Wild-type amyloid beta 1-40 peptide induces vascular smooth muscle cell death independently from matrix metalloprotease activity Re ´ gis Blaise, 1 Ve ´ ronique Mateo, 1 * Clotilde Rouxel, 1 Franc ¸ ois Zaccarini, 1 Martine Glorian, 1 Gilbert Be ´re ´ ziat, 1 Vladislav S. Golubkov 2 and Isabelle Limon 1 1 University Paris 6, UR4, Vieillissement, Stress et Inflammation 7 quai Saint-Bernard, 75252 Paris, France 2 Cancer Research Center, Sanford-Burnham Medical Research Institute, La Jolla, CA 92037, USA Summary Cerebral amyloid angiopathy (CAA) is an important cause of intracerebral hemorrhages in the elderly, characterized by amy- loid-b (Ab) peptide accumulating in central nervous system blood vessels. Within the vessel walls, Ab-peptide deposits [composed mainly of wild-type (WT) Ab 1-40 peptide in sporadic forms] induce impaired adhesion of vascular smooth muscle cells (VSMCs) to the extracellular matrix (ECM) associated with their degeneration. This process often results in a loss of blood vessel wall integrity and ultimately translates into cerebral ischemia and microhemorrhages, both clinical features of CAA. In this study, we decipher the molecular mechanism of matrix metalloprotease (MMP)-2 activation in WT-Ab 1-40 -treated VSMC and provide evidence that MMP activity, although playing a critical role in cell detachment disrupting ECM components, is not involved in the WT-Ab 1-40 -induced degeneration of VSMCs. Indeed, whereas this peptide clearly induced VSMC apoptosis, neither preventing MMP-2 activity nor hampering the expres- sion of membrane type1-MMP, or preventing tissue inhibitors of MMPs-2 (TIMP-2) recruitment (two proteins evidenced here as involved in MMP-2 activation), reduced the number of dead cells. Even the use of broad-range MMP inhibitors (GM6001 and Batimastat) did not affect WT-Ab 1-40 -induced cell apoptosis. Our results, in contrast to those obtained using the Ab 1-40 Dutch variant suggesting a link between MMP-2 activity, VSMC mortality and degradation of specific matrix components, indi- cate that the ontogenesis of the Dutch familial and sporadic forms of CAAs is different. ECM degradation and VSMC degen- eration would be tightly connected in the Dutch familial form while being two independent processes in sporadic forms of CAA. Key words: amyloid beta peptide; cell death; matrix metal- loproteases; vascular smooth muscle cell. Introduction Cerebral amyloid angiopathy (CAA) refers to sporadic and hereditary cerebrovascular disorders frequently associated with cognitive impair- ment in the elderly, including Alzheimer’s disease. From a histopathologi- cal point of view, it is characterized by the amyloid accumulation in the media and adventitia of small and large arteries irrigating the central ner- vous system and ⁄ or amyloid deposition of Ab peptides around the capil- laries perfusing the cerebellum, cerebral cortex and leptomeninges (Smith & Greenberg, 2009). The Ab-peptide accumulation within arteries induces vascular smooth muscle cell (VSMC) death (referred to, in the lit- erature, as VSMC degeneration) and results in a loss of blood vessel wall integrity. This possibly translates into cerebral ischemia and microhemor- rhages, both clinical features of CAA (Knudsen et al., 2001; Maia et al., 2007). The Ab-induced VSMC degeneration is associated with impaired VSMC adhesion to the extracellular matrix (ECM) because of elevated pericellular proteolysis of the ECM components (Maruyama et al., 1990; Kawai et al., 1993; Mok et al., 2006). The human matrix metalloprotease (MMP) family encoded by 24 genes is composed of Zn 2+ -dependent proteases known to degrade a large vari- ety of ECM components and a number of bioactive molecules at the prox- imity of the cell surface. Among all the MMPs, six members are anchored to the cell membrane (membrane-type MMPs, MT-MMPs), whereas the other members are soluble and secreted into the extracellular space. Of note, there is increasing evidence that soluble MMPs (such as MMP-2) are recruited to the local cell environment by interacting with cell surface pro- teins (including MT-MMPs) and via the pericellular matrix (Murphy & Nagase, 2011). MMP activity is regulated at transcriptional and post- translational levels. All MMPs are synthesized as inactive zymogens. The cysteine switch containing N-terminal propeptide of the latent pro- enzyme interacts with the Zn 2+ at the active site, blocking proteolytic activity (Van Wart & Birkedal-Hansen, 1990). The first post-translational modification cleaves the propeptide allowing the latent proenzyme to become activated, enabling proteolysis of its substrate molecules. The second one involves the broad-spectrum proteinase inhibitor b2-macro- globulin and four specific inhibitors named tissue inhibitors of MMPs (TIMPs-1 to 4, Visse & Nagase, 2003). A 1:1 stoichiometric interaction of MMPs with TIMPs inhibits the enzyme activity, whereas unbalancing this ratio in favor of the proteases results in increasing MMP activity. Ab-peptides are produced by proteolytic processing of the amyloid pro- tein precursor (APP) by b- and c-secretases. Amyloid deposits within the vessel walls are mainly composed of wild-type (WT) in sporadic CAAs or mutated forms in familial CAAs of the 40 amino acids Ab-peptide species (Ab 1-40 ) (Alonzo et al., 1998). Previous studies have demonstrated that a mutated form, the Dutch mutant E22Q Ab 1-40 (Ab 1-40 D) involved in a rare but severe hereditary CAA (Levy et al., 1990), triggers the expression and activation of MMP-2 in human smooth muscle cells (HSMC, Davis & Van Nostrand, 1996; Jung et al., 2003); they also suggest that it may contrib- ute to the Ab 1-40 D-induced HSMC death. However, the pathogenicity of WT Ab 1-40 (WT-Ab 1-40 ) and the possible role of MMPs in sporadic CAAs (which represent more than 90% of CAAs) have not been defined. In this study, we demonstrate that WT-Ab 1-40 induces both VSMC apoptosis Correspondence Isabelle Limon, University Paris 6, UR4, Vieillissement, Stress et Inflammation 7 quai Saint-Bernard, Bat A 5eme etage, 75252 Paris, France. Tel.: +33 144 273 716; fax: +33 144 274 140; e-mail: isabelle.limon@snv. jussieu.fr *Present address: Paris 6 Pierre et Marie Curie University, CNRS UMR 7211 and CNRS U959, 75651 Paris, France. Accepted for publication 23 December 2011 384 ª 2012 The Authors Aging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland Aging Cell (2012) 11, pp384–393 Doi: 10.1111/j.1474-9726.2012.00797.x Aging Cell

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Wild-type amyloid beta 1-40 peptide induces vascular smoothmuscle cell death independently from matrixmetalloprotease activity
Regis Blaise,1 Veronique Mateo,1* Clotilde Rouxel,1
Francois Zaccarini,1 Martine Glorian,1 Gilbert Bereziat,1
Vladislav S. Golubkov2 and Isabelle Limon1
1University Paris 6, UR4, Vieillissement, Stress et Inflammation 7 quai
Saint-Bernard, 75252 Paris, France2Cancer Research Center, Sanford-Burnham Medical Research Institute,
La Jolla, CA 92037, USA
Summary
Cerebral amyloid angiopathy (CAA) is an important cause of
intracerebral hemorrhages in the elderly, characterized by amy-
loid-b (Ab) peptide accumulating in central nervous system
blood vessels. Within the vessel walls, Ab-peptide deposits
[composed mainly of wild-type (WT) Ab1-40 peptide in sporadic
forms] induce impaired adhesion of vascular smooth muscle
cells (VSMCs) to the extracellular matrix (ECM) associated with
their degeneration. This process often results in a loss of blood
vessel wall integrity and ultimately translates into cerebral
ischemia and microhemorrhages, both clinical features of CAA.
In this study, we decipher the molecular mechanism of matrix
metalloprotease (MMP)-2 activation in WT-Ab1-40-treated VSMC
and provide evidence that MMP activity, although playing a
critical role in cell detachment disrupting ECM components, is
not involved in the WT-Ab1-40-induced degeneration of VSMCs.
Indeed, whereas this peptide clearly induced VSMC apoptosis,
neither preventing MMP-2 activity nor hampering the expres-
sion of membrane type1-MMP, or preventing tissue inhibitors
of MMPs-2 (TIMP-2) recruitment (two proteins evidenced here
as involved in MMP-2 activation), reduced the number of dead
cells. Even the use of broad-range MMP inhibitors (GM6001
and Batimastat) did not affect WT-Ab1-40-induced cell apoptosis.
