Versuchsanleitung zum Praktikum Allgemeine Chemie für Studierende des Lehramts Chemie (Realschule und Gymnasium)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Versuchsanleitung zum Praktikum Allgemeine Chemie für Studierende des Lehramts Chemie
(Realschule und Gymnasium)

Inhaltsverzeichnis Vorbemerkungen
Sicherheit im Labor 5 Bekleidung und Verhalten im Labor 5 Entsorgung von Abfällen 6 Bitte an den Praktikumstagen mitbringen 6
1. Kurstag: Bunsenbrenner, Aggregatzustände, Analysenwaage
1.1 Bunsenbrenner 7 1.2 Erhitzen im Reagenzglas 7 1.3 Siedeverzug 7 1.4 Verdunsten 7 1.5 Erhitzen von Feststoffen 8 1.6 Analysenwaage 8
2. Kurstag: Chemische Formel, Phasentrennung, Gravimetrie
2.1 Synthese von Kupfersulfid 9 2.2 Abtrennung von Bariumsulfat 10 2.3 Gravimetrische Bestimmung von Eisen (1. Teil: Fällen und Filtrieren des Niederschlags) 10
3. Kurstag: Gravimetrie, Wasserstoff, Reduktion von Kupferoxid
3.1 Gravimetrische Bestimmung von Eisen (2. Teil: Glühen und Wiegen des Niederschlags) 11 3.2 Wasserstoffentwicklung 11 3.3 Knallgasprobe 11 3.4 Reduktion von CuO bzw. Cu2O 12
4. Kurstag: Gasversuche
4.1 Gaswägung (Molmassenbestimmung) von CO2 und NH3 13 4.2 Volumetrische Analyse des Ammoniaks 13 4.3 Molmassenbestimmung von Magnesium 14
5. Kurstag: Flüssigkeiten, Verteilungssatz, Kristallisation
5.1 Mischbarkeit von Flüssigkeiten verschiedener Polarität 16 5.2 Netzwirkung und Grenzflächenspannung 16 5.3 Lichtabsorption, Verteilungssatz 16 5.4 Polare Verbindungen im inhomogenen elektrischen Feld 16 5.5 Übersättigte Lösung 17 5.6 Umkristallisieren 17

6. Kurstag: Löslichkeit, Lösungswärme, homogenes und heterogenes Gleichgewicht 6.1 Temperaturabhängigkeit der Löslichkeit 18 6.2 Lösungswärme von CaCl2 und CaCl2 · 6 H2O 18 6.3 Bestimmung der Löslichkeit von Kaliumperchlorat 18 6.4 Verschiebung eines homogenen Gleichgewichts 19 6.5 Verschiebung eines heterogenen Gleichgewichts 19
7. Kurstag: Komplexe
7.1 Fällung von Silberhalogeniden 20 7.2 Verhalten der Silberhalogenide gegenüber Ammoniak 20 7.3 Komplexe des Kupfers 21 7.4 Wassernachweis 21 7.5 Gravimetrische Bestimmung von Nickel 21
8. Kurstag: Leitfähigkeit von Lösungen
8.1 Leitfähigkeit von Lösungen 22 8.2 Konzentrationsabhängigkeit der Leitfähigkeit 22 8.3 Abhängigkeit der Leitfähigkeit von der Ionenladung 22 8.4 Leitfähigkeit schwacher Säuren und ihrer Salze 22 8.5 Aktivität 22
9. Kurstag: Säuren, Basen, Titration
9.1 Indikatoren 23 9.2 Bestimmung des pH-Werts mit Indikatorpapier 23 9.3 Titrationskurve einer starken Säure 23 9.4 Titrationskurve einer schwachen Säure 23 9.5 Titrationskurve einer mehrwertigen Säure 24 9.6 Sauer und basisch reagierende Salzlösungen 24 9.7 Titration verschiedener Säuren 24 9.8 Herstellen einer NaOH-Normallösung 25
10. Kurstag: Ionenaustauscher, Pufferlösungen
10.1 Einstellen einer NaOH-Normallösung 26 10.2 Quantitative Bestimmung einer H2SO4/Na2SO4-Lösung 26 10.3 Pufferwirkung 27 10.4 Bestimmung der Dissoziationskonstante über die Puffergleichung 27 10.5 Herstellen einer Pufferlösung mit definiertem pH-Wert 28
11. Kurstag: Leitfähigkeitstitration
11.1 Leitfähigkeitstitration 29 11.2 Quantitative Bestimmung von Phosphorsäure 30

12. Kurstag: Reaktionsgeschwindigkeit, Katalyse, Reaktionen von Metallen 12.1 Geschwindigkeit der Reaktion von Fe3+ mit Thiosulfat 31 12.2 Heterogene Katalyse: Katalytische Zersetzung von H2O2 31 12.3 Autokatalyse 31 12.4 Reaktionen von Metallen mit Säuren 32 12.5 Spannungsreihe 32 12.6 Korrosion von Eisen (1. Teil: Durchführung) 32
13. Kurstag: Redoxreaktionen
13.1 Korrosion von Eisen (2. Teil: Auswertung) 33 13.2 Reaktionen von Natrium 33 13.3 Silberspiegel 34 13.4 Oxidation von Fe2+ 34 13.5 Reduktion von Kupferoxid 34 13.6 Oxidation von Alkohol mit Chromat 34 13.7 Redox-Amphoterie 34 13.8 Dis- und Synproportionierung 34
14. Kurstag: Elektrochemie
14.1 Galvanisches Element 55 14.2 Konzentrationskette 35 14.3 Zersetzungsspannung von CuSO4 35
Hinweise zum Protokoll
Aufbau und Inhalt des Protokolls 36

Sicherheit im Labor
Informieren Sie sich zu Beginn des Praktikums über Standort und Funktion wichtiger
Sicherheitseinrichtungen im Labor:
• Feuerlöscher
• Löschdecken
• Löschsand
• Augenduschen
• Notduschen
• Feuermelder
• Erste-Hilfe-Kasten
• Notausgänge
• Fluchtwege
• Not-Aus-Schalter für die elektrische Anlage
Bekleidung und Verhalten im Labor
• Beim Aufenthalt im Labor sind geschlossener Laborkittel, geschlossenes, trittsicheres Schuhwerk,
langes Beinkleid und Schutzbrille zu tragen. Lange Haare dürfen nicht offen getragen werden.
• Essen, Trinken und Rauchen sowie die Aufbewahrung von Lebensmitteln sind im Labor nicht
erlaubt.
• Das Tragen von Kontaktlinsen sowie die Benutzung von Handys sind im Laborbereich untersagt.
• Der Verbrauch von Gas, Wasser, Elektrizität und Chemikalien ist auf das Notwendigste zu
beschränken. Brenner und Gas sind vor Verlassen des Labors abzustellen. Der Arbeitsplatz ist
sauber und aufgeräumt zu hinterlassen. Verbleibende Gerätschaften sind schriftlich zu
kennzeichnen.
• Den Anweisungen der Praktikumsbetreuer ist unbedingt Folge zu leisten. Wiederholte Verstöße
gegen die Laborordnung können den Ausschluss von der Nutzung des Labors zur Folge haben.
Die Gewährleistung der Sicherheit für alle am Praktikum beteiligten Personen ist oberster
Grundsatz. Aus diesem Grund wird nachdrücklich auf folgende Gebote hingewiesen:
NOTRUF – TELEFONNUMMER: 112

• Der Genuss von alkoholischen Getränken unmittelbar vor oder während des Praktikums führt zum
Ausschluss vom Praktikum.
• Achten Sie auf äußerste Sauberkeit an Ihrem Arbeitsplatz und den Gemeinschaftseinrichtungen
(Abzüge, Ausgüsse, Waagen etc.). Von Ihnen verursachte Verunreinigungen etc. sind unverzüglich
zu beseitigen.
• Schwangere, stillende Mütter und Personen, die der Sicherheit nicht belehrt wurden (z.B.
Besucher), haben keinen Zutritt zu den Praktikumsräumen.
• Chronische Krankheiten sowie Allergien gegen bestimmte Substanzen sind vor Beginn des
Praktikums anzuzeigen.
• Auch bei kleinsten Verletzungen und Unfällen ist sofort ein Assistent zu verständigen.
• Das Tragen von Schutzhandschuhen empfiehlt sich in manchen Fällen. Achten Sie jedoch darauf,
dass Sie nichts mit Schutzhandschuhen anfassen, was von anderen Studierenden ohne
Schutzhandschuhe angefasst werden könnte, beispielsweise ein Wasserhahn.
• Versuche, bei denen übel riechende oder giftige Gase entstehen, müssen unter allen Umständen
unter dem Abzug ausgeführt werden. Die Frontschieber unbenutzter Abzüge sind geschlossen zu
halten, weil die Entlüftungswirkung in den anderen sonst verringert wird.
Entsorgung von Abfällen
• In die Ausgüsse dürfen keine festen Gegenstände wie Filter, Glasscherben usw. geworfen werden.
Konzentrierte Säuren und Laugen sowie Schwermetallabfälle sind in die dafür vorhandenen
Behälter zu entsorgen.
• Wässrige Schwermetallabfälle werden in Abfallbehältern (braune Flaschen oder Kanister unter
den Abzügen) gesammelt. Die Abfallbehälter dürfen nicht vollständig gefüllt werden, sondern
müssen vom Labordienst rechtzeitig in den Sammelkanister entleert werden.
• Feste schwermetallhaltige oder kontaminierte Abfälle werden in der blauen Feststofftonne
gesammelt.
• In die Hausmülltonnen dürfen nur nicht kontaminierte Abfälle entsorgt werden.
• Alle scharfkantigen Abfälle (Glas, Keramik) gehören in den gesonderten Sammelbehälter.
Bitte an den Praktikumstagen mitbringen:
Skript, Spiralblock als Laborjournal, Taschenrechner, wasserfesten Stift, Streichhölzer oder
Feuerzeug, am 11. Praktikumstag Millimeterpapier

