Aggregate formation and phosphorylation of neurofilament-L Pro22 Charcot – Marie – Tooth disease mutants Takahiro Sasaki 1,{ , Takahiro Gotow 2 , Motoko Shiozaki 2 , Fumika Sakaue 1 , Taro Saito 1 , Jean-Pierre Julien 3 , Yasuo Uchiyama 4 and Shin-ichi Hisanaga 1, * 1 Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Minami-ohsawa, Hachiohji, Tokyo 192-0397, Japan, 2 Laboratory of Cell Biology, College of Nutrition, Koshien University, Takarazuka, Hyogo 665-0006, Japan, 3 Centre Hospitalier de l’Universite Laval Research Center, Quebec City, Quebec, Canada and 4 Department of Cell Biology and Neuroscience, Osaka University Graduate School of Medicine, Suita, Osaka 565-0871, Japan Received December 4, 2005; Revised and Accepted January 30, 2006 Charcot–Marie–Tooth disease (CMT) is the most common inherited peripheral nerve disorder. The causative gene for axonal type CMT2E has been identified as neurofilament light (NF-L) chain. Using cultured cells and in vitro assays, we analyzed the filament formation ability of Pro22 CMT mutant proteins of NF-L, P22S and P22T. NF-L Pro22 mutant proteins formed large aggregates in SW132 cells and cortical neurons and assembled into short twisty threads thinner than 10 nm filaments in vitro. Those threads associated with each other at their ends and entangled into large aggregates, also abnormalities, were detected at steps in oligomer formation. Pro22 mutations abolished Thr21 phosphorylation by cyclin-dependent kinase 5 and external signal regulated kinase, which suppressed filament assembly, but phosphorylation by protein kinase A (PKA) inhibited aggregate formation in vitro and alleviated aggregates in cortical neur- ons. These results indicate that the Pro22 CMT mutation induces abnormal filament aggregates by disrupting proper oligomer formation and the aggregates are mitigated by phosphorylation with PKA, which makes it a viable target for the development for therapeutics. INTRODUCTION Charcot–Marie–Tooth disease (CMT) is the most common inherited peripheral nerve disease (1–3). CMT is classified into two main types on the basis of nerve conduction velocity, demyelinating CMT1 and axonal CMT2. Each class of CMT is further divided into several subgroups by genetic locus. Pathological features of CMT1 are characterized by segmental demyelination and remyelination, and proteins involved in myelin formation have been identified as gene products for CMT1. Mutations in CMT2, which are accompanied by axonal degeneration, are found in proteins involved in axonal transport and integrity. The neurofilament light (NF-L) chain gene has been ident- ified as the causative gene for CMT2E (4). Neurofilaments (NFs) are the most abundant cytoskeletal element in axons (5,6) and NF-L is the basic subunit of NFs, which are hetero- polymers composed of neurofilament middle (NF-M) chain and neurofilament heavy (NF-H) chain (7,8). The three NF sub- units consist of the N-terminal head, a-helical central rod and C-terminal tail domains. The rod domain is involved in the formation of coiled-coil dimers, an essential step for filament assembly. The head domain is a region regulating filament assembly through phosphorylation by second-messenger- dependent protein kinases (5). NF-M and NF-H have a longer C-terminal tail domain protruding from the core filaments (9) with multiple Lys–Ser–Pro repeat sequences which can be phosphorylated by proline-directed protein kinases (PDPKs). Several missense mutations of NF-L have been reported for CMT (4,10–15). There have been four reports describing the # The Author 2006. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] { Present address: Nathan Kline Institute, New York University School of Medicine, Orangeburg, NY 10962, USA. *To whom correspondence should be addressed. Tel: þ81 426772577; Fax: þ81 426772559; Email: [email protected] Human Molecular Genetics, 2006, Vol. 15, No. 6 943–952 doi:10.1093/hmg/ddl011 Advance Access published on February 1, 2006 by guest on March 20, 2016 http://hmg.oxfordjournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Aggregate formation and phosphorylation ofneurofilament-L Pro22 Charcot–Marie–Toothdisease mutants
Takahiro Sasaki1,{, Takahiro Gotow2, Motoko Shiozaki2, Fumika Sakaue1, Taro Saito1,
Jean-Pierre Julien3, Yasuo Uchiyama4 and Shin-ichi Hisanaga1,*
1Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Minami-ohsawa,
Hachiohji, Tokyo 192-0397, Japan, 2Laboratory of Cell Biology, College of Nutrition, Koshien University, Takarazuka,
Hyogo 665-0006, Japan, 3Centre Hospitalier de l’Universite Laval Research Center, Quebec City, Quebec, Canada
and 4Department of Cell Biology and Neuroscience, Osaka University Graduate School of Medicine, Suita, Osaka
565-0871, Japan
Received December 4, 2005; Revised and Accepted January 30, 2006
Charcot–Marie–Tooth disease (CMT) is the most common inherited peripheral nerve disorder. The causativegene for axonal type CMT2E has been identified as neurofilament light (NF-L) chain. Using cultured cells andin vitro assays, we analyzed the filament formation ability of Pro22 CMT mutant proteins of NF-L, P22S andP22T. NF-L Pro22 mutant proteins formed large aggregates in SW132 cells and cortical neuronsand assembled into short twisty threads thinner than 10 nm filaments in vitro. Those threads associatedwith each other at their ends and entangled into large aggregates, also abnormalities, were detected atsteps in oligomer formation. Pro22 mutations abolished Thr21 phosphorylation by cyclin-dependentkinase 5 and external signal regulated kinase, which suppressed filament assembly, but phosphorylationby protein kinase A (PKA) inhibited aggregate formation in vitro and alleviated aggregates in cortical neur-ons. These results indicate that the Pro22 CMT mutation induces abnormal filament aggregates by disruptingproper oligomer formation and the aggregates are mitigated by phosphorylation with PKA, which makes it aviable target for the development for therapeutics.
INTRODUCTION
Charcot–Marie–Tooth disease (CMT) is the most commoninherited peripheral nerve disease (1–3). CMT is classifiedinto two main types on the basis of nerve conduction velocity,demyelinating CMT1 and axonal CMT2. Each class of CMTis further divided into several subgroups by genetic locus.Pathological features of CMT1 are characterized by segmentaldemyelination and remyelination, and proteins involved inmyelin formation have been identified as gene products forCMT1. Mutations in CMT2, which are accompanied byaxonal degeneration, are found in proteins involved in axonaltransport and integrity.
The neurofilament light (NF-L) chain gene has been ident-ified as the causative gene for CMT2E (4). Neurofilaments
(NFs) are the most abundant cytoskeletal element in axons(5,6) and NF-L is the basic subunit of NFs, which are hetero-polymers composed of neurofilament middle (NF-M) chainand neurofilament heavy (NF-H) chain (7,8). The three NF sub-units consist of the N-terminal head, a-helical central rod andC-terminal tail domains. The rod domain is involved in theformation of coiled-coil dimers, an essential step for filamentassembly. The head domain is a region regulating filamentassembly through phosphorylation by second-messenger-dependent protein kinases (5). NF-M and NF-H have a longerC-terminal tail domain protruding from the core filaments (9)with multiple Lys–Ser–Pro repeat sequences which can bephosphorylated by proline-directed protein kinases (PDPKs).
