FEBRUARY 2019 VOLUME 21 ® 1 VIDEOS ONLINE Price per issue : 112 €

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

F E B R UA RY 2 0 1 9
V O L U M E 2 1
®
1VIDEOS ONLINE
Price per issue : 112 €
VIDEOS
ImpactFactor 2017:
1.500
Subscribe to
Online Only SUBSCRIPTION
See subscription terms and conditions
* The rates above are available for institutions with less than 20 users or less than 1,000 full time employees
** These special rates apply for members of all national ILAE chapters
• The reference website in epileptology • Access to over 450 videos, EEG
sequences, ...
Please enter my subscription to Epileptic Disorders Online Only for one year
The Educational Journal of the International League Against Epilepsy
BUL.
ABO
.ED/
2019
APE
: 581
4Z •
SIR
ET 3
28 1
95 9
04 0
0037
PERSONAL DETAILS
NAME: ————————————————————————— First Name: ——————————————————————
Department/Hospital: —————————————————————————————————————————————
Address: ————————————————————————————————————————————————————
————————————————————————————————————————————————————————
Postcode: —————————— City: ——————————————— Country: ———————————————————
Tel: ——————————————————————
E-mail: (mandatory) ———————————————————————————————————————————————
Under articles 38 to 40 of the French law of January 6th 1978 addressing computing, files and personal information, you are entitled to access any information referring to you. If you want to modify, edit or delete it, you can contact us.
“Institutions“ subscription: Establishment of less than 20 users or less than 1,000 Full Time Employees
Contact email address (mandatory): ————————————————————————————————————————
Choose your access: r login/passwordr IP recognition
IP address: ——————————————————————————————————————
PAYMENT DETAILS
Please find enclosed my payment for the amount of: —————————— €
r By check payable to John Libbey Eurotext
r By credit card:
r Visa r Eurocard / Mastercard
N°:
Expiry date:
Cryptogram: Signature:
r Please send me an invoicemarked «Paid» for mystatement of professionalexpenses.
• VAT number(compulsory forinstitutions):
———————————————
TO BE COMPLETED AND RETURNED l l l
to Éditions John Libbey Eurotext - 127, Avenue de la République / 92120 Montrouge - France Tel.: +33 1 46 73 06 60 • [email protected] • www.jle.com
The Educational Journal of the INTERNATIONAL LEAGUE AGAINST EPILEPSY
SUBSCRIPTION TERMS AND CONDITIONS6 issues per year in 2019
• Individual subscriptionIndividual subscription must be made in the nameof the recipient and at the personal address withfull advance payment. The payment cannot bemade by an institution, commercial entity orassociation.> The access to the electronic version is
included in your subscription. You will receive your access codes (login and password) by email (please indicate your email address on the subscription form).
• Members subscriptionPlease enclose a proof of your membership.
• Institutional subscriptionInstitutional rates given here apply solelyto non-university hospital centers (HC), clinics,medical centers, associations…The access is limited to a sole establishment of lessthan 20 users or less than 1,000 Full Time Employees on a single location.
For those institutions, the access is possibleat your convenience, either by login and passwordor by IP recognition (you will receive your accesscode to the email address specified in thesubscription form)
A specific license is required for teaching hospitals,universities, research centers or private companies(pharmaceutical industry, etc...). The completeversion of the license is available online on ourwebsite: www.jle.com (heading: Services/License).
To get a customized quotation, institutions shouldcontact specialized agencies or our sales service:[email protected]
www.epilepticdisorders.com
2019RATES
Epileptic Disorders
ISSUES per year + videos
+ 20 years of complete archives
state-of-the-art
tool
6
r 318 € r 671€ r 40 €
Individuals Institutions* Members**
France/EUOther
countries
ONLINE ONLY e-journal only

The Educational Journal of the International League Against Epilepsy
http://www.epilepticdisorders.com
EDITORIAL & PRODUCTION STAFF
MANAGING EDITOR Oliver Gubbay
PUBLICATIONS DIRECTORGilles Cahn
DESK EDITORMarine Rivière
PRODUCT MANAGERArnaud Cobo
ADVERTISING DIRECTORBrigitte Chantrelle-Olliveaud
EDITORS-IN-CHIEF
Alexis A. ArzimanoglouProfessor, Epilepsy Research Coordinator,
Hospital Sant Joan de Déu, Universitat de Barcelona, SpainHead of Department of Paediatric Clinical Epileptology,
Sleep Disorders and Functional Neurology, University Hospitals of Lyon, France
Sándor Beniczky Professor, Aarhus University Hospital, Aarhus, Denmark
Head of Clinical Neurophysiology Department, Danish Epilepsy Centre, Dianalund, Denmark
FOUNDING EDITOR
Jean AicardiParis, France
ILAE EXECUTIVE COMMITTEE
Samuel Wiebe, PresidentCalgary, CanadaAlla Guekht, Vice PresidentMoscow, Russian FederationEdward H. Bertram, Secretary GeneralCharlottesville, VA, USAJ. Helen Cross, TreasurerLondon, UKEmilio Perucca, Past PresidentPavia, Italy
Angelina KakoozaKampala, UgandaAkio IkedaKyoto, JapanEugen TrinkaSalzburg, AustriaChahnez TrikiSfax, TunisiaRoberto CaraballoBuenos Aires, Argentina
Nathalie JettéNew York, NY, USAAstrid NehligParis, FranceMichael SperlingPhiladelphia, PA, USAAlexis ArzimanoglouLyon, FranceAristea GalanopoulouNew York, NY, USA
Shichuo LiBeijing, ChinaNicola MaggioTel Aviv, IsraelXuefeng WangChongqing, ChinaJean GotmanMontreal, CanadaMartin Brodie, IBE PresidentGlasgow, Scotland
Mary Secco, IBE Secretary GeneralLondon, Ontario, CanadaAnthony Zimba, IBE TreasurerLusaka, Zambia
ASSOCIATE EDITORS
Ingmar BlümckeErlangen, GermanyMichael DuchownyMiami, USAYushi InoueShizuoka, Japan
Philippe KahaneGrenoble, FranceRüdiger KöhlingRostock, GermanyMichalis KoutroumanidisLondon, UK
Doug NordliLos Angeles, USALieven LagaeLeuven, BelgiumGuido RubboliDianalund, Denmark
Graeme SillsLiverpool, UKPierre ThomasNice, FranceTorbjörn TomsonStockholm, Sweden
Sarah WilsonMelbourne, Australia
EpilepticDisorders
EDITORIAL BOARD
Nadia Bahi-BuissonParis, FranceCarmen BarbaFlorence, ItalyFabrice BartolomeiMarseille, FranceThomas BastKork, GermanyPatricia BragaMontevideo, UruguayKees BraunUtrecht, The NetherlandsRoberto CaraballoBuenos Aires, ArgentinaMar CarrenoBarcelona, Spain
Francine ChassouxParis, FrancePetia DimovaSofi a, BulgariaDavid DunnIndianapolis, USAAndras FogarasiBudapest, HungaryGiuseppe GobbiBologna, ItalyJean GotmanMontreal, CanadaGregory HolmesVermont, USAHans HolthausenVogtareuth, Germany
Andres KannerMiami, USAKatsuhiro KobayashiOkayama, JapanGaetan LescaLyon, FranceShih-Hui LimSingaporeAndrew LuxBristol, UKStefano MelettiModena, ItalyMohamad MikatiDurham, USAFàbio A. NascimentoTexas, USA
André PalminiPorto Alegre, BrazilGeorgia RamantaniZürich, SwitzerlandAleksandar RisticBelgrade, SerbiaIngrid SchefferMelbourne, AustraliaSanjay SisodiyaLondon, UKMary Lou SmithToronto, CanadaLaura TassiMilan, ItalyChong Tin TanKuala Lumpur, Malaysia
Pierangelo VeggiottiPavia, ItalyAnna Maria VezzaniMilan, ItalyFlavio VillaniMilan, ItalyJo WilmshurstCape Town, South Africa

II
Review article
1 Classifi cation of paroxysmal events and the four-dimensional epilepsy classifi cation systemHans Lüders, Guadalupe Fernandez-Baca Vaca, Naoki Akamatsu, Shahram Amina,Alexis Arzimanoglou, Christoph Baumgartner, Selim R. Benbadis, Andrew Bleasel,Adriana Bermeo-Ovalle, Alireza Bozorgi, Mar Carreño, Michael Devereaux,Stefano Francione, Naiara García Losarcos, Hajo Hamer, Hans Holthausen, Shirin Jamal-Omidi, Giri Kalamangalam, Andrés M. Kanner, Susanne Knake, Nuria Lacuey,Samden Lhatoo, Shih Hui Lim, Luisa V. Londoño, Jayanti Mani, Riki Matsumoto,Jonathan P. Miller, Soheyl Noachtar, André Palmini, Jun Park, Felix Rosenow, Asim Shahid, Stephan Schuele, Bernhard J. Steinhoff, Charles Ákos Szabó, Nitin Tandon, Kiyohito Terada,Walter van Emde Boas, Peter Widdess-Walsh, Philippe Kahane
Original articles
30 EEG of asymptomatic fi rst-degree relatives of patients with juvenile myoclonic, childhood absence and rolandic epilepsy: a systematic review and meta-analysisMariam Tashkandi, Duaa Baarma, Andrea C. Tricco, Cyrus Boelman, Reem Alkhater,Berge A. Minassian
42 DEPDC5 mutation and familial focal epilepsy with variable foci: genotype and phenotype of a familyMarina Aberastury, Romina Fernández, Marta Córdoba, Betiana Comas, Martín Peralta, Guillermo Agosta, Marcelo Kauffman, Walter Silva
48 Quinidine therapy and therapeutic drug monitoring in four patients with KCNT1 mutationsShinsaku Yoshitomi, Yukitoshi Takahashi, Tokito Yamaguchi, Taikan Oboshi, Asako Horino,Hiroko Ikeda, Katsumi Imai, Tohru Okanishi, Mitsuko Nakashima, Hirotomo Saitsu, Naomichi Matsumoto, Jun Yoshimoto, Takako Fujita, Atsushi Ishii, Shinichi Hirose, Yushi Inoue
55 Functional brain connectivity in electrical status epilepticus in sleepSteven H. Mott, Richard P. Morse, Scott A. Burroughs, Ashura W. Buckley, Cristan A. Farmer,Audrey E. Thurm, Susan E. Swedo, Amara L. Krag, Gregory L. Holmes
65 A comprehensive clinico-pathological and genetic evaluation of bottom-of-sulcus focal cortical dysplasia in patients with diffi cult-to-localize focal epilepsyZhong Ying, Irene Wang, Ingmar Blümcke, Juan Bulacio, Andreas Alexopoulos, Lara Jehi,William Bingaman, Jorge Gonzalez-Martinez, Katja Kobow, Lisa Marie Niestroj, Dennis Lal,Konrad Koelble, Imad Najm
78 Neuropsychological correlates of obstructive sleep apnea severity in patients with epilepsyVéronique Latreille, Kim C. Willment, Rani A. Sarkis, Milena Pavlova
VIDEOS ONLINE
VIDEO ONLINE
PublisherÉditions John Libbey Eurotext127, avenue de la République92120 Montrouge, FranceTel.: (+33) (0)1 46 73 06 60Fax: (+33) (0)1 40 84 09 [email protected]://www.jle.com
Publication directorGilles Cahn
Editors-in-ChiefAlexis ArzimanoglouSándor Beniczky
Desk editorMarine Riviè[email protected]
Advertising directorsBrigitte [email protected].: (+33) (0)1 46 73 06 77Noëlle [email protected].: (+33) (0)1 46 73 06 78
Electronic PublishingThomson Digital (Mauritius) Ltd,Île Maurice
PrinterCorlet Imprimeur S.A.Z.I., route de Vire14110 Condé-en-Normandie, FranceN°SubscriptionsABORISISService abonnements John LibbeyBP 5391540 MennecyFranceTél. : (+33) (0)1 84 18 10 [email protected]
Editorial Offi ce SecretariatMr Oliver Gubbayc/o Pr A. ArzimanoglouUniversity Hospitals of Lyon (HCL)Hôpital Femme-Mère-EnfantEpilepsy, Sleep and PediatricNeurophysiology Department59, boulevard Pinel69677 [email protected]
6 issues per year, Institutions France: 671 € TTC.Please fi nd on the order form enclosed,the subscription price list.
Epileptic Disorders is indexed/included in:Index Medicus MEDLINE,EMBASE/Excerpta Medica, Google Scholar, the Science Citation Index ExpandedTN, ISI Alerting Services,Neuroscience Citation Index®, CurrentContents®/Clinical Medicine, and Index Copernicus.
CCPAP: 0920T78801ISSN: 1294-9361ISSN (on line): 1950-6945 Legal deposit: at publication
Papier certifi é PEFC (fi bres issues de forêts gérées durablement)
Origine du papier : France
Taux de fi bres recyclées : 0 %
Eutrophisation : 0,02 kg/T II
Contents Vol. 21, No. 1, February 2019

III
Cover page figure: Cover figure is Figure 4 from the original article by Ying et al. on A comprehensive clinico-pathological and genetic evaluation of bottom-of-sulcus focal cortical dysplasia in patients with difficult-to-localize focal epilepsy (this issue, p. 65–77).
Contents Vol. 21, No. 1, February 2019
Informations for authors: http://www.epilepticdisorders.com
Clinicals commentaries
87 A novel mutation in KCNQ3-related benign familial neonatal epilepsy: electroclinical features and neurodevelopmental outcomeEttore Piro, Rosaria Nardello, Elena Gennaro, Antonina Fontana, Maurizio Taglialatela,Giuseppe Donato Mangano, Giovanni Corsello, Salvatore Mangano
92 Tonic status epilepticus in a centenarian womanJosé L. Fernández-Torre, Javier Riancho, María Martín-García, Gonzalo Martínez-de las Cuevas,Pilar Bosque-Varela
97 Absence status induced by lacosamide adjunctive therapyCharles Ákos Szabó, Lola C. Morgan, Suzanne Sonnenberg, Kameel M. Karkar
102 Focal visual status epilepticusCaspar Stephani, Walter Paulus, Niels K. Focke
108 Rasmussen syndrome: absence seizures may be induced by oxcarbazepineRoberto H. Caraballo, Pedro Cachia, Gabriela Reyes Valenzuela, Agustin Calvo
112 A Rasmussen encephalitis, autoimmune encephalitis, and mitochondrial disease mimicker: expanding the DNM1L-associated intractable epilepsy and encephalopathy phenotypeDanielle A. Nolan, Baibing Chen, Anne Marie Michon, Emily Salatka, Daniel Arndt
117 Berardinelli-Seip syndrome and progressive myoclonus epilepsyDomenico Serino, Chiara Davico, Nicola Specchio, Carlo Efisio Marras, Franco Fioretto
122 Epilepsy surgery in the fi rst months of life: a large type IIb focal cortical dysplasia causing neonatal drug-resistant epilepsyIngo Borggraefe, Moritz Tacke, Lucia Gerstl, Steffen Leiz, Roland Coras, Ingmar Blümcke ,Armin Giese, Birgit Ertl-Wagner, Christian T. Thiel, Soheyl Noachtar, Aurelia Peraud
VIDEO ONLINE
VIDEO ONLINE

The role of EEGin the diagnosis and classification of the epilepsy syndromes:
ROL_
EEG1
8
APE
: 58
14Z
/ SIR
ET :
328
195
904
0003
7
• July 2018
• 17 x 24 cm • 180 pages
• ISBN: 978-2-7420-1562-7
• 56e
This book, written by international experts in clinical epileptology and EEG, comprehensively covers the clinical and EEG features of all paediatric and adult epilepsy syndromes, as reco-gnized by the ILAE.
Each syndrome-chapter provides detailed description of the associated seizure types and the characteristic interictal findings in wakefulness and sleep, illustrated by a plethora of EEG plates. It also includes recording protocols that, adapted to available resources and complete with practical information to improve recording strategies, are designed to maximize diagnostic yield. Finally, the diagnostic confidence of the EEG report is rated according to the findings in hand and the available clinical information.
A fully informative, but concise and easy-to-use, companion in the daily clinical practice for electroencephalographers and EEG technologists, but also a reference guide for epilep- tologists and general neurologists who care for children and adults with epilepsy.
EDITOR
• Michalis Koutroumanidis GSTT, Clin Neurophysiology and Epilepsy, Kings College, London, UK
a tool for clinical practice by the ILAE Neurophysiology Task Force
On the webwww.jle.com(secured payment)
By fax+33 (0) 1 40 84 09 99
By mail
Éditions John Libbey Eurotext127, avenue de la République92120 Montrouge - France
BookshopAt your usual bookseller
For any further information+33 (0) 1 46 73 06 62
To be returned to John Libbey Eurotext - 127, avenue de la République - 92120 Montrouge - France
Please send me
The role of EEG in the diagnosisand classification of the epilepsysyndromes
Handling & postage charges + 6
56
TOTAL
Name ———————————————————————— First name ——————————————————————————
Address ————————————————————————————————————————————————————————
Zip code City——————————————————————— Country ———————————————————
Tel.: ——————————————— E-mail ———————————————————————————————————————
Please send me an invoice marked “Paid” for my statement of professional expensesIn accordance with the French law No. 78-17 of 6 January 1978 relative to information technology, files and freedom ("Loi Informatique et Libertés du 06/01/78"), you are required to providesome personal information for us to process your order. You are free to access, modify, rectify and delete this information. To do so, send us an email at: [email protected] or write to:John Libbey Eurotext, 127 avenue de la République, 92120 Montrouge, France.
PaymentPlease find enclosed my payment for the amount of
By cheque payable to John Libbey Eurotext
By credit card
Visa Eurocard/Mastercard American Express
Carte N°
Last 3 digits on the back of your card
Expiry date Signature :
VAT n° (compulsory for institutions):
ORDER FORM
Book available onwww.jle.com kindle
Ebook available on

do
i:10.
1684
/ep
d.2
019.
1033
Epileptic Disord, Vol. 21, No. 1, February 2019 1
VIDEOS ONLINE
Correspondence:Hans LüdersCase medical Center - Neurology,11100 Euclid Ave Cleveland,Ohio 44106-6058, USA<[email protected]>
Review articleEpileptic Disord 2019; 21 (1): 1-29
Classification of paroxysmalevents and thefour-dimensional epilepsyclassification system
Hans Lüders 1, Guadalupe Fernandez-Baca Vaca 2,Naoki Akamatsu 3, Shahram Amina 4, Alexis Arzimanoglou 5,Christoph Baumgartner 6, Selim R. Benbadis 7,Andrew Bleasel 8, Adriana Bermeo-Ovalle 9, Alireza Bozorgi 10,Mar Carreno 11, Michael Devereaux 12, Stefano Francione 13,Naiara García Losarcos 14, Hajo Hamer 15, Hans Holthausen 16,Shirin Jamal-Omidi 17, Giri Kalamangalam 18,Andrés M. Kanner 19, Susanne Knake 20, Nuria Lacuey 21,Samden Lhatoo 22, Shih Hui Lim 23, Luisa V. Londono 24,Jayanti Mani 25, Riki Matsumoto 26, Jonathan P. Miller 27,Soheyl Noachtar 28, André Palmini 29, Jun Park 30,Felix Rosenow 31, Asim Shahid 32, Stephan Schuele 33,Bernhard J. Steinhoff 34, Charles Ákos Szabó 35, NitinTandon 36, Kiyohito Terada 37, Walter van Emde Boas 38,Peter Widdess-Walsh 39, Philippe Kahane 40
1 Case medical Center - Neurology, Cleveland, Ohio, USA2 University Hospitals Ringgold standard institution - Neurology, Cleveland, Ohio, USA3 International University of Health and Welfare School of Medicine, Department ofNeurology, Narita, Japan4 Kaiser Permanente Northern California, Neuroscience Department, Redwood City,California, USA5 Department of Clinical Epileptology, Sleep Disorders and Functional PediatricNeurology, University Hospitals of Lyon; Member of the European Reference Networkon Rare and Complex epilepsies, ERN EpiCARE, Lyon, France6 Sigmund Freud Privat Universitat Wien Paris Ringgold standard institution, Departmentfor Epileptology and Clinical Neurophysiology, and General Hospital Hietzing withNeurological Center Rosenhuegel, Department of Neurology, Vienna, Austria7 University of South Florida, Department of Neurology, Tampa, Florida, USA8 Westmead Hospital-Neurology, Wentworthville, New South Wales, Australia9 Rush University Medical Center - Department of Neurological Sciences Section ofEpilepsy, Chicago, Illinois, USA10 St. Elizabeth Mercy hospital, - Neurology, Youngstown, Ohio, USA11 Hospital Clínic - Epilepsy Unit, Department of Neurology; Member of the EuropeanReference Network on Rare and Complex epilepsies, ERN EpiCARE, Barcelona, Spain12 Case medical Center – Neurology, Cleveland, Ohio, USA13 Claudio Munari Epilepsy Surgery Centre - Department of Neuroscience, Milan, Italy14 University Hospitals Cleveland Medical Center – Neurology, Cleveland, Ohio, USA15 Epilepsy Center – Neurology, Erlangen, Germany16 Schoen-Klinik Vogtareuth - Neuropediatric Clinic and Clinic for Neurorehabilitation,Epilepsy Center for Children and Adolescents, Vogtareuth, Germany

2
H. Lüders, et al.
17 University Hospital Cleveland Medical Center - Neurology, Epilepsy Center,Cleveland, Ohio, USA18 University of Florida - Department of Neurology, Gainesville, Florida, USA19 University of Miami, Miller School of Medicine - Department of Neurology, Miami,Florida, USA20 Universitatsklinikum Giessen und Marburg - Standort Marburg Ringgold standardinstitution - Epilepsy Center, Neurology, Marburg, Hessen, Germany21 University Hospitals - Neurology (Epilepsy), Cleveland Heights, Ohio, USA22 UT Health Memorial Hermann Hospital, Texas Medical Center - Texas Epilepsy,Neurotechnologies and Neuroinformatics Institute, Houston, Texas, USA23 National Neuroscience Institute Ringgold standard institution – Neurology, SingaporeGeneral Hospital Academia, and Duke-NUS Medical School Ringgold standardinstitution - Academic Development Department, Office of Academic and ClinicalDevelopment, Singapore24 Neuromédica IPS – Epilepsy, Medillin, Colombia25 Kokilaben Dhirubhai Ambani Hospital - Department of Brain and Nervous System,Mumbai, India26 Kobe University Graduate School of Medicine - Division of Neurology, Kobe, Japan27 University Hospitals Case Medical Center/Case Western Reserve University –Neurosurgery, Cleveland, USA28 Ludwig Maximilians University, Munich - Department of Neurology, Epilepsy Center,Munich, Germany29 School of Medicine, Pontificia Universidade Católica do Rio Grande do Sul (PUCRS) -Neurology Service, Porto Alegre Epilepsy Surgery Program, Porto Alegre, Brazil30 Case Medical Center – Pediatrics, Cleveland, Ohio, USA31 Hospital of the Goethe-University Frankfurt am Main - Epilepsy Center FrankfurtRhine-Main, Frankfurt, Germany32 Rainbow Babies & Children’s Hospital, Case Western University School of Medicine –Pediatrics, Cleveland, Ohio, USA33 Northwestern University Feinberg School of Medicine Ringgold standard institution –Neurology, Chicago, Illinois, USA34 Kork - Epilepsy Center, Kehl-Kork, Germany35 UTHSCSA - Neurology, San Antonio and South Texas Comprehensive Epilepsy Center,San Antonio, Texas36 University of Texas Health Science Center – Neurosurgery, Houston, Texas, USA37 National Hospital Organization Shizuoka Institute of Epilepsy and NeurologicalDisorders, Department of Epileptology, Urushiyama, Japan38 Stichting Epilepsie Instellingen Nederland Ringgold standard institution – Neurology,Hoofddorp, Noord-Holland, The Netherlands39 Beaumont Hospital - Department of Neurology, Dublin, Ireland40
Grenoble-Alpes Hospital and University - Neurology Department and GIN INSERMU-1216, Grenoble, FranceReceived October 19, 2018; Accepted January 02, 2019
ABSTRACT – This educational review describes the classification ofparoxysmal events and a four-dimensional epilepsy classification system.Paroxysmal events are classified as epileptic and non-epileptic paroxys-mal events. Non-epileptic events are, in turn, classified as psychogenic andorganic paroxysmal events. The following four dimensions are used to clas-sify epileptic paroxysmal events: ictal semiology, the epileptogenic zone,etiology, and comorbidities. Efforts are made to keep these four dimen-sions as independent as possible.The review also includes 12 educational vignettes and three more detailedcase reports classified using the 2017 classification of the ILAE and thefour-dimensional epilepsy classification. In addition, a case is describedwhich is classified using the four-dimensional epilepsy classification with
Epileptic Disord, Vol. 21, No. 1, February 2019
different degrees of precision by an emergency department physician, aneurologist, and an epileptologist. [Published with video sequences onwww.epilepticdisorders.com]
Key words: classification, semiology, epileptogenic zone, etiology

E
Pw“emeeynOecoOpoanptcreonscFodtteFeits(eafiwg
Go
(mdbt(
ibtbfmhffat
(tl(biaofiaT4F(tspapTio
(keceihia
PSEC
Td
hysicians are frequently called upon to see patientsith paroxysmal events. We use the non-specific term
paroxysmal events” when we do not have sufficientvidence to diagnose with certainty whether a paroxys-al event is epileptic or non-epileptic. The paroxysmal
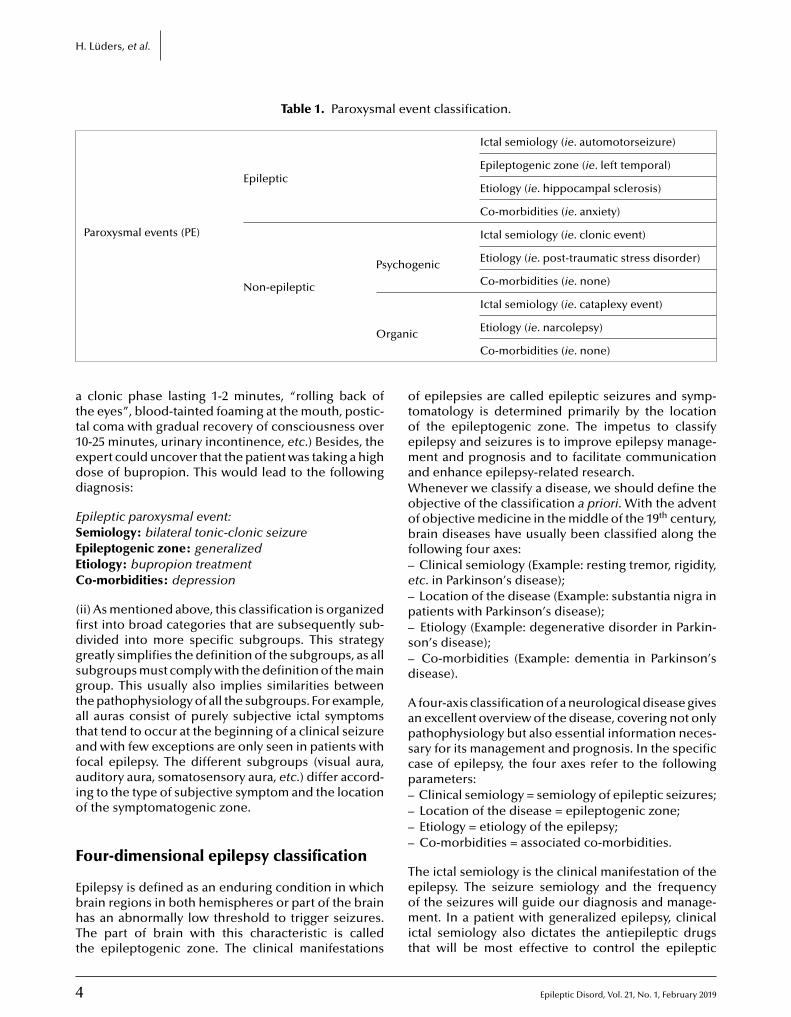
vents we evaluate as physicians, however, may bepileptic or non-epileptic. For this, we divide the parox-smal events into epileptic paroxysmal events andon-epileptic paroxysmal events (table 1).nce we have diagnosed that a paroxysmal event is
pileptic in nature, we define the four dimensions thatharacterize epileptic paroxysmal events: ictal semiol-gy, epileptogenic zone, etiology, and comorbidities.n the other hand, if we diagnose non-epileptic
aroxysmal events, we classify these as psychogenicr organic paroxysmal events. Once we confirm thatnon-epileptic paroxysmal event is psychogenic in
ature, we define the following three dimensions:aroxysmal event semiology, etiology, and comorbidi-
ies. In this case, we use the same semiological seizurelassification used for epileptic events (see below) buteplace the expression “seizure” by “event” and thexpression “aura” by “aura event.” The classificationf non-specific “paroxysmal events” (physician doesot know if the event is epileptic or not) follows theame system as the classification of non-epileptic psy-hogenic paroxysmal events.inally, if we diagnose a patient with a non-epilepticrganic paroxysmal event, we also specify the threeimensions: semiology, etiology, and comorbidities. In
his case, however, the semiology is defined by theype of non-epileptic, non-psychogenic event as, forxample, syncope, resting tremor, cataplexy, etc.ollowing a detailed description of the 4-dimensionalpilepsy classification presented below, we also
ncluded 12 educational vignettes (Appendix 1) andhree more detailed case reports (Appendix 2) clas-ified using the 2017 classifications of the ILAEFisher et al. 2017; 2017b) and the four-dimensionalpilepsy classification described below. In addition,
case is described (Appendix 3) which is classi-ed using the four-dimensional epilepsy classificationith different degrees of precision by an emer-ency department physician, a neurologist, and an
epileptologist.
eneral organizationf the classification system
1) The dimensions that characterize all the paroxys-al events are independent and defined by different
pileptic Disord, Vol. 21, No. 1, February 2019
iagnostic methods. For example, a patient may haveilateral clonic seizures (defined by semiology), but
he MRI shows an extensive left fronto-temporal tumorepileptogenic lesion) and the epileptogenic zone
tcsi
Four-dimensional epilepsy classification
s most likely the left frontal lobe (mainly definedy interictal and ictal EEG). The independence of
he different dimensions allows precise correlationsetween the different dimensions. For example, the
our-dimensional classification of the epilepsies per-its us to calculate the percentage of patients who
ave no focal ictal findings by semiology but have aocal epilepsy. Because of the independence of theour dimensions, the classification system permits anlmost infinite number of correlation studies betweenhe different subgroups included in each dimension.
2) The classifications of the paroxysmal events andhe four dimensions that classify the epilepsies fol-ow the same hierarchal system. The target parameternamely one of the dimensions) is first subdivided intoroad categories and each of them is again subdivided
nto more specific subcategories. In many cases, thesegain are subdivided into even smaller categories. Inther words, as we move from “left to right”, wend that the dimension is progressively defined moreccurately.he tables that follow provide a global overview of the-dimensional classification.or example, in the classification of paroxysmal eventstable 1), the broadest category is “paroxysmal event”hat includes all the subcategories mentioned, and theecond broadest category is epileptic vs non-epilepticaroxysmal events. Non-epileptic paroxysmal eventsre, in turn, subdivided into psychogenic and organicaroxysmal events.he purpose of organizing the different categories
nto progressively smaller subgroups has the followingbjectives:
i) Non-specialists who do not have the tools andnowledge to make a very precise classification of thepilepsies or other paroxysmal events can still use thislassification system by just defining the broadest cat-gories (“on the left hand of the table”). For example,
f they just know that the patient was depressed andad a paroxysmal episode with generalized “twitch-
ng”, unresponsiveness, and no memory for the eventfterwards, they can classify the event as follows:
aroxysmal event:emiology: bilateral clonic event (LOC)tiology: unknowno-morbidities: depression
he same patient seen by an expert might obtain aetailed history from a family member who witnessed
3
he seizure. The expert could elucidate semiologi-al details that make the probability of an epilepticeizure much more likely (initial ictal cry, tonic phasen decerebrate posture lasting 30 seconds followed by
4
H. Lüders, et al.
Table 1. Paroxysmal event classification.
Paroxysmal events (PE)
Epileptic
Ictal semiology (ie. automotorseizure)
Epileptogenic zone (ie. left temporal)
Etiology (ie. hippocampal sclerosis)
Co-morbidities (ie. anxiety)
Non-epileptic
Psychogenic
Ictal semiology (ie. clonic event)
Etiology (ie. post-traumatic stress disorder)
Co-morbidities (ie. none)
rgan
Ictal semiology (ie. cataplexy event)
att1edd
ESEEC
(fidgsgtatafaio
F
EbhTt
otoemaWoobf–e–p–s–d
Aapscp––––
T
O
clonic phase lasting 1-2 minutes, “rolling back ofhe eyes”, blood-tainted foaming at the mouth, postic-al coma with gradual recovery of consciousness over0-25 minutes, urinary incontinence, etc.) Besides, thexpert could uncover that the patient was taking a highose of bupropion. This would lead to the followingiagnosis:
pileptic paroxysmal event:emiology: bilateral tonic-clonic seizurepileptogenic zone: generalizedtiology: bupropion treatmento-morbidities: depression
ii) As mentioned above, this classification is organizedrst into broad categories that are subsequently sub-ivided into more specific subgroups. This strategyreatly simplifies the definition of the subgroups, as allubgroups must comply with the definition of the mainroup. This usually also implies similarities betweenhe pathophysiology of all the subgroups. For example,ll auras consist of purely subjective ictal symptomshat tend to occur at the beginning of a clinical seizurend with few exceptions are only seen in patients withocal epilepsy. The different subgroups (visual aura,uditory aura, somatosensory aura, etc.) differ accord-ng to the type of subjective symptom and the locationf the symptomatogenic zone.
our-dimensional epilepsy classification
pilepsy is defined as an enduring condition in whichrain regions in both hemispheres or part of the brainas an abnormally low threshold to trigger seizures.he part of brain with this characteristic is calledhe epileptogenic zone. The clinical manifestations
eomit
ic Etiology (ie. narcolepsy)
Co-morbidities (ie. none)
f epilepsies are called epileptic seizures and symp-omatology is determined primarily by the locationf the epileptogenic zone. The impetus to classifypilepsy and seizures is to improve epilepsy manage-ent and prognosis and to facilitate communication
nd enhance epilepsy-related research.henever we classify a disease, we should define the
bjective of the classification a priori. With the adventf objective medicine in the middle of the 19th century,rain diseases have usually been classified along the
ollowing four axes:Clinical semiology (Example: resting tremor, rigidity,
tc. in Parkinson’s disease);Location of the disease (Example: substantia nigra in
atients with Parkinson’s disease);Etiology (Example: degenerative disorder in Parkin-
on’s disease);Co-morbidities (Example: dementia in Parkinson’s
isease).
four-axis classification of a neurological disease givesn excellent overview of the disease, covering not onlyathophysiology but also essential information neces-ary for its management and prognosis. In the specificase of epilepsy, the four axes refer to the followingarameters:Clinical semiology = semiology of epileptic seizures;Location of the disease = epileptogenic zone;Etiology = etiology of the epilepsy;Co-morbidities = associated co-morbidities.
he ictal semiology is the clinical manifestation of the
Epileptic Disord, Vol. 21, No. 1, February 2019
pilepsy. The seizure semiology and the frequencyf the seizures will guide our diagnosis and manage-ent. In a patient with generalized epilepsy, clinical
ctal semiology also dictates the antiepileptic drugshat will be most effective to control the epileptic

E
Four-dimensional epilepsy classification
stpc
Dm–qm–oi
Eg
Feearee
Adcwnps“n“dddlsgifi2aebo
C(
Ilat
Table 2. Semiology classification of low complexity.
Auras*
Autonomic seizures*
Dyscognitive seizures
Motor seizures*
Special seizures
Tu
gtiscotwieptcgcvs
Ts–cc–tatcpgsg–acct
eizures. In a patient with focal seizures refractoryo medical treatment, ictal semiology is an importantiece of information to decide if a patient is a surgicalandidate.
efinition of the epileptogenic zone is essential in theanagement of the epilepsy:Focal epilepsies and generalized epilepsies fre-
uently respond best to different types of antiepilepticedication.For surgical treatment of epilepsy, precise definition
f the location and extent of the epileptogenic zone isndispensable.
tiology in most patients is another essential factor thatuides treatment and prognosis.
inally, knowledge of the main co-morbidities isssential to get a complete picture of the patient’s dis-ase, particularly cognitive impairment and psychiatricbnormalities. Besides, comorbidities such as severeenal, hepatic or psychiatric disease may greatly influ-nce the type and dose of antiepileptic drug to use, orven the patient’s candidacy for surgery.
s already mentioned above, it is essential that theifferent dimensions in a multi-dimensional classifi-ation system be as independent as possible. In otherords, classifying one category in a dimension shouldot automatically define another dimension. For exam-le, classifying the epileptogenic zone as “regional”hould not force a classification of the seizure asfocal” and classifying the seizure as “focal” shouldot force classification of the epileptogenic zone asregional.” Ideally, to achieve independence in eachimension, the tests and criteria we use to define eachimension should not overlap. Independence of theifferent dimensions also allows us to evaluate corre-
ations between them. Example: type of semiologicaleizures in patients with temporal or frontal epilepto-enic zones. A typical example of violating this rule of
ndependence is the latest version of the ILAE classi-cation of epilepsies and seizures (Fisher et al., 2017a,017b; Berg et al., 2010). In this classification system,ll test results are used to classify both seizures andpilepsies. Therefore, no correlation studies are possi-le, and defining the seizures already defines the typef epilepsy.
lassification of epileptic seizuresRefer to tables 2, 3, 4, 5, 6, 7)
pileptic Disord, Vol. 21, No. 1, February 2019
nvestigators have taken the highly successful and bio-ogically relevant classification of plants and animalss a template to create a similarly relevant classifica-ion of epileptic seizures. Plants and animals contain
saiwc
Asymptomatic EEG seizure
he asterisk (*) indicates that a somatotopic modifier may besed to specify the category with more precision (see Table 6).
enetic evolutionary information that naturally leadso a biologically relevant classification. However, it ismpossible to develop a “biologically relevant clas-ification” system for objects, since the informationontained within varies, for example, wooden boxesf different shapes, sizes, and colors. This fundamen-
al difference in the subject matter largely explainshy the ILAE’s Committees have been unsuccessful
n developing a biologically relevant classification ofpileptic seizures that is similar to the classification oflants and animals by Linnaeus. On the other hand,
he semiological characteristics of epileptic seizuresontain highly relevant information regarding the ori-in and spread of epileptic discharges. Therefore, anylassification of epileptic seizures should maximize thealue of seizure semiology regarding the origin andpread of epileptic discharges.
he following principles guide the semiologicaleizure classification:
Epileptic seizures are broken down into “seizureomponents”. Each seizure consists of 1-4 seizureomponents.
Seizure components consist of sets of ictal symp-oms that have semiological similarities and frequentlycommon pathophysiology, such as a common symp-
omatogenic zone. In other words, each seizureomponent tends to be triggered by a defined patho-hysiology, i.e. activation of a defined symptomato-enic zone in most cases. For example: a left-handomatosensory aura corresponds to a symptomato-enic zone in the right hemispheric hand S1 area.
The different seizure components are linked insequence by arrows. The “sequence of seizure
omponents”, with each seizure component usuallyorresponding to a more or less clearly defined symp-omatogenic zone, elucidate the most likely seizure
5
pread. For example, the seizure of a patient having anura of flashing lights in the left visual field, progress-ng to a sensation of nausea, chewing automatismsith loss of contact, and eventually becoming bilateral
lonic, would be classified as follows:

6 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
Table 3. Semiology classification of moderate complexity.
Auras *
Auditory aura*
Autonomic aura Abdominal Aura
Gustatory aura
Olfactory aura
Psychic aura
Somatosensory aura*
Vestibular aura
Visual aura*
Autonomic seizure*
Bradycardic seizure
Emetic seizure
Sialorrheic seizure
Tachycardic seizure
Urinary seizure
Dyscognitive seizure
Aphasic seizure
Akinetic seizure
Dialeptic seizure
Motor seizure*
Simple motor seizures*
Clonic seizure*
Epileptic spasm*
Myoclonic seizure*
Tonic seizure*
Tonic-clonic seizure*
Versive seizure*
Complex motor seizures
Automotor seizure
Gelastic seizure
Hypermotor seizure
Special seizures
Astatic seizure
Atonic seizure
Central apneic seizure
Hypnopompic seizure
Hypomotor seizure
Negative myoclonic seizure*
Asymptomatic EEG seizure
The asterisk (*) indicates that a somatotopic modifier may be used to specify the category with more precision (see Table 6).

Epileptic Disord, Vol. 21, No. 1, February 2019 7
Four-dimensional epilepsy classification
Table 4. Semiology classification of high complexity.
Aura* Auditory aura
Autonomic aura
Abdominal aura Choking aura
Diaphoretic aura *
Dipsosic aura
Pilomotor aura *
Sialorrheic aura
Tachycardic aura
Urinary aura
Vasomotor aura*
Gustatory aura
Olfactory aura
Psychic aura Affective aura Pleasure aura
Ecstasy aura
Religious aura
Sexual aura
Unpleasant aura
Anger aura
Depression/Sadness aura
Embarrassment aura
Fear/Panic aura
Guilt aura
Cognitive aura
Experiential aura
Familiarity auraDéjà-vu aura
Jamais-vu aura
lllusionary aura
Auditory aura
Body aura
Time aura
Visual aura
Somatosensory aura*
Vestibular aura
Visual aura* Ictal blindness*
Autonomicseizures*
Abdominal seizure
Anisocoric seizure*
Bradycardic seizure
Emetic seizure
Fecal incontinence seizure

8 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
Table 4. Semiology classification of high complexity. (Continued)
Autonomic seizure* Hippus seizure
Hyperhydrotic seizure*
Hypertensive seizure
Lacrimatory seizure
Pilomotor seizure*
Sexual seizure
Sialorrheic seizure
Tachycardic seizure
Urinary seizure
Vasomotore seizure*
Dyscognitive seizures
Amnestic seizure
Aphasic seizure
Akinetic seizure
Dialeptic seizure
Motor seizures*
Simple motor seizure*
Clonic seizure*
Epileptic spasm*
Myoclonic seizure*
Nystagmoid seizure*
Tonic seizure*
Tonic-clonic seizure*
Versive seizure*
Vocalization seizure
Complex motor seizure
Alien limb seizure
Automotor seizure
Dacrystic seizure
Gelastic seizure
Hypermotor seizure Emotional hypermotor seizure
Kissing seizure
Singing seizure
Spitting seizure
Verbalization seizure

E
Four-dimensional epilepsy classification
Table 4. Semiology classification of high complexity. (Continued)
Special seizures
Astatic seizure
Atonic seizure*
Central apneic seizure
Fear facies seizure
Hypnopompic seizure
Hypomotor seizure
Negative myoclonic seizure*
Water drinking seizure
T d to specify the category with more precision (see Table 6).
(t
FssiTo“w1(
Tttpclbd
IcFm
Eenbstpee
Table 5. Lateralizing signs.
Automotor seizures with no dialepsis
Asymmetric ending seizure (1)Clonic seizure*
Versive seizure*
Early head deviation*
Figure of 4*
Ictal dystonia*
Ictal speech
Ictal unilateral automatisms*
Ictal unilateral blinking*
Immediate postictal speech
M2e sign*
Postictal aphasia
Postictal hemiparesis*
Postiictal nose wiping*
Unilateral pupillary dilation*
The asterisk (*) indicates that a somatotopic modifier may beused to specify the category with more precision (see Table 6).(oe
Asymptomatic EEG seizure
he asterisk (*) indicates that a somatotopic modifier may be use
1) left visual aura → (2) abdominal aura → (3) automo-or (LOC) → (4) bilateral clonic seizure
rom this classification, we would assume that theeizure started in the right calcarine gyrus and thenpread into the mesial temporal region before becom-ng generalized.he concept that seizures spread and that the semi-logical evolution of epileptic seizures reflects themarch” of the epilepsy over the cortical surfaceas already applied first by Bravais in 1827 (Bravais,
827) and was later adopted and expanded by JacksonTaylor, 1958).
he semiological seizure classification assumes alsohat clinical seizures may sometimes remain limitedo the first component (left visual aura in the exam-le given above, under Point 3), occasionally spread toomponent 2 or 3 (left hand clonic component andeft versive component in the example above), andecome bilateral only rarely. If needed, this can beocumented as follows:
ctal semiology: (1) left visual aura → (2) left handlonic → (3) left versive → (4) bilateral clonic seizurerequency: (1) one/week; (2) one/month; (4) one/sixonths
xclusively subjective components are followed by thexpression “aura” (examples: left visual aura, abdomi-al aura). All the objective components are expressedy adjectives (example: left hand clonic, right ver-
pileptic Disord, Vol. 21, No. 1, February 2019
ive) except the last component that is followed byhe expression “seizure” when classifying an epilepticaroxysmal event, or “event” when classifying a non-pileptic event or a paroxysmal event that could bepileptic or non-epileptic.
Ifafia
1) Asymmetric ending seizures refer to an asymmetric endingf bilateral tonic-clonic seizure or bilateral clonic seizure withither unilateral clonic jerks or a version.
9
n the semiological seizure classification, a total ofour seizure components are allowed. This restrictionvoids excessive detail that might make the classi-cation impractical. However, additional signs thatdd lateralizing/localizing power may be added to the

1
H. Lüders, et al.
Table 6. Somatotopic modifiers.
Bilateral
Bilateral asymmetric
Left
Right
Axial
Throat
Head
Face
Eyes
Eyelid
Lips
Tongue
Hand
Arm
Trunk
Abdomen
Leg
ccBcpuie
IcFmL
Lsotwosa
Table 7. Seizure triggering factors.
Alcohol withdrawal
Auditory
Music
Sounds
Voices
Complex cognitive
Eating
Hypoglycemia
Hyperventilation
MovementActive movement
Passive movement
Reading
Somatosensory
Sleep
Sleep deprivation
Startle
Visual
Flash
Pattern evoked
csaccl
IcsFm
Aoosbe identified. Triggers that can elicit epileptic seizures
Foot
lassification. In the example of Point 3 above, weould add a “left Todd’s paralysis” as a lateralizing sign.esides, if a seizure consists of more than four seizureomponents and some of the redundant seizure com-onents have lateralizing value, they can be listednder “lateralizing signs”. Table 5 shows the lateral-
zing signs that can be identified during epileptic ictalvents.
ctal semiology: (1) left visual aura → (2) left handlonic → (3) left versive → (4) bilateral clonic seizurerequency: (1) one/week; (2) one/month; (4) one/sixonths
ateralizing signs: left Todd’s paralysis; left face tonic
oss of consciousness (LOC), defined as relative unre-ponsiveness associated with amnesia for the episodef unresponsiveness, is an essential semiological fea-
ure. In previous classifications, loss of consciousness
0
as the main factor dividing focal seizures into simpler complex partial seizure (Bancaud et al., 1981). In theemiological seizure classification, LOC is indicated bydding the notation “(LOC)” following the first seizure
accf
Eye closure
Sensitivity-off
omponent for which the patient is relatively unre-ponsive and amnestic. In the example shown above“(LOC)” will be inserted after the left hand clonic
omponent if the patient was unresponsive during thelonic seizure component and does not remember theeft clonic movements:
ctal semiology: (1) left visual aura → (2) left handlonic (LOC) → (3) left versive → (4) bilateral cloniceizurerequency: (1) one/week; (2) one/month; (4) one/sixonths
ll epileptic seizures develop as the consequence ofne or more triggers that lower the epileptic thresh-ld. In most cases, these triggers are unknown. Inome patients, however, a clearly defined trigger may
Epileptic Disord, Vol. 21, No. 1, February 2019
re shown in table 7. Triggers are listed in the seizurelassification; as shown below. The approximate per-entage of seizures provoked by the trigger is listedollowing each trigger.

Epileptic Disord, Vol. 21, No. 1, February 2019
Table 8. Epileptogenic zone classification.
Generalized
FocalHemis-phere* Temporal*
Lateral temporal*
Mesial temporal*
Temporal pole*
Basal temporal*
Frontal*
Prefrontal lateral*
Prefrontal mesial*
Basal frontal*
Premotor lateral*
Premotor mesial*
Central*Centro-temporal*
Mesial central*
Parietal*Mesial parietal*
Lateral parietal*
Occipital*Lateral occipital*
Mesial occipital*
Cingulate*
Anterior cingulate*
Mid cingulate*
Posterior cingulate*
Insula*Anterior insula*
Posterior insula*
Multifocal
Unknown
The asterisk (*) implies that a left or right modifier can be addedto the specified brain area (example: right mesial occipital).
Table 9. Etiology classification.
Structural*
Genetic
Inflammatory
Infectious
Unknown
The asterisk (*) indicates that a brain region may be defined tospecify the category with more precision (see Table 8).
EIFT
TscssgatsLgf(triwgcm–s–cjmtp–bEsnsa(–cpcaav
Mrohto
Four-dimensional epilepsy classification
xample:ctal semiology: automotor seizure (LOC)requency: one/monthrigger: music (100%)
ables 2, 3, 4 show the seizure classification. Table 2hows the broadest categories, which are less pre-ise and more useful to non-neurologists. Table 3hows more detailed seizure components that can beeen during epileptic ictal events. General neurolo-ists should have enough ictal semiology training topply this degree of semiological precision. Finally,able 4 shows the maximum semiological detail andhould be used primarily by epileptologists.ike other dimensions, the seizure components arerouped in major sets that share similar semiologicaleatures and frequently also a similar pathophysiologyexample: auras that all consist of subjective symptomsend to occur at the beginning of a seizure and are theesult of epileptiform dysfunction of a relatively lim-ted cortical territory). As we move from left to right
ithin the table, we find that the dimension is pro-ressively defined more accurately. Having “seizureomponents” that cover all possible ictal semiologicalanifestations has significant advantages:By design of the seizure components, any epileptic
eizure can be classified semiologically.Breaking down the seizure symptoms into seizure
omponents and expressing the seizure evolution byoining different seizure components by an arrow
akes it possible not only to classify the seizure symp-omatology, but also to express the infinite possibleatterns of evolution.
The same semiological seizure classification cane applied to classify newborns, children, and adults.pileptologists only need to be aware that certaineizure components do not occur or cannot be diag-osed for certain age groups (for example, automotoreizures do not occur until age three years, and aurasre not, or cannot, be reported until age 3-5 years)Fernandez-Baca Vaca et al., 2018).
The same classification system can also be used tolassify other paroxysmal episodes and non-epilepticsychogenic paroxysmal episodes. However, in thisase, the expression “aura” is replaced by “aura event”,nd the expression “seizure” is replaced by “event”t the end of the sequence of components (example:isual aura (bilateral clonic event).
any of the auras, seizure components or seizuresequire a somatotopic modifier to define the semiol-
11
gy precisely. Examples include left visual aura, rightand somatosensory aura, and bilateral asymmetric
onic seizure component. Auras, seizure components,r seizures that may be modified by a somatotopic

1
H
mTu“msta
Tasdas
C
TctnfdttfTftlvfscpcrt
E
Itmmc2eeatStan
GdIoImsU
Gs–togarb–ioi(m
EEo––––
SwepdtaioilosAebcias precisely and objectively as possible, the character-
. Lüders, et al.
odifier are indicated by an asterisk in tables 2, 3, 4.able 6 shows the somatotopic modifiers that aresed. For some auras, seizure components or seizures,left” or “right” may only be used as a somatotopicodifier (example: left auditory aura). Other auras,
eizure components or seizures allow a detailed soma-otopic modifier (example: left hand somatosensoryura).
ables 2, 3, 4 also include a sixth category, labelleds “asymptomatic EEG seizures.” In the epilepsy clas-ification, the “epileptogenic zone” will essentiallyefine the location of the EEG seizure. This categorylso allows specification of the frequency of the EEGeizures.
lassification of the epileptogenic zone (table 8)
he epileptogenic zone is defined as the minimalortical region that must be resected, disconnected,hermo-coagulated, thermo-ablated or desynchro-ized by multiple transections to produce seizure
reedom. It cannot be determined directly but it iseduced by outlining related cortical areas, including
he irritative zone, the seizure onset zone, the epilep-ogenic lesion, the symptomatogenic zone, and theunctional deficit zone.he epileptogenic zone can also be defined with dif-erent degrees of precision; for example, by just listinghe abnormal hemisphere (left or right), one or twoobes (left fronto-temporal, right occipital) or subdi-ision of one lobe (left mesial temporal lobe, rightusiform gyrus, etc.). Obviously, without performingurgery, the exact location of the epileptogenic zoneannot be determined with certainty. Besides, if aatient becomes seizure-free after surgery, it only indi-ates that the epileptogenic zone is a subset of theesected cortex; it does not mean that all the resectedissue is part of the epileptogenic cortex.
tiological classification (table 9)
n this classification system, special attention is giveno classification of the etiology of each epilepsy with
aximum precision depending on the available infor-ation. The etiology is subdivided into five broad
ategories, as suggested by the ILAE (Scheffer et al.,017). For each patient, however, the most detailedtiology is indicated in parenthesis. It is the detailedtiological classification that permits the clinician tossociate a specific etiology with a specific medicalherapy or epilepsy surgery.
2
tructural refers to causes for which the seizures arehe direct result of an abnormal underlying brainnatomy. Structural lesions are usually diagnosed byeuroimaging, commonly high-resolution MRI.
ifHa
enetic refers to causes for which the seizures are airect result of a known or presumed genetic error.
nfectious refers to causes for which the developmentf seizures is the result of post-infectious processes.
nflammatory refers to causes for which the develop-ent of seizures is immune-mediated central nervous
ystem inflammation.nknown.
eneral principles guiding the etiology of epilepticeizures:In all patients, the etiology of the seizures is multifac-
orial, including at least one (and sometimes more thanne) main etiological factor (example: left parietal gan-lioglioma) and a number of contributing factors, suchs susceptibility genes. As genetic testing becomesoutine, the multi-etiological nature of epilepsy willecome more evident.
In general, for patient management, just specify-ng the broad main etiological category is of no ornly minimal value. Therefore, we encourage the spec-
fication of the most precise category in each caseexample: left middle cerebral artery infarction; SCN1A
utation).
pileptic syndromespilepsy syndromes consist of specific constellationsf:semiologiesEEG abnormalitiescomorbiditiesetiologies
yndromes were defined by astute epileptologistsho realized that the correct identification of anpilepsy syndrome was often helpful to determinerognosis and treatment. Syndromes, however, are, byefinition, empirical and artificial. Modern diagnostic
echniques including MRI and genetic testing nowllow precise diagnosis of epilepsy causes, thereforedentification of syndromes is less important than itnce was (Kellinghaus et al., 2004), although several still
mpact therapy decisions (e.g. West syndrome, self-imited Rolandic epilepsy, juvenile myoclonic epilepsy)r have relevance to genetic research (e.g. Dravetyndrome).s diagnostic technology and knowledge aboutpilepsy improve, it is likely that more syndromes willecome obsolete in the near future. The emphasis of alassification scheme should not be to preserve a set ofncreasingly archaic conventions, but rather to define,
Epileptic Disord, Vol. 21, No. 1, February 2019
stics of each individual case of epilepsy in order toacilitate discovery of new etiologies.owever, for many decades, classic epileptology
ssumed that identification of an epilepsy syndrome

E
wipmwroe
EESbTEeEC
DN
R
BDeFo1
Barn
Bl
FJE
FfE
FopT
KetE
Sof the epilepsies: position paper of the ILAE Commis-sion for Classification and Terminology. Epilepsia 2017; 58:512-21.
Taylor J. Selected Writings of John Hughlings Jackson. NewYork: Basic books, Inc, 1958.
as the diagnostic gold standard. Besides, there arennumerable publications on study treatment andrognosis for different epileptic syndromes. This infor-ation is useful for management of epileptic patientsho have a well-defined epilepsy syndrome. This is the
eason why we decided to include the syndrome as anption in parenthesis following the definition of thepileptogenic zone.
xample:pileptic paroxysmal eventemiology: bilateral myoclonic → bilateral clonic →ilateral tonic-clonic seizurerigger: sleep deprivation, alcohol withdrawalpileptogenic zone: generalized (juvenile myoclonicpilepsy)tiology: geneticomorbidities: none �
isclosures.one of the authors have any conflict of interest to declare.
Legend for video sequences
– Video Case 1 (Appendix 2)– Video Case 2 (Appendix 2)– Video Case 3 (Appendix 2)
Key words for video research onwww.epilepticdisorders.com
Video case 1
Phenomenology: bilateral tonic-clonic seizureLocalisation: insula (right)Syndrome: focal structuralAetiology: unknown
Video case 2
Phenomenology: hypermotor, emotionalLocalisation: frontal (left)Syndrome: focal (structural and genetic)Aetiology: cortical dysplasia
Video case 3
pileptic Disord, Vol. 21, No. 1, February 2019
Phenomenology: tonic (bilateral asymmetric);clonic (bilateral)Localisation: unknownSyndrome: neonatal (familial); generalisedAetiology: genetic
Four-dimensional epilepsy classification
eferences
ancaud J, Henriksen O, Rubio-Donnadieu F, Seino M,reifuss FE, Penry JK. Proposal for revised clinical andlectroencephalographic classification of epileptic seizures.rom the Commission on Classification and Terminologyf the International League Against Epilepsy. Epilepsia981; 22: 489-501.
erg AT, Berkovic SF, Brodie MJ, et al. Revised terminologynd concepts for organization of seizures and epilepsies:eport of the ILAE Commission on Classification and Termi-ology, 2005-2009. Epilepsia 2010; 51: 676-85.
ravais F. Recherches sur les symptomes et le traitment de’epilepsie hemiplegique. Faculte de Medicine de Paris, 1827.
ernandez-Baca Vaca G, Mayor C, Garcia Losarcos N, Park, Lüders HO. Seizure semiology in different age groups.pileptic Disord 2018; 20: 179-88.
isher RS, Cross JH, D’souza C, et al. Instruction manualor the ILAE 2017 operational classification of seizure types.pilepsia 2017a; 58: 531-42.
isher RS, Cross JH, French JA, et al. Operational classificationf seizure types by the International League Against Epilepsy:osition paper of the ILAE Commission for Classification anderminology. Epilepsia 2017b; 58: 522-30.
ellinghaus C, Loddenkemper T, Najm IM, et al. Specificpileptic syndromes are rare even in tertiary epilepsy cen-ers: a patient-oriented approach to epilepsy classification.pilepsia 2004; 45: 268-75.
cheffer IE, Berkovic S, Capovilla G, et al. ILAE classification
13

14 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
Appendix 1. PRACTICAL EXERCISE: CLASSIFICATION USING THE ILAE ANDPAROXYSMAL EVENT FOUR-DIMENSIONAL EPILEPSY CLASSIFICATION
In this exercise, we replicate the 12 vignettes that Fisher et al. (2017a) included in their “Instructor manual forthe ILAE operational classification of sensory types”. The exercise consists of classifying the epilepsy in eachcase using the ILAE system and the paroxysmal event and four-dimensional system.
When classifying using the ILAE system, the following four levels must be specified:a. Seizure typeb. Epilepsy typec. Epilepsy syndromed. Etiology
When classifying using the paroxysmal event and four-dimensional epilepsy classification, the followingdimensions must be defined:Paroxysmal event type:I. Ictal semiology
II. Epileptogenic zoneIII. EtiologyIV. Co-morbidities
The answers for each case, under both systems of classification, with comments can be found after eachvignette.
CASE 1: Unknown-onset tonic-clonic
A woman awakens to find her husband having a seizure in bed. The onset is not witnessed, but she is ableto describe bilateral stiffening followed by bilateral shaking. EEG and MRI findings are normal. This seizure isclassified as unknown-onset tonic-clonic. There is no supplementary information to determine whether theonset was focal or generalized. Under the old classification, this seizure would have been unclassifiable withno further qualifiers.
ILAE classification
a. Seizure type: unknown onset tonic-clonic seizureb. Epilepsy type: unknownc. Epilepsy syndrome: N/Ad. Etiology: unknown
Paroxysmal event and four-dimensional epilepsy classification
Paroxysmal eventI. Event semiology: bilateral tonic-clonic event
II. Etiology: unknownIII. Co-morbidities: none
Comments
The vignette does not contain sufficient information to reliably diagnose epilepsy. A detailed anamnesis mostlikely would have been sufficient to make a reliable diagnosis if the patient had an epileptic seizure or not.The interview should provide an answer to the following questions:
� Duration of “stiffening” and of “bilateral shaking”?
� Eyes open or closed?
� Did the eyes “roll back”?
� Was there foaming at the mouth?

Epileptic Disord, Vol. 21, No. 1, February 2019 15
Four-dimensional epilepsy classification
� Was there blood anywhere?
� Did he wet himself?� What happened after the shaking was over? Was there stertorous hyperventilation? How long did it takehim to recover consciousness?� Did he complain of muscle ache the following day? Did his tongue hurt? Where did he bite his tongue?
CASE 2: Focal-onset bilateral tonic-clonic
In an alternate scenario of Case 1, the EEG shows a clear right parietal slow-wave focus. The MRI shows aright parietal region of cortical dysplasia. In this circumstance, the seizure can be classified as focal to bilateraltonic-clonic, despite the absence of an observed onset, because a focal etiology has been identified, and theoverwhelming likelihood is that the seizure had a focal onset. According to the old classification, this seizurewould have been classified as partial onset, secondarily generalized.
ILAE classification
a. Seizure type: focal to bilateral tonic-clonic seizureb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: genetic and structural
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: bilateral tonic-clonic seizure
II. Epileptogenic zone: right parietalIII. Etiology: right parietal cortical dysplasiaIV. Co-morbidities: none
Comments
Now even without a detailed clinical history, the chances that the patient has an epileptic seizure are extremelyhigh. Therefore, we now classify the event as an epileptic paroxysmal event.
CASE 3: Absence
A child is diagnosed with Lennox-Gastaut syndrome of unknown etiology. EEG shows runs of slow spike-waves.Seizure types include absence, tonic, and focal motor seizures. The absence seizures are prolonged, haveindistinct onset and cessation, and sometimes result in falls. In this case, the absence seizures are classified asatypical absence due to their characteristics, the EEG pattern, and underlying syndrome. The absence seizureswould have had the same classification in the old system.
ILAE classification
a. Seizure type: atypical absence, tonic seizure, focal motor seizureb. Epilepsy type: combined generalized and focalc. Epilepsy syndrome: Lennox-Gastaut syndromed. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: tonic seizure, dialeptic seizure, motor seizure
II. Epileptogenic zone: generalized (Lennox-Gastaut syndrome)III. Etiology: unknownIV. Co-morbidities: intellectual disability

16 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
Comments
Again, the amnestic information provided in the vignette is inadequate to properly classify the epilepsy.
� Duration and somatotopic distribution of the tonic seizure.
� What do you mean by “focal motor seizure”? In the ILAE classification, a focal motor seizure means thatthe epileptogenic zone for the motor seizure is focal (or regional). Besides, motor may imply automatismsor atonic, tonic, clonic, hyperkinetic, myoclonic manifestations as also epileptic spams! A good anamnesiscertainly can resolve this dilemma.
� In the vignette, it is mentioned that the patient has Lennox-Gastaut syndrome. This is the reason why weadded intellectual disability as a comorbidity.
� As we mentioned in the main text, selected syndromes can be useful in the management of epilepticpatients. Therefore the four-dimensional classification leaves the option open to include it in parenthesis afterlisting the epileptogenic zone.
CASE 4: TonicA child has brief seizures with stiffening of the right arm and leg, during which responsiveness and awarenessare retained. This seizure is a focal aware tonic seizure (the words “motor onset” can be assumed). In the oldsystem, the seizure would have been called tonic, with a perhaps incorrect assumption of generalized onset.
ILAE classification
a. Seizure type: focal aware tonic seizureb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: right tonic seizureII. Epileptogenic zone: left hemisphereIII. Etiology: unknownIV. Co-morbidities: none
Comments
� The information provided in the vignette is insufficient to establish the epileptic nature of the symptoma-tology. With the available information listed in the vignette, we would classify this as a “paroxysmal event”,before additional data may confirm that the symptoms are epileptic. The classification listed above is basedon the assumption that the epileptic nature of the symptoms has been provided.
� Above is a preliminary classification. Neurological examination and neuroimaging would be essential toproperly classify the epilepsy.
CASE 5: Focal impaired awareness
A 25-year-old woman describes seizures beginning with 30 seconds of an intense feeling that “familiar musicis playing”. She can hear other people talking, but afterwards realizes that she could not determine what theywere saying. After an episode, she is mildly confused, and has to “reorient herself”. The seizure would beclassified as focal impaired awareness. Even though the patient is able to interact with her environment, shecannot interpret her environment, and is mildly confused. Prior classification would have been complex partialseizure.

