282 P eripheral arterial disease (PAD) is a complication of sys- temic atherosclerosis that affects >10 million people in the United States alone, where occlusions reduce perfusion to the leg(s) causing pain with walking, pain at rest, and ischemic ulcers that put the limb at risk for amputation. 1,2 Surgical and catheter-based revascularization therapies are preferred first line of treatment for patients with the most extreme form of PAD, but many patients are poor candidates or have no revas- cularization option. 1,2 Thus, ≈200 000 amputations/yr occur in the United States alone, with PAD being the major cause, and no medical therapies are available to increase leg perfusion. 3 In symptomatic PAD patients, a total occlusion in the inflow vessels means that resting or maximal leg blood flow is de- pendent on the extent of the angiogenic response to ischemia. 4 Vascular endothelial growth factor-A (VEGF-A) is a key member in the VEGF superfamily that can bind and activate vascular endothelial growth factor receptor (VEGFR)1 and VEGFR2, to modulate physiological and pathological angio- genesis. 5 VEGF-A–mediated VEGFR2-signaling activation is largely viewed as the dominant receptor tyrosine kinase signal- ing to induce angiogenesis. 6,7 VEGFR1 plays important roles in several cardiovascular diseases including experimental PAD 8– 10 ; however, the processes that regulate VEGFR1 activation or VEGFR1-specific downstream signaling events are not clear. Human clinical trials aimed at inducing VEGF-A– mediated VEGFR2 signaling in PAD via VEGF-A delivery to ischemic muscle were not successful. 11–13 Many factors may have contributed for this lack of beneficial effect, but it is Molecular Medicine © 2016 American Heart Association, Inc. Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.116.309516 Rationale: Atherosclerotic-arterial occlusions decrease tissue perfusion causing ischemia to lower limbs in patients with peripheral arterial disease (PAD). Ischemia in muscle induces an angiogenic response, but the magnitude of this response is frequently inadequate to meet tissue perfusion requirements. Alternate splicing in the exon- 8 of vascular endothelial growth factor (VEGF)-A results in production of proangiogenic VEGF xxx a isoforms (VEGF 165 a, 165 for the 165 amino acid product) and antiangiogenic VEGF xxx b (VEGF 165 b) isoforms. Objective: The antiangiogenic VEGF xxx b isoforms are thought to antagonize VEGF xxx a isoforms and decrease activation of VEGF receptor-2 (VEGFR2), hereunto considered the dominant receptor in postnatal angiogenesis in PAD. Our data will show that VEGF 165 b inhibits VEGFR1 signal transducer and activator of transcription (STAT)-3 signaling to decrease angiogenesis in human and experimental PAD. Methods and Results: In human PAD versus control muscle biopsies, VEGF 165 b: (1) is elevated, (2) is bound higher (versus VEGF 165 a) to VEGFR1 not VEGFR2, and (3) levels correlated with decreased VEGFR1, not VEGFR2, activation. In experimental PAD, delivery of an isoform-specific monoclonal antibody to VEGF 165 b versus control antibody enhanced perfusion in animal model of severe PAD (Balb/c strain) without activating VEGFR2 signaling but with increased VEGFR1 activation. Receptor pull-down experiments demonstrate that VEGF 165 b inhibition versus control increased VEGFR1–STAT3 binding and STAT3 activation, independent of Janus-activated kinase-1)/Janus-activated kinase-2. Using VEGFR1 +/− mice that could not increase VEGFR1 after ischemia, we confirm that VEGF 165 b decreases VEGFR1–STAT3 signaling to decrease perfusion. Conclusions: Our results indicate that VEGF 165 b prevents activation of VEGFR1–STAT3 signaling by VEGF 165 a and hence inhibits angiogenesis and perfusion recovery in PAD muscle. (Circ Res. 2017;120:282-295. DOI: 10.1161/CIRCRESAHA.116.309516.) Key Words: alternative splicing ■ amputation ■ anti-angiogenic VEGF-A isoforms ■ ischemia ■ peripheral artery disease Original received July 8, 2016; revision received November 22, 2016; accepted December 14, 2016. In November 2016, the average time from submission to first decision for all original research papers submitted to Circulation Research was 15.7 days. From the Cardiovascular Research Center (V.C.G., M.C., B.H.A.), Department of Biology (A.K.), and Department of Cardiovascular Medicine, University of Virginia, Charlottesville (B.H.A.). Presented in part at the Scientific Sessions of the American Heart Association, Orlando, FL, November 7–11, 2015. The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA. 116.309516/-/DC1. Correspondence to Dr Brian H. Annex, Cardiovascular Research Center, Division of Cardiovascular Medicine, University of Virginia Health System, PO Box 800158, Charlottesville, VA 22908. E-mail [email protected] VEGF 165 b Modulates Endothelial VEGFR1–STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease Vijay Chaitanya Ganta, Min Choi, Anna Kutateladze, Brian H. Annex by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from by guest on September 1, 2017 http://circres.ahajournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

282
Peripheral arterial disease (PAD) is a complication of sys-temic atherosclerosis that affects >10 million people in the
United States alone, where occlusions reduce perfusion to the leg(s) causing pain with walking, pain at rest, and ischemic ulcers that put the limb at risk for amputation.1,2 Surgical and catheter-based revascularization therapies are preferred first line of treatment for patients with the most extreme form of PAD, but many patients are poor candidates or have no revas-cularization option.1,2 Thus, ≈200 000 amputations/yr occur in the United States alone, with PAD being the major cause, and no medical therapies are available to increase leg perfusion.3 In symptomatic PAD patients, a total occlusion in the inflow vessels means that resting or maximal leg blood flow is de-pendent on the extent of the angiogenic response to ischemia.4
Vascular endothelial growth factor-A (VEGF-A) is a key member in the VEGF superfamily that can bind and activate vascular endothelial growth factor receptor (VEGFR)1 and VEGFR2, to modulate physiological and pathological angio-genesis.5 VEGF-A–mediated VEGFR2-signaling activation is largely viewed as the dominant receptor tyrosine kinase signal-ing to induce angiogenesis.6,7 VEGFR1 plays important roles in several cardiovascular diseases including experimental PAD8–
10; however, the processes that regulate VEGFR1 activation or VEGFR1-specific downstream signaling events are not clear.
Human clinical trials aimed at inducing VEGF-A–mediated VEGFR2 signaling in PAD via VEGF-A delivery to ischemic muscle were not successful.11–13 Many factors may have contributed for this lack of beneficial effect, but it is
Molecular Medicine
© 2016 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.116.309516
Rationale: Atherosclerotic-arterial occlusions decrease tissue perfusion causing ischemia to lower limbs in patients with peripheral arterial disease (PAD). Ischemia in muscle induces an angiogenic response, but the magnitude of this response is frequently inadequate to meet tissue perfusion requirements. Alternate splicing in the exon-8 of vascular endothelial growth factor (VEGF)-A results in production of proangiogenic VEGFxxxa isoforms (VEGF165a, 165 for the 165 amino acid product) and antiangiogenic VEGFxxxb (VEGF165b) isoforms.
Objective: The antiangiogenic VEGFxxxb isoforms are thought to antagonize VEGFxxxa isoforms and decrease activation of VEGF receptor-2 (VEGFR2), hereunto considered the dominant receptor in postnatal angiogenesis in PAD. Our data will show that VEGF165b inhibits VEGFR1 signal transducer and activator of transcription (STAT)-3 signaling to decrease angiogenesis in human and experimental PAD.
Methods and Results: In human PAD versus control muscle biopsies, VEGF165b: (1) is elevated, (2) is bound higher (versus VEGF165a) to VEGFR1 not VEGFR2, and (3) levels correlated with decreased VEGFR1, not VEGFR2, activation. In experimental PAD, delivery of an isoform-specific monoclonal antibody to VEGF165b versus control antibody enhanced perfusion in animal model of severe PAD (Balb/c strain) without activating VEGFR2 signaling but with increased VEGFR1 activation. Receptor pull-down experiments demonstrate that VEGF165b inhibition versus control increased VEGFR1–STAT3 binding and STAT3 activation, independent of Janus-activated kinase-1)/Janus-activated kinase-2. Using VEGFR1+/− mice that could not increase VEGFR1 after ischemia, we confirm that VEGF165b decreases VEGFR1–STAT3 signaling to decrease perfusion.
Conclusions: Our results indicate that VEGF165b prevents activation of VEGFR1–STAT3 signaling by VEGF165a and hence inhibits angiogenesis and perfusion recovery in PAD muscle. (Circ Res. 2017;120:282-295. DOI: 10.1161/CIRCRESAHA.116.309516.)
Key Words: alternative splicing ■ amputation ■ anti-angiogenic VEGF-A isoforms ■ ischemia ■ peripheral artery disease
Original received July 8, 2016; revision received November 22, 2016; accepted December 14, 2016. In November 2016, the average time from submission to first decision for all original research papers submitted to Circulation Research was 15.7 days.
From the Cardiovascular Research Center (V.C.G., M.C., B.H.A.), Department of Biology (A.K.), and Department of Cardiovascular Medicine, University of Virginia, Charlottesville (B.H.A.).
Presented in part at the Scientific Sessions of the American Heart Association, Orlando, FL, November 7–11, 2015.The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.
116.309516/-/DC1.Correspondence to Dr Brian H. Annex, Cardiovascular Research Center, Division of Cardiovascular Medicine, University of Virginia Health System, PO
Box 800158, Charlottesville, VA 22908. E-mail [email protected]
VEGF165b Modulates Endothelial VEGFR1–STAT3 Signaling Pathway and Angiogenesis in Human and Experimental
Peripheral Arterial DiseaseVijay Chaitanya Ganta, Min Choi, Anna Kutateladze, Brian H. Annex
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 283
clear that induction of functional blood vessel formation in ischemic muscle is a formidable challenge, and an inadequate understanding of VEGF–VEGFR signaling is one major pos-sible explanation. Our understanding of the VEGF ligands has become more complex with the recognition of antiangiogenic VEGF-A isoforms family, termed VEGF
xxxb (VEGF
165b),
which occurs from alternate splicing in exon-8 of VEGF-A (a 6 amino acid frame shift from CDKPRR [proangiogenic] to PLTGKD [antiangiogenic]).14,15 Replacement of positively charged arginine residues in proangiogenic isoforms with neutral aspartic acid and lysine in antiangiogenic isoforms is predicted to decrease VEGFR2 activation16 and angiogenesis. On the basis of the existing paradigm on VEGF
165b–VEGFR2
interactions, it was predicted that VEGF165
b inhibition would increase the bioavailability of proangiogenic VEGF-A isoforms to VEGFR2 for receptor-mediated angiogen-esis. However, our data will show that VEGF
165b modulates
VEGFR1 and a novel VEGFR1 signal transducer and activa-tor of transcription (STAT3) signaling pathway that promotes angiogenesis and perfusion recovery in PAD.