Several missense mutations of NF-L have been reported forCMT (4,10–15). There have been four reports describing the
# The Author 2006. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
{Present address: Nathan Kline Institute, New York University School of Medicine, Orangeburg, NY 10962, USA.
*To whom correspondence should be addressed. Tel: þ81 426772577; Fax: þ81 426772559; Email: [email protected]
Human Molecular Genetics, 2006, Vol. 15, No. 6 943–952doi:10.1093/hmg/ddl011Advance Access published on February 1, 2006
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

effects of NF-L CMT mutations on filamentous network for-mation in cultured cells. Those mutations are E7K, P8L,P8Q, P8R, P22S, P22T, E89K, N97S, Q333P and D469N(16–19). All the mutants, except for E7K and D469N,display aggregate formation when expressed in culturedcells, but it is not known how the mutations affect filamentassembly. We have an interest in mutations at Pro22, asmutation at this site abolishes the Thr–Pro PDPK consensusphosphorylation sequence in the head domain of NF-L. Weexpect that this mutation would disrupt the regulation offilament assembly by phosphorylation.
In this study, we analyzed the assembly and phosphoryla-tion of NF-L P22S and P22T mutants in cultured cells andin vitro. The mutants formed large aggregates in cells andabnormal filaments in vitro. They were not phosphorylatedat Thr21 by cyclin-dependent kinase 5 (Cdk5) or externalsignal regulated kinase 2 (ERK2), which inhibits assemblyof the filaments. However, they retained the phosphorylationsite for protein kinase A (PKA), which diminished aggregatesin vitro and in neurons. Thus, the phosphorylation in the headdomain could be used as a therapeutic strategy for CMT2Epatients.
RESULTS
Aggregate formation of NF-L mutants,P22S and P22T, in SW132 cells
SW132 cl.2/Vim2 cells (here referred to as SW132), whichlack endogenous cytoplasmic intermediate filaments, are oftenused to assess the assembly of intermediate filament proteins.Several CMT mutants of NF-L, including the mutation atPro22, have been reported to display defects in filamentousnetwork formation in transfected SW132 cells (16–19). Wefirst tested the filament formation ability of Pro22 mutants in
SW132 cells in our experimental system. Human NF-L wild-type (WT) assembled into a network of filaments (Fig. 1A).Both NF-L P22S and P22T formed large irregular aggregatesin SW132 cells (Fig. 1F and K), confirming the previousresults by Perez-Olle et al. (19).
NFs are obligate heteropolymers of NF-L, NF-M and NF-Hin neurons (7,8). To examine whether co-expression of NF-Mor NF-H suppresses aggregate formation or whether they arealso incorporated into the aggregates, NF-L WT, P22S orP22T was cotransfected with NF-M or NF-H in SW132cells. NF-L WT co-assembled into filaments with NF-M orNF-H (Fig. 1B–E). NF-L P22S and P22T mutants not onlyformed aggregates when they were co-expressed with NF-Mor NF-H but also incorporated NF-M or NF-H into the aggre-gates (Fig. 1G–J and L–O). We did not notice any differencein aggregates formed between NF-L/NF-M and NF-L/NF-H atleast under fluorescent microscopy. These results suggest thatNF-L P22S and P22T can form aggregates even in the pre-sence of NF-M and NF-H in neurons. In contrast, Perez-Olleet al. (19) describe that co-transfection with NF-M results inheteropolymeric filamentous networks in some cells. Thisdifference may be due to differences in expression levels ofproteins and using different expression vectors.
Aggregate formation of NF-L P22S and P22Tin cultured cortical neurons
To examine aggregate formation in a neuronal cellularenvironment, we expressed NF-L P22S and P22T in culturedcortical neurons. NF-L WT, P22S or P22T was expressed incultured cortical neurons together with enhanced green fluor-escent protein (EGFP) to help identify transfected neurons(Fig. 2A, E and I). NF-L WT was observed as a network offilaments in the cell body (Fig. 2D) and its staining was alsodetected in neurites (Fig. 2B). However, the distribution of
Figure 1. Aggregate formation of NF-L CMT mutants, P22S and P22T, in SW132 cells. Immunofluorescent staining of SW132 cells expressing human NF-LWT (A–E), P22S (F–J) or P22T (K–O) alone or in combination with the other NF subunit of NF-M or NF-H. The cells were transfected with pCMV5 vectorsencoding NF-L (A, F and K), NF-L and NF-M (B, C, G, H, L and M), NF-L and NF-H (D, E, I, J, N and O) and then were cultured for 24 h. The expression ofNF-L (A, B, D, F, G, I, K, L and N), NF-M (C, H and M) and NF-H (E, J and O) was detected by immunostaining with the anti-NF-L rabbit polyclonal antibody,the anti-NF-M mouse monoclonal antibody NN18 and the anti-NF-H mouse monoclonal antibody SMI32, respectively. Scale bar represents 10 mm.
944 Human Molecular Genetics, 2006, Vol. 15, No. 6
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

NF-L P22S and P22T was restricted to the cell body (Fig. 2Fand J), and those in the cell body formed large aggregates, aswas seen in SW132 cells (Fig. 2H and L). EGFP was incor-porated into these aggregates of NF-L P22S and P22T (arrowsin Fig. 2G and K), in contrast to the case of NF-L WT, whichdid not show colocalization with EGFP even in a highly con-centrated region of NF-L WT filaments (Fig. 2C).
Large aggregates were grown from smaller punctatestructures in SW132 cells
How NF-L P22S or P22T form large aggregates was nextexamined by observing NF-L staining in SW132 cells at anearlier time after transfection (Fig. 3). At 10 h after transfec-tion, the cells were fixed and stained with anti-NF-L antibody.These cells exhibited various stages of filament assembly,which was most likely dependent upon the expression levelsof NF-L protein. Three images of NF-L WT and P22S areshown in Figure 3 and may represent the process of filamen-tous network formation (Fig. 3A–C) and aggregate growth(Fig. 3D–F), respectively. In the case of NF-L WT, fibrilsappeared to elongate gradually from smaller punctate struc-tures, as has been reported for other IF proteins (20). Aggre-gates of NF-L P22S appear to become larger directly fromthe dotted structures. The results indicate that the large aggre-gates of NF-L P22S and P22T were formed by growth of thesmaller aggregates rather than bundling of previously formedintermediate-size filaments.
In vitro assembly of NF-L P22S and P22T
The Pro22 mutation of NF-L seemed to affect filament assem-bly rather than interfilament interactions. Thus, to understandthe molecular mechanism of aggregate formation, we exam-ined the assembly of NF-L in vitro with purified proteins.NF-L WT, P22S and P22T, which had been tagged with ahistidine hexamer at the C-terminus, were purified fromEscherichia coli inclusion bodies (Fig. 4A). Purified NF-Lproteins were assembled by dialyzing against the assemblybuffer at 378C for 3 h. Filament assembly was first checkedby a sedimentation assay (Fig. 4B). NF-L P22S and P22T,as well as NF-L WT, were found in the pellets, indicatingthat NF-L P22S and P22T can form aggregates that sedimentduring ultracentrifugation at 100 000g for 20 min. Polymerscould be observed by negative staining electron microscopy(Fig. 4C–H). NF-L WT polymerized into long intermediate-size filaments (Fig. 4C and D); however, no such long fila-ments were observed with NF-L P22S and P22T (Fig. 4Eand G). Instead, short, thinner filaments and various sizes ofintertwined filaments were observed with both NF-L P22Sand P22T (Fig. 4F and H).
