Algorithms for testing that sets of DNA words concatenate without secondary structure ∗ Mirela Andronescu University of British Columbia, The Department of Computer Science Danielle Dees Georgia Institute of Technology, College of Computing Laura Slaybaugh Rose-Hulman Institute of Technology, Computer Science Department Yinglei Zhao and Anne Condon † University of British Columbia, The Department of Computer Science Barry Cohen and Steven Skiena State University of New York at Stony Brook, Computer Science Department Abstract. We present an efficient algorithm for determining whether all molecules in a combinatorial set of DNA or RNA strands are structure free, and thus available for bonding to their Watson-Crick complements. This work is motivated by the goal of testing whether strands used in DNA computations or as molecular bar-codes are structure free, where the strands are concatenations of short words. We also present an algorithm for determining whether all words in S ∗ , for some finite set S of equi-length words, are structure free. Keywords: RNA secondary structure, testing DNA word sets 1. Introduction In search-and-prune DNA computations, many long DNA strands are created from a small number of short strands, called words. For exam- ple, Braich et al. (Braich et al., 2001) use the words pairs (TATTCTCACCCATAA, CTATTTATATCCACC), (ACACTATCAACATCA, ACACCTAACTAAACT), (CCTTTACCTCAATAA, CTACCCTATTCTACT), (CTCCCAAATAACATT, ATCTTTAAATACCCC), (AACTTCACCCCTATA, TCCATTTCTCCATAT), (TCATATCAACTCCAC, TTTCTTCCATCACAT) ∗ This material is based upon work supported by the U.S. National Science Foundation under Grant No. 0130108, by the National Sciences and the Engi- neering Research Council of Canada, and by the Defense Advanced Research Projects Agency (DARPA) and Air Force Research Laboratory, Air Force Material Command, USAF, under agreement number F30602-01-2-0555. † Contact author: [email protected] c 2003 Kluwer Academic Publishers. Printed in the Netherlands. journal.tex; 26/05/2003; 11:50; p.1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Algorithms for testing that sets of DNA words concatenate
without secondary structure ∗
Mirela AndronescuUniversity of British Columbia, The Department of Computer Science
Danielle DeesGeorgia Institute of Technology, College of Computing
Laura SlaybaughRose-Hulman Institute of Technology, Computer Science Department
Yinglei Zhao and Anne Condon†University of British Columbia, The Department of Computer Science
Barry Cohen and Steven SkienaState University of New York at Stony Brook, Computer Science Department
Abstract. We present an efficient algorithm for determining whether all moleculesin a combinatorial set of DNA or RNA strands are structure free, and thus availablefor bonding to their Watson-Crick complements. This work is motivated by the goalof testing whether strands used in DNA computations or as molecular bar-codesare structure free, where the strands are concatenations of short words. We alsopresent an algorithm for determining whether all words in S∗, for some finite set Sof equi-length words, are structure free.
Keywords: RNA secondary structure, testing DNA word sets
1. Introduction
In search-and-prune DNA computations, many long DNA strands arecreated from a small number of short strands, called words. For exam-ple, Braich et al. (Braich et al., 2001) use the words pairs
(TATTCTCACCCATAA, CTATTTATATCCACC), (ACACTATCAACATCA, ACACCTAACTAAACT),
(CCTTTACCTCAATAA, CTACCCTATTCTACT), (CTCCCAAATAACATT, ATCTTTAAATACCCC),
(AACTTCACCCCTATA, TCCATTTCTCCATAT), (TCATATCAACTCCAC, TTTCTTCCATCACAT)
∗ This material is based upon work supported by the U.S. National ScienceFoundation under Grant No. 0130108, by the National Sciences and the Engi-neering Research Council of Canada, and by the Defense Advanced ResearchProjects Agency (DARPA) and Air Force Research Laboratory, Air Force MaterialCommand, USAF, under agreement number F30602-01-2-0555.
† Contact author: [email protected]
c© 2003 Kluwer Academic Publishers. Printed in the Netherlands.
journal.tex; 26/05/2003; 11:50; p.1

2 Andronescu et al.
to construct 26 strands, namely those obtainable by taking one of thewords from each pair, and concatenating these in pair order. (Sincethere are two strands per pair and 6 pairs in total, the number ofstrands obtainable in this way is 26.)Word sets are carefully designed using computational or information-
theoretic methods, so that the resulting long strands behave well inthe computation. One property of well-designed words is that the longstrands formed from these words do not form secondary structures.For example, the design of Braich et al. uses a 3-letter alphabet andensures that no sequence of length 8 appears more than once in anystrand. The success of the design rests (in part) on the hypothesis thatwords satisfying these design criteria form strands with no secondarystructure. While this hypothesis seems plausible, to our knowledgethere is currently no rigorous argument that supports the hypothesis,either for the design of Braich et al. or for any other word designcurrently employed in DNA computing. For this reason, an efficientalgorithm that can test whether a given word set produces strandswith no secondary structure would be valuable. With this motivation,we consider the following problem.
Simple Structure Freeness Problem for Combinatorial Sets.Given a list of t pre-designed pairs of words {w(i), w̄(i), 1 ≤ i ≤ t}(here the bar notation is not intended to represent Watson-Crickcomplementation). All words have the same length l, and are stringsover {A,C,G, T} representing DNA strands with the start (left end)of the string corresponding to the 5’ end of the strand.Determine whether all of the 2t strands in the set S denoted bythe regular expression (w(1) + w̄(1))(w(2) + w̄(2)) . . . (w(t) + w̄(t))are structure free, that is, are predicted to have no secondary struc-ture, according to the standard, pseudoknot-free, thermodynamicmodels, at a given temperature.
Zuker and Steigler (Zuker and Steigler, 1981) developed an efficientalgorithm, based on dynamic programming, for determining the opti-mal (lowest energy) secondary structure of an RNA molecule. Theirmethod can also be applied to DNA molecules, given suitable ther-modynamic parameters (SantaLucia, 1998). The running time of theiralgorithm is O(n4) on a strand of length n, and an improved algorithm(Lyngso et al., 1999) runs in time O(n3). If the algorithm of Lyngso etal. were applied to each of the strands in S independently, the runningtime would be O(2tn3), where n = tl is the length of the strands in S.
journal.tex; 26/05/2003; 11:50; p.2

Testing sets concatenate without secondary structure 3
In Section 3 we describe an algorithm for the simple structure free-ness problem for combinatorial sets that runs in time O(n3). Thealgorithm is a simple generalization of the algorithms of Zuker andSteigler and Lyngso et al. The algorithm can easily be adapted to reportwhich strand in the combinatorial set has the minimum free energystructure, or a list of the strands with the low free energy structures ata given fixed temperature. We also present experimental results thatcompare the performance of our algorithm with an exhaustive searchapproach to the structure freeness problem for combinatorial sets, anddescribe cases where we have found structures in previously reportedword designs. Our algorithm easily generalizes to solve the followingproblem.
Structure Freeness Problem for Combinatorial Sets.Given sets of strands S1, S2, . . . , St, such that for all i, 1 ≤ i ≤ t, allstrands in Si have the same length li (where the li are not restrictedto be of the same length).Determine whether all strands in the set S = S1 × S2 × . . . × St
are structure free.
Our algorithm for this problem runs in time O(maxi|Si|2n3), wherehere n = l1+ l2+ . . .+ lt is the length of strands in S. The generalizedalgorithm can be used to verify structure freeness of strands in thecombinatorial sets of Faulhammer et al. (Faulhammer et al., 2000),and others.Our algorithm can also be used to verify that sets of molecular tags,
or bar-codes (Brenner et al., 1999) are structure free. In this design,tags are constructed from the set W of 8 4mer words
{TTAC, AATC, TACT, ATCA, ACAT, TCTA, CTTT, CAAA}.
This set of words, or tags, is constructed over a 3-letter alphabet (G’somitted), each word has exactly one “C”, and each word differs fromthe others in three of the four positions. This set S of tags contains allof the 88 strands in the set W 8. That is, S contains strands of the form5’-w-3’, where w ∈ W 8.This example motivates the second problem studied in this paper.
Suppose that in the future, Brenner et al. need more tags, and usestrands from the set S9 or S10. Even if all strands in the set S8 arestructure free, it might be possible that some strand in S9 or S10 hasstructure. Ideally, one would like to know that all words in S∗ arestructure free. Here, S∗ is the set of strands obtained by concatenatingzero or more copies of strands in S together. Note that S∗ contains an
journal.tex; 26/05/2003; 11:50; p.3

