Yoga Windhu Wardhana

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Yoga Windhu Wardhana

DUA PRODUK OBAT, memiliki :
Zat Aktif sama
Kadar Zat Aktif sama Bentuk sediaan sama (misalnya tablet)
APAKAH KHASIAT/EFEK TERAPI SAMA ???

Sebagian besar obat memperlihatkan gambaran laju
disolusi yang terbatas dan absorpsi in vivo yang tidak lengkap
Beberapa obat diabsorpsi secara terbatas pada saluran cerna
Formulasi obat kemungkinan mengubah laju dan jumlah absorpsi, sehingga menghasilkan kegagalan terapi atau menimbulkan efek samping.
Banyak obat-obat yang mengalami peristiwa extensive first pass effect yang menyebabkan variasi kadar darah yang tinggi antar individu.
Alasan Uji Bioavailabilitas
Metode Uji Biovailabilitas
Metode in Vivo
Melalui demonstrasi efek klinis yang
signifikan
Kompleks, mahal, memakan waktu
Memerlukan ukuran yang sensitif dan
kuantitatif dari respon yang diinginkan

Istilah yang menyatakan jumlah/proporsi (extent)
obat yang diabsorpsi dan kecepatan (rate) absorpsi itu terjadi. Extent biasanya dinyatakan dalam ‘F’.
Biasanya diukur dari perkembangan kadar obat (senyawa aktif) dalam darah atau dari ekskresinya dalam urin.
Bioavaibilitas (Ketersediaan Hayati)

Pengembangan senyawa baru Eksplorasi/pengembangan ilmu Pengembangan produk/formulasi Jaminan mutu produk (quality control)
Tujuan penelitian ketersediaan hayati

Faktor obat Faktor subyek Faktor rute pemberian Faktor antaraksi obat/makanan
Faktor yang mempengaruhi bioavailabilitas

Sifat fisikokimia obat Faktor Formulasi Faktor Teknologi Pembuatan
Faktor obat

Karakteristik subyek (umur, bobot badan,
jenis kelamin, gol. darah, ras,dll) Kondisi pato-fisiologis Aktivitas dan posisi tubuh (pada subyek
yang sama) dll.
Faktor subyek

Makanan / Minuman obat-obatan lain
Interferensi bahan/ senyawa lain

Absolut : dibandingkan dengan rute i.v. Relatif : antar sediaan/rute
Perhitungan ‘F’

Perhitungan ‘F’ absolut
Data darah: Data urin:
..100%
.uji iv
iv uji
AUC DF
AUC D=
..100%
.uji iv
iv uji
Qe DF
Qe D=

Perhitungan ‘F’ relatif
Data darah: Data urin:
..100%
.uji p
p uji
AUC DF
AUC D=
..100%
.uji p
p uji
Qe DF
Qe D=
AUC adalah luas di bawah kurva (area under the curve) Qe adalah jumlah obat yang diekskresikan secara kumulatif
dalam urin

Dua produk obat disebut bioekivalen jika keduanya pada pemberian dengan dosis yang sama akan menghasilkan bioavailabilitas yang setara/ekivalen (dengan kondisi yang dibakukan) sehingga efeknya diharapkan akan sama (interchangeable).
Bioekivalen

Kriteria Produk yang diuji Bioekivalensi
1. Produk yang tidak memerlukan uji ekivalensi
2. Produk yang cukup dilakukan uji ekivalensi in vitro (uji disolusi terbanding)
3. Produk yang memerlukan uji ekivalensi in vivo.

