shown in human vascular smooth muscle cells (26). It is well appreciated that superoxide anion is capable of initiating NF-κB activation (38). Another recent finding that has a bearing on inflammation is the fact that Ang II activates NF-κB through both Ang receptors (AT1 and AT2) (4, 39). We documented a chain of inflammatory events involving NF-κB activation in our dTGR model and showed that the cascade could be modified by treating the rats with a NF-κB inhibitor, pyrrolidine dithiocarbamate (28). NF-κB plays an important role in the pathogenesis of numerous cardiovascular diseases with an activated renin-angiotensin system, including myocardial infarction, renal disease, stroke, restenosis after balloon injury, reperfusion injury after transplantation and atherosclerosis (6, 9–13, 27, 28, 40, 41). Myocardial infarction, renal disease, and reperfusion injury after transplantation develop to a large extent subsequent to the complex interaction of multiple cytokines and activated adhesion molecules leading to inflammatory damage (10, 12, 13). Morishita et al. (10) demonstrated that specific NF-κB inhibition by a decoy technique reduced the extent of myocardial infarction following reperfusion. Recently, two other groups used the NF-κB decoy technique to demonstrate the importance of NF-κB in cardiovascular disease (12, 13). In renal allografts, pretreatment of donor kidneys with NF-κB decoys led to reduced NF-κB activity and expression of VCAM-1, leading to reduced cell infiltration (12). Inhibition of NF-κB in various models with renal damage resulted in decreased expression of adhesion molecules with reduced inflammatory response, leading to improved renal function (13, 28). In this study, only high-dose aspirin treatment was sufficient to reduce NF-κB DNA binding activity, decreased mortality, cardiac hypertrophy, albuminuria, and inflammation. Inhibition of cyclooxygenase activity was not sufficient to reduce progression of the disease.

Kopp et al. (20) and Pierce et al. (19) showed that sodium salicylate and aspirin can both inhibit the activation of NF-κB by preventing the phosphorylation and degradation of IκBα. Furthermore, Pierce et al. also reported that sodium salicylate inhibited NF-κB-regulated adhesion molecules and neutrophil transmigration in endothelial cells. Our data are in agreement with and extend the observation by Pierce from the cell culture to the in vivo situation. Recently, cell culture studies of Yin et al. (21) provided a cellular target for the aspirin-NF-κB intervention. They identified the ATP binding site of IKKβ as a target for reversible, competitive aspirin binding, whereas IKKα was not affected. However, the half-maximal inhibitory concentration IC50 for aspirin required to inhibit endogenous IKKβ was relatively high, ranging from 50 to 100 µM. In contrast, various other groups observed only inhibitory effects at even higher micromolar concentrations (19, 20, 23). Therefore, the in vivo relevance of their observations is still unclear. Although serum aspirin levels of 1–2 mM seem to be too low for NF-κB inhibition, local concentrations at the site of inflammation may reach sufficient levels for IKK/NF-κB inhibition. Salicylates are organic acids and could accumulate at the mildly acidic environments occurring at sites of inflammation (42–44). At low pH, salicylates are uncharged and can enter the cell membrane with subsequent deprotonation in the cell. In their anion form, salicylates are trapped in a more neutral environment and could reach concentrations that exceed the IC50 for NF-κB inhibition in vivo. Our results show that chronic high-dose aspirin treatment was sufficient to inhibit NF-κB. However, the inhibitory effects of both low and high aspirin concentrations on IKK activity shown in our study primarily reflects lowered IKKβ amounts, presumably caused by impaired lymphocyte infiltration. Direct aspirin effects on IKK activity are less likely detectable due to the excess of ATP present in the in vitro kinase reaction. Yin et al. (21) and Kwak et al. (45) both showed a direct aspirin-IKKβ interaction. Kwak et al. showed that the presence of the leucin zipper and helix-loop-helix domain of IKKβ, but not the corresponding domains of IKKα, were associated with inhibited IKK kinase activity. However, other investigators (22–24) have questioned the specificity of any aspirin-NF-κB relationship. Recently, Alpert et al. (23) demonstrated that in cell culture, sodium salicylate preincubation inhibited only TNF-α, but not IL-1-induced IKK kinase activity. In contrast, under in vitro conditions, IKK kinase activity of both cytokines was reduced. In addition, they presented data that p38 MAP kinase is an essential cofactor for the salicylate-IKK kinase inhibition. Frantz et al. (24) demonstrated the effect of sodium salicylate on CRE and AP-1 promoter activity and kinase activity. Because AP-1 is also activated by Ang II, we addressed the possibility that aspirin may also inhibit AP-1 DNA binding activity. Our data clearly show that aspirin also reduced AP-1 activity and AP-1-regulated fibrosis in kidney and heart. The effect of high-dose aspirin on AP-1 may also in part explain the marked reduction in cardiac hypertrophy. In our earlier study, chronic PDTC treatment in dTGR affected primarily NF-κB, and not AP-1. The treatment led only to a partial reduction in cardiac hypertrophy (28). In this study, high-dose aspirin was more effective in ameliorating hypertrophy. Recently, two groups showed that Ang II induced c-fos, c-jun, and AP-1 activity in vascular smooth muscle cells (25, 26). We can only speculate whether aspirin inhibited AP-1 directly or whether NF-κB inhibition affected AP-1 activation (46). In light of earlier data (24), the first explanation appears more likely. Very recently, Marra et al. (16) have shown that high-dose salicylates inhibited smooth