Epileptic Disord, Vol. 21, No. 1, February 2019 17
Four-dimensional epilepsy classification
ILAE classification
a. Seizure type: focal impaired awareness seizureb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: déjà-vu aura→ dialeptic seizureII. Epileptogenic zone: temporal lobeIII. Etiology: unknownIV. Co-morbidities: unknown
Comments
� The information provided in the vignette is insufficient to establish the epileptic nature of the symptoma-tology. With the available information listed in the vignette, we would classify this as a “paroxysmal event”,before additional data may confirm that the symptoms are epileptic. The classification listed above is basedon the assumption that the epileptic nature of the symptoms has been provided
� The temporal lobe was identified as the epileptogenic zone because déjà-vu aura(dialeptic seizures almostalways originate in the temporal lobe.
CASE 6: Autonomic
A 22-year-old man has seizures during which he remains fully aware, with “hair on my arms standing on edge”and a feeling of being flushed. These are classified as focal aware nonmotor autonomic seizures, or moresuccinctly, focal aware autonomic seizures. Based on the old classification, these would have been referred toas simple partial autonomic seizures.
ILAE classification
a. Seizure type: focal aware autonomic seizuresb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: vasomotor aura → pilomotor auraII. Epileptogenic zone: temporal lobeIII. Etiology: unknownIV. Co-morbidities: none
Comments
� The information provided in the vignette is insufficient to establish the epileptic nature of the symptoma-tology. With the available information listed in the vignette, we would classify this as a “paroxysmal event”before additional data may confirm that the symptoms are epileptic. The classification listed above is basedon the assumption that the epileptic nature of the symptoms has been provided
� The temporal lobe was identified as the epileptogenic zone because vasomotor aura (pilomotor auras almostalways originate in the temporal lobe. In many cases, the epileptogenic zone is an inference from the semiology,until additional investigations provide more information. This may not be accurate as the epileptogenic zonemay be a non-eloquent area from where the seizure spreads to a symptomatogenic zone.

18 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
CASE 7: Focal clonicA one-month-old boy has rhythmic jerking of the left arm that does not remit when repositioning the arm.Corresponding EEG shows right frontal ictal rhythms. These seizures are focal motor onset clonic seizures, ormore parsimoniously, focal clonic seizures. Because the level of awareness cannot be ascertained, awarenessis not involved in classifying this seizure. No appropriate term exists under the old classification.
ILAE classification
a. Seizure type: focal aware tonic seizureb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: right tonic seizure
II. Epileptogenic zone: left hemisphereIII. Etiology: unknownIV. Co-morbidities: none
Comments
None
CASE 8: Sequential seizure manifestationsA seizure begins with tingling in the right arm of a 75-year-old man. The patient says that it then progressesto rhythmic jerking of the right arm, lasting for about 30 seconds. He retains awareness and memory for theevent. This seizure is a focal (non-motor-onset) sensory seizure. Additional description would be useful, namelyfocal sensory seizure with somatosensory features progressing to right arm clonic activity. If the sensory andmotor events were to be discontinuous or the clinician had reason to consider the event to be two separate(bifocal or multifocal) seizures, then each component would be classified as a separate seizure. Under theold classification, this would have been called a simple partial sensorimotor seizure. An advantage of the 2017classification is specification of the sensory onset, which may have clinical importance.
ILAE classificationa. Seizure type: focal impaired awareness seizureb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: déjà-vu aura→ dialeptic seizure
II. Epileptogenic zone: temporal lobeIII. Etiology: unknownIV. Co-morbidities: unknown
Comments
� The information provided in the vignette is insufficient to establish the epileptic nature of the symptoma-tology. With the available information listed in the vignette, we would classify this as a “paroxysmal event”,before additional data may confirm that the symptoms are epileptic. The classification listed above is basedon the assumption that the epileptic nature of the symptoms has been provided
� The left parietal lobe was identified as the epileptogenic zone because right arm somatosensory aura →right arm clonic seizures almost always originate from the left parietal lobe.

Epileptic Disord, Vol. 21, No. 1, February 2019 19
Four-dimensional epilepsy classification
CASE 9: Myoclonic-atonic
A four-year-old boy with Doose syndrome has seizures with a few arm jerks and then a rapid drop with lossof tone. These are now classified as myoclonic-atonic seizures. Based on prior unofficial usage, these wouldhave been called myoclonic-astatic seizures.
ILAE classification
a. Seizure type: focal aware autonomic seizuresb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: bilateral myoclonic axial atonic seizure
II. Epileptogenic zone: generalized (Doose syndrome)III. Etiology: unknownIV. Co-morbidities: none
Comments
None
CASE 10: Myoclonic-tonic-clonic seizures
A 13-year-old with juvenile myoclonic epilepsy has seizures beginning with a few jerks, followed by stiffeningof all limbs and then rhythmic jerking of all limbs. These would be classified as myoclonic-tonic-clonic seizures.No corresponding single seizure type exists in the old classification, but they might have been called myoclonicor clonic seizures followed by tonic-clonic seizures.
ILAE classification
a. Seizure type: myoclonic-tonic-clonic seizuresb. Epilepsy type: generalizedc. Epilepsy syndrome: juvenile myoclonic epilepsyd. Etiology: genetic
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: bilateral myoclonic → bilateral tonic-clonic seizuresII. Epileptogenic zone: generalized (juvenile myoclonic epilepsy)III. Etiology: geneticIV. Co-morbidities: none
Comments
None.

20 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
CASE 11: Focal epileptic spasms
A 14-month-old girl has sudden extension of both arms and flexion of the trunk for about 2 seconds. Theseseizures repeat in clusters. EEG shows hypsarrhythmia with bilateral spikes, most prominent over the leftparietal region. MRI shows a left parietal dysplasia. Resection of the dysplasia terminated the seizures. Becauseof the ancillary information, the seizure type would be considered as focal epileptic spasms (the term “motoronset” can be assumed). Based on the previous classification, these would have been called infantile spasms,with information on focality not included. The term “infantile” can still be used when spasms occur in infancy.
ILAE classification
a. Seizure type: focal epileptic spasmb. Epilepsy type: focalc. Epilepsy syndrome: West syndromed. Etiology: genetic and structural
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: bilateral epileptic spasmII. Epileptogenic zone: left parietal (West syndrome)III. Etiology: left parietal dysplasiaIV. Co-morbidities: none
Comments
None.
CASE 12: Unclassified
A 75-year-old man, known to have epilepsy, reports an internal sense of body trembling and a sense ofconfusion. No other information is available. EEG and MRI are normal. This event is unclassified.
ILAE classification
a. Seizure type: unclassifiedb. Epilepsy type: unknownc. Epilepsy syndrome: unknownd. Etiology: unknown
Four-dimensional epilepsy classification
Paroxysmal eventI. Event semiology: aura → dialeptic eventII. Etiology: unknownIII. Co-morbidities: unknown
Comments
� The vignette is very confusing. It indicates that the patient has epilepsy but does not indicate the seizuresemiology of “known” epileptic seizures.
� The paroxysmal events are very non-specific and could well be non-epileptic paroxysmal events.
� The patient requires additional testing to elucidate the nature of the symptomatology (MRI and video EEG)

Epileptic Disord, Vol. 21, No. 1, February 2019 21
Four-dimensional epilepsy classification
Appendix 2. CASE STUDIES: AN EXAMPLE OF THREE PATIENTS WITH PAROXYSMALEVENTS
CASE 1A 20-year-old, right-handed woman presents for evaluation of paroxysmal events. Onset of the events was atage 15.
(1) ANAMNESISPer patient: The last thing she remembers before her events is a “weird feeling”, which she cannot furtherdescribe, and then she knows that the seizure is coming. This feeling just lasts for few seconds (∼20 seconds).The next thing she remembers is laying on the floor, being surrounded by people and feeling confused. Shefeels tired and she goes back to sleep until the next day. She does not recall any particular difficulty talkingor understanding after her events. She denies any particular pain, such as muscular pain, jaw pain or tonguesoreness. She does not recall any episode with urinary incontinence.Per witness: The mother hears a loud cry at the onset of the episode. Then the patient is unresponsive andturns her head to one side (the mother recalls “to the left”), while her eyes are open and “rolled back”. This isfollowed by bilateral shaking with arms and legs extended; lasting for about a minute. She does not recall anyfoaming at the mouth. After the episode, the patient is unresponsive and her breathing is deep and stertorousfor several seconds. She is then confused for about 20-30 minutes. No urinary incontinence. When asked, thepatient denies recalling any pulling of her head towards one side or the other.She is currently having 1-2 events a month.
Classification after clinical history
2017 ILAE classification system
a. Seizure type: focal to bilateral tonic-clonic seizureb. Epilepsy type: focalc. Epilepsy syndrome: NAd. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: aura → left versive (LOC) → bilateral tonic-clonic seizureII. Epileptogenic zone: right hemisphereIII. Etiology: unknownIV. Co-morbidities: noneComment: The description of the mother is consistent with generalized tonic-clonic seizures. This is alsoconsistent with the history obtained by the patient; no recollection of the generalized convulsions. These factssupport the conclusion that the patient has “epileptic paroxysmal events”. Not infrequently, the observersare relatively inaccurate when lateralizing versions. Therefore, after obtaining the clinical history, the seizurescould also be classified as follows:Aura→versive (LOC) →bilateral tonic-clonic seizure.The epileptogenic zone would be “focal”.
(2) EMU EVALUATION (figure 1A, B and video 1).Interictal: sharp waves, left frontal (F3-C3)Ictal: right versive (LOC)→ right face tonic → bilateral asymmetric tonic clonic seizureLateralizing signs: right M2e, right sign of 4
(3) NEUROIMAGING (figure 2).Small focus of abnormal signal in the left frontal periventricular white matter.

22 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
1 Fp1-F7
2 F7- T7
3 T7-P7
4 P7-O1
5 Fp2-F8
6 F8-T8
7 T8-P8
8 P8-O2
9 Fp1-F3
10 F3-C3
11 C3-P3
12 P3-O1
13 Fp2-F4
14 F4-C4
15 C4-P4
16 P4-O2
17 Fz-Cz
18 Cz-Pz
22 EKG1-EKG3
M
1 Fp1-F7
[SENS 7 HF 70 TC 0.1 CAL 50]
2 F7- T7
3 T7-P7
4 P7-O1
5 Fp2-F8
6 F8-T8
7 T8-P8
8 P8-O2
9 Fp1-F3
10 F3-C3
11 C3-P3
12 P3-O1
13 Fp2-F4
14 F4-C4
15 C4-P4
16 P4-O2
17 Fz-Cz
18 Cz-Pz
22 EKG1-EKG3
M
A
B[SENS *5 HF *70 TC *0.1 CAL *50]
Figure 1. (A) Sharp waves, left fronto-central. (B) EEG seizure pattern, left fronto-central.

Epileptic Disord, Vol. 21, No. 1, February 2019 23
Four-dimensional epilepsy classification
1/2
48F
1
P
1/21
L
34
F
Figure 2. Small focus of abnormal signal in the left frontal periventricular white matter.
(4) FINAL CLASSIFICATION
2017 ILAE classification system
a. Seizure type: focal to bilateral tonic-clonic seizureb. Epilepsy type: focalc. Epilepsy syndrome: NAd. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: aura→ right versive (LOC) → right face tonic → bilateral asymmetric tonic clonic seizureII. Epileptogenic zone: left frontalIII. Etiology: unknownIV. Co-morbidities: MRI shows a small focus of abnormal signal in the left frontal periventricular white matter
CASE 2
(1) ANAMNESISA 53-year-old, left-handed woman with dyslipidemia and hypothyroidisms presents to the epilepsy clinic forevaluation of paroxysmal events that started at age eight.Per patient: Her episodes are nocturnal. She wakes up with a feeling “the seizure is coming and I am losingcontrol”. This feeling last for just “a second”. Then, she remembers her left arm shakes uncontrollably. She triesto stop it by grabbing her left arm with the right hand, but she cannot control it. Occasionally, she also feels herlegs moving up and down. The episodes last 2-3 minutes. After the seizure, she feels slightly confused, tired,and she has difficulty talking, but she feels she is able to understand. She denies biting her tongue and sheonly recalls urinary incontinence on one occasion. She feels she is probably aware during the entire episode,but she is not totally sure.Per witness (husband): The patient suddenly wakes up and yells “help”. Then, she starts moving uncontrollablyall over. Her eyes are open. Her arms and legs move up and down. There is no foaming at the mouth, nor eyeor head deviation that he recalls. Eyes do not roll up. After the seizure, she seems awake but confused andtired. He is not sure whether she would be able to follow any commands during this time, but she would knowthat she just had an episode.On presentation, she was having one episode per week while on two AEDs.

24 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
Classification after clinical history
2017 ILAE classification system
a. Seizure type: focal aware non-motor onsetb. Epilepsy type: focalc. Epilepsy syndrome: N/Ad. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: aura → left arm clonic → hypermotor seizureII. Epileptogenic zone: right frontalIII. Etiology: unknownIV. Co-morbidities: noneComment: The duration and stereotypy of the episodes strongly suggests an epileptic paroxysmal episode.Besides, in this case, the patient herself lateralized the clonic seizure. This is usually a reliable lateralizing sign.
(2) EMU EVALUATION (figure 3A, B, C and video 2)Interictal: sharp waves, maximum at right temporal electrodes (F8 and Sp2) (figure 3).Ictal:-Seizure semiology: emotional hypermotor seizure (video 2)-EEG seizure pattern: right temporal (figure 3).Comment: After the EMU evaluation, the seizure semiology classification was adjusted. The “aura” and the“left arm clonic” components were removed from the classification. Her subjective sensation started at thesame time as the emotional hypermotor seizure, therefore it was felt to be most likely caused by it. Also, her leftarm never moved in a clonic fashion, but rather exhibited complex movements, as expected in a hypermotorseizure. This exemplifies how the four-dimensional epilepsy classification may change overtime as the patientmay undergo further investigations.
(3) NEUROIMAGING (figure 4)Axial and coronal FLAIR MRI shows a “comet-like” high signal in the right anterior insula, consistent with acortical dysplasia. The main juxtacortical lesion has a “tail”, tracking along the expected course of the radialglial fibers to the subependimal margin.
(4) FINAL CLASSIFICATION
2017 ILAE classification system
a. Seizure type: focal aware hyperkinetic seizureb. Epilepsy type: focalc. Epilepsy syndrome: NAd. Etiology: structural and genetic
Four-dimensional epilepsy classification
I. Ictal semiology: emotional hypermotor seizureII. Epileptogenic zone: right insulaIII. Etiology: structural and genetic (right insular cortical dysplasia)IV. Co-morbidities: none

Epileptic Disord, Vol. 21, No. 1, February 2019 25
Four-dimensional epilepsy classification
1 Fp1-F7
[SENS *7 HF *70 TC *0.1 CAL *50]
2 F7-SP1
3 SP1-T7
4 T7-P7
5 P7-O1
6 Fp2-F8
7 F8-SP2
9 T8-P8
10 P8-O2
11 *SP1-*SP2
12 *A1-*A2
13 *Fp1-*F3
14 *F3-*C3
15 *C3-*P3
16 *P3-*O1
17 *Fp2-*F4
18 *F4-*C4
19 *C4-*P4
20 *P4-*O2
21 *Fz-*Cz
22 *Cz-*Pz
26 EKG1-EKG4
M
8 SP2-T8
1 Fp1-F7
[SENS *10 HF *70 TC *0.1 CAL *50]
2 F7-SP1
3 SP1-T7
4 T7-P7
5 P7-O1
6 Fp2-F8
7 F8-SP2
9 T8-P8
10 P8-O2
11 *SP1-*SP2
23 EKG1-EKG3
M
8 SP2-T8
A
B
1 Fp1-F7
[SENS *15 HF *70 TC *0.1 CAL *50]
2 F7-SP1
3 SP1-T7
4 T7-P7
5 P7-O1
6 Fp2-F8
7 F8-SP2
9 T8-P8
10 P8-O2
11 *SP1-*SP2
23 EKG1-EKG3
M
8 SP2-T8
C
Figure 3. (A) Sharp waves, F8 Sp2. (B) EEG seizure onset. (C) EEG seizure evolution (1 minute from onset). EEGseizure pattern, right temporal.
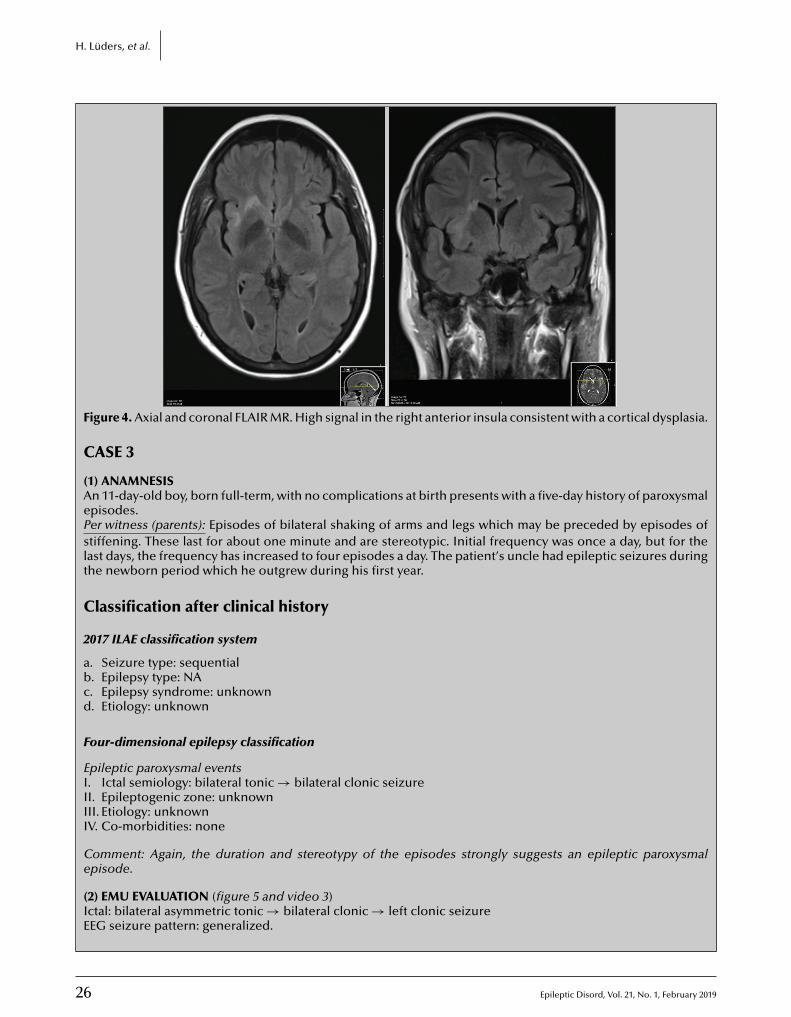
26 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
1/3
P
Figure 4. Axial and coronal FLAIR MR. High signal in the right anterior insula consistent with a cortical dysplasia.
CASE 3
(1) ANAMNESISAn 11-day-old boy, born full-term, with no complications at birth presents with a five-day history of paroxysmalepisodes.Per witness (parents): Episodes of bilateral shaking of arms and legs which may be preceded by episodes ofstiffening. These last for about one minute and are stereotypic. Initial frequency was once a day, but for thelast days, the frequency has increased to four episodes a day. The patient’s uncle had epileptic seizures duringthe newborn period which he outgrew during his first year.
Classification after clinical history
2017 ILAE classification system
a. Seizure type: sequentialb. Epilepsy type: NAc. Epilepsy syndrome: unknownd. Etiology: unknown
Four-dimensional epilepsy classification
Epileptic paroxysmal eventsI. Ictal semiology: bilateral tonic → bilateral clonic seizureII. Epileptogenic zone: unknownIII. Etiology: unknownIV. Co-morbidities: none
Comment: Again, the duration and stereotypy of the episodes strongly suggests an epileptic paroxysmalepisode.
(2) EMU EVALUATION (figure 5 and video 3)Ictal: bilateral asymmetric tonic → bilateral clonic → left clonic seizureEEG seizure pattern: generalized.

Epileptic Disord, Vol. 21, No. 1, February 2019 27
Four-dimensional epilepsy classification
Fp1-C3
30 HF *70 TC *0.1 CAL *50
Fp2-C4
C4-O2
Fp1-T7
T7-O1
Fp2-T8
T8-O2
A1-C3
C3-Cz
Cz-C4
C4-A2
C3-O1
Figure 5. Case 3. EEG seizure pattern, generalized. Diffuse suppression followed by generalized rhythmic deltamaximum over frontal and central regions.
(3) EVOLUTIONDuring his initial evolution, blood and urine and metabolic work-up was negative. Lumbar puncture wasnormal. Seizures responded to phenobarbital. At seven months, he had developed normally and no furtherseizures were noticed. Therefore, phenobarbital was stopped.He is currently eight years old. He has growth and development appropriate for his age. He has been seizure-free, off phenobarbital, since age seven months old.
(4) FINAL CLASSIFICATION
2017 ILAE neonatal classification system
a. Seizure type: sequential seizureb. Epilepsy type: NAc. Epilepsy syndrome: self-limited neonatal or familial neonatal epilepsyd. Etiology: genetic
Four-dimensional epilepsy classification
Epileptic paroxysmal eventI. Ictal semiology: bilateral asymmetric tonic → bilateral clonic → left clonic seizureII. Epileptogenic zone: generalized (self-limited neonatal or familial neonatal epilepsy)III. Etiology: geneticIV. Co-morbidities: none

28 Epileptic Disord, Vol. 21, No. 1, February 2019
H. Lüders, et al.
Appendix 3. CASE STUDY: A PRACTICAL EXAMPLE OF USING THE EPILEPSYCLASSIFICATION WITH DIFFERENT DEGREES OF PRECISION
Case studyA 35-year-old man presents to the emergency department (ED) for a new-onset paroxysmal event that occurredearlier that day. The ED physician gathers some information from the patient’s wife who describes the patienthaving violent shaking of all limbs. The patient now feels that his condition corresponds to baseline. He hasalso had anxiety and depression for the last five years.Based on the information gathered by the ED, the physician could try to classify the patient’s epilepsy usingthe epilepsy classification and semiological classification with low or moderate complexity.
Paroxysmal eventIctal semiology: bilateral motor eventFrequency: one event, hours agoEtiology: unknownComorbidities: anxiety and depression
The patient is seen by a junior neurology resident who has rotated in the epilepsy service a few times. He asksadditional questions to the patient’s wife. The resident finds out the patient has been having brief episodes ofstaring and unresponsiveness for the last year, with an approximate frequency of one every month. In addition,he had urinary incontinence and has bitten his tongue today.Based on the additional information, the resident suspects an epileptic paroxysmal event and he can furtherdevelop the epilepsy classification using the more complex semiological classification. At this moment, he isunsure whether the patient may have a focal vs a generalized epileptogenic zone.
Epileptic paroxysmal eventIctal semiology: (1) dialeptic → (2) bilateral clonic seizureFrequency: (1) one/month; (2) once todayEpileptogenic zone: unknown.Etiology: unknown.Comorbidities: anxiety and depression
The patient is later seen by the epilepsy faculty who accompanies the resident to see the patient again. Thepart of the interview that focuses on the ictal semiology is outlined below:Epilepsy doctor: Mr. S, what is the last thing you remember before the episode? I would like you to tell me onlyyour own experience, not what you have been told.Mr. S: I just recall waking up this morning feeling fine and the last thing I remember is going into the kitchento get some coffee.Epilepsy doctor: Anything else unusual preceding the episode that you may remember?Mr. S: I may have felt nauseated for a second, but I am not sure.Epilepsy doctor: What is the next thing you remember?Mr. S: I just remember hearing my wife, asking me questions, like “How are you feeling? Can you stand up?”Epilepsy doctor: How were you feeling at that point?Mr. S: I was feeling confused, but otherwise OK.Epilepsy doctor: Any particular pain?Mr. S: Not that I can recall.Epilepsy doctor: What about now? Any particular pain now?Mr. S: Well... yes, my tongue, my tongue feels swollen and painful. My jaw is sore and I have some muscleaches around my shoulders, but overall, I feel fine. Just tired.Epilepsy doctor: Your wife has mentioned you may have been having other episodes where you would stare.Were you aware of these?Mr. S: She has told me before, but I am really not sure what she means.Epilepsy doctor: Have you been feeling anything else unusual?

Epileptic Disord, Vol. 21, No. 1, February 2019 29
Four-dimensional epilepsy classification
Mr. S: Now that you mention... I have been getting this feeling in my stomach... It is like an anxiety or nauseafeeling or like being in a roller coaster. And I get this sensation of déjà vu or familiarity, as if I am experiencingsomething that has happened before. I mentioned this because lately it has gotten quite strong, and makesme even a little scared.Epilepsy doctor: For how long does it last and how often do you get these sensations?Mr. S: It is just a matter of seconds. I get these maybe once or twice a week, but for the last few days, I havethem almost daily. It was really striking.Epilepsy doctor: Mrs. S, I would like to ask you a few questions regarding the episodes you have seen. Can youstart by describing these staring spells? Can you give me an example?Mrs. S: Well, the last one I saw, we were seating on the couch, just watching TV, and I just saw him staring.Epilepsy doctor: But what caught your attention? Anything in particular that made you look towards yourhusband while watching TV?Mrs. S: He makes these chewing sounds with his mouth. And I try calling him, but he does not respond.Epilepsy doctor: For how long does it last for? And what happens afterwards?Mrs. S: It usually lasts for 1-2 minutes and afterwards he is somewhat confused.
After further questioning the patient’s wife, the Epilepsy doctor can gather additional information that isconvincing for a generalized tonic-clonic seizure: ictal cry, blood-tainted foaming at the mouth, “eyes rolledback”, tonic phase in decerebrate posture lasting 15 seconds followed by a clonic phase lasting approximatelyone minute, all followed by postictal coma for 5-10 minutes and gradual recovery.Based on the additional information, the epilepsy faculty can further develop the epilepsy classification usingthe semiological classification of moderate complexity.
Epileptic paroxysmal eventIctal semiology: (1) abdominal aura→ (2) psychic aura → (3) automotor (LOC) → (4) bilateral clonic seizureFrequency: (1) (2) one/day; (3) one/month; (4) once todayEpileptogenic zone: temporal lobeEtiology: unknownComorbidities: anxiety and depression
We know now that the patient has focal epilepsy, and among the focal epilepsies that can cause this seizuretype, the most likely is temporal lobe epilepsy. This maybe correct for now, nevertheless this classification canbe further developed and confirmed over time with additional testing, such as EEG, epilepsy monitoring unit(EMU) admission, and MRI of the brain.

do
i:10.1684/epd
.2019.1024
30 Epileptic Disord, Vol. 21, No. 1, February 2019
Correspondence:Berge MinassianUniversity of Texas SouthwesternMedical Center,5323 Harry Hines Blvd.,Dallas TX 75390,USA<[email protected]>
Original articleEpileptic Disord 2019; 21 (1): 30-41
EEG of asymptomaticfirst-degree relativesof patients with juvenilemyoclonic, childhood absenceand rolandic epilepsy:a systematic review andmeta-analysis
Mariam Tashkandi 1, Duaa Baarma 2, Andrea C. Tricco 3,Cyrus Boelman 1, Reem Alkhater 4, Berge A. Minassian 1,5
1 Program in Genetics and Genome Biology, The Hospital for Sick Children, Institute ofMedical Science, University of Toronto, Toronto2 Paediatric Neurology Division, Department of Paediatrics, King Abdullah SpecialistChildren Hospital, Riyadh3 Knowledge translation program, Li Ka Shing Knowledge Institute of St Michael’shospital, Epidemiology division, Dalla Lana school of public health, University ofToronto, Canada4 Johns’ Hopkins Aramco Healthcare, Dhahran, Saudi Arabia5 Division of Neurology, Department of Pediatrics, University of Texas Southwestern,Dallas, USA
Received August 20, 2018; Accepted October 18, 2018
ABSTRACT – Aims. Rolandic (RE), childhood absence (CAE) and juvenilemyoclonic (JME) epilepsy encompass centrotemporal sharp waves, 3-Hzspike waves and >3-Hz spike or polyspike waves, respectively. Evidenceabounds for genetic roles in all three syndromes, yet involved genes forthe vast majority of patients remain unknown. It has long been proposedthat while each disease is genetically complex, its specific EEG trait mayrepresent a genetically simpler endophenotype. This meta-analysis of theliterature focuses on the frequency of EEG traits in clinically unaffectedfirst-degree relatives towards determining inheritance patterns of the EEGendophenotypes.Methods. We used the Preferred Reporting Items for Systematic Reviewand Meta-Analysis for protocols (PRISMA-P) and searched Medline,EMBASE, CINHAL and the Cochrane Central Register of Controlled Trials.Results. Following extensive screening, 15 studies were included witha total of 3,858 asymptomatic relatives. The prevalence of ‘abnormal’EEG waves was 21%, 42% and 33% for JME, CAE and RE, respectively,close to what would be expected based on Mendelian inheritance.

E
JME, CAE and RE: EEG of asymptomatic relatives
However, breaking down the reported EEG abnormalities, most consistednot of the respective EEG signature traits -prevalences of which were as lowas 5%- but of non-specific EEG ‘abnormalities’/variants.Conclusions. Prevalence of non-specific EEG ‘abnormalities’/variants in thegeneral population ranges from 0.1 to 10%. Underlying this 100-fold-widerange is a spectrum of what is considered ‘abnormal’ or variant. The preva-lences of ‘abnormalities’/variants in asymptomatic siblings in RE, CAE andJME significantly exceed even the highest value in the general populationand fall within Mendelian expectations. These results suggest that EEG‘abnormalities’/variants shared with the general population are enrichedin the three syndromes and are endophenotypes inherited in a geneti-cally simple near-Mendelian fashion. Future work with modern EEG variantdefinitions should uncover genetic variants contributing to neuronal hyper-synchrony in epilepsy.
Key words: Rolandic epilepsy, childhood absence epilepsy, juvenile myo-ndo
TRn1aM2demfp22taoTrweMifieatfoaJsctAMa
irWhtiwctRawAbociEpoftattfiC
M
Protocol
clonic epilepsy, e
he three most common childhood epilepsies areolandic (RE), childhood absence (CAE) and juve-ile myoclonic (JME) epilepsy, accounting for 15%,0-15% and 5-10% of cases, respectively (Avanzinind Noebels, 2009; Panayiotopoulos, 2010; Berg andillichap, 2013; Camfield et al., 2013; Pal et al.,
016; Verrotti et al., 2017). There is abundant evi-ence that genetic factors play important roles inach of these conditions though none (in the vastajority of families) is inherited in a Mendelian
ashion, and all three are therefore genetically com-lex (Anderman and Metrakos, 1969; Delgado-Escueta,007; Panayiotopoulos, 2010; Panayiotopoulos et al.,012). Despite the genetic and genomic revolutions ofhe last three decades, only a few genes have beenssociated with these very common diseases, and thennly in a small minority of patients.he genetic complexities of CAE and JME were alreadyecognized even prior to the two being carved out ofhat was called in the early 1950s, ‘centrencephalic’pilepsy (Penfield, 1952). In their seminal work,etrakos and Metrakos (1961a) reported that approx-
mately 50% of clinically unaffected, age-matchedrst-degree relatives of patients with centrencephalicpilepsy had the same age-dependent generalized EEGbnormalities as the latter, and suggested that whilehe epilepsy itself was not inherited in a Mendelianashion, the EEG trait, present as it is in nearly 50%f young adolescent relatives, may well be (Metrakosnd Metrakos, 1961a). Following the spinoff of CAE andME from the parent ‘centrencephalic’ concept, EEGtudies of relatives of these patients continued to be
pileptic Disord, Vol. 21, No. 1, February 2019
arried out, but results usually showed rates substan-ially lower than 50%.
decade following the work of Metrakos andetrakos, studies in RE also reported a rate of EEG
bnormality in clinically unaffected siblings of approx-
AiPt
phenotype, EEG trait, spike wave, sibling, unaffected
mately 50% when the siblings were studied during theange of childhood years in which RE occurs (Bray and
iser, 1964; Heijbel et al., 1975). More recent work,owever, questioned the role of genes in RE, based on
he rate of non-concordance for the clinical syndromen monozygotic twins (Valdamudi et al., 2006). Mean-
hile, ongoing EEG studies of relatives of RE patientsontinued to show rates of EEG abnormalities substan-ially higher than in children in the general population.E, CAE and JME are not only common, they alsore ‘pure’ epilepsies in which the CNS is other-ise grossly morphologically and functionally intact.s such, understanding the pathogeneses of theseenign conditions will be highly insightful to theverall understanding of epilepsy. Solving the geneticomplexities of RE, CAE and JME would be greatly aidedf any aspect of these conditions, e.g. their specificEG traits, were endophenotypes inherited in simpler,erhaps a Mendelian, fashion. Given the opaquenessf, and contradictions in the literature regarding, the
requencies of EEG abnormalities in unaffected rela-ives of patients with RE, CAE and JME, we conductedsystematic review and meta-analysis of this literature
o clarify the current state of knowledge. It is hopedhat this work will serve as a basis and springboardor additional studies that will resolve the genet-cs of the epileptiform abnormalities underlying RE,AE and JME.
ethods
31
protocol was developed using the Preferred Report-ng Items for Systematic Review and Meta-Analysis forrotocols (PRISMA-P) (Moher et al., 2015) and regis-ered with the PROSPERO database (CRD42013005615).

3
M
E
WcteEw
S
MtJlieeaiepArbaS2srcoc
S
A2wdoac(
D
Aesfddl(lt
M
Ws(tctordcfstedoe
S
WmtFftavTifyDa
R
S
Tefte1asplbeen conducted in Germany (Tsuboi and Christian,1973; Doose et al., 1973; Degen and Degen, 1992;
. Tashkandi, et al.
ligibility criteria
e included studies using cohort, case-control orross-sectional methodology examining EEG in asymp-omatic relatives (parents, siblings or offspring) ofpileptic patients of all ages. Both English and non-nglish language, published and unpublished, reportsere included.
earch
edline, EMBASE, CINAHL, and the Cochrane Cen-ral Register of Controlled Trials were searched onuly 5, 2013. Searches were performed with no year oranguage restrictions, using the Medical Subject Head-ngs and text words and phrases: Juvenile myoclonicpilepsy, Janz syndrome, idiopathic epilepsy, geneticpilepsy, electroencephalograph, humans, childhoodbsence epilepsy, pyknolepsy, idiopathic general-zed epilepsy, centrencephalic epilepsy, Rolandicpilepsy, benign childhood epilepsy with centrotem-oral spikes, epilepsy syndrome, and Sylvian seizures.ppropriate wildcards were used to account for plu-
als and spelling variations. This search was conductedy an experienced librarian and peer-reviewed bynother librarian using the Peer Review of Electronicearch Strategies (PRESS) checklist (McGowan et al.,010). The electronic search was supplemented bycanning the reference lists of included studies andelevant reviews. The full search strategy for MEDLINEan be found in the supplementary material and thethers are available upon reasonable request from theorresponding author.
tudy selection
pilot test was conducted on a random sample of5 titles and abstract citations. After 94% agreementas achieved, two reviewers (MT and DB) indepen-ently screened the search results for inclusion. Webtained the full-text of potentially relevant articlesnd assessed them in a similar manner. Discrepan-ies were resolved by discussion with a third reviewerBAM).
ata collection process
fter a pilot test of 25 articles, two independent review-rs (MT and DB) performed data extraction on all theelected articles using the standardized data extractionorm. To ensure accuracy, the reviewers extracted allata in duplicate and conflicts were resolved throughiscussion amongst the team. When multiple pub-
ications reported data from the same population
2
companion reports), we considered the study with theargest sample size as the major publication, and usedhe other report(s) for supplementary material only.
Wea
ethodological quality
e assessed methodological quality of individualtudies using the Newcastle-Ottawa Scale (NOS)Wells et al., 2014), which consists of eight items per-aining to selection (representativeness of the exposedohort, selection of the non-exposed cohort, ascer-ainment of exposure, demonstration that outcomef interest was not present at start of study), compa-ability (comparability of cohorts on the basis of theesign or analysis), and outcome (assessment of out-ome, sufficient duration of follow-up, adequacy ofollow-up). We modified the NOS for cross-sectionaltudies to include the following five items: represen-ativeness of the exposed cohort, ascertainment ofxposure, comparability of cohorts on the basis of theesign or analysis, assessment of outcome, adequacyf response rate (Higgins and Thompson, 2002; Fnaist al., 2014; Wells et al., 2014).
ynthesis of results
e described the results narratively and conducted aeta-analysis using a random effects model, as statis-
ical heterogeneity was expected across the studies.or the meta-analysis, we combined the extracted datarom the studies to calculate a pooled estimate ofhe proportion of abnormal EEG in each populationlong with the corresponding 95% confidence inter-al (CI) based on a normal distribution (Higgins andhompson, 2002). We assessed statistical heterogene-
ty using the I2 statistic and depicted the studies in aorest plot to examine heterogeneity visually. All anal-ses were conducted using the R statistical program (Revelopment Core Team 2010) with the metafor pack-
ge (Viechtbauer, 2010).
esults
tudy selection and study characteristics
he literature search yielded 10,223 citations. Afterxcluding 255 duplicates, 9,968 studies were screenedor eligibility. A total of 211 potentially relevant full-ext articles met a preliminary screen for epilepsy, EEGxaminations, and mention of relatives. From these,91 were excluded because they clearly did not includesymptomatic relatives (86), did not study the epilep-ies in question or their EEGs (77), did not providerimary data (10), or were only abstracts or could not be
ocated (18). A total of 15 studies remained. These had
Epileptic Disord, Vol. 21, No. 1, February 2019
andschneider et al., 2010), the United States (Alonsot al., 2005a, 2005b; Bali et al., 2007), Turkey (Atakli etl., 1999; Akgun et al., 2009), Italy (Serra et al., 2001;

E
VSa2wOacsotrt(oigsaigeamp
JSJaeAtOi(dNloe>jofemrwwsr2Tsu
Aw2aipps
CF1A1msaaaaswsmfttnwts(TaalaHo(
RF12bw(
M
errotti et al., 2013), India (Jayalakshmi et al., 2006),weden (Heijbel et al., 1975), and Canada (Metrakosnd Metrakos, 1961a) and published between 1961 and013. EEG recording times ranged from 20 to 60 minutesith varying capture of sleep.f the 15 studies, 11 were included in the meta-
nalysis. The design of two studies by Alonso andolleagues (cohort) differed from all the others (cross-ectional) and thus could not be combined with thethers in the meta-analysis. Their results are never-
heless shown (table 1 ), as they represent valuableelevant data. The two other studies not included inhe meta-analysis are those of Metrakos and Metrakos1961a) and Tsuboi and Christian (1973) (table 1), bothf which did not formally specify that the relatives stud-
ed were asymptomatic. However, this information wasleaned from details in their papers, including theirelection of any-comer (and not multiplex) patients,nd the sheer number of families and relatives stud-ed. The vast majority of these families and relatives,iven what we know of these epilepsies, would bexpected to be asymptomatic. As such, we conductedsensitivity analysis including these two studies in theeta-analysis, in addition to the 11 studies that were
reviously combined.
uvenile Myoclonic Epilepsyix studies specified that their epileptic patients hadME and reported on EEG abnormalities in the patientsnd their relatives (Tsuboi and Christian, 1973; Ataklit al., 1999; Alonso et al., 2005a; Jayalakshmi et al., 2006;kgun et al., 2009; Wandschneider et al., 2010). Two of
hese studies were not included in the meta-analysis.ne (Alonso et al., 2005a) was not included because
t differed in design (cohort) from the other studiescross-sectional). This study also did not specify theegree of relatedness between relatives and patients.otwithstanding, the results of this study are tabu-
ated: there were 186 JME patients and 1,756 relatives,f whom 24 (1%) had EEG findings that were consid-red abnormal. These abnormalities were: generalized3-Hz spike or polyspikes and waves (SPSW) in 15 sub-
ects, 3-Hz spike waves (SW) in three, bursts of focalr diffuse slowing in four, and bursts of focal or dif-
use sharp waves in two (table 1). The study by Tsuboit al. was the other JME study not included in theeta-analysis, because it did not specify whether the
elatives studied were clinically affected or not. Thereere 136 JME patients and 370 first-degree relatives ofhom 262 (i.e. 71%) had EEG findings that were con-
idered potentially abnormal. These were SPSW in 57
pileptic Disord, Vol. 21, No. 1, February 2019
elatives and paroxysmal sharp waves in the remaining05 (table 1).he remaining four JME studies were all cross-ectional and specified the relatives as first-degree andnaffected (Atakli et al., 1999; Jayalakshmi et al., 2006;
EDA2
JME, CAE and RE: EEG of asymptomatic relatives
kgun et al., 2009; Wandschneier et al., 2010). Thereas a total of 108 JME patients in these studies and
06 first-degree relatives, of whom 39 (19%) had EEGbnormalities. These were SPSW in 18, ‘theta waves’ orntermittent generalized or paroxysmal slowing in 13,hotoparoxysmal response (PPR) in three, centrotem-oral spikes in three, bifrontal sharp waves in one, andingle spikes in one (table 1).
hildhood Absence Epilepsyour studies looked at CAE (Metrakos and Metrakos,961b; Doose et al., 1973; Degen and Degen, 1990a;lonso et al., 2005b). Two (Metrakos and Metrakos,961b; Alonso et al., 2005b) were not included in theeta-analysis. The Metrakos and Metrakos (1961b)
tudy did not specify the affected status of the rel-tives. The patients had ‘centrencephalic’ epilepsy,nd were likely to be predominantly a mix of CAEnd JME cases. There were 211 patients and 418 rel-tives. Of the latter, 145 had EEG abnormalities (35%),even 3-Hz SW, and 138 with an unspecified mix ofhat were considered abnormalities (table 1). The
tudy by Alonso et al. (2005b) was not included in theeta-analysis because of its different (cohort) design
rom the other studies (cross-sectional). In addition,heir CAE cases were ones that evolved into JME andhus diverge from the common remitting CAE phe-otype. There were 45 patients and 541 relatives ofhom 38 (7%) had EEG abnormalities, which included
hree SPSW, 15 3-Hz SW, and the remainder, a mix oflow waves and isolated generalized or focal spikestable 1).he remaining two studies (Doose et al., 1973; Degennd Degen, 1990a) were included in the meta-analysisnd together encompassed 274 patients and 292 sib-ings of whom 104 (36%) were considered to have EEGbnormalities. Of these abnormalities, only 12 were 3-z SW and the remainder were a mix of runs of focalr generalized slow waves or sharp waves or spikes
table 1).
olandic Epilepsyive RE studies (Heijbel et al., 1975; Degen and Degen,990b; Serra et al., 2001; Bali et al., 2007; Verrotti et al.,013) reported on EEG in unaffected relatives. All coulde included in the meta-analysis. Overall, 275 relativesere studied of whom 82 (30%) had EEG abnormalities
table 1).
eta-analysis
33
leven studies (Doose et al., 1973; Heijbel et al., 1975;egen and Degen, 1990a; Degen and Degen, 1990b;takli et al., 1999; Serra et al., 2001; Jayalakshmi et al.,006; Bali et al., 2007; Akgun et al., 2009; Wandschneider
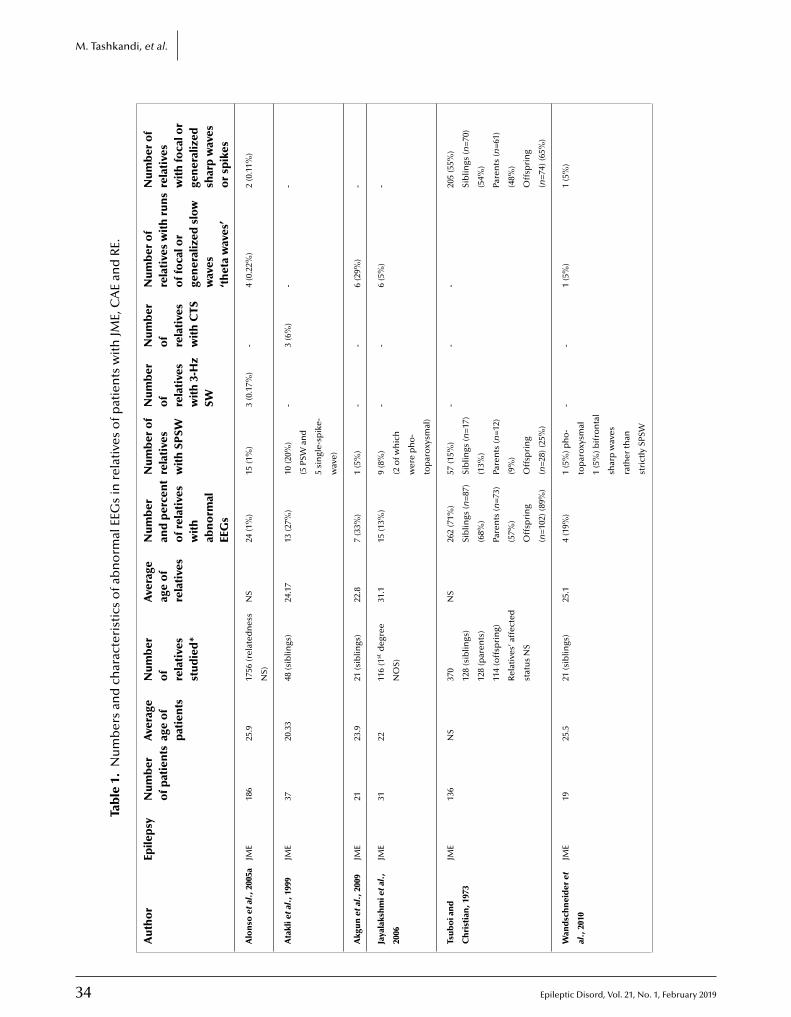
34 Epileptic Disord, Vol. 21, No. 1, February 2019
M. Tashkandi, et al.
Tab
le1.
Nu
mb
ers
and
char
acte
rist
ics
ofa
bn
orm
alEE
Gs
inre
lati
ves
ofp
atie
nts
wit
hJM
E,C
AE
and
RE.
Au
tho
rEp
ilep
syN
um
ber
ofp
atie
nts
Ave
rage
age
of
pat
ien
ts
Nu
mb
ero
fre
lati
ves
stu
die
d*
Ave
rage
age
of
rela
tive
s
Nu
mb
eran
dp
erce
nt
ofr
elat
ives
wit
hab
no
rmal
EEG
s
Nu
mb
ero
fre
lati
ves
wit
hSP
SW
Nu
mb
ero
fre
lati
ves
wit
h3-
Hz
SW
Nu
mb
ero
fre
lati
ves
wit
hC
TS
Nu
mb
ero
fre
lati
ves
wit
hru
ns
off
oca
lor
gen
eral
ized
slow
wav
es‘t
het
aw
aves
’
Nu
mb
ero
fre
lati
ves
wit
hfo
calo
rge
ner
aliz
edsh
arp
wav
eso
rsp
ikes
Alo
nso
etal
.,20
05a
JME
186
25.9
1756
(rel
ated
nes
s
NS)
NS
24(1
%)
15(1
%)
3(0
.17%
)-
4(0
.22%
)2
(0.1
1%)
Ata
klie
tal.
,199
9JM
E37
20.3
348
(sib
lings
)24
.17
13(2
7%)
10(2
0%)
(5PS
Wan
d
5si
ngl
e-sp
ike-
wav
e)
-3
(6%
)-
-
Akg
un
etal
.,20
09JM
E21
23.9
21(s
iblin
gs)
22.8
7(3
3%)
1(5
%)
--
6(2
9%)
-
Jaya
laks
hm
ieta
l.,
2006
JME
3122
116
(1st
deg
ree
NO
S)
31.1
15(1
3%)
9(8
%)
(2o
fwh
ich
wer
ep
ho
-
top
aro
xysm
al)
--
6(5
%)
-
Tsu
bo
ian
d
Ch
rist
ian
,197
3
JME
136
NS
370
128
(sib
lings
)
128
(par
ents
)
114
(off
spri
ng)
Rel
ativ
es’a
ffec
ted
stat
us
NS
NS
262
(71%
)
Sib
lings
(n=
87)
(68%
)
Pare
nts
(n=
73)
(57%
)
Off
spri
ng
(n=
102)
(89%
)
57(1
5%)
Sib
lings
(n=
17)
(13%
)
Pare
nts
(n=
12)
(9%
)
Off
spri
ng
(n=
28)(
25%
)
--
-20
5(5
5%)
Sib
lings
(n=
70)
(54%
)
Pare
nts
(n=
61)
(48%
)
Off
spri
ng
(n=
74)(
65%
)
Wan
dsc
hn
eid
eret
al.,
2010
JME
1925
.521
(sib
lings
)25
.14
(19%
)1
(5%
)ph
o-
top
aro
xysm
al
1(5
%)b
ifro
nta
l
shar
pw
aves
rath
erth
an
stri
ctly
SPSW
--
1(5
%)
1(5
%)
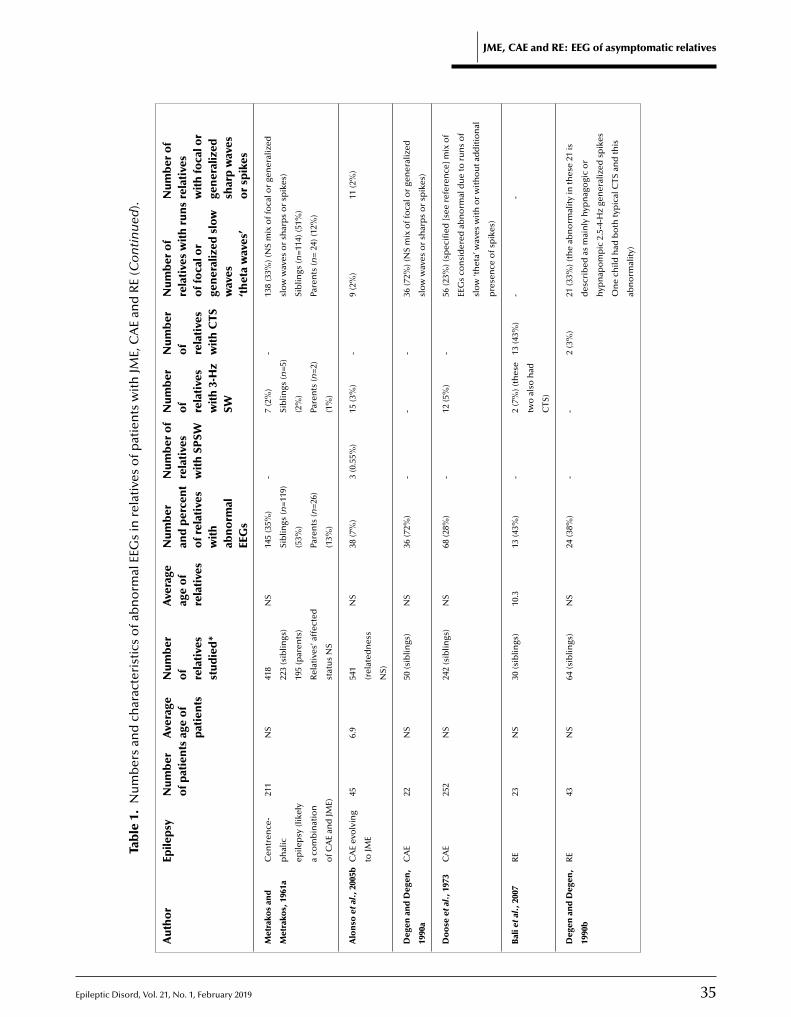
Epileptic Disord, Vol. 21, No. 1, February 2019 35
JME, CAE and RE: EEG of asymptomatic relatives
Tab
le1.
Nu
mb
ers
and
char
acte
rist
ics
ofa
bn
orm
alEE
Gs
inre
lati
ves
ofp
atie
nts
wit
hJM
E,C
AE
and
RE
(Co
nti
nu
ed).
Au
tho
rEp
ilep
syN
um
ber
ofp
atie
nts
Ave
rage
age
of
pat
ien
ts
Nu
mb
ero
fre
lati
ves
stu
die
d*
Ave
rage
age
of
rela
tive
s
Nu
mb
eran
dp
erce
nt
ofr
elat
ives
wit
hab
no
rmal
EEG
s
Nu
mb
ero
fre
lati
ves
wit
hSP
SW
Nu
mb
ero
fre
lati
ves
wit
h3-
Hz
SW
Nu
mb
ero
fre
lati
ves
wit
hC
TS
Nu
mb
ero
fre
lati
ves
wit
hru
ns
off
oca
lor
gen
eral
ized
slow
wav
es‘t
het
aw
aves
’
Nu
mb
ero
fre
lati
ves
wit
hfo
calo
rge
ner
aliz
edsh
arp
wav
eso
rsp
ikes
Met
rako
san
d
Met
rako
s,19
61a
Cen
tren
ce-
ph
alic
epile
psy
(lik
ely
aco
mb
inat
ion
ofC
AE
and
JME)
211
NS
418
223
(sib
lings
)
195
(par
ents
)
Rel
ativ
es’a
ffec
ted
stat
us
NS
NS
145
(35%
)
Sib
lings
(n=
119)
(53%
)
Pare
nts
(n=
26)
(13%
)
-7
(2%
)
Sib
lings
(n=
5)
(2%
)
Pare
nts
(n=
2)
(1%
)
-13
8(3
3%)(
NS
mix
off
oca
lor
gen
eral
ized
slo
ww
aves
or
shar
ps
or
spik
es)
Sib
lings
(n=
114)
(51%
)
Pare
nts
(n=
24)(
12%
)
Alo
nso
etal
.,20
05b
CA
Eev
olv
ing
toJM
E
456.
954
1
(rel
ated
nes
s
NS)
NS
38(7
%)
3(0
.55%
)15
(3%
)-
9(2
%)
11(2
%)
Deg
enan
dD
egen
,
1990
a
CA
E22
NS
50(s
iblin
gs)
NS
36(7
2%)
--
-36
(72%
)(N
Sm
ixo
ffo
calo
rge
ner
aliz
ed
slo
ww
aves
or
shar
ps
or
spik
es)
Do
ose
etal
.,19
73C
AE
252
NS
242
(sib
lings
)N
S68
(28%
)-
12(5
%)
-56
(23%
)(sp
ecifi
ed[s
eere
fere
nce
]mix
of
EEG
sco
nsi
der
edab
no
rmal
du
eto
run
so
f
slo
w‘th
eta’
wav
esw
ith
or
wit
ho
uta
dd
itio
nal
pre
sen
ceo
fsp
ikes
)
Bal
ieta
l.,2
007
RE
23N
S30
(sib
lings
)10
.313
(43%
)-
2(7
%)(
thes
e
two
also
had
CTS
)
13(4
3%)
--
Deg
enan
dD
egen
,
1990
b
RE
43N
S64
(sib
lings
)N
S24
(38%
)-
-2
(3%
)21
(33%
)(th
eab
no
rmal
ity
inth
ese
21is
des
crib
edas
mai
nly
hyp
nag
ogi
co
r
hyp
nap
om
pic
2.5-
4-H
zge
ner
aliz
edsp
ikes
On
ech
ildh
adb
oth
typ
ical
CTS
and
this
abn
orm
alit
y)
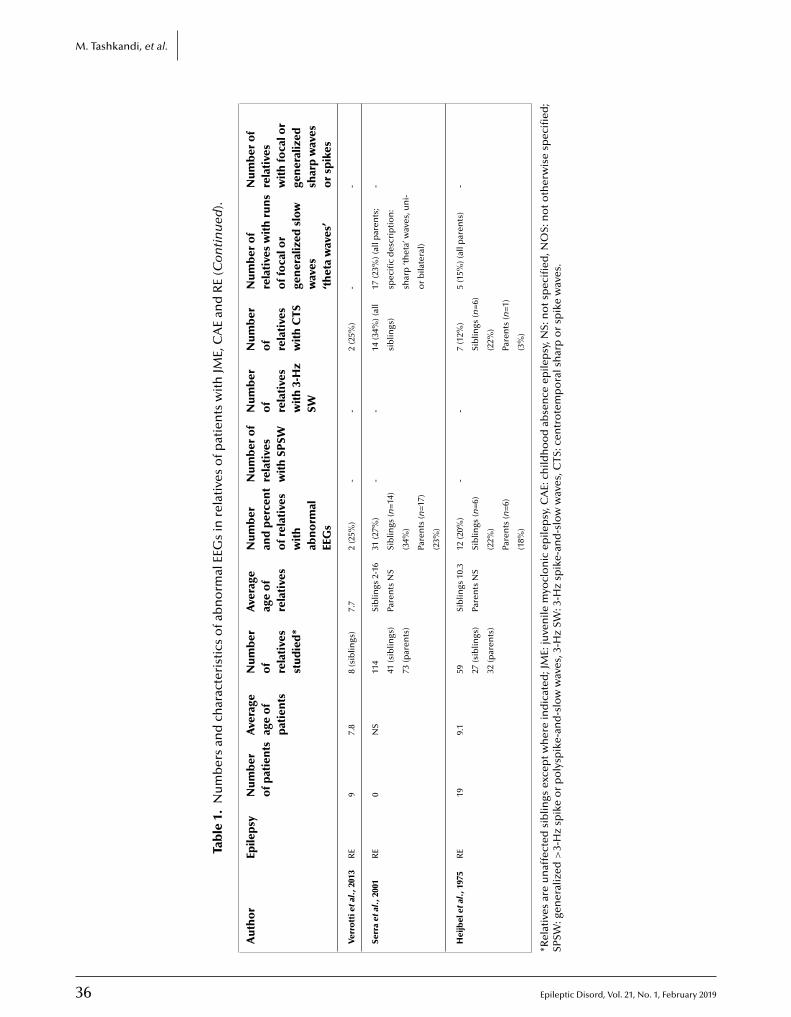
36 Epileptic Disord, Vol. 21, No. 1, February 2019
M. Tashkandi, et al.
Tab
le1.
Nu
mb
ers
and
char
acte
rist
ics
ofa
bn
orm
alEE
Gs
inre
lati
ves
ofp
atie
nts
wit
hJM
E,C
AE
and
RE
(Co
nti
nu
ed).
Au
tho
rEp
ilep
syN
um
ber
ofp
atie
nts
Ave
rage
age
of
pat
ien
ts
Nu
mb
ero
fre
lati
ves
stu
die
d*
Ave
rage
age
of
rela
tive
s
Nu
mb
eran
dp
erce
nt
ofr
elat
ives
wit
hab
no
rmal
EEG
s
Nu
mb
ero
fre
lati
ves
wit
hSP
SW
Nu
mb
ero
fre
lati
ves
wit
h3-
Hz
SW
Nu
mb
ero
fre
lati
ves
wit
hC
TS
Nu
mb
ero
fre
lati
ves
wit
hru
ns
off
oca
lor
gen
eral
ized
slow
wav
es‘t
het
aw
aves
’
Nu
mb
ero
fre
lati
ves
wit
hfo
calo
rge
ner
aliz
edsh
arp
wav
eso
rsp
ikes
Verr
ott
ieta
l.,2
013
RE
97.
88
(sib
lings
)7.
72
(25%
)-
-2
(25%
)-
-
Serr
aet
al.,
2001
RE
0N
S11
4
41(s
iblin
gs)
73(p
aren
ts)
Sib
lings
2-16
Pare
nts
NS
31(2
7%)
Sib
lings
(n=
14)
(34%
)
Pare
nts
(n=
17)
(23%
)
--
14(3
4%)(
all
sib
lings
)
17(2
3%)(
allp
aren
ts;
spec
ific
des
crip
tio
n:
shar
p‘th
eta’
wav
es,u
ni-
or
bila
tera
l)
-
Hei
jbel
etal
.,19
75R
E19
9.1
59 27(s
iblin
gs)
32(p
aren
ts)
Sib
lings
10.3
Pare
nts
NS
12(2
0%)
Sib
lings
(n=
6)
(22%
)
Pare
nts
(n=
6)
(18%
)
--
7(1
2%)
Sib
lings
(n=
6)
(22%
)
Pare
nts
(n=
1)
(3%
)
5(1
5%)(
allp
aren
ts)
-
*Rel
ativ
esar
eu
naf
fect
edsi
blin
gsex
cep
tw
her
ein
dic
ated
;JM
E:ju
ven
ilem
yocl
on
icep
ilep
sy,C
AE:
child
ho
od
abse
nce
epile
psy
,NS:
no
tsp
ecifi
ed,N
OS:
no
to
ther
wis
esp
ecifi
ed;
SPSW
:gen
eral
ized
>3-
Hz
spik
eo
rp
oly
spik
e-an
d-s
low
wav
es,3
-Hz
SW:3
-Hz
spik
e-an
d-s
low
wav
es,C
TS:c
entr
ote
mp
ora
lsh
arp
or
spik
ew
aves
.