4 Andronescu et al.
infinite number of strands if S is not empty. The set S∗ is often calledthe Kleene closure of S. Thus, we define the following problem.
Structure Freeness Problem for Kleene Sets.Given a set S of words.Determine whether all words in S∗ are structure free.
In Section 4 we show that there is a constant m (depending on S)such that if some word in S∗ has structure, then some word in Sm
has structure. This reduces the structure freeness problem for Kleenesets to the structure freeness problem for combinatorial sets. However,our bound on m is exponential in the size of S and the length of thewords in S. Whether or not a better bound on m can be obtained isan interesting open question.Before getting to our algorithms in Sections 3 and 4, we provide
background on thermodynamic models for calculating the free energyof RNA and DNA secondary structures in Section 2 along with anoverview of the secondary structure prediction algorithm of Zuker andSteigler (Zuker and Steigler, 1981).
1.1. Related Work
Our algorithm for determining structure freeness of combinatorial setswas already developed by Cohen and Skiena (Cohen and Skiena, 2002),but applied to a different problem in that work. They were interestedin determining, among the RNA sequences coding for a given proteinP , which has the most stable secondary structure.Other important related work concerns algorithms for predicting the
secondary structure of an RNA or DNA strand. In contrast with theZuker-Steigler algorithm which is the basis for our work and returns thefree energy of the most stable structure accessible to a given strand, thepartition function approach of McCaskill (McCaskill, 1990) measuresthe propensity of a strand to fold in terms of the sum of the Gibb’sfactors (Z =
∑S exp(−∆GS/RT )), taken over all secondary structures
S. The quantity Z can be calculated in O(n3) time, using a dynamicprogramming algorithm. An extension of our approach that uses thepartition function of McCaskill would provide more insight on thepropensity of a strand to fold than does the “all or nothing” approxima-tion of our current algorithm. We believe that the techniques describedin our paper can be extended to the partition function approach, andview this as an important next step for this work.Other thermodynamic models for DNA and RNA structure folding,
that could be used as a basis for approaching the structure freeness
journal.tex; 26/05/2003; 11:50; p.4

Testing sets concatenate without secondary structure 5
problem for combinatorial sets, have been proposed by Hartemink etal. (Hartemink and Gifford, 1997) and by Rose et al. (Rose et al.,1999)). In addition, Rose et al. (Rose and Deaton, 2000) describe athermodynamic model (based on a statistical zipper model) for esti-mating folding propensity of a mixture of DNA strands. Our algorithmcan incorporate the model of Hartemink et al., but not that of Rose etal., which is based on statistical distributions of folding states.
2. Background and Notation
Consider an RNA molecule, or strand, to be a sequence of bases, whereeach base is either Adenine, Cytosine, Guanine, or Uracil. A single-stranded DNA molecule is similar, except that Thymine replaces Uracilas the fourth possible base. The sequence has two chemically distinctends, known as the 5′ and 3′ ends.Under the appropriate chemical conditions, an RNA or DNA strand
may fold upon itself by forming intramolecular bonds between pairsof its bases, as illustrated in Figure 1 below. In what follows, if s =s1s2 . . . sn is an RNA sequence and 1 ≤ i, j ≤ n, then i.j denotes thepairing of bases si with sj . A secondary structure of s is a set of basepairs such that each base is paired at most once. More precisely, forall i.j and i′.j′ in the set, i = i′ if and only if j = j′. A pseudoknot ina secondary structure is a pair of base pairs i.j, i′.j′ in the structurewith i < i′ < j < j′. Throughout this work, we restrict our attentionto secondary structures that have no pseudoknots.
Hairpin loop
Bulge
Multibranched loop
Stacked pairs
Internal loopExternal base
Figure 1. Secondary structure of an RNA strand of length 43. The thick black lineindicates the backbone and the thin lines indicate paired bases.
A pseudoknot free secondary structure naturally groups the bases ofan RNA strand into loops, as illustrated in Figure 1. For more details,see (Zuker et al., 1999). We classify such loops according to the number
journal.tex; 26/05/2003; 11:50; p.5

6 Andronescu et al.
of base pairs that they contain. A hairpin loop contains exactly one basepair. An internal loop contains exactly two base pairs. A bulge is aninternal loop with one base from each of its two base pairs adjacent.A stacked pair is a loop formed by two consecutive base pairs i.j and(i + 1).(j − 1). A multibranched loop, or multiloop, is a loop thatcontains more than two base pairs. An external base is a base notcontained in any loop. One base pair in any given loop is closest to theends of the RNA strand. This is known as the exterior or closing pair.All other pairs are interior. More precisely, the exterior pair is the onethat maximizes j − i over all pairs i.j in the loop.Speaking qualitatively, bases that are bonded tend to stabilize the
RNA molecule, whereas unpaired bases form destabilizing loops. Theso-called free energy of a secondary structure, measures (in kcal/mol)the stability of a secondary structure at fixed temperature - the lowerthe free energy, the more stable the structure. The free energy of afolded RNA or DNA strand can be estimated as the sum of the freeenergies of its component loops, if the secondary structure contains nopseudoknots. Through thermodynamic experiments, it has been possi-ble to estimate the free energy of common types of loops (SantaLucia,1998; Serra et al., 1995).
2.1. Standard Free Energy Model and Notation
Computational methods for predicting the secondary structure of anRNA or DNA molecule are based on models of the free energy of loops.The parameters of these models are driven in part by current under-standing of experimentally determined free energies, and in part bywhat can be incorporated into an efficient algorithm. The free energyof a loop depends on temperature; throughout we assume that thetemperature is fixed. We next summarize the notation used to refer tothe free energy of loops, along with some standard assumptions thatare incorporated into loop free energy models. We refer to a model thatsatisfies all of our assumptions as a standard free energy model.
− eSs(i, j). This function gives the free energy of a stacked pairthat consists of i.j and (i + 1).(j − 1), when i < j − 2. eSs(i, j)depends on the bases involved in the stack. We use the notatione-Stack(W,X, Y, Z), where W,X, Y, Z ∈ {A,C,G, T} in the caseof DNA, and W,X, Y, Z ∈ {A,C,G,U} in the case of RNA, todenote the free energy of a stacked pair in which base W is pairedwith base Z, X is paired with Y , and for some i and j withi < j − 2, the positions of W,X, Y , and Z in the strand arei, i+1, j− 1, and j respectively. Thus, for a strand s = s1s2 . . . sn,eSs(i, j) = e-Stack(si, si+1, sj−1, sj).
journal.tex; 26/05/2003; 11:50; p.6

Testing sets concatenate without secondary structure 7
− eHs(i, j). This function gives the free energy of a hairpin loopclosed by i.j. We assume that for all but a small number of cases,eHs(i, j) depends only on the length of the loop, si and sj , and onthe unpaired bases adjacent to si and sj on the loop. In particular,if j − i is greater than some constant,
eH(i, j) = eH-Stack(si, si+1, sj−1, sj) + eH-Length(j − i− 1),for some functions eH-Stack and eH-Length. Moreover, we assumethat the function eH-Length(m) is O(m).
− eLs(i, j, i′, j′). This function gives the free energy of an internalloop or bulge with exterior pair i.j and interior pair i′.j′. When i′ >i+1, we assume that eLs(i, j, i′, j′) is eL-Stack(si, si+1, sj−1, sj) +eL-Stack(sj′ , sj′+1, si′−1, si′) +eL-Length(i′ − i− 1, j − j′ − 1), forsome functions eL-Stack and eL-length with eL-Length(m,m′) =O(m+m′). When i′ = i+1, the expression of eLs(i, j, i′, j′) is thesame, except that si+1 and si′−1 are replaced by ε. Similarly, whenj′ = j − 1, sj′+1 and sj−1 are replaced by ε.
− eMs(i, j, i1, j1, . . . ik, jk). This function gives the free energy of amultibranched loop with interior pairs i1.j1, . . . ik.jk that is closedby i.j. This function is the least well understood at this time.For approximation, it is often assumed that the free energy ofa multibranched loop is given by an affine linear function of thenumber k + 1 of branches and the size of the loop (measured asthe number of unpaired bases):
eMs(i, j, i1, j1, . . . ik, jk) = a+ b(k + 1)+c((i1 − i− 1) + (j − jk − 1) +∑k−1
h=1(ih+1 − jh − 1))where a, b, and c are constants.
The free energy of a strand s with respect to a fixed sec-ondary structure F is the sum of the free energies of the loops of F .Sometimes when the strand s is fixed, it is convenient to refer simplyto the free energy of the structure F . In this paper, we define the freeenergy of a strand s to be the minimum free energy of the strand,with respect to all structures F (we note that this is a simplification;a better approach would be to define the free energy in terms of thesum of Gibb’s factors (McCaskill, 1990)). We say that a strand s isstructure free (at a given fixed temperature) if its free energy isgreater than or equal to 0. If the free energy of s is less than 0, wesay that the strand s has structure. We note that this definition ofstructure freeness is rather restrictive, but our algorithm can trivially
journal.tex; 26/05/2003; 11:50; p.7

