Amino acids 1–20 of the hepatitis C virus (HCV) core protein specifically inhibit HCV IRES- dependent translation in HepG2 cells, and inhibit both HCV IRES- and cap-dependent translation in HuH7 and CV-1 cells Dongsheng Li, 1,2 3 Seyed Taghi Takyar, 1,2 4 William B. Lott 1,2 and Eric J. Gowans 1,2 3 Correspondence Eric Gowans (at Macfarlane Burnet Institute) [email protected] 1 Sir Albert Sakzewski Virus Research Centre, Royal Children’s Hospital, Brisbane, QLD 4029, Australia 2 Clinical Medical Virology Research Centre, University of Queensland, St Lucia, QLD 4067, Australia Received 8 July 2002 Accepted 26 November 2002 A self-modulating mechanism by the hepatitis C virus (HCV) core protein has been suggested to influence the level of HCV replication, but current data on this subject are contradictory. We examined the effect of wild-type and mutated core protein on HCV IRES- and cap-dependent translation. The wild-type core protein was shown to inhibit both IRES- and cap-dependent translation in an in vitro system. This effect was duplicated in a dose-dependent manner with a synthetic peptide representing amino acids 1–20 of the HCV core protein. This peptide was able to bind to the HCV IRES as shown by a mobility shift assay. In contrast, a peptide derived from the hepatitis B virus (HBV) core protein that contained a similar proportion of basic residues was unable to inhibit translation or bind the HCV IRES. A recombinant vaccinia–HCV core virus was used to examine the effect of the HCV core protein on HCV IRES-dependent translation in cells and this was compared with the effects of an HBV core-recombinant vaccinia virus. In CV-1 and HuH7 cells, the HCV core protein inhibited translation directed by the IRES elements of HCV, encephalomyocarditis virus and classical swine fever virus as well as cap-dependent translation, whereas in HepG2 cells, only HCV IRES-dependent translation was affected. Thus, the ability of the HCV core protein to selectively inhibit HCV IRES-dependent translation is cell-specific. N-terminal truncated (aa 1–20) HCV core protein that was expressed from a novel recombinant vaccinia virus in cells abrogated the inhibitory phenotype of the core protein in vivo, consistent with the above in vitro data. INTRODUCTION It is estimated that about 3 % of the worldwide population is infected with hepatitis C virus (HCV) and more than 80 % of infected individuals develop persistent infection (World Health Organisation, 1999). It is unclear why such a high proportion of individuals fails to resolve the infection. However, several studies suggest that multiple factors might be involved, including the high genetic variability of the genome (Ray et al., 1999; Simmonds, 1995), extrahepatic replication (Laskus et al., 1998; Muller et al., 1993) and the lack of an effective early immune response (Minton et al., 1998; Zibert et al., 1997). No consensus has emerged. Similar to many other viruses, it is most likely that HCV has deve- loped a strategy to evade the host defence to ensure survival. HCV is a member of the Flaviviridae, along with the pesti- viruses and flaviviruses (Robertson et al., 1998). The genome is a single-strand positive-sense RNA molecule of approxi- mately 9600 nucleotides that contains a single long open reading frame (ORF) which is flanked by untranslated regions (UTR) at the 59 and 39 ends. The 59 UTR is a highly conserved region that contains an internal ribosome entry site (IRES), which initiates translation by a cap-independent mechanism (Rijnbrand & Lemon, 2000). Translation yields a polyprotein, which is cleaved into three structural (core, E1, E2/p7) and six non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A, NS5B). 3Present address: Macfarlane Burnet Institute for Medical Research and Public Health, Cnr Punt & Commercial Roads, Prahran, VIC 3181, Australia. 4Present address: Sinsheimer Laboratories, University of California, Santa Cruz, CA 95064, USA. 0001-8697 G 2003 SGM Printed in Great Britain 815 Journal of General Virology (2003), 84, 815–825 DOI 10.1099/vir.0.18697-0

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Amino acids 1–20 of the hepatitis C virus (HCV)core protein specifically inhibit HCV IRES-dependent translation in HepG2 cells, and inhibitboth HCV IRES- and cap-dependent translation inHuH7 and CV-1 cells
Dongsheng Li,1,23 Seyed Taghi Takyar,1,24 William B. Lott1,2
and Eric J. Gowans1,23
Correspondence
Eric Gowans (at Macfarlane
Burnet Institute)
1Sir Albert Sakzewski Virus Research Centre, Royal Children’s Hospital, Brisbane, QLD 4029,Australia
2Clinical Medical Virology Research Centre, University of Queensland, St Lucia, QLD 4067,Australia
Received 8 July 2002
Accepted 26 November 2002
A self-modulating mechanism by the hepatitis C virus (HCV) core protein has been suggested to
influence the level of HCV replication, but current data on this subject are contradictory. We
examined the effect of wild-type and mutated core protein on HCV IRES- and cap-dependent
translation. The wild-type core protein was shown to inhibit both IRES- and cap-dependent
translation in an in vitro system. This effect was duplicated in a dose-dependent manner with a
synthetic peptide representing amino acids 1–20 of the HCV core protein. This peptide was able to
bind to the HCV IRES as shown by a mobility shift assay. In contrast, a peptide derived from the
hepatitis B virus (HBV) core protein that contained a similar proportion of basic residues was unable
to inhibit translation or bind the HCV IRES. A recombinant vaccinia–HCV core virus was used
to examine the effect of the HCV core protein on HCV IRES-dependent translation in cells and
this was compared with the effects of an HBV core-recombinant vaccinia virus. In CV-1 and HuH7
cells, the HCV core protein inhibited translation directed by the IRES elements of HCV,
encephalomyocarditis virus and classical swine fever virus as well as cap-dependent translation,
whereas in HepG2 cells, only HCV IRES-dependent translation was affected. Thus, the ability of the
HCV core protein to selectively inhibit HCV IRES-dependent translation is cell-specific. N-terminal
truncated (aa 1–20) HCV core protein that was expressed from a novel recombinant vaccinia virus
in cells abrogated the inhibitory phenotype of the core protein in vivo, consistent with the above
in vitro data.
INTRODUCTION
It is estimated that about 3% of the worldwide population isinfected with hepatitis C virus (HCV) andmore than 80% ofinfected individuals develop persistent infection (WorldHealth Organisation, 1999). It is unclear why such a highproportion of individuals fails to resolve the infection.However, several studies suggest that multiple factors mightbe involved, including the high genetic variability of thegenome (Ray et al., 1999; Simmonds, 1995), extrahepaticreplication (Laskus et al., 1998; Muller et al., 1993) and the
lack of an effective early immune response (Minton et al.,1998; Zibert et al., 1997). No consensus has emerged. Similarto many other viruses, it is most likely that HCV has deve-loped a strategy to evade the host defence to ensure survival.
HCV is a member of the Flaviviridae, along with the pesti-viruses and flaviviruses (Robertson et al., 1998). The genomeis a single-strand positive-sense RNA molecule of approxi-mately 9600 nucleotides that contains a single long openreading frame (ORF) which is flanked by untranslatedregions (UTR) at the 59 and 39 ends. The 59 UTR is a highlyconserved region that contains an internal ribosome entrysite (IRES), which initiates translation by a cap-independentmechanism (Rijnbrand & Lemon, 2000). Translation yields apolyprotein, which is cleaved into three structural (core, E1,E2/p7) and six non-structural proteins (NS2, NS3, NS4A,NS4B, NS5A, NS5B).
3Present address: Macfarlane Burnet Institute for Medical Researchand Public Health, Cnr Punt & Commercial Roads, Prahran, VIC 3181,Australia.