1. Produk sediaan parenteral (iv, im, sk) dengan kadar sama.
Untuk rute ekstravaskular, boleh menggunakan eksipien berbeda asalkan tidak mempengaruhi keamanan & efikasi.
2. Produk sediaan larutan dengan kadar sama. 3. Produk sediaan serbuk untuk dilarutkan. 4. Produk sediaan gas / aerosol untuk inhalasi. 5. Produk sediaan untuk tetes mata / telinga yang larut air. 6. Produk sediaan topikal.
Kecuali bila produk tanpa pembanding (baru) Perlu pengujian secara in-vivo & in-vitro
Produk yang tidak perlu Uji Ekivalensi

In Vitro Bioekivalen (Disolusi Terbanding)
Prinsip Dua atau lebih produk atau bets yang terdiri dari zat aktif
yang sama dibandingkan Kekuatan dosis produk / bets mungkin atau tidak memiliki
kesamaan (bergantung pada tujuan pengujian) Kondisi pelaksanaan disolusi mirip, seperti :
• Apparatus, medium, volume, kecepatan rotasi & temperatur. • Meminimalkan perbedaan kondisi pengujian sekecil mungkin

Prinsip (cont’d) Sampel diambil pada saat titik yang sama dan
kemudian data (dissolution profiles) dibandingkan Perhitungan: Perubahan volume harus sama pada
medium disolusi
In Vitro Bioekivalen

Dua kondisi tersebut dapat ditentukan jika profile disolusi
kedua produk/bets pada setiap pengujian dengan medium disolusi memiliki kesamaan :
1. Jika kedua hasil uji dan produk referen (baku) menunjukkan lebih dari 85% disolusi selama 15 min, profile demikian dipertimbangkan memiliki kemiripan • Tidak diperlukan perhitungan
jika tidak seperti diatas, gunakan poin 2 2. Hitung nila f2 (faktor similaritas):
• Jika f2 ≥ 50, Profile pada normalnya memiliki kemiripan (similar)
In Vitro Bioekivalen

Similarity Factor (f2) n = number of time points R(t) = mean % API dissolved of reference product at time point x T(t) = mean % API dissolved of test product at time point x Minimum of 3 time points (zero excluded) 12 units (each in own dissolution vessel) for each product (for
“official” purposes) Only one measurement should be considered after both products
have reached 85 % dissolution RSD at higher time points ≤ 10%

Test Condition
Apparatus (choice)
• Paddle, 50 (75) rpm or • Basket, 100 rpm
Dissolution media All three media for full comparison
1. Buffer pH 6.8 or simulated intestinal fluid without enzymes
2. Buffer pH 4.5 3. 0.1 M HCl or buffer pH 1.2 or simulated gastric
fluid without enzymes
Volume of media 900 ml or less
Temperature 37°C ± 0.5°C
Sampling points 10, 15, 20, 30, 45, (60, 120) min. (typical)
Units (individual) 12 for “official” studies

Example Lamivudine 150 mg & zidovudine 300 mg tablets
Source, WHO publication: Ongoing Monitoring of Antiretroviral Products as Part of
WHO’s Prequalification Project. Journal of Generic Medicines (accepted for publication, January 2006 edition)
Samples from PQ project or bought/requested
Apparatus: paddle at 75 rpm Medium: 900 ml 0.1 M hydrochloric acid, 37°C Sample times: 5, 10, 15, 20, 30 and 45 minutes Analysis: HPLC
Data presented for individual APIs in next tables

Example Lamivudine 150 mg & zidovudine 300 mg tablets
Time (min)
% Lamivudine of label claim dissolved Combivir® Gen-1 Gen-2 Gen-3 Gen-4
5 85 25 92 65 45 10 96 46 96 85 81 15 97 65 98 95 92 20 97 80 98 98 95 30 97 97 98 98 96 45 97 97 98 98 97
≥ 85 in 15 min ?
✔ Reference
no ✔ ✔ ✔
f2 21

Time (min)
% Zidovudine of label claim dissolved Combivir® Gen-1 Gen-2 Gen-3 Gen-4
5 85 22 74 68 45 10 97 44 90 88 83 15 98 64 97 96 95 20 98 81 99 100 98 30 98 100 101 99 99 45 99 100 100 99 100
≥ 85 in 15 min ?
✔ Reference
no ✔ ✔ ✔
f2 20

