ORIGINAL PAPERS Alpha-catenin is required for IGF-I-induced cellular migration but not invasion in human colonic cancer cells Fre´ de´ ric Andre´ 1,4 , Barbara Janssens 2 , Erik Bruyneel 1 , Frans van Roy 2 , Christian Gespach 3 , Marc Mareel 1 and Marc Bracke* ,1 1 Laboratory of Experimental Cancerology, Ghent University Hospital, De Pintelaan 185, Ghent B-9000, Belgium; 2 Molecular Cell Biology Unit, Department of Molecular Biology, VIB, Ghent University, Ghent, Belgium; 3 Inserm U482, Hopital Saint-Antoine, Paris, France The mechanisms by which growth factors cooperate with cell adhesion molecules to modulate epithelial cell motility remain poorly understood. Here, we investigated the role of the E-cadherin/catenin complex in insulin-like growth factor (IGF-I)-dependent cell migration and invasion. We used variants of the HCT-8 colon cancer family that differ in their expression of aE-catenin, an intracellular molecule that links the E-cadherin/catenin complex to the actin cytoskeleton. Migration was determined using a mono- layer wound model and cell invasion by the penetration of the cells into type-I collagen gels. We showed that a- catenin-deficient cells were not able to migrate in cohort upon IGF-I stimulation. Transfection of these cells with a- catenin isoforms (aN- or aT-catenin) restored migratory response IGF-I. These results suggest that a-catenins are involved in the signal issued from the E-cadherin/catenin complex to regulate IGF-I-stimulated migration. In contrast, IGF-I promoted invasion of both a-catenin- deficient and a-catenin-expressing cells, indicating that a- catenin did not participate in the regulation of IGF-I- induced invasion. Inhibition of E-cadherin function by treatment with MB-2 monoclonal antibodies inhibited both IGF-I-dependent cell migration and invasion. Taken together, our results indicate that functional a-catenin is essential for migration but not for invasion, while E-cadherin is involved in both phenomena. Oncogene (2004) 23, 1177–1186. doi:10.1038/sj.onc.1207238 Keywords: colorectal cancer; E-cadherin/catenin com- plex; IGF-IR; type-I collagen; woundhealing Introduction To colonize new organ sites, cancer cells must access the lymphatic and blood vessels, disseminate, extravasate and invade the new organ’s parenchyma. All these processes require events involving cell adhesion, migra- tion, local and distant invasion. Although the detailed mechanisms of cell motility are not yet understood, it is now clear that dynamic and reciprocal interactions between cell adhesion molecules, extracellular matrix and soluble factors are essential (Conacci-Sorrell et al., 2002; Schwartz and Ginsberg, 2002). Insulin-like growth factors (IGF-I and IGF-II) are multifunctional regulatory peptides that share structural homology with proinsulin and mediate several signaling pathways involved in cell proliferation, differentiation and survival. There is increasing evidence that IGFs are also able to stimulate cell motility (Leventhal and Feldman, 1997). IGFs exert these effects through the IGF-I receptor (IGF-IR), a tyrosine kinase heterote- tramer homologous to the insulin receptor (Furstenber- ger and Senn, 2002). An early step in signal transduction by IGF-IR is the extensive tyrosine phosphorylation of IRS-1, which initiates several distinct signaling path- ways such as phosphatidyl inositol-3 kinase (PI-3K) and mitogen-activated protein kinase cascades (Baserga, 2000). In addition, the action of IGF-I is regulated by interactions with IGF-binding proteins (IGFBPs) that modulate the IGF bioavailability to the cell surface IGF-IR (Furstenberger and Senn, 2002). Converging data from recent clinical and laboratory studies clearly indicate that the IGF system is involved in the pathogenesis of colorectal cancers (Furstenberger and Senn, 2002; Sandhu et al., 2002). In epithelial cells, intercellular adhesion is primarily mediated by E-cadherin, a 120-kDa calcium-dependent transmembrane glycoprotein. Extracellularly, E-cadher- in molecules associate in a homophilic way to promote specific cell–cell adhesion (Angst et al., 2001). The cytoplasmic domain of E-cadherin binds noncovalently to b-catenin or g-catenin (Angst et al., 2001). Both b- and g-catenins bind a-catenin that can link directly or indirectly the actin cytoskeleton. To date, three a- catenin subtypes have been described: aE-catenin, aN- catenin and aT-catenin (Janssens et al., 2001). The dysfunction and the loss of the E-cadherin/catenin complex have been strongly implicated in carcinogenesis and invasiveness of epithelial tumor cells (Mareel and Leroy, 2002). The E-cadherin/catenin complex can be regulated at multiple levels, for example, mutations in cadherin or catenin genes vs functional inactivation of Received 16 June 2003; revised 10 September 2003; accepted 25 September 2003 *Correspondence: M Bracke; E-mail: [email protected] 4 Current address: UMR CNRS 6032, Faculte´ de Pharmacie, 27 Bvd Jean Moulin, Marseille F-13385, France Oncogene (2004) 23, 1177–1186 & 2004 Nature Publishing Group All rights reserved 0950-9232/04 $25.00 www.nature.com/onc

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL PAPERS
Alpha-catenin is required for IGF-I-induced cellular migration but not
invasion in human colonic cancer cells
Frederic Andre1,4, Barbara Janssens2, Erik Bruyneel1, Frans van Roy2, Christian Gespach3,Marc Mareel1 and Marc Bracke*,1
1Laboratory of Experimental Cancerology, Ghent University Hospital, De Pintelaan 185, Ghent B-9000, Belgium; 2Molecular CellBiology Unit, Department of Molecular Biology, VIB, Ghent University, Ghent, Belgium; 3Inserm U482, Hopital Saint-Antoine,Paris, France
The mechanisms by which growth factors cooperate withcell adhesion molecules to modulate epithelial cell motilityremain poorly understood. Here, we investigated the roleof the E-cadherin/catenin complex in insulin-like growthfactor (IGF-I)-dependent cell migration and invasion. Weused variants of the HCT-8 colon cancer family that differin their expression of aE-catenin, an intracellular moleculethat links the E-cadherin/catenin complex to the actincytoskeleton. Migration was determined using a mono-layer wound model and cell invasion by the penetration ofthe cells into type-I collagen gels. We showed that a-catenin-deficient cells were not able to migrate in cohortupon IGF-I stimulation. Transfection of these cells with a-catenin isoforms (aN- or aT-catenin) restored migratoryresponse IGF-I. These results suggest that a-catenins areinvolved in the signal issued from the E-cadherin/catenincomplex to regulate IGF-I-stimulated migration. Incontrast, IGF-I promoted invasion of both a-catenin-deficient and a-catenin-expressing cells, indicating that a-catenin did not participate in the regulation of IGF-I-induced invasion. Inhibition of E-cadherin function bytreatment with MB-2 monoclonal antibodies inhibitedboth IGF-I-dependent cell migration and invasion. Takentogether, our results indicate that functional a-cateninis essential for migration but not for invasion, whileE-cadherin is involved in both phenomena.Oncogene (2004) 23, 1177–1186. doi:10.1038/sj.onc.1207238
Keywords: colorectal cancer; E-cadherin/catenin com-plex; IGF-IR; type-I collagen; woundhealing
Introduction
To colonize new organ sites, cancer cells must access thelymphatic and blood vessels, disseminate, extravasateand invade the new organ’s parenchyma. All theseprocesses require events involving cell adhesion, migra-
tion, local and distant invasion. Although the detailedmechanisms of cell motility are not yet understood, it isnow clear that dynamic and reciprocal interactionsbetween cell adhesion molecules, extracellular matrixand soluble factors are essential (Conacci-Sorrell et al.,2002; Schwartz and Ginsberg, 2002).
