Why Does Polyglutamine Aggregate? Insights from studies of monomers Xiaoling Wang, Andreas Vitalis, Scott Crick, Rohit Pappu Biomedical Engineering & Center for Computational Biology, Washington University in St.Louis [email protected] http://lima.wustl.edu

Why Does Polyglutamine Aggregate? Insights from studies of monomers Xiaoling Wang, Andreas Vitalis, Scott Crick, Rohit Pappu Biomedical Engineering & Center.
Dec 20, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Why Does Polyglutamine Aggregate? Insights from studies of monomers
Xiaoling Wang, Andreas Vitalis, Scott Crick, Rohit PappuBiomedical Engineering & Center for Computational Biology,
Washington University in [email protected]
http://lima.wustl.edu

DISEASE GENE PRODUCT
NORMAL CAG
REPEAT RANGE
MUTANT CAG REPEAT RANGE
Huntington’s huntingtin 6 - 39 36-200 DRPLA atrophin 1 3 – 35 49 - 88 SBMA androgen rec. 9 – 33 38 - 65 SCA1 ataxin -1 6 – 44 39 - 83 SCA2 ataxin -2 13 – 33 32 - 200 SCA3/MJD ataxin -3 3 – 40 54 - 89 SCA6
CACNA1A 4 – 19 20 - 33 SCA7 ataxin - 7 4 – 35 37 - 306 SCA17 TBP 24 – 44 46 - 63
Expanded CAG Repeat Diseases and Proteins
Bates, et al., Eds. (2002) Huntington's Disease, Oxford University Press

Basic physics of aggregation
: Free energy of soluble monomer
: Free energy of aggregate
Aggregation is spontaneous if:0
M
A
A MA M
G
G
G G
n
n: denotes the number of peptide molecules in the system (concentration)
N: Length of each peptide molecule in the system

Work done to grow a cluster
Cluster excess or interfacial free energy
For *, 1 0
For *, 1 0
ex
ex
W n n G n
G n
n n W n W n
n n W n W n
In vitro aggregation studies of synthetic polyglutamine peptides1. Evidence for nucleation-dependent polymerization2. Rates of elongation versus concentration are fit to a pre-equilibrium model 3. And fits to the model suggests that n*=1 for Q28, Q36, Q47
4. See Chen, Ferrone, Wetzel, PNAS, 2002
n*

UV-CD data: Q5(-), D2Q15K2(-.-), Q28(…), Q45(---); Chen et al. JMB, 311, 173 (2001)
1. No major difference between different chain lengths2. CD spectra for polyglutamine resemble those of denatured proteins

For given N, there is a concentration (n) for which ∆ < 0. Why?
Hypothesis: Water is a poor solvent for polyglutamine: Chain flexibility and attractions overwhelm chain-solvent
interactions Polymers form internally solvated collapsed globules
Rg and other properties scale with chain length as N0.34
Most chains aggregate and fall out of solution
CD data and heuristics counter our hypothesis: For denatured proteins, Rg~ N0.59 - polymers in good solvents
Polyglutamine is polar – suggests that water is a good solvent Requires new physics to explain polyglutamine aggregation
Let’s test our hypothesis

MRMD – the “algorithm”
1. Using a series of “short” simulations, estimate the time scale over which : Autocorrelation of “soft” modes decay There are recurrent transitions between compact and
swollen conformations
2. Use the estimate for , the time scale for each “elementary simulation” is tS~10 60-100 independent simulations, each of “length” ts
3. Pool data from all simulations and construct conformational distributions using bootstrap methods

Simulation engine
Forcefield: OPLSAA for peptides and TIP4P for water Constant pressure (P), constant temperature (T): NPT T = 298K, P = 1atm Thermostat and barostat: Berendsen weak coupling Long-range interactions: Twin range spherical cutoffs Periodic boundary conditions in boxes that contain > 4000 water
molecules Peptides: ace-(Gln)N-nme, N=5,15,20,… Cumulative simulation times > 5s We have an internal control – the excluded volume (EV) limit – to
quantify conformational equilibria in good solvents

