699 ANNALS OF GEOPHYSICS, VOL. 48, N. 4/5, August/October 2005 Key words diffusion – silicate melts – volatiles – water – carbon dioxide – sulfur – fluorine – chlorine – melt inclusion – igneous processes 1. Introduction Volatiles play an enormously important role in igneous processes. They are not only respon- sible for dramatic volcanic eruptions, but also af- fect the transport (e.g., diffusion and viscosity) and equilibrium (e.g., phase equilibria) proper- ties of magmas. Understanding the diffusion of volatiles in silicate magmas provides the frame- work necessary for the understanding of process- es such as bubble formation and growth that can ultimately lead to violent volcanic eruptions. The volatiles commonly found at the highest concentrations in magmatic systems are H 2 O, CO 2 , S-species, F, and Cl. The dominant volatile in magmas is H 2 O; evidence of its abundance is manifested in measured volcanic emissions (Symonds et al., 1994) and the presence of hy- drous minerals (e.g., amphibole, biotite, mus- Volatile diffusion in silicate melts and its effects on melt inclusions Don R. Baker ( 1 )( 2 ), Carmela Freda ( 2 ), Richard A. Brooker ( 3 ) and Piergiorgio Scarlato ( 2 ) ( 1 ) Earth and Planetary Sciences, McGill University, Montreal, Quebec, Canada ( 2 ) Istituto Nazionale di Geofisica e Vulcanologia, Roma, Italy ( 3 ) Department of Earth Sciences, University of Bristol, U.K. Abstract A compendium of diffusion measurements and their Arrhenius equations for water, carbon dioxide, sulfur, flu- orine, and chlorine in silicate melts similar in composition to natural igneous rocks is presented. Water diffusion in silicic melts is well studied and understood, however little data exists for melts of intermediate to basic com- positions. The data demonstrate that both the water concentration and the anhydrous melt composition affect the diffusion coefficient of water. Carbon dioxide diffusion appears only weakly dependent, at most, on the volatile- free melt composition and no effect of carbon dioxide concentration has been observed, although few experi- ments have been performed. Based upon one study, the addition of water to rhyolitic melts increases carbon dioxide diffusion by orders of magnitude to values similar to that of 6 wt% water. Sulfur diffusion in intermedi- ate to silicic melts depends upon the anhydrous melt composition and the water concentration. In water-bearing silicic melts sulfur diffuses 2 to 3 orders of magnitude slower than water. Chlorine diffusion is affected by both water concentration and anhydrous melt composition; its values are typically between those of water and sulfur. Information on fluorine diffusion is rare, but the volatile-free melt composition exerts a strong control on its dif- fusion. At the present time the diffusion of water, carbon dioxide, sulfur and chlorine can be estimated in silicic melts at magmatic temperatures. The diffusion of water and carbon dioxide in basic to intermediate melts is on- ly known at a limited set of temperatures and compositions. The diffusion data for rhyolitic melts at 800°C to- gether with a standard model for the enrichment of incompatible elements in front of growing crystals demon- strate that rapid crystal growth, greater than 10 −10 ms −1 , can significantly increase the volatile concentrations at the crystal-melt interface and that any of that melt trapped by the formation of melt inclusions may not be rep- resentative of the bulk melt. However, basaltic melt inclusions trapped at 1300°C are more likely to contain bulk melt concentrations of water and carbon dioxide. Mailing address: Dr. Don R. Baker, Earth and Planetary Sciences, McGill University, 3450 Rue University, Montreal, QC H3A 2A7 Quebec, Canada; e-mail: [email protected]

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

699
ANNALS OF GEOPHYSICS, VOL. 48, N. 4/5, August/October 2005
Key words diffusion – silicate melts – volatiles –water – carbon dioxide – sulfur – fluorine – chlorine –melt inclusion – igneous processes
1. Introduction
Volatiles play an enormously important rolein igneous processes. They are not only respon-
sible for dramatic volcanic eruptions, but also af-fect the transport (e.g., diffusion and viscosity)and equilibrium (e.g., phase equilibria) proper-ties of magmas. Understanding the diffusion ofvolatiles in silicate magmas provides the frame-work necessary for the understanding of process-es such as bubble formation and growth that canultimately lead to violent volcanic eruptions.
The volatiles commonly found at the highestconcentrations in magmatic systems are H2O,CO2, S-species, F, and Cl. The dominant volatilein magmas is H2O; evidence of its abundance ismanifested in measured volcanic emissions(Symonds et al., 1994) and the presence of hy-drous minerals (e.g., amphibole, biotite, mus-
Volatile diffusion in silicate melts and its effects on melt inclusions
Don R. Baker (1)(2), Carmela Freda (2), Richard A. Brooker (3) and Piergiorgio Scarlato (2)(1) Earth and Planetary Sciences, McGill University, Montreal, Quebec, Canada
(2) Istituto Nazionale di Geofisica e Vulcanologia, Roma, Italy
(3) Department of Earth Sciences, University of Bristol, U.K.
AbstractA compendium of diffusion measurements and their Arrhenius equations for water, carbon dioxide, sulfur, flu-orine, and chlorine in silicate melts similar in composition to natural igneous rocks is presented. Water diffusionin silicic melts is well studied and understood, however little data exists for melts of intermediate to basic com-positions. The data demonstrate that both the water concentration and the anhydrous melt composition affect thediffusion coefficient of water. Carbon dioxide diffusion appears only weakly dependent, at most, on the volatile-free melt composition and no effect of carbon dioxide concentration has been observed, although few experi-ments have been performed. Based upon one study, the addition of water to rhyolitic melts increases carbondioxide diffusion by orders of magnitude to values similar to that of 6 wt% water. Sulfur diffusion in intermedi-ate to silicic melts depends upon the anhydrous melt composition and the water concentration. In water-bearingsilicic melts sulfur diffuses 2 to 3 orders of magnitude slower than water. Chlorine diffusion is affected by bothwater concentration and anhydrous melt composition; its values are typically between those of water and sulfur.Information on fluorine diffusion is rare, but the volatile-free melt composition exerts a strong control on its dif-fusion. At the present time the diffusion of water, carbon dioxide, sulfur and chlorine can be estimated in silicicmelts at magmatic temperatures. The diffusion of water and carbon dioxide in basic to intermediate melts is on-ly known at a limited set of temperatures and compositions. The diffusion data for rhyolitic melts at 800°C to-gether with a standard model for the enrichment of incompatible elements in front of growing crystals demon-strate that rapid crystal growth, greater than 10−10 m s−1, can significantly increase the volatile concentrations atthe crystal-melt interface and that any of that melt trapped by the formation of melt inclusions may not be rep-resentative of the bulk melt. However, basaltic melt inclusions trapped at 1300°C are more likely to contain bulkmelt concentrations of water and carbon dioxide.
Mailing address: Dr. Don R. Baker, Earth and PlanetarySciences, McGill University, 3450 Rue University, Montreal,QC H3A 2A7 Quebec, Canada; e-mail: [email protected]