E
emiRrh(ttIatGip72S1
S
Twasa4wbw(l(THfCwPp2
Q
Ttiq
D
To3
tersiddAlccps1tccCeascmelredfHnTtwtttriiiAlt(oha(bm
t al., 2010; Verrotti et al., 2013) were included in theeta-analysis. The pooled prevalence of abnormal EEG
n asymptomatic relatives of patients with JME, CAE andE was 30.51% (95% CI: 20.70, 40.33; I2=87.9%). Sepa-ating according to epilepsy syndromes showed theighest prevalence in CAE (41.82%), followed by RE
30.42%) and JME (21.10%). Grouping based on asymp-omatic siblings only (i.e. excluding other relatives),he overall prevalence was 34.76% (95% CI: 24.79, 44.73;2=79.61%), and by syndromes: CAE 41.8%, RE 33.76%,nd JME 26.57% (tabulated and detailed in supplemen-ary table 1 and figure 1).rouping according to characteristic EEG abnormal-
ties (SPSW, 3-Hz SW or CTS) or ‘other’, the pooledrevalences in asymptomatic relatives were: SPSW.14%, 3-Hz SW 5.40%, CTS 14.39%, ‘other’ waves3.56%, and PPR 9.04%. Restricting to siblings alone:PSW 7.74%, 3-Hz SW 5.40%, CTS 25.55%, and PPR4.13% (supplementary table 2 and figures 2-5).
ensitivity analysis
he sensitivity analysis included the 11 studies asell as the results reported in the large Metrakos
nd Metrakos (1961a) and Tsuboi and Christian (1973)tudies. The pooled prevalence of abnormal EEG insymptomatic relatives was 37.15% (95% CI: 25.53,8.76; I2=95.03%). In this case, the highest prevalenceas in relatives of patients with JME (42.41%) followedy CAE (38.43%), and RE (28.55%). When only siblingsere considered, the pooled prevalence was 41.80%
95% CI: 31.24, 52.35; I2=88.78%), divided between sib-ings of JME (43.66%), RE (33.76%), and CAE (46.41%)supplementary figure 6).he pooled prevalence for SPSW was 10.97%, for 3-z SW 3.57%, and CTS 14.39%. The pooled prevalence
or ‘other’ abnormalities was 31% and for PPR 9.04%.onsidering only siblings, the pooled prevalencesere SPSW 10.97%, 3-Hz SW 4.09%, CTS 25.55%, andPR 14.13% (supplementary figures 7-10). Finally, theooled prevalence of abnormal EEG in parents was8.79%.
uality of included studies
he quality of the included studies is provided inhe supplementary material. More than 50% of thencluded studies failed to ascertain exposure ade-uately.
pileptic Disord, Vol. 21, No. 1, February 2019
iscussion
he 15 studies reviewed in this work comprised a totalf 4,912 subjects including 1,054 epileptic patients and,858 relatives; large numbers that would be difficult
siato
JME, CAE and RE: EEG of asymptomatic relatives
o obtain in any one independent study. The high-st percentages of ‘abnormal’ EEG in asymptomaticelatives are obtained by combining all 15 studies (sen-itivity analysis) and focusing on siblings alone, whichs important given the age dependency of the syn-romes studied. The pooled number in that case is 42%istributed as 44% for JME, 34% for RE, and 47% for CAE.ccounting for missed abnormalities due to the short
ength of routine EEGs, the numbers are sufficientlylose to 50% to suggest that EEG abnormalities in theseommon syndromes are autosomal dominant traits, asroposed by the authors of the earliest and largesttudies (Metrakos and Metrakos, 1961a; Doose et al.,973; Tsuboi and Christian, 1973; Heijbel et al., 1975). Ifhese syndromes indeed include dominant Mendelianontributions to their EEG endophenotypes, the locusould possibly be shared across two (e.g. JME andAE) or more of the syndromes, or be different inach. But even in the latter case, if each of JME, CAEnd RE has an underlying dominant locus, it would beurprising that the mutations in these loci have notome to light in the current genomic era, in whichany hundreds of these patients have had whole-
xome or genome sequencing. It is possible that theseoci are in yet to be clarified non-coding genomicegions, or that in each case, there is wide genetic het-rogeneity with numerous loci separately acting as aominant predisposition for the EEG trait in separate
amilies.owever, when certain studies are excluded, theumbers change. For JME, if one excludes the largesuboi and Christian (1973) study (506 subjects) onhe grounds that the authors never quite specifiedhether the relatives were clinically affected or not,
he prevalence of EEG ‘abnormality’ drops to closeo 27%. Such a number, close to 25%, might suggesthat the EEG endophenotype of JME is an autosomalecessive trait (or a series of separate recessive traitsn different families). But if one looks closely at whats meant by EEG abnormalities in the different stud-es, the picture becomes even blurrier. For JME, in thetakli et al. (1999) study, 20% of siblings (10 of 48 sib-
ings studied) had SPSW, a number that approacheshe overall ∼25% figure. However, in the Akgun et al.2009) study, the percentage for SPSW was only 5% (onef 21 siblings studied), while another 29% of siblingsad ‘theta’ waves (table 1). What are the latter? Theyre bursts of slowing that are not quite epileptiformi.e. lack spikes), but are unexpected enough to haveeen labelled as an abnormality, or potential abnor-ality. This raises a major question. To what extent are
37
uch irregularities, which in the present age, for clin-cal purposes, would not be considered epileptiformctual subtle endophenotypes of potential relevanceowards understanding the genetic underpinningsf JME?

3
M
THtt3oa(iTatstsfaDthmidpirsmutTgfesabfeeiuttttlriififJbt
diigpda‘‘eiioCtncoJaun
SSa
ATCEehN
References
. Tashkandi, et al.
he above issues are even more pronounced in CAE.ere, the meta-analysis provides a figure of 42% and
he sensitivity analysis 46%. However, if one looks athe numbers of siblings of CAE patients who have-Hz SW, it is no more than 5%, the remainderf the percentage being made up of ‘theta’ wavesnd other non-specific abnormalities/irregularitiestable 1). Again, to what extent the latter constitutencomplete parts of the syndrome remains unknown.he situation is slightly clearer in RE. The percent-ges of EEG abnormalities in unaffected siblings inhe five RE studies range from 22 to 43% (table 1). Inome studies, the entire percentage is composed ofhe syndrome-specific CTS trait, while in the others,ubstantial portions of the percentages are derivedrom non-specific abnormalities such as ‘theta’ wavesnd generalized sharp waves (table 1). In the largeegen and Degen (1990b) study (64 siblings studied),
he vast majority of abnormalities are hypnagogic orypnapompic 2.5-4-Hz generalized spikes. This abnor-ality is not commonly discussed in RE, especially
n clinical practice, where the CTS is considered theefining feature. However, it has been reported as aarticularity of RE by other authors. Not all the RE stud-
es reviewed in the present paper included sleep EEGecording, and none performed overnight EEGs. Asuch, it is likely that the percentages of CTS or abnor-alities related to progression into or out of sleep are
nderestimated, suggesting a high EEG endopheno-ype(s) heritability in RE.he reported incidence of EEG abnormalities in theeneral non-epileptic population varies drasticallyrom les than 0·1% to 10% (Gibbs et al., 1943; Cavazzutit al., 1980). This 100-fold range is emblematic of theame issue as in the above studies of epileptic rel-tives, namely of the question as to what is meanty ‘abnormal’. Is uncommon ‘abnormal’, and by what
old should the frequency of a finding be higher inpileptic versus non-epileptic families to be consid-red ‘abnormal’? Clearly, the EEG in epileptic families
s substantially ‘different’ to that in the general pop-lation, with rates of ‘abnormality’ ranging from ∼25
o 50% in the former versus a maximum of 10% inhe latter, and therefore there is important informa-ion on the genetics of epilepsy within the EEG. Arait occurring at a frequency of 25% in siblings wouldikely be considered to be inherited in an autosomalecessive manner, and at 50% in an autosomal dom-nant, Mendelian manner. It is possible that ‘defects’n a single gene inherited in a recessive or dominantashion underlie the constellation of EEG ‘abnormal-
8
ties’ in each of the above epilepsies (i.e. one geneor RE-associated EEG ‘abnormalities’ and one each forME and CAE related-‘abnormalities’). It is also possi-le that defects in any number of single genes underlie
he set of ‘abnormalities’ associated with each syn-
Ans2
rome (in other words, that the EEG trait is inheritedn a Mendelian fashion but with genetic heterogeneity,.e. different JME families with, for example, segre-ating ‘defects’ in different single genes). Anotherossibility is that variants in different genes underlieifferent EEG ‘abnormalities’. Yet another is that vari-nts in multiple genes summate to result in a range of
abnormality’ from simply ‘uncommon’ features (e.g.theta waves’) to frank epileptiform spike waves. How-ver, it is important to note that a multiplicity of genes
nvolved cannot be very large, because otherwise ratesn the ‘Mendelian’ range of ∼25 to 50% would not bebserved.learly, much work lies ahead, but the genetic tools
hat were not available in the previous century of EEG,ow are. Future studies should carefully describe andorrelate EEG irregularities of age-appropriate relativesf epileptic patients with their genome sequences.
ME, CAE and RE families are highly likely to yield a rel-tively small number of genes that are important fornderstanding why and how otherwise, by and large,ormally developed brains seize.
Key points
• EEG ‘abnormalities’/variants in JME, CAE and REextend beyond their signature EEG traits and areshared with the general population.• EEG ‘abnormalities’/variants in JME, CAE and REare genetically less complex than the clinical syn-dromes and are useful endophenotypes.• Prevalences of EEG ‘abnormalities’/variants in JME,CAE and RE (21%, 42% and 33%, respectively) arewithin the Mendelian inheritance range.• EEG endophenotypes of JME, CAE and RE shouldfacilitate identification of genes contributing tohypersynchrony in these common epilepsies.
upplementary data.ummary didactic slides and supplementary material are avail-ble on the www.epilepticdisorders.com website.
cknowledgements and disclosures.his work was funded by the Ontario Brain Institute, Genomeanada and the University of Toronto Michael Bahen Chair inpilepsy Research. BAM holds the University of Texas Southwest-rn Jimmy Elizabeth Westcott Chair in Pediatric Neurology. ACTolds a Tier 2 Canada Research Chair in Knowledge Synthesis.one of the authors have any conflict of interest to declare.
Epileptic Disord, Vol. 21, No. 1, February 2019
kgun Y, Soysal A, Atakli D, et al. Cortical excitability in juve-ile myoclonic epileptic patients and their asymptomaticiblings: a transcranial magnetic stimulation study. Seizure009; 18: 387-91.

E
Ai2
Ai2
Apc
Ape
Ae
BoE
Bs
Bt1
Ce1
Cm
DpE
De1
Dr1
Dm
Ds
Fcm
GcN
Hh1
Hm
Jaa2
Mc(
MIc
Mi1
Mi(
Pl
Pa
PpiGE
Pi1
Soi
Tam
Vel
Vlt
Vct2
Vm
W
lonso ME, Medina MT, Martínez-Juárez IE, et al. Famil-al juvenile myoclonic epilepsy. Adv Neurol 2005a; 95:27-43.
lonso ME, Medina MT, Martínez-Juárez IE, et al. Famil-al juvenile myoclonic epilepsy. Adv Neurol 2005b; 95:27-43.
nderman E, Metrakos JD. EEG studies of relatives ofrobands with focal epilepsy who have been treated surgi-ally. Epilepsia 1969; 10: 415.
takli D, Soysal A, Atay T, et al. Somatosensory evokedotentials and EEG findings in siblings of juvenile myoclonicpilepsy patients. Epileptic Disord 1999; 1: 173-7.
vanzini G, Noebels JL. Genetics of epilepsy and geneticpilepsies. Montrouge: John Libbey Eurotext, 2009.
ali B, Kull LL, Strug LJ, et al. Autosomal dominant inheritancef centrotemporal sharp waves in rolandic epilepsy families.pilepsia 2007; 48: 2266-72.
erg AT, Millichap JJ. The 2010 revised classification ofeizures and epilepsy. Continuum 2013; 19: 571-97.
ray PF, Wiser WC. Evidence for a genetic etiology ofemporal-central abnormalities in focal epilepsy. N Engl J Med964; 271: 926-33.
avazzuti GB, Capella L, Nalin A. Longitudinal study ofpileptiform EEG patterns in normal children. Epilepsia980; 21: 43-55.
amfield CS, Striano P, Camfield PR. Epidemiology of juvenileyoclonic epilepsy. Epilepsy Behav 2013; 28: S2-7.
egen R, Degen HE, Roth C. Some genetic aspects of idio-athic and symptomatic absence seizures: waking and sleepEGS in siblings. Epilepsia 1990a; 31: 784-94.
egen R, Degen HE. Some genetic aspects of rolandicpilepsy: waking and sleep EEG in siblings. Epilepsy990b; 31: 795-801.
egen R, Degen HE. Contribution to the genetics ofolandic epilepsy: waking and sleep EEGs in siblings. Epilepsy992; 6: 49-52.
elgado-Escueta AV. Advances in genetics of juvenileyoclonic epilepsies. Epilepsy Curr 2007; 7: 61-7.
oose H, Gerken H, Horstmann T, et al. Genetic factors inpike-wave absences. Epilepsia 1973; 14: 57-75.
nais N, Soobiah C, Chen MH, et al. Harassment and dis-rimination in medical training: a systematic review andeta-analysis. Acad Med 2014; 89: 817-21.
ibbs FA, Gibbs EL, Lennox WG. Electroencephalographiclassification of epileptic patients and control subjects. Archeurol Psychiatry 1943; 50: 111-28.
pileptic Disord, Vol. 21, No. 1, February 2019
eijbel J, Blom S, Rasmuson M. Benign epilepsy of child-ood with centrotemporal EEG foci: a genetic study. Epilepsia975; 16: 285-93.
iggins JP, Thompson SG. Quantifying heterogeneity in aeta-analysis. Stat Med 2002; 21: 1539-58.
mt
Ss
JME, CAE and RE: EEG of asymptomatic relatives
ayalakshmi SS, Mohandas S, Sailaja S, et al. Clinicalnd electroencephalographic study of first-degree relativesnd probands with juvenile myoclonic epilepsy. Seizure006; 15: 177-83.
cGowan J, Sampson M, Lefebvre C. An evidence basedhecklist for the peer review of electronic search strategiesPRESS EBC). Evid Based Libr Inf Pract 2010; 5: 149-54.
etrakos K, Metrakos JD. Genetics of convulsive disorders.I. Genetic and electroencephalographic studies in centren-ephalic epilepsy. Neurology 1961a; 11: 474-83.
etrakos K, Metrakos JD. Is the centrencephalic EEG inher-ted as a dominant? Electroencephalogr Clin Neurophysiol961b; 13: 289.
oher D, Shamseer L, Clarke M, et al. Preferred report-ng items for systematic review and meta-analysis protocolsPRISMA-P) 2015 statement. Syst Rev 2015; 4: 1.
al DK, Ferrie C, Addis L, et al. Idiopathic focal epilepsies: “theost tribe”. Epileptic Disord 2016; 18: 1-37.
anayiotopoulos CP. A clinical guide to epileptic syndromesnd their treatment. New York: Springer Publishing, 2010.
anayiotopoulos CP, Bureau M, Caraballo RH, et al. Idio-athic focal epilepsies in childhood. In: Epileptic syndromes
n infancy, childhood and adolescence. 5th Ed. Bureau M,enton P, Dravet C, et al. Montrouge, France: John Libbey
urotext, 2012.
enfield W. Epileptic automatism and centrencephalicntegrating system. Res Publ Assoc Res Nerv Ment Dis952; 30: 513-28.
erra D, Congiu R, Minafra L, et al. EEG trait analysis in siblingsf patients with benign rolandic epilepsy. Bollettino-Lega Ital-
ana Contro L’Epilessia 2001; 113-4: 267-8.
suboi T, Christian W. On the genetics of the primary gener-lized epilepsy with sporadic myoclonias of impulsive petital type. Humangenetik 1973; 19: 155-82.
aldamudi L, Kjeldsen MJ, Corey LA, et al. Analyzing thetiology of benign rolandic epilepsy: a multicenter twin col-
aboration. Epilepsia 2006; 47: 550-5.
errotti A, Matricardi S, Di Giacomo DL, et al. Neuropsycho-ogical impairment in children with rolandic epilepsy and inheir siblings. Epilepsy Behav 2013; 28: 108-12.
errotti A, Casciato S, Spalice A, et al. Coexistence ofhildhood absence epilepsy and benign epilepsy with cen-rotemporal spikes: a case series. Eur J Paediatr Neurol017; 21: 570-5.
iechtbauer W. Conducting meta-analyses in R with theetafor package. J Stat Softw 2010; 36: 1-48.
andschneider B, Kopp UA, Kliegel M, et al. Prospectiveemory in patients with juvenile myoclonic epilepsy and
39
heir healthy siblings. Neurology 2010; 75: 2161-7.
Wells GA, Shea B, O’Connell D, et al. The Newcastle-Ottawacale (NOS) for assessing the quality of nonrandomizedtudies in meta-analysis. 2014.
40 Epileptic Disord, Vol. 21, No. 1, February 2019
M. Tashkandi, et al.
TEST YOURSELFEDUCATION
(1) What is the approximate reported prevalence of EEG abnormalities in first-degree relatives of patients withjuvenile myoclonic, childhood absence and Rolandic epilepsies?A. 0-1%B. 10-20%C. 20-50%
(2) Are the reported EEG abnormalities in first-degree relatives of patients with juvenile myoclonic, childhoodabsence and Rolandic epilepsies true abnormalities?
(3) Are EEG changes found in relatives of patients with juvenile myoclonic, childhood absence and Rolandicepilepsies developmental stage specific?
Note: Reading the manuscript provides an answer to all questions. Correct answers may be accessed on thewebsite, www.epilepticdisorders.com, under the section “The EpiCentre”.
Appendix 1. Quality of included studies.
First author Year Representativenessof the exposedcohort
Ascertainmentof exposure
Comparabilityof cohorts on thebasis of the designor analysis
Assessmentof outcome
Adequacyof response rate
Alonso 2005 B A C A A
Atakli 1999 B A A D A
Akgun 2009 B A A D A
Jayalakshmi 2006 B D C A A
Tsuboi 1973 A C A B A
Wandschneider 2010 C D A D A
Metrakos 1961 A A B A
Degen 1990 B D C D A
Doose 1973 C D B A A
Bali 2007 B A A A A
Degen 1990 B D C D A
Verrotti 2013 B D A A B
Serra 2001 D D C D D
Heijbel 1975 A A C B A

E
JME, CAE and RE: EEG of asymptomatic relatives
Newcastle Ottawa Scale
Selection1) Representativeness of the exposed cohort
a) Truly representative of the average individualb) Somewhat representative of the average individualc) Selected group of usersd) No description of the derivation of the cohort
2) Ascertainment of exposurea) Secure record (e.g. surgical records)b) Structured interviewc) Written self reportd) No description
Comparability1) Comparability of cohorts on the basis of the design or analysis
a) Study controls for age or genderb) Study controls for any additional factor (e.g. body mass index, comorbidity)c) No control
Outcome1) Assessment of outcome
a) Independent blind assessmentb) Record linkagec) Self reportd) No description
2) Adequacy of response ratea) All subjects accounted forb) Subjects lost unlikely to introduce bias - small number lost (<10%)
pileptic Disord, Vol. 21, No. 1, February 2019 41
c) Subject loss >10%d) No statement

4
V
CMDI4<
Original articleEpileptic Disord 2019; 21 (1): 42-7
DEPDC5 mutation and familialfocal epilepsy with variablefoci: genotype and phenotypeof a family*
Marina Aberastury 1, Romina Fernández 1, Marta Córdoba 2,3,Betiana Comas 1, Martín Peralta 4, Guillermo Agosta 5,Marcelo Kauffman 2, Walter Silva 1
1 Division of Neuropediatrics, Italian Hospital of Buenos Aires, CABA2 Consultorio y laboratorio de Neurogenética, Centro Universitario de Neurología “J.M.Ramos Mejía” and División Neurología, Hospital JM Ramos Mejía, Facultad de Medicina,UBA, CABA3 IBCN Eduardo de Robertis, Facultad de Medicina, UBA-CONICET, CABA4 Bahía Blanca Italian Hospital, Bahía Blanca, Buenos Aires5 Neurología Infantil, Hospital Italiano de Buenos Aires, CABA, Argentina
Received April 14, 2018; Accepted November 01, 2018
ABSTRACT – Aims. Familial focal epilepsy with variable foci is a relativelyrare autosomal disease with an unclear incidence, which is characterizedby focal seizures arising from different cortical regions in different familymembers.Methods. We describe three members of a two-generation Argentine fam-ily with familial focal epilepsy with variable foci syndrome and a DEPDC5gene mutation.Results. The mean onset age was nine years old. The father experiencedepisodes with occipital semiology and both siblings exhibited frontal lobeseizures. Their neurological examination and neuroimaging studies werenormal. All three patients are currently seizure-free, in spite of initially expe-riencing frequent seizures. Complete exome sequencing revealed a newDEPDC5 gene mutation (NM_001242896: c.4718T>C; p.L1573P).Conclusions. This study of a family with clinical characteristics that metall the criteria for familial focal epilepsy with variable foci demonstrates theusefulness of exome sequencing as a diagnostic tool. [Published with video
do
i:10.1684/epd
.2019.1025
2 Epileptic Disord, Vol. 21, No. 1, February 2019
IDEO ONLINE
orrespondence:arina Aberasturyivision of Neuropediatrics,
talian Hospital of Buenos Aires,135 Potosí St., CABA [email protected]>
sequence on www.epilepticdisorders.com]
Key words: familial focal epilepsy with variable foci, DEPDC5, semiology,occipital seizure semiology, frontal seizures
*This work was presented at the 31st International Epilepsy Congress, Istanbul, 2015 andCongreso de Neurología Infantil, 2016, SANI, Buenos Aires, Argentina.

E
FigAesfs1FwttloeMlrdcNpdDcaiteteimoieSDeaw
P
C
WfteAtm
Ifl
E
WpAlsf(wdi
C
ThosidvtsflTiTtleomaAeilsasfTeAa
amilial focal epilepsy with variable foci (FFEVF) isncluded among epilepsy syndromes of genetic ori-in (Dibbens et al., 2013). It was first reported in anustralian family in 1998 by Ingrid Scheffer (Scheffert al., 1998), who initially associated it with chromo-ome 2. Later, in 1999, it was reported in two Canadianamilies, suggesting linkage to chromosome 22 andhowing probable genetic heterogeneity (Xiong et al.,999; Klein et al., 2012).FEVF is characterized by a wide range of onset age,ith an average of 13 years. Epileptic seizures may
ake place during daytime, night-time, or both, andhey frequently originate in the frontal or temporalobes, although they may occasionally be of occipitalr parietal origin (Morales-Corraliza et al., 2010; Ishidat al., 2013).ost patients with this syndrome have a normal neuro-
ogical examination, although there have been isolatedeports in which autistic spectrum disorder, psychiatricisorders, and intellectual disability were present asomorbidities (Klein et al., 2012).euroimaging studies are also often normal and mostatients show an excellent response to antiepilepticrugs (AEDs) (Morales-Corraliza et al., 2010).EPDC5 is an important gene in focal epilepsy, espe-
ially in patients with a positive family history (Tsai etl., 2017). Mutations in this gene have been identifiedn more than 8% of families with FFEVF, causing activa-ion of the downstream mTOR pathway (Weckhuysent al., 2016). Reports suggest that DEPDC5 is not onlyhe most common gene associated with familial focalpilepsy but also could be a significant gene involved
n sporadic focal epilepsy (Tsai et al., 2017). DEPDC5utation has also been linked to an increased risk
f sudden unexpected death in epilepsy (SUDEP), ast is described in one family with DEPDC5-relatedpilepsy which included two family members withUDEP (Nascimento et al., 2015). The significance ofEPDC5 mutations in patients with sporadic focalpilepsy has yet to be characterized. Here, we describen Argentine family meeting all the criteria for FFEVFith DEPDC5 gene mutation.
atients and methods
linical studies
e studied two generations of a non-consanguineousamily. All three affected members had focal epilepsy:
pileptic Disord, Vol. 21, No. 1, February 2019
wo had frontal epilepsy and one had occipitalpilepsy.32-channel video-EEG recording was carried out for
he proband and an EEG for the other two affectedembers of the family.
TiaTa
DEPDC5 mutation and familial focal epilepsy
nformed consent was obtained from each participantamily member or, in the case of the two children, theiregal guardian.
xome sequencing and Sanger sequencing
hole-exome sequencing (WES) was performed onurified DNA samples from the patient using thegilent SureSelect Human All Exon V5 Kit (Agi-
ent Technologies, Santa Clara, CA) with an Illuminaequencing system. Bioinformatic analysis was per-ormed following procedures described by our groupKoile et al., 2018). The identified variant in DEPDC5as validated by Sanger sequencing following stan-ard procedures. The presence of this variant was
nvestigated in affected members of the family.
linical description
he proband was a 10-year-old male who experiencedis first seizure at the age of eight. At first, episodesccurred more than 20 times per day, during bothleep and awake states. The episodes were character-zed by eye opening, followed by ocular and cephaliceviation to the left, associated with monosyllabicocalization. He then presented right hand automa-isms associated with left upper limb flexion. Duringome episodes, he extended the lower limbs andexed the upper limbs, as if he was stretching out.hese events were occasionally associated with smil-
ng (mainly while awake).he patient was initially treated with valproic acid,hen clobazam and topiramate. While receiving theatter, he experienced visual hallucinations and hyper-xcitability. An increase in seizure frequency wasbserved when levetiracetam was added to this treat-ent regimen. He was then started on carbamazepine
nd became seizure-free.24-hour video telemetry recording revealed seven
vents. The ictal EEG showed sharp rhythmic spikesn the right fronto-central region, followed by fastow-voltage activity and, four or five seconds later, aharp-and-slow-wave bilateral fronto-central temporalctivity (figure 1, video sequence). The interictal EEGhowed frequent spike-and-sharp-wave dischargesrom the right fronto-central temporal region (figure 2).hese observations led to the hypothesis that thepisodes originated in the right fronto-temporal area.3-tesla MRI brain scan was performed showing no
bnormalities.
43
he neuropsychological assessment showed normalntellectual performance with language difficulties andttention deficit.he proband’s father presented with episodes withsemiology suggesting occipital lobe involvement;
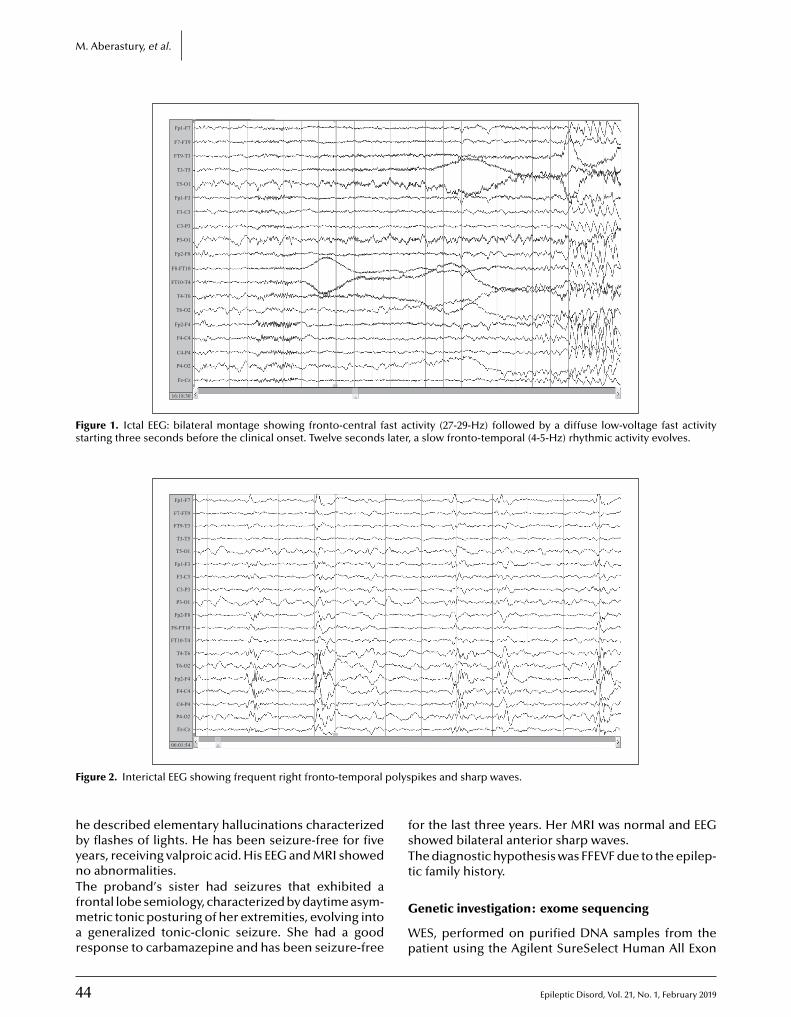
4
M. Aberastury, et al.
Fp1-F7
F7-FT9
FT9-T3
T3-T5
T5-O1
Fp1-F3
F3-C3
C3-P3
P3-O1
Fp2-F8
F8-FT10
FT10-T4
T4-T6
T6-O2
Fp2-F4
F4-C4
C4-P4
P4-O2
Fz-Cz
16:18:30
Figure 1. Ictal EEG: bilateral montage showing fronto-central fast activity (27-29-Hz) followed by a diffuse low-voltage fast activitystarting three seconds before the clinical onset. Twelve seconds later, a slow fronto-temporal (4-5-Hz) rhythmic activity evolves.
Fp1-F7
F7-FT9
FT9-T3
T3-T5
T5-O1
Fp1-F3
F3-C3
C3-P3
P3-O1
Fp2-F8
F8-FT10
FT10-T4
T4-T6
T6-O2
Fp2-F4
F4-C4
C4-P4
P4-O2
Fz-Cz
06:01:54
F polys
hbynTfmar
fsTt
igure 2. Interictal EEG showing frequent right fronto-temporal
e described elementary hallucinations characterizedy flashes of lights. He has been seizure-free for fiveears, receiving valproic acid. His EEG and MRI showedo abnormalities.
4
he proband’s sister had seizures that exhibited arontal lobe semiology, characterized by daytime asym-
etric tonic posturing of her extremities, evolving intogeneralized tonic-clonic seizure. She had a good
esponse to carbamazepine and has been seizure-free
G
Wp
pikes and sharp waves.
or the last three years. Her MRI was normal and EEGhowed bilateral anterior sharp waves.he diagnostic hypothesis was FFEVF due to the epilep-ic family history.
Epileptic Disord, Vol. 21, No. 1, February 2019
enetic investigation: exome sequencing
ES, performed on purified DNA samples from theatient using the Agilent SureSelect Human All Exon

E
Vaogtpa
D
TocntppFowh(FesfFlooPieidnrarisAKnwoeleTwddwt
IvfteTtrwfTFwwgpfmisepgaowufTmaDma(AlDaRamDpprhnmNb
5 Kit (Agilent Technologies, Santa Clara, CA) withn Illumina sequencing system, led to identificationf a likely pathogenic novel mutation in the DEPDC5ene (NM_001242896: c.4718T>C; p.L1573P). Segrega-ion analysis by Sanger sequencing confirmed theresence of this variant in the proband and in the otherffected relatives.
iscussion
he main familial focal epilepsies of known geneticrigin with specific age-related and electroclinicalharacteristics include: autosomal dominant noctur-al frontal lobe epilepsy (ADNFLE), familial mesial
emporal lobe epilepsy (FMTLE), familial lateral tem-oral lobe epilepsy TLE (FLTLE) or autosomal dominantartial epilepsy with auditory features (ADPEAF), andFEVF. These familial syndromes show phenotypicverlap and small families may be initially labelledith ADNFLE or FLTLE/ADPEAF, and later recognized toave FFEVF when new affected members are identified
Dibbens et al., 2013).FEVF is a relatively rare autosomal dominant dis-ase with an unclear incidence, characterized by focaleizures arising from different cortical regions in dif-erent family members (Klein et al., 2012). Reports ofFEVF describe a mean age at onset of 13 years with aarge range extending from one month to 52 years. Inur study, the proband had his first seizure at the agef eight, the father at ten, and the sibling at age nine.atients with FFEVF usually have normal neurolog-cal examination and normal neuroimaging (Kleint al., 2012). However, based on studies published
n 2015, patients with FFEVF and focal neurologicaleficit (hemiparesis) were reported, together witheuroimaging findings of focal cortical dysplasia. Theesponse to AEDs is variable. While some individu-ls respond well to first-line AEDs, others are moreefractory to treatment. All members of the study fam-ly had a normal neurological examination and becameeizure-free on AEDs.DNFLE (involving CHRNA4, CHRNA2, CHRNB2, andCNT1 gene mutations) is characterized by shortocturnal episodes, usually presenting as a clusterith hypermotor seizures, which are also commonlybserved in FFEVF (Callenbach et al., 2003; Dibbenst al., 2013; Ishida et al., 2013). As a consequence, the
atter is often misdiagnosed as the former, althoughpisodes during daytime are rare in this epilepsy type.he patient we studied presented with focal episodesith and without loss of conscience, which took place
pileptic Disord, Vol. 21, No. 1, February 2019
uring the day and the night, initially with multipleaily seizures. Therefore, video telemetry monitoringas performed, allowing us to hypothesize a fron-
otemporal epileptogenic origin.
wtNca
DEPDC5 mutation and familial focal epilepsy
n contrast to other FFEVF families which include indi-iduals with nocturnal frontal lobe epilepsy (NFLE), ouramily could easily be distinguished from NFLE dueo the predominant diurnal seizures and a posteriorpilepsy in the other member of the family.here have been reports of EEG studies with interic-al focal discharges, such as in our patients and oneeport from a French-Canadian family who presentedith normal interictal EEG (Xiong et al., 1999), as in the
ather.he penetrance of DEPDC5 mutation associated withFEVF was estimated at 66%, and obligate gene carriersithout a history of seizures can often be identifiedithin a family (Dibbens et al., 2013). In this study,enetic testing (PCR followed by LOD scores) was onlyerformed in the three symptomatic members of the
amily. In all of them, FFEVF was associated with chro-osome 22q12. We could not perform genetic testing
n other family members. In 2012, 16 families with auto-omal dominant focal epilepsy were reassessed andxome sequencing was carried out in all cases. Twoatients with FFEVF presented with the same DEPDC5ene mutation (deletion), which was also detected insymptomatic relatives. This strengthens the theoryf incomplete penetrance and expression variabilityithin a family, suggesting that the phenotype is mod-lated by other genes or environmental and epigenetic
actors (Ishida et al., 2013).he mutation described here would introduce a pre-ature stop, causing loss of DEPDC5 gene function
nd the subsequent epilepsy phenotype. Neither theEPDC5 nor the LGI1 gene (associated with autoso-al dominant lateral temporal lobe epilepsy) encodetransmembrane receptor subunit or ion channel
Ishida et al., 2013). In another study, the exomes of anustralian family and a Dutch family were sequenced,
eading to the detection of a nonsense mutation of theEPDC5 gene. Thus, different DEPDC5 gene mutations
re associated with FFEVF (Dibbens et al., 2013).ecently, in 2014, Ingrid Scheffer confirmed the vari-bility of phenotypes associated with DEPDC5 geneutations. She identified a nonsense variant of theEPDC5 gene in two siblings with focal cortical dys-lasia type IIA by exome sequencing, which hadreviously been associated only with normal neu-oimaging studies. The father and paternal uncle iner study had the same mutation, but had normaleuroimaging studies and good response to carba-azepine (Scerri et al., 2015). A missense variant of theF1 and DEPTOR genes was also found, but only inoth siblings with focal cortical dysplasia. In our study,e did not find other gene mutations in any of the
45
hree patients. The three genes, DEPDC5, DEPTOR, andF1, encode components of the mTOR pathway, which
ould contribute to the phenotype variability associ-ted with the DEPDC5 gene (which inhibits mTORC1),

4
M
cagtRsDDedacdbiTtroveW
C
Fdltfa
DN
R
Bf2
CFcm
DDG
Ia5
Kwo
KYsm
MGwc
NDe2
Rme
StT
Sew1
Sme7
Tf4
W
. Aberastury, et al.
ausing both lesional and non-lesional epilepsy. Thessociation between DEPDC5 and the mTOR pathwayenes and the presence of cortical malformations inhese patients is still unclear.ecently, with regards to FFEVF, advances in exomeequencing have revealed an association betweenEPDC5 gene mutation and other genes such asEPTOR and NF1, which could be linked to severepilepsy with focal cortical malformations (corticalysplasia type IIA and focal heterotopia) (Scerri etl., 2015; Baulac et al., 2015; Tsai et al., 2017). Nolear genotype-phenotype correlations have beenescribed, although, to date, missense variants haveeen reported mostly in small families including
ndividuals with apparently non-lesional epilepsies.he family presented here had a missense muta-ion with normal MRI. Moreover, all individuals witheported brain malformations (focal cortical dysplasiar hemimegalencephaly) had nonsense or frameshiftariants leading to a premature stop codon (Scheffert al., 2014; Scerri et al., 2015; Ricos et al., 2016;eckhuysen et al., 2016).
onclusion
FEVF is a genetic epilepsy syndrome with autosomalominant inheritance, incomplete penetrance, and
arge phenotypic variability. We emphasize the impor-ance of the patient’s family medical history as a basisor selecting relevant diagnostic testing which leads toccurate diagnosis and subsequent management. �
Legend for video sequenceA typical seizure of the patient. The seizure semi-ology starts three seconds from the first change inEEG with eye opening, followed by ocular and leftversive cephalic deviation, associated with vocal-ization. The patient then presents with right handautomatisms associated with left lower limb flexion,followed by bilateral manual automatisms.
Key words for video research onwww.epilepticdisorders.com
Phenomenology: focal seizureLocalisation: variable fociSyndrome: familial focal epilepsy with variable foci
6
Aetiology: DEPDC5 mutation
isclosures.one of the authors have any conflict of interest to declare.
Gc
Xi2
eferences
aulac S, Shida S, Marsan E, et al. Familial focal epilepsy withocal cortical displasia due to DEPDC5 mutations. Ann Neurol015; 77: 675-83.
allenbach P, Van den Maagdenberg A, Hottenga J, et al.amilial partial epilepsy with variable foci in a Dutch family:linical characteristics and confirmation of linkage to chro-osome 22q. Epilepsia 2003; 44: 1298-305.
ibbens L, De Vries B, Donatello S, et al. Mutations inEPDC5 cause familial focal epilepsy with variable foci. Natenet 2013; 45: 546-51.
shida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 causeutosomal dominant focal epilepsies. Nat Genet 2013; 45:52-5.
lein KM, O’Brien TJ, Praveen K, et al. Familial focal epilepsyith variable foci mapped to chromosome 22q12: expansionf the phenotypic spectrum. Epilepsia 2012; 53: e151-5.
oile D, Cordoba M, De Sousa Serro M, Kauffman MA,ankilevich P. GenIO: a phenotype-genotype analysis weberver for clinical genomics of rare diseases. BMC Bioinfor-atics 2018; 19: 25.
orales-Corraliza J, Gomez-Garre P, Sanz R, Diaz Otero G,utierrez Delicado E, Serratosa JM. Familial focal epilepsyith variable foci: a new family with suggestion of linkage to
hromosome 22q12. Epilepsia 2010; 51: 1910-4.
ascimento FA, Borlot F, Cossette P, Minassian BA, AndradeM. Two definite cases of sudden unexpected death inpilepsy in a family with a DEPDC5 mutation. Neurol Genet015; 1: e28.
icos MG, Hodgson BL, Pippucci T, et al. Mutations in theTOR pathway regulators NPRL2 and NPRL3 cause focal
pilepsy. Ann Neurol 2016; 79: 120-31.
cerri T, Riseley J, Gillies G, et al. Familial cortical displasiaype IIA caused by a germline mutation in DEPDC5. Ann Clinransl Neurol 2015; 2: 575-80.
cheffer IE, Phillips HA, O’Brien CE, et al. Familial partialpilepsy with variable foci: a new partial epilepsy syndromeith suggestion of linkage to chromosome 2. Ann Neurol
998; 44: 890-9.
cheffer IE, Heron SE, Regan BM, et al. Mutations in mam-alian target of rapamycin regulator DEPDC5 cause focal
pilepsy with brain malformations. Ann Neurol 2014; 75:82-7.
sai MH, Chan CK, Chang YC, et al. DEPDC5 mutations inamilial and sporadic focal epilepsy. Clin Genet 2017; 92: 397-04.
eckhuysen S, Marsan E, Lambrecq V, et al. Involvement ofATOR complex genes in familial focal epilepsies and focal
ortical dysplasia. Epilepsia 2016; 57: 994-1003.
iong L, Labuda M, Li D, et al. Mapping of a gene determin-
Epileptic Disord, Vol. 21, No. 1, February 2019
ng familial focal epilepsy with variable foci to chromosome2q11-q12. Am J Hum Genet 1999; 65: 1698-710.

Epileptic Disord, Vol. 21, No. 1, February 2019 47
DEPDC5 mutation and familial focal epilepsy
TEST YOURSELFEDUCATION
(1) In what other epileptic syndromes can DEPDC5 mutations be found?
(2) Can patients with DEPDC5 mutations have brain malformations?
(3) What genetic counselling would you give to a patient with DEPDC5-related epilepsy?
Note: Reading the manuscript provides an answer to all questions. Correct answers may be accessed on thewebsite, www.epilepticdisorders.com, under the section “The EpiCentre”.

do
i:10.1684/epd
.2019.1026
48 Epileptic Disord, Vol. 21, No. 1, February 2019
Correspondence:Shinsaku YoshitomiNational Epilepsy Center,NHO Shizuoka Institute of Epilepsyand Neurological Disorder,886 Urushiyama, Aoi-ku,Shizuoka 420-8688, Japan<[email protected]>
Original articleEpileptic Disord 2019; 21 (1): 48-54
Quinidine therapyand therapeutic drugmonitoring in four patientswith KCNT1 mutations
Shinsaku Yoshitomi 1, Yukitoshi Takahashi 1,Tokito Yamaguchi 1, Taikan Oboshi 1, Asako Horino 1,Hiroko Ikeda 1, Katsumi Imai 1, Tohru Okanishi 2,Mitsuko Nakashima 3,4, Hirotomo Saitsu 3,4,Naomichi Matsumoto 3, Jun Yoshimoto 5, Takako Fujita 6,Atsushi Ishii 6, Shinichi Hirose 6, Yushi Inoue 1
1 National Epilepsy Center, NHO Shizuoka Institute of Epilepsy and NeurologicalDisorders, Shizuoka2 Seirei Hamamatsu General Hospital, Department of Child Neurology, Hamamatsu3 Yokohama City University Graduate School of Medicine, Department of HumanGenetics, Yokohama4 Hamamatsu University School of Medicine, Department of Biochemistry, Hamamatsu,5 Shizuoka Children’s Hospital, Department of Cardiology, Shizuoka6 Department of Pediatrics School of Medicine, Fukuoka University, Fukuoka, Japan
Received December 23, 2017; Accepted November 06, 2018
ABSTRACT – Aims. Several recent studies have reported potassiumsodium-activated channel subfamily T member 1 (KCNT1) mutations inepilepsy patients on quinidine therapy. The efficacy and safety of quinidinefor epilepsy treatment, however, remains controversial.Methods. We herein report the cases of four patients with KCNT1 muta-tions treated with quinidine.Results. A reduction in seizures of more than 50% after quinidine treat-ment was observed in one patient with epilepsy of infancy with migratingfocal seizures (EIMFS), whereas two patients with EIMFS and one withfocal epilepsy did not achieve apparent seizure reduction. The rela-tionship between quinidine dose and serum quinidine concentrationwas inconsistent, particularly at high quinidine doses. One patient withEIMFS developed ventricular tachycardia the day after an increase inquinidine dose from 114 to 126 mg/kg/day. The serum trough quinidineconcentration and the corrected QT interval (QTc) before arrhythmiaonset were 2.4 �g/ml and 420 ms, respectively, and peak serum quinidineconcentration after arrhythmia onset was 9.4 �g/ml. Another patient withEIMFS showed aberrant intraventricular conduction with a quinidine doseof 74.5 mg/kg/day and a serum trough concentration of 3.2 �g/ml.Conclusions. Given that serum quinidine levels may elevate sharply aftera dose increase, careful monitoring of electrocardiographs and serumconcentrations is required. Based on a review of previous reports and ourexperience with this case, quinidine should be considered as a promising

E
Quinidine drug monitoring and KCNT1 mutation
drug for patients with EIMFS harbouring KCNT1 mutations, however, its effi-cacy remains controversial due to the limited number of cases, and moreinformation on optimal serum concentrations and appropriate titrationmethods is required.
T1,eizu
PmpcBtatotw(tnRbseFedotiqdmpGptcIhac
C
P
PgepabE
cbnpTiCtedde(icmptqToQtaa7fcm4dTtlihw
P
Poo
Key words: KCNmigrating focal s
otassium sodium-activated channel subfamily Tember 1 (KCNT1) encodes a sodium-activated
otassium channel that is highly expressed in theentral nervous system (Bhattacharjee et al., 2002;hattacharjee and Kaczmarek, 2005). KCNT1 con-
ributes to neuronal excitability and subsequent firings well as modulation of the resting membrane poten-ial (Bhattacharjee et al., 2005). The precise functionsf KCNT1, however, remain unclear. KCNT1 muta-
ions have been described in 39-50% of patientsith epilepsy of infancy with migrating focal seizures
EIMFS) (Ohba et al., 2015; Lim et al., 2016) and in lesshan 5% of patients with autosomal dominant noctur-al frontal lobe epilepsy (Heron et al., 2012).ecently, several studies have reported patients har-ouring KCNT1 mutations with intractable epilepticeizures who received quinidine treatment (Beardent al., 2014; Mikati et al., 2015; Chong et al., 2016;ukuoka et al., 2017; Abdelnour et al., 2018; Madaant al., 2018; Mullen et al., 2018). Although accumulatingata from multiple cases have enabled the elucidationf prognostic factors in patients with EIMFS, such as
he type of epilepsy and the age at which quinidines administered, few studies have investigated serumuinidine levels and association between serum quini-ine concentration and other antiepileptic drugs thatight hinder quinidine, such as phenobarbital in these
atients.iven that quinidine is one of the few drugs withotential as a treatment for patients with KCNT1 muta-
ions and EIMFS, determining optimal serum quinidineoncentration is essential for its safe and effective use.n this report, we present the cases of four patientsarbouring KCNT1 mutations who developed seizuresnd were treated with quinidine, with the aim of elu-idating its efficacy and utility.
ase reports
atient 1
atient 1 was a 20-month-old male born at 39 weeks ofestation without distress after in vitro fertilisation and
pileptic Disord, Vol. 21, No. 1, February 2019
mbryo transfer. He developed focal seizures com-rising asymmetric tonic posturing with eye deviationt one month of age and was diagnosed with EIMFSased on seizure symptoms and migrating foci on ictalEG. His seizures were unresponsive to phenobarbital,
casst
EIMFS, quinidine, serum concentration, arrhythmia,res
lonazepam, clobazam, levetiracetam or potassiumromide. Whole-exome sequencing revealed a deovo heterozygous mutation in KCNT1 (c.1283G>A:.Arg428Gln).he patient was administered quinidine at a start-
ng dose of 2 mg/kg/day at the age of nine months.ardiological evaluation prior to quinidine adminis-
ration, including Holter electrocardiography (ECG),chocardiography, and chest X-ray by a paediatric car-iologist, revealed no abnormalities. The antiepilepticrugs used in combination with quinidine were lev-tiracetam (48 mg/kg/day) and potassium bromide44 mg/kg/day). Although the dose of quinidine wasncreased gradually, the frequency of seizures did nothange significantly for approximately five months. Sixonths after the initiation of quinidine therapy, the
atient developed ventricular tachycardia and a clus-er of focal tonic seizures the day after the increase inuinidine dose from 114 mg/kg/day to 126 mg/kg/day.he peak serum quinidine concentration at the timef the arrhythmic event was 9.4 �g/ml. The correctedT interval (QTc), which was 362 ms before the initia-
ion of quinidine therapy, was longer at 420 ms duringquinidine dose of 114 mg/kg/day. Arrhythmia dis-
ppeared following a reduction in quinidine dose to3 mg/kg/day. Thereafter, a mild decrease in seizurerequency was observed despite no changes in medi-ation. The average seizure frequency during the threeonths before the initiation of quinidine therapy was
3.3 times/day, and was 62.4% lower at 16.3 times/dayuring the last three months (figure 1A).he relationship between serum quinidine concentra-ion and quinidine dose, based on peak and troughevels, is shown in figure 2A. Briefly, there was a lim-ted association between the trough level and dose,owever, the peak quinidine level was not associatedith quinidine dose between 40 and 50 mg/kg/day.
atient 2
atient 2 was a three-year-old female born at term with-ut distress. Her first seizure occurred at two monthsf age, and her seizures comprised asymmetrical tonic
49
onvulsions with cyanosis, eye deviation, and oralutomatism. She was diagnosed with EIMFS based oneizure symptoms and migrating foci on ictal EEG. Theeizures were refractory to conventional antiepilep-ic drugs. Whole-exome sequencing revealed a de
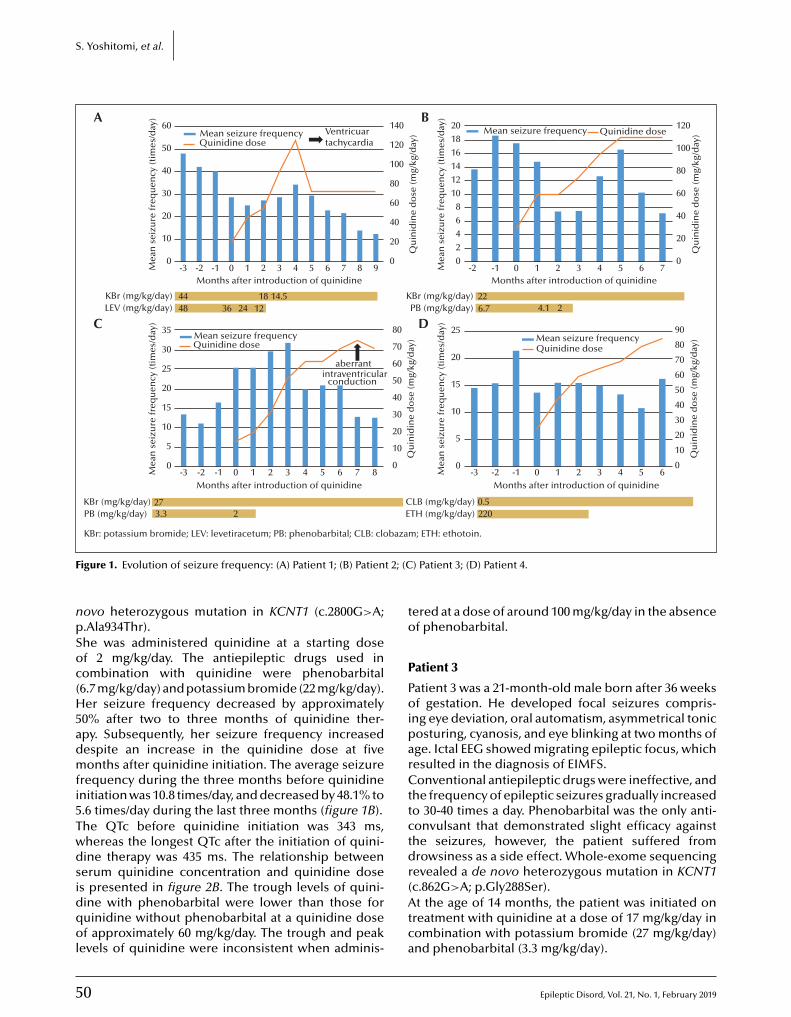
5
S. Yoshitomi, et al.
141210
Mea
n s
eizu
re fr
equ
ency
(tim
es/d
ay)
Qu
inid
ine
do
se (m
g/kg
/day
)
8
246
0-2
226.7 4.1
Mean seizure frequency
2KBr (mg/kg/day)PB (mg/kg/day)
-1 0 1 2 3 4Months after introduction of quinidine
5 6 70
20
40
60
80
100
120201816
Quinidine dose
15
Mea
n s
eizu
re fr
equ
ency
(tim
es/d
ay)
Qu
inid
ine
do
se (m
g/kg
/day
)
10
5
0-3
0.5220
Mean seizure frequency
CLB (mg/kg/day)ETH (mg/kg/day)
-2 -1 10 2 3 4Months after introduction of quinidine
5 60
20
30
10
40
50
60
80
70
9025
20Quinidine dose
50
40
30
Mea
n s
eizu
re fr
equ
ency
(tim
es/d
ay)
Qu
inid
ine
do
se (m
g/kg
/day
)
20
10
0-3
4448
18 14.536 24
Mean seizure frequencyQuinidine dose
12KBr (mg/kg/day)LEV (mg/kg/day)
-2 -1 0 1 2 3 4 5Months after introduction of quinidine
6 7 8 90
20
40
60
80
100
120Ventricuartachycardia
14060
25
20
Mea
n s
eizu
re fr
equ
ency
(tim
es/d
ay)
Qu
inid
ine
do
se (m
g/kg
/day
)15
5
10
0-3
273.3
Mean seizure frequency
2KBr (mg/kg/day)PB (mg/kg/day)
KBr: potassium bromide; LEV: levetiracetum; PB: phenobarbital; CLB: clobazam; ETH: ethotoin.
-2 -1 10 2 3 4Months after introduction of quinidine
5 6 870
20
30
10
40
50
60
70
8035
A
C
B
D
30 Quinidine dose
aberrantintraventricular
conduction
F t 2; (C
npSoc(H5admfi5Twdsidqol
to
P
PoiparCttctdr
igure 1. Evolution of seizure frequency: (A) Patient 1; (B) Patien
ovo heterozygous mutation in KCNT1 (c.2800G>A;.Ala934Thr).he was administered quinidine at a starting dosef 2 mg/kg/day. The antiepileptic drugs used inombination with quinidine were phenobarbital6.7 mg/kg/day) and potassium bromide (22 mg/kg/day).er seizure frequency decreased by approximately
0% after two to three months of quinidine ther-py. Subsequently, her seizure frequency increasedespite an increase in the quinidine dose at fiveonths after quinidine initiation. The average seizure
requency during the three months before quinidinenitiation was 10.8 times/day, and decreased by 48.1% to.6 times/day during the last three months (figure 1B).he QTc before quinidine initiation was 343 ms,hereas the longest QTc after the initiation of quini-ine therapy was 435 ms. The relationship betweenerum quinidine concentration and quinidine dose
0
s presented in figure 2B. The trough levels of quini-ine with phenobarbital were lower than those foruinidine without phenobarbital at a quinidine dosef approximately 60 mg/kg/day. The trough and peak
evels of quinidine were inconsistent when adminis-
(Atca
) Patient 3; (D) Patient 4.
ered at a dose of around 100 mg/kg/day in the absencef phenobarbital.
atient 3
atient 3 was a 21-month-old male born after 36 weeksf gestation. He developed focal seizures compris-
ng eye deviation, oral automatism, asymmetrical tonicosturing, cyanosis, and eye blinking at two months ofge. Ictal EEG showed migrating epileptic focus, whichesulted in the diagnosis of EIMFS.onventional antiepileptic drugs were ineffective, and
he frequency of epileptic seizures gradually increasedo 30-40 times a day. Phenobarbital was the only anti-onvulsant that demonstrated slight efficacy againsthe seizures, however, the patient suffered fromrowsiness as a side effect. Whole-exome sequencingevealed a de novo heterozygous mutation in KCNT1
Epileptic Disord, Vol. 21, No. 1, February 2019
c.862G>A; p.Gly288Ser).t the age of 14 months, the patient was initiated on
reatment with quinidine at a dose of 17 mg/kg/day inombination with potassium bromide (27 mg/kg/day)nd phenobarbital (3.3 mg/kg/day).
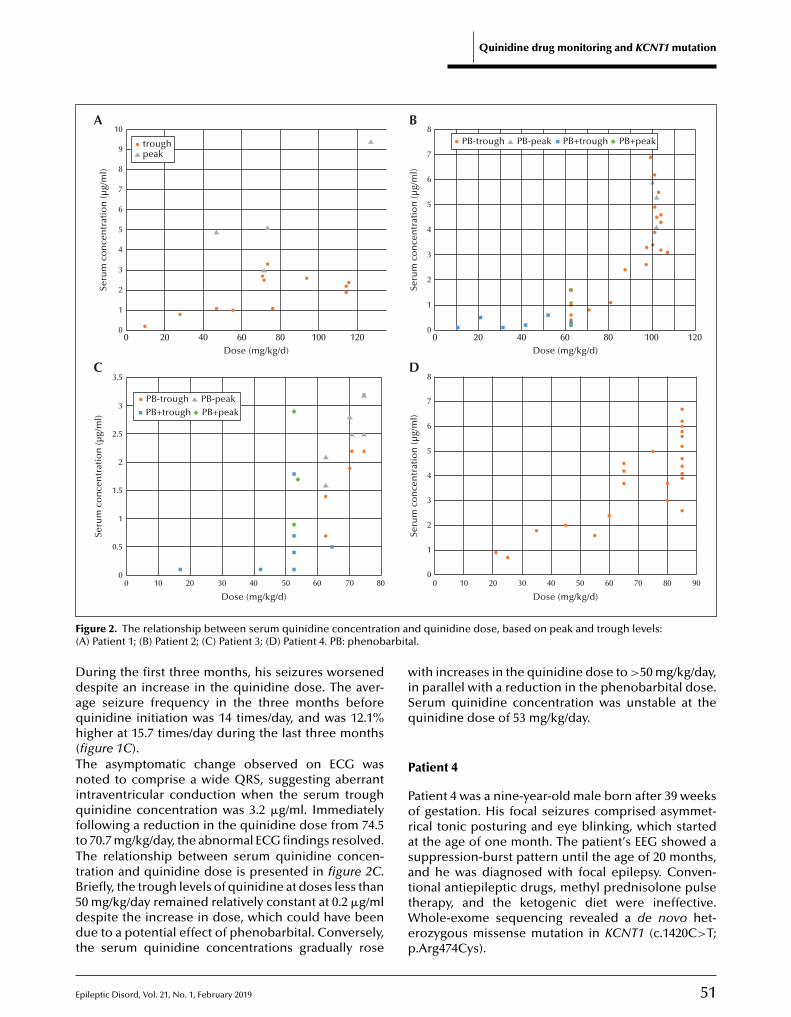
E
Quinidine drug monitoring and KCNT1 mutation
10A B
C D
00 20 40 60 80 100 120
1
2
3
4
5
6
7
Seru
m c
on
cen
trat
ion
(µg/
ml) 8
9
Dose (mg/kg/d)
troughpeak
8
7
6
5
4
3
2
1
00 20 40 60 80 100 120
Seru
m c
on
cen
trat
ion
(µg/
ml)
Dose (mg/kg/d)
PB-trough PB-peak PB+peakPB+trough
3.5
3
2.5
2
1.5
1
0.5
00 20 3010 40 50 60 70 80
Seru
m c
on
cen
trat
ion
(µg/
ml)
PB-trough PB-peakPB+peakPB+trough
8
00 10 20 30 40 50 60 70 80 90
1
2
3
4
5
6
Seru
m c
on
cen
trat
ion
(µg/
ml)
7
F on an( arbita
Ddaqh(TniqftTtB5ddt
wiSq
P
Porasa
Dose (mg/kg/d)
igure 2. The relationship between serum quinidine concentratiA) Patient 1; (B) Patient 2; (C) Patient 3; (D) Patient 4. PB: phenob
uring the first three months, his seizures worsenedespite an increase in the quinidine dose. The aver-ge seizure frequency in the three months beforeuinidine initiation was 14 times/day, and was 12.1%igher at 15.7 times/day during the last three months
figure 1C).he asymptomatic change observed on ECG wasoted to comprise a wide QRS, suggesting aberrant
ntraventricular conduction when the serum troughuinidine concentration was 3.2 �g/ml. Immediately
ollowing a reduction in the quinidine dose from 74.5o 70.7 mg/kg/day, the abnormal ECG findings resolved.he relationship between serum quinidine concen-ration and quinidine dose is presented in figure 2C.
pileptic Disord, Vol. 21, No. 1, February 2019
riefly, the trough levels of quinidine at doses less than0 mg/kg/day remained relatively constant at 0.2 �g/mlespite the increase in dose, which could have beenue to a potential effect of phenobarbital. Conversely,
he serum quinidine concentrations gradually rose
ttWep
Dose (mg/kg/d)
d quinidine dose, based on peak and trough levels:l.
ith increases in the quinidine dose to >50 mg/kg/day,n parallel with a reduction in the phenobarbital dose.erum quinidine concentration was unstable at theuinidine dose of 53 mg/kg/day.
atient 4
atient 4 was a nine-year-old male born after 39 weeksf gestation. His focal seizures comprised asymmet-ical tonic posturing and eye blinking, which startedt the age of one month. The patient’s EEG showed auppression-burst pattern until the age of 20 months,nd he was diagnosed with focal epilepsy. Conven-
51
ional antiepileptic drugs, methyl prednisolone pulseherapy, and the ketogenic diet were ineffective.
hole-exome sequencing revealed a de novo het-rozygous missense mutation in KCNT1 (c.1420C>T;.Arg474Cys).

