Antiviral Therapies and Prospects for a Cure of Chronic Hepatitis B Fabien Zoulim and David Durantel INSERM U1052, Cancer Research Center of, University of Lyon, Hospices Civils de Lyon, Lyon, France Correspondence: [email protected] Current therapies of chronic hepatitis B remain limited to either pegylated interferon-a (Peg- IFN-a), or one of the five approved nucleoside analog (NA) treatments. Although viral sup- pression can be achieved in the majority of patients with high-barrier-to-resistance new- generation NAs (i.e., entecavir and tenofovir), HBsAg loss is achieved in only 10% of patients with both classes of drugs aftera follow-up of 5 years. Attempts to improve the response by administering two different NAs or a combination of NA and Peg-IFN-a have been unsuc- cessful. Therefore, there is a renewed interest to investigate a numberof steps in the HBV replication cycle and specific virus–host cell interactions as potential targets for new anti- virals. Novel targets and compounds could readily be evaluated using both relevant in vitro and newly developed in vivo models of HBVinfection. The addition of one or several new drugs to current regimens should offer the prospect of markedly improving the response to therapy, thus reducing the burden of drug resistance, as well as the incidence of cirrhosis and hepatocellular carcinoma. BACKGROUND—BASIS OF ANTI-HBV THERAPY E ffective therapies have been developed for chronic hepatitis B (CHB) infection. Hence, interferon-a (IFN-a) and its pegylated form (Peg-IFN-a), and five other drugs that belong to the class of nucleos(t)ide analogs (NAs), have been approved for this indication in most parts of the world (EASL 2012; Lampertico and Liaw 2012; Scaglione and Lok 2012; Jordheim et al. 2013; Buti 2014; Kao 2014). IFN-a is an im- mune modulator that induces, in a nonspecific manner, the expression of interferon-stimulated genes (ISGs) encoding intracellular or secreted proteins with direct or indirect antiviral proper- ties in both infected and noninfected cells, and promotes the differentiation/activation of im- mune cells (Samuel 2001; Sadler and Williams 2008). In the HBV setting, the IFN-a antiviral activity results from a complex mode of action including the activation of natural killer (NK)/ NKT cells, inhibition of viral genome transcrip- tion, destabilization of viral nucleocapsid, but also, as recently suggested, degradation of co- valently closed circular DNA (cccDNA) Q1 via the activation of APOBEC3A in infected cells (Micco et al. 2013; Thimme and Dandri 2013; Lucifora et al. 2014). NAs directly inhibit the reverse transcriptase activity of the HBV polymerase. The approved NAs include lamivudine (LMV), a deoxycyti- Editors: Christoph Seeger and Stephen Locarnini Additional Perspectives on Hepatitis B and Delta Viruses available at www.perspectivesinmedicine.org Copyright # 2014 Cold Spring Harbor Laboratory Press; all rights reserved. Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 1

Zoulim article cshperspectmed-hep-a021501-proof seen d dfz copie
Jul 14, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Antiviral Therapies and Prospects for a Cureof Chronic Hepatitis B
Fabien Zoulim and David Durantel
INSERM U1052, Cancer Research Center of, University of Lyon, Hospices Civils de Lyon, Lyon, France
Correspondence: [email protected]
Current therapies of chronic hepatitis B remain limited to either pegylated interferon-a (Peg-IFN-a), or one of the five approved nucleoside analog (NA) treatments. Although viral sup-pression can be achieved in the majority of patients with high-barrier-to-resistance new-generation NAs (i.e., entecavir and tenofovir), HBsAg loss is achieved in only 10% of patientswith both classes of drugs after a follow-up of 5 years. Attempts to improve the response byadministering two different NAs or a combination of NA and Peg-IFN-a have been unsuc-cessful. Therefore, there is a renewed interest to investigate a number of steps in the HBVreplication cycle and specific virus–host cell interactions as potential targets for new anti-virals. Novel targets and compounds could readily be evaluated using both relevant in vitroand newly developed in vivo models of HBV infection. The addition of one or several newdrugs to current regimens should offer the prospect of markedly improving the response totherapy, thus reducing the burden of drug resistance, as well as the incidence of cirrhosis andhepatocellular carcinoma.
BACKGROUND—BASIS OF ANTI-HBVTHERAPY
Effective therapies have been developed forchronic hepatitis B (CHB) infection. Hence,
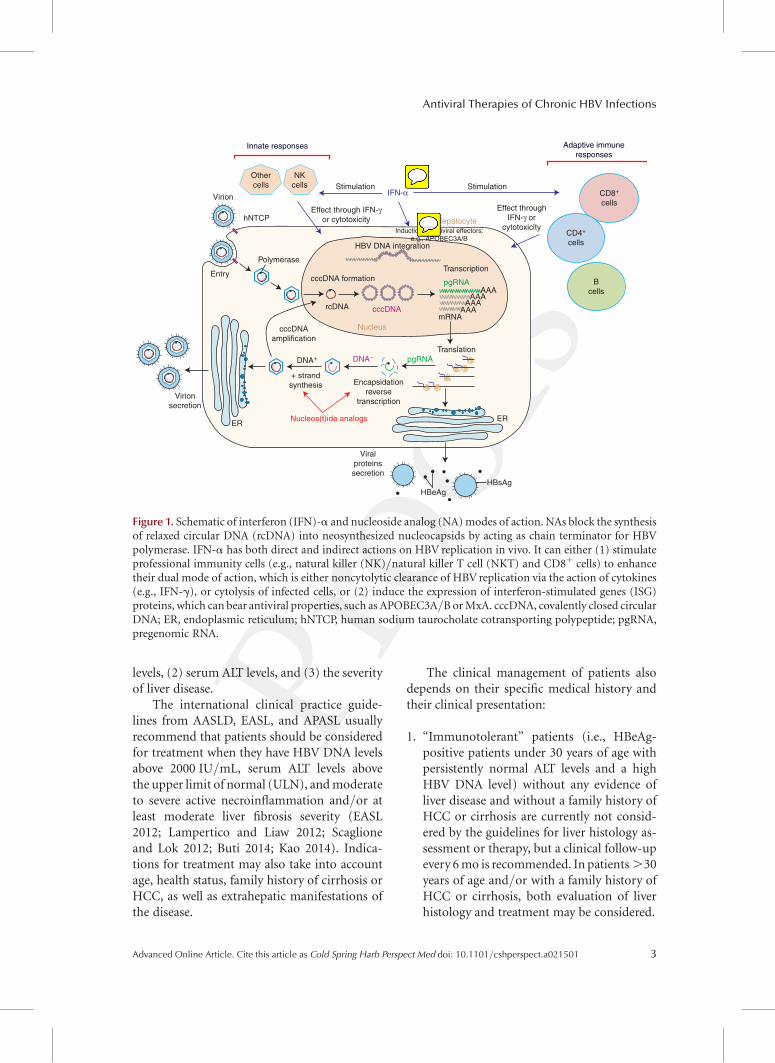
interferon-a (IFN-a) and its pegylated form(Peg-IFN-a), and five other drugs that belongto the class of nucleos(t)ide analogs (NAs), havebeen approved for this indication in most partsof the world (EASL 2012; Lampertico and Liaw2012; Scaglione and Lok 2012; Jordheim et al.2013; Buti 2014; Kao 2014). IFN-a is an im-mune modulator that induces, in a nonspecificmanner, the expression of interferon-stimulatedgenes (ISGs) encoding intracellular or secretedproteins with direct or indirect antiviral proper-
ties in both infected and noninfected cells, andpromotes the differentiation/activation of im-mune cells (Samuel 2001; Sadler and Williams2008). In the HBV setting, the IFN-a antiviralactivity results from a complex mode of actionincluding the activation of natural killer (NK)/NKT cells, inhibition of viral genome transcrip-tion, destabilization of viral nucleocapsid, butalso, as recently suggested, degradation of co-valently closed circular DNA (cccDNA) Q1via theactivation of APOBEC3A in infected cells(Micco et al. 2013; Thimme and Dandri 2013;Lucifora et al. 2014).
NAs directly inhibit the reverse transcriptaseactivity of the HBV polymerase. The approvedNAs include lamivudine (LMV), a deoxycyti-
Editors: Christoph Seeger and Stephen Locarnini
Additional Perspectives on Hepatitis B and Delta Viruses available at www.perspectivesinmedicine.org
Copyright # 2014 Cold Spring Harbor Laboratory Press; all rights reserved.
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
1
david-durantel-inserm
Note
covalently closed circular DNA

dine analog with an unnatural L conforma-tion, and the related L-nucleoside, telbivudine(LdT; b-L-thymidine). A second group, the acy-clic phosphonates, includes adefovir dipivoxil(ADV), a prodrug for the acyclic 20-deoxy aden-osine monophosphate analog adefovir, andthe structurally similar tenofovir (TFV). A thirdgroup contains a D-cyclopentane sugar moietyand has the most potent anti-HBV drug dis-covered to date, the deoxyguanosine analog en-tecavir (ETV). This structural classification ofNAs is useful clinically because it helps predictpathways of NA drug resistance (Zoulim andLocarnini 2009; Gish et al. 2012). In chronicallyHBV-infected hepatocytes, NAs inhibit the viralpolymerase activity resulting in a decreased pro-duction of virions, a reduced recycling of viralnucleocapsids to the nucleus of infected cells,and theoretically a decline of viral cccDNA, al-though the latter can only be observed aftermany years of treatment (Zoulim and Locarnini2009; Gish et al. 2012; Buti 2014). NAs do notinhibit the de novo formation of cccDNA innewly infected cells, implying that persistent re-sidual viremia during antiviral therapy can leadto infection of new hepatocytes and reestablish-ment of viral cccDNA reservoir. A decrease ofthe total amount of intrahepatic cccDNA isobserved during long-term therapy as a conse-quence of (1) the inhibition of the intracellularrecycling pathway, (2) dilution of cccDNA viahepatocyte turnover, as cccDNA may be lostthrough cell division, and (3) decreased rate ofinfection of new cells (Moraleda et al. 1997; LeGuerhier et al. 2000, 2001; Zhu et al. 2001;Werle-Lapostolle et al. 2004).
The therapeutic efficacy of these treatmentscan be affected by factors such as the develop-ment of adverse effects, poor patient compli-ance, previous treatment with suboptimal regi-mens, infection with drug-resistant viral strains,inadequate drug exposure because of pharma-cologic properties of particular drug(s), and in-dividual genetic variation (Zoulim 2011; EASL2012; Gish et al. 2012; Lampertico and Liaw2012; Scaglione and Lok 2012; Buti 2014; Kao2014). A simplified view of the mode of actionsof the approved antiviral agents in the HBV lifecycle is shown in Figure 1.
GOALS OF THERAPY AND TREATMENTEND POINTS
The goal of therapy for CHB is to improvethe quality of life and survival by preventingor significantly delaying progression of the dis-ease toward cirrhosis, decompensated cirrho-sis, end-stage liver disease, and hepatocellularcarcinoma (HCC). This goal can be achievedif HBV replication is suppressed in a sustainedmanner. It is accompanied by a reduction in thehistological activity of CHB and a decreased riskof developing cirrhosis and HCC, particularlyin noncirrhotic patients (EASL 2012; Lamper-tico and Liaw 2012; Scaglione and Lok 2012).Several recent studies in large cohorts haveshown that the risk of HCC development issignificantly decreased by successful antiviraltherapy compared with untreated historical pa-tient cohorts, but is not abated (Hosaka et al.2013; Lai and Yuen 2013; Cho et al. 2014;Wu et al. 2014). Chronic HBV infection cannotbe completely eradicated owing to the persis-tence of cccDNA in the nucleus of infected he-patocytes, which explains HBV reactivation, forinstance in patients who receive immunosup-pressive therapy or chemotherapy (Werle-La-postolle et al. 2004; Maynard et al. 2005; Wonget al. 2013; Seeger et al. 2014). Thus, therapy atleast to ensure a degree of viral suppression (i.e.,undetectable blood viremia) that will then leadto biochemical remission, histological improve-ment, and prevention of complications. This isthe currently achievable end point, which can beeither maintained during therapy or sustainedafter treatment cessation. However, the idealend point is HBsAg loss (i.e., HBsAg seroclear-ance) and/or anti-HBs antibody (i.e., HBsAb)seroconversion, which is currently infrequentlyachievable with the available anti-HBV agents(EASL 2012; Lampertico and Liaw 2012; Sca-glione and Lok 2012; Buti 2014; Kao 2014).
TREATMENT INDICATIONS
The indications for treatment are generally thesame for both HBeAg-positive and HBeAg-neg-ative CHB. This is based mainly on the combi-nation of three criteria: (1) serum HBV DNA
F. Zoulim and D. Durantel
2 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
admin1
Inserted Text
should
admin1
Cross-Out
levels, (2) serum ALT levels, and (3) the severityof liver disease.
The international clinical practice guide-lines from AASLD, EASL, and APASL usuallyrecommend that patients should be consideredfor treatment when they have HBV DNA levelsabove 2000 IU/mL, serum ALT levels abovethe upper limit of normal (ULN), and moderateto severe active necroinflammation and/or atleast moderate liver fibrosis severity (EASL2012; Lampertico and Liaw 2012; Scaglioneand Lok 2012; Buti 2014; Kao 2014). Indica-tions for treatment may also take into accountage, health status, family history of cirrhosis orHCC, as well as extrahepatic manifestations ofthe disease.
The clinical management of patients alsodepends on their specific medical history andtheir clinical presentation:
1. “Immunotolerant” patients (i.e., HBeAg-positive patients under 30 years of age withpersistently normal ALT levels and a highHBV DNA level) without any evidence ofliver disease and without a family history ofHCC or cirrhosis are currently not consid-ered by the guidelines for liver histology as-sessment or therapy, but a clinical follow-upevery 6 mo is recommended. In patients .30years of age and/or with a family history ofHCC or cirrhosis, both evaluation of liverhistology and treatment may be considered.
Innate responses
Othercells
NKcells Stimulation
Virion
hNTCP
Entry
Polymerase
cccDNA formation
cccDNAamplification
DNA+
+ strandsynthesis
Virionsecretion
Viralproteinssecretion
HBeAgHBsAg
ERERNucleos(t)ide analogs
pgRNA
rcDNA cccDNAmRNA
TranslationDNA– pgRNA
Encapsidationreverse
transcription
Bcells
CD4+
cells
CD8+
cells
Adaptive immuneresponses
Stimulation
HepatocyteInduction of antiviral effectors:
e.g., APOBEC3A/B
Effect throughIFN-γ or
cytotoxicity
Nucleus
Effect through IFN-γor cytotoxicity
Transcription
HBV DNA integration
AAAAAA
AAAAAA
IFN-α
Figure 1. Schematic of interferon (IFN)-a and nucleoside analog (NA) modes of action. NAs block the synthesisof relaxed circular DNA (rcDNA) into neosynthesized nucleocapsids by acting as chain terminator for HBVpolymerase. IFN-a has both direct and indirect actions on HBV replication in vivo. It can either (1) stimulateprofessional immunity cells (e.g., natural killer (NK)/natural killer T cell (NKT) and CD8þ cells) to enhancetheir dual mode of action, which is either noncytolytic clearance of HBV replication via the action of cytokines(e.g., IFN-g), or cytolysis of infected cells, or (2) induce the expression of interferon-stimulated genes (ISG)proteins, which can bear antiviral properties, such as APOBEC3A/B or MxA. cccDNA, covalently closed circularDNA; ER, endoplasmic reticulum; hNTCP, human sodium taurocholate cotransporting polypeptide; pgRNA,pregenomic RNA.
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 3
admin1
Cross-Out
admin1
Sticky Note
in red colour
admin1
Sticky Note
text not really visible on the figure
admin1
Cross-Out