Our results, in contrast to those obtained using the Ab1-40
Dutch variant suggesting a link between MMP-2 activity, VSMC
mortality and degradation of specific matrix components, indi-
cate that the ontogenesis of the Dutch familial and sporadic
forms of CAAs is different. ECM degradation and VSMC degen-
eration would be tightly connected in the Dutch familial form
while being two independent processes in sporadic forms of
CAA.
Key words: amyloid beta peptide; cell death; matrix metal-
loproteases; vascular smooth muscle cell.
Introduction
Cerebral amyloid angiopathy (CAA) refers to sporadic and hereditary
cerebrovascular disorders frequently associated with cognitive impair-
ment in the elderly, including Alzheimer’s disease. From a histopathologi-
cal point of view, it is characterized by the amyloid accumulation in the
media and adventitia of small and large arteries irrigating the central ner-
vous system and ⁄ or amyloid deposition of Ab peptides around the capil-
laries perfusing the cerebellum, cerebral cortex and leptomeninges
(Smith & Greenberg, 2009). The Ab-peptide accumulation within arteries
induces vascular smooth muscle cell (VSMC) death (referred to, in the lit-
erature, as VSMC degeneration) and results in a loss of blood vessel wall
integrity. This possibly translates into cerebral ischemia and microhemor-
rhages, both clinical features of CAA (Knudsen et al., 2001; Maia et al.,
2007). The Ab-induced VSMC degeneration is associated with impaired
VSMC adhesion to the extracellular matrix (ECM) because of elevated
pericellular proteolysis of the ECM components (Maruyama et al., 1990;
Kawai et al., 1993; Mok et al., 2006).
The human matrix metalloprotease (MMP) family encoded by 24 genes
is composed of Zn2+-dependent proteases known to degrade a large vari-
ety of ECM components and a number of bioactive molecules at the prox-
imity of the cell surface. Among all the MMPs, six members are anchored
to the cell membrane (membrane-type MMPs, MT-MMPs), whereas the
other members are soluble and secreted into the extracellular space. Of
note, there is increasing evidence that soluble MMPs (such as MMP-2) are
recruited to the local cell environment by interacting with cell surface pro-
teins (including MT-MMPs) and via the pericellular matrix (Murphy &
Nagase, 2011). MMP activity is regulated at transcriptional and post-
translational levels. All MMPs are synthesized as inactive zymogens. The
cysteine switch containing N-terminal propeptide of the latent pro-
enzyme interacts with the Zn2+ at the active site, blocking proteolytic
activity (Van Wart & Birkedal-Hansen, 1990). The first post-translational
modification cleaves the propeptide allowing the latent proenzyme to
become activated, enabling proteolysis of its substrate molecules. The
second one involves the broad-spectrum proteinase inhibitor b2-macro-
globulin and four specific inhibitors named tissue inhibitors of MMPs
(TIMPs-1 to 4, Visse & Nagase, 2003). A 1:1 stoichiometric interaction of
MMPs with TIMPs inhibits the enzyme activity, whereas unbalancing this
ratio in favor of the proteases results in increasing MMP activity.
Ab-peptides are produced by proteolytic processing of the amyloid pro-
tein precursor (APP) by b- and c-secretases. Amyloid deposits within the
vessel walls are mainly composed of wild-type (WT) in sporadic CAAs or
mutated forms in familial CAAs of the 40 amino acids Ab-peptide species
(Ab1-40) (Alonzo et al., 1998). Previous studies have demonstrated that a
mutated form, the Dutch mutant E22Q Ab1-40 (Ab1-40D) involved in a rare
but severe hereditary CAA (Levy et al., 1990), triggers the expression and
activation of MMP-2 in human smooth muscle cells (HSMC, Davis & Van
Nostrand, 1996; Jung et al., 2003); they also suggest that it may contrib-
ute to the Ab1-40D-induced HSMC death. However, the pathogenicity of
WT Ab1-40 (WT-Ab1-40) and the possible role of MMPs in sporadic CAAs
(which represent more than 90% of CAAs) have not been defined. In this
study, we demonstrate that WT-Ab1-40 induces both VSMC apoptosis
Correspondence
Isabelle Limon, University Paris 6, UR4, Vieillissement, Stress et Inflammation 7
quai Saint-Bernard, Bat A 5eme etage, 75252 Paris, France.
Tel.: +33 144 273 716; fax: +33 144 274 140; e-mail: isabelle.limon@snv.
jussieu.fr
*Present address: Paris 6 Pierre et Marie Curie University, CNRS UMR 7211 and
CNRS U959, 75651 Paris, France.
Accepted for publication 23 December 2011
384 ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
Aging Cell (2012) 11, pp384–393 Doi: 10.1111/j.1474-9726.2012.00797.xAg
ing
Cell

and the MT1-MMP-dependent MMP-2 activation in vitro and decipher
the molecular mechanism of this activation. We also prove that there is
no cause-to-effect relationship between WT-Ab1-40-induced cell death
and MMP activity suggesting that MMP proteolytic activity is not the pri-
mary cause of VSMC apoptosis observed in sporadic CAA.
Results
WT-Ab1-40 decreases VSM cells viability
Comparing cell morphology of WT-Ab1-40 with that of inverted Ab1-40-
treated VSMC revealed modifications of their adhesion to the ECM evi-
dent by cell shape alteration (Fig. 1A). Hoechst nuclei labeling followed
by immunofluorescence microscopy evidenced pycnotic nuclei, character-
istic of cells undergoing apoptosis. Consistently, the percentage of hypo-
diploid nuclei [measured by propidium iodide flow cytometric assay
(Mateo et al., 2007)] was more than 70% after a 72 h-treatment with
WT-Ab1-40 peptide; inverted Ab40-1 peptide has no effect on spontaneous
cell death (Fig. 1B). Apoptosis induced by WT-Ab1-40 was also measured
by evaluating phosphatidylserine (PS) exposure in the cytoplasmic outer
leaflet membrane using APC-conjugated Annexin-V in combination with
7-amino-actinomycin D exclusion dye (data not shown).
WT-Ab1-40 induces MT1-MMP-dependent MMP-2 activation
MT1-MMP and MMP-2 activity are known to be essential for pericellular
proteolysis (Itoh & Seiki, 2006), whereas MMP-9 is rather involved in
inflammatory diseases, including peripheral arterial diseases (Busti et al.,
2010). MMP-2 and MMP-9 are the predominant soluble MMPs in VSMCs
(Newby, 2006). As both processes, pericellular proteolysis and inflamma-
tion, are the major pathogenic features in CAA, we next studied the
expression and activity of these MMPs in WT-Ab1-40-treated VSMCs. As
shown in Fig. 2A, left panel, MMP-2 mRNA level (estimated by quantita-
tive RT–PCR) was more than 6-fold increased (P < 0.001) in cells treated
with WT-Ab1-40, when compared to cells incubated with vehicle or the
inverted Ab40-1 control peptide. MMP-9 mRNA was undetectable either
in control, inverted Ab40-1 or WT-Ab1-40-treated cells (Fig. 2A, right
panel), while IL-1b induced the expression of MMP-9 transcripts in agree-
ment with earlier reports (Delbosc et al., 2008). The active form of the
MMP-2 protein (referred to as Act. MMP-2, Fig. 2B) could only be
detected in WT-Ab1-40-treated VSMCs. In vehicle or inverted Ab40-1-trea-
ted cells, the observed bands corresponded to nonmature forms of the
enzyme (Fig. 2B). Of note, the lower unidentified band (*) was always
present independently of the MMP-2 antibody used. Accordingly, MMP-
2 activity (revealed by a 68-kDa zymographic band referred to as Act.
MMP-2 in Fig. 2C) was only detectable in media conditioned by cells trea-
ted with WT-Ab1-40. The identity of the zymographic bands was con-
trolled by immunoprecipitation using the anti-MMP-2 antibody and naıve
Ig on conditioned media derived from rat lung incubated in serum-free
medium. The active form of MMP-2 was identified as the lowest band by
(aminomethyl) phosphinic acid (AMPA)-induced activation of a control
sample prior to zymography (data not shown). According to the level of
MMP-9 transcripts, WT-Ab1-40 did not trigger MMP-9 activity, although
IL-1b treatment significantly increased it.