Top row in water, bottom row in EV limit
Q5 Q15 Q20
EV Limit
In water

Scaling of internal distances is consistent with behavior of chain in a poor solvent
Data for polyglutamine in EV limitData for polyglutamine in water
Q5 Q15 Q20

Can we test our “prediction”? Yes
Using Fluorescence Correlation Spectroscopy (FCS)
Peptides studied: -Gly-(Gln)N-Cys*-Lys2
* indicates fluorescent label, which is Alexa488 Solution conditions:
PBS: pH 7.3, 8.0g NaCl, 0.2g KCl, 1.15g Di-sodium orthophosphate, 0.2g Potassium di-hydrogen orthophosphate, dissolved in pure H2O
Approximately one molecule in beam volume
Is diffusion time, D N0.33 or is ln(D ) 0.33ln(N)?

Evidence for poor solvent scaling

Polyglutamine: Compact albeit disordered
Observation of disorder is consistent with CD data

Quantifying topology
N i
Ci
C i+1
N n
Cj C j+1
θ
residue i
residue j
What is the length scale over which spatial correlations decay? Compute <cos(θij)> as a function of |j-i|
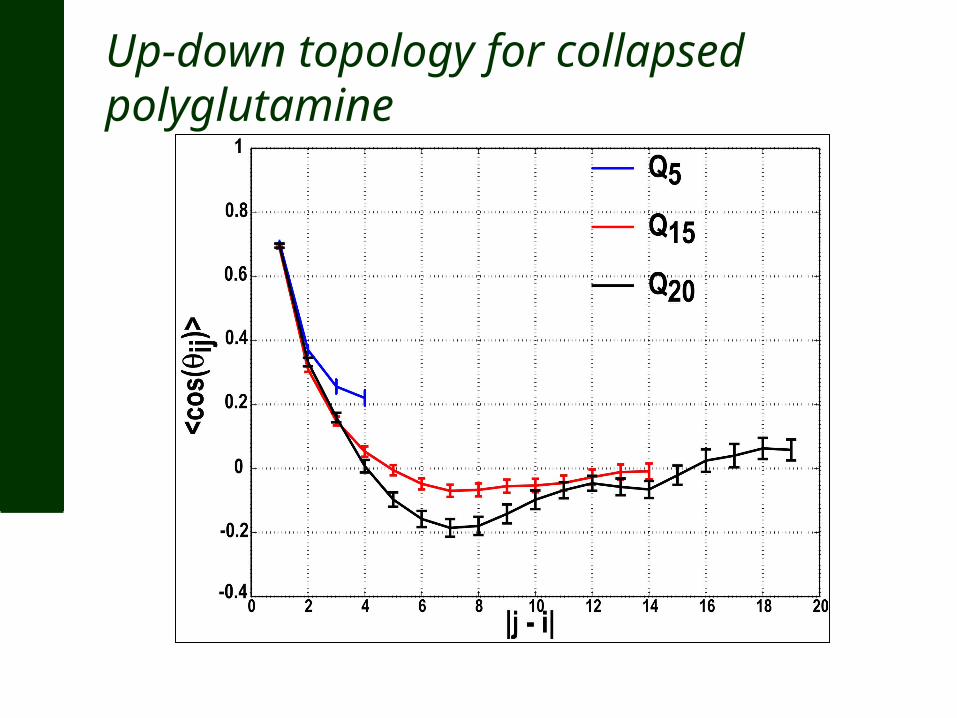
Up-down topology for collapsed polyglutamine