2. Patients with obviously active CHB (i.e.,HBeAg-positive and HBeAg-negative pa-tients with ALT above 2 times ULN and se-rum HBV DNA above 2000 IU/mL) maystart treatment even without a liver biopsy,as it would not be mandatory for treatmentdecision. A noninvasive method for the esti-mation of the extent of fibrosis/cirrhosis isextremely useful in patients who start treat-ment without liver biopsy, to implementscreening of HCC and portal hypertension.
3. HBeAg-negative patients with persistentlynormal ALT levels and HBV DNA levelsabove 2000 but below 20,000 IU/mL, with-out any clinical evidence of liver disease, arecurrently not considered for liver biopsy ortherapy. However, a close follow-up of ALTand HBV DNA is recommended.
4. Patients with compensated cirrhosis and de-tectable HBV DNA must be considered fortreatment even if ALT levels are normal.
5. Patients with decompensated cirrhosis anddetectable HBV DNA require urgent antivi-ral treatment with NAs. Significant clinicalimprovement can be associated with controlof viral replication (Liaw et al. 2011a,b).However, antiviral therapy may not be suf-ficient to rescue some patients with veryadvanced liver disease who should be con-sidered for liver transplantation at the sametime.
6. Inactive carriers receiving chemotherapyor other immune suppressant treatmentsneed to receive preemptive antiviral therapyto prevent viral reactivation, implying thatHBV screening is mandatory for these pa-tients before starting immune suppressanttherapy.
7. HIV coinfected patients should be treatedwith treatment regimens including a highbarrier-to-resistance NA active on both HIVand HBV (i.e., Tenofovir). The antiviral reg-imen should meet current highly activeantiretroviral therapy (HAART) criteria foreffective HIV management including ade-quate viral suppression.
RESULTS OF INTERFERON AND NATHERAPIES
The efficacy of antiviral drugs has been assessedmainly at 1 yr in large randomized controlledtrials and has been reviewed recently (EASL2012; Lampertico and Liaw 2012; Scaglioneand Lok 2012; Buti 2014; Kao 2014). Longer-term results are now available from extension ofrandomized trials, sometime in patient sub-groups and from several cohort studies (Hosakaet al. 2013; Lai and Yuen 2013; Cho et al. 2014;Wu et al. 2014). Table 1 shows the response rateswith Peg-IFN-a, TFV, and ETV from differenttrials. These trials used different HBV DNA as-says and there are no head-to-head compari-sons for all the drugs.
HBeAg-Positive Patients
Response rates, including HBV DNA undetect-ability and anti-HBe seroconversion, at 6 mofollowing 48 wk of Peg-IFN-a and at 1 yr ofNA therapy are given in Table 1. Anti-HBe se-roconversion rates were of the order of 30%with Peg-IFN-a and �20% with NAs after 1yr of therapy (Buti 2014; Kao 2014). In adher-ent-to-treatment patients, a virological remis-sion rate of .90% can be maintained with ei-ther entecavir or tenofovir with prolongedtherapy (Gish et al. 2007; Heathcote et al.2011; Lok et al. 2012; Ono et al. 2012; Gordonet al. 2013; Fung et al. 2014; Kitrinos et al. 2014).Rates of HBsAg loss following 12 mo of treat-ment were of 3%–7% with Peg-IFN-a, 1% withlamivudine, 0% with adefovir, 2% with enteca-vir, 0.5% with telbivudine, and 3% with teno-fovir (Buti 2014; Kao 2014). HBsAg loss ratesincrease after the end of Peg-IFN-a therapy inpatients with sustained off-treatment virologi-cal response and with prolongation of NA ther-apy, and reach approximately 10% after 5 yr offollow-up for Peg-IFN-a or of continuous NAtreatment (Kao 2014).
HBeAg-Negative Patients
Response rates at 6 mo following 48 wk of Peg-IFN-a and at 12 mo of NA therapy are given in
F. Zoulim and D. Durantel
4 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
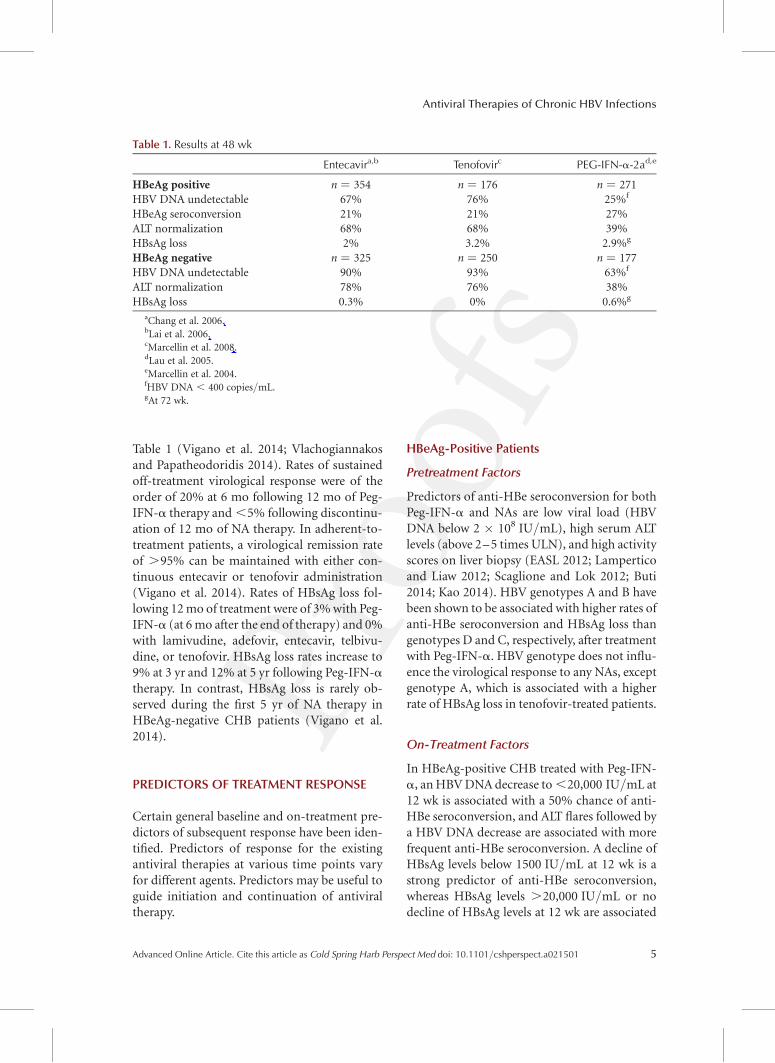
Table 1 (Vigano et al. 2014; Vlachogiannakosand Papatheodoridis 2014). Rates of sustainedoff-treatment virological response were of theorder of 20% at 6 mo following 12 mo of Peg-IFN-a therapy and ,5% following discontinu-ation of 12 mo of NA therapy. In adherent-to-treatment patients, a virological remission rateof .95% can be maintained with either con-tinuous entecavir or tenofovir administration(Vigano et al. 2014). Rates of HBsAg loss fol-lowing 12 mo of treatment were of 3% with Peg-IFN-a (at 6 mo after the end of therapy) and 0%with lamivudine, adefovir, entecavir, telbivu-dine, or tenofovir. HBsAg loss rates increase to9% at 3 yr and 12% at 5 yr following Peg-IFN-atherapy. In contrast, HBsAg loss is rarely ob-served during the first 5 yr of NA therapy inHBeAg-negative CHB patients (Vigano et al.2014).
PREDICTORS OF TREATMENT RESPONSE
Certain general baseline and on-treatment pre-dictors of subsequent response have been iden-tified. Predictors of response for the existingantiviral therapies at various time points varyfor different agents. Predictors may be useful toguide initiation and continuation of antiviraltherapy.
HBeAg-Positive Patients
Pretreatment Factors
Predictors of anti-HBe seroconversion for bothPeg-IFN-a and NAs are low viral load (HBVDNA below 2 � 108 IU/mL), high serum ALTlevels (above 2–5 times ULN), and high activityscores on liver biopsy (EASL 2012; Lamperticoand Liaw 2012; Scaglione and Lok 2012; Buti2014; Kao 2014). HBV genotypes A and B havebeen shown to be associated with higher rates ofanti-HBe seroconversion and HBsAg loss thangenotypes D and C, respectively, after treatmentwith Peg-IFN-a. HBV genotype does not influ-ence the virological response to any NAs, exceptgenotype A, which is associated with a higherrate of HBsAg loss in tenofovir-treated patients.
On-Treatment Factors
In HBeAg-positive CHB treated with Peg-IFN-a, an HBV DNA decrease to ,20,000 IU/mL at12 wk is associated with a 50% chance of anti-HBe seroconversion, and ALT flares followed bya HBV DNA decrease are associated with morefrequent anti-HBe seroconversion. A decline ofHBsAg levels below 1500 IU/mL at 12 wk is astrong predictor of anti-HBe seroconversion,whereas HBsAg levels .20,000 IU/mL or nodecline of HBsAg levels at 12 wk are associated
Table 1. Results at 48 wk
Entecavira,b Tenofovirc PEG-IFN-a-2ad,e
HBeAg positive n ¼ 354 n ¼ 176 n ¼ 271HBV DNA undetectable 67% 76% 25%f
HBeAg seroconversion 21% 21% 27%ALT normalization 68% 68% 39%HBsAg loss 2% 3.2% 2.9%g
HBeAg negative n ¼ 325 n ¼ 250 n ¼ 177HBV DNA undetectable 90% 93% 63%f
ALT normalization 78% 76% 38%HBsAg loss 0.3% 0% 0.6%g
aChang et al. 2006.bLai et al. 2006.cMarcellin et al. 2008.dLau et al. 2005.eMarcellin et al. 2004.fHBV DNA , 400 copies/mL.gAt 72 wk.
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 5
admin1
Inserted Text
Chang, T. T., R. G. Gish, R. de Man, A. Gadano, J. Sollano, Y. C. Chao, A. S. Lok, K. H. Han, Z. Goodman, J. Zhu, A. Cross, D. DeHertogh, R. Wilber, R. Colonno, and D. Apelian. 2006. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med 354:1001-1010.
admin1
Inserted Text
Lai, C. L., D. Shouval, A. S. Lok, T. T. Chang, H. Cheinquer, Z. Goodman, D. DeHertogh, R. Wilber, R. C. Zink, A. Cross, R. Colonno, and L. Fernandes. 2006. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med 354:1011-1020.
admin1
Inserted Text
Marcellin, P., E. J. Heathcote, M. Buti, E. Gane, R. A. de Man, Z. Krastev, G. Germanidis, S. S. Lee, R. Flisiak, K. Kaita, M. Manns, I. Kotzev, K. Tchernev, P. Buggisch, F. Weilert, O. O. Kurdas, M. L. Shiffman, H. Trinh, M. K. Washington, J. Sorbel, J. Anderson, A. Snow-Lampart, E. Mondou, J. Quinn, and F. Rousseau. 2008. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med 359:2442-2455.