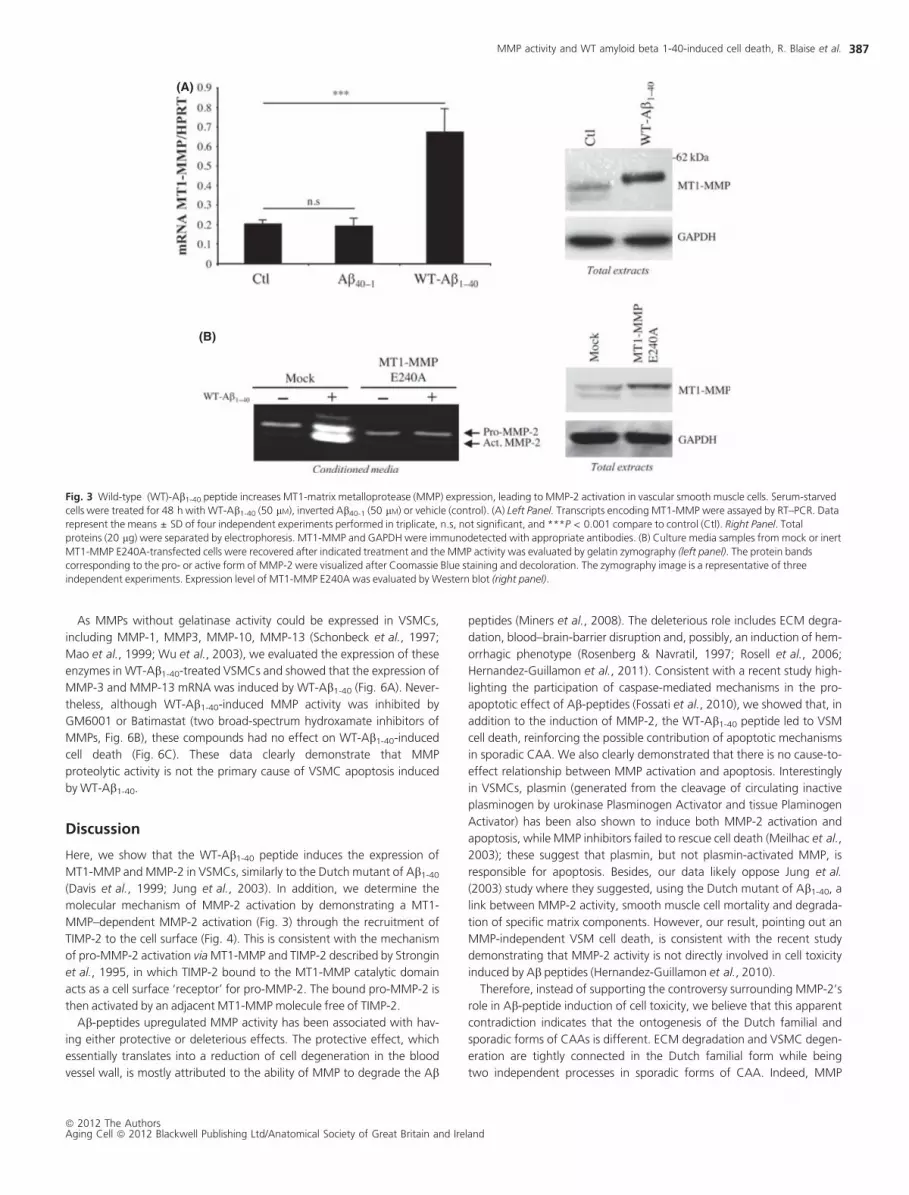
We also demonstrated an increase of MT1-MMP expression in WT-Ab1-
40-treated cells at both mRNA and protein levels by quantitative RT–PCR
(Fig. 3A, left panel) and Western blot (Fig. 3A, right panel). Because
MMP-2 activation is dependent on MT1-MMP activity (Strongin et al.,
1995), we next examined whether WT-Ab1-40-induced MT1-MMP
expression is responsible for MMP-2 activation. To do so, VSMCs were
transfected with a plasmid encoding the catalytically inert E240A mutant
of MT1-MMP (Fig. 3B). MMP-2 status was analyzed by gelatin zymogra-
phy. The overexpression of MT1-E240A protein was confirmed by Wes-
tern blot (Fig. 3B, right panel). As expected, the forced expression of
MT1-MMP E240A completely blocked the WT-Ab1-40-induced MMP-2
activity (Fig. 3B, left panel). MMP-2 mRNA expression was not affected
by the expression of MT1-E240A constructs whether with or without WT-
Ab1-40 treatment (data not shown). Altogether, these data highlighted a
cause-to-effect relationship between the WT-Ab1-40 induction of MT1-
MMP expression and MMP-2 activity.
(A)
(B)
Fig. 1 Wild-type (WT)-Ab1-40 peptide induces vascular smooth muscle cells
apoptosis. Serum-starved cells were treated for 48 h with WT-Ab1-40 (50 lM),
inverted Ab40-1 (50 lM) or vehicle (control). (A) Cell morphology was evaluated by
phase contrast microscopy, and nuclei were stained with Hoechst (blue). Images are
representative of three independent experiments. (B) Apoptosis was determined as
described in Materials and methods section by propidium iodide staining. Data
represent means ± SD of four independent experiments performed in triplicate;
n.s, not significant and ***P < 0.001 compared with control (Ctl).
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
385

WT-Ab1-40-induced MMP-2 activation is regulated by
TIMP-2 accumulation at the cell surface
It has been shown that the MT1-MMP–dependent MMP-2 activation
requires strictly regulated amounts of TIMP-2 molecules (Strongin et al.,
1995). In this activation mechanism, TIMP-2 bound to the catalytic
domain of MT1-MMP acts as a ‘receptor’ for pro-MMP-2 at the cell sur-
face. Within this protein complex, the hemopexin domain of pro-MMP-2
binds to the C-terminal domain of TIMP-2 allowing proteolytic activation
of pro-MMP-2 by an adjacent molecule of MT1-MMP that is free of TIMP-
2 (see Fig. S1). In this scenario, the TIMP-2 molecule, part of the trimolec-
ular complex (composed of MT1-MMP ⁄ TIMP-2 ⁄ pro-MMP-2), acts as an
MMP-2 activator. Here, TIMP-2 implication in WT-Ab1-40-induced MMP-2
activation was evidenced by the drastic reduction of MMP-2 active forms
(Fig. 4A, left panel) visualized in cells transfected with TIMP-2 siRNA com-
pared with control siRNA-transfected cells. TIMP-2 siRNA efficiency was
attested by RT–PCR, Fig. 4A, left panel. To study the mechanism of MMP-
2 activation, we analyzed TIMP-2 protein levels in control and WT-Ab1-40-
treated whole cell extracts (as opposed to cell media) assuming that it
would reflect TIMP-2 recruitment to the cell surface. We also defined
TIMP-2 subcellular localization. TIMP-2 protein was not present in con-
trols (whether in vehicle- or inverted peptide-treated VSMCs), but highly
accumulated in the WT-Ab1-40 treated VSMCs (Fig. 4B left panel). Data
obtained from immunocytochemistry experiments evidenced that TIMP-2
staining defines the plasma membrane in WT-Ab1-40-treated cells
(Fig. 4C). Altogether, these results strongly suggest the WT-Ab1-40-
dependent accumulation of TIMP-2 at the cell surface. Because WT-Ab1-
40 activates MMP-2 through MT1-MMP, we next examined whether
TIMP-2 could be recruited at the VSMC surface by silencing MT1-MMP
expression. As shown in Fig. 4D, the intensity of the TIMP-2 band was
strongly reduced in MT1-MMP-silenced cells compared with control siR-
NA-treated cells. As neither the amount of TIMP-2 mRNA nor that of
secreted protein (Fig. 4B, right panel) was modified by the WT-Ab1-40
treatment, we concluded that the WT-Ab1-40-dependent accumulation
of TIMP-2 takes place at the cell surface, because of its interaction with
the upregulated membrane type 1-MMP (MT1-MMP).