Insulin-like growth factors (IGF-I and IGF-II) aremultifunctional regulatory peptides that share structuralhomology with proinsulin and mediate several signalingpathways involved in cell proliferation, differentiationand survival. There is increasing evidence that IGFs arealso able to stimulate cell motility (Leventhal andFeldman, 1997). IGFs exert these effects through theIGF-I receptor (IGF-IR), a tyrosine kinase heterote-tramer homologous to the insulin receptor (Furstenber-ger and Senn, 2002). An early step in signal transductionby IGF-IR is the extensive tyrosine phosphorylation ofIRS-1, which initiates several distinct signaling path-ways such as phosphatidyl inositol-3 kinase (PI-3K) andmitogen-activated protein kinase cascades (Baserga,2000). In addition, the action of IGF-I is regulated byinteractions with IGF-binding proteins (IGFBPs) thatmodulate the IGF bioavailability to the cell surfaceIGF-IR (Furstenberger and Senn, 2002). Convergingdata from recent clinical and laboratory studies clearlyindicate that the IGF system is involved in thepathogenesis of colorectal cancers (Furstenberger andSenn, 2002; Sandhu et al., 2002).
In epithelial cells, intercellular adhesion is primarilymediated by E-cadherin, a 120-kDa calcium-dependenttransmembrane glycoprotein. Extracellularly, E-cadher-in molecules associate in a homophilic way to promotespecific cell–cell adhesion (Angst et al., 2001). Thecytoplasmic domain of E-cadherin binds noncovalentlyto b-catenin or g-catenin (Angst et al., 2001). Both b-and g-catenins bind a-catenin that can link directly orindirectly the actin cytoskeleton. To date, three a-catenin subtypes have been described: aE-catenin, aN-catenin and aT-catenin (Janssens et al., 2001). Thedysfunction and the loss of the E-cadherin/catenincomplex have been strongly implicated in carcinogenesisand invasiveness of epithelial tumor cells (Mareel andLeroy, 2002). The E-cadherin/catenin complex can beregulated at multiple levels, for example, mutations incadherin or catenin genes vs functional inactivation of
Received 16 June 2003; revised 10 September 2003; accepted 25September 2003
*Correspondence: M Bracke; E-mail: [email protected] address: UMR CNRS 6032, Faculte de Pharmacie, 27 BvdJean Moulin, Marseille F-13385, France
Oncogene (2004) 23, 1177–1186& 2004 Nature Publishing Group All rights reserved 0950-9232/04 $25.00
www.nature.com/onc
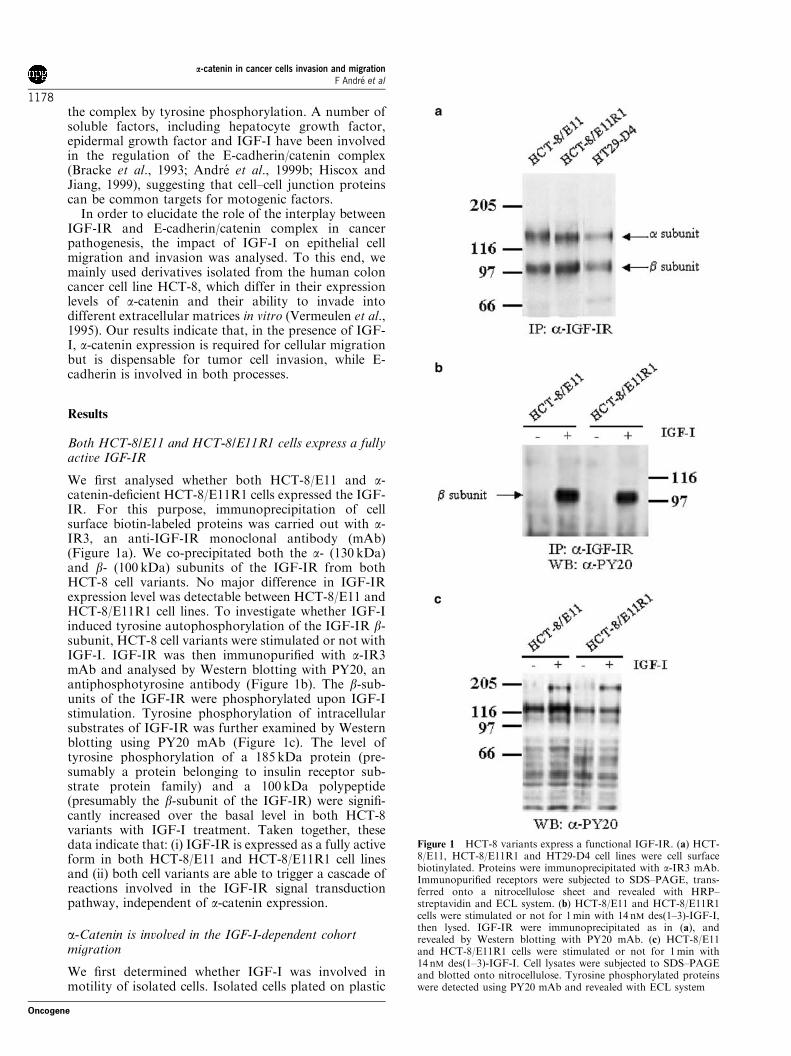
the complex by tyrosine phosphorylation. A number ofsoluble factors, including hepatocyte growth factor,epidermal growth factor and IGF-I have been involvedin the regulation of the E-cadherin/catenin complex(Bracke et al., 1993; Andre et al., 1999b; Hiscox andJiang, 1999), suggesting that cell–cell junction proteinscan be common targets for motogenic factors.
In order to elucidate the role of the interplay betweenIGF-IR and E-cadherin/catenin complex in cancerpathogenesis, the impact of IGF-I on epithelial cellmigration and invasion was analysed. To this end, wemainly used derivatives isolated from the human coloncancer cell line HCT-8, which differ in their expressionlevels of a-catenin and their ability to invade intodifferent extracellular matrices in vitro (Vermeulen et al.,1995). Our results indicate that, in the presence of IGF-I, a-catenin expression is required for cellular migrationbut is dispensable for tumor cell invasion, while E-cadherin is involved in both processes.