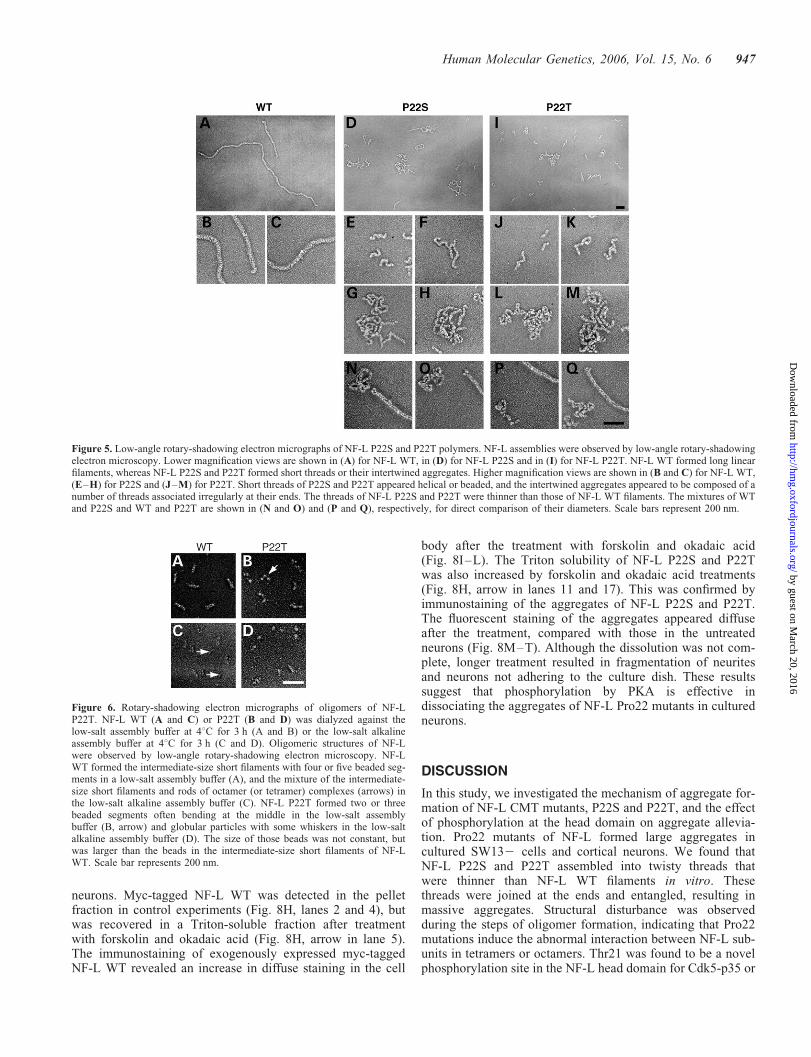
A more detailed analysis of the filamentous structures wasobserved by low-angle rotary-shadowing electron microscopy.Long, straight filaments, typical for the intermediate filament,were observed with NF-L WT (Fig. 5A–C). However, no suchfilaments were found in the samples assembled from NF-LP22S and P22T, which produced only mixtures of shortthreads and masses of intertwined filaments (Fig. 5D and I).Higher magnification views are shown in Fig. 5E–H and J–M for NF-L P22S and P22T, respectively. Similar structureswere observed with NF-L P22S and P22T. Some of theshort threads appeared to be helical (Fig. 5E and J) andsome appeared to be beaded and attached at their ends toeach other (Fig. 5F and K). Larger masses were composedof several intertwined longer threads (Fig. 5G, H, l and m).The size of the aggregates was smaller than those observedby negative staining electron microscopy, but this was dueto fragmentation of the aggregates when the samples weresprayed onto mica. The diameter of NF-L P22S or P22T
Figure 2. Aggregate formation of NF-L CMT mutants, P22S and P22T, in ratcortical neurons. Human NF-L WT (A–D), P22S (E–H) or P22T (I–L) wasexpressed in rat cortical neurons. Transfected neurons were identified by flu-orescence of co-expressed EGFP (A, C, E, G, I and K). NF-L was stained byanti-human NF-L-specific antibody 24 h after transfection (B, D, F, H, J andL). NF-L WT was detected in the cell body and neurites (A and B),whereas NF-L P22S (E and F) and P22T (I and J) were localized only inthe cell body. The higher magnification micrographs of the cell body revealeda filamentous network of NF-L WT (D), but large aggregates for both P22S(H) and P22T (L). Incorporation of EGFP in the aggregates was observed inthese mutants (arrows in G, H, K and L). Scale bars represent 10 mm.
Figure 3. Aggregates of NF-L P22S are observed soon or shortly after trans-fection in SW132 cells. The SW132 cells were transfected with NF-L WT(A–C) or P22S (D–F), and their immunofluorescent images were obtainedat 10 h after transfection, that is, 14 h earlier than those shown in Figure 1.Cells displayed various assembly or aggregation stages of NF-L. Threetypical micrographs are shown to represent the growing process of filaments(A–C) or aggregates (D–F). Higher magnification micrographs are shownin inset. Scale bars in (F) and its inset represent 1 and 10 mm, respectively.
Human Molecular Genetics, 2006, Vol. 15, No. 6 945
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

threads was smaller than that of NF-L WT filaments. To verifythis, assemblies of NF-L WT and Pro22 mutants wereobserved in identical specimens (Fig. 5 N–Q). The threadsof Pro22 mutants were evidently thinner than NF-L WT fila-ments. The diameter of NF-L P22S and P22T threads was18 nm, three-fourths of the wild-type filaments (24 nm),when measured with rotary-shadowed samples.
Several intermediate assemblies of NF-L can be formedstably through the choice of dialysis solution (21). After dialy-sis against the low-salt assembly buffer, human NF-L WTformed the short intermediate-size filaments consisting offour or five beaded segments (Fig. 6A). NF-L P22S displayedstructures of two or three beads connected by a thin fibril(Fig. 6B). Some of them appeared to be boomerang-like struc-tures, with a bend at the middle bead (Fig. 6B, arrow). Whendialyzed against the low salt, alkaline assembly solution, NF-LWT assembled into short intermediate-size filaments and thinrods (arrows in Fig. 6C). Under the same conditions, NF-LP22S showed globular particles with several whisker-likeshort filaments around them (Fig. 6D). Although we do notknow how these structures are constructed from mutantNF-L, these results indicate that disruption in filament assem-bly of the Pro22 mutant resides in a step of oligomerformation.
Thr21 phosphorylation by PDPKs and its loss in NF-LP22S and P22T mutants
Pro22 is the amino acid C-terminal next to Thr21, constitutingthe Thr–Pro consensus phosphorylation sequence for PDPKs.The mutation of Pro22 results in the abolition of thisconsensus sequence in NF-L. As there has been no reporton Thr21 phosphorylation, we examined phosphorylation at
Thr21 using Cdk5-p35 as a PDPK. Phosphorylation of NF-LWT and T21A is shown in Figure 7A. NF-L WT was phos-phorylated by Cdk5-p35, and this phosphorylation was mark-edly decreased in the T21A mutant. A single major spot on the2D-phosphopeptide map disappeared with T21A phosphory-lated by Cdk5-p35 (Fig. 7B). These results indicate thatThr21 is the major Cdk5 phosphorylation site in NF-L.