4Present address: Sinsheimer Laboratories, University of California,Santa Cruz, CA 95064, USA.
0001-8697 G 2003 SGM Printed in Great Britain 815
Journal of General Virology (2003), 84, 815–825 DOI 10.1099/vir.0.18697-0

The core protein, which is thought to form the viral capsid,is located in the most N-terminal portion of the polyprotein.Evidence has accumulated to suggest that the core proteinmay inhibit the host response to virus infection throughmultiple mechanisms (Lai & Ware, 1999; McLauchlan,2000). In addition, the core protein has been shown to bindheterogeneous nuclear ribonucleoprotein K (hnRNP K),which is involved in cellular pre-mRNA splicing and nuclearRNA transport, and modulate cellular RNA transcription(Ray et al., 1995, 1997; Shrivastava et al., 1998).
It has also been suggested that HCV has a self-modulatingmechanism to maintain a low level of replication andexpression that may promote virus persistence. To accountfor this, it was speculated that stem–loop IV of the HCVIRES might be stabilized by interaction with a viral protein,to result in inhibition of translation (Honda et al., 1996).The core protein was later shown to bind positive- but notnegative-strand HCV RNA, an interaction that resulted insuppression of translation (Shimoike et al., 1999). Aminoacids (aa) 1–75 of the core protein were previously reportedto be responsible for the interaction with the viral RNA(Santolini et al., 1994). In addition to binding the viral RNA,the core protein can interact with itself and with the E1 andE2 proteins (Lo et al., 1996). It was also reported that theHCV core protein reduced the efficiency of HCV translationby binding to the IRES (Shimoike et al., 1999) and aa 33–44were recently shown to interact with the IRES andcontribute to the inhibition of translation (Zhang et al.,2002). In contrast, a previous study suggested that the coreprotein did not appear to have any specific effect on HCVIRES-directed translation, and instead, it was reported thatsuppression of IRES-directed translation resulted from anRNA–RNA interaction (Wang et al., 2000).
The potential role of the HCV core protein in the effi-ciency of HCV IRES-directed translation is a key issue inunderstanding the replication and expression of HCV.Consequently, the aim of this study was to clarify the natureof the HCV core–IRES interaction. We examined the effectof the wild-type and mutated core protein on the expressionof reporter genes in an in vitro system and the effect of theprotein expressed from a recombinant vaccinia virus onHCV IRES-directed translation in different cell lines.
METHODS
Cells. Monkey kidney (CV-1) cells and the human hepatocellularcarcinoma cells, HuH7 and HepG2, were maintained in Dulbecco’smodified Eagle’s medium (DMEM) containing 5% or 10% foetalcalf serum (FCS) with penicillin and streptomycin.
Plasmids. The plasmids pCore and pD20, which encode aa 1–167and aa 21–167 of the HCV core protein respectively, were synthesizedby insertion of the appropriate PCR-generated fragment fromp59UTR-A2 (Trowbridge & Gowans, 1998) into pcDNA3 (Invitrogen).pD20 was engineered to encode a methionine at the start of the ORFand a stop codon at the end. Plasmid pD1, in which the start codonof the core protein was deleted from pCore, was made using theQuikchange Site-Directed Mutagenesis Kit (Stratagene), following
the manufacturer’s instructions. A series of C-terminal truncatedcore proteins was generated by appropriate restriction enzyme diges-tion of pCore. The reporter plasmid pIRES-CAT (Lott et al., 2001),which contains the HCV 59 UTR sequence and 27 nt of the down-stream core protein-coding sequence ligated in-frame with the chlor-amphenicol acetyltransferase (CAT) gene, was constructed inpGEM-T (Promega). pCAP-LUC was constructed by inserting thefirefly luciferase gene into pGEM-T. Transcription from these plas-mids is controlled by the T7 promoter and expression controlled bythe HCV IRES and a cap-dependent mechanism respectively.
A bicistronic reporter pcCAT, from which expression of fireflyluciferase and CAT were controlled by cap- and IRES-dependentmechanisms respectively, was constructed by ligating pCAP-LUC andpIRES-CAT in pcDNA3. Three additional bicistronic constructs, whichcontained the IRES elements from HCV, encephalomyocarditis virus(EMCV) and classical swine fever virus (CSFV) (referred to as HIRES,EIRES and CIRES) inserted between theRenilla luciferase (R-LUC) andCAT genes (Lott et al., 2001), were also used as reporter molecules.Thus, the expression of R-LUC and CAT was directed by cap- andIRES-dependent mechanisms respectively.
In vitro transcription of RNA. The plasmids were linearized by theappropriate restriction enzyme and then purified by phenol/chloro-form extraction followed by isopropanol precipitation or by theBRESAclean DNA purification system (Bresatec). RNA was synthe-sized from each plasmid by T7 RNA polymerase with or without theaddition of RNA capping analogue (Gibco-BRL) as appropriate. TheRNAs were purified by phenol/chloroform extraction and isopropanolprecipitation followed by two washes with 70% ethanol. The RNApellets were dissolved in RNase-free water. The quality and quantityof the RNAs were checked by agarose gel electrophoresis and the con-centration was determined by optical density measurement.
In vitro translation. Wild-type core protein and the pD1 and pD20protein products were expressed in a 25 ml rabbit reticulocyte lysatetranslation reaction (RRL; Promega) loaded with 500 ng of the respec-tive RNA. This reaction will be referred to as RRL1. The productswere analysed by SDS-PAGE and immunoblot as previouslydescribed (Wang et al., 1997). Radiolabelled products were visualizedby SDS-PAGE followed by PhosphorImager analysis (MolecularDynamics) and by immunoblot to confirm the authenticity of thesynthesized protein. To examine the effect of the expressed proteinon IRES- or cap-dependent translation, an aliquot of the aboveRRL1 was then added to a second RRL containing 500 ng of pIRES-CAT RNA and/or 100 ng of pCAP-LUC RNA. This reaction will bereferred to as the reporter translation reaction (RRL2). All transla-tion reactions were carried out at 30˚C for 90 min unless notedotherwise. The products of RRL2 were analysed as described aboveand all experiments were carried out in triplicate.
In some experiments, synthetic peptides were added to the RRL2.The peptides were synthesized by Mimotopes (Australia) to >90%purity. The sequences of the peptides are: (1) HCV core aa1-20-MSTNPKPQRKTKRNTNRRPQ; (2) HCV E2 HVR1 aa384-419-DTHTTGGVAGRDTLRFTGFFSFGPKQK; (3) HBV coreaa141-160-STLPETTVVRRRGRSPRRRT; (4) HBsAg aa 202-213-IPQSLDSWWTSL.
RNA–protein binding assay. The RNA–protein binding assay wascarried out essentially as described (Furuya & Lai, 1993) with slightmodifications. Briefly, 32P-labelled RNA was mixed with peptide in10 ml of binding buffer (10 mM HEPES, 2?5 mM MgCl2, 40 mMHCl, 5% glycerol, 2?5mM DTT, 20 U RNase inhibitor). The bindingreaction was incubated at 30 ˚C for 10 min and then mixed with 56loading buffer (50% glycerol, 0?05% bromophenol blue, 0?05% xylenecyanol, 26 TBE). The samples were analysed by electrophoresis ona 4% native polyacrylamide gel. The gel was pre-electrophoresed at
816 Journal of General Virology 84
D. Li and others

80 V for 1 h prior to loading the samples in 0?56 TBE and electro-phoresis was performed at 80 V for 1?5 h. Labelled RNA wasdetected by PhosphorImager analysis of the dried gel.