5
S
Q2b(ifdtqwotaqd
D
Qpatftte(att2eforSewWCeAEtsfpiQw(sseo
AraKd1aPbn(rsitAmdfqndrtao(atmAuWpfptewdfttBdtiaAbe
. Yoshitomi, et al.
uinidine therapy was initiated at a dose of1 mg/kg/day at seven years of age and was com-ined with clobazam (0.5 mg/kg/day) and ethotoin
220 mg/kg/day). Although the quinidine dose wasncreased to 85 mg/kg/day, the average seizurerequency in the three months before quinidine intro-uction was 17.3 times/day, and was 23.1% lower at 13.3
imes/day during the last three months (figure 1D). Nouinidine-related side effects, such as ECG changes,ere observed. The serum quinidine concentrationsf the patient are shown in figure 2D. Although
he trough levels of quinidine were generally associ-ted with quinidine doses <80 mg/kg/day, the serumuinidine concentration was inconsistent at quinidineoses >80 mg/kg/day.
iscussion
uinidine was effective in only one of the four patientsresented herein, based on >50% seizure reductions the definition of quinidine efficacy. The most effec-ive trough and peak serum quinidine concentrationsor Patient 1 were 2.2-3.3 �g/ml and 5.1 �g/ml, respec-ively. The quinidine levels of Patient 1 were close tohe previously reported effective serum quinidine lev-ls for epilepsy that ranged between 0.4 and 5 �g/mlBearden et al., 2014; Mikati et al., 2015; Fukuoka etl., 2017; Abdelnour et al., 2018). The timing of initia-ion of treatment in Patient 1, however, was far laterhan that reported in previous reports (Bearden et al.,014; Mikati et al., 2015; Fukuoka et al., 2017; Abdelnourt al., 2018; Mullen et al., 2018). Additionally, seizurerequency often fluctuates during the natural coursef EIMFS. Therefore, it is unclear whether the seizureeduction in Patient 1 was indeed due to quinidine.even studies published to date include a total of 15pilepsy patients treated with quinidine: four patientsith EIMFS, 10 with other focal epilepsy, and one withest syndrome (Bearden et al., 2014; Mikati et al., 2015;hong et al., 2016; Fukuoka et al., 2017; Abdelnourt al., 2018; Madaan et al., 2018; Mullen et al., 2018).mong these patients, three of the four patients withIMFS responded well to quinidine (3/4; 75%). Withhe inclusion of our three patients with EIMFS, whosetatuses improved, quinidine was overall effective inour out of seven patients (4/7; 57.1%). In contrast, noatients with other focal epilepsies, including Patient 4
n the current study, responded to quinidine (0/11; 0%).uinidine was effective in the only reported patientith West syndrome who was treated with quinidine
2
1/1; 100%) (Fukuoka et al., 2017). Overall, these resultsuggest quinidine as a promising treatment option forome patients with EIMFS and West syndrome, how-ver, quinidine may not be beneficial for patients withther focal epilepsies.
caHii
ll KCNT1 mutations in the current four patients wereeported previously (Bearden et al., 2014; Mikati etl., 2015; Chong et al., 2016; Fukuoka et al., 2017). TheCNT1 mutation c.1283G>A (p.Arg428Gly) has beenetected in a total of three patients, including Patientin the current study (Bearden et al., 2014; Chong et
l., 2016). Although quinidine was partially effective foratient 1 in the current study and the patient reportedy Bearden et al., both of whom had EIMFS, it wasot beneficial in patients suffering from focal epilepsy
Chong et al., 2016). Although not conclusive, theseesults based on the available reports and our casesuggest that quinidine therapy should be consideredn patients with EIMFS who harbour the KCNT1 muta-ion, c.1283G>A (p.Arg428Gly).
previous study suggested that age of the patientsight be an important factor for the efficacy of quini-
ine therapy (Abdelnour et al., 2018). The authorsound that all patients who showed good response touinidine therapy were under the age of four and thato patient over four years of age responded to quini-ine. Although the response of Patient 1 in the currenteport is consistent with their finding, the outcomes ofhe remaining three patients do not lend support. Innother report, a patient with EIMFS who was startedn quinidine at six months of age also failed to respond
Madaan et al., 2018). Importantly, factors other thange should be considered which could account forhe observation of Abelnour et al. and the disgree-
ent between their study and ours. In the study bybdelnour et al., the seizure types of the four patientsnder four years of age corresponded to EIMFS orest syndrome, whereas the seizure types of the four
atients over four years of age corresponded to otherocal epilepsies (Abdelnour et al., 2018). Given that theatients with EIMFS and West syndrome showed a bet-
er response to quinidine than those with other focalpilepsies in the study of Abdelnour et al. (2018) asell as in the current study, age at the time of quini-ine treatment initiation may not be a good prognostic
actor. Future studies are warranted to clarify impor-ant prognostic factors for good response to quinidineherapy.ased on previous studies comparing psychomotorevelopment before and after quinidine adminis-
ration, psychomotor development was reported tomprove perceptibly in two patients with EIMFS whochieved complete or partial seizure suppression.
three-year-old male with 80% seizure reductionecame more alert and more interactive (Beardent al., 2014), whereas a three-year-old female withomplete seizure suppression began to utter words
Epileptic Disord, Vol. 21, No. 1, February 2019
fter initiating quinidine therapy (Mikati et al., 2015).owever, none of the patients in the current study,
ncluding those with seizure reduction, showedmprovement in their development after initiating

E
qmAqwtdicawsIwwttomeaictcqAtdwaaitoegtddtaetnQoe2wa((drQ
qocectqmeBtKdtibrcTwbodertise
SSw
ATNNh
R
AatS
Bbi
Bn
BSlack potassium channel in the rat central nervous system. J
uinidine therapy, and their psychomotor develop-ent remains severely delayed.ll patients in the current study were administereduinidine three or four times a day. In this study, troughas defined as the timepoint immediately before
he second administration of the day, and peak wasefined as the timepoint 2.5 hours after the first admin-
stration. The relationship between serum quinidineoncentration and quinidine dose was inconsistentmong the patients, and increased concentrationsere reported even though the dose remained the
ame.n the current study, two out of the four patientsere administered phenobarbital in combinationith quinidine. During the clinical course for these
wo patients, the elevation in quinidine concentra-ion was significantly hindered due to the inductionf cytochrome P450 3A4 by phenobarbital, whichetabolises quinidine (figures 2B, C). It took sev-
ral weeks for quinidine concentrations to increasefter the discontinuation of phenobarbital, suggest-ng that the aftereffect of phenobarbital on quinidineoncentration lingers. Therefore, careful quinidineitration should be planned when drugs that induceytochrome P450 3A4 are used in combination withuinidine.t the time of ventricular tachycardia in Patient 1,
he peak serum quinidine concentration with a quini-ine dose of 126 mg/kg was 9.4 �g/ml, both of whichere higher than previously reported for this patient
nd the other patients in the current study. Moreover,rrhythmia appeared within one day after the increasen quinidine dose. Several studies have also reportedhat cardiac arrhythmias generally occur within daysf quinidine administration (Cohen et al., 1977; Rodent al., 1986; Hohnloser et al., 1995). These results sug-est that careful patient monitoring, particularly afterhe administration of, and an increase in, the quini-ine dose, is critical. Additionally, a previous reportescribed a small number of patients who developed
orsade de pointes during long-term quinidine ther-py, usually in association with hypokalaemia (Rodent al., 1986). The serum potassium level of Patient 1 athe onset of arrhythmia was 4.4 mEq/l, which was withinormal limits.T elongation with quinidine was observed in seven
ut of the 15 patients reported in the literature (Mikatit al., 2015; Fukuoka et al., 2017; Abdelnour et al.,018; Mullen et al., 2018). Notably, one patient treatedith quinidine developed QT elongation despitelow serum quinidine concentration of 0.4 �g/ml
Abdelnour et al., 2018) or a low dose of 34.4 mg/kg/day
pileptic Disord, Vol. 21, No. 1, February 2019
Mikati et al., 2015). Clearly, instances of QT elongationo not translate to an increased risk of arrhythmia orequire urgent discontinuation of quinidine, becauseT elongation itself only reflects the primary action of
C
Bin
Quinidine drug monitoring and KCNT1 mutation
uinidine on ion channels. To achieve a truly safe rangef serum quinidine level and dose is considerably diffi-ult because its toxic levels depend on genetic factors,lectrolytes, and other complications such as serumoncentration and dose. However, it remains certainhat a quinidine dose over 74.5 mg/kg/day or a serumuinidine concentration above 9.4 �g/ml during treat-ent can lead to issues that are more serious than QT
longation.ecause quinidine remains one of the few promising
herapeutic drugs for patients with EIMFS harbouringCNT1 mutations, elucidating optimal serum quini-ine concentrations and appropriate methods of
itration is essential for its safe and effective use. Theres a possibility that serum quinidine levels may note directly related to efficacy or side effects, whichequires additional case reports and case series forlarification.he current report has several limitations. First, thisas a small, open-label study. Additionally, the possi-ility remains that the seizure reduction observed inur patients after the introduction of quinidine wasue to the natural history of the disease, and the truefficacy of quinidine in patients with KCNT1 mutationsemains to be elucidated. Finally, it should be notedhat quinidine therapy has not yet been approvedn patients with EIMFS and KCNT1 mutations andhould be prescribed with caution due to serious sideffects. �upplementary data.ummary didactic slides are available on theww.epilepticdisorders.com website.
cknowledgements and disclosures.his study was approved by the Institutional Review Board of theational Epilepsy Center, NHO Shizuoka Institute of Epilepsy andeurological Disorders, Shizuoka, Japan. None of the authorsave any conflict of interest to declare.
eference
bdelnour E, Gallentine W, McDonald M, et al. Does ageffect response to quinidine in patients with KCNT1 muta-ions? Report of three new cases and review of the literature.eizure 2018; 55: 1-3.
earden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Gold-erg EM. Targeted treatment of migrating partial seizures of
nfancy with quinidine. Ann Neurol 2014; 76: 457-61.
hattacharjee A, Kaczmarek LK. For K+ channels, Na+ is theew Ca2+. Trends Neurosci 2005; 28: 422-8.
hattacharjee A, Gan L, Kaczmarek LK. Localization of the
53
omp Neurol 2002; 454: 241-54.
hattacharjee A, von Hehn CA, Mei X, Kaczmarek LK. Local-zation of the Na+-activated K+ channel Slick in the rat centralervous system. J Comp Neurol 2005; 484: 80-92.

5
S
CRt2
ChCD
FWD
HsaG
Hrfid
. Yoshitomi, et al.
hong PF, Nakamura R, Saitsu H, Matsumoto N, Kira. Ineffective quinidine therapy in early onset epilep-
ic encephalopathy with KCNT1 mutation. Ann Neurol016; 79: 502-3.
ohen IS, Jick H, Cohen SI. Adverse reactions to quinidine inospitalized patients: findings based on data from the Bostonollaborative Drug Surveillance Program. Prog Cardiovascis 1977; 20: 151-63.
ukuoka M, Kuki I, Kawawaki H, et al. Quinidine therapy forest syndrome with KCNT1 mutation: a case report. Brainev 2017; 39: 80-3.
eron SE, Smith KR, Bahlo M, et al. Missense mutations in theodium-gated potassium channel gene KCNT1 cause severeutosomal dominant nocturnal frontal lobe epilepsy. Natenet 2012; 44: 1188-90.
ohnloser SH, van de Loo A, Baedeker F. Efficacy and proar-hythmic hazards of pharmacologic cardioversion of atrialbrillation: prospective comparison of sotalol versus quini-ine. J Am Coll Cardiol 1995; 26: 852-8.
TEST YOURSELFEDUCATION
(1) Which epilepsy syndromes are believed to show a response to quinidine therapy?
(2) Which antiepileptic drugs suppress serum quinidine concentration?
Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutationsin seizure disorders: the phenotypic spectrum and functionaleffects. J Med Genet 2016; 53: 217-25.
Madaan P, Jauhari P, Gupta A, Chakrabarty B, Gulati S. Aquinidine non-responsive novel KCNT1 mutation in an Indianinfant with epilepsy of infancy with migrating focal seizures.Brain Dev 2018; 40: 229-32.
Mikati MA, Jiang YH, Carboni M, et al. Quinidine inthe treatment of KCNT1-positive epilepsies. Ann Neurol2015; 78: 995-9.
Mullen SA, Carney PW, Roten A, et al. Precision therapy forepilepsy due to KCNT1 mutations: a randomized trial of oralquinidine. Neurology 2018; 90: e67-72.
Ohba C, Kato M, Takahashi N, et al. De novo KCNT1mutations in early-onset epileptic encephalopathy. Epilepsia2015; 56: 121-8.
Roden DM, Woosley RL, Primm RK. Incidence and clini-cal features of the quinidine-associated long QT syndrome:implications for patient care. Am Heart J 1986; 111: 1088-93.
4
Note: Reading the manuscript provides an answer to all qwebsite, www.epilepticdisorders.com, under the section
Epileptic Disord, Vol. 21, No. 1, February 2019
uestions. Correct answers may be accessed on the“The EpiCentre”.

do
i:10.
1684
/ep
d.2
019.
1027
Epileptic Disord, Vol. 21, No. 1, February 2019 55
Correspondence:Gregory L. HolmesDepartment of Neurological Sciences,Larner College of Medicine,University of Vermont,Stafford Hall 118C,Burlington, VT 05405, USA<[email protected]>
Original articleEpileptic Disord 2019; 21 (1): 55-64
Functional brain connectivityin electrical status epilepticusin sleep
Steven H. Mott 1, Richard P. Morse 2, Scott A. Burroughs 2,Ashura W. Buckley 3, Cristan A. Farmer 3, Audrey E. Thurm 3,Susan E. Swedo 3, Amara L. Krag 4, Gregory L. Holmes 4
1 Department of Pediatrics, University of California, Irvine2 Department of Neurology, Geisel School of Medicine at Dartmouth, Hanover,New Hampshire3 National Institute of Mental Health, National Institutes of Health4 Department of Neurological Sciences, Larner College of Medicine,University of Vermont, Burlington, Vermont, USA
Received March 25, 2018; Accepted November 12, 2018
ABSTRACT – Aims. Electrical status epilepticus in sleep (ESES) is anage-related, self-limited epileptic encephalopathy. The syndrome is charac-terized by cognitive and behavioral abnormalities and a specific EEG patternof continuous spikes and waves during slow-wave sleep. While spikes andsharp waves are known to result in transient cognitive impairment duringlearning and memory tasks performed during the waking state, the effect ofepileptiform discharges during sleep on cognition and behavior is unclear.There is increasing evidence that abnormalities of coherence, a measureof the consistency of the phase difference between two EEG signals whencompared over time, is an important feature of brain oscillations and playsa role in cognition and behavior. The objective of this study was to deter-mine whether coherence of EEG activity is altered during slow-wave sleepin children with ESES when compared to typically developing children.Methods. We examined coherence during epochs of ESES versus epochswhen ESES was not present. In addition, we compared coherence duringslow-wave sleep between typically developing children and children withESES.Results. ESES was associated with remarkably high coherences at all band-widths and most electrode pairs. While the high coherence was largelyattributed to the spikes and spike-and-wave discharge, activity betweenspikes and spike-and-wave discharge also demonstrated high coherence.Conclusions. This study indicates that EEG coherence during ESES is rel-atively high. Whether these increases in coherence correlate with thecognitive and behavioral abnormalities seen in children with this EEG pat-tern remains to be determined.
Key words: electrical status epilepticus in sleep (ESES), EEG, coherence,oscillations, phase lag, continuous spike and waves during slow wave sleep(CSWS)

5
S
Eatbcw12ScmcTpetsmoWtCnEf–pf–s–dNc(Tcdicppl2tt211miHSare
Tuts2ddfitcnrwmrirmiRcoqcsisrvbEtsapmWcppcceeo
M
S
Twenty-four-hour inpatient EEGs were documented
.H. Mott, et al.
lectrical status epilepticus in sleep (ESES) is defineds an age-related, self-limited epileptic encephalopa-hy. The condition is characterized by cognitive andehavioral abnormalities and a specific electroen-ephalographic (EEG) pattern of continuous spike andaves during slow-wave sleep (CSWS) (Patry et al.,
971; Galanopoulou et al., 2000; Scheltens-de Boer,009; Sanchez Fernandez et al., 2012, 2014; Singhal andullivan, 2014; Gencpinar et al., 2016). While the clini-al presentation of children with ESES is variable, theost severe clinical syndrome presents with global
ognitive regression in addition to clinical seizures.he age at onset ranges from one to 14 years, with aeak between four and eight years (van den Munckhoft al., 2015). Although seizures may be absent in upo 20% of cases, they are most often the presentingymptom, after which developmental delay, develop-ental arrest, or regression in cognitive performance
r behavior becomes evident (Tassinari et al., 2000).hile CSWS and ESES are used interchangeably, ESES
ypically is used to describe the EEG pattern whileSWS is used to describe the clinical syndrome of cog-itive and behavioral abnormalities associated with theSES pattern (Gencpinar et al., 2016). The hallmark EEGeatures of ESES are:
a spike and wave occurring “during a significant pro-ortion” of non-REM sleep with a threshold ranging
rom 25% to 85%;continuous or nearly-continuous, bilateral, or occa-
ionally lateralized slow spikes and waves;and marked potentiation of epileptiform discharges
uring non-REM sleep (Sanchez Fernandez et al., 2013).ear-continuous epileptiform discharges have been
ausally related to neurocognitive regression in CSWSTassinari et al., 2000; Holmes and Lenck-Santini, 2006).he pathophysiologic mechanisms underlying thisondition are still incompletely understood. Recentata suggest that the abnormal epileptic EEG activ-
ty occurring during sleep might cause the typicallinical symptoms by interfering with sleep-relatedhysiologic functions, and possible neuroplasticityrocesses mediating higher cortical functions such as
earning and memory consolidation (Tassinari et al.,000; Holmes and Lenck-Santini, 2006). It is knownhat spikes and spike-and-wave discharges can leado cognitive impairment in both animals (Kleen et al.,010) and humans (Aarts et al., 1984; Binnie et al., 1987,990, 1991; Shewmon and Erwin, 1989; Krauss et al.,997; Ung et al., 2017). However, the cognitive impair-ent seen with interictal spikes is transient in nature
n both humans (Aarts et al., 1984; Nair et al., 2014;orak et al., 2017) and rodents (Holmes and Lenck-
6
antini, 2006; Zhou et al., 2007; Kleen et al., 2010)nd it has been difficult to link the neurocognitiveegression in CSWS solely to nocturnal spikes (Ebust al., 2011).
f(ys
here is increasing evidence that abnormalities innderlying oscillatory activity may play an impor-
ant role in cognitive impairment in children witheizures (Holmes and Lenck-Santini, 2006; Holmes,014; Barry and Holmes, 2016). For example, in chil-ren with epilepsy, neither spikes nor spike-and-waveischarges correlate with the neuropsychological pro-le, whereas slow-wave activity on the EEG is related
o memory impairment (Koop et al., 2005). In a study ofhildren with Dravet syndrome, it was found that cog-itive outcome was related more to preserved alphahythm of the EEG than seizures or generalized spike-ave discharges on the EEG. Likewise, in an animalodel of Dravet syndrome, cognitive impairment was
elated to altered theta rather than seizures or inter-ctal spikes (Bender et al., 2013, 2016). These studiesaise the question of whether EEG background abnor-
alities are related more to cognitive impairment thannterictal spikes.ecent work in humans has demonstrated thatoherence is a valuable marker of functional brainrganization and connectivity. On a frequency by fre-uency basis, EEG spectral coherence represents theonsistency of the phase difference between two EEGignals when compared over time. EEG coherence isnterpreted as a measure of “coupling” and as a mea-ure of the functional association between two brainegions (Thatcher et al., 1987, 2012). High coherencealues are taken as a measure of strong connectivityetween the brain regions that produce the comparedEG signals (Srinivasan et al., 2007). In both autis-ic spectrum disorder (Buckley et al., 2015) and Westyndrome (Burroughs et al., 2014), EEG coherencesre abnormally high. Remarkably, there have been noapers to date assessing coherence as a functionaleasure of brain connectivity in ESES.e hypothesized that ESES during SWS has high
oherence values. To address this hypothesis, we com-ared coherences across bandwidths and electrodeairs during SWS in children with ESES and normalhildren. In children with ESES, we also comparedoherences during SWS during non-ESES and ESESpochs. Finally, to determine the “driver” of coher-nce during ESES, we examined epochs containingnly spikes with epochs not containing spikes.
ethods
tudy design and participants
Epileptic Disord, Vol. 21, No. 1, February 2019
rom 29 neurotypically developing (TYP) childrenmean±SD: 4.18±1.70 years) and 18 children (5.37±1.85ears) with ESES, as defined as an EEG with generalizedpikes, sharp waves, spike and wave or polyspikes and
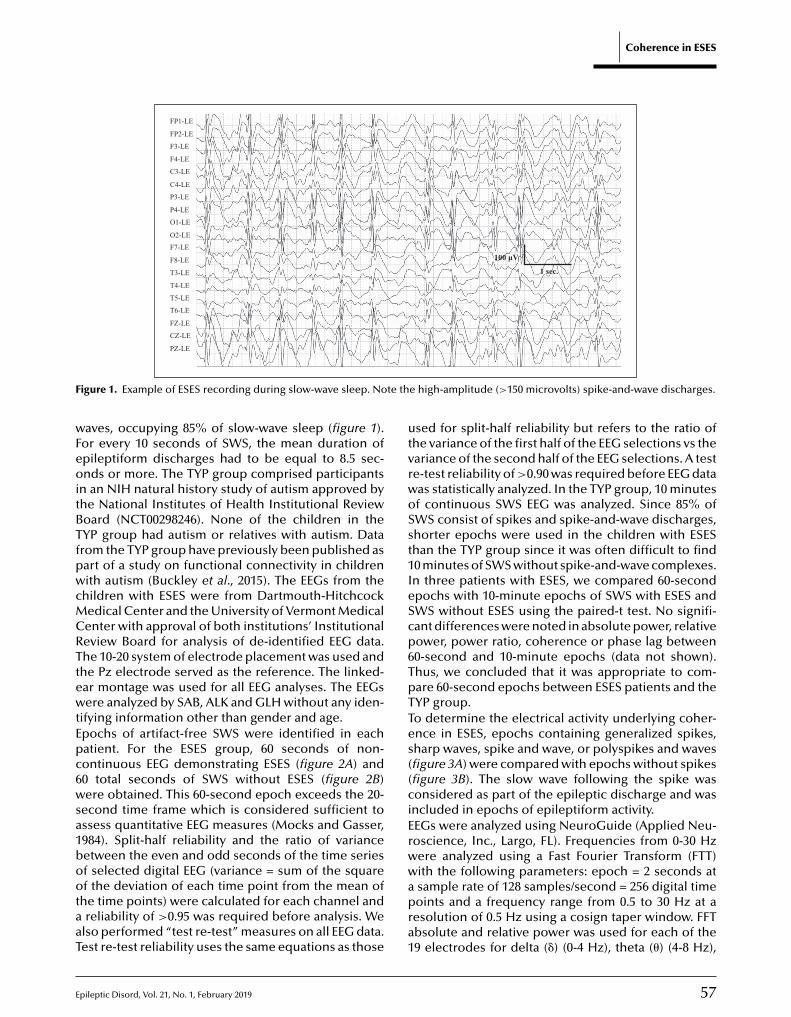
E
Coherence in ESES
FP1-LE
FP2-LE
F3-LE
F4-LE
C3-LE
C4-LE
P3-LE
P4-LE
O1-LE
O2-LE
F7-LE
F8-LE
T3-LE
T4-LE
T5-LE
100 μV
1 sec.
T6-LE
FZ-LE
CZ-LE
PZ-LE
F te th
wFeoitBTfpwcMCRTtewtEpc6wsa1bootaaT
utvrwoSst1IeScp6TpTTes((ciErww
igure 1. Example of ESES recording during slow-wave sleep. No
aves, occupying 85% of slow-wave sleep (figure 1).or every 10 seconds of SWS, the mean duration ofpileptiform discharges had to be equal to 8.5 sec-nds or more. The TYP group comprised participants
n an NIH natural history study of autism approved byhe National Institutes of Health Institutional Reviewoard (NCT00298246). None of the children in theYP group had autism or relatives with autism. Datarom the TYP group have previously been published asart of a study on functional connectivity in childrenith autism (Buckley et al., 2015). The EEGs from the
hildren with ESES were from Dartmouth-Hitchcockedical Center and the University of Vermont Medicalenter with approval of both institutions’ Institutionaleview Board for analysis of de-identified EEG data.he 10-20 system of electrode placement was used andhe Pz electrode served as the reference. The linked-ar montage was used for all EEG analyses. The EEGsere analyzed by SAB, ALK and GLH without any iden-
ifying information other than gender and age.pochs of artifact-free SWS were identified in eachatient. For the ESES group, 60 seconds of non-ontinuous EEG demonstrating ESES (figure 2A) and0 total seconds of SWS without ESES (figure 2B)ere obtained. This 60-second epoch exceeds the 20-
econd time frame which is considered sufficient tossess quantitative EEG measures (Mocks and Gasser,984). Split-half reliability and the ratio of varianceetween the even and odd seconds of the time seriesf selected digital EEG (variance = sum of the square
pileptic Disord, Vol. 21, No. 1, February 2019
f the deviation of each time point from the mean ofhe time points) were calculated for each channel andreliability of >0.95 was required before analysis. We
lso performed “test re-test” measures on all EEG data.est re-test reliability uses the same equations as those
apra1
e high-amplitude (>150 microvolts) spike-and-wave discharges.
sed for split-half reliability but refers to the ratio ofhe variance of the first half of the EEG selections vs theariance of the second half of the EEG selections. A teste-test reliability of >0.90 was required before EEG dataas statistically analyzed. In the TYP group, 10 minutesf continuous SWS EEG was analyzed. Since 85% ofWS consist of spikes and spike-and-wave discharges,horter epochs were used in the children with ESEShan the TYP group since it was often difficult to find0 minutes of SWS without spike-and-wave complexes.n three patients with ESES, we compared 60-secondpochs with 10-minute epochs of SWS with ESES andWS without ESES using the paired-t test. No signifi-ant differences were noted in absolute power, relativeower, power ratio, coherence or phase lag between0-second and 10-minute epochs (data not shown).hus, we concluded that it was appropriate to com-are 60-second epochs between ESES patients and theYP group.o determine the electrical activity underlying coher-nce in ESES, epochs containing generalized spikes,harp waves, spike and wave, or polyspikes and wavesfigure 3A) were compared with epochs without spikesfigure 3B). The slow wave following the spike wasonsidered as part of the epileptic discharge and wasncluded in epochs of epileptiform activity.EGs were analyzed using NeuroGuide (Applied Neu-oscience, Inc., Largo, FL). Frequencies from 0-30 Hzere analyzed using a Fast Fourier Transform (FTT)ith the following parameters: epoch = 2 seconds at
57
sample rate of 128 samples/second = 256 digital timeoints and a frequency range from 0.5 to 30 Hz at aesolution of 0.5 Hz using a cosign taper window. FFTbsolute and relative power was used for each of the9 electrodes for delta (�) (0-4 Hz), theta (�) (4-8 Hz),
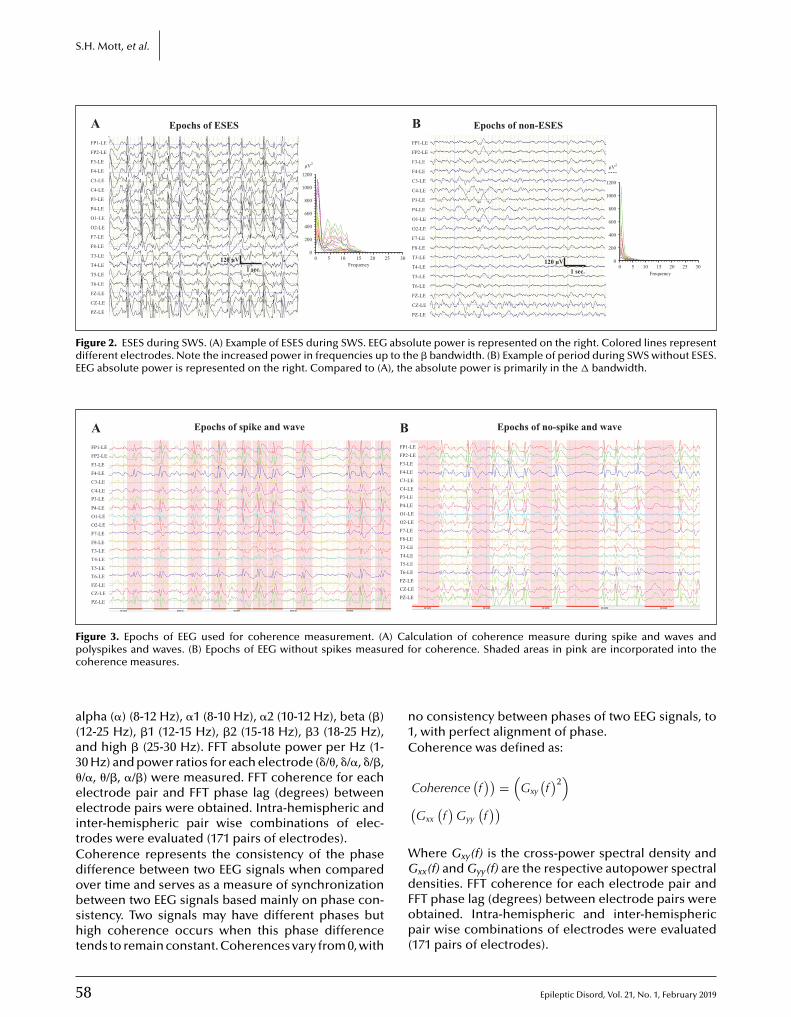
5
S.H. Mott, et al.
FP1-LE
FP2-LE
F3-LE
F4-LE
C3-LE
C4-LE
P3-LE
P4-LE
O1-LE
O2-LE
F7-LE
F8-LE
T3-LE
T4-LE
T5-LE
T6-LE
FZ-LE
CZ-LE
PZ-LE
FP1-LE
Epochs of ESES
FP2-LE
F3-LE
F4-LE
C3-LE1200
μV2
1000
800
600
400
200
00 5 10 15
Frequency20 25 30
C4-LE
P3-LE
P4-LE
O1-LE
O2-LE
F7-LE
F8-LE
T3-LE
T4-LE
T5-LE
T6-LE
FZ-LE
CZ-LE
PZ-LE
A Epochs of non-ESESB
120 μV1 sec.
120 μV1 sec.
1200
μV2
1000
800
600
400
200
00 5 10 15
Frequency20 25 30
Figure 2. ESES during SWS. (A) Example of ESES during SWS. EEG absolute power is represented on the right. Colored lines representdifferent electrodes. Note the increased power in frequencies up to the � bandwidth. (B) Example of period during SWS without ESES.EEG absolute power is represented on the right. Compared to (A), the absolute power is primarily in the � bandwidth.
FP1-LE
Epochs of spike and wave Epochs of no-spike and wave
FP2-LE
F3-LE
F4-LE
C3-LE
C4-LEP3-LE
P4-LE
O1-LE
O2-LE
F7-LE
F8-LE
T3-LE
T4-LE
T5-LE
T6-LE
FZ-LECZ-LE
PZ-LE
A BFP1-LE
FP2-LE
F3-LE
F4-LE
C3-LE
C4-LE
P3-LE
P4-LE
O1-LE
O2-LE
F7-LE
F8-LE
T3-LE
T4-LE
T5-LE
T6-LE
FZ-LE
CZ-LE
PZ-LE
Figure 3. Epochs of EEG used for coherence measurement. (A) Calculation of coherence measure during spike and waves andp red fc
a(a3�eeitCdobsht
n1C
WG
olyspikes and waves. (B) Epochs of EEG without spikes measuoherence measures.
lpha (�) (8-12 Hz), �1 (8-10 Hz), �2 (10-12 Hz), beta (�)12-25 Hz), �1 (12-15 Hz), �2 (15-18 Hz), �3 (18-25 Hz),nd high � (25-30 Hz). FFT absolute power per Hz (1-0 Hz) and power ratios for each electrode (�/�, �/�, �/�,/�, �/�, �/�) were measured. FFT coherence for eachlectrode pair and FFT phase lag (degrees) betweenlectrode pairs were obtained. Intra-hemispheric and
nter-hemispheric pair wise combinations of elec-rodes were evaluated (171 pairs of electrodes).oherence represents the consistency of the phaseifference between two EEG signals when compared
8
ver time and serves as a measure of synchronizationetween two EEG signals based mainly on phase con-istency. Two signals may have different phases butigh coherence occurs when this phase difference
ends to remain constant. Coherences vary from 0, with
dFop(
or coherence. Shaded areas in pink are incorporated into the
o consistency between phases of two EEG signals, to, with perfect alignment of phase.oherence was defined as:
Coherence(f)) =
(Gxy
(f)2
)
(Gxx
(f)
Gyy
(f))
here Gxy(f) is the cross-power spectral density andxx(f) and Gyy(f) are the respective autopower spectral
Epileptic Disord, Vol. 21, No. 1, February 2019
ensities. FFT coherence for each electrode pair andFT phase lag (degrees) between electrode pairs werebtained. Intra-hemispheric and inter-hemisphericair wise combinations of electrodes were evaluated
171 pairs of electrodes).

E
S
Hucpassi–lts–swt
R
Dc4bt(itdtc
cwai((chlati�etgdcsttwfTcod
FEs
tatistical analysis
ypotheses were proven or discarded based onnpaired t tests for comparisons between the TYPhildren and ESES children and paired t tests for com-arisons within the same patient for SWS with ESESnd SWS without ESES using Neurostat EEG statisticaloftware. The t test was used since the data demon-trated a normal distribution. The p values are shownn two ways:
electrode maps with color and thickness of theines connecting electrodes, reflecting direction ofhe differences between groups and the degree ofignificance;
p value heat maps with degree of significance inelected color-coded electrode pairs. Although dataere reviewed from 171 electrode pairs, selected elec-
rodes were chosen for illustration.
esults
uring ESES, there was a marked increase in coherenceompared to the SWS segments without ESES (figures, 5). This increase in coherence occurred across allandwidths and many electrode pairs. Of the 62 elec-
rode pairs demonstrated in the heat map in figure 5, 1219.3%) in the � range, 36 (58%) in the � range, 49 (79%)
pileptic Disord, Vol. 21, No. 1, February 2019
n the � range, and 49 (79%) in the � range showed sta-istically increased coherences. In no electrode pairsid the ESES epochs show lower coherences than
he non-ESES epochs. Likewise, there were signifi-ant increases in coherence in the EEGs with ESES
ctssa
Delta (1.0 - 4.0 Hz) Theta (4.0 - 8.0 Hz)
High Beta (25.0 - 30.0 Hz)
- +P-Value <= 0.050
Beta 1 (12.0 - 15.0 Hz)
L
L
-P-Value <= 0.
igure 4. Coherence during ESES. Marked increases in coherence weSES periods. Red lines indicate that the ESES segments had higher coignificance values are illustrated by weight of the lines. L/R refer to th
Coherence in ESES
ompared to the TYP group (figures 6, 7). Coherencesere significantly increased in the ESES group across
ll bandwidths. Of the 62 electrode pairs demonstratedn the heat map in figure 8, 16 (25.8) in the � range, 3150%) in the � range, 54 (87%) in the � range, and 5283.8%) in the � range showed statistically increasedoherences, other than a few electrode pairs in theigh � (25-30-Hz) bandwidth where coherences were
ower in the TYP group than the ESES group. Whilell bandwidths demonstrated increases in coherence,he � frequencies were less likely to be significantlyncreased than the other major bandwidths (�, � and). In the � bandwidth, some asymmetries in coher-nce were seen, with higher coherences noted overhe left hemisphere, when compared with the TYProup. The composite coherence score showed thaturing ESES, coherence was substantially increasedompared to non-ESES periods and with slow waveleep (SWS) in the TYP group (table 1). In additiono the mean differences between groups shown inable 1, for each individual child, the mean coherencesere higher in the ESES patient than the mean score
or the TYP group.o determine the component of the ESES that wasontributing to the increased coherences, periodsf ESES with and without spikes were compared. Asemonstrated in figure 8, coherences were signifi-
59
antly higher during the spike component of the ESEShan the non-spike component. Likewise, the compo-ite coherence score during spikes was higher duringpikes versus no-spike epochs (table 1). This waslso true for each individual patient. Also, coherence
Alpha (8.0 - 12.0 Hz) Beta (12.0 - 25.0 Hz)
Beta 2 (15.0 - 18.0 Hz) Beta 3 (18.0 - 25.0 Hz)
R
R
+025
- +P-Value <= 0.010
re seen at most electrode pairs during ESES compared to non-herences than during the non-ESES segments during SWS. Thee left and right side of the head.
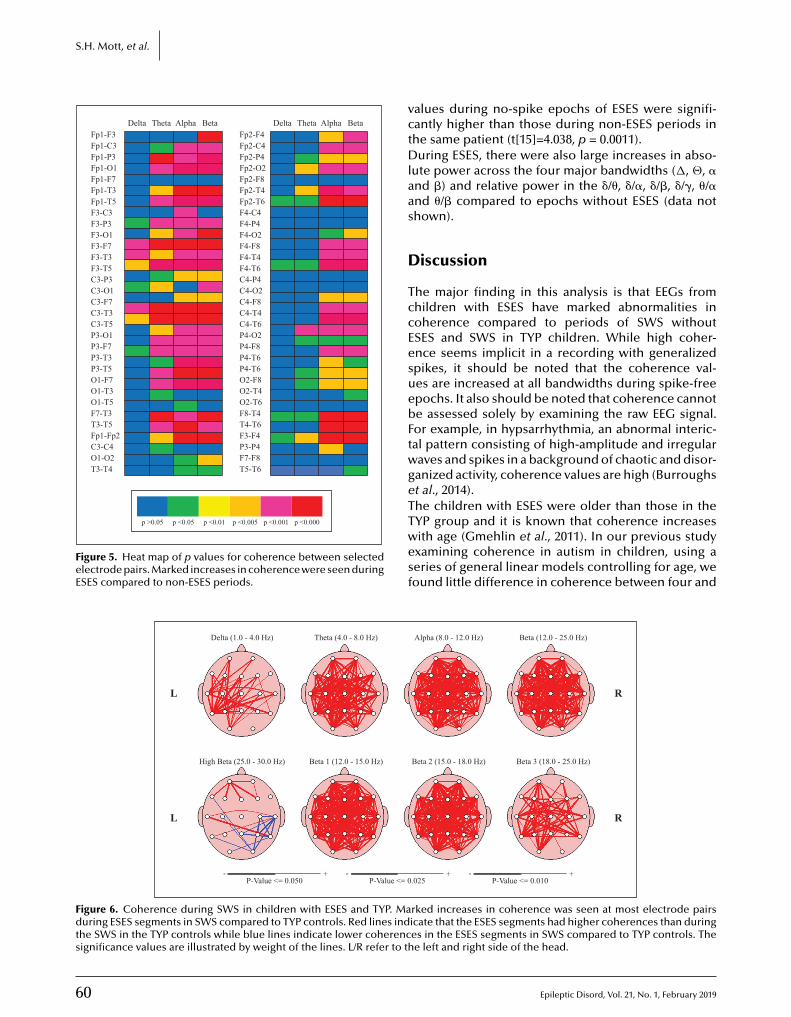
60
S.H. Mott, et al.
DeltaFp1-F3Fp1-C3Fp1-P3Fp1-O1Fp1-F7Fp1-T3Fp1-T5F3-C3F3-P3F3-O1F3-F7F3-T3F3-T5C3-P3C3-O1C3-F7C3-T3C3-T5P3-O1P3-F7P3-T3P3-T5O1-F7O1-T3O1-T5F7-T3T3-T5Fp1-Fp2C3-C4O1-O2T3-T4
Theta Alpha Beta
p >0.05 p <0.05 p <0.01 p <0.005 p <0.001 p <0.000
DeltaFp2-F4Fp2-C4Fp2-P4Fp2-O2Fp2-F8Fp2-T4Fp2-T6F4-C4F4-P4F4-O2F4-F8F4-T4F4-T6C4-P4C4-O2C4-F8C4-T4C4-T6P4-O2P4-F8P4-T6P4-T6O2-F8O2-T4O2-T6F8-T4T4-T6F3-F4P3-P4F7-F8T5-T6
Theta Alpha Beta
Figure 5. Heat map of p values for coherence between selectedelectrode pairs. Marked increases in coherence were seen duringESES compared to non-ESES periods.
vctDlaas
D
TccEesuebFtwgeTTwesf
Delta (1.0 - 4.0 Hz) Theta (4.0 - 8.0 Hz)
High Beta (25.0 - 30.0 Hz)
- +P-Value <= 0.050
Beta 1 (12.0 - 15.0 Hz)
L
L
-P-Value <= 0
Figure 6. Coherence during SWS in children with ESES and TYP. Maduring ESES segments in SWS compared to TYP controls. Red lines indthe SWS in the TYP controls while blue lines indicate lower coherencsignificance values are illustrated by weight of the lines. L/R refer to th
alues during no-spike epochs of ESES were signifi-antly higher than those during non-ESES periods inhe same patient (t[15]=4.038, p = 0.0011).uring ESES, there were also large increases in abso-
ute power across the four major bandwidths (�, �, �nd �) and relative power in the �/�, �/�, �/�, �/�, �/�nd �/� compared to epochs without ESES (data nothown).
iscussion
he major finding in this analysis is that EEGs fromhildren with ESES have marked abnormalities inoherence compared to periods of SWS withoutSES and SWS in TYP children. While high coher-nce seems implicit in a recording with generalizedpikes, it should be noted that the coherence val-es are increased at all bandwidths during spike-freepochs. It also should be noted that coherence cannote assessed solely by examining the raw EEG signal.or example, in hypsarrhythmia, an abnormal interic-al pattern consisting of high-amplitude and irregularaves and spikes in a background of chaotic and disor-anized activity, coherence values are high (Burroughst al., 2014).he children with ESES were older than those in the
Epileptic Disord, Vol. 21, No. 1, February 2019
YP group and it is known that coherence increasesith age (Gmehlin et al., 2011). In our previous studyxamining coherence in autism in children, using aeries of general linear models controlling for age, weound little difference in coherence between four and
Alpha (8.0 - 12.0 Hz) Beta (12.0 - 25.0 Hz)
Beta 2 (15.0 - 18.0 Hz) Beta 3 (18.0 - 25.0 Hz)
R
R
+.025
- +P-Value <= 0.010
rked increases in coherence was seen at most electrode pairsicate that the ESES segments had higher coherences than duringes in the ESES segments in SWS compared to TYP controls. Thee left and right side of the head.
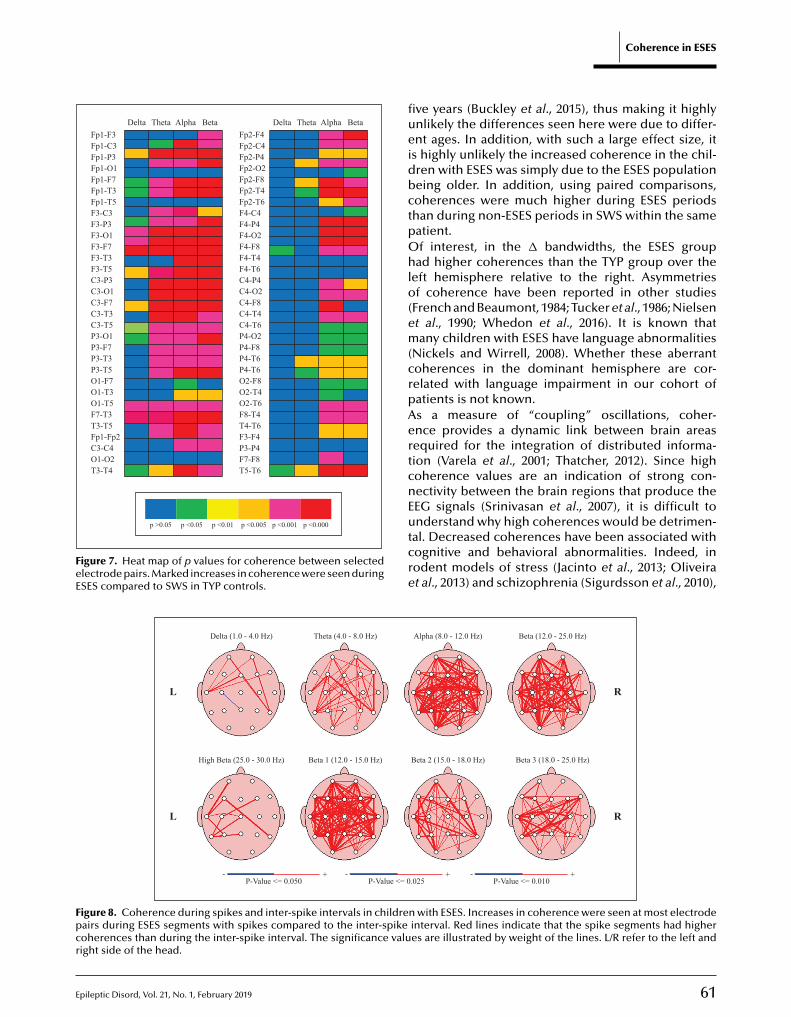
Epileptic Disord, Vol. 21, No. 1, February 2019
DeltaFp1-F3Fp1-C3Fp1-P3Fp1-O1Fp1-F7Fp1-T3Fp1-T5F3-C3F3-P3F3-O1F3-F7F3-T3F3-T5C3-P3C3-O1C3-F7C3-T3C3-T5P3-O1P3-F7P3-T3P3-T5O1-F7O1-T3O1-T5F7-T3T3-T5Fp1-Fp2C3-C4O1-O2T3-T4
Theta Alpha Beta DeltaFp2-F4Fp2-C4Fp2-P4Fp2-O2Fp2-F8Fp2-T4Fp2-T6F4-C4F4-P4F4-O2F4-F8F4-T4F4-T6C4-P4C4-O2C4-F8C4-T4C4-T6P4-O2P4-F8P4-T6P4-T6O2-F8O2-T4O2-T6F8-T4T4-T6F3-F4P3-P4F7-F8T5-T6
p >0.05 p <0.05 p <0.01 p <0.005 p <0.001 p <0.000
Theta Alpha Beta
Figure 7. Heat map of p values for coherence between selectedelectrode pairs. Marked increases in coherence were seen duringESES compared to SWS in TYP controls.
fiueidbctpOhlo(em(crpAertcnEutcre
Delta (1.0 - 4.0 Hz) Theta (4.0 - 8.0 Hz)
High Beta (25.0 - 30.0 Hz)
- +P-Value <= 0.050
Beta 1 (12.0 - 15.0 Hz)
L
L
-P-Value <= 0
Figure 8. Coherence during spikes and inter-spike intervals in childrepairs during ESES segments with spikes compared to the inter-spikecoherences than during the inter-spike interval. The significance valuright side of the head.
Coherence in ESES
ve years (Buckley et al., 2015), thus making it highlynlikely the differences seen here were due to differ-nt ages. In addition, with such a large effect size, it
s highly unlikely the increased coherence in the chil-ren with ESES was simply due to the ESES populationeing older. In addition, using paired comparisons,oherences were much higher during ESES periodshan during non-ESES periods in SWS within the sameatient.f interest, in the � bandwidths, the ESES group
ad higher coherences than the TYP group over theeft hemisphere relative to the right. Asymmetriesf coherence have been reported in other studies
French and Beaumont, 1984; Tucker et al., 1986; Nielsent al., 1990; Whedon et al., 2016). It is known thatany children with ESES have language abnormalities
Nickels and Wirrell, 2008). Whether these aberrantoherences in the dominant hemisphere are cor-elated with language impairment in our cohort ofatients is not known.s a measure of “coupling” oscillations, coher-nce provides a dynamic link between brain areasequired for the integration of distributed informa-ion (Varela et al., 2001; Thatcher, 2012). Since highoherence values are an indication of strong con-ectivity between the brain regions that produce theEG signals (Srinivasan et al., 2007), it is difficult to
61
nderstand why high coherences would be detrimen-al. Decreased coherences have been associated withognitive and behavioral abnormalities. Indeed, inodent models of stress (Jacinto et al., 2013; Oliveirat al., 2013) and schizophrenia (Sigurdsson et al., 2010),
Alpha (8.0 - 12.0 Hz) Beta (12.0 - 25.0 Hz)
Beta 2 (15.0 - 18.0 Hz) Beta 3 (18.0 - 25.0 Hz)
R
R
+.025
- +P-Value <= 0.010
n with ESES. Increases in coherence were seen at most electrodeinterval. Red lines indicate that the spike segments had higheres are illustrated by weight of the lines. L/R refer to the left and
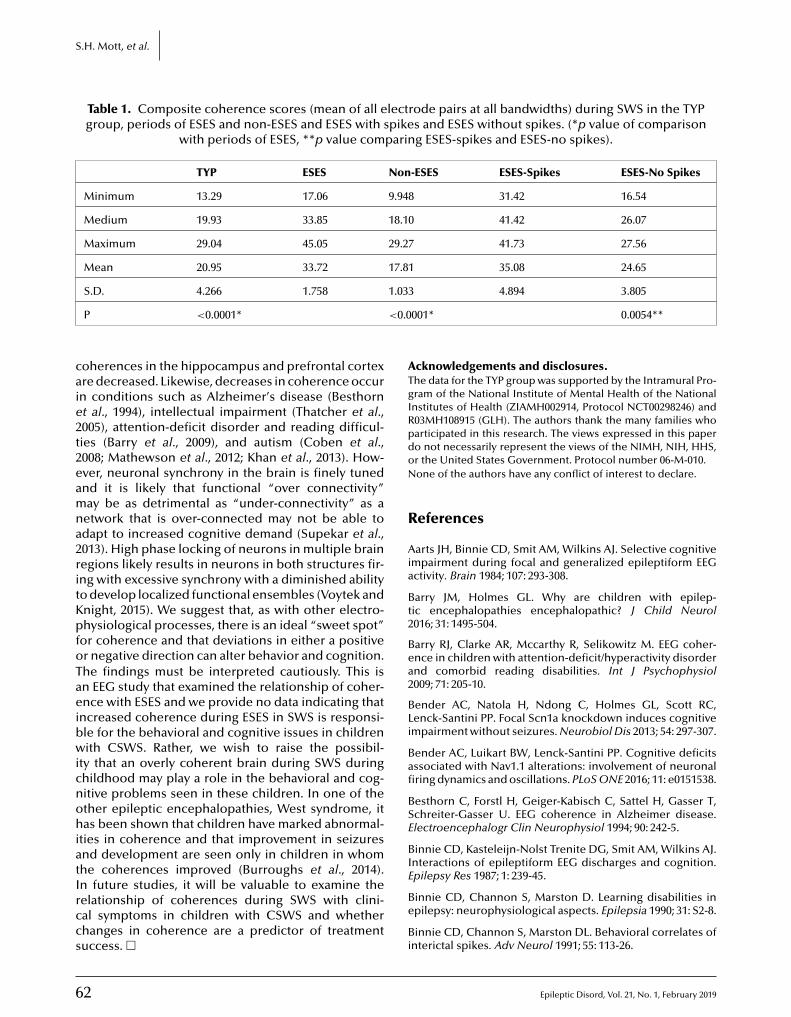
6
S.H. Mott, et al.
Table 1. Composite coherence scores (mean of all electrode pairs at all bandwidths) during SWS in the TYPgroup, periods of ESES and non-ESES and ESES with spikes and ESES without spikes. (*p value of comparison
with periods of ESES, **p value comparing ESES-spikes and ESES-no spikes).
TYP ESES Non-ESES ESES-Spikes ESES-No Spikes
Minimum 13.29 17.06 9.948 31.42 16.54
Medium 19.93 33.85 18.10 41.42 26.07
Maximum 29.04 45.05 29.27 41.73 27.56
Mean 20.95 33.72 17.81 35.08 24.65
1.03
<0.0
caie2t2eamna2ritKpfoTaeibwicnohiatIrccs
ATgIRpdoN
R
Aia
Bt2
Bea2
BLi
Bafi
BSE
BIEpilepsy Res 1987; 1: 239-45.
S.D. 4.266 1.758
P <0.0001*
oherences in the hippocampus and prefrontal cortexre decreased. Likewise, decreases in coherence occurn conditions such as Alzheimer’s disease (Besthornt al., 1994), intellectual impairment (Thatcher et al.,005), attention-deficit disorder and reading difficul-ies (Barry et al., 2009), and autism (Coben et al.,008; Mathewson et al., 2012; Khan et al., 2013). How-ver, neuronal synchrony in the brain is finely tunednd it is likely that functional “over connectivity”ay be as detrimental as “under-connectivity” as a
etwork that is over-connected may not be able todapt to increased cognitive demand (Supekar et al.,013). High phase locking of neurons in multiple brainegions likely results in neurons in both structures fir-ng with excessive synchrony with a diminished abilityo develop localized functional ensembles (Voytek andnight, 2015). We suggest that, as with other electro-hysiological processes, there is an ideal “sweet spot”
or coherence and that deviations in either a positiver negative direction can alter behavior and cognition.he findings must be interpreted cautiously. This isn EEG study that examined the relationship of coher-nce with ESES and we provide no data indicating that
ncreased coherence during ESES in SWS is responsi-le for the behavioral and cognitive issues in childrenith CSWS. Rather, we wish to raise the possibil-
ty that an overly coherent brain during SWS duringhildhood may play a role in the behavioral and cog-itive problems seen in these children. In one of thether epileptic encephalopathies, West syndrome, itas been shown that children have marked abnormal-
ties in coherence and that improvement in seizuresnd development are seen only in children in whomhe coherences improved (Burroughs et al., 2014).
2
n future studies, it will be valuable to examine theelationship of coherences during SWS with clini-al symptoms in children with CSWS and whetherhanges in coherence are a predictor of treatmentuccess. �
Be
Bi
3 4.894 3.805
001* 0.0054**
cknowledgements and disclosures.he data for the TYP group was supported by the Intramural Pro-ram of the National Institute of Mental Health of the Nationalnstitutes of Health (ZIAMH002914, Protocol NCT00298246) and03MH108915 (GLH). The authors thank the many families whoarticipated in this research. The views expressed in this papero not necessarily represent the views of the NIMH, NIH, HHS,r the United States Government. Protocol number 06-M-010.one of the authors have any conflict of interest to declare.
eferences
arts JH, Binnie CD, Smit AM, Wilkins AJ. Selective cognitivempairment during focal and generalized epileptiform EEGctivity. Brain 1984; 107: 293-308.
arry JM, Holmes GL. Why are children with epilep-ic encephalopathies encephalopathic? J Child Neurol016; 31: 1495-504.
arry RJ, Clarke AR, Mccarthy R, Selikowitz M. EEG coher-nce in children with attention-deficit/hyperactivity disordernd comorbid reading disabilities. Int J Psychophysiol009; 71: 205-10.
ender AC, Natola H, Ndong C, Holmes GL, Scott RC,enck-Santini PP. Focal Scn1a knockdown induces cognitivempairment without seizures. Neurobiol Dis 2013; 54: 297-307.
ender AC, Luikart BW, Lenck-Santini PP. Cognitive deficitsssociated with Nav1.1 alterations: involvement of neuronalring dynamics and oscillations. PLoS ONE 2016; 11: e0151538.
esthorn C, Forstl H, Geiger-Kabisch C, Sattel H, Gasser T,chreiter-Gasser U. EEG coherence in Alzheimer disease.lectroencephalogr Clin Neurophysiol 1994; 90: 242-5.
innie CD, Kasteleijn-Nolst Trenite DG, Smit AM, Wilkins AJ.nteractions of epileptiform EEG discharges and cognition.
Epileptic Disord, Vol. 21, No. 1, February 2019
innie CD, Channon S, Marston D. Learning disabilities inpilepsy: neurophysiological aspects. Epilepsia 1990; 31: S2-8.
innie CD, Channon S, Marston DL. Behavioral correlates ofnterictal spikes. Adv Neurol 1991; 55: 113-26.

E
Bfw1
Bt
Cc2
Epwa
Fe1
Gos
Gtsc2
GOse1
Ha2
Hf2
HfE
JNnN
Kfs3
Kp2
Kle
KMr
MJbC
Mq1
NIi2
NS
NEr
OnN
Pt1
SSs3
SRdm
Sms2
St1
ScE
Sdg7
Ss2
Sc
uckley AW, Scott R, Tyler A, et al. State-dependent dif-erences in functional connectivity in young childrenith autism spectrum disorder. EBioMedicine 2015; 2:
905-15.
urroughs SA, Morse RP, Mott SH, Holmes GL. Brain connec-ivity in West syndrome. Seizure 2014; 23: 576-9.
oben R, Clarke AR, Hudspeth W, Barry RJ. EEG power andoherence in autistic spectrum disorder. Clin Neurophysiol008; 119: 1002-9.
bus SC, Overvliet GM, Arends JB, Aldenkamp AP. Readingerformance in children with rolandic epilepsy correlatesith nocturnal epileptiform activity, but not with epileptiform
ctivity while awake. Epilepsy Behav 2011; 22: 518-22.
rench CC, Beaumont JG. A critical review of EEG coher-nce studies of hemisphere function. Int J Psychophysiol984; 1: 241-54.
alanopoulou AS, Bojko A, Lado F, Moshe SL. The spectrumf neuropsychiatric abnormalities associated with electricaltatus epilepticus in sleep. Brain Dev 2000; 22: 279-95.
encpinar P, Dundar NO, Tekgul H. Electrical status epilep-icus in sleep (ESES)/continuous spikes and waves duringlow sleep (CSWS) syndrome in children: an electroclini-al evaluation according to the EEG patterns. Epilepsy Behav016; 61: 107-11.
mehlin D, Thomas C, Weisbrod M, Walther S, Resch F,elkers-Ax R. Development of brain synchronisation within
chool-age: individual analysis of resting (alpha) coher-nce in a longitudinal data set. Clin Neurophysiol 2011; 122:973-83.
olmes GL. What is more harmful, seizures or epileptic EEGbnormalities? Is there any clinical data? Epileptic Disord014; 16: 12-22.
olmes GL, Lenck-Santini PP. Role of interictal epilepti-orm abnormalities in cognitive impairment. Epilepsy Behav006; 8: 504-15.
orak PC, Meisenhelter S, Song Y, et al. Interictal epilepti-orm discharges impair word recall in multiple brain areas.pilepsia 2017; 58: 373-80.
acinto LR, Reis JS, Dias NS, Cerqueira JJ, Correia JH, Sousa. Stress affects theta activity in limbic networks and impairsovelty-induced exploration and familiarization. Front Behaveurosci 2013; 7: 127.
han S, Gramfort A, Shetty NR, et al. Local and long-rangeunctional connectivity is reduced in concert in autismpectrum disorders. Proc Natl Acad Sci USA 2013; 110:107-12.
leen JK, Scott RC, Holmes GL, Lenck-Santini PP. Hippocam-al interictal spikes disrupt cognition in rats. Ann Neurol010; 67: 250-7.
oop JI, Fastenau PS, Dunn DW, Austin JK. Neuropsycho-
pileptic Disord, Vol. 21, No. 1, February 2019
ogical correlates of electroencephalograms in children withpilepsy. Epilepsy Res 2005; 64: 49-62.
rauss GL, Summerfield M, Brandt J, Breiter S, Ruchkin D.esial temporal spikes interfere with working memory. Neu-
ology 1997; 49: 975-80.
s2
StC
Coherence in ESES
athewson KJ, Jetha MK, Drmic IE, Bryson SE, GoldbergO, Schmidt LA. Regional EEG alpha power, coherence, andehavioral symptomatology in autism spectrum disorder.lin Neurophysiol 2012; 123: 1798-809.
ocks J, Gasser T. How to select epochs of the EEG at rest foruantitative analysis. Electroencephalogr Clin Neurophysiol984; 58: 89-92.
air A, Keown CL, Datko M, Shih P, Keehn B, Muller RA.mpact of methodological variables on functional connectiv-ty findings in autism spectrum disorders. Hum Brain Mapp014; 35: 4035-48.
ickels K, Wirrell E. Electrical status epilepticus in sleep.emin Pediatr Neurol 2008; 15: 50-60.
ielsen T, Abel A, Lorrain D, Montplaisir J. InterhemisphericEG coherence during sleep and wakefulness in left- andight-handed subjects. Brain Cogn 1990; 14: 113-25.
liveira JF, Dias NS, Correia M, et al. Chronic stress disruptseural coherence between cortico-limbic structures. Fronteural Circuits 2013; 7: 10.
atry G, Lyagoubi S, Tassinari CA. Subclinical “electrical sta-us epilepticus” induced by sleep in children. Arch Neurol971; 24: 242-52.
anchez Fernandez I, Loddenkemper T, Peters JM, KothareV. Electrical status epilepticus in sleep: clinical pre-entation and pathophysiology. Pediatr Neurol 2012; 47:90-410.
anchez Fernandez I, Chapman KE, Peters JM, Harini C,otenberg A, Loddenkemper T. Continuous spikes and wavesuring sleep: electroclinical presentation and suggestions foranagement. Epilepsy Res Treat 2013; 2013: 583531.
anchez Fernandez I, Chapman K, Peters JM, et al. Treat-ent for continuous spikes and waves during sleep (CSWS):
urvey on treatment choices in North America. Epilepsia014; 55: 1099-108.
cheltens-de Boer M. Guidelines for EEG in encephalopa-hy related to ESES/CSWS in children. Epilepsia 2009; 50:3-7.
hewmon DA, Erwin RJ. Transient impairment of visual per-eption induced by single interictal occipital spikes. J Clinxp Neuropsychol 1989; 1: 675-91.
igurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gor-on JA. Impaired hippocampal-prefrontal synchrony in aenetic mouse model of schizophrenia. Nature 2010; 464:63-7.
inghal NS, Sullivan JE. Continuous spike-wave duringlow wave sleep and related conditions. ISRN Neurol014; 2014: 619079.
rinivasan R, Winter WR, Ding J, Nunez PL. EEG and MEGoherence: measures of functional connectivity at distinct
63
patial scales of neocortical dynamics. J Neurosci Methods007; 166: 41-52.
upekar K, Uddin LQ, Khouzam A, et al. Brain hyperconnec-ivity in children with autism and its links to social deficits.ell Rep 2013; 5: 738-47.