with a very low probability of subsequent anti-HBe seroconversion.
Virological response (undetectable HBVDNA) at 24 wk during treatment with lamivu-dine or telbivudine and at 48 wk during treat-ment with adefovir is associated with a lowerincidence of resistance (i.e., an improved chanceof maintained virological response) in bothHBeAg-positive and HBeAg-negative patientsand with a higher chance of anti-HBe serocon-version in HBeAg-positive patients. A declineof HBsAg during NA treatment in HBeAg-pos-itive patients may identify cases with subse-quent HBeAg or HBsAg loss.
HBeAG-NEGATIVE PATIENTS
Pretreatment Factors
In HBeAg-negative CHB, there are no strongpretreatment predictors of virological responsefor Peg-IFN-a and NAs (EASL 2012; Lamper-tico and Liaw 2012; Vigano et al. 2014; Vlacho-giannakos and Papatheodoridis 2014).
On-Treatment Factors
In HBeAg-negative CHB treated with Peg-IFN-a, HBV DNA decrease to ,20,000 IU/mL at 12wk has been reported to be associated with a50% chance of sustained off-treatment re-sponse. A combination of no HBsAg declineand ,2 log10 IU/mL decline of HBV DNAseems to be a predictor of nonresponse in Eu-ropean HBeAg-negative patients with genotypeD. Several recent reports showed that HBsAgdecline is predictive of sustained off-treatmentvirological response and HBsAg loss. However,further studies are needed to clarify how to op-timize the use of HBsAg levels in the manage-ment of patients in clinical practice. In patientsreceiving NAs, maintained viral suppression isrequired to prevent the emergence of antiviraldrug-resistant strains.
Treatment Strategies
Currently, there are two different treatmentstrategies for both HBeAg-positive and HBeAg-negative CHB patients: (1) treatment of finiteduration with Peg-IFN-a or a NA, and (2)
long-term treatment with NAs (EASL 2012;Lampertico and Liaw 2012; Scaglione and Lok2012; Buti 2014; Kao 2014; Vigano et al. 2014;Vlachogiannakos and Papatheodoridis 2014).
The main theoretical advantages of Peg-IFN-a are the absence of resistance and the po-tential for immune-mediated control of HBVinfection with an opportunity to obtain a sus-tained virological response off-treatment and achance of HBsAg loss in patients who achieveand maintain undetectable HBV DNA. Fre-quent side effects and subcutaneous injectionare the main disadvantages of Peg-IFN-a treat-ment. Peg-IFN-a is contraindicated in patientswith decompensated HBV-related cirrhosis orautoimmune disease, in patients with uncon-trolled severe depression or psychosis, and infemale patients during pregnancy. Entecavirand tenofovir are potent HBV inhibitors witha high barrier to resistance. Thus, they can beconfidently used as first-line monotherapies(Zoulim and Locarnini 2009; Gish et al. 2012).
The other three NAs may only be used in thetreatment of CHB if more potent drugs withhigh barrier to resistance are not available. Lam-ivudine is an inexpensive agent, but engendersvery high rates of resistance with long-termmonotherapy. Adefovir is less efficacious andmore expensive than tenofovir, leading to high-er rates of resistance. Telbivudine is a potentinhibitor of HBV (Sun et al. 2014), but owingto a lower barrier to resistance, a high incidenceof resistance has been observed in patients withhigh baseline HBV DNA levels and in those withdetectable HBV DNA after 6 mo of therapy;resistance rates to telbivudine are relatively lowin patients who achieve undetectable HBV DNAafter 6 mo of therapy. Recent retrospective stud-ies suggest that long-term telbivudine therapymay improve kidney functions assessed by theestimated glomerular filtration rate (Gane et al.2014).
Treatment of Finite Durationwith PEG-IFN or a NA
This strategy is intended to achieve a sustainedoff-treatment virological response. A 48- wkcourse of Peg-IFN-a is mainly recommended
F. Zoulim and D. Durantel
6 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
david-durantel-inserm
Note
HBeAg-negative

for HBeAg-positive patients with the bestchance of anti-HBe seroconversion. It is practi-cally the only option that may offer a chance forsustained off-treatment response after a finiteduration of therapy. In HBeAg-negative pa-tients, Peg-IFN-a therapy can achieve on-treat-ment viral suppression, but is followed by viro-logical relapse after treatment cessation in manypatients.
Finite-duration treatment with a NA isachievable for HBeAg-positive patients who se-roconvert to anti-HBe on treatment. However,treatment duration is unpredictable before ther-apy as it depends on the timing of anti-HBeseroconversion and the treatment continua-tion after anti-HBe seroconversion. Anti-HBeseroconversion may not be durable after NAsdiscontinuation in a substantial proportion ofthese patients, therefore requiring close virolog-ic monitoring after treatment cessation. Evenafter NA treatment prolongation for an addi-tional 12 mo after anti-HBe seroconversion, adurable off-treatment response can be expectedin 40%–80% of these patients.
Long-Term Treatment with NAs
This strategy is necessary for patients who arenot expected to or failed to achieve a sustainedoff-treatment virological response and require
extended therapy (i.e., for HBeAg-positive pa-tients who do not develop anti-HBe seroconver-sion and HBeAg-negative patients) (Buti 2014;Vigano et al. 2014). This strategy is also recom-mended in patients with cirrhosis irrespectiveof HBeAg status or anti-HBe seroconversionon treatment. The most potent drugs with theoptimal resistance profile (i.e., tenofovir or en-tecavir), should be used as first-line monothera-pies. It is optimal to achieve and maintain anundetectable HBV DNA level tested by real-time polymerase chain reaction (PCR), what-ever the drug used. Treatment with either teno-fovir or entecavir monotherapy for �5 yearsachieves maintained virological remission inthe vast majority of patients.
RESISTANCE TO ANTIVIRAL DRUGS ANDTREATMENT FAILURE
Main Concepts
The development of drug resistance begins withmutations in the polymerase gene, followed byan increase in viral load, an increase in serumalanine aminotransferase (ALT) levels severalweeks to months later, and progression of liverdisease (Zoulim and Locarnini 2009; Gish et al.2012). The main mutations associated with re-sistance to a given NA are listed in Figure 2. The
Terminalprotein Spacer POL/RT RNaseH
845 a.a692 (rt 344)349 (rt)183
I(G) II(F)
LVD resistance
LdT resistance
rtL801
rtN236T
rtM2041
rtM204V/IrtL180MrtA181T/VrtL180mrtA181T/VrtA181T/V
ADV resistanceTDF resistance
rtM2041/VrtS202**
rtM2501/V
*S/A/I/L/G/C/M **C/G/I
A B
F_V_LLAQ_YMDD
C D E
1
ETV resistancertL180MrtT184*rtl169T
To be determined
Figure 2. Position of resistance mutations within HBV polymerase. Mutations conferring resistance to the fiveapproved NAs are located within the subdomains A, B, C, and D of HBV polymerase. Some mutations conferresistance to different NAs; this cross-resistance profile is to be taken into consideration for the clinical man-agement of patients Q2.
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 7
david-durantel-inserm
Note
POL/RT = reverse transcriptase domain of HBV polymerase
admin1
Cross-Out
admin1
Inserted Text
I (capital "I")
admin1
Cross-Out
admin1
Inserted Text
I (capital "I")
admin1
Cross-Out
admin1
Inserted Text
M
admin1
Cross-Out
admin1
Inserted Text
I
admin1
Cross-Out
admin1
Inserted Text
I

appearance of mutation in vivo can be moni-tored by direct sequencing (including next-gen-eration sequencing), sequencing after cloning,or by hybridization techniques, and mutantscan be phenotyped in vitro by previously de-scribed techniques (Durantel et al. 2005; Liuand Kitrinos 2013). In patients with LMV resis-tance, the risk of increased serum ALT is usuallycorrelated with the duration of detectability ofthe resistant strain (Lok et al. 2003). These pa-tients are also at significant risk of ALT flare,which may be accompanied by hepatic decom-pensation (Lok et al. 2003). The detrimentaleffect of HBV drug resistance on liver histology(Dienstag et al. 2003) and then on clinical out-come was shown by a trial of LMV in patientswith advanced fibrosis (Liaw et al. 2004). Incontrast to LMV, the kinetics of emergence ofresistance to ADVare typically slower, but followthe same sequence of events (Hadziyannis et al.2006). In some cases, the emergence of ADVresistance is also associated with acute exacer-bation of disease and liver failure (Fung et al.2005). Only limited data are available on theclinical outcome of patients who are infectedwith LdT-, ETV-, or TFV-resistant HBV, mainlybecause treatment adaptation, usually based onin vitro cross-resistance data, has been initiatedmuch earlier.
The availability of antiviral drugs with com-plementary cross-resistance profiles (Fig. 2) al-lows physicians to adapt antiviral therapy ac-cording to the virological situation to preventthe clinical deterioration resulting from theemergence of resistance. There are several clin-ical risk factors associated with the developmentof NA resistance, including high levels of serumHBV DNA, high serum ALT levels, and highbody mass index. Prior therapy with NAs, andinadequate viral suppression during therapyalso predict drug resistance.
Typically, the development of NA resistancedepends on the following factors (Zoulim andLocarnini 2009; Gish et al. 2012): (1) rate ofvirus replication; (2) complexity and diversityof the viral quasispecies; (3) selective pressureexerted by the NA (potency); (4) viral repli-cation space in the liver; (5) replication fitnessof the emerging NA-resistant HBV; (6) genetic
barrier to resistance of the NA; (7) previoustreatment history and archiving of drug-resis-tant strains; and (8) treatment observance/compliance. This explains that sequential ther-apy with low-barrier-to-resistance NA sharingcross-resistance may favor the emergence ofmultiresistant strains that may explain, for in-stance, failure to ETV therapy after an initialresistance to LMV or LdT (Villet et al. 2007;Liu et al. 2010).
Clinical Aspects of Resistanceand Treatment Failure
All patients receiving NA therapy for CHBshould be closely monitored for virologic re-sponse and breakthrough during treatment.Serum HBV DNA should be tested every 3 moduring treatment; however, if the patient iscompliant and a high genetic barrier, high po-tency drug (ETV and TFV) is used, then thisfrequency can be reduced to 6 mo. In a compli-ant patient, it is important to distinguish be-tween primary nonresponse, partial virologicresponse, and virologic breakthrough (viralrebound) owing to underlying antiviral drugresistance, as it has implications for treatmentadaptation (Zoulim and Locarnini 2009; Gishet al. 2012).
1. Primary nonresponse: The failure to achievea 1.0 log10 IU/mL decline in viral load after12 wk of therapy is considered as a primarynonresponse. It may be owing to a lack ofcompliance or the medication may not useits antiviral activity in a particular individual.Suboptimal response was often seen withADV and was shown to be unrelated to a re-duced drug susceptibility of viral strains asmeasured in vitro by phenotypic assay (Car-rouee-Durantel et al. 2008). With the adventof more potent antiviral drugs, such as TFVand ETV, this phenomenon is now much lessfrequent. When a primary nonresponse isidentified, treatment should be modified toprevent disease progression and subsequentrisk of emergence of drug-resistant mutants.
2. Partial response: The recommendations ofinternational clinical practice guidelines are
F. Zoulim and D. Durantel
8 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501