Results
Both HCT-8/E11 and HCT-8/E11R1 cells express a fullyactive IGF-IR
We first analysed whether both HCT-8/E11 and a-catenin-deficient HCT-8/E11R1 cells expressed the IGF-IR. For this purpose, immunoprecipitation of cellsurface biotin-labeled proteins was carried out with a-IR3, an anti-IGF-IR monoclonal antibody (mAb)(Figure 1a). We co-precipitated both the a- (130 kDa)and b- (100 kDa) subunits of the IGF-IR from bothHCT-8 cell variants. No major difference in IGF-IRexpression level was detectable between HCT-8/E11 andHCT-8/E11R1 cell lines. To investigate whether IGF-Iinduced tyrosine autophosphorylation of the IGF-IR b-subunit, HCT-8 cell variants were stimulated or not withIGF-I. IGF-IR was then immunopurified with a-IR3mAb and analysed by Western blotting with PY20, anantiphosphotyrosine antibody (Figure 1b). The b-sub-units of the IGF-IR were phosphorylated upon IGF-Istimulation. Tyrosine phosphorylation of intracellularsubstrates of IGF-IR was further examined by Westernblotting using PY20 mAb (Figure 1c). The level oftyrosine phosphorylation of a 185 kDa protein (pre-sumably a protein belonging to insulin receptor sub-strate protein family) and a 100 kDa polypeptide(presumably the b-subunit of the IGF-IR) were signifi-cantly increased over the basal level in both HCT-8variants with IGF-I treatment. Taken together, thesedata indicate that: (i) IGF-IR is expressed as a fully activeform in both HCT-8/E11 and HCT-8/E11R1 cell linesand (ii) both cell variants are able to trigger a cascade ofreactions involved in the IGF-IR signal transductionpathway, independent of a-catenin expression.
a-Catenin is involved in the IGF-I-dependent cohortmigration
We first determined whether IGF-I was involved inmotility of isolated cells. Isolated cells plated on plastic
Figure 1 HCT-8 variants express a functional IGF-IR. (a) HCT-8/E11, HCT-8/E11R1 and HT29-D4 cell lines were cell surfacebiotinylated. Proteins were immunoprecipitated with a-IR3 mAb.Immunopurified receptors were subjected to SDS–PAGE, trans-ferred onto a nitrocellulose sheet and revealed with HRP–streptavidin and ECL system. (b) HCT-8/E11 and HCT-8/E11R1cells were stimulated or not for 1min with 14 nM des(1–3)-IGF-I,then lysed. IGF-IR were immunoprecipitated as in (a), andrevealed by Western blotting with PY20 mAb. (c) HCT-8/E11and HCT-8/E11R1 cells were stimulated or not for 1min with14 nM des(1–3)-IGF-I. Cell lysates were subjected to SDS–PAGEand blotted onto nitrocellulose. Tyrosine phosphorylated proteinswere detected using PY20 mAb and revealed with ECL system
a-catenin in cancer cells invasion and migrationF Andre et al
1178
Oncogene

were treated for 5 h with or without des(1–3)IGF-I, atruncated analogue that binds with high affinity to theIGF-IR but not IGFBPs. The positions of the cell nucleiwere analysed every 5min. Using this assay, wedetermined that both HCT-8 cell variants exhibitedsame velocity rates (23.6 vs 24.1 mM/h for HCT-8/E11R1and HCT-8/E11 respectively). Moreover, IGF-I did notaffect the motility of any of the two HCT-8/E11 cellvariants (25.8 vs 24.3 mM/h for HCT-8/E11R1 and HCT-8/E11, respectively), indicating that a-catenin is notnecessary for random motility.
We next examined the function of a-catenin in cohortmigration using a woundhealing assay (Figure 2a). BothHCT-8/E11 and HCT-8/E11R1 cells migrated sponta-neously, without exogeneous stimulus. IGF-I did notincrease the number of HCT-8/E11R1 migrating cells,whereas it stimulated HCT-8/E11 cell migration in adose-dependent manner. This effect could be observed
for a concentration as low as 1 nM and reached a plateaufor a concentration of 14 nM.
To check whether IGFBPs could modulate IGFbioavailability, we used des(1–3)-IGF-I. As shown inFigure 2a, des(1–3)-IGF-I had no effect on the HCT-8/E11R1 cell migration whatever the concentration tested,whereas it increased the number of HCT-8E/11 migrat-ing cells. This des(1–3)-stimulated migration was alwayshigher than that observed using IGF-I, suggesting thatIGFBPs partially inhibited IGF-I-stimulated cell migra-tion. For subsequent experiments, des(1–3)-IGF-I wastherefore used at the concentration of 14 nM, that is, theconcentration inducing the maximal stimulation ofHCT-8/E11 cells.
To delineate whether IGF-I-dependent cell migrationwas directly mediated through IGF-IR, we preincubatedwounded monolayers with 10 mg/ml a-IR3 (Figure 2b).a-IR3 dramatically reduced the HCT-8/E11 migratoryresponse to IGF-I, indicating that the effect of IGF-I oncell migration was mediated through IGF-IR. Thespecificity was tested with an irrelevant mouse antibody(data not shown).
The wounded monolayers also exhibited differencesof morphology, as shown in Figure 3. The leading edgeof HCT-8/E11 monolayer had a relatively smooth shapereflecting a directional migration, whereas jagged wound
Figure 2 IGF-I and des(1–3)-IGF-I induce HCT-8/E11 cellmigration. (a) Confluent monolayers of HCT-8/E11 (squares) orHCT-8/E11R1 (circles) were wounded and incubated for 24 h withIGF-I concentrations (open squares and open circles) or des(1–3)-IGF-I (dark squares and dark circles) ranging from 0 to 70 nM.Migrating cells were counted as described in the Materials andmethods section. (b) Confluent HCT-8/E11R1 (open bars) andHCT-8/E11 (dashed bars) were wounded, treated with (þ ) andwithout (�) 10 mg/ml a-IR3 mAb and allowed to migrate for 24 hwith (þ ) or without (�) 14 nM des(1–3)-IGF-I. Results areexpressed as the average number of cells (þ s.d.) from threeindependent experiments performed in duplicate
Figure 3 Morphological features of HCT-8 cell migration.Monolayers of HCT-8/E11 (a) or HCT-8/E11R1 (b) cells werewounded and incubated for 24 h with 14 nM des(1–3)-IGF-I.Arrowheads indicate the original edges of the wounds. Scale bar:50mm
a-catenin in cancer cells invasion and migrationF Andre et al
1179
Oncogene

borders were observed for HCT-8/E11R1 cells, indicat-ing that these cells move without coordination. More-over, the actin staining patterns suggest that a contactinhibition of membrane ruffling occurs in HCT-8/E11cells but not or less so in HCT-8/E11R1 cells (notshown).