MMPs activity is not involved in WT-Ab1-40 induced
cell death
Anoıkis is a form of programmed cell death induced by cell detach-
ment from the ECM. As MMP activity contributes to ECM degradation,
we questioned whether WT-Ab1-40-induced VSMC death is linked to
MT1-MMP ⁄ MMP-2 activation. We analyzed the effect of MMP-2 or
MT1-MMP silencing with siRNA on the WT-Ab1-40-induced VSMC
apoptosis. The efficiency of MMP-2 and MT1-MMP silencing was esti-
mated by RT–PCR and gelatin zymography (Fig. 5A,C). To our surprise,
MMP-2 silencing did not change the rate of apoptosis in cells treated
with the WT-Ab1-40 (Fig. 5B), neither did MT1-MMP siRNA (Fig. 5D), as
the percentage of apoptotic cells was very similar whether MT1-MMP
expression was silenced or not (siRNA MT1-MMP:79.2 ± 7.4; siRNA
control: 69.3 ± 5.5). Similar results were obtained in the MCF-7 breast
carcinoma cell subline deficient in MT1-MMP and MMP-2 expression
(Rozanov et al., 2001) (Fig. S2). Altogether, these experiments demon-
strated that MMP-2 and MT1-MMP are not involved in the WT-Ab1-40-
induced cell death.
(A)
(B) (C)
Fig. 2 Wild-type (WT)-Ab1-40 peptide increases expression and activation of matrix metalloprotease (MMP)-2, but not MMP-9 in vascular smooth muscle cells. Serum-starved
cells were treated for 48 h with IL-1b (10 ng mL)1), WT-Ab1-40 (50 lM), inverted Ab40-1 (50 lM), or vehicle (control). (A) Transcripts encoding MMP-2 (left panel) and MMP-9
(right panel) were assayed by RT–PCR. Data represent means ± SD of four independent experiments performed in triplicate; n.s, not significant, n.d, not detectable, and
***P < 0.001 compared with control (Ctl). (B) MMP-2 immunoblot on total proteins (20 lg). The Western blot is representative of two independent experiments. Int. MMP-
2, intermediate form of MMP-2; Act. MMP-2, Active form of MMP-2; *Nonspecific band. (C) Culture medium was recovered after treatment; 45 lL were used for
determination of secreted MMP-2 and MMP-9 activities on gelatin zymography as described in Materials and methods. The lysis bands corresponding to the pro- or active
form (Act.) of MMP-2 and MMP-9 were visualized after Coomassie Blue staining and decoloration. The gelatin zymography image is representative of four independent
experiments.
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
386
As MMPs without gelatinase activity could be expressed in VSMCs,
including MMP-1, MMP3, MMP-10, MMP-13 (Schonbeck et al., 1997;
Mao et al., 1999; Wu et al., 2003), we evaluated the expression of these
enzymes in WT-Ab1-40-treated VSMCs and showed that the expression of
MMP-3 and MMP-13 mRNA was induced by WT-Ab1-40 (Fig. 6A). Never-
theless, although WT-Ab1-40-induced MMP activity was inhibited by
GM6001 or Batimastat (two broad-spectrum hydroxamate inhibitors of
MMPs, Fig. 6B), these compounds had no effect on WT-Ab1-40-induced
cell death (Fig. 6C). These data clearly demonstrate that MMP
proteolytic activity is not the primary cause of VSMC apoptosis induced
by WT-Ab1-40.
Discussion
Here, we show that the WT-Ab1-40 peptide induces the expression of
MT1-MMP and MMP-2 in VSMCs, similarly to the Dutch mutant of Ab1-40
(Davis et al., 1999; Jung et al., 2003). In addition, we determine the
molecular mechanism of MMP-2 activation by demonstrating a MT1-
MMP–dependent MMP-2 activation (Fig. 3) through the recruitment of
TIMP-2 to the cell surface (Fig. 4). This is consistent with the mechanism
of pro-MMP-2 activation via MT1-MMP and TIMP-2 described by Strongin
et al., 1995, in which TIMP-2 bound to the MT1-MMP catalytic domain
acts as a cell surface ‘receptor’ for pro-MMP-2. The bound pro-MMP-2 is
then activated by an adjacent MT1-MMP molecule free of TIMP-2.
Ab-peptides upregulated MMP activity has been associated with hav-
ing either protective or deleterious effects. The protective effect, which
essentially translates into a reduction of cell degeneration in the blood
vessel wall, is mostly attributed to the ability of MMP to degrade the Ab
peptides (Miners et al., 2008). The deleterious role includes ECM degra-
dation, blood–brain-barrier disruption and, possibly, an induction of hem-
orrhagic phenotype (Rosenberg & Navratil, 1997; Rosell et al., 2006;
Hernandez-Guillamon et al., 2011). Consistent with a recent study high-
lighting the participation of caspase-mediated mechanisms in the pro-
apoptotic effect of Ab-peptides (Fossati et al., 2010), we showed that, in
addition to the induction of MMP-2, the WT-Ab1-40 peptide led to VSM
cell death, reinforcing the possible contribution of apoptotic mechanisms
in sporadic CAA. We also clearly demonstrated that there is no cause-to-
effect relationship between MMP activation and apoptosis. Interestingly
in VSMCs, plasmin (generated from the cleavage of circulating inactive
plasminogen by urokinase Plasminogen Activator and tissue Plaminogen
Activator) has been also shown to induce both MMP-2 activation and
apoptosis, while MMP inhibitors failed to rescue cell death (Meilhac et al.,
2003); these suggest that plasmin, but not plasmin-activated MMP, is
responsible for apoptosis. Besides, our data likely oppose Jung et al.
(2003) study where they suggested, using the Dutch mutant of Ab1-40, a
link between MMP-2 activity, smooth muscle cell mortality and degrada-
tion of specific matrix components. However, our result, pointing out an
MMP-independent VSM cell death, is consistent with the recent study
demonstrating that MMP-2 activity is not directly involved in cell toxicity
induced by Ab peptides (Hernandez-Guillamon et al., 2010).
Therefore, instead of supporting the controversy surrounding MMP-2’s
role in Ab-peptide induction of cell toxicity, we believe that this apparent
contradiction indicates that the ontogenesis of the Dutch familial and
sporadic forms of CAAs is different. ECM degradation and VSMC degen-
eration are tightly connected in the Dutch familial form while being
two independent processes in sporadic forms of CAA. Indeed, MMP
(A)
(B)
Fig. 3 Wild-type (WT)-Ab1-40 peptide increases MT1-matrix metalloprotease (MMP) expression, leading to MMP-2 activation in vascular smooth muscle cells. Serum-starved
cells were treated for 48 h with WT-Ab1-40 (50 lM), inverted Ab40-1 (50 lM) or vehicle (control). (A) Left Panel. Transcripts encoding MT1-MMP were assayed by RT–PCR. Data
represent the means ± SD of four independent experiments performed in triplicate, n.s, not significant, and ***P < 0.001 compare to control (Ctl). Right Panel. Total
proteins (20 lg) were separated by electrophoresis. MT1-MMP and GAPDH were immunodetected with appropriate antibodies. (B) Culture media samples from mock or inert
MT1-MMP E240A-transfected cells were recovered after indicated treatment and the MMP activity was evaluated by gelatin zymography (left panel). The protein bands
corresponding to the pro- or active form of MMP-2 were visualized after Coomassie Blue staining and decoloration. The zymography image is a representative of three
independent experiments. Expression level of MT1-MMP E240A was evaluated by Western blot (right panel).
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
387

(A)
(C)
(D)
(B)
Fig. 4 Matrix metalloprotease (MMP)-2 activation is dependent on the accumulation of TIMP-2 at the cell surface induced by wild-type (WT)-Ab1-40 in vascular smooth
muscle cells. Serum-starved cells were treated for 48 h with WT-Ab1-40 (50 lM), inverted Ab40-1 (50 lM) or vehicle (control). (A) Culture media obtained from cells previously
transfected with control siRNA or TIMP-2 siRNA were collected after indicated treatment and MMP activities were evaluated by gelatin zymography (left panel). The gelatin
zymography image shown is representative of three independent experiments. TIMP-2 siRNA efficiency was evaluated by RT–PCR (right panel); data represent the
means ± SD of three independent experiments performed in triplicate, ***P < 0.001 compared with control siRNA related to each treatment. (B) Left Panel. Cell lysate
samples (20 lg) were separated by electrophoresis; TIMP-2 and GAPDH were immunodetected with the appropriate antibodies; the western blot shown is representative of
five independent experiments. Right Panel. Transcripts encoding TIMP-2 were assayed by RT–PCR (upper right panel). The data represent the means ± SD of four independent
experiments performed in triplicate, n.s, not significant compared with control (Ctl). TIMP-2 immunoblot was performed on 40 lL of conditioned culture medium separated
by electrophoresis (lower right panel). The western blot is representative of two independent experiments. (C) Immunostaining of TIMP-2 on PFA-fixed cells was performed
after 24 h-treatment using monoclonal antibody against TIMP-2 and a secondary antibody coupled to FITC (Green Stain, 20·). Cell nuclei were stained with Hoechst (blue,
20· same microscopic field as FITC). (D) Total proteins (20 lg) obtained from treated cells previously transfected with control or MT1-MMP siRNA were separated by
electrophoresis; TIMP-2 and GAPDH were immunodetected with appropriate antibodies. The western blot shown is representative of three independent experiments.