8 Andronescu et al.
be modified to test whether any strand in a combinatorial set has freeenergy below any other threshold, too.
2.2. An Algorithm for Secondary Structure Prediction
Let Ws(j) be the minimum free energy taken over all structures of thestrand s1s2 . . . sj . Zuker and Steigler (Zuker and Steigler, 1981) de-scribed a dynamic programming algorithm for computing Ws(j); theiralgorithm can easily be extended to produce a structure whose freeenergy is Ws(n). The algorithm is based on the following recurrences:
Ws(j) =
{0, for j = 0,min
1≤i≤j(Vs(i, j) +Ws(i− 1)), for j > 0.
Here, Vs(i, j) is the free energy of the optimal structure for si . . . sj ,assuming i.j forms a base pair in that structure, also expressible as arecurrence:
Vs(i, j) =
+∞, for i ≥ j,min(eHs(i, j), eSs(i, j) + Vs(i+ 1, j − 1),
V BIs(i, j), V Ms(i, j)), for i < j.
In turn, V BIs(i, j) is the free energy of the optimal structure forsi . . . sj , assuming i.j closes a bulge or internal loop:
V BIs(i, j) =
+∞, for j − i ≤ 1,mini′.j′
i<i′<j′<j
(eLs(i, j, i′, j′) + Vs(i′, j′)), otherwise.
Using the approximation for the free energy of a multiloop of Section2.1, we can obtain a recurrence that calculates the loop cost as the sumof two subparts:
V Ms(i, j) = mini+1<h≤j−1
(WMs(i+ 1, h− 1) +WMs(h, j − 1) + a)
Here we introduce an additional functionWMs, whereWMs(i, j) givesthe free energy of an optimal structure for si . . . sj , assuming that si andsj are on a multibranched loop. Now WMs can be defined as follows:
WMs(i, j) =
c, for i = j,min(Vs(i, j) + b,mini<h≤j(WMs(i, h− 1)+
WMs(h, j))), for i < j.
In the equation above, the term Vs(i, j)+ b accounts for the case wheresi and sj forms a base pair and defines one of the k branches, whosefree energy is Vs(i, j). The other term in the min expression accounts
journal.tex; 26/05/2003; 11:50; p.8

Testing sets concatenate without secondary structure 9
for the case where si and sj are not paired with each other, so thefree energy is given by the minimum partition of the sequence into twocontiguous subsequences.The running time of the resulting algorithm is O(n4). It can be
improved to O(n3) using the method of Lyngso et al. (Lyngso et al.,1999).
3. Structure Freeness for Combinatorial Sets
In this section, we describe an algorithm for the simple structure free-ness problem for combinatorial sets. The algorithm can easily be gener-alized to the more general structure freeness problem. The input to ouralgorithm is a list of words w(1), w(1), w(2), w(2), . . . w(t), w(t), eachword has length l. Let
S = {z1z2 . . . zt|zk ∈ {w(k) or w(k)}}.Intuitively, the set S is the set of strands of length n = lt obtainedby following a path in the graph of Figure 2 (first suggested by Lipton(Lipton, 1995)) from the left end to the right end and concatenatingthe edge labels along the path.
Figure 2. “Diamond Graph”: paths in the graph correspond to strands of S.
While conceptually the algorithm is quite simple, the descriptionuses a lot of notation that blurs the intuition. In Section 3.1 we firstdescribe an algorithm for a very simple model of secondary structureformation, which captures the intuition. The details of our algorithmfor the standard free energy model are presented in Section 3.2.
3.1. Algorithm for the No Repeated k-Strings Model
A simple model of RNA folding is the no repeated k-strings model(simplified from the staggered zipper model). In this model, a sequencehas non-empty secondary structure if there is a string of length k thatrepeats without overlaps anywhere in the string. This captures thenotion that long runs of stacked pairs are a primary source of secondary
journal.tex; 26/05/2003; 11:50; p.9

10 Andronescu et al.
structure, and correspond to occurrences of a given substring and itsreverse complement in sequence S. Avoiding the reverse complementinstead of a repetition does not introduce any important algorithmiccomplexity.Cohen and Skiena (Cohen and Skiena, 2002) prove, under the no
repeated k-strings model, that it is NP-complete to verify that at leastone sequence is structure free, in a combinatorial set generated in a sim-ilar fashion to the diamond graph above. Here, we show that verifyingall sequences are structure free can be done in polynomial time.We can verify that all 2t sequences of length n = lt contain no
repeated k-string in O(kn2) time. For each of the n starting positionsin the sequence, there are at most two such positions in the “diamondgraph” which generate the sequence starting from this position (eitherw(i) or w̄(i)). Given any two word/position pairs, we now walk forwarda total of k characters, comparing the equality of the symbol on eachpath at every character. At most two such paths are active in eachwalk, because the paths meet at in-degree-2 vertices of the diamondgraph. The existence of a length-k shared walk represents a repeatedk-string in at least one sequence, and terminates the algorithm.We next enhance this basic approach for the more general class of
secondary structures of the standard model.
3.2. Algorithm for the Free Energy Model
Let W ′S(n) be the minimum of Ws(n) (as defined in Section 2.2), taken
over all s ∈ S. In this section we present an algorithm to computeW ′
S(n). The algorithm runs in time O(n4). Using techniques of Lyngsoet al. (Lyngso et al., 1999) the running time can be improved to O(n3).In describing our algorithm, we follow the approach of Section 2.2. LetSj be the set of prefixes of length j of strands in S and letW ′
S(j) be theminimum of Ws(j) taken over all s ∈ Sj . That is, W ′
S(j) is the optimalenergy of the strand in Sj , with the lowest energy secondary structure.Formally,
W ′S(j) =
{0, for j = 0,mins∈Sj
Ws(j), for 0 < j ≤ n.
Roughly speaking, we would like to be able to express W ′S(j) in terms
ofW ′S(j−1). It turns out to be more convenient to introduce some new
functions to work with in our recurrences. We use word#(j) to refer tothe index of the word to which base j of a string in S belongs. Thus,word#(j) = �j/l�, where l is the word length.Let S(T, j) be the subset of S in which the word containing the
jth base is w(word#(j)). Intuitively, S(T, j) corresponds to the set of
journal.tex; 26/05/2003; 11:50; p.10