Time-lapse microscopy was used to detail some eventsinvolved in IGF-I-stimulated migration. As observed inFigure 4a, nuclei tracks of HCT-8/E11 cells wereroughly parallel, indicating that cells migrate unidir-ectionally towards the center of the wound withoutloosing contact with their neighbors. Des(1–3)-IGF-Ienhanced the migration velocity rate from 8 to 12–15 mM/h (Figure 4b). In contrast, nuclei tracks of HCT-8E11R1 cells were not parallel, indicating that cellsmoved in various directions without close cell–cellcontacts (Figures 4c and 4d). Using this assay, we werenot able to detect any effect of des(1–3)-IGF-I on HCT-8/E11R1 movement, since migration velocity rates were5 and 4.5 mM/h for control and des(1–3)-IGF-I-treatedcells, respectively.
Taken together, these results suggest that a-catenin isinvolved in IGF-I-induced directional migration. Toconfirm this hypothesis, a-catenin-deficient HCT-8/E11R1 cells were transfected with aT- or aN-cateninexpression vectors, then wounded and treated or notwith des(1–3)-IGF-I (Figure 5). Both parental andHCT-8/R1/1743 control cells were insensitive to des(1–3)-IGF-I. In line with our observations on HCT-8/E11cells, re-expression of aT- or aN-catenin in a-catenin-deficient cells restored the migratory response to des(1–3)-IGF-I. These results clearly suggest that a-cateninexpression is necessary for IGF-I-induced cohortmigration.
IGF-I-induced invasion is independent of a-cateninexpression
We examined the possibility that a-catenin may parti-cipate in the regulation of invasion (Figure 6). Both
isolated HCT-8 variants did not exhibit spontaneousinvasiveness into type-I collagen gels. However, treat-ment of cells with des(1–3)-IGF-I strongly promotedinvasion of the two HCT-8 variants. Invasion wasdramatically inhibited by a-IR3 mAb. We verified thatirrelevant mouse IgG1 did not affect the IGF-I-inducedinvasion. As a negative control, we showed that des(1–3)-IGF-I did not induce invasion of LoVo-C5 cells, acolon cell line that does not express an active IGF-IR.These data clearly indicate that (i) IGF-I inducedinvasion of both HCT-8 variants into type-I collagenthrough the activation of the IGF-IR, and (ii) a-catenin
Figure 4 Des(1–3)-IGF-I increases directional migration. HCT-8/E11 (a and b) and HCT-8/E11R1 (c and d) were wounded andincubated with (b and d) or without (a and c) 14 nM des(1–3)-IGF-I. Migration was recorded by time-lapse videomicroscopy for 6 h.Tracks of cell nuclei were drawn using position parameters. Thestraight dotted lines indicate the edge of the wounds. Scale bar:10mm
Figure 5 Transfection of cDNAs encoding various a-catenin-isoforms restores IGF-I-induced cell migration. Alpha-catenin-negative cells (HCT-8/E11R1 and cells transfected with controlvector), HCT-8/E11R1 cells expressing exogenous aN- or aT-catenin (aN-ctn and aT-ctn) and cells expressing endogenous aE-catenin (HCT-8/E11) were wounded and incubated for 24 h with(dashed bars) or without (open bars) 14 nM des(1–3)-IGF-I. Resultsare expressed as the average number of migrating cells (þ s.d.)from two independent experiments performed in duplicate
Figure 6 Induction of HCT-8/E11 and HCT-8/E11R1 cellinvasion into type-I collagen by des(1–3)-IGF-I. HCT-8/E11R1(open bars), HCT-8/E11 (dashed bars) and LoVo-C5 cells (blackbars) were seeded on top of collagen gel and incubated for 24 h inserum-free medium (Ctrl) containing 14 nM des(1–3)-IGF-I (IGF-I), or with 14 nM des(1–3)-IGF-I plus 10mg/ml a-IR3 mAb (IGF-Ia-IR3). A mouse IgG1 antibody was used as a control for a-IR3specificity. Invading cells were scored as described in Materials andmethods section. Bars represent mean (þ s.d.) from threeindependent experiments
a-catenin in cancer cells invasion and migrationF Andre et al
1180
Oncogene

expression is not required for cell invasion into type-Icollagen.
Signaling pathways involved in IGF-I-induced invasionand migration
We determined the possible contribution of PI-3K, Rho-like GTPases and heterotrimeric G-proteins in themigratory and proinvasive activity of IGF-I in HCT-8/E11 and HCT-8/E11R1 colonic cells. Wortmannin(10 nM) and LY29004 (10mM), C3 exotransferase(5 mg/ml) and pertussis toxin (200 ng/ml) almost com-pletely inhibited cellular invasion induced by des(1–3)-IGF-I in both HCT-8 variants (Table 1). Thoseinhibitors also dramatically reduced the IGF-I-stimu-lated HCT-8/E11 directional migration as assessed bythe monolayer-wounding assay. To confirm these
observations, we analysed whether dominant-negativemutants of PI-3K and rhoGTPases inhibited IGF-I-dependent cell migration. As observed in Table 2, theexpression of dominant-negative forms of N19 RhoA orN17 Rac in kidney MDCK T23 cells completelyabrogated the IGF-I-dependent cell migration. Intro-duction of the dominant-negative mutant of the PI-30Kp110 in kidney MDCK ts.src Cl2 cells also stronglyinhibited IGF-I-dependent cell migration.
IGF-I-induced cohort migration and invasion requireE-cadherin
We evaluated whether E-cadherin was involved in bothinvasion and migration by using three function-blockinganti-E-cadherin mAbs. Both MB2 and HECD-1 reactwith human E-cadherin, whereas DECMA-1 is specificfor canine E-cadherin. As observed in Figure 7a, the soleaddition of anti-E-cadherin mAbs MB2 or HECD-1 didnot decrease the number of migrating HCT-8 cellvariants. However, both MB2 and HECD-1 mAbsmarkedly inhibited des(1–3)-IGF-I-stimulated HCT-8/E11 cell migration. To consolidate this observation, wetested the effect of DECMA-1 on the migration ofMDCK T23 cell line (Figure 7b). This mAb inhibitedthe des(1–3)-IGF-I-stimulated MDCK T23 cell migra-tion, while MB2 mAb had no effect (not shown),confirming the specificity of the inhibition in presence ofDECMA-1.