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
388

broad-range inhibitor GM6001 can significantly reduce the apoptosis
induced by a 48 h Ab1-40Dutch-treatment (Fig S3B), but did not have this
effect on WT-Ab1-40-treated cells (Fig. 6). A likely divergence between
WT, Dutch and Arctic Ab-peptides has already been reported regarding
anti-angiogenic effects in brain endothelium (Solito et al., 2009). How-
ever, although several data argue for distinct mechanisms between WT
and Dutch Ab1-40 peptides (including ours, demonstrating that the
WT-Ab1-40- but not the Ab1-40Dutch-induced apoptosis is inhibited by
fetal calf serum, see Fig S3C), one might be caution with comparing
these two peptide effects as they are differently solubilized and display
distinct secondary structures and oligomerization ⁄ fibrillization kinetics
(Solito et al., 2009; Fossati et al., 2010).
It is not clear whether or not WT-Ab peptide-induced MMP activity has
a beneficial role (protecting the cells by degrading the WT-Ab peptides) in
our experimental conditions. If it was the case, inhibition of MMPs should
have translated into an increased Ab-induced apoptosis (Hernandez-
Guillamon et al., 2010). A possible explanation for not being able to
detect such an increase could be the dose of peptide used (50 lM). As a
matter of fact, such a high concentration could prevent the effect of a
minor Ab-proteolytic degradation. Consistent with this, degradation of
WT-Ab peptides in a cellular context was only observed at a low dose
(< 1 lM) using MMP overexpressing cells (Liao & Van Nostrand, 2010). In
addition, because our cell culture condition medium does not contain
plasminogen, it is also conceivable that the Ab-dependent MMP activa-
tion (and therefore Ab-peptide degradation) was limited. Indeed, plasmin
activates MMP in VSMCs (Galis & Khatri, 2002). Interestingly, plasmin
activators (uPA and tPA) are induced by Ab-peptides (Davis et al., 2003
and Fig. S4), whereas the expression of the tPA ⁄ uPA inhibitor, PAI-1, is
decreased (Fig. S4), suggesting that Ab-treated VSMCs could convert
plasminogen into plasmin. It is important to note that plasmin is also
capable of degrading Ab peptides (Tucker et al., 2000; Jacobsen et al.,
2008) similar to other proteases including neprisylin, insulin-degarding
enzyme, endothelin-converting enzyme, angiotensin-converting enzyme
(Miners et al., 2008). Besides, plasmin, similar to MMP-2, can have both
protective and deleterious effects, whereas plasmin activity can protect
Ab-peptide-treated VSMCs from apoptosis and can also lead to cell
(A)
(B)
(C)
(D)
Fig. 5 Effect of matrix metalloprotease (MMP)-2 and MT1-MMP silencing on wild-type (WT)-Ab1-40-induced cell death. Serum-starved cells were treated for 48 h with WT-
Ab1-40 (50 lM), or vehicle (control). (A,C) upper panel. siRNAs efficiency was evaluated by RT–PCR on transcripts obtained from cells transfected with control, MMP-2 siRNA
(A) or MT1-MMP siRNA (C). Data represents the means ± SD of three independent experiments performed in triplicate, ***P < 0.001 compared with vehicle (Ctl)-treated
cells transfected with control siRNA. ###P < 0.001 compared with WT-Ab1-40-treated cells transfected with control siRNA. (A,C) lower panel. Culture medium samples issued
from MMP-2 siRNA (A) or MT1-MMP siRNA (C) -transfected cells were collected after indicated treatment; MMP activities were evaluated by gelatin zymography. The
zymography shown are representative of three independent experiments. (B,D) the apoptosis of MMP-2 siRNA (B) or MT1-MMP siRNA (D) -transfected cells was determined
by Propidium Iodide staining. Data represents the means ± SD of three independent experiments performed in triplicate. n.s, not significant; ***P < 0.001; **P < 0.01 and
*P < 0.05 compared with related control (Ctl).
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
389

detachment (Davis et al., 2003), the process resulting in cell death by
anoıkis (Michel, 2003).
Although the expression of MMP-2 was upregulated by WT-Ab1-40
within a 72-h time frame, no effect could be detected on MMP-9. As
expected, the pro-inflammatory cytokine IL-1b triggered its expres-
sion ⁄ activity (Fig. 2). Taking into account that MMP-9 is an inflammatory
marker (del Zoppo, 2010), we suggest that it is unlikely that WT-Ab1-40
initiates VSMC inflammation directly. Interestingly, Ab1-40 peptides
(Dutch or WT) induce the expression of adhesion molecules and pro-
inflammatory cytokines in human arterial endothelial cells (Suo et al.,
1998) arguing for an inflammatory context present in CAA (Vukic et al.,
2009). Because MMP-9 (as well as MMP-2) has been extensively involved
in blood–brain barrier breakdown and intracerebral hemorrhage (Rosen-
berg & Navratil, 1997; Rosell et al., 2006; Hernandez-Guillamon et al.,
2011), two main features of CAA, it would be of interest to re-evaluate
the WT-Ab1-40 capacity of inducing MMP activity within the inflammatory
context of CAA. This could first be approached in vitro on IL-1b-treated
VSMCs.
In summary, our results tend to prove that the deposition of WT-Ab1-40
(present in sporadic CAA) induces VSMC apoptosis independently from
MMP activity. One might suggest that the WT-Ab1-40-induced MMPs
(because of Ab accumulation) should actively participate in the destruc-
tion of the blood–brain barrier and the alteration of vessel wall integrity
characteristics of the CAAs. Nevertheless, in vivo data should be obtained
on relevant CAA models to further support our hypothesis.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium, type I collagen from calf skin, glu-
tamine, penicillin, streptomycin, fatty acid-free bovine serum albumin
were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France).
Fetal calf serum and collagenase were from Gibco BRL (Cergy Pontoise,
France). Elastase, protease inhibitors, LightCycler-DNA Master Plus SYBR
Green and Fugene HD transfection reagent were obtained from Roche
Diagnostics (Meylan, France). Oligonucleotides were sourced from
MWG Biotech AG (Courtaboeuf, France). Kits for RNA extraction
(RNeasy Mini kit) were obtained from Qiagen (Courtaboeuf, France).
RT-MMLV, RNAsin, Lipofectamine RNAi Max transfection reagent were
obtained from Invitrogen (Cergy Pontoise, France). siRNA were from
Ambion (Invitrogen, Cergy Pontoise, France) or Qiagen (Courtaboeuf,
France). Nitrocellulose membranes were from Schleicher and Schuell
(Dassel, Germany). ECL reagent kit was from Amersham Pharmacia
Biotech (les Ulis, France). Interleukin-1b (IL-1b) was from Santa Cruz
Biotechnology (Heildberg, Germany). GM6001 and Batimastat broad-
range MMP inhibitors were from Millipore (Molsheim, France) and
Santa cruz Biotechnology, respectively. The FRET MMPs substrate
Mca-Arg-Pro-Lys-Pro-Tyr-Ala-Nva-Trp-Met-Lys(Dnp)-NH2 was purchased
from Bachem. Ethidium Homodimer III (ETHD-III) DNA dye was from
Promocell (Heildberg, Germany).
(A)
(B) (C)
Fig. 6 Effect of matrix metalloprotease (MMP) inhibitors on wild-type (WT)-Ab1-40-induced cell death. Serum-starved cells were treated for 48 h with WT-Ab1-40 (50 lM), or
vehicle (control), with or without MMP inhibitors (GM6001 or Batimastat). (A) Transcripts encoding MMP-3 (left panel) and MMP-13 (right panel) were assayed by RT–PCR.