1. Kurstag: Bunsenbrenner, Aggregatzustände, Analysenwaage
1.1 Bunsenbrenner
a) Der Bunsenbrenner wird bei geschlossener Luftzufuhr angezündet. In die leuchtende Flamme hält
man kurzzeitig einen trockenen, sauberen Porzellantiegel.
Man öffnet die Luftzufuhr, bis eine geteilte, ruhig brennende, nicht leuchtende Flamme entsteht,
und hält den Porzellantiegel wieder in die Flamme. Schließlich glüht man einen mit LA
beschrifteten Porzellantiegel ohne Deckel auf einem Dreifuß mit Tondreieck aus und stellt ihn nach
kurzem Abkühlen zum vollständigen Erkalten mit Hilfe einer Tiegelzange in einen Exsikkator.
b) Temperaturverteilung. Ein Holzstab (Streichholz oder Holzspan) wird quer in die nicht
leuchtende Flamme direkt über das Brennrohr gehalten.
Zusätzlich wird die Temperaturverteilung in mindestens fünf einzelnen Zonen des Bunsenbrenners
mit einem Thermoelement gemessen.
! Anmerkung: Zur Gasersparnis und zur besseren Erkennung der Flamme ist der Brenner nach
Gebrauch stets auf Sparflamme zu stellen.
! Aufgabe: Skizzieren Sie einen Bunsenbrenner mit nicht leuchtender Flamme und seine
unterschiedlichen Temperaturzonen.
1.2 Erhitzen im Reagenzglas
a) Ein Reagenzglas wird zur Hälfte mit Wasser gefüllt und mittels einer Reagenzglasklammer
schräg in die nicht leuchtende Flamme gehalten.
Die Öffnung des Reagenzglases richte man beim Erhitzen weder auf sich noch auf den Nachbarn !
b) Ein zweites Reagenzglas wird ca. 2 cm hoch mit Wasser gefüllt, mit der Reagenzglasklammer
gehalten und unter stetem Schütteln in der Flamme erhitzt.
!Anmerkung: Nur Reagenzgläser in der Flamme erhitzen, niemals Zentrifugengläser!
1.3 Siedeverzug
In einem auf einem Drahtnetz stehenden Erlenmeyerkolben bringt man etwa 40 mL Wasser zum
Sieden. Man entfernt die Flamme und wirft einige Körner eines porösen Materials (Siedesteine) in
den Kolben.
Die Siedesteine werden anschließend im Hausmüll entsorgt.
1.4 Verdunsten
Man verdunstet 3 Tropfen Leitungswasser und 3 Tropfen destilliertes Wasser auf je einem
Objektglasträger durch vorsichtiges Erhitzen.

1.5 Erhitzen von Feststoffen
a) Man erhitzt jeweils eine kleine Menge Ammoniumchlorid, Zucker, Natriumchlorid und Schwefel
im Reagenzglas unter dem Abzug. Reagenzgläser, in denen Zucker oder Schwefel erhitzt wurde,
werden nach dem Abkühlen im Glasabfall entsorgt.
b) Schwefel soll zunächst so geschmolzen werden, dass er nicht dunkelrot wird. Die honiggelbe
Schmelze lässt man erstarren, erhitzt wiederum und steigert die Temperatur allmählich, bis beim
Neigen des Reagenzglases der Schwefel nicht mehr ausfließt. Dann wird bis zum Sieden erhitzt und
zuletzt neigt man das Reagenzglas und lässt den Dampf auf ein Papierblatt fließen.
1.6 Analysenwaage
Die Benutzung ist erst nach erfolgter Einweisung erlaubt. Die Einweisung erfolgt durch den
Betreuer.
Der im Versuch 1.1 ausgeglühte und im Exsikkator erkaltete Tiegel wird zur Vorbereitung für den
zweiten Kurstag auf der Analysenwaage gewogen (alle angezeigten Stellen notieren) und in einem
Exsikkator aufbewahrt.
? Fragen: Was versteht man unter den Begriffen "Aggregatzustand" und "Phase" ?
Wodurch unterscheiden sich Feststoffe und Flüssigkeiten ?
Welche Produkte erhält man bei der vollständigen Verbrennung von Erdgas,
welche bei der unvollständigen Verbrennung ?
Welchen Kurvenverlauf sollte man erwarten, wenn man ein Stück Eis von 253 K
durch stetige Energiezufuhr auf 413 K erwärmt und die Temperatur als Funktion
der Energie darstellt ?
Vorbereitung für den 2. Kurstag
Bitte einen gespülten und mit der Platznummer und LA beschrifteten 100 mL-Messkolben für die
Gravimetrie ausstellen.

2. Kurstag: Chemische Formel, Phasentrennung, Gravimetrie
2.1 Synthese von Kupfersulfid
Zur quantitativen Synthese des Kupfersulfids benutzt man ca. 1 g Kupferblech (Schablonenkupfer
0,1 mm), das zu einer offenen Spirale zusammengerollt wird. Die Kupferspirale wird in den am 1.
Kurstag ausgeglühten und gewogenen Tiegel gegeben und beides zusammen auf der Analysenwaage
ausgewogen (alle angezeigten Stellen notieren). Anschließend wird in den Tiegel 1-2 g elementarer
Schwefel auf der oberschaligen Waage so eingewogen, dass die Kupferspirale gut mit Schwefel
bedeckt ist. Der Tiegel wird mit einem Deckel bedeckt und im Tondreieck über einem
Bunsenbrenner in der nicht leuchtenden Flamme vorsichtig erhitzt (Abzug!). Wenn die blaue
Flamme im Tiegel verschwunden ist, wird stärker erhitzt, bis der Tiegelboden anfängt zu glühen.
Um zu verhindern, dass sich Schwefel am Deckel niederschlägt, wird dieser zwischendurch direkt
mit dem Brenner erhitzt. Nach beendeter Reaktion lässt man den Tiegel etwas abkühlen und stellt
ihn zum vollständigen Erkalten in den Exsikkator. Nach dem Erkalten wird der Tiegel mit dem
Kupfersulfid ohne Deckel auf der Analysenwaage ausgewogen (wieder alle angezeigten Stellen
notieren).
Den Tiegel nach Abschluss des Versuchs gut reinigen.
Messergebnisse:
Masse des Tiegels ...................... g
Masse des Tiegels mit Kupfer ...................... g
Masse des Tiegels mit Kupfersulfid ...................... g
Masse des Kupfers ...................... g
Masse des Kupfersulfid ...................... g
Masse des Sulfids ...................... g
1. Berechnung der Formel
Aus dem Massenverhältnis
(Cu = Kupfer, S = Sulfid)
erhält man das Stoffmengenverhältnis
m(Cu)/m(S) =
n(Cu)/n(S) =
Chemische Formel des Kupfersulfids:
Massenverhältnis in Prozent:
Das experimentell bestimmte Massenverhältnis wird in Prozent ausgedrückt.

2.2 Abtrennung von Bariumsulfat
Man verdünnt 2-3 mL der ausstehenden Bariumchloridlösung auf etwa das Zehnfache und versetzt
mit 5 mL verdünnter Schwefelsäure. Von der entstehenden Suspension von Bariumsulfat filtriert
man je ca. ein Drittel
a) durch ein gewöhnliches Filterpapier / Platzausrüstung
b) durch ein gehärtetes Filterpapier (Blauband) / Chemikalienregal
c) den Rest klärt man durch Zentrifugieren
! Aufgabe: Vergleichen Sie die Trennwirkung fest/flüssig.
! Anmerkung: Für a) und b) verwendet man ein Filtriergestell und einen Analysentrichter, in den
man das gefaltete Filterpapier einlegt und mit destilliertem Wasser befeuchtet.
Für c) füllt man die Suspension in ein Zentrifugenglas, füllt ein zweites
Zentrifugenglas gleich hoch mit Wasser und lässt sich die Funktionsweise der
Zentrifuge von einem Assistenten erklären. Suspensionen bzw. Filtrate gehören in
den Schwermetallabfall.
2.3 Gravimetrische Bestimmung von Eisen (1. Teil: Fällen und Filtrieren des Niederschlags)
Die Analysenlösung wird im Messkolben mit destilliertem Wasser auf genau 100 mL aufgefüllt und
gut durchgeschüttelt. Mit der trockenen Vollpipette werden genau 25 mL entnommen und in einem
großen Becherglas mit destilliertem Wasser auf etwa 100 mL verdünnt. Man versetzt mit einigen
Tropfen konzentrierter HNO3 und H2O2 und kocht die Lösung kurz auf (Abzug!). Die noch warme
Lösung versetzt man unter Rühren mit einem Glasstab mit verdünntem Ammoniak, wodurch ein
voluminöser Niederschlag von Eisen(III)-hydroxid ausfällt.
Wenn auf weiteren Zusatz von Ammoniak nichts mehr ausfällt, erhitzt man bis zum Sieden, lässt
absinken und dekantiert (dekantieren = abgießen vom Bodensatz) die noch warme Flüssigkeit durch
ein Filter (Schwarzband, Chemikalienregal). Den Niederschlag wirbelt man mit heißem Wasser auf,
lässt absinken, dekantiert erneut und spült schließlich den gesamten Niederschlag auf das Filter
(Filter nur zu etwa 3/4 füllen, damit der Niederschlag beim späteren Falten nicht herausquillt).
Zur Prüfung des Waschwassers auf Chloridfreiheit gibt man etwa 1 mL des zuletzt erhaltenen
Waschwassers (nicht der Gesamtlösung) in ein Reagenzglas, versetzt mit verdünnter Salpetersäure
bis zur sauren Reaktion (mit Indikatorpapier prüfen) und gibt einige Tropfen Silbernitrat-Lösung
hinzu. Der Niederschlag ist vollständig ausgewaschen, wenn keine Trübung auftritt. Die Silberreste
gehören in den Schwermetall-Abfall.
Das noch feuchte Filterpapier wird vorsichtig so gefaltet, dass es mit dem gesamten Niederschlag
am nächsten Kurstag in einen Porzellantiegel passt und auf einem beschrifteten Uhrglas aufbewahrt.
? Frage: Weshalb benutzt man hier keine Siedesteine?
Welchen Sinn hat die Prüfung des Waschwassers auf Chloridfreiheit?