Phosphorylation at the head domain of NF-L is known toinduce filament disassembly or prevent filament assembly(22–24). We examined whether Thr21 phosphorylation alsoaffects filament assembly. NF-L WT or T21A phosphorylatedby Cdk5-p35 was assembled by dialyzing against the assemblybuffer and assembled filaments were examined by sedimen-tation assay (Fig. 7C) and electron microscopy (Fig. 7D–I).Unphosphorylated NF-L was sedimented by centrifugation(Fig. 7C) and formed long filaments (Fig. 7D and E) (seealso Supplementary Material, Fig. S1). However, about ahalf of phosphorylated NF-L WT remained in the supernatantafter centrifugation (Fig. 7C), and only short filaments weredetected under electron microscopy (Fig. 7F and G), indicat-ing that the phosphorylation at Thr21 suppresses filamentassembly. This was further demonstrated by the decreasedeffect of phosphorylation on the filament assembly of NF-LT21A (Fig. 7C and F–I).
To examine whether the Pro22 mutation effects Thr21 phos-phorylation and which PDPK phosphorylates the site, P22S orP22T was phosphorylated by three different PDPKs: glycogensynthase kinase 3b (GSK3b), ERK and Cdk5-p35 (Fig. 7J).ERK and Cdk5-p35 phosphorylated NF-L WT strongly, andthe phosphorylation decreased considerably with NF-L P22Sand P22T mutants. In contrast, a slight phosphorylation ofNF-L WT was observed when GSK3b was used, and theextent of phosphorylation was not significantly changed bymutation at Pro22. These results suggest that Pro22 mutationabolishes a regulatory mechanism of filament disassembly,i.e. the phosphorylation at Thr21 by Cdk5-p35 or ERK.
Alleviation of NF-L P22S and P22T aggregatesby PKA-dependent phosphorylation
Aggregates of NF-L P22S and P22T could be induced byabnormal interaction between mutant subunits during assem-bly (Figs 4–6), and the loss of Cdk5-p35 or ERK-dependentphosphorylation at Thr21 in the head domain could enhanceaggregate formation by eliminating a disassembly pathwayin neurons. However, other phosphorylation sites besidesThr21 in the head domain might still function to regulate theassembly of mutant NF-L. PKA is a well-known headdomain kinase (22,23,25–27) and its phosphorylation, forexample, at Ser41, Ser55 and Ser62, may not be affected bythe Pro22 mutation. This was confirmed by examining phos-phorylation of NF-L Pro22 mutants by PKA. NF-L P22Sand P22T were phosphorylated to the same extent as NF-LWT and were disassembled as efficiently as NF-L WT invitro by PKA phosphorylation (Fig. 8A–G).
Next, we examined the effect of PKA-dependent phos-phorylation on alleviation of the aggregates of NF-L Pro22mutants in cultured neurons. We first made sure that the acti-vation of PKA by forskolin and okadaic acid increased theTriton solubility of NF-L WT when transfected into cultured
Figure 4. Assembly of NF-L P22S and P22T mutants in vitro. (A) Coomassiebrilliant blue (CBB) staining of an SDS–PAGE gel of NF-L proteins purifiedfrom E. coli. (B) The sedimentation assay of assembled NF-L proteins. NF-LWT, P22S or P22T was assembled in vitro by dialysis against the assemblybuffer at 378C for 3 h. Assembled NF-L was sedimented by centrifugationat 100 000g for 20 min and the supernatant and pellet were subjected toSDS–PAGE. (C–H) Negative staining electron micrographs of assembledNF-L proteins. Micrographs are NF-L WT in (C and D), P22S in (E and F)and P22T in (G and H), respectively, in low and higher magnifications.Scale bars represent 200 nm.
946 Human Molecular Genetics, 2006, Vol. 15, No. 6
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from
neurons. Myc-tagged NF-L WT was detected in the pelletfraction in control experiments (Fig. 8H, lanes 2 and 4), butwas recovered in a Triton-soluble fraction after treatmentwith forskolin and okadaic acid (Fig. 8H, arrow in lane 5).The immunostaining of exogenously expressed myc-taggedNF-L WT revealed an increase in diffuse staining in the cell
body after the treatment with forskolin and okadaic acid(Fig. 8I–L). The Triton solubility of NF-L P22S and P22Twas also increased by forskolin and okadaic acid treatments(Fig. 8H, arrow in lanes 11 and 17). This was confirmed byimmunostaining of the aggregates of NF-L P22S and P22T.The fluorescent staining of the aggregates appeared diffuseafter the treatment, compared with those in the untreatedneurons (Fig. 8M–T). Although the dissolution was not com-plete, longer treatment resulted in fragmentation of neuritesand neurons not adhering to the culture dish. These resultssuggest that phosphorylation by PKA is effective indissociating the aggregates of NF-L Pro22 mutants in culturedneurons.
DISCUSSION
In this study, we investigated the mechanism of aggregate for-mation of NF-L CMT mutants, P22S and P22T, and the effectof phosphorylation at the head domain on aggregate allevia-tion. Pro22 mutants of NF-L formed large aggregates incultured SW132 cells and cortical neurons. We found thatNF-L P22S and P22T assembled into twisty threads thatwere thinner than NF-L WT filaments in vitro. Thesethreads were joined at the ends and entangled, resulting inmassive aggregates. Structural disturbance was observedduring the steps of oligomer formation, indicating that Pro22mutations induce the abnormal interaction between NF-L sub-units in tetramers or octamers. Thr21 was found to be a novelphosphorylation site in the NF-L head domain for Cdk5-p35 or
Figure 5. Low-angle rotary-shadowing electron micrographs of NF-L P22S and P22T polymers. NF-L assemblies were observed by low-angle rotary-shadowingelectron microscopy. Lower magnification views are shown in (A) for NF-L WT, in (D) for NF-L P22S and in (I) for NF-L P22T. NF-L WT formed long linearfilaments, whereas NF-L P22S and P22T formed short threads or their intertwined aggregates. Higher magnification views are shown in (B and C) for NF-L WT,(E–H) for P22S and (J–M) for P22T. Short threads of P22S and P22T appeared helical or beaded, and the intertwined aggregates appeared to be composed of anumber of threads associated irregularly at their ends. The threads of NF-L P22S and P22T were thinner than those of NF-L WT filaments. The mixtures of WTand P22S and WT and P22T are shown in (N and O) and (P and Q), respectively, for direct comparison of their diameters. Scale bars represent 200 nm.