700
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
covite, apatite) in many igneous rocks. The sec-ond most abundant volatile in magmas is CO2,whose presence can be found in volcanic gases(Symonds et al., 1994) and as fluid inclusions inminerals (Roedder, 1965, 1979). The existenceof rare carbonate magmas also testifies to theimportance of carbon in igneous petrogenesis,but these unique magmas are not discussed fur-ther in this review. The presence of S in mag-matic gases is often obvious; quantitative meas-urements demonstrate that volcanoes can ventthousands of tonnes of S per day (Symonds etal., 1994; Wallace, 2001). Additional evidenceof sulfur’s role in magmatic processes is thepresence of common magmatic sulfides and raremagmatic sulfates (Luhr et al., 1984). The halo-gens, F and Cl, have been measured in volcanicgases and plumes (Symonds et al., 1994), andthey are important constituents of hydrous min-erals and occasionally are found as halogenminerals in igneous rocks.
We begin with a selected review of diffu-sion measurements of H2O, CO2, S, F, and Clthat we believe are most germane to the studyof volatile transport in magmatic silicate melts.We then apply simple models to explore the ef-fects of volatile diffusion on the compositionsof melt inclusions trapped in minerals.
2. Volatile diffusion measurements
The first study of volatile diffusion in mag-matic melts of which we are aware is Shaw’s(1974) measurements of water diffusion in arhyolitic melt. Since that time more studies ofwater, carbon dioxide, sulfur and halogens havebeen completed; for an excellent summary ofexperimental techniques and results obtainedbefore the mid-1990’s the interested reader isreferred to the review by Watson (1994).
All studies to date demonstrate Arrhenianbehaviour of volatile diffusivity; thus the result-ing diffusion coefficients can be succinctly ex-pressed by equations of the general form
where D is the diffusion coefficient, in m2 s−1,Do is the pre-exponential factor, also in m2 s−1,
expD DRT
Eo
a=-b l
Ea is the activation energy, in Jmol−1, R is thegas constant, 8.3143 JK−1 mol−1, and T is thetemperature, in K.
Broadly speaking, volatile diffusion ismeasured in two diffusive regimes, which aredistinguished by the absence or presence of achemical potential gradient. The first regime isthat of tracer diffusion where the volatile of in-terest is only present in trace amounts and itsmovement through the melt can be modelled asa random walk in the absence of a chemical po-tential gradient. Experiments that study tracerdiffusion typically utilize radioactive isotopesof the element of interest applied as a thin lay-er to one end of the sample and then allowed todiffuse through a vanishingly small chemicalpotential gradient. The second regime is that ofchemical diffusion, which occurs in the pres-ence of a significant chemical potential gradi-ent. One experiment of this type is the diffusioncouple where a cylinder of glass with weightpercent abundances of a selected element or el-ements is juxtaposed against a glass cylinderwith low concentrations of the selected elementor elements, and the elements diffuse from thehigh to the low concentration melts during theexperiment. Another experimental techniquefor chemical diffusion allows a mineral with theelement of interest to partially dissolve into themelt releasing the element and creating a diffu-sion profile away from the mineral; however,this technique often results in multicomponentdiffusion, making data analysis difficult.
A third technique is to expose samples to afluid phase with a low concentration of thevolatile of interest resulting in devolatilization, orexposing the sample to a fluid phase with a highconcentration of the volatile resulting in diffusionof the volatile into the sample. Researchers agreethat tracer diffusion is a highly improbablyprocess in magmatic systems, whereas chemicaldiffusion occurs whenever two melts, or a meltand a crystal, with differing chemical potentialsof a chemical component come into contact.
2.1. Water diffusion
Water is present in silicate melts in both themolecular form and as hydroxyl (e.g., Stolper,

701
Volatile diffusion in silicate melts and its effects on melt inclusions
1982). Understanding this speciation is impor-tant for thermodynamic modelling and for themicroscopic modelling of diffusion in the melt.However, for the purposes of this work we areinterested in only the bulk water diffusion coef-ficients and refer the interested reader to the re-view by McMillan (1994) of water speciation insilicate melts.
For all of the volatiles considered in this re-view the diffusion data is provided for thetransport of the bulk volatile, not for a particu-lar species. A comparison of the water diffusionmeasurements discussed below are summarizedin fig. 1.
Shaw (1974) found that water diffusion in arhyolitic melt is a function of water concentra-tion at 750 to 850°C and 200 MPa. Using atechnique based upon weighing glass cylindersbefore and after hydration, and after subse-quent dehydration, he determined the water dif-fusion coefficient. Shaw found that the diffu-sion coefficient for water was a function of thewater concentration and estimated the activa-tion energy for water diffusion at weight per-cent concentrations to be approximately 67000Jmol−1. Watson (1994) used Shaw’s data to es-timate an Arrhenius equation water diffusion at
concentrations near 6 wt%
Watson (1994) also extracted an Arrheniusequation from Shaw (1974) for water diffusionat low, nominally anhydrous, concentrations
Shaw’s (1974) diffusion measurements at 10 to200 MPa, 850°C, demonstrated no measurablepressure effect on water diffusion.
Karsten et al. (1982) and Lapham et al.(1984) applied ion microprobe techniques tothe study of water diffusion in rhyolitic meltsand found similar values and clearly demon-strated the dependence of water diffusion uponits concentration. Karsten et al. (1982) meas-ured water diffusion at 70 MPa and 650 to950°C in rhyolitic melts. They found that theArrhenius equations describing water diffusionchanged with water content; for rhyolitic meltswith 1, 2, and 3 wt% water the activation ener-gies were 82000, 80000, and 78000 Jmol−1, re-
. expDRT
1 100
674
water10#=
-- 00b l
5. expD
RT1 8 10
6 0water
8#=-- 00b l
Fig. 1. Diffusion of water in magmatic silicate melts. Unless otherwise noted the measurements are made inmelts of either rhyolitic or haplogranitic composition. Where calculations are pressure dependent a pressure of200 MPa has been used.

702
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
spectively, and the pre-exponential factors were1.44×10−8, 2.65×10−8, and 4.78×10−8 m2 s−1, re-spectively. Although these results are slightlydifferent from those of Shaw (1974) they arestill consistent with his experimental measure-ments. Lapham et al. (1984) extended the workof Karsten et al. (1982) by investigating the ef-fect of water concentration, from 0.42 to 3.48wt%, and pressure, from 70 to 500 MPa, on thediffusion of water in rhyolite at 850°C. Laphamet al. (1984) further documented the effects ofwater concentration on water diffusion in rhy-olitic melts and demonstrated that increasingthe pressure from 70 to 500 MPa had no meas-urable effect on water diffusion at 850°C.
Zhang et al. (1991) utilized infra-red spec-troscopy to characterize the diffusion of OH− andH2O species in rhyolitic melts and glasses. Thesemeasurements demonstrate that water diffusespredominantly as molecular H2O and that thechange from a rhyolitic glass to a melt had no ef-fect on water diffusion. Combining their resultswith the previous measurements discussedabove, Zhang et al. (1991) were able to constructa general Arrhenius equation for the diffusion ofmolecular, not bulk, water in rhyolitic glasses andmelts from approximately 400 to 950°C and wa-ter concentrations between 1 and 3 wt%
Nowak and Behrens (1997) investigated waterdiffusion in a haplogranitic melt at tempera-tures between 800 and 1200°C, pressures of 50to 500 MPa, and water concentrations between0 and 9 wt%. They found that the activation en-ergy for water diffusion drops from 64000Jmol−1 for 0.5 wt% water to 46000 Jmol−1 formelts with 4 wt% water and remained at the lat-ter value for high water concentrations. Theyobserved a small effect of pressure on waterdiffusion and constructed a general equation forthe diffusion of water, where H2O is in weightpercent and P is the pressure in MPa
+)
)
logD
T
P
O O)
3378 483 O 46.9 O) 0.475
water
2 22
2 22
=
- +
(
(.
8.81 0.045H 0.027(H
H (H
- - +
- +
. .expDRT
4 6 10103 0 5000
.H O 3 318 7
2 #!
=-
-+ - 00b l
The most general expression for water diffusionin rhyolitic melts is that of Zhang and Behrens(2000) who present an equation for the diffu-sion of water at concentrations from 0 to 7.7wt%, temperatures from 400 to 1200°C, andpressures from 0.1 to 810 MPa
where X is the mole fraction of total water inthe melt based upon a one-oxygen mole(Stolper, 1982), and
and
For water contents between 0 and 2 wt% (inclu-sive) Zhang and Behrens (2000) provide a sim-pler equation
where in this case water is measured in weightpercent. Although these expressions are onlybased upon the results of Zhang et al. (1991)and Zhang and Behrens (2000), they accuratelydescribe the results of Karsten et al. (1982) andNowak and Behrens (1997).
Only one study on the diffusion of water inbasaltic melts has been published. Zhang andStolper (1991) measured water diffusion in abasaltic melt at 1.0 GPa, 1300 to 1500°C, andfound that for 0.2 wt% H2O
Freda et al. (2003) studied water diffusion in a
. .exp3 8 10#=26 2
DRT
1 0 3 0.water 3 4
35 6 !--+ - 0 00 0b l
0O( ) .
.
D
T
P
T10 H exp 10 49
1 1
1 772
water12
2= - +
-
- 66bl
..
.b 0 0914 77 106#
= +T 2
..
aT T
P34 1
44 0 57 3=- + +
26
..
mT T
P20 79
5030 1 4- - -=
expD m Xa
X b
X10 e 1 56waterm12= + + + +
-
- ]^h i
6
A