Conclusion (considering only 0.1 M HCl as medium)
1. 3 products show profile similarity with Combivir® (≥ 85% in 15 minutes)
2. The profiles of Combivir® and Gen-1 are not similar • The products may still show bio-equivalency
The dissolution profiles of the APIs in a particular product are
similar (true for all 5 products) Examples: see profiles of Combivir® and Gen-1
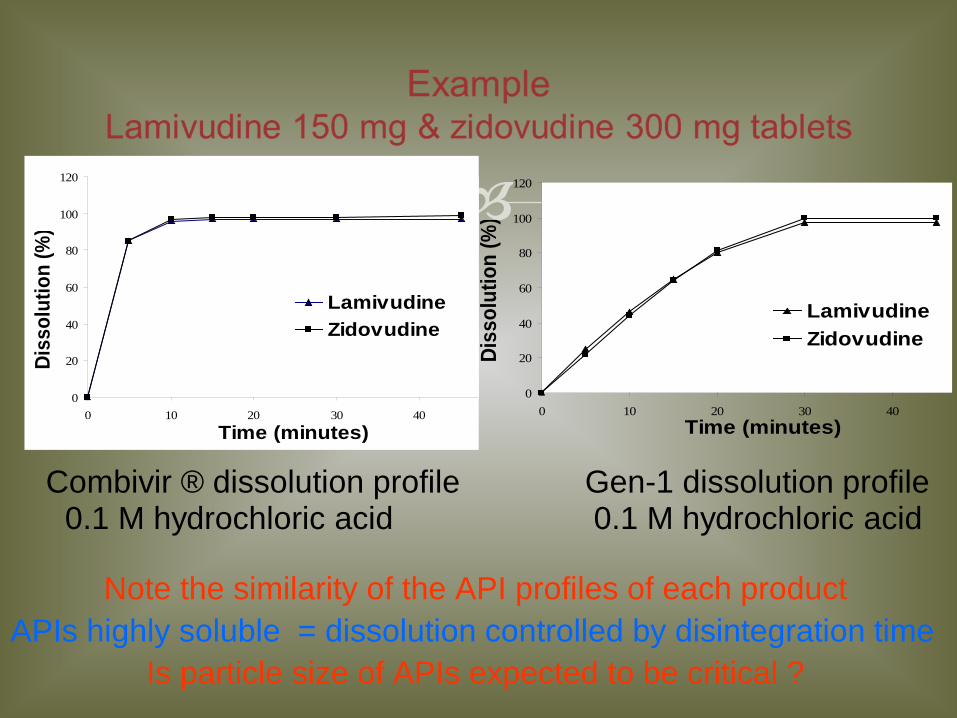
Combivir ® dissolution profile Gen-1 dissolution profile 0.1 M hydrochloric acid 0.1 M hydrochloric acid
Note the similarity of the API profiles of each product APIs highly soluble = dissolution controlled by disintegration time
Is particle size of APIs expected to be critical ?
0
20
40
60
80
100
120
0 10 20 30 40
Time (minutes)
Diss
olut
ion
(%)
LamivudineZidovudine
0
20
40
60
80
100
120
0 10 20 30 40Time (minutes)
Diss
olut
ion
(%)
LamivudineZidovudine
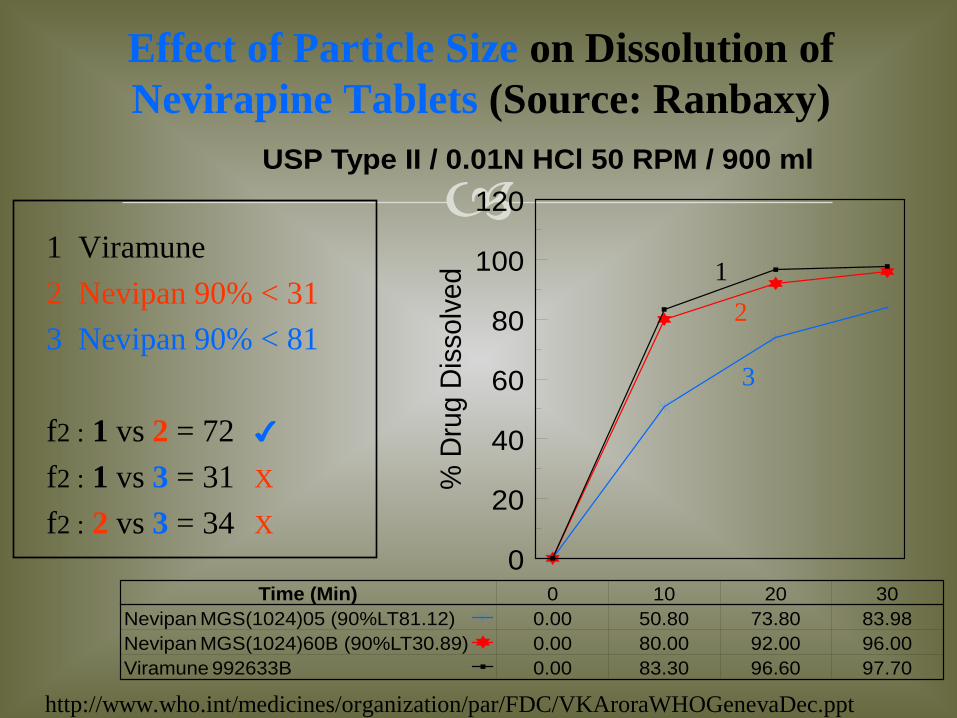
USP Type II / 0.01N HCl 50 RPM / 900 ml
0
20
40
60
80
100
120
Time (Min)Nevipan MGS(1024)05 (90%LT81.12)Nevipan MGS(1024)60B (90%LT30.89)Viramune 992633B
0 10 20 300.00 50.80 73.80 83.980.00 80.00 92.00 96.000.00 83.30 96.60 97.70
% D
rug
Dis
solv
ed
http://www.who.int/medicines/organization/par/FDC/VKAroraWHOGenevaDec.ppt
1 Viramune 2 Nevipan 90% < 31 3 Nevipan 90% < 81 f2 : 1 vs 2 = 72 ✔ f2 : 1 vs 3 = 31 X
f2 : 2 vs 3 = 34 X
1 2
3
Effect of Particle Size on Dissolution of Nevirapine Tablets (Source: Ranbaxy)