Figure 6. Rotary-shadowing electron micrographs of oligomers of NF-LP22T. NF-L WT (A and C) or P22T (B and D) was dialyzed against thelow-salt assembly buffer at 48C for 3 h (A and B) or the low-salt alkalineassembly buffer at 48C for 3 h (C and D). Oligomeric structures of NF-Lwere observed by low-angle rotary-shadowing electron microscopy. NF-LWT formed the intermediate-size short filaments with four or five beaded seg-ments in a low-salt assembly buffer (A), and the mixture of the intermediate-size short filaments and rods of octamer (or tetramer) complexes (arrows) inthe low-salt alkaline assembly buffer (C). NF-L P22T formed two or threebeaded segments often bending at the middle in the low-salt assemblybuffer (B, arrow) and globular particles with some whiskers in the low-saltalkaline assembly buffer (D). The size of those beads was not constant, butwas larger than the beads in the intermediate-size short filaments of NF-LWT. Scale bar represents 200 nm.
Human Molecular Genetics, 2006, Vol. 15, No. 6 947
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

ERK, which inhibited filament assembly, and the phosphoryla-tion was abolished by the Pro22 mutation, suggesting thepossibility that the Pro22 mutation causes more severe symp-toms by disrupting a phosphorylation-dependent aggregateeliminating pathway in neurons. However, phosphorylationby PKA was not affected by the Pro22 mutation. Theincreased PKA-phosphorylation of NF-L Pro22 mutants incultured neurons reduced the aggregates.
Abnormal filament assembly of NF-L P22Sor P22T mutants
In this study, NF-L P22S and P22T mutants formed largeaggregates in SW132 cells (19) and cortical neurons. Other
Figure 8. Phosphorylation of NF-L P22S or P22T by PKA and its effect onaggregate alleviation in neurons. (A) Phosphorylation of NF-L WT, P22S orP22T by PKA in vitro. NF-L proteins were phosphorylated by the catalyticsubunit of PKA. The time course of phosphorylation detection was 0, 5, 15, 30and 60 min, shown by autoradiography after SDS–PAGE. (B–G) Electronmicrographs of NF-L WT, P22S or P22T before (B–D) and after (E–G) phos-phorylation. Scale bar in (G) represents 200 nm. (H) The Triton solubility ofphosphorylated NF-L Pro22 mutants in cortical neurons. Rat cortical neuronswere transfected with myc-tagged human NF-L WT (Myc-WT), P22S(Myc-P22S) or P22T (Myc-P22T) and were treated with 15 mM forskolin and0.1 mM okadaic acid for 2 h. Mock transfection (Mock) is shown in right sidefour lanes. The Triton-soluble (S) and -insoluble (P) fractions were subjectedto western blotting with anti-NF-L rabbit polyclonal antibody. Myc-NF-L isan upper band indicated by white arrowhead, and endogenous NF-L is a lowerband indicated by black arrowhead. The soluble Myc-NF-L proteins are indi-cated by arrows in the Triton-soluble fractions (lanes 5, 11 and 17). Smearedbands may represent highly phosphorylated NF-L species. (I–T) The forskolinand okadaic acid treatment alleviated aggregates formed from NF-L P22S orP22T mutants in cultured neurons. NF-L WT (I–L), P22S (M–P) or P22T(Q–T) was expressed in rat cortical neurons. The neurons were treated with15 mM forskolin and 0.1 mM okadaic acid for 2 h (K, L, O, P, S and T). As acontrol, untreated neurons are shown in (I, M and Q). Neurons treated withDMSO are shown in (J, N and R). Human NF-L exogenously expressed wasdetected by immunostaining with anti-human NF-L-specific monoclonal anti-body. Note that diffuse staining in the cytoplasm was increased and the aggre-gates of NF-L P22S or P22T appeared to be dispersed after the treatment withforskolin and okadaic acid. Scale bar represents 10 mm.
Figure 7. Phosphorylation of NF-L at Thr21 by PDPKs. (A) Phosphorylationof NF-L WT and T21A by Cdk5-p35. NF-L WT and T21A (CBB) were phos-phorylated in vitro by Cdk5-p35 using [g-32P]ATP. Phosphorylation wasdetected by autoradiograph after SDS–PAGE (ARG). (B) PhosphorylatedNF-L was subjected to 2D-phosphopeptide mapping after trypsin digestion.The major spot (arrow in WT) disappeared in the T21A mutant. (C) Sedimen-tation assay of assembled filaments from NF-L phosphorylated by Cdk5-p35.Rat NF-L WT or T21A was incubated with Cdk5-p35 in the presence orabsence of ATP. The reaction was stopped by addition of 6 M urea and thenthe NF-L proteins were assembled into filaments by dialysis against the assem-bly buffer at 378C for 3 h. The assembled filaments were pelleted by centrifu-gation at 100 000g for 20 min and the supernatant and pellet were examinedby SDS–PAGE. (D–I) The assembled filaments were observed by negativestaining electron microscopy; unphosphorylated NF-L WT in (D and E), phos-phorylated NF-L WT in (F and G) and phosphorylated NF-L T21A in (H andI), respectively, in low and higher magnifications. Scale bars represent200 nm. (J) Phosphorylation of NF-L WT, P22S and P22T by PDPKsin vitro. NF-L proteins were phosphorylated by GSK3b, ERK2 or Cdk5-p35using [g-32P]ATP. The time course of phosphorylation detection was 0, 5,15, 30 and 60 min, shown by autoradiography after SDS–PAGE.
948 Human Molecular Genetics, 2006, Vol. 15, No. 6
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

CMT mutants, E7K, P8L, P8Q, P8R, E89K, N97S, Q333P andD469N, had also been analyzed for aggregate formation in thesame or similar types of cells (16–19). These mutants, exceptfor E7K and D469N, were not able to form filamentous net-works, but instead formed aggregates, although the aggregatedstructures differed depending on the mutation sites. NF-LQ333P formed small dot-like aggregates with a disruption offilaments. The mutants at Pro8 showed a similar phenotypeto P22S and P22T, which gave one huge aggregate. AndNF-L E89K and N97S showed ‘sheet-like’ non-filamentousstructures (18). Q333P is located in the rod domain, E89Kand N97S are at the border between the head and rod andPro8 and Pro22 mutations are in the N-terminal region ofthe head domain of NF-L. Thus, the position of the mutationmay determine the phenotype of the assembly defect. Bycarrying out a thorough study of all the mutants, it could bepossible to predict the CMT phenotypes from the positionand amino acid of mutation.
Pro8 and Pro22 have been suggested to be hot spots forCMT2E mutations (11–13,15). The head domain of NF-L,where both residues are located, is the domain that regulatesfilament assembly through its phosphorylation. The effect ofphosphorylation could result in the introduction of negativecharges rather than conformation changes, because the headdomain is assumed to be globular with no secondary structure(28). However, proline is a unique amino acid residue that dis-rupts secondary structures by providing the fixed bendingangle to a polypeptide chain. The fact that mutations at Proresidues make NF-L unable to form proper filaments indicatesthat there might be some structural entities required for properassembly of filaments.
Low-angle rotary-shadowing electron microscopy is auseful method to analyze the assembly process at a molecularlevel (21). We found several structural abnormalities in poly-mers formed from Pro22 mutants using this technique. NF-LP22S and P22T assembled into short threads that werethinner than NF-L WT filaments (32 subunits per cross-section) and thicker than protofibrils (eight subunits percross-section). Two types of short threads were observed,helical and beaded. The massive aggregates appeared to becomposed of intertwined threads attached to each other attheir ends. The diameter of the threads was about half thediameter of the intermediate-size filaments of NF-L WTwhen observed by negative staining electron microscopy.Assuming that the subunit arrangement in the threads issimilar to that of the normal filaments, the threads of Pro22mutants might be composed of around 16 subunits per cross-section. The structural abnormality of NF-L Pro22 mutantswas also detected in 4- or 8-mer complexes, which aremainly formed by the coiled-coil interaction of thea-helical rod domain between NF subunits, indicating thatthe structural abnormality of the head domain would affectthe coiled-coil interaction in the 4- or 8-mer complex. Thismay be crucial to determine what role the head domainplays during filament assembly.