703
Volatile diffusion in silicate melts and its effects on melt inclusions
potassium-rich, trachytic melt at 1.0 Gpa, 1100to 1400°C. They measured water diffusion atconcentrations between 0.25 and 2 wt% H2O;their results are described by
During their study of water solubility and D/Hfractionation between fluid and a basaltic an-desite melt, Pineau et al. (1998) incidentallyfound an apparent diffusion coefficient for afew weight percent water at 1250°C: 5.1×10−8
m2 s−1. This value is 2 orders of magnitude high-er than all other measurements of water diffu-sion at similar temperatures and indicates theneed for more experiments designed to accu-rately measure water diffusion in basic melts.
The best constrained data for water diffu-sion in melts with 1 to 3 wt% H2O at tempera-tures between approximately 1000 and 1300°Cspans only about 1/2 an order of magnitude(fig. 1). At higher water concentrations and at
O
O
( . . ( ))
. . ( ).
exp ln
expexp ln
D
RT
11 924 1 003
11 836 0 139
water 2
2
$= - -
- -
H
H^c hm
magmatic temperatures significantly below1000°C important differences in water diffu-sion coefficients are observed. On the otherhand, the effect of pressure on water diffusionis a second order effect and can often be neg-lected, but clearly needs more investigation.
2.2. Carbon dioxide diffusion
Carbon is found as both carbonate groupsand molecular CO2 in silicate melts and glasses(see review by Blank and Brooker, 1994), al-though a variety of different carbonate types mayexist (Brooker et al., 2001). In the same way aswe treated water above we will only consider thebulk diffusion of carbon dioxide, which is themost important measurement for our applica-tions. Figure 2 presents a summary of the diffu-sion results available for carbon dioxide.
The first experiments on carbon dioxide dif-fusion were tracer diffusion studies by Watsonet al. (1982) on a sodium alumino-silicate meltwith 60 wt% SiO2 and a haplobasaltic melt with53 wt% SiO2. Watson et al. (1982) found car-
Fig. 2. Diffusion of carbon dioxide in magmatic silicate melts. Note that the variation of measurements in thework of Sierralta et al. (2002) is due to the variety of compositions used in their experiments, see text for fur-ther discussion.

704
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
bon dioxide diffusion in both melts to be simi-lar and defined the Arrhenius relation for car-bon dioxide diffusion in the sodium alumino-silicate melt between 800 and 1500°C at 500MPa to be
Watson et al. (1982) demonstrated that at 1200°Cthe effect of increasing pressure by 2.5 GPa di-minished the diffusion coefficient by approxi-mately an order of magnitude. Carbon dioxidediffusion in the haplobasaltic melt at 1.5 GPa pro-duced an activation energy 249000 Jmol−1 and apre-exponential factor of 2.12×10−3 m2 s−1. Fogeland Rutherford’s (1990) study of carbon dioxidesolubility in a rhyolitic melt produced chemicaldiffusion measurements at 1050°C and pressuresbetween 100 and 295 MPa.
The pressure effect on diffusion at theseconditions was not measurable and at this tem-perature the diffusion coefficient of CO2 in rhy-olite was 2.4×10−12 m2 s−1, approximately a fac-tor of two less than that found by Watson et al.(1982) in compositionally different melts. Wat-son (1991) investigated the effect of 8 wt% wa-ter on the tracer diffusion of carbon dioxide inrhyolitic melt at 1 GPa and found that carbondioxide diffusion was significantly higher andthe activation energy lower than in dry rhyoliticmelts
Furthermore, Watson (1991) measured similardiffusion coefficients for carbon dioxide in adacitic melt and estimated that addition of 5wt% water to a dry rhyolitic melt would resultin a 10-fold increase in CO2 diffusion.
Watson (1994) reports the results of CO2 dif-fusion experiments in rhyolitic melts by Blank et al. (1991) and Blank (1993). These measure-ments were an extension of earlier infra-red in-vestigations by E. Stolper’s group at CalTech,who demonstrated the presence of both molecu-lar CO2 and carbonate groups in silicate glasses.These chemical diffusion experiments were per-formed at pressures from 50 to 105 MPa and
.exp10#.6 5=5
DRT
7 0 21 08carbon dioxide
!-- 0 00 0b l
95. .expD
RT3 5 10
1 04carbon dioxide #=
-- 00b l
temperatures of 450 to 1050°C. Fourier-Trans-form Infrared (FTIR) measurements demonstrat-ed that the only species present in the quenchedglasses was CO2 and that the measured diffusioncoefficients can be described by
Sierralta et al. (2002) investigated carbon diox-ide diffusion at 500 MPa and 1250°C in hy-drous albite and albitic melts modified throughvariation of the Na/Al ratio. These authors doc-umented the presence of both molecular CO2
and carbonate in their glasses and determinedthat both diffused at similar rates. Additionally,they determined that the effect of added alkaliesand water on carbon dioxide diffusion wereequivalent when expressed as the ratio of non-bridging oxygens to tetrahedral cations. Theseauthors suggested that the small compositionaleffect on CO2 diffusion was proportional tochanges in the ratio of non-bridging oxygens totetrahedral cations, NBO/T, as the alkalies wereadded to an albitic melt. However, new data byNowak et al. (2004) failed to identify any com-positional effect for a wide range of NBO/T inanalogues of natural melts. One other explana-tion for the effect observed by Sierralta et al.(2002) is peralkalinity, as their diffusion coeffi-cients also increase as a function of this param-eter and extrapolate to agree with the highlyperalkaline Na-aluminosilicate data of Watsonet al. (1982). In contrast, the lack of an NBO/Teffect observed by Nowak et al. (2004) is fornon-peralkaline compositions. Brooker et al.(2001) have suggested that a very isolated car-bonate group exists in peralkaline melts, andwe suggest it is this complex that is diffusing ata high rate. Such effects are only likely to beimportant in rare, highly peralkaline magmas.
Zhang and Stolper’s (1991) study of waterdiffusion in basalt at 1.0 GPa also produced asingle measurement of carbon dioxide chemi-cal diffusion in dry basaltic melt at 1300°C of1.3×10−11 m2 s−1.
We performed reconnaissance experiments at1.0 GPa in which calcite partially dissolved into
= .
exp
D
RT
6 2 10
0114
.
.2 64 3 7
carbon dioxide # $
$-
-+ -
.041060 !b l