Data represent the means ± SD of four independent experiments performed in triplicate, ***P < 0.001 and *P < 0.05 compared with control (Ctl). (B) Inhibition of
WT-Ab1-40-induced MMP activities by GM6001 (25 lM) or Batimastat (2.5 lM) treatment was evaluated by adding the MMP fluorescent substrate (40 lM) to the collected
culture medium. The fluorescence of MMP-cleaved substrate (kex = 320 nm, kem = 390 nm) was measured with a fluorescence multiplate reader. Data represent the
means ± SD of three independent experiments performed in triplicate, n.s, not significant; ***P < 0.001, ###P < 0.001 and ���P < 0.001 compared with related control (Ctl).
(C) Apoptosis of treated cells was determined by Eth-DIII staining. Data represent the means ± SD of three independent experiments performed in triplicate; n.s, not
significant and ***P < 0.001 compared with related control (Ctl). The fluorescence of DNA-bound EthD-III (kex = 515 nm, kem = 620 nm) was measured with a fluorescence
multiplate reader. The results are expressed in percentage of dead cells compared with cells treated by absolute ethanol (referred to as 100% dead cells). Data represent the
means ± SD of six independent experiments performed in triplicate; n.s, not significant; ***P < 0.001, ###P < 0.001 and ���P < 0.001 compared with related control (Ctl)-
treated cells.
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
390

Wild-type amyloid beta 1-40 (WT-Ab1-40) (DAEFRHDSGYEVHHQKL
VFFAEDVGSNKGAIIGLMVGGVV), Dutch mutant (E22Q) amyloid beta
peptide (Ab1-40 Dutch) (DAEFRHDSGYEVHHQKLVFFAQDVGSNKGAIIGL
MVGGVV), or control inverted 40-1 (Ab40-1) (VVGGVMLGIIAGKNSG
VDEAFFVLKQHHVEYGSDHRFEAD) peptides were synthesized by Gene-
Cust (Dudelange, Luxembourg) and analyzed by HPLC and mass spectrom-
etry with a purity ranged between 94% and 98%. WT-Ab1-40 and
scrambled Ab40-1 peptides were resuspended in ultrapure water at 2 mM
and immediately frozen. WT-Ab1-40 and scrambled Ab40-1 peptides were
directly added to culture medium at 50 lM. Ab1-40 Dutch was resuspended
at 1 mM in 1,1,1,3,3,3,-hexafluoro-2-propanol (Sigma-Aldrich) and incu-
bated at room temperature until obtaining a clear solution. Ab1-40 Dutch
peptides were aliquoted and allowed to evaporate over night in a fume
hood. Dried peptide films were stored at )20 �C. Prior to use, Ab1-40 Dutch
peptides films were resuspended at 5 mM into DMSO (Sigma-Aldrich) and
diluted to 1 mM with PBS before added to cell culture at 50 lM.
Isolation and culture of smooth muscle cells
Rat vascular smooth muscle cells (VSMC) were isolated by enzymatic
digestion of aortic media from male Wistar rats and cultured as described
previously (Clement et al., 2006). All experiments were performed on cells
at passages ranging from two to six. Twenty-four hours before any treat-
ment, confluent cells were made quiescent by culturing them in a serum-
free medium. All experiments were performed in serum-free medium. Of
note, basal cell death 72 h after serum starvation is very similar to that of
measured in presence of 10% fetal calf serum within this time frame
(approximately 10%). Conditions of cell treatment are as indicated in the
figure legends. All incubations were performed at 37 �C, 5% CO2. Cells
were exposed to peptides diluted in culture medium at 50 lM for 24–72 h.
Real-time quantitative reverse transcription–polymerase
chain reaction (RT–PCR) assays
Total RNA was extracted from VSMCs using the RNeasy kit (Qiagen,
Hilden, Germany) according to the manufacturer’s instructions. Con-
centrations were determined spectrophotometrically. After annealing
oligodT (1 lM) to template RNAs (0.5 lg) at 70 �C for 5 min, primer
extension was initiated by adding the RT-MMLV enzyme plus 0.5 mM
dNTP, 1U RNAsin, and 10 mM dithiothreitol (DTT) and carried out for
45 min at 37 �C. Quantitative PCR was performed using the LightCycler
LC480 (Roche Diagnostics). The PCR mix included 5 lL of each reverse
transcriptase (diluted 1:25) and 300 nM of each primer in 1 · LightCy-
cler-DNA SYBR Green 1 Master Mix. Specific primers for complemen-
tary DNA (cDNA) were chosen with the LightCycler Probe Design2
program according to European Molecular Biology Laboratory accession
numbers. The forward and reverse primers used to selectively amplify
the cDNA encoding rat hypoxanthine phosphoribosyltransferase (HPRT),
MMP-2, MMP-3, MMP-9, MMP-13, MT1-MMP and TIMP-2 are pre-
sented in Table 1.
The PCRs were performed using the following thermal settings: dena-
turation and enzyme activation at 95 �C for 5 min, with cycles at 95 �Cfor 10 s, 60 �C for 15 s, and 72 �C for 10 s. Amplification was followed
up online, and the PCRs were stopped after the logarithmic phase. Melt-
ing curve analyses were also performed after PCR to check the reaction
specificity. Controls and water blanks were included in each run; they
were negative in all cases.
Real-time quantitative PCR data represent the amount of each tar-
get messenger RNA (mRNA) relative to the amount of a housekeeping
reference gene mRNA, the HPRT, estimated in the logarithmic phase
of the PCR. Serial dilutions were used to determine the fit coefficients
of the relative standard curve. When the PCR target efficiencies were
similar, individual cultures could be compared. If not, an internal cali-
brator was used.
Plasmid preparation and transient transfections
Expression plasmid for MT1-MMP mutant E240A was made as described
in Rozanov et al., 2001. For plasmid expression, cells were transfected at
70–80% confluency by adding 10 lg DNA and 15 lL Fugene HD (ratio
2:3) per 10-cm dish according to the manufacturer’s instructions (Roche
Diagnostics). Plasmids were inert MT1-MMP E240A mutant or control
(pCDNA3, invitrogen). Twenty-four to 48 h post-transfection, cells were
starved overnight and treated as indicated in the figure legends.
siRNA were transfected with Lipofectamine RNAiMAX reagent (Invitro-
gen) according to the manufacturer’s instructions. Briefly, 50% confluent
cells were cultured for 4 h in complete medium without antibiotics and
transfected by siRNA (5 nM final for 5 lL of reagent per well in 6-wells
plate) for 24–48 h. After 12-h serum starvation, the cells were treated as
indicated in the figure legends. The siRNA forward and reverse strand
sequences are, respectively, 5¢-CCAUCGAGACCAUGCGGAATT-3¢ and
5¢-UUCCGCAUGGUCUCGAUGGTG-3¢ for MMP-2, 5¢-CCAGUGUUUCA
UUUGCCUATT3¢ and 5¢- UAGGCAAAUGAAACACUGGTT-3¢ for TIMP-2;
5¢-CUUCUACAAAGGGAACAAATT-3¢ and 5¢-UUUGUUCCCUUUGUAGA
AGTA-3¢ for TIMP-2.
Protein extraction and western blot analysis
Cells were washed with ice-cold phosphate-buffered saline (PBS) and
resuspended in lysis buffer (20 mM Tris–HCl, pH 7.6, 150 mM NaCl, 1 mM
EDTA, 1% Triton, 1% sodium deoxycholate) plus Complete Protease
Inhibitor mixture (Roche Diagnostics), for 10 min on ice. The lysate was
centrifuged at 13 000 g for 10 min at 4 �C, and the supernatant (cell
lysate) was collected. Cell lysates (20–40 lg of protein) were analyzed by
Table 1 Summary of PCR primers
Target gene Forward primer (5¢-3¢) Reverse primer (5¢-3¢) Accession number
HPRT aggacctctcgaagtgt atccctgaagtgctcattata NM_012583.2
MMP-2 atcgcccatcatcaagttc catgttctcgatggtgttc NM_031054.2
MMP-3 atgtagatggtattcaatccctctat aaagcagagctacacatt NM_133523.2
MMP-9 ttcgagggagcgtcctat ttagtggtgcaggcagagta NM_031055.1
MMP-13 tggtcttctggcacacg gagtggtccagaccg NM_133530.1
MT1-MMP tgggaaggaatccctgagt ctctaccttcagcttctggtt NM_031056.1
TIMP-2 gacctgacaaggacatcgaa tcccagggcacaataaagt NM_021989.2
HPRT, hypoxanthine phosphoribosyltransferase; MMP, matrix metalloprotease.