to achieve undetectable HBV DNA duringtherapy; therefore, partial response is de-fined by detectable HBV DNA using a real-time PCR assay during continuous therapy(EASL 2012; Lampertico and Liaw 2012;Scaglione and Lok 2012). With antiviraldrugs that have a low genetic barrier to resis-tance (LMV, LdT), the lack of complete an-tiviral response at week 24 of therapy wasshown to predict the subsequent resistancerate. With the more potent and high geneticbarrier drugs such as ETV and TFV, the rateof undetectable HBV DNA after 1 yr of ther-apy is significantly improved, reachingaround 70% in HBeAg-positive patientsand 90% in HBeAg-negative patients (Buti2014; Vigano et al. 2014). Because the rate ofviral suppression continues to increase overtime with ETV and TFV, the timing of treat-ment adaptation mainly depends on the ki-netics of viral load decay, especially in pa-tients starting from a very high viral loadwho may just need additional weeks of ther-apy to reach undetectable HBV DNA by PCRtesting (Buti 2014; Vigano et al. 2014).Therefore, the pattern of viral load declineis more useful than a single assessment, be-cause the latter may result in a misleadinginterpretation of treatment response. Whenusing drugs with a low barrier to resistance,it is recommended that in cases of persistinglow viremia, treatment be adapted to maxi-mize viral suppression and minimize thesubsequent risk of emergence of resistance(Zoulim and Locarnini 2009; Gish et al.2012). In the case of NA with high barrierto resistance (TFV and ETV), the continua-tion of the same treatment associated withcounseling on treatment compliance may al-low one to reach complete virological re-sponse several weeks later (Zoulim and Lo-carnini 2009; Gish et al. 2012).
3. Virologic breakthrough and rebound: Virolog-ic breakthrough typically results from theemergence of drug-resistant viral strains. Itis defined by an increase of at least 1.0 log10
IU/mL compared with the lowest valueachieved during treatment, confirmed by a
second test, in a treatment-compliant pa-tient. It usually follows the detection of resis-tance mutations (Zoulim and Locarnini2009; Gish et al. 2012). In the absence oftreatment adaptation, the increase in viremiamay be followed by an increase in ALT levels(biochemical breakthrough) and subse-quently progression of liver disease (clinicalbreakthrough) (Zoulim and Locarnini 2009;Gish et al. 2012). The increase of viral loadassociated with the emergence of resistancemutations depends on the fitness of the mu-tants; interestingly it was shown that resis-tance mutations in the polymerase gene af-fecting the overlapping surface gene (e.g.,rtA181T/sW172�) may affect both their ca-pacity to be secreted from infected hepato-cytes and their infectivity (Warner and Lo-carnini 2008; Billioud et al. 2011). This mayresult in a slow increase of viral load forwhich the identification of a 1 log10 IU/mLincrease may be difficult.
Management of Treatment Failure
Assessment of Treatment Adherence
Good adherence to anti-HBV therapies is im-portant for maintaining maximal suppressionof HBV replication (Table 2). Poor adherencecan result in substantially reduced plasma druglevels, depending on the number of dosesmissed and the half-life of the drug, and canresult in increased viral replication (Zoulimand Locarnini 2009; Gish et al. 2012). Investi-gation of adherence to NA therapy in patientswith CHB has shown that nearly 40% may notbe fully adherent; this significantly impacts onthe rates of viral suppression (Sogni et al. 2012).Low-level viral replication associated with non-adherence increases the pressure on the potencyof the NA, and consequently increases the riskof selecting for resistance. Specific treatmentadherence questionnaires and drug concentra-tion monitoring can be useful for the manage-ment of patients. The level of education, type ofhealth insurance, cultural factors, as well as lowcopayment for medications can significantlyimpact medication adherence. Thus, programson patient counseling and on medication ad-
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 9

herence to improve effectiveness of antiviraltherapy in clinical practice are recommended.
Treatment Adaptation Accordingto Cross-Resistance
Cross-resistance is defined as resistance to drugsto which a virus has never been exposed as aresult of changes that have been selected for bythe use of another drug (Zoulim and Locarnini2009; Gish et al. 2012). The resistance-associat-ed mutations selected by a particular NA conferat least some degree of cross-resistance to othermembers of its structural group but may alsodiminish the sensitivity to NAs from a differentchemical group. The initial drug choice andsubsequent rescue therapies should be basedon the knowledge of cross-resistance, so thatthe second agent has a different resistance pro-file to the initial failing agent. This is particu-larly important because drug-resistant mutantsthat have been selected by previous treatmentsare thought to be archived in viral cccDNA res-ervoirs in the liver (Zoulim and Locarnini 2009;Gish et al. 2012). The add-on strategy with NAshaving complementary cross-resistance profilesis mandatory when using drugs with a low bar-rier to resistance.
Management of Antiviral Drug Resistance
Virologic breakthrough in compliant patients isrelated to viral resistance. Resistance should be
identified as early as possible, before ALT levelsincrease, by monitoring HBV DNA levels andif possible identifying the NA resistance profile;the best therapeutic strategy can then be deter-mined based on this information. Clinical andvirological studies have shown the benefit of anearly adaptation of treatment (Zoulim and Lo-carnini 2009; Gish et al. 2012). In case of resis-tance, an appropriate rescue therapy should beinitiated as soon as possible. Adding a seconddrug that is not in the same cross-resistancegroup as the first (i.e., L-nucleoside vs. acyclicphosphonate vs. D-cyclopentane) is recom-mended at least for drugs with a low barrier toresistance. However, although there is a strongvirologic rationale for an add-on strategy with acomplementary drug to prevent the emergenceof multidrug-resistant strains and raise the bar-rier to resistance, there is a current trend to rec-ommend a switch to a complementary drughaving a high barrier to resistance such as TFV(Berg et al. 2014). This critical point will need aprecise evaluation by long-term clinical andmolecular virology studies, as some mutantsare associated with slow decline of viral load(Villet et al. 2008; Patterson et al. 2011; Lavocatet al. 2013). Furthermore, the switch strategydoes not apply to patients who have been ex-posed to multiple alternating monotherapies;these patients should be enrolled in add-onstrategies to minimize the risk of subsequenttreatment failure, especially in the presence of
Table 2. Management of treatment failure
Type of failure Treatment adaptation
Lamivudineresistance
1. Add TFV (add ADV if TFV not available). 2. A switch to TFV is also advised by someguidelines. 3. A switch to ADV is not recommended owing to a high rate of resistanceand its low potency.
Adefovir resistance 1. Switch to TFV if available and add a second drug without cross-resistance. 2. If nohistory of LMV, switching to ETV is also effective. 3. If rtN236Tsubstitution, consideradding LMV, ETV, or LdT to the TFVor switch to TFV plus FTC; if no history of LMVprior, consider switching to ETV. 4.If rtA181 V/T substitution, alone or incombination with rtN236T, switch to TFV plus ETV; as before, if no history of LMV,consider switching to ETV.
Telbivudineresistance
1. Add TFV. 2. A switch to TFV has also been considered in some guidelines. 3. A switchto ADV is not recommended.
Entecavir resistance 1. Add TFV. 2. A switch to TFV can also be considered.Tenofovir resistance 1. Not been confirmed so far. 2. Genotyping and phenotyping required. 3. May add ETV.
F. Zoulim and D. Durantel
10 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501

underlying cirrhosis. Figure 2 shows the majorresistance substitutions; cross-resistance canbe inferred from these profiles for the most fre-quent resistant HBV variants and treatmentadaptation should be performed accordingly(Zoulim and Locarnini 2009; Gish et al. 2012).
REASONS TO CONSIDER “EARLY”TREATMENT INTERVENTION
Current international treatment guidelines rec-ommend delaying therapy until patients showclear signs of active liver disease extending overseveral months, including persistent ALTeleva-tions and, when biopsies are available, evidenceof inflammation and/or fibrosis (EASL 2012;Lampertico and Liaw 2012; Scaglione and Lok2012). These guidelines, if rigorously applied,should identify patients entering the immunereactive phase, when synergy with the host re-sponse can maximize therapeutic outcomes ofantiviral therapy, hopefully with HBeAg and,ideally, HBsAg seroconversion. Application ofthe guidelines can block the progression of fi-brosis and cirrhosis and may reduce the rate ofprogression to HCC (Hosaka et al. 2013; Lai andYuen 2013; Cho et al. 2014; Wu et al. 2014).
An important placebo control trial wasperformed with LMV to determine its efficacyon clinical end points. Patients enrolled for thisstudy had significant liver disease and advancedfibrosis. A .50% reduction in liver disease pro-gression including HCC was found after 36 moof therapy (Liaw et al. 2004). This study was afirst proof of concept that antiviral therapy ofCHB even at late stages can decrease the majorcomplications of chronic infection (Liaw et al.2004). Other studies suggested a trend for alower incidence of HCC in patients treatedwith lamivudine for chronic hepatitis comparedwith those treated at the stage of cirrhosis (Pa-patheodoridis et al. 2011). It is important toremember, however, that the HCC incidencein these chronic hepatitis B patients treatedwith NAs was significantly decreased but noteliminated (Papatheodoridis et al. 2011; Hosakaet al. 2013; Lai and Yuen 2013; Cho et al. 2014;Wu et al. 2014). Strict adherence to clinicalguidelines requires a level of clinical monitor-
ing, public awareness, and case ascertainmentthat will be difficult to achieve as long as youngadults think they are not yet at risk and possiblyin need of treatment to reduce the incidence ofcirrhosis and HCC later in life. Another impor-tant concern is that HCC risk factors in HBVcarriers are not well understood. Current think-ing favors the notion that the HCC risk begins,in the vast majority of cases, with the immunereactive phase, but there is no proof that it doesnot begin much earlier.
The current information on long-term an-tiviral treatment efficacy and safety allows oneto consider earlier treatment intervention inpatients with chronic HBV infection. A changein treatment practices is much more feasiblethan it was even a few years ago, as much betterdrugs have become available with a better anti-viral potency and a higher barrier to resistance.It is interesting to see that the results of the firstclinical trial of NAs in immune-tolerant pa-tients has recently been published (Chan et al.2014) and showed a significant drop in viremialevels in the majority of patients, although noHBsAg seroconversion occurred and the im-pact on HCC development could not be deter-mined owing to the short duration of follow-up.In theory, it would be best to initiate NA treat-ment in all immune-tolerant patients. However,a more conservative approach which would beone step beyond the current guidelines wouldbe to propose therapy in all patients with per-sistently high-normal ALT levels, or with nor-mal ALTs who show relatively low levels ofviremia (e.g., .104 but �108 copies per mL),including patients in their 20s, not just thosebeyond 40 years of age (Lai et al. 2007; Zoulimand Mason 2012). When biopsies are avail-able, attempts should be made to establishhepatocyte infection levels and identify low-level inflammatory activity. The presence ofsome degree of inflammatory activity associatedwith a reduction of HBV capsid/core protein(HBc)Ag-positive Q3hepatocytes, and lower levelsof HBV DNA in serum (,8 log10 IU/mL, but.4 log10 IU/mL) would suggest a high level ofaccumulated hepatocyte damage/change evenin the absence of other indicators of histologi-cal change, and treatment would seem strongly
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 11
david-durantel-inserm
Note
OK!HBc = core/capsid protein