The large aggregates detected in SW13– cells and neuronsmay correspond to the mass of the intertwined threadsobserved under electron microscopy, although the exactrelationship must await further analysis of the intracellularaggregates. The aggregates appeared to grow directly from
smaller punctate structures, but not from an accumulation ofintermediate-size filaments. This was consistent with ourin vitro observation that aggregates were not made ofpacked intermediate-size filaments, but comprised manythreads associated mainly in an end-to-end fashion, indicatingthe gradual growth of aggregates without assembling intointermediate-size filaments. Thus, the mutation at Pro22 isthe primary cause for aggregate formation of NF-L.
The relationship of in vitro reconstructed abnormalfilaments and axonal NFs in a Pro22 CMT patient
Three individual families have been reported to have thePro22 mutation: P22S for a Slovenian family (12), and anItalian family (13), and P22T for a Japanese family (11).However, pathological features are only available on a suralnerve biopsy sample from the Italian family (13). The trans-verse section of sural nerve showed the loss of large myeli-nated axons and a few swollen axons encased in a thinmyelin sheath. The authors noted that NFs were grouped ina random orientation in swollen axons on electron micro-graphs. As there was no description of large aggregates orthinner and twisted filaments, such abnormal filaments mightnot be detected in those axons. NFs in patient axons shouldbe heteropolymers of NF-L P22S with wild-type NF-L,NF-M and NF-H. Assuming that the subunit ratio in NFs is3:1:1 for NF-L:NF-M:NF-H, as it is for bovine spinal cordNFs (29), and that mutant NF-L is also incorporated intoNFs in the same manner, mutant NF-L would account onlyfor 30% of total NF subunits. In contrast, the aggregates weobserved in vitro and in SW132 cells were composed exclu-sively of mutant NF-L and aggregates in cortical neuronswould also be formed mainly of over-expressed Pro22mutants. It has been reported that co-expression of othermutants with WT NF proteins in SW132 cells would attenuateaggregate formation (19). It would be important to investigateNF assembly at the ratio of mutant NF-L found in the axonsof patients.
Phosphorylation of NF-L Pro22 mutants and its possibleuse as a therapeutic strategy
The relationship between mutation and phosphorylation sitesand the effect of phosphorylation on filament assembly oraggregate formation are schematically shown in Figure 9.Thr21–Pro22 is the only PDPK consensus phosphorylationsequence in NF-L. We found Thr21 as a novel phosphoryl-ation site on NF-L for Cdk5 or ERK. The mutation at Pro22resulted in the loss of phosphorylation at Thr21. Like otherphosphorylation sites in the head domain (24–27), phos-phorylation at Thr21 suppressed filament assembly in vitro.Thus, the phosphorylation at the head domain could be amodulator of aggregate formation. The abolition of phos-phorylation at Thr21 by Pro22 mutation could enhance aggre-gate formation of NF-L Pro22 mutants more than other NF-LCMT mutants. The symptoms of Pro22 mutations are indeedmore severe than other CMT mutants (11–13). These resultsmay also suggest that the phosphorylation at Thr21 wouldwork in vivo in regulating NF organization.
Human Molecular Genetics, 2006, Vol. 15, No. 6 949
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

Defects in axonal transport are thought to be a cause ofdistal axonal atrophy in CMT patients. The aggregatesformed by NF-L CMT mutants inhibit the supply of cyto-skeletal proteins of NFs themselves and tubulin (19), inhibitother axonal proteins by trapping them into aggregates aswas observed with EGFP and perturb the transport of otheraxonal components such as mitochondria (16–19). The alle-viation of aggregated NFs in the cell body would be oneway to resume axonal transport. We showed that NF aggrega-tions were reduced to some extent following head domainphosphorylation by PKA. Looking at the mutation sites andPKA phosphorylation sites, PKA could act as a disassemblyfactor even for other CMT mutants (Fig. 9A), although thishas to be examined in future. Our results may suggest theuse of head domain phosphorylation as a potential therapeuticapproach to dissociate NF aggregates seen in CMT.
MATERIALS AND METHODS
Construction of plasmids
The mammalian expression plasmids of human NF-L, ratNF-M and rat NF-H were constructed by cloning the respec-tive cDNAs into the pCMV5 vector. The bacterial expression
plasmids of rat NF-L and human NF-L were constructed bycloning the respective cDNAs into the pET23a (þ) vector.To tag a histidine hexamer at the C-terminus of humanNF-L, human NF-L expression plasmid was fused to the his-tidine hexamer of pET23a (þ) by deleting the nucleotidesequence of the stop codon from human NF-L cDNA byPCR. NF-L T21A, P22S and P22T mutants were generatedusing the Quick Change site-directed mutagenesis kit (Strata-gene, La Jolla, CA, USA) according to the manufacturer’sinstructions.
Cell culture and transfection
SW132 cells were cultured in Dulbecco’s modified Eagle’smedium with 10% fetal bovine serum and 100 U/ml penicillinand 0.1 mg/ml streptomycin. Transfection into SW132 cellswas performed using the Lipofectamine 2000 (InvitrogenCorp., Carlsbad, CA, USA) according to the manufacturer’sinstructions. Cerebral cortical neurons were prepared from17-day-old embryonic rat brains (30). Cortical neurons cul-tured for three days were transfected with a calcium phosphateProfection kit (Promega, Madison, WI, USA).
Immunofluorescence staining of transfected cells
SW132 cells and cortical neurons were fixed with 4% para-formaldehyde in phosphate buffered saline (PBS) for 10 min,and then permeabilized in 0.1% Triton X-100 in PBS for1 min. After blocking with 1% skim milk in PBS for30 min, cells were probed with anti-NF-L rabbit polyclonalantibody NA1214 (500-fold dilution, Affiniti, Nottingham,UK), anti-human-specific NF-L mouse monoclonal antibodyNF70 (200-fold dilution, Chemicon, Temecula, CA, USA),anti-NF-M mouse monoclonal antibody NN18 (500-dilution,Sigma-Aldrich, St Louis, MO, USA) or anti-NF-H mousemonoclonal antibody SMI32 (2000-fold dilution, SternbergerMonoclonal Inc., Baltimore, MD, USA) for 1 h at room tempe-rature. After being washed with PBS, they were stained withFITC-conjugated secondary anti-mouse IgG or TRITC-conjugated secondary anti-rabbit IgG for 1 h at room tempera-ture. After washing, specimens were mounted with 90%glycerol containing 1 mg/ml p-phenylenediamine in PBS.Images were observed under an Axioskop fluorescencemicroscopy or an LSM 410 laser scanning confocal micro-scope (Carl Zeiss Inc., Oberkochen, Germany) and processedwith Adobe Photoshop software.