MethodsPlease see the Materials and Methods section in the Online Data Supplement.
ResultsIn Human and Experimental PAD Muscle, VEGF165b Induction Occurs With Ischemia and Is Associated With Lower VEGFR1, Not VEGFR2 ActivationIn a previous study of gastrocnemius skeletal muscle from PAD and non-PAD control subjects (age and sex matched), we reported that VEGF
165b was higher in PAD muscle using an
ELISA.14 Kikuchi et al17 more recently showed that peripheral blood monocytes from PAD versus control patients express significantly higher VEGF
165b by immunoblotting.17 We first
confirmed that VEGF-A antibody, raised against full-length VEGF-A protein, detects both proangiogenic (VEGF
165a) and
antiangiogenic (VEGF165
b) VEGF-A isoforms, and an iso-form-specific VEGF
xxxb antibody raised against the 6 amino
acids of exon8b in VEGFxxx
b isoforms is extremely specific to recombinant VEGF
165b and does not detect recombinant
VEGF165
a (Online Figure IA), which was in accordance to previous publications.15,18 In cell-free ELISA, although VEGF-A was able to detect both recombinant VEGF
165a and
VEGF165
b isoforms (at equal concentrations) with similar af-finity, VEGF
165b antibody was not able to detect recombinant
Nonstandard Abbreviations and Acronyms
EC endothelial cells
HEK293-VR1 VEGFR1-expressing HEK293 cells
HEK293-VR2 VEGFR2-expressing HEK293 cell
HLI hindlimb ischemia
HSS hypoxia serum starvation
HUVEC human umbilical vein endothelial cells
IGA ischemic gastrocnemius muscle
Jak Janus-activated kinase
NAM nonischemic adductor muscle
NGA nonischemic gastrocnemius muscle Nor
PAD peripheral arterial disease
STAT3 signal transductor and activator of transcription-A
V165b-Ab VEGF165b antibody
VEGFR1 vascular endothelial growth factor receptor 1
VEGFR2 vascular endothelial growth factor receptor 2
WT wild type
What Is Known?
• Alternate splicing in exon-8 of vascular endothelial growth factor (VEGF)-A gene produces 2 isoform families that are typically described as proangiogenic (C-terminal amino acid sequence CDKPRR) and anti-angiogenic (C-terminal amino acid sequence SLTRKD).
• The antiangiogenic VEGF-A isoforms were predicted to inhibit proan-giogenic VEGF-A isoforms’ ability to activate VEGFR2.
What New Information Does This Article Contribute?
• Decreasing the antiangiogenic VEGF-A levels increased VEGFR1 acti-vation with no change in VEGFR2 activation in experimental peripheral arterial disease (PAD).
• Increasing the antiangiogenic VEGF-A isoform decreased VEGFR1 acti-vation and increased VEGFR2 activation.
• Inhibition of antiangiogenic VEGF-A isoforms increased VEGFR1–STAT3 binding interactions to enhance STAT3 activation that was independent of Janus-activated kinase-1/Janus-activated kinase-2 activation.
The antiangiogenic VEGF-A isoforms exist in human muscle, but how these antiangiogenic VEGF-A isoforms modulate ischemic muscle recovery in PAD is not clear. In human and experimental
PAD, increased levels and binding of antiangiogenic VEGF-A iso-forms to VEGFR1 correlated with decreased VEGFR1 activation. Inhibition of antiangiogenic VEGF-A isoforms in preclinical PAD models increased binding of proangiogenic VEGF-A to VEGFR1 to increase VEGFR1–STAT3 interactions and signaling result-ing in enhanced ischemic muscle perfusion. VEGFR2 activation is necessary to revascularize ischemic muscle, and the antian-giogenic VEGF-A isoforms were predicted to inhibit VEGFR2. Our work alters current thinking about VEGFR-mediated angiogenesis by showing that antiangiogenic VEGF-A isoforms are inhibitors/blockers for VEGFR1 (not VEGFR2) and that removal of the in-hibitor increased VEGFR1 activation to improve ischemic muscle perfusion. Our data provide the first evidence that strategies de-signed to inhibit the antiangiogenic isoforms activate VEGFR1–STAT3 signaling. Furthermore, our finding that the antiangiogenic VEGF-A isoforms can inhibit proangiogenic VEGF-A isoforms even at 10× lower levels may also explain the failure of previous hu-man trials designed to increase the proangiogenic VEGF-A in ischemic muscle. Hence, therapies aimed at inhibiting antian-giogenic VEGF-A isoforms may indeed provide a better strategy to promote perfusion in PAD through VEGFR1 and not through VEGFR2 activation.
Novelty and Significance
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

284 Circulation Research January 20, 2017
VEGF165
a even at 20× higher concentration than VEGF165
b (data not shown), indicating that VEGF
165b antibody is highly
specific for VEGFxxx
b isoforms and hence was used to exam-ine VEGF
165b levels and function in our experiments.
We next quantified total VEGF-A and VEGF165
b levels in PAD and normal muscle biopsies by ELISA. In PAD muscle bi-opsies, we observed a decrease in total VEGF-A levels (normal: 166.3±27.8 versus PAD: 135.6±5.5 pg/mg; Figure 1A, left), with an increase in the VEGF
xxxb fraction (normal: 81.6±9.5 ver-
sus PAD: 98.1±12.7 pg/mg; Figure 1A, middle) compared with normal muscle biopsies. Subtracting VEGF
xxxb fraction from
total VEGF-A showed that the VEGFxxx
a fraction was signifi-cantly reduced (≈2×; P=0.04) in PAD muscle biopsies compared with normal (normal: 84.7±21.6 versus PAD: 37.5±8.0 pg/mg; Figure 1A, right). We confirmed the specificity of our VEGF-A and VEGF
xxxb ELISA data from PAD and normal muscle bi-
opsies by immunoblot analysis of VEGF-A and VEGF165
b. In immunoblot analysis, although no significant differences were observed in total VEGF-A levels between PAD and normal muscle biopsies, VEGF
165b levels were significantly induced
(P<0.03) in PAD muscle biopsies compared with normal (Online Figure IB). Because VEGF
165b antibody detects only
VEGF165
b and the VEGF-A antibody detects both pro- and anti-angiogenic isoforms, we derived a ratio of VEGF
165b:VEGF-A,
which showed that VEGF165
b is induced ≈3X in PAD muscle versus normal (Online Figure IB), indicating that in human PAD muscle, total VEGF-A includes ≈75% VEGF
165b fraction and
≈25% VEGF165
a fraction. The ability of VEGF165
b ELISA to de-tect other VEGF
xxxb isoforms,19 including VEGF
165b, offers one
of the possible explanations for differences in VEGF165
b levels in our ELISA and immunoblot analysis. On the basis of our ELISA and immunoblot analysis, we conclude that in PAD muscle a tilt in the balance of proangiogenic versus antiangiogenic VEGF-A isoforms toward antiangiogenic VEGF
165b isoforms results in
decreased angiogenesis.Because VEGFR2 is the dominant proangiogenic receptor,
we next examined the degree of VEGFR2 activation (Y1175) in human PAD versus control. Replacement of Y1173/Y1175 residue with phenylalanine results in diminished en-dothelial cell development and embryonic death20 similar to
Figure 1. A, Vascular endothelial growth factor (VEGF)-A and VEGF165b ELISA in normal (NL) and peripheral arterial disease (PAD) muscle biopsies. Table on the right shows the pg/mg of total protein levels of VEGF-A, VEGF165b, and VEGF165a in normal (n=5) and PAD (n=7) muscle biopsies. B, VEGFR2 was immunoprecipitated from normal and PAD tissue biopsies and analyzed for bound total VEGF-A and VEGF165b by immunoblotting. C, VEGFR1 was immunoprecipitated from normal (n=6) and PAD (n=10) tissue biopsies and analyzed for bound VEGF-A and VEGF165b by immunoblotting. Unpaired t test. Full-length Western blot images are presented in Online Figure XII.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 285
embryonic lethality in VEGFR2 global knockout mice,21 indi-cating a critical role for Y1175 in regulating VEGFR2 down-stream signaling. Hence, we focused on examining the status of VEGFR2-Y1175 activation in our experiments. Immunoblot of the VEGFR2-immunoprecipitated fraction showed signifi-cantly higher VEGFR2 activation (Y1175) in the PAD muscle biopsies versus control (P=0.009; Figure 1B). To examine whether increased VEGFR2 phosphorylation correlates with changes in binding of VEGF-A and VEGF
165b to VEGFR2, we
used the same VEGFR2-immunoprecipitated samples from normal and PAD muscle biopsies and analyzed for VEGF
165b
and total VEGF-A. Despite lower total VEGF-A (VEGF165
a) and higher VEGF
165b levels in PAD versus normal muscle bi-
opsies, no significant differences in VEGF165
b or total VEGF-A binding was observed in VEGFR2-immunoprecipitated com-plexes between PAD and normal muscle biopsies (Figure 1B), suggesting that higher VEGF
165b levels in PAD versus normal
do not inhibit VEGFR2 activation.Using a similar strategy and the same cohort, we next
examined the degree of VEGFR1 activation in human PAD versus control. Immunoblot of VEGFR1-immunoprecipitated fraction showed significantly decreased VEGFR1 activation (Y1333) in PAD muscle biopsies versus normal (P=0.003; Figure 1C). To examine whether decreased VEGFR1-phosphorylation correlates with changes in binding of
VEGF-A and VEGF165
b to VEGFR1, we used the same VEGFR1-immunoprecipitated samples and analyzed for bound VEGF
165b and total VEGF-A. VEGFR1 pull-down
experiments from PAD and normal muscle biopsies showed a significant increase in VEGF
165b levels bound to VEGFR1
with no significant change in total VEGF-A in PAD muscle biopsies compared with normal (P<0.03; Figure 1C). Thus, VEGFR1 activation inversely correlated with increased VEGF
165b binding to VEGFR1.