USA • In vitro dissolution test can give a theoretical basis to
ensure BE based on IVIVC consideration.
BCS Japan • In vitro dissolution test can ensure BE by enhancing the
sensitivity to the differences in dissolution. Discriminative dissolution test
Dissolution test in BE assessment

IR products (No dose-dumping) • Low mechanical stress (e.g. Paddle method) • Low agitation (e.g. 50 rpm) • Multiple pHs in a physiological pH range • No surfactant
CR products (Dose-dumping) o Low and high mechanical stress (Paddle and
Disintegration apparatus) o Low and high agitation (50 and 200 rpm) o Multiple pHs in a physiological pH range o Low and high concentrations of surfactant o Low and high ionic strength
Discriminative dissolution test

Dissolution test for IR products - Paddle method -
Acidic drug Basic drug Neutral drug Coated product
poorly-soluble drug
50 rpm pH 1.2 pH 1.2 pH 1.2 pH 5.5-6.5 pH 3.0-5.0 pH 4.0 pH 6.8-7.5 pH 6.8 pH 6.8 Water Water Water each medium
+ surfactant 100 rpm A discriminative pH between pH 1.2 – 7.5

In Japan, more than 60 % of aged people (over 60 years
old) are achlorhydric subjects and show almost the neutral gastric pH.
Multiple pHs are necessary to ensure BE in normal and
achlorhydric subjects since the oral products showing the similar dissolution at one pH point do not always show the same dissolution at another pH.
Why multiple-pH media ?