Recombinant vaccinia viruses. Two recombinant vaccinia viruses(RecVV), RecVV-HCC and RecVV-HBC, were used to express eitherthe full-length HCV core protein (aa 1–191) or the full-length HBVcore protein (aa 1–183), respectively. These were synthesized andsupplied by J. Hammond and B. Couper (Australian Animal HealthLaboratory, Geelong, Australia). Expression of the core proteins fromthe recombinant viruses was controlled by the VVp7.5 promoter inthe thymidine kinase (TK) gene locus of Western Reserve (WR). Thevirus stocks were prepared and titrated as previously described(Boyle et al., 1985).
To construct the N-terminal truncated version of RecVV-HCC(RecVV-D20), the PCR product, encoding aa 21–191 of the HCVcore protein, was inserted into the pBCB06 transfer vector (Boyle et al.,1985). The recombinant plasmid was transfected into CV1 cells,previously infected with WR, then followed by two cycles of TK pheno-type selection in 143B tk2 cells. The DNA sequence of the recombinanttransfer vector was confirmed and the authenticity of the recom-binant virus was further confirmed by PCR amplification of theinserted fragment.
Infection and transfection. Confluent monolayers of CV-1, HuH7and HepG2 cells were prepared in 6-well plates (TPP) and infectedwith trypsinized VV at an m.o.i. of 10 in 0?5 ml of DMEM + 1%FCS). The inoculum was replaced with fresh culture medium afterincubation for 1 h in a humidified CO2 incubator at 37˚C and thecells were subsequently incubated overnight (18–24 h). Before trans-fection, the cells were washed twice with 1 ml OptiMEM (Gibco-BRL). 1 mg each of pIRES-CAT and pCAP-LUC RNA, or 2 mg of eachbicistronic reporter RNA, were prepared with Lipofectin reagent(Gibco-BRL) per well according to the manufacturer’s protocol.
CAT and luciferase assays. The cells were washed once with ice-cold PBS, and then lysed with 0?3 ml of buffer provided with thedual luciferase assay (DLA; Promega) for 10 min. The cell debris wasremoved by centrifugation and the supernatant transferred to a freshtube. CAT activity was determined by ELISA (Roche) and LUC andR-LUC activities were measured by the DLA, following the instruc-tions of the manufacturer.
Relative translational efficiency (RTE). The RTE was calculatedby normalizing the activities of the reporter molecules in the RRL2containing the RNA(2)Control and in RecVV-HBC-infected cellsto 100%.
RESULTS
Core protein inhibits both IRES- andcap-directed translation in a dose-dependentmanner in an in vitro system
To investigate the effect of the HCV core protein on HCVIRES- and cap-directed translation, the core protein was pre-synthesized and then added to a reporter translation reaction(RRL2). In this experiment, we used aa 1–167 of the protein,which is similar in size to the mature core protein (Santoliniet al., 1994). The protein was produced in an RRL1 reactionby translation of pCore RNA and an aliquot of this reactionwas then transferred to the RRL2. An aliquot of RRL1containing pD1 RNA and one without the addition of anyRNA [RNA(2) control] represented controls. Core protein
expressed in this manner did not require purification, as theprotein was contained in the same medium as the reportertranslation reaction. The pCore translation product wasdetected as an 18 kDa polypeptide by immunoblot and theproduct expressed from pD1 appeared as a 6 kDa proteinwhen it was labelled with [35S]methionine (data not shown).This product is consistent with initiation at the methioninein the21 ORF (nt position 257) of an ORF which extends tont position 431 (i.e. 59 aa) of the Australian HCV isolate(Trowbridge & Gowans, 1998). Five ml of the respectiveRRL1 was added to the pIRES-CAT and/or pCAP-LUC RNAprogrammedRRL2 and the effect on translationmeasured byquantification of the CAT and luciferase products. The levelsof CAT and luciferase expression in the RNA(2) controlreaction were normalized to 100%. The addition of the coreprotein to the reporter translation reaction resulted in aninhibition of CAT and luciferase expression (Fig. 1A) thatwas found to be 60% and 56% respectively by Phosphor-Imager analysis (Fig. 1B). In contrast, the pD1 control onlyshowed a slight inhibition of translation (10% and 5%respectively). Translation inhibition by the pCore product
Fig. 1. The inhibition of cap- and HCV IRES-dependent transla-tion by the HCV core protein. The HCV core protein productswere synthesized in respective RRL1 reactions and a 5 ml aliquotof these RRL1s was transferred to a second RRL2 reaction.These were previously loaded with RNA from the reporter plas-mids pIRES-CAT and pCAP-LUC. (A) The reporter products luci-ferase and CAT were analysed by SDS-PAGE and (B) quantifiedby PhosphorImager analysis. The density of the reporter productsfrom the reaction which contained the RNA(2) control (track 1)was adjusted to 100 % and the RTE was measured in the reac-tions which contained the pCore aa 1–167 (track 2) and pD1products (track 3).
http://vir.sgmjournals.org 817
HCV core modulation of translation
was dose-dependent (data not shown). These results indicatethat translation was inhibited by the pCore translationproduct and not by RNA including the pD1RNA, whichis identical to pCore RNA (except for the removal ofthe initiation codon), or any component of the RRL.Furthermore, in order to generate similar levels of protein, itwas necessary to add 5-fold more IRES RNA compared withcap-dependent RNA (see Materials and Methods) becausecap-dependent translation is more efficient in our hands;consequently, the inhibitory effect of the core protein isactually 5-fold more specific for the HCV IRES.
The N-terminal 21 aa of core protein aresufficient to produce a similar inhibition to core1–167 in the in vitro system
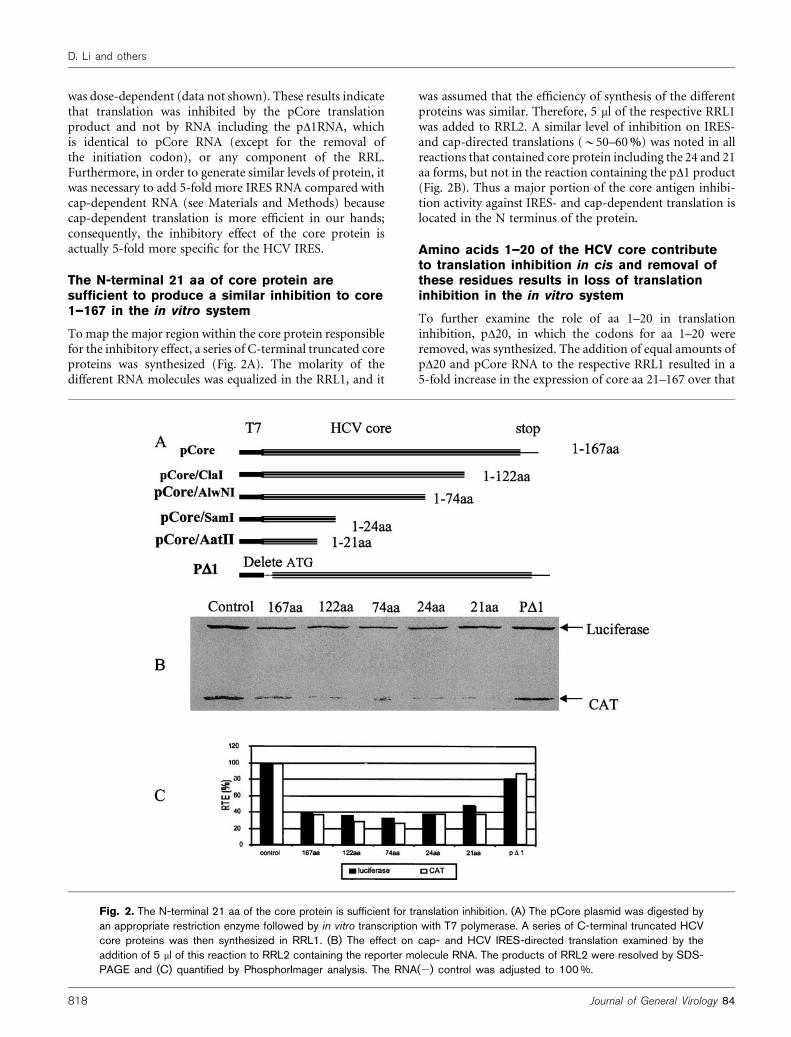
Tomap the major region within the core protein responsiblefor the inhibitory effect, a series of C-terminal truncated coreproteins was synthesized (Fig. 2A). The molarity of thedifferent RNA molecules was equalized in the RRL1, and it
was assumed that the efficiency of synthesis of the differentproteins was similar. Therefore, 5 ml of the respective RRL1was added to RRL2. A similar level of inhibition on IRES-and cap-directed translations (~50–60%) was noted in allreactions that contained core protein including the 24 and 21aa forms, but not in the reaction containing the pD1 product(Fig. 2B). Thus a major portion of the core antigen inhibi-tion activity against IRES- and cap-dependent translation islocated in the N terminus of the protein.