We then used unilateral femoral artery ligation and resec-tion (hindlimb ischemia [HLI]) in Balb/c mice, as a preclini-cal experimental model for severe PAD (critical limb ischemia PAD).22–26 To determine whether VEGF
165b induction correlates
with decreased activation of VEGFR1 versus VEGFR2 in isch-emic muscle compared with nonischemic muscle, we analyzed cells from nonischemic and ischemic whole muscle tissue by flow cytometry (see Online Figure II for VEGF
165b, pVEG-
FR1Y1333
/VEGFR1, pVEGFR2Y1175
/VEGFR2). We observed ≈4× increase (P<0.05) in VEGF
165b-expressing cells in ischemic
gastrocnemius muscle (IGA; nonischemic gastrocnemius mus-cle [NGA]: 0.9±0.5% versus IGA: 3.8±1.0% of total live cells) compared with NGA (Figure 2A) and ≈2× increase (P<0.05) in adductor muscle from ischemic leg (IAM) compared with adductor muscle from nonischemic leg (NAM; NAM: 4.3±2.1 versus IAM: 9.2±1.9%; Online Figure IIIA). We also observed
Figure 2. A, Flow cytometry of vascular endothelial growth factor (VEGF)165b expression in total live cells from NGA (nonischemic gastrocnemius muscle) and IGA (ischemic gastrocnemius muscle), n=4. B, Flow cytometry of VEGFR1 activation (mean fluorescence intensity [MFI] of PVR1Y1333/VR1) in total live cells from NGA and IGA, n=4. C, Flow cytometry of VEGFR2 activation (MFI of PVR2Y1175/VR2) in total live cells from NGA and IGA, n=4. D, Flow cytometry of VEGF165b expression in endothelial cells (ECs; gated on CD31+CD45−) from NGA and IGA, n=4. E, Flow cytometry of VEGFR1 activation in ECs (gated on CD31+CD45−) from NGA and IGA. n=4. F, Flow cytometry of VEGF165b-expressing ECs (CD31+CD45−VEGF165b
+ve) with VEGFR1 activation, n=4. G, Flow cytometry of VEGF165b nonexpressing ECs (CD31+CD45−VEGF165b
−ve) with VEGFR1 activation, n=4. H, VEGFR1 was immunoprecipitated from NGA and IGA and examined for bound VEGF-A and VEGF165b by immunoblotting, n=4. Full-length Western blot images are presented in Online Figure XII. A–H, Unpaired t test. I, Necrosis scores. Necrosis was evaluated according to previously established necrosis-scoring system ranging from 0 to 4 at days 1 and 3. Unpaired, nonparametric Mann–Whitney test. J, Perfusion recovery measured noninvasively by quantifying microvascular blood flow by laser Doppler. Repeated-measures ANOVA with Dunnett post-test. K, CD31 immunostaining in Balb/c IGA treated with VEGF165b antibody (Ab; n=7) or IgG (n=5). Vascular density was calculated as CD31+ cells per muscle fiber. Unpaired t test.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

286 Circulation Research January 20, 2017
significantly lower VEGFR1 activation (determined as mean fluorescence intensity of pVEGFR1
Y1333/VEGFR1) in both IGA
(≈3×; P=0.01; Figure 2B) and IAM (≈3×; P=0.007; Online Figure IIIB) than nonischemic muscles but no difference in VEGFR2 activation (Figure 2C; Online Figure IIIC).
We then correlated endothelium-specific VEGF165
b ex-pression with VEGFR1 activation in ischemic muscle. CD31+CD45− endothelial fraction had ≈6× higher VEGF
165b
expression in IGA (P<0.0001; NGA: 10.9±2.1 versus IGA: 68.4±2.6%; Figure 2D) and ≈4× higher VEGF
165b expression in
IAM than in NAM (P<0.05; NAM: 2.2±0.4 versus 9.2±1.9%; Online Figure IIID) and significantly lower VEGFR1 activation than in nonischemic muscles (IGA ≈2×; P=0.01; Figure 2E and IAM ≈2×; P=0.004; Online Figure IIIE).
VEGF165
b-expressing (VEGF165
b+ve) endothelial cells (ECs) had significantly lower VEGFR1 activation in isch-emic muscle (IGA ≈6×, P=0.0003; Figure 2F and IAM ≈2×; P=0.01; Online Figure IIIF) than in nonischemic muscles, whereas ECs that do not express VEGF
165b (VEGF
165b−ve)
showed significantly higher VEGFR1 phosphorylation in ischemic muscle (IGA ≈2×; P<0.03; Figure 2G and IAM; P<0.03; Online Figure IIIG) than in nonischemic muscles. These data showed an inverse correlation between VEGF
165b
induction and VEGFR1 activation in experimental PAD.To determine whether changes in VEGFR1 activation
are because of the receptor-bound VEGF165
b in experimental PAD, we performed VEGFR1 pull-down experiments and im-munoblotted for VEGF
165b and VEGF-A. Although there was
no significant difference in total VEGF-A bound to VEGFR1 between NGA and IGA, we observed a significant increase in VEGF
165b bound to VEGFR1-immunoprecipitated frac-
tions (P<0.04; Figure 2H) in ischemic muscle compared with nonischemic muscle. These data demonstrated that with isch-emia, there is increased production and binding of VEGF
165b to
VEGFR1 with decreased VEGFR1 activation in ischemic mus-cle compared with nonischemic muscle in experimental PAD.
We next examined the role of VEGF165
b in modulating outcomes in experimental PAD by inhibiting VEGF
165b in
ischemic muscle. Consistent with the previous findings from Kikuchi et al,17 after HLI, intramuscular injection of VEGF
165b
isoform–specific monoclonal antibody significantly decreased necrosis (P<0.03) and improved perfusion recovery at day 14 post HLI (P<0.04; IgG: 44.0±6.0 versus VEGF
165b antibody:
65.7±5.8, measured by noninvasively quantifying microvascu-lar blood flow by laser Doppler) compared with IgG (Figure 2I and 2J). VEGF
165b antibody treatment showed a ≈2.5× induc-
tion (P=0.004) in angiogenesis, evaluated by CD31+ immunos-taining (% average of CD31+cells/muscle fiber) in ischemic muscle, compared with IgG treatment (Figure 2K).
VEGF165b Inhibition Does Not Activate the Classical Proangiogenic VEGFR2 Signaling but Activates VEGFR1 to Promote Angiogenesis and Perfusion Recovery in Ischemic MuscleOn the basis of the well-established role of VEGFR2 signaling in angiogenesis, we first examined whether increased angio-genesis and perfusion post VEGF
165b inhibition is because of
the activation of VEGFR2 signaling in ischemic muscle treated with VEGF
165b antibody compared with IgG. Immunoblotting
of VEGFR2 and its key signaling intermediates7,19 showed no significant differences in VEGFR2 activation (pVEGFR2
Y1175/
VEGFR2; Figure 3A) or activation of VEGFR2 downstream signaling including Akt, Erk, or eNos (data not shown) in VEGF
165b antibody–treated ischemic muscle compared with
IgG-treated ischemic muscle, suggesting that VEGF165
b inhi-bition did not activate VEGFR2 signaling in ischemic muscle.
Thus, we next examined the status of VEGFR1 activation post VEGF
165b inhibition in ischemic muscle. Immunoblotting
showed that VEGF165
b antibody treatment significantly induced VEGFR1 activation by increasing Y1333 phos-phorylation compared with IgG treatment in ischemic muscle (≈3×; Figure 3B). VEGFR1 activation in ischemic muscle ECs post VEGF
165b inhibition was confirmed visu-
ally by performing double immunofluorescence analysis for pVEGFR1
Y1333(AlexaFluor555) and CD31(AlexaFluor488;
Figure 3C). The extent of endothelium-specific VEGFR1 ac-tivation was quantified by flow cytometry in ischemic mus-cle treated with VEGF
165b antibody or IgG. Flow cytometry
showed a significant increase in VEGFR1Y1333
activation in ischemic endothelium treated with VEGF
165b antibody com-
pared with IgG (P=0.0003; Figure 3D).We next examined whether VEGF
165b inhibition can in-
duce endothelium-specific VEGFR1 activation in vitro. Flow cytometry showed that VEGF
165b inhibition in human
umbilical vein endothelial cells (HUVECs) significantly in-duced VEGFR1 activation (P=0.0004) compared with IgG (Figure 3E), confirming our in vivo data. The specificity of VEGF
165b antibody was validated by treating HUVECs with
VEGF165
b antibody preadsorbed with VEGF165
b peptide (1:1) overnight at 4°C. Preadsorbed VEGF
165b antibody did
not induce VEGFR1 activation, confirming the specificity of VEGF
165b antibody (Online Figure IV). Our data demon-
strating that VEGF165
b inhibition activates VEGFR1 but not VEGFR2 in ischemic muscle to induce a proangiogenic phe-notype clearly suggest that VEGFR1 activation also plays a major role in modulating angiogenesis and perfusion recovery in experimental critical limb ischemia PAD.
To confirm that VEGFR1 plays a role in regulating angio-genesis post VEGF
165b inhibition, we first treated HUVECs
with recombinant VEGF165
b ligand or VEGF165
b antibody under normal or hypoxia serum starvation (HSS) conditions. VEGF
165b ligand treatment showed no significant difference in
capillary tube formation on growth factor–reduced matrigel, under normal or HSS conditions (Online Figure VA and VB). However, VEGF
165b inhibition significantly induced capil-
lary-like tube structures on growth factor–reduced matrigel in HUVECs under normal (P=0.004; Online Figure VA) and HSS (P=0.003; Online Figure VB) conditions compared with IgG. To understand whether VEGF
165b antibody inhibits secreted
or endogenous VEGF165
b, conditioned media and cell lysates from normal and HSS HUVECs were examined for VEGF
165b
expression and levels by ELISA and immunoblot analysis, respectively. Immunoblot analysis of VEGF
165b in normal
and HSS HUVECs showed that HUVECs express VEGF165
b under normal conditions, and the expression of VEGF
165b is
significantly induced in HSS HUVECs (Online Figure VC). However, we were not able to detect VEGF
165b signal from
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 287
HUVEC-conditioned medium in VEGF165
b ELISA (data not shown), indicating that the levels of VEGF
165b in culture me-
dia are below the detection limit of the ELISA or VEGF165
b in secreted form is bound to other carrier molecules resulting in its inability to be detected by conventional ELISA techniques. Hence, we think that VEGF
165b antibody inhibits not only
endogenous VEGF165
b but also secreted VEGF165
b to induce endothelial angiogenesis.