warranted. This is also supported by the obser-vation that HBV-specific T-cell functions areconserved in patients in the so-called “immunetolerance” phase (Kennedy et al. 2012). An un-appreciated cause of clonal hepatocyte repop-ulation occurs in noncirrhotic liver as well.Immune killing of infected hepatocytes is thestrongest known pressure on the infected he-patocyte population in the noncirrhotic liverand, analogous to cirrhosis, should lead to theemergence of HBV-resistant hepatocytes thatare able, in this example, to avoid immune kill-ing. Indeed, most analyses of long-term carrierssuggest that 50% or more of hepatocytes nolonger support HBV infection and/or supportmuch reduced levels of replication (Mason et al.2008). Therefore, although it may seem para-doxical at first glance, any reduction in HBVtiters in HBV carriers may warrant initiationof antiviral therapy, even if biopsy does notreveal histologically detectable active hepatitis(Zoulim and Mason 2012).
TOWARD A CURE OF HBV INFECTION WITHNOVEL COMBINATION STRATEGIES?
One of the major questions regarding antiviraltherapy of CHB was whether the combinationof Peg-IFN-a with NAs could improve the off-treatment response rate and the rate of HBsAgseroconversion to shorten treatment duration.However, despite the observation that thecombination of Peg-IFN-a with LMV or LdTshowed a higher on-treatment virological re-sponse, it did not show a higher rate of sus-tained off-treatment virological or serologicalresponse (Marcellin et al. 2004; Janssen et al.2005; Lau et al. 2005). Several studies are on-going with the combination of Peg-IFN-a andETV or TFV (Kao 2014), but presently thistype of combination is not yet recommended.Furthermore, there are no data to indicate anadvantage of de novo combination with ETVand TFV in NA-naıve patients, although morestudies in patients with high baseline viremia(HBV DNA .108 IU/mL) are required.
Current treatments for CHB based on NAsallow one to control viral replication and liverdisease in the majority of patients (EASL 2012;
Lampertico and Liaw 2012; Scaglione and Lok2012; Buti 2014). However, because NAs arenot able to clear cccDNA, lifelong therapiesare required to maintain the antiviral effect.To define new therapeutic options and headtoward treatments with finite duration, it istherefore necessary to develop new moleculesacting on novel targets to set true combinationtherapies (Zoulim 2012). The persistence ofHBV infection and the maintenance of the he-patocytes harboring cccDNA mainly result froma weak HBV-specific immune response. In thisrespect, strategies directly or indirectly target-ing cccDNA, as well as the stimulation of theimmune response against HBV-infected cellsmight represent a relevant approach. An efficientcontrol of viral infections requires a concertedaction of both innate and adaptive immuneresponses, as observed in the case of self-resolv-ing HBV infection, which occurs in around90% of “immune-competent adults” exposedto the virus (Bertoletti and Ferrari 2012). Re-storing such responses in the chronic infectionsetting could help in reaching an immune con-trol status similar to that observed in anti-HBsseroconverted patients or “inactive carriers.”
Definitions of a “Cure” of HBV Infection
There are several concepts around the defini-tion of a “cure of HBV infection.” The ultimategoal of treatment would be to eradicate viralcccDNA from the liver leading to a completeand definite clearance (i.e., “absolute cure”) ofinfected hepatocytes, thereby preventing therisk of reactivation in case of a loss of immunecontrol. However, it is worth noting that thiswould not abolish the consequences of viral ge-nome integration in the host chromosomes ofinfected cells, as this event could occur earlyafter the onset of infection (Seeger et al. 2014).On the other hand, in patients who spontane-ously resolved viral infection with HBsAg clear-ance and anti-HBs (HBs antibody, i.e., HBsAb)seroconversion, cccDNA might not be com-pletely eradicated, and the few persisting in-fected cells are supposed to be under the con-trol of the host immune response. Therefore, a“clinical or functional cure” of infection could
F. Zoulim and D. Durantel
12 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
admin1
Inserted Text
in
be defined by HBsAg clearance and HBsAbseroconversion, despite the lack of completecccDNA eradication. The “functional cure”would be considered as long as the host immuneresponse controls the infection and could bedefined by the absence of relapse after treatmentcessation. Another end point, which could beenvisaged, is the control of infection, as ob-served in inactive carriers—defined by the per-sistence of low levels of serum HBsAg and HBVDNA levels with normal ALT levels—in whomthe HBV-specific immune response wouldbe strong enough to keep viral replication undercontrol, thereby allowing antiviral treatmentcessation.
Identification of Novel Drug Targets
New drugs targeting novel targets are needed todevelop true combination therapies and steptoward a cure of HBV infection. Several targetsand novel compounds are currently being eval-uated in in vitro and in vivo experimental mod-
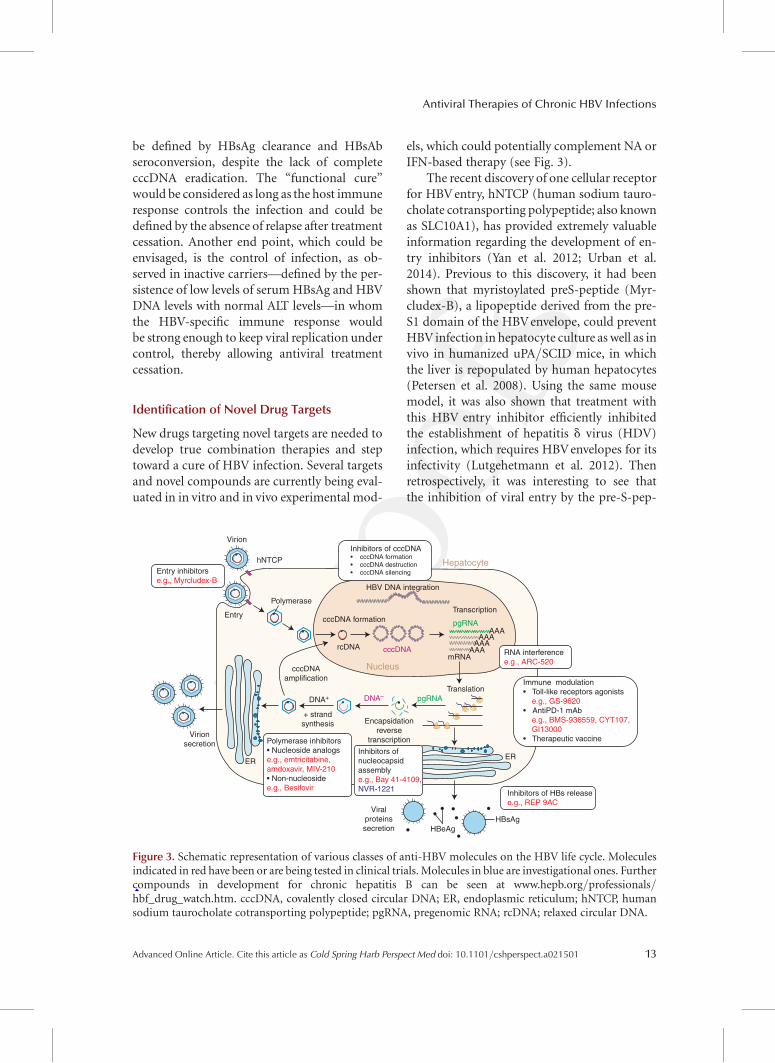
els, which could potentially complement NA orIFN-based therapy (see Fig. 3).
The recent discovery of one cellular receptorfor HBV entry, hNTCP (human sodium tauro-cholate cotransporting polypeptide; also knownas SLC10A1), has provided extremely valuableinformation regarding the development of en-try inhibitors (Yan et al. 2012; Urban et al.2014). Previous to this discovery, it had beenshown that myristoylated preS-peptide (Myr-cludex-B), a lipopeptide derived from the pre-S1 domain of the HBV envelope, could preventHBV infection in hepatocyte culture as well as invivo in humanized uPA/SCID mice, in whichthe liver is repopulated by human hepatocytes(Petersen et al. 2008). Using the same mousemodel, it was also shown that treatment withthis HBV entry inhibitor efficiently inhibitedthe establishment of hepatitis d virus (HDV)infection, which requires HBV envelopes for itsinfectivity (Lutgehetmann et al. 2012). Thenretrospectively, it was interesting to see thatthe inhibition of viral entry by the pre-S-pep-
Virion
hNTCP
Entry
Polymerase
cccDNA formation
cccDNAamplification
DNA+
+ strandsynthesis
Virionsecretion
Viralproteinssecretion HBeAg
HBsAg
ERER
rcDNA cccDNAmRNA
TranslationDNA– pgRNA
Encapsidationreverse
transcription
Hepatocyte
Nucleus
Transcription
HBV DNA integration
Inhibitors of HBs releasee.g., REP 9AC
Inhibitors ofnucleocapsidassemblye.g., Bay 41-4109,NVR-1221
Polymerase inhibitors• Nucleoside analogse.g., emtricitabine,amdoxavir, MIV-210• Non-nucleosidee.g., Besifovir
RNA interferencee.g., ARC-520
Entry inhibitorse.g., Myrcludex-B
Immune modulation• Toll-like receptors agonists e.g., GS-9620• AntiPD-1 mAb e.g., BMS-936559, CYT107, GI13000• Therapeutic vaccine
Inhibitors of cccDNA• cccDNA formation• cccDNA destruction• cccDNA silencing
pgRNAAAA
AAAAAA
AAA
Figure 3. Schematic representation of various classes of anti-HBV molecules on the HBV life cycle. Moleculesindicated in red have been or are being tested in clinical trials. Molecules in blue are investigational ones. Furthercompounds in development for chronic hepatitis B can be seen at www.hepb.org/professionals/hbf_drug_watch.htm. cccDNA, covalently closed circular DNA; ER, endoplasmic reticulum; hNTCP, humansodium taurocholate cotransporting polypeptide; pgRNA, pregenomic RNA; rcDNA; relaxed circular DNA.
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 13
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Cross-Out
admin1
Inserted Text
C
admin1
Cross-Out

tide was indeed owing to its interaction withhNTCP (Ni et al. 2014). Furthermore, drugsthat inhibit hNTCP function, such as cyclospor-ine, also decrease viral infectivity in cell-culturemodels (Nkongolo et al. 2014; Watashi et al.2014). As hepatocyte turnover and reinfectioncycles might be needed to maintain persistentinfection, this could make a reasonable case forthe evaluation of such an entry inhibitor in thecontext of chronic infections. The specific valueof entry inhibitors in the treatment of chronichepatitis B will need to be shown in clinicaltrials. Because there is currently no specific an-tiviral for HDV infection except IFN-a, whichprovides sustained virologic response in only25% of patients (Heidrich et al. 2014), the clin-ical evaluation of entry inhibitors in patientswho are coinfected with HBV and HDV is alsowarranted.
The initial formation (and maintenance byrecycling of nucleocapsid) of cccDNA, from re-laxed circular DNA (rcDNA) genome nucleardelivery, represents a very important antiviraltarget (Zoulim et al. 2013b; Seeger et al. 2014).The cellular and biochemical events required forthis process involve the transport of nucleocap-sid to the nucleus, and the transformation of thercDNA genome into cccDNAvia the removal ofthe viral polymerase covalently linked to viralminus-strand DNA, the removal of the shortRNA primer for plus-strand DNA synthesis,the completion of plus-strand DNA, and theremoval of the viral minus-strand DNA redun-dancy (Zoulim et al. 2013b; Seeger et al. 2014).These steps seem to involve several nuclearenzymes, including TDP2 and endonucleases,for which it may be difficult to target a func-tion specific to the viral life cycle (Sohn et al.2009). Administration of NAs failed to preventthe initial formation of cccDNA after de novoinfection of hepatocytes in animal models ofinfection, whereas their long-term administra-tion to already infected individuals seems todecrease the pool of already established cccDNAby the potential inhibition of the recycling ofnucleocapsids containing viral genomes to thenucleus; but one cannot exclude that this mightalso be owing to the clearance of infected cellsfrom the liver by programmed cell death or im-
mune killing. Interestingly, it was recently re-ported that small molecules might specificallytarget cccDNA formation. Two structurally re-lated disubstituted-sulfonamide compoundswere identified and may potentially serve asproof-of-concept (POC) drug candidates toeliminate cccDNA from chronic HBV infectionby preventing initial formation and/or main-tenance by nucleocapsid recycling, but not bydegrading already formed cccDNA (Cai et al.2012). On the other hand, it was recently shownthat IFN-a and lymphotoxin-b receptor activa-tion up-regulated APOBEC3A and APOBEC3Bcytidine deaminases, respectively, and inducednonhepatotoxic degradation of nuclear hepati-tis B virus cccDNA. Interestingly, HBc couldmediate APOBEC3A/B interaction with nucle-ar cccDNA, resulting in cytidine deamination,apurinic/apyrimidinic site formation, and fi-nally cccDNA degradation that prevented HBVreactivation (Lucifora et al. 2014). This opensnew avenues to achieve cccDNA degradationby novel strategies. In this respect, the use ofcccDNA-specific meganuclease (or related se-quence-specific homing endonucleases) de-livered to infected cells by gene therapy couldalso be an interesting approach to degradecccDNA. Because the degradation of cccDNAfrom all cells remains a difficult goal to reach,the transcriptional silencing of cccDNA activityrepresents a step forward in developing originalstrategies. A first POC approach consisted in theexpression of zinc-finger proteins able to bindthe duck hepatitis B virus regulatory geneticsequence (enhancer), in infected cells. After co-transfection of vectors encoding these proteinsand DHBV in cultured cells, it was shown thatzinc-finger proteins are able to bind to theDHBV enhancer and interfere with viral tran-scription, resulting in decreased productionof viral products and progeny virus genomes(Zimmerman et al. 2008). Yet the delivery ofsuch targeted proteins to infected hepatocytesin vivo remains a challenge. Interfering withcccDNA-associated chromatin proteins is an-other exciting approach. Indeed, the acetyla-tion and/or methylation status of the histonesbound to cccDNA affect its transcriptional ac-tivity. It was shown in cell-culture and in hu-
F. Zoulim and D. Durantel
14 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
admin1
Cross-Out
admin1
Inserted Text
the result of
admin1
Inserted Text
Koniger, C., I. Wingert, M. Marsmann, C. Rosler, J. Beck, and M. Nassal. 2014. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci U S A 111:E4244-4253