Preparation of NF-L proteins expressed in E. coli
Rat NF-L proteins were expressed in E. coli BL21 (DE3)pLysS in the presence of 0.5 mM IPTG for 3 h at 378C.NF-L recovered in inclusion bodies was solubilized in 8 M
urea in 10 mM sodium phosphate (pH 7.5), 1 mM EGTA and0.5 mM dithiothreitol and was purified by columns ofDEAE–cellulose and Mono-Q (Amersham PharmaciaBiotech., Tokyo, Japan) in the presence of 6 M urea. HumanNF-L proteins were purified with Ni-NTA agarose beads(Qiagen, Valencia, CA, USA) and Mono-Q and SephacrylS-200 gel filtration columns (Amersham Pharmacia Biotech.)in the presence of 6 M urea.
Figure 9. A scheme displaying CMT mutation sites and phosphorylation sitesin NF-L and their effects on NF-L assembly. (A) CMT mutation sites whoseeffects on filamentous network formation in cultured cells have been studiedare shown above molecular structure of NF-L. CMT mutations and PKA orCdk5 (or ERK) phosphorylation sites in the head domain are shown aboveor below the amino acid sequence of the NF-L head domain. Pro22 mutationstudied here is indicated in bold. The Thr21 Cdk5 (or ERK) phosphorylationsite is indicated by a thin arrow. The PKA phosphorylation sites are shown byshort arrows. (B) Effect of phosphorylation on filament assembly of NF-L WTor aggregate formation of Pro22 CMT mutants. PKA and Cdk5 (or ERK)inhibit assembly of NF-L by phosphorylation of the head domain. Pro22mutation to Ser or Thr changed the assembly property of NF-L to form aggre-gates. Abolishment of Thr21 Cdk5 (or ERK1/2) phosphorylation by themutations may exacerbate aggregate formation, but PKA phosphorylationwas not affected by the Pro22 mutation.
950 Human Molecular Genetics, 2006, Vol. 15, No. 6
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

Assembly of NF-L proteins in vitro
Assembly of NF-L (0.3 mg/ml) was performed by dialyzingagainst assembly buffer [20 mM Pipes (pH 6.8), 0.1 mM
EDTA, 0.15 M NaCl, 2 mM MgCl2, 0.5 mM dithiothreitol,0.4 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF),10 mg/ml leupeptin and 1 mg/ml pepstatin A] at 378C for3 h. Filament assembly was assessed by centrifugation at100 000g for 20 min or by electron microscopic observation.Short filaments were reconstituted by dialyzing against thelow-salt assembly buffer, the assembly buffer without 0.15 M
NaCl, at 48C for 3 h. NF-L oligomers were reconstituted bydialyzing against the low salt, alkaline assembly buffer[5 mM Tris–HCl (pH 8.5), 0.1 mM EDTA, 0.1 mM EGTA,0.5 mM dithiothreitol, 0.4 mM AEBSF, 10 mg/ml leupeptinand 1 mg/ml pepstatin A] at 48C for 3 h.
Electron microscopy analysis
Assembled filaments were diluted to 0.1 mg/ml and stainednegatively with 1.5% uranyl acetate or processed for low-angle rotary-shadowing as described previously (31). Speci-mens were examined with a JEM-1010 electron microscope(JEOL, Tokyo, Japan).
Phosphorylation of NF-L proteins
NF-L was phosphorylated by Cdk5-p35, ERK2, GSK3b orPKA using [g-32P]ATP (32). The reaction was stopped bythe addition of 4� Laemmli’s sample buffer. After sodiumdodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) on a 10% polyacrylamide gel, phosphorylation wasdetected with a BAS 2000 Bioimage Analyzer (Fuji Film,Tokyo, Japan). Two-dimensional phosphopeptide map ana-lyses were performed as described previously (32).
Triton-solubility assay of NFs in cultured cortical neurons
Cortical neurons were treated with 15 mM forskoline and0.1 mM okadaic acid for 2 h, collected by brief centrifugation,and suspended in Triton buffer [100 mM Pipes (pH 6.8), 5 mM
EGTA, 2 mM MgCl2, 50 mM KCl, 1% Triton X-100, 5 mM
NaF, 10 mM b-glycerophosphate, 0.4 mM AEBSF and10 mg/ml leupeptin]. Triton-soluble and -insoluble fractionswere separated by centrifugation at 100 000g for 20 min.
SDS–PAGE, western blotting and proteinconcentration determination
SDS–PAGE was performed according to Laemmli (33). Sep-arated proteins were transferred to Immobilon membranes(Millipore, Bedford, MA, USA). Blots were probed withanti-NF-L rabbit polyclonal antibody NA1214 (1000-folddilution), followed by an alkaline phosphatase-conjugated sec-ondary antibody (1000-fold dilution, DAKO, Glostrup,Denmark). The reaction was developed using a BCIP/NBTphosphatase system (KPL, Gaithersburg, MD, USA). Proteinconcentrations were determined with the Coomassie proteinassay reagent using bovine serum albumin as a standard(Pierce Biotechnology, Inc., Rockford, IL, USA).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We would like to express our thanks to Dr Robert Evans atUniversity of Colorado Health Science Centre for providingSW132 cells, Dr Masahiko Yamamoto for discussion andDr Pavan Krishnamurthy for reading the manuscript. Thiswork was supported by grants-in-aid from the Ministry ofEducation, Culture, Sports, Science and Technology ofJapan to T.G., Y.U. and S.H.
Conflict of Interest statement. None declared.
REFERENCES
1. Reilly, M.M. (2000) Classification of the hereditary motor and sensoryneuropathies. Curr. Opin. Neurol., 13, 561–564.
2. Shy, M.E., Garbern, J.Y. and Kamholz, J. (2002) Hereditary motor andsensory neuropathies: a biological perspective. Lancet Neurol., 1, 110–118.
3. Suter, U. and Scherer, S.S. (2003) Disease mechanisms in inheritedneuropathies. Nat. Rev. Neurosci., 4, 714–726.
4. Mersiyanova, I.V., Perepelov, A.V., Polyakov, A.V., Sitnikov, V.F.,Dadali, E.L., Oparin, R.B., Petrin, A.N. and Evgrafov, O.V. (2000) A newvariant of Charcot–Marie–Tooth disease type 2 is probably the result of amutation in the neurofilament-light gene. Am. J. Hum. Genet., 67, 37–46.
5. Pant, H.C. and Veeranna (1995) Neurofilament phosphorylation. Biochem.Cell Biol., 73, 575–592.
6. Julien, J.P. and Mushynski, W.E. (1998) Neurofilaments in health anddisease. Prog. Nucleic Acid Res. Mol. Biol., 61, 1–23.
7. Ching, G.Y. and Liem, R.K. (1993) Assembly of type IV neuronalintermediate filaments in nonneuronal cells in the absence of preexistingcytoplasmic intermediate filaments. J. Cell Biol., 122, 1323–1335.
8. Lee, M.K., Xu, Z., Wong, P.C. and Cleveland, D.W. (1993) Neurofilamentsare obligate heteropolymers in vivo. J. Cell Biol., 122, 1337–1350.
9. Hisanaga, S. and Hirokawa, N. (1988) Structure of the peripheral domainsof neurofilaments revealed by low angle rotary shadowing. J. Mol. Biol.,202, 297–305.