Next, we transfected HUVECs with scrambled SiRNA or VEGFR1 SiRNA with or without VEGF
165b inhibition un-
der normal and HSS conditions and examined the ability of HUVECs to form capillary-like tubes on matrigel. VEGFR1
Figure 3. A, Immunoblot analysis of pVEGFR2Y1175 and VEGFR2. B, Immunoblot analysis of pVEGFR1Y1333 and VEGFR1 (immunoprecipitated fraction) in Balb/c IGA (ischemic gastrocnemius muscle) treated with VEGF165b antibody (Ab; n=7) or IgG (n=5). C, Double immunofluorescence analysis of pVEGFR1 (Y1333; AlexaFluor-555) and CD31 (AlexaFluor 488) pVEGFR1+ endothelial cells (ECs) in VEGF165b-Ab–treated IGA. No negative staining was observed in sections stained with secondary antibody only. D, Flow cytometry of VEGFR1 activation in CD31+CD45− ECs from Balb/c IGA treated with VEGF165b-Ab vs IgG, n≥5. E, Flow cytometry of VEGFR1 activation in human umbilical vein endothelial cells (HUVECs) treated with VEGF165b-Ab or IgG (10 µg/mL) for 24 h, n=4. A, B, D, and E, Unpaired t test. Full-length Western blot images are presented in Online Figure XII.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

288 Circulation Research January 20, 2017
inhibition was confirmed by quantitative polymerase chain reaction of VEGFR1 expression (Online Figure VD). A sig-nificant decrease in the number of capillary-like tubes was observed in HUVECs treated with VEGFR1 SiRNA com-pared with scrambled SiRNA under normal (P=0.002; Online Figure VE) and HSS (P<0.0001; Online Figure VF) condi-tions. VEGF
165b inhibition did not induce capillary-like tube
formation on matrigel in HUVECs transfected with VEGFR1 SiRNA compared with scrambled SiRNA, indicating that VEGF
165b regulates angiogenesis partly through modulating
VEGFR1 in endothelial cells.VEGF
165b has been classically considered an antiangio-
genic ligand that functions by decreasing VEGFR2 activa-tion. Limited literature describes the function of VEGF
165b in
modulating VEGFR1 signaling and function.27,28 Because we observed that VEGF
165b inhibition induces VEGFR1 activation
but not VEGFR2 activation, we next wanted to examine wheth-er VEGF
165b delivery has differential effects on VEGFR1 ver-
sus VEGFR2 activation in ECs. Thus, we treated HUVECs with recombinant VEGF
165a and recombinant VEGF
165b in time-de-
pendent (5, 15, and 30 minutes) and dose-dependent (5, 25 and 50ng) conditions and examined for VEGFR1 and VEGFR2 activation (calculated as % mean fluorescence intensity of baseline pVEGFR expression). We observed that VEGF
165a
significantly increased VEGFR1 activation at 5, 25, and 50 ng concentrations at 5, 15, and 30 minutes compared with untreat-ed HUVECs at 0 minutes. VEGFR1 activation by VEGF
165b
was significantly lower than that of VEGF165
a at 15 and 30 minutes at all concentrations (P<0.05; Figure 4A). However, no significant difference in VEGFR2 activation was observed between VEGF
165a and VEGF
165b treatments at any concentra-
tion or time points (Figure 4C), indicating that VEGF165
b can decrease VEGF-A–mediated VEGFR1 activation.
To examine whether VEGF165
b has the ability to block VEGF
165a-mediated VEGFR1 versus VEGFR2 activa-
tion, we expressed VEGFR1 and VEGFR2 in HEK293 cells (VEGFR1-expressing HEK293 cells [HEK293-VR1; Online Figure VIA] and VEGFR2-expressing HEK293 cell [HEK293-VR2; Online Figure VIB]) and treated them with VEGF
165a, VEGF
165b alone, or in combination. Consistent
with our findings in HUVECs (Figure 4A), we observed that although VEGF
165a significantly induced VEGFR1
activation in HEK293-VR1 cells, the extent of VEGFR1 activation induced by VEGF
165b was significantly lower
(Figure 4B). Furthermore, a combination of VEGF165
a and VEGF
165b at varying concentrations revealed that VEGF
165b
even at 10× lower quantity has the ability to block VEGF165
a-mediated VEGFR1 activation compared with VEGF
165a
alone. Furthermore, similar to the data from HUVECs that showed VEGF
165b activated VEGFR2 (Figure 4C), treatment
of HEK293-VR2 cells with VEGF165
b induced VEGFR2 ac-tivation albeit slightly lower than VEGF
165a. Because both
ligands function as agonists to VEGFR2, combinations of VEGF
165a and VEGF
165b induced VEGFR2 activation equal
Figure 4. A and C, Flow cytometry of VEGFR1Y1333 and VEGFR2Y1175 phosphorylation in human umbilical vein endothelial cells (HUVECs), n=3. Two-way ANOVA with Sidak multiple comparison test was used to test the significance between VEGF165a- and VEGF165b-treated samples at the same time points. B and D, Flow cytometry of VEGFR1 and VEGFR2 activation in HEK293-VR1 and HEK293-VR2 cells, n=3. Two-way ANOVA with Dunnett multiple comparison test was used to test the significance between VEGF165a-treated group at specific time point with other treatment groups at that specific time points. *The time points that are significantly different from VEGF165a at that time point. E, Flow cytometry of VEGF165b levels in Balb/c nonischemic gastrocnemius muscle Nor (NGA) that received VEGF165b-expressing or scrambled plasmid 7 d post electroporation. F, Flow cytometry of VEGFR1Y1333 activation (normalized to VEGFR1) in Balb/c NGA treated with VEGF165b-expressing or scrambled plasmid. G, Flow cytometry of VEGFR2Y1175 activation in Balb/c NGA treated with VEGF165b-expressing or scrambled plasmid. E–G, n=4, unpaired t test. by guest on Septem
ber 1, 2017http://circres.ahajournals.org/
Dow
nloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 289
to or higher than VEGF165
a and VEGF165
b individual treat-ments (Figure 4D). These data show that although VEGF
165b
functions as an agonist for VEGFR2, it is a competitive in-hibitor for VEGF
165a-meditated VEGFR1 activation.
To obtain a direct correlation between VEGF165
b and VEGFR1 activation in vivo, we next induced VEGF
165b lev-
els in Balb/c normal skeletal muscle in the hind limbs by electroporation and examined the extent of endothelium-specific VEGFR1 activation by flow cytometry. We found that gastrocnemius muscle that received VEGF
165b-expressing
plasmid showed a significant increase in VEGF165
b levels (P=0.003; Figure 4C) correlating with decreased endothelial (CD31+CD45−) VEGFR1 activation compared with gastroc-nemius muscle that received scrambled plasmid (P<0.05; Figure 4D). However, no changes in endothelium-specific VEGFR2 activation were observed in skeletal muscle that re-ceived VEGF
165b-expressing plasmid compared with scram-
bled plasmid (Figure 4E). These data showed that increased VEGF
165b levels could decrease VEGFR1 activation indepen-
dent of the ischemic state of tissue.Although extensive data exist on VEGFR2 signaling in
angiogenesis, information on VEGFR1 signaling is extreme-ly sparse. On the basis of the previous reports demonstrat-ing that ischemic myocardium from VEGFR1+/− mice had lesser STAT3 binding to DNA than VEGFR1+/+ mice29 and that VEGFR1 associated with STAT3 in cancer models,30,31 we examined the status of STAT3 activation post VEGF
165b
inhibition. Immunoblotting showed that VEGF165
b inhibition significantly induced STAT3 activation (≈3×; P<0.05) in isch-emic muscle compared with IgG (Figure 5A). CD31+CD45− ECs had significantly higher STAT3 activation in VEGF
165b
antibody–treated ischemic muscle (P=0.005; Figure 5B) compared with IgG-treated ischemic muscle. STAT3 activa-tion also correlated with significantly decreased apoptosis (P<0.04, assayed by counting terminal deoxy uridine nick end labeling–positive cells in at least 3 random images/section) and P53 inhibition (P<0.04, by immunoblotting)9 in VEGF
165b antibody–treated ischemic muscle compared
with IgG-treated ischemic muscle (Online Figure VIIA and VIIB). Because Janus-activated kinase (Jak)1/Jak2 are key kinases in STAT3 activation, we examined Jak1/Jak2 acti-vation in VEGF
165b antibody–treated versus IgG-treated
ischemic muscle by immunoblotting. STAT3 activation post VEGF
165b inhibition occurred without Jak1/Jak2 activation
(Online Figure VIII). We next wanted to confirm whether VEGF
165b inhibition induces STAT3 activation in ECs in
vitro and correlates with VEGFR1 activation. Flow cytom-etry showed that VEGF
165b inhibition significantly induced
STAT3 activation in HUVECs (≈10×, mean fluorescence intensity pSTAT3/STAT3; P=0.0005; Figure 5C) compared with IgG.
VEGF165b Inhibition, In Vivo, Increases the Bioavailability of VEGF-A to Bind and Activate VEGFR1 in Ischemic MuscleTo determine whether VEGFR1–STAT3 activation post VEGF
165b inhibition is because of increased binding of
VEGF165
a to VEGFR1, we performed VEGFR1 pull-down assays from VEGF
165b antibody–treated and IgG-treated
ischemic muscle and assayed for bound VEGF165
b and total VEGF-A. Immunoblotting showed that VEGFR1-immunoprecipitated complexes have significantly higher VEGF
165a fraction (≈3×, P=0.0002) in VEGF
165b antibody–
treated ischemic muscle compared with IgG-treated ischemic muscle (Figure 5D), indicating that VEGF
165b antibody treat-
ment increased the binding of proangiogenic VEGF165
a to VEGFR1 in ischemic muscle compared with IgG treatment. In vitro, VEGFR1 was immunoprecipitated from HUVECs and immunoblotted for VEGF
165b and total VEGF-A under normal
and HSS conditions. VEGFR1-immunoprecipitated complex-es from VEGF
165b antibody–treated HUVECs showed signifi-
cantly higher VEGF165
a bound to VEGFR1 than IgG-treated HUVECs under normal (P=0.007; Online Figure IXA) and HSS (P=0.002; Figure 5E) conditions. Increased VEGF
165a
binding over VEGF165
b to VEGFR1 in vivo and in vitro cor-relates with enhanced VEGFR1 and STAT3 activation post VEGF
165b inhibition compared with IgG.
VEGF165b Inhibition Induces VEGFR1–STAT3 Interactions to Promote STAT3 Activation in Ischemic MuscleAs shown in Figure 3D and Online Figure IV, STAT3 acti-vation post VEGF
165b inhibition occurred without changes
in Jak1/Jak2 activation, and a recent report showed that VEGFR1 is physically associated with STAT3 in can-cer models.30,31 Hence, we sought to determine whether VEGFR1 could bind and activate STAT3 on VEGF
165b in-
hibition. In vivo, VEGFR1 was immunoprecipitated from VEGF
165b antibody–treated and IgG-treated ischemic mus-
cle samples and examined for physical interactions between VEGFR1 and STAT3. In VEGFR1-immunoprecipitated fractions, immunoblotting of STAT3 showed significantly higher STAT3 binding (≈2×; P<0.03) after VEGF
165b inhibi-
tion than that after IgG inhibition (Figure 5F). In vitro, im-munoblotting of VEGFR1-immunoprecipitated complexes from VEGF
165b antibody–treated HUVECs showed no sig-
nificant changes in endothelial STAT3 binding compared with IgG-treated HUVECs under normal conditions (Online Figure IXB). However, VEGFR1-immunoprecipitated com-plexes from HSS HUVECs (P=0.0002; Figure 5G) treated with VEGF
165b antibody showed a significant increase in
STAT3 binding to VEGFR1 compared with those treated with IgG.
Activation of VEGFR1–STAT3 signaling in ischemic muscle post VEGF
165b inhibition was visually confirmed
by double immunofluorescence analysis of pVEGFR1Y1333
(AlexaFluor-555) and pSTAT3 (AlexaFluor-488), which showed extensive colocalization of pVEGFR1 and pSTAT3 (Figure 5H). Flow cytometry of CD31+CD45− ECs showed that VEGF
165b inhibition induced a significant increase in the
numbers of pVEGFR1+pSTAT3+ ECs (IgG: 2.1±0.8% versus VEGF
165b antibody: 5.0±0.8%; P<0.02; Figure 5I) in isch-
emic muscle compared with IgG.To confirm that VEGFR1 has the ability to activate
STAT3, HEK293 cells (deficient in VEGFR1 and VEGFR2) were transfected with a VEGFR1-expressing plasmid (Online Figure X) and assayed for STAT3 activation. Immunoblotting showed that VEGFR1 expression in HEK293 significantly
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from
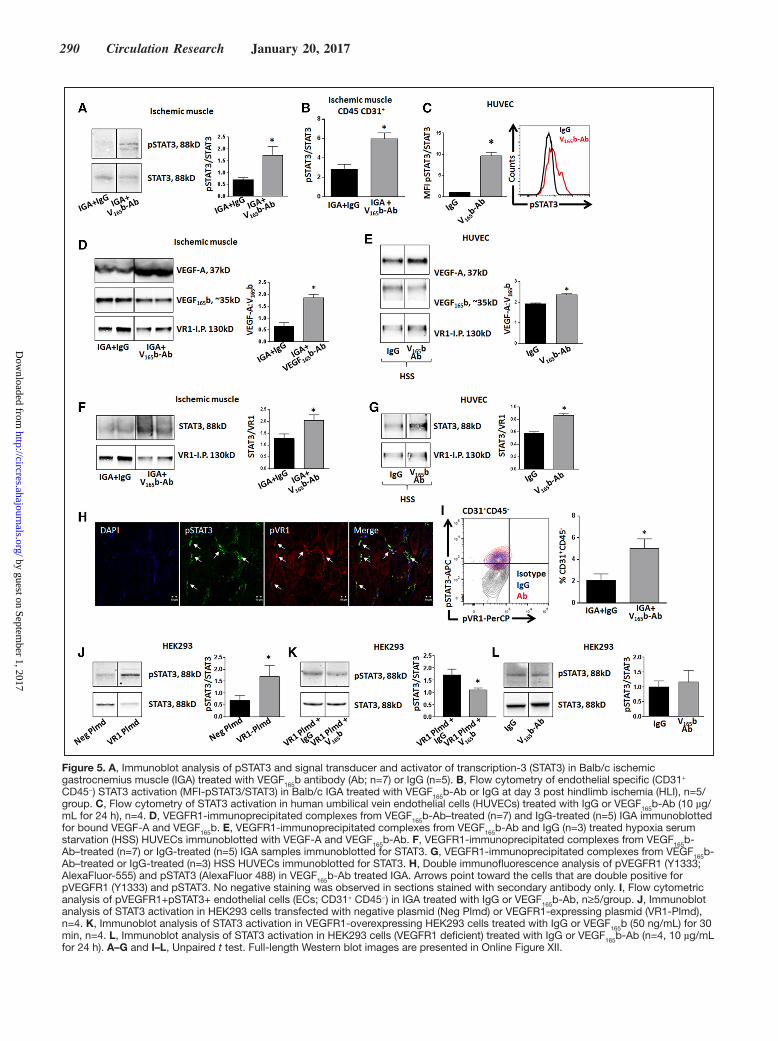
290 Circulation Research January 20, 2017
Figure 5. A, Immunoblot analysis of pSTAT3 and signal transducer and activator of transcription-3 (STAT3) in Balb/c ischemic gastrocnemius muscle (IGA) treated with VEGF165b antibody (Ab; n=7) or IgG (n=5). B, Flow cytometry of endothelial specific (CD31+ CD45−) STAT3 activation (MFI-pSTAT3/STAT3) in Balb/c IGA treated with VEGF165b-Ab or IgG at day 3 post hindlimb ischemia (HLI), n=5/group. C, Flow cytometry of STAT3 activation in human umbilical vein endothelial cells (HUVECs) treated with IgG or VEGF165b-Ab (10 µg/mL for 24 h), n=4. D, VEGFR1-immunoprecipitated complexes from VEGF165b-Ab–treated (n=7) and IgG-treated (n=5) IGA immunoblotted for bound VEGF-A and VEGF165b. E, VEGFR1-immunoprecipitated complexes from VEGF165b-Ab and IgG (n=3) treated hypoxia serum starvation (HSS) HUVECs immunoblotted with VEGF-A and VEGF165b-Ab. F, VEGFR1-immunoprecipitated complexes from VEGF165b-Ab–treated (n=7) or IgG-treated (n=5) IGA samples immunoblotted for STAT3. G, VEGFR1-immunoprecipitated complexes from VEGF165b-Ab–treated or IgG-treated (n=3) HSS HUVECs immunoblotted for STAT3. H, Double immunofluorescence analysis of pVEGFR1 (Y1333; AlexaFluor-555) and pSTAT3 (AlexaFluor 488) in VEGF165b-Ab treated IGA. Arrows point toward the cells that are double positive for pVEGFR1 (Y1333) and pSTAT3. No negative staining was observed in sections stained with secondary antibody only. I, Flow cytometric analysis of pVEGFR1+pSTAT3+ endothelial cells (ECs; CD31+ CD45−) in IGA treated with IgG or VEGF165b-Ab, n≥5/group. J, Immunoblot analysis of STAT3 activation in HEK293 cells transfected with negative plasmid (Neg Plmd) or VEGFR1-expressing plasmid (VR1-Plmd), n=4. K, Immunoblot analysis of STAT3 activation in VEGFR1-overexpressing HEK293 cells treated with IgG or VEGF165b (50 ng/mL) for 30 min, n=4. L, Immunoblot analysis of STAT3 activation in HEK293 cells (VEGFR1 deficient) treated with IgG or VEGF165b-Ab (n=4, 10 µg/mL for 24 h). A–G and I–L, Unpaired t test. Full-length Western blot images are presented in Online Figure XII.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 291
induced STAT3 activation (≈3×; P=0.007) compared with control, suggesting that VEGFR1 has the ability to activate STAT3 (Figure 5J). We next examined whether VEGF
165b can
inhibit STAT3 activation in HEK293-VR1 cells. Immunoblot analysis demonstrated that VEGF
165b treatment significantly
decreased (P<0.05) STAT3 activation in HEK293-VR1 cells
Figure 6. A, VEGFR1 expression in VEGFR1+/− (VEGFR1+/−) mice. Quantitative polymerase chain reaction (qPCR) of wild-type littermates (WT) and VEGFR1+/− normal and ischemic muscle at day 3 post hindlimb ischemia (HLI). One-way ANOVA with Bonferroni select pair comparison. n=4/group. B, VEGFR1 immunofluorescence in VEGFR1+/− ischemic gastrocnemius muscle (IGA) and WT IGA. C, Necrosis scores at day 3 post HLI in WT+IgG-HLI (n=9), VEGFR1+/−+IgG-HLI (n=8), and VEGFR1+/−+VEGF165b-Ab-HLI mice (n=6). Nonparametric Kruskal–Wallis test with Dunnetts post-test. D, Terminal deoxy uridine nick end labeling (TUNEL) assay in WT+IgG-HLI (n=9), VEGFR1+/−+IgG-HLI (n=8), and VEGFR1+/−+VEGF165b-Ab-HLI IGA (n=6) at day 3 post HLI. E, Immunoblot analysis of STAT3 activation in WT littermates, VEGFR1+/− IGA treated with IgG or VEGFR1+/− IGA treated with VEGF165b antibody (Ab), n=5. F, Flow cytometric analysis of STAT3 activation in WT littermates, VEGFR1+/− IGA treated with IgG or VEGFR1+/− IGA treated with VEGF165b-Ab, n=4. G, Flow cytometric analysis of STAT3 activation in CD31+ CD45− ECs in WT littermates, VEGFR1+/− IGA treated with IgG or VEGFR1+/− IGA treated with VEGF165b-Ab, n=4. D–G, One-way ANOVA with Dunnett post-test. Full-length Western blot images for all the figures are presented in Online Figure XII.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

292 Circulation Research January 20, 2017
compared with IgG-treated HEK293-VR1 cells (Figure 5K). In nontransfected HEK293 cells, VEGF
165b inhibition did not
significantly change STAT3 phosphorylation compared with IgG (Figure 5L), indicating that VEGFR1 can directly en-hance STAT3 activation.