manized mice that IFN administration inducescccDNA-bound histone hypoacetylation, aswell as active recruitment to the cccDNA of tran-scriptional corepressors (Belloni et al. 2012).IFN-a treatment also reduced binding of theSTAT1 and STAT2 transcription factors to activecccDNA. This may represent a molecular mech-anism whereby IFN-a mediates epigenetic re-pression of cccDNA transcriptional activity,which may assist in the development of noveltherapeutics. In this respect, the research aim-ing at developing epigenome-modifying en-zyme inhibitors for cancer indications (BojangJr and Ramos 2014; Jones 2014), has to be scru-tinized to potentially apply this knowledge toCHB treatment.
Beside the polymerase protein, HBVs donot encode for other proteins bearing enzymat-ic activities, thus rendering difficult the devel-opment of other direct acting agents (DAA), assuccessfully shown for in the HCV field (Welschet al. 2012). Yet other HBV protein functionscould be targeted. In this respect HBx proteinrepresents an interesting theoretical target. In-deed, it was shown that this protein is requiredfor viral infection in vivo (Zoulim et al. 1994;Seeger et al. 2014). More recently, it was shownin primary hepatocyte cultures as well as Hep-aRG cells (Gripon et al. 2002) that the HBxprotein is necessary to initiate and maintainviral replication via cccDNA transcriptional reg-ulation (Lucifora et al. 2011). This may be con-sistent with the observation that nuclear HBxbinds the HBV minichromosome and modifiesthe epigenetic regulation of cccDNA function(Belloni et al. 2009). HBx has also been shownto interact with DDB1, an adaptor protein,which on binding to CUL4 proteins, formscullin-RING ligase complexes involved in majorcellular functions (i.e., DNA repair, DNA tran-scription, DNA replication, etc.) (Angers et al.2006). This interaction would prevent HBx deg-radation (i.e., favor HBx stabilization), and alsoinduce the degradation of host factors, whichcould have antiviral functions, with both ac-tions contributing to better HBV replication.The domain of interaction between HBx andDDB1 has been mapped (Li et al. 2010), andcould serve to design molecules interfering
with this protein–protein interaction and lead-ing to an antiviral phenotype. A more detailedknowledge of HBx function should help to tar-get this key regulator of viral replication.
Several attempts have been made to developinhibitors of nucleocapsid assembly or stabili-ty. A few non-nucleosidic molecules, belongingto the family of phenylpropenamides (AT-61and AT-130) and heteroaryldihydropyrimidines(BAY41–4109) (Delaney et al. 2002; Deres etal. 2003), could prevent RNA encapsidation ordestabilize nucleocapsids, respectively. Theseantiviral compounds were shown to inhibit thereplication of wild-type HBV as well as HBVmutants resistant to NAs (Billioud et al. 2011).Their molecular mechanism of action is to bindto the HBc and either induce its misdirectionor speed up its multimerization in a way thatpregenomic RNA (pgRNA) Q4is not incorporatedanymore (Zlotnick and Mukhopadhyay 2011).Besides their effect on viral DNA synthesis andvirion production, these agents may poten-tially inhibit the intracellular amplification ofcccDNA via the inhibition of nucleocapsid re-cycling to the nucleus, and may have other ben-eficial effects by modulating interactions be-tween HBV and its hosts, for which the exactmechanisms need to be unraveled. For instance,results of recent studies suggest that HBV coremay activate the transcriptional activity of viralcccDNA and repress the transcription of someISGs (Belloni et al. 2013; Gruffaz et al. 2013).Targeting these specific viral functions may re-sult in mutual beneficial antiviral effects.
A more general manner to inhibit HBV pro-tein functions would be to prevent their trans-lation by degrading viral RNAs. In this respectthe use of antisense or small interfering RNAs(siRNAs) could represent a POC approach toshow that inhibiting the expression of viral pro-teins in the first place could inhibit viral repli-cation or restore functions otherwise inhibitedby viral protein (Wooddell et al. 2013). Hence,one could inhibit the production of HBx, HBc,as well as viral secreted protein (HBeAg andHBsAg, which may have immunomodulatoryfunctions and contribute to HBV immune es-cape [Bertoletti and Ferrari 2012]) and obtainmultiple antiviral effects. But using siRNAs in
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 15
david-durantel-inserm
Note
yes OKcould be written pre-genomic
admin1
Cross-Out
admin1
Inserted Text
es
admin1
Cross-Out

vivo and delivering them to the entire liver, totarget all infected cells, remains a therapeuticchallenge, although major progress has recentlybeen made in that area.
Interfering with other steps of viral mor-phogenesis and virion infectivity through themodulation of viral envelope glycosylation bya-glucosidase inhibitors represent other rele-vant approaches to be developed (Block et al.1998; Lazar et al. 2007). Other groups have alsotried to use triazolopyrimidine derivatives todecrease viral envelope protein secretion in ex-perimental models in the perspective of restor-ing specific immune responses against viral en-velope epitopes (Yu et al. 2011).
Besides the inhibition of viral replication,other antiviral strategies consist in the boost-ing of specific immune responses against HBV.Based on recent knowledge of the role of innateresponses in the control of HBV infection (Zou-lim et al. 2013a), several approaches have beenevaluated to determine, among others, the ef-fect of TLR2 or TLR7 stimulation in the wood-chuck and chimpanzee models, respectively. Forinstance, it was shown that a TLR7 agonist caninduce IFN-a and ISG expression in chimpan-zees, which was associated with reduced serumand liver viral load (Lanford et al. 2013). Tran-sient elevations of serum transaminase levelswere observed. The data were consistent withimmune elimination of infected hepatocytes.Another recent study showed that ETV admin-istration can restore TLR2 expression in infectedcells, and that administration of TLR2 ligandsinhibited viral replication (Zhang et al. 2012). Itwould be interesting to test whether the com-bination of NA with a TLR2 or TLR7 agonistresults in an enhanced antiviral effect (Duranteland Zoulim 2012). Targeting viral determi-nants, which are responsible for defective innateimmune responses, could specifically restore in-nate immunity that would be restricted to in-fected cells and not to all cells expressing innatesensors.
In chronic HBV infection, defective T-cellfunction is probably maintained by the effectof the prolonged exposure of T cells to largequantities of viral antigens and by the tolero-genic features of both liver cells and liver resi-
dent cells (Bertoletti and Ferrari 2012; Knolleand Thimme 2014). These two combinedmechanisms can result in the deletion of HBV-specific T cells or in their functional inactivation(exhaustion), which is characterized by an in-creased expression of negative costimulatorymolecules and dysregulation of costimulatorypathways, which affect antiviral T-cell responses.In principle, restoration of immune controlcould follow different strategies (Bertoletti andFerrari 2012; Knolle and Thimme 2014). Theinhibition of viral replication and decline inHBV antigens could lead to partial restorationof antiviral HBV-specific T-cell functions andinhibition of HBV suppressive effects (Boni etal. 2012). Blockade of negative regulatory path-ways could be effective, by partially restoringHBV-specific T-cell functions. Antiapoptoticdrugs may reduce HBV-specific T-cell apoptosisand fight against T-cell exhaustion. The de novoreconstitution of functionally active HBV-spe-cific T cells or activation of heterologous T cellsis also another potential strategy. Besides thesetargeted immune strategies, attempts to delivertherapeutic vaccines (with recombinant pro-teins, specific peptides, DNA vaccine, or DNAdelivered by viral vectors) have been evalu-ated in chronically infected patients or animals,and may represent an interesting treatment op-tion to be further evaluated in association withNAs, at least in selected patient populations;these different studies have been reviewed re-cently (Michel et al. 2011).
These new strategies should be evaluatedin the most relevant experimental models. In-fectious cell-culture models for HBV rely onprimary human hepatocyte culture and theHepaRG cell line (Gripon et al. 1988, 2002),which are the only robust models used to studythe entire HBV life cycle, but are still tediousto work with, compared with the traditionalHepG2 or Huh7 hepatoma cell lines, whichare used to study the late stages of HBV replica-tion after transfection of replication-competentconstructs. These cell-culture models are nowimproved with the reconstitution of hNTCP ex-pression allowing viral infection and the studyof critical steps just as cccDNA formation (Yanet al. 2012; Urban et al. 2014). Because access to
F. Zoulim and D. Durantel
16 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501
admin1
Inserted Text
(Kosinska, A. D., E. Zhang, L. Johrden, J. Liu, P. L. Seiz, X. Zhang, Z. Ma, T. Kemper, M. Fiedler, D. Glebe, O. Wildner, U. Dittmer, M. Lu, and M. Roggendorf. 2013. Combination of DNA prime--adenovirus boost immunization with entecavir elicits sustained control of chronic hepatitis B in the woodchuck model. PLoS Pathog 9:e1003391.)
admin1
Inserted Text
Kosinska, A. D., E. Zhang, L. Johrden, J. Liu, P. L. Seiz, X. Zhang, Z. Ma, T. Kemper, M. Fiedler, D. Glebe, O. Wildner, U. Dittmer, M. Lu, and M. Roggendorf. 2013. Combination of DNA prime--adenovirus boost immunization with entecavir elicits sustained control of chronic hepatitis B in the woodchuck model. PLoS Pathog 9:e1003391.

chimpanzees is restricted, human HBV replica-tion is currently studied in vivo in humanizeduPA/SCID mice (Petersen et al. 2008). Howev-er, these mouse models have the disadvantage ofusing an immune-deficient host. Nevertheless,these experimental models should be useful forvalidating new targets and elucidating the modeof action of new antiviral compounds. Improve-ment of these models is now in progress withgenetic engineering of mouse lineage to expresshNTPC, as well as to engineer doubly human-ized mice for both human hepatocytes and thehuman immune system. The development ofmore robust small primate models to recapitu-late HBV infection and its pathogenesis is alsohighly desirable (Dupinay et al. 2013).
CONCLUSION
The field of anti-HBV therapy is entering a newera with a renewed interest of the scientific,medical, and industrial communities to developnew treatment concepts toward a cure of HBVinfection. The better knowledge of the viral lifecycle and its interaction with the liver microen-vironment and host immune responses, togeth-er with the development of new study modelswill provide the right momentum for upfrontresearch in this area. The better understandingand measurement of the major clinical endpoints also provide better guidance for the pre-clinical and early clinical evaluation of treat-ment concepts, which should translate into im-proved treatment outcomes in the future.
REFERENCES
Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. 2006.Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443: 590–593.
Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G,Fanciulli M, Raimondo G, Levrero M. 2009. NuclearHBx binds the HBV minichromosome and modifiesthe epigenetic regulation of cccDNA function. Proc NatlAcad Sci 106: 19975–19979.
Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Polli-cino T, Petersen J, Raimondo G, Dandri M, Levrero M.2012. IFN-a inhibits HBV transcription and replicationin cell culture and in humanized mice by targeting theepigenetic regulation of the nuclear cccDNA minichro-mosome. J Clin Invest 122: 529–537.
Belloni L, Li LC, Palumbo GA, Chirapu SR, Calvo L, Finn M,Lopatin U, Zlotnick A, Levrero M. 2013. HAPs hepatitis Bvirus (HBV) capsid inhibitors block core protein inter-action with the viral minichromosome and host cellgenes and affect cccDNA transcription and stability. Hep-atology 58: 277A.
Berg T, Zoulim F, Moeller B, Trinh H, Marcellin P, Chan S,Kitrinos KM, Dinh P, Flaherty JF Jr, McHutchison JG,et al. 2014. Long-term efficacy and safety of emtricitabineplus tenofovir DF vs. tenofovir DF monotherapy in ade-fovir-experienced chronic hepatitis B patients. J Hepatol60: 715–722.
Bertoletti A, Ferrari C. 2012. Innate and adaptive immuneresponses in chronic hepatitis B virus infections: Towardsrestoration of immune control of viral infection. Gut61: 1754–1764.
Billioud G, Pichoud C, Puerstinger G, Neyts J, Zoulim F.2011. The main hepatitis B virus (HBV) mutants resis-tant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antiviral Res92: 271–276.
Block TM, Lu X, Mehta AS, Blumberg BS, Tennant B, EblingM, Korba B, Lansky DM, Jacob GS, Dwek RA. 1998.Treatment of chronic hepadnavirus infection in a wood-chuck animal model with an inhibitor of protein foldingand trafficking. Nat Med 4: 610–614.
Bojang P Jr, Ramos KS. 2014. The promise and failures ofepigenetic therapies for cancer treatment. Cancer TreatRev 40: 153–169.
Boni C, Laccabue D, Lampertico P, Giuberti T, Vigano M,Schivazappa S, Alfieri A, Pesci M, Gaeta GB, BrancaccioG, et al. 2012. Restored function of HBV-specific T cellsafter long-term effective therapy with nucleos(t)ide ana-logues. Gastroenterology 143: 963–973.e9.
Buti M. 2014. HBeAg-positive chronic hepatitis B: Why do Itreat my patients with nucleos(t)ide analogs? Liver Int34: 108–111.
Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, MasonWS, Xu X, Guo JT, Block TM, et al. 2012. Identificationof the disubstituted sulfonamide compounds as specificinhibitors of hepatitis B virus covalently closed circularDNA formation. Antimicrob Agents Chemother 56: 4277–4288.
Carrouee-Durantel S, Durantel D, Werle-Lapostolle B, Pi-choud C, Naesens L, Neyts J, Trepo C, Zoulim F. 2008.Suboptimal response to adefovir dipivoxil therapy forchronic hepatitis B in nucleoside-naıve patients is notdue to pre-existing drug-resistant mutants. Antivir Ther13: 381–388.
Chan HL, Chan CK, Hui AJ, Chan S, Poordad F, Chang TT,Mathurin P, Flaherty JF, Lin L, Corsa A, et al. 2014. Effectsof tenofovir disoproxil fumarate in hepatitis B e antigen-positive patients with normal levels of alanine amino-transferase and high levels of hepatitis B virus DNA. Gas-troenterology 146: 1240–1248.
Cho J-Y, Paik Y-H, Sohn W, Cho HC, Gwak G-Y, Choi MS,Lee JH, Koh KC, Paik SW, Yoo BC. 2014. Patients withchronic hepatitis B treated with oral antiviral therapyretain a higher risk for HCC compared with patientswith inactive stage disease. Gut 63: 1943–1950.
Delaney WE, Edwards R, Colledge D, Shaw T, Furman P,Painter G, Locarnini S. 2002. Phenylpropenamide deriv-
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 17
admin1
Inserted Text
Shlomai, A., R. E. Schwartz, V. Ramanan, A. Bhatta, Y. P. de Jong, S. N. Bhatia, and C. M. Rice. 2014. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc Natl Acad Sci U S A 111:12193-12198.
admin1
Inserted Text
cell culture models and