3. Kurstag: Gravimetrie, Wasserstoff, Reduktion von Kupferoxid
3.1 Gravimetrische Bestimmung von Eisen (2. Teil: Glühen und Wiegen des Niederschlags)
Das Filterpapier mit dem Eisenhydroxid-Niederschlag vom 2. Kurstag wird in den am 1. Kurstag
ausgeglühten und gewogenen Porzellantiegel gegeben, mit einem Deckel bedeckt und über kleiner
Flamme allmählich verkohlt und ohne Feuer zu fangen verglimmt (Abzug!). Schließlich verbrennt
man die Filterkohle durch stärkeres Erhitzen und glüht den Niederschlag ca. 10 min. bei ca. 700°C
(Rotglut). Nach dem Erkalten im Exsikkator (Tiegel nicht glühend in den Exsikkator stellen!) wird
der Tiegel ohne Deckel erneut gewogen.
! Aufgabe: Berechnen Sie den Eisengehalt (mg) in 100 mL der Analysenlösung und sagen Sie
das Ergebnis beim Assistenten an.
3.2 Wasserstoffentwicklung
In einem Reagenzglas werden 3 Zink-Granalien (nicht mit Sn verwechseln) mit halbkonzentrierter
Salzsäure (konzentrierte Salzsäure vorsichtig in das gleiche Volumen Wasser einfließen lassen)
versetzt, in einem zweiten Reagenzglas gibt man zu Zink und halbkonzentrierter Salzsäure einen
Tropfen Kupfersulfatlösung.
Das zweite Reagenzglas (das zu etwa 2/3 mit halbkonzentrierter Salzsäure gefüllt ist) wird mit
einem durchbohrten Stopfen verschlossen, in dessen Bohrung ein Glasrohr mit Spitze steckt. Gegen
Rückschlag der Flamme befindet sich im Glasrohr eine Stahl- oder Glaswollsicherung (vorbereitet
im Regal).
Nach Verdrängung der Luft entzündet man das austretende Gas und hält über die Flamme einen
Trichter oder eine Porzellanschale. Wenn die Gasentwicklung zu schwach ist, fügt man etwas
konzentrierte Salzsäure hinzu.
Entsorgung im Schwermetall-Abfall!
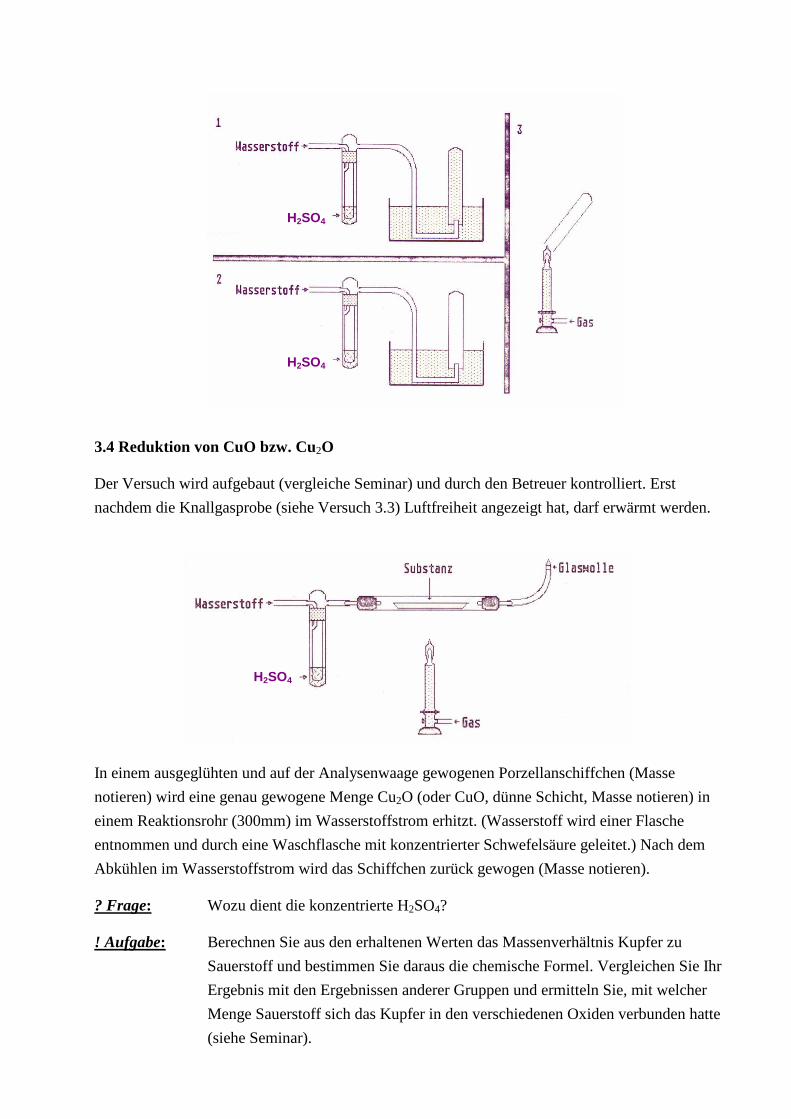
3.3 Knallgasprobe
Ein Reagenzglas wird in einer pneumatischen Wanne mit Wasserstoff gefüllt, den man einer
Wasserstoffflasche entnimmt, und mit dem Daumen verschlossen. Der Wasserstoff wird an der
Bunsenbrennerflamme entzündet, nachdem man unmittelbar vorher den Daumen entfernt hatte. Den
gleichen Versuch wiederholt man, wobei das Reagenzglas zu 2/3 bzw. 1/2 mit Luft gefüllt ist.
3.4 Reduktion von CuO bzw. Cu2O
Der Versuch wird aufgebaut (vergleiche Seminar) und durch den Betreuer kontrolliert. Erst
nachdem die Knallgasprobe (siehe Versuch 3.3) Luftfreiheit angezeigt hat, darf erwärmt werden.
In einem ausgeglühten und auf der Analysenwaage gewogenen Porzellanschiffchen (Masse
notieren) wird eine genau gewogene Menge Cu2O (oder CuO, dünne Schicht, Masse notieren) in
einem Reaktionsrohr (300mm) im Wasserstoffstrom erhitzt. (Wasserstoff wird einer Flasche
entnommen und durch eine Waschflasche mit konzentrierter Schwefelsäure geleitet.) Nach dem
Abkühlen im Wasserstoffstrom wird das Schiffchen zurück gewogen (Masse notieren).
? Frage: Wozu dient die konzentrierte H2SO4?
! Aufgabe: Berechnen Sie aus den erhaltenen Werten das Massenverhältnis Kupfer zu
Sauerstoff und bestimmen Sie daraus die chemische Formel. Vergleichen Sie Ihr
Ergebnis mit den Ergebnissen anderer Gruppen und ermitteln Sie, mit welcher
Menge Sauerstoff sich das Kupfer in den verschiedenen Oxiden verbunden hatte
(siehe Seminar).
H2SO4
H2SO4
H2SO4

4. Kurstag: Gasversuche
4.1 Gaswägung (Molmassenbestimmung) von CO2 und NH3
Zur Wägung dient eine Doppelhahnkugel (Gasmaus) mit etwa 250 mL Inhalt (genaues Volumen ist
markiert). Sie wird an der Vakuumpumpe evakuiert und auf einer oberschaligen Waage gewogen.
Der Versuch findet im Abzug statt. Nach dem Öffnen beider Hähne wird zuerst CO2, das schwerer
als Luft ist, von unten her etwa 2 Minuten lang über eine Waschflasche in die Gasmaus gefüllt.
Nach dem Schließen der Hähne wird wieder gewogen. NH3, das leichter als Luft ist, wird nach
gründlichem Evakuieren von oben her in die Gasmaus gefüllt und die Gasmaus erneut gewogen.
Anschließend wird die Gasmaus mit geöffneten Hähnen unter dem Abzug gelagert.
! Aufgabe: Berechnen Sie die Molmasse (g/mol) und die Dichte (g/L) der Gase mit Hilfe des
idealen Gasgesetzes
4.2 Volumetrische Analyse des Ammoniaks
Hinweis zur Benutzung: Kolbenprober dürfen nie fest eingespannt werden.
Vorversuch: Dichtigkeitsprüfung des Kolbenprobers. Etwa 20 mL Luft werden in den Kolbenprober
eingesaugt. Nach dem Schließen des Einsaugrohrs mit dem Hahn (bzw. dem Finger) wird die Luft
im Kolbenprober durch mehrmaligen Druck am Kolben verdichtet. Das Volumen wird vorher und
nachher bestimmt.
? Fragen: Kann man Kolbenprober bei größeren Druckdifferenzen einsetzen?
Welche Nachteile könnte ein Dichtungsmittel haben?
a) Trockenes Ammoniak, das einer Stahl-Druckgasflasche entnommen wird, füllt man über einen
Dreiweghahn in einen Kolbenprober. (Vorsicht - Kolben nicht völlig aus der Hülse drücken!) Zur
Regelung des Gasstromes leitet man das Ammoniak durch konzentrierte Natronlauge oder
Paraffinöl.

Das Hahnvolumen wird zunächst mit dem Gas gespült. Dann werden ca. 80 mL Ammoniak
eingefüllt und durch den Hahn abgesperrt. An den Dreiweghahn wird über ein Quarzrohr (8 mm
Durchmesser), das zwischen Quarzwatte Stahlwolle enthält, ein zweiter Kolbenprober
angeschlossen. Nach dem Umschalten des Hahns wird aus dem ersten Kolbenprober so viel
Ammoniak in den zweiten gedrückt, bis ersterer genau 45 mL enthält. Nach dem Umschalten des
Hahns wird der zweite Kolbenprober entleert.
Nach erneutem Umschalten des Hahns wird das Quarzrohr erhitzt und das Gas mehrere Male
langsam hindurch geleitet. Nach dem Erkalten wird das Gasvolumen abgelesen.
b) Das erhaltene Gasgemisch wird mit dem Dreiweghahn im ersten Kolbenprober abgesperrt und
das Quarzrohr mit Stahlwolle durch ein Quarzrohr mit Kupferoxid (CuO/Cu2O zur
Elementaranalyse!) ersetzt.
Nach erneuter Einstellung des Dreiweghahnes wird das Quarzrohr erhitzt und das Gasgemisch
wieder langsam über das erhitzte Kupferoxid geleitet. Nach dem Erkalten wird wieder das Volumen
des Gases abgelesen.
! Aufgabe: Formulieren Sie die Volumengleichung der Ammoniakzersetzung.
Geben Sie mit Hilfe des Satzes von Avogadro die Reaktionsgleichung und damit
die chemische Formel des Ammoniaks an.
4.3 Molmassenbestimmung von Magnesium
Ein Stück blankes Magnesiumband (etwa 40 mg = 0,04 g), das auf der Analysenwaage genau
gewogen wurde, wird an die Endfläche des Kolbens eines Kolbenprobers geklebt. Der
Kolbenprober wird dann so weit in die Hülse geschoben, dass noch etwa 10 mL Luft eingeschlossen
bleiben. Aus einem Becherglas werden 10 mL Salzsäure (wird durch Zugabe von 10 mL
konzentrierter Salzsäure zu 60 mL Wasser hergestellt.) in den senkrecht gehaltenen Kolbenprober
gesaugt und das Anfangsvolumen notiert. Anschließend wird der Kolbenprober umgedreht, so dass
das Magnesium mit der Salzsäure reagieren kann. Die Menge des entwickelten Wasserstoffs wird
gemessen (Endvolumen nach vollständiger Reaktion notieren).
! Aufgabe: Berechnen Sie die molare Masse von Magnesium.
(Gleichgewichtsdampfdruck von Wasser bei 20 °C: p(H2O) = 2335 Pa)