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
391

sodium dodecyl sulfate–polyacrylamide gel electrophoresis on 12%
resolving gels or 4–12% gradient gels (Invitrogen NuPAGE system) fol-
lowed by transfer to nitrocellulose membranes. After blocking 1 h at
room temperature with a Tris-buffered saline containing 0.1% Tween 20
(TBS-T) (20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20) and 5%
fat-free milk or bovine serum albumin, membranes were incubated over-
night with primary antibodies in TBS-T with 5% of milk or 5% bovine
serum albumin, at 4 �C, washed in TBS-T, and incubated with horseradish
peroxidase (HRP)-conjugated secondary antibodies for 1 h at room tem-
perature. Signals were detected with the ECL detection system and
exposed to Fujifilm LAS-300 (Fujifilm Medical Systems, Stamford, CT,
USA). Equal protein loading and transfer efficiency were determined by
Ponceau Red staining after electrotransfer and GAPDH detection. The
densitometry patterns were analyzed with ImageGauge software (Sci-
ence Lab 2004; Fujifilm) and normalized to the density of GAPDH.
Antibodies
Anti-MMP-2 (mouse monoclonal; Santa Cruz), Anti-TIMP-2 (rabbit mono-
clonal; Cell signaling, Ozyme, St Quentin Yvelines, France), Anti MT1-
MMP (mouse monoclonal; Millipore), anti-GAPDH (goat polyclonal;
Sigma-Aldrich) were purchased. HRP-coupled secondary antibodies were
from P.A.R.I.S Ltd, Compiegne, France.
Determination of MMP-2 ⁄ -9 activity by
gelatin zymography assay
Zymography of conditioned media samples was performed as previously
described (Delbosc et al., 2008). Briefly, equal volume of sample issued
from conditioned medium was loaded onto an 10% polyacrylamide gel
containing 1% gelatin. After electrophoresis, gels were washed in a
2.5% Triton X-100 solution (2 · 30 min), incubated for 20 h in buffer
containing 50 mM Tris and 2.5 mM CaCl2, stained with Coomassie Blue,
and destained in methanol ⁄ acetic acid buffer until the bands could be
detected.
Apoptosis detection
Cell death was determined as the percentage of hypodiploid nuclei (HN)
assessed by propidium iodide staining (Mateo et al., 2007). Briefly, cells
were resuspended in a hypotonic solution containing 0.1% sodium cit-
rate, 0.1% Triton X-100 (Sigma-Aldrich), and 50 lg mL)1 propidium
iodide (Sigma). Cell nuclei were analyzed on EPICS XL instrument using
Expo32 software (Becton Coulter). The percentage of induced apoptosis
was calculated as 100 · (percentage of experimental HN)percentage of
spontaneous HN) ⁄ (100)% of spontaneous HN). Cell death was also
determined by fluorescent ethidium homodimer (EthD-III) assay that pro-
duces a bright red fluorescence in dying cells. VSMCs were plated in black
with clear bottom 96-well microplates at 10 000 cells ⁄ well 2 days prior
treatment. After 48–72 h of treatment, the medium was removed and
replaced by a volume of 100 lL per well of PBS containing 5 lM of ethidi-
um homodimer III (EthD-III). After 30 min incubation, fluorescence was
evaluated on a microplate reader (Infinite F500; Tecan France S.A.S, Lyon,
France) at ex ⁄ em 520 nm ⁄ 630 nm. One hundred percent dead cells were
determined by measuring the fluorescence emitted by an absolute
ethanol-treated well. Percentage of cell death was determined as
100 · (Fluorescence of experimental condition)background fluorescence) ⁄(Fluorescence of ethanol-treated cells)background fluorescence). Broad-
range MMP inhibitors GM6001 (25 lM) or Batimastat (2.5 lM) were added
simultaneously to Ab-peptide and renewed after 24 h.
Immunocytochemistry and microscopy
Vascular smooth muscle cells were seeded directly onto glass coverslips
placed in 24-well culture plates. After treatment (always indicated in fig-
ure legends), cells were washed twice with PBS, fixed in 20 min at room
temperature in 4% paraformaldehyde (neutralized with 50 mM of NH4Cl
for 10 min), permeabilized in PBS+0.2% Triton X100, and incubated for
1 h in PBS+10% FCS with TIMP-2 monoclonal antibody. The incubation
with DyLight 488-Conjugated Goat Anti-Rabbit secondary antibody was
performed during 1 h and Hoechst staining for 5 min. Coverslips were
mounted with Dako fluorescent mounting medium (Dako, Carpinteria,
CA, USA). Cells were examined by phase contrast or fluorescent micros-
copy using a Nikon Diaphoto 300 microscope equipped with a mercury
lamp (Nikon, Tokyo, Japan).
Statistical analysis
The data are reported as the mean ± SD. The numbers of independent
experiments are reported in figure legends. Values were compared
between groups with the Welch’s unpaired, corrected t-test.
Acknowledgments
We thank Dr. Xavier Houard for helpful discussion and advice and
Dr. Alex Strongin for providing reagents. This project has been finan-
cially supported by ‘Pierre Fabre Innovation’.
Authors contribution
Isabelle Limon had full access to all of the data in the study and takes
responsibility for the integrity of the data and the accuracy of the
data analysis. All the authors approved the final manuscript version
to be published. Regis Blaise, Veronique Mateo, Vladislav Golubkov,
and Isabelle Limon contributed to the study conception and design.
Regis Blaise, Veronique Mateo, Clotilde Rouxel, Francois Zaccarini,
Martine Glorian, and Vladislav Golubkov contributed to the acquisi-
tion of data. Regis Blaise, Veronique Mateo, Francois Zaccarini, Gil-
bert Bereziat, Vladislav Golubkov, and Isabelle Limon carried out the
analysis and interpretation of data. Regis Blaise, Veronique Mateo,
Vladislav Golubkov, and Isabelle Limon contributed to the manu-
script preparation. Regis Blaise and Isabelle Limon performed the
statistical analysis.
References
Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cere-
bral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels.
J. Neuropathol. Exp. Neurol. 57, 353–359.
Busti C, Falcinelli E, Momi S, Gresele P (2010) Matrix metalloproteinases and
peripheral arterial disease. Intern. Emerg. Med. 5, 13–25.
Clement N, Glorian M, Raymondjean M, Andreani M, Limon I (2006) PGE2
amplifies the effects of IL-1beta on vascular smooth muscle cell de-differentia-
tion: a consequence of the versatility of PGE2 receptors 3 due to the emerging
expression of adenylyl cyclase 8. J. Cell. Physiol. 208, 495–505.
Davis J, Van Nostrand WE (1996) Enhanced pathologic properties of Dutch-type
mutant amyloid beta-protein. Proc. Natl. Acad. Sci. U S A 93, 2996–3000.
Davis J, Cribbs DH, Cotman CW, Van Nostrand WE (1999) Pathogenic amyloid
beta-protein induces apoptosis in cultured human cerebrovascular smooth
muscle cells. Amyloid 6, 157–164.
Davis J, Wagner MR, Zhang W, Xu F, Van Nostrand WE (2003) Amyloid beta-
protein stimulates the expression of urokinase-type plasminogen activator
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
392

(uPA) and its receptor (uPAR) in human cerebrovascular smooth muscle cells.
J. Biol. Chem. 278, 19054–19061.
Delbosc S, Glorian M, Le Port AS, Bereziat G, Andreani M, Limon I (2008) The
benefit of docosahexanoic acid on the migration of vascular smooth muscle
cells is partially dependent on Notch regulation of MMP-2 ⁄ -9. Am. J. Pathol.
172, 1430–1440.
Fossati S, Cam J, Meyerson J, Mezhericher E, Romero IA, Couraud PO, Weksler
BB, Ghiso J, Rostagno A (2010) Differential activation of mitochondrial apop-
totic pathways by vasculotropic amyloid-beta variants in cells composing the
cerebral vessel walls. FASEB J. 24, 229–241.
Galis ZS, Khatri JJ (2002) Matrix metalloproteinases in vascular remodeling and
atherogenesis: the good, the bad, and the ugly. Circ. Res. 90, 251–262.
Hernandez-Guillamon M, Mawhirt S, Fossati S, Blais S, Pares M, Penalba A, Bo-
ada M, Couraud PO, Neubert TA, Montaner J, Ghiso J, Rostagno A (2010)
Matrix metalloproteinase 2 (MMP-2) degrades soluble vasculotropic amyloid-
beta E22Q and L34V mutants, delaying their toxicity for human brain micro-
vascular endothelial cells. J. Biol. Chem. 285, 27144–27158.