Amino acids 1–20 of the HCV core contributeto translation inhibition in cis and removal ofthese residues results in loss of translationinhibition in the in vitro system
To further examine the role of aa 1–20 in translationinhibition, pD20, in which the codons for aa 1–20 wereremoved, was synthesized. The addition of equal amounts ofpD20 and pCore RNA to the respective RRL1 resulted in a5-fold increase in the expression of core aa 21–167 over that
Fig. 2. The N-terminal 21 aa of the core protein is sufficient for translation inhibition. (A) The pCore plasmid was digested byan appropriate restriction enzyme followed by in vitro transcription with T7 polymerase. A series of C-terminal truncated HCVcore proteins was then synthesized in RRL1. (B) The effect on cap- and HCV IRES-directed translation examined by theaddition of 5 ml of this reaction to RRL2 containing the reporter molecule RNA. The products of RRL2 were resolved by SDS-PAGE and (C) quantified by PhosphorImager analysis. The RNA(2) control was adjusted to 100 %.
818 Journal of General Virology 84
D. Li and others
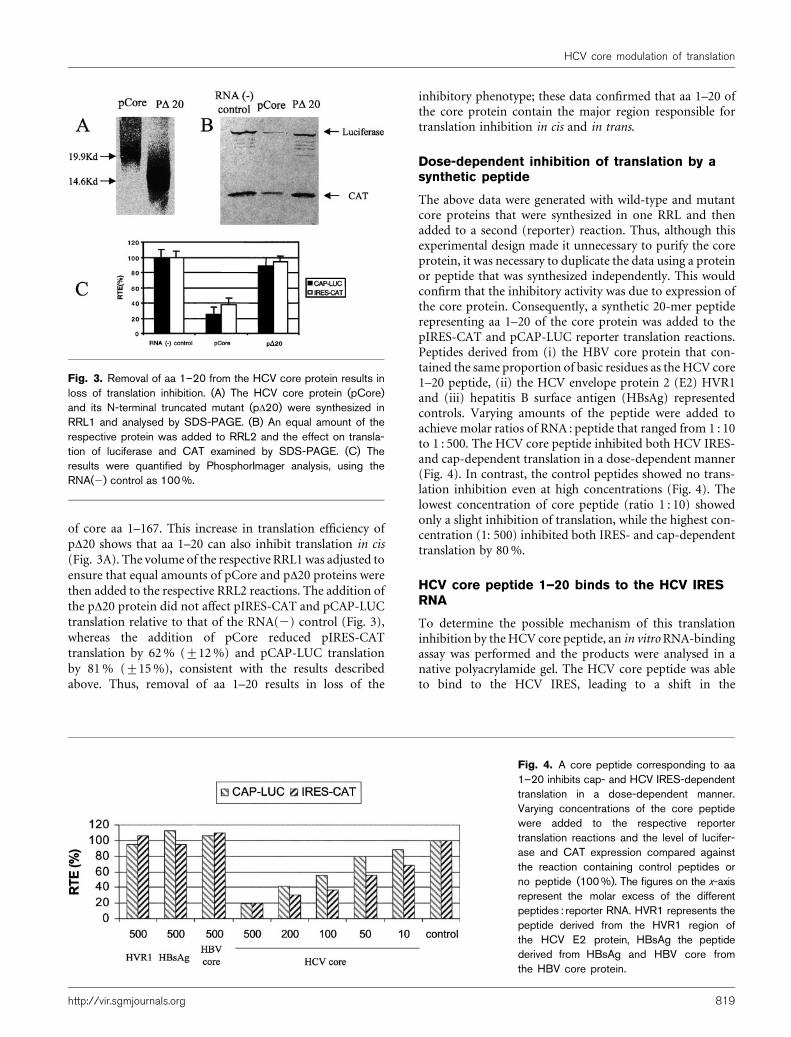
of core aa 1–167. This increase in translation efficiency ofpD20 shows that aa 1–20 can also inhibit translation in cis(Fig. 3A). The volume of the respective RRL1 was adjusted toensure that equal amounts of pCore and pD20 proteins werethen added to the respective RRL2 reactions. The addition ofthe pD20 protein did not affect pIRES-CAT and pCAP-LUCtranslation relative to that of the RNA(2) control (Fig. 3),whereas the addition of pCore reduced pIRES-CATtranslation by 62% (±12%) and pCAP-LUC translationby 81% (±15%), consistent with the results describedabove. Thus, removal of aa 1–20 results in loss of the
inhibitory phenotype; these data confirmed that aa 1–20 ofthe core protein contain the major region responsible fortranslation inhibition in cis and in trans.
Dose-dependent inhibition of translation by asynthetic peptide
The above data were generated with wild-type and mutantcore proteins that were synthesized in one RRL and thenadded to a second (reporter) reaction. Thus, although thisexperimental design made it unnecessary to purify the coreprotein, it was necessary to duplicate the data using a proteinor peptide that was synthesized independently. This wouldconfirm that the inhibitory activity was due to expression ofthe core protein. Consequently, a synthetic 20-mer peptiderepresenting aa 1–20 of the core protein was added to thepIRES-CAT and pCAP-LUC reporter translation reactions.Peptides derived from (i) the HBV core protein that con-tained the same proportion of basic residues as the HCV core1–20 peptide, (ii) the HCV envelope protein 2 (E2) HVR1and (iii) hepatitis B surface antigen (HBsAg) representedcontrols. Varying amounts of the peptide were added toachieve molar ratios of RNA : peptide that ranged from 1 : 10to 1 : 500. The HCV core peptide inhibited both HCV IRES-and cap-dependent translation in a dose-dependent manner(Fig. 4). In contrast, the control peptides showed no trans-lation inhibition even at high concentrations (Fig. 4). Thelowest concentration of core peptide (ratio 1 : 10) showedonly a slight inhibition of translation, while the highest con-centration (1: 500) inhibited both IRES- and cap-dependenttranslation by 80%.