Auswertungshilfe zum Versuch: Molmassenbestimmung von Mg
Bei der Durchführung des Versuches ergeben sich die folgenden Zustände des Kolbenprobers (als
Anfangs- und Endzustand bezeichnet):
a) Näherung ohne Berücksichtigung des Wasserdampfs:
Anfangsvolumen VA = VFlüssigkeit + VLuft (das Volumen von Mg wird vernachlässigt)
Endvolumen VE = VFlüssigkeit + VLuft + VH2
Volumen des Wasserstoffs (Reaktionsvolumen): VH2 = VE - VA
p · VH2 = nH2 · R · T (p in Pa, V in m3, T in K, R = 8,314 Pa m3 mol-1 K-1)
nH2 entspricht nMg
b) Näherung mit Berücksichtigung des Wasserdampfs:
Anfangsvolumen: VA = VFlüssigkeit + VLuft + VAH2O(g) (das Volumen von Mg wird vernachlässigt)
Endvolumen: VE = VFlüssigkeit + VLuft + VEH2O(g) + VH2
wichtig: VAH2O(g) ? VE
H2O(g) wir nennen: VH2O(g) = VEH2O(g) - V
AH2O(g)
Reaktionsvolumen: Vges = VE - VA = VH2 + VH2O(g)
Stoffmenge im Reaktionsvolumen: nges = nH2 + nH2O(g)
Druck: pges = pH2 + pH2O es folgt: pH2 = pges - pH2O
pges · Vges = nges · R · T bzw. (pH2 + pH2O) · Vges = (nH2 + nH2O) · R · T
pH2 · Vges = nH2 · R · T bzw. (pges - pH2O) · Vges = nH2 · R · T
nH2 entspricht nMg

5. Kurstag: Flüssigkeiten, Verteilungssatz, Kristallisation
5.1 Mischbarkeit von Flüssigkeiten verschiedener Polarität
In einem Reagenzglas mischt man Wasser und Ethanol in etwa gleichen Mengen. Der Versuch wird
mit Wasser/Petrolether und Ethanol/Petrolether wiederholt.
5.2 Netzwirkung und Grenzflächenspannung
Eine Nadel, Büroklammer oder ähnliches legt man mit einer Pinzette oder einem Papierstreifen
vorsichtig auf die Wasseroberfläche in einer Petrischale oder einem breiten Becherglas.
Am Rand lässt man anschließend einige Tropfen einer Seifen- oder Spülmittellösung einfließen.
5.3 Lichtabsorption, Verteilungssatz
Man gibt eine kleine Spatelspitze! Iod in ein Reagenzglas und gibt 3 mL Wasser dazu. Dann fügt
man einige Tropfen KI-Lösung hinzu und schüttelt (Beobachtung?).
Nernstscher Verteilungssatz: Die Hälfte der Lösung (Iod muss vollständig gelöst sein!) wird in ein
zweites Reagenzglas überführt (gleiche Füllhöhe in beiden Gläsern). Reagenzglas 1 wird mit
Wasser auf die doppelte Füllhöhe verdünnt. Beide Reagenzgläser werden mit je 1 mL Petrolether
versetzt und geschüttelt (Korkstopfen verwenden). Die (untere) wässrige Phase wird jeweils mit
einer Pasteurpipette abgenommen und in je ein weiteres Reagenzglas überführt. Diese beiden
Lösungen werden nochmals mit je 1 mL Petrolether ausgeschüttelt und die wässrige Phase
abgenommen. Vergleichen Sie die Farbintensität der vier Petrolether-Lösungen vor einem weißen
Hintergrund, indem Sie einmal quer und einmal längs zum Reagenzglas blicken.
Beersches Gesetz: Nehmen Sie die organische Lösung mit der intensivsten Farbe und verteilen Sie
diese Lösung zu gleichen Teilen auf drei Reagenzgläser. Die zweite dieser Proben versetzen Sie mit
dem gleichen Volumen Petrolether, die dritte mit dem doppelten Volumen Petrolether und schütteln
jeweils gut durch. Betrachten Sie die Proben quer und längs zum Reagenzglas vor einem weißen
Hintergrund. Beobachtung? Schlussfolgerung?
5.4 Polare Verbindungen im inhomogenen elektrischen Feld
Aus einer Bürette lässt man Wasser in dünnem Strahl in ein breites Becherglas ausfließen. Dicht
unterhalb des Hahns hält man einen elektrisch aufgeladenen Stab (geriebener Hartgummi- oder
Glasstab) in die Nähe des Wasserstrahls.
Der Versuch wird jeweils mit Ethanol und Petrolether wiederholt.

5.5 Übersättigte Lösungen
In drei sauberen Reagenzgläsern werden je 1,5 g Natriumacetat-Trihydrat in 0,5 mL Wasser durch
Aufkochen gelöst, wobei durch den aufsteigenden Dampf auch alle Kristalle, die etwa im oberen
Teil des Reagenzglases haften, gelöst und herabgespült werden müssen. Die Reagenzgläser lässt
man mit Wattepropfen verschlossen im Reagenzglasgestell erschütterungsfrei erkalten. Im ersten
Reagenzglas löst man die Kristallisation durch Einwerfen eines kleinen Natriumacetat-Kristalls aus,
im zweiten durch Reiben eines Glasstabs an der Innenwand (Reagenzglas während der
Kristallisation in der Hand behalten). Im dritten, das als Kontrolle dient, muss die Kristallisation
ausbleiben.
5.6 Umkristallisieren
(Vierergruppe)
Ein durch Mischkristallbildung mit Kaliumpermanganat violett gefärbtes Kaliumperchlorat soll
durch Umkristallisieren gereinigt werden.
Vorbereitung: Ein mit der Platznummer beschrifteter Analysentrichter wird in den Trockenschrank
gestellt.
Etwa 10-15 g des Salzes werden in der eben ausreichenden Menge kochenden Wassers in einem
400 mL Becherglas gelöst und von evtl. ungelösten Resten durch einen angewärmten Trichter
(Trockenschrank) rasch abfiltriert. Nach dem Abkühlen saugt man das auskristallisierte Salz auf
einem Glasfilter ab. Das Filtrat wird dunkelviolett, das Salz heller sein als die Ausgangssubstanz.
Eine kleine Probe des Salzes bewahrt man in einem Rollrandglas auf, die Hauptmenge löst man wie
oben in kochendem Wasser und filtriert nach dem Abkühlen erneut durch den Glasfilter. Man
entnimmt wieder eine kleine Salzprobe und wiederholt das Verfahren noch zweimal.
Die vier erhaltenen Präparate sind vorzuzeigen.
Geräte, die durch Braunstein (MnO2) verunreinigt sind, werden mit schwefeliger Säure (H2SO3)
gereinigt.
! Anmerkung: Mit diesem Versuch bereits früh beginnen.
Zum Lösen kochendes Wasser nach und nach zugeben und nicht mehr Wasser
verwenden, als zum Auflösen gerade eben ausreicht!

6. Kurstag: Löslichkeit, Lösungswärme, homogenes und heterogenes Gleichgewicht
6.1 Temperaturabhängigkeit der Löslichkeit
In einem Zentrifugenglas wird eine kleine Menge Bleinitrat (Spatelspitze, ca. 1 mm) in Wasser
gelöst. Dazu gibt man solange tropfenweise Kaliumiodid-Lösung, bis kein weiterer PbI2-
Niederschlag mehr entsteht. Der Niederschlag wird abzentrifugiert, die überstehende Lösung wird
verworfen. Man füllt das Zentrifugenglas zur Hälfte mit Wasser, schüttelt durch und erhitzt es im
Wasserbad, bis sich das PbI2 gelöst hat. Dann lässt man die Lösung in Ruhe abkühlen.
Beobachtung?
Bleireste in den Schwermetall-Abfall geben.
6.2 Lösungswärme von CaCl2 und CaCl2 · 6 H2O
Zwei 100 mL-Bechergläser beschickt man mit je 25 mL Wasser, dessen Temperatur man misst. In
das eine bringt man 14 g wasserfreies CaCl2, in das andere 28 g CaCl2 · 6 H2O (auf der
Oberschalenwaage abwiegen) und sorgt durch Umrühren für möglichst rasches Auflösen der Salze.
Dann misst man erneut die Temperatur.
! Aufgabe: Erklären Sie die Beobachtungen.
6.3 Bestimmung der Löslichkeit von Kaliumperchlorat
Je ca. 60 mL einer bei 20°C gesättigten Lösung von KClO4 (steht aus) werden in kleine
Bechergläser gefüllt und eines der Bechergläser in ein Eisbad von 0°C gestellt. Die Lösung im
zweiten Becherglas wird auf ca. 80°C erwärmt und so lange festes KClO4 zugefügt, bis die Lösung
gesättigt ist. Diese Lösung wird anschließend in den auf 60°C temperierten Trockenschrank gestellt.
Beide Lösungen lässt man längere Zeit unter gelegentlichem Schütteln stehen, wobei sich ein festes
Salz ausscheidet. Ist vollständiger Temperatur- und Löslichkeitsausgleich eingetreten, dekantiert
man von beiden Lösungen etwa 10 g in vorher gekennzeichnete und gewogene Porzellanschalen
und wiegt die Lösungen aus (Oberschalenwaage!). Eine evtl. entstandene Kristallhaut auf der
Oberfläche der Lösungen wird vorher mit einem Filterpapier entfernt bzw. beim Dekantieren
zurückgehalten. Die beiden Proben werden im Trockenschrank ohne Spritzverluste bei 120°C
eingedampft und getrocknet und wieder gewogen. Der Massenverlust gibt die Wassermenge, die
Masse des Rückstandes die gelöste KClO4-Menge an.
! Aufgabe: Man gebe den KClO4-Gehalt beider Lösungen in Milligramm pro 100 g Lösung
an und vergleiche die erhaltenen Werte mit denen der anderen Studierenden.
Berechnen Sie aus Ihren Messwerten das Löslichkeitsprodukt von KClO4 bei 0°C
und bei 60°C.