10. De Jonghe, P., Mersivanova, I., Nelis, E., Del Favero, J., Martin, J.J.,Van Broeckhoven, C., Evgrafov, O.C. and Timmerman, V. (2001) Furtherevidence that neurofilament light chain gene mutations can causeCharcot–Marie–Tooth disease type 2E. Ann. Neurol., 49, 245–249.
11. Yoshihara, T., Yamamoto, M., Hattori, N., Misu, K., Mori, K., Koike, H.and Sobue, G. (2002) Identification of novel sequence variants in theneurofilament-light gene in a Japanese population: analysis ofCharcot–Marie–Tooth disease patients and normal individuals.J. Peripher. Nerv. Syst., 7, 221–224.
12. Georgiou, D.M., Zidar, J., Korosec, M., Middleton, L.T., Kyriakides, T. andChristodoulou, K. (2002) A novel NF-L mutation Pro22Ser is associatedwith CMT2 in a large Slovenian family. Neurogenetic, 4, 93–96.
13. Fabrizi, G.M., Cavallaro, T., Angiari, C., Bertolasi, L., Cabrini, I.,Ferrarini, M. and Rizzuto, N. (2004) Giant axon and neurofilamentaccumulation in Charcot–Marie–Tooth disease type 2E. Neurology,62, 1429–1431.
14. Zuchner, S., Vorgerd, M., Sindern, E. and Schroder, J.M. (2004) Thenovel neurofilament light (NEFL) mutation Glu397Lys is associated witha clinically and morphologically heterogeneous type of Charcot–Marie–Tooth neuropathy. Neuromuscul. Disord., 14, 147–157.
15. Jordanova, A., De Jonghe, P., Boerkoel, C.F., Takashima, H.,De Vriendt, E., Ceuterick, C., Martin, J.J., Butler, I.J., Mancias, P.,Papasozomenos, S.Ch. et al. (2003) Mutations in the neurofilament lightchain gene (NEFL) cause early inset severe Charcot–Marie–Toothdisease. Brain, 126, 590–597.
16. Brownlees, J., Ackerley, S., Grierson, A.J., Jacobsen, N.J., Shea, K.,Anderton, B.H., Leigh, P.N., Shaw, C.E. and Miller, C.C. (2002)Charcot–Marie–Tooth disease neurofilament mutations disrupt
Human Molecular Genetics, 2006, Vol. 15, No. 6 951
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

neurofilament assembly and axonal transport. Hum. Mol. Genet.,11, 2837–2844.
17. Perez-Olle, R., Leung, C.L. and Liem, R.K. (2002) Effects ofCharcot–Marie–Tooth-linked mutations of the neurofilamentlight subunit on intermediate filament formation. J. Cell Sci., 115,4937–4946.
18. Perez-Olle, R., Jones, S.T. and Liem, R.K. (2004) Phenotypic analysis ofneurofilament light gene mutations linked to Charcot–Marie–Toothdisease in cell culture models. Hum. Mol. Genet., 13, 2207–2220.
19. Perez-Olle, R., Lopez-Toledano, M.A., Goryunov, D., Cabrera-Poch, N.,Stefanis, L., Brown, K. and Liem, R.K. (2005) Mutations in theneurofilament light gene linked to Charcot–Marie–Tooth disease causedefects in transport. J. Neurochem., 93, 861–874.
20. Prahlad, V., Yoon, M., Moir, R.D., Vale, R.D. and Goldman, R.D.(1998) Rapid movements of vimentin on microtubule tracks:kinesin-dependent assembly of intermediate filament networks.J. Cell Biol., 143, 159–170.
21. Hisanaga, S., Ikai, A. and Hirokawa, N. (1990) Molecular architecture ofthe neurofilament. I. Subunit arrangement of neurofilament L protein inthe intermediate-sized filament. J. Mol. Biol., 211, 857–869.
22. Hisanaga, S., Gonda, Y., Inagaki, M., Ikai, A. and Hirokawa, N. (1990)Effects of phosphorylation of the neurofilament L protein on filamentousstructures. Cell. Regul., 1, 237–248.
23. Nakamura, Y., Takeda, M., Angelides, K.J., Tanaka, T., Tada, K. andNishimura, T. (1990) Effect of phosphorylation on 68 kDa neurofilamentsubunit protein assembly by the cyclic AMP dependent protein kinasein vitro. Biochem. Biophys. Res. Commun., 169, 744–750.
24. Tokui, T., Yamauchi, T., Yano, T., Nishi, Y., Kusagawa, M., Yatani, R.and Inagaki, M. (1990) Ca2(þ)-calmodulin-dependent protein kinase IIphosphorylates various types of non-epithelial intermediate filamentproteins. Biochem. Biophys. Res. Commun., 169, 896–904.
25. Sihag, R.K. and Nixons, R.A. (1991) Identification of Ser-55 as a major
protein kinase A phosphorylation site on the 70-kDa subunit ofneurofilaments. Early turnover during axonal transport. J. Chem. Biol.,
266, 18861–18867.
26. Hisanaga, S., Matsuoka, Y., Nishizawa, K., Saito, T., Inagaki, M. and
Hirokawa, N. (1994) Phosphorylation of native and reassembledneurofilaments composed of NF-L, NF-M, and NF-H by the catalytic
subunit of cAMP-dependent protein kinase. Mol. Biol. Cell, 5, 161–172.
27. Nakamura, Y., Hashimoto, R., Kashiwagi, Y., Aimoto, S., Fukusho, E.,
Matsumoto, N., Kudo, T. and Takeda, M. (2000) Major phosphorylationsite (Ser55) of neurofilament L by cyclic AMP-dependent protein kinase
in rat primary neuronal culture. J. Neurochem., 74, 949–959.
28. Shaw, G. (1998) Neurofilament. Springer-Verlag, Berlin, Germany.
29. Hisanaga, S. and Hirokawa, N. (1990) Dephosphorylation-induced
interactions of neurofilaments with microtubules. J. Biol. Chem., 265,21852–21858.
30. Saito, T., Ishiguro, K., Uchida, T., Miyamoto, E., Kishimoto, T. andHisanaga, S. (1995) In situ dephosphorylation of tau by proteinphosphatase 2A and 2B in fetal rat primary cultured neurons. FEBS Lett.,
376, 238–242.
31. Gotow, T., Tanaka, T., Nakamura, Y. and Takeda, M. (1994)Dephosphorylation of the largest neurofilament subunit protein influencesthe structure of crossbridges in reassembled neurofilaments. J. Cell Sci.,
107, 1949–1957.
32. Sasaki, T., Taoka, M., Ishiguro, K., Uchida, A., Saito, T., Isobe, T. andHisanaga, S. (2002) In vivo and in vitro phosphorylation at Ser-493 in theglutamate (E)-segment of neurofilament-H subunit by glycogen synthase
kinase 3b. J. Biol. Chem., 277, 36032–36039.
33. Laemmli, U.K. (1970) Cleavage of structural proteins during the assemblyof the head of bacteriophage T4. Nature, 227, 680–685.
952 Human Molecular Genetics, 2006, Vol. 15, No. 6
by guest on March 20, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from
Related Documents