705
Volatile diffusion in silicate melts and its effects on melt inclusions
the same potassium-rich, trachyte melts we usedin our study of water diffusion (Freda et al.,2003). The melts were either anhydrous or con-tained 5 wt% H2O and experiments were per-formed at 1.0 GPa, 1200 and 1100°C. We deter-mined carbon dioxide concentrations in the meltusing the electron microprobe and the «differ-ence from 100%» technique. Because of the lowquality of this analytical technique these meas-urements must be considered only as estimatesof carbon dioxide diffusion. At 1200°C the diffu-sion of carbon dioxide in dry and hydrous meltsappears similar within experimental uncertaintyand equaled 2×10−12 m2 s−1; at 1100°C the diffu-sion profile in the anhydrous melt was too shortto measure, but the hydrous melt produced adiffusion coefficient of 7×10−13 m2 s−1. Interest-ingly, these experiments plot along an exten-sion of an Arrhenius relationship defined bycombining the data of Watson et al. (1982) andZhang and Stolper (1991) as shown in fig. 2. Asthere is a large increase in the Ca content of themelt (and therefore in NBO/T) near the inter-face of the dissolving calcite, but the CO2 pro-file is consistent with a single diffusion value,our data appear to confirm the conclusions ofNowak and Screen (2003): there is little meltcompositional effect on bulk CO2 diffusion.
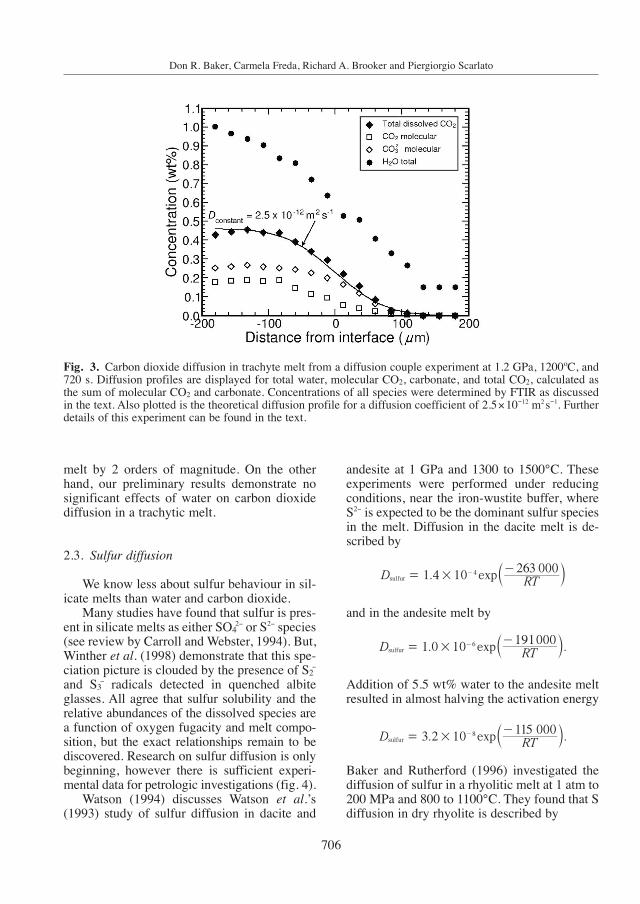
Initially surprised by these results we per-formed diffusion couple experiments for CO2 at1.2 GPa in the same trachytic melt using the dif-fusion couple technique and analyzing thequenched glass using FTIR spectroscopy. Car-bon dioxide-bearing trachytic glass was pre-pared by creating a mixture of oxides and car-bonates equivalent in composition to the tra-chyte. A portion of this mixture was de-volatilized and melted at 1400ºC, 1 atm and thenmixed with a small fraction of the oxide+ +car-bonate mix; this mix was melted at 0.9 GPa,1350 ºC for 2 h in a piston-cylinder apparatus toproduce a carbon-bearing trachyitic glass foruse in the diffusion couple experiments. Duringthis synthesis a small amount of water, approxi-mately 1 wt%, was absorbed by the melt. Thiscarbon-bearing glass was powdered and juxta-posed against a glass cylinder of the same natu-ral trachyte composition. We performed experi-ments at 1.2 GPa in a piston-cylinder for 720 sat 1200ºC and for 600 s at 1300ºC; after each
experiment the diffusion couple was ground andpolished to approximately 50 µm thickness foranalysis by FTIR spectroscopy. The extinctioncoefficients necessary to quantify the FTIR spec-tra were determined using trachyte glasses syn-thesized at high pressure with differing amountsof dissolved carbon dioxide. These glasses wereprepared by mixing different ratios of the tra-chytic oxide+carbonate mix and the glass madefrom this mix followed by melting at 0.9 GPa,1350ºC; the bulk CO2 concentration in aliquotsof these glasses was determined with a LECOcarbon analyzer (Brooker et al., 1999), and theabsorption spectra of polished samples wasmeasured. Based upon this calibration, the ex-tinction coefficient for the molecular CO2 bandat 2348 cm−1 is 975 L mol−1cm−1, and for thecarbonate bands at 1550 and 1412 cm−1 are191±4 L mol−1cm−1 and 193±6 L mol−1cm−1, re-spectively.
The diffusion of bulk carbon dioxide in tra-chytic melt at 1.2 GPa is 2.5×10−12 m2 s−1 at1200ºC and 4.2×10−12 m2 s−1 at 1300ºC. Overthe range of concentrations along the diffusionprofiles a small variation in DCO2 was observed,but it is less than a factor of 2 and probably dueto the small amounts of water present in the car-bon-bearing trachytic glass that diffused togeth-er with the carbon into the volatile-free meltduring the experiment (fig. 3). Although the da-ta are limited in temperature, they define an Ea
of 100000 Jmol−1 and a Do of 8.8×10−9 m2 s−1.Remarkably, the CO2 diffusion coefficients ex-tracted from the calcite dissolution experimentsare consistent with the diffusion couple resultsand support the former’s validity.
With the exception of Watson’s measure-ments of carbon dioxide diffusion in wet rhyo-lite, all CO2 diffusion measurements in naturalmelt compositions are similar at magmatic tem-peratures. These results demonstrate the verysmall dependence of carbon dioxide diffusionupon melt composition. At magmatic tempera-tures the carbon dioxide diffusion measure-ments in all the dry melts can be combined toyield a single Arrhenius relation. However, wa-ter has a significant effect on carbon dioxidediffusion in rhyolite melts. Watson’s results in-dicate the ability of 8 wt% water addition to in-crease carbon dioxide diffusion in rhyolitic
706
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
melt by 2 orders of magnitude. On the otherhand, our preliminary results demonstrate nosignificant effects of water on carbon dioxidediffusion in a trachytic melt.
2.3. Sulfur diffusion
We know less about sulfur behaviour in sil-icate melts than water and carbon dioxide.
Many studies have found that sulfur is pres-ent in silicate melts as either SO4
2− or S2− species(see review by Carroll and Webster, 1994). But,Winther et al. (1998) demonstrate that this spe-ciation picture is clouded by the presence of S2
−
and S3− radicals detected in quenched albite
glasses. All agree that sulfur solubility and therelative abundances of the dissolved species area function of oxygen fugacity and melt compo-sition, but the exact relationships remain to bediscovered. Research on sulfur diffusion is onlybeginning, however there is sufficient experi-mental data for petrologic investigations (fig. 4).
Watson (1994) discusses Watson et al.’s(1993) study of sulfur diffusion in dacite and
andesite at 1 GPa and 1300 to 1500°C. Theseexperiments were performed under reducingconditions, near the iron-wustite buffer, whereS2− is expected to be the dominant sulfur speciesin the melt. Diffusion in the dacite melt is de-scribed by
and in the andesite melt by
Addition of 5.5 wt% water to the andesite meltresulted in almost halving the activation energy
Baker and Rutherford (1996) investigated thediffusion of sulfur in a rhyolitic melt at 1 atm to200 MPa and 800 to 1100°C. They found that Sdiffusion in dry rhyolite is described by
51. .expD
RT2 10
03
1sulfur
8#=-- 00b l
. .expDRT
1 0 10191 0
sulfur6#=
-- 00b l
36. expD
RT1 4 10
2 0sulfur
4#=-- 00b l
Fig. 3. Carbon dioxide diffusion in trachyte melt from a diffusion couple experiment at 1.2 GPa, 1200ºC, and720 s. Diffusion profiles are displayed for total water, molecular CO2, carbonate, and total CO2, calculated asthe sum of molecular CO2 and carbonate. Concentrations of all species were determined by FTIR as discussedin the text. Also plotted is the theoretical diffusion profile for a diffusion coefficient of 2.5×10−12 m2 s−1. Furtherdetails of this experiment can be found in the text.

707
Volatile diffusion in silicate melts and its effects on melt inclusions
and that addition of 7 wt% water increased sul-fur diffusion by 1.5 to 2 orders of magnitude.Unfortunately, the hydrous experiments cov-ered too small a temperature range to define anArrhenius relationship, but the sulfur diffusiondata of Baker and Rutherford (1996) in hydrousrhyolite all plot close to the low temperature ex-trapolation of Watson et al.’s (1993) Arrheniusline for sulfur diffusion in a hydrous andesite(fig. 4).
Baker and Rutherford performed experi-ments at geological oxygen fugacities rangingfrom the fayalite-quartz-magnetite buffer (~1order of magnitude below the nickel-nickel ox-ide buffer) to the MnO-Mn3O4 buffer (~2 or-ders of magnitude above the nickel-nickel ox-ide buffer) and found no oxygen fugacity ef-fects on S diffusion. Baker and Rutherford ar-gued that this insensitivity to oxygen fugacityreflected the presence of a single diffusing Sspecies, which they identified as S2−. Their ar-gument rests upon a comparison of their resultsto other diffusion experiments, theoretical cal-culations of how the size of the diffusing
0. expD
RT10
2 0 8 05 0
21sulfur
6#!
=-- 0 00 0b l species affects the diffusion coefficient, and
their experiments in air, where virtually no S2−
should be dissolved in the melt, which demon-strated significantly lower diffusion coeffi-cients than the other experiments at lower oxy-gen fugacities.
Winther et al.’s (1998) measurements of Sdiffusion in dry albitic melt at 1.0 GPa, 1300 to1500°C, yielded
This activation energy is surprisingly largewhen compared to those for sulfur diffusion inother anhydrous melts. Using a variety of spec-troscopic methods Winther et al. (1998) foundthat sulfur is present in the quenched glasses asSO4
2−, SO2− and SO3
−. They provided evidencethat at least some of the species previouslythought to be S2−, based upon the sulfur peak lo-cation during electron microprobe analysis,were instead a combination of the radicals S2
−
and S3−. These authors found that the reduced
species S2− and S3
− diffuse more rapidly thanSO4
2−, consistent with the findings of Baker andRutherford (1996).
. .expDRT
70
14458
sulfur =- 01b l
Fig. 4. Diffusion of sulfur in magmatic silicate melts.