6.4 Verschiebung eines homogenen Gleichgewichts
Zu 5 mL Wasser, die in einem kleinen Becherglas mit einem Tropfen Salzsäure angesäuert werden,
gibt man 0,5 mL FeCl3-Lösung und 1,5 mL NH4SCN-Lösung (beide ca. 1 mol/L). Man verdünnt die
tiefrote Lösung mit Wasser, bis eine in ein Reagenzglas umgefüllte Probe in der Durchsicht quer zur
Längsachse nur noch schwach gelb aussieht. Man verteilt die Lösung auf zwei Reagenzgläser und
fügt zur ersten Probe einige Tropfen der Eisen(III)-Salzlösung, zur zweiten einige Tropfen der
Ammoniumthiocyanatlösung hinzu. Die Lösungen werden anschließend im Schwermetallabfall
entsorgt.
! Aufgabe: Erklärung des Versuchsergebnisses:
a) nach dem Prinzip von Le Chatelier
b) mit dem Massenwirkungsgesetz
6.5 Verschiebung eines heterogenen Gleichgewichts
Man verteilt eine gesättigte Lösung von KClO4 auf drei Reagenzgläser. Zum ersten gibt man eine
kleine Menge gesättigte NaCl-Lösung, zum zweiten ein Viertel des Volumens an gesättigter KCl-
Lösung und zum dritten einige Tropfen 60 bis 70%ige Perchlorsäure.
Löslichkeitsprodukt von KClO4 bei Zimmertemperatur: 9 · 10-3 mol2/L2.
? Frage: Wie viel Gramm KClO4 befinden sich in 100 mL gesättigter Lösung?
Vorbereitung für den 7. Kurstag
Bitte einen gespülten und mit der Platznummer und LA beschrifteten 100 mL-Messkolben für die
Gravimetrie ausstellen.

7. Kurstag: Komplexe
7.1 Fällung von Silberhalogeniden
7.1.1 Zu je einer mit HNO3 angesäuerten HCl-*, NH4Cl- und BaCl2-Lösung gibt man einige
Tropfen AgNO3-Lösung. Ferner versetzt man stark verdünnte KBr-* bzw. KI-Lösung* mit AgNO3-
Lösung.
! Anmerkung: * HCl, KBr und KI im Zentrifugenglas statt im Reagenzglas umsetzen,
Niederschläge aufheben und gleich Versuch 7.2 anschließen.
7.1.2 Außerdem gibt man zu je einer Lösung von Chloralhydrat (Cl3C-CH(OH)2) und
Kaliumchlorat (KClO3) Silbernitrat-Lösung.
7.1.3 Zu einer mit HNO3 angesäuerten AgNO3-Lösung gibt man etwas K2CrO4-Lösung und
anschließend NaCl-Lösung.
7.1.4 Zu einer neutralen AgNO3-Lösung (evtl. mit etwas Hydrogencarbonat versetzen) gibt man
K2CrO4-Lösung und anschließend NaCl-Lösung.
7.2 Verhalten der Silberhalogenide gegenüber Ammoniak
7.2.1 Man fällt jeweils etwas AgCl, AgBr und AgI im Zentrifugenglas, zentrifugiert ab und wäscht
mit Wasser aus; dies wiederholt man einmal und zentrifugiert zum Schluss wieder ab. Man schüttelt
die Niederschläge mit etwa 1 mL verdünntem NH3 einige Zeit auf.
Man zentrifugiert die verbliebenen Niederschläge ab und übergießt sie mit konzentriertem NH3.
Während AgI auch hier praktisch unlöslich ist, löst sich das AgBr auf. Diese Proben hebt man für
den nächsten Versuch auf.
! Vorsicht: Ammoniakalische Silbersalzlösungen dürfen niemals längere Zeit stehen bleiben!
Explosionsgefahr!
7.2.2 Zerstörung des Silberdiamminkomplexes
a) Der Silberdiamminkomplex kann durch Säure zerstört werden. Hierfür säuert man die
ammoniakalische AgCl-Lösung vorsichtig mit verdünnter Salpetersäure an.
b) Alternativ kann der Silberdiamminkomplex durch Erhitzen zerstört werden. Einen Teil der
ammoniakalischen Silberchlorid-Lösung lässt man im heißen Wasserbad stehen, bis die
Hauptmenge des Ammoniaks vertrieben und die Lösung getrübt ist.
c) Des Weiteren kann der Silberdiamminkomplex durch Zugabe von Iodid zerstört werden. Zu
einem Teil der ammoniakalischen Silberbromid-Lösung gibt man einige Tropfen KI-Lösung, wobei
sich wieder gelbes AgI bildet.

7.3 Komplexe des Kupfers
7.3.1 Einige Tropfen Kupfersulfatlösung gibt man im Reagenzglas unter Umschütteln zu
Ammoniak-Lösung. Ebenso gibt man Kupfersulfat-Lösung zu konzentrierter Salzsäure.
7.3.2 Man schüttelt Kupfercarbonat (CuCO3 · Cu(OH)2) mit Wasser und verteilt die
Aufschlämmung auf zwei Reagenzgläser. Man versetzt die eine Probe mit Ammoniak, die andere
mit Salzsäure.
7.3.3 Je eine Spatelspitze weißes, wasserfreies CuSO4 bringe man in Wasser und Ammoniak-
Lösung.
? Frage: Durch welche Partikel werden die Farb- bzw. Löslichkeitsänderungen verursacht?
7.4 Wassernachweis
Im Reagenzglas erwärmt man vorsichtig festes CoCl2 · 6 H2O. Beim Liegenlassen in feuchter
Atmosphäre oder durch Zugabe von Wasser wird das Salz wieder rot. Man löst es in möglichst
wenig Wasser auf und tränkt damit ein Stück Filterpapier. Das Papier wird über der
Bunsenbrennerflamme (Abstand halten, Papier nicht anzünden!) vollständig getrocknet.
7.5 Gravimetrische Bestimmung von Nickel
Ein mit der Platznummer beschrifteter Glasfiltertiegel wird eine halbe Stunde bei 120°C im
Trockenschrank getrocknet und nach dem Abkühlen im Exsikkator auf der Analysenwaage
gewogen.
Die Analysenlösung wird im Messkolben auf genau 100 mL aufgefüllt und gut durchgeschüttelt.
Mit der trockenen Vollpipette werden genau 25 mL entnommen, in einem Becherglas mit
destilliertem Wasser auf 150 mL verdünnt und zum Sieden erhitzt. Man nimmt vom Feuer und gibt
unter dem Abzug ca. 50 mL 1%ige alkoholische Diacetyldioxim-Lösung hinzu. Dann wird
tropfenweise verdünnte NH3-Lösung zugesetzt bis die Lösung schwach ammoniakalisch ist
(pH 8-9) und etwa eine halbe Stunde stehen gelassen. Anschließend wird der Niederschlag durch
den zuvor gewogenen Glasfiltertiegel filtriert und mit heißem Wasser nachgewaschen. Falls
Niederschlag durch den Filter läuft, wird die Lösung mit etwas Ammoniak versetzt und erneut
filtriert.
Der Niederschlag (Ni(C4H7O2N2)2) wird etwa zwei Stunden bei 120°C im Trockenschrank
getrocknet und nach dem Erkalten (im Exsikkator) auf der Analysenwaage ausgewogen
(Massenanteil des Ni im Niederschlag: w(Ni) = 0,2032). Der Nickelgehalt (mg) in 100 mL
Analysenlösung ist beim Assistenten anzusagen.
Danach den Tiegel mit warmer Salzsäure reinigen!

8. Kurstag: Leitfähigkeit von Lösungen
8.1 Leitfähigkeit von Lösungen
Der Leitfähigkeitsprüfer wird grundsätzlich mit destilliertem Wasser gespült. Anschließend wird die
Leitfähigkeit (in der angegebenen Reihenfolge) von jeweils einer 1 M Lösung folgender Substanzen
bestimmt: Harnstoff, Rohrzucker (oder Glucose), Essigsäure*, Ammoniak, Natriumacetat*,
Natriumchlorid*, Salzsäure und Natronlauge. Beim Wechsel der Lösung ist der Leitfähigkeitsprüfer
jedes Mal gründlich mit destilliertem Wasser zu spülen. Man notiere die gemessenen Stromstärken.
! Anmerkung: * Diese Lösungen für den folgenden Versuch aufheben
8.2 Konzentrationsabhängigkeit der Leitfähigkeit
Man bestimmt die Leitfähigkeit von jeweils einer Natriumchlorid-, einer Natriumacetat- und einer
Essigsäurelösung der Konzentration 0,5 mol/L (die Lösungen werden durch Verdünnen der
entsprechenden Lösungen aus Versuch 8.1 im Verhältnis 1:1 hergestellt). Anschließend wird weiter
bis zur Konzentration 0,1 mol/L verdünnt und erneut die Leitfähigkeit bestimmt.
8.3 Abhängigkeit der Leitfähigkeit von der Ionenladung
Man misst die Leitfähigkeit von Natriumchlorid- und Magnesiumsulfat-Lösungen der
Konzentration 0,001 mol/L. Die NaCl-Lösung wird durch weiteres Verdünnen der bereits
hergestellten Lösung bereitet. Zur Herstellung der MgSO4-Lösung verdünnt man eine 0,01 mol/L-
Lösung, für die man die entsprechende Menge MgSO4 · 7 H2O (Bittersalz) einwiegt.
8.4 Leitfähigkeit schwacher Säuren und ihrer Salze
Es wird die Leitfähigkeit von Ammoniak- und Essigsäure-Lösungen der Konzentration 2 mol/L
gemessen. Dann mischt man gleiche Volumina dieser Lösungen und misst erneut.
? Frage: Muss man die Volumenänderung berücksichtigen?
8.5 Aktivität
In einem Gefäß (rechteckige pneumatische Wanne oder großes Becherglas) sollen zwei zueinander
parallele Elektroden (Platten oder Kohlestäbe) bis zum Boden führen. Man verbindet die eine
Elektrode direkt, die andere über ein Amperemeter mit einer Wechselstromquelle.
Das Gefäß wird zu ca. 1/4 mit NaCl-Lösung, c(NaCl) = 1 mol/L, gefüllt. Nach dem Ablesen der
Stromstärke wird (bei konstanter Spannung) mit destilliertem Wasser auf etwa das doppelte
Volumen verdünnt, gemischt und erneut abgelesen. Man setzt nochmals ungefähr die gleiche Menge
destilliertes Wasser zu und liest wieder ab.
? Frage: In welcher Weise sollte sich die Stromstärke ändern?