6
S
Ted2
Ta2
Th1
TbN
Tam1
Tcc
UaB
VcE
VpN
Vud
WE
.H. Mott, et al.
assinari CA, Rubboli G, Volpi L, et al. Encephalopathy withlectrical status epilepticus during slow sleep or ESES syn-rome including the acquired aphasia. Clin Neurophysiol000; 111: S94-102.
hatcher RW. Coherence, phase differences, phase shift,nd phase lock in EEG/ERP analyses. Dev Neuropsychol012; 37: 476-96.
hatcher RW, Walker RA, Giudice S. Human cerebralemispheres develop at different rates and ages. Science987; 236: 1110-3.
hatcher RW, North D, Biver C. EEG and intelligence: relationsetween EEG coherence, EEG phase delay and power. Clineurophysiol 2005; 116: 2129-41.
hatcher RW, Krause PJ, Hrybyk M. Cortico-corticalssociations and EEG coherence: a two-compartmental
4
odel. Electroencephalogr Clin Neurophysiol 2012; 64:23-43.
ucker DM, Roth DL, Bair TB. Functional connections amongortical regions: topography of EEG coherence. Electroen-ephalogr Clin Neurophysiol 1986; 63: 242-50.
a2
ZiE
ng H, Cazares C, Nanivadekar A, et al. Interictal epileptiformctivity outside the seizure onset zone impacts cognition.rain 2017; 140: 2157-68.
an den Munckhof B, Van D, Sagi L, et al. Treatment of electri-al status epilepticus in sleep: a pooled analysis of 575 cases.pilepsia 2015; 56: 1738-46.
arela F, Lachaux JP, Rodriguez E, Martinerie J. The brainweb:hase synchronization and large-scale integration. Nat Reveurosci 2001; 2: 229-39.
oytek B, Knight RT. Dynamic network communication as anifying neural basis for cognition, development, aging, andisease. Biol Psychiatry 2015; 77: 1089-97.
hedon M, Perry NB, Calkins SD, Bell MA. Changes in frontalEG coherence across infancy predict cognitive abilities at
Epileptic Disord, Vol. 21, No. 1, February 2019
ge 3: the mediating role of attentional control. Dev Psychol016; 52: 1341-52.
hou JL, Lenck-Santini PP, Zhao Q, Holmes GL. Effect of inter-ctal spikes on single-cell firing patterns in the hippocampus.pilepsia 2007; 48: 720-31.

do
i:10.
1684
/ep
d.2
019.
1028
Epileptic Disord, Vol. 21, No. 1, February 2019 65
Correspondence:Zhong YingS51 Epilepsy Center,Cleveland Clinic,9500 Euclid Ave,Cleveland, OH 44195, USA<[email protected]>
Original articleEpileptic Disord 2019; 21 (1): 65-77
A comprehensiveclinico-pathologicaland genetic evaluationof bottom-of-sulcus focalcortical dysplasia in patientswith difficult-to-localizefocal epilepsyZhong Ying 1, Irene Wang 1, Ingmar Blümcke 1,2, Juan Bulacio 1,Andreas Alexopoulos 1, Lara Jehi 1, William Bingaman 1,Jorge Gonzalez-Martinez 1, Katja Kobow 2, Lisa MarieNiestroj 3, Dennis Lal 1,3,4, Konrad Koelble 2, Imad Najm 1
1 Epilepsy Center, Cleveland Clinic, Cleveland, Ohio, USA2 Neuropathological Institute, University Hospitals Erlangen, Erlangen3 Cologne Center for Genomics, University of Cologne, Cologne, Germany4 Stanley Center for Psychiatric Research, Broad Institute of Harvard & MIT, Cambridge,Massachusetts, USA
Received October 30, 2017; Accepted November 18, 2018
ABSTRACT – Aims. We comprehensively studied the clinical presentation,stereo-EEG and MRI findings, histopathological diagnosis, and brainsomatic mutations in a retrospective series of drug-resistant patients withdifficult-to-localize epilepsy due to focal cortical dysplasia at the bottomof a sulcus (BOS-FCD).Methods. We identified 10 patients with BOS-FCD from the ClevelandClinic epilepsy surgery database submitted for intracranial video-EEGmonitoring. Brain MRI, including voxel-based morphometric analysisand surgical tissue submitted for histopathology, was reviewed. Paraf-fin tissue samples from five patients were made available for targetednext-generation sequencing. Postsurgical follow-up was available in ninepatients.Results. BOS-FCD was identified in the superior frontal sulcus in sixpatients, inferior frontal sulcus in one patient, central sulcus in one patient,and intraparietal sulcus in two patients. All patients had stereotypedseizures. Intracranial EEG recordings identified ictal onset at the BOS-FCDin all 10 patients, whereas ictal scalp EEG had a localizing value in only sixpatients. Complete resection was achieved by lesionectomy or focal cor-ticectomy in nine patients. Histopathologically, six patients had FCD typeIIb and three had FCD type IIa. Next-generation sequencing analysis of DNA

6
Z. Ying, et al.
extracted from lesion-enriched (micro-dissected) tissue from five patientswith FCD type II led to the identification of a germline frameshift insertionin DEPDC5, introducing a premature stop in one patient. Eight out of ninepatients with available follow-up were completely seizure-free (Engel ClassIA) after a mean follow-up period of six years.Conclusion. Our results confirm previous studies classifying difficult-to-localize BOS-FCD into the emerging spectrum of FCD ILAE type IImTORopathies. Further studies with large patient numbers and ultra-deep
ay hD.
, sei
FlrhsatwoiLaiass(eWttti2hcrFttefboMiWtt2ctis
hsFBM2lBtFtwobe
MS
TecEu2Rf––s–––s–t–m
genetic testing maetiologies of FC
Key words: brain
ocal cortical dysplasia (FCD) are common histopatho-ogical lesions in children and adults with drug-esistant focal epilepsy (Blümcke et al., 2017), anditherto classified into separate clinico-pathologicalubtypes (Blümcke et al., 2011). However, the aetiologynd pathogenesis of most of these subtypes remaino be clarified (Najm et al., 2018). Such knowledgeill be mandatory to also understand their variableccurrence in size, cellular phenotypes, brain local-
zation and clinical presentation (Krsek et al., 2008;erner et al., 2009; Blümcke et al., 2010; Chassoux etl., 2012; Harvey et al., 2015). Continuous improvementn magnetic field strength for MRI diagnosis and thepplication of advanced post-processing analyses hasignificantly enhanced clinical identification of FCDubtypes in vivo, in particular, of FCD ILAE type IIUrbach et al., 2002; Besson et al., 2008; Bernasconit al., 2011; Wagner et al., 2011; Mellerio et al., 2014;ang et al., 2014). As a pertinent example, hyperin-
ense MRI signalling from the lateral ventricle towardshe crown of the gyrus was described as a “transman-le sign” (Barkovich et al., 1997), and mostly confirmedn FCD IIb and in the frontal lobe (Colombo et al.,009; Colombo et al., 2012). Not all FCD II present,owever, with a transmantle sign suggesting a largerlinico-pathological spectrum disorder or even sepa-ate disease entities. Two previous reports focused onCD II located at the bottom of sulcus and highlightedhese challenges in neuroimaging, clinical, and elec-roclinical presentation (Chassoux et al., 2012; Harveyt al., 2015). Tailored surgical resection was particularavourable, with 87-94% of reported patients (n=57)eing completely seizure-free. Intriguingly, about 25%f patients did not reveal abnormal signals at initialRI examination, and the combination of PET with MRI
ncreased the detection rate (Chassoux et al., 2010).ith few exceptions, current research has failed
o establish pathology-specific molecular biomarkershat clearly distinguish FCD subtypes (Guerrini et al.,
6
015). In the absence of adequate animal models, surgi-al brain tissue samples open the unique opportunityo further study tissue-specific signatures. A milestonen FCD research represented the identification of brainomatic mutations, germline mutations, or second-
M
FT
elp to bridge the current knowledge gap in genetic
zure, mTOR, epilepsy surgery, outcome
it mosaic mutations activating the mTOR pathway inurgical brain specimens with histopathology-provenCD type II (Jamuar et al., 2014; Scheffer et al., 2014;aulac et al., 2015; D’Gama et al., 2015; Lim et al., 2015;irzaa et al., 2016; Moller et al., 2016; D’Gama et al.,
017; Ribierre et al., 2018). With only a third of pub-ished cases showing a genetic lesion (Marsan andaulac, 2018), however, continuous efforts are required
o improve our understanding of clinically-meaningfulCD subtypes and successful treatment strategies inhe near future. The integration of clinical phenotypesith histopathology and genetic analysis is a powerfulption, as recently proposed and already implementedy the WHO for the diagnosis of malignant gliomas andmbryonal brain tumours (Louis et al., 2016).
ethodselection of patients
o investigate patients with difficult-to-localizepilepsy due to cortical dysplasia at the bottom of sul-us, we retrospectively reviewed the Cleveland Clinicpilepsy Center’s surgery database with patients whonderwent invasive intracranial studies from 2004 to014 (as approved by the Cleveland Clinic Institutionaleview Board). Inclusion of patients was based on the
ollowing criteria:drug-resistant focal epilepsy;a single MRI lesion restricted to the bottom of a
ulcus;no previous epilepsy surgery;intracranial video-EEG monitoring prior to surgery;no concomitant other diagnosis, such as tuberous
clerosis or brain tumour;post-operative MRI available to assess the extent of
he resection;and histopathology slides available for post hoc
icroscopic review.
Epileptic Disord, Vol. 21, No. 1, February 2019
agnetic resonance imaging (MRI)
our patients were imaged with a 3 T Siemensrio/Skyra scanner (Erlangen, Germany) and six
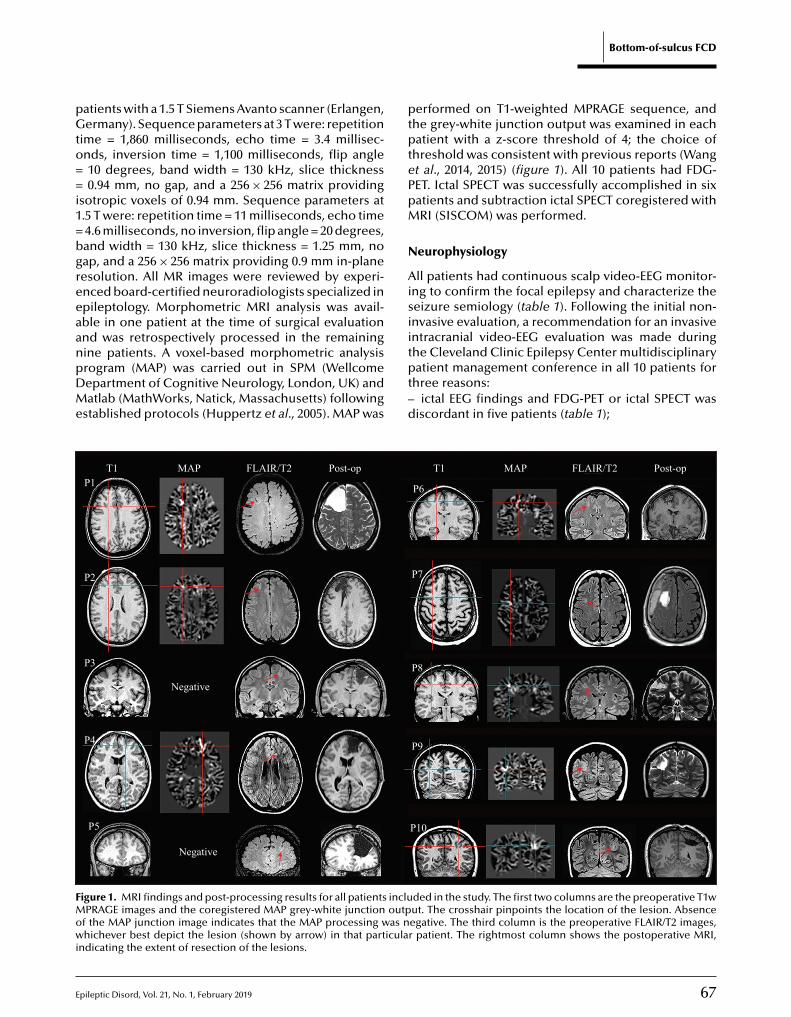
E
pGto==i1=bgreeaanpDMe
ptptePpM
N
Aisii
FMowi
atients with a 1.5 T Siemens Avanto scanner (Erlangen,ermany). Sequence parameters at 3 T were: repetition
ime = 1,860 milliseconds, echo time = 3.4 millisec-nds, inversion time = 1,100 milliseconds, flip angle10 degrees, band width = 130 kHz, slice thickness0.94 mm, no gap, and a 256 × 256 matrix providing
sotropic voxels of 0.94 mm. Sequence parameters at.5 T were: repetition time = 11 milliseconds, echo time4.6 milliseconds, no inversion, flip angle = 20 degrees,and width = 130 kHz, slice thickness = 1.25 mm, noap, and a 256 × 256 matrix providing 0.9 mm in-planeesolution. All MR images were reviewed by experi-nced board-certified neuroradiologists specialized inpileptology. Morphometric MRI analysis was avail-ble in one patient at the time of surgical evaluationnd was retrospectively processed in the remaining
pileptic Disord, Vol. 21, No. 1, February 2019
ine patients. A voxel-based morphometric analysisrogram (MAP) was carried out in SPM (Wellcomeepartment of Cognitive Neurology, London, UK) andatlab (MathWorks, Natick, Massachusetts) following
stablished protocols (Huppertz et al., 2005). MAP was
tpt–d
T1 MAP FLAIR/T2 Post-opP1
P2
P3
P4
P5
Negative
Negative
igure 1. MRI findings and post-processing results for all patients incluPRAGE images and the coregistered MAP grey-white junction outp
f the MAP junction image indicates that the MAP processing was nhichever best depict the lesion (shown by arrow) in that particula
ndicating the extent of resection of the lesions.
Bottom-of-sulcus FCD
erformed on T1-weighted MPRAGE sequence, andhe grey-white junction output was examined in eachatient with a z-score threshold of 4; the choice of
hreshold was consistent with previous reports (Wangt al., 2014, 2015) (figure 1). All 10 patients had FDG-ET. Ictal SPECT was successfully accomplished in sixatients and subtraction ictal SPECT coregistered withRI (SISCOM) was performed.
europhysiology
ll patients had continuous scalp video-EEG monitor-ng to confirm the focal epilepsy and characterize theeizure semiology (table 1). Following the initial non-nvasive evaluation, a recommendation for an invasiventracranial video-EEG evaluation was made during
67
he Cleveland Clinic Epilepsy Center multidisciplinaryatient management conference in all 10 patients for
hree reasons:ictal EEG findings and FDG-PET or ictal SPECT was
iscordant in five patients (table 1);
T1 MAP FLAIR/T2 Post-op
P6
P7
P8
P9
P10
ded in the study. The first two columns are the preoperative T1wut. The crosshair pinpoints the location of the lesion. Absenceegative. The third column is the preoperative FLAIR/T2 images,r patient. The rightmost column shows the postoperative MRI,
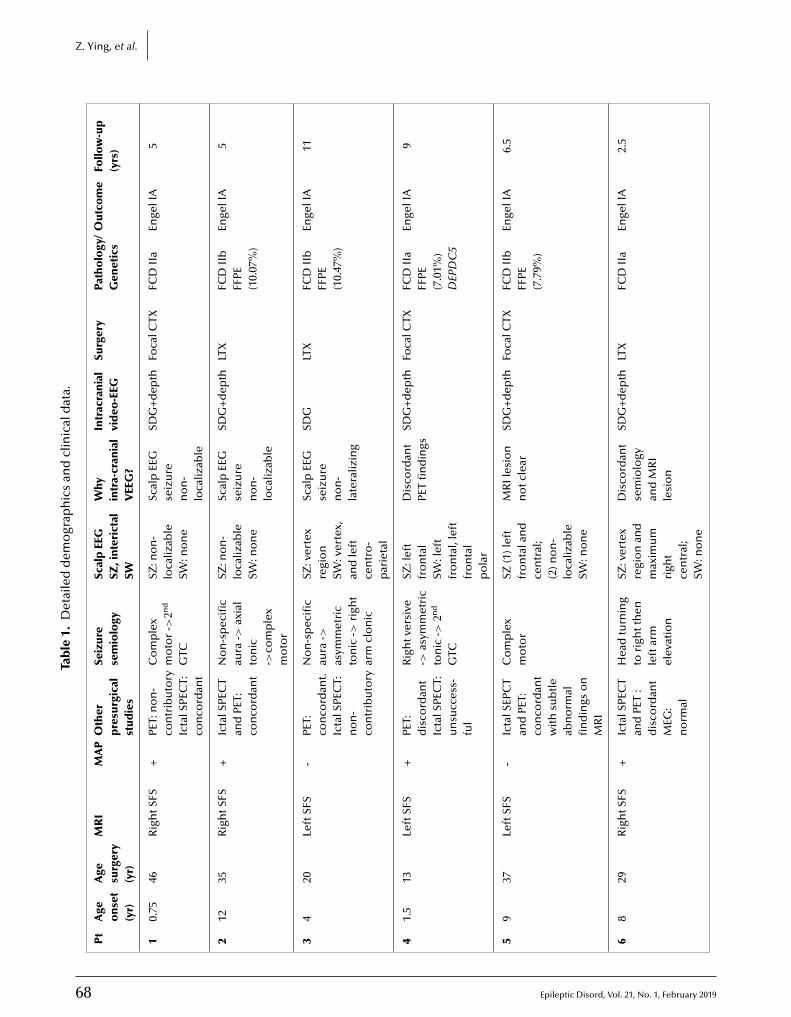
68 Epileptic Disord, Vol. 21, No. 1, February 2019
Z. Ying, et al.
Tab
le1.
Det
aile
dd
emo
grap
hic
san
dcl
inic
ald
ata.
Pt
Age
on
set
(yr)
Age
surg
ery
(yr)
MR
IM
AP
Oth
erp
resu
rgic
alst
ud
ies
Seiz
ure
sem
iolo
gySc
alp
EEG
SZ,i
nte
rict
alSW
Why
intr
a-cr
ania
lV
EEG
?
Intr
acra
nia
lvi
deo
-EEG
Surg
ery
Path
olo
gy/
Gen
etic
sO
utc
om
eFo
llow
-up
(yrs
)
10.
7546
Rig
htS
FS+
PET:
no
n-
con
trib
uto
ryIc
talS
PEC
T:co
nco
rdan
t
Co
mp
lex
mo
tor
->2n
d
GTC
SZ:n
on
-lo
caliz
able
SW:n
on
e
Scal
pEE
Gse
izu
ren
on
-lo
caliz
able
SDG
+d
epth
Foca
lCTX
FCD
IIa
Enge
lIA
5
212
35R
igh
tSFS
+Ic
talS
PEC
Tan
dPE
T:co
nco
rdan
t
No
n-s
pec
ific
aura
->ax
ial
ton
ic->
com
ple
xm
oto
r
SZ:n
on
-lo
caliz
able
SW:n
on
e
Scal
pEE
Gse
izu
ren
on
-lo
caliz
able
SDG
+d
epth
LTX
FCD
IIb
FFPE
(10.
07%
)
Enge
lIA
5
34
20Le
ftSF
S-
PET:
con
cord
ant.
Icta
lSPE
CT:
no
n-
con
trib
uto
ry
No
n-s
pec
ific
aura
->as
ymm
etri
cto
nic
->ri
ght
arm
clo
nic
SZ:v
erte
xre
gio
nSW
:ver
tex,
and
left
cen
tro
-p
arie
tal
Scal
pEE
Gse
izu
ren
on
-la
tera
lizin
g
SDG
LTX
FCD
IIb
FFPE
(10.
47%
)
Enge
lIA
11
41.
513
Left
SFS
+PE
T:d
isco
rdan
tIc
talS
PEC
T:u
nsu
cces
s-fu
l
Rig
htv
ersi
ve->
asym
met
ric
ton
ic->
2nd
GTC
SZ:l
eft
fro
nta
lSW
:lef
tfr
on
tal,
left
fro
nta
lp
ola
r
Dis
cord
ant
PET
fin
din
gsSD
G+
dep
thFo
calC
TXFC
DII
aFF
PE(7
.01%
)D
EPD
C5
Enge
lIA
9
59
37Le
ftSF
S-
Icta
lSEP
CT
and
PET:
con
cord
ant
wit
hsu
btl
eab
no
rmal
fin
din
gso
nM
RI
Co
mp
lex
mo
tor
SZ(1
)lef
tfr
on
tala
nd
cen
tral
;(2
)no
n-
loca
lizab
leSW
:no
ne
MR
Iles
ion
no
tcle
arSD
G+
dep
thFo
calC
TXFC
DII
bFF
PE(7
.79%
)
Enge
lIA
6.5
68
29R
igh
tSFS
+Ic
talS
PEC
Tan
dPE
T:
dis
cord
ant
MEG
:n
orm
al
Hea
dtu
rnin
gto
righ
tth
enle
ftar
mel
evat
ion
SZ:v
erte
xre
gio
nan
dm
axim
um
righ
tce
ntr
al;
SW:n
on
e
Dis
cord
ant
sem
iolo
gyan
dM
RI
lesi
on
SDG
+d
epth
LTX
FCD
IIa
Enge
lIA
2.5
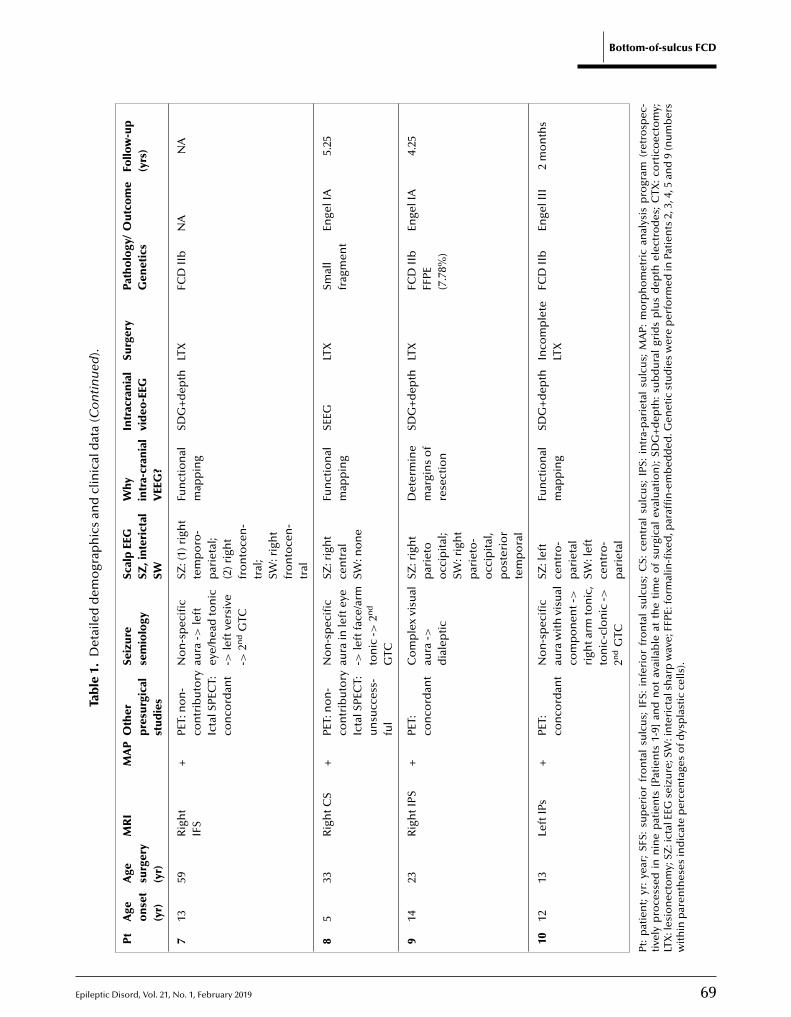
Epileptic Disord, Vol. 21, No. 1, February 2019 69
Bottom-of-sulcus FCD
Tab
le1.
Det
aile
dd
emo
grap
hic
san
dcl
inic
ald
ata
(Co
nti
nu
ed).
Pt
Age
on
set
(yr)
Age
surg
ery
(yr)
MR
IM
AP
Oth
erp
resu
rgic
alst
ud
ies
Seiz
ure
sem
iolo
gySc
alp
EEG
SZ,i
nte
rict
alSW
Why
intr
a-cr
ania
lV
EEG
?
Intr
acra
nia
lvi
deo
-EEG
Surg
ery
Path
olo
gy/
Gen
etic
sO
utc
om
eFo
llow
-up
(yrs
)
713
59R
igh
tIF
S+
PET:
no
n-
con
trib
uto
ryIc
talS
PEC
T:co
nco
rdan
t
No
n-s
pec
ific
aura
->le
ftey
e/h
ead
ton
ic->
left
vers
ive
->2n
dG
TC
SZ:(
1)ri
ght
tem
po
ro-
par
ieta
l;(2
)rig
ht
fro
nto
cen
-tr
al;
SW:r
igh
tfr
on
toce
n-
tral
Fun
ctio
nal
map
pin
gSD
G+
dep
thLT
XFC
DII
bN
AN
A
85
33R
igh
tCS
+PE
T:n
on
-co
ntr
ibu
tory
Icta
lSPE
CT:
un
succ
ess-
ful
No
n-s
pec
ific
aura
inle
ftey
e->
left
face
/arm
ton
ic->
2nd
GTC
SZ:r
igh
tce
ntr
alSW
:no
ne
Fun
ctio
nal
map
pin
gSE
EGLT
XSm
all
frag
men
tEn
gelI
A5.
25
914
23R
igh
tIPS
+PE
T:co
nco
rdan
tC
om
ple
xvi
sual
aura
->d
iale
pti
c
SZ:r
igh
tp
arie
too
ccip
ital
;SW
:rig
ht
par
ieto
-o
ccip
ital
,p
ost
erio
rte
mp
ora
l
Det
erm
ine
mar
gin
so
fre
sect
ion
SDG
+d
epth
LTX
FCD
IIb
FFPE
(7.7
8%)
Enge
lIA
4.25
1012
13Le
ftIP
s+
PET:
con
cord
ant
No
n-s
pec
ific
aura
wit
hvi
sual
com
po
nen
t->
righ
tarm
ton
ic,
ton
ic-c
lon
ic->
2nd
GTC
SZ:l
eft
cen
tro
-p
arie
tal
SW:l
eft
cen
tro
-p
arie
tal
Fun
ctio
nal
map
pin
gSD
G+
dep
thIn
com
ple
teLT
XFC
DII
bEn
gelI
II2
mo
nth
s
Pt:
pat
ien
t;yr
:ye
ar;
SFS:
sup
erio
rfr
on
tal
sulc
us;
IFS:
infe
rio
rfr
on
tal
sulc
us;
CS:
cen
tral
sulc
us;
IPS:
intr
a-p
arie
tal
sulc
us;
MA
P:m
orp
ho
met
ric
anal
ysis
pro
gram
(ret
rosp
ec-
tive
lyp
roce
ssed
inn
ine
pat
ien
ts[P
atie
nts
1-9]
and
no
tav
aila
ble
atth
eti
me
of
surg
ical
eval
uat
ion
);SD
G+
dep
th:
sub
du
ral
grid
sp
lus
dep
thel
ectr
od
es;
CTX
:co
rtic
oec
tom
y;LT
X:l
esio
nec
tom
y;SZ
:ict
alEE
Gse
izu
re;S
W:i
nte
rict
alsh
arp
wav
e;FF
PE:f
orm
alin
-fixe
d,p
araf
fin
-em
bed
ded
.Gen
etic
stu
die
sw
ere
per
form
edin
Pati
ents
2,3,
4,5
and
9(n
um
ber
sw
ith
inp
aren
thes
esin
dic
ate
per
cen
tage
so
fdys
pla
stic
cells
).

7
Z. Ying, et al.
Interictal EEG Ictal EEG
450 uV
A6-A5
a4-a5
a5-a6
a6-a7
a7-a8
b1-b2
b2-b3
A6-A7
A7-A8
798
6543
21
b4b1
a5a3a1
9 54 2
a5-a6
a6-a7
a7-a8
a4-a5
a5-a6
a6-a7
a7-a8
a6-a7
a7-a8
a8-a9
a9-a10
310 8 6
a6 a7
a8 a9a7a6
17
a7a6b2
PT5
PT6
PT8
PT9
PT10
1 second
Interictal EEG Ictal EEG
Interictal EEG Ictal EEG
Interictal EEG Ictal EEG
Interictal EEG Ictal EEG
450 uV 1 second
450 uV1 second
450 uV1 second
450 uV1 second
Figure 2. Location of the depth electrode contacts and their anatomic relation to BOS lesions as co-registered on T1-weighted MRIcoronal cuts in five patients are shown on the left. The electrode contacts recording ictal EEG onset are labelled in red. The samplesof EEG channels recording ictal EEG onset from the depth electrodes (bipolar montage) are shown on the right. The red bars pointto the ictal EEG onsets that were preceded by the preictal repetitive spikes/polyspikes in each of the patients. Patients (PT) 6 and 9showed interictal rhythmic polyspikes and wave discharges that became more frequent immediately prior to the ictal onset. Patients5 d wit re che
–i–tEeeptvvh(
tsi(
N
S
and 10 showed interictal repetitive spikes that were intermixeonic fast-frequency discharges. Seizures in Patients 6 and 10 wemergence of the low-amplitude fast activities.
eloquent cortical areas had to be mapped for the def-nition of surgical resection borders in four patients;
and BOS-FCD was not unambiguously accepted byhe group in one patient (Patient 5) (table 1).ight patients had an implantation of subdural gridlectrodes (SDG) together with intracerebral depthlectrodes targeting the BOS-FCD of interest. Thelacement of depth electrodes and their 3D spa-
0
ial correlation with the MRI-identified lesion waserified through co-registration of post-implantationolume acquisition CT scans and preimplantationigh-resolution MRI volume acquisition sequences
figure 2). One patient had SDGs without depth elec-
icahw
th low-voltage fast activities. In all patients, ictal EEGs showedaracterized by a brief initial attenuation of the EEG prior to the
rode implantation (Patient 3) (table 1). Patient 8 hadtereotactic implantation of depth electrodes accord-ng to the SEEG methodology, as previously describedGonzalez-Martinez et al., 2014).
eurosurgery
urgical resection strategies were discussed follow-
Epileptic Disord, Vol. 21, No. 1, February 2019
ng the invasive evaluation at our patient managementonference, integrating all available data from MRInalysis and neurophysiological recordings. Postoc analysis of the extent of surgical resectionas obtained from post-surgical MRI. Post-operative

E
Bottom-of-sulcus FCD
A1
A2
B
C
BC
Vimentin
pS6
NFP
DN
HE
D
E
Figure 3. (A-C) H&E staining: whole-slide imaging of Patient 9 (table 1) before (A1) and after (A2) microdissection with a 2-mm diameterpunching device (scale bar = 4 mm); (B) higher magnification of area indicated by arrow in (A) and used for DNA extraction fol-lowing micro-dissection (shown in [A’]). (C) Vimentin immunohistochemistry highlighting balloon cells (BC); serial section from (B).( g boc mistr( 8% o
sta
I
FwhitdaaaSiDIe
UpwlspFpspppdHw
D) phosphorylated S6 (pS6) immunohistochemistry highlightinells (arrowheads); serial section from (B). (E) ImmunohistocheDN); serial section from (B). Microscopic measurements reveal
eizure outcome was assessed during regular outpa-ient visits using Engel’s classification scale (Engel etl., 1993).
mmunohistochemistry
or histopathological diagnosis, surgical specimensere formalin-fixed and paraffin-embedded. Postoc review of all surgical tissue was based on
mmunohistochemical stainings to visualize architec-ural dysplasia (Blümcke et al., 2016) using antibodiesirected against NeuN (clone A60, Chemicon, USA)nd MAP2 (clone HM2, DAKO, Denmark), to visu-lize dysmorphic neurons with antibodies directedgainst non-phosphorylated neurofilaments (clone
pileptic Disord, Vol. 21, No. 1, February 2019
MI32, Covance, USA) or balloon cells with antibod-es directed against vimentin (polyclonal antibody V9,AKO, Denmark) (see also Blümcke et al. [2016]).
mmunohistochemical detection of the phospho-S6pitope (clone ser235/236, Cell Signaling Technology,
6nI(a
th FCD II cell types, dysmorphic neurons (arrow), and balloony for neurofilament protein highlighting dysmorphic neurons
f cells with a FCDII phenotype. Scale bar in (B-E) = 50 �m.
SA) was used to demonstrate an activated mTORathway. Areas with highest content of abnormal cellsere identified on the H&E section, and the same area
abelled in the FFPE bloc. In nine patients, FFPE tis-ue was micro-dissected using a 2-mm diameter largeunching needle (figure 3). DNA extraction from theFPE tissue punch was performed using customizedrotocols for small FFPE tissue (Qiagen, Germany) withufficient DNA available for deep sequencing in fiveatients. Semi-quantitative cell measurements wereerformed as following: a HE stained section was pre-ared before and after the tissue punch and fullyigitized using whole slide digital imaging (3DHistech,ungary). All cells within 1 mm2 of the punched areaere counted from the computer screen (range: 171-
73 cells). FCD-specific cells referred to dysmorphiceurons in FCD IIa and IIb and balloon cells in FCD
71
Ib and were counted from the same region of interestrange: 18-80 cells). Results were expressed as percent-ge of FCD-specific from total cells.

7
Z
G
TAprpiTtggp(dubtCEiFrhDrmSDtvrafhcf>ViiebibcafW
R
P
Ta
np(seey(ahp7ot
I
BewitFwaiFdoi1ptp>wrsmsfi
S
HshBwe
. Ying, et al.
enetic analysis
o perform the targeted sequencing, we used thegilent SureSelect Custom Enrichment Kit for libraryreparation of 166 self-selected genes. Library prepa-ation was conducted according to the manufacturer’srotocols and subsequent paired-end library sequenc-
ng was performed using the Illumina HiSeq4000.he targeted genes included those encoding pro-eins of the mTOR and PI3K-AKT signalling pathway,enes associated with low-grade brain tumours, andenes associated with epilepsy. The list of mTORathway genes was derived from the Kegg Pathway
ID: hsa04150). The PI3K-AKT pathway genes wereerived from RT2 Profiler PCR Array (Qiagen, Prod-ct no.: 330231). Genes associated with low-graderain tumours were derived from the recent litera-
ure and epilepsy genes were derived from the EpiPMonsortium review in 2015 (Vogelstein et al., 2013;piPMConsortium, 2015). The full list of included geness disclosed in supplementary table 1.or bioinformatic analysis, we generated analysis-eady bam files using BWA to map reads to theuman genome reference build GRCh37 (Li andurbin, 2009). GATK was used to mark duplicated
eads (McKenna et al., 2010), perform local realign-ent, recalibrate the base quality scores, and call
NPs and short indels together with SAM tools andindel (Li et al., 2009; Albers et al., 2011). In addi-
ion, we used Platypus to call low allele frequencyariants (Rimmer et al., 2014). We used the humaneference genome build GRCh37 and annotated vari-nt functional consequences and population allelerequencies using wANNOVAR (excessed: 12/2016;ttp://wannovar.wglab.org). We removed non-protein-oding variants and variants present in individualsrom the general population with allele frequency0.1% to enrich for rare variants of large effect.ariants passing our applied filters, were manually
nspected for sequencing and variant calling qual-ty using the Integrative Genomics Viewer (Robinsont al., 2011). The manual evaluation was conductedy three independent scientists. Variant pathogenic-
ty was assessed in accordance with 28 criteria definedy guidelines of the American College of Medi-al Genetics and Genomics (ACMG) (Richards etl., 2015). We used the online tool, InterVar, toacilitate the variant interpretation process (Li and
ang, 2017).
esults
2
atient data
en patients fulfilled our inclusion criteria; four malesnd six females. None of the patients had any
bFpta
eurological deficit at clinical examination. Oneatient had mild developmental delay (Patient 4)
table 1). All patients presented with stereotypedeizures (see table 1), without a history of statuspilepticus, infantile spasms or febrile seizures. Age atpilepsy onset ranged from 0.75 to 14 years (mean: 7.2ears). Age at time of surgery ranged from 13 to 59 yearsmean: 33.2 years). Epilepsy duration was between onend 46 years (mean: 23.4 years). None of the patientsad a history of pre- or perinatal injuries except for oneatient who was born two months premature (Patient) (table 1). Clinical histories and seizure descriptionf all patients included in the study are summarized in
able 1.
maging data
OS-FCD was identified in all patients by epilepsyxpert neuroradiology review (figure 1). Six lesionsere located in the superior frontal sulcus, one lesion
n the inferior frontal sulcus, one lesion in the cen-ral sulcus, and two lesions in the intraparietal sulcus.ocal cortical thickening and blurring of the grey-hite matter junction at the bottom of a sulcus wascommon MRI finding in all patients, as illustrated
n figure 1. These abnormalities were best visible onLAIR sequences. Eight out of 10 patients had concor-ant signal changes on T1w images (figure 1). Blurringf the grey-white matter junction was evident on T1w
mages in four patients by visual inspection (Patients, 4, 9, and 10) (table 1), and eight patients showedositive MAP foci. Blurring of the grey-white mat-
er junction on T1w images became evident in fouratients only after MAP analysis with a z-score set4 (Patients 2, 6, 7, and 8). No T1w signal changesere observed visually or by MAP analysis in the
emaining two patients (Patients 3 and 5). A funnel-haped, subcortical hyper-intensity tapering abnor-ality towards the ventricular surface (transmantle
ign) was seen in three patients (Patients 1, 8 and 9) (seegure 1).
calp video-EEG monitoring
abitual seizures were documented in all patients bycalp video-EEG-monitoring. Semiology could not be,owever, correlated simply with lobar location of theOS-FCD (table 1). As an example, most of the aurasere non-specific. Secondly, presence of auras withye involvement could be anatomically misleading in
Epileptic Disord, Vol. 21, No. 1, February 2019
oth patients (Patients 9 and 10) with intraparietal BOS-CD. Ictal EEG patterns were non-localizable in twoatients (Patients 1 and 2) (table 1), regional lobar for
wo patients in one brain region (Patients 4 and 8)nd for two patients with two adjacent brain regions

E
(twv
F
Ipidhwpav
I
ApdtpivhpHbmBe(iSfhTvOeovIco(sfEctofi
pfr
S
Noitptp1yfpwbt(
H
AiFppimiipuwsarls
G
Tagaso
Patients 9 and 10), and at the midline vertex region inwo patients (Patients 3 and 6). Interictal sharp wavesere absent in five patients and did not add localizing
alue in non-localizing ictal EEGs.
DG-PET and ictal SPECT
nterictal FDG-PET studies were performed in allatients (table 1); only five out of 10 patients exhib-
ted focal hypometabolism concordant with the lesionetected by MRI, whereas the remaining five patientsad non-contributory results, including two patientsith discordant PET localization. In four out of sixatients with successful ictal SPECT and SISCOM, therea of hyperperfusion was concordant with the MRI-isible BOS-FCD (table 1).
nvasive video-EEG monitoring
t the time of surgical evaluation, all 10 patientsroceeded to invasive EEG evaluation based on theecision of the patient management conference at
hat time (table 1): non-localizing ictal EEG in threeatients, atypical semiology associated with the lesion
n one patient, MRI lesion was not unanimously con-inced by the committee in one patient, discordant PETypometabolism in one patient, and functional map-ing was recommended in four patients.abitual seizures were documented in all patients alsoy invasive video-EEG monitoring. Ictal EEG onset wasapped to depth electrode contacts localized in the
OS-FCD or in areas sampled adjacent to the lesion inight patients (figure 2). In the remaining two patientsPatient 2 with no depth electrode contacts in the prox-mity of the lesion or depth of sulcus, and Patient 3 withDG electrodes only), ictal EEG onset was recordedrom electrodes placed in the gyral crown of the sulciarbouring the lesion.he depth electrodes were located within or in closeicinity to the BOS-FCD in seven patients (table 1).nly in Patient 2, co-registration revealed the depth
lectrode not inside or close to the lesion. Histopathol-gy analysis confirmed the iEEG trajectory in closeicinity to the FCD in one patient (data not shown).ntracranial EEG recordings with depth electrodeontacts in bottom-of-sulcus lesions showed continu-us interictal discharge patterns consisting of rhythmic
1-3-Hz) spikes/polyspikes and waves. The rhythmicpiking pattern was intermixed with low-amplitudeast discharges (figure 2). Extraoperative video and
pileptic Disord, Vol. 21, No. 1, February 2019
EG recordings captured the patients’ typical clini-al seizures with stereotyped ictal EEG onset patternshat were localized at the depth electrode contacts inr near the BOS-FCD (figure 2, also in reference togure 1). All EEG seizures transitioned from continuous
sdhva
Bottom-of-sulcus FCD
re-ictal rhythmic epileptiform discharges to tonic fastrequency discharges of variable voltage for durationsanging from 15 to 25 seconds (figure 2).
urgical resection and seizure outcome
eurosurgical resection of the lesion was limited toverlying and surrounding cortex (i.e. lesionectomy)
n seven patients and corticoectomy with resection ofhe lesion plus adjacent gyri was performed in threeatients (table 1). Figure 1 illustrates the anatomy of
he resection in all patients (post-operative MRI). Eightatients became free of seizures and auras (Engel ClassA) (table 1) with a mean follow-up time of six years (2.5ears to 11 years). One patient (Patient 7) did not returnor follow-up. One patient (Patient 10) had incom-lete resection due to the epileptic zone overlappingith eloquent cortex, as determined by extraoperativerain stimulation via subdural grids and depth elec-
rodes, and this patient continued to have seizuresEngel Class III).
istopathological findings
ll surgical specimens were histopathologically andmmunohistochemically reviewed and classified asCD ILAE type II, with a combination of dysmor-hic neurons and balloon cells (FCD IIb) in sixatients and dysmorphic neurons only (FCD IIa)
n three patients (table 1). Only small tissue frag-ents were submitted for pathological examination
n one patient and microscopic review remainednconclusive (Patient 8). The gross neuroanatomicalresentation of FCD IIb can be demonstrated bestsing immunohistochemical labelling of surgicallyell-preserved specimens with the characteristic pre-
entation of vimentin-immunoreactive balloon cellsnd neurofilament-accumulating dysmorphic neu-ons (figures 3, 4). In addition, immunohistochemicalabelling of the phospho-S6 epitope revealed specifictaining in all specimens.
enetic findings
argeted next-generation sequencing (NGS) achievedmean coverage of 245 × (SD = 60) across the tar-
et genes, with 96.3% (SD = 0.95) of bases coveredt 50 × (supplementary table 1). The microdis-ected patient samples harboured a mean fractionf dysplastic cells of 8.6% (SD=1.4). Given our
73
equencing coverage and the assumption that allysplastic cells should carry the variant, we wouldave been able to detect 99% of all brain somaticariants with Platypus in these cells (Richards etl., 2015). We screened for coding variants in 166

7
Z. Ying, et al.
A
*
B
BC
Vimentin DN
SMI32
Figure 4. Surgical histopathology of Patient 9 (table 1). (A) Concentration of dysmorphic neurons (arrow) decorated with anti-non-phosphorylated neurofilament H-specific antibodies (clone SMI32, Alexa555-labeled anti-mouse IgG1 secondary antibody, orangepseudocolour) at the bottom of a sulcus (sulcal surface indicated by small arrowheads), with concomitant accumulation of vimentin-positive balloon cells (clone SP20, Alexa488-labeled anti-rabbit IgG secondary antibody, green pseudocolour; arrowheads) in theunderlying white matter. In addition, vascular myocytes expressing smooth muscle actin (clone 1A4, Alexa647-labelled anti-murineIgG2a secondary antibody, magenta pseudocolour) and nuclei (Hoechst 33342, blue pseudocolour) are visualized (multichannel-i (B) Hs e balS r. Sca
cnfioavacasapiwar2
DOdit––––––c
mmunofluorescence whole slide imaging, 3DHistech MIDI).howing predominant localization of vimentin-immunopositivMI32-immunopositive dysmorphic neurons (DN) in grey matte
andidate genes (supplementary table 1) and didot identify brain somatic variants. DNA obtained
rom blood leucocytes was, however, not availablen this retrospective analysis to confirm germlinerigin. The variants were identified in eight genesnd comprised nine exonic heterozygous missenseariants (mean allele frequency = 47%; SD = 3.6)nd one frameshift insertion introducing a stopodon. All nine missense variants were classifieds ‘variants of uncertain significance’ (VUS) usingtate-of-the-art guidelines in the field (Richards etl., 2015). The frameshift insertion (NM_001136029,.Asp1075Glufs*3) introduces a premature stop codon
4
n DEPDC5, likely leading to haploinsufficiency andas classified as ‘likely pathogenic’ for the epilepsy
ccording to recommended ACMG guidelines and cur-ent epilepsy literature (Ishida et al., 2013; Lal et al.,014; Epi4Kconsortium, 2017).
–cmTt
igh-power magnification of area indicated by asterisk in (A)loon cells (BC) in white matter (as indicated by arrows) andle bar in (A) = 2 mm, in (B) = 50 �m.
iscussionur comprehensive analysis of 10 patients with
ifficult-to-localize BOS-FCD confirms previous stud-es with a “syndromic description” of FCD ILAEype II:
seizure onset at preschool or school age;mostly of frontal localization;stereotyped seizures;distinct MRI features;intrinsic epileptogenicity;favourable postsurgical seizure outcome following
omplete resection of the epileptic region;
Epileptic Disord, Vol. 21, No. 1, February 2019
and exclusivity of FCD type II with immunohisto-hemical or genetic evidence for activation of theTOR pathway.
he fact that FCD-BOS lesions are small and localizedo the bottom of sulcus suggested, however, a later

E
optihaNeeufismo(hqMLDDsemtIDIhpftpoies8bppapstIpcpwvrgspc
btpaDncpicdattprgevsr2
SSw
AT#RN
R
AWd
Btd
Bf2
BA2
Bcs
Bi
ccurrence of a (presumably) genetically acquiredathogen during the estimated 32 mitotic cycles of cor-
ical brain development, compared to FCD II lesionsnvolving a larger cortical area or extending even toemimegalencephaly (D’Gama et al., 2017; Blümckend Sarnat, 2016).GS analysis of a panel of 166 mTor, PIK3/Akt, and otherpilepsy-related genes detected a likely pathogenic,pilepsy-associated variant in DEPDC5 (terminologysed according to ACMG guidelines) in only one out ofve patients studied. This is consistent with previoustudies, which reported ‘likely pathogenic’ variants inTOR pathway-associated genes in only 25% (SD=40)
f patients with histopathologically confirmed FCD IIMarsan and Baulac, 2018). The majority of variantsave been reported as brain somatic with allele fre-uencies of 1-12.6%, and predominantly affecting theTOR gene (Baulac et al., 2015; D’Gama et al., 2015;
im et al., 2015; Mirzaa et al., 2016; Moller et al., 2016;’Gama et al., 2017). One study reported germlineEPDC5 mutations in cases of BOS-FCD, which further
tressed the association with mTORopathies (Scheffert al., 2014). Recently described second-hit mosaicutations may be another etiologic pathomechanism
o be taken into consideration (Ribierre et al., 2018).n our present study, we detected a new pathogenicEPDC5 heterozygous variant in one patient with FCD
Ia. Stop codon-inducing germline variants in DEPDC5ave recently been shown to be present in 3% ofatients (cohort: n=1187) with familial non-acquired
ocal epilepsy without cortical structural abnormali-ies and only in 0.05% of controls (cohort: n=3877;=9.6 × 10−12) (Epi4Kconsortium, 2017). In addition,ne study identified a somatic mutation in DEPDC5
n addition to an existing germline mutation (Baulact al., 2015). The DEPDC5 variant identified in thistudy had an allele frequency of 41%. However, only.6% (SD=1.4) of cells shared a dysplastic phenotypey microscopic review, indicating that the identified.Asp1075Glufs*3 DEPDC5 variant is unlikely to be onlyresent in dysplastic cells. Unfortunately, we were notble to validate this prediction because blood sam-les were not available retrospectively. Further studieshould clarify whether this DEPDC5 variant is causal forhe epilepsy or if the epilepsy is secondary to FCDII.n all other BOS-FCD patients, no likely pathogenic orathogenic variant was identified. However, our NGSoverage was, for the majority of samples, higher com-ared to previous reports (median: 243.72x vs. 180x),hich should enable us to detect 99% of all somatic
ariants with variant allele frequencies of > 8%. Our
pileptic Disord, Vol. 21, No. 1, February 2019
esults call for extended molecular/genetic investi-ations integrating ultra-deep exome-wide DNA andingle-cell RNA sequencing, as well as methylome androteomic analysis to identify a possible pathogenicause(s). Future progress in precision medicine will
t
Bcis
Bottom-of-sulcus FCD
uild on such analysis to develop a targeted drugreatment for specific mTOR signalling molecules, inarticular, when epilepsy surgery is not an option forgiven patient.espite the fact that our study addressed only a smallumber of patients and any conclusion would needonfirmation by larger and prospectively collectedatient series, the comprehensive approach integrat-
ng genotype with phenotype analysis will help toonsolidate the recognition of FCD-BOS in focal andifficult-to-localize epilepsies. Re-review of MRI andpplication of post-processing methodologies led tohe identification of cortical dysplasia at BOS localiza-ion in all our patients, most often in the frontal orarietal lobes. Favourable outcome after neurosurgicalesection, histopathological diagnosis of FCD II, andenetic testing helped to validate the clinical hypoth-sis. No other diagnostic modality added significantalue in clinical management, as seen from a retro-pective angle. In the future, MRI fingerprinting is theesolution for this population of patients (Ma et al.,013). �upplementary data.upplementary table is available on theww.epilepticdisorders.com website.
cknowledgements and disclosures.he work was supported by the European Union (FP7 DESIRE GA602531). We are thankful to Emily Kiefer (Cleveland) and Birteings (Erlangen) for their expert technical assistance.one of the authors have any conflict of interest to declare.
eferences
lbers CA, Lunter G, MacArthur DG, McVean G, OuwehandH, Durbin R. Dindel: accurate indel calls from short-read
ata. Genome Res 2011; 21: 961-73.
arkovich AJ, Kuzniecky RI, Bollen AW, Grant PE. Focalransmantle dysplasia: a specific malformation of corticalevelopment. Neurology 1997; 49: 1148-52.
aulac S, Ishida S, Marsan E, et al. Familial focal epilepsy withocal cortical dysplasia due to DEPDC5 mutations. Ann Neurol015; 77: 675-83.
ernasconi A, Bernasconi N, Bernhardt BC, Schrader D.dvances in MRI for ’cryptogenic’ epilepsies. Nat Rev Neurol011; 7: 99-108.
esson P, Andermann F, Dubeau F, Bernasconi A. Small focalortical dysplasia lesions are located at the bottom of a deepulcus. Brain 2008; 131: 3246-55.
lümcke I, Sarnat HB. Somatic mutations rather than viralnfection classify focal cortical dysplasia type II as mTORopa-
75
hy. Curr Opin Neurol 2016; 29: 388-95.
lümcke I, Pieper T, Pauli E, et al. A distinct variant of focalortical dysplasia type I characterised by magnetic resonancemaging and neuropathological examination in children withevere epilepsies. Epileptic Disord 2010; 12: 172-80.

7
Z
BstM
Bmef2
BfiN
Csd
Ccr
CID
CIo
Dra
Dmcp
Eco
Ee
Ee2
Ged2
Ga2
Hrs
Htd2
Ia5
Jt2
Khs
Lt2
LstE
LB
La2
La2
Lii
LHn8
Mg
M(r
MAi2
Mtp
Moim
M
. Ying, et al.
lümcke I, Thom M, Aronica E, et al. The clinico-pathologicalpectrum of focal cortical dysplasias: a consensus classifica-ion proposed by an ad hoc Task Force of the ILAE Diagnostic
ethods Commission. Epilepsia 2011; 52: 158-74.
lümcke I, Aronica E, Miyata H, et al. International recom-endation for a comprehensive neuropathologic workup of
pilepsy surgery brain tissue: a consensus Task Force reportrom the ILAE Commission on Diagnostic Methods. Epilepsia016; 57: 348-58.
lümcke I, Spreafico R, Haaker G, et al. Histopathologicalndings in brain tissue obtained during epilepsy surgery.ew Engl J Med 2017; 377: 1648-56.
hassoux F, Rodrigo S, Semah F, et al. FDG-PET improvesurgical outcome in negative MRI Taylor-type focal corticalysplasias. Neurology 2010; 75: 2168-75.
hassoux F, Landre E, Mellerio C, et al. Type II focal corti-al dysplasia: electroclinical phenotype and surgical outcomeelated to imaging. Epilepsia 2012; 53: 349-58.
olombo N, Salamon N, Raybaud C, Ozkara C, Barkovich AJ.maging of malformations of cortical development. Epilepticisord 2009; 11: 194-205.
olombo N, Tassi L, Deleo F, et al. Focal cortical dysplasia typeIa and IIb: MRI aspects in 118 cases proven by histopathol-gy. Neuroradiology 2012; 54: 1065-77.
’Gama AM, Geng Y, Couto JA, et al. Mammalian target ofapamycin pathway mutations cause hemimegalencephalynd focal cortical dysplasia. Ann Neurol 2015; 77: 720-5.
’Gama AM, Woodworth MB, Hossain AA, et al. Somaticutations activating the mtor pathway in dorsal telen-
ephalic progenitors cause a continuum of cortical dys-lasias. Cell Rep 2017; 21: 3754-66.
ngel J Jr, Van Ness PC, Rasmussen TB, Ojemann LM. Out-ome with respect to epileptic seizures. In: Surgical treatmentf the epilepsies. Engel J Jr. New York: Raven Press, 1993.
piPMConsortium. A roadmap for precision medicine in thepilepsies. Lancet Neurol 2015; 14: 1219-28.
pi4Kconsortium. Ultra-rare genetic variation in commonpilepsies: a case-control sequencing study. Lancet Neurol017; 16: 135-43.
onzalez-Martinez J, Mullin J, Bulacio J, et al. Stereo-lectroencephalography in children and adolescents withifficult-to-localize refractory focal epilepsy. Neurosurgery014; 75: 258-68, discussion: 67-8.
uerrini R, Duchowny M, Jayakar P, et al. Diagnostic methodsnd treatment options for focal cortical dysplasia. Epilepsia015; 56: 1669-86.
arvey AS, Mandelstam SA, Maixner WJ, et al. The surgicallyemediable syndrome of epilepsy associated with bottom-of-
6
ulcus dysplasia. Neurology 2015; 84: 2021-8.
uppertz HJ, Grimm C, Fauser S, et al. Enhanced visualiza-ion of blurred gray-white matter junctions in focal corticalysplasia by voxel-based 3D MRI analysis. Epilepsy Res005; 67: 35-50.
ss
Ncu
shida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 causeutosomal dominant focal epilepsies. Nat Genet 2013; 45:52-5.
amuar SS, Lam AT, Kircher M, et al. Somatic muta-ions in cerebral cortical malformations. New Engl J Med014; 371: 733-43.
rsek P, Maton B, Korman B, et al. Different features ofistopathological subtypes of pediatric focal cortical dyspla-ia. Ann Neurol 2008; 63: 758-69.
al D, Reinthaler EM, Schubert J, et al. DEPDC5 muta-ions in genetic focal epilepsies of childhood. Ann Neurol014; 75: 788-92.
erner JT, Salamon N, Hauptman JS, et al. Assessment andurgical outcomes for mild type I and severe type II cor-ical dysplasia: a critical review and the UCLA experience.pilepsia 2009; 50: 1310-35.
i H, Durbin R. Fast and accurate short read alignment withurrows-Wheeler transform. Bioinformatics 2009; 25: 1754-60.
i Q, Wang K. InterVar: clinical interpretation of genetic vari-nts by the 2015 ACMG-AMP Guidelines. Am J Hum Genet017; 100: 267-80.
i H, Handsaker B, Wysoker A, et al. The sequencelignment/map format and SAMtools. Bioinformatics009; 25: 2078-9.
im JS, Kim WI, Kang HC, et al. Brain somatic mutationsn MTOR cause focal cortical dysplasia type II leading tontractable epilepsy. Nature Med 2015; 21: 395-400.
ouis DN, Perry A, Reifenberger G, et al. The 2016 Worldealth Organization classification of tumors of the centralervous system: a summary. Acta Neuropathol 2016; 131:03-20.
a D, Gulani V, Seiberlich N, et al. Magnetic resonance fin-erprinting. Nature 2013; 495: 187-92.
arsan E, Baulac S. Review: Mechanistic target of rapamycinmTOR) pathway, focal cortical dysplasia and epilepsy. Neu-opathol Appl Neurobiol 2018; 44: 6-17.
cKenna A, Hanna M, Banks E, et al. The Genomenalysis Toolkit: a MapReduce framework for analyz-
ng next-generation DNA sequencing data. Genome Res010; 20: 1297-303.
ellerio C, Labeyrie MA, Chassoux F, et al. 3T MRI improveshe detection of transmantle sign in type 2 focal cortical dys-lasia. Epilepsia 2014; 55: 117-22.
irzaa GM, Campbell CD, Solovieff N, et al. Associationf MTOR mutations with developmental brain disorders,
ncluding megalencephaly, focal cortical dysplasia, and pig-entary mosaicism. JAMA Neurol 2016; 73: 836-45.
oller RS, Weckhuysen S, Chipaux M, et al. Germline and
Epileptic Disord, Vol. 21, No. 1, February 2019
omatic mutations in the MTOR gene in focal cortical dyspla-ia and epilepsy. Neurol Genet 2016; 2: e118.
ajm IM, Sarnat HB, Blümcke I. Review: the internationalonsensus classification of focal cortical dysplasia - a criticalpdate 2018. Neuropathol Appl Neurobiol 2018; 44: 18-31.

E
Rtd
RfsGP
Rav2
Rg
Sme7
Udei2
VJ2
Wpd
Wp
ibierre T, Deleuze C, Bacq A, et al. Second-hit mosaic muta-ion in mTORC1 repressor DEPDC5 causes focal corticalysplasia-associated epilepsy. J Clin Invest 2018; 128: 2452-8.
ichards S, Aziz N, Bale S, et al. Standards and guidelinesor the interpretation of sequence variants: a joint consen-us recommendation of the American College of Medicalenetics and Genomics and the Association for Molecular
athology. Genet Med 2015; 17: 405-24.
immer A, Phan H, Mathieson I, et al. Integrating mapping-,ssembly- and haplotype-based approaches for callingariants in clinical sequencing applications. Nat Genet014; 46: 912-8.
obinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative
pileptic Disord, Vol. 21, No. 1, February 2019
enomics viewer. Nature Biotech 2011; 29: 24-6.
cheffer IE, Heron SE, Regan BM, et al. Mutations in mam-alian target of rapamycin regulator DEPDC5 cause focal
pilepsy with brain malformations. Ann Neurol 2014; 75:82-7.
e
Wpc1
Bottom-of-sulcus FCD
rbach H, Scheffler B, Heinrichsmeier T, et al. Focal corticalysplasia of Taylor’s balloon cell type: a clinicopathologicalntity with characteristic neuroimaging and histopatholog-
cal features, and favorable postsurgical outcome. Epilepsia002; 43: 33-40.
ogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diazr. LA, Kinzler KW. Cancer genome landscapes. Science013; 339: 1546-58.
agner J, Weber B, Urbach H, Elger CE, Huppertz HJ. Mor-hometric MRI analysis improves detection of focal corticalysplasia type II. Brain 2011; 134: 2844-54.
ang ZI, Alexopoulos AV, Jones SE, et al. Linking MRI post-rocessing with magnetic source imaging in MRI-negative
77
pilepsy. Ann Neurol 2014; 75: 759-70.
ang ZI, Jones SE, Jaisani Z, et al. Voxel-based mor-hometric magnetic resonance imaging (MRI) postpro-essing in MRI-negative epilepsies. Ann Neurol 2015; 77:060-75.

do
i:10.1684/epd
.2019.1029
78 Epileptic Disord, Vol. 21, No. 1, February 2019
Correspondence:Department of Neurology, Brighamand Women’s Hospital,60 Fenwood Road,Boston, MA 02115, USA<[email protected]>
Original articleEpileptic Disord 2019; 21 (1): 78-86
Neuropsychologicalcorrelates of obstructivesleep apnea severityin patients with epilepsy
Véronique Latreille, Kim C. Willment,Rani A. Sarkis, Milena PavlovaDepartment of Neurology, Brigham and Women’s Hospital, Harvard Medical School,Boston, USA
Received July 06, 2018; Accepted December 12, 2018
ABSTRACT – Aims. Obstructive sleep apnea affects up to 30% of patientswith epilepsy. As obstructive sleep apnea represents a clinical risk factorfor cognitive deficits, its occurrence in epilepsy patients may exacerbatecognitive deficits associated with this condition. However, the cognitiveburden of obstructive sleep apnea in epilepsy remains poorly understood.We conducted a retrospective record review of adults with epilepsy whounderwent a polysomnography and a neuropsychological assessment atBrigham and Women’s Hospital.Methods. We examined the relationship between obstructive sleep apneaseverity and cognitive functioning, particularly attention/executive func-tions, memory, and processing speed in untreated obstructive sleep apneapatients with epilepsy. Twenty patients with epilepsy and mild-to-severeobstructive sleep apnea were included in the analyses.Results. We found significant positive correlations between the oxygen sat-uration levels during rapid-eye-movement sleep and attention/executivetests (p<0.05), as well as time spent with saturation levels ≤90% and exec-utive functioning (p=0.008). Similarly, worse verbal memory performanceswere associated with lower oxygen levels (p=0.003). In addition, more severerespiratory events during rapid-eye-movement sleep were associated withworse performances on attention tests (p=0.03).Conclusions. Our findings indicate that more severe obstructive sleepapnea-related hypoxemia during sleep is associated with poorer cognitiveperformances on tests that assess attention/executive functions and ver-bal memory in patients with epilepsy. Overall, these results are consistentwith the sleep apnea literature, and suggest that patients with epilepsy arealso vulnerable to the effects of obstructive sleep apnea. Future prospec-tive studies will help in determining whether treatment of obstructive sleepapnea may help improve cognitive functioning in patients with epilepsy.
Key words: epilepsy, polysomnography, obstructive sleep apnea, neuropsy-chology, cognition

E
Ste2awa(m2otaWht((n2tcmpattmBbfoeceCwlstpaicmtusaWethaop
M
P
WiapcobrDeasamaFi((t(qdAsS
P
Artdcsbgrisua31daosBaseline PSG variables included total sleep time,
leep-related breathing disorders, such as obstruc-ive sleep apnea (OSA), are common in adults withpilepsy, affecting up to one third of patients (Lin et al.,017). OSA is one of the most common reported factorsssociated with reduced quality of life in individualsith epilepsy (Piperidou et al., 2008). These patients
lso frequently report excessive daytime sleepinessManni et al., 2003; Gammino et al., 2016) and are
ore likely to have seizures during sleep (Malow et al.,000; Manni et al., 2003). OSA may exacerbate seizureccurrence by causing nocturnal episodes of intermit-
ent hypoxemia and electroencephalographic (EEG)rousals (Devinsky et al., 1994).
hen left untreated, OSA can have major negativeealth consequences; it increases the risk of hyper-
ension, type 2 diabetes, and cardiovascular diseasesShahar et al., 2001); for a review see Maeder et al.2016). OSA is also a well-known risk factor for cog-itive deficits (Yaffe et al., 2011; Rosenzweig et al.,015). Indeed, accumulating evidence demonstrateshe negative impacts of OSA on nearly all domains ofognition, though larger effect sizes are more com-only found for attention, executive functions, and
sychomotor speed (Rosenzweig et al., 2015; Stranksnd Crowe, 2016). Several facets of executive func-ioning are impaired in adults with OSA as comparedo healthy controls, including set-shifting, working
emory, inhibition, and problem-solving (Olaithe anducks, 2013; Bucks et al., 2017). The mechanismsy which OSA may impair cognition are not yet
ully clear, but it is postulated that the combinationf sleep fragmentation, cyclical intermittent hypox-mia, and hypercapnia, as well as ensuing metaboliconsequences, may all play a role (Rosenzweigt al., 2015).ognitive problems are also often reported in patientsith various epilepsy syndromes. On neuropsycho-
ogical testing, many studies have demonstratedignificant impairments in cognitive flexibility, atten-ion, psychomotor speed, and memory functions inatients with epilepsy (Elger et al., 2004; Hermann etl., 2007; Loughman et al., 2014). As OSA is frequentn epilepsy and represents a clinical risk factor forognitive deficits, its occurrence in epilepsy patientsight worsen initial cognitive impairments. However,
he cognitive burden of OSA in epilepsy is poorlynderstood, and only one study to date investigatedubjective cognitive functioning in epilepsy patientst risk of OSA (Piperidou et al., 2008).e conducted a retrospective study to examine the
ffects of sleep apnea severity on cognitive func-ioning in untreated OSA patients with epilepsy. Weypothesized that more severe OSA would be associ-
pileptic Disord, Vol. 21, No. 1, February 2019
ted with poorer cognitive performances, particularlyn tests assessing attention/executive functions androcessing speed.
s(a
Sleep apnea, cognition, and epilepsy
aterial and methods
articipants
e retrospectively reviewed clinical data of all adultndividuals with epilepsy seen in the neurology clinict Brigham and Women’s Hospital who underwent aolysomnography (PSG) for evaluation of OSA andomplete neuropsychological testing (with an intervalf less than 18 months), from May 2012 to Novem-er 2017. The study was approved by the institutionaleview board.iagnosis of epilepsy was confirmed by expertpileptologists using history, seizure semiology, EEG,nd neuroimaging data. Patients with non-epilepticeizures were not included in the study. Subjects werelso excluded from the analysis if they received treat-ent for OSA at the time of the neuropsychological
ssessment, and if they were diagnosed with dementia.or each subject, we examined demographic and clin-cal data, including education level, body mass indexBMI), neck circumference, cardiovascular risk factorssuch as hypertension, diabetes, and hypercholes-erolemia), smoking status, epilepsy refractorinessdefined as the persistence of seizures despite ade-uate trials of two antiepileptic drugs [AEDs]), epilepsyuration, seizure characteristics, and the number ofEDs. We also collected data on subjective daytimeleepiness, as measured by the Epworth Sleepinesscale (ESS), when available.
olysomnographic recordings
ll subjects underwent in-laboratory overnight sleepecordings. The PSG montage included EEG, elec-rooculography, electromyography, and electrocar-iography recordings. Respiration was monitoredontinuously using nasal thermistor flow, nasal pres-ure, pharyngeal snoring, and thoracic and abdominalelts. Oxygen saturation was monitored using fin-er pulse oximetry sensors. Leg movements wereecorded using surface electrodes on anterior tib-alis muscles. Sleep stages were visually scored pertandard criteria (Iber et al., 2007). Apneas were doc-mented if they occurred for 10 seconds or longernd hypopneas were scored when there was at least0% decrement in nasal pressure signal for at least0 seconds in combination with either a 3% oxygenesaturation or EEG arousal (Berry et al., 2012). Thepnea-hypopnea index (AHI) was defined as the sumf all apnea and hypopnea events divided by totalleep time.
79
leep latency and efficiency, rapid-eye-movementREM) sleep latency, wakefulness after sleep onset,pnea-hypopnea arousal index (number of apnea and

8
V
hhasaauSbentiis
N
Catttcfscsi(wtMoitwofiMfoFtmtsuweTLta(
(trrradAtdee
S
AM(OtvFa(a(isicnswuvan
R
AwwHphrmp
. Latreille, et al.
ypopnea events associated with an EEG arousal perour), periodic limb movement during sleep index,nd duration of sleep stages. In addition to the well-tudied AHI in OSA research, we also included fornalysis variables that reflected the severity of OSAssociated hypoxemia, such as the nadir oxygen sat-ration (SaO2) levels and percent of time spent withaO2 levels lower or equal to 90%. Such variables haveeen suggested to be more sensitive measures of theffects of OSA on cognition, as compared to solely theumber of apnea events (Quan et al., 2011). Thus, in
he present analysis, OSA-related variables of interestncluded total and REM AHI, apnea-hypopnea arousalndex, total and REM nadir SaO2, and percent of totalleep time with SaO2 ≤90%.
europsychological assessment
omplete neuropsychological testing was performedt Brigham and Women’s Hospital either as part ofhe pre-surgical assessment or upon referral fromhe treating physician (as part of the epilepsy evalua-ion). The neuropsychological battery assessed severalognitive domains, including attention, executiveunctions, episodic memory, language, visuospatialkills, and speed processing. To focus the number ofomparisons, and given that previous studies havehown more consistently that OSA has a negativempact on attention, executive, and speed functionsRosenzweig et al., 2015; Stranks and Crowe, 2016),e included tests that assess attention/executive func-
ions, episodic memory, and processing speed.oreover, because the testing was performed as part
f a clinical investigation, and therefore individual-zed for each patient, we selected for analysis onlyhe tests for which sufficient data (>50% of patients)ere available (with the exception of episodic mem-ry tests; see details below). Converted z-scores of the
ollowing neuropsychological variables were includedn the analyses: (1) Attention/Executive functions: Trail
aking Test Part A and B (time), Digit Span subtestrom the Wechsler Adult Intelligence Scale (WAIS-IIIr IV editions), and Phonemic (F, A, and S) Verballuency; and (2) Speed processing: the Coding sub-est from the WAIS-III or IV editions. For episodic
emory tests, composite scores were computed forhe verbal and non-verbal domains using averaged z-cores to account for the large heterogeneity of testssed to assess memory processes. The following testsere included in the composite scores for (3) Verbalpisodic memory: the Rey Auditory Verbal Learning
0
est (learning trials, immediate and delayed recalls),ogical Memory (immediate and delayed recalls) sub-est from the Wechsler Memory Scale-Third Edition,nd California Verbal Learning Test-Second editionlearning trials, short and long delay free recalls); and
iaeys
4) Non-verbal episodic memory: the Brief Visuospa-ial Memory Test-Revised (learning trials and delayedecall), 7/24 Spatial Recall Test (immediate and delayedecalls), Visual Reproduction (immediate and delayedecalls) from the Wechsler Memory Scale-IV Edition,nd Rey-Osterrieth Complex Figure (immediate andelayed recalls).ll the neuropsychological test scores were converted
o age-corrected z-scores using standard normativeata. The Trail Making Test, Verbal Fluency, andpisodic memory scores were also corrected forducation.
tatistical analyses
ll neuropsychological z-scores, except the Trailaking Test Part B, followed a normal distribution
Shapiro-Wilk test; p>0.05). However, none of theSA-related variables were normally distributed, and
herefore non-parametric tests were used for theseariables.irst, to identify potential clinical confounding vari-bles, we performed correlations between clinical dataage, BMI, number of AEDs, and duration of epilepsy)nd our variables of interest, including both OSAtotal AHI, REM sleep AHI, apnea-hypopnea arousalndex, nadir SaO2, REM sleep nadir SaO2, and timepent in SaO2 ≤90%) and neuropsychological variablesn all patients. Partial correlation was then used toontrol for potential confounding factors when a sig-ificant relationship was found, and this was doneeparately for each analysis. If no confounding factorsere identified, Pearson or Spearman correlation wassed to assess the relationship between OSA-relatedariables and neuropsychological scores. Statisticalnalyses were performed using SPSS, version 24. Sig-ificance was set at p<0.05.
esults
total of 34 adults diagnosed with epilepsy under-ent a PSG and a neuropsychological assessmentithin an 18-month interval at Brigham and Women’sospital between May 2012 and November 2017. Allatients reported complaints of sleep apnea, includingypersomnolence and snoring, and one patient alsoeported insomnia symptoms. Twenty-eight patients
et OSA criteria (AHI ≥5). Of that sample, eightatients were excluded because of dementia (n=1),
Epileptic Disord, Vol. 21, No. 1, February 2019
nitiation of OSA therapy at the time of testing (n=6),nd invalid test results (n=1). Thus, 20 patients withpilepsy and comorbid OSA were included in the anal-sis. Demographic, clinical, and sleep data of our studyample of epilepsy patients are presented in table 1.

