Geoderma, 53 (1992) 45-63 45 Elsevier Science Publishers B.V., Amsterdam Weathering of silicate minerals by organic acids. I. Nature of cation solubilisation Aloke K. Barman ~, Chandrika Varadachari 2 and Kunal Ghosh Department of Agricultural Chemistry and Soil Science, University of Calcutta, 35 Ballygunge Circular Road, Calcutta 700 019, India (Received April 10, 1991; accepted after revision September 30, 1991 ) ABSTRACT Barman, A.K., Varadachari, C. and Ghosh, K., 1992. Weathering of silicate minerals by organic acids. I. Nature of cation solubilisation. Geoderma, 53: 45-63. The solubilisation of olivine, epidote, hornblende, tourmaline, biotite and microcline by oxalic, citric, salicylic acids and glycine were studied. The results indicate that organic acids dissolve minerals by a combined action of complexation and acid attack. Relative solubilities of the cations from a mineral are greatly dependent on their positions within the crystal and the crystal structure as a whole. These factors determine the accessibility of the ligand ions to the cations and also the extent to which the removal of one ion affects the neighbouring ions. Ions in polymeric chains or in sterically hindered positions are most resistant to dissolution. There is no unique stability sequence of the silicate min- erals in different acids. The sequences are determined by the combined effect of two factors, viz., the crystal structure and the forces of interaction of the constituent ions with the surrounding medium. Kinetic studies indicate that the rate determining step at the initial stages of reaction is surface de- tachment of cation-ligand complexes; subsequently, the creation of fresh surfaces by fragmentation, etching, etc., may become rate-limiting. INTRODUCTION Fundamentally, all chemical weathering of silicates, whether inorganic or biochemical, involves solubilisation of the structural ions at the initial stage of the process. Studies on various aspects of the mechanism of mineral dis- solution have, consequently, been the focus of a great deal of attention. In a purely aqueous inorganic environment, it appears that the exchange of pro- tons in water with the cations on the mineral surface is the rate-determining step of the transformation (Correns, 1961; Loughnan, 1969 ). More recently, 1present address: All India Soil & Land Use Survey, 424/95/2 6th Main Road, 15th Cross, Malleswaram, Bangalore 560 003, India. 2present address: Polymer Science Unit, Indian Association for the Cultivation of Science, Ja- davpur, Calcutta 700 032, India.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Geoderma, 53 (1992) 45-63 45 Elsevier Science Publishers B.V., Amsterdam
Weathering of silicate minerals by organic acids. I. Nature of cation solubilisation
Aloke K. Barman ~, Chandrika Varadachari 2 and Kunal Ghosh Department of Agricultural Chemistry and Soil Science, University of Calcutta, 35 Ballygunge
Circular Road, Calcutta 700 019, India
(Received April 10, 1991; accepted after revision September 30, 1991 )
ABSTRACT
Barman, A.K., Varadachari, C. and Ghosh, K., 1992. Weathering of silicate minerals by organic acids. I. Nature of cation solubilisation. Geoderma, 53: 45-63.
The solubilisation of olivine, epidote, hornblende, tourmaline, biotite and microcline by oxalic, citric, salicylic acids and glycine were studied. The results indicate that organic acids dissolve minerals by a combined action of complexation and acid attack. Relative solubilities of the cations from a mineral are greatly dependent on their positions within the crystal and the crystal structure as a whole. These factors determine the accessibility of the ligand ions to the cations and also the extent to which the removal of one ion affects the neighbouring ions. Ions in polymeric chains or in sterically hindered positions are most resistant to dissolution. There is no unique stability sequence of the silicate min- erals in different acids. The sequences are determined by the combined effect of two factors, viz., the crystal structure and the forces of interaction of the constituent ions with the surrounding medium. Kinetic studies indicate that the rate determining step at the initial stages of reaction is surface de- tachment of cation-ligand complexes; subsequently, the creation of fresh surfaces by fragmentation, etching, etc., may become rate-limiting.
INTRODUCTION
Fundamentally, all chemical weathering of silicates, whether inorganic or biochemical, involves solubilisation of the structural ions at the initial stage of the process. Studies on various aspects of the mechanism of mineral dis- solution have, consequently, been the focus of a great deal of attention. In a purely aqueous inorganic environment, it appears that the exchange of pro- tons in water with the cations on the mineral surface is the rate-determining step of the transformation (Correns, 1961; Loughnan, 1969 ). More recently,
1present address: All India Soil & Land Use Survey, 424/95/2 6th Main Road, 15th Cross, Malleswaram, Bangalore 560 003, India. 2present address: Polymer Science Unit, Indian Association for the Cultivation of Science, Ja- davpur, Calcutta 700 032, India.

46 A.K. BARMAN ET AL.
a number of hypotheses have been proposed, on the mechanism of dissolu- tion of silicates in water. None of these, however, have found unanimous acceptance. The hypotheses are (i) dissolution rate is controlled by the rate of reaction at the interface (Petrovi6 et al., 1976; Schott et al., 1981 ), (ii) release of cations is regulated by the rate at which the cations can diffuse through an altered surface layer (Luce et al., 1972; Pa6es, 1973), (iii) disso- lution rate is controlled by diffusion through a surface layer of amorphous silica-alumina precipitate (Wollast, 1967 ), or (iv) dissolution is controlled by diffusion through a layer of crystalline precipitate (Helgeson, 1971; Bus- enberg and Clemency, 1976). Other investigators, however, point out that even though an altered surface layer of 1 nm thickness (Schott et al., 1981; Schott and Berner, 1983 ) or a surface coating (Petrovi6 et al., 1976) may be present, these do not appear to hinder the normal movement of ions and, therefore, are not rate-inhibiting. Some workers (Berner and Holdren, 1979; Holdren and Berner, 1979 ) concluded that reaction occurs preferably along dislocations and other such sites of high energy and leads to the development of distinctive etch pits at the surface of the grains.
In the presence of organic acids, chelating ability of the acids for the cations in the minerals becomes an important factor of cation release (Schatz et al., 1957; Henderson and Duff, 1963; Stumm et al., 1985). This, however, may be true in some instances but not in others (Schalscha et al., 1967; Ponomar- eva, 1969; Tan, 1980). Dissolution is mostly incongruent but this depends on the nature of reacting acid and mineral (Huang and Keller, 1970; Schnitzer and Kodama, 1976; Song and Huang, 1988). Regarding the mechanism of organic acid dissolution, it has been suggested that the rate of reaction is con- trolled by surface processes (Zutic and Stumm, 1984; Song and Huang, 1988 ). The weakening of the metal-oxygen bond in the mineral is probably brought about by the formation of inner sphere complexes (Motschi, 1983; Stumm et al., 1985 ). According to Zutic and Stumm (1984), dissolution is a function of the degree of surface protonation and the concentration of surface ligand complexes.
In this work, an attempt has been made to obtain a more detailed picture of the nature of silicate dissolution by organic acids. The following aspects are particularly emphasised: (i) The order of cation release as influenced by the positions of the ions in the crystals, (ii) effect ofligands as well as crystal structure on mineral stability sequence, (iii) patterns of congruency of cation dissolution, and (iv) kinetics of dissolution with reference to the mechanism of the process. For this purpose, representative silicates from each of the six classes of silicates (from the neso to the tecto ) have been taken. Reactions of these silicates with four different organic acids of varying complexing abili- ties have been studied. Various aspects of the solubilisation of the major con- stituent cations have been determined after suitable modification of some of the analytical procedures.

WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS I 47
MATERIALS AND METHODS
The minerals used in this study were olivine, epidote, tourmaline, horn- blende, biotite and microcline, obtained from the Geological Survey of India, Calcutta. They were ground in an agate mortar, sieved to obtained the 80- 150 B.S. mesh (0.2 to 0.1 m m ) size fraction and subsequently washed and dried. XRD of the samples was recorded on a Philips PW 1140 diffractometer using Ni-filtered Cu-K~ radiation at a scanning speed of 1 ° 20/min; XRD photograph was obtained on a Philips PW 1730 instrument fitted with a Gui- nier Camera.
The organic acids used were oxalic acid, citric acid, salicylic acid and gly- cine (all of analytical grade); the acids were chosen to represent decreasing acid strength (in the order listed).
Chemical analyses of the samples were done according to a combined scheme of Shapiro and Brannock and of Riley (Maxwell, 1968 ). A portion of the sample, fused in NaOH was analysed for Si 4+ as the molybdenum blue complex using 1-amino-2-naphthol-4-sulphonic acid as reductant (Maxwell, 1968 ). Another portion of the sample fused in HF-HC104 was analysed for AP + as the calcium a luminum alizarin-red S complex (Maxwell, 1968), Fe 2+/3+ as the o-phenanthroline complex (Black, 1965), Ti 4+ as the H202 complex (Jeffery, 1970), Na + and K + by flame photometry, Ca 2 + and Mg a÷ by EDTA titration (Black, 1965) and Mn 2+ as HMnO4 (Maxwell, 1968). Since tourmaline is not fusable in HF-HC104, it was fused in NH4F (Max- well, 1968 ) instead. The determination of B 3+ in tourmaline was done on a NaOH-fused sample as the 1-1' dianthrimide complex in H2SO4 medium (Vogel, 1961 ).
The methods for the determination of Si 4+, AP + and Fe 3+/2+ in the solu- tions obtained after mineral-organic acid reactions, had to be slightly modi- fied since organic acids were observed to cause serious interference in the colorimetric procedures. For the determination of AP + and Fe 3 +/2+, the so- lutions (5 ml or less) were treated with 1 ml of 30% H202, 2 drops of 2N HC1, kept for 24 h and then treated with 3 ml of 26% Na2SO3 and allowed to stand for 30 min to decompose the excess H202. This process was adopted for all solutions except those containing citric acid, which is not readily decomposed by H202. In such solutions, the amount of HaO2 was doubled and the solu- tions allowed to stand for 10 days before treatment with Na2SO3. Colorimet- ric determinations were then done as described earlier.
For the determination of Si 4÷, the H202 treatment could not be adopted since no method for the removal of excess H202 could be found that would not interfere with the colour development. However, it was found experimen- tally that if the amount of molybdate reagent added was increased to four times the recommended value (Maxwell, 1968), then interference from the organic acids could be completely removed (provided less than 3 meq or-
48 A.K. BARMAN ET AL.
ganic acid is present). For higher amounts of organic acid, more molybdate must be added.
Determinations of Ca z+, Mg 2+, Na + and K + were done as described ear- lier without any modification since organic acids were observed to cause no interference in these methods.
Rates of release of various cations from the silicate minerals were studied in the following manner: To 0.5 g of the minerals in plastic bottles, 25 ml of 0.5M oxalic acid (pH, 0.5), 0.5M citric acid (pH, 1.0), 0.01M salicylic acid (pH, 2.3 ) or 0.5M glycine (pH, 5.6) were pipetted in. These were shaken on a horizontal shaker at ambient temperature (27 °C) for 5 h each day and al- lowed to equilibriate for the remaining 19 h. At the end of the reaction period, viz., 24, 48, 72, 96 or 120 h, the solutions were filtered, washed and made to volume. All solutions were stored in an asceptic chamber under UV light to prevent microbial growth till the analyses were complete.
RESULTS AND DISCUSSION
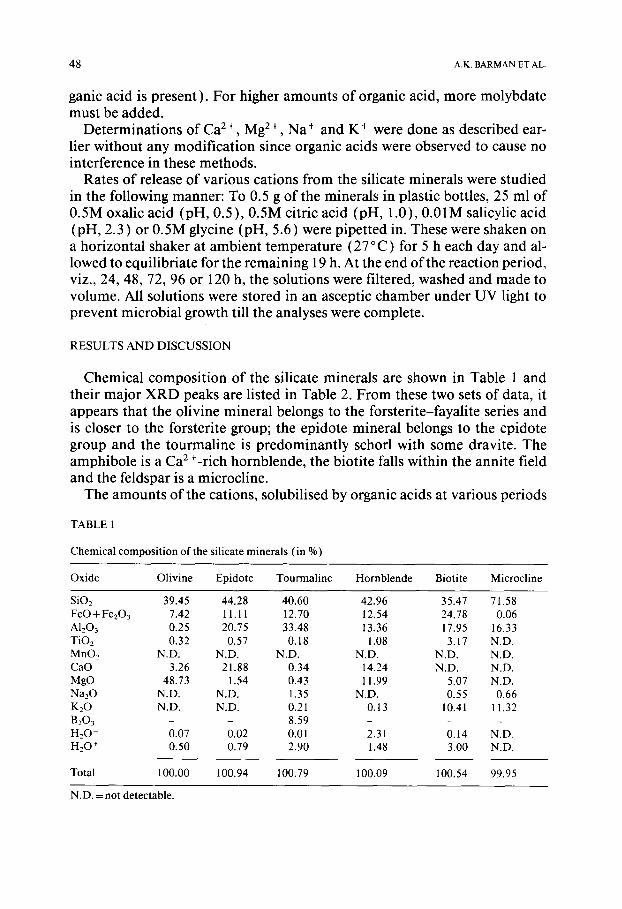
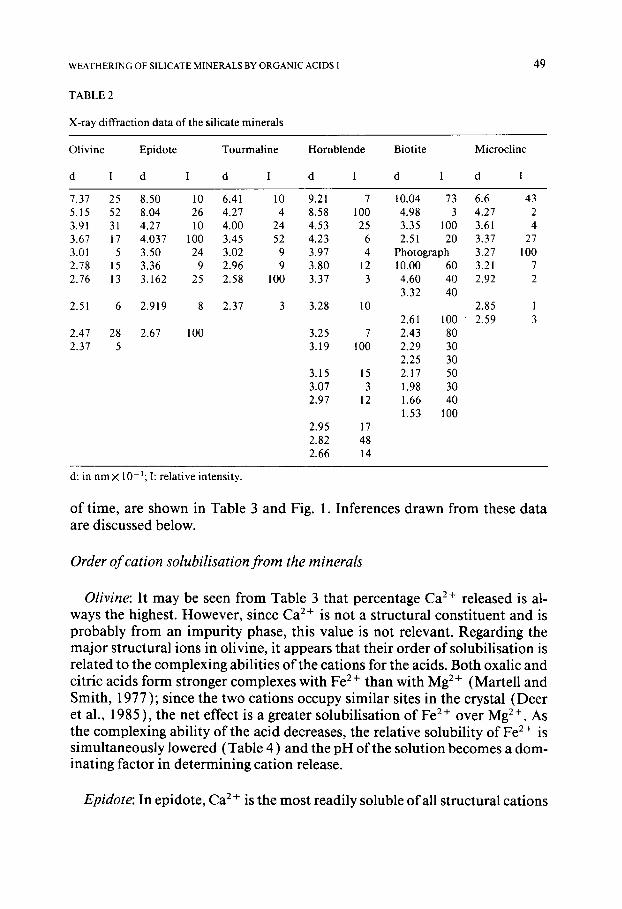
Chemical composit ion of the silicate minerals are shown in Table 1 and their major XRD peaks are listed in Table 2. From these two sets of data, it appears that the olivine mineral belongs to the forsterite-fayalite series and is closer to the forsterite group; the epidote mineral belongs to the epidote group and the tourmaline is predominantly schorl with some dravite. The amphibole is a Ca2+-rich hornblende, the biotite falls within the annite field and the feldspar is a microcline.
The amounts of the cations, solubilised by organic acids at various periods
TABLE 1
Chemical composition of the silicate minerals (in %)
Oxide Olivine Epidote Tourmaline Hornblende Biotite Microcline
SiO2 39.45 44.28 40.60 42.96 35.47 71.58 FeO + Fe203 7.42 11.11 12.70 12.54 24.78 0.06 A1203 0.25 20.75 33.48 13.36 17.95 16.33 TiO2 0.32 0.57 0.18 1.08 3.17 N.D. MnO2 N.D. N.D. N.D. N.D. N.D. N.D. CaO 3.26 21.88 0.34 14.24 N.D. N.D. MgO 48.73 1.54 0.43 11.99 5.07 N.D. Na20 N.D. N.D. 1.35 N.D. 0.55 0.66 K20 N.D. N.D. 0.21 0.13 10.41 11.32 B203 - - 8.59 - - - H20- 0.07 0.02 0.01 2.31 0.14 N.D. H20 + 0.50 0.79 2.90 1.48 3.00 N.D.
Total 100.00 100.94 100.79 100.09 100.54 99.95
N.D. = not detectable.
WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS 1 49
TABLE 2
X-ray diffraction data of the silicate minerals
Olivine Epidote Tourmaline Hornblende
d I d I d I d I
Biotite Microcline
d I d I
7.37 25 8.50 10 6.41 10 9.21 7 10.04 73 6.6 43 5.15 52 8.04 26 4.27 4 8.58 100 4.98 3 4.27 2 3.91 31 4.27 10 4.00 24 4.53 25 3.35 100 3.61 4 3.67 17 4.037 100 3.45 52 4.23 6 2.51 20 3.37 27 3.01 5 3.50 24 3.02 9 3.97 4 Photograph 3.27 100 2.78 15 3.36 9 2.96 9 3.80 12 10.00 60 3.21 7 2.76 13 3.162 25 2.58 100 3.37 3 4.60 40 2.92 2
3.32 40 2.51 6 2.919 8 2.37 3 3.28 10 2.85 1
2.61 100 ' 2.59 3 2.47 28 2.67 100 3.25 7 2.43 80 2.37 5 3.19 100 2.29 30
2.25 30 3.15 15 2.17 50 3.07 3 1.98 30 2.97 12 1.66 40
1.53 100 2.95 17 2.82 48 2.66 14
d: in nm × 10- ~; I: relative intensity.
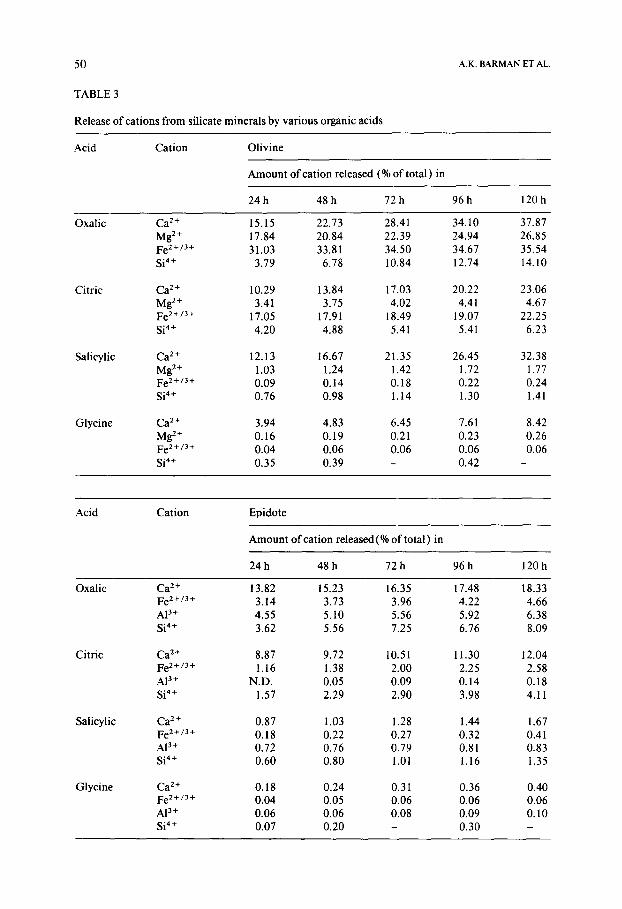
of time, are shown in Table 3 and Fig. 1. Inferences drawn from these data are discussed below.
Order of cation solubilisation from the minerals
Olivine: It may be seen from Table 3 that percentage Ca 2+ released is al- ways the highest. However, since Ca 2 ÷ is not a structural constituent and is probably from an impurity phase, this value is not relevant. Regarding the major structural ions in olivine, it appears that their order of solubilisation is related to the complexing abilities of the cations for the acids. Both oxalic and citric acids form stronger complexes with Fe 2 ÷ than with Mg 2+ (Martell and Smith, 1977 ); since the two cations occupy similar sites in the crystal (Deer et al., 1985 ), the net effect is a greater solubilisation of Fe 2÷ over Mg 2÷. As the complexing ability of the acid decreases, the relative solubility of Fe z + is simultaneously lowered (Table 4 ) and the pH of the solution becomes a dom- inating factor in determining cation release.
Epidote: In epidote, C a 2+ is the most readily soluble of all structural cations
50
TABLE 3
Release of cations from silicate minerals by various organic acids
A.K. BARMAN ET AL
Acid Cation Olivine
Amount of cation released (% of total) in
24 h 48 h 72 h 96 h 120 h
Oxalic Ca 2+ Mg 2+ Fe2+/3+ Si 4+
Citric Ca 2+ Mg 2+ Fe2+/3+ Si 4+
Salicylic Ca 2÷ Mg 2+ Fe2+/3+ Si 4+
Glycine Ca 2+ Mg 2+ Fe2+/3+ Si 4+
15.15 22.73 28.41 34.10 37.87 17.84 20.84 22.39 24.94 26.85 , 31.03 33.81 34.50 34.67 35.54
3.79 6.78 10.84 12.74 14.10
10.29 13.84 17.03 20.22 23.06 3.41 3.75 4.02 4.41 4.67
17.05 17.91 18.49 19.07 22.25 4.20 4.88 5.41 5.41 6.23
12.13 16.67 21.35 26.45 32.38 1.03 1.24 1.42 1.72 1.77 0.09 0.14 0.18 0.22 0.24 0.76 0.98 1.14 1.30 1.41
3.94 4.83 6.45 7.61 8.42 0.16 0.19 0.21 0.23 0.26 0.04 0.06 0.06 0.06 0.06 0.35 0.39 - 0.42 -
Acid Cation Epidote
Amount of cation released (% of total ) in
24 h 48 h 72 h 96 h 120 h
Oxalic Ca 2+ Fe2+/3+ AI 3+ Si 4+
Citric Ca 2+ Fe2+/3+ A13+ Si 4+
Salicylic Ca 2÷ Fe2+/3+ A13+ Si 4+
Glycine Ca 2+ Fe2+/3+ AI 3+ Si 4+
13.82 15.23 16.35 17.48 18.33 3.14 3.73 3.96 4.22 4.66 4.55 5.10 5.56 5.92 6.38 3.62 5.56 7.25 6.76 8.09
8.87 9.72 10.51 11.30 12.04 1.16 1.38 2.00 2.25 2.58
N.D, 0.05 0.09 0.14 0.18 1.57 2.29 2.90 3.98 4.11
0.87 1.03 1,28 1.44 1.67 0.18 0.22 0,27 0.32 0.41 0.72 0.76 0,79 0.81 0.83 0.60 0.80 1,01 1.16 1.35
0.18 0.24 0.31 0.36 0.40 0.04 0.05 0.06 0.06 0.06 0.06 0.06 0.08 0.09 0.10 0.07 0.20 - 0.30 -
WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS I
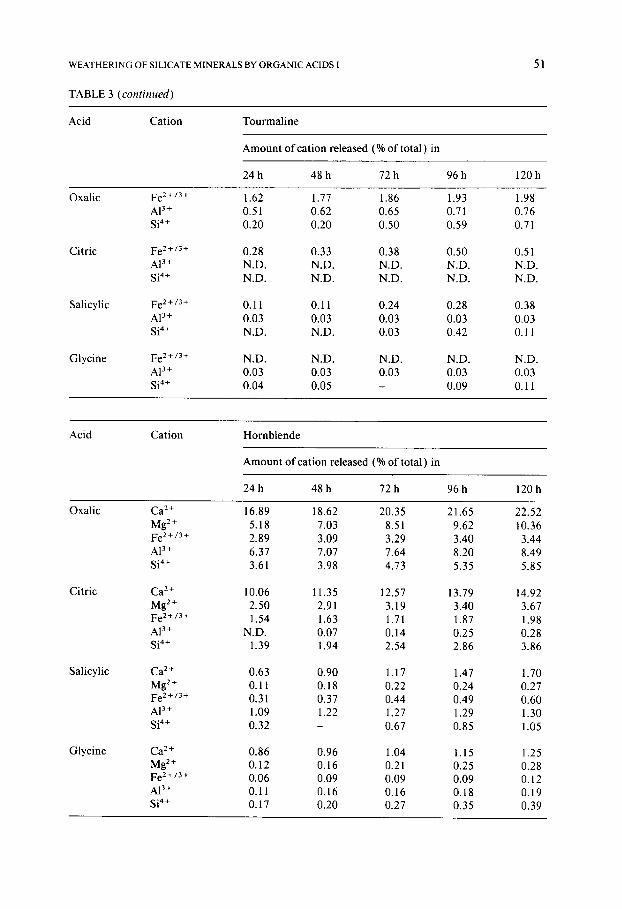
TABLE 3 (continued)
51
Acid Cation Tourmaline
Amount of cation released (% of total ) in
2 4 h 4 8 h 72h 9 6 h 120h
Oxalic Fe 2 +/3 + AI 3+ Si 4+
Citric Fe2+/3+ A13+ Si 4+
Salicylic Fe 2 +/3 + A13+
Si a+
Glycine Fe 2+/3+ A13+ Si 4+
1.62 1.77 1.86 1.93 1.98 0.51 0.62 0.65 0.71 0.76 0.20 0.20 0.50 0.59 0.71
0.28 0.33 0.38 0.50 0.51 N.D. N.D. N.D. N.D. N.D. N.D. N.D. N.D. N.D. N.D.
0.11 0.11 0.24 0.28 0.38 0.03 0.03 0.03 0.03 0.03 N.D. N.D. 0.03 0.42 0.11
N.D. N.D. N.D. N.D. N.D. 0.03 0.03 0.03 0.03 0.03 0.04 0.05 - 0.09 0.11
Acid Cation Hornblende
Amount of cation released (% of total) in
24 h 48 h 72 h 96 h 120 h
Oxalic
Citric
Salicylic
Glycine
Ca 2÷ 16.89 18.62 20.35 21.65 22.52 Mg 2÷ 5.18 7.03 8.51 9.62 10.36 Fe 2+/3+ 2.89 3.09 3.29 3.40 3.44 A13÷ 6.37 7.07 7.64 8.20 8.49 Si 4+ 3.61 3.98 4.73 5.35 5.85
Ca 2+ 10.06 11.35 12.57 13.79 14.92 Mg 2÷ 2.50 2.91 3.19 3.40 3.67 Fe 2+/3÷ 1.54 1.63 1.71 1.87 1.98 AP ÷ N.D. 0.07 0.14 0.25 0.28 Si 4+ 1.39 1.94 2.54 2.86 3.86
Ca ~+ 0.63 0.90 1.17 1.47 1.70 Mg 2÷ 0.11 0.18 0.22 0.24 0.27 Fe2+/34 0.31 0.37 0.44 0.49 0.60
AP + 1.09 1.22 1.27 1.29 1.30 Si 4+ 0.32 - 0.67 0.85 1.05
Ca e+ 0.86 0.96 1.04 1.15 1.25 Mg 2÷ 0.12 0.16 0.21 0.25 0.28 Fe 2+/3 + 0.06 0.09 0.09 0.09 0.12 AP + 0.11 0.16 0.16 0.18 0.19 Si 4+ 0.17 0.20 0.27 0.35 0.39
52
TABLE 3 (continued)
A.K. BARMAN ET AL
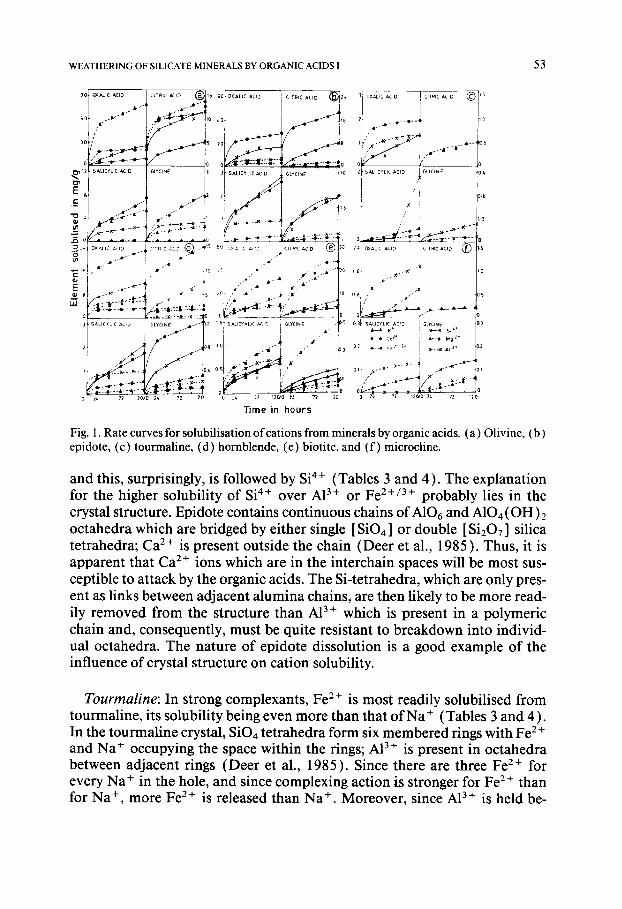
Acid Cation Biotite
Amount of cation released (% of total) in
24 h 48 h 72 h 96 h 120 h
Oxalic Mg 2+ K + Fe2+/3+ A! 3+
Sp +
Citric Mg 2+ K + Fe2+/3+ AI 3+
Sp +
Salicylic Mg 2 + K + Fe2+/3+ A13+ S i 4 +
Glycine Mg 2+ K + Fe2+/3+ A13+ S i 4 +
21.83 27.07 30.56 34.05 37.54 16.66 24.07 30.55 35.16 38.88 12.33 16.87 21.29 24.40 28.55 8.43 9.27 9.90 10.32 10.53 2.86 3.77 6.03 9.35 11.16
32.95 45.46 54.29 62.38 69.00 2.60 2.90 3.49 4.34 4.34 6.49 9.35 9.60 12.20 12.46 - 3.31 3.90 4.58 4.90 1.21 1.99 2.89 3.47 4.67
0.89 1.18 1.56 1.78 2.06 0.39 0.50 0.57 0.65 0.69 0.29 0.34 0.45 0.47 0.57 0.42 0.57 0.69 0.75 0.76 0.15 0.21 - 0.36 0.48
0.67 1.03 1.24 1.45 1.73 0.08 0.10 0.10 0.12 0.16 0.02 0.03 0.03 0.03 0.03 0.08 0.11 0.11 0.12 0.14 0.04 0.05 0.03 0.10 -
Acid Cation Microcline
Amount of cation released (% of total) in
24 h 48 h 72 h 96 h 120 h
Oxalic
Citric
Salicylic
Glycine
K + 0.13 0.19 0.24 0.29 0.31 A13+ 1.16 1.45 1.68 1.85 1.97 Si a+ 0.02 0.02 0.13 0.19 0.28
K +
A13+ S i 4 +
K +
AI 3+ Si 4+
0.15 0.18 0.19 0.20 0.20 N.D. N.D. N.D. N.D. N.D. N.D. N.D. N.D. N.D. N.D.
0.04 0.04 0.05 0.05 0.05 0.10 0.11 0.12 0.12 0.15 N.D. N.D. N.D. N.D. N.D.
K ÷ 0.02 0.02 0.02 0.03 0.04 A13+ 0.06 0.06 0.06 0.06 0.06 Si 4+ 0.02 0.03 0.03 0.04 0.04
N.D.: Not detectable.
WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS I 53
. . . . . . . . . . l . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ~ . . v " 4 " j " ~ ' " " o In
,0 2 . - . . . . . . , 0 1 " ,7 i
/ D"
3o [ ' s ~o t~ ~ " " . . " ' " ' . . - J ~ - ' ~ - , . e ' " 6 6 - * s
0," 0 ~ 2 SAL'C'~L'CAC'0 3 ~ ,o 2Fs~,c"uc,~c,~ / c'L~'cr"E ~o.9
,_c s i i I P
= "~ ',L:x '
2L~ | / @ l ~
/
1 ~- S,~LIZ'fALI C ~ ID i 6LYCINE ~ , / F " S O] SALICYLIC ACID i ~LYCINE Ot~
0 2/. "]2 IZ010 24 72 0 ~7, - r ~ ~ 1 2 ~ 0 )~ 7] Tl°
Time i n h o u r s
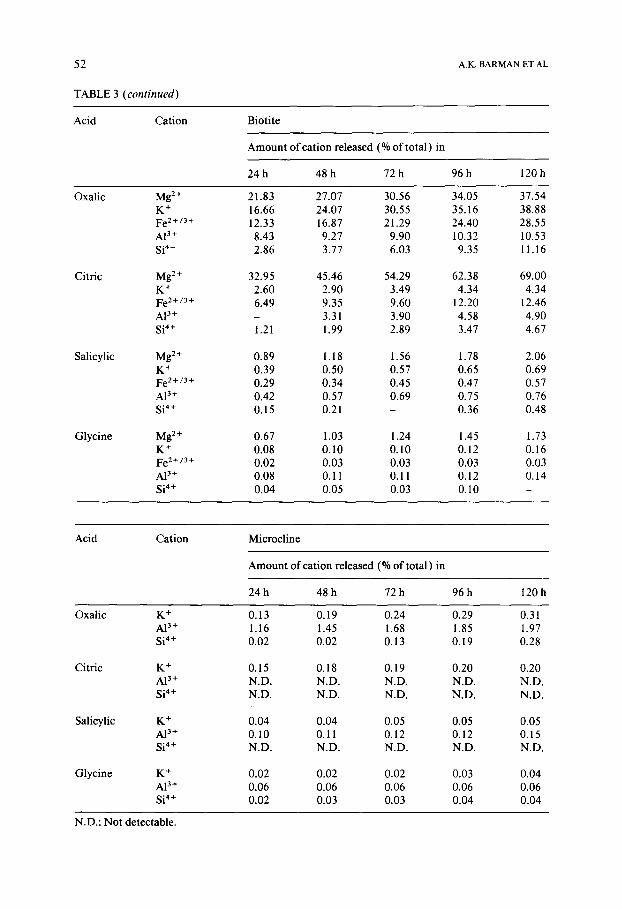
Fig. 1. Rate curves for solubilisation of cations from minerals by organic acids. (a) Olivine, (b) epidote, (c) tourmaline, (d) hornblende, (e) biotite, and (f) microcline.
and this, surprisingly, is followed by Si 4+ (Tables 3 and 4). The explanat ion for the higher solubility of Si 4+ over A13+ or Fe z+/3+ probably lies in the crystal structure. Epidote contains cont inuous chains of A 1 0 6 and AIO4 (OH)2 octahedra which are br idged by either single [ SiO4 ] or double [ Si207 ] silica tetrahedra; Ca 2+ is present outside the chain (Deer et al., 1985). Thus, it is apparent that Ca 2+ ions which are in the interchain spaces will be most sus- ceptible to at tack by the organic acids. The Si-tetrahedra, which are only pres- ent as links between adjacent a lumina chains, are then likely to be more read- ily r emoved f rom the structure than AP ÷ which is present in a polymeric chain and, consequently, mus t be quite resistant to breakdown into individ- ual octahedra. The nature of epidote dissolution is a good example of the influence of crystal structure on cation solubility.
Tourmaline: In strong complexants , Fe z+ is mos t readily solubilised f rom tourmal ine , its solubility being even more than that o f N a ÷ (Tables 3 and 4). In the tourmal ine crystal, SiO4 te t rahedra form six m e m b e r e d rings with Fe 2 ÷ and Na + occupying the space within the rings; A13+ is present in octahedra between adjacent rings (Deer et al., 1985). Since there are three Fe 2÷ for every Na + in the hole, and since complexing ac t ion is stronger for Fe 2 ÷ than for Na ÷, more Fe z+ is released than Na +. Moreover , since A13+ is held be-

5 4
T A B L E 4
Solubilisation of the minerals after reaction with various acids for 120 h
A.K. BARMAN ET AL.
Ac id Mineral Total major % solubilisation cations of major solubilised cations in the in m g / g of mineral mineral
Order of cation solubilised
Oxal ic
Ci t r i c
Salicylic
Glycine
Oliv ine 134.24 24 .00
Epidote 56.02 10.50
Tourmaline 4.65 1.00
Hornblende 51.29 9.56
Biotite 128.60 22 .54
Mic roc l ine 2 .94 0 .55
Ol iv ine 43 .43 7.76
Epidote 29 .53 5.53
Tourmaline 0.50 0.11
Hornblende 27 .60 5.12
Biotite 61 .29 10.74
Microcline 0.19 0 .03
Ol iv ine 15.47 2.77
Epidote 6.63 1.24
Tourmaline 2.24 0 .48
Hornblende 5.49 1.02
Biotite 3.86 0 .68
Microcline 0.18 0 .03
Ol iv ine 3.35 0 .60
Ep ido t e 1.14 0.21
Tourmaline 0.25 0 .05
Hornblende 2.49 0 .46
Biotite 1.09 1.09
Microcline 0.24 0 .05
Fe2+/3+ ~. Mg2+ > Si4+
C a 2+ > Si 4+ > A P + > Fe 2+/3+
FeZ+/3+ )~ A p + > Si4+
Ca2+ > Mg2+ > AI3+ > Si4+ > Fe2+/3+
Mg2+ i> K + > Fe2+/3+ > AI3+ > Si4+
A P + > K + > S i 4+
Fe 2+/3+ > Si 4+ > M g 2+
Ca2+ > Si4+ > Fe2+/3+ > AI3+
Fe2+/3+ >A13+, Si 4+
Ca2+ > Mg2+ > Si4+ > Fe2+/3+ > A p +
Mg2+ > Fe2+/3+ > A13+ > K + > Si4+
K + > A I 3+, Si 4+
M g 2+ > Si 4+ > Fe 2+/3+
Ca2+ > Si4+ > AI3+ > Fe2+/3+
Si4+ > Fe2+/3+ > A13+
CaZ+ > AI3+ > Si4+ > Fe2+/3+ > M g 2 .
M g 2+ > A I 3+ > K + > Fe 2+/3+ > S i 4+
A p + > K + > Si *+
Si 4+ > M g 2+ > Fe 2+/3+
C a 2+ > Si 4+ > AI 3+ > Fe 2+/3+
Si 4+ > A I 3+ > Fe 2+/3+
Ca 2+ > Si 4+ > M g 2+ > AI 3+ > Fe 2+/3+
Mg2+ > A13+ i> K + > Si4+ >/Fee+/3+
A P + > K +, Si 4+
tween the rings, it is more susceptible to complexation than Si 4+ which is part of a chain. In weak complexants like glycine, the pH of the solution becomes the determining factor. Thus in glycine solutions, which have pH close to neu- tral, Fe 2÷ and AI 3+ are much less soluble than Si 4+ (Table 4).
Hornblende: The most notable feature of hornblende dissolution is the dif- ference in the amounts of Ca 2+ and Mg 2+ dissolved (Table 3); the Ca 2+ is always much more rapidly solubilised than Mg 2÷ irrespective of the nature of the acid. As before, a consideration of the crystal structure provides the clue to this difference in the behaviour of the two alkaline earth metal ions. The Mg 2÷ ions are present in talc-like strips which are sandwiched between two bands of silica tetrahedra in the crystallographic sites M1, M2 and M3. The

WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS I 5 5
Ca 2 + ion occupies site M 4 which serves to link the above units (Deer et al., 1985 ). Therefore, the Ca 2+ ions will be more exposed to attack by organic acids than the Mg 2÷ ions which are better protected by being sandwiched between the silica tetrahedra. The A13+ is present mostly as a substituent for Si 4+ in the tetrahedral chain and also to some extent as a substituent for Mg 2+ in the octahedral positions. Therefore, depending on the complexing action of the organic acids, A13÷ is solubilised to a greater or lesser extent than Si 4÷. Unlike A13÷, Fe 2+/3+ is present only in the octahedral positions; conse- quently, its solubility is in general less than that of the former ion.
Biotite: From biotite, Mg 2+ is observed to be the most soluble in all organic acids (Tables 3 and 4). Surprisingly, K + is much less soluble than Mg 2+ and in some instances even less than A13 +.
In the biotite structure, both Mg 2+ and Fe z+ are present in an octahedral sheet which is sandwiched between two sheets of silica tetrahedra. The K ÷ ions are firmly wedged between adjacent silica planes and A13+ is present both in the tetrahedral as well as in the octahedral sheet (Deer et al., 1985 ). Since the Mg 2+ and Fe 2÷ ions are not readily accessible to the solution, it may be inferred that the attack takes place from the edges. The complexant may be visualised as slowly boring its way into the octahedral sheet, from the edge towards the interior. The K ÷ interlayer would also be similarly tunneled into, most likely by the H30 + ions. Moreover, once a K ÷ ion is removed from a hexagonal cavity, three octahedral ions directly below it, will be free to diffuse out. When this is possible, Mg 2÷ may diffuse more rapidly than Fe 2÷ from the octahedral positions, because of its greater solubility. The A13 ÷ ion can be solubilised both during the tunneling of the octahedral layer by the complex- ants as well as from the tetrahedral sheets after removal of the K + ion. Thus, although the solubility of A13÷ is lower than that of Mg 2+, its position with respect to Fe 2+ is variable; when complexation is strong, Fe 2÷ is more soluble than A13÷ (as in oxalic acid) whereas in a weaker complexant such as glycine Fe 2÷ may become less soluble. The solubilisation of Si 4+ is probably a sec- ondary effect; breakdown of the octahedral sheet and the depletion of the K + interlayer would leave an unstable residual silica framework from which the individual tetrahedra may break away and be removed as soluble ions. This argument is further supported by the fact that Si 4+ is the least soluble of the ionic species present (Table 4).
Microcline: Dissolution of all cations is very low; microcline has a very compact structure (Deer et al., 1985 ) which greatly limits the accessibility of the complexants to the ions in the interior. Since all ions are equally inacces- sible, their extent of release depends on the solubilising effect of the medium (Tables 3 and 4 ). Thus in a strongly complexing medium (oxalic acid), A13 + is most soluble whereas with a weak complexant at neutral pH, Si 4+ is the

56 A.K. BARMAN ETAL.
most soluble ion. The low solubilisation of K ÷ may be due to the predomi- nating influence of structural factors.
Relative stabilities of the minerals
Table 4 shows the total amount of the major constituent cations dissolved from the minerals as well as their percentage relative to the total amounts of the same cations therein. It may be observed that the relative stabilities of the minerals varies with the nature of the acid. In fact, solubilisation appears to be the resultant of three simultaneous factors, viz., the nature of the organic acid, the chemical composition of the mineral and the structure of the mineral.
The order of stabilities of the minerals in different organic acids can also be seen from Table 4. From the position of olivine in the series, it may be inferred that olivine is both chemically as well as structurally unstable. Ap- parently, in the presence ofcomplexing agents and in acid solutions, the Mg 2+ and Fe 2 + ions in the crystal are susceptible to attack; in neutral solutions, the Si 4+ ions can also solubilise readily. These processes are facilitated by the fact that (a) silica is present as discrete anions and not as a polymer, (b) the balancing cations are not interlinked, and (c) the structure is held together predominantly by ionic forces.
The fact that polymeric units increase the stability of minerals is best illus- trated by the difference between epidote and olivine. Epidote also contains loosely held balancing cations (Ca 2 ÷ ) as well as monomeric and dimeric sil- ica units; the latter are, however, bound at two comers by chains of AIO6 octahedra (Deer et al., 1985). Therefore, its resistance to breakdown is al- ways much more than that of olivine. Thus, oxalic acid can dissolve 14.10% o f t h e Si 4+ in olivine in 120 h, whereas the figure for epidote is only 8.09% (Table 3 ).
Hornblende appears to be relatively more resistant than epidote in strongly complexing acids but is less stable than epidote in weakly complexing solu- tions. Since the Si 4+ in hornblende is present as a double chain, it is more resistant than the Si 4+ in olivine or epidote (Table 3 ). The other ions in horn- blende which are likely to be susceptible to attack by organic acids, such as Mg 2 + and Fe z +, are fairly well guarded in a talc-like structure between silicate polyhedra. Only the position of the Ca 2+ ion appears to be the weakest spot in the crystal, as a result of which this ion is more readily removed from horn- blende than from epidote by all organic acids. Probably it is because of the ease of removal of Ca 2+ ions from hornblende that the structure is weakened, relative to epidote, in glycine solutions.
The position of biotite in the stability series is very interesting. Whereas in the weaker complexants, its stability is only next to tourmaline and microc- line, in the stronger complexants, its stability decreases to such an extent that it becomes only next to olivine. The low stability of biotite in oxalic and citric

WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS 1 57
acids is indeed remarkable. It may be conjectured that complexation and acid attack together with the unique crystal structure are responsible for this phe- nomenon. To elucidate further, two major constituents of biotite, viz., Mg 2÷ and Fe 2 + are susceptible to dissolution by complexation whereas another im- portant constituent, viz., K +, may be dissolved by H3 O+ ions. Moreover, the structure of the mineral is such that the cations in the octahedral sheet can be fairly readily removed by the tunneling processes explained earlier. The com- bined effect of these factors, therefore, produces rapid dissolution. However, when any one of these factors is altered, dissolution decreases significantly, e.g., in salicylic acid solution which is much less acidic (0.01M) than oxalic or citric acids (0.5M) the release of K ÷ is significantly lower thereby result- ing in decreased structural breakdown. This effect is further magnified in gly- cine solution which is not only weakly acidic but also weakly complexing.
The very high stability, of both tourmaline and microcline, is in significant contrast to the behaviour of the other minerals. Both these minerals have very compact cross-linked structures which are formed by predominantly covalent linkages. Moreover although alkali and alkaline earth metal ions are present, these are not the only ions serving to hold the units together, as does K ÷ in biotite. Thus, removal of small amounts of these most readily soluble cations does not cause any serious weakening of the structure. An additional factor is that, since accessibility of such ions to water is sterically hindered by the alu- minosilicate framework, their dissolution is probably limited to those at the surface.
Congruency of dissolution
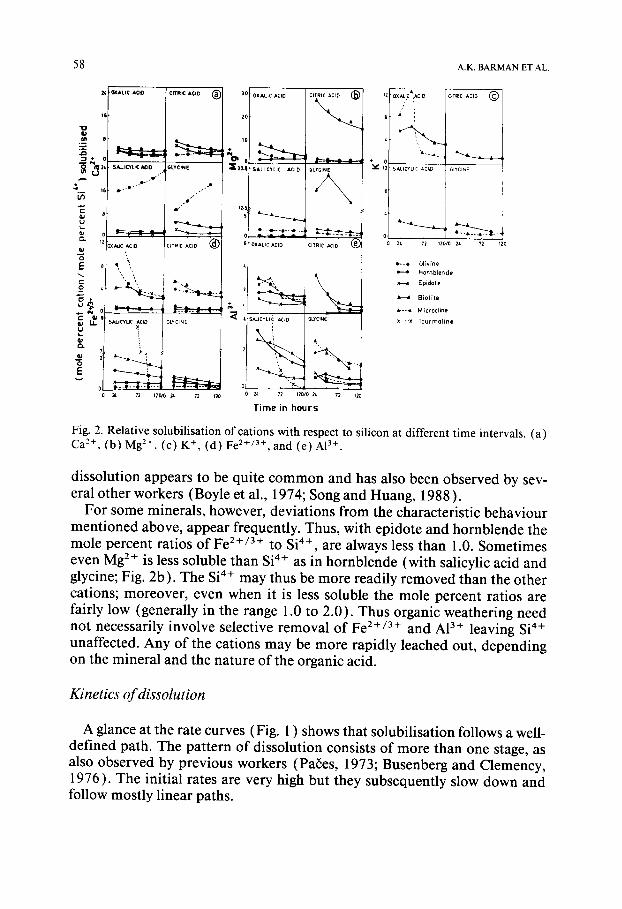
Figure 2 shows the changes with time, in the ratios of mole percent of var- ious elements solubilised relative to that of Si 4+ solubilised. All the minerals studied here, are observed to show incongruent dissolution behaviour. This is in line with expectations since congruency can only be a general character- istic of simple solubilisation mechanisms; incongruency indicates sequential release of the components by complex processes and is therefore consistent with the reaction mechanisms envisaged earlier.
It may be observed from Fig. 2 that in most cases the ratio, which is high at the initial stages of the reaction, decreases with time and then attains a fairly constant value. Moreover the ratios are usually greater than 1.0. Both these observations indicate that as the mineral dissolves, the cations at the surfaces are more rapidly removed than the Si 4+ ions. Thus the surface becomes en- riched in silica. However, such a cation-depleted siliceous layer is not a stable phase (Schott and Berner, 1983); it must consequently disintegrate slowly and dissolve. The period of constancy in the ratio of cation to Si 4+ released, therefore, represents the stage where the ratio of cation release more or less equals the rate at which the silica residue disintegrates. This pattern of silicate
58 A.K. BARMAN ET AL.
2t. ~(ALIC ACiD
~6
Qa
8
el,RE ACID ( ~
SALICYL~ ACID ~LYCINE
- .e"" . 'O / . ' "
a e '"
~(,,,uC ,,,C,D CJTR,C AC,D (~ )
u ¢ .
0 ~ 72 120'0 2z, 72 120
OXALIC ACID CITRIC ACID ( ~
SALICYLIC ACID G LYGINE
~- - 4 - o - D o - - e - . . , n
OXAUCAC,D CTR,C ~,o @
s a . ~ l ~ C,L C E
2t, 72 120/0 24 72 120
Time in hours
• 0XALICA I~C~D CIT~C ACID ( ~
i
SALICYLIC ACkD GLY CIN E
24, 7l 120/0 3.~ 72 120
e - - o Olivine H Hornblende
Epidot e
Biotite
A- - - ,1 Microcllne
~ - - - i ( Tour mallne
Fig. 2. Relative solubilisation of cations with respect to silicon at different time intervals. (a) Ca 2+, (b) Mg 2+, (c) K +, (d) Fe 2+/3+, and (e) A13+.
dissolution appears to be quite common and has also been observed by sev- eral other workers (Boyle et al., 1974; Song and Huang, 1988 ).
For some minerals, however, deviations from the characteristic behaviour mentioned above, appear frequently. Thus, with epidote and hornblende the mole percent ratios of Fe 2+/3+ to Si 4+, are always less than 1.0. Sometimes even Mg 2÷ is less soluble than S i 4+ a s in hornblende (with salicylic acid and glycine; Fig. 2b). The Si 4+ may thus be more readily removed than the other cations; moreover, even when it is less soluble the mole percent ratios are fairly low (generally in the range 1.0 to 2.0). Thus organic weathering need not necessarily involve selective removal of Fe 2+/3+ and AI 3+ leaving Si 4+ unaffected. Any of the cations may be more rapidly leached out, depending on the mineral and the nature of the organic acid.
Kinetics of dissolution
A glance at the rate curves (Fig. 1 ) shows that solubilisation follows a well- defined path. The pattern of dissolution consists of more than one stage, as also observed by previous workers (Pa~es, 1973; Busenberg and Clemency, 1976 ). The initial rates are very high but they subsequently slow down and follow mostly linear paths.

WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS I 59
In comparison to reactions in solution the interpretation of kinetic data, for reactions involving solid phases, is extremely complex relative to that for reactions in solution. According to Lerman (1979), dissolution may be influ- enced by (a) the formation of a protective coating on the surface, (b) disin- tegration of the particle into smaller fragments, and (c) pitting and etching of the surface. Lattice imperfectious and lattice strain also play an important role by facilitating diffusion of structural ions (Laidler, 1963 ).
The nature of the curves (Fig. 1 ) at the initial stages of the reaction has been observed to generally follow the parabolic law, i.e., the rate of dissolu- tion is proportional to (time)½. According to Dibble and Tiller ( 1981 ), who developed a new model for mineral-water interactions, a parabolic time de- pendence indicates a rate-determining step involving diffusion but not in the usual sense of mass transfer of components through solid or solution phases as hypothesised by earlier workers (Helgeson, 1971; Pa~es, 1973 ). Thus dif- fusion of a reaction product to or from the interface could become rate limit- ing if adsorption retards formation of fresh surfaces. This also results if sur- face reconstruction limited by surface diffusion dominates the reaction (Dibble and Tiller, 1981 ). Holdren and Berner (1979), however, offered a different explanation. According to them, the dissolution of ultrafine parti- cles which adhere tenaciously to the surface of the larger grains (and which can only be removed by treatment with HF) , results in such non-linear dis- solution patterns.
It thus appears that the initial high rates of release may be attributed to two factors, namely (a) the dissolution of ultrafine particles, and (b) a diffusion controlled process involving dissolution of cations at the surface and close to the surface of the grains. The former may be an important factor in the more resistant minerals like microcline and tourmaline. However, with minerals like olivine or epidote which are themselves rapidly solubilised and, perhaps, fragmented as well, the dissolution of smaller particles cannot, logically, be solely responsible for the initial high rates. The cations at the surface will also be rapidly solubilised, together with some sub-surface cations which are read- ily accessible to the solution. The rate limiting step of this stage has been en- visaged to involve diffusion of the reaction product as explained earlier. Therefore, in such cases diffusion of the cation-organic complex formed at the surface of the particle is the probable rate limiting step. This may be rep- resented as:
M + fast M + - L - slow M + - L - Mineral M ÷ + 3L- . • Mineral M + - L - . " Mineral + M + - L -
M ÷ M+_L - M÷_L -
In the next stage, mineral dissolution curves show mostly linear patterns (Fig. 1 ). Linear kinetics implies that fresh sites are created at a rate equalling the rate of removal of cations from the mineral surface. In the case of olivine,

60 A.K. BARMAN ET AL.
epidote, hornblende (and to some extent, biotite) it appears, from the fairly rapid rates, that the progressive dissolution of structural cations causes a frag- mentation of the mineral to smaller units, thereby exposing fresh sites for reaction. With the more resistant minerals like tourmaline and microcline, the creation of fresh sites is probably along the edges, cracks and other regions of high energy such as along defect planes. In fact, the selective dissolution of feldspar grains, along dislocations and at points where fluid inclusions inter- sect the surface, has been observed (Holdren and Berner, 1979). However, all minerals including those of the olivine group must be most susceptible to attack along defect planes and other regions of high energy. The difference is that in the minerals which are more readily dissolved, these planes may be severely attacked thereby sometimes causing fragmentation. Such an attack can also occur along planes containing specific cations which are most readily dissolved. Thus, acid attack on biotite causes rapid dissolution of K + as a result of which separation of the flakes occurs along this plane. Similarly in epidote, cleavage probably occurs most readily along planes containing Ca 2+ ions, in addition to defect regions. Due to the rapid removal of C a 2÷ by or- ganic acids, cleavage along these planes may prevail over that in the regions of defect. Such a situation may be reversed in microcline or tourmaline since no cation shows any marked tendency to be solubilised and, consequently, defect planes may be the regions most affected by weathering.
In short, the kinetic data indicate that at the initial stages of reaction of minerals with organic acids, only surface detachment parameters control the rate of the reaction. Once the surface is depleted of most of its cations, then the availability of fresh surfaces by fragmentation, formation of defect planes, etching etc., may become the rate limiting step of dissolution.
CONCLUSIONS
The order of solubilisation of the major cations from silicate minerals weathered by organic acids suggests that the position of the cations in the crystals is a dominant factor. Complexing abilities of the acids for different cations also determines to some extent the order of cation release. Ions in interchain spaces such as the Ca 2 ÷ ions in epidote and hornblende are usually the most soluble species. These are followed by ions present as short chain units or as links between long chains and sheets; for example, the Si207 dou- ble tetrahedra sandwiched between alumina octahedral chains in epidote and the Mg 2÷ strips between double silica chains in hornblende. Finally, the least soluble are ions in polymeric units (A13+ in octahedral sheets of biotite) and also those in sterically hindered positions (K ÷ in feldspar and Na ÷ in tourmaline ).
Stability sequence of the minerals in various organic acids, is not unique; it varies with the nature of the acid. The observed sequences have been ex-

WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS I 61
plained on the basis of cation-ligand stabilities as well as the arrangement of the silica tetrahedra, the chain length of the octahedral coordinated ions such as Mg 2÷, Fe 2+/3÷ and AI 3+, and the accessibility of the solution phase to the most labile ions like Na +, K +, Ca 2÷ etc., in the crystal. In general, increased polymerisation of the silica tetrahedra increases the stability of the mineral. Stability in strongly complexing media also increases if Mg 2+ and the transi- tion metal ions are present in octahedral or tetrahedral chains rather than in interchain spaces. Thus, stability increases in the series olivine-epidote- hornblende when strong complexants are present; in such media biotite be- comes very unstable and may even equal olivine in its unstability.
All the minerals studied, showed incongruent dissolution behaviour. It has been envisaged that initially a cation depleted siliceous surface layer is formed which starts to disintegrate after attaining a certain thickness. In some min- erals, the mole percent dissolution of Si 4+ may be higher than Fe 2+/3+ or Mg 2+ suggesting that organic acid weathering need not necessarily lead to a silica enriched residue.
Rates of cation dissolution from the minerals exhibit a well-defined pat- tern. The initial rates of reaction follow the parabolic law and subsequently linear rates are observed. Parabolic rates have been suggested to result from a diffusion-controlled, rate-limiting step of reaction involving the transfer of cation-ligand complexes from the surface of the solid to the solution. Linear dissolution in the later stages of reaction has been interpreted as the creation of fresh sites at rates equalling the rate of removal of cations from the surface. With olivine, epidote, hornblende and biotite fresh sites are probably created predominantly by fragmentation along the planes containing the most readily soluble cations and along cleavage planes. With microcline and tourmaline, however, fragmentation is unlikely and new sites are most probably formed along high-energy regions such as along crystal edges, defects and dislocations resulting in pitting and etching of the surface and breaking of edges.
REFERENCES
Berner, R.A. and Holdren, G.R., 1979. Mechanism of feldspar weathering. II. Observations of feldspars from soils. Geochim. Cosmochim. Acta, 43:1173-1186.
Black, C.A., 1965. Methods of Soil Analysis. American Society of Agronomy, Madison, WI, pp. 999-1010.
Boyle, J.R., Voigt, G.K. and Sawhney, B.L., 1974. Chemical weathering of biotite by organic acids. Soil Sci., 117: 42-45.
Busenberg, E. and Clemency, C.V., 1976. The dissolution kinetics of feldspars at 25 °C and 1 atm CO2 partial pressure. Geochim. Cosmochim. Acta, 40: 41-49.
Correns, C.W., 1961. The experimental chemical weathering of silicates. Clay Miner. Bull., 4: 249-265.
Deer, W.A., Howie, R.A. and Zussman, J., 1985. An Introduction to the Rock-forming Min- erals. ELBS/Longman, London, pp. 2, 63, 90, 148-150, 193-195.

62 A.K. BARMAN ET AL.
Dibble, W.E. and Tiller, W.A., 1981. Non-equilibrium water/rock interactions. I. Model for interface-controlled reactions. Geochim. Cosmochim. Acta, 45: 79-92.
Helgeson, H.C., 1971. Kinetics of mass transfer among silicates and aqueous solutions. Geo- chim. Cosmochim. Acta, 35: 421-469.
Henderson, M.E.K. and Duff, R.B., 1963. The release of metallic and silicate ions from min- erals, rocks and soils by fungal activity. J. Soil Sci., 14: 236-246.
Holdren, G.R. and Berner, R.A., 1979. Mechanism of feldspar weathering. I. Experimental studies. Geochim. Cosmochim. Acta, 43:116 i - 1171.
Huang, W.H. and Keller, W.D., 1970. Dissolution of rock-forming silicate minerals in organic acids: simulated first-stage weathering of fresh mineral surfaces. Am. Miner., 55: 2076-2094.
Jeffery, P.G., 1970. Chemical Methods of Rock Analysis. Pergamon, Oxford, pp. 447-451. Loughnan, F.C., 1969. Chemical Weathering of the Silicate Minerals. Elsevier, New York, pp.
27-66. Laidler, K.J., 1963. Reaction Kinetics, I. Pergamon, Oxford, 316 pp. Lerman, A., 1979. Geochemical processes: water and sediment environments. Wiley-Intersci-
ence, New York, pp. 227-229. Luce, R.W., Barlett, R.W. and Parks, G.A., 1972. Dissolution kinetics of magnesium silicates.
Geochim. Cosmochim. Acta, 36: 35-50. Martell, A.E. and Smith, R.M., 1977. Critical Stability Constants, 3. Plenum, New York, pp.
93, 161. Maxwell, J.A., 1968. Rock and Mineral Analysis. Interscience, New York, pp. 101,336-357,
387-389, 540-541. Motschi, W., 1983. Cu (II) bound to hydrous surfaces, EPR measurements characterize surface
coordination. Naturwissenschaften, 70:519-520. Pates, T., 1973. Steady-state kinetics and equilibrium between ground water and granitic rock.
Geochim. Cosmochim. Acta, 37: 2641-2663. Petrovi6, R., Berner, R.A. and Goldhaber, M.B., 1976. Rate control in dissolution of alkali
feldspars. I. Study of residual feldspar grains by X-ray photoelectron spectroscopy. Geochim. Cosmochim. Acta, 40: 537-548.
Ponomareva, V.V., 1969. Theory of Podzolisation. Academy of Sciences of USSR, Moscow, pp. 92-94 (English translation).
Schalscha, E.B., Appelt, H. and Schlatz, A., 1967. Chelation as a weathering mechanism. I. Effect of complexing agents on the solubilization of iron from granodiorite. Geochim. Cos- mochim. Acta, 31: 587-596.
Schatz, A., Schatz, V. and Martin, J.J., 1957. Chelation as a biochemical weathering factor. Am. Geol. Soc. Bull., 68:1792-1793.
Schnitzer, M. and Kodama, H., 1976. The dissolution of micas by fulvic acid. Geoderma, 15: 381-391.
Schott, J. and Berner, R.A., 1983. X-ray photoelectron studies of the mechanism of iron silicate dissolution during weathering. Geochim. Cosmochim. Acta, 47: 2233-2240.
Schott, J., Berner, R.A. and Sj/Sberg, E.L., 1981. Mechanism ofpyroxene and amphibole weath- ering. I. Experimental studies of iron-free minerals. Geochim. Cosmochim. Acta, 45:2123- 2135.
Song, S.K. and Huang, P.M., 1988. Dynamics of potassium release from potassium-bearing minerals as influenced by oxalic and citric acids. Soil Sci. Soc. Am. J., 52: 383-390.
Stumm, W., Furrer, G., Wieland, E. and Zinder, B., 1985. The effects of complex-forming li- gands on the dissolution of oxides and alumino-silicates. In: J.I. Drever (Editor), The Chemistry of Weathering. Reidel, Dordrecht, pp. 55-74.
Tan, K.H., 1980. The release of silicon, aluminium and potassium during decomposition of soil minerals by humic acid. Soil Sci., 129:5-11.

WEATHERING OF SILICATE MINERALS BY ORGANIC ACIDS 1 63
Vogel, A.I., 1961. A Textbook of Quantitative Inorganic Analysis. ELBS & Longmans Green, London, pp. 810-811.
Wollast, R., 1967. Kinetics of the alteration of K-feldspar in buffered solutions at low tempera- ture. Geochim. Cosmochim. Acta, 31: 635-648.
Zutic, V. and Stumm, Z., ! 984. Effect of organic acids and fluoride on the dissolution kinetics of hydrous alumina. A model study using the rotating disc electrode. Geochim. Cosmochim. Acta, 48: 1493-1503.
Related Documents