9. Kurstag: Säuren, Basen, Titration
9.1 Indikatoren
9.1.1 In einem Reagenzglas werden einige Tropfen Bromthymolblau nacheinander mit wenig
verdünnter HCl, NaOH, HNO3 und NH3 versetzt.
9.1.2 Bromthymolblau, Methylorange und Phenolphthalein werden in verschiedenen
Reagenzgläsern jeweils mit Säure, Lauge, destilliertem Wasser und Leitungswasser versetzt. Die
Ergebnisse werden in einer Tabelle zusammengefasst.
9.2 Bestimmung des pH-Werts mit Indikatorpapier
Man bestimmt mit Universalindikatorpapier den pH-Wert von (selbst hergestellten) Lösungen mit
folgenden Konzentrationen:
c(HCl) = 0,1 mol/L, c(HCl) = 0,0001 mol/L, c(KNO3) = 1 mol/L, destilliertes Wasser,
Leitungswasser, c(NaOH) = 0,001 mol/L, c(NH4Cl) = 1 mol/L und c(CH3COONa) = 1 mol/L
Die entsprechenden Lösungen werden durch Verdünnen der ausstehenden 2M HCl bzw. 2M NaOH
und durch Berechnung der Einwaagen der Salze hergestellt.
9.3 Titrationskurve einer starken Säure
Man titriert in folgender Weise: Eine trockene oder mit der Lauge mehrfach gespülte fettfreie
Bürette wird mit verdünnter Natronlauge (ausstehende 2 N NaOH) bis etwas über den Null-
Teilstrich gefüllt. Weil der Flüssigkeitsspiegel durch langsam an der Wandung herabfließende
Lauge noch 1 bis 2 Minuten lang steigt ("Nachlauf"), wartet man etwas, ehe man die überschüssige
Lauge durch den Hahn in ein kleines Becherglas abfließen lässt. Hierbei achtet man darauf, dass die
gesamte Bürette inklusive Hahn und Auslaufspitze luftblasenfrei mit Lauge gefüllt ist.
10 mL verdünnte Salzsäure werden in einen Erlenmeyerkolben pipettiert und auf etwa 100 mL
verdünnt. Der pH-Wert wird mit Indikatorpapier bestimmt. Aus einer Bürette versetzt man die
Säure mit 1 mL verdünnter Natronlauge, schwenkt gut um und bestimmt erneut den pH-Wert.
Diesen Vorgang wiederholt man bis zu einem Zusatz von insgesamt 15 mL NaOH. Die erhaltenen
Wertepaare werden in eine Tabelle eingetragen und auf Millimeterpapier (evtl. kariertem Papier)
grafisch dargestellt.
9.4 Titrationskurve einer schwachen Säure
Wie im vorherigen Versuch werden 10 mL verdünnte Essigsäure auf 100 mL verdünnt und mit
insgesamt 15 mL Natronlauge titriert. Ist der pH-Wert = 5,5 erreicht, fügt man die Lauge nur noch
in Anteilen von 0,5 mL hinzu. Ab pH-Wert = 10 wird die Lauge wieder in Anteilen von 1 mL
zugefügt. Die erhaltenen Wertepaare werden wieder in eine Tabelle eingetragen und auf
Millimeterpapier grafisch dargestellt.

! Aufgabe: Erläutern Sie den Verlauf der Titrationskurve der Essigsäure!
9.5 Titrationskurve einer mehrwertigen Säure
Eine 10 mL-Phosphorsäureprobe wird in einem 400 mL Becherglas auf etwa 100 mL verdünnt und
mit Natronlauge titriert. Die Natronlauge wird solange in Anteilen von jeweils 0,5 mL zugesetzt, bis
der pH-Wert nicht mehr steigt sondern praktisch konstant bleibt. Der pH-Wert wird
potentiometrisch mit einer Glaselektrode gemessen. Zum Durchmischen der Lösung benutzt man
einen Magnetrührer mit Rührfisch. Die Ergebnisse werden grafisch dargestellt und diskutiert.
! Achtung: Glaselektrode nicht längere Zeit in stark alkalischen Lösungen stehen lassen!
9.6 Sauer und basisch reagierende Salzlösungen
Untersuchen Sie den pH-Wert der Lösungen von Natriumacetat, Ammoniumchlorid,
Natriumcarbonat und Natriumhydrogencarbonat sowie einer Aluminiumsalz-Lösung.
9.7 Titration verschiedener Säuren
Je eine 10 mL-Probe verdünnter H2SO4, HAc (CH3COOH) und HCl (ausstehende Lösungen) wird
mit verdünnter NaOH (ausstehend 2 N NaOH) gegen Bromthymolblau oder Phenolphthalein als
Indikator titriert.
Die Bürette wird wie unter 9.3 mit Natronlauge gefüllt. Man pipettiert jeweils eine 10 mL-Probe der
verdünnten Säuren in einen Erlenmeyerkolben, verdünnt mit destilliertem Wasser auf etwa 100 mL,
gibt 2 bis 3 Tropfen Bromthymolblau oder Phenolphthalein zu und lässt nunmehr die Natronlauge
in dünnem Strahl aus der Bürette in das darunter gehaltene Gefäß mit der Säureprobe einfließen (die
Pipette soll trocken sein oder mehrfach mit der jeweiligen Säure ausgespült werden).
Durch dauerndes Umschwenken vermischt man die Flüssigkeiten unmittelbar beim Einfließen. Das
nahe Ende der Reaktion erkennt man daran, dass sich in der gelb gefärbten Lösung um die
Eintrittsstelle der Lauge ein kleiner, allmählich größer werdender Hof von blauer Farbe bildet, bei
Phenolphthalein als Indikator entsteht ein magentafarbener Hof in einer farblosen Lösung. Jetzt gibt
man die Natronlauge nur noch tropfenweise unter gutem Umschütteln hinzu, spült die Wandungen
des Gefäßes mit destilliertem Wasser ab, um anhaftende Laugentropfen mit der Hauptmenge zu
vereinigen, und titriert die Lösung bis zum Farbumschlag. Nun liest man den Verbrauch an
Natronlauge an der Bürette ab.
Tragen Sie die ermittelten und die zu erwartenden Werte in eine Tabelle ein; notieren Sie außerdem,
ob der Farbumschlag gegen Ende der Titration schnell oder langsam erfolgte.

9.8 Herstellen einer NaOH-Normallösung
Man kocht 600 mL destilliertes Wasser aus und lässt abkühlen. Man wiegt soviel festes NaOH ab
(Oberschalenwaage), wie zur Herstellung von 500 mL mit c(NaOH) = 0,1 mol/L notwendig ist.
Man löst die NaOH mit dem abgekochten Wasser in einem 500 mL Messkolben und füllt bis zur
Eichmarke mit Wasser auf (gut durchmischen!).
Die hergestellte Lösung wird in einer Polyethylen-Flasche aufbewahrt (beschriftet mit Laborplatz-
Nummer und NaOH ca. 0,1 mol/L, genaue Konzentration im Protokoll).
Vorbereitung für den 10. Kurstag
Bitte einen gespülten und mit der Platznummer und LA beschrifteten 100 mL-Messkolben für die
H2SO4/Na2SO4-Analyse ausstellen.

10. Kurstag: Ionenaustauscher, Pufferlösungen
10.1 Einstellen einer NaOH-Normallösung
Zur genauen Bestimmung der Konzentration der unter 9.8 hergestellten NaOH-Lösung werden ca.
120 mg Oxalsäure-Dihydrat (C2O4H2·2 H2O) auf der Analysenwaage exakt abgewogen (alle
angezeigten Stellen notieren). Die Oxalsäure wird in einem 300 mL Erlenmeyerkolben in ca.
100 mL Wasser gelöst. Die zu bestimmende Natronlauge wird in eine trockene (oder mit der Lauge
2 mal ausgewaschene) Bürette gegeben und die Oxalsäure gegen Phenolphthalein titriert. Die
Bestimmung gegen Oxalsäure wird einmal wiederholt (Doppelbestimmung). Die Ergebnisse beider
Bestimmungen müssen innerhalb einer Abweichung von 1% übereinstimmen; wenn nicht, sind die
Bestimmungen zu wiederholen.
Berechnung der NaOH-Konzentration:
Molmasse
Masse abgewogene
)OH2HO(C)OH2HO(C
= )HO(C2242
2242242 ←
←⋅⋅
M
mn
volumenTitrations
(NaOH))HO(C 2
= )NaOH( 242
←V
nc
10.2 Quantitative Bestimmung einer H2SO4/Na2SO4-Lösung
10.2.1 Bestimmung des H2SO4-Gehaltes
Zunächst wird die im Messkolben vorliegende Analysenlösung mit destilliertem Wasser auf genau
100 mL aufgefüllt und durch Schütteln gut homogenisiert. Man pipettiert zwei 25 mL-Proben
(Vollpipette) in je einen 300 mL Erlenmeyerkolben und füllt mit destilliertem Wasser auf etwa
100 mL auf. Man gibt etwas Phenolphthalein als Indikator hinzu und titriert die Proben sorgfältig
gegen die eingestellte Natronlauge.
10.2.2 Bestimmung des Na2SO4-Gehaltes durch Ionenaustausch
Aus dem 100 mL Messkolben pipettiert man eine dritte 25 mL-Probe (Vollpipette) in ein kleines
Becherglas. Zur Bestimmung des Na-Gehaltes lässt man das überstehende Waschwasser der
Ionenaustauschersäule bis dicht über das obere Ende des Füllmaterials ab (Achtung, Füllmaterial
nicht trocken laufen lassen!) und schließt den Hahn. Man gibt die dritte 25 mL-Probe auf die Säule,
spült mit etwas destilliertem Wasser das Becherglas und die Wandung des Ionenaustauschers nach
und lässt die überstehende Wassermenge tropfenweise in einen 300 mL Erlenmeyerkolben ab. Die
Durchlaufgeschwindigkeit soll ungefähr 3-5 Tropfen pro Sekunde betragen.
Man gibt wieder etwas destilliertes Wasser auf die Säule und lässt die überstehende Wassermenge