E
Sleep apnea, cognition, and epilepsy
Table 1. Demographic, clinical, and sleep data of all epilepsy patients.
Clinical characteristics Patients,n = 20
Polysomnographic data Patients,n = 20
Age (years) 50.3 ± 15.1 Total AHI 22.4 ± 20.3
Gender (M/F) 13/7 REM sleep AHI 26.5 ± 28.5
Education (years) 15.1 ± 2.9 Total nadir SaO2 85.8 ± 5.4
Body mass index 30.8 ± 8.9 REM sleep nadir SaO2 88.6 ± 5.4
Neck circumference (inches) 15.4 ± 1.7 % total sleep time with SaO2 ≤90% 4.1 ± 5.2
ESS score 10.4 ± 6.9 PLMS index 8.7 ± 23.3
Cardiovascular risk factors (n [%]) Total sleep time (minutes) 293.5 ± 95.401-2>3
13 (65%)6 (30%)1 (5%)
Sleep latency (minutes)
Sleep efficiency (%)
41.9 ± 69.4
73.6 ± 19.2
Active smoking status (n [%]) 2 (10%) Wakefulness after sleep onset (minutes) 76.0 ± 55.5
Duration of epilepsy (years) 22.9 ± 20.0 Apnea-hypopnea arousal index 14.6 ± 14.7
Drug-resistant epilepsy (n [%]) 6 (30%) Stage N1 (%) 15.6 ± 12.0
Seizure frequency per month (range) 3.8 ± 9.1 (0-30) Stage N2 (%) 54.9 ± 13.9
Nocturnal seizures (n [%]) 7 (35%) Stage N3 (%) 9.0 ± 8.8
Number of antiepileptic drugs 1.7 ± 0.8 Stage REM (%) 18.6 ± 8.5
Epilepsy type (n [%])Focal 16 (80%)Generalized 4 (20%)
E and iR perio
Mapa≥(dTspP(
An
Stw
Table 2. Neuropsychological z-scores of all epilepsypatients.
Neuropsychological variables Z-score
Trail Making Test Part A -0.86 ± 1.66
Trail Making Test Part B -3.81 ± 4.76
Digit Span -0.32 ± 1.11
Phonemic Verbal Fluency -0.72 ± 1.51
Coding -0.60 ± 1.03
SS: Epworth Sleepiness Score; ED: epileptiform discharges (ictalEM: rapid eye movement; OSA: obstructive sleep apnea; PLMS:
ost patients had left temporal lobe epilepsy (60%)nd seizures were medically well-controlled in 70% ofatients. Nine (45%) patients had mild OSA (AHI ≥5nd <15), five (25%) patients had moderate OSA (AHI15 and <30), and six (30%) patients had severe OSA
AHI ≥30). Of note, none of the patients had a seizureuring the PSG recordings.able 2 shows the averaged neuropsychological z-cores of all epilepsy patients. Overall, worse grouperformances were observed for the Trail Making Testart B and the non-verbal memory composite scoremean <1 standard deviation).
ssociation of OSA severity with
pileptic Disord, Vol. 21, No. 1, February 2019
europsychological variables
ignificant negative correlations were found betweenhe BMI and REM sleep nadir SaO2 (r=-0.52, p=0.02), asell as between the number of AEDs and scores on
nterictal); AHI: apnea/hypopnea index; SaO2: oxygen saturation;dic limb movement during sleep.
81
Verbal memory composite score -0.48 ± 1.16
Non-verbal memory composite score -1.22 ± 1.37
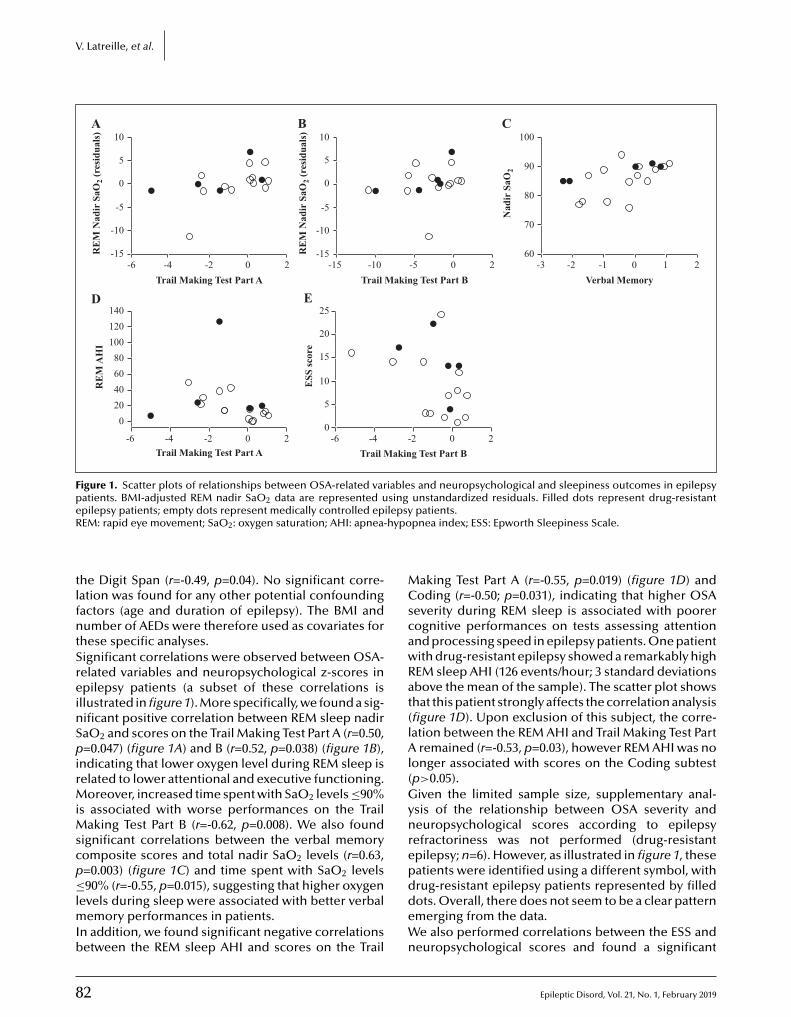
8
V. Latreille, et al.
100C
90
Nad
ir S
aO2
80
70
60-3 -2 -1
Verbal Memory10 2
10B
5
0
RE
M N
adir
SaO
2 (re
sidu
als)
-5
-10
-15-15 -10 -5
Trail Making Test Part B0 2
25E
20
15
ESS
scor
e
10
5
0-6 -4 -2
Trail Making Test Part B0 2
10A
5
0
RE
M N
adir
SaO
2 (re
sidu
als)
-5
-10
-15-6 -4 -2
Trail Making Test Part A0 2
D
RE
M A
HI
020406080
100120140
-6 -4 -2Trail Making Test Part A
0 2
Figure 1. Scatter plots of relationships between OSA-related variables and neuropsychological and sleepiness outcomes in epilepsypatients. BMI-adjusted REM nadir SaO2 data are represented using unstandardized residuals. Filled dots represent drug-resistante ilepsyR hypo
tlfntSreinSpirMiMscp≤lmIb
MCscawRat(lAl(Gynrep
pilepsy patients; empty dots represent medically controlled epEM: rapid eye movement; SaO2: oxygen saturation; AHI: apnea-
he Digit Span (r=-0.49, p=0.04). No significant corre-ation was found for any other potential confoundingactors (age and duration of epilepsy). The BMI andumber of AEDs were therefore used as covariates for
hese specific analyses.ignificant correlations were observed between OSA-elated variables and neuropsychological z-scores inpilepsy patients (a subset of these correlations is
llustrated in figure 1). More specifically, we found a sig-ificant positive correlation between REM sleep nadiraO2 and scores on the Trail Making Test Part A (r=0.50,=0.047) (figure 1A) and B (r=0.52, p=0.038) (figure 1B),
ndicating that lower oxygen level during REM sleep iselated to lower attentional and executive functioning.
oreover, increased time spent with SaO2 levels ≤90%s associated with worse performances on the Trail
aking Test Part B (r=-0.62, p=0.008). We also foundignificant correlations between the verbal memoryomposite scores and total nadir SaO2 levels (r=0.63,=0.003) (figure 1C) and time spent with SaO2 levels
2
90% (r=-0.55, p=0.015), suggesting that higher oxygenevels during sleep were associated with better verbal
emory performances in patients.n addition, we found significant negative correlationsetween the REM sleep AHI and scores on the Trail
ddeWn
patients.pnea index; ESS: Epworth Sleepiness Scale.
aking Test Part A (r=-0.55, p=0.019) (figure 1D) andoding (r=-0.50; p=0.031), indicating that higher OSA
everity during REM sleep is associated with poorerognitive performances on tests assessing attentionnd processing speed in epilepsy patients. One patientith drug-resistant epilepsy showed a remarkably highEM sleep AHI (126 events/hour; 3 standard deviationsbove the mean of the sample). The scatter plot showshat this patient strongly affects the correlation analysisfigure 1D). Upon exclusion of this subject, the corre-ation between the REM AHI and Trail Making Test Part
remained (r=-0.53, p=0.03), however REM AHI was noonger associated with scores on the Coding subtestp>0.05).iven the limited sample size, supplementary anal-
sis of the relationship between OSA severity andeuropsychological scores according to epilepsyefractoriness was not performed (drug-resistantpilepsy; n=6). However, as illustrated in figure 1, theseatients were identified using a different symbol, with
Epileptic Disord, Vol. 21, No. 1, February 2019
rug-resistant epilepsy patients represented by filledots. Overall, there does not seem to be a clear patternmerging from the data.e also performed correlations between the ESS and
europsychological scores and found a significant

E
nBsiFAoantelAvnpan2twirsb
D
OateglastfteTefcapcasCfewae
OrmtetfrwaawsdwrdeeTateswwohwaetitiitlstltvactIcpmO
egative correlation with the Trail Making Test Part(r=-0.49, p=0.041), indicating that higher daytime
leepiness is associated with worse executive function-ng in epilepsy patients (figure 1E).inally, although we initially included the number ofEDs as a potential confounding factor in our analyses,ur sample size limited specific analysis of AED typesnd dosages, particularly the AEDs with known cog-itive side effects (such as phenobarbital, phenytoin,
opiramate, valproic acid, and benzodiazepines) (Eddyt al., 2011; Witt and Helmstaedter, 2017). Neverthe-
ess, we examined the number of patients taking theseEDs, and found that only a small number were takingalproic acid (n=6) and benzodiazepines (for anxiety,=2). None of the subjects were taking phenobarbital,henytoin, or topiramate at the time of testing, whichre the ones associated with the most negative cog-itive profile (Eddy et al., 2011; Witt and Helmstaedter,017). Again, given the limited sample size, supplemen-ary analysis according to polytherapy (n=13) statusas not performed, but data are presented visually
n supplementary figure 1. Patients on polytherapy areepresented by the filled stars. Overall, there does noteem to be a clear pattern, with patients on polytherapyeing spread at both ends of the data spectrum.
iscussion
ur findings indicate that higher OSA severity andssociated intermittent nocturnal hypoxemia is linkedo worse cognitive performances in adults withpilepsy. More specifically, we found that lower oxy-en levels across all sleep stages are associated with
ower scores on tests assessing executive functioningnd verbal memory. Impaired breathing during REMleep appears to have a strong relationship with cogni-ion, affecting predominantly attention and executiveunctions. Epilepsy patients who reported more day-ime sleepiness were also more likely to have lowerxecutive functioning.o our knowledge, this is the first study that hasxamined the effects of OSA severity on cognitiveunctioning as assessed by comprehensive neuropsy-hological testing in adults with epilepsy. Our resultsre consistent with the literature findings in the OSAopulation, showing that OSA is linked with poorerognitive performances, especially based on testsssessing attention, and executive and psychomotorpeed functions (Rosenzweig et al., 2015; Stranks androwe, 2016). Although larger effect sizes are usually
pileptic Disord, Vol. 21, No. 1, February 2019
ound for the above-mentioned cognitive domains,pisodic verbal memory (mainly retrieval processes,hich are closely related to executive capacity) has
lso been reported to be impacted by OSA (Buckst al., 2017).
ivaia
Sleep apnea, cognition, and epilepsy
ur results also extend previous reports that OSA-elated hypoxemia variables may be more sensitiveeasures of the effects of OSA on cognition, by con-
rast to the frequency of apnea events per hour (Quant al., 2011). Similarly, in older adults with OSA, noc-urnal hypoxemia was found to be a significant riskactor of future cognitive decline, while the number ofespiratory events was not (Yaffe et al., 2011). Besides,e found no significant relationship between OSA-
ssociated arousals (apnea-hypopnea arousal index)nd any cognitive measure in our patients. Althoughe did not have a group of healthy controls as compari-
on, sleep architecture variables such as sleep latency,uration of sleep stages, and number of awakeningsere overall within the normal range. Therefore, our
esults suggest that nocturnal hypoxemia may be moreebilitating for cognition than the number of apneavents and global sleep architecture in adults withpilepsy.he mechanisms by which OSA may impair cognitionre not fully understood. Yet, it has been proposedhat both sleep fragmentation and intermittent hypox-mia may play a role (Rosenzweig et al., 2015). Severaltudies have demonstrated that OSA is associatedith structural and functional cerebral abnormalities,hich are thought to underlie the cognitive deficitsbserved in these patients. Indeed, adults with OSAave reduced grey matter volume (Shi et al., 2017),hite matter fiber integrity (Castronovo et al., 2014),
nd cerebral glucose metabolism (Yaouhi et al., 2009; Jut al., 2012) in multiple areas, including the frontal andemporal lobes. These areas are known to be involvedn executive functions and memory processes, andhus may explain why these are particularly impairedn OSA individuals. Although no study to date hasnvestigated the effects of OSA on specific areas ofhe brain in patients with epilepsy, it may be postu-ated that these patients may be more vulnerable andhow more pronounced brain abnormalities relativeo individuals with OSA but without epilepsy. Futurearge case-control prospective studies will be neededo examine whether epilepsy patients are indeed moreulnerable to the effects of OSA from a cognitivend neuronal standpoint. Moreover, whether a spe-ific seizure onset zone has a differential vulnerabilityo the effects of OSA will require further investigation.mportantly, these brain abnormalities and asso-iated cognitive consequences can be, at leastartially, reversed by consistent and accurate treat-ent. Indeed, meta-analytic studies in patients withSA have shown that treatment with continuous pos-
83
tive airway pressure (CPAP) therapy may improveigilance, attention, and executive functions (Olaithend Bucks, 2013; Pan et al., 2015). These improvementsn cognitive functioning in OSA patients, compli-ntly treated with CPAP, were paralleled by positive

8
V
cbastd(amrnTsisctm
L
SrtswioowtcvwWbmiOdtetrctOicotphmt
esrfcfpO(tafnisvtiptltForipipcfs(aAecofAfitqsrsaTrt
. Latreille, et al.
hanges in grey and white matter integrity and cere-ral glucose metabolism (Canessa et al., 2011; Ju etl., 2012; Castronovo et al., 2014). Other treatmentsuch as mandibular advancement has also been foundo improve executive functioning, psychomotor skills,aytime sleepiness, and quality of life in OSA patients
Galic et al., 2016). In epilepsy patients, studies havelso shown that patients treated with CPAP wereore than five times more likely to have a significant
eduction in seizure frequency and daytime sleepi-ess compared to untreated patients (Lin et al., 2017).hese results suggest that CPAP may help reduceleep apnea-related hypoxemia and arousals, furthermproving sleep stability, and thereby reducing seizureusceptibility. CPAP might also help in reducing OSAonsequences in epilepsy patients such as cogni-ive impairment, but also apnea-related cardiovascular,
etabolic, and neuronal dysfunctions.
imitations
ome limitations of our study should be noted. It is aetrospective chart review and uses a patient popula-ion that is seen in the regular epilepsy clinic, and asuch, our inclusion criteria were more limited. Thus,e cannot exclude that some potential confound-
ng factors such as other OSA-related comorbiditiesr the effects of AEDs could have had an impactn our sleep and cognitive measurements. However,e have included the number of AEDs as a poten-
ial confounding factor in our analysis so that it wasontrolled for when significantly associated with ourariables of interest. Moreover, a minority of subjectsere taking AEDs with known cognitive side effects.hile this does not preclude any potential contri-
ution of drug-related cognitive side effects on ourain results, it is unlikely to explain all of our find-
ngs. Visual analysis of the relationships between ourSA and neuropsychological variables according to
rug polytherapy status also revealed no clear pat-ern, with patients on polytherapy being spread at bothnds of the OSA or cognitive spectrum. Additionally,he retrospective nature of the study (medical charteview) limited extensive evaluation of OSA-relatedlinical outcomes, such as duration of OSA symp-oms. One could hypothesize that longer duration of
SA symptoms would lead to more severe cognitivempairment in the long-term. However, based on thelinical notes, patients usually reported unclear onsetf symptoms (‘for several years’), long-standing day-
4
ime sleepiness (‘always been sleepy’), and/or no bedartner to confirm snoring or witness apneas. We alsoad a small number of patients, which precluded aore detailed analysis of the effects of OSA on cogni-
ion in relation to epilepsy types (e.g. temporal versus
C
Ot
xtra-temporal) or seizure characteristics, in particular,eizure frequency. Since only six patients had drug-esistant epilepsy (with large heterogeneity in seizurerequency), we lacked statistical power to performorrelations between seizure frequency and cognitiveunctioning/OSA-related variables. Yet, it was shownreviously in a cohort of older adults with epilepsy thatSA was associated with a higher seizure frequency
Chihorek et al., 2007). In this study, it was not justhe hypoxemia, but also probably the arousals frompnea/hypopnea that lead to the worsening of seizurerequency (Chihorek et al., 2007). Moreover, noctur-al seizures were reported in some patients (7/20). It
s likely that nocturnal seizures contribute to worsenleep quality (sleep fragmentation, lighter sleep), andice-versa. Yet it is still undetermined whether cogni-ive dysfunction in epilepsy patients is driven by thempact of nocturnal seizures on sleep quality. It is notossible with our current sample to examine this ques-
ion, but future work using a mediation model with aarge sample of patients could help better understandhese mechanisms.inally, we acknowledge that the use of a delay intervalf up to 18 months between the PSG and neu-opsychological testing constitutes a limitation. Ideally,n a prospective study, the PSG would have beenerformed at the same time as the neuropsycholog-
cal testing (and review of epilepsy-related data). Foratients with the longest intervals, it is possible that thelinical profile (OSA severity, cognition, and seizurerequency) changed, thereby modifying the relation-hips we observed. Yet, only a minority of patientsn=3) had more than 12 months delay between the PSGnd neuropsychological examination.s far as we know, this is the first study that hasxamined the relationship between OSA severity andognitive dysfunctions in patients with epilepsy usingbjective measures. These results remain to be tested
or replication in larger cohorts of epilepsy patients.lthough no comparison group was included, this is arst step towards a better understanding of the poten-
ial consequences of OSA in epilepsy. In fact, we wereuite surprised that from our chart review, only amall number of patients who were referred for a neu-opsychological assessment also underwent PSG. Yetleep disorders are very common in epilepsy, with OSAffecting up to one third of patients (Lin et al., 2017).his highlights the need for clinicians to screen, on aegular basis, their patients at high risk of OSA so thathey can be referred and treated accordingly.
Epileptic Disord, Vol. 21, No. 1, February 2019
onclusions
SA is frequent in adults with epilepsy and is one ofhe most common reported factors associated with

E
raetcpomc
SSw
DDIsrbh
R
BrfM
Btg
Cat2
Cim
Caw
DL1
Eo3
Ec
GErts
Gsw7
HCr
IuT2
Jcss
Lw
Lim
Mod
MaN
MTeE
Ob1
Ppam2
Pee
QtpS
RSt
Sb
educed quality of life. Our study shows that OSA islso associated with worse cognitive functioning inpilepsy, affecting primarily attention, executive func-ions, and verbal memory processes. These results areonsistent with the OSA literature and suggest thatatients with epilepsy are also vulnerable to the effectsf OSA. Future prospective studies will help in deter-ining whether treatment of OSA may help improve
ognitive functioning in patients with epilepsy. �
upplementary data.upplementary figure is available on theww.epilepticdisorders.com website.
isclosures.r. Latreille is supported by a scholarship from the Canadian
nstitutes of Health Research. Dr. Sarkis has received compen-ation for activities with DigiTrace/SleepMed. Dr. Pavlova haseceived research funding from Lundbeck Inc. and from Biomo-ie Inc., neither of which is relevant to this study. Dr. Willmentas no conflict of interest to declare.
eferences
erry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoringespiratory events in sleep: update of the 2007 AASM manualor the scoring of sleep and associated events. J Clin Sleep
ed 2012; 8: 597-619.
ucks RS, Olaithe M, Rosenzweig I, Morrell MJ. Reviewinghe relationship between OSA and cognition: where do weo from here? Respirology 2017; 22: 1253-61.
anessa N, Castronovo V, Cappa SF, et al. Obstructive sleeppnea: brain structural changes and neurocognitive func-ion before and after treatment. Am J Respir Crit Care Med011; 183: 1419-26.
astronovo V, Scifo P, Castellano A, et al. White matterntegrity in obstructive sleep apnea before and after treat-
ent. Sleep 2014; 37: 1465-75.
hihorek AM, Abou-Khalil B, Malow BA. Obstructive sleeppnea is associated with seizure occurrence in older adultsith epilepsy. Neurology 2007; 69: 1823-7.
evinsky O, Ehrenberg B, Barthlen GM, Abramson HS,uciano D. Epilepsy and sleep apnea syndrome. Neurology994; 44: 2060-4.
ddy CM, Rickards HE, Cavanna AE. The cognitive impactf antiepileptic drugs. Ther Adv Neurol Disord 2011; 4:85-407.
lger CE, Helmstaedter C, Kurthen M. Chronic epilepsy andognition. Lancet Neurol 2004; 3: 663-72.
pileptic Disord, Vol. 21, No. 1, February 2019
alic T, Bozic J, Ivkovic N, Gunjaca G, Ticinovic TK, Dogas Z.ffects of mandibular advancement device treatment on arte-ial stiffness and glucose metabolism in patients with mildo moderate obstructive sleep apnea: a prospective 1-yeartudy. Sleep Breath 2016; 20: 69-77.
o2
SbR
Sleep apnea, cognition, and epilepsy
ammino M, Zummo L, Bue AL, et al. Excessive daytimeleepiness and sleep disorders in a population of patientsith epilepsy: a case-control study. J Epilepsy Res 2016; 6:
9-86.
ermann B, Seidenberg M, Lee EJ, Chan F, Rutecki P.ognitive phenotypes in temporal lobe epilepsy. J Int Neu-
opsychol Soc 2007; 13: 12-20.
ber C, Ancoli-Israel S, Chesson A, Quan S. The AASM Man-al for the Scoring of Sleep and Associates Events: Rules.erminology and technical specifications. Sleep (Rochester)007: 59.
u G, Yoon IY, Lee SD, Kim YK, Yoon E, Kim JW. Modesthanges in cerebral glucose metabolism in patients withleep apnea syndrome after continuous positive airway pres-ure treatment. Respiration 2012; 84: 212-8.
in Z, Si Q, Xiaoyi Z. Obstructive sleep apnoea in patientsith epilepsy: a meta-analysis. Sleep Breath 2017; 21: 263-70.
oughman A, Bowden SC, D’Souza W. Cognitive functioningn idiopathic generalised epilepsies: a systematic review and
eta-analysis. Neurosci Biobehav Rev 2014; 43: 20-34.
aeder MT, Schoch OD, Rickli H. A clinical approach tobstructive sleep apnea as a risk factor for cardiovascularisease. Vasc Health Risk Manag 2016; 12: 85-103.
alow BA, Levy K, Maturen K, Bowes R. Obstructive sleeppnea is common in medically refractory epilepsy patients.eurology 2000; 55: 1002-7.
anni R, Terzaghi M, Arbasino C, Sartori I, Galimberti CA,artara A. Obstructive sleep apnea in a clinical series of adultpilepsy patients: frequency and features of the comorbidity.pilepsia 2003; 44: 836-40.
laithe M, Bucks RS. Executive dysfunction in OSAefore and after treatment: a meta-analysis. Sleep 2013; 36:297-305.
an YY, Deng Y, Xu X, Liu YP, Liu HG. Effects of continuousositive airway pressure on cognitive deficits in middle-ged patients with obstructive sleep apnea syndrome: aeta-analysis of randomized controlled trials. Chin Med J
015; 128: 2365-73.
iperidou C, Karlovasitou A, Triantafyllou N, et al. Influ-nce of sleep disturbance on quality of life of patients withpilepsy. Seizure 2008; 17: 588-94.
uan SF, Chan CS, Dement WC, et al. The associa-ion between obstructive sleep apnea and neurocognitiveerformance-the Apnea Positive Pressure Long-term Efficacytudy (APPLES). Sleep 2011; 34: 303-14.
osenzweig I, Glasser M, Polsek D, Leschziner GD, WilliamsC, Morrell MJ. Sleep apnoea and the brain: a complex rela-ionship. Lancet Respir Med 2015; 3: 404-14.
hahar E, Whitney CW, Redline S, et al. Sleep-disorderedreathing and cardiovascular disease: cross-sectional results
85
f the Sleep Heart Health Study. Am J Respir Crit Care Med001; 163: 19-25.
hi Y, Chen L, Chen T, et al. A meta-analysis of voxel-basedrain morphometry studies in obstructive sleep apnea. Sciep 2017; 7: 10095.

8
V
Ssc
WcP
. Latreille, et al.
tranks EK, Crowe SF. The cognitive effects of obstructiveleep apnea: an updated meta-analysis. Arch Clin Neuropsy-hol 2016; 31: 186-93.
itt JA, Helmstaedter C. How can we overcome neuropsy-hological adverse effects of antiepileptic drugs? Expert Opinharmacother 2017; 18: 551-4.
TEST YOURSELFEDUCATION
(1) When left untreated, obstructive sleep apnea can have major impact on cognition. Which cognitive domainsare particularly affected by obstructive sleep apnea in the general population?
(2) According to this study, which cognitive domains seem particularly affected by more severe obstructivesleep apnea in epilepsy?
(3) When designing a prospective study to examine the effects of obstructive sleep apnea on cognitive func-tioning in patients with epilepsy and whether treatment of obstructive sleep apnea may help improve cognition,what would be the most sensitive OSA-related measure?
Yaffe K, Laffan AM, Harrison SL, et al. Sleep-disorderedbreathing, hypoxia, and risk of mild cognitive impairment anddementia in older women. JAMA 2011; 306: 613-9.
Yaouhi K, Bertran F, Clochon P, et al. A combined neu-ropsychological and brain imaging study of obstructive sleepapnea. J Sleep Res 2009; 18: 36-48.
6
Note: Reading the manuscript provides an answer to all qwebsite, www.epilepticdisorders.com, under the section
Epileptic Disord, Vol. 21, No. 1, February 2019
uestions. Correct answers may be accessed on the“The EpiCentre”.

do
i:10.
1684
/ep
d.2
019.
1030
Epileptic Disord, Vol. 21, No. 1, February 2019 87
VIDEO ONLINE
Correspondence:Salvatore ManganoDepartment of Sciences for HealthPromotion and Mother and Child Care“G.D’Alessandro”,University of Palermo,Palermo, Italy<[email protected]>
Clinical commentaryEpileptic Disord 2019; 21 (1): 87-91
A novel mutation inKCNQ3-related benign familialneonatal epilepsy:electroclinical features andneurodevelopmental outcomeEttore Piro 1, Rosaria Nardello 1, Elena Gennaro 2, AntoninaFontana 1, Maurizio Taglialatela 3, Giuseppe DonatoMangano 1, Giovanni Corsello 1, Salvatore Mangano 1
1 Department of Sciences for Health Promotion and Mother andChild Care “G. D’Alessandro,” University of Palermo, Palermo2 Laboratory of Human Genetics, Galliera Hospital, Genoa3 Unit of Pharmacology, Department of Neuroscience, Reproductive andOdontostomatological Sciences, University of Naples Federico II, Naples, Italy
Received July 11, 2018; Accepted November 06, 2018
ABSTRACT – Benign familial neonatal epilepsy (BFNE) is caused, in about5% of families, by mutations in the KCNQ3 gene encoding voltage-gatedpotassium channel subunits. Usually, newborns with BFNE show a normalneurological outcome, but recently, refractory seizures and/or develop-mental disability have been reported suggesting phenotype variabilityassociated with KCNQ3-related BFNE. Here, we describe a proband from aBFNE family carrying a novel variant in the KCNQ3 gene. Regarding thepaucity of data in the literature, we describe the presented case witha view to further establishing: (1) a genotype/phenotype correlation inorder to define a BFNE phenotype associated with favourable outcome;(2) an electroclinical pattern associated with BFNE based on video-EEGrecording; (3) appropriate first-line AEDs; and (4) the duration of AEDtreatment. The presented case from Day 3 exhibited a cluster of ictalevents, identified as epileptic seizures on Day 10 based on continuousvideo-EEG polygraphy. The seizures were characterized by asymmetrictonic posturing, associated with a generalized decrease in EEG amplitude,and followed by bilateral asynchronous clonic movements associated withbicentral sharp-wave discharges. The seizures were refractory to intra-venous pyridoxine, whereas levetiracetam resulted in rapid total seizurecontrol which has remained to date. This study demonstrates that the novelheterozygous KCNQ3 (c. 914A>T; p.Asp305Val) variant, affecting residuesin the pore region, is associated with a specific electroclinical pattern andfavourable neurodevelopmental outcome. [Published with video sequenceon www.epilepticdisorders.com]
Key words: benign familial neonatal epilepsy, KCNQ, voltage-gated potas-sium channels, genotype-phenotype correlations, electroclinical features

8
E
Bsbswnl3wstBtgsc1Affifwertftagctcmfc
C
TohtpmitAa(cFerapr
snCnwOvoesbdtuccstcAtisathlLrBnOwMbc2AsstaHgsttIs1tm
. Piro, et al.
enign familial neonatal epilepsy (BFNE) is an auto-omal dominant epilepsy syndrome characterizedy frequent unprovoked focal or generalized toniceizures, followed by apnoea and clonic movementsith oculofacial features often associated with auto-omic signs, starting around the second/third day of
ife and occurring during wakefulness and sleep, up to0 times a day. The seizures often remit spontaneouslyithin weeks or months, but some patients become
eizure-free only after trials with different antiepilep-ic drugs (AEDs) (Ryan et al., 1991; Ronen et al., 1993).FNE is caused in >80% of families by mutations in
he KCNQ2 and KCNQ3 genes encoding for voltage-ated potassium channel subunits, which underlie alowly activating, non-inactivating potassium currentalled M-current (Biervert et al., 1998; Charlier et al.,998; Singh et al., 1998; Wang et al., 1998).
reduction in M-current emerged as the commonactor underlying neonatal seizures and haploinsuf-ciency, as the primary pathogenetic mechanism
or BFNE (Soldovieri et al., 2007). Usually, newbornsith BFNE show normal neurological and physicalxamination and unremarkable laboratory and neu-oradiological investigations. Follow-up studies revealhat about 10-15% of patients develop a form of benignocal or generalized epilepsy later in life within a con-ext of normal neurocognitive development (Ronen etl., 1993; Singh et al., 2003). Given the recent emer-ence of phenotype variability concerning the clinicalourse, the sensitivity to AEDs, and the treatment dura-ion, in the present study, we describe the clinicalourse, electroclinical pattern revealed by video-EEGonitoring, response to AEDs, and the apparently
avourable outcome of a proband from a BFNE familyarrying a novel variant in the KCNQ3 gene.
ase study
he proband was a 13-month-old male; the sec-nd born to non-consanguineous and apparentlyealthy parents at 38 + 6 weeks of gestation by elec-
ive Caesarean section following an uncomplicatedregnancy. The study was approved by the Ethics Com-ittee “Palermo 1” of the University Hospital. Written
nformed consent for publication was obtained fromhe parents.pgar score at birth was 9 and 10 at one minutend five minutes, respectively. His weight was 3,240 g36th centile), length 50 cm (44th centile), and head cir-umference 35 cm (63th centile).rom Day 3, he exhibited recurrent postprandialpisodes of jerking involving the upper limbs, perio-
8
al cyanosis, and crying; symptoms considered to bessociated with gastroesophageal reflux disease. Asaroxysmal events recurred, on Day 9, the patient waseferred to our NICU. On admission, the new-born
Tnmn
howed mild axial hypotonia and reduced sponta-eous motility. Complete blood count, procalcitonin,RP, blood cultures, glucose, serum electrolytes,eonatal metabolic screening, and a head ultrasoundere normal.n Day 10, during a conventional EEG polygraphy (see
ideo), the new-born showed, in active sleep, a clusterf three similar ictal events characterized by gen-ralized or focal tonic (asymmetric tonic posturing)eizures with shallow breathing, mild desaturation, eyelinking, and tachycardia associated with a generalizedecrease in EEG amplitude. The tonic component of
he seizure progressed during a vibratory phase, grad-ally evolving into unilateral or bilateral asynchronouslonic movements, lasting for about a minute and asso-iated with bicentral sharp-wave discharges. The focaleizures involved both sides, varying from one seizureo another one. The seizures ended without focal clini-al and EEG signs or postictal EEG depression (figure 1).fter careful clinical and EEG evaluation suggesting
he epileptic nature of paroxysmal events, we admin-stered pyridoxine (100 mg; intravenous). Anotherimilar seizure occurred three hours later. Therefore,fter parental informed consent, we shifted to leve-iracetam (LEV) (20 mg/kg; intravenous), repeated 12ours later. A third brief seizure occurred 13 hours
ater; treatment with pyridoxine was interrupted andEV was increased to 60 mg/Kg/day in two doses,esulting, after the first dose, in full seizure control.y Day 14, axial tone and spontaneous motility wereormalised.n Day 18, the infant was discharged, seizure-free,ith oral LEV (200 mg/day). At one month of age, brainRI and EEG were normal. At eight months of age, the
aby showed normal global development and socialontact. Oral LEV dosage (22 mg/Kg/day, equivalent to00 mg/day) was then gradually withdrawn.t clinical evaluation performed at 13 months andeven days of age, the child was alert, perceptive, andensitive. He demonstrated a healthy interest in theesting materials with an appropriate level of activity,ttention, adaptation to changes, and task persistence.e was socially engaged with the examiner, showingood communicative intent and reciprocity. He couldtand without support. He vocalized in response tohe examiner and to express attitude. His developmen-al functioning, assessed using the Bayley Scales ofnfant and Toddler Development (Bayley III), showedcores in the average/upper average range (cognitive:25; language: 94; motor: 91; social-emotional: 95; adap-ive behaviour: 90). He walked without support at 13
onths and 15 days of age.
Epileptic Disord, Vol. 21, No. 1, February 2019
he neurological signs of the proband observed in theeonatal period, the family history with three previousiscarriages, and the occurrence of uncertain paternal
eonatal clinical events suggested that genetic testing

E
A novel mutation in KCNQ3-related BFNE
Fp2-C4
A B
C D
Fp2-T4
Fp1-C3
Fp1-T3
C4-O2
C3-O1
T4-O2
T3-O1
FNG1-FNG1
ENG1-ENG1
MOR-MOR
Fp2-C4
Fp2-T4
Fp1-C3
Fp1-T3
C4-O2
C3-O1
T4-O2
T3-O1
FNG1-FNG1
ENG1-ENG1
MOR-MOR
Fp2-C4
Fp2-T4
Fp1-C3
Fp1-T3
C4-O2
C3-O1
T4-O2
T3-O1
FNG1-FNG1
ENG1-ENG1
MOR-MOR
Fp2-C4
Fp2-T4
Fp1-C3
Fp1-T3
C4-O2
C3-O1
T4-O2
T3-O1
FNG1-FNG1
ENG1-ENG1
MOR-MOR
-C4-C4-C4C4-C4C4-C4-C4C4-C4C4C4C4
--T-T-T-TTT44TT4
-C-C-C-CC-CC3C33C3C3C3
-T3-T3-T-T3-T3T3T3T3T3TTTTT3
OOO2OO222O
OOOOO111O
OO2OOO222O22
OO1O1O1O1O111
Figure 1. Electroclinical phenotype of the presented patient with BFNE. The seizure, following active sleep and arousal(A), is characterized by tonic extension and adduction of the upper limbs, flexion of the lower limbs, left trunk and head rotationa decreT radua scha
ssndtb(trvsmctPdpdtwauno
D
Ag
(atsrensaiceBisvgtaeamtT
ssociated with shallow breathing, tachycardia, and generalizedhe tonic seizure evolves into a vibratory phase (C), and then gnd ocular-facial regions associated with bicentral sharp-wave di
hould be performed. Based on a next-generationequencing panel, not including the KCNQ3 gene,o pathogenetically-relevant gene variants wereetected. In close analogy to KCNQ2 variants, pheno-
ypic heterogeneity during the clinical course has alsoeen reported in families carrying variants in KCNQ3
Miceli et al., 2017), therefore direct sequencing ofhe KCNQ3 gene (NM_004519.3) was also performed,evealing the occurrence of a c.914A>T heterozygousariant. This nucleotide substitution, inherited by theymptomatic father, is responsible for the missenseutation, p.Asp305Val. The variant, affecting a highly
onserved residue located in the S5-S6 pore region ofhe protein, is predicted to be “pathogenic” based onolyPhen2 and Mutation Taster with a very high confi-ence score (>0.999). Multiplex ligation-dependentrobe amplification of KCNQ2 and KCNQ3 did notisclose indels (for more details, see the supplemen-
ary material). The same variant was found in the fatherho likely suffered from neonatal epileptic seizures
nd in an asymptomatic sister. In addition, his paternalncle, whose genetic data were not available, hadeonatal seizures and an isolated seizure at 13 yearsld (figure 2).
pileptic Disord, Vol. 21, No. 1, February 2019
iscussion
bout 5% of families with BFNE carry KCNQ3 patho-enetic variants with incomplete (0.8-0.85) penetrance
vtrdl
ase in EEG amplitude (B).ally into bilateral asynchronous clonic movements of the limbsrges (D).
Miceli et al., 2017). KCNQ3 mutations have been forlong time considered to cause a typical pheno-
ype characterized by neonatal seizures that remitpontaneously after a few months with normal neu-ocognitive development (Charlier et al., 1998; Singht al., 2003). Instead, in close analogy to the wide phe-otypic spectrum associated with KCNQ2 mutations,ome KCNQ3 mutations were recently found to bessociated with more severe phenotypes character-zed by refractory seizures and variable motor andognitive impairment (Soldovieri et al., 2014; Micelit al., 2015). The rare occurrence of KCNQ3-relatedFNE and the uncertainty about the electroclin-
cal phenotype, outlined mainly by retrospectivetudies or, sometimes, by incomplete clinical obser-ations, has hampered the identification of accurateenotype-phenotype correlations. Thus, we believehat video-EEG monitoring is the best method to definespecific BFNE electroclinical pattern. We present thelectroclinical, genetic, and developmental data fromfamily with neonatal seizures and a novel KCNQ3utation identified in three affected individuals over
wo generations (figure 2).he ictal electroclinical features, documented by
89
ideo-EEG monitoring, showed initial asymmetriconic posturing, shallow breathing, inconstant desatu-ation, and tachycardia, associated with a generalizedecrease in EEG amplitude. The tonic phase was fol-
owed by unilateral or asynchronous bilateral clonic

9
E. Piro, et al.
I
II? +/-
“-”
= BFNE Phenotype;= wild-type KCNQ3 allele;= mutant KCNQ3 allele;
“+”
+/- +/-III
F
mdaw(2otaptteiiobaopcNRpcacrnUwapoasp
OLsduceamopowpSdtodcaIKedgte
SSa
igure 2. Pedigree of the family with BFNE.
ovements associated with generalized sharp-waveischarges without postictal depression. Both clinicalnd EEG data of our patient seem to be consistentith the electroclinical pattern previously reported
Hirsch et al., 1993; Ronen et al., 1993; Maljevic et al.,016; Sands et al., 2016). The KCNQ3 variant found inur family is the substitution c.914A>T, which leads to
he p.Asp305Val missense mutation. This substitutionffects a residue located in the S5-S6 pore region of therotein, likely altering the potassium channel struc-
ure at this functionally-critical region. A mutation athe same codon (c.914A>G), but resulting in a differ-nt amino acid substitution (p.Asp305Gly), was found
n a patient described by Ryan et al. (1991) exhibit-ng a typical BFNE phenotype, characterized by onsetf seizures at two days, positive response to pheno-arbital, seizure freedom from the third week of life,nd normal psychomotor development at 10 monthsf age. When expressed with KCNQ2 subunits, incor-oration of KCNQ3 mutant subunits into heteromerichannels decreased the maximal M-current by ∼40%.otably, both Asp305Gly found in the family studied byyan et al. and the Asp305Val variant described in theresent study cause the replacement of a negativelyharged amino acid with a smaller, non-polar aminocid. Such structural similarities likely translate into aomparable degree of channel dysfunction and cur-ent decrease, although functional studies would beeeded to confirm such a hypothesis.ntil now, seizures in BFNE patients have been treatedith various conventional AEDs (Miceli et al., 2015),
nd only recently have first-line drugs emerged. In
0
articular, evidence indicates that carbamazepine orxcarbazepine are safe and more effective, providingrapid response, seizure control, shorter hospitali-
ation, and favourable long-term outcome for BFNEatients (Sands et al., 2016).
AWN
“?” indicates DNA not available.
ur patient showed a rapid and effective response toEV and remained seizure-free from the third dose,imilar to the patient of Maljevic et al. (2016). Theuration of treatment reported in previous studies isnclear, ranging from a few weeks to 18 months. In ourase, we are unable to state whether the child recov-red from active epilepsy before eight months of age,s a gradual reduction in LEV was initiated with ter-ination of treatment at 10 months following a lack
f seizure relapse. The electroclinical outcome of ouratient seems to be consistent with the typical coursef the disorder. The composite scores of Bayley III wereithin normal range with cognitive performance beingarticularly strong.imilar to a previous report (Ryan et al., 1991), theevelopmental follow-up of our patient was limited
o the first 13 months of age, however, the lackf seizure relapse without evidence of significantevelopmental delay suggests that the novel variant,.914A>T (p.Asp305Val), is likely to be associated withfavourable outcome.
n conclusion, we demonstrate a novel variant in theCNQ3 gene within a family with rather typical BFNElectroclinical features, consistent with the previouslyescribed benign outcome associated with heterozy-ous variants affecting residues in the pore region ofhis voltage-gated potassium channel subunit (Micelit al., 2017). �
upplementary data.ummary didactic slides and supplementary materials are avail-ble on the www.epilepticdisorders.com website.
Epileptic Disord, Vol. 21, No. 1, February 2019
cknowledgements and disclosures.e thank the children and parents who participated in this study.one of the authors have any conflict of interest to declare.

E
Legend for video sequenceOn Day 10, the newborn showed, in active sleep,a cluster of three similar ictal events character-ized by generalized or focal tonic (asymmetric tonicposturing) seizures with shallow breathing, milddesaturation, eye blinking, and tachycardia associ-ated with a generalized decrease in EEG amplitude.The tonic component of the seizure progressed intoa vibratory phase, gradually evolving into unilateralor bilateral asynchronous clonic movements, last-ing for about a minute, and associated with bicentralsharp-wave discharges. The focal seizures involvedboth sides, varying from one seizure to another one.
Key words for video research onwww.epilepticdisorders.com
Phenomenology: neonatal seizureLocalisation: focal seizure not otherwise specified
RBc1
Cae
Hn
Mme2
Mtd
MTAs
RMn
RMd1
St2
S
Syndrome: benign familial neonatal epilepsy (bfne)Aetiology: KCNQ3 mutationeferencesiervert C, Schroeder BC, Kubisch C, et al. A potassiumhannel mutation in neonatal human epilepsy. Science998; 279: 403-6.
harlier C, Singh NA, Ryan SG, et al. A pore mutation innovel KQT-like potassium channel gene in an idiopathic
pilepsy family. Nat Genet 1998; 18: 53-5.
irsch E, Velez A, Sellal F, et al. Electroclinical signs of benigneonatal familial convulsions. Ann Neurol 1993; 34: 835-41.
aljevic S, Vejzovic S, Bernhard MK, et al. Novel KCNQ3utation in a large family with benign familial neonatal
pilepsy: a rare cause of neonatal seizures. Mol Syndromol016; 7: 189-96.
TEST YOURSELFEDUCATION
(1) Which of the following statements about families with BFNE carrying KCNQ3 pathogenic variants is correct?A. About 15% show complete penetranceB. About 10% show incomplete penetranceC. About 5% show incomplete penetrance
(2) Which of the following statements about BFNE associated with KCNQ3 mutations is not correct?
channel gene, KCNQ2, is mutated in an inherited epilepsy ofnewborns. Nat Genet 1998; 18: 25-9.
Singh NA, Westenskow P, Charlier C, et al. KCNQ2 andKCNQ3 potassium channel genes in benign familial neona-tal convulsions: expansion of the functional and mutationspectrum. Brain 2003; 126: 2726-37.
Soldovieri MV, Miceli F, Bellini G, Coppola G, Pascotto A,Taglialatela M. Correlating the clinical and genetic features ofbenign familial neonatal seizures (BFNS) with the functionalconsequences of underlying mutations. Channels (Austin)2007; 1: 228-33.
Soldovieri MV, Boutry-Kryza N, Milh M, et al. Novel KCNQ2and KCNQ3 mutations in a large cohort of families withbenign neonatal epilepsy: first evidence for an altered chan-nel regulation by Syntaxin-1A. Human Mutat 2014; 35: 356-67.
Wang HS, Pan Z, Shi W, et al. KCNQ2 and KCNQ3 potassiumchannel subunits: molecular correlates of the M-channel. Sci-ence 1998; 282: 1890-3.
pileptic Disord, Vol. 21, No. 1, February 2019
A. BFNE associated with KCNQ3 mutation is an autosomaB. KCNQ3 mutations are always associated with severe pC. The phenotypic spectrum associated with KCNQ3 and
Note: Reading the manuscript provides an answer to all qwebsite, www.epilepticdisorders.com, under the section
A novel mutation in KCNQ3-related BFNE
iceli F, Striano P, Soldovieri MV, et al. A novel KCNQ3 muta-ion in familial epilepsy with focal seizures and intellectualisability. Epilepsia 2015; 56: e15-20.
iceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper EC,aglialatela M. KCNQ3-related disorders. In: GeneReviews®.dam MP, Ardinger HH, Pagon RA, et al. Seattle (WA): Univer-ity of Washington, 2017.
onen GM, Rosales TO, Connolly M, Anderson VE, Leppert. Seizure characteristics in chromosome 20 benign familial
eonatal convulsions. Neurology 1993; 43: 1355-60.
yan SG, Wiznitzer M, Hollman C, Torres MC, Szekeresova, Schneider S. Benign familial neonatal convulsions: evi-
ence for clinical and genetic heterogeneity. Ann Neurol991; 29: 469-73.
ands TT, Balestri M, Bellini G, et al. Rapid and safe responseo low-dose carbamazepine in neonatal epilepsy. Epilepsia016; 57: 2019-30.
ingh NA, Charlier C, Stauffer D, et al. A novel potassium
91
l dominant epilepsy syndromehenotypesKCNQ2 mutations is similar.
uestions. Correct answers may be accessed on the“The EpiCentre”.

do
i:10.
9
V
CJDMAC<<
Clinical commentaryEpileptic Disord 2019; 21 (1): 92-6
Tonic status epilepticusin a centenarian womanJosé L. Fernández-Torre 1,2,3, Javier Riancho 2,3,María Martín-García 1, Gonzalo Martínez-de las Cuevas 4,Pilar Bosque-Varela 5
1 Department of Clinical Neurophysiology, Marqués de Valdecilla University Hospital,Santander2 Department of Physiology and Pharmacology, University of Cantabria (UNICAN),Santander3 Biomedical Research Institute (IDIVAL)4 Department of Internal Medicine, Marqués de Valdecilla University Hospital, Santander5 Department of Neurology, Marqués de Valdecilla University Hospital, Santander, Spain
Received September 09, 2018; Accepted November 18, 2018
ABSTRACT – Generalized tonic status epilepticus (TSE) is a rare epilep-tic condition. It occurs usually in the context of symptomatic generalizedepilepsy, in particular, in subjects with a diagnosis of Lennox-Gastaut syn-drome, atypical forms of idiopathic (genetic) generalized epilepsy, or asa paradoxical effect during treatment with diverse antiepileptic drugs.Herein, we describe the case of an elderly woman on chronic treatmentwith psychotropic drugs who developed an episode of generalized TSE.Motor manifestations were subtle and difficult to recognize as seizures,and a detailed video-EEG importantly contributed to accurate and promptdiagnosis. TSE was initially refractory to conventional anti-seizure drug ther-apy including levetiracetam and valproate but was finally controlled withlacosamide. Our case indicates a potential therapeutic effect of lacosamideon TSE in the elderly after treatment failure with first-line anti-seizure drugs.
ww.epilepticdisorders.com]
ideo-EEG, elderly patient, lacosamide
generalized TSE, possibly secondaryto chronic use of psychotropicdrugs.
Case study
A 102-year-old woman, partiallydependent in daily-life activities,with antecedents of hyperten-sion and atrial fibrillation, wasadmitted to our hospital because
IDEO ONLINE
orrespondence:osé L. Fernández-Torre
[Published with video sequence on w
Key words: tonic status epilepticus, v
Tonic status epilepticus (TSE) is arare epileptic condition. It occursusually in the context of symp-tomatic generalized epilepsy, inparticular, in subjects with a diagno-sis of Lennox-Gastaut syndrome oratypical forms of idiopathic general-ized epilepsy (IGE) (Kobayashi et al.,2005). Moreover, TSE has been alsodescribed as a paradoxical effectduring treatment with diverse
1684/epd
.2019.1031
2 Epileptic Disord, Vol. 21, No. 1, February 2019
epartment of Clinical Neurophysiology,arqués de Valdecilla University Hospital,
vda. Valdecilla, 25, 39008 Santander,antabria, [email protected]>[email protected]>
antiepileptic drugs (Prior et al., 1972;Capocchi et al., 1998; Grande-Martínet al., 2016). Herein, we describethe case of a centenarian womanwho developed an episode of
of general deterioration anddyspnoea. She was on chronic treat-ment with trazodone, amiloride,hydrochlorothiazide, pantropa-zole, and bromazepam. On general

E
pbtesosfiolareeitsmscstweOma(sastnbintacslifvttaOawtcnc(f
D
IoseeepsTaneaemntiMai(cipgactsTSsSmnwemcecWcTtmiwthe authors did not find reports or studies confirminga seizure threshold-lowering effect of amoxicillin-
hysical examination, she was haemodynamically sta-le with a temperature of 36.0◦ C. Routine laboratory
ests were within normal limits except for the pres-nce of severe bacteriuria. With these results in mind,he was diagnosed with urinary tract infection, andral treatment with amoxicillin-clavulanic acid wastarted for five days. On Day 5 of admission, afternishing the antibiotic treatment, she complainedf “funny” movements as a “tremor” of both lower
imbs. These episodes were interpreted as anxietynd nervousness, and treatment with clonazepam andisperidone was initiated. The next day, these “funny”pisodes in both legs persisted and the patient wasvaluated by our neurologists. On neurological exam-
nation, she was awake and alert, but disorientedo time. No focal motor or sensory deficits wereeen. She reported self-limited involuntary move-ents in the lower limbs. These movements were
ynchronous, fast, and were not accompanied byognitive disconnection, automatisms or loss of con-ciousness. On some occasions, she acquired a certainonic aspect in both feet, but “funny” movementsere not clearly tremulous. At that moment, a video-lectroencephalogram (v-EEG) study was requested.n v-EEG, recurrent generalized bursts of rhyth-ic, high-voltage, sharp, well-defined, sinusoidal, beta
ctivity, lasting from 2 to 6 seconds, were observedfigure 1). Simultaneously, the patient experienced aubtle increase in tone and stiffness involving thexial musculature and both lower limbs (see videoequence). Occasionally, she also experienced sub-le jerks in both feet. Autonomic manifestations wereot evident. Irregular theta and delta waves were seenetween bursts. The episodes recurred continuously
n clusters along the recording and, therefore, a diag-osis of generalized TSE was suggested. A computed
omography (CT) scan of the brain disclosed diffusend symmetric cortico-subcortical atrophy and sub-ortical hypodense lesions compatible with chronicmall vessel ischaemia. Treatment with intravenousevetiracetam (LEV) (1,000 mg/day) was initiated. Dur-ng the following four days, tonic seizures were lessrequent but were not completely controlled and intra-enous valproate (VPA) (800 mg/24 hours) was addedo her anti-seizure drug (ASD) therapy. On Day 11,onic seizures worsened involving also upper limbsnd the dose of VPA was increased (1,000 mg/24 hours).ne day later, the episodes of stiffness persisted
nd intravenous lacosamide (LCM) (100 mg/24 hours)as included in the ASD therapy. After the onset of
reatment with LCM, tonic seizures were completelyontrolled, and clinical improvement persisted for theext days. On Day 14, the patient was discharged under
pileptic Disord, Vol. 21, No. 1, February 2019
hronic ASD therapy with LEV (750 mg/24 hours), VPA1,200 mg/24 hours), and LCM (100 mg/24 hours) withollow-up via neurology consultation.
cwc
Tonic status epilepticus in an elderly woman
iscussion
t is well known that tonic seizures and TSE typicallyccur in patients with intellectual disability and severeymptomatic epilepsy. However, the occurrence ofpisodes of generalized TSE is fairly rare in adults andlderly subjects without antecedents of epilepsy. Nev-rtheless, Kobayashi et al. (2005) drew attention to theossibility that status epilepticus (SE) with minor toniceizures may occur in IGE.he case of our centenarian patient withoutntecedents of epilepsy shows a different sce-ario. Previously, Garmel et al. (1992) described anpisode of unresponsiveness in a 73-year-old womans secondary to generalized TSE. These authorsmphasized the complexity of diagnosis since motoranifestations may be subtle and difficult to recog-
ize. Our findings support this conclusion because inhe case report described here, tonic seizures werenitially interpreted to result from the patient’s anxiety.
ore recently, Ostrow and Kaplan (2011) reportedyoung woman with prolonged TSE which evolved
nto a stimulus-induced diffuse voltage attenuationSIDVA) pattern in the setting of aseptic meningoen-ephalitis. This was the first report of a SIDVA patternn an adult without a history of epilepsy. The ictal EEGattern observed in our patient consisted of recurrenteneralized bursts of rhythmic, sinusoidal, beta-likectivity, lasting several seconds without spike-waveomplexes. This ictal EEG pattern resembled thatypically observed in tonic seizures of Lennox-Gastautyndrome (Prior et al., 1972).SE is more difficult to recognize than other forms ofE. This is because, firstly, TSE is rare in adults or elderlyubjects, in comparison with generalized tonic-clonicE or partial motor SE, and secondly, motor symptomsay be subtle and the fact that increased tone or stiff-
ess in isolation may have an epileptic origin is notidely known. Thus, fine tonic seizures may only bevident with careful video analysis or with the use ofuscle recording electrodes. The description of our
ase may help to emphasize that TSE may occur in thelderly population and, in particular, in patients withhronic psychotropic drug therapy.e cannot rule out that the treatment with amoxicillin-
lavulanic acid could have precipitated the episode ofSE. However, we believe that this was unlikely becausehe clinical manifestations occurred once the treat-
ent was completed. Moreover, in a recent articlen which the current evidence for seizures associatedith the use of antibiotics was systematically reviewed,
93
lavulanic, and an association with seizures or SEas not demonstrated (Sutter et al., 2015). The
onfluence of multiple factors seems more plausible.
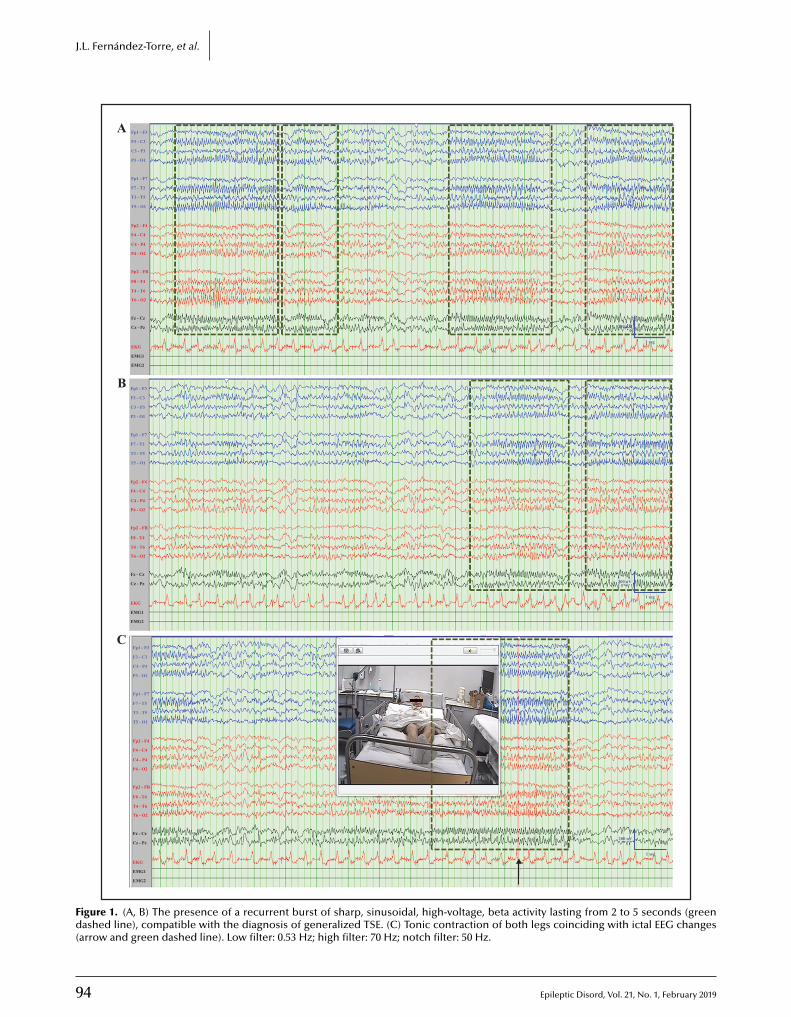
94 Epileptic Disord, Vol. 21, No. 1, February 2019
J.L. Fernández-Torre, et al.
Fp1 - F3
F3 - C3
C3 - P3
P3 - O1
Fp1 - F7
F7 - T3
T3 - T5
T5 - O1
Fp2 - F4
F4 - C4
C4 - P4
200 uV
1 seg
P4 - O2
Fp2 - FB
F8 - T4
T4 - T6
T6 - O2
EKG
Fz - Cz
A
B
C
Cz - Pz
EMG1
EMG2
Fp1 - F3
F3 - C3
C3 - P3
P3 - O1
Fp1 - F7
F7 - T3
T3 - T5
T5 - O1
Fp2 - F4
F4 - C4
C4 - P4
P4 - O2
Fp2 - FB
F8 - T4
T4 - T6
T6 - O2
EKG
Fz - Cz
Cz - Pz
EMG1
EMG2
Fp1 - F3
F3 - C3
C3 - P3
P3 - O1
Fp1 - F7
F7 - T3
T3 - T5
T5 - O1
Fp2 - F4
F4 - C4
C4 - P4
P4 - O2
Fp2 - FB
F8 - T4
T4 - T6
T6 - O2
EKG
Fz - Cz
Cz - Pz
EMG1
EMG2
200 uV
1 seg
200 uV
1 seg
Figure 1. (A, B) The presence of a recurrent burst of sharp, sinusoidal, high-voltage, beta activity lasting from 2 to 5 seconds (greendashed line), compatible with the diagnosis of generalized TSE. (C) Tonic contraction of both legs coinciding with ictal EEG changes(arrow and green dashed line). Low filter: 0.53 Hz; high filter: 70 Hz; notch filter: 50 Hz.