Q15 Q20
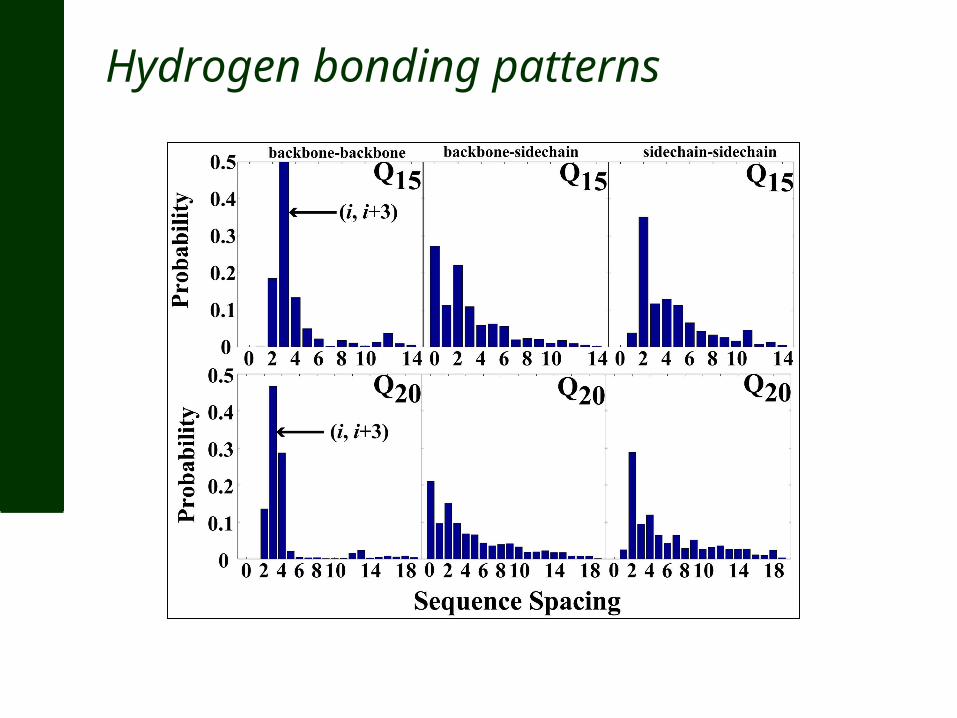
Hydrogen bonding patterns

Why collapse and what does it mean?
1. Summary – The ensemble for polyglutamine in water: Is disordered albeit collapsed Has a preferred up-down average topology With a strong propensity for forming beta turns And little to no long-range backbone hydrogen bonds
2. What drives collapse in water: Generic backbone?3. Is there anything special about polyglutamine?4. What does all this mean for nucleation of aggregation?

Distributions for polyglycine
Mimics of polypeptide backbones prefer to be collapsed in water, which appears to be a universal poor solvent for polypeptides
Polyglutamine is a chain of two types of amides: secondary and primary
Water 8M Urea EV Limit

Primary and secondary amides
Propanamide (PPA) N-methylformamide (NMF)

Amides in water
Pure (primary or secondary) Amides in water: N =nW + nA
NPT Simulations with varying nA implies varying A
T=300K, P = 1atm OPLSAA forcefield for amides, TIP4P for H2O nA = 16, 32, 64, etc. for 1, 2, 3, … molal solutions; nW = 800
Amide (ternary) mixtures: Primary and secondary amides N = nW + nP + nS Keep nW and nP fixed and vary nS or nW and nS fixed, vary nP
Will show data for nP = nS = 32

Pair correlations
1. NMF prefers water-separated contacts over hydrogen bonded contacts2. PPA prefers hydrogen bonded contacts over water-separated contacts3. PPA donor - NMF acceptor hydrogen bonds are preferred in mixtures