708
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
Measurements of sulfur diffusion in silicatemelts of geological interest are rare and muchmore work is needed. However, most of the re-sults to date are consistent and suggest thatwithin the range of anhydrous andesite to rhyo-lite there is little effect of melt composition onsulfur diffusion. The effect of water is signifi-cant, increasing S diffusion by up to 2 orders ofmagnitude in rhyolitic melts, but Arrhenius re-lations for most hydrous melts remain to be ac-curately determined.
2.4. Fluorine and Chlorine
Fluorine and chlorine are not uncommonmagmatic components (Carroll and Webster,1994). Both elements are found at concentra-tions of 1000’s of ppm and in some graniticrocks F can reach weight percent concentra-tions. Studies of the diffusion of these volatilesin magmatic melts are quite rare, but still suffi-cient to provide an idea of the diffusion coeffi-cients of these elements (fig. 5).
Dingwell and Scarfe (1984, 1985) were thefirst to measure fluorine diffusion in a geologi-cally relevant melt composition. They measuredF and oxygen interdiffusion in anhydrous jadeite
melt at high pressure using a diffusion couple inwhich one half of the couple was composed ofjadeite melt and the other half was composed ofa jadeite melt in which some of the oxygen wasreplaced by fluorine. Over the temperature rangeof 1200 to 1400°C and pressure range of 1.0 to1.5 GPa, Dingwell and Scarfe found that F diffu-sion in jadeite melt is pressure insensitive andfollows the Arrhenius equation:
These authors also measured fluorine-oxygendiffusion in albite at 1 atm at the same tempera-tures (Dingwell and Scarfe, 1985) by exposing afluoridated albitic melt to air at high tempera-ture, which resulted in the diffusion of fluorineout of and oxygen in to the sample. Interesting-ly, this study produced a lower activation energy
Chlorine tracer diffusion in a geological meltwas first measured by Watson and Bender (1980)in a simple Na-Ca-Al-Si-O melt with 56 wt% sil-ica at temperatures from 1100 to 1300°C, and
.. expDRT
101 0
1 821
fluorine9#=
-- 00b l
59. .expD
RT5 9 10
1 0fluorine
6#=-- 00b l
Fig. 5. Diffusion of halogens in magmatic silicate melts.

709
Volatile diffusion in silicate melts and its effects on melt inclusions
pressures from 0.6 to 1.8 GPa. At 0.6 GPa theyfound
Comparison of diffusion coefficients measuredat different pressures indicated no pressure de-pendence of Cl diffusion.
Watson (1991) investigated Cl tracer diffu-sion in a rhyolitic and a dacitic melt with 8 wt%water at 1.0 GPa. The temperature of these ex-periments spanned 850 to 1100°C, which wasnot sufficient to define an Arrhenius relation, butthe diffusion coefficients ranged from 3×10−12 to4×10−11 m2s−1.
Bai and Koster van Groos (1994) measuredchlorine chemical diffusion in a haplograniticmelt with 75 wt% SiO2 and a natural obsidianmelt with 78 wt% SiO2 at pressures from 1 atmto 460 MPa. Their anhydrous experiments atlow pressure produced an Arrhenius relation of
70.. expD
RT10
03 4
2chlorine
4#=-- 00b l for temperatures between 850 and 1400°C.
Chemical diffusion of chlorine in a hydrous,granitic melt saturated with a NaCl brine at 200MPa, 650 to 900°C is described by
These authors demonstrate that over the pressurerange of 200 to 460 MPa the Cl diffusion coeffi-cient decreases by approximately a factor of 2.
The results for chlorine diffusion are excit-ing in that the few data available from differentlabs using differing techniques on rhyoliticcompositions are very consistent, with the inter-esting exception of the pressure dependence forCl diffusion. Regrettably there are not sufficientdata to better evaluate fluorine diffusion. Clear-ly, more research in this area is urgently needed.
01. .expD
RT6 46 10
1 07chlorine #=
-- 09b l
. expDRT
3 16 1086
chlorine9#=
-- 091b l
Fig. 6. Comparison of volatile diffusion in hydrous silicic (almost exclusively rhyolitic) melts. Water diffusion,for 2 and 6 wt% total, is calculated from Zhang and Behrens (2000). CO2 diffusion is calculated from Watson(1991) in a melt with 8 wt% dissolved water. S diffusion is a combination of the high-temperature results of Wat-son et al. (1993), see also Watson (1994), and the low-temperature results of Baker and Rutherford (1996). Cldiffusion is a combination of the high temperature results of Watson (1991) and the low temperature results ofBai and Koster van Groos (1994). Note that the apparent activation energies for S and Cl diffusion may be anartifact of combining data from two different studies. Lines for Si-Al interdiffusion in silicic melts containingeither 3 or 6 wt% H2O are extrapolated using the Arrhenius relationships determined by Baker (1991).

710
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
2.5. Comparison of the different volatiles
Figure 6 presents a comparison of the meas-ured diffusion coefficients of water, carbon diox-ide, sulfur and chlorine in a hydrous rhyoliticmelt. At this time there are insufficient data todepict the diffusion of fluorine. The lines shownfor sulfur and for chlorine are constructed bycombining the high-temperature results of onestudy with the low-temperature results of anoth-er; thus, they only represent the diffusion ofthese volatiles in a general way. Nevertheless,the data demonstrate that over a broad range ofmagmatic temperatures, from ∼1200 to 600ºC ,the diffusion coefficients of water, carbon diox-ide, and chlorine are expected to be within an or-der of magnitude of each other. Sulfur, diffusesmuch more slowly, and its diffusion coefficientis approximately 2, or more at lower tempera-tures, orders of magnitude less than those of theother dominant magmatic volatiles. As we shallsee this difference in diffusion coefficients canresult in some interesting effects.
3. Effects of crystal growth rate anddiffusion on the composition of meltinclusions
It is well known that the dynamics of bub-ble growth controls the physics of volcaniceruptions (cf., Sparks et al., 1994). In turn, bub-ble growth is mainly controlled by two process-es: diffusion of volatiles from the melt into thebubbles and the expansion of the bubbles due todecompression (Sparks et al., 1994). Since theformulation of Scriven’s (1959) growth law forbubbles in a superheated one-phase system
where R is the radius of the bubble, t the time,D the diffusion coefficient and β is a growthrate constant, many researchers created morecomplex models taking into account additionalparameters such as advection, viscosity, pres-sure and variable volatile concentration (e.g.,Proussevitch et al., 1993; Proussevitch and Sa-hagian, 1998; Blower et al., 2001). It is appar-ent that knowledge of volatile diffusion coeffi-
R Dt2= b
cients in silicate melts is a fundamental neces-sity for the modelling of magma vesiculation.
It is probably less well known that knowl-edge of volatile diffusivity aids the interpreta-tion of data obtained studying melt inclusions incrystals. Melt inclusions are small (tens of mi-crons) samples of melt trapped by magmaticcrystals during their growth and are consideredwitnesses of petrologic processes, thus they areused as petrogenetic indicators (cf., Hauri et al.,2002). Because some melt inclusions aretrapped at relatively high pressure, they are auseful tool to estimate volatile contents in mag-mas (e.g., Roedder, 1979; Hauri, 2002). Impor-tantly, melt inclusions might form at differentstages of crystal growth and they record only thecomposition of the melt at the crystal-melt inter-face. If the diffusion coefficient in the melt of anincompatible element in the crystal is slow com-pared to the crystal growth rate, the melt-crystalinterface will become enriched in the incompat-ible element (e.g., Smith et al., 1955; Watson etal., 1982; Harrison and Watson, 1984); this phe-nomenon is often referred to as pile-up or thesnowplow effect. Because of the common use ofmelt inclusions as volatile records (e.g., Mas-sare et al., 2002), we performed calculations toestimate the volatile (H2O, CO2, Cl, and S) con-centration curve in front of a growing crystal.Our conceptual model is that of a crystal whosegrowth rate suddenly increases for a short dura-tion and then decreases, trapping melt at thecrystal-melt interface forming melt inclusions.
We modelled the pile-up of volatiles at thecrystal-melt interface for both rhyolitic andbasaltic melts. All volatiles are incompatible el-ements with a crystal-liquid partition coefficientof 0.001. Decreasing this partition coefficientby one order of magnitude insignificantly af-fects the results. Two crystal growth rates wereused in the calculations, 10−8 and 10−10 m s−1.These values are based upon the results ofSwanson (1977) who investigated plagioclase,alkali feldspar and quartz growth in a graniticmelt containing 3.5 wt% H2O at temperaturesbetween 690 and 810ºC and measured maxi-mum growth rates of 10−8, 10−9, and 10−10 m s−1,respectively. At temperatures between 600 and1000ºC, plagioclase growth rates varied from10−9 to 10−8 m s−1, alkali feldspar growth rates