HCV core peptide 1–20 binds to the HCV IRESRNA
To determine the possible mechanism of this translationinhibition by theHCV core peptide, an in vitroRNA-bindingassay was performed and the products were analysed in anative polyacrylamide gel. The HCV core peptide was ableto bind to the HCV IRES, leading to a shift in the
Fig. 3. Removal of aa 1–20 from the HCV core protein results inloss of translation inhibition. (A) The HCV core protein (pCore)and its N-terminal truncated mutant (pD20) were synthesized inRRL1 and analysed by SDS-PAGE. (B) An equal amount of therespective protein was added to RRL2 and the effect on transla-tion of luciferase and CAT examined by SDS-PAGE. (C) Theresults were quantified by PhosphorImager analysis, using theRNA(2) control as 100 %.
Fig. 4. A core peptide corresponding to aa1–20 inhibits cap- and HCV IRES-dependenttranslation in a dose-dependent manner.Varying concentrations of the core peptidewere added to the respective reportertranslation reactions and the level of lucifer-ase and CAT expression compared againstthe reaction containing control peptides orno peptide (100 %). The figures on the x-axisrepresent the molar excess of the differentpeptides : reporter RNA. HVR1 represents thepeptide derived from the HVR1 region ofthe HCV E2 protein, HBsAg the peptidederived from HBsAg and HBV core fromthe HBV core protein.
http://vir.sgmjournals.org 819
HCV core modulation of translation
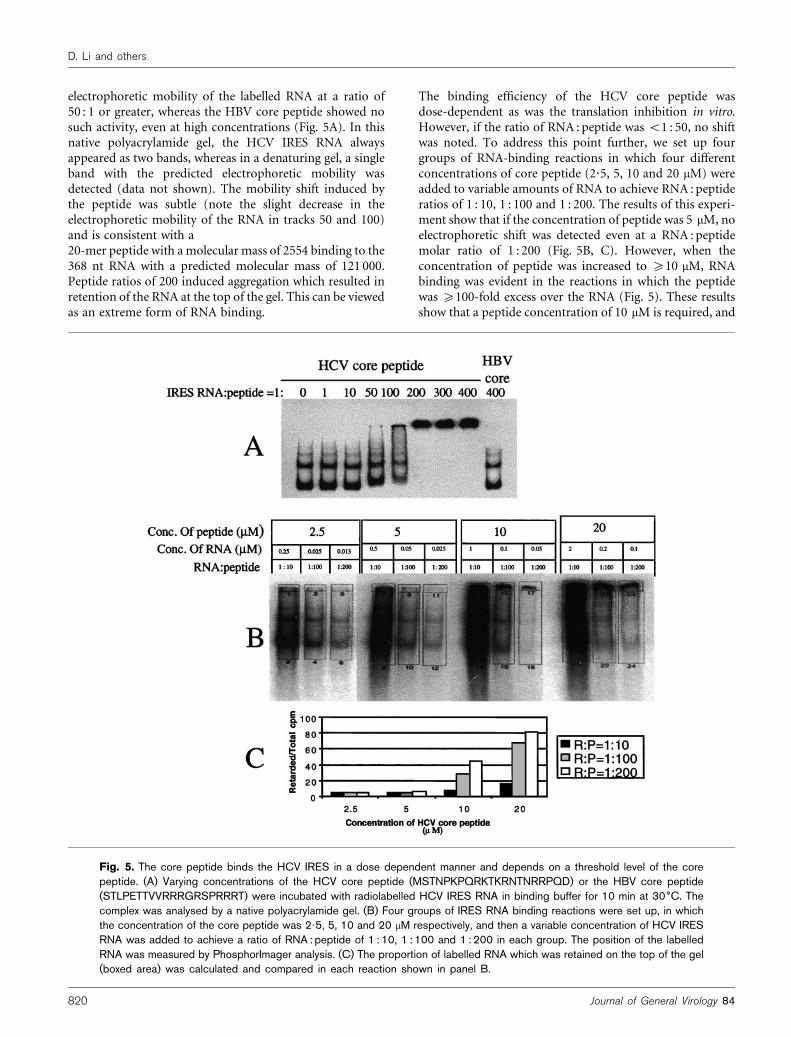
electrophoretic mobility of the labelled RNA at a ratio of50 : 1 or greater, whereas the HBV core peptide showed nosuch activity, even at high concentrations (Fig. 5A). In thisnative polyacrylamide gel, the HCV IRES RNA alwaysappeared as two bands, whereas in a denaturing gel, a singleband with the predicted electrophoretic mobility wasdetected (data not shown). The mobility shift induced bythe peptide was subtle (note the slight decrease in theelectrophoretic mobility of the RNA in tracks 50 and 100)and is consistent with a20-mer peptide with a molecular mass of 2554 binding to the368 nt RNA with a predicted molecular mass of 121 000.Peptide ratios of 200 induced aggregation which resulted inretention of the RNA at the top of the gel. This can be viewedas an extreme form of RNA binding.
The binding efficiency of the HCV core peptide wasdose-dependent as was the translation inhibition in vitro.However, if the ratio of RNA : peptide was <1 : 50, no shiftwas noted. To address this point further, we set up fourgroups of RNA-binding reactions in which four differentconcentrations of core peptide (2?5, 5, 10 and 20 mM) wereadded to variable amounts of RNA to achieve RNA : peptideratios of 1 : 10, 1 : 100 and 1 : 200. The results of this experi-ment show that if the concentration of peptide was 5 mM, noelectrophoretic shift was detected even at a RNA : peptidemolar ratio of 1 : 200 (Fig. 5B, C). However, when theconcentration of peptide was increased to ¢10 mM, RNAbinding was evident in the reactions in which the peptidewas ¢100-fold excess over the RNA (Fig. 5). These resultsshow that a peptide concentration of 10 mM is required, and
Fig. 5. The core peptide binds the HCV IRES in a dose dependent manner and depends on a threshold level of the corepeptide. (A) Varying concentrations of the HCV core peptide (MSTNPKPQRKTKRNTNRRPQD) or the HBV core peptide(STLPETTVVRRRGRSPRRRT) were incubated with radiolabelled HCV IRES RNA in binding buffer for 10 min at 30˚C. Thecomplex was analysed by a native polyacrylamide gel. (B) Four groups of IRES RNA binding reactions were set up, in whichthe concentration of the core peptide was 2?5, 5, 10 and 20 mM respectively, and then a variable concentration of HCV IRESRNA was added to achieve a ratio of RNA : peptide of 1 : 10, 1 : 100 and 1 : 200 in each group. The position of the labelledRNA was measured by PhosphorImager analysis. (C) The proportion of labelled RNA which was retained on the top of the gel(boxed area) was calculated and compared in each reaction shown in panel B.
820 Journal of General Virology 84
D. Li and others

provided that this minimum concentration is achieved, thenthe core peptide can bind to the HCV IRES.
HCV core protein inhibits HCV IRES-directedtranslation in vivo in a cell-specific manner
To examine the effect of the HCV core protein on HCVIRES-directed translation in vivo, the HCV core protein wasexpressed from a recombinant vaccinia virus (RecVV-HCC),and an HBV core recombinant vaccinia virus (RecVV-HBC)represented a control. We have shown that the level ofexpression from pIRES-CAT and pCAP-LUC in RecVV-HBC-infected cells was similar to that in cells infected withthe WR strain of vaccinia virus (data not shown). The effectof the expression of the core protein was examined in threedifferent cell lines, CV-1, HuH7 and HepG2. The cells wereinfected with the respective viruses at an m.o.i. of 10,transfected with the different RNA reporter molecules afterovernight infection and the expression of the reporterproteins was measured 5 h later.
Initially, we co-transfected the RecVV-infected cells withpIRES-CAT and pCAP-LUC RNA. The levels of CAT andluciferase expressed in the RecVV-HBC-infected cells werenormalized to 100% (Fig. 6A). By comparison, the expres-sion of CAT in all three cell lines infected with RecVV-HCCwas reduced by 45–70%. The expression of luciferase inRecVV-HCC-infected CV-1 and HuH7 cells showed asimilar reduction, but no such reduction was noted in theRecVV-HCC-infected HepG2 cells. Thus, expression of theHCV core protein appeared to inhibit both HCV IRES- andcap-dependent translation in the CV-1 and HuH7 cells, butwas specific for the HCV IRES in HepG2 cells
We then tested the effect of the HCV core protein on CATand luciferase expression from a bicistronic reporter mole-cule, pcCAT, in which the luciferase and CAT genes wereseparated by the HCV IRES. The results obtained using thisbicistronic RNA duplicated those obtained after transfectionwith the monocistronic RNAs (data not shown).
The above data were derived using the HCV IRES from agenotype 1b virus (Trowbridge & Gowans, 1998). We thenextended the study to examine the effect of expression of the
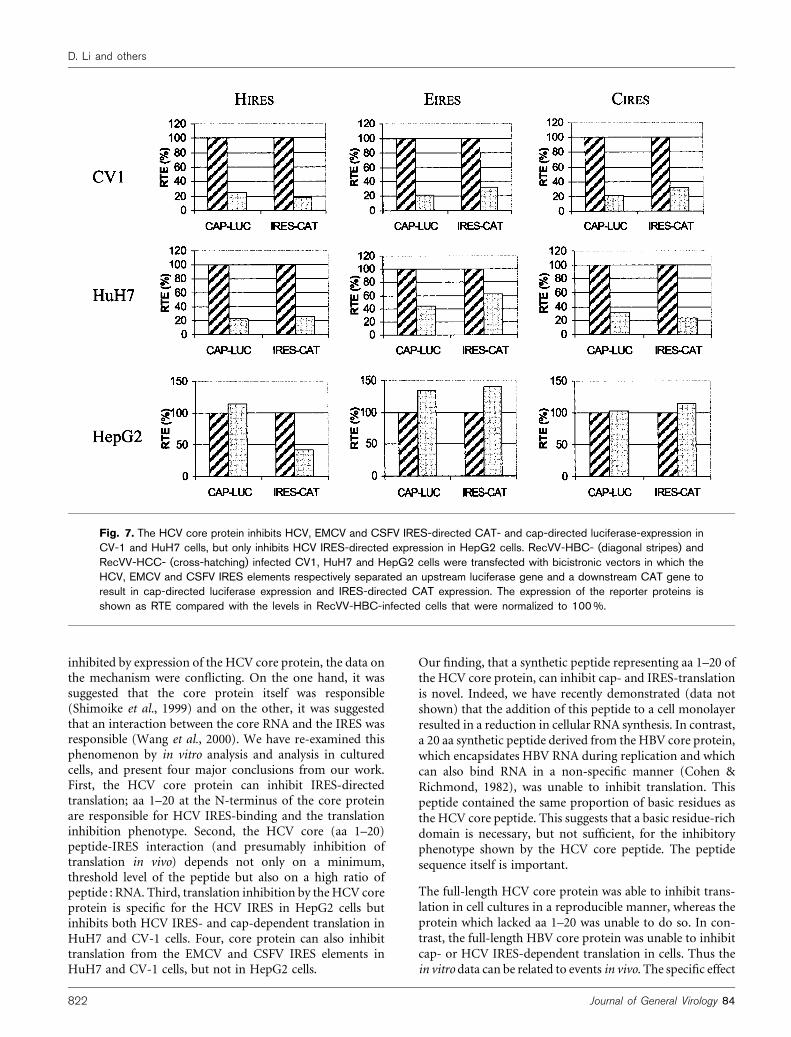
HCV core protein (1b) on an IRES element derived from agenotype 1a virus and compared this against IRES elementsderived from EMCV and CSFV. These bicistronic vectorshave been described previously (Lott et al., 2001). As des-cribed above, RecVV-HBC was used as a negative control.In CV-1 and HuH7 cells, the HCV core protein appearedto inhibit translation from all three IRES elements andcap-dependent translation to similar degrees (Fig. 7). Incontrast, in HepG2 cells, the HCV core protein onlyinhibited translation from the HCV IRES and had noeffect on the EMCV and CSFV IRES elements, or on cap-dependent translation.
Amino acids 1–20 of the HCV core protein isimportant for translation inhibition in vivo
Since in vitro translation inhibition was lost when aa 1–20 ofthe HCV core protein was removed, a RecVV construct wassynthesized that encoded aa 21–191, to evaluate this observ-ation in vivo. This virus was used to infect HuH7 and HepG2cells and the cells were then transfected with the bicistronicpcCAT RNA. CAT and luciferase activities were assayed asdescribed above. Consistent with the above results, infectionof both cell types with RecVV-HCC resulted in a 60%inhibition of CAT activity (Fig. 8). However, in cells infectedwith RecVV-D20, CAT expression was only reduced by~15% in both cell lines. The activity of luciferase in cellsinfected with RecVV-HCC or RecVV-D20 was similar to theabove results (Fig. 6A), i.e. HCV core protein showed noinhibitory effect on cap-directed translation in HepG2 cellsbut inhibited cap-directed translation by 60% inHuH7 cells.Similarly, and consistent with the IRES-CAT data, deletionof aa 1–20 from the core protein virtually ablated theinhibitory effect on cap-directed expression in the HuH7cells (Fig. 8, left panel). The results clearly demonstratethat the bulk of the activity associated with the HCV coreprotein-related inhibition of IRES- or cap-directed transla-tion resides within aa 1–20 of the protein.
DISCUSSION
Although previous studies (Shimoike et al., 1999;Wang et al.,2000) agreed that translation from the HCV IRES could be
Fig. 6. The HCV core protein inhibits expression of the reporter in a cell-specific manner. CV-1, HuH7 and HepG2 cells wereinfected with RecVV-HCV (cross-hatching) or RecVV-HBV (diagonal stripes), and then after overnight incubation, transfectedwith monocistronic pIRES-CAT and pCAP-LUC RNA. The cells were harvested 5 h later and the expression of the reporterproteins assayed. The RTE was calculated relative to the level of the reporter protein in RecVV-HBC-infected cells,determined as 100 %.
http://vir.sgmjournals.org 821
HCV core modulation of translation
inhibited by expression of the HCV core protein, the data onthe mechanism were conflicting. On the one hand, it wassuggested that the core protein itself was responsible(Shimoike et al., 1999) and on the other, it was suggestedthat an interaction between the core RNA and the IRES wasresponsible (Wang et al., 2000). We have re-examined thisphenomenon by in vitro analysis and analysis in culturedcells, and present four major conclusions from our work.First, the HCV core protein can inhibit IRES-directedtranslation; aa 1–20 at the N-terminus of the core proteinare responsible for HCV IRES-binding and the translationinhibition phenotype. Second, the HCV core (aa 1–20)peptide-IRES interaction (and presumably inhibition oftranslation in vivo) depends not only on a minimum,threshold level of the peptide but also on a high ratio ofpeptide : RNA. Third, translation inhibition by theHCV coreprotein is specific for the HCV IRES in HepG2 cells butinhibits both HCV IRES- and cap-dependent translation inHuH7 and CV-1 cells. Four, core protein can also inhibittranslation from the EMCV and CSFV IRES elements inHuH7 and CV-1 cells, but not in HepG2 cells.
Our finding, that a synthetic peptide representing aa 1–20 ofthe HCV core protein, can inhibit cap- and IRES-translationis novel. Indeed, we have recently demonstrated (data notshown) that the addition of this peptide to a cell monolayerresulted in a reduction in cellular RNA synthesis. In contrast,a 20 aa synthetic peptide derived from the HBV core protein,which encapsidates HBV RNA during replication and whichcan also bind RNA in a non-specific manner (Cohen &Richmond, 1982), was unable to inhibit translation. Thispeptide contained the same proportion of basic residues asthe HCV core peptide. This suggests that a basic residue-richdomain is necessary, but not sufficient, for the inhibitoryphenotype shown by the HCV core peptide. The peptidesequence itself is important.
The full-length HCV core protein was able to inhibit trans-lation in cell cultures in a reproducible manner, whereas theprotein which lacked aa 1–20 was unable to do so. In con-trast, the full-length HBV core protein was unable to inhibitcap- or HCV IRES-dependent translation in cells. Thus thein vitro data can be related to events in vivo. The specific effect
Fig. 7. The HCV core protein inhibits HCV, EMCV and CSFV IRES-directed CAT- and cap-directed luciferase-expression inCV-1 and HuH7 cells, but only inhibits HCV IRES-directed expression in HepG2 cells. RecVV-HBC- (diagonal stripes) andRecVV-HCC- (cross-hatching) infected CV1, HuH7 and HepG2 cells were transfected with bicistronic vectors in which theHCV, EMCV and CSFV IRES elements respectively separated an upstream luciferase gene and a downstream CAT gene toresult in cap-directed luciferase expression and IRES-directed CAT expression. The expression of the reporter proteins isshown as RTE compared with the levels in RecVV-HBC-infected cells that were normalized to 100 %.
822 Journal of General Virology 84
D. Li and others

of the HCV core protein in HepG2 cells as opposed to HuH7and CV-1 cells may be related to differences in the intra-cellular distribution of the protein (Wang et al., 2000),although we were unable to demonstrate any difference inthe localization patterns in the three cell lines used in ourstudy (data not shown). If the HCV core protein inhibitstranslation and/or transcription (Ray et al., 1995, 1997;Shrivastava et al., 1998) in human hepatocytes, then this mayinfluence cell viability. A recent study (Bantel et al., 2001)reported that a high proportion of hepatocytes in HCV-infected livers showed evidence of apoptosis, although it isnot known if this is related to the expression of core or otherHCV proteins. It has been suggested that the HCV coreprotein can sensitize the cell to apoptosis, although this maynot necessarily be linked to reduced translation efficiency(Lai & Ware, 1999).
In HepG2 cells, the HCV core protein specifically inhibitedtranslation from the HCV IRES, irrespective of whether theIRES element was derived from a 1a or 1b genotype, but hadno effect on translation from the EMCV and CFSV IRESelements. These results are consistent with previous data inwhich the translation from the HCV IRES was specificallyinhibited by the HCV core protein in HepG2 and HepT cells(Shimoike et al., 1999; Zhang et al., 2002). Our data also
highlight yet another difference between HepG2 and HuH7cells, as it has previously been demonstrated that the HuH7cells but not the HepG2 cells support replication of the HCVreplicon (Lohmann et al., 1999; Blight et al., 2000). We havesought an explanation for our results which differ to thoseshowing that the effect is mediated by RNA (Wang et al.,2000). In our study, a ratio of 1 : 100 of RNA : peptide wasnecessary for binding, while 1 : 500 was necessary for com-plete inhibition of translation in vitro. In contrast, theconcentration of HCV core used in vitro in the Wang studywas much lower (Wang et al., 2000). There is an additionalimportant difference between the two studies; in the Wangstudy, the bicistronic reporter plasmids were transfected24 h prior to infection of the cells with recombinant baculo-virus which encoded the HCV core protein, whereas wetransfected our reporter plasmids after overnight infectionof the cells with a recombinant vaccinia virus. Thus, it ispossible that the level of expression of the HCV core proteinin the Wang study was insufficient to inhibit HCV IRESfunction. Indeed, our study shows that a high concentrationof core protein and a high ratio of protein : RNA (Fig. 5) arenecessary for an interaction between the HCV core and theHCV IRES. These features are consistent with our under-standing of HCV replication (Bartenschlager & Lohmann,2000). Our in vitro data showed that a similar concentrationof the core peptide was required for translation inhibitionand RNA binding (Figs 4 and 5 respectively). Furthermore,this concentration is similar to that necessary for HCV coreprotein to induce assembly of an HCV nucleocapsid-likeparticle in vitro (Kunkel et al., 2001). Extrapolation of thesedata suggests that a high threshold concentration of the coreprotein, as might be found in the typical punctate stainingpatterns (Gowans, 2000), is required for RNA bindingin vivo. This may either inhibit translation per se or reducetranslation by removal of the RNA template through pack-aging. The latter mechanism may account for the difficultiesin detecting HCV products in naturally infected liversamples (Gowans, 2000). In this respect, HCV resemblesbovine viral diarrhoea virus because the bulk of the virus israpidly secreted and very little remains cell-associated (Gonget al., 1996).
Other circumstantial evidence supports our conclusion thatthe HCV core protein inhibits HCV IRES activity; first, theefficiency of colony formation by the full-length repliconwhich encodes the structural proteins was reported to be 3–4logs lower than that of the subgenomic replicons (Ikedaet al., 2002). Second, the level of replication of the full-lengthreplicon in the selected cell lines is around 5-fold lower thanthat of the subgenomic replicon (Pietschmann et al., 2002).Together, these data suggest that one or more of the struc-tural proteins regulates HCV replication. As E1/E2 havebeen shown to have no effect on HCV IRES function(Shimoike et al., 1999), it can be concluded that the coreprotein itself inhibits HCV IRES-directed translation.Finally, we have recently expressed the HCV core, E1 andE2 proteins from a Semliki Forest virus replicon (Greive
Fig. 8. Removal of aa 1–20 from the HCV core protein resultsin loss of the translation inhibition phenotype in vivo. HuH7 andHepG2 cells were infected with RecVV-HBC, RecVV-HCC orRecVV-D20, and then transfected with the bicistronic pcCATreporter RNA. The RTE was determined as described in thelegend of figure 6.
http://vir.sgmjournals.org 823
HCV core modulation of translation

et al., 2002). In similar studies, the level of expression of theHCV structural proteins was reduced by >100-fold whenthe expression was controlled by the HCV IRES (Greive,2001), again suggesting that one or more of the HCVstructural proteins inhibited HCV IRES-related translation.It is likely that the HCV IRES–core interaction has evolvedto reduce the level of HCV replication consistent withpersistent infection.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Health &Medical Research Council of Australia and from the Royal Children’sHospital Foundation. This is manuscript number 147 from the SirAlbert Sakzewski Virus Research Centre. We thank Elizabeth Grgacic,David Anderson and Hans Netter for helpful discussion and review ofthe manuscript.
REFERENCES
Bantel, H., Lugering, A., Poremba, C., Lugering, N., Held, J.,Domscheke, W. & Schulze-Osthoff, K. (2001). Caspase activationcorrelates with the degree of inflammatory liver injury in chronichepatitis C virus infection. Hepatology 34, 758–767.
Bartenschlager, R. & Lohmann, V. (2000). Replication of hepatitis Cvirus. J Gen Virol 81, 1631–1648.
Blight, K. J., Kolykhalov, A. A. & Rice, C. M. (2000). Efficientinitiation of HCV RNA replication in cell culture. Science 290,1972–1974.
Boyle, D. B., Coupar, B. E. & Both, G. W. (1985). Multiple-cloning-site plasmids for the rapid construction of recombinant poxviruses.Gene 35, 169–177.