We also analysed the contribution of E-cadherin oninvasion in type-I collagen (Figure 8a). Treatment ofboth HCT-8/E11 and HCT-8/E11R1 cells with MB2mAb dramatically inhibited the des(1–3)-stimulatedinvasion. The impact of the mAb was also tested onHCT-8 cell spreading on type-I collagen, an initial step
Table 1 Effect of synthetic inhibitors on cellular migration and invasion into type-I collagen
Cell migration
HCT-8/E11 HCT-8/E11R1
Inhibitor �des(1–3)-IGF-I +des(1–3)-IGF-I �des(1–3)-IGF-I +des(1–3)-IGF-I
None 98 (6) 240 (15) 102 (7) 95 (3)LY29004 102 (4) 122 (8) 100 (6) 102 (5)C3T 92 (8) 95 (13) 103 (8) 100 (2)Pertussis toxin 103 (9) 110 (4) 101 (5) 95 (8)
Cell invasion
HCT-8/E11 HCT-8/E11R1
Inhibitor �des(1–3)-IGF-I +des(1–3)-IGF-I �des(1–3)-IGF-I +des(1–3)-IGF-I
None 0.5 (0.3) 7.0 (1.3) 0.3 (0.1) 7.2 (0.2)LY29004 0.3 (0.1) 0.4 (0.1) 0.2 (0.1) 0.3 (0.1)C3T 0.5 (0.3) 0.2 (0.1) 0.2 (0.1) 0.4 (0.1)Pertussis toxin 0.7 (0.5) 1.0 (0.2) 0.4 (0.2) 0.7 (0.4)
For migration: monolayers were wounded and incubated for 90min in the presence or absence of inhibitors. Monolayers were then cultured for anadditional 24 h period in the presence or absence of inhibitors or 14 nM des(1–3)-IGF-I. Migrating cells were counted as described in the Materialsand methods section. [2]For invasion: cells were seeded on top of the collagen in the presence or absence of 14 nM des(1–3)-IGF-I or inhibitors andallowed to invade for 24 h. Invading cells were scored as described in Materials and methods section. [3]Data represent mean of three separateexperiments. Numbers in parentheses represent s.d.
Table 2 Des(1–3)-IGF-I-dependent migration is abrogated bydominant-negative mutants of the rho-GTPases rho and Rac and
p110 PI3-K
Cell line �des(1–3)-IGF-I +des(1–3)-IGF-I
MDCKT23 203 (35) 309 (23)MDCKT23 N19 rhoA 103 (31) 96 (15)MDCKT23 N17 rac-1 85 (25) 79 (29)351C MDCKts.src 193 (37) 272 (53)401C MDCKts.src 190 (41) 276 (42)351C MDCKts.src P110DN 74 (28) 69 (30)401C MDCKts.src P110DN 102 (22) 105 (21)
Cell monolayers were wounded, then incubated for 3.5 h in serum-freemedium without or with 14 nM des(1–3)-IGF-I. The width of thewound was measured at time zero and after the incubation period.Migration was expressed as the difference between the two measure-ments. Results represent distance of migration (in mM) from threeindependent assays. Numbers in parentheses represent s.d.
a-catenin in cancer cells invasion and migrationF Andre et al
1181
Oncogene

of invasion (Figure 8b). MB2 mAb abrogated des(1–3)-IGF-I-induced spreading of both HCT-8/E11R1 andHCT-8/E11 cells. Taken together, these data suggestthat E-cadherin is involved in both directional migrationand invasion.
Discussion
We have analysed the interplay between IGF-IR and theE-cadherin/catenin complex during cohort migrationand invasion of colonic cells. To this end, we have used acouple of variants of the HCT-8 cell line: HCT-8/E11cells have a functional E-cadherin/catenin complex,while HCT-8/E11R1 cells are deficient in a-cateninprotein (Vermeulen et al., 1995). According to our data,
the role of a-catenin and E-cadherin in IGF-I-stimulatedcohort migration and invasion can be summarized asfollows: (i) isolated cells on tissue culture plastic are notsensitive to IGF-I stimulation; (ii) when cell–cellcontacts are established, directional migration is stimu-lated by IGF-I and this effect is E-cadherin and a-catenin dependent; and (iii) isolated cells seeded on type-I collagen are induced by IGF-I to spread and invade,this effect being E-cadherin dependent but not requiringa-catenin.In vivo and in vitro studies indicated that carcinoma
cells frequently invade the surrounding tissue ascoherent clusters or nests of cells, a process termedcohort migration (Nabeshima et al., 1999). This migra-tion requires the coordination of several mechanismsincluding directional guidance and modulation of cell–cell adhesion within the migrating cell groups. Bothactivities can be studied using the monolayer-woundingassay. We have previously shown that IGF-I is able to
Figure 7 Anti-E-cadherin mAbs inhibit IGF-I-induced migration.(a) Confluent monolayers of HCT-8/E11 (dashed bars) and HCT-8/E11R1 (open bars) were pretreated for 90min with MB2 (0.1mg/ml) or HECD-1 (1mg/ml) mAbs. Cell monolayers were thenwounded and incubated with or without 14 nM des(1–3)-IGF-I for24 h in presence of the mAbs. (b) Confluent monolayers ofMDCKT23 cells were pretreated for 90min with 1mg/ml DEC-MA-1 mAb as indicated. Cell monolayers were then wounded andincubated with or without 14 nM des(1–3)-IGF-I. Results areexpressed as the average number of cells (þ s.d.) from threeindependent experiments performed in duplicate
Figure 8 MB2 mAb inhibits cellular IGF-I-induced invasion intotype-I collagen and cell spreading. (a) HCT-8/E11R1 (open bars),HCT-8/E11 (dashed bars) cells were seeded on top of type-Icollagen gel and incubated for 24 h in serum-free medium with orwithout 14 nM des(1–3)-IGF-I and in the presence or the absence ofMB2 mAb as indicated. Invading cells were scored as described inMaterials and methods section. (b) HCT-8/E11R1 (open bars),HCT-8/E11 (dashed bars) cells were seeded on top of type-Icollagen and incubated for 2 h with or without 14 nM des(1–3)-IGF-I and in the presence or the absence of MB2 mAb asindicated. Spreading cells were scored as described in Materials andmethods section. Bars represent mean þ s.d. (n¼ 3 assay)
a-catenin in cancer cells invasion and migrationF Andre et al
1182
Oncogene

induce cohort migration of HT29-D4 colonic epithelialcells (Andre et al., 1999a). We report here that thisability can be extended to other colonic (HCT-8/E11) orkidney (MDCK) epithelial cell lines. Interestingly, IGF-I increases migration of a-catenin-expressing cells(HCT-8/E11 cells) but not of a-catenin-deficient cells(HCT-8/E11R1 cells). The inability of HCT-8/E11R1cells to migrate in cohort in response to IGF-I isobviously not due to a defect in the IGF-I system.Indeed, (i) these cells express IGF-IR, (ii) binding ofIGF-I to IGF-IR leads to IGF-IR b-subunit tyrosinephosphorylation and activation of IGF-IR downstreamsignaling molecules, and (iii) IGF-I induces HCT-8/E11R1 cell invasion into type-I collagen gels. Thus, thispaper reports for the first time a crucial role for a-catenin during IGF-I-induced cohort migration.According to this, introduction of aT- or aN-catenin-in a-catenin-deficient cells restored the migratoryresponse to IGF-I. This finding is in agreement withrecent reports, suggesting that a-catenin is required fordistinct biological activities including reduction of celldeath and alteration of keratinocyte proliferation andcell polarity (Matsubara and Osawa, 2001; Vasioukhinet al., 2001).
The mechanism(s) by which a-catenin expressionmodulates IGF-I-induced cohort migration needs to beexplored in more depth. Firstly, one may speculate thata-catenin is directly involved in the IGF-I signalingpathway. Consistent with this hypothesis, studies havesuggested a physical interaction between IGF-IR and E-cadherin/catenin complex (Mauro et al., 2001; Moraliet al., 2001). Moreover, loss of a-catenin alters theresponse of keratinocytes to several growth factors(Vasioukhin et al., 2001). Therefore, the defect in a-catenin expression or molecular structure, leading to anuncoupling between the IGF-I signaling pathway andthe E-cadherin/catenin complex, may abolish thecapacity of HCT-8/E11R1 cells to migrate upon IGF-Istimulation. In agreement with this, inactivation ofcell–cell adhesion with anti-E-cadherin mAbs alters theIGF-I-induced tyrosine phosphorylation of someproteins (not shown). Secondly, cohort migration couldbe impaired by alteration between cell–cell contactsand the actin cytoskeleton. Since a-catenin is a keymolecule that links the E-cadherin/b-catenin complexto the actin cytoskeleton (Angst et al., 2001), lack ofa-catenin could cause alteration of the actin cytoskele-ton and therefore coordinated migration. Since IGF-IRactivation affects the actin cytoskeletal organization(Guvakova and Surmacz, 1999), further studiesare needed to explore the relationship between theIGF-IR and the actin cytoskeleton during cohortmigration.
Invasion of cancer cells from various tissue origins isoften inversely related to the expression of E-cadherin(Van Hoorde et al., 2000). According to this, neitherLoVo cells nor HCT-8 cells invade type-I collagenwithout the addition of external stimuli other than thecollagen itself. In addition, since neither HCT-8/E11 norHCT-8/E11R1 showed autonomous invasion into type-Icollagen, we postulated that loss of a-catenin is neither
sufficient nor necessary to stimulate invasion of epithe-lial colonic cells into substrata.
In our studies, IGF-I promoted cellular invasion inboth HCT-8/E11 and HCT-8/E11R1 cell variants. Thus,we can conclude that a-catenin is not required for IGF-I-induced invasion. Our finding that cells, when droppedon type-I collagen gels react to IGF-I even in theabsence of a-catenin, is in line with the concept thattype-I collagen functions not only as a substratum butalso as a proinvasive signaling substratum (Van Hoordeet al., 2000). Along with this notion, pancreatic cancercells dramatically reduce E-cadherin gene expressionwhen cultured on type-I collagen, leading to increasedproliferation and migration behavior (Menke et al.,2001). Moreover, when exposed to extracellular matrixduring invasion, carcinoma cells do not possess theadhesion apparatus characteristic for normal cells, andcan invade despite a low level of a-catenin (Glukhovaet al., 1995). It is clear that IGF-I is not the uniquecofactor necessary to stimulate invasion, since variousextrinsic factors have similar effects on some epithelialcancer cells from various origins (Van Hoorde et al.,2000; Steelant et al., 2001). However, our experimentsclearly indicate that IGF-I should be considered as apotent and efficient paracrine host factor triggeringinvasion during colon cancer progression.
Our data clearly showed that E-cadherin plays anessential role in both IGF-I-induced cohort migrationand invasion. Inhibition of cell–cell contacts using anti-E-cadherin mAb MB2 inhibited the IGF-I-inducedcohort cell migration. This confirms other workssuggesting that colon adenocarcinoma cells can moveas coherent cell nests, while maintaining E-cadherin-based cell–cell adhesion links in some parts andextending filopodia or lamellipodia in other parts(Andre et al., 1999b; Nabeshima et al., 1999). Wedemonstrated here that inactivation of E-cadherin byMB2 mAb prevents IGF-I-induced invasion in type-Icollagen, and abrogated IGF-I-induced cell spreadingon type-I collagen. This discrepancy can be related tothe cell types considered since the MB2 mAb preventedcellular invasion induced by IGF-I in HT29D4 cells,another colonic cancer cell line (not shown). On theother hand, it has been shown that IGF-IR belongs tothe same membrane complex as E-cadherin (Moraliet al., 2001). We can thus hypothesize that MB2 mAbinterfere with this membrane complex, leading to theinhibition of IGF-I-induced cellular invasion in type-Icollagen. Consistent with this hypothesis, we observedthat MB2 mAb alters the IGF-I-induced tyrosinephosphorylation of some proteins (not shown).
No obvious difference seems to exist between thesignaling pathways used by IGF-I to convey eithermigratory or cellular invasion responses. Pharmacolo-gical and dominant-negative mutants of PI-3K abro-gated both IGF-I-induced migration and invasion, asreported previously (Barbier et al., 2001). Similardependence was noticed regarding the implication ofthe small GTPases Rho-A and Rac-1. In line with thisfinding, other report indicate that the Rho-like smallGTPases regulate cell motility, cell–cell adhesion and
a-catenin in cancer cells invasion and migrationF Andre et al
1183
Oncogene

invasion (Mareel and Leroy, 2002). Moreover, we canpostulate that the IGF-R signal through heterotrimeric-dependent G-proteins Gao/i and Gbg since PTx inhibitsIGF-induced invasion. Using MDCK cells, we demon-strated that the signaling pathways involved in IGF-I-induced cell migration can be extended to kidneyepithelial cells. Indeed, (1) pharmacological inhibitorsabrogated kidney cell migration and (2) expression ofdominant-negative mutants of RhoA, Rac1 or PI-3K inMDCK cells reduced the IGF-I-induced MDCK cellmigration.
The microenvironment of the local host tissue can bean active participant of cellular invasion. Indeed, both invivo and in vitro studies have demonstrated a continuouscrosstalk between cancer cells, host cells (fibroblasts,myofibroblasts, endothelial and inflammatory cells) andextracellular matrix components. In this connection,previous studies demonstrated that both HCT-8/E11and HCT-8/E11R1 cells line differed in invasiveness intodifferent substrates and proinvasive factors. For in-stance, we have shown that HCT-8/E11 cells arespontaneously invasive into precultured chick heartfragments and Matrigel (Van Hoorde et al., 2000).Furthemore, our data clearly establish that IGF-I-induced colonic cancer cell invasion in type-I collagenis not determined by the expression of a-catenin, whilethe motile phenotype is a-catenin dependent. Incontrast, a-catenin-deficient HCT-8/E11R1 cells arenot invasive in collagen gels after the addition of bileacids and other effectors (Debruyne et al., 2002).Therefore, IGF-I is a very pejorative proinvasive factoracting as a dominant paracrine/autocrine host factorthat overcome the depletion of a-catenin in HCT-8/E11R1 human colon cancer cells. Accordingly, we haveshown that a-catenin expression in orthotopicallyimplanted HCT-8 cells in nude mice did not influencethe invasive phenotype of the tumor xenografts (VanHoorde et al., 2000). Further studies are needed toestablish whether paracrine/autocrine IGF-I secretion inhuman solid tumors can be a target for future anti-invasive and -metastatic therapy.