E
OwIocItTduptrtAcsTppwcacoawwGiIctccTA
SSw
DN
R
Ce
Gu1
GMv2
HlA
KcD
Ot2
Pew
RfaN
Sevents of antibiotic drugs. A systematic review. Neurology2015; 85: 1332-41.
Thompson AGB, Cock HR. Successful treatment of super-refractory tonic status epilepticus with rufinamide: firstclinical report. Seizure 2016; 39: 1-4.
ur patient had a chronic vascular disorder and sheas a chronic consumer of psychotropic medication.
t is possible that under these circumstances, a home-static imbalance secondary to urinary tract infectionould account for the occurrence of SE.n this case, TSE was refractory to conventional ASDreatment. It is well known that tonic seizures andSE, particularly associated with Lennox-Gastaut syn-rome, can be aggravated by benzodiazepines. Wesed VPA and LEV as first-line treatment in order torevent this potential complication. Although initially,
onic seizures improved after several days, the seizuresemained uncontrolled and we therefore added LCMo her antiepileptic treatment. LCM is a relatively newSD therapy that has become a well-established anti-onvulsive medication for the treatment of focal-onseteizures, with and without secondary generalization.reatment with LCM has demonstrated efficacy inatients with absence SE in whom VPA and LEV havereviously failed (Reif et al., 2018). Moreover, LCMas noninferior to fosphenytoin as treatment for non-
onvulsive seizures in critically ill patients (Hussain etl., 2018). Conversely, the use of high doses of LCMombined with other antiepileptic drugs in a casef super-refractory TSE was ineffective (Thompsonnd Cock, 2016). The patient was a 24-year-old manith autistic spectrum disorder and learning disabilityith an electroclinical picture reminiscent of Lennox-astaut syndrome, who responded to a rapid increase
n rufinamide.n summary, TSE may occur in elderly patients underhronic psychotropic drug treatment. Motor manifes-ations can be subtle and, therefore, a detailed v-EEGontributes to accurate and prompt diagnosis. Ourase indicates a potential therapeutic effect of LCM onSE in the elderly after treatment failure with first-lineSDs. �
Legend for video sequenceRecurrent and continuous episodes in which thereis flexion of the patient’s entire body along with hip,knee and ankle flexion of both lower limbs. Note theflexion of the left arm during the second sequence.
Key words for video research onwww.epilepticdisorders.com
Phenomenology: status epilepticus(convulsive)/tonic seizureLocalisation: generalized
pileptic Disord, Vol. 21, No. 1, February 2019
Syndrome: not applicableAetiology: toxics abuse
Tonic status epilepticus in an elderly woman
upplementary data.ummary didactic slides are available on theww.epilepticdisorders.com website.
isclosures.one of the authors have any conflict of interest to declare.
eferences
apocchi G, Balducci A, Cecconi M, et al. Valproate-inducedpileptic tonic status. Seizure 1998; 7: 237-41.
armel GM, Jacobs AK, Eilers MA. Tonic status epilepticus: annusual presentation of unresponsiveness. Ann Emerg Med992; 21: 223-7.
rande-Martín A, Pardal-Fernández JM, Carrascosa-RomeroC, De Cabo C. Tonic seizure status epilepticus triggered by
alproate in a child with Doose syndrome. Neuropediatrics016; 47: 187-9.
ussain AM, Lee JW, Kolls BJ, et al. Randomized trial ofacosamide versus fosphenytoin for nonconvulsive seizures.nn Neurol 2018; 83: 1174-85.
obayashi E, Thomas P, Andermann F. Tonic status epilepti-us in patients with idiopathic generalized epilepsy. Epilepticisord 2005; 7: 327-31.
strow LW, Kaplan PW. Tonic status and electrodecremen-al paroxysms in an adult without epilepsy. Epileptic Disord011; 13: 99-101.
rior PF, Maclaine GN, Scott DF, Laurance BM. Tonic statuspilepticus precipitated by intravenous diazepam in a childith petit mal status. Epilepsia 1972; 13: 467-72.
eif PS, Männer A, Willems LM, et al. Intravenous lacosamideor treatment of absence status epilepticus in genetic gener-lized epilepsy: a case report and review of literature. Actaeurol Scand 2018; 138: 259-62.
utter R, Rüegg S, Tschudin-Sutter S. Seizures as adverse
95

9
J
.L. Fernández-Torre, et al.TEST YOURSELFEDUCATION
(1) List three epileptic conditions associated with tonic status epilepticus.
(2) What is the most frequent ictal EEG pattern during tonic status epilepticus in patients with Lennox-Gastautsyndrome?
(3) Which antiepileptic drug can trigger tonic status epilepticus in generalized epilepsy?
6 Epileptic Disord, Vol. 21, No. 1, February 2019
Note: Reading the manuscript provides an answer to all questions. Correct answers may be accessed on thewebsite, www.epilepticdisorders.com, under the section “The EpiCentre”.

do
i:10.
1684
/ep
d.2
019.
1032
E
CCDU7S<
Clinical commentaryEpileptic Disord 2019; 21 (1): 97-101
Absence status induced bylacosamide adjunctive therapyCharles Ákos Szabó, Lola C. Morgan, Suzanne Sonnenberg,Kameel M. KarkarDepartment of Neurology and South Texas Comprehensive Epilepsy Center, UT HealthSan Antonio, San Antonio, Texas
Received August 16, 2018; Accepted November 01, 2018
ABSTRACT – Since lacosamide was approved as an adjuvant agent for thetreatment of medically refractory focal epilepsy over ten years ago, it isbecoming more widely used for the treatment of idiopathic (genetic) gen-eralized epilepsies. Several studies have demonstrated efficacy in reducingprimary generalized tonic-clonic seizures (GTCS), but efficacy is less well-characterized for myoclonic and absence seizures. A 29-year-old man withjuvenile myoclonic epilepsy and medically refractory GTCS on a combina-tion of levetiracetam and topiramate was started on lacosamide adjunctivetherapy with the plan to replace topiramate. While his GTCS became con-trolled, he was witnessed to have confusional episodes, with waxing andwaning responsiveness, lasting a few days, several times a month. After eightmonths of adjunctive lacosamide therapy, he was admitted to the epilepsymonitoring unit, where paroxysms of generalized spike-and-wave comple-xes, lasting for 30-90 minutes, were recorded, interrupted only by sleep.During these periods, he demonstrated psychomotor slowing and disori-entation on examination. The absence status was successfully broken bylorazepam, and lacosamide was discontinued. The patient had no further
ent follow-up visit, four months after
tus, idiopathic generalized epilepsy,ravation
are reports regarding the efficacyof lacosamide for the treatmentof idiopathic generalized epilepsysyndromes, juvenile myoclonicepilepsy in particular (Afra andAdamolekun, 2012). A recent studydemonstrated short-term efficacyfor the treatment of GTCS, whichwas further improved during thecourse of a 59-week open-labelextension of this study (Wechsleret al., 2017). Absence and myoclonic
orrespondence:confusional episodes at the most recdischarge.
Key words: lacosamide, absence staantiepileptic medications, seizure agg
Lacosamide was initially approvedas an adjunctive agent for thetreatment of medically refrac-tory focal-onset seizures in adultsin 2007 (Ben-Menachem et al.,2007; Chung et al., 2010). Sinceit was introduced, its indicationshave increased to include chil-dren and adolescents, and even asmonotherapy in adults (Vimpat USPrescribing Information, 2018). Sim-ilar to carbamazepine, phenytoin
pileptic Disord, Vol. 21, No. 1, February 2019 97
harles Ákos Szabóepartment of Neurology,T Health San Antonio,
703 Floyd Curl Drive,an Antonio, TX, [email protected]>
or lamotrigine, lacosamide targetsvoltage-gated sodium channels, butinstead of blocking rapidly depo-larizing currents, it enhances slowinactivation of the channels. There
seizures were also reduced in theopen-label phase, as well as adecrease in the overall burden ofgeneralized spike-and-wave dis-charges. Nonetheless, five patients

9
C
iososwlhrnhdc
C
TsetmGawHmpnftOwaihHCtadfihogLt5f3tnAll1
twfiAHodaotbfitawbatadcaatdLiHbdtdwh2lrpew
D
Tmoclsr
. Ákos Szabó, et al.
n the pilot study experienced an increase in absencer myoclonic seizures, two of whom exhibited absenceeizures for the first time. In the extended study,nly one patient experienced an increase in absenceeizures at a dose of 800 mg/day, but these resolvedith dose reduction, and another demonstrated a self-
imited period of increased myoclonic seizures. Thereave been no reports of absence status to date. In thiseport, we present a patient with a history of juve-ile myoclonic epilepsy who experienced control ofis GTCS with lacosamide adjunctive therapy, but alsoeveloped frequently recurring confusional episodesonsistent with absence status.
ase study
he patient was a 29-year-old right-handed man pre-enting for video-EEG evaluation, with a history ofpilepsy beginning at age six years old. He ini-ially presented with absence seizures, developing
yoclonic seizures, and GTCS in adolescence. TheTCS occurred only a few times per year, but were
lways followed by a prolonged period of confusion,hich could last from hours to days.e had no family history of epilepsy but was born pre-aturely in the 28th week of gestation, weighing two
ounds and seven ounces. He denied a history of focaleurological abnormalities postnatally but suffered
rom mild developmental delay and was eventuallyreated for attention deficit disorder.
ther co-morbidities included hyperlipidaemia whichas treated with fenofibrate and obstructive sleep
pnoea with CPAP. He had chronic insomnia requir-ng a combination of 100 mg trazodone and 50 mgydroxyzine at night.is most recent brain MRI was normal except for ahiari I malformation. His EEG was normal just prior
o starting lacosamide but generalized 3-5-Hz spike-nd-wave as well as generalized polyspike-and-waveischarges resurfaced once started on lacosamide;ndings that were mirrored by earlier EEG reports. Head failed topiramate, zonisamide, valproic acid, lam-trigine, phenytoin, carbamazepine, clonazepam, andabapentin.acosamide was introduced to replace topiramate ashe adjunctive agent for levetiracetam and titrated to00 mg daily. This combination controlled his GTCSor eight months, but he was witnessed as having 2--day periods of waxing and waning confusion. Ashese were occurring several times a month, he was
8
o longer able to be gainfully employed.t the time of his admission, his random levetiracetam
evel was 17 mcg/ml (normal range: 15-40 mcg/ml) andacosamide level was 12.3 mcg/ml (normal range: 5-0 mcg/ml). He did not complain of any side effects on
seiTm
his regimen, and previous trough lacosamide levelsere within the normal range. His metabolic pro-le demonstrated mild elevation of ALT/SGPT andST/SGOT, but no other abnormalities.is EEG at admission indicated prolonged paroxysmsf 3-5-Hz generalized spike- and polyspike-and-waveischarges, occurring in runs of 20-30 seconds, withbrief 1-2-second interruption with transient return
f his posterior background (figure 1). This EEG pat-ern lasted from 9:30 am into early afternoon, resolvingriefly whenever he fell asleep. The absence status wasnally aborted by two doses of lorazepam at 1 mg, with
he EEG pattern responding within 10 minutes of itsdministration. During his absence status, he under-ent bedside testing and was only oriented to placeut not to person or the current date. He was notble to solve simple single-digit mathematical addi-ions, suffered from short-term memory impairment,nd could not recall what he had eaten for lunch thatay. His partner confirmed that these symptoms wereonsistent with the confusional episodes he witnessedt home. Repeat testing after the absence status wasborted, with subsequent resolution of his disorien-ation to person and place and improvement in hisyscalculia and short-term memory deficits.acosamide was held, while levetiracetam wasncreased to 1,000 mg twice daily to prevent GTCS.
is EEG reverted to his normal awake and sleepackground with brief generalized spike-and-waveischarges, not lasting longer than a second in dura-
ion (figure 2). Ethosuximide was also added prior toischarge to help control the absence seizures, butas poorly tolerated by the patient due to nausea andiccoughs, requiring a dose reduction of 500 mg to50 mg twice daily. His levetiracetam and ethosuximideevels were 11 mcg/ml and 26 mcg/ml (therapeuticange: 40-100 mcg/ml), respectively. According to theatient and his partner, he had no further confusionalpisodes in the four months since lacosamide wasithdrawn.
iscussion
his case report describes a patient with juvenileyoclonic epilepsy presenting with recurrent bouts
f absence status on lacosamide, despite improvedontrol of his GTCS. While there is concern thatacosamide can aggravate absence and myocloniceizures in some patients, absence status has not beeneported to date (Wechsler et al., 2017). The patient
Epileptic Disord, Vol. 21, No. 1, February 2019
tarted experiencing 2-3-day periods of confusion, sev-ral times each month, soon after lacosamide was
ntroduced, despite complete control of his GTCS.he episodes did not recur afterwards, within fouronths following his discharge from hospital. Other
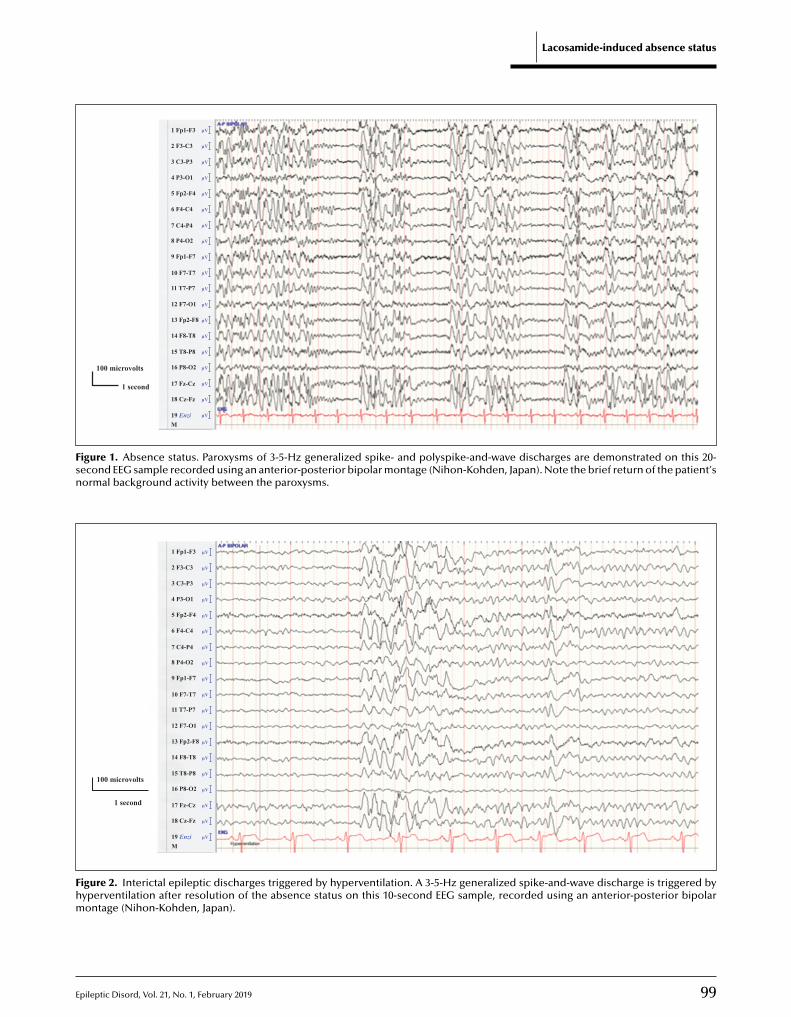
E
Lacosamide-induced absence status
100 microvolts
1 second
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
1 Fp1-F3
2 F3-C3
3 C3-P3
4 P3-O1
5 Fp2-F4
6 F4-C4
7 C4-P4
8 P4-O2
9 Fp1-F7
10 F7-T7
11 T7-P7
12 F7-O1
13 Fp2-F8
14 F8-T8
15 T8-P8
16 P8-O2
17 Fz-Cz
18 Cz-Fz
19 EnziM
Figure 1. Absence status. Paroxysms of 3-5-Hz generalized spike- and polyspike-and-wave discharges are demonstrated on this 20-second EEG sample recorded using an anterior-posterior bipolar montage (Nihon-Kohden, Japan). Note the brief return of the patient’snormal background activity between the paroxysms.
1 Fp1-F3
2 F3-C3
3 C3-P3
4 P3-O1
5 Fp2-F4
6 F4-C4
7 C4-P4
8 P4-O2
9 Fp1-F7
10 F7-T7
11 T7-P7
12 F7-O1
13 Fp2-F8
14 F8-T8
15 T8-P8
16 P8-O2
17 Fz-Cz
18 Cz-Fz
100 microvolts
1 second
19 EnziM
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
µV
Fhm
pileptic Disord, Vol. 21, No. 1, February 2019
igure 2. Interictal epileptic discharges triggered by hyperventilationyperventilation after resolution of the absence status on this 10-secontage (Nihon-Kohden, Japan).
99
. A 3-5-Hz generalized spike-and-wave discharge is triggered byond EEG sample, recorded using an anterior-posterior bipolar

1
C
twsfttbwdtblTmms(at1tdtAilipeif
SSw
ATiaILspsppn
R
Am
BRt2
Ctc
GMi2
Gar
Hasb1
KTe
PPt
V
. Ákos Szabó, et al.
han the discontinuation of lacosamide, levetiracetamas increased by 500 mg a day at discharge, and etho-
uximide was added. However, at the most recentollow-up visit, the levels of both of these medica-ions were subtherapeutic, therefore less likely to behe cause of the prevention of absence status. Hence,ased on the evidence that absence status startedhen lacosamide was introduced and resolved with itsiscontinuation, this is the most likely explanation of
his adverse effect. Future case reports or series maye helpful to better characterize whether or not this
acosamide effect is dose-dependent.he mechanism underlying the paroxysmal enhance-ent of spike-and-wave discharges is unclear. Severaledications have been reported to trigger absence
tatus including carbamazepine and oxcarbazepineGenton et al., 2000; Gelisse et al., 2004), putativelys sodium-channel blockers, as well as vigabatrin andiagabine (Panayiotopoulos et al., 1997; Knake et al.,999) due to potentiation of GABAB-receptor activa-ion. While lacosamide’s action on the sodium channeliffers from that of carbamazepine and oxcarbazepine,
he overall effect may be similar (Hebeisen et al., 2015).s in the case of carbamazepine and oxcarbazepine, it
s still unclear why absence seizures would respond toacosamide adjunctive therapy in most patients withdiopathic generalized epilepsy, yet worsen in a fewatients, and, in this case, even evolve to absencepilepsy (Wechsler et al., 2017). The answers could
nclude clinical, electrophysiological or even geneticactors. �
Key Points
In addition to focal-onset seizures, lacosamide maybe helpful for the treatment of primary GTCS.The efficacy of lacosamide for the treatmentof other generalized seizure types is still beingevaluated.We report exacerbation of absence status epilepti-cus in a person with lacosamide adjunctive therapy.Caution is advised when using lacosamide to treatidiopathic generalized epilepsy.
00
upplementary data.ummary didactic slides are available on theww.epilepticdisorders.com website.
v
Wfa2
cknowledgements and disclosures.he first author (CAS) is currently supported through an
nvestigator-initiated grant funded by LivaNova, Inc. Two co-uthors (LCM and KMK) serve as consultants to Brain Sentinel,nc. The first author (CAS) is a speaker for UCB Pharma.CM (site PI) and CAS (site co-PI) are participating in thetudy entitled “A double-blind, randomized, placebo-controlled,arallel-group, multicenter study to evaluate the efficacy andafety of lacosamide as adjunctive therapy for uncontrolledrimary generalized tonic-clonic seizures in subjects with idio-athic generalized epilepsy (SP0982)”, however, the patient wasot screened for this study.
eferences
fra P, Adamolekun B. Lacosamide treatment of juvenileyoclonic epilepsy. Seizure 2012; 21: 2024.
en-Menachem E, Biton V, Jatuzis D, Abou-Khalil B, Doty P,udd GD. Efficacy and safety of oral lacosamide as adjunc-
ive therapy in adults with partial onset seizures. Epilepsia007; 48: 1308-17.
hung S, Sperling MR, Biton V, et al. Lacosamide as adjunc-ive therapy for partial-onset seizures: a randomized andontrolled trial. Epilepsia 2010; 51: 958-67.
elisse P, Genton P, Kuate C, Pesenti A, Baldy-Moulinier, Crespel A. Worsening of seizures by oxcarbazepine
n juvenile idiopathic generalized epilepsies. Epilepsia004; 45: 1282-6.
enton P, Gelisse P, Thomas P, Dravet C. Do carbamazepinend phenytoin aggravate juvenile myoclonic epilepsy? Neu-ology 2000; 55: 1106-9.
ebeisen S, Pires N, Louriero AI, et al. Eslicarbazepinend the enhancement of slow inactivation of voltage-gatedodium channels: a comparison with carbamazepine, oxcar-azepine and lacosamide. Neuropharmacology 2015; 89:22-35.
nake S, Hamer HM, Schomburg U, Oertel WH, Rosenow F.iagabine-induced absence status in idiopathic generalizedpilepsy. Seizure 1999; 8: 314-7.
anayiotopoulos CP, Agathonikou A, Ahmed-Sharoqi I,arker APJ. Vigabatrin aggravates absences and absence sta-us. Neurology 1997; 49: 1467.
impat US prescribing information: Available at: https://www.impat.com/vimpat-prescribing-information.pdf.
echsler RT, Yates SL, Messenheimer J, et al. Lacosamideor uncontrolled primary generalized tonic-clonic seizures:
Epileptic Disord, Vol. 21, No. 1, February 2019
n open pilot study with 59-week extension. Epilepsy Res017; 130: 13-20.

Epileptic Disord, Vol. 21, No. 1, February 2019 101
Lacosamide-induced absence status
TEST YOURSELFEDUCATION
(1) Which of the following medications can cause an aggravation of absence seizures in people with idiopathicgeneralized epilepsy?A. Vigabatrin and tiagabineB. LacosamideC. Carbamazepine and phenytoinD. All of the above
(2) Lacosamide’s main mechanism of action is due to enhancement of __________ of sodium channels.A. Rapid activationB. Slow inactivationC. Rapid inactivationD. Slow activation
(3) Lacosamide is a promising treatment for idiopathic generalized epilepsy, but caution is advised in patientswith ________ seizures.A. Bilateral convulsive seizuresB. Myoclonic seizuresC. Absence seizuresD. All of the above
Note: Reading the manuscript provides an answer to all questions. Correct answers may be accessed on thewebsite, www.epilepticdisorders.com, under the section “The EpiCentre”.

1
CCDUR3<
Clinical commentaryEpileptic Disord 2019; 21 (1): 102-7
Focal visual status epilepticus*
Caspar Stephani, Walter Paulus, Niels K. FockeDepartment for Clinical Neurophysiology, University Medical Center Göttingen,Göttingen, Germany
Received August 19, 2018; Accepted November 06, 2018
ABSTRACT – Epileptic visual auras are elementary to complex and some-times occur as colourful visual phenomena located close to or within thecentral part of the contralateral hemi-field. They typically last from secondsto a few minutes, which discriminates them from the usually longer-lastingvisual auras (5-30 minutes) of patients suffering from migraine. We presentan adult patient with occipital lobe epilepsy whose visual aura underepilepsy monitoring lasted for more than 30 minutes with almost no prop-agation, demonstrating a rare, but remarkable, sustained local epilepticnetwork activity associated with resection of an occipital arterio-venousmalformation.
icus, visual aura, occipital epilepsy,n
alternatively termed “aura con-tinua”, lasting for at least 60 minutes(Mameniskiene et al., 2011).
Case studyA right-handed, 41-year-old womanhad suffered an atypical intracere-bral haemorrhage from an arterio-venous malformation (AVM) in theright occipital lobe two years previ-ously. She reported a left paracentralvisual scotoma but no other seque-lae. Half a year after the event andafter resection of the AVM, she expe-rienced a visual aura that evolvedinto a tonic-clonic seizure.
Key words: epilepsy, status epileptmigraine, arterio-venous malformatio
The majority of epileptic seizureslast from seconds to a few min-utes. A longer duration may pointto an alternative cause such as psy-chogenic non-epileptic seizuresor indicate status epilepticus,operationally defined as the per-sistence of a (tonic-clonic) seizurefor more than five minutes orthe occurrence of more thanone (tonic-clonic) seizure withoutrestoration of consciousness inbetween (Trinka et al., 2015). How-ever, such pathophysiology-basedconventions have not been fullyestablished for non-convulsive sta-tus epilepticus without alteration of
do
i:10.1684/epd
.2019.1034
02 Epileptic Disord, Vol. 21, No. 1, February 2019
orrespondence:aspar Stephaniepartment for Clinical Neurophysiology,niversity Medical Center Göttingen,obert-Koch-Strasse 40,7075 Göttingen, [email protected]>
consciousness (Trinka et al., 2015).In addition, “epilepsia partialis con-tinua” refers to a subgroup of focalstatus epilepticus with motor ornon-motor phenomena, the latter
Despite antiepileptic treatment, shereported experiencing visual aurason a weekly basis. She describedthem as bright, at times rotat-ing, otherwise largely immobile,
*This case has been presented as a poster and in an oral form at the meeting of the GermanSociety for Clinical Neurophysiology in March 2018.

E
Ft
sfindststwaptttTwt2ttwbari
D
DvtmdE
owTwawtcfat(tuaPieuhwetiBwiP“ir(ArhevoEai(pacsrooot
igure 1. T1-weighed MRI demonstrating a right occipital scarwo years after resection of an arterio-venous malformation.
ometimes colourful spots in the lower left visualeld. These would often last for minutes, and she hadoticed a duration of 15 minutes. She then occasionallyeveloped epigastric and/or unpleasant olfactory sen-ations that could last for more than a minute. Everywo months, on average, she suffered a tonic-cloniceizure. Several days after withdrawing her medica-ion under epilepsy monitoring during pre-surgicalorkup, such a visual aura, lasting for 40 minutes and
rising within her left lower visual field close to theermanent scotoma, occurred. The EEG seizure pat-
ern remained highly localized to electrode O2 andhus close to the right occipital scar throughout most ofhe seizure (figures 1, 2). After it spread to electrodes6 and P4, clonazepam (1 mg; IV) was administeredhich quickly terminated the seizure. Source localiza-
ion (solely based on a routine electrode montage with0 electrodes) placed the epileptogenic source right ofhe pole of the right calcarine cortex, as expected byhe highly localized seizure pattern. fMRI-retinotopy asell as tractography demonstrated a close relationshipetween epileptic scar and visual tract, indicating thatvisual field defect may be regarded as an inevitable
isk of potential occipital lobe epilepsy surgeryn our case.
iscussion
ue to their duration, visual status epilepticus and
pileptic Disord, Vol. 21, No. 1, February 2019
isual epilepsia partialis continua may be difficulto differentiate from similar phenomena arising with
igraine. In the clinical context, however, a definitiveiagnosis is usually possible (Panayiotopoulos, 1999a;riksen et al., 2005; Hartl et al., 2017). Indeed, there are
ssmwe
Focal visual status epilepticus
nly very few reports of focal visual status epilepticusith symptoms lasting for longer than five minutes.hese are summarized in table 1. Epileptic amaurosishich is poorly distinguished from postictal deficits
nd status epilepticus originating in the occipital lobe,here visual hallucinations are only the initial and not
he predominating symptom, were excluded. In suchases, the criteria reported to be most reliable in dif-erentiating between an epileptic and a migraine aurare the unilaterality of the former and the longer dura-ion of the latter (Panayiotopoulos, 1999a). Eriksen et al.2005) proposed a five-item score that assesses dura-ion, symptom-dynamic, scotoma, fortification, andnilaterality in order to recognize visual auras associ-ted with migraine and aid in clinical decision-making.anayiotopoulos (1999b) found a prevalence of occip-tal lobe epilepsy of about 5%; 63 of his 1,360pilepsy patients had occipital lobe epilepsy. Thenderlying conditions were early onset, benign child-ood epilepsy (40%), idiopathic occipital epilepsyithout photosensitivity, symptomatic occipital lobepilepsy (25% each), and idiopathic photosensi-ive epilepsy (10%). Importantly, the prevalence ofnterictal epileptiform activity is particularly low.ased on neurosurgical series, only 20% of patientsith symptomatic occipital lobe epilepsy exhibit
nterictal occipital epileptiform activity (Adcock andanayiotopoulos, 2012). The same authors stated thata well-localized unifocal rhythmic ictal discharge dur-ng occipital seizures is infrequent”, indicating theather unusual presentation in the current reportAdcock and Panayiotopoulos, 2012).
search of the literature for similar cases yielded aeport nearly identical to ours; a 42-year-old, right-anded woman with a history of epilepsy aftermbolization of a right parieto-occipital arterio-enous malformation, who had prolonged visual auras,ne lasting for 13 minutes, documented during video-EG monitoring. The lesion was eventually resected,nd the patient remained seizure-free, albeit withmpaired consciousness and occasional visual seizuresHartl et al., 2015). Another similar case was recentlyublished with an arterio-venous malformation agains the epileptogenic lesion. After embolization andomplete resection of the lesion, the patient had beeneizure-free for three years without any further neu-ological deficits (Strzelczyk et al., 2017). This may bef relevance to the issue of the long-lasting focalityf the ictal discharges. While in our case, withdrawalf the antiepileptic medication certainly did increase
he chance of occurrence and a longer duration of
103
eizures, it did not induce rapid propagation as ofteneen after withdrawal of anticonvulsive therapy. Theechanisms by which epileptic activity is maintainedithin a focal network are not well defined. Apparently,pileptogenic tissue in these cases is, or becomes,
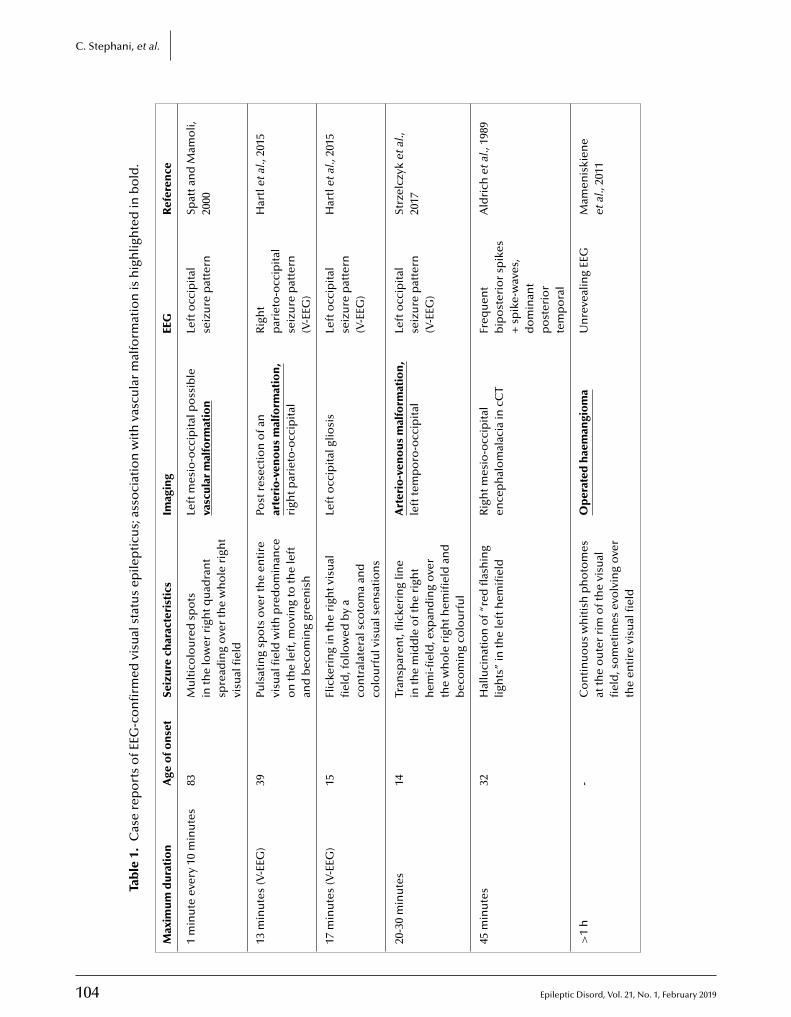
104 Epileptic Disord, Vol. 21, No. 1, February 2019
C. Stephani, et al.
Tab
le1.
Cas
ere
po
rts
ofE
EG-c
on
firm
edvi
sual
stat
us
epile
pti
cus;
asso
ciat
ion
wit
hva
scu
lar
mal
form
atio
nis
hig
hlig
hte
din
bo
ld.
Max
imu
md
ura
tio
nA
geo
fon
set
Seiz
ure
char
acte
rist
ics
Imag
ing
EEG
Ref
eren
ce
1m
inu
teev
ery
10m
inu
tes
83M
ult
ico
lou
red
spo
tsin
the
low
erri
ghtq
uad
ran
tsp
read
ing
ove
rth
ew
ho
leri
ght
visu
alfi
eld
Left
mes
io-o
ccip
ital
po
ssib
leva
scu
lar
mal
form
atio
nLe
fto
ccip
ital
seiz
ure
pat
tern
Spat
tan
dM
amo
li,20
00
13m
inu
tes
(V-E
EG)
39Pu
lsat
ing
spo
tso
ver
the
enti
revi
sual
fiel
dw
ith
pre
do
min
ance
on
the
left
,mo
vin
gto
the
left
and
bec
om
ing
gree
nis
h
Post
rese
ctio
no
fan
arte
rio
-ven
ou
sm
alfo
rmat
ion
,ri
ghtp
arie
to-o
ccip
ital
Rig
ht
par
ieto
-occ
ipit
alse
izu
rep
atte
rn(V
-EEG
)
Har
tlet
al.,
2015
17m
inu
tes
(V-E
EG)
15Fl
icke
rin
gin
the
righ
tvis
ual
fiel
d,f
ollo
wed
by
aco
ntr
alat
eral
sco
tom
aan
dco
lou
rfu
lvis
ual
sen
sati
on
s
Left
occ
ipit
algl
iosi
sLe
fto
ccip
ital
seiz
ure
pat
tern
(V-E
EG)
Har
tlet
al.,
2015
20-3
0m
inu
tes
14Tr
ansp
aren
t,fl
icke
rin
glin
ein
the
mid
dle
oft
he
righ
th
emi-
fiel
d,e
xpan
din
go
ver
the
wh
ole
righ
them
ifiel
dan
db
eco
min
gco
lou
rfu
l
Art
erio
-ven
ou
sm
alfo
rmat
ion
,le
ftte
mp
oro
-occ
ipit
alLe
fto
ccip
ital
seiz
ure
pat
tern
(V-E
EG)
Strz
elcz
yket
al.,
2017
45m
inu
tes
32H
allu
cin
atio
no
f“re
dfl
ash
ing
ligh
ts”
inth
ele
fth
emifi
eld
Rig
htm
esio
-occ
ipit
alen
cep
hal
om
alac
iain
cCT
Freq
uen
tb
ipo
ster
ior
spik
es+
spik
e-w
aves
,d
om
inan
tp
ost
erio
rte
mp
ora
l
Ald
rich
etal
.,19
89
>1
h-
Co
nti
nu
ou
sw
hit
ish
ph
oto
mes
atth
eo
ute
rri
mo
fth
evi
sual
fiel
d,s
om
etim
esev
olv
ing
ove
rth
een
tire
visu
alfi
eld
Op
erat
edh
aem
angi
om
aU
nre
veal
ing
EEG
Mam
enis
kien
eet
al.,
2011

Epileptic Disord, Vol. 21, No. 1, February 2019 105
Focal visual status epilepticus
Tab
le1.
Cas
ere
po
rts
ofE
EG-c
on
firm
edvi
sual
stat
us
epile
pti
cus;
asso
ciat
ion
wit
hva
scu
lar
mal
form
atio
nis
hig
hlig
hte
din
bo
ld(C
on
tin
ued
).
Max
imu
md
ura
tio
nA
geo
fon
set
Seiz
ure
char
acte
rist
ics
Imag
ing
EEG
Ref
eren
ce
>1
h-
Flic
keri
ng
inth
ele
ftvi
sual
fiel
d-
Rig
hto
ccip
ital
epile
pti
form
EEG
acti
vity
Mam
enis
kien
eet
al.,
2011
>1
h-
Flic
keri
ng
inth
ele
ftvi
sual
fiel
d,r
epet
itiv
e1-
2-h
epis
od
esN
on
-sp
ecifi
edo
ccip
ital
lesi
on
No
n-s
pec
ific
foca
lic
talE
EGab
no
rmal
itie
s
Mam
enis
kien
eet
al.,
2011
>1
h-
Vis
ual
hal
luci
nat
ion
so
flin
esan
dci
rcle
s-
Left
occ
ipit
alru
ns
ofs
pik
esM
amen
iski
ene
etal
.,20
11
60-9
0m
inu
tes
42Ye
llow
,red
and
blu
esp
ots
Rig
hto
ccip
ital
lesi
on
afte
rm
yco
pla
smic
men
ingo
ence
ph
alit
is
Rig
hto
ccip
ital
inte
rict
alsh
arp
wav
es;r
igh
to
ccip
ital
seiz
ure
pat
tern
Job
stet
al.,
2009
Afe
wh
ou
rs60
Palin
op
sia
or
abn
orm
ally
recu
rrin
gvi
sual
imag
ery,
mac
rop
sia,
un
form
edh
allu
cin
atio
ns,
hem
ian
op
ia
Left
tem
po
roo
ccip
ital
cave
rno
us
hae
man
gio
ma
Left
occ
ipit
alse
izu
rep
atte
rnK
awai
etal
.,20
06
Up
to2
day
s3
Flic
keri
ng
ligh
ts,o
bfu
scat
ion
ofv
isio
nb
yre
dan
dgr
een
ligh
ts;o
rsh
imm
erin
gel
lipso
id,s
ilver
ligh
ts(“
like
aca
mer
afl
ash
”)
Hem
isp
her
icas
ymm
etry
Rig
hto
ccip
ital
lob
ese
izu
rep
atte
rn;a
bn
orm
alp
ho
tic
stim
ula
tio
nre
spo
nse
Wal
ker
etal
.,19
95
>3
year
s10
Vis
ual
lear
nin
gd
iso
rder
;no
visu
alh
allu
cin
atio
nM
RIn
orm
al;b
utF
DG
-PET
wit
hp
rom
inen
tlef
tocc
ipit
alh
yper
met
abo
lism
Rig
hto
ccip
ital
stat
us
epile
pti
cus
Shet
han
dR
iggs
,19
99

1
C. Stephani, et al.
Fp1 - F7
F7 - Ft9
Ft9 - T3
T3 - T5
T5 - O1
Fp2 - F8
F8 - Ft10
Ft10 - T4
T4 - T6
T6 - O2
Fp1 - F3
F3 - C3
C3 - P3
P3 - O1
Fp2 - F4
F4 - C4
C4 - P4
P4 - O2
Fz - Cz
Cz - Pz
EKG1 - EKG2
F
eiilitttaamtbdlsLsgm
DN
R
Ae
AlE
Ba
Ei2
Hm
HSc
Jnei
KlN
Mveo
Pnd1
P
igure 2. Well-localized EEG seizure pattern at electrode O2.
lectrically isolated. Isolation in such cases may benduced by proliferation of non-neurogenic tissue ofnflammatory, glial or vascular origin. The high preva-ence of vascular malformations in the cases presentedn table 1, as well as in cases of amaurotic status epilep-icus (Barry et al., 1985), indicate that such tissue, or scarissue after its resection, may provide suitable condi-ions for focal status epilepticus. On the other hand,nd taking into account the long duration of visualuras in migraine, conditions in the striate cortex itselfay render spreading of electric activity less likely
han in other cortices. Whether this property coulde related to the macroscopic “stria” of Gennari -theense stripe of highly myelinated horizontal fibres in
ayer IV of the primary visual cortex- remains to behown.ong-lasting epileptic visual auras illustrate the widepectrum of propagation dynamics within epilepto-enic networks and can be associated with vascularalformations. �
isclosures.one of the authors have any conflict of interest to declare.
06
eferences
dcock JE, Panayiotopoulos P. Occipital lobe seizures andpilepsies. J Clin Neurophysiol 2012; 29: 397-407.
ldrich MS, Vanderzant CW, Alessi AG, Abou-Khalil B, Sackel-ares JC. Ictal cortical blindness with permanent visual loss.pilepsia 1989; 30: 116-20.
ot
Se
Sh
arry E, Sussman NM, Bosley TM, Harner RN. Ictal blindnessnd status epilepticus amauroticus. Epilepsia 1985; 26: 577-84.
riksen MK, Thomsen LL, Olesen J. The Visual Aura Rat-ng Scale (VARS) for migraine aura diagnosis. Cephalalgia005; 25: 801-10.
artl E, Rémi J, Noachtar S. Two patients with visual aura -igraine, epilepsy or migralepsy? Headache 2015; 55: 1148-51.
artl E, Gonzalez-Victores JA, Rémi J, Schankin CJ, Noachtar. Visual auras in epilepsy and migraine - an analysis of clinicalharacteristics. Headache 2017; 57: 908-16.
obst BC, Roberts DW, Williamson PD. Occipital lobeonconvulsive status epilepticus. In: Nonconvulsive statuspilepticus. Kaplan PW, Drislane FW. New York: Demos Med-
cal, 2009.
awai M, Cherches IM, Goldsmith IL. Visual illusory and hal-ucinatory phenomena in a patient with left occipital seizures.
eurology 2006; 67: 1457.
ameniskiene R, Bast T, Bentes C, et al. Clinical course andariability of non-Rasmussen, nonstroke motor and sensorypilepsia partialis continua: a European survey and analysisf 65 cases. Epilepsia 2011; 52: 1168-76.
anayiotopoulos CP. Elementary visual hallucinations, blind-ess, and headache in idiopathic occipital epilepsy:ifferentiation from migraine. J Neurol Neurosurg Psychiatry999a; 66: 536-40.
anayiotopoulos CP. Visual phenomena and headache inccipital epilepsy: a review, a systematic study and differen-
iation from migraine. Epileptic Disord 1999b; 1: 205-16.
Epileptic Disord, Vol. 21, No. 1, February 2019
heth RD, Riggs JE. Persistent occipital electrographic statuspilepticus. J Child Neurol 1999; 14: 334-6.
patt J, Mamoli B. Ictal visual hallucinations and post-ictalemianopia with anosognosia. Seizure 2000; 9: 502-4.

E
SAE
TfiC
Focal visual status epilepticus
trzelczyk A, Gaul C, Rosenow F, Kurlemann G. Visuelleuren im Grenzgebiet zwischen Epilepsie und Migräne. Zpileptol 2017; 20: 21-7.
rinka E, Cock H, Hesdorffer D, et al. A definition and classi-cation of status epilepticus-report of the ILAE Task Force onlassification of Status Epilepticus. Epilepsia 2015; 56: 1515-23.
TEST YOURSELFEDUCATION
(1) Which clinical criteria differentiate best between a migrainous and an epileptic visual aura?
(2) Which clinical criteria currently define convulsive status epilepticus?
Walker MC, Smith SJ, Sisodiya SM, Shorvon SD. Case ofsimple partial status epilepticus in occipital lobe epilepsymisdiagnosed as migraine: clinical, electrophysiological,and magnetic resonance imaging characteristics. Epilepsia1995; 36: 1233-6.
pileptic Disord, Vol. 21, No. 1, February 2019
Note: Reading the manuscript provides an answer to all qwebsite, www.epilepticdisorders.com, under the section
107
uestions. Correct answers may be accessed on the“The EpiCentre”.

do
i:10.
1
CRHC“B<
Clinical commentaryEpileptic Disord 2019; 21 (1): 108-11
Rasmussen syndrome:absence seizures may beinduced by oxcarbazepine
Roberto H. Caraballo 1, Pedro Cachia 2,Gabriela Reyes Valenzuela 1, Agustin Calvo 1
1 Department of Neurology, Hospital de Pediatría “Prof Dr Juan P Garrahan”,Buenos Aires2 Department of Neurology, Hospital de Pediatría “Victor J. Vilela”, Rosario, Argentina
Received August 15, 2018; Accepted November 12, 2018
ABSTRACT – A female patient with electroclinical and neuroradiologicalfeatures compatible with Rasmussen syndrome developed a particularclinical and EEG pattern. As the seizures were refractory to valproate at750 mg/kg/day, oxcarbazepine (OXC) at 30 mg/kg/day was added. Seizuresbecame more frequent and on neurological examination, no hemiparesiswas detected. The interictal EEG showed focal spikes and diffuse paroxysmsin the right fronto-temporal regions. Brain MRI revealed right hemiatro-phy, mainly at the Sylvian fissure. After initiating OXC daily, brief absenceseizures, lasting less than 20 seconds and associated with bilateral and syn-chronous 2.5-3-Hz spike-and-waves compatible with typical absences, wereobserved. OXC was discontinued and the typical absences disappeared.Treatment with intravenous gammaglobulin was started. At the last controlvisit, at nine years of age, no absence seizures were observed either by theparents or on the EEG recording. Our patient who met the diagnostic cri-teria for Rasmussen syndrome presented with absence seizures that mayhave been induced by OXC. The absence seizures disappeared after OXC
alis continua, Rasmussen syndrome,
patients may manifest with absenceor delayed-onset seizures, unusualevents such as epileptic spasms andhemidystonic episodes, headacheas the initial manifestation, dualpathology, or bilateral brain involve-ment. A dual pathology is seen in10% of patients and varies from low-
orrespondence:
was discontinued.
Key words: absences, epilepsia partioxcarbazepine
Rasmussen syndrome (RS) is a rareand severe immune-mediated braindisorder resulting in unilateral brainatrophy and leading to progres-sive neurological dysfunction andrefractory seizures (Bien et al., 2005).Different mechanisms have beensuggested, however, the aetiopatho-
1684/epd
.2019.1035
08 Epileptic Disord, Vol. 21, No. 1, February 2019
oberto Caraballoospital de Pediatría, Neurología,ombate de los Pozos 1881, CP 1245,Prof Dr Juan P Garrahan”,uenos Aires, [email protected]>
genesis is not fully understood (Bienet al., 2005; Caraballo et al., 2013).In the literature, patients withatypical features have been pub-lished (Granata et al., 2012). These
grade tumour, cortical dysplasia,tuberous sclerosis, mesial temporalsclerosis, vascular abnormalities, toold ischaemic lesions (Bien et al.,2007; Granata et al., 2012).
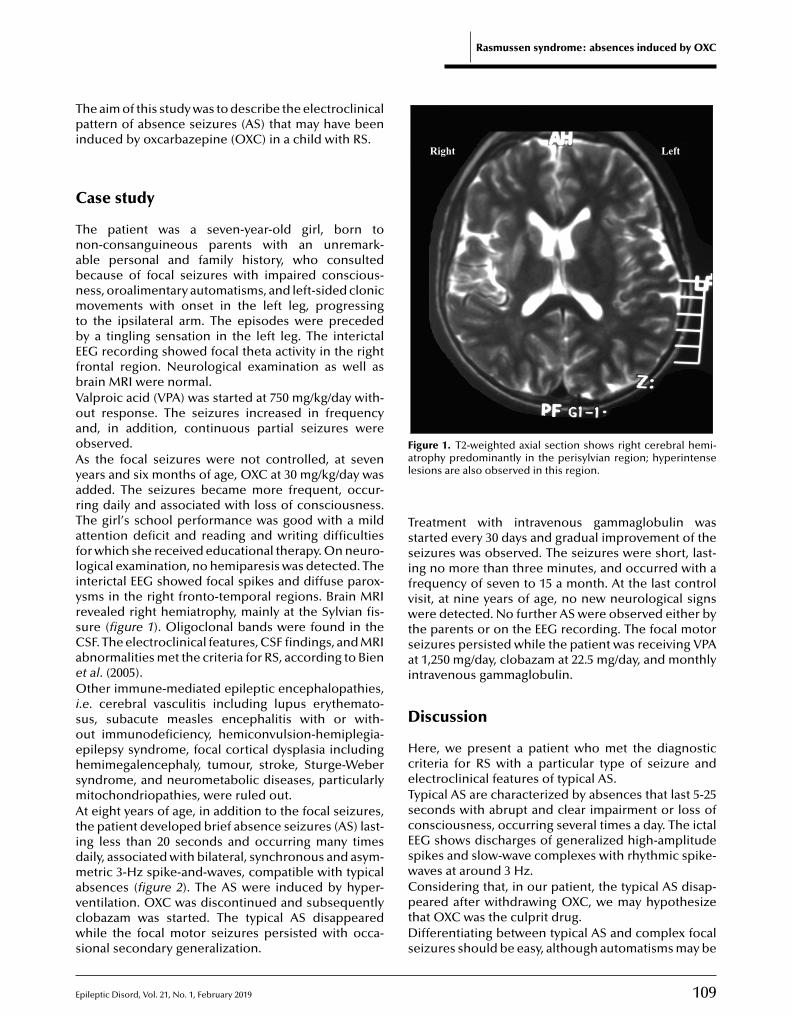
E
Rasmussen syndrome: absences induced by OXC
Tpi
C
TnabnmtbEfbVoaoAyarTafliyrsCaeOisoehsmAtidmavcws
Right Left
Figure 1. T2-weighted axial section shows right cerebral hemi-al
Tssifvwtsai
D
HceTscEsw
he aim of this study was to describe the electroclinicalattern of absence seizures (AS) that may have been
nduced by oxcarbazepine (OXC) in a child with RS.
ase study
he patient was a seven-year-old girl, born toon-consanguineous parents with an unremark-ble personal and family history, who consultedecause of focal seizures with impaired conscious-ess, oroalimentary automatisms, and left-sided clonicovements with onset in the left leg, progressing
o the ipsilateral arm. The episodes were precededy a tingling sensation in the left leg. The interictalEG recording showed focal theta activity in the rightrontal region. Neurological examination as well asrain MRI were normal.alproic acid (VPA) was started at 750 mg/kg/day with-ut response. The seizures increased in frequencynd, in addition, continuous partial seizures werebserved.s the focal seizures were not controlled, at sevenears and six months of age, OXC at 30 mg/kg/day wasdded. The seizures became more frequent, occur-ing daily and associated with loss of consciousness.he girl’s school performance was good with a mildttention deficit and reading and writing difficultiesor which she received educational therapy. On neuro-ogical examination, no hemiparesis was detected. Thenterictal EEG showed focal spikes and diffuse parox-sms in the right fronto-temporal regions. Brain MRIevealed right hemiatrophy, mainly at the Sylvian fis-ure (figure 1). Oligoclonal bands were found in theSF. The electroclinical features, CSF findings, and MRIbnormalities met the criteria for RS, according to Bient al. (2005).ther immune-mediated epileptic encephalopathies,
.e. cerebral vasculitis including lupus erythemato-us, subacute measles encephalitis with or with-ut immunodeficiency, hemiconvulsion-hemiplegia-pilepsy syndrome, focal cortical dysplasia includingemimegalencephaly, tumour, stroke, Sturge-Weberyndrome, and neurometabolic diseases, particularlyitochondriopathies, were ruled out.t eight years of age, in addition to the focal seizures,
he patient developed brief absence seizures (AS) last-ng less than 20 seconds and occurring many timesaily, associated with bilateral, synchronous and asym-etric 3-Hz spike-and-waves, compatible with typical
pileptic Disord, Vol. 21, No. 1, February 2019
bsences (figure 2). The AS were induced by hyper-entilation. OXC was discontinued and subsequentlylobazam was started. The typical AS disappearedhile the focal motor seizures persisted with occa-
ional secondary generalization.
CptDs
trophy predominantly in the perisylvian region; hyperintenseesions are also observed in this region.
reatment with intravenous gammaglobulin wastarted every 30 days and gradual improvement of theeizures was observed. The seizures were short, last-ng no more than three minutes, and occurred with arequency of seven to 15 a month. At the last controlisit, at nine years of age, no new neurological signsere detected. No further AS were observed either by
he parents or on the EEG recording. The focal motoreizures persisted while the patient was receiving VPAt 1,250 mg/day, clobazam at 22.5 mg/day, and monthlyntravenous gammaglobulin.
iscussion
ere, we present a patient who met the diagnosticriteria for RS with a particular type of seizure andlectroclinical features of typical AS.ypical AS are characterized by absences that last 5-25econds with abrupt and clear impairment or loss ofonsciousness, occurring several times a day. The ictalEG shows discharges of generalized high-amplitudepikes and slow-wave complexes with rhythmic spike-aves at around 3 Hz.onsidering that, in our patient, the typical AS disap-
109
eared after withdrawing OXC, we may hypothesizehat OXC was the culprit drug.ifferentiating between typical AS and complex focal
eizures should be easy, although automatisms may be

1
R.H. Caraballo, et al.
Fp1-F7
F7-T3
T3-O1
O1-O2
O2-T4
T4-F8
F8-Fp2
Fp2-Fp1
Fp1-F3
F3-C3
C3-P3
100 uV 1 sec.
P3-O1
Fp2-F4
F4-C4
C4-P4
P4-O2
F d asy
cteHmsdtdTtmfsait2IsoftIOttga(
2idoiIaoamIAsIth
DN
R
Auvin S, Chhun S, Berquin P, Ponchel E, Delanoe C, Chiron
igure 2. The ictal EEG recording shows bilateral, synchronous an
ommon in both. One of the main problems involvesypical AS of frontopolar lobe origin that may alsoxhibit concomitant, more or less, regular bilateral 3-z spike-wave discharges (Medina et al., 2012). Focalotor components, asymmetric ictal discharges, or
table interictal frontal foci on the EEG may help toistinguish them. MRI may show frontal abnormali-
ies. Ferrie et al. (1995) listed diffuse and focal brainisorders in which AS have been reported.ypical AS should be distinguished from atypical AShat occur in children with epileptic encephalopathies,
ainly Lennox-Gastaut syndrome. These are distinctrom typical absences in that onset and termination islow, impairment of consciousness is mild, and theyre often associated with loss of muscle tone. On thectal EEG, the diffuse spikes and waves are slower thanhose observed in typical AS, usually between 1.5 and.5 Hz.n our case, considering the presence of focal frontalpikes and the focal brain lesion in the insular regionn brain MRI, the AS may have arisen from the right
rontal lobe, triggering a thalamo-cortical system dueo secondary bilateral synchrony (Medina et al., 2012).n our patient, the AS may have been induced by
XC, since upon discontinuation of this antiepilep-
10
ic drug (AED), the AS disappeared. It is widely knownhat many AEDs, such as carbamazepine (CBZ), OXC,abapentin, vigabatrine, and tiagabine, may aggravatebsence epilepsies (Genton et al., 2012). PhenytoinPHT) seems to be less aggravating for AS (Genton et al.,
CE
Bsc
mmetric spike-waves at 3 Hz, associated with an absence seizure.
012). Phenobarbital may have a dual effect by increas-ng absences at high doses and decreasing them at lowoses (Genton et al., 2012). In one study, aggravationf AS was reported in eight cases within days of VPA
ntroduction. All improved after VPA discontinuation.n five, VPA was reintroduced, resulting in new seizureggravation (Lerman-Sagie et al., 2001). An aggravationf AS was reported in three adolescents with juvenilebsence epilepsy by levetiracetam at a daily dose ofore than 1,750 mg/day (Auvin et al., 2011).
n rat models of genetic absence epilepsy, certainEDs, such as CBZ and PHT, have been found to worsenpiking (Depaulis and Van Luijteaar, 2006).n our case, the AS may have resulted from the rela-ionship between RS and the reaction to OXC or mayave occurred as a coincidence. �
isclosures.one of the authors have any conflict of interest to declare.
eferences
Epileptic Disord, Vol. 21, No. 1, February 2019
. Aggravation of absence seizure related to levetiracetam.ur J Paediatr Neurol 2011; 15: 506-11.
ien CG, Granata T, Antozzi C, et al. Pathogenesis, diagno-is and treatment of Rasmussen encephalitis: a Europeanonsensus statement. Brain 2005; 128: 454-71.