711
Volatile diffusion in silicate melts and its effects on melt inclusions
were from 10−11 to 10−9 m s−1, and quartz growthrates spanned 10−11 to 10−10 m s−1 (Swanson1977). We previously studied crystal growth inthe system orthoclase-quartz-H2O and meas-ured similar growth rates, between 1×10−10 and5×10−9 m s−1 (Baker and Freda, 2001). Growthrates of ferromagnesian minerals in composi-tionally simple, basic melts range from 1×10−4
to 1×10−6 m s−1 (see summary in Lasaga, 1998).Recently, Simakin et al. (2003) have measuredclinopyroxene and olivine growth rates near1100ºC in a hawaiite melt at undercoolings of25 and 45ºC, respectively, and found them to beboth approximately 1×10−8 m s−1. The highgrowth rate used in our modelling, 10−8 m s−1,appears to be close to the maximum expectedgrowth rate in magmatic systems, whereas thelow growth rate used in the models, 10−10 m s−1,is probably more common in nature, but stillpotentially rapid compared to normal growthrates in igneous systems (cf., Lu et al., 1995).
We modelled linear crystal growth of 10, 50and 100 µm and calculated the volatile profile inthe melt following eq. 26 of Smith et al. (1955).The diffusion coefficients for volatiles ingranitic melts were extracted from fig. 5 for cal-culations at 800°C (DS=2.5×10−14, D2 wt%H2O==2.6 × 10−12, DCl=6.6 × 10−12, DCO2=2.0×10−11,D6 wt%H2O=2.0×10−11 m2 s−1). Crystal growth ratesare affected by diffusion of their components inthe melt, in this case primarily silicon and alu-minum (Lasaga, 1998); at these conditions thechemical interdiffusion of Si and Al in silicicmelts (fig. 6) is approximately 1×10−14 m2s−1 at2 wt% H2O in the melt, and 1×10−13 m2s−1 at 6wt% H2O (Baker, 1991). The diffusion coeffi-cients of water and CO2 measured by Zhangand Stolper (1991) in a basaltic melt were usedin modelling the effects of crystal growth at1300°C (DH2O=2.5×10−10 and DCO2=1.3×10−11
m2 s−1). Although no measurements of Si and Aldiffusion in hydrous basaltic melts are avail-able, water is unlikely to significantly affectdiffusion in this composition and we expect thatSi-Al interdiffusion in hydrous basaltic melts isprobably at most an order of magnitude abovethat measured in dry melts at 1300ºC, ∼1×10−12
m2 s−1 (Watson, 1982).We do not consider changes in the volatile
diffusion coefficients due to changing water
concentrations because once melts attain ap-proximately 3 to 4 wt% water the diffusion co-efficients of all elements are relatively insensi-tive to the exact water concentration (Watson,1994). The only diffusion profile this simplifi-cation might significantly affect is that of 2wt% H2O. However, because of the effect ofwater concentration on water diffusion, our re-sults at 2 and 6 wt% H2O bracket the possibleenrichment profiles for water concentrations inthis range, and probably at water concentrationsup to 8 wt%. In all cases we ignore the possibil-ity of reaching fluid saturation and subsequentvesiculation. In order to compare calculationsfor different volatiles, we plot the results of thecalculations as the enrichment factor of the meltnear the crystal-melt interface relative to thebulk concentration of the volatile in the melt(figs. 7a-d and 8).
The volatile enrichment in front of a crystalgrowing from a hydrous rhyolitic melt at 800ºCis displayed in fig. 7a-d. If 10 µm of crystalgrow at 10−8 ms−1 (fig. 7a) the maximumvolatile enrichment factor is 6x; this is the en-richment factor for sulfur and is due to its lowdiffusion coefficient. However, only 10 µmaway from the crystalmelt interface the S reach-es the bulk concentration in the melt. The max-imum enrichment of the other volatiles is 1.25x.In contrast to sulfur, these volatiles only reachthe bulk melt concentrations at 100 to 275 µmfrom the crystal-melt interface. At 800°C thediffusion coefficients of 6 wt% H2O and CO2
are the same, which results in overlapping en-richment profiles in fig. 7a-d. If the crystalgrew 50 µm (fig. 7b), the S enrichment reaches22x in this case, and for a bulk melt with 2 wt%water the concentration at the interface is en-riched 1.6x (3.2 wt% H2O). Chlorine is en-riched by 1.35x and both CO2 and water for amelt with a bulk water concentration of 6 wt%are enriched 1.2x. The region of enriched meltextends to 20 µm in front of the crystalmelt in-terface for sulfur and 300 to 600 µm away forthe other volatiles. Allowing the crystal to grow100 µm (fig. 7c) results in a sulfur enrichmentof 42x, but the 20 µm-wide enriched region infront of the crystal is approximately the samelength as in the case of 50 µm of growth. Theenrichment factor for a melt with a bulk compo-

712
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
sition of 2 wt% water is 1.9x at the interface; forchlorine the enrichment is 1.55x and for 6 wt%water and CO2 is 1.28x. The profiles for the 2and 6 wt% H2O, Cl and CO2 extend from ap-proximately 400 to 1100 µm into the melt.
If the growth rate is decreased two orders ofmagnitude to 10−10 m s−1, the volatile enrichmentat the crystal-melt interface is significantly less(fig. 7d). For 50 µm of growth the enrichmentof sulfur is only a factor of 1.6x (compared to
Fig. 7a,b. Volatile concentration enrichment profiles in front of a crystal growing in a rhyolitic melt at 800°C. Thediffusion coefficients for volatiles are from fig. 5 and the sources of the crystal growth rates are discussed in the text.Note that composition enrichment profiles for 6 wt% H2O and CO2 plot on top of one another because their diffu-sion coefficients are identical at the conditions studied (see fig. 5). The effects of crystal growth on volatile concen-tration enrichment profiles relative to the concentration at infinite distance near the crystal-melt interface are mod-elled at a growth rate of 10−8 m s−1 for a total growth of 10 µm (a), 50 µm (b). Further discussion is found in the text.
a
b

713
Volatile diffusion in silicate melts and its effects on melt inclusions
22x at a growth rate of 10−8 m s−1) and all of theother volatiles are enriched by less than a factorof 1.06x, which is below analytical uncertaintyin many cases.
These calculations for crystal growth from arhyolitic melt at 800°C demonstrate that at highgrowth rates any melts trapped from the crystal-melt interface into melt inclusions may not con-
Fig. 7c,d. Volatile concentration enrichment profiles in front of a crystal growing in a rhyolitic melt at 800°C. Thediffusion coefficients for volatiles are from fig. 5 and the sources of the crystal growth rates are discussed in the text.Note that composition enrichment profiles for 6 wt% H2O and CO2 plot on top of one another because their diffu-sion coefficients are identical at the conditions studied (see fig. 5). The effects of crystal growth on volatile concen-tration enrichment profiles relative to the concentration at infinite distance near the crystal-melt interface are mod-elled at a growth rate of 10−8 m s−1 for a total growth of 100 µm (c). The volatile concentration enrichment profilesafter 50 µm of crystal growth at 10−10 m s−1 are also modelled to demonstrate the effect of crystal growth rate onvolatile enrichment (d). Further discussion is found in the text.
c
d

714
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
tain volatile concentrations representative ofthe bulk melt. In fact, our one-dimensionalmodel provides a conservative estimate of en-richment that would occur during growth andentrapment of a spherical melt inclusion. Amore positive way of considering the results ofthese calculations is that concentrations of H2O,CO2 and Cl measured in melt inclusions are un-likely to be more than twice the bulk melt con-centration of these volatiles. The existing dataon crystal growth in silicic melts (discussedabove) indicate that quartz is the slowest grow-ing silicate crystal, with a maximum of approx-imately 10−10 m s−1, and should contain melt in-clusions representative of the bulk melt. Ourstudy reinforces the conclusions of Lu et al.(1995) who through their study of melt inclu-sions in crystals from the Bishop Tuff, Califor-nia, found that melt inclusions larger than 50µm in diameter should be compositionallyequivalent to the bulk melt. If melt inclusionscan be trapped near 800ºC at what appear to becrystal growth rates below 10−10 m s−1 (cf., Lu et al., 1995) their volatile concentrations of
H2O, CO2 and Cl almost certainly will be repre-sentative of the bulk melt, but the minimumgrowth rate necessary for a crystal-melt inter-face to become unstable and potentially lead tothe entrapment of melt inclusions remains un-known. For example, our experiments on crys-tal growth in the orthoclase-quartz-water sys-tem (Baker and Freda, 2001) did not trap meltinclusions even at a growth rate 5×10−9 m s−1.
Modelling the effects of a 10−8 ms−1 growthrate on H2O and CO2 concentration profiles ina basaltic melt at 1300°C (fig. 8) demonstratesthat the maximum enrichment, in this case forCO2, is only a factor of 1.35x for 100 µm ofgrowth. The water concentration is insignifi-cantly enriched by a factor of less than 1.08 forthe conditions investigated. Despite the smallvolatile enrichment factors at the crystal-meltinterface the concentration profiles do notreach the bulk melt composition until at least300 µm away from the crystal. This modellingsuggests that melt basaltic inclusions trappedat 1300°C contain concentrations of CO2 andH2O similar to those of the bulk melt.
Fig. 8. Volatile concentration enrichment profiles in front of a crystal growing in a basaltic melt at 1300°C. Dif-fusion coefficients of H2O and CO2 are from Zhang and Stolper (1991) and crystal growth rates are based on da-ta in Lasaga (1998) and Simakin et al. (2003). For further discussion please see the text.