muscle cell proliferation by cell cycle arrest at the G1-S phase. Thus, salicylate action might might also contribute to the antihypertrophic/antiproliferative effect observed in high aspirin treated dTGR. In addition, Wang and Brecher have shown that salicylate inhibits phosphorylation of the nonreceptor tyrosine kinases, proline-rich tyrosine kinase 2 and c-src. Because c-src plays an important role in Ang II signal transduction (47), chronic high-dose aspirin might also inhibit c-src phosphorylation in vivo. Our results also indicate that blood pressure reduction was not necessary to protect the organs from damage. Rats treated with high-dose aspirin still had systolic blood pressure levels between 170 and 200 mm Hg. We showed earlier (48) that complete normalization of blood pressure with reserpine, hydralazine, and hydrochlorothiazide only delayed endorgan damage by 1 wk. Treatment of dTGR with a soluble TNF-α receptor antagonist also reduced cardiac and renal damage independent of blood pressure reduction (unpublished data). These results underscore the effects of local Ang II on cytokines and adhesion molecules in mediating organ damage. In summary, our results are the first to demonstrate that IKK/NF-κB, as well as AP-1, inhibition by aspirin leads to organ protection in vivo. High-dose aspirin reduced mortality, cardiac hypertrophy, fibrosis, and albuminuria independent of blood pressure. Aspirin itself or its derivatives may have therapeutic utility in ameliorating Ang II-related effects. In addition, more potent IKKβ inhibitors may be an important therapeutic option in cardiovascular disease. ACKNOWLEDGMENTS This study was supported by a grant-in-aid from Hoffmann-La Roche, Basel, Switzerland. D.N.M. was supported by the Klinisch Pharamakologische Verbund Berlin-Brandenburg. Karin Dressler, Mathilde Schmidt, Christel Lipka, Monika Schlöter, Michaela Beese, and Petra Berkefeld gave expert technical assistance. REFERENCES
1. Ingelfinger, J. R., and Dzau, V. J. (1991) Molecular biology of renal injury: emphasis on the role of the renin-angiotensin system. J. Am. Soc. Nephrol. 2, S9–20
2. Malik, F. S., Lavie, C. J., Mehra, M. R., Milani, R. V., and Re, R. N. (1997) Renin-angiotensin
system: genes to bedside. Am. Heart J. 134, 514–526 3. Weir, M. R., and Dzau, V. J. (1999) The renin-angiotensin-aldosterone system: a specific target
for hypertension management. Am. J. Hypertens. 12, 205S–213S 4. Ruiz-Ortega, M., Lorenzo, O., Ruperez, M., Konig, S., Wittig, B., and Egido, J. (2000)
Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: molecular mechanisms. Circ. Res. 86, 1266–1272
5. Sadoshima, J. (2000) Cytokine actions of angiotensin II. Circ. Res. 86, 1187–1189