Cluster statistics

Typical large cluster in PPA:NMF mixtures
Consistent with data of Eberhardt and Raines, JACS, 1994
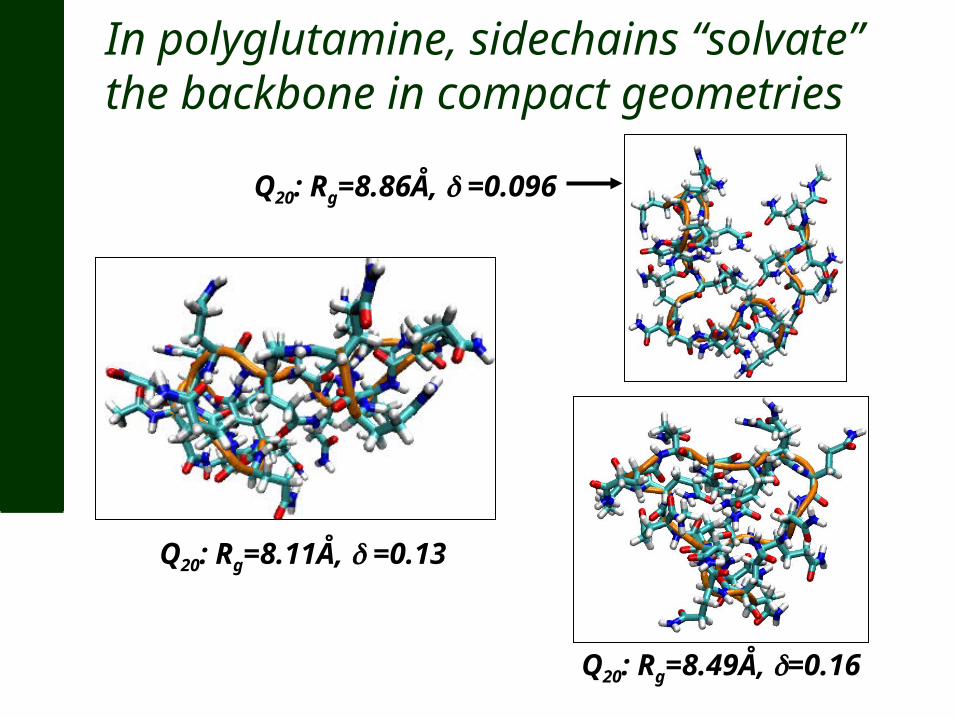
In polyglutamine, sidechains “solvate” the backbone in compact geometries
Q20: Rg=8.11Å, =0.13
Q20: Rg=8.49Å, =0.16
Q20: Rg=8.86Å, =0.096

Hypothesis – part I: Why is aggregation spontaneous?
For a system of peptides of length N: There is a finite concentration (n) for which ∆ < 0
∆ < 0 if: Aggregated state of intermolecular solvation via
glutamine sidechains is preferred to the disordered state of intramolecular solvation whereby sidechains solvate their own backbones
It is our hypothesis that: Peptide concentration at which ∆ becomes negative
will decrease “rapidly” with increasing chain length

Hypothesis – part II: Nucleation
Ensemble of nucleus is species of highest free energy for monomer Nucleation must involve the following penalties:
DESOLVATION: Replace favorable sidechain-backbone contacts and residual water-backbone contacts with unfavorable backbone-backbone contacts
ENTROPIC BOTTLENECK: Replace disordered ensemble with ordered nucleus
Conformations in the nucleus ensemble? 1. β-helix-like (see work of Dokholyan group, PLoS, 2005)
2. -pleated sheet (see work of Daggett group, PNAS, 2005)
3. Antiparallel β-sheet (see fiber diffraction data)

Thanks to…
THE LAB Xiaoling Wang Andreas Vitalis Scott Crick Hoang Tran Alan Chen Matthew Wyczalkowski
Collaborations Ron Wetzel – UTK Murali Jayaraman – UTK Carl Frieden – WUSTL

Ongoing work…
1. Monomer distributions for N > 25 2. Free energies of nucleating intramolecular beta sheets3. Influence of sequence context: In vivo, its not just a
polyglutamine 4. Quantitative characterization of oligomer landscape 5. Generalizations to aggregation of other intrinsically
disordered proteins rich in polar amino acids6. Experiments: New FCS methods to study oligomers
and nucleation kinetics
Related Documents