715
Volatile diffusion in silicate melts and its effects on melt inclusions
The calculations highlight the need to knowboth the diffusion coefficients for volatiles (andother elements for that matter) and crystal growthrates in order to assess whether or not melt inclu-sions are representative of the bulk melt. Differ-ences of approximately 4 orders of magnitudebetween diffusion coefficients and crystal growthrates are sufficient to enrich the crystal-melt in-terface significantly, greater than 10 relative per-cent, above the bulk melt. The conclusion thatmelt inclusions formed in basaltic systems trapsamples that may be compositionally representa-tive is based only upon calculations for water andcarbon dioxide at an unrealistically high temper-ature of crystallization.
Although the calculations above purposelyignore the possibility of volatile saturation andexsolution due to rapid crystal growth, it isworthwhile to qualitatively consider this possibil-ity. Even though the bulk melt may not be volatilesaturated, rapid crystal growth could cause theconcentration of a volatile at the crystal-melt in-terface to reach saturation and exsolve a fluidphase. The concentration necessary to reach satu-ration in magma chambers at a few kilometresdepth varies from the weight percent level forH2O to only hundreds to thousands of ppm forCO2, S, and Cl (Carroll and Holloway, 1994).Thermodynamics demands that once onevolatile reaches saturation and exsolves the oth-er components in the system must partition be-tween the melt and the fluid phase in order toreach, or attempt to reach, equilibrium. Obvi-ously this process affects volatile concentra-tions in the melt at the crystal-melt interfaceand causes them to deviate from those in thebulk melt. If these bubbles were able to sepa-rate from the interface before the melt wastrapped, there might be no record of exsolution(presuming these bubbles ascend into the bulk,fluid undersaturated, melt they would be re-sorbed). Only recently are we beginning tomeasure the equilibrium partitioning of volatilecomponents, primarily H2O and CO2 (e.g.,Tamic et al., 2001), between silicate melts andfluids, so that we are at the beginning stage ofquantitatively modelling this process.
Anomalous volatile enrichment of melt inclu-sions might be detected by measuring volatileconcentrations in contemporaneous melt inclu-
sions of differing sizes. Modelling suggests thatslowly diffusing sulfur is only enriched withinapproximately 20 µm of the crystal-melt inter-face in rhyolitic melts at 800°C (fig. 6), which isconsistent with the results of Lu et al. (1995);thus if anomalous enrichment due to pile-up dur-ing crystal growth occurred, smaller inclusionsare expected to be significantly enriched in sulfurcompared to larger ones. Once the size of themelt inclusion significantly exceeds the enrichedregion at the crystal-melt interface, the sulfurconcentration should reach a constant, bulk, val-ue. However, our models demonstrate that evenwhen sulfur reaches the bulk melt concentrationin large melt inclusions, the concentrations ofother volatiles in the inclusion may still be signif-icantly higher than the bulk melt concentrations.
The measurement of volatile diffusion in sil-icate melts has advanced significantly sinceShaw published his work in 1974. Our knowl-edge of water diffusion in silicic melts is partic-ularly extensive. However, we still need addi-tional measurements of CO2, S, F and Cl in sili-cic melts and of the diffusion of all volatiles inintermediate and basic compositions. Addition-ally, we need to know how water concentrationand pressure affect diffusion of other volatiles inall magmatic silicate melts. Our simple modelsindicate that under conditions of rapid crystalgrowth diffusion pile-up can occur at crystal-melt interfaces and the melts trapped as melt in-clusions may not contain volatile concentrationsrepresentative of the bulk melt; this problem ap-pears particularly severe for rhyolitic systems athigh crystal growth rates and probably minor forhigh-temperature basaltic systems.
Acknowledgements
We thank the two anonymous reviewerswhose insightful comments spurred us to im-prove the presentation of this paper. We alsothank Mike Carroll for his work in editing thisvolume. We particularly thank M. Nowak forproviding us with a preprint of his paper onCO2 diffusion. Funding for the experiments andanalyses reported in this work came from:NSERC Discovery grant to D.R.B.; Italian Civ-il Protection (GNV project N. 2000-02/17) to