atives AT-61 and AT-130 inhibit replication of wild-typeand lamivudine-resistant strains of hepatitis B virus invitro. Antimicrob Agents Chemother 46: 3057–3060.
Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ,Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J,et al. 2003. Inhibition of hepatitis B virus replication bydrug-induced depletion of nucleocapsids. Science 299:893–896.
Dienstag JL, Goldin RD, Heathcote EJ, Hann HW, WoessnerM, Stephenson SL, Gardner S, Gray DF, Schiff ER. 2003.Histological outcome during long-term lamivudine ther-apy. Gastroenterology 124: 105–117.
Dupinay T, Gheit T, Roques P, Cova L, Chevallier-Queyron P,Tasahsu SI, Le Grand R, Simon F, Cordier G, Wakrim L,et al. 2013. Discovery of naturally occurring transmissiblechronic hepatitis B virus infection among Macaca fasci-cularis from Mauritius Island. Hepatology 58: 1610–1620.
Durantel D, Zoulim F. 2012. Interplay between hepatitis Bvirus and TLR2-mediated innate immune responses: Canrestoration of TLR2 functions be a new therapeutic op-tion? J Hepatol 57: 486–489.
Durantel D, Brunelle M-N, Gros E, Carrouee-Durantel S,Pichoud C, Villet S, Trepo C, Zoulim F. 2005. Resistanceof human hepatitis B virus to reverse transcriptase inhib-itors: From genotypic to phenotypic testing. J Clin Virol34: S34–S43.
EASL. 2012. EASL clinical practice guidelines: Managementof chronic hepatitis B virus infection. J Hepatol 57: 167–185.
Fung SK, Andreone P, Han SH, Rajender Reddy K, Regev A,Keeffe EB, Hussain M, Cursaro C, Richtmyer P, MarreroJA, et al. 2005. Adefovir-resistant hepatitis B can be asso-ciated with viral rebound and hepatic decompensation. JHepatol 43: 937–943.
Fung S, Kwan P, Fabri M, Horban A, Pelemis M, Hann H-W,Gurel S, Caruntu FA, Flaherty JF, Massetto B, et al. 2014.Randomized comparison of tenofovir disoproxil fuma-rate vs emtricitabine and tenofovir disoproxil fumarate inpatients with lamivudine-resistant chronic hepatitis B.Gastroenterology 146: 980–988.
Gane EJ, Deray G, Liaw Y-F, Lim SG, Lai C-L, Rasenack J,Wang Y, Papatheodoridis G, Di Bisceglie A, Buti M, et al.2014. Telbivudine improves renal function in patientswith chronic hepatitis B. Gastroenterology 146: 138–146.e5.
Gish RG, Lok AS, Chang TT, de Man RA, Gadano A, SollanoJ, Han KH, Chao YC, Lee SD, Harris M, et al. 2007.Entecavir therapy for up to 96 weeks in patients withHBeAg-positive chronic hepatitis B. Gastroenterology133: 1437–1444.
Gish R, Jia JD, Locarnini S, Zoulim F. 2012. Selection ofchronic hepatitis B therapy with high barrier to resis-tance. Lancet Infect Dis 12: 341–353.
Gordon SC, Krastev Z, Horban A, Petersen J, Sperl J, Dinh P,Martins EB, Yee LJ, Flaherty JF, Kitrinos KM, et al. 2013.Efficacy of tenofovir disoproxil fumarate at 240 weeks inpatients with chronic hepatitis B with high baseline viralload. Hepatology 58: 505–513.
Gripon P, Diot C, Theze N, Fourel I, Loreal O, Brechot C,Guguen-Guillouzo C. 1988. Hepatitis B virus infection of
adult human hepatocytes cultured in the presence of di-methyl sulfoxide. J Virol 62: 4136–4143.
Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I,Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C.2002. Infection of a human hepatoma cell line by hepa-titis B virus. Proc Natl Acad Sci 99: 15655–15660.
Gruffaz M, Testoni B, Luangsay S, Fusil F, Malika AG, Man-cip J, Petit MA, Javanbakht H, Cosset FL, Zoulim F, et al.2013. The nuclear function of hepatitis B capsid (HBc)protein is to inhibit IFN response very early after infec-tion of hepatocytes. Hepatology 58: 276A.
Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT,Kitis G, Rizzetto M, Marcellin P, Lim SG, Goodman Z,Ma J, et al. 2006. Long-term therapy with adefovir dipi-voxil for HBeAg-negative chronic hepatitis B for up to5 years. Gastroenterology 131: 1743–1751.
Heathcote EJ, Marcellin P, Buti M, Gane E, De Man RA,Krastev Z, Germanidis G, Lee SS, Flisiak R, Kaita K,et al. 2011. Three-year efficacy and safety of tenofovirdisoproxil fumarate treatment for chronic hepatitis B.Gastroenterology 140: 132–143.
Heidrich B, Yurdaydin C, Kabacam G, Ratsch BA, Zachou K,Bremer B, Dalekos GN, Erhardt A, Tabak F, Yalcin K, et al.2014. Late HDV RNA relapse after peginterferona-basedtherapy of chronic hepatitis delta. Hepatology 60: 87–97.
Hosaka T, Suzuki F, Kobayashi M, Seko Y, Kawamura Y,Sezaki H, Akuta N, Suzuki Y, Saitoh S, Arase Y, et al.2013. Long-term entecavir treatment reduces hepatocel-lular carcinoma incidence in patients with hepatitis Bvirus infection. Hepatology 58: 98–107.
Janssen HLA, van Zonneveld M, Senturk H, Zeuzem S,Akarca US, Cakaloglu Y, Simon C, So TMK, Gerken G,de Man RA, et al. 2005. Pegylated interferon alfa-2b aloneor in combination with lamivudine for HBeAg-positivechronic hepatitis B: A randomised trial. Lancet 365: 123–129.
Jones PA. 2014. At the tipping point for epigenetic therapiesin cancer. J Clin Invest 124: 14–16.
Jordheim LP, Durantel D, Zoulim F, Dumontet C. 2013.Advances in the development of nucleoside and nucleo-tide analogues for cancer and viral diseases. Nat Rev DrugDiscov 12: 447–464.
Kao JH. 2014. HBeAg-positive chronic hepatitis B: Why do Itreat my patients with pegylated interferon? Liver Int 34:112–119.
Kennedy PT, Sandalova E, Jo J, Gill U, Ushiro-Lumb I, TanAT, Naik S, Foster GR, Bertoletti A. 2012. Preserved T-cellfunction in children and young adults with immune-tol-erant chronic hepatitis B. Gastroenterology 143: 637–645.
Kitrinos KM, Corsa A, Liu Y, Flaherty J, Snow-Lampart A,Marcellin P, Borroto-Esoda K, Miller MD. 2014. No de-tectable resistance to tenofovir disoproxil fumarate after 6years of therapy in patients with chronic hepatitis B.Hepatology 59: 434–442.
Knolle PA, Thimme R. 2014. Hepatic immune regulationand its involvement in viral hepatitis infection. Gastroen-terology 146: 1193–1207.
Lai CL, Yuen MF. 2013. Prevention of hepatitis B virus-re-lated hepatocellular carcinoma with antiviral therapy.Hepatology 57: 399–408.
F. Zoulim and D. Durantel
18 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501

Lai M, Hyatt BJ, Nasser I, Curry M, Afdhal NH. 2007. Theclinical significance of persistently normal ALT in chronichepatitis B infection. J Hepatol 47: 760–767.
Lampertico P, Liaw YF. 2012. New perspectives in the ther-apy of chronic hepatitis B. Gut 61: 18–24.
Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL,Brasky KM, Fosdick A, Frey CR, Zheng J, Wolfgang G,et al. 2013. GS-9620, an oral agonist of toll-like receptor-7, induces prolonged suppression of hepatitis B virus inchronically infected chimpanzees. Gastroenterology 144:1508–1517.
Lau GKK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S,Cooksley G, Gane E, Fried MW, Chow WC, Paik SW, et al.2005. Peginterferon alfa-2a, lamivudine, and the combi-nation for HBeAg-positive chronic hepatitis B. N Engl JMed 352: 2682–2695.
Lavocat F, Deny P, Pichoud C, Al Hawajri N, Kitrinos K,Borroto-Esoda K, Zoulim F. 2013. Similar evolution ofhepatitis B virus quasispecies in patients with incompleteadefovir response receiving tenofovir/emtricitabinecombination or tenofovir monotherapy. J Hepatol 59:684–695.
Lazar C, Durantel D, Macovei A, Zitzmann N, Zoulim F,Dwek RA, Branza-Nichita N. 2007. Treatment of hepatitisB virus-infected cells with a-glucosidase inhibitors re-sults in production of virions with altered molecularcomposition and infectivity. Antiviral Res 76: 30–37.
Le Guerhier F, Pichoud C, Guerret S, Chevallier M, JamardC, Hantz O, Li XY, Chen SH, King I, Trepo C, et al. 2000.Characterization of the antiviral effect of 20,30-dideoxy-20, 30-didehydro-b-L-5-fluorocytidine in the duck hepa-titis B virus infection model. Antimicrob Agents Chemo-ther 44: 111–122.
Le Guerhier F, Pichoud C, Jamard C, Guerret S, ChevallierM, Peyrol S, Hantz O, King I, Trepo C, Cheng YC, et al.2001. Antiviral activity of b-L-20,30-dideoxy-20,30-dide-hydro-5-fluorocytidine in woodchucks chronically in-fected with woodchuck hepatitis virus. Antimicrob AgentsChemother 45: 1065–1077.
Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. 2010.A promiscuous a-helical motif anchors viral hijackersand substrate receptors to the CUL4-DDB1 ubiquitinligase machinery. Nat Struct Mol Biol 17: 105–111.
Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H,Tanwandee T, Tao QM, Shue K, Keene ON, et al. 2004.Lamivudine for patients with chronic hepatitis B andadvanced liver disease. N Engl J Med 351: 1521–1531.
Liaw YF, Raptopoulou-Gigi M, Cheinquer H, Sarin SK, Tan-wandee T, Leung N, Peng CY, Myers RP, Brown RS Jr,Jeffers L, et al. 2011a. Efficacy and safety of entecavirversus adefovir in chronic hepatitis B patients with he-patic decompensation: A randomized open-label study.Hepatology 54: 91–100.
Liaw YF, Sheen IS, Lee CM, Akarca US, Papatheodoridis GV,Suet-Hing Wong F, Chang TT, Horban A, Wang C, KwanP, et al. 2011b. Tenofovir disoproxil fumarate (TDF), em-tricitabine/TDF, and entecavir in patients with decom-pensated chronic hepatitis B liver disease. Hepatology 53:62–72.
Liu Y, Kitrinos KM. 2013. In vitro phenotyping of recom-binant hepatitis B virus containing the polymerase/
reverse transcriptase gene from clinical isolates. MethodsMol Biol 1030: 163–181.
Liu Y, Wang C, Zhong Y, Chen L, Li X, Ji D, Wang H, Xin S,Zoulim F, Xu D. 2010. Evolution and suppression of HBVstrains with multidrug resistance to lamivudine, adefovirdipivoxil and entecavir in a patient with chronic hepatitisB. Antivir Ther 15: 1185–1190.
Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, Dien-stag JL, Heathcote EJ, Little NR, Griffiths DA, et al. 2003.Long-term safety of lamivudine treatment in patientswith chronic hepatitis B. Gastroenterology 125: 1714–1722.
Lok AS, Trinh H, Carosi G, Akarca US, Gadano A, Haber-setzer F, Sievert W, Wong D, Lovegren M, Cohen D,et al. 2012. Efficacy of entecavir with or without teno-fovir disoproxil fumarate for nucleos(t)ide-naıve patientswith chronic hepatitis B. Gastroenterology 143: 619–628.e1.
Lucifora J, Arzberger S, Durantel D, Belloni L, StrubinM, Levrero M, Zoulim F, Hantz O, Protzer U. 2011.Hepatitis B virus X protein is essential to initiate andmaintain virus replication after infection. J Hepatol 55:996–1003.
Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X,Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, et al.2014. Specific and nonhepatotoxic degradation of nucle-ar hepatitis B virus cccDNA. Science 343: 1221–1228.
Lutgehetmann M, Mancke LV, Volz T, Helbig M, Allweiss L,Bornscheuer T, Pollok JM, Lohse AW, Petersen J, Urban S,et al. 2012. Humanized chimeric uPA mouse model forthe study of hepatitis B and D virus interactions andpreclinical drug evaluation. Hepatology 55: 685–694.
Marcellin P, Lau GKK, Bonino F, Farci P, Hadziyannis S, JinR, Lu Z-M, Piratvisuth T, Germanidis G, Yurdaydin C,et al. 2004. Peginterferon alfa-2a alone, lamivudine alone,and the two in combination in patients with hbeag-neg-ative chronic hepatitis B. N Engl J Med 351: 1206–1217.
Mason WS, Litwin S, Jilbert AR. 2008. Immune selectionduring chronic hepadnavirus infection. Hepatol Int 2:3–16.
Maynard M, Parvaz P, Durantel S, Chevallier M, ChevallierP, Lot M, Trepo C, Zoulim F. 2005. Sustained HBs sero-conversion during lamivudine and adefovir dipivoxilcombination therapy for lamivudine failure. J Hepatol42: 279–281.
Micco L, Peppa D, Loggi E, Schurich A, Jefferson L, CursaroC, Panno AM, Bernardi M, Brander C, Bihl F, et al. 2013.Differential boosting of innate and adaptive antiviral re-sponses during pegylated-interferon-a therapy of chron-ic hepatitis B. J Hepatol 58: 225–233.
Michel ML, Deng Q, Mancini-Bourgine M. 2011. Thera-peutic vaccines and immune-based therapies for thetreatment of chronic hepatitis B: Perspectives and chal-lenges. J Hepatol 54: 1286–1296.
Moraleda G, Saputelli J, Aldrich CE, Averett D, Condreay L,Mason WS. 1997. Lack of effect of antiviral therapy innondividing hepatocyte cultures on the closed circularDNA of woodchuck hepatitis virus. J Virol 71: 9392–9399.
Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, FalthM, Stindt J, Koniger C, Nassal M, Kubitz R, et al. 2014.Hepatitis B and D viruses exploit sodium taurocholate
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 19