6. Klahr, S., and Morrissey, J. J. (2000) The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int. 57 Suppl 75, S7–14
7. Barnes, P. J., and Karin, M. (1997) Nuclear factor-kappaB: a pivotal transcription factor in
chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071 8. Karin, M., and Ben-Neriah, Y. (2000) Phosphorylation meets ubiquitination: the control of NF-
[kappa]B activity. Annu. Rev. Immunol. 18, 621–663 9. Hernandez Presa, M. A., Bustos, C., Ortego, M., Tunon, J., Ortega, L., and Egido, J. (1998) ACE
inhibitor quinapril reduces the arterial expression of NF-kappaB-dependent proinflammatory factors but not of collagen I in a rabbit model of atherosclerosis. Am. J. Pathol. 153, 1825–1837
10. Morishita, R., Sugimoto, T., Aoki, M., Kida, I., Tomita, N., Moriguchi, A., Maeda, K., Sawa, Y.,
Kaneda, Y., Higaki, J., and Ogihara, T. (1997) In vivo transfection of cis element "decoy" against nuclear factor- kappaB binding site prevents myocardial infarction. Nat. Med. 3, 894–899
11. Morrissey, J. J., and Klahr, S. (1997) Rapid communication. Enalapril decreases nuclear factor
kappa B activation in the kidney with ureteral obstruction. Kidney Int. 52, 926–933 12. Vos, I. H., Govers, R., Grone, H. J., Kleij, L., Schurink, M., De Weger, R. A., Goldschmeding,
R., and Rabelink, T. J. (2000) NFkappaB decoy oligodeoxynucleotides reduce monocyte infiltration in renal allografts. FASEB J. 14, 815–822
13. Tomita, N., Morishita, R., Lan, H. Y., Yamamoto, K., Hashizume, M., Notake, M., Toyosawa,
K., Fujitani, B., Mu, W., Nikolic-Paterson, D. J., Atkins, R. C., Kaneda, Y., Higaki, J., and Ogihara, T. (2000) In vivo administration of a nuclear transcription factor-kappaB decoy suppresses experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 11, 1244–1252
14. Israel, A. (2000) The IKK complex: an integrator of all signals that activate NF-kappaB? Trends
Cell Biol. 10, 129–133 15. Vane, J. R. (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like
drugs. Nat. New Biol. 231, 232–235 16. Marra, D. E., Simoncini, T., and Liao, J. K. (2000) Inhibition of vascular smooth muscle cell
proliferation by sodium salicylate mediated by upregulation of p21(Waf1) and p27(Kip1). Circulation 102, 2124–2130
17. Xu, X. M., Sansores-Garcia, L., Chen, X. M., Matijevic-Aleksic, N., Du, M., and Wu, K. K.
(1999) Suppression of inducible cyclooxygenase 2 gene transcription by aspirin and sodium salicylate. Proc. Natl. Acad. Sci. USA 96, 5292–5297
18. Wu, K. K. (2000) Aspirin and salicylate: An old remedy with a new twist. Circulation 102,
2022–2023

19. Pierce, J. W., Read, M. A., Ding, H., Luscinskas, F. W., and Collins, T. (1996) Salicylates
inhibit I kappa B-alpha phosphorylation, endothelial- leukocyte adhesion molecule expression, and neutrophil transmigration. J. Immunol. 156, 3961–3969
20. Kopp, E., and Ghosh, S. (1994) Inhibition of NF-kappa B by sodium salicylate and aspirin.
Science 265, 956–959 21. Yin, M. J., Yamamoto, Y., and Gaynor, R. B. (1998) The anti-inflammatory agents aspirin and
salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396, 77–80 22. O'Neill, E. A. (1998) A new target for aspirin. Nature 396, 15, 17 23. Alpert, D., and Vilcek, J. (2000) Inhibition of IkappaB kinase activity by sodium salicylate in
vitro does not reflect its inhibitory mechanism in intact cells. J. Biol. Chem. 275, 10925–10929 24. Frantz, B., and O'Neill, E. A. (1995) The effect of sodium salicylate and aspirin on NF-kappa B.
Science 270, 2017–2019 25. Moriguchi, Y., Matsubara, H., Mori, Y., Murasawa, S., Masaki, H., Maruyama, K., Tsutsumi,
Y., Shibasaki, Y., Tanaka, Y., Nakajima, T., Oda, K., and Iwasaka, T. (1999) Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ. Res. 84, 1073–1084
26. Viedt, C., Soto, U., Krieger-Brauer, H. I., Fei, J., Elsing, C., Kubler, W., and Kreuzer, J. (2000)
Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 20, 940–948
27. Muller, D. N., Mervaala, E. M., Dechend, R., Fiebeler, A., Park, J. K., Schmidt, F., Theuer, J.,
Breu, V., Mackman, N., Luther, T., Schneider, W., Gulba, D., Ganten, D., Haller, H., and Luft, F. C. (2000) Angiotensin II (AT(1)) receptor blockade reduces vascular tissue factor in angiotensin II-induced cardiac vasculopathy. Am. J. Pathol. 157, 111–122
28. Muller, D. N., Dechend, R., Mervaala, E. M., Park, J. K., Schmidt, F., Fiebeler, A., Theuer, J.,
Breu, V., Ganten, D., Haller, H., and Luft, F. C. (2000) NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35, 193–201
29. Aarons, L., Hopkins, K., Rowland, M., Brossel, S., and Thiercelin, J. F. (1989) Route of
administration and sex differences in the pharmacokinetics of aspirin, administered as its lysine salt. Pharm. Res. 6, 660–666.
30. Wientjes, M. G., and Levy, G. (1988) Nonlinear pharmacokinetics of aspirin in rats. J
Pharmacol. Exp. Ther. 245, 809–815.

31. Heissmeyer, V., Krappmann, D., Wulczyn, F. G., and Scheidereit, C. (1999) NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. Embo. J. 18, 4766–4778
32. Krappmann, D., Hatada, E. N., Tegethoff, S., Li, J., Klippel, A., Giese, K., Baeuerle, P. A., and
Scheidereit, C. (2000) The Ikappa B Kinase (IKK) Complex Is Tripartite and Contains IKKgamma but Not IKAP as a Regular Component. J. Biol. Chem. 275, 29779–29787
33. Kriz, W., Hahnel, B., Rosener, S., and Elger, M. (1995) Long-term treatment of rats with FGF-2
results in focal segmental glomerulosclerosis. Kidney Int. 48, 1435–1450 34. Khoshnan, A., Kempiak, S. J., Bennett, B. L., Bae, D., Xu, W., Manning, A. M., June, C. H., and
Nel, A. E. (1999) Primary human CD4+ T cells contain heterogeneous I kappa B kinase complexes: role in activation of the IL-2 promoter. J. Immunol. 163, 5444–5452
35. Zandi, E., Rothwarf, D. M., Delhase, M., Hayakawa, M., and Karin, M. (1997) The IkappaB
kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91, 243–252
36. Muller, D. N., Mervaala, E. M., Schmidt, F., Park, J. K., Dechend, R., Genersch, E., Breu, V.,
Loffler, B. M., Ganten, D., Schneider, W., Haller, H., and Luft, F. C. (2000) Effect of bosentan on NF-kappaB, inflammation, and tissue factor in angiotensin II-induced end-organ damage. Hypertension 36, 282–290
37. Nakazono, K., Watanabe, N., Matsuno, K., Sasaki, J., Sato, T., and Inoue, M. (1991) Does
superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci.USA 88, 10045–10048
38. Li, N., and Karin, M. (1999) Is NF-kappaB the sensor of oxidative stress? FASEB J. 13, 1137–
1143 39. Klahr, S., and Morrissey, J. J. (1997) Comparative study of ACE inhibitors and angiotensin II
receptor antagonists in interstitial scarring. Kidney Int. Suppl. 63, S111–114. 40. Ruiz-Ortega, M., Bustos, C., Hernandez-Presa, M., Lorenzo, O., Plaza, J., and Edigo, J. (1998)
Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor -kB activation and monocyte chemoattractant protein-1 synthesis. J. Immunol. 161, 430–439.
41. Autieri, M. V., Yue, T. L., Ferstein, G. Z., and Ohlstein, E. (1995) Antisense oligonucleotides to
the p65 subunit of NF-kB inhibit human vascular smooth muscle cell adherence and proliferation and prevent neointima formation in rat carotid arteries. Biochem. Biophys. Res. Commun. 213, 827–836
42. Abramson, S. B., and Weissmann, G. (1989) The mechanisms of action of nonsteroidal
antiinflammatory drugs. Arthritis Rheum. 32, 1–9

43. Brooks, P. M., and Day, R. O. (1991) Nonsteroidal antiinflammatory drugs--differences and
similarities. N. Engl. J. Med. 324, 1716–1725 44. Weissmann, G. (1991) Aspirin. Sci. Am. 264, 84–90 45. Kwak, Y. T., Guo, J., Shen, J., and Gaynor, R. B. (2000) Analysis of domains in the IKKalpha
and IKKbeta proteins that regulate their kinase activity. J. Biol. Chem. 275, 14752–14759 46. Shapiro, V. S., Mollenauer, M. N., Greene, W. C., and Weiss, A. (1996) c-rel regulation of IL-2
gene expression may be mediated through activation of AP-1. J. Exp. Med. 184, 1663–1669 47. Berk, B. C. (1999) Angiotensin II signal transduction in vascular smooth muscle: pathways
activated by specific tyrosine kinases. J. Am. Soc. Nephrol. 10 Suppl 11, S62–68. 48. Mervaala, E., Muller, D. N., Schmidt, F., Park, J. K., Gross, V., Bader, M., Breu, V., Ganten, D.,
Haller, H., and Luft, F. C. (2000) Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension 35, 587–594
Received December 20, 2000; revised April 5, 2001.

Fig. 1
Figure 1. High-dose aspirin (ASA 600) reduced mortality and ameliorated end-organ damage independent of blood pressure in dTGR. A) Renal glomerulus of untreated 7-wk-old dTGR shows mesangial expansion and podocyte damage. Renal dTGR arterioles exhibit severe changes in wall structure, including proliferation of smooth muscle cells and deposition of dense vacuoles. In contrast, nontransgenic Sprague-Dawley (SD) rats showed no vascular and glomerular damage (1-µm semithin Epon section, toluidine blue; light micrograph, magnification 800-fold). B) Untreated dTGR and low-dose aspirin (ASA 25) groups had >50% mortality. In contrast, high-dose aspirin significantly reduced mortality. C) Neither high- nor low-dose aspirin affected blood pressure. SD control rats showed significantly lower blood pressure levels. D) GR showed a 150-fold increased 24-h albuminuria compared with rats. High-dose aspirin reduced albuminuria by 85%. E) Serum thromboxane levels were increased in dTGR compared with all other groups. Thromboxane levels after low-dose aspirin were not different from high-dose and SD rats. Results are mean ±SE. (*P<0.05 high-dose aspirin vs. untreated and low-dose aspirin treated dTGR, #P<0.05 SD vs. all other groups).

Fig. 2
Figure 2. Untreated dTGR showed increased cardiac fibrosis and hypertrophy. A) Representative immunofluorescence staining of cardiac fibronectin expression. High-dose aspirin (ASA 600) markedly reduced fibronectin. B) M-mode echocardiography showed markedly reduced left ventricle diameters with massive left ventricular hypertrophy in untreated dTGR. High-dose aspirin reduced left hypertrophy and increased ventricular volume toward Sprague-Dawley controls. C) Heart weight to body weight ratio confirmed the matrix and echocardiography data.