Use of Dissolution test in BE assessment
Similar dissolution at all pHs by discriminative in vitro tests
Likely bioequivalent without subject-formulation interaction
No need of strict human study decrease of sample size waiver of human study

Use of Dissolution test in BE assessment
1. Generic products Exemption from 90 % confidence interval (C.I.)
If dissolutions are similar, test products will be approved even if 90 % C.I. of Cmax or AUC is out of 80 - 125 %.
Strict human test using specific subjects
If dissolutions differ at pH 6.8, achlorhydric subjects are required in human test.
2. Minor change in formulation or strengths
If dissolutions are similar, human test can be waived for all drugs.

Guidance in USA (SUPAC-IR : changes in components and composition, wide therapeutic range)

Guidance in JAPAN (IR, minor changes in Excipients)

BCS is not yet introduced in BE tests in Japan although its advantage and disadvantage have been discussed in many opportunities.
Why ?
Comments from Japanese regulatory are…

Reasons of No BCS in Japan
Permeability and solubility of drugs are not the direct causes of Bioinequivalence in oral drug products. Bioinequivalence is caused mainly by the differences in formulation and/or manufacturing processes.
BE of most IR products is well assured by the multiple-pH
media dissolution test in Japan in which classification of drugs is not necessary.

Reasons of No BCS in Japan
BCS is employed to simplify dissolution tests in U.S.A. However, multiple-pH media dissolution test must be performed in Japan because of the high % achlorhydric subjects.
Permeability is still unknown for many drugs, that makes it
difficult to introduce BCS in regulation.

Let’s see our BPOM legislation

Diperlukan bila :
Ada risiko bahwa perbedaan bioavailabilitas dapat menyebabkan
inekivalensi terapi.
In Vivo Bioekivalensi

studi bioekivalensi farmakokinetik studi farmakodinamik komparatif uji klinik komparatif
Pelaksanaan In Vivo Bioekivalen

1. IR, dengan kriteria : Respons terapi yang pasti (critical use drugs), seperti :
antituberkulosis, antiretroviral, antimalaria, antibakteri, antihipertensi, antiangina, obat gagal jantung, antiepilepsi, antiasma.
Batas keamanan/indeks terapi yang sempit; kurva dosis-respons yang curam, seperti : digoksin, antiaritmia, antikoagulan, obat - obat sitostatik, litium, fenitoin, siklosporin, sulfonilurea, teofilin.
Produk yang diuji in vivo bioekivalen

1. IR, dengan kriteria : Terbukti ada masalah bioavailabilitas atau bioinekivalensi
dengan obat yang bersangkutan atau obat-obat dengan struktur kimia atau formulasi yang mirip (tidak berhubungan dengan masalah disolusi), misal : absorpsi bervariasi atau tidak lengkap; eliminasi presistemik yang tinggi; farmakokinetik nonlinear; sifat-sifat fisiokimia yang tidak menguntungkan (misal :
kelarutan rendah, permeabilitas rendah, tidak stabil, dsb.).
Produk yang diuji in vivo bioekivalen

1. IR, dengan kriteria eksipien dan proses pembuatannya diketahui mempengaruhi
bioekivalensi
2. Produk obat non-oral dan non-parenteral yang didesain untuk bekerja sistemik, misal : sediaan transdermal, supositoria, permen karet nikotin, gel testosteron dan kontraseptif bawah kulit.
Produk yang diuji in vivo bioekivalen

3. Produk obat lepas lambat atau termodifikasi yang
bekerja sistemik.
4. Produk kombinasi tetap untuk bekerja sistemik, yang paling sedikit salah satu zat aktifnya memerlukan studi in vivo.
Produk yang diuji in vivo bioekivalen

5. Produk bekerja lokal, bukan larutan (oral, nasal, okular,
dermal, rektal, vaginal, dsb.) studi klinik atau farmakodinamik, dermatofarmakokinetik komparatif dan/atau studi in vitro.
Pada kasus-kasus tertentu, pengukuran kadar obat dalam darah masih diperlukan dengan alasan keamanan untuk melihat adanya absorpsi yang tidak diinginkan
Produk yang diuji in vivo bioekivalen

Kam Sia
Related Documents