Materials and methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), RPMI, Ham’sF12, and McCoy’s 5A modified medium and fetal calf serum(FCS) were purchased from Life Technologies (Ghent,Belgium). Wortmannin, LY29004, C3 exoenzyme and pertus-sis toxin were from Sigma (St Louis, MO, USA). Humanrecombinant IGF-I and des(1–3)-IGF-I were from BachemAG (Bubendorf, Switzerland) and Gropep (Adelaide, Austra-lia), respectively.MB2 is a mAb against human E-cadherin with neutralizing
effects on E-cadherin functions (Bracke et al., 1993). Rat anti-murine E-cadherin mAb DECMA-1 was from Sigma, whereasmouse mAb against E-cadherin was purchased from Takara(Shuzo, Kyoto, Japan). Mouse PY20 mAb antiphosphotyr-osine was from Transduction Laboratories (Lexington,KY, USA). Mouse mAb against IGF-IR (a-IR3 clone)was purchased from Oncogene Science (Uniondale,
NY, USA). Horseradish peroxidase sheep anti-mouse Igwas from Amersham. Mouse mAb anti-transferrinreceptor (clone YDJ1.2.2) was from Immunotech (Marseille,France).
Cell lines
HCT-8/E11 and HCT-8/E11R1 are subcloned from the HCT-8human colon carcinoma cell line (Vermeulen et al., 1995).HCT-8/E11R1 cells are a-catenin-deficient due to a constitu-tive nonsense mutation in CTNNA1 and a putative secondmutation causing alternative splicing (Vermeulen et al., 1999).HCT-8 cells were cultured in RPMI containing 10% FCS.HCT-8/R1/T31 and HrpCaN1 were HCT-8/E11R1 cellstransfected with a-T- and a-N-catenin, respectively (vanHengel et al., 1997; Janssens et al., 2001). HCT-8/R1/1743cells are HCT-8/E11R1 cells transfected with the emptyPEF6MH vector and were used as a negative control. Thehuman colonic adenocarcinoma cell lines HT29-D4 and LoVo-C5 were cultured in, respectively, DMEM and McCoy’s 5Amodified medium containing 10% FCS.MDCKts.srcCl2 are Madin–Darby canine kidney cells
transfected with a temperature-sensitive mutant for the srconcogene (Behrens et al., 1993). The MDCKts.src-p110DNcell line were MDCKts.srcCl2 cells transfected with thedominant-negative mutant p110 a EcoS of PI-3K (Kotelevetset al., 1998). The MDCKT23 cell lines expressing themutant G-proteins Rho-A (N19 Rho) and Rac-1 (N17Rac) were a generous gift from Dr J Nelson (Jou and Nelson,1998).
Cohort migration
Wounding assays were essentially performed with a razorblade as described previously (Andre et al., 1999a), thenincubated for 24 h in serum-free medium supplemented with0.1% BSA containing test substances or not. Cell migrationwas assessed by counting the cell nuclei observed across thewound borders. When appropriate function-blocking mAbswere added 90min prior to wounding and during the timeof migration. Migration results are expressed as theaverage7s.d. of the number of migrating cells per microscopicfield (� 200).Owing to an higher migration velocity rate, MDCK cell
monolayers were wounded with an asepticized plastic tooth-pick. The width of the wound was measured with an invertedmicroscope (� 200) at time zero and after a 3.5 h incubationperiod. For each wound, 10 measures were performed.Migration was expressed as the difference between the twomeasurements.
Video registration
Migration of cells after monolayer wounding was recorded byvideomicroscopy. Time-lapse images were recorded with aninverted microscope (phase contrast; objective � 32) con-nected with an MTI CCD72 camera (Dage MTI, MichiganCity, IN, USA) and a U-matic VO-5850P videorecorder (Sony,Tokyo, Japan) at 30 s intervals. Track lengths of nucleardisplacements were drawn from the display screen ontransparencies for a total observation time of 6 h.
Cell motility assay
Isolated cells were allowed to attach on the substratum for1 h in DME/10% FCS. Cells were washed three timeswith DME and then incubated in DME/0.1% BSA withor without 14 nM des(1–3)-IGF-I at 371C in a 5% CO2
a-catenin in cancer cells invasion and migrationF Andre et al
1184
Oncogene

atmosphere. Image acquisition was performed every 5min for6 h using the Vision Explorer VA application from Graftek(Mirmande, France). ETC 300 software (Graftek) was used toanalyse nuclear trajectories and to measure the nuclearvelocity.
Collagen invasion assay
Collagen type-I gels (Upstate Biotechnologies; Lake Placid,NY, USA) were prepared in DMEM (0.22% w/v) using six-well plates, as described (Vleminckx et al., 1991). Cells wereseeded on top of the collagen gels and incubated for 24 h at371C in the presence or absence of 14 nM des(1–3)-IGF-I aloneor combined with function-blocking antibodies or appropriateinhibitors of signal transduction pathways. The invasion indexwas calculated from the mean number of invasive cells over thetotal number of cells, counted in at least 12 microscopic fields.
Cell spreading assay
Overnight 24-well plates were coated at 41C with 250 ml oftype-I collagen at 10mg/ml. Coated wells were blocked with0.5% BSA in phosphate-buffered saline (PBS) for 30min andthen washed twice with 0.1% PBS. Single-cell suspensions(25 000 cells/0.5ml) were seeded in substratum-coated wellsand allowed to adhere for 2 h at 371C. Spreading cells werecounted microscopically.
Cell surface labeling and IGF-IR immunoprecipitation
Subconfluent cells were washed twice with PBS and incubatedwith 1mg/ml EZ-Link Sulfo-NHS-LC-biotin (Pierce; Rock-ford, IL,USA) in PBS for 20min at 41C under shaking. Cellswere then washed with PBS and solubilized in RIPA buffer asdescribed previously (Andre et al., 1999a). Clarified cell lysateswere incubated with 10 mg/ml mAb a-IR3 overnight at 41C.
After adding Sepharose protein A (Pharmacia; North Pea-pack, NJ, USA) for 45min, the suspension was centrifugedand pellets were extensively washed. Immunoprecipitatedproteins were solubilized in Laemmli sample buffer, heatedat 951C for 5min, submitted to SDS–PAGE (7.5% acryla-mide) under reducing conditions and blotted onto nitrocellu-lose sheets. Membranes were saturated with 50mM Tris-HCl(pH 8.0), 10% glycerol, 1M glucose, 0.5% BSA and 0.1%Tween 20 for 1 h at room temperature. Biotinylated proteinswere labeled with HRP–streptavidin (Amersham; Roosendaal,The Netherlands) in the same buffer for 45min at roomtemperature and were detected by the ECL system accordingto the manufacturer’s instructions.
IGF-I-stimulated tyrosine phosphorylation
Cells were serum starved in DME containing 0.1% BSA for 2 hat 371C, then washed and stimulated with 14 nM des(1–3)-IGF-I for 1min in the same buffer. Cells were lysed with RIPAbuffer containing 5mM sodium orthovanadate, 100mM NaFplus the cocktail of proteases inhibitors mentioned above.IGF-IR was immunopurified from cell lysates with a-IR3 mAbas described above. Equal amounts of proteins were subjectedto 7.5% acrylamide gel and transferred onto a nitrocellulosesheet. Tyrosine phosphorylated IGF-IR was probed withPY20 mAb and detected by chemiluminescence. In someexperiments, tyrosine phosphorylation of cellular proteins wasanalysed by immunoblotting with PY 20 mAb.
AcknowledgementsThis work was supported by the Association de la Recherchecontre le Cancer (France), the Societe de Secours des Amis desSciences (France), the Fortis-Bank/Verzekeringen and theFund for Scientific Research (FWO-Vlaanderen), Brussels,Belgium.
References
Andre F, Rigot V, Remacle-Bonnet M, Luis J, Pommier G andMarvaldi J. (1999a). Gastroenterology, 116, 64–77.
Andre F, Rigot V, Thimonier J, Montixi C, Parat F, PommierG, Marvaldi J and Luis J. (1999b). Int. J. Cancer, 83,
497–505.Angst BD, Marcozzi C and Magee AI. (2001). J. Cell Sci., 114,629–641.
Barbier M, Attoub S, Calvez R, Laffargue M, Mareel M,Altruda F, Gespach C, Hirsch E and Wymann M. (2001).Nature, 413, 796.
Baserga R. (2000). Oncogene, 19, 5574–5581.Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F,Mareel MM and Birchmeier W. (1993). J. Cell Biol., 120,757–766.
Bracke ME, Vyncke BM, Bruyneel EA, Vermeulen SJ, DeBruyne GK, Van Larebeke NA, Vleminckx K, Van Roy FMand Mareel MM. (1993). Br. J. Cancer, 68, 282–289.
Conacci-Sorrell M, Zhurinsky J and Ben-Ze’ev A. (2002). J.Clin. Invest., 109, 987–991.
Debruyne P, Bruyneel E, Karaguni I-M, Li X, Flatau G,Muller O, Zimber A, Gespach C and Mareel M. (2002).Oncogene, 21, 7640–7650.
Furstenberger G and Senn HJ. (2002). Lancet Oncol., 3,
298–302.Glukhova M, Koteliansky V, Sastre X and Thiery JP. (1995).
Am. J. Pathol., 146, 706–716.
Guvakova MA and Surmacz E. (1999). Exp. Cell Res., 251,
244–255.Hiscox S and Jiang WG. (1999). Biochem. Biophys. Res.
Commun., 261, 406–411.Janssens B, Goossens S, Staes K, Gilbert B, van Hengel J,Colpaert C, Bruyneel E, Mareel M and van Roy F. (2001).J. Cell Sci., 114, 3177–3188.
Jou TS and Nelson WJ. (1998). J. Cell Biol., 142, 85–100.Kotelevets L, Noe V, Bruyneel E, Myssiakine E, Chastre E,Mareel M and Gespach C. (1998). J. Biol. Chem., 273,
14138–14145.Leventhal P and Feldman E. (1997). Trends Endocrinol.
Metab., 8, 1–6.Mareel M and Leroy A. (2002). Physiol. Rev., 83,
337–376.Matsubara S and Osawa M. (2001). J. Cell Biol., 154,573–584.
Mauro L, Bartucci M, Morelli C, Ando S and Surmacz E.(2001). J. Biol. Chem., 276, 39892–39897.
Menke A, Philippi C, Vogelmann R, Seidel B, Lutz MP,Adler G and Wedlich D. (2001). Cancer Res., 61,
3508–3517.Morali O, Delmas V, Moore R, Jeanney C, Thiery J and LarueL. (2001). Oncogene, 20, 4942–4950.
Nabeshima K, Inoue T, Shimao Y, Kataoka H and Koono M.(1999). Histol. Histopathol., 14, 1183–1197.
a-catenin in cancer cells invasion and migrationF Andre et al
1185
Oncogene

Sandhu MS, Dunger DB and Giovannucci EL. (2002). J. Natl.Cancer Inst., 94, 972–980.
Schwartz MA and Ginsberg MH. (2002). Nat. Cell Biol., 4,
E65–E68.Steelant W, Goeman J, Philippe J, Oomen L, Hilkens J,Krzewinski-Recchi M, Huet G, Van der Eycken J, DelannoyP, Bruyneel E and Mareel M. (2001). Int. J. Cancer, 92,
527–536.van Hengel J, Gohon L, Bruyneel E, Vermeulen S, CornelissenM,Mareel M and van Roy F. (1997). J. Cell Biol., 137, 1103–1116.
Van Hoorde L, Van Aken E and Mareel M. (2000). Prog. Mol.Subcell. Biol., 25, 105–134.
Vasioukhin V, Bauer C, Degenstein L, Wise B and Fuchs E.(2001). Cell, 104, 605–617.
Vermeulen SJ, Bruyneel EA, Bracke ME, De BruyneGK, Vennekens KM, Vleminckx KL, Berx GJ, vanRoy FM and Mareel MM. (1995). Cancer Res., 55,4722–4728.
Vermeulen SJ, Nollet F, Teugels E, Vennekens KM,Malfait F, Philippe J, Speleman F, Bracke ME,van Roy FM and Mareel MM. (1999). Oncogene, 18,905–915.
Vleminckx K, Vakaet Jr L, Mareel M, Fiers W and van Roy F.(1991). Cell, 66, 107–119.
a-catenin in cancer cells invasion and migrationF Andre et al
1186
Oncogene
Related Documents





![WallFlex Colonic Stent - Boston Scientific- US · WallFlex ™ Colonic Stent Visualization Expertise in combining stent materials has resulted ... (BTS). “The WallFlex™ [Colonic]](https://static.cupdf.com/doc/110x72/5ae601bc7f8b9a8b2b8ca931/wallflex-colonic-stent-boston-scientific-us-colonic-stent-visualization-expertise.jpg)