Testing sets concatenate without secondary structure 11
paths that go through the top of the diamond containing the jthbase in Figure 2. Then,
S(T, j) ={z1z2 . . . zt|zk ∈ {w(k), w(k)} for k = word#(j)and zword#(j) = w(word#(j))}.
Similarly, let S(B, j) be the subset of S in which the word containingthe jth base is w(word#(j)). Intuitively, S(B, j) corresponds to theset of paths that go through the bottom of the diamond containingthe jth base in Figure 2. Formally,
S(B, j) ={z1z2 . . . zt|zk ∈ {w(k), w(k)} for k = word#(j)and zword#(j) = w(word#(j))}.
LetW ′S(bj , j) be the minimum ofWs(j) for all s ∈ S(bj , j). Formally:
W ′S(bj , j) = min
s∈S(bj ,j)Ws(j), for bj ∈ {T,B}.
Also, W ′S(j) = min
bj∈{T,B}W ′
S(bj , j). So if we can calculate the W ′S(bj , j),
we are done. It turns out to be easier to obtain a recurrence forW ′S(bj , j)
than for W ′S(j).
Similarly, we use generalizations of the functions Vs, V BIs, V Ms
and WMs in our recurrences. For example, we use V ′S(bi, bj , i, j) to
denote mins∈S(bi,bj ,i,j) Vs(i, j), where S(T, T, i, j) is the subset of S cor-responding to the set of paths that go through the top of the diamondcontaining the ith base and the diamond containing the jth base. Thatis, S(T, T, i, j) =
{z1z2 . . . zt|zk ∈ {w(k), w(k)} for k = word#(i), k = word#(j),zword#(i) = w(word#(i)), and zword#(j) = w(word#(j))}.
S(bi, bj , i, j) is defined similarly for other values of bi and bj .Also,
V BI ′S(bi, bj , i, j) = mins∈S(bi,bj ,i,j) V BIs(i, j),V M ′
S(bi, bj , i, j) = mins∈S(bi,bj ,i,j) V Ms(i, j), andWM ′
S(bi, bj , i, j) = mins∈S(bi,bj ,i,j) WMs(i, j).
We now describe a recurrence for W ′S(bj , j), bj ∈ {T,B}. The other
needed recurrences are in the appendix to the paper.
W ′S(bj , j) =
0, for j = 0,
min(bi,bi−1)∈X(i,j),
1≤i≤j
(V ′S(bi, bj , i, j) +W ′
S(bi−1, i− 1)), for j > 0,
where X(i, j) is given in Table I.
journal.tex; 26/05/2003; 11:50; p.11

12 Andronescu et al.
Table I. X(i, j). In the entries of the right handcolumn, each ∗ may be replaced by a T or B; thus{(bj , ∗)} denotes the set {(bj , B), (bj , T )}.
word#(i) word#(i) X(i, j)
= =
word#(i − 1) word#(j)
yes yes {(bj , bj)}yes no {(T, T ), (B, B)}no yes {(bj , ∗)}no no {(∗, ∗)}
3.3. Experimental Results
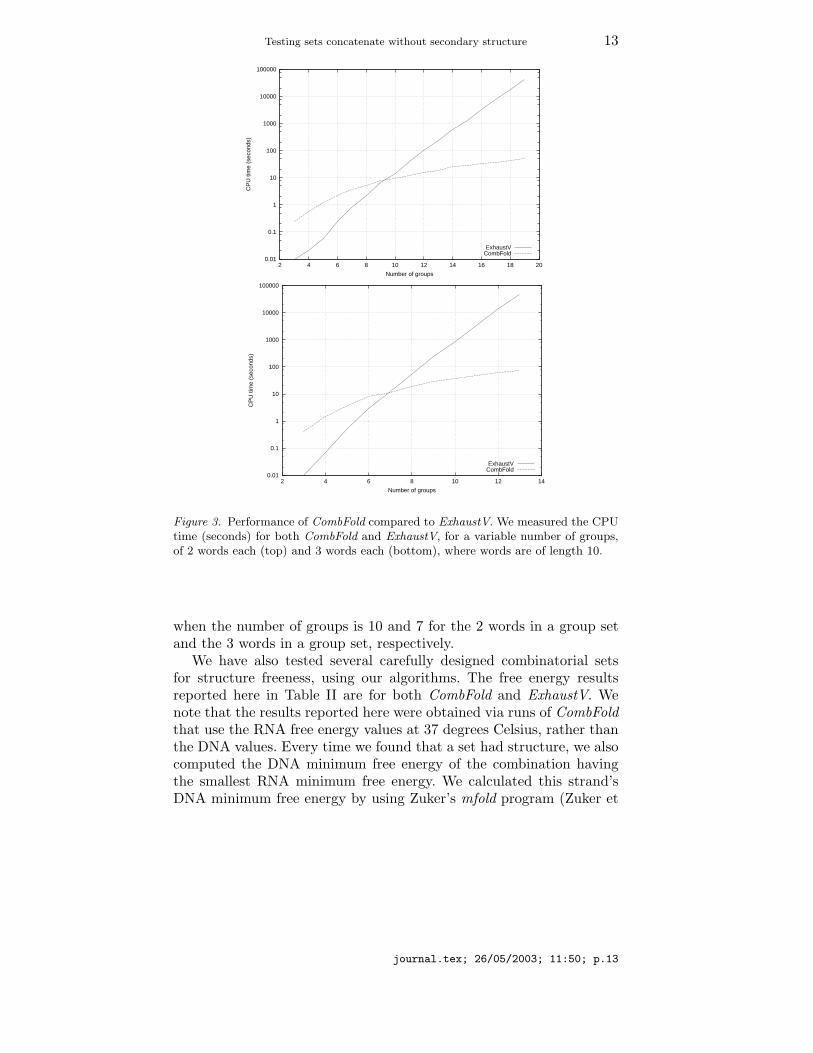
We have implemented our algorithm for the structure freeness problemon combinatorial sets (Andronescu et al., 2003), which we refer to asCombFold in what follows1. Our implementation uses Turner’s freeenergy parameters (Serra et al., 1995), as incorporated in the mfoldalgorithm (Zuker et al., 1999). There are some small differences inthe calculated energy values, compared with Zuker’s mfold or Viennapackage’s RNAfold (Hofacker et al., 1994). Also, CombFold does notreturn suboptimal energies, as mfold does.We compared the running time performance of CombFold with that
of a simple (exponential time) exhaustive search algorithm, which wecall ExhaustV. The ExhaustV algorithm uses the Vienna package’slibrary to fold each of the possible combinations of words, computesthe energy for each, and then returns the combination that folds intothe smallest energy. Figure 3 shows the running time of CombFoldcompared to ExhaustV, plotted on a semi-logarithmic scale. Our re-sults were obtained on a dual Pentium III 2GHz processor machine,with 4GB of RAM. We measured the CPU time in seconds (y axis)when running CombFold and ExhaustV, for different number of sets (xaxis) of randomly generated strands, i.e different values of t as in thestatement of the structure freeness problem in Section 1. For the leftgraph, we used 2 words of length 10 in each set and for the right graphwe used 3 words of length 10 in each set. The graphs indicate that therunning time of ExhaustV grows exponentially, while that of CombFoldgrows polynomially. At this version of CombFold, ExhaustV is faster fora small number of groups. However, it is outperformed by CombFold
1 CombFold is accessible on line at http://www.rnasoft.ca
journal.tex; 26/05/2003; 11:50; p.12
Testing sets concatenate without secondary structure 13
0.01
0.1
1
10
100
1000
10000
100000
2 4 6 8 10 12 14 16 18 20
CP
U ti
me
(sec
onds
)
Number of groups
ExhaustVCombFold
0.01
0.1
1
10
100
1000
10000
100000
2 4 6 8 10 12 14
CP
U ti
me
(sec
onds
)
Number of groups
ExhaustVCombFold
Figure 3. Performance of CombFold compared to ExhaustV. We measured the CPUtime (seconds) for both CombFold and ExhaustV, for a variable number of groups,of 2 words each (top) and 3 words each (bottom), where words are of length 10.
when the number of groups is 10 and 7 for the 2 words in a group setand the 3 words in a group set, respectively.We have also tested several carefully designed combinatorial sets
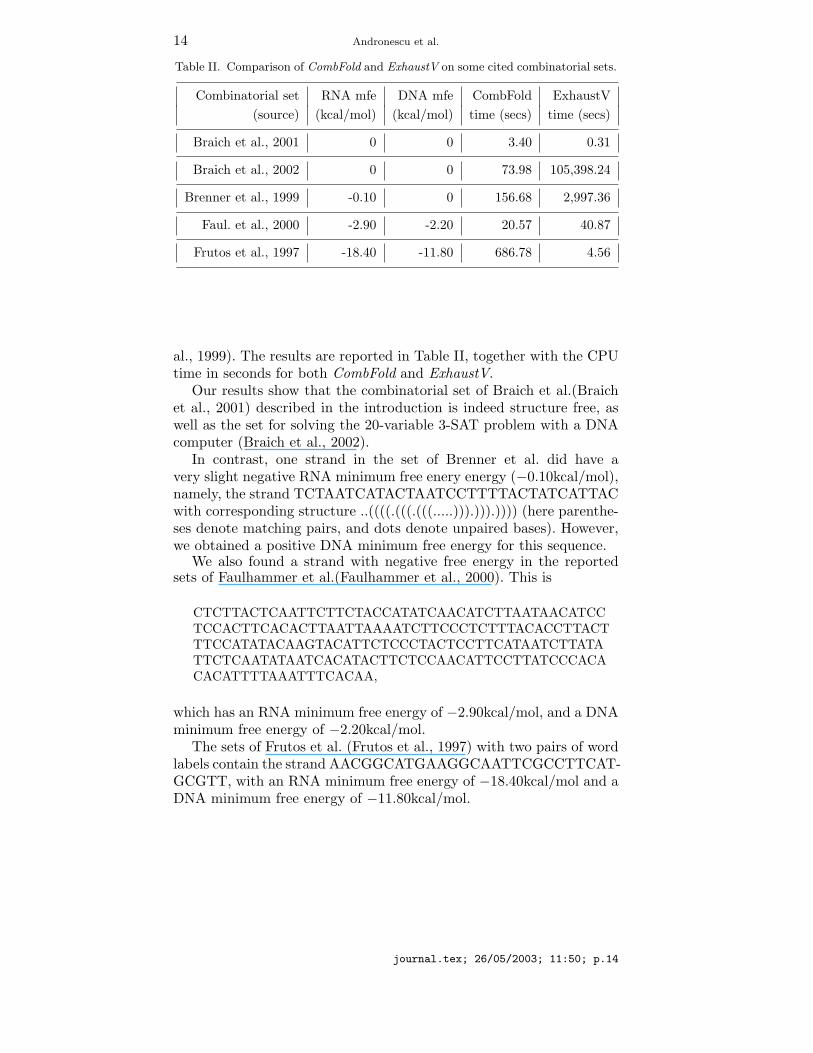
for structure freeness, using our algorithms. The free energy resultsreported here in Table II are for both CombFold and ExhaustV. Wenote that the results reported here were obtained via runs of CombFoldthat use the RNA free energy values at 37 degrees Celsius, rather thanthe DNA values. Every time we found that a set had structure, we alsocomputed the DNA minimum free energy of the combination havingthe smallest RNA minimum free energy. We calculated this strand’sDNA minimum free energy by using Zuker’s mfold program (Zuker et
journal.tex; 26/05/2003; 11:50; p.13
14 Andronescu et al.
Table II. Comparison of CombFold and ExhaustV on some cited combinatorial sets.
Combinatorial set RNA mfe DNA mfe CombFold ExhaustV(source) (kcal/mol) (kcal/mol) time (secs) time (secs)
Braich et al., 2001 0 0 3.40 0.31
Braich et al., 2002 0 0 73.98 105,398.24
Brenner et al., 1999 -0.10 0 156.68 2,997.36
Faul. et al., 2000 -2.90 -2.20 20.57 40.87
Frutos et al., 1997 -18.40 -11.80 686.78 4.56
al., 1999). The results are reported in Table II, together with the CPUtime in seconds for both CombFold and ExhaustV.Our results show that the combinatorial set of Braich et al.(Braich
et al., 2001) described in the introduction is indeed structure free, aswell as the set for solving the 20-variable 3-SAT problem with a DNAcomputer (Braich et al., 2002).In contrast, one strand in the set of Brenner et al. did have a
very slight negative RNA minimum free enery energy (−0.10kcal/mol),namely, the strand TCTAATCATACTAATCCTTTTACTATCATTACwith corresponding structure ..((((.(((.(((.....))).))).)))) (here parenthe-ses denote matching pairs, and dots denote unpaired bases). However,we obtained a positive DNA minimum free energy for this sequence.We also found a strand with negative free energy in the reported
sets of Faulhammer et al.(Faulhammer et al., 2000). This is
CTCTTACTCAATTCTTCTACCATATCAACATCTTAATAACATCCTCCACTTCACACTTAATTAAAATCTTCCCTCTTTACACCTTACTTTCCATATACAAGTACATTCTCCCTACTCCTTCATAATCTTATATTCTCAATATAATCACATACTTCTCCAACATTCCTTATCCCACACACATTTTAAATTTCACAA,
which has an RNA minimum free energy of −2.90kcal/mol, and a DNAminimum free energy of −2.20kcal/mol.The sets of Frutos et al. (Frutos et al., 1997) with two pairs of word
labels contain the strand AACGGCATGAAGGCAATTCGCCTTCAT-GCGTT, with an RNA minimum free energy of −18.40kcal/mol and aDNA minimum free energy of −11.80kcal/mol.
journal.tex; 26/05/2003; 11:50; p.14