Cohen, B. & Richmond, J. E. (1982). Electron microscopy ofhepatitis B virus core antigen synthesized in E. coli. Nature 296,677–678.
Furuya, T. & Lai, M. M. C. (1993). Three different cellular proteinsbind to complementary sites on the 59-end-positive and 39-end-negative strands of mouse hepatitis virus RNA. J Virol 67,7215–7222.
Gong, Y., Trowbridge, R., Macnaughton, T. B., Westaway, E. G.,Shannon, A. D. & Gowans, E. J. (1996). Characterization of RNAsynthesis during a one-step growth curve and of the replicationmechanism of bovine viral diarrhoea virus. J Gen Virol 77, 2729–2736.
Gowans, E. J. (2000). Distribution of markers of hepatitis C virusinfection throughout the body. Semin Liver Dis 20, 85–102.
Greive, S. J. (2001). Studies on the expression of the structural proteinsof hepatitis C virus. PhD thesis, University of Queensland, Australia.
Greive, S. J., Webb, R. I., Mackenzie, J. M. & Gowans, E. J. (2002).Expression of the hepatitis C virus structural proteins in mammaliancells induces morphology similar to that in natural infection. J ViralHepat 9, 9–17.
Honda, M., Brown, E. A. & Lemon, S. M. (1996). Stability of a stem-loop involving the initiator AUG controls the efficiency of internalinitiation of translation on hepatitis C virus RNA. RNA 2, 955–968.
Ikeda, M., Yi, M., Li, K. & Lemon, S. M. (2002). Selectable subgenomicand genome-length dicistronic RNAs derived from an infectiousmolecular clone of the HCV-N strain of hepatitis C virus replicateefficiently in cultured Huh7 cells. J Virol 76, 2997–3006.
Kunkel, M., Lorinczi, M., Rijnbrand, R., Lemon, S. M. & Watowich, S. J.(2001). Self-assembly of nucleocapsid-like particles from recombinanthepatitis C virus core protein. J Virol 75, 2119–2129.
Lai, M. M. C. & Ware, C. F. (1999). Hepatitis C virus core protein:possible roles in viral pathogenesis. Curr Top Microbiol Immunol 242,117–134.
Laskus, T., Radkowski, M., Wang, L. F., Vargas, H. & Rakela, J.(1998). Search for hepatitis C virus extrahepatic replication sites inpatients with acquired immunodeficiency syndrome: specific detec-tion of negative-strand viral RNA in various tissues. Hepatology 28,1398–1401.
Lo, S. Y., Selby, M. J. & Ou, J. H. (1996). Interaction betweenhepatitis C virus core protein and E1 envelope protein. J Virol 70,5177–5182.
Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L. &Bartenschlager, R. (1999). Replication of subgenomic hepatitis Cvirus RNAs in a hepatoma cell line. Science 285, 110–113.
Lott, W. B., Takyar, S. S., Tuppen, J., Crawford, D. H., Harrison, M.,Sloots, T. P. & Gowans, E. J. (2001). Vitamin B12 and hepatitis C:molecular biology and human pathology. Proc Natl Acad Sci U S A98, 4916–4921.
McLauchlan, J. (2000). Properties of the hepatitis C virus coreprotein: a structural protein that modulates cellular processes. J ViralHepat 7, 2–14.
Minton, E. J., Smillie, D., Neal, K. R., Irving, W. L., Underwood, J. C. &James, V. (1998). Association between MHC class II alleles andclearance of circulating hepatitis C virus. Members of the TrentHepatitis C Virus Study Group. J Infect Dis 178, 39–44.
Muller, H. M., Pfaff, E., Goeser, T., Kallinowski, B., Solbach, C. &Theilmann, L. (1993). Peripheral blood leukocytes serve as a possibleextrahepatic site for hepatitis C virus replication. J Gen Virol 74,669–676.
Pietschmann, T., Lohmann, V., Kaul, A., Krieger, N., Rinck, G.,Rutter, G., Strand, D. & Bartenschlager, R. (2002). Persistent andtransient replication of full-length hepatitis C virus genomes in cellculture. J Virol 76, 4008–4021.
Ray, R. B., Lagging, L. M., Meyer, K., Steele, R. & Ray, R. (1995).Transcriptional regulation of cellular and viral promoters by thehepatitis C virus core protein. Virus Res 37, 209–220.
Ray, R. B., Steele, R., Meyer, K. & Ray, R. (1997). Transcriptionalrepression of p53 promoter by hepatitis C virus core protein. J BiolChem 272, 10983–10986.
Ray, S. C., Wang, Y. M., Laeyendecker, O., Ticehurst, J. R., Villano, S. A.& Thomas, D. L. (1999). Acute hepatitis C virus structural genesequences as predictors of persistent viremia: hypervariable region 1 asa decoy. J Virol 73, 2938–2946.
Rijnbrand, R. C. & Lemon, S. M. (2000). Internal ribosome entry site-mediated translation in hepatitis C virus replication. Curr TopMicrobiol Immunol 242, 85–116.
Robertson, B., Myers, G., Howard, C. & 14 other authors. (1998).Classification, nomenclature, and database development for hepatitisC virus (HCV) and related viruses: proposals for standardization. Inter-national Committee on Virus Taxonomy. Arch Virol 143, 2493–2503.
Santolini, E., Migliaccio, G. & La Monica, N. (1994). Biosynthesis andbiochemical properties of the hepatitis C virus core protein. J Virol68, 3631–3641.
Shimoike, T., Mimori, S., Tani, H., Matsuura, Y. & Miyamura, T.(1999). Interaction of hepatitis C virus core protein with viral senseRNA and suppression of its translation. J Virol 73, 9718–9725.
Shrivastava, A., Manna, S. K., Ray, R. & Aggarwal, B. B. (1998).Ectopic expression of hepatitis C virus core protein differentiallyregulates nuclear transcription factors. J Virol 72, 9722–9728.
824 Journal of General Virology 84
D. Li and others

Simmonds, P. (1995). Variability of hepatitis C virus. Hepatology 21,570–583.
Trowbridge, R. & Gowans, E. J. (1998). Molecular cloning of anAustralian isolate of hepatitis C virus. Arch Virol 143, 501–511.
Wang, Y. H., Trowbridge, R. & Gowans, E. J. (1997). Expression andinteraction of the hepatitis C virus structural proteins and the 59untranslated region in baculovirus infected cells. Arch Virol 142,2211–2223.
Wang, T. H., Rijnbrand, R. C. & Lemon, S. M. (2000). Core protein-coding sequence, but not core protein, modulates the efficiency ofcap-independent translation directed by the internal ribosome entrysite of hepatitis C virus. J Virol 74, 11347–11358.
World Health Organisation. (1999). Global surveillance and control
of hepatitis C. Report of a WHO Consultation organized in
collaboration with the Viral Hepatitis Prevention Board, Antwerp,
Belgium. J Viral Hepat 6, 35–47.
Zhang, J., Yamada, O., Yoshida, H., Iwai, T. & Araki, H. (2002).Autogenous translation inhibition of core protein: implication for
switch from translation to RNA replication in hepatitis C virus.Virology 293, 141–150.
Zibert, A., Meisel, H., Kraas, W., Schulz, A., Jung, G. & Roggendorf, M.(1997). Early antibody response against hypervariable region 1 is
associated with acute self-limiting infections of hepatitis C virus.Hepatology 25, 1245–1249.
http://vir.sgmjournals.org 825
HCV core modulation of translation
Related Documents