To understand the causal role of VEGFR1 in regulat-ing STAT3 activation in ischemic muscle, we developed VEGFR1+/− mice on Balb/c background (VEGFR1−/− are embryonic lethal), which enabled us not only to understand the role of VEGFR1 in regulating STAT3 activation but also to further confirm the causal role of VEGFR1 in promoting perfusion recovery post VEGF
165b inhibition. Quantitative
polymerase chain reaction and VEGFR1 immunofluores-cence analysis showed that VEGFR1+/− mice have compa-rable VEGFR1 levels in normal skeletal muscle, but these mice cannot upregulate VEGFR1 in ischemic muscle com-pared with wild-type (WT) littermates (Figure 6A and 6B). These VEGFR1+/− HLI mice have significantly higher necro-sis scores (P<0.05) and apoptosis (P<0.01, analyzed by deoxy uridine nick end labeling) versus WT; and VEGF
165b antibody
did not improve necrosis scores (Figure 6C) or apoptosis (Figure 6D) compared with IgG treatment in VEGFR1+/− HLI mice, indicating that perfusion recovery post VEGF
165b inhibi-
tion is VEGFR1 dependent.We next examined the status of STAT3 activation in
VEGFR1+/− HLI mice IGA. Immunoblotting for STAT3 ac-tivation in VEGFR1+/− HLI mice IGA showed a significant decrease in STAT3 activation (P<0.03) compared with WT IGA (Figure 6E), and STAT3 was not activated by VEGF
165b
inhibition in VEGFR1+/− HLI mice compared with IgG (Figure 6E). Furthermore, flow cytometry also demonstrated a significant decrease in total STAT3 activation (IGA ≈5×; P<0.04; Figure 6F and IAM ≈2×; P<0.04; Online Figure XIA) in VEGFR1+/− IGA compared with WT IGA. CD31+CD45− endothelial fraction showed a significant decrease in endothe-lium-specific STAT3 activation (IGA ≈5×; P=0.01; Figure 6G and IAM ≈5×; P=0.03; Online Figure XIB) in VEGR1+/− IGA compared with WT IGA. VEGF
165b inhibition did not modu-
late STAT3 activation in VEGFR1+/− IGA. These data clearly indicated that VEGFR1 has the ability to regulate STAT3 acti-vation in ischemic muscle.
DiscussionOur knowledge of the VEGF superfamily continues to in-crease. Although the totality of data to date have led to the conclusion that VEGF
165b antagonizes VEGF
165a to decrease
VEGFR2 activation, our study demonstrates that VEGF165
b does not inhibit VEGFR2 in endothelial cells and depletion/displacement of VEGF
165b in ischemic muscle did not result
in more VEGFR2 activation. Rather, we found an increased bioavailability of VEGF
165a to bind and activate VEGFR1.
Furthermore, VEGF165
b inhibition increased VEGFR1–STAT3 interactions to promote angiogenesis and enhance perfusion recovery. Our study demonstrates, for the first time, that VEGF
165b inhibits VEGFR1 signaling in ischemic
muscle and depletion of VEGF165
b enhances an underappre-ciated VEGFR1 activation to promote previously unknown
VEGFR1–STAT3 signaling in ischemic muscle and increases perfusion recovery.
Although extensive literature exists on VEGFR2 signal-ing networks in PAD,7,32 information on VEGFR1 activa-tion and downstream signaling events is sparse. Several of the VEGFR1 functions that have been identified are in non-ECs,33–38 and endothelium-specific VEGFR1 functions remain uncertain. Our experimental data showed that VEGF
165b inhi-
bition induces VEGFR1 activation and not VEGFR2 activa-tion or its downstream signaling in Balb/c ischemic muscle. Failure to upregulate VEGFR1 resulted in a loss of this ef-fect. Our in vitro experiments with VEGF
165a and VEGF
165b
ligand treatments in time- and dose-dependent manner showed that VEGF
165b has the ability to induce VEGFR2
Y1175
phosphorylation almost to a similar extent as VEGF165
a in endothelial cells. However, although VEGF
165a significantly
induced VEGFR1Y1333
activation, VEGF165
b failed to induce VEGFR1
Y1333 activation in ECs. Consistent with our in vitro
experimental findings, VEGF165
b delivery into nonischemic muscle also decreased endothelium-specific VEGFR1
Y1333 ac-
tivation but not VEGFR2Y1175
activation.Kawamura et al16 has demonstrated that pulmonary arteri-
al endothelial cells that express VEGFR2 (pulmonary arterial endothelial VEGFR2) or VEGFR2-NRP1 (PAE-VEGFR2-NRP1) treated with VEGF
165b show increased VEGFR2 activa-
tion (Y1052/Y1057) compared with untreated controls but not to the extent induced by VEGF
165a.16 Another report by Catena
et al39 demonstrated that recombinant human VEGF165
b-PP (produced in Pichia Pastoris) was able to induce VEGFR2
Y1175
phosphorylation even more than that of VEGF165
a, and recom-binant human VEGF
165b-HS (produced in Chinese Hamster
Ovarian cells) was able to induce VEGFR2Y1175
to the same ex-tent as VEGF
165a in HUVECs.39 In our current study, we show
that VEGF165
b functions as a blocker of VEGF165
a-mediated VEGFR1
Y1333 activation (in HEK293-VR1 cells, Figure 4B)
and VEGF165b VEGF165
b-induced VEGFR2Y1175
activation (in HEK293-VR2 cells, Figure 4D) almost to the same ex-tent as VEGF
165a. However, Kikuchi et al17 (using HUVECs
in vitro) and Ngo et al40 (in ex vivo cultured visceral adipose tissue) have demonstrated that antibody-medicated VEGF
165b
inhibition induced VEGFR2Y951
activation. Taken together, these findings indicate that VEGF
165b can differentially modu-
late site-specific phosphorylation on VEGFR1 and VEGFR2 and puts forward the requirement for an in-depth analysis of the specific phosphorylation sites modulated by VEGF
165b in
VEGFR1 and VEGFR2.Although we show that VEGF
165b decreases VEGFR1,
but not VEGFR2, activation elucidating the molecular mecha-nisms that regulate VEGF
165b-selective inhibitory effect toward
VEGFR1 is important. Previous studies by Waltenberger et al32 showed that the binding affinity of VEGF
165a-VEGFR1 is Kd
≈16 pmol/L and for VEGF165
a-VEGFR2 is Kd ≈760 pmol/L, indicating that VEGF
165a binding affinity for VEGFR1 is sev-
eral fold higher than its binding affinity for VEGFR2. Sawano et al41 also reported similar findings that the binding affinity of VEGF
165a to VEGFR1 is Kd 1 to 16 pmol/L, whereas for
VEGFR2 it is Kd 410 pmol/L in porcine aortic endothelial cells expressing VEGFR1 or VEGFR2. However, the extent
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 293
of VEGFR1 autophosphorylation that follows ligand bind-ing and receptor dimerization by VEGF
165a is significantly
weaker compared with VEGFR232. Because the binding sites for VEGFR1 (in exon 3) and VEGR2 (in exon 4) are same in VEGF
165a and VEGF
165b isoforms,19 VEGF
165b binding af-
finity to VEGFR142 and VEGFR2 is similar to VEGF165
a.19,42 Our data from HEK293 cells with forced expression of ei-ther VEGFR1 or VEGFR2, and then treated with VEGF
165a,
VEGF165
b or in combinations show that VEGF165
b can block VEGF
165a-mediated VEGFR1 activation (not VEGFR2) even
when present at 10-fold lower levels than VEGF165
a. On the basis of these previously published reports19,32,41 and our current data, we predict that the replacement of positively charged arginine residues in VEGF
165a isoforms with neutral
lysine and aspartic acid residues in VEGF165
b isoforms results in an inhibitory effect toward VEGFR1 but not VEGFR2. In totality, VEGF
165a and VEGF
165b binding versus activation of
VEGFR1 and VEGFR2 may not be straightforward. Further experiments at the protein structural changes and binding affinities are needed to get more defined information on the function of VEGF
165b in regulating VEGFR1 and VEGFR2
activation at molecular level.We, for the first time, show that activation of VEGFR1
Y1333
is involved in STAT3 activation in ischemic muscle. However, interestingly, we observed that STAT3 activation occurred without any changes in key STAT3 activation kinases, Jak1/Jak2). Recent report by Lee et al30 has shown that VEGFR1 is physically associated with STAT3 in cancer cells, and another report by Zhao et al31 has shown VEGF-A drives breast and lung cancer stem cells self-renewal by increasing VEGFR2/Jak/STAT3 interactions. VEGFR1 pull-down assays clearly showed that VEGF
165b inhibition can increase the binding of
VEGFR1 to STAT3 resulting in increased STAT3 activation. We further confirmed that VEGFR1 has the ability to regulate STAT3 activation in VEGFR1+/− mice (on Balb/c background) in ischemic muscle. Our experiments conclude that VEGFR1 binding to STAT3 can increase STAT3 activation post VEGF
165b
inhibition, indicating that a novel VEGFR1–STAT3 signaling is activated in ischemic muscle to promote perfusion recovery. However, the potential mechanisms that regulate VEGFR1–STAT3 interactions to induce STAT3 activation need to be fur-ther investigated. One possibility is that the kinase activity of VEGFR1 is responsible for STAT3 activation, and additional binding and adaptor molecules might also be involved in me-diating VEGFR1–STAT3 interactions. STAT3 activation can result in the induction of several STAT3 gene targets that have well-documented functions43,44 in inhibiting apoptosis and in-ducing angiogenesis to revascularize ischemic muscle. Our data do not exclude that VEGFR2 activation is important to promote angiogenesis in ischemic muscle in PAD but rather demonstrate that VEGFR1 activation and the resulting STAT3 activation also play a key role in improving perfusion recovery.45
Our study was largely, but not exclusively, based on data obtained from the use of antibody-mediated approach to in-hibit VEGF
165b. To confirm that the responses observed post
VEGF165
b antibody treatment are specific, we performed sev-eral experiments that included (1) specificity of VEGF
165b an-
tibody for VEGF165
b isoform but not VEGF165
a by immunoblot
analysis (Online Figure I); (2) inability of VEGF165
b antibody preadsorbed to VEGF
165b lignad to activate VEGFR1 (Online
Figure IV); and (3) VEGF165
b antibody decreases the bind-ing of VEGF
165b to VEGFR1 in vivo and in vitro versus IgG
control (Figure 5D and 5E; Online Figure VIIIA). Separate from the antibody data, we also confirmed that VEGF
165b-
expressing plasmid delivery (gain of function) decreases VEGFR1 activation, which is consistent with increased VEGFR1 activation with VEGF
165b antibody treatment (loss
of function). These data strongly suggest that the outcomes observed by VEGF
165b antibody are specific rather than non-
specific events induced by antibody.