E
BheN
Cd2
DePA
FSaC
GdlM
GE5J
LP
ien CG, Elger CE, Leitner Y, et al. Slowly progressiveemiparesis in childhood as a consequence of Rasmussenncephalitis without or with delayed-onset seizures. Eur Jeurol 2007; 14: 387-90.
araballo RH, Fortini S, Cersósimo R, et al. Rasmussen syn-rome: an Argentinean experience in 32 patients. Seizure013; 22: 360-7.
epaulis A, Van Luijteaar G. Genetic models of absencepilepsy in the rat. In: Models of seizures and epilepsy.itkanen A, Schwartzkroin P, Moshé S. Burlington: Elsevier
pileptic Disord, Vol. 21, No. 1, February 2019
cademic Press, 2006.
errie C, Giannakodimo S, Robinson R, Panayiotopoulos C.ymptomatic typical absences seizures. In: Typical absencesnd related epileptic syndromes. Duncan JS, Panayiotopoulos. Edinburgh: Churchill Livingstone, 1995.
2
MhcP
Rasmussen syndrome: absences induced by OXC
enton P, Fejerman N, Gelisse P. Syndromes and antiepilepsyrugs. In: Epileptic syndromes in infancy, childhood and ado-
escence. 5th Edition. Bureau M, Genton P, Dravet C, et al.ontrouge: John Libbey Eurotext, 2012.
ranata T, Hart Y, Andermann F. Rasmussen’s encephalitis. In:pileptic syndromes in infancy, childhood and adolescence.th Edition. Bureau M, Genton P, Dravet C, et al. Montrouge:ohn Libbey Eurotext, 2012.
erman-Sagie T, Watemberg N, Kramer U, Shahar E, Lerman. Absence seizures aggravated by valproic acid. Epilepsia
111
001; 42: 941-3.
edina M, Bureau M, Hirsch E, Panayiotopoulos C. Child-ood absence epilepsy. In: Epileptic syndromes in infancy,hildhood and adolescence. 5th Edition. Bureau M, Genton, Dravet C, et al. Montrouge: John Libbey Eurotext, 2012.

do
i:10.1684/epd
.2019.1036
112 Epileptic Disord, Vol. 21, No. 1, February 2019
Correspondence:Danielle A. NolanBeaumont Children’s,Neuroscience Center,3555 West 13 Mile Rd,Suite N120, Royal Oak,MI 48073, USA<[email protected]>
Clinical commentaryEpileptic Disord 2019; 21 (1): 112-6
A Rasmussen encephalitis,autoimmune encephalitis,and mitochondrial diseasemimicker: expanding theDNM1L-associated intractableepilepsy and encephalopathyphenotype
Danielle A. Nolan 1, Baibing Chen 2, Anne Marie Michon 1,Emily Salatka 1, Daniel Arndt 1
1 Department of Pediatric Epilepsy, Beaumont Health, Royal Oak, MI 480732 Oakland University William Beaumont School of Medicine, Rochester, MI 48309, USA
Received November 05, 2018; Accepted December 01, 2018
ABSTRACT – Dynamin-1-like protein (DNM1L) gene variants have beenlinked to childhood refractory epilepsy, developmental delay, encephalopa-thy, microcephaly, and progressive diffuse cerebral atrophy. However, onlya few cases have been reported in the literature and there is still a limitedamount of information about the symptomatology and pathophysiologyassociated with pathogenic variants of DNM1L. We report a 10-year-oldgirl with a one-year history of mild learning disorder and absence seizureswho presented with new-onset focal status epilepticus which progressedto severe encephalopathy and asymmetric hemispheric cerebral atro-phy. Differential diagnosis included mitochondrial disease, Rasmussen’sencephalitis, and autoimmune encephalitis. Disease progressed from onehemisphere to the other despite anti-seizure medications, hemispherec-tomy, vagus nerve stimulator, ketogenic diet, and immunomodulators.Continued cerebral atrophy and refractory seizures evolved until deathfour years after initial presentation. Post-mortem whole-exome sequencingrevealed a pathogenic DNM1L variant. This paper presents a novel case ofadolescent-onset DNM1L-related intractable epilepsy and encephalopathy.
Key words: developmental delay, seizure, refractory epilepsy, cerebral atro-phy, encephalopathy congenital, DNM1L

E
TehEmtDdrc2dmheDtpoadaaaeHteasdhfcceieDdwd#
C
Twonbsdta
poifScatwcHwismeatIstvlcewstttdacanUstctpbatCachtfm
he advent of next-generation sequencing is rapidlyxpanding the already genetically and phenotypicallyeterogenous category of mitochondrial disorders.pilepsy gene panels are now identifying novel nuclearitochondrial pathologic variants, including those in
he DNM1L (dynamin-1-like, MIM*603850) gene.NM1L is a nuclear mitochondrial gene that encodesynamic-related protein 1 (DRP1) and plays a criticalole in mediating the mitochondrial fission pro-ess and in regulating peroxisomal fission (Archer,013; Lackner, 2014). DNM1L-related disorders presenturing infancy with early-onset diffuse cerebral abnor-alities, microcephaly, optic atrophy, lactic acidosis,
ypotonia, and death within early infancy (Waterhamt al., 2007). Recently, there have been reports ofNM1L-related disorders that include: developmen-
al delay, refractory epilepsy, normal brain MRI, androlonged survival (Vanstone et al., 2016); childhood-nset epileptic encephalopathy with diffuse cerebraltrophy (Fahrner et al., 2016); postnatal microcephaly,evelopmental delay, and pain insensitivity (Sheffer etl., 2016); or progressive neurological disease, char-cterized by mild cognitive impairment, cerebellarnd pyramidal signs, and ocular involvement (Nascat al., 2016).ere, we report a patient with disease progression
hat mimicked Rasmussen encephalitis, autoimmunencephalitis, and mitochondrial disease. Onset ofbsence seizures at age nine was followed by focaltatus epilepticus in early adolescence at age 10, withevelopment of severe encephalopathy, progressiveemiparesis, developmental regression, refractory
ocal epilepsy, and notably asymmetric progressiveerebral atrophy. The asymmetric atrophy was con-erning for Rasmussen encephalitis, autoimmunencephalitis, or mitochondrial disease that did not
mprove with typical therapies. Post-mortem whole-xome sequencing was diagnostic for a pathogenicNM1L variant (c.1207C>T [p.R403C]; a heterozygouse novo mutation) (Fahrner et al., 2016) associatedith DNM1L-associated lethal encephalopathy due toefective mitochondrial peroxisomal fission 1 (OMIM614388).
ase study
he proband was a right-handed Caucasian femaleho had an uneventful birth and appropriate devel-pment, aside from a mild learning disorder. She hado prior history of traumatic brain injury, abnormal
pileptic Disord, Vol. 21, No. 1, February 2019
irth history/development, personal/family history ofeizures, nor family history of epilepsy. She wasiagnosed with typical absence seizures at age nine
hat initially responded to ethosuximide. One yearfter her initial diagnosis, at 10 years of age, the
apbfm
DNM1L-associated intractable epilepsy and encephalopathy
roband experienced new-onset frontal headache forne week before suddenly developing left neck twitch-
ng. This progressed to involve her left arm and leg,ollowed by sudden left hemibody loss of sensation.he presented to the emergency department (ED) andlinically demonstrated non-resolving rhythmic leftrm and leg twitching with altered awareness. Con-inuous EEG revealed electroclinical status epilepticusith right hemispheric 2-Hz spike and wave dis-
harges, alternating with diffuse background slowing.er seizures continued despite escalating treatmentith lorazepam, diazepam, and levetiracetam; she was
ntubated and placed on a propofol infusion. Hereizures continued and she was further treated with aidazolam infusion, fosphenytoin, phenobarbital, and
thosuximide. Pentobarbital infusion was started andburst suppression pattern was obtained with resolu-
ion of electrographic seizures.nitial magnetic resonance imaging (MRI) demon-trated extensive areas of restricted diffusion withinhe right cerebral hemisphere, predominantly at theertex and involving the right frontal and parietalobes, right insular cortex, and right thalamus withorresponding increased T2/FLAIR signal and corticaldema. The findings were suggestive of changes seenith status epilepticus (figure 1). Magnetic resonance
pectroscopy demonstrated decreased n-acetyl aspar-ate (NAA) and minimally increased choline peaks ofhe right cerebral hemisphere compared to the con-ralateral side. Magnetic resonance perfusion imagesemonstrated overall symmetric perfusion, bilater-lly. At this time, hemispheric presentation was mostoncerning for a mitochondrial disorder versus anutoimmune etiology, such as Rasmussen-type phe-omena. Initial CSF studies were unrevealing.nfortunately, right hemispheric electroclinical
eizures returned following pentobarbital discon-inuation despite treatment with IVIG. An outsideenter evaluated and performed a right hemispherec-omy 13 days after her initial presentation, sparingortions of the right thalamus and medial rightasal ganglia. Seizures reoccurred almost immedi-tely post-operatively, presenting with right facialwitching, nystagmus, and right hemibody twitching.ontinuous EEG now demonstrated left hemisphericnd multi-focal left hemispheric epileptiform dis-harges, in addition to expected post-surgical rightemispheric attenuation. She exhibited epilepsia par-
ialis continua clinically as well as electroclinical leftrontotemporal focal seizures. At that time, she was
aintained on levetiracetam, lacosamide, clobazam,
113
nd topiramate. An autoimmune encephalopathyanel revealed elevated anti-glutamic acid decar-oxylase (GAD) antibodies, furthering the concern
or autoimmune encephalitis. She was started ononthly IVIG and high-dose steroids as well as
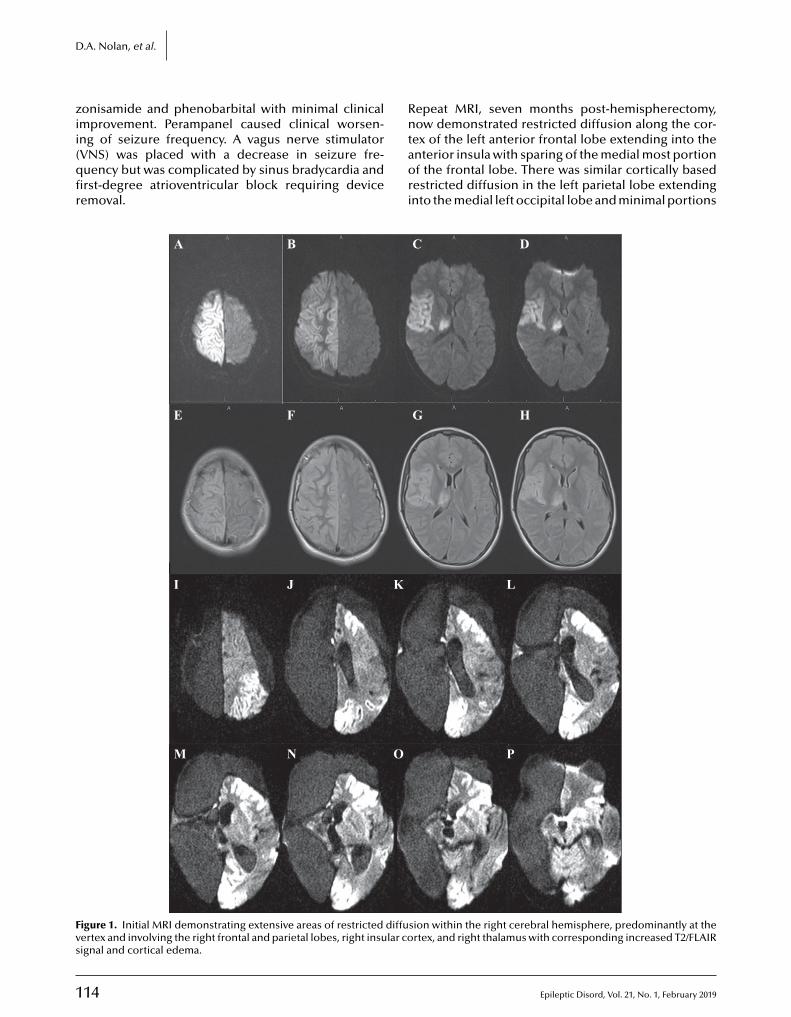
1
D
zii(qfir
Rn
Fvs
.A. Nolan, et al.
onisamide and phenobarbital with minimal clinicalmprovement. Perampanel caused clinical worsen-
14
ng of seizure frequency. A vagus nerve stimulatorVNS) was placed with a decrease in seizure fre-uency but was complicated by sinus bradycardia andrst-degree atrioventricular block requiring deviceemoval.
taori
A B
E F
I J K
M N O
igure 1. Initial MRI demonstrating extensive areas of restricted diffuertex and involving the right frontal and parietal lobes, right insular coignal and cortical edema.
epeat MRI, seven months post-hemispherectomy,ow demonstrated restricted diffusion along the cor-
Epileptic Disord, Vol. 21, No. 1, February 2019
ex of the left anterior frontal lobe extending into thenterior insula with sparing of the medial most portionf the frontal lobe. There was similar cortically basedestricted diffusion in the left parietal lobe extendingnto the medial left occipital lobe and minimal portions
C D
G H
L
P
sion within the right cerebral hemisphere, predominantly at thertex, and right thalamus with corresponding increased T2/FLAIR
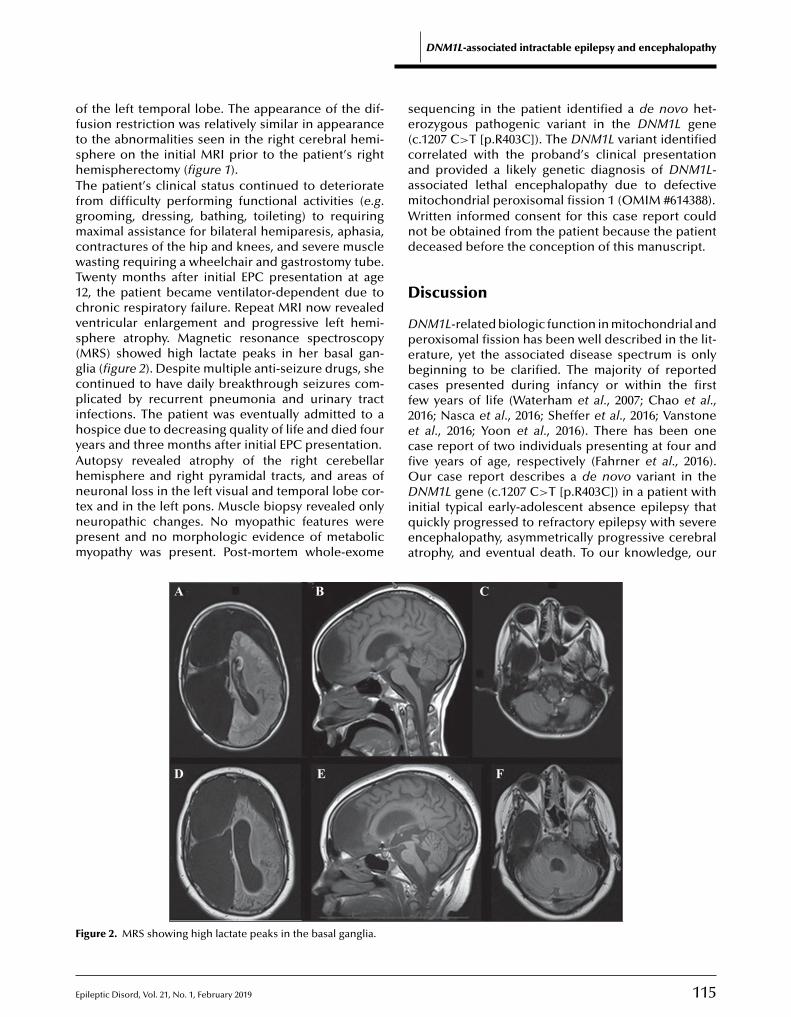
E
oftshTfgmcwT1cvs(gcpihyAhntnpm
se(caamWnd
D
Dpebcf2ecfiO
F
f the left temporal lobe. The appearance of the dif-usion restriction was relatively similar in appearanceo the abnormalities seen in the right cerebral hemi-phere on the initial MRI prior to the patient’s rightemispherectomy (figure 1).he patient’s clinical status continued to deterioraterom difficulty performing functional activities (e.g.rooming, dressing, bathing, toileting) to requiringaximal assistance for bilateral hemiparesis, aphasia,
ontractures of the hip and knees, and severe muscleasting requiring a wheelchair and gastrostomy tube.
wenty months after initial EPC presentation at age2, the patient became ventilator-dependent due tohronic respiratory failure. Repeat MRI now revealedentricular enlargement and progressive left hemi-phere atrophy. Magnetic resonance spectroscopyMRS) showed high lactate peaks in her basal gan-lia (figure 2). Despite multiple anti-seizure drugs, sheontinued to have daily breakthrough seizures com-licated by recurrent pneumonia and urinary tract
nfections. The patient was eventually admitted to aospice due to decreasing quality of life and died fourears and three months after initial EPC presentation.utopsy revealed atrophy of the right cerebellaremisphere and right pyramidal tracts, and areas of
pileptic Disord, Vol. 21, No. 1, February 2019
euronal loss in the left visual and temporal lobe cor-ex and in the left pons. Muscle biopsy revealed onlyeuropathic changes. No myopathic features wereresent and no morphologic evidence of metabolicyopathy was present. Post-mortem whole-exome
Diqea
A B
D E
igure 2. MRS showing high lactate peaks in the basal ganglia.
DNM1L-associated intractable epilepsy and encephalopathy
equencing in the patient identified a de novo het-rozygous pathogenic variant in the DNM1L genec.1207 C>T [p.R403C]). The DNM1L variant identifiedorrelated with the proband’s clinical presentationnd provided a likely genetic diagnosis of DNM1L-ssociated lethal encephalopathy due to defectiveitochondrial peroxisomal fission 1 (OMIM #614388).ritten informed consent for this case report could
ot be obtained from the patient because the patienteceased before the conception of this manuscript.
iscussion
NM1L-related biologic function in mitochondrial anderoxisomal fission has been well described in the lit-rature, yet the associated disease spectrum is onlyeginning to be clarified. The majority of reportedases presented during infancy or within the firstew years of life (Waterham et al., 2007; Chao et al.,016; Nasca et al., 2016; Sheffer et al., 2016; Vanstonet al., 2016; Yoon et al., 2016). There has been onease report of two individuals presenting at four andve years of age, respectively (Fahrner et al., 2016).ur case report describes a de novo variant in the
115
NM1L gene (c.1207 C>T [p.R403C]) in a patient withnitial typical early-adolescent absence epilepsy thatuickly progressed to refractory epilepsy with severencephalopathy, asymmetrically progressive cerebraltrophy, and eventual death. To our knowledge, our
C
F

1
D
paod#fDmeIcprdtaptcgmfinwsttcapccTaeebeo
.A. Nolan, et al.
atient had the latest onset of encephalopathy atge 10, compared to the described infantile onsetf DNM1L-associated lethal encephalopathy due toefective mitochondrial peroxisomal fission 1 (OMIM614388), expanding the described age of presentationor this disorder. This case report also suggests thatNM1L-related disorders may initially present with aore seemingly benign presentation, such as absence
pilepsy, which is not atypical in this age range.n addition to this patient’s relatively late onset, thisase was notable due to the shifting focal hemisphericredominance of the status epilepticus. This is the firsteport of DNM1L-associated lethal encephalopathyue to defective mitochondrial peroxisomal fission 1
hat presented with focal hemispheric findings initially,s opposed to global atrophy. This suggests that inatients with rapid progression of hemispheric refrac-
ory epilepsy, an underlying genetic etiology should beonsidered in conjunction with autoimmune etiolo-ies. Typical first-step genetic testing, chromosomalicroarray (CMA), and epilepsy panels are not suf-
cient in such cases. Indeed, in our case, reportedegative first-line genetic testing and the etiologyas not revealed until post-mortem whole-exome
equencing was performed. If DNM1L-related refrac-ory epilepsy had been identified during that initialesting, the expected prognosis may have drasticallyhanged management or provided parental guid-nce during a challenging time. In refractory epilepsyatients, especially those who demonstrate rapid clini-al deterioration, whole-exome sequencing should beonsidered early in the diagnostic process.o conclude, this case report presents a patient withnovel DNM1L R403C variant and clinical findings ofarly-adolescent-onset refractory seizures with severencephalopathy and progressive asymmetric cere-ral atrophy associated with DNM1L-associated lethalncephalopathy due to defective mitochondrial per-xisomal fission 1. This patient’s presentation not only
TEST YOURSELFEDUCATION
(1) Name a disorder that can mimic DNM1L-associated lethal encephalopathy.
(2) If chromosomal microarray and a gene panel are negative, what genetic test should be considered next inthe investigation?
expands the phenotypic age range of the disorder butalso demonstrates how the initial presentation canshow focal findings that may mimic an autoimmunedisorder such as Rasmussen’s encephalopathy. �
Disclosures.None of the authors have any conflict of interest to declare.
References
Archer SL. Mitochondrial dynamics-mitochondrial fissionand fusion in human diseases. N Engl J Med 2013; 369: 2236-51.
Chao YH, Robak LA, Xia F, et al. Missense variants in themiddle domain of DNM1L in cases of infantile encephalopa-thy alter peroxisomes and mitochondria when assayed inDrosophila. Hum Mol Genet 2016; 25: 1846-56.
Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novelde novo dominant negative mutation in DNM1L impairsmitochondrial fission and presents as childhood epilepticencephalopathy. Am J Med Genet A 2016; 170: 2002-11.
Lackner LL. Shaping the dynamic mitochondrial network.BMC Biol 2014; 12: 35.
Nasca A, Legati A, Baruffini E, et al. Biallelic mutations inDNM1L are associated with a slowly progressive infantileencephalopathy. Hum Mutat 2016; 37: 898-903.
Sheffer R, Douiev L, Edvardson S, et al. Postnatal micro-cephaly and pain insensitivity due to a de novo heterozygousDNM1L mutation causing impaired mitochondrial fission andfunction. Am J Med Genet A 2016; 170: 1603-7.
Vanstone JR, Smith AM, Mcbride S, et al. DNM1L-relatedmitochondrial fission defect presenting as refractoryepilepsy. Eur J Hum Genet 2016; 24: 1084-8.
Waterham HR, Koster J, Van Roermund CW, et al. A lethaldefect of mitochondrial and peroxisomal fission. N Engl J Med2007; 356: 1736-41.
Yoon G, Malam Z, Paton T, et al. Lethal disorder of mito-chondrial fission caused by mutations in DNM1L. J Pediatr2016; 171: 313-6.e1-2.
16
Note: Reading the manuscript provides an answer to all qwebsite, www.epilepticdisorders.com, under the section
Epileptic Disord, Vol. 21, No. 1, February 2019
uestions. Correct answers may be accessed on the“The EpiCentre”.

do
i:10.
1684
/ep
d.2
019.
1038
E
CDC“V1<
Clinical commentaryEpileptic Disord 2019; 21 (1): 117-21
Berardinelli-Seip syndromeand progressive myoclonusepilepsyDomenico Serino 1, Chiara Davico 2, Nicola Specchio 3,Carlo Efisio Marras 4, Franco Fioretto 1
1 Child Neurology and Psychiatry Unit, “Regina Montis Regalis” Hospital, Mondovì2 Child Neurology and Psychiatry Unit, “Regina Margherita” Pediatric Hospital, Turin3 Child Neurology Unit, “Bambino Gesù” Pediatric Hospital, Rome4 Child Neurosurgery Unit, “Bambino Gesù” Pediatric Hospital, Rome, Italy
Received August 28, 2018; Accepted December 01, 2018
ABSTRACT – Berardinelli-Seip syndrome, or congenital generalized lipodys-trophy type 2 (CGL2), is characterized by a lack of subcutaneous adiposetissue and precocious metabolic syndrome with insulin resistance, result-ing in diabetes, dyslipidaemia, hepatic steatosis, cardiomyopathy, andacanthosis nigricans. Most reported mutations are associated with mild,non-progressive neurological impairment. We describe the clinical and EEGdata of a patient with progressive myoclonus epilepsy (PME), CGL2, and pro-gressive neurological impairment, carrying a homozygous BSCL2 nonsensemutation. The patient had epilepsy onset at the age of two, characterized bymonthly generalized tonic-clonic seizures. By the age of three, he presentedwith drug-resistant ongoing myoclonic absence seizures, photosensitiv-ity, progressive neurological degeneration, and moderate cognitive delay.Molecular analysis of the BSCL2 gene yielded a homozygous c.(1076dupC)p.(Glu360*) mutation. Application of a vagus nerve stimulator led to tempo-rary improvement in seizure frequency, general neurological condition, andEEG background activity. Specific BSCL2 mutations may lead to a peculiarCGL2 phenotype characterized by PME and progressive neurodegenera-tion. Application of a vagus nerve stimulator, rarely used for PMEs, mayprove beneficial, if only temporarily, for both seizure frequency and generalneurological condition.
Berardinelli-Seip syndrome, BSCL2,eurodegenerative encephalopathy,
phenotype correlations and an auto-somal recessive inheritance wereproposed with regards to a num-ber of mutations in the BSCL2
orrespondence:Key words: lipodystrophy type 2,progressive myoclonus epilepsy, nEEG, vagus nerve stimulator
Berardinelli-Seip syndrome, or con-genital generalized lipodystrophytype 2 (CGL2), is characterized bya lack of subcutaneous adipose
pileptic Disord, Vol. 21, No. 1, February 2019 117
omenico Serinohild Neurology and Psychiatry Unit,Regina Montis Regalis” Hospital,ia S. Rocchetto, 99,2084 Mondovì, [email protected]>
tissue and precocious metabolicsyndrome with insulin resistance,resulting in diabetes, dyslipidaemia,hepatic steatosis, cardiomyopathy,and acanthosis nigricans. Genotype-
gene, that encodes Seipin. Mostreported mutations are associatedwith mild, non-progressive neuro-logical impairment. In 2016, Opriet al. published a small case series

1
D
opgogeaah
M
ApBeBscg
C
TPthtisasdiHhwMaSsecrc(Siidfoaan
taechsptdteseiis(poi3tfbsatsaw(
D
Eta2ahgma“pfrr2d
. Serino, et al.
f three patients with a rare association between CGL2,rogressive myoclonus epilepsy (PME) and severe pro-ressive neurological impairment (Opri et al., 2016). Inne patient, a novel compound heterozygous BSCL2ene mutation was found, resulting in two differ-nt frameshift mutations. Here, we describe clinicalnd EEG data of a further patient with PME, CGL2,nd progressive neurological impairment, carrying aomozygous BSCL2 nonsense mutation.
ethods
fter obtaining informed consent from the patient’sarents, genetic molecular analysis was performed.lood genomic DNA was extracted from whole periph-ral blood. Exons and exon-intron boundaries ofSCL2 were analysed by PCR amplification and directequencing (the first nucleotide of the ATG initiationodon was considered as the first nucleotide of theene).
ase study
he patient was a male born in Macedonia in 2013.regnancy was reported as uneventful and psychomo-or development as normal. At the age of 12 months,e developed monthly tonic seizures and antiepilep-
ic drug (AED) therapy with valproic acid (VPA) wasntroduced in his home country, resulting in goodeizure control. He moved to Italy at the age of twond a new medical evaluation was undertaken. Lack ofubcutaneous adipose tissue, moderate psychomotorelay with speech impairment, and severe hyperactiv-
ty prompted metabolic testing.ypertriglyceridaemia, hypertransaminasaemia, andepatic steatosis were documented and a low fat dietas implemented. Cerebral MRI was normal.olecular analysis of the BSCL2 gene yieldedhomozygous c.(1076dupC)/p.(Glu360*) mutation.
eizure recurrence followed the parents’ autonomoususpension of VPA, and was characterized by gen-ralized tonic seizures and episodes of loss ofonsciousness, perioral cyanosis, and bilateral eyeetropulsion. The EEG correlate of such episodes washaracterized by brief diffuse discharges of spike-waveSW) complexes, followed by slow waves (figure 1).eizure control was briefly obtained after gradual
ntroduction of lamotrigine (LTG). During the follow-ng three months, the patient developed numerousaily absence seizures with eyelid myoclonias and
18
orward head tilt, and VPA was reintroduced as add-n. After one month, there was a recurrence ofbsence seizures and an appearance of frequent dropttacks. The patient was eventually hospitalized foron-convulsive status epilepticus. A significant, albeit
PdBNO
emporary, reduction in absence seizure and dropttack frequency was obtained with introduction ofthosuximide (ESM) and substitution of LTG withlonazepam (CZP). CPZ, however, severely worsenedyperactivity which led to accidental head trauma withubdural haemorrhage and transitory left facial nervealsy. At this stage, ataxic gate and severe psychomo-
or delay were also evident. Serial brain MRI scansid not show signs of cerebral atrophy. Worsening of
he general neurological condition paralleled degen-ration of EEG background activity, especially duringleep, and an increase in photosensitivity, which wasvident even at 1-Hz photic stimulation. A steady
ncrease in seizure frequency gave way to relaps-ng refractory non-convulsive status, controlled withecond-line IV administration of phenobarbital (PB)figure 2). Introduction of PB therapy resulted in tem-orary improvement in seizure control but worseningf ataxic gait. A vagal nerve stimulator (VNS) was
mplanted (cyclic stimulation: 30 sec on; 5 min off;0 Hz; 500 msec). The VNS was gradually calibratedo 1.2 mA with temporary improvement in seizurerequency, general neurological condition, and EEGackground activity. After three months, PB was sub-tituted with perampanel because of a relapse in dropttacks and the appearance of prolonged generalizedonic seizures. After two months of follow-up, toniceizures disappeared and drop attack frequency andbsence seizure frequency and duration were reducedith improvement in general neurological condition
gait, speech, and social interaction).
iscussion
vidence that a form of PME exists in the con-ext of CGL2 has already been suggested (Tseng etl., 2009; Guillén-Navarro et al., 2013; Opri et al.,016). Guillèn-Navarro et al. described six patientsffected by a fatal neurodegenerative syndrome whoad homozygous or compound heterozygous BSCL2ene mutations. Five out of six patients developedyoclonic seizures between two and four years of
ge. This peculiar clinical presentation was namedCelia’s encephalopathy” (CE), characterized by theresence of intranuclear aggregates of mutated, mis-
olded Seipin which are thought to have a pathogenicole in neurodegeneration by inducing endoplasmiceticulum stress in neurons (Ruiz-Riquelme et al.,015). Even though there were no available data toemonstrate a pathogenic role for Seipin aggregates,
Epileptic Disord, Vol. 21, No. 1, February 2019
AS-positive inclusions were also found in the patientsescribed by Tseng et al. and Opri et al. While theSCL2 transcripts of the patients described by Guillèn-avarro et al. lacked exon 7, those described bypri et al. presented with a heterozygous mutation

E
Berardinelli-Seip syndrome and progressive myoclonus epilepsy
Fp2-C4
Fp2-T4
C4-O2
T4-O2
T4-C4
C4-C3
C3-T3
Fp1-C3
Fp1-T3
C3-O1
T3-O1
Fp2-C4
Fp2-T4
C4-O2
T4-O2
T4-C4
C4-C3
C3-T3
Fp1-C3
Fp1-T3
C3-O1
T3-O1
Fp2-C4
Fp2-T4
C4-O2
T4-O2
T4-C4
C4-C3
C3-T3
Fp1-C3
Fp1-T3
C3-O1
T3-O1
Fp2-C4
Fp2-T4
C4-O2
T4-O2
T4-C4
C4-C3
C3-T3
Fp1-C3
Fp1-T3
C3-O1
T3-O1
A
B
C
D
Fw
pileptic Disord, Vol. 21, No. 1, February 2019
igure 1. (A-D). Continuous EEG recording showing the EEG correlateave (SW) complexes, followed by slow waves.
119
of episodes characterized by brief diffuse discharges of spike-
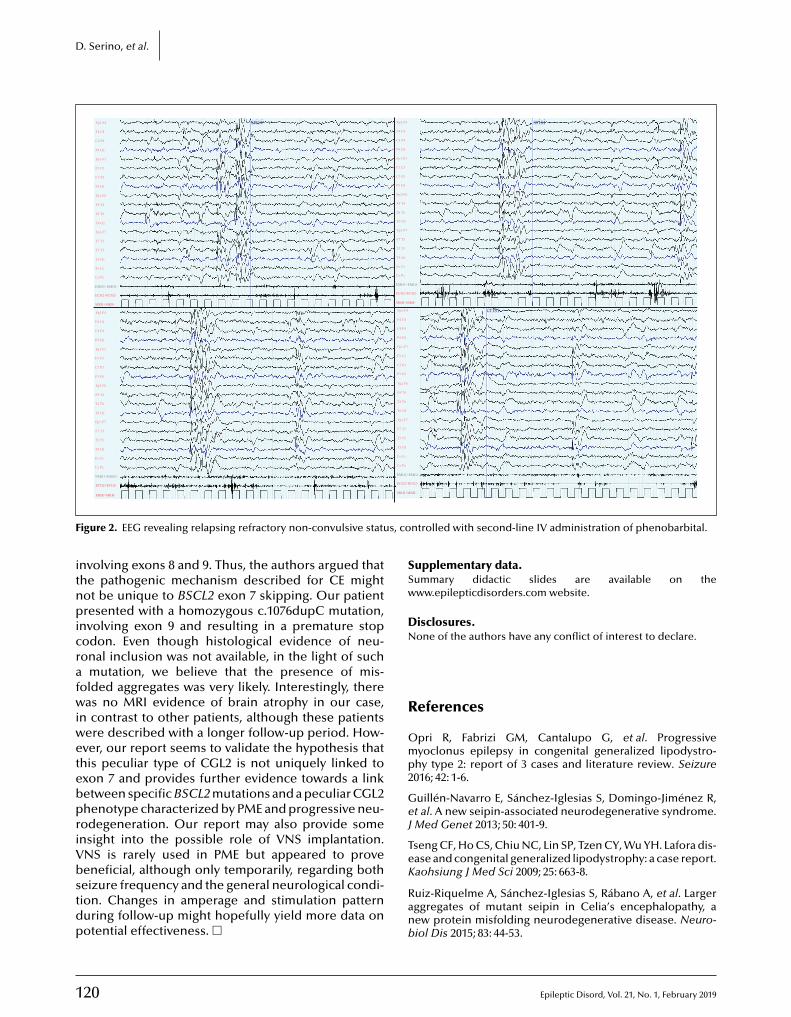
1
D. Serino, et al.
Fp2-F4
C4 P4
P4 O2
F4 C4
Fp1-F3
C3 P3
P3 O1
F3 C3
Fp2-F8
T4 T6
T6 O2
F8 T4
Fp1-F7
T3 T5
T5 O1
Fz Cz
Cz Pz
EMG1+EMG1-
ECG2+ECG2-
MKR+MKR-
F7 T3
Fp2-F4
C4 P4
P4 O2
F4 C4
Fp1-F3
C3 P3
P3 O1
F3 C3
Fp2-F8
T4 T6
T6 O2
F8 T4
Fp1 F7
T3 T5
T5 O1
Fz Cz
Cz Pz
EMG1+EMG1-
ECG2+ECG2-
MKR+MKR-
F7 T3
Fp2 F4
C4 P4
P4 O2
F4 C4
Fp1 F3
C3 P3
P3 O1
F3 C3
Fp2 F8
T4 T6
T6 O2
F8 T4
Fp1-F7
T3 T5
T5 O1
Fz Cz
Cz Pz
EMG1+EMG1-
ECG2+ECG2-
F7 T3
Fp2-F4
C4 P4
P4 O2
F4 C4
Fp1-F3
C3 P3
P3 O1
F3 C3
Fp2 F8
T4 T6
T6 O2
F8 T4
Fp1 F7
T3 T5
T5 O1
Fz Cz
Cz Pz
EMG1+EMG1-
ECG2+ECG2-
MKR+M
F7 T3
srisi srisi
crisi
F s, co
itnpicrafwiwetebpriVbstdp
SSw
DN
R
Omp2
GeJ
TeK
MKR+MKR-
igure 2. EEG revealing relapsing refractory non-convulsive statu
nvolving exons 8 and 9. Thus, the authors argued thathe pathogenic mechanism described for CE mightot be unique to BSCL2 exon 7 skipping. Our patientresented with a homozygous c.1076dupC mutation,
nvolving exon 9 and resulting in a premature stopodon. Even though histological evidence of neu-onal inclusion was not available, in the light of such
mutation, we believe that the presence of mis-olded aggregates was very likely. Interestingly, thereas no MRI evidence of brain atrophy in our case,
n contrast to other patients, although these patientsere described with a longer follow-up period. How-ver, our report seems to validate the hypothesis thathis peculiar type of CGL2 is not uniquely linked toxon 7 and provides further evidence towards a linketween specific BSCL2 mutations and a peculiar CGL2henotype characterized by PME and progressive neu-odegeneration. Our report may also provide somensight into the possible role of VNS implantation.NS is rarely used in PME but appeared to proveeneficial, although only temporarily, regarding both
20
eizure frequency and the general neurological condi-ion. Changes in amperage and stimulation patternuring follow-up might hopefully yield more data onotential effectiveness. �
Ranb
KR-
ntrolled with second-line IV administration of phenobarbital.
upplementary data.ummary didactic slides are available on theww.epilepticdisorders.com website.
isclosures.one of the authors have any conflict of interest to declare.
eferences
pri R, Fabrizi GM, Cantalupo G, et al. Progressiveyoclonus epilepsy in congenital generalized lipodystro-
hy type 2: report of 3 cases and literature review. Seizure016; 42: 1-6.
uillén-Navarro E, Sánchez-Iglesias S, Domingo-Jiménez R,t al. A new seipin-associated neurodegenerative syndrome.Med Genet 2013; 50: 401-9.
seng CF, Ho CS, Chiu NC, Lin SP, Tzen CY, Wu YH. Lafora dis-ase and congenital generalized lipodystrophy: a case report.aohsiung J Med Sci 2009; 25: 663-8.
Epileptic Disord, Vol. 21, No. 1, February 2019
uiz-Riquelme A, Sánchez-Iglesias S, Rábano A, et al. Largerggregates of mutant seipin in Celia’s encephalopathy, aew protein misfolding neurodegenerative disease. Neuro-iol Dis 2015; 83: 44-53.

E
Berardinelli-Seip syndrome and progressive myoclonus epilepsy
TEST YOURSELFEDUCATION
(1) Is CGL2 usually associated with progressive neurological impairment?
(2) Is CGL2 usually associated with epilepsy?
(3) What seems to be the pathogenetic mechanism behind the neurodegenerative CGL2 phenotype associatedwith epilepsy?
pileptic Disord, Vol. 21, No. 1, February 2019 121
Note: Reading the manuscript provides an answer to all questions. Correct answers may be accessed on thewebsite, www.epilepticdisorders.com, under the section “The EpiCentre”.

do
i:10.1684/epd
.2019.1037
122 Epileptic Disord, Vol. 21, No. 1, February 2019
Correspondence:Ingo BorggraefeDr. von Hauner‘s Childens Hospital,University of Munich, Germany<[email protected]>
Clinical commentaryEpileptic Disord 2019; 21 (1): 122-7
Epilepsy surgery in the firstmonths of life:a large type IIb focal corticaldysplasia causing neonataldrug-resistant epilepsyIngo Borggraefe 1,2, Moritz Tacke 1, Lucia Gerstl 1,Steffen Leiz 3, Roland Coras 4, Ingmar Blümcke 4,Armin Giese 5, Birgit Ertl-Wagner 6, Christian T. Thiel 7,Soheyl Noachtar 2,8, Aurelia Peraud 9
1 Divison of Pediatric Neurology, Developmental Neurology and Social Pediatrics,Department of Pediatrics, University Hospital LMU Munich2 Epilepsy Center for children, adolescents and adults, University Hospital LMU Munich3 Department of Pediatrics and Adolescent Medicine, Hospital Dritter Orden, Munich4 Department of Neuropathology, Neuropathological Reference Center for EpilepsySurgery, University Hospital Erlangen, Erlangen5 Department of Neuropathology, University Hospital LMU, Munich6 Department of Radiology, University Hospital LMU, Munich7 Institute of Human Genetics, Friedrich-Alexander-Universität Erlangen-Nürnberg,Erlangen8 Department of Neurology, University Hospital LMU, Munich9 Department of Neurosurgery, Section for Pediatric Neurosurgery, University of Ulm,Germany
Received April 30, 2018; Accepted December 01, 2018
ABSTRACT – Focal cortical dysplasia is a common cause of medically refrac-tory epilepsy in infancy and childhood. We report a neonate with seizuresoccurring within the first day of life. Continuous video-EEG monitoring ledto detection of left motor seizures and a right frontal EEG seizure pattern.Brain MRI revealed a lesion within the right frontal lobe without contrastenhancement. The patient was referred for epilepsy surgery due to drugresistance to vitamin B6 and four antiepileptic drugs. Lesionectomy wasperformed at the age of two and a half months, and histopathological eval-uation confirmed the diagnosis of focal cortical dysplasia type IIb (FCDIIb). The patient is free of unprovoked seizures without medication (EngelClass I) and is normally developed at 36 months after surgery. The casestudy demonstrates that FCD IIb may cause seizures within the first day oflife and that epilepsy surgery can be successfully performed in medicallyintractable patients with a clearly identifiable seizure onset zone within thefirst three months of life. Although radical surgery such as hemispherec-tomy and multi-lobar resections are over-represented in early infancy, thiscase also illustrates a favourable outcome with a more limited resection inthis age group.
Key words: neonate, seizure, focal cortical dysplasia, type IIb, surgery

E
FcHcpphp2iTwwbwcit
C
P
TawtsptidnSort
Ra
RXEutRirisSlww
sgaanlssasnAhTrs(rhT
E
TosofttbccooTI(imumtarof
D
ocal cortical dysplasia type IIb (FCD IIb) is a majorause of drug-resistant focal epilepsy (Palmini andolthausen, 2013). The pathogenesis has not yet been
ompletely unravelled, but dysregulation of the mTORathway appears to play a role in the formation of dys-lastic neurons (Crino, 2015). The histopathologicalallmark of FCD IIb is the presence of dysmor-hic neurons and balloon cells (Blümcke et al., 2009,011). Typical magnetic resonance imaging (MRI) find-ngs consist of an increased subcortical signal on2-weighted (w) and FLAIR sequences, often in aedge-shape configuration with a blurring of the grey-hite matter interface. FCD IIb arises during foetalrain development. However, seizures rarely occurithin the neonatal period but typically in infancy and
hildhood. Here, we report a patient with seizure man-festation as early as within the first 24 hours of life dueo right frontal FCD IIb.
ase study
atient history and clinical findings
he patient was a female newborn with a history ofn uneventful pregnancy and vaginal delivery at 39eeks of gestational age. After normal postnatal adap-
ation (APGAR: 10/10; umbilical cord: pH 7.35), seizurestarted 16 hours after birth. Seizure semiology com-rised left motor seizures of the arm and leg evolving
o generalized tonic-clonic seizures. There was no fam-ly history of epilepsy, stillbirths, or neurodegenerativeisorders of early infancy. An extensive work-up foreurometabolic diseases revealed no abnormalities.eizures were refractory to age-appropriate dosagesf vitamin B6, phenobarbitone, levetiracetam, topi-amate, and oxcarbazepine. None of the criteria foruberous sclerosis complex were met.
outine EEG, continuous video-EEG monitoring,nd brain MRI
outine and continuous video-EEG monitoring usingltek hard- and software equipment (Natus DBA,xcel-Tech Corp., Oakville, Canada) were performedsing standard adjustments (0.5-Hz low-frequency fil-
er, 70-Hz high-frequency filter; resistance <10 k�).outine EEG revealed subclinical seizure patterns,
ntermittent slow activity, and sharp waves over theight frontal region. Continuous video-EEG monitor-ng detected right frontal seizure patterns, 13 to 30
pileptic Disord, Vol. 21, No. 1, February 2019
econds prior to clinical seizure onset (figure 1A, B).eizure semiology comprised clonic seizures of the
eft extremities and bilateral clonic seizures. Brain MRIas obtained using a 1.5-Tesla scanner (3D sequencesith an isotropic resolution of 1.0 to 1.5 mm: T1w
WoeTb
Neonatal drug-resistant epilepsy due to type IIb FCD
equences before and after the administration of aadolinium-based contrast agent and T2w and fluidttenuated inversion recovery [FLAIR] sequences, withxial, sagittal, and coronal reformations; Siemens Mag-etom Aera, Munich, Germany). A well delineated
esion of the right frontal lobe was observed, mea-uring 2.0 × 2.5 × 2.6 cm (figures 2A, B). The MRIignal intensity was hyperintense on T1w imagingnd hypointense on T2w imaging compared to theurrounding unmyelinated white matter. There waseither contrast enhancement nor perifocal oedema.s a reference, the MRI of an eight-year-old boy withistologically proven FCD IIb is shown (figures 2C, D).his reference patient was operated due to medicallyefractory epilepsy at the age of eight years and iseizure-free after an observation period of five yearsEngel Class I). The boundaries of the FCD IIb of theeference patient appear much less sharp and revealypointensity on T1w imaging and hyperintensity on2w imaging.
pilepsy surgery, genetics, and outcome
he patient was referred for epilepsy surgery basedn concordant results from presurgical evaluation ([1]eizure semiology: left motor seizure; [2] EEG seizurenset zone: right frontal; and [3] MRI lesion: right
rontal). Lesionectomy was performed at the age ofwo and a half months and histopathological inves-igation of the surgical specimen revealed FCD withalloon cells fulfilling criteria for FCD type IIb (Blüm-ke, Thom et al., 2011). The specimen revealed higherellularity (figures 3A-D) compared to the specimenf the reference patient mentioned above who wasperated on at the age of eight years (figures 3E-G).rio exome sequencing on a HighSeq2500 (Illuminanc., San Diego, USA) after SureSelect v6 enrichmentAgilent Technologies Inc., Santa Clara, USA) for thendex patient and both parents revealed no pathogenic
utations of proteins involved in mTOR pathway reg-lation. The antiepileptic medication was tapered sixonths after surgery. During a post-surgical observa-
ional period of 36 months, the patient suffered fromfever-associated seizure at the age of two years. She
eached age-appropriate milestones for infant devel-pment (i.e. motor skills and speech) and revealed no
unctional deficits.
iscussion
123
e report a patient with type IIb FCD and seizure onsetn Day 1 of life who successfully underwent resectivepilepsy surgery at two and a half months of life.he epileptogenic zone may significantly extendeyond the visible lesion on brain imaging (Bouet et
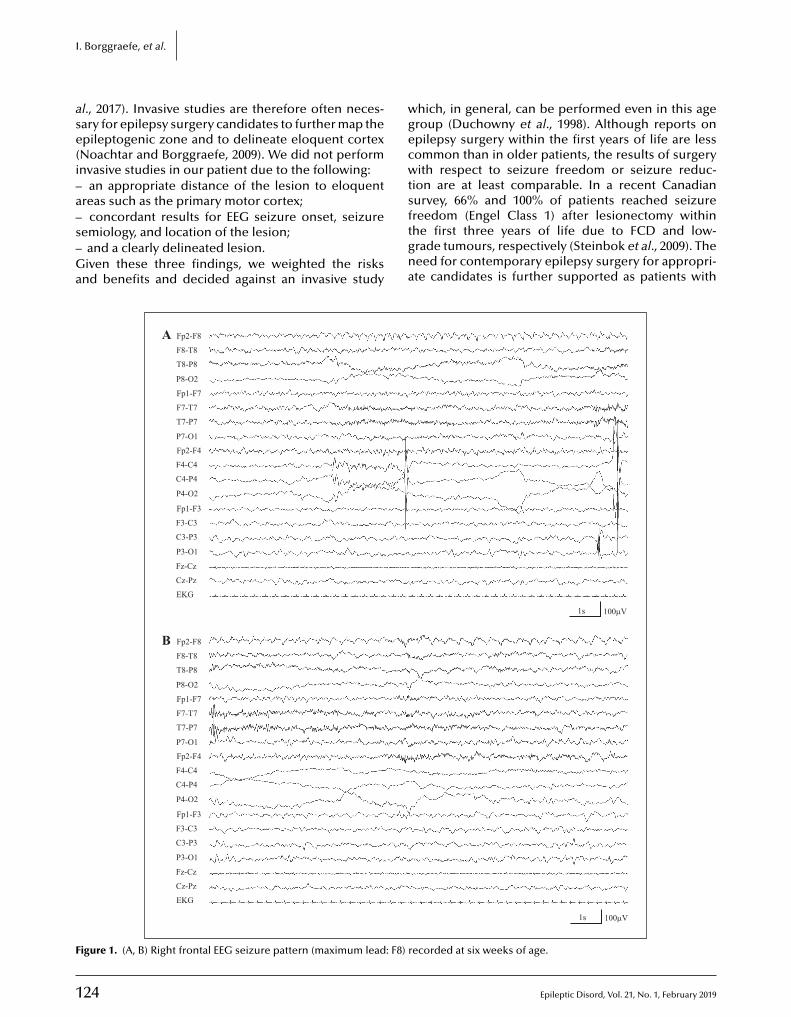
1
I
ase(i–a–s–Ga
wgecwts
F
. Borggraefe, et al.
l., 2017). Invasive studies are therefore often neces-ary for epilepsy surgery candidates to further map thepileptogenic zone and to delineate eloquent cortexNoachtar and Borggraefe, 2009). We did not performnvasive studies in our patient due to the following:
an appropriate distance of the lesion to eloquentreas such as the primary motor cortex;
24
concordant results for EEG seizure onset, seizureemiology, and location of the lesion;and a clearly delineated lesion.iven these three findings, we weighted the risks
nd benefits and decided against an invasive study
ftgna
Fp2-F8AF8-T8
T8-P8
P8-O2
Fp1-F7
F7-T7
T7-P7
P7-O1
Fp2-F4
F4-C4
C4-P4
P4-O2
Fp1-F3
F3-C3
C3-P3
P3-O1
Fz-Cz
Cz-Pz
EKG
Fp2-F8BF8-T8
T8-P8
P8-O2
Fp1-F7
F7-T7
T7-P7
P7-O1
Fp2-F4
F4-C4
C4-P4
P4-O2
Fp1-F3
F3-C3
C3-P3
P3-O1
Fz-Cz
Cz-Pz
EKG
igure 1. (A, B) Right frontal EEG seizure pattern (maximum lead: F8) r
hich, in general, can be performed even in this ageroup (Duchowny et al., 1998). Although reports onpilepsy surgery within the first years of life are lessommon than in older patients, the results of surgeryith respect to seizure freedom or seizure reduc-
ion are at least comparable. In a recent Canadianurvey, 66% and 100% of patients reached seizure
Epileptic Disord, Vol. 21, No. 1, February 2019
reedom (Engel Class 1) after lesionectomy withinhe first three years of life due to FCD and low-rade tumours, respectively (Steinbok et al., 2009). Theeed for contemporary epilepsy surgery for appropri-te candidates is further supported as patients with
1s 100µV
1s 100µV
ecorded at six weeks of age.
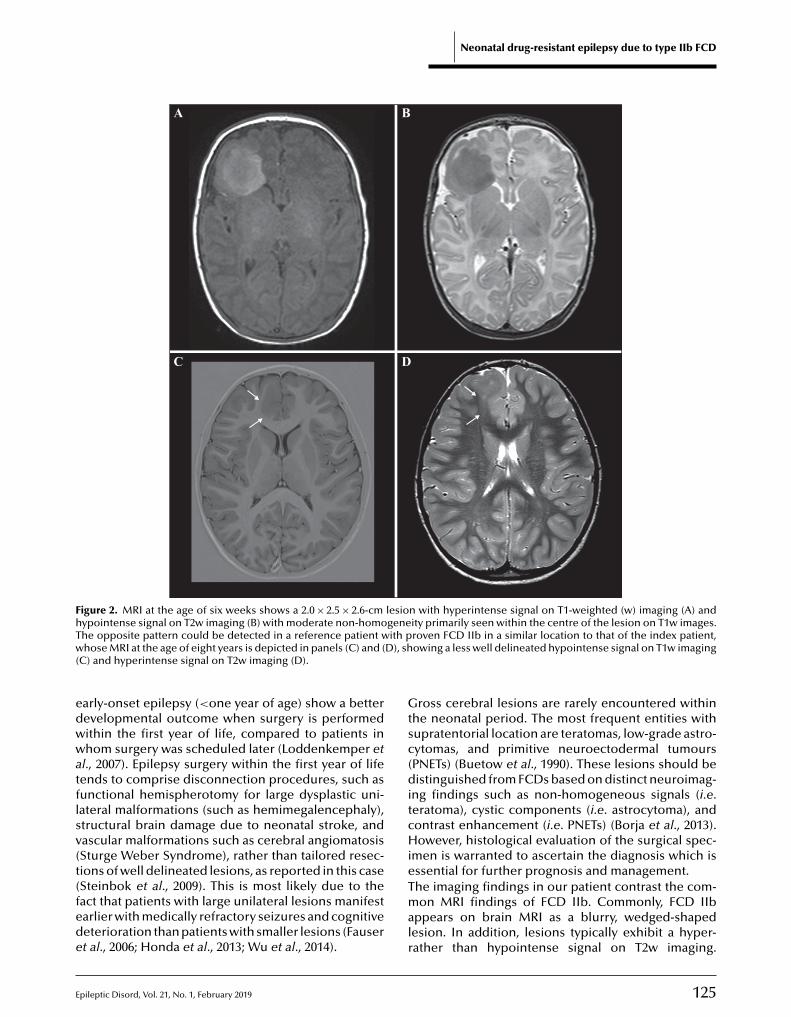
E
Neonatal drug-resistant epilepsy due to type IIb FCD
A B
C D
Figure 2. MRI at the age of six weeks shows a 2.0 × 2.5 × 2.6-cm lesion with hyperintense signal on T1-weighted (w) imaging (A) andhypointense signal on T2w imaging (B) with moderate non-homogeneity primarily seen within the centre of the lesion on T1w images.T ith pw D), sh(
edwwatflsv(t(fede
Gtsc(ditcHie
he opposite pattern could be detected in a reference patient whose MRI at the age of eight years is depicted in panels (C) and (
C) and hyperintense signal on T2w imaging (D).
arly-onset epilepsy (<one year of age) show a betterevelopmental outcome when surgery is performedithin the first year of life, compared to patients inhom surgery was scheduled later (Loddenkemper et
l., 2007). Epilepsy surgery within the first year of lifeends to comprise disconnection procedures, such asunctional hemispherotomy for large dysplastic uni-ateral malformations (such as hemimegalencephaly),tructural brain damage due to neonatal stroke, andascular malformations such as cerebral angiomatosisSturge Weber Syndrome), rather than tailored resec-ions of well delineated lesions, as reported in this case
pileptic Disord, Vol. 21, No. 1, February 2019
Steinbok et al., 2009). This is most likely due to theact that patients with large unilateral lesions manifestarlier with medically refractory seizures and cognitiveeterioration than patients with smaller lesions (Fausert al., 2006; Honda et al., 2013; Wu et al., 2014).
Tmalr
roven FCD IIb in a similar location to that of the index patient,owing a less well delineated hypointense signal on T1w imaging
ross cerebral lesions are rarely encountered withinhe neonatal period. The most frequent entities withupratentorial location are teratomas, low-grade astro-ytomas, and primitive neuroectodermal tumoursPNETs) (Buetow et al., 1990). These lesions should beistinguished from FCDs based on distinct neuroimag-
ng findings such as non-homogeneous signals (i.e.eratoma), cystic components (i.e. astrocytoma), andontrast enhancement (i.e. PNETs) (Borja et al., 2013).owever, histological evaluation of the surgical spec-
men is warranted to ascertain the diagnosis which isssential for further prognosis and management.
125
he imaging findings in our patient contrast the com-on MRI findings of FCD IIb. Commonly, FCD IIb
ppears on brain MRI as a blurry, wedged-shapedesion. In addition, lesions typically exhibit a hyper-ather than hypointense signal on T2w imaging.
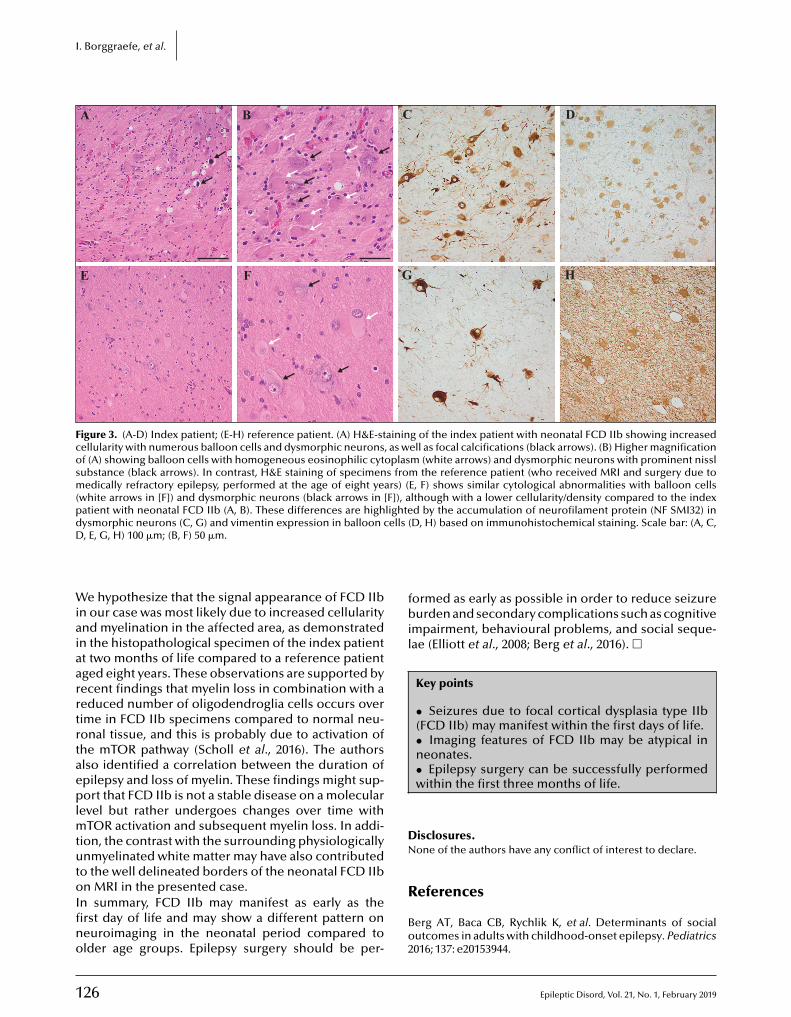
1
I. Borggraefe, et al.
A B C D
E F G H
Figure 3. (A-D) Index patient; (E-H) reference patient. (A) H&E-staining of the index patient with neonatal FCD IIb showing increasedcellularity with numerous balloon cells and dysmorphic neurons, as well as focal calcifications (black arrows). (B) Higher magnificationof (A) showing balloon cells with homogeneous eosinophilic cytoplasm (white arrows) and dysmorphic neurons with prominent nisslsubstance (black arrows). In contrast, H&E staining of specimens from the reference patient (who received MRI and surgery due tomedically refractory epilepsy, performed at the age of eight years) (E, F) shows similar cytological abnormalities with balloon cells(white arrows in [F]) and dysmorphic neurons (black arrows in [F]), although with a lower cellularity/density compared to the indexpatient with neonatal FCD IIb (A, B). These differences are highlighted by the accumulation of neurofilament protein (NF SMI32) ind ells (D
WiaiaarrtrtaeplmtutoIfino
fbil
DN
ysmorphic neurons (C, G) and vimentin expression in balloon c, E, G, H) 100 �m; (B, F) 50 �m.
e hypothesize that the signal appearance of FCD IIbn our case was most likely due to increased cellularitynd myelination in the affected area, as demonstratedn the histopathological specimen of the index patientt two months of life compared to a reference patientged eight years. These observations are supported byecent findings that myelin loss in combination with aeduced number of oligodendroglia cells occurs overime in FCD IIb specimens compared to normal neu-onal tissue, and this is probably due to activation ofhe mTOR pathway (Scholl et al., 2016). The authorslso identified a correlation between the duration ofpilepsy and loss of myelin. These findings might sup-ort that FCD IIb is not a stable disease on a molecular
evel but rather undergoes changes over time withTOR activation and subsequent myelin loss. In addi-
ion, the contrast with the surrounding physiologicallynmyelinated white matter may have also contributed
o the well delineated borders of the neonatal FCD IIb
26
n MRI in the presented case.n summary, FCD IIb may manifest as early as therst day of life and may show a different pattern oneuroimaging in the neonatal period compared tolder age groups. Epilepsy surgery should be per-
R
Bo2
D, H) based on immunohistochemical staining. Scale bar: (A, C,
ormed as early as possible in order to reduce seizureurden and secondary complications such as cognitive
mpairment, behavioural problems, and social seque-ae (Elliott et al., 2008; Berg et al., 2016). �
Key points
• Seizures due to focal cortical dysplasia type IIb(FCD IIb) may manifest within the first days of life.• Imaging features of FCD IIb may be atypical inneonates.• Epilepsy surgery can be successfully performedwithin the first three months of life.
isclosures.one of the authors have any conflict of interest to declare.
Epileptic Disord, Vol. 21, No. 1, February 2019
eferences
erg AT, Baca CB, Rychlik K, et al. Determinants of socialutcomes in adults with childhood-onset epilepsy. Pediatrics016; 137: e20153944.

E
BofiD
BstM
Bas5
Bbat
Bt1
CmM
Dt
Eic
Fae1
HocB
Lm2
NE
Pce5
SdfP
S
lümcke I, Vinters HV, Armstrong D, et al. Malformationsf cortical development and epilepsies: neuropathologicalndings with emphasis on focal cortical dysplasia. Epilepticisord 2009; 11: 181-93.
lümcke I, Thom M, Aronica E, et al. The clinicopathologicpectrum of focal cortical dysplasias: a consensus classifica-ion proposed by an ad hoc Task Force of the ILAE Diagnostic
ethods Commission. Epilepsia 2011; 52: 158-74.
orja MJ, Plaza MJ, Altman N, et al. Conventional anddvanced MRI features of pediatric intracranial tumors:upratentorial tumors. AJR Am J Roentgenol 2013; 200: 483-03.
ouet R, Mauguiere F, Daligault S, et al. The relationshipetween morphological lesion, magnetic source imaging,nd intracranial stereo-electroencephalography in focal cor-ical dysplasia. Neuroimage Clin 2017; 15: 71-9.
uetow PC, Smirniotopoulos JG, Done S. Congenital brainumors: a review of 45 cases. AJR Am J Roentgenol990; 155: 587-93.
rino PB. mTOR signaling in epilepsy: insights from malfor-ations of cortical development. Cold Spring Harb Perspected 2015; 5: a022442.
pileptic Disord, Vol. 21, No. 1, February 2019
uchowny M, Jayakar P, Resnick T, et al. Epilepsy surgery inhe first three years of life. Epilepsia 1998; 39: 737-43.
lliott IM, Lach L, Kadis DS, et al. Psychosocial outcomesn children two years after epilepsy surgery: has anythinghanged? Epilepsia 2008; 49: 634-41.
i2
Wh2
Neonatal drug-resistant epilepsy due to type IIb FCD
auser S, Huppertz HJ, Bast T, et al. Clinical char-cteristics in focal cortical dysplasia: a retrospectivevaluation in a series of 120 patients. Brain 2006; 129:907-16.
onda R, Kaido T, Sugai K, et al. Long-term developmentalutcome after early hemispherotomy for hemimegalen-ephaly in infants with epileptic encephalopathy. Epilepsyehav 2013; 29: 30-5.
oddenkemper T, Holland KD, Stanford LD, et al. Develop-ental outcome after epilepsy surgery in infancy. Pediatrics
007; 119: 930-5.
oachtar S, Borggraefe I. Epilepsy surgery: a critical review.pilepsy Behav 2009; 15: 66-72.
almini A, Holthausen H. Focal malformations ofortical development: a most relevant etiology ofpilepsy in children. Handb Clin Neurol 2013; 111:49-65.
choll T, Muhlebner A, Ricken G, et al. Impaired oligoden-roglial turnover is associated with myelin pathology in
ocal cortical dysplasia and tuberous sclerosis complex. Brainathol 2016; 27: 770-80.
teinbok P, Gan PY, Connolly MB, et al. Epilepsy surgery
127
n the first 3 years of life: a Canadian survey. Epilepsia009; 50: 1442-9.
u N, Borlot F, Ali A, et al. Hemimegalencephaly: whatappens when children get older? Dev Med Child Neurol014; 56: 905-9.

Epilepsy surgery is an effective treatment for children with structural epilep-sies. Seizure freedom rates vary from 50% to over 80%. Early and appropriate selection of candidates for surgery highly depends upon the multidisciplinary competences of teams with expertise in pediatric epilepsies and child neurology.
In this comprehensive first edition of PEDIATRIC EPILEPSY SURGERY, the au-thors, all members of the Task Force of the International League Against Epilepsy, offer an up-to-date critical review of available data with respect to etio-
logy. Modalities of pre-surgical evaluation and post-surgical follow-up are at the center of each chapter. Both frequent (focal cortical dysplasias; WHO Grade I and II Benign Tumors; Mesial temporal lobe epilepsy; MRI-negative focal epilepsies) and rare etiologies (Sturge-We-ber; Hamartomas; Hemimegalencephaly; Tuberous Sclerosis; …) are covered, focusing exclu-sively on children.
Other sections of the book thoroughly review the indications and limits of currently available diagnostic tools, focal seizure semiology in children, and neuropsychological evaluation modalities. The section on surgical techniques for the neurologist is a unique compilation of valuable information on all aspects a clinician needs to know to better inform his/her patient prior to and after surgery. Future perspectives are also outlined in this rapidly expanding field. In summary, the authors have created a reference book for child neurologists, neurologists, and neurosurgeons involved in epilepsy care.
• December 2016• ISBN: 978-2-7420-1424-8• 568 pages• 170 euros
Book available onwww.jle.com
EDITORS:
l Alexis Arzimanoglou is a neuro- pediatrician, Director of the Pediatric Epileptology Department at the University Hospitals of Lyon, France; Research Coordi-nator of the Pediatric Epilepsy Unit, Univer-sity Hospital San Juan de Dios in Barcelona, Spain; Professor of child neurology, member of the Management committee of the International League Against Epilepsy (ILAE) and Editor-in-Chief of its educational journal Epileptic Disorders.
l J. Helen Cross is a professor at the Institute of Child Health in London and secretary general of the International League Against Epilepsy (ILAE).
l William D. Gaillard is a professor of neurology and pediatrics and division chief at the Children Research Institute in Washington D.C.
l Hans Holthausen is a neurologist at Schon Kliniken in Vogtareuth, Germany.
l Prasanna Jayakar is a neurologist and director of Niklaus Children’s Hospital in Miami.
l Philippe Kahane is a professor of neu-rology, director of the Hospital-University Neuroscience Division and president of the Scientific Council of the Fédération pour la Recherche sur le Cerveau (Brain Research Foundation) in Lyon, France.
l Gary Mathern is a professor of neurology at the Brain Research Institute of the University of California, Los Angele.
The “White Guide” of pediatric epilepsy surgery
Pediatric Epilepsy Surgery
On the webwww.jle.com(secured payment)
By fax+33 (0) 1 40 84 09 99
By mailÉditions John Libbey Eurotext127, avenue de la République92120 Montrouge - France
BookshopAt your usual bookseller
For any further information+33 (0) 1 46 73 06 62
To be returned to John Libbey Eurotext - 127, avenue de la République - 92120 Montrouge - France
Please send me
Pediatric Epilepsy Surgery
Handling & postage charges + 6
170
TOTAL
Name ———————————————————————— First name ——————————————————————————
Address ————————————————————————————————————————————————————————
Zip code City——————————————————————— Country ———————————————————
Tel.: ——————————————— E-mail ———————————————————————————————————————
Please send me an invoice marked “Paid” for my statement of professional expensesIn accordance with the French law No. 78-17 of 6 January 1978 relative to information technology, files and freedom ("Loi Informatique et Libertés du 06/01/78"), you are required to providesome personal information for us to process your order. You are free to access, modify, rectify and delete this information. To do so, send us an email at: [email protected] or write to:John Libbey Eurotext, 127 avenue de la République, 92120 Montrouge, France.
Pub_
Pedi
atric
Epi
l.Sur
g_20
16 -
AP
E : 5
814Z
/ SI
RET
: 328
195
904
000
37
PaymentPlease find enclosed my payment for the amount of
By cheque payable to John Libbey Eurotext
By credit card
Visa Eurocard/Mastercard American Express
Carte N°
Last 3 digits on the back of your card
Expiry date Signature :
VAT n° (compulsory for institutions):
ORDER FORM
Related Documents