erneut in den Erlenmeyerkolben ab. Diesen Vorgang wiederholt man, bis die Lösung neutral
ausläuft (auslaufende Tropfen mit pH-Papier prüfen). Dies ist nach etwa 200 mL Eluat
(durchgelaufene Flüssigkeit) der Fall. Bitte anschließend die Säule mit einem Strich markieren
(Anzahl der aufgebrachten Proben). Das gesamte Eluat wird anschließend mit der eingestellten
NaOH sorgfältig gegen Phenolphthalein titriert.
Berechnen Sie die Konzentration und die Stoffmenge sowie die Masse (bezogen auf den
Kolbeninhalt) der H2SO4/Na2SO4-Probe.
Die bestimmten Massen (in mg) an H2SO4 und Na2SO4 sind beim Assistenten anzusagen.
10.3 Pufferwirkung
Zwei Reagenzgläser füllt man mit je 15 mL Wasser, zwei weitere mit je 15 mL einer Mischung aus
etwa gleichen Volumenteilen verd. Essigsäure und Natriumacetat-Lösung. Alle Lösungen versetzt
man mit je 2 bis 3 Tropfen Methylrot-Lösung und bestimmt ihren pH-Wert mit Indikatorpapier. Zu
einer Wasserprobe und einer Essigsäure/Acetat-Mischung gibt man je einen Tropfen Salzsäure;
entsprechend versetzt man die beiden anderen Proben mit je einem Tropfen Natronlauge und
bestimmt jeweils wieder den pH-Wert. Notieren Sie neben den pH-Werten auch die Farben der
Lösungen.
10.4 Bestimmung der Dissoziationskonstante über die Puffergleichung
10.4.1 25 mL verdünnte Essigsäure titriert man mit (nicht eingestellter) verdünnter Natronlauge
(Phenolphthalein als Indikator). Eine weitere Probe von 25 mL versetzt man mit genau der Hälfte
der bei der Titration verbrauchten Lauge. Nach dem Durchmischen bestimmt man den pH-Wert
dieser Lösung.
10.4.2 25 mL verdünnte Ammoniak-Lösung titriert man mit verdünnter Salzsäure (Methylrot als
Indikator). Eine zweite 25 mL-Probe versetzt man mit genau der Hälfte der Säuremenge, die bei der
Titration bis zum Umschlag verbraucht wurde. In dieser Lösung bestimmt man nach dem
Durchmischen ebenfalls den pH-Wert.
! Aufgabe: Wie groß sind die pK-Werte von Essigsäure und Ammoniak (Puffergleichung)?

10.5 Herstellen einer Pufferlösung mit definiertem pH-Wert
Eine Pufferlösung mit einem auf eine Dezimale genauen pH-Wert wird wie beim im Seminar
berechneten Beispiel hergestellt. Jede Gruppe bekommt einen eigenen pH-Wert zugewiesen.
Bei der Herstellung eines Phosphatpuffers sollten die Konzentrationen nicht viel mehr als 0,1 mol/L
betragen.
100 mL der hergestellten Pufferlösung werden dem Assistenten zur Kontrolle in einem Becherglas
abgegeben.
! Aufgabe: Vergleichen Sie den Umschlagbereich eines pH-Indikators mit einem
Puffersystem.
? Frage: Welchen Wirkungsbereich besitzt ein Puffer ungefähr?
Vorbereitung für den 11. Kurstag
Bitte zwei gespülte und mit der Platznummer und LA beschriftete 100 mL-Messkolben für die
HCl/CH3COOH-Analyse und die H3PO4-Analyse ausstellen.

11. Kurstag: Leitfähigkeitstitration
11.1 Leitfähigkeitstitration
11.1.1 Man pipettiert 5 mL verdünnte Salzsäure in ein 400 mL-Becherglas, verdünnt mit 100 mL
destilliertem Wasser und durchmischt die Lösung während des Versuches mit einem Magnetrührer.
Nach dem Eintauchen des Leitfähigkeitsprüfers (Stromstärke notieren) gibt man aus einer mit
verdünnter NaOH gefüllten Bürette die Lauge in Anteilen von genau 1 mL zu und misst nach jedem
Laugenzusatz die Stromstärke. Die erhaltenen Werte werden in eine Tabelle eingetragen und auf
Millimeterpapier grafisch dargestellt. Nach zehnmaligem Zusatz von 1 mL Lauge bricht man den
Versuch ab.
Eine zweite 5 mL-Probe der verdünnten Salzsäure wird im Erlenmeyerkolben gegen
Bromthymolblau oder Phenolphthalein mit NaOH titriert. Vergleichen Sie den erhaltenen Wert mit
der grafischen Darstellung.
11.1.2 Der Versuch 11.1.1 wird entsprechend mit Essigsäure durchgeführt.
11.1.3 Der Messkolben mit der Analysenlösung (HCl/CH3COOH-Gemisch) wird mit destilliertem
Wasser auf genau 100 mL aufgefüllt und gut durchmischt.
Zwei 25 mL-Proben (Vollpipette) werden in jeweils einen 300 mL-Erlenmeyerkolben gegeben, auf
etwa 100 mL verdünnt und gegen einen geeigneten Indikator mit der eingestellten NaOH (vom
10. Kurstag) titriert.
Eine weitere 25 mL-Probe (Vollpipette) wird in ein 400 mL-Becherglas pipettiert, mit ausreichend
destilliertem Wasser aufgefüllt und wie in den Versuchen 11.1.1. und 11.1.2 ebenfalls mit der
eingestellten NaOH titriert. Man gibt so lange jeweils 1 mL Natronlauge zu, bis man etwa 5-7 mL
über dem vorher ermittelten Titrationsvolumen liegt. Die erhaltenen Werte werden in eine Tabelle
eingetragen und auf Millimeterpapier grafisch aufgetragen. Bestimmen Sie aus dieser Grafik die
Anteile an Salzsäure und Essigsäure in Ihrer Probe (vergleiche Seminar).
Berechnen Sie aus den erhaltenen Daten die Stoffmenge sowie die Masse (bezogen auf den
Kolbeninhalt) der HCl/CH3COOH-Probe.
Die bestimmten Massen (in mg) an HCl und CH3COOH sind beim Assistenten anzusagen.

11.2 Quantitative Bestimmung von Phosphorsäure
Der Messkolben mit der Analysen-Lösung wird auf genau 100 mL aufgefüllt und gut durchmischt.
Eine 25 mL-Probe (Vollpipette) wird in einen 300 mL-Erlenmeyerkolben pipettiert, mit
destilliertem Wasser auf etwa 100 mL verdünnt und mit der eingestellten Natronlauge gegen
Methylorange oder Bromphenolblau titriert. Eine zweite 25 mL-Probe wird in gleicher Weise gegen
Thymolphthalein als Indikator titriert.
Umschlagbereiche:
Methylorange: pH 3,6 - 4,3; Bromphenolblau: pH 3,0 - 4,6; Thymolphthalein: pH 9,3 - 10,5.
Vergleichen Sie die Ergebnisse der beiden Titrationen und berechnen Sie die Masse der
Phosphorsäure in 100 mL der Probenlösung. Das Ergebnis ist beim Assistenten anzusagen.
? Frage: Wonach richtet sich die Wahl des Indikators? (Vergleiche Versuch 9.5)

12. Kurstag: Reaktionsgeschwindigkeit, Katalyse, Reaktionen von Metallen
12.1 Geschwindigkeit der Reaktion von Fe3+ mit Thiosulfat
12.1.1 Gleiche Volumenteile einer FeCl3-Lösung und einer Na2S2O3-Lösung mit jeweils
c = 1 mol/L werden im Reagenzglas vermischt. Die Zeit, die bis zum Verschwinden der rotvioletten
Farbe vergeht, wird notiert.
2 Fe3+ + 2 S2O32- → 2 Fe2+ + S4O6
2-
12.1.2 Versuch 12.1.1 wird wiederholt, jedoch im Wasserbad bei 50°C.
? Frage: Welchen Einfluss hat die Temperatur auf die Reaktionsgeschwindigkeit?
12.1.3 Homogene Katalyse
Versuch 12.1.1 wird wiederholt, jedoch wird der FeCl3-Lösung zuvor ein Tropfen CuSO4-Lösung
zugesetzt.
12.2 Heterogene Katalyse: Katalytische Zersetzung von H2O2
Zerfallsreaktion: 2 H2O2(aq) → 2 H2O + O2(g)
H2O2 zersetzt sich nur langsam. Die Reaktion wird jedoch durch Katalysatoren (z.B. MnO2)
beschleunigt.
Zu je 50 mL H2O2-Lösung (3 %), die sich in der Saugflasche befindet, fügt man in getrennten
Versuchen 0,1, 0,5 und 1,0 g MnO2-Pulver und einmal 1,0 g MnO2 gekörnt hinzu, verschließt sofort
das Reaktionsgefäß mit Manschette und Stopfen und fängt den sich entwickelnden Sauerstoff auf
(Messzylinder, pneumatische Wanne). Ermittelt wird die Zeit t, die benötigt wird, um ein
bestimmtes Volumen Sauerstoff (z.B. 30 mL) freizusetzen. Pro Ansatz werden zwei Messungen
durchgeführt. Die Versuchsergebnisse werden in einer Tabelle zusammengestellt.
12.3 Autokatalyse
Eine Lösung von Natriumoxalat, c(Na2C2O4) ≈ 0,05 mol/L, wird mit verdünnter Schwefelsäure
angesäuert. Die Lösung wird auf zwei Reagenzgläser verteilt. In einem Reagenzglas löst man eine
kleine Spatelspitze MnSO4. Zu beiden Lösungen gibt man aus einer Tropfpipette je einen Tropfen
KMnO4-Lösung. Beobachten Sie die Geschwindigkeit der Entfärbung des Permanganats. Zu der
Lösung ohne MnSO4-Zusatz wird nochmals ein Tropfen KMnO4-Lösung gegeben. Beobachtung?

12.4 Reaktionen von Metallen mit Säuren
12.4.1 Im Reagenzglas werden ein Stück Magnesiumband, eine Zinkgranalie, etwas Nickelpulver
und einige blanke Kupferspäne jeweils mit verdünnter Salzsäure übergossen.
12.4.2 Einige Kupferspäne werden im Reagenzglas mit konzentrierter Salpetersäure versetzt
(Abzug!).
12.5 Spannungsreihe
In Reagenzgläsern stellt man sich je drei bzw. vier Lösungen der unten angegebenen Metallsalze her
(jeweils eine Spatelspitze). Diese Lösungen prüft man auf ihre Reaktion mit Eisendraht (oder einem
Eisennagel), Kupferblech (oder Kupferdraht), Magnesiumband und Zinkgranalien. Man vermerke
die Ergebnisse in einer Tabelle:
AgNO3 FeSO4 CuSO4 MgCl2 ZnSO4
________________________________________________________________
Fe —
Cu —
Mg —
Zn —
Man ordne die Metalle nach zunehmender Oxidationswirkung ihrer Ionen.
12.6 Korrosion von Eisen (1. Teil: Durchführung)
Chemikalien: KNO3(s), ethanolische Phenolphthalein-Lösung, K3[Fe(CN)6]-Lösung (1 %),
Gelatine (Agar), 5 Eisennägel, Kupferdraht, 1 Stück Zinkblech. Ein Eisennagel wird rechtwinklig
gebogen, ein anderer wird zur Hälfte verzundert, d.h. im Brenner kurzzeitig zum Glühen erhitzt.
Die Metallteile dürfen nicht mit den Händen berührt werden!
Durchführung: Im Becherglas fügt man zu 100 mL destilliertem Wasser 1 g KNO3, 2 mL
Phenolphthalein-Lösung, 4 mL K3[Fe(CN)6]-Lösung und 4 g Gelatine und erhitzt unter Rühren
vorsichtig, bis gerade eben eine klare Lösung entstanden ist. In eine Petrischale legt man die fünf
Eisennägel, ohne dass sie sich berühren. Von den geraden, unbehandelten Nägeln wird einer
teilweise mit Kupferdraht umwickelt und ein zweiter über einen kurzen Cu-Draht mit einem
Stückchen Zinkblech verbunden. Die abgekühlte, nur noch leicht warme Gelatine-Lösung wird in
die Petrischale gegossen. Die Petrischale wird abgedeckt, beschriftet und an einen ruhigen Ort
gestellt.
Am 13. Kurstag wird der Versuch ausgewertet (Beschreibung und Auswertung des Versuchs im
Protokoll zum 13. Versuchstag).