Hernandez-Guillamon M, Martinez-Saez E, Delgado P, Domingues-Montanari S,
Boada C, Penalba A, Boada M, Pagola J, Maisterra O, Rodriguez-Luna D,
Molina CA, Rovira A, Alvarez-Sabin J, Ortega-Aznar A, Montaner J (2011)
MMP-2 ⁄ MMP-9 plasma level and brain expression in cerebral amyloid angiop-
athy-associated hemorrhagic stroke. Brain Pathol., doi: 10.1111/j.1750-3639.
2011.00512.x.
Itoh Y, Seiki M (2006) MT1-MMP: a potent modifier of pericellular microenviron-
ment. J. Cell. Physiol. 206, 1–8.
Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, As-
chmies S, Kirksey Y, Gonzales C, Xu J, Zhou H, Atchison K, Wagner E, Zaleska
MM, Das I, Arias RL, Bard J, Riddell D, Gardell SJ, Abou-Gharbia M, Robichaud
A, Magolda R, Vlasuk GP, Bjornsson T, Reinhart PH, Pangalos MN (2008)
Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis
cascade. Proc. Natl. Acad. Sci. U S A 105, 8754–8759.
Jung SS, Zhang W, Van Nostrand WE (2003) Pathogenic A beta induces the
expression and activation of matrix metalloproteinase-2 in human cerebrovas-
cular smooth muscle cells. J. Neurochem. 85, 1208–1215.
Kawai M, Kalaria RN, Cras P, Siedlak SL, Velasco ME, Shelton ER, Chan HW,
Greenberg BD, Perry G (1993) Degeneration of vascular muscle cells in cere-
bral amyloid angiopathy of Alzheimer disease. Brain Res. 623, 142–146.
Knudsen KA, Rosand J, Karluk D, Greenberg SM (2001) Clinical diagnosis of
cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 56,
537–539.
Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen
SG, Bots GT, Luyendijk W, Frangione B (1990) Mutation of the Alzheimer’s
disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science
248, 1124–1126.
Liao MC, Van Nostrand WE (2010) Degradation of soluble and fibrillar amyloid
beta-protein by matrix metalloproteinase (MT1-MMP) in vitro. Biochemistry 49,
1127–1136.
Maia LF, Mackenzie IR, Feldman HH (2007) Clinical phenotypes of cerebral amy-
loid angiopathy. J. Neurol. Sci. 257, 23–30.
Mao D, Lee JK, VanVickle SJ, Thompson RW (1999) Expression of collagenase-3
(MMP-13) in human abdominal aortic aneurysms and vascular smooth muscle
cells in culture. Biochem. Biophys. Res. Commun. 261, 904–910.
Maruyama K, Ikeda S, Ishihara T, Allsop D, Yanagisawa N (1990) Immunohisto-
chemical characterization of cerebrovascular amyloid in 46 autopsied cases
using antibodies to beta protein and cystatin C. Stroke 21, 397–403.
Mateo V, Menager M, de Saint-Basile G, Stolzenberg MC, Roquelaure B, Andre
N, Florkin B, le Deist F, Picard C, Fischer A, Rieux-Laucat F (2007)
Perforin-dependent apoptosis functionally compensates Fas deficiency in
activation-induced cell death of human T lymphocytes. Blood 110, 4285–4292.
Meilhac O, Ho-Tin-Noe B, Houard X, Philippe M, Michel JB, Angles-Cano E
(2003) Pericellular plasmin induces smooth muscle cell anoikis. FASEB J. 17,
1301–1303.
Michel JB (2003) Anoikis in the cardiovascular system: known and unknown
extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 23, 2146–2154.
Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S (2008) Abeta-degrad-
ing enzymes in Alzheimer’s disease. Brain Pathol. 18, 240–252.
Mok SS, Losic D, Barrow CJ, Turner BJ, Masters CL, Martin LL, Small DH (2006)
The beta-amyloid peptide of Alzheimer’s disease decreases adhesion of vascu-
lar smooth muscle cells to the basement membrane. J. Neurochem. 96, 53–
64.
Murphy G, Nagase H (2011) Localizing matrix metalloproteinase activities in the
pericellular environment. Febs J. 278, 2–15.
Newby AC (2006) Matrix metalloproteinases regulate migration, proliferation,
and death of vascular smooth muscle cells by degrading matrix and non-
matrix substrates. Cardiovasc. Res. 69, 614–624.
Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina
CA, Lo EH, Montaner J (2006) Increased brain expression of matrix metallopro-
teinase-9 after ischemic and hemorrhagic human stroke. Stroke 37, 1399–
1406.
Rosenberg GA, Navratil M (1997) Metalloproteinase inhibition blocks edema in
intracerebral hemorrhage in the rat. Neurology 48, 921–926.
Rozanov DV, Deryugina EI, Ratnikov BI, Monosov EZ, Marchenko GN, Quigley JP,
Strongin AY (2001) Mutation analysis of membrane type-1 matrix metallopro-
teinase (MT1-MMP). The role of the cytoplasmic tail Cys(574), the active site
Glu(240), and furin cleavage motifs in oligomerization, processing, and self-
proteolysis of MT1-MMP expressed in breast carcinoma cells. J. Biol. Chem.
276, 25705–25714.
Schonbeck U, Mach F, Sukhova GK, Murphy C, Bonnefoy JY, Fabunmi RP, Libby
P (1997) Regulation of matrix metalloproteinase expression in human vascular
smooth muscle cells by T lymphocytes: a role for CD40 signaling in plaque
rupture? Circ. Res. 81, 448–454.
Smith EE, Greenberg SM (2009) Beta-amyloid, blood vessels, and brain function.
Stroke 40, 2601–2606.
Solito R, Corti F, Fossati S, Mezhericher E, Donnini S, Ghiso J, Giachetti A, Rost-
agno A, Ziche M (2009) Dutch and Arctic mutant peptides of beta amyloid
(1–40) differentially affect the FGF-2 pathway in brain endothelium. Exp. Cell
Res. 315, 385–395.
Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI (1995)
Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation
of the activated form of the membrane metalloprotease. J. Biol. Chem. 270,
5331–5338.
Suo Z, Tan J, Placzek A, Crawford F, Fang C, Mullan M (1998) Alzheimer’s beta-
amyloid peptides induce inflammatory cascade in human vascular cells: the
roles of cytokines and CD40. Brain Res. 807, 110–117.
Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, Walker
D, Scheff S, McGillis JP, Rydel RE, Estus S (2000) The plasmin system is
induced by and degrades amyloid-beta aggregates. J. Neurosci. 20, 3937–
3946.
Van Wart HE, Birkedal-Hansen H (1990) The cysteine switch: a principle of regu-
lation of metalloproteinase activity with potential applicability to the entire
matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. U S A 87, 5578–
5582.
Visse R, Nagase H (2003) Matrix metalloproteinases and tissue inhibitors of me-
talloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827–839.
Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO, Romero IA, Weks-
ler B, Stanimirovic DB, Zhang W (2009) Expression of inflammatory genes
induced by beta-amyloid peptides in human brain endothelial cells and in
Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol.
Dis. 34, 95–106.
Wu L, Tanimoto A, Murata Y, Sasaguri T, Fan J, Sasaguri Y, Watanabe T (2003)
Matrix metalloproteinase-12 gene expression in human vascular smooth mus-
cle cells. Genes Cells 8, 225–234.
del Zoppo GJ (2010) The neurovascular unit, matrix proteases, and innate inflam-
mation. Ann. N Y Acad. Sci. 1207, 46–49.
Supporting information
Additional supporting information may be found in the online version of
this article:
Fig. S1 TIMP-2 ⁄ MT1-MMP-dependent MMP-2 activation.
Fig. S2 WT-Ab1-40 induces cell death in MCF-7.
Fig. S3 WT-Ab1-40 and Ab1-40 Dutch-induced cell-death are differentially
affected by GM6001 and Serum.
Fig. S4 WT-Ab1-40 peptide increases tPA and decreases PAI-1 expression in
VSMCs.
As a service to our authors and readers, this journal provides supporting
information supplied by the authors. Such materials are peer-reviewed and
may be re-organized for online delivery, but are not copy-edited or typeset.
Technical support issues arising from supporting information (other than
missing files) should be addressed to the authors.
MMP activity and WT amyloid beta 1-40-induced cell death, R. Blaise et al.
ª 2012 The AuthorsAging Cell ª 2012 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland
393
Related Documents