ConclusionsVEGFR2 is widely regarded as the dominant VEGF receptor in postnatal/ischemia-mediated angiogenesis. However, our data in both mouse and human PAD showed an inverse corre-lation between VEGF
165b binding to VEGFR1, and VEGFR1
activation and depletion of VEGF165
b from ischemic muscle activates VEGFR1–STAT3 signaling to promote perfusion. Importantly, in addition to increased endothelial VEGFR1–STAT3 activation, increased VEGFR1–STAT3 activation in nonendothelial sources including monocyte/macrophages could also contribute to increased VEGFR1–STAT3 signal-ing in ischemic muscle. Data from VEGFR1+/− PAD mice that are unable to upregulate VEGFR1 in ischemic muscle not only confirmed that VEGFR1 plays important role in perfu-sion but also confirmed that VEGF
165b modulates VEGFR1
to decrease therapeutic angiogenesis and perfusion in PAD. Our data provide evidence to the theoretical hypothesis that removal of an angiogenesis inhibitor by monoclonal antibody approach may be a superior strategy than delivery of an an-giogenic activator to treat ischemic cardiovascular diseases especially PAD.
AcknowledgmentsWe thank Dr John Lye for the technical assistance in preparing and expanding VEGF
165b and VEGFR1 plasmids and maintaining
VEGFR1+/− mice colony. V.C. Ganta and B.H. Annex researched, designed, analyzed, discussed, wrote, and reviewed the article. V.C. Ganta, M. Choi, and A. Kutateladze contributed to discussions and experimental work.
Sources of FundingB.H. Annex is supported by 1R01 HL116455, 1R01 HL121635, and 2R01 HL101200. V.C. Ganta thanks American Heart Association for scientist development grant 16SDG30340002.
DisclosuresNone.
References 1. Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev
Cardiol. 2013;10:387–396. doi: 10.1038/nrcardio.2013.70. 2. Elsayed S, Clavijo LC. Critical limb ischemia. Cardiol Clin. 2015;33:37–
47. doi: 10.1016/j.ccl.2014.09.008. 3. Olin JW, Sealove BA. Peripheral artery disease: current insight into the
disease and its diagnosis and management. Mayo Clin Proc. 2010;85:678–692. doi: 10.4065/mcp.2010.0133.
4. Chu LH, Vijay CG, Annex BH, Bader JS, Popel AS. PADPIN: protein-pro-tein interaction networks of angiogenesis, arteriogenesis, and inflammation
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

294 Circulation Research January 20, 2017
in peripheral arterial disease. Physiol Genomics. 2015;47:331–343. doi: 10.1152/physiolgenomics.00125.2014.
5. Shibuya M. VEGF-VEGFR signals in health and disease. Biomol Ther (Seoul). 2014;22:1–9. doi: 10.4062/biomolther.2013.113.
6. Shibuya M. Differential roles of vascular endothelial growth fac-tor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–478.
7. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF recep-tor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911.
8. Amano H, Kato S, Ito Y, Eshima K, Ogawa F, Takahashi R, Sekiguchi K, Tamaki H, Sakagami H, Shibuya M, Majima M. The role of vascular en-dothelial growth factor receptor-1 signaling in the recovery from ischemia. PLoS One. 2015;10:e0131445. doi: 10.1371/journal.pone.0131445.
9. Nishi J, Minamino T, Miyauchi H, Nojima A, Tateno K, Okada S, Orimo M, Moriya J, Fong GH, Sunagawa K, Shibuya M, Komuro I. Vascular en-dothelial growth factor receptor-1 regulates postnatal angiogenesis through inhibition of the excessive activation of Akt. Circ Res. 2008;103:261–268. doi: 10.1161/CIRCRESAHA.108.174128.
10. Yang KS, Lim JH, Kim TW, Kim MY, Kim Y, Chung S, Shin SJ, Choi BS, Kim HW, Kim YS, Chang YS, Kim HW, Park CW. Vascular en-dothelial growth factor-receptor 1 inhibition aggravates diabetic ne-phropathy through eNOS signaling pathway in db/db mice. PLoS One. 2014;9:e94540. doi: 10.1371/journal.pone.0094540.
11. Rajagopalan S, Mohler ER 3rd, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, Blebea J, Macko J, Kessler PD, Rasmussen HS, Annex BH. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, con-trolled study of adenoviral delivery of vascular endothelial growth fac-tor 121 in patients with disabling intermittent claudication. Circulation. 2003;108:1933–1938. doi: 10.1161/01.CIR.0000093398.16124.29.
12. Rajagopalan S, Mohler E 3rd, Lederman RJ, Saucedo J, Mendelsohn FO, Olin J, Blebea J, Goldman C, Trachtenberg JD, Pressler M, Rasmussen H, Annex BH, Hirsch AT; Regional Angiogenesis With Vascular Endothelial Growth Factor trial. Regional angiogenesis with vascular endothelial growth factor (VEGF) in peripheral arterial disease: design of the RAVE trial. Am Heart J. 2003;145:1114–1118. doi: 10.1016/S0002-8703(03)00102-9.
13. Rasmussen HS, Rasmussen CS, Macko J. VEGF gene therapy for coro-nary artery disease and peripheral vascular disease. Cardiovasc Radiat Med. 2002;3:114–117.
14. Jones WS, Duscha BD, Robbins JL, Duggan NN, Regensteiner JG, Kraus WE, Hiatt WR, Dokun AO, Annex BH. Alteration in angiogenic and anti-angiogenic forms of vascular endothelial growth factor-A in skeletal mus-cle of patients with intermittent claudication following exercise training. Vasc Med. 2012;17:94–100. doi: 10.1177/1358863X11436334.
15. Woolard J, Wang WY, Bevan HS, et al. VEGF165b, an inhibitory vascu-lar endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64:7822–7835. doi: 10.1158/0008-5472.CAN-04-0934.
16. Kawamura H, Li X, Harper SJ, Bates DO, Claesson-Welsh L. Vascular en-dothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of ki-nase activity. Cancer Res. 2008;68:4683–4692. doi: 10.1158/0008-5472.CAN-07-6577.
17. Kikuchi R, Nakamura K, MacLauchlan S, et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat Med. 2014;20:1464–1471. doi: 10.1038/nm.3703.
18. Bates DO, Mavrou A, Qiu Y, Carter JG, Hamdollah-Zadeh M, Barratt S, Gammons MV, Millar AB, Salmon AH, Oltean S, Harper SJ. Detection of VEGF-A(xxx)b isoforms in human tissues. PLoS One. 2013;8:e68399. doi: 10.1371/journal.pone.0068399.
19. Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic thera-peutics? Nat Rev Cancer. 2008;8:880–887. doi: 10.1038/nrc2505.
20. Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci USA. 2005;102:1076–1081. doi: 10.1073/pnas.0404984102.
21. Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0.
22. Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol Genomics. 2007;30:179–191. doi: 10.1152/physiolgenomics.00047.2007.
23. Chalothorn D, Faber JE. Strain-dependent variation in collateral circula-tory function in mouse hindlimb. Physiol Genomics. 2010;42:469–479. doi: 10.1152/physiolgenomics.00070.2010.
24. Dokun AO, Keum S, Hazarika S, Li Y, Lamonte GM, Wheeler F, Marchuk DA, Annex BH. A quantitative trait locus (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation. 2008;117:1207–1215. doi: 10.1161/CIRCULATIONAHA.107.736447.
25. Dokun AO, Chen L, Okutsu M, Farber CR, Hazarika S, Jones WS, Craig D, Marchuk DA, Lye RJ, Shah SH, Annex BH. ADAM12: a genetic modi-fier of preclinical peripheral arterial disease. Am J Physiol Heart Circ Physiol. 2015;309:H790–H803. doi: 10.1152/ajpheart.00803.2014.
26. Hazarika S, Farber CR, Dokun AO, Pitsillides AN, Wang T, Lye RJ, Annex BH. MicroRNA-93 controls perfusion recovery after hindlimb ischemia by modulating expression of multiple genes in the cell cycle pathway. Circulation. 2013;127:1818–1828. doi: 10.1161/CIRCULATIONAHA.112.000860.
27. Glass CA, Harper SJ, Bates DO. The anti-angiogenic VEGF isoform VEGF165b transiently increases hydraulic conductivity, probably through VEGF receptor 1 in vivo. J Physiol. 2006;572:243–257. doi: 10.1113/jphysiol.2005.103127.
28. Bills VL, Salmon AH, Harper SJ, Overton TG, Neal CR, Jeffery B, Soothill PW, Bates DO. Impaired vascular permeability regulation caused by the VEGF₁₆₅b splice variant in pre-eclampsia. BJOG. 2011;118:1253–1261. doi: 10.1111/j.1471-0528.2011.02925.x.
29. Thirunavukkarasu M, Juhasz B, Zhan L, Menon VP, Tosaki A, Otani H, Maulik N. VEGFR1 (Flt-1+/-) gene knockout leads to the disruption of VEGF-mediated signaling through the nitric oxide/heme oxygenase pathway in ischemic preconditioned myocardium. Free Radic Biol Med. 2007;42:1487–1495. doi: 10.1016/j.freeradbiomed.2007.02.011.
30. Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells inter-act with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19:513–523. doi: 10.1038/sj.leu.2403667.
31. Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, Picon-Ruiz M, Kim M, Ullmer W, El-Ashry D, Creighton CJ, Slingerland JM. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2015;34:3107–3119. doi: 10.1038/onc.2014.257.
32. Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995.
33. Selvaraj D, Gangadharan V, Michalski CW, Kurejova M, Stösser S, Srivastava K, Schweizerhof M, Waltenberger J, Ferrara N, Heppenstall P, Shibuya M, Augustin HG, Kuner R. A functional role for VEGFR1 expressed in peripheral sensory neurons in cancer pain. Cancer Cell. 2015;27:780–796. doi: 10.1016/j.ccell.2015.04.017.
34. Fong GH, Zhang L, Bryce DM, Peng J. Increased hemangioblast commit-ment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development. 1999;126:3015–3025.
35. d’Audigier C, Gautier B, Yon A, et al. Targeting VEGFR1 on endothe-lial progenitors modulates their differentiation potential. Angiogenesis. 2014;17:603–616. doi: 10.1007/s10456-013-9413-2.
36. Muramatsu M, Yamamoto S, Osawa T, Shibuya M. Vascular endothe-lial growth factor receptor-1 signaling promotes mobilization of macro-phage lineage cells from bone marrow and stimulates solid tumor growth. Cancer Res. 2010;70:8211–8221. doi: 10.1158/0008-5472.CAN-10-0202.
37. Beck H, Raab S, Copanaki E, Heil M, Scholz A, Shibuya M, Deller T, Machein M, Plate KH. VEGFR-1 signaling regulates the homing of bone marrow-derived cells in a mouse stroke model. J Neuropathol Exp Neurol. 2010;69:168–175. doi: 10.1097/NEN.0b013e3181c9c05b.