13. Kurstag: Redoxreaktionen
13.1 Korrosion von Eisen (2. Teil: Auswertung)
Der am 12. Kurstag angesetzte Versuch wird ausgewertet.
Anmerkungen:
Blaue Zonen zeigen Bereiche der Fe-Korrosion; die bei der Primäroxidation Fe → Fe2+ + 2 e-
entstandenen Fe2+ -Ionen geben mit K3[Fe(CN)6] die Berliner-Blau Reaktion.
Mit Cu bzw. Zn entstehen galvanische Elemente; Fe-Cu = Lokalelement. Bei Fe-Cu-Zn dient Zn als
Opferanode; die bei der Opferreaktion Zn → Zn2+ + 2 e- entstandenen Zn2+ -Ionen bilden gelbes
Zn3[Fe(CN)6]2, die zum edleren Fe (Kathode) geflossenen Elektronen reduzieren Sauerstoff:
O2(aq) + 2 H2O + 4e- → 4 OH- (aq). Die OH--Ionen werden durch das Phenolphthalein
(Rotfärbung) angezeigt.
Was beobachtet man am geknickten und am verzunderten Nagel?
Schritte der Korrosion:
Primäroxidation (Fe als Lokalanode):
Fe(s) → Fe2+(aq) + 2 e-
Reduktion von O2 (Lokalkathode):
O2(aq) + 2 H2O + 4 e- → 4 OH- (aq)
Fällung:
2 Fe2+(aq) + 2 OH- (aq) → Fe(OH)2(s)
Sekundärreaktion:
2 Fe(OH)2(s) + O2 → 2 FeO(OH) + H2O
↓
Fe2O3·H2O (Rost)
13.2 Reaktionen von Natrium
Vorsicht: Schutzbrille, Na nicht mit den Händen berühren!
13.2.1 Von dem unter Petroleum aufbewahrten Natrium-Metall schneidet man auf trockener
Filtrierpapier-Unterlage ein maximal linsengroßes Stück ab, den Rest bringt man sofort wieder in
das Vorratsgefäß. Das abgeschnittene Stück wirft man in eine mit Wasser gefüllte große
Porzellanschale (Abzug!).Nach Ablauf der Reaktion prüft man den pH-Wert des Wassers.
Formulieren Sie die Reaktionsgleichung.
13.2.2 Versuch 12.3.1 wird wiederholt, jedoch mit Ethanol an Stelle von Wasser.

13.3 Silberspiegel
In einem Reagenzglas gibt man zu Silbernitratlösung (AgNO3) tropfenweise Ammoniak (NH3), bis
sich der entsprechende Niederschlag (Trübung) gerade gelöst hat. Dann versetzt man mit Glucose
oder Haushaltszucker, erhitzt das Reagenzglas im Wasserbad und lässt es einige Zeit stehen. Nicht
vergessen! Weshalb? Sobald sich der Silberspiegel gebildet hat, säuert man die Lösung an und
entsorgt sie im Schwermetallabfall.
13.4 Oxidation von Fe2+
In einem Reagenzglas wird eine Spatelspitze FeSO4. in Wasser gelöst und die Lösung auf drei
Reagenzgläser verteilt.
13.4.1 Zu einer der Lösungen gibt man Thiocyanat-Lösung (SCN-). Zur zweiten Lösung gibt man
etwas Chlor- oder Bromwasser, verkocht den Überschuss an Chlor bzw. Brom unter dem Abzug
und fügt dann Thiocyanat-Lösung hinzu.
13.4.2 Die dritte Lösung versetzt man mit verdünnter Schwefelsäure, erhitzt und fügt dann
tropfenweise Kaliumpermanganatlösung hinzu, bis eine rötliche Farbe bestehen bleibt.
! Anmerkung: Der letzte Versuch eignet sich zur oxidimetrischen Titration einer Eisenlösung.
Wie erkennt man den Äquivalenzpunkt (Endpunkt)?
13.5 Reduktion von Kupferoxid
Ein schmaler Streifen Kupferblech wird in der Brennerflamme erhitzt und heiß in ein Reagenzglas
mit etwas Ethanol getaucht.
13.6 Oxidation von Alkohol mit Chromat
Im Reagenzglas versetzt man etwa 0,5 mL Ethanol mit Kaliumchromatlösung und verdünnter
H2SO4 und erwärmt im Wasserbad.
13.7 Redox-Amphoterie
In zwei Reagenzgläsern säuert man H2O2 mit etwas verdünnter Schwefelsäure an und versetzt
a) mit Kaliumiodidlösung und Stärke-Lösung
b) mit Kaliumpermanganat-Lösung
13.8 Dis- und Synproportionierung
Wenig Bromwasser versetzt man im Reagenzglas tropfenweise bis zur Entfärbung mit verdünnter
Natronlauge, anschließend gibt man tropfenweise verdünnte Schwefelsäure hinzu.

14. Kurstag: Elektrochemie
14.1 Galvanisches Element
Man stellt sich ein galvanisches Element aus Zn/Zn2+ und Cu/Cu2+ her. Die Spannung wird mit
einem empfindlichen Voltmeter gemessen. Zur Herstellung der Halbzellen werden die Schenkel
eines U-Rohres (mit Fritte) mit den jeweiligen Lösungen (c = 1 mol/L) gefüllt, in die man die
entsprechenden Elektroden eintaucht.
14.2 Konzentrationskette
In einen Schenkel eines U-Rohres mit Fritte füllt man CuSO4-Lösung ein (c(CuSO4) = 1 mol/L), in
den anderen eine ammoniakalische Lösung (etwas 1 mol/L CuSO4 mit konzentriertem Ammoniak
verdünnt), taucht jeweils Kupfer-Elektroden ein und misst die Spannung.
! Aufgabe: Erklären Sie die Funktionsweise einer Konzentrationskette und berechnen Sie aus
der gemessenen Spannung das Konzentrationsverhältnis der beiden
Kupferlösungen.
14.3 Zersetzungsspannung von CuSO4
Man ermittelt das Stromstärke-Spannungsdiagramm einer CuSO4-Lösung (c(CuSO4) = 1 mol/L)
unter Verwendung einer Platinnetzelektrode. Schaltung zuerst überprüfen lassen: Man misst bei
vorgegebener Spannung (Gleichspannung) die resultierende Stromstärke.

Hinweise zum Protokoll
Zu jedem Kurstag ist ein Protokoll anzufertigen. Die Protokolle müssen spätestens eine Woche nach
dem entsprechenden Kurstag abgegeben werden. Die testierten Protokolle für alle Kurstage sind die
Voraussetzung für die Anerkennung der Studienleistung „Praktikum“ im Modul NBP1.
Aufbau und Inhalt des Protokolls
• Die Protokolle sind in Einzelarbeit zu erstellen (sauber und lesbar).
• Jedes Protokoll muss ein Deckblatt enthalten, auf dem Kapitel und Datum des Praktikumstags,
Name des/der Studierenden und Laborplatznummer verzeichnet sind.
• Alle Versuche, Aufgaben und Fragen müssen in der Reihenfolge des Skripts vollständig und
sorgfältig bearbeitet werden.
• Die Bearbeitung eines Versuchs umfasst die Angabe der Versuchsnummer, die Durchführung, die
Auswertung und die Diskussion des Versuchs.
• Unter Durchführung des Versuchs genügt es, sich auf das Praktikumsskript zu beziehen, falls Sie
den Versuch exakt nach Anleitung durchgeführt haben. Wurde die Versuchsdurchführung in
einzelnen Punkten abgewandelt, müssen diese Punkte im Protokoll ausgeführt werden.
• Unter Auswertung des Versuchs werden die Versuchsergebnisse beschrieben. Die Auswertung
enthält die Beobachtungen, die während des Versuchs gemacht wurden, alle Reaktionen inklusive
Reaktionsgleichungen sowie (falls gefordert) die Rechenergebnisse mit den zugehörigen Formeln.
Rechenergebnisse sind mit einer plausiblen Anzahl von signifikanten Stellen anzugeben!
• Unter Diskussion des Versuchs werden Versuchsteile miteinander verglichen, die Ergebnisse mit
Hilfe von Literaturstudien begründet und diskutiert sowie mögliche Fehler analysiert.
• Bei quantitativen Analysen müssen unter dem Punkt Auswertung und Diskussion jeweils das von
Ihnen gefundene Analysenergebnis, der zugehörige Rechengang, das korrekte (vom Assistenten
mitgeteilte) Ergebnis sowie eine Fehlerbetrachtung enthalten sein.
• Die entsprechenden Seiten aus dem Laborjournal mit den Versuchsbeobachtungen und dem
Vortestat sind mit dem jeweiligen Protokoll zusammen abzugeben.
• Die Protokolle sind in unpersönlicher Schreibweise zu formulieren.
• Auf jeder Seite müssen 5 cm Rand für die Anmerkungen bei der Korrektur freigehalten werden.
• Alle Abbildungen und Tabellen sind mit Bild- bzw. Tabellenunterschriften zu versehen. Werden
im Protokoll nicht selbst erstellte Abbildungen verwendet, muss ihre Quelle zitiert werden.
• Am Ende der Protokolle sind die verwendeten Informationsquellen mit den vollständigen
bibliografischen Angaben zu zitieren.
• Achten Sie auf korrekte Grammatik und Rechtschreibung!
• Nur vollständige Protokolle werden testiert!
Related Documents