Testing sets concatenate without secondary structure 15
4. Structure Freeness for Kleene Sets
In this section we provide an algorithm for the structure freeness prob-lem for Kleene sets. We show that there is a constant m, which de-pends on S, such that if there is a word with structure in S∗, thenthere is a word with structure in Sm. The constant m is bounded byexp(O((|S|l)3)), where l is the number of words in S. With the constantm in hand, the structure freeness problem for Kleene sets is solvableusing the method of Section 3. We prove the existence of m as follows.
1. There is a strand with structure in S∗ if and only if there is a strandin S∗ with a so-called energy bounded structure.We define what is an energy bounded structure in detail in Section4.1, but the intuition is as follows. If there is a strand with structurein S∗ that has a very long hairpin loop, we can remove whole wordsfrom the unpaired bases of the loop to obtain a shorter strand inS∗ which also has structure, and the energy of the shorter hairpinis bounded. Other parts of the structure that contribute a highpositive free energy to the structure can similarly be replaced by ahairpin with bounded energy.
2. The set of strands of S∗ that have energy bounded structures iscontext free.
Moreover, the size of the context free grammar that generates thisset is O((|S|l)3) where l is the maximum length of the words in S.We note that for a given strand with an energy bounded structure,the optimal structure for this strand may not be an energy boundedstructure.
From this, in Theorem 4.1 we apply standard results on context freelanguages to show that if there is a strand in S∗ that has structure,then there is such a strand of length exp(O((|S|l)3)).
4.1. Energy Bounded Structures
A secondary structure F is E-energy bounded for a strand s andE > 0 if, with respect to s, no substructure of F has negative energy,and no substructure of F is expensive, i.e. has energy above somethreshold E. Here, a substructure of F is any proper subset of F thatcontains all base pairs i′.j′ of F with i ≤ i′ ≤ j′ ≤ j, for some i.j in F ,and contains no other base pairs. We call this the substructure of F thatis bounded by i.j. We say that a strand s has E-energy boundedstructure if the free energy of some E-energy bounded structure isless than 0.
journal.tex; 26/05/2003; 11:50; p.15

16 Andronescu et al.
LEMMA 4.1. Let S be a set of strands. There exists a constant Ethat grows at most linearly in the length l of words in S such that thefollowing is true: There is a strand with structure in S∗ if and only ifthere is a strand with E-energy bounded structure in S∗.
Proof One direction of the proof is easy: regardless of the value ofE, if there is a strand with E-energy bounded structure in S∗, thentrivially this strand is a strand with structure in S∗. The proof ofthe other direction is based on the fact that for suitable E, expensivesubstructures can be “cut” out of a structure, and replaced by E-energybounded structures.In what follows, we assume without loss of generality that there are
complementary bases in the words of S. Otherwise, all words in S∗ aretrivially structure free, and we are done. Also, we assume that l ≥ 4. Ifl < 4, we can let S′ = S4, and work with S′ instead of S. Clearly, (S′)∗is structure free if and only if S∗ is.Let E be the maximum free energy of a hairpin loop (of finite cost)
that could be formed by concatenating any three words of S and form-ing a base pair between any two complementary bases in these words.(Such a hairpin must be possible, since it is possible to construct ahairpin of finite cost when there are four unpaired bases in the loop,and from the assumptions of the previous paragraph, the unpairedbases include at least four bases from the middle word of the threewords.) Let s be a strand with structure in S∗. Let F be a structurefor which the free energy of s with respect to F is less than 0. Supposethat F is not E-energy bounded.We first handle expensive substructures of F . We say that an expen-
sive substructure is maximal if it is not properly contained in any otherexpensive substructure of F . Intuitively, for each maximal substructureof F , we “cut out” most or all of this substructure, and replace by ahairpin whose energy is bounded by E. This serves to reduce the totalenergy of F , while removing the offending expensive substructures.More precisely, for each expensive maximal substructure, if the struc-
ture is bounded by i.j, we perform a Cut(s, F ) operation at i.j:
− Let i′ ≥ i be the end of the word containing i, that is, i′ = �i/l�l,where l is the length of the words in S. Let j′ ≤ j be the start ofthe word containing j, that is, j′ = �j/l�(l − 1) + 1. Replace thebases in the range [i′ + 1, j′ − 1] from s by an arbitrary word wof S; thus if s = s1s2 . . . sk before the replacement, then after thereplacement s = s1s2 . . . si′wsj′ . . . sk.
Now, update the base pairs in the secondary structure F accord-ingly. First, for all base pairs a.b with a > i and b < j, removea.b from F . Then, replace each remaining base pair a.b in F with
journal.tex; 26/05/2003; 11:50; p.16