co-transporting polypeptide for species-specific entryinto hepatocytes. Gastroenterology 146: 1070–1083.
Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, Lohmann V, Mier W, Mehrle S, Urban S. 2014.Cyclosporin A inhibits hepatitis B and hepatitis D virusentry by cyclophilin-independent interference with theNTCP receptor. J Hepatol 60: 723–731.
Ono A, Suzuki F, Kawamura Y, Sezaki H, Hosaka T, Akuta N,Kobayashi M, Suzuki Y, Saitou S, Arase Y, et al. 2012.Long-term continuous entecavir therapy in nucleos(t)ide-naıve chronic hepatitis B patients. J Hepatol 57:508–514.
Papatheodoridis GV, Manolakopoulos S, Touloumi G,Vourli G, Raptoppoulou-Gigi M, Vasiladis T, MimidisK, Gogos C, Ketikoglou I, Manesis EK, et al. 2011. Viro-logical suppression does not prevent the development ofhepatocellular carcinoma in HBeAg-negative chronichepatitis B patients with cirrhosis receiving oral antivi-ral(s) starting with lamivudine monotherapy: Results ofthe nationwide HEPNET. Greece cohort study. Gut 60:1109–1116.
Patterson SJ, George J, Strasser SI, Lee AU, Sievert W, NicollAJ, Desmond PV, Roberts SK, Locarnini S, Bowden S,et al. 2011. Tenofovir disoproxil fumarate rescue therapyfollowing failure of both lamivudine and adefovir dipi-voxil in chronic hepatitis B. Gut 60: 247–254.
Petersen J, Dandri M, Mier W, Lutgehetmann M, Volz T, vonWeizsacker F, Haberkorn U, Fischer L, Pollok JM, Erbes B,et al. 2008. Prevention of hepatitis B virus infection invivo by entry inhibitors derived from the large envelopeprotein. Nat Biotechnol 26: 335–341.
Sadler AJ, Williams BRG. 2008. Interferon-inducible antivi-ral effectors. Nat Rev Immunol 8: 559–568.
Samuel CE. 2001. Antiviral actions of interferons. Clin Mi-crobiol Rev 14: 778–809.
Scaglione SJ, Lok AS. 2012. Effectiveness of hepatitis B treat-ment in clinical practice. Gastroenterology 142: 1360–1368.e1.
Seeger C, Zoulim F, Mason WS. 2014. Hepadnaviruses. InField’s virology (ed. Knipe DM, Howley PM), p. 2185.Lippincott Williams & Wilkins, Philadelphia.
Sogni P, Carrieri MP, Fontaine H, Mallet V, Vallet-Pichard A,Trabut JB, Meritet JF, Pol S. 2012. The role of adherence invirological suppression in patients receiving anti-HBVanalogues. Antivir Ther 17: 395–400.
Sohn JA, Litwin S, Seeger C. 2009. Mechanism for CCCDNA synthesis in hepadnaviruses. PLoS ONE 4: e8093.
Sun J, Xie Q, Tan D, Ning Q, Niu J, Bai X, Fan R, Chen S,Cheng J, Yu Y, et al. 2014. The 104-week efficacy andsafety of telbivudine-based optimization strategy inchronic hepatitis B patients: A randomized, controlledstudy. Hepatology 59: 1283–1292.
Thimme R, Dandri M. 2013. Dissecting the divergent effectsof interferon-a on immune cells: Time to rethink com-bination therapy in chronic hepatitis B? J Hepatol 58:205–209.
Urban S, Bartenschlager R, Kubitz R, Zoulim F. 2014. Strat-egies to inhibit entry of HBVand HDV into hepatocytes.Gastroenterology 147: 48–64.
Vigano M, Mangia G, Lampertico P. 2014. HBeAg-negativechronic hepatitis B: Why do I treat my patients withnucleos(t)ide analogues? Liver Int 34: 120–126.
Villet S, Ollivet A, Pichoud C, Barraud L, Villeneuve JP,Trepo C, Zoulim F. 2007. Stepwise process for the devel-opment of entecavir resistance in a chronic hepatitis Bvirus infected patient. J Hepatol 46: 531–538.
Villet S, Pichoud C, Billioud G, Barraud L, Durantel S, TrepoC, Zoulim F. 2008. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J Hepatol48: 747–755.
Vlachogiannakos J, Papatheodoridis GV. 2014. HBeAg-neg-ative chronic hepatitis B: Why do I treat my patients withpegylated interferon-alfa? Liver Int 34: 127–132.
Warner N, Locarnini S. 2008. The antiviral drug selectedhepatitis B virus rtA181T/sW172� mutant has a domi-nant negative secretion defect and alters the typical pro-file of viral rebound. Hepatology 48: 88–98.
Watashi K, Sluder A, Daito T, Matsunaga S, Ryo A, Naga-mori S, Iwamoto M, Nakajima S, Tsukuda S, Borroto-Esoda K, et al. 2014. Cyclosporin A and its analogs inhibithepatitis B virus entry into cultured hepatocytes throughtargeting a membrane transporter, sodium taurocholatecotransporting polypeptide (NTCP). Hepatology 59:1726–1737.
Welsch C, Jesudian A, Zeuzem S, Jacobson I. 2012.New direct-acting antiviral agents for the treatment ofhepatitis C virus infection and perspectives. Gut 61:i36–i46.
Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K,Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z,Delaney WE, et al. 2004. Persistence of cccDNA duringthe natural history of chronic hepatitis B and declineduring adefovir dipivoxil therapy. Gastroenterology 126:1750–1758.
Wong DK, Seto WK, Fung J, Ip P, Huang FY, Lai CL, YuenMF. 2013. Reduction of hepatitis B surface antigen andcovalently closed circular DNA by nucleos(t)ide ana-logues of different potency. Clin Gastroenterol Hepatol11: 1004–1010.
Wooddell CI, Rozema DB, Hossbach M, John M, HamiltonHL, Chu Q, Hegge JO, Klein JJ, Wakefield DH, OropezaCE, et al. 2013. Hepatocyte-targeted RNAi therapeuticsfor the treatment of chronic hepatitis B virus infection.Mol Ther 21: 973–985.
Wu C-Y, Lin J-T, Ho HJ, Su C-W, Lee T-Y, Wang S-Y, Wu C,Wu J-C. 2014. Association of nucleos(t)ide analoguetherapy with reduced risk of hepatocellular carcinomain patients with chronic hepatitis B—A Nationwide Co-hort Study. Gastroenterology 147: 143–151.e5.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y,Peng B, Wang H, et al. 2012. Sodium taurocholate co-transporting polypeptide is a functional receptor for hu-man hepatitis B and D virus. Elife 1: e00049.
Yu W, Goddard C, Clearfield E, Mills C, Xiao T, Guo H,Morrey JD, Motter NE, Zhao K, Block TM, et al. 2011.Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis Bvirus surface antigen (HBsAg) secretion. J Med Chem 54:5660–5670.
Zhang X, Ma Z, Liu H, Liu J, Meng Z, Broering R, Yang D,Schlaak JF, Roggendorf M, Lu M. 2012. Role of Toll-like
F. Zoulim and D. Durantel
20 Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501

receptor 2 in the immune response against hepadnaviralinfection. J Hepatol 57: 522–528.
Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, MillerDS, Litwin S, Furman PA, Jilbert AR, Mason WS. 2001.Kinetics of hepadnavirus loss from the liver during inhi-bition of viral DNA synthesis. J Virol 75: 311–322.
Zimmerman KA, Fischer KP, Joyce MA, Tyrrell DL. 2008.Zinc finger proteins designed to specifically target duckhepatitis B virus covalently closed circular DNA inhibitviral transcription in tissue culture. J Virol 82: 8013–8021.
Zlotnick A, Mukhopadhyay S. 2011. Virus assembly, alloste-ry and antivirals. Trends Microbiol 19: 14–23.
Zoulim F. 2011. Hepatitis: Treatment failure in chronic hep-atitis B. Nat Rev Gastroenterol Hepatol 8: 366–367.
Zoulim F. 2012. Are novel combination therapies needed forchronic hepatitis B? Antiviral Res 96: 256–259.
Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance tonucleos(t)ide analogues. Gastroenterology 137: 1593–1608. e1591–e1592.
Zoulim F, Mason WS. 2012. Reasons to consider earliertreatment of chronic HBV infections. Gut 61: 333–336.
Zoulim F, Saputelli J, Seeger C. 1994. Woodchuck hepatitisvirus X protein is required for viral infection in vivo. JVirol 68: 2026–2030.
Zoulim F, Luangsay S, Durantel D. 2013a. Targeting innateimmunity: A new step in the development of combina-tion therapy for chronic hepatitis B. Gastroenterology 144:1342–1344.
Zoulim F, Testoni B, Lebosse F. 2013b. Kinetics of intrahe-patic covalently closed circular DNA and serum hepatitisB surface antigen during antiviral therapy for chronichepatitis B: Lessons from experimental and clinical stud-ies. Clin Gastroenterol Hepatol 11: 1011–1013.
Antiviral Therapies of Chronic HBV Infections
Advanced Online Article. Cite this article as Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021501 21

cshperspectmed-HEP-a021501Queries
Fabien Zoulim and David Durantel
Q1 Please check definition of cccDNA.
Q2 Please define POL/RT, which appears in figure 2.
Q3 Please check definition of HBc.
Q4 Please verify definition of pgRNA.
Antiviral Therapies and Prospects for a Cure of Chronic Hepatitis B
Fabien Zoulim and David Durantel
TOC Blurb: The ideal goal for hepatitis B (HBV) treatment (HBsAg clearance and/or HBsAbseroconversion) is rarely achieved with current anti-HBVagents. Improved knowledge of theHBV life cycle and HBV–host interactions is therefore needed.
Related Documents



![1. k]π$Nr[ L°$ ]y$NÆr[ dp h p A¨[fd ° Apcpfu R>° · 3. dfZ ∆h__y¨ Ad©[ R>°. Mf°Mf iy¨ dfZ Mfpb gpN° R>° ? A°L$ fu[° rhQpfuA° [p° dfZ Mfpb _\u gpN[y¨. Ofdp¨ Ap`Zp¨](https://static.cupdf.com/doc/110x72/5f0e6bc07e708231d43f297b/1-knr-l-ynr-dp-h-p-afd-apcpfu-r-3-dfz-ahy-ad.jpg)