Fig. 3
Figure 3. DNA binding nuclear factors in the kidney and heart. A and C) Electrophoretic mobility shift assay (EMSA) for the detection of NF-κB showed increased activity in dTGR kidney and heart compared with Sprague-Dawley rats. High-dose aspirin (ASA 600) reduced this activation markedly, whereas low-dose aspirin (ASA 25) did not. Nuclear extracts were supershifted with antibodies against the p50 and p65 NF-κB subunits. NF-κB DNA binding was blocked by excess unlabeled oligonucleotides containing the kB site from the MHC-enhancer (H2K). B and D) EMSA for AP-1. High-dose aspirin reduced AP-1 activation in kidney and heart. c-fos and c-jun were supershifted. Binding specificity was demonstrated by competition of excess unlabeled oligonucleotides containing the AP-1 site. Each lane represents a separate animal. E and F) Semiquantification for NF-κB and AP-1. The densitometric signal is expressed as a percentage of untreated dTGR.
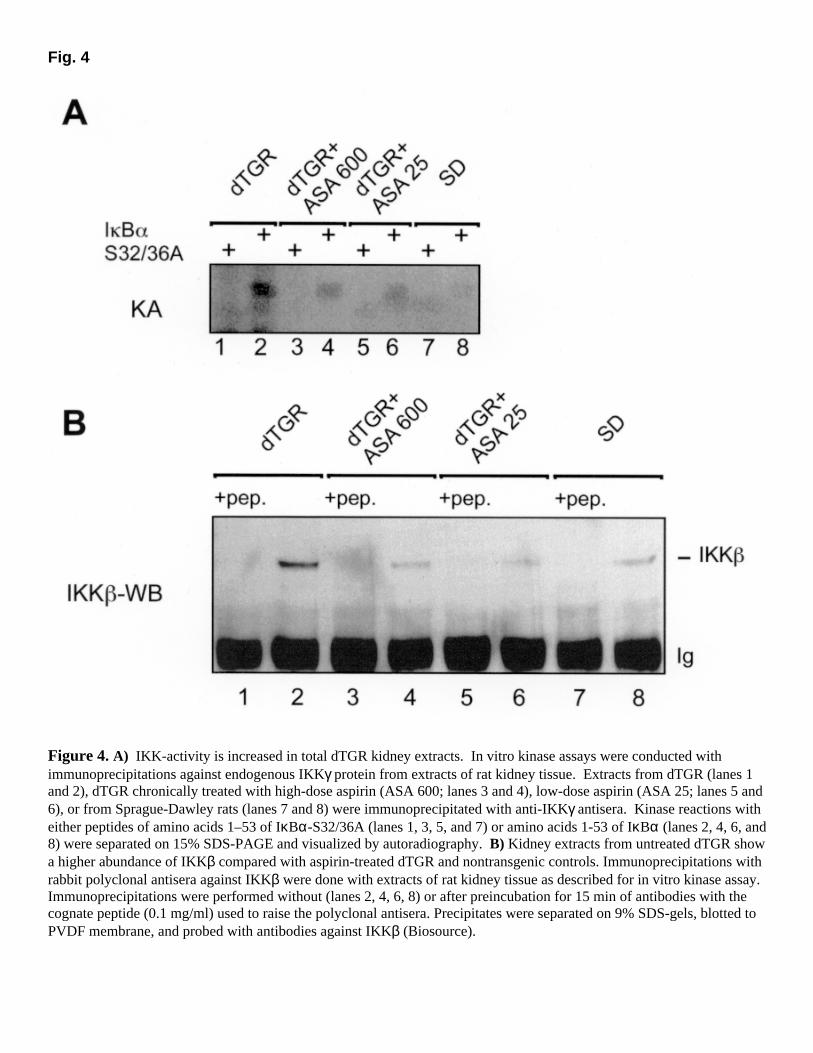
Fig. 4
Figure 4. A) IKK-activity is increased in total dTGR kidney extracts. In vitro kinase assays were conducted with immunoprecipitations against endogenous IKKγ protein from extracts of rat kidney tissue. Extracts from dTGR (lanes 1 and 2), dTGR chronically treated with high-dose aspirin (ASA 600; lanes 3 and 4), low-dose aspirin (ASA 25; lanes 5 and 6), or from Sprague-Dawley rats (lanes 7 and 8) were immunoprecipitated with anti-IKKγ antisera. Kinase reactions with either peptides of amino acids 1–53 of IκBα-S32/36A (lanes 1, 3, 5, and 7) or amino acids 1-53 of IκBα (lanes 2, 4, 6, and 8) were separated on 15% SDS-PAGE and visualized by autoradiography. B) Kidney extracts from untreated dTGR show a higher abundance of IKKβ compared with aspirin-treated dTGR and nontransgenic controls. Immunoprecipitations with rabbit polyclonal antisera against IKKβ were done with extracts of rat kidney tissue as described for in vitro kinase assay. Immunoprecipitations were performed without (lanes 2, 4, 6, 8) or after preincubation for 15 min of antibodies with the cognate peptide (0.1 mg/ml) used to raise the polyclonal antisera. Precipitates were separated on 9% SDS-gels, blotted to PVDF membrane, and probed with antibodies against IKKβ (Biosource).

Fig. 5
Figure 5. A) Leukocytes attached to the endothelial surface of a renal arcuate artery (scanning electron micrograph, magnification 12,000-fold). Semi-quantification of infiltrated leukocytes revealed that high-dose aspirin (ASA 600) reduced ED-1+ (B), CD4+ (C), and CD8+ (D) cells in heart and kidney. Low-dose aspirin (ASA 25) led to more pronounced reduction of CD4+; there was only a slight effect on CD8+ and no reduction of Mo/Ma. High-dose aspirin reduced all infiltrated cell types further, compared with low-dose aspirin. Fifteen different areas of each kidney and heart were analyzed. Results are mean ± SE of five animals per group (*P<0.05).

Fig. 6
Figure 6. A) VCAM-1 staining (red) in renal vessels of untreated dTGR, high-dose and low-dose aspirin groups, and SD rats. The sections were counterstained with an anti-laminin antibody to visualize the internal elastic lamina (green). VCAM-1 expression was increased in the intima. VCAM-1 was reduced only with high-dose aspirin. B) VCAM-1 Western blots from kidney and heart homogenates. Both kidney and heart showed high VCAM-1 expression in untreated and low-dose aspirin-treated dTGR. High-dose aspirin reduced VCAM-1 expression. C) Increased VLA-4 integrin expression on infiltrated cells. High-dose aspirin decreased VLA-4 integrin expression. Semiquantitative scoring was performed using a computerized cell-counting program. Fifteen different areas of each kidney and heart were analyzed. Results are mean ±SE of five animals per group. (*P<0.05).
Related Documents