Testing sets concatenate without secondary structure 17
a ≤ i and b ≥ j by the pair a.b′, where b′ = b − (j′ − i′ − 1) + l.Finally, replace each base pair a.b in F with a > j and b > j bya′.b′, where a′ = a− (j′ − i′ − 1) + l and b′ = (b− j′ − i′ − 1) + l).
It remains to handle the situation where some substructure of F hasnegative energy. This is easy: simply replace F by any of the smallestsubstructure F ′ of F which has negative energy. By construction, F ′cannot have a substructure with negative energy.
4.2. The Language of Strands with Energy BoundedStructures is Context Free
In this section, we show that there is a context free grammar thatgenerates the language of strands with energy bounded structures.
LEMMA 4.2. The language of strands with E-energy bounded struc-tures, when the energy is measured according to the standard energyassumptions, is context free. There is a context free grammar that gen-erates this language which has a number of nonterminals that is linearin E, in which each production has bounded length (independent of E).
Proof We describe a context free grammar for the language ofstrands with energy bounded structures. The terminals of the grammarare {A,C,G, T}, and the nonterminals generate structures with fixedenergy values. For simplicity, we assume that base pairs can form onlybetween a base and its Watson-Crick complement, but the grammarcan easily be extended to handle non-Watson-Crick pairs.First, we consider a simple case, namely how to generate energy
bounded hairpins. Specifically, we develop a context free grammar thatgenerates the set of all possible strings that can form a hairpin loop(with no dangling ends) that is closed by the base pair (t, t̄) and hasenergy e. Call this set H(t, e). We will use the nonterminal Ht,e togenerate the set H(t, e). Since H(t, e) is finite, we could simply haveone production from Ht,e that directly generates each string in H(t, e).However, according to the standard energy model, other than a few spe-cial cases (such as tri-loops or tetra-loops), the free energy of a hairpinformed by pairing the outside bases in the string tau1u2 . . . ujbt̄ (wheret, a, b and ui, 1 ≤ i ≤ j are in {A,C,G, T}) can be expressed as thesum of two terms, namely a stacking energy term, eH-Stack(t, a, b, t̄),and a length term eH-Length(j + 2), where j + 2 is the number ofunpaired bases in the hairpin. By taking advantage of this, fewer rulesare needed. Specifically, from Ht,e, we add rules of the form:
Ht,e → taUjbt̄
journal.tex; 26/05/2003; 11:50; p.17

18 Andronescu et al.
for each a, b ∈ {A,C,G, T} for which there exists a j for which e −eH-Stack(t, a, b, t̄) = eH-Length(j + 2). Here, Uj is a nonterminal thatgenerates all sequences of j bases: for each t ∈ {A,C,G, T} and 1 <i ≤ j, we add the rules
Ui → tUi−1 and U1 → t.
The total number of rules generated from Ht,e is a constant indepen-dent of e. In addition, the number of nonterminals Ui is approximatelyeH-Length−1(e). Fewer nonterminals could be used by using rules ofthe form Ui → Ui/2Ui/2 for i even and similar rules for i odd, but thiswould not ultimately lead us to a better bound for m.Generalizing from the case of hairpins, we need other new non-
terminals in our grammar that generate different types of structures.Mirroring the notation used in the recurrence relations of Section 2.2,we use the following nonterminals:
− We: generates the set of strings that can form anE-energy-boundedstructure with energy e.
− Vt,e: generates the set of strings that can form an E-energy boundedstructure that is closed by the base pair (t, t̄) and has energy e.
− It,e: generates the set of strings that can form an E-energy-boundedstructure with energy e, the outermost loop of which is an interiorloop or stacked pair that is closed by the base pair (t, t̄).
− Mt,e: generates the set of strings that can form an E-energy boundedstructure with energy e, the outermost loop of which is a multi-loopthat is closed by the base pair (t, t̄).
We first consider productions from these nonterminals for the casewhen e ≥ 0. The productions from We are as follows:
We → Wet|We′Vt,e−e′
for all t ∈ {A,C,G, T}, and all e′ ≥ 0 for which also e − e′ ≥ 0. Here,we need e′ ≥ 0 and e− e′ ≥ 0 because by definition of energy boundedstructures, it must be possible for strings generated from We to foldinto a structure (of energy e) which has no substructures of negativeenergy. For the case e = 0 we also need W0 → t, so that from W0, anystrand can be generated.The productions from Vt,e are as follows:
Vt,e → Ht,e|It,e|Mt,e
journal.tex; 26/05/2003; 11:50; p.18

Testing sets concatenate without secondary structure 19
The productions from It,e are of two types:
It,e → tVt′,e′ t̄
for all t, where here, e′ = e− e-Stack(t, t′, t̄′, t̄), andIt,e → taUj−2a
′Vt′,e′b′Uj′−2bt̄
for all t ∈ {A,C,G, T}, all a, b ∈ {A,C,G, T, ε} where ε denotes theempty string, and
e′ = e−eL-Stack(t, a, b, t̄)−eL-Stack(t̄′, b′, a′, t′)−eL-Length(j, j′) ≥ 0.Finally, the productions from Mt,e are as follows, where recall that
according to the standard model, the free energy of a multi-loop isa plus b times the number of branches plus c times the number ofunpaired bases:
Mt,e = tWMe′WMe′′ t̄
for all t and pairs e′, e′′ for which e′ + e′′ = e − a and e′, e′′ ≥ 0. Here,WMe is an additional nonterminal, with productions as follows:
WMe → Vt,e−b | tWMe−c
for all t ∈ {A,C,G, T}. To generate all strands with E-energy boundedstructures, we need nonterminals of the above types for values of e inthe range [0, E]. The free energy values given by the functions e-Stack,eH-Stack, e-Length, eL-Stack, eH-Length, and eL-Length and the threeconstants a, b, and c for multiloops are typically decimal numbers ofthe form ±d.d′, where d, d′ are decimal digits. So the total number ofnonterminals needed for values of e in the range [0, E] is bounded by10 times E, and therefore is linear in E.In addition to all of these nonterminals and rules, we need a few
additional starting nonterminals that generate strands with structure.We introduce nonterminals Be and rules
Be → W0tVt′,e′ t̄W0
for all e < 0 and t, t′ ∈ {A,C,G, T} for which e′ = e−e-Stack(t, t′, t̄′t̄) ≥0. Finally we introduce one start (or beginning) nonterminal B, with
B → Be
for each nonterminal Be already introduced.There is a maximum of nine symbols on the right hand side of any
rule, and so all of the productions above have bounded length.
journal.tex; 26/05/2003; 11:50; p.19