38. Ohkubo H, Ito Y, Minamino T, Eshima K, Kojo K, Okizaki S, Hirata M, Shibuya M, Watanabe M, Majima M. VEGFR1-positive macrophages fa-cilitate liver repair and sinusoidal reconstruction after hepatic ischemia/reperfusion injury. PLoS One. 2014;9:e105533. doi: 10.1371/journal.pone.0105533.
39. Catena R, Larzabal L, Larrayoz M, Molina E, Hermida J, Agorreta J, Montes R, Pio R, Montuenga LM, Calvo A. VEGF₁₂₁b and VEGF₁₆₅b are weakly angiogenic isoforms of VEGF-A. Mol Cancer. 2010;9:320. doi: 10.1186/1476-4598-9-320.
40. Ngo DT, Farb MG, Kikuchi R, Karki S, Tiwari S, Bigornia SJ, Bates DO, LaValley MP, Hamburg NM, Vita JA, Hess DT, Walsh K, Gokce N. Antiangiogenic actions of vascular endothelial growth factor-A165b, an inhibitory isoform of vascular endothelial growth factor-A, in human obesity. Circulation. 2014;130:1072–1080. doi: 10.1161/CIRCULATIONAHA.113.008171.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Ganta et al VEGF165b Inhibition Modulates VEGFR1 Signaling 295
41. Sawano A, Takahashi T, Yamaguchi S, Aonuma M, Shibuya M. Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ. 1996;7:213–221.
42. Hua J, Spee C, Kase S, Rennel ES, Magnussen AL, Qiu Y, Varey A, Dhayade S, Churchill AJ, Harper SJ, Bates DO, Hinton DR. Recombinant human VEGF165b inhibits experimental choroidal neovasculariza-tion. Invest Ophthalmol Vis Sci. 2010;51:4282–4288. doi: 10.1167/iovs.09-4360.
43. Lee SH, Lee KB, Lee JH, Kang S, Kim HG, Asahara T, Kwon SM. Selective interference targeting of Lnk in umbilical cord-derived late en-dothelial progenitor cells improves vascular repair, following hind limb
ischemic injury, via regulation of JAK2/STAT3 signaling. Stem Cells. 2015;33:1490–1500. doi: 10.1002/stem.1938.
44. Wang T, Cunningham A, Dokun AO, Hazarika S, Houston K, Chen L, Lye RJ, Spolski R, Leonard WJ, Annex BH. Loss of interleukin-21 recep-tor activation in hypoxic endothelial cells impairs perfusion recovery after hindlimb ischemia. Arterioscler Thromb Vasc Biol. 2015;35:1218–1225. doi: 10.1161/ATVBAHA.115.305476.
45. Babiak A, Schumm AM, Wangler C, Loukas M, Wu J, Dombrowski S, Matuschek C, Kotzerke J, Dehio C, Waltenberger J. Coordinated activa-tion of VEGFR-1 and VEGFR-2 is a potent arteriogenic stimulus leading to enhancement of regional perfusion. Cardiovasc Res. 2004;61:789–795. doi: 10.1016/j.cardiores.2003.12.014.
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

Vijay Chaitanya Ganta, Min Choi, Anna Kutateladze and Brian H. Annexin Human and Experimental Peripheral Arterial Disease
STAT3 Signaling Pathway and Angiogenesis−b Modulates Endothelial VEGFR1165VEGF
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2016 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.116.3095162017;120:282-295; originally published online December 14, 2016;Circ Res.
http://circres.ahajournals.org/content/120/2/282World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2016/12/14/CIRCRESAHA.116.309516.DC1Data Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 1, 2017
http://circres.ahajournals.org/D
ownloaded from

1
MATERIALS AND METHODS
Human normal and PAD muscle biopsies: Detailed description of the demographic and clinical
characteristics of the PAD and control patients used in this study was summarized in our previous
publications1-4. The Institutional Review Boards at Duke University and the University of Colorado
approved the research protocols.
ELISA: Total VEGF-A (VEGFxxxa and VEGFxxxb isoforms) and VEGF165b (detects VEGFxxxb isoforms)
levels in normal and PAD human muscle tissue were estimated using ELISA kits (VEGF-A ELISA Cat
No: DY293B, VEGF165b ELISA Cat No: DY3045) from R&D according to manufacturer’s instructions.
Tissues were homogenized in RIPA buffer, total protein concentration measured and equal amounts
(200µg) of protein from normal and PAD muscle tissue suspended in 100µL RIPA were used for VEGF-
A and VEGF165b detection. Obtained VEGF-A and VEGF165b concentration were normalized to 1 mg
of total protein.
Mice: Animal studies were approved by the University of Virginia Institutional Animal Care Committee
and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National
Institutes of Health. A combination of ketamine and Xylazine (ketamine 90 mg/kg and xylazine 10
mg/kg) was used to induce anesthesia and perform unilateral femoral artery ligation and excision as a
model of experimental PAD in 8- to 12-week-old BALB/cJ, C.129-Flt1tm1Jrt/BannJ (VEGFR1+/- or
littermates controls VEGFR1+/-) mice (male or female, number is indicated for each experiment).
Parental VEGFR1+/- mice on C57BL/6 background were backcrossed to Balb/c mice for more than 10
generations (donated to Jackson labs, C.129-Flt1tm1Jrt/BannJ, https://www.jax.org/strain/022541). Sex
and age of mice for control and treated group were matched in all experiments.
Murine Model of Hind limb Ischemia (HLI) and quantifying microvascular blood flow: HLI was
performed as described previously5. Briefly, femoral artery was ligated and resected from just above the
inguinal ligament to its bifurcation at the origin of saphenous and popliteal arteries. Perfusion recovery

2
was measured by quantifying microvascular blood flow by laser Doppler imaging (Perimed, Inc,
Ardmore, PA) on days 0, 3, 7, and 14 post HLI. Perfusion in the ischemic limb was normalized to non-
ischemic limb for each mouse.
In vivo VEGF165b-expressing plasmid delivery by electroporation: For VEGF165b expressing plasmid
delivery into mouse hindlimbs, electric-pulse-mediated gene transfer was performed as described
previously6, 7. Briefly, under isoflurane anesthesia 150µg of VEGF165b-expressing plasmid 50μg (TA) and
100μg (GA) at a concentration of 2μg/μl in normal saline was injected in mouse hind limbs using a 0.5-
ml syringe with a 28-gauge needle in 3 sites (1 site in TA and 2 non-overlapping sites in GA, 25μL per
site). Eight electric pulses (100 ms, 1 Hz, and 100 V) were delivered immediately to the injected muscle
using a S88K square-pulse stimulator (Grass-Telefactor, West Warwick, RI) through a model 533, 2-
Needle Array (BTX Instrument Division, Harvard Apparatus, Holliston, MA), placed on the medial and
lateral sides of the muscle so that the electrical field is perpendicular to the long axis of the myofibers.
Mice were allowed to recover for 7 days before use in experiments6, 7.
Necrosis scores: The extent of necrosis was scored as follows, Grade I: involving only toes, Grade II:
extending to dorsum pedis, Grade III: extending to crus, and Grade IV: extending to thigh or complete
necrosis8, 9.
Antibodies: Antibodies against pVEGFR1 (Y1333-cat No: SAB4504006, Y1048-Cat No: SAB4504649)
and pVEGFR2 (Y1175, Cat No: SAB4504567) were purchased from Sigma-Aldrich, St. Louis, MO; Flt1
(Cat No: SC-316, raised against C-terminus amino acids between 1288-1338; hence cannot bind to
soluble Flt1 ), VEGF-A (Cat No: SC-7269) and pSTAT3 (Cat No: SC-7993) purchased from Santa Cruz
biotechnology Inc, Dallas, TX; VEGFR1 (Cat No: PA5-16493) purchased from ThermoFisher Scientific,
Grand Island, NY; VEGFR2 (Cat No: 55B11), pAkt (Cat No: 4060), Akt (Cat No: 4691), pErk1/2 (Cat No:
4370) , Erk1/2 (Cat No: 9102), pSTAT3 (Cat No: 9145), STAT3 (Cat No: 12640), Jak1 and Jak2 (Cat
N0: 9945), pJak1 (Cat No: 3331) and pJak2 (Cat No: 8082) from Cell Signaling and Technology,
Danvers, MA; eNos (Cat No: 610299) and CD31 (Cat No: 558736) from BD Pharmingen, San Jose, CA;

3
peNos (Cat No: 66127) from Abcam, Cambridge, MA; VEGF165b (Cat No: MAB3045) used in western,
flow cytometry as well as to block VEGF165b in animal experiments was purchased from R&D,
Minneapolis, MN. CD; F4/80 (Cat No: MCA497) from Bio-Rad, Hercules, CA.
Antibody conjugation for flow cytometry: pVEGFR1 and pVEGFR2 were conjugated to PerCP (Cat
No: 718-0015); pSTAT3 and VEGF165b to APC (Cat No: 705-0005) using fluorescent antibody
conjugation kits from Innova Biosciences, Babraham, Cambridge, UK according to manufacturer’s
instructions.
Cell isolation and flow cytometry: GA and AM were collected from mice. Leukocyte and endothelial
cells were from GA and AM were prepared by digestion of finely minced muscle with RPMI 1640
medium/2% FBS and containing 1mg/mL of collagenase type 2 (Worthington, Lakewood, NJ), and
50U/mL DNase I (Sigma-Aldrich) for 40 min in a 37°C orbital shaker at 250rpm. For flow cytometry
staining, ∼106 cells were placed in individual wells of a 96-well plate, incubated first with FcR block
(CD16/CD32), stained with fixable viability dye (Invitrogen, ThermoFisher Scientific, Grand Island, NY)
and appropriate Ab mixtures, fixed for 20 min on ice in Fixation/Permeabilization Buffer (BD Bioscience,
San Diego, CA) . After the fixation, cells were either stained for intracellular proteins or additionally
permeabilized for 1hr in 1X Permeabilization Buffer (BD Bioscience) at RT and then stained with
phospho antibodies for 1hr at 37C and analyzed the same day on FACSCalibur (Cytek Development,
Fremont, CA). All data were analyzed using FlowJo software (Version 10.0.8 for PC; Tree Star, Ashland,
OR, USA). Cells were stained using following antibodies: anti-CD45-PeCy7 (clone 104) -CD31-
FITC/PE/PerCP eFluor 710 (clone 390) (purchased from BD Bioscience or eBiosciences, San Diego,
CA); pVEGFR1 (Y1333) and -pVEGFR2 (Y1175)-PerCP (Sigma-Aldrich/Innova Biosