39 2 PHARMACOVIGILANCE Médicaments génériques et médicaments originaux Par Jacinthe Leclerc, inf., M.Sc., Ph.D.(Pharm.)(c.), Claudia Blais, Ph.D., Line Guénette, B.Pharm., Ph.D., et Paul Poirier, M.D., Ph.D., FAHA, FACC, FRCPC, FCCS Faire la différence OBJECTIFS PÉDAGOGIQUES Quelles sont les différences entre médicaments originaux et médicaments génériques. Qu’en est-il de leur biodisponibilité? Sont-ils vraiment bioéquivalents? Cet article contribue à améliorer la pharmacovigilance infirmière en comparant les processus d’homologation des médicaments originaux et génériques tout en expliquant les concepts de base de la pharmacocinétique. MISE EN SITUATION Depuis une dizaine d’années, M. Tremblay, 75 ans, souffre d’hypertension artérielle (> 140/90 mmHg) contrôlée grâce à un antihypertenseur de marque originale. Lors de son dernier renouvellement, il remarque que ses pilules ne sont plus de la même couleur. Le pharmacien lui explique qu’il s’agit d’un médicament générique moins dispendieux qui contient les mêmes ingrédients. Quelques jours plus tard, M. Tremblay a un étourdissement et chute. Infirmière assignée au triage à l’urgence, vous le recevez et procédez à son évaluation. Il associe ses symptômes au récent remplacement de ses pilules par une version générique. Il vous demande si cela est possible. Qu’en pensez-vous? Les médicaments génériques sont-ils réellement identiques aux originaux? Après l’évaluation de M. Tremblay, il semble que sa chute ait possiblement été causée par une hypotension artérielle. Vous aviez d’ailleurs noté que sa pression était de 100/80 mmHg au triage. En supposant que le remplacement par la version générique soit en cause, que proposerez-vous? S anté Canada définit un médicament comme une drogue servant à diagnostiquer, traiter, atténuer ou prévenir une maladie (Santé Canada, 2015a). Un médicament peut aussi servir à restaurer, corriger ou modifier des fonctions organiques. Dans le marché canadien comme ailleurs dans le monde, il y a des médicaments dits « originaux » dont le composé actif innovateur a été breveté par son inventeur. Ils se distinguent des autres produits par l’exclusivité relative à leur formule chimique. Il existe également des médicaments dits « génériques » que Santé Canada définit comme « une reproduction d’un médicament d’origine » (Santé Canada, 2012c). Les médicaments génériques peuvent être commercialisés après l’expiration du brevet du produit de référence. Ils contiennent les mêmes ingrédients médicinaux et sont considérés équivalents au plan thérapeutique (Santé Canada, 2012c). Toutefois, ils se distinguent du médicament d’origine entre autres par leur nom, la composition de leurs ingrédients non médicamenteux et l’homologation faite par Santé Canada. Ils sont aussi moins dispendieux. © A-papantoniou / Dreamstime.com Qu’est-ce qui les distingue? Quelles sont les étapes d’homologation des médicaments au Canada? Comment le remplacement d’un médicament d’origine par un générique peut-il modifier la thérapie d’un patient?

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

39
2
PHARMACOVIGILANCE
Médicaments génériques et
médicaments originaux
Par Jacinthe Leclerc, inf., M.Sc., Ph.D.(Pharm.)(c.), Claudia Blais, Ph.D., Line Guénette, B.Pharm., Ph.D., et Paul Poirier, M.D., Ph.D., FAHA, FACC, FRCPC, FCCS
Faire la différence
OBJECTIFS PÉDAGOGIQUESQuelles sont les différences entre médicaments originaux et médicaments génériques. Qu’en est-il de leur biodisponibilité? Sont-ils vraiment bioéquivalents? Cet article contribue à améliorer la pharmacovigilance infirmière en comparant les processus d’homologation des médicaments originaux et génériques tout en expliquant les concepts de base de la pharmacocinétique.
MISE EN SITUATION Depuis une dizaine d’années, M. Tremblay, 75 ans, souffre d’hypertension artérielle (> 140/90 mmHg) contrôlée grâce à un antihypertenseur de marque originale. Lors de son dernier renouvellement, il remarque que ses pilules ne sont plus de la même couleur. Le pharmacien lui explique qu’il s’agit d’un médicament générique moins dispendieux qui contient les mêmes ingrédients. Quelques jours plus tard, M. Tremblay a un étourdissement et chute. Infirmière assignée au triage à l’urgence, vous le recevez et procédez à son évaluation. Il associe ses symptômes au récent remplacement de ses pilules par une version générique. Il vous demande si cela est possible. Qu’en pensez-vous? Les médicaments génériques sont-ils réellement identiques aux originaux?
Après l’évaluation de M. Tremblay, il semble que sa chute ait possiblement été causée par une hypotension artérielle. Vous aviez d’ailleurs noté que sa pression était de 100/80 mmHg au triage. En supposant que le remplacement par la version générique soit en cause, que proposerez-vous?
Santé Canada définit un médicament comme une drogue servant à diagnostiquer, traiter, atténuer ou prévenir une maladie (Santé Canada, 2015a). Un médicament peut aussi servir à restaurer,
corriger ou modifier des fonctions organiques. Dans le marché canadien comme ailleurs dans le monde, il y a des médicaments dits « originaux » dont le composé actif innovateur a été breveté par son inventeur. Ils se distinguent des autres produits par l’exclusivité relative à leur formule chimique. Il existe également des médicaments dits « génériques » que Santé Canada
définit comme « une reproduction d’un médicament d’origine » (Santé Canada, 2012c).
Les médicaments génériques peuvent être commercialisés après l’expiration du brevet du produit de référence. Ils contiennent les mêmes ingrédients médicinaux et sont considérés équivalents au plan thérapeutique (Santé Canada, 2012c). Toutefois, ils se distinguent du médicament d’origine entre autres par leur nom, la composition de leurs ingrédients non médicamenteux et l’homologation faite par Santé Canada. Ils sont aussi moins dispendieux.
© A
-pa
pa
nto
nio
u /
Dre
am
stim
e.c
om
Qu’est-ce qui les distingue? Quelles sont les étapes d’homologation des médicaments au Canada? Comment le remplacement d’un médicament d’origine par un générique peut-il modifier la thérapie d’un patient?
novembre / décembre / 2016 / vol . 13 / n° 5 40
PHARMACOVIGILANCE MÉDICAMENTS GÉNÉRIQUES ET MÉDICAMENTS ORIGINAUX
Dans leur pratique, les infirmières administrent aux patients des médicaments originaux et génériques. Il importe qu’elles puissent les distinguer si des changements cliniques surviennent dans l’état de santé d’un patient après une substitution.
Le nom
Un médicament original, aussi nommé médicament d’origine, de marque, novateur ou de référence, se définit comme un produit breveté et innovateur par sa formulation ou sa classe thérapeutique. Un médicament générique, quant à lui, est un produit pharmaceutique dont l’ingrédient actif est la copie d’une molécule innovatrice. Il est fabriqué par un promoteur après l’expiration du brevet de la molécule originale (Santé Canada, 2015a). Plusieurs versions génériques d’un médicament d’origine peuvent être commercialisées.
Les ouvrages de référence comme le Compendium of Pharmaceuticals and Specialities (CPS), les bases de données des produits pharmaceutiques (Santé Canada, 2015d) ou la liste des médicaments assurés par le régime général d’assurance médicaments de la RAMQ (2016) utilisent plusieurs dénominations.
Le nom commercial : Aussi appelé marque de commerce, nom de marque ou nom de spécialité, le nom commercial est un nom enregistré portant généralement le symbole ® dans la documentation et la publicité. Il doit être approuvé par Santé Canada (Santé Canada, 2015c). Coumadin®, Diovan® ou Cordarone® sont des exemples de marques de commerce choisies par les sociétés les ayant commercialisées. Ces noms sont réservés et ne peuvent être repris par d’autres. Ils peuvent varier d’un pays à l’autre. Par exemple, un biphosphonate intraveineux composé d’acide zolédronique est nommé Aclasta® au Canada et Reclast® aux États-Unis. Enfin, un nom commercial est facile à retenir pour des raisons de marketing.
Le nom générique ou la dénomination commune internationale (DCI) : Attribué par l’Organisation mondiale de la Santé (OMS) à la suite d’une demande déposée par le fabricant, le nom générique ou DCI identifie la ou les substances actives contenues dans le médicament (OMS, 2016). Mentionnons par exemple la warfarine sodique, le valsartan ou l’amiodarone. Reconnue mondialement, la DCI est identique dans tous les pays.
Les fabricants de médicaments génériques intègrent fréquemment la DCI dans le nom commercial de leurs produits (ex. : Apo-valsartan®, Gen-amiodarone®, etc.). Il y a donc une différence entre « nom générique » et « médicament générique ».
Le nom chimique : Cette désignation est plus complexe que celle des noms commerciaux ou génériques. Le nom chimique correspond à la formule chimique de l’ingrédient actif, soit l’ingrédient médicamenteux. Pour le valsartan par exemple, ce nom est (S)-N-valeryl-N-([2’-(1H-tetrazol-5-yl) biphenyl-4-yl] methyl)-valine (Novartis, 2015). On le retrouve dans la monographie du médicament. Il demeure le même tant pour les originaux que pour les génériques.
En résumé, tous les médicaments ont un nom commercial qui leur est propre. Tous ont aussi un nom
Encadré 1 Les médicaments pseudo-génériques
Il arrive que la version générique, y compris ses ingrédients non médicamenteux, soit identique à l’originale, voire produite dans la même usine. On parle alors de « pseudo-générique » ou d’« ultra-génériques » (Hollis, 2003).
Deux exemples de ces médicaments « pseudo-génériques » sont la digoxine en comprimés de la compagnie Pharmascience inc. (pms-DIGOXIN®) et le salbutamol en vaporisation dosée de la compagnie Sanis Health Inc. (Salbutamol HFA®). Leur composition respective est identique à celle de leur produit de référence, soit le Lanoxin® et le Ventolin®. Selon leurs monographies spécifiques, seul le nom de la compagnie apparaissant sur le comprimé est modifié pour désigner celle qui les commercialise (Novartis, 2013; 2015; Sandoz Canada, 2015; 2016b). Les emballages peuvent toutefois différer et bien sûr, le coût.
Tableau 1 Différences et similarités entre les médicaments génériques et originaux
Nom
Homologation
Coût
Obligations de pharmaco- vigilance
Différences
Nom commercial
Ingrédients inactifs (mais pourraient être identiques si médicament est « pseudo-générique »)
Étude de biodisponibilité comparative (sauf si « pseudo-générique »)
Générique généralement trois fois plus économique
Similarités
Dénomination commune internationale (DCI)
Nom chimique
Ingrédient actif : Formule chimique et structure moléculaire. Toutefois, le processus de synthèse peut varier selon le fabricant.
Nécessité de rapporter à Santé Canada les événements indésirables.
Composition

41
chimique (formule) et un nom générique appelé aussi DCI qui indique leur substance active. La DCI apparaît dans le nom commercial de nombreux médicaments génériques, soit telle quelle ou avec un préfixe désignant la compagnie (« Apo » pour Apotex inc., « Mylan » pour Mylan Pharmaceuticals ULC), ou encore avec un suffixe précisant la dose (Simvastatin-10® de la compagnie Pro Doc Limitée).
Les brevets
Selon les ententes internationales, le droit de propriété intellectuelle protège toute nouvelle invention et accorde à ses créateurs un pouvoir exclusif de fabrication et de commercialisation dans le pays où un brevet a été délivré, et ce, pour une durée de 20 ans à partir de la date de demande du brevet (Bélanger, 2005; OPIC, 2013).
Tous les médicaments contiennent au moins un ingré-dient actif capable de provoquer une action pharma-cologique dans le but de révéler un diagnostic, de guérir, d’atténuer, de traiter ou de prévenir une maladie chez l’humain ou chez l’animal (Santé Canada, 2015c). Aus-sitôt découvert par un fabricant, l’ingrédient actif d’un médicament original est breveté et ce, bien avant d’être testé sur les humains, donc bien avant sa mise en marché.
Au Canada, les brevets de nouvelles molécules ou composés médicamenteux sont enregistrés au Bureau des médicaments brevetés et de la liaison. Débute ensuite le processus d’homologation par Santé Canada. La démarche peut prendre de huit à dix ans avant que la nouvelle molécule soit approuvée et commercialisée. Durant cette période, le promoteur doit fournir des preuves relativement à son efficacité et à son innocuité. Au moment où la molécule est offerte aux citoyens, il ne reste qu’une douzaine d’années au droit exclusif de commercialisation accordé au développeur.
La composition
Le fabricant de médicament générique, quant à lui, attend que le brevet du médicament original expire avant de pouvoir acheter son ingrédient actif ou encore le reproduire en laboratoire afin de l’inclure à un produit commercialisable qui sera considéré équivalent au médicament original (Santé Canada, 2012d).
Dans ce cas, la formule chimique de l’ingrédient actif doit être en tous points identique à celle du médicament original (Santé Canada, 2012c). Seuls les ingrédients non médicamenteux comme les agents de conservation, les colorants ou les produits de remplissage peuvent différer (Santé Canada, 2012c; CGPA, 2014). Étant considérés inactifs, ces ingrédients sont documentés mais non réglementés (Rowe et al., 2009). Leur concentration et leur nature dans le médicament original et dans le médicament générique peuvent donc varier grandement (voir Tableau 2).
De plus, une formule chimique identique de la substance active ne garantit pas que le processus de synthèse du médicament sera identique. Ce processus est propre à chaque fabricant. Ainsi, la forme finale de l’ingrédient actif, ou de la pilule, peut différer. Par exemple, la solidité d’un comprimé influence sa dissolution et son absorption et peut entraîner des écarts sur le plan pharmacocinétique (Singhal et Curatolo, 2004).
L’homologation
L’homologation se définit notamment comme l’action de déclarer quelque chose, ici un médicament, conforme aux règlements en vigueur.
Le médicament d’origine
Depuis la tragédie liée aux effets tératogènes de la thalidomide au début des années soixante, les autorités réglementaires ont resserré les exigences concernant l’approbation de nouveaux produits pharmaceutiques (Santé Canada, 2007b; L’Encyclopédie canadienne, 2015).
La thalidomide, anciennement prescrite comme antiémétique, a causé de sévères anomalies congénitales à des bébés nés de femmes qui en avaient consommé pendant leur grossesse. Depuis, les sociétés doivent fournir davantage de preuves relativement à l’innocuité et à l’efficacité de leur produit avant de parvenir à l’étape 5 où leur produit est homologué et obtient un numéro d’identification (Règlement sur les aliments et drogues, 2016; Santé Canada, 2009). Bien que ces exigences augmentent les délais et les coûts, elles assurent une meilleure sécurité aux patients (Bélanger, 2005; Santé Canada, 2007b; Chow, 2013).
Le médicament générique
Au Canada, les lignes directrices intitulées Conduite et analyse des études de biodisponibilité comparatives et Normes en matière d’études de biodisponibilité comparatives : Formes pharmaceutiques de médicaments à effet systémique dictent administrativement les preuves d’équivalences biologiques que les promoteurs de médicaments génériques doivent fournir pour recevoir leur homologation (Santé Canada, 2012b; 2012d).
Tableau 2 Comparaison des ingrédients non médicamenteux du DIOVAN® (valsartan original) et du RAN-VALSARTAN®(générique)
DIOVAN®
(original)
1. Colloidal silicone dioxyde
2. Crospovidone
3. Stéarate de magnésium
4. Cellulose microcristalline
5. Méthylcellulose hydroxypropyl
6. Polyéthylène glycol
7. Oxyde de fer noir
8. Oxyde de fer rouge
9. Oxyde de fer jaune
10. Dioxyde de titane
RAN-VALSARTAN®
(générique)
1. Crospovidone
2. Hypromellose
3. Oxyde de fer noir
4. Oxyde de fer rouge
5. Oxyde de fer jaune
6. Stéarate de magnésium
7. Cellulose microcristalline
8. Polyéthylène glycol
9. Amidon prégélatinisé
10. Colloidal silice anhydre
11. Talc
12. Dioxyde de titane
Note : Les produits en gras diffèrent.

novembre / décembre / 2016 / vol . 13 / n° 5 42
PHARMACOVIGILANCE MÉDICAMENTS GÉNÉRIQUES ET MÉDICAMENTS ORIGINAUX
Les étapes : Puisqu’ils arrivent plusieurs années après le produit novateur, les génériques n’ont pas à franchir toutes les étapes d’homologation. Avant d’être commercialisés, ils devront se conformer à l’une des sections de l’étape 2 et aux étapes 3 à 6 (Santé Canada, 2012d).
Aussi, les études cliniques pour un médicament générique sont conduites sur un petit nombre de sujets en bonne santé. Elles s’apparentent à une version allégée des études de Phase I requises pour les homologations de produits novateurs.
Les études in vitro comprennent notamment des profils comparatifs de dissolution. C’est au moment des études in vivo que seront considérés bioéquivalents les produits dont la forme pharmaceutique (ex. : orale) et la quantité d’ingrédients médicinaux (ex. : teneur en mg) sont identiques. Les sociétés devront démontrer que la copie produite est équivalente au médicament original par des études comparatives de biodisponibilité.
Il convient ici de définir certains termes pharmacologiques. La pharmacocinétique se définit comme la façon dont le corps absorbe, distribue, métabolise et excrète les médicaments (Hollinger, 2007). C’est, en d’autres termes, ce que le corps fait au médicament. Ceci est différent de la pharmacodynamie, qui est l’étude de l’action du médicament et indirectement de sa puissance. C’est donc le contraire, soit ce que le médicament fait au corps.
La biodisponibilité : Elle mesure la fraction du médicament atteignant la circulation sanguine sous sa forme initiale, soit le degré de passage et de vitesse, peu importe la voie d’administration (Santé Canada, 2012b; Holford, 2015).
En mesurant la quantité du médicament qui atteint la circulation sanguine et la vitesse à laquelle elle l’atteint, la biodisponibilité évalue la partie « absorption » de la pharmacocinétique. Ces données sont la pierre angulaire des normes encadrant l’équivalence des médicaments génériques avec le produit d’origine.
Des dosages sériques en série de la substance, effectués le plus souvent après l’administration d’une dose unique à jeun, permettent de calculer trois paramètres pharmacocinétiques selon la concentration plasmatique de l’ingrédient actif :
1. l’aire (ou surface) sous la courbe (ASC) : Area under the curve (AUC),
2. la concentration maximale (Cmax), et
3. le temps nécessaire qu’il faudra pour atteindre la concentration maximale (Tmax) (Santé Canada, 2012b).
L’AUCT et la Cmax sont les paramètres qui serviront à déterminer la bioéquivalence entre deux produits (Santé Canada, 2012d). Les valeurs moyennes obtenues avec le produit générique (numérateur) sont comparées à celles du produit original (dénominateur) (Santé Canada, 2012c; ACMTS, 2012), le tout nous fournissant un ratio estimé, nommé aussi moyenne relative.
Encadré 2 Étapes d'homologation d'un produit novateur
Figure 1 Démarche d’homologation des médicaments par Santé Canada
Source : Santé Canada, 2007a (reproduction autorisée).
Pouvoir de réglementation de Santé Canada
1. Le promoteur/créateur procède aux études pré-cliniques. Elles sont le plus souvent conduites in vitro sur des cellules ou des échantillons de tissus ou in vivo sur des animaux dans le but d’obtenir davantage d’information sur le ou les dosages et d’établir un profil d’innocuité du médicament, y compris sa toxicité.
2. Le promoteur procède aux essais cliniques chez les humains :
Phase I. Ils sont conduits pour la première fois auprès d’un petit nombre de sujets en santé dans le but d’évaluer l’innocuité du médicament, de déterminer les intervalles posologiques sécuritaires et de détecter les effets indésirables.
Phase II. Ils sont conduits sur un nombre de sujets (> 100) atteints de la maladie à traiter dans le but d’obtenir des premières données d’efficacité, davantage d’information sur l’innocuité et de déterminer la ou les meilleures doses de traitement.
Phase III. Ils sont conduits généralement sur plus de 1 000 sujets atteints de la maladie à traiter dans le but de confirmer l’efficacité du médicament, de comparer les données à des traitements reconnus et de surveiller les effets indésirables en vue de la commercialisation.
3. Le promoteur dépose son application d’homologation à Santé Canada, y compris la monographie du produit.
4. Les scientifiques de Santé Canada examinent l’application et demandent davantage de données au besoin.
5. Santé Canada accorde l’homologation en émettant l’avis de conformité (AC) et en accordant un numéro d’identification au médicament (Drug Identification Number [DIN]). Le médicament peut être mis en marché.
6. Le public accède au produit selon la province dans laquelle il se trouve.
7. Le promoteur exerce des activités de surveillance, d’inspection et d’enquête tant que le produit demeure sur le marché. Il poursuit des études de Phase IV et des programmes de pharmacovigilance. Les professionnels de la santé et le public peuvent faire des déclarations.
Les modifications après-
commercialisation
Autorisation d’essai clinique
Évaluation de la présentation
Activités d’après-commercialisation
Études précliniques
Les essais cliniques
La présentation réglementaire
du produit
L’évaluation de l’innocuité, de l’efficacité et
de la qualité du médicament
La décision d’autorisation de
mise sur le marché
Accès public
La surveillance, l’inspection et les
enquêtes
43
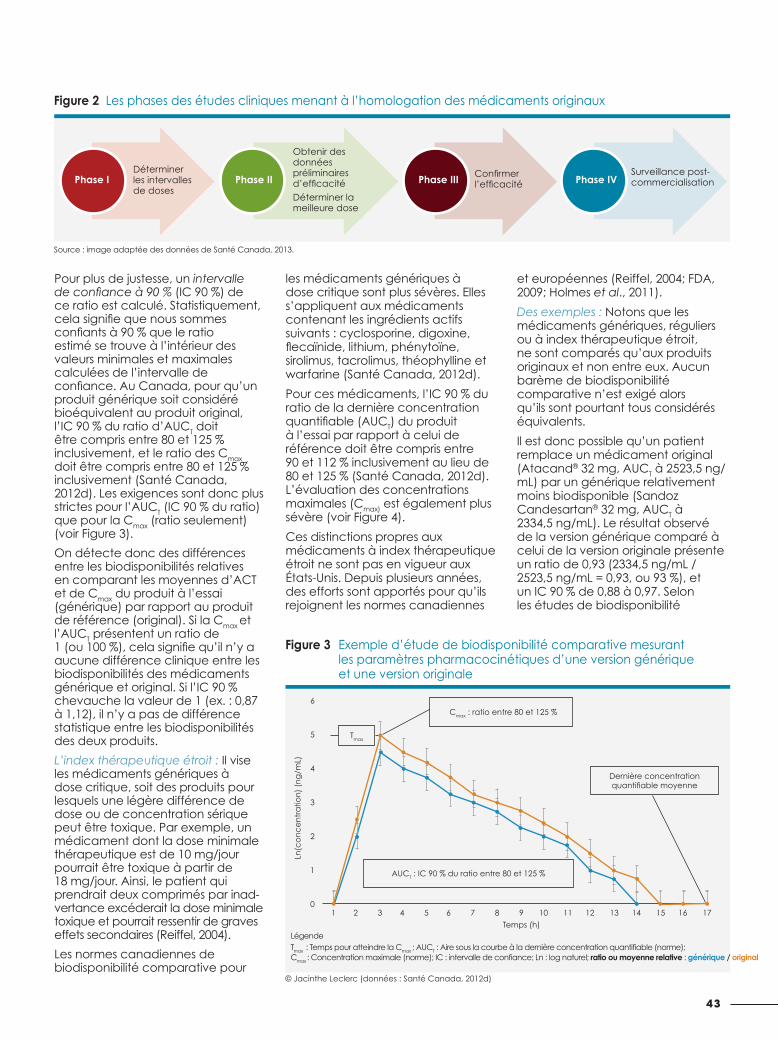
Pour plus de justesse, un intervalle de confiance à 90 % (IC 90 %) de ce ratio est calculé. Statistiquement, cela signifie que nous sommes confiants à 90 % que le ratio estimé se trouve à l’intérieur des valeurs minimales et maximales calculées de l’intervalle de confiance. Au Canada, pour qu’un produit générique soit considéré bioéquivalent au produit original, l’IC 90 % du ratio d’AUCT doit être compris entre 80 et 125 % inclusivement, et le ratio des Cmax doit être compris entre 80 et 125 % inclusivement (Santé Canada, 2012d). Les exigences sont donc plus strictes pour l’AUCT (IC 90 % du ratio) que pour la Cmax (ratio seulement) (voir Figure 3).
On détecte donc des différences entre les biodisponibilités relatives en comparant les moyennes d’ACT et de Cmax du produit à l’essai (générique) par rapport au produit de référence (original). Si la Cmax et l’AUCT présentent un ratio de 1 (ou 100 %), cela signifie qu’il n’y a aucune différence clinique entre les biodisponibilités des médicaments générique et original. Si l’IC 90 % chevauche la valeur de 1 (ex. : 0,87 à 1,12), il n’y a pas de différence statistique entre les biodisponibilités des deux produits.
L’index thérapeutique étroit : Il vise les médicaments génériques à dose critique, soit des produits pour lesquels une légère différence de dose ou de concentration sérique peut être toxique. Par exemple, un médicament dont la dose minimale thérapeutique est de 10 mg/jour pourrait être toxique à partir de 18 mg/jour. Ainsi, le patient qui prendrait deux comprimés par inad-vertance excéderait la dose minimale toxique et pourrait ressentir de graves effets secondaires (Reiffel, 2004).
Les normes canadiennes de biodisponibilité comparative pour
les médicaments génériques à dose critique sont plus sévères. Elles s’appliquent aux médicaments contenant les ingrédients actifs suivants : cyclosporine, digoxine, flecaïnide, lithium, phénytoïne, sirolimus, tacrolimus, théophylline et warfarine (Santé Canada, 2012d).
Pour ces médicaments, l’IC 90 % du ratio de la dernière concentration quantifiable (AUCT) du produit à l’essai par rapport à celui de référence doit être compris entre 90 et 112 % inclusivement au lieu de 80 et 125 % (Santé Canada, 2012d). L’évaluation des concentrations maximales (Cmax) est également plus sévère (voir Figure 4).
Ces distinctions propres aux médicaments à index thérapeutique étroit ne sont pas en vigueur aux États-Unis. Depuis plusieurs années, des efforts sont apportés pour qu’ils rejoignent les normes canadiennes
et européennes (Reiffel, 2004; FDA, 2009; Holmes et al., 2011).
Des exemples : Notons que les médicaments génériques, réguliers ou à index thérapeutique étroit, ne sont comparés qu’aux produits originaux et non entre eux. Aucun barème de biodisponibilité comparative n’est exigé alors qu’ils sont pourtant tous considérés équivalents.
Il est donc possible qu’un patient remplace un médicament original (Atacand® 32 mg, AUCT à 2523,5 ng/mL) par un générique relativement moins biodisponible (Sandoz Candesartan® 32 mg, AUCT à 2334,5 ng/mL). Le résultat observé de la version générique comparé à celui de la version originale présente un ratio de 0,93 (2334,5 ng/mL / 2523,5 ng/mL = 0,93, ou 93 %), et un IC 90 % de 0,88 à 0,97. Selon les études de biodisponibilité
Figure 3 Exemple d’étude de biodisponibilité comparative mesurant les paramètres pharmacocinétiques d’une version générique et une version originale
Figure 2 Les phases des études cliniques menant à l’homologation des médicaments originaux
Source : image adaptée des données de Santé Canada, 2013.
© Jacinthe Leclerc (données : Santé Canada, 2012d)
LégendeTmax : Temps pour atteindre la Cmax ; AUCT : Aire sous la courbe à la dernière concentration quantifiable (norme);Cmax : Concentration maximale (norme); IC : intervalle de confiance; Ln : log naturel; ratio ou moyenne relative : générique / original
Phase I Phase II Phase III Phase IVDéterminer les intervalles de doses
Obtenir des données préliminaires d’efficacitéDéterminer la meilleure dose
Confirmer l’efficacité
Surveillance post-commercialisation
Tmax
Cmax : ratio entre 80 et 125 %
Dernière concentration quantifiable moyenne
AUCT : IC 90 % du ratio entre 80 et 125 %
Ln(c
on
ce
ntr
atio
n)
(ng
/mL)
Temps (h)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
6
5
4
3
2
1
0

novembre / décembre / 2016 / vol . 13 / n° 5 44
PHARMACOVIGILANCE MÉDICAMENTS GÉNÉRIQUES ET MÉDICAMENTS ORIGINAUX
comparative, cela signifie que la version de Sandoz Candesartan est cliniquement 7 % moins biodisponible que la version originale. Par ailleurs, le fabricant ayant conduit l’étude est confiant à 90 % que ce ratio estimé se situe entre les bornes de 88 % et 97 %, ce qui veut dire que le générique est également statistiquement plus faible que l’original (Sandoz Canada, 2016a). Théoriquement, cela pourrait en réduire la puissance.
L’inverse est également possible si la concentration maximale du médicament générique est supérieure à celle de l’original, par exemple la Cmax du générique Ran-valsartan® (4417,0 ng/mL) comparativement à celle de l’original Diovan® (3966,8 ng/mL) [ratio de Cmax de 1,11, IC 90 % : 0,99 1,25]. Selon l’étude de biodisponibilité comparative, cela signifie que la version de Ran-valsartan est 11 % plus biodisponible que la version originale. Le fabricant ayant conduit l’étude est confiant à 90 % que ce ratio estimé se situe entre les bornes de 99 % et 125 % (Sandoz Canada, 2016a). Théoriquement, cela pourrait augmenter la puissance.
Il peut aussi arriver qu’un patient passe d’un médicament original à une version générique plus comparable au plan de la biodisponibilité. Par exemple, la Cmax du générique Mylan-valsartan® (6,08 µg/mL) est similaire à celle du valsartan original (Diovan®) (5,79 µg/mL), le ratio étant de 1,05, donc cliniquement plus fort de 5 %, mais statistiquement similaire car il chevauche le 1 (IC 90 % : 0,93-1,18) (Mylan Pharmaceuticals, 2015). Ceci ne devrait théoriquement pas avoir d’impact clinique sur le patient.
Commentaires : Les études de biodisponibilité comparative sont menées sur un petit nombre de sujets (n < 50), ce qui rend les valeurs relatives de biodisponibilité plus sensibles aux écarts. C’est pourquoi les intervalles de confiance peuvent être aussi étendus. De plus, les participants doivent être en bonne santé et âgés de 18 à 55 ans. Dans le cas où le même fabricant propose un médicament offert en plusieurs dosages, seul celui avec la concentration la plus élevée de l’ingrédient actif sera généralement testé (Holmes et al., 2011).
D’autres exigences s’appliquent aux médicaments à libération prolongée ou dont la demi-vie est supérieure à 24 heures. Par ailleurs, aux exigences d’AUCT et de Cmax s’ajoutent celles demandant que les formes pharmaceutiques et les teneurs en ingrédient médicinal soient absolument identiques (Santé Canada, 2012d).
Les coûts
Une fois commercialisé, le médicament générique devient disponible dans les pharmacies et les établissements de santé. Il y a quelques années, ils représentaient de 43 à 67 % de l’ensemble du marché canadien (ACMG, 2016b; BCC, 2007). Cette proportion atteint près de 100 % dans les hôpitaux.
Parce que le générique est moins cher, il est économiquement avantageux pour le patient de le substituer à son médicament d’origine (Gumbs et al., 2007; RAMQ, 2016). Le prix des génériques est en effet un atout considérable puisqu’il est généralement trois fois moins dispendieux que l’original. Il n’est donc pas surprenant que les assureurs privés et publics, tout comme le citoyen, choisissent une option thérapeutique équivalente offerte à moindre coût (voir Figure 5).
Qu’est-ce qui explique une telle différence? Tout d’abord, le fabricant n’a pas à investir pour découvrir un nouvel ingrédient actif (ACMG, 2010). Ses investissements initiaux et le coût des études pré-commercialisation sont moins élevés. Et ces sociétés ne prennent pas le risque que le résultat de leur recherche ne parvienne pas à obtenir une homologation, ce qui arrive une fois sur dix aux fabricants de médicaments originaux (Bélanger, 2005).
Enfin, les développeurs doivent poursuivre des études de Phase IV et instaurer des programmes de pharmacovigilance pendant la commercialisation de leur médicament d’origine. Peu de sociétés génériques dépensent pour de tels programmes.
Pharmacovigilance
La surveillance des événements indésirables à la suite de l’administration de médicaments commercialisés est un enjeu de santé publique. La pharmacovigilance est une science visant à identifier les risques potentiels de l’utilisation des médicaments dans un contexte de vie réelle, hors d’études cliniques encadrées, et à prévenir les effets indésirables aux patients actuels ou futurs utilisateurs des médicaments (Santé Canada, 2016).
Dans le cadre de son Programme Canada Vigilance, Santé Canada maintient MedEffetmc, une banque de données exhaustive qui offre un accès centralisé aux informations sur les produits de santé. Les professionnels ont la responsabilité de rapporter les effets indésirables dont ils ont connaissance selon des normes précises, y compris l’identité du patient, le nom du déclarant (qu’il soit ou non professionnel), l’effet indésirable, les conditions et évidemment, le produit. Les fabricants de médicaments originaux doivent déclarer les effets indésirables de leur produit selon les normes de « bonnes pratiques cliniques » encadrant les études cliniques pré- et post-commercialisation (Santé Canada, 2012a).
Quant aux médicaments génériques, les effets indésirables relatifs à leurs produits sont
Figure 4 Normes canadiennes de biodisponibilités équivalentes pour les médicaments génériques
Aire sous la courbe (AUCT)
n Médicament générique régulier : IC 90 % de la moyenne relative compris entre 80 et 125 %
n Médicament générique à dose critique : IC 90 % de la moyenne relative compris entre 90 et 112 %
Concentration maximale (Cmax)
n Médicament générique régulier : moyenne relative des Cmax comprise entre 80 et 125 %
n Médicament générique à dose critique : IC 90 % de la moyenne relative des Cmax compris entre 80 et 125 %
IC : Intervalle de confiance
Produit générique (testé) Produit original (référence)
Source : adapté de Santé Canada, 2012d.
= Moyenne relative (ratio)

45
vraisemblablement sous-rapportés (ISMP, 2015). D’ailleurs, il est difficile de les identifier dans MedEffet parce qu’ils sont peu souvent déclarés ou encore, qu’ils l’ont été sous le nom de leur DCI et non de leur nom commercial. Ainsi, davantage de vigilance relativement au nom commercial du médicament, qu’il soit original ou générique, améliorerait la déclaration d’événements indésirables et rendrait les soins pharmaceutiques plus sécuritaires. De plus, si un événement indésirable est possiblement associé à la substitution médicamenteuse, le médecin et le pharmacien du patient doivent en être informés.
Les sept BONS
Lorsque l’infirmière se prépare à administrer des médicaments, la démarche standardisée nommée « Les sept Bons » est recommandée (Fortin, 2010) (voir Encadré 4).
À l’étape du BON médicament, elle s’assure du nom du produit et peut voir s’il est d’origine ou générique. Elle vérifie le nom inscrit sur la feuille d’administration des médicaments (FADM) et sur l’emballage où les noms commerciaux et génériques (DCI) devraient obligatoirement apparaître. En milieu hospitalier et en pharmacie communautaire, le nom commercial du produit original est inscrit entre parenthèses lorsqu’il a été remplacé par un médicament générique afin que les cliniciens et les patients le reconnaissent plus facilement.
Pour confirmer s’il s’agit d’un produit original ou générique, l’infirmière peut consulter le CPS ou encore la base de données sur les produits pharmaceutiques (BDPP) de Santé Canada où les fabricants sont clairement identifiés. Cette étape est particulièrement importante si un effet indésirable survient puisqu’il devra être déclaré à
MedEffet. Les données des études de biodisponibilités comparatives sont également publiées dans les monographies des médicaments génériques. L’infirmière peut donc vérifier si le médicament du patient est plus ou moins biodisponible comparativement à l’original, surtout dans un contexte de substitution.
Dans leur pratique quotidienne, les infirmières administrent des médicaments de toutes sortes et plusieurs d’entre elles sont autorisées à les prescrire dans certaines situations cliniques ou selon une ordonnance collective. Les patients les questionnent fréquemment sur l’efficacité des médicaments et leurs effets secondaires. Bien que le médicament générique offre des avantages indéniables sur le plan des coûts, il importe de connaître les différences et les similitudes entre un médicament d’origine et un médicament générique afin de pouvoir répondre au patient. La réponse la plus courante consiste à dire que l’unique différence est le prix (ACMG, 2016a). Pourtant, quelques études récentes qui
comparent les effets indésirables et l’efficacité remettent en cause leur équivalence clinique, notamment dans le traitement de l’ostéoporose (Van den Bergh et al., 2013; Ringe et Moller, 2009). Des études sont en cours au Québec, particulièrement en cardiologie.
Il convient donc de porter une attention particulière au statut générique ou original d’un médicament et de déclarer tout événement indésirable à Santé Canada. Ainsi, dans le cas de M. Tremblay présenté en introduction, l’infirmière devra d’abord informer son médecin et son pharmacien que son nouvel antihypertenseur générique est potentiellement associé à son hypotension artérielle. Possiblement qu’il conviendra de rétablir la thérapie à la version originale. Ensuite, même si le lien de causalité n’est pas clairement établi, l’hypotension artérielle (événement indésirable) doit être rapportée à Santé Canada via la base de données MedEffet.
Figure 5 Exemple d’un contexte de remplacement par le médicament générique en pharmacie
Le patient malade désire consulter son médecin
À la maison
Dans le bureau du médecin
« Nous avons le médicament générique Apo-valsartan® que je peux vous proposer. D’ailleurs, la RAMQ ne
rembourse que le prix du générique. Vous devrez débourser un supplément de 7,23 $ si vous désirez tout de même
le Diovan®. »
À la pharmacie
Encadré 4 Les sept Bons
1. Bon client2. Bon médicament3. Bon dosage4. Bonne voie d’administration5. Bon moment6. Bonne documentation 7. Bonne surveillance des effets attendus/indésirables
Source : Fortin, 2010.
© Jacinthe Leclerc
Médecin : « Je vous prescris Diovan®
80 mg/comprimé, 1 comprimé une fois par jour pour 30 jours, dans le but de traiter votre hypertension. Nous nous reverrons dans un mois pour
ré-évaluer votre condition. »

novembre / décembre / 2016 / vol . 13 / n° 5 46
PHARMACOVIGILANCE MÉDICAMENTS GÉNÉRIQUES ET MÉDICAMENTS ORIGINAUX
BibliographieAgence canadienne des médicaments et des technologies de la santé (ACMTS). « Biodisponibilité et bioéquivalence », Ottawa, ACMTS, 2012. [En ligne : https://www.cadth.ca/sites/default/files/pdf/What_Are_Bioavailability_and_Bioequivalence_f.pdf] (Page consultée le 29 août 2016.)Association canadienne du médicament générique (ACMG). « Comparer les médica-ments génériques et les médicaments de marque », 2016a. [En ligne : www.canadian-generics.ca/fr/resources/if_brandvsgeneric.asp] (Page consultée le 4 août 2016.)Association canadienne du médicament générique (ACMG). « Le marché des mé-dicaments d’ordonnance du Québec 2015 », 2016b. [En ligne : www.canadiangener-ics.ca/fr/advocacy/quebec_market_share.asp] (Page consultée le 4 août 2016.)Association canadienne du médicament générique (ACMG). Développement des médicaments génériques, 2010, 10 p. [En ligne : www.canadiangenerics.ca/fr/re-sources/docs/GenericDrugDevelFr10-03-25.pdf] (Page consultée le 4 août 2016.)Bélanger, G. L’économique de la santé et l’État providence, Montréal, Éditions Varia, 2005, 280 p.Bureau de la concurrence du Canada (BCC). « Étude du secteur canadien des médicaments génériques », octobre 2007. [En ligne : www.bureaudelaconcurrence.gc.ca/eic/site/cb-bc.nsf/fra/02495.html] (Page consultée le 4 août 2016.)Canadian Generic Pharmaceutical Association (CGPA). « Generic drugs versus brand-name – what’s the difference? », 2014. [En ligne : www.canadiangenerics.ca/en/resources/docs/GenericsVSBrandWhat’stheDifference_Eng_2016.pdf]Chow, S.C. Biosimilars design and analysis of follow-on biologics, Londres (UK), Chapman and Hall, 2013, 444 p.Food and Drug Administration (FDA). « Therapeutic equivalence of generic drugs – response to national association of boards of pharmacy », 21 avril 2009. [En ligne : www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandAp-proved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/ucm073224.htm] (Page consultée le 31 août 2016.)Fortin, M. Math et méd : guide pour une administration sécuritaire des médicaments, Montréal, Chenelière Éducation, 2010, 272 p.Gumbs, P.D., W.M. Verschuren, P.C. Souverein, A.K. Mantel-Teeuwisse, G.A. de Wit, A. de Boer et al. « Society already achieves economic benefits from generic substitution but fails to do the same for therapeutic substitution », British Journal of Clinical Pharmacology, vol. 64, n° 5, nov. 2007, p. 680-685.Holford, N.H.G. « Pharmacokinetics and pharmacodynamics: rational dosing and the time course of drug action », in B.G. Katzung et A.J. Trevor (ss la dir. de) Basic and Clinical Pharmacology (12e éd.), New York (NY), McGraw Hill, 2015. [En ligne : http://accessmedicine.mhmedical.com/book.aspx?bookid=388] (Page consultée le 31 août 2016.)Hollinger, M.A. « Chapter 1 : Introduction », in M.A. Hollinger (ss la dir. de), Introduction to Pharmacology (3e éd.), Boca Raton (FL), CRC Press, 2007.Hollis, A. « The anti-competitive effects of brand-controlled "pseudo-generics" in the Canadian pharmaceutical market », Canadian Public Policy – Analyse de Politiques, vol. 29, n° 1, mars 2003, p. 21-31.Holmes, D.R., J.A. Becker, C.B. Granger, M.C. Limacher, R.L. Page et C. Sila. « ACCF/AHA 2011 health policy statement on therapeutic interchange and substitution: a re-port of the American College of Cardiology Foundation Clinical Quality Committee », Circulation, vol. 124, n° 11, 13 sept. 2011, p. 1290-1310.Institute for Safe Medication Practices (ISMP). « A critique of a key drug safety report-ing system », Quarter Watch–Monitoring FDA MedWatch Reports, 28 janv. 2015 (Data from 2014 Quarter 1 / 2013 Quarter 4), 22 p. [En ligne : https://www.ismp.org/quarter-
Cet article est accompagné d’un post-test en ligne donnant droit à des heures admissibles dans la catégorie formation accréditée. Il sera en ligne sur la plateforme de téléapprentissage Mistral.
watch/pdfs/2014Q1.pdf] (Page consultée le 31 août 2016.)L’Encyclopédie canadienne. « Industrie pharmaceutique », 3 avril 2015. [En ligne : www.encyclopediecanadienne.ca/fr/article/industrie-pharmaceutique-1/] (Page consultée le 31 août 2016.)Mylan Pharmaceuticals. Monographie de produit – Mylan-Valsartan (valsartan), 31 juillet 2015, 40 p. [En ligne : http://www.webprod5.hc-sc.gc.ca/dpd-bdpp/item-iteme.do?pm-mp=00031690] (Page consultée le 29 août 2016.)Novartis. Monographie de produit : Lopresor (tartrate de métoprolol), 9 janvier 2013, 43 p. [En ligne : https://www.novartis.ca/sites/www.novartis.ca/files/lopresor_scrip_f.pdf] (Page consultée le 31 août 2016.)Novartis. Monographie de produit : Lopresor (tartrate de métoprolol), 9 janvier 2013, 43 p. [En ligne : https://www.novartis.ca/sites/www.novartis.ca/files/lopresor_scrip_f.pdf] (Page consultée le 31 août 2016.)Office de la propriété intellectuelle du Canada (OPIC). « Base de données sur les brevets canadiens », 2013. [En ligne : www.ic.gc.ca/opic-cipo/cpd/fra/introduction.html] (Page consultée le 31 août 2016.)Organisation mondiale de la Santé (OMS). « Les dénominations communes inter-nationales (DCI) », 2016. [En ligne : www.who.int/medicines/technical_briefing/tbs/inn_rdg_prs/fr/] (Page consultée le 31 août 2016.)Ranbaxy Pharmaceuticals Canada. Monographie de produit Ran-valsartan (valsartan), Mississauga (ON), 22 juillet 2015 , 41 p. [En ligne : http://webprod5.hc-sc.gc.ca/dpd-bdpp/item-iteme.do?pm-mp=00031725] (Page consultée le 29 août 2016.)Régie de l’assurance maladie du Québec (RAMQ). « Liste des médicaments », 2016. [En ligne : www.ramq.gouv.qc.ca/fr/regie/publications-legales/Pages/liste-medica-ments.aspx] (Page consultée le 31 août 2016.)Règlement sur certaines activités professionnelles qui peuvent être exercées par une infirmière et un infirmier, c. M-9, r. 12.001, 1er juin 2016. [En ligne : http://legisquebec.gouv.qc.ca/fr/ShowDoc/cr/M-9,%20r.%2012.001/]Règlement sur les aliments et drogues, C.R.C., c. 870, 26 août 2016. [En ligne : http://laws-lois.justice.gc.ca/fra/reglements/C.R.C.,_ch._870/] (Page consultée le 15 août 2016.)Reiffel, J.A. « Formulation substitution: a frequently overlooked variable in cardiovas-cular drug management », Progress in Cardiovascular Diseases, vol. 47, n° 1, juill./août 2004, p. 3-10.Ringe, J.D. et G. Moller. « Differences in persistence, safety and efficacy of generic and original branded once weekly bisphosphonates in patients with postmenopausal osteoporosis: 1-year results of a retrospective patient chart review analysis », Rheumatology International, vol. 30, n° 2, déc. 2009, p. 213-221.Rowe, R.C., P.J. Sheskey et M.E. Quinn. Handbook of Pharmaceutical Excipients (6e éd.), Londres (UK), Pharmaceutical Press, 2009, 944 p.Sandoz Canada. Monographie de produit Sandoz – Candesartan (candésartan), 4 mai 2016a, 40 p. [En ligne : www.sandoz.ca/cs/groups/public/@sge_ca/documents/document/n_prod_1782725.pdf] (Page consultée le 31 août 2016.).Sandoz Canada. Monographie de produit Sandoz – Valsartan (valsartan), 13 mai 2016b, 40 p. [En ligne : www.sandoz.ca/cs/groups/public/@sge_ca_sandoz/documents/document/n_prod_1726925.pdf] (Page consultée le 31 août 2016)Sandoz Canada. Monographie de produit : Metoprolol (métoprolol), 28 avril 2015, 29 p. [En ligne : www.sandoz.ca/cs/groups/public/@sge_ca/documents/document/n_prod_1301726.pdf] (Page consultée le 31 août 2016.)Santé Canada. « Programme Canada Vigilance », 19 février 2016. [En ligne : www. hc-sc.gc.ca/dhp-mps/medeff/vigilance-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Ligne directrice : Règlement sur les médicaments brevetés », 3 février 2015a. [En ligne : www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/patmedbrev/pmreg3_mbreg3-fra.php#definition] (Page consultée le 31 août 2016.)Santé Canada. « Médicaments et produits de santé. Base de données sur les produits pharmaceutiques (BDPP) », 18 juin 2015b. [En ligne : www.hc-sc.gc.ca/dhp-mps/prod-pharma/databasdon/index-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Médicaments et produits de santé. Base de données sur les produits pharmaceutiques (BDPP). Terminologie », 18 juin 2015c. [En ligne : www.hc-sc.gc.ca/dhp-mps/prodpharma/databasdon/terminolog-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Recherche de produits pharmaceutiques en ligne », 17 juillet 2015d. [En ligne : http://webprod5.hc-sc.gc.ca/dpd-bdpp/index-fra.jsp] (Page consultée le 31 août 2016.)Santé Canada. « Les essais cliniques et l’innocuité des médicaments », 29 mai 2013. [En ligne : www.hc-sc.gc.ca/hl-vs/iyh-vsv/med/clinical_trials-essais_cliniques-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Bonnes pratiques cliniques », 28 mars 2012a. [En ligne : www.hc-sc.gc.ca/dhp-mps/compli-conform/clini-pract-prat/index-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Conduite et analyse des études de biodisponibilité comparatives », 6 juin 2012b. [En ligne : www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/bio/gd_cbs_ebc_ld-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Innocuité et efficacité des médicaments génériques », 23 avril 2012c. [En ligne : www.hc-sc.gc.ca/hl-vs/iyh-vsv/med/med-gen-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Ligne directrice – Normes en matière d’études de biodisponibilité comparatives : Formes pharmaceutiques de médicaments à effets systémiques », 22 mai 2012d. [En ligne : www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-fra.php] (Page consultée le 29 août 2016.)Santé Canada. « Numéro d’identification d’un médicament (DIN) », 5 juin 2009. [En ligne : www.hc-sc.gc.ca/dhp-mps/prodpharma/activit/fs-fi/dinfs_fd-fra.php] (Page consultée le 31 août 2016.)Santé Canada. MedEffet Canada. La déclaration des effets indésirables et les renseignements concernant l’innocuité des produits de santé. Guide à l’intention des professionnels de la santé, 2008, 12 p. [En ligne : http://rqasf.qc.ca/files/Guide%20des%20professionnelles%20-%20Effets%20secondaires.pdf]Santé Canada. « Démarche d’homologation des médicaments », 24 avril 2007a. [En ligne : www.hc-sc.gc.ca/dhp-mps/homologation-licensing/system/map-carte/index-fra.php] (Page consultée le 31 août 2016.)Santé Canada. « Bref historique de la réglementation des médicaments au Canada », 11 avril 2007b. [En ligne : www.hc-sc.gc.ca/dhp-mps/homologation-licensing/info-renseign/hist-fra.php] (Page consultée le 31 août 2016.)Singhal, D. et W. Curatolo. « Drug polymorphism and dosage form design: a practical perspective », Advanced Drug Delivery Reviews, vol. 56, n° 3, 23 févr. 2004, p. 335-347.Van den Bergh, J.P., M.E. Bouts, E. van der Veer, R.Y. van der Velde, M.J. Janssen, P.P. Geusens et al. « Comparing tolerability and efficacy of generic versus brand alendronate: a randomized clinical study in postmenopausal women with a recent fracture », Public Library of Science One, vol. 8, n° 10, oct. 2013, p. e78153.
Les auteurs
Jacinthe Leclerc est candidate au doctorat en sciences pharmaceutiques – pharmaco-épidémiologie de la Faculté de pharmacie de l’Université Laval. Ses études sont conduites à l’Institut national de santé publique du Québec.
Elle est professeure au Département des sciences infirmières de l’Université du Québec à Trois-Rivières.
Claudia Blais est chercheuse à l'Institut national de santé publique du Québec et professeure associée à la Faculté de pharmacie de l’Université Laval.
Line Guénette est pharmacienne. Elle est professeure à la Faculté de pharmacie de l’Université Laval et chercheuse au Centre de recherche du CHU de Québec, Axe Santé des populations et pratiques optimales en santé.
Dr Paul Poirier est professeur à la Faculté de pharmacie de l’Université Laval ainsi que cardiologue et chercheur à l’Institut universitaire de cardiologie et de pneumologie de Québec.
47
30 janvier 2017
1er mars 2017
10 janvier 2017
FORMATION COMPLÈTE160 heures
Reconnue par l’AIISPQ
HYBRIDE
Enseignement en groupe restreint de 12 à 15personnes permet un meilleur suivi.
SURPIED est un membre institutionnelde la SOFEDUC. Par cette accréditation, les formations de SURPiED donnent droit à des Unités d’Éducation Continue (UEC)
HEBDOMADAIRE
INTENSIF
nboels
Rectangle
Related Documents