20 Andronescu et al.
LEMMA 4.3. Let S be a set of strands, each of length l. Let L be thelanguage of strands in S∗ with E-energy bounded structures. Then Lis context free, and there is a context free grammar that generates L,for which the number of nonterminals is O(E(l|S|)2) and each rule hasbounded length (independent of E, l, and S).
Proof A classical result of automata theory is that the intersec-tion of a context free language and a regular language is context free(Hopcroft and Ullman, 1979)[Theorem 6.5]. Let LE be the languageof strands with E-energy bounded structures, which is context free byLemma 4.2. The language L is the intersection of S∗, a regular language,with LE , and therefore is context free.We now show how to obtain a context free grammar G for L − {ε}
from a context free grammar GE for LE and a nondeterministic finitestate automaton (nfa) for S∗. We assume that the only final state ofthis nfa is also the unique start state, of the nfa, and refer to this stateas q0. Also, we can assume that the size (number of states) of the nfais bounded by |S|l.It is also convenient to assume that GE is a grammar in Chomsky
Normal Form (CNF), that is, each production is of the formX → Y Z orX → t, where Y, Z are nonterminals and t is a terminal. The grammarof Lemma 4.2 is not in CNF, but because all of its productions havebounded length (independent of E), it can be converted to CNF whilekeeping the size (number of nonterminals) of the grammar linear in E(Hopcroft and Ullman, 1979).For each nonterminal X of GE , we add nonterminals of the form
X(q, q′) to G for every pair of states q, q′ of the nfa. The startingnonterminal of G is B(q0, q0), where B is the start nonterminal of GE .Therefore, the total number of nonterminals is O(|E|(l|S|)2).The rules of the grammar are constructed so that if X generates
the set of strands L(X) in GE , then X(q, q′) generates the subset ofstrands of L(X) that cause the nfa for S∗ to reach q′ from q. For eachproduction of the form X → Y Z of GE , the grammar G contains theproductions:
X(q, q′)→ Y (q, q′′)Z(q′′, q′)
for all q, q′ and q′′ in the nfa for S∗. Also, for each rule of the formX → t in GE , the rule X(q, q′) → t is added to G for each pair (q, q′)with a transition from q to q′ on symbol t in the nfa.It can be shown by induction that each nonterminal X(q, q′) gen-
erates exactly the strings that can be generated by X and in additiontake the nfa for S∗ from state q to state q′.The main theorem of this section follows from the previous lemmas,
together with classical results from automata theory.
journal.tex; 26/05/2003; 11:50; p.20

Testing sets concatenate without secondary structure 21
THEOREM 4.1. There is a constant m, which depends on S, suchthat if there is a word with structure in S∗, then there is a word withstructure in Sm. The constant m is bounded by exp(O(l3|S|2)), wherel is the length of words in S.
Proof This result follows directly from the pumping lemma forcontext free grammars, which states that if the language generatedby a context free language is nonempty, then there is a string in thelanguage whose length is bounded by an exponential in the number ofnonterminals of a grammar for the language that is in Chomsky NormalForm (Hopcroft and Ullman, 1979).By Lemma 4.1, if there is a word with structure in S∗, then there
is a word with E-energy bounded structure in S∗, where E grows atmost linearly with l. By Lemma 4.3, there is a context free grammarin Chomsky Normal Form that generates the language of strands withE-energy bounded structure in S∗, and the number of nonterminals ofthis grammar is O(E(l|S|)2) = O(l3|S|2), since E = O(l).Therefore, we can choose m which is exp(O(l3|S|2)), for which the
theorem is true.
5. Conclusions and Future Work
We present an efficient algorithm for determining structure freeness ofcombinatorial sets. The algorithm should prove useful in verifying thequality of word sets designed for DNA and RNA computations.An important direction for future work is to use partition functions
to cover the range of alternative structures. In addition, the abilityto test the fidelity of a library in the presence of hybridization part-ners would be valuable. Another direction for future work is to studywhether our algorithm can be generalized to determine structure free-ness of more general sets of strands, such as those used in Adleman’sHamiltonian graph experiment (Adleman, 1994). We would also like tosee if the method could be used to prove that certain word design prin-ciples guarantee structure freeness. For example, is every strand over3-letter alphabet {A, T,C}, in which a “C” appears in every windowof length 4, structure free? Answering this would require more generaltechniques than those reported here.
Acknowledgement.We thank the anonymous reviewers for their veryvaluable suggestions on a draft of this paper.
journal.tex; 26/05/2003; 11:50; p.21

22 Andronescu et al.
References
Adleman, L. M. Molecular computation of solutions to combinatorial problems.Science, Vol 266(11):1021–1024, 1994.
Andronescu, M., Aguirre-Hernandez, R., Condon, A., and Hoos, H. H. RNAsoft: asuite of RNA secondary structure prediction and design software tools. NucleicAcids Research, 31:13, to appear 2003.
Braich, R. S., Johnson, C., Rothemund, P. W. K., Hwang, D., Chelyapov, N., andAdleman, L. M. Solution of a satisfiability problem on a gel-based DNA com-puter. In Proceedings of the 6th International Conference on DNA Computation,Springer-Verlag LNCS, 2054:27–41, 2000.
Braich, R. S., Chelyapov, N., Johnson, C., Rothemund, P.W.K., and Adleman, L. M.Solution of a 20-variable 3-SAT Problem on a DNA computer. Science, 296:499–502, 2002.
Brenner, S., Williams, R. S., Vermaas, E. H., Storck, T., Moon, K., McCollum,C., Mao, J-I., Luo, S., Kirchner, J. J., Eletr S., DuBridge, R. B., Burcham, T.,and Albrecht. G. In vitro cloning of complex mixtures of DNA on microbeads:Physical separation of deferentially expressed cDNAs. Proc. Natl. Acad. Sci.USA, 97(4):1665–1670, February 15, 2000.
Cohen, B. and Skiena, S. Designing RNA Sequences: Natural and Artificial Selection.In Proc. 6th Int. Conf. Computational Molecular Biology (RECOMB), 109–116,2002.
Faulhammer, D., Cukras, A. R., Lipton, R. J., and Landweber, L. F. Molecularcomputation: RNA solutions to chess problems. In Proc. Natl. Acad. Sci. USA,97:1385–1389, 2000.
Frutos, A. G., Liu, Q., Thiel, A. J., Sanner, A. M. W., Condon, A. E., Smith, L. M.,and Corn, R. M. Demonstration of a Word Design Strategy for DNA Computingon Surfaces. Nucleic Acids Research, 25:4748-4757, 1997.
Hartemink, A. J. and Gifford, D. K. Thermodynamic simulation of deoxyoligonu-cleotide hybridization. Prel. Proc. 3rd DIMACS Workshop on DNA BasedComputers, 15–25, June 23-27, 1997.
Hofacker, I. L., Fontana, W., Stadler, P. F., Bonhoeffer, L. S., Tacker, M., andSchuster, P. Fast Folding and Comparison of RNA Secondary Structures.Monatsh.Chem., 125:167–188, 1994.
Hopcroft, J. E. and Ullman, J. D. Introduction to Automata Theory, Languages,and Computation. Addison-Wesley, 1979.
Lipton, R. DNA solution of hard computational problems. Science 268:542–545,1995.
Lyngso, R. B., Zuker, M., and Pedersen, C. N. S. Internal Loops in RNA SecondaryStructure Prediction. In Proc. Third International Conference in ComputationalMolecular Biology, 260–267, April 1999.
McCaskill, J. S. The equilibrium partition function and base pair bindingprobabilities for RNA secondary structure. Biopolymers, 29:1105–1119, 1990.
Rose, J. A., Deaton, R., Franceschetti, D. R. Garzon, M., and Stevens, Jr., S. E.A statistical mechanical treatment of error in the annealing biostep of DNAcomputation. Special program in DNA and Molecular Computing, Genetic andEvolutionary Computation Conference (GECCO-99), Morgan Kaufmann, July13-17, 1999.
Rose, J. A. and Deaton, R. J. The fidelity of Annealing-ligation: a theoreticalanalysis. In Proc. 6th Intl. Workshop on DNA-Based Computers, Springer-VerlagLNCS, 2054:231–246, June, 2000.
journal.tex; 26/05/2003; 11:50; p.22

Testing sets concatenate without secondary structure 23
SantaLucia, Jr., J. A Unified View of Polymer, Dumbell, and Oligonucleotide DNANearest-Neighbor Thermodynamics. Proc. Natl. Acad. Sci. USA, 95:1460–1465,1998.
Serra, M. J., Turner, D. H., and Freier, S. M, Predicting thermodynamic propertiesof RNA. Meth. Enzymol., 259:243–261, 1995.
Zuker, M. and Steigler, P. Optimal Computer Folding of Large RNA Sequences usingThermodynamics and Auxiliary Information. Nucleic Acids Research, 9:133–148,1981.
Zuker, M., Mathews, D. H. and Turner, D. H. Algorithms and Thermodynamics forRNA Secondary Structure Prediction: A Practical Guide. In RNA Biochemistryand Biotechnology, J. Barciszewski and B.F.C. Clark, eds., NATO ASI Series,Kluwer Academic Publishers, Dordrecht, NL, 11–43, 1999.
Appendix
The remaining recurrences for our algorithm of Section 3 are presentedhere.
A1. V ′S and V BI ′S
We next give recurrences for V ′S(bi, bj , i, j) and V BI ′S(bi, bj , i, j). When
i = j, V ′S(bi, bj , i, j) =∞. When i < j, V ′
S(bi, bj , i, j) =
min
eH ′S(bi, bj , i, j),
eS′S(bi, bj , i, j)+
min(bi+1,bj−1)∈X(i,i+1,j,j−1)
V ′S(bi+1, bj−1, i+ 1, j − 1),
V BI ′S(bi, bj , i, j),V M ′
S(bi, bj , i, j).
V BI ′S(bi, bj , i, j) =
+∞, for j < i+ 4,
min(bi′ ,bj′ )∈X(i,i′,j′,j)
i<i′<j′<j
(eL(bi, bj , bi′ , bj′ , i, j, i′, j′)
+ V ′S(bi′ , bj′ , i
′, j′)), otherwise,
where X(i, i′, j′, j) is given in Table III and X(i, i+1, j, j− 1) can alsobe deduced from this table.
journal.tex; 26/05/2003; 11:50; p.23

24 Andronescu et al.
Table III. Table for X(i, i′, j′j). In the entries of the righthand column, each ∗ may be replaced by a T or B.
word#(i) word#(i′) word#(j′) X(i, j)
= = =
word#(i′) word#(j′) word#(j)
yes yes yes {(bi, bj)}yes yes no {(bi, bi)}yes no yes {(bi, bj)}yes no no {(bi, ∗)}no yes yes {(bj , bj)}no yes no {(T, T ), (B, B)}no no yes {(∗, bj)}no no no {(∗.∗)}
A2. V M ′S
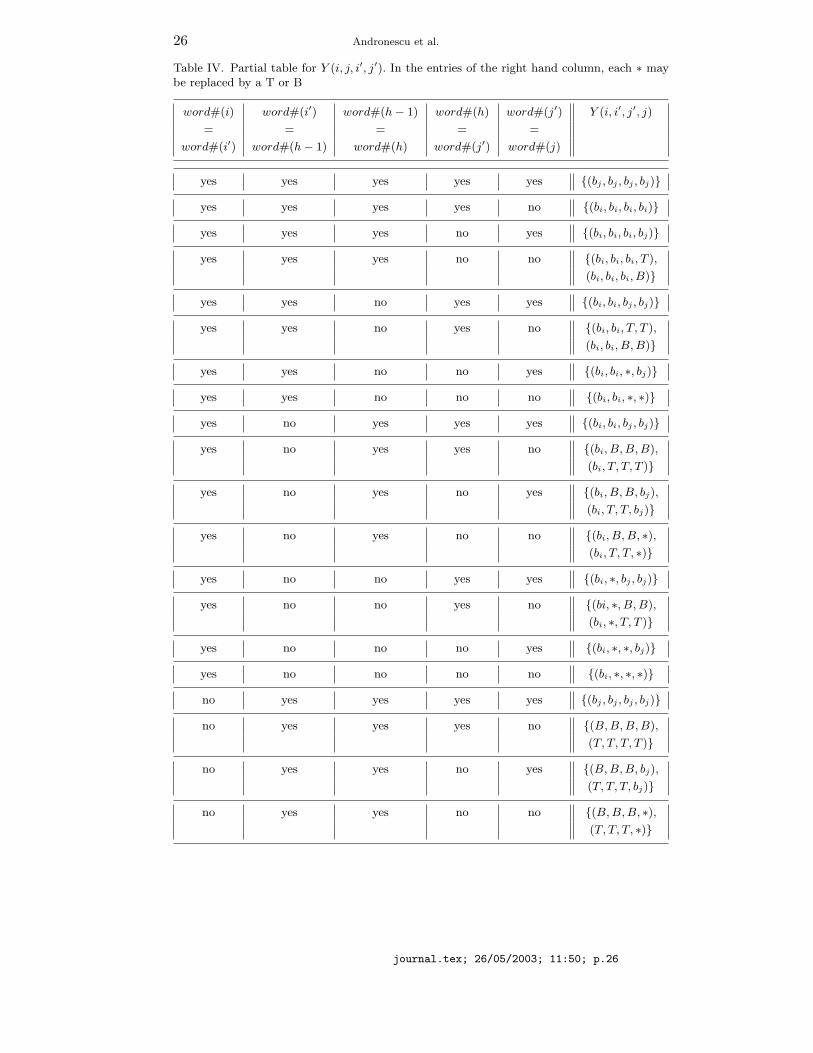
We next give recurrences for V M ′S(bi, bj , i, j) and WM ′
S(bi, bj , i, j).V M ′
S(bi, bj , i, j) =
mini+1<h≤j−1
(bi+1,bh−1,bh,bj−1)∈Y (i,j,h−1,h)
(WM ′S(bi+1, bh−1, i+1, h−1)+WM ′
S(bh, bj−1, h, j−1)+a).
WM ′S(bi, bj , i, j) =
min
V ′S(bi, bj , i, j) + b,
mini<h≤j
(bh−1,bh)∈X(i,j,h−1,h)
(WM ′S(bi, bh−1, i, h− 1) +WM ′
S(bh, bj , h, j)),
where Y (i, j, i′, j′) is given by Table IV.
A3. Base Cases
This algorithm depends on eH ′S , eS
′S , and eL′
S which are the recur-rences for a hairpin loop, a stacked pair, and an internal loop respec-tively, generalized to combinatorial sets. These quantities are definedbelow, and each can be calculated in constant time.
eH ′S(bi, bj , i, j)
= mins∈S(bi,bj ,i,j)
eHs(i, j)
= mins∈S(bi,bj ,i,j)
(eH-Stack(si, si+1, sj−1, sj) + eH-Length(j − i− 1))
journal.tex; 26/05/2003; 11:50; p.24

Testing sets concatenate without secondary structure 25
Since the eH-Stack function only depends on the four bases si, si+1,sj−1, and sj , the min taken over all s ∈ S(bj , bj , i, j), can be calculatedin constant time.
eL′S(bi, bi′ , bj′ , bj , i, i
′, j′, j)= mins∈S(bi,bi′ ,bj′ ,bj ,i,j) eLs(i, j, i′, j′)= mins∈S(bi,bi′ ,bj′ ,bj ,i,i′,j′,j)
(eL-Stack(si, si+1, sj−1, sj) + eL-Stack(sj′ , sj′+1, si′−1, si′)+ eL-Length(i′ − i− 1, j′ − j − 1)),
where the definition of S(bi, bi′ , bj′ , bj , i, i′, j′, j) is the natural general-
ization of the definition of S(bi, bj , i, j).
journal.tex; 26/05/2003; 11:50; p.25
26 Andronescu et al.
Table IV. Partial table for Y (i, j, i′, j′). In the entries of the right hand column, each ∗ maybe replaced by a T or B
word#(i) word#(i′) word#(h − 1) word#(h) word#(j′) Y (i, i′, j′, j)
= = = = =
word#(i′) word#(h − 1) word#(h) word#(j′) word#(j)
yes yes yes yes yes {(bj , bj , bj , bj)}
yes yes yes yes no {(bi, bi, bi, bi)}
yes yes yes no yes {(bi, bi, bi, bj)}
yes yes yes no no {(bi, bi, bi, T ),
(bi, bi, bi, B)}
yes yes no yes yes {(bi, bi, bj , bj)}
yes yes no yes no {(bi, bi, T, T ),
(bi, bi, B, B)}
yes yes no no yes {(bi, bi, ∗, bj)}
yes yes no no no {(bi, bi, ∗, ∗)}
yes no yes yes yes {(bi, bi, bj , bj)}
yes no yes yes no {(bi, B, B, B),
(bi, T, T, T )}
yes no yes no yes {(bi, B, B, bj),
(bi, T, T, bj)}
yes no yes no no {(bi, B, B, ∗),(bi, T, T, ∗)}
yes no no yes yes {(bi, ∗, bj , bj)}
yes no no yes no {(bi, ∗, B, B),
(bi, ∗, T, T )}
yes no no no yes {(bi, ∗, ∗, bj)}
yes no no no no {(bi, ∗, ∗, ∗)}
no yes yes yes yes {(bj , bj , bj , bj)}
no yes yes yes no {(B, B, B, B),
(T, T, T, T )}
no yes yes no yes {(B, B, B, bj),
(T, T, T, bj)}
no yes yes no no {(B, B, B, ∗),(T, T, T, ∗)}
journal.tex; 26/05/2003; 11:50; p.26
Related Documents