716
Don R. Baker, Carmela Freda, Richard A. Brooker and Piergiorgio Scarlato
C.F. and P.S.; European Community-Access toResearch Infrastructure Action of the Improv-ing Human Potential Programme to C.F. andR.A.B.
REFERENCES
BAI, T.B. and A. KOSTER VAN GROOS (1994): Diffusion ofchlorine in granitic melts, Geochim. Cosmochim. Acta,58, 113-123.
BAKER, D.R. (1991): Interdiffusion of hydrous dacitic andrhyolitic melts and the efficacy of rhyolite contamina-tion of dacitic enclaves, Contrib. Mineral. Petrol., 106,462-473.
BAKER, D.R. and C. FREDA (2001): Eutectic crystallization inthe undercooled orthoclase-quartz-H2O system: experi-ments and simulations, Eur. J. Mineral., 13, 453-466.
BAKER, L.L. and M.J. RUTHERFORD (1996): Sulfur diffusionin rhyolite melts, Contrib. Mineral. Petrol., 123, 335-344.
BLANK, J.G. (1993): An experimental investigation of thebehavior of carbon dioxide in rhyolitic melt, Ph.D. The-sis (California Institute of Technology, Pasadena, CA).
BLANK, J.G. and R.A. BROOKER (1994): Experimental stud-ies of carbon dioxide in silicate melts: solubility, speci-ation, and stable carbon isotope behavior, in Volatilesin Magmas, edited by M.R. CARROLL and J.R. HOL-LOWAY, Rev. Mineral., 30, 157-186.
BLANK, J.G., E.M. STOLPER and Y. ZHANG (1991): Diffusionof CO2 in rhyolitic melt, Eos Trans. Am. Geophys. Un.,72, 312 (abstract).
BLOWER, J.D., H.M. MADER and S.D.R. WILSON (2001):Coupling of viscous and diffusive controls on bubblegrowth during explosive volcanic eruptions, EarthPlanet. Sci. Lett., 193, 47-56.
BROOKER, R.A., S.C. KOHN, J.R. HOLLOWAY, P.F. MCMIL-LAN and M.R. CARROLL (1999): Solubility, speciation,and dissolution mechansims for CO2 in melts on theNaAlO2-SiO2 join, Geochim. Cosmochim. Acta, 63,3549-3565.
BROOKER, R.A., S.C. KOHN, J.R. HOLLOWAY and P.F.MCMILLAN (2001): Structural controls on the solubili-ty of CO2 in silicate melts, Part II. IR characteristics ofcarbonate groups in silicate glasses, Chem. Geol., 174,241-254.
CARROLL, M.R. and J.R. HOLLOWAY (Editors) (1994):Volatiles in magmas, Rev. Mineral., 30, pp. 517.
CARROLL, M.R. and J.D. WEBSTER (1994): Solubilities ofsulfur, noble gases, nitrogen, chlorine, and fluorine inmagmas, in Volatiles in Magmas, edited by M.R. CAR-ROLL and J.R. HOLLOWAY, Rev. Mineral., 30, 231-279.
DINGWELL, D.B. and C.M. SCARFE (1984): Chemical diffu-sion of fluorine in jadeite melt at high pressure,Geochim. Cosmochim. Acta, 48, 2517-2525.
DINGWELL, D.B. and C.M. SCARFE (1985): Chemical diffu-sion of fluorine in melts in the system Na2O-Al2O3-SiO2, Earth Planet. Sci. Lett., 73, 377-384.
FOGEL, R.A. and M.J. RUTHERFORD (1990): The solubilityof carbon dioxide in rhyolitic melts: a quantitativeFTIR study, Am. Mineral., 75, 1311-1326.
FREDA, C., D.R. BAKER, C. ROMANO and P. SCARLATO
(2003): Water diffusion in natural potassic melts, inVolcanic Degassing, edited by C. OPPENHEIMER, D.M.PYLE and J. BARCLAY, Geol. Soc., London, Spec. Publ.213, 53-62.
HARRISON, T.M. and E.B. WATSON (1984): The behavior ofapatite during crustal anatexis: equilibrium and kineticconsiderations, Geochim. Cosmochim. Acta, 48, 1467-1477.
HAURI, E. (2002): SIMS analysis of volatiles in silicateglasses, 2. Isotopes and abundances in Hawaiian meltinclusions, Chem. Geol., 183, 115-141.
HAURI, E., A.J.R. KENT and N. ARNDT (2002): Melt inclu-sions at the millennium; toward a deeper understandingof magmatic processes, Chem. Geol., 183, 1-3.
KARSTEN, J.L., J.R. HOLLOWAY and J.R. DELANEY (1982):Ion microprobe studies of water in silicate melts: tem-perature-dependent water diffusion in obsidian, EarthPlanet. Sci. Lett., 59, 420-428.
LAPHAM, K.E., J.R. HOLLOWAY and J.R. DELANEY (1984):Diffusion of H2O and D2O in obsidian at elevated tem-peratures and pressures, J. Non-Cryst. Solids, 67, 179-191.
LASAGA, A. (1998): Kinetic Theory in the Earth Sciences(Princeton University Press, Princeton, New Jersey),pp. 810.
LU, F., A.T. ANDERSON and A.M. DAVIS (1995): Diffusionalgradients at the crystal-melt interface and their effecton the composition of melt inclusions, J. Geol., 103,591-597.
LUHR, J.F., I.S.E. CARMICHAEL and J.C. VAREKAMP (1984):The 1982 eruption of El Chichon volcano, Chiapas,Mexico: mineralogy and petrology of the anhydrite-bearing pumices, J. Volcanol. Geotherm. Res., 23, 69-108.
MASSARE, D., N. MÉTRICH and R. CLOCCHIATTI (2002):High-temperature experiments on silicate melt inclu-sions in olivine at 1 atm: inference on temperatures ofhomogenization and H2O concentrations, Chem. Geol.,182, 87-98.
MCMILLAN, P.F. (1994): Water solubility and speciationmodels, in Volatiles in Magmas, edited by M.R. CAR-ROLL and J.R. HOLLOWAY, Rev. Mineral., 30, 131-156.
NOWAK, M. and H. BEHRENS (1997): An experimental in-vestigation of diffusion of water in haplogranitic melts,Contrib. Mineral. Petrol., 126, 365-376.
NOWAK, M., D. SCHREEN and K. SPICKENBOM (2004): Argonand CO2 on the race track in silicate melts: a tool for thedevelopment of a CO2 speciation and diffusion model,Geochim. Cosmochim. Acta, 68 (24), 5127-5138.
PINEAU, F., S. SHILOBREEVA, A. KADIK and M. JAVOY (1998)Water solubility and D/H fractionation in the systembasaltic andesite-H2O at 1250°C and between 0.5 and3 kbar, Chem. Geol., 147, 173-184.
PROUSSEVITCH, A.A. and D.L. SAHAGIAN (1998): Dynamicsand energetics of bubble growth in magmas: Analyticalformulation and numerical modeling, J. Geophys. Res.,103B, 18,223-18,251.
PROUSSEVITCH, A.A., D.L. SAHAGIAN and A.T. ANDERSON
(1993): Dynamics of diffusive bubble growth in mag-mas: isothermal case, J. Geophys. Res., 98B, 22,283-22,307.
ROEDDER, E. (1965): Liquid CO2 inclusions in olivine-bear-

717
Volatile diffusion in silicate melts and its effects on melt inclusions
ing nodules and phenocrysts from basalts, Am. Miner-al., 50, 1746-1782.
ROEDDER, E. (1979): Origin and significance of magmaticinclusions, Bull. Mineralogie, 102, 487-510.
SCRIVEN, L.E. (1959): On the dynamics of phase growth,Chem. Eng. Sci., 10, 1-13.
SHAW, H.R. (1974): Diffusion of H2O in granitic liquids:Part I. Experimental data, Part II. Mass transfer in mag-ma chambers, in Geochemical Transport and Kinetics,edited by W. HOFMANN, B.J. GILETTI, H.S. YODER JR.and R.A. YUND (Carnegie Institution of Washington,Washington), 139-170.
SIERRALTA, M., M. NOWAK and H. KEPPLER (2002): The in-fluence of bulk composition on the diffusivity of car-bon dioxide in Na aluminosilicate melts, Am. Mineral.,87, 1710-1716.
SIMAKIN, A.G., T.P. SALOVA and P. ARMIENTI (2003): Kinet-ics of clinopyroxene growth from a hydrous hawaiitemelt, Geochem. Int., 41, 1165-1175.
SMITH, V.G., W.A. TILLER and J.W. RUTTER (1955): A math-ematical analysis of solute redistribution during solidi-fication, Can. J. Phys., 33, 723-744.
SPARKS, R.S.J., J. BARCLAY, C. JAUPART, H.M. MADER and J.C.PHILLIPS (1994): Physical aspects of magma degassing, I.Experimental and theoretical constrains on vesiculation,in Volatiles in Magmas, edited by M.R. CARROLL and J.R.HOLLOWAY, Rev. Mineral., 30, 413-445.
STOLPER, E. (1982): The speciation of water in silicatemelts, Geochim. Cosmochim. Acta, 46, 2609-2620.
SWANSON, S.E. (1977): Relation of nucleation and crystal-growth rate to the development of granitic textures,Am. Mineral., 62, 966-978.
SYMONDS, R.B., W.I. ROSE, G.J.S. BLUTH and T.M. GERLACH
(1994): Volcanic-gas studies: methods, results, and ap-plications, in Volatiles in Magmas, edited by M.R. CAR-ROLL and J.R. HOLLOWAY, Rev. Mineral., 30, 1-66.
TAMIC, N., H. BEHRENS and F. HOLTZ (2001): The solubili-
ty of H2O and CO2 in rhyolitic melts in equilibriumwith a mixed H2O-CO2 fluid phase, Chem. Geol., 174,333-347.
WALLACE, P.J. (2001): Volcanic SO2 emissions and theabundance and distribution of exsolved gas in magmabodies, J. Volcanol. Geotherm. Res., 108, 85-106.
WATSON, E.B. (1982) Basalt contamination by continentalcrust: some experiments and models, Contrib. Mineral.Petrol., 80, 73-87.
WATSON, E.B. (1991): Diffusion of dissolved CO2 and Cl inhydrous silicic to intermediate magmas, Geochim.Cosmochim. Acta, 55, 1897-1902.
WATSON, E.B. (1994): Diffusion in volatile-bearing mag-mas, in Volatiles in Magmas, edited by M.R. CARROLL
and J.R. HOLLOWAY, Rev. Mineral., 30, 371-411.WATSON, E.B. and J.F. BENDER (1980): Diffusion of cesium,
samarium, strontium, and chlorine in molten silicate athigh temperatures and pressures, Geol. Soc. Am. Abstr.Program, 12, 545.
WATSON, E.B., M.A. SNEERINGER and A. ROSS (1982): Dif-fusion of dissolved carbonate in magmas: experimentalresults and applications, Earth Planet. Sci. Lett., 61,346-358.
WATSON, E.B., D.A. WARK and J.W. DELANO (1993): Initialreport on sulfur diffusion in magmas, Eos, Trans. Am.Geophys. Un., 74, 620.
WINTHER, K.T., E.B WATSON and G.M. KORENOWSKI
(1998): Magmatic sulfur compounds and sulfur diffu-sion in albite melt at 1 GPa and 1300-1500°C, Am.Mineral., 83, 1141-1151.
ZHANG, Y. and H. BEHRENS (2000): H2O diffusion in rhy-olitic melts and glasses, Chem. Geol., 169, 243-262.
ZHANG, Y. and E.M. STOLPER (1991): Water diffusion inbasaltic melts, Nature, 351, 306-309.
ZHANG, Y., E.M. STOLPER and G.J. WASSERBURG (1991):Diffusion of water in rhyolitic glasses, Geochim. Cos-mochim. Acta, 55, 441-456.
Related Documents











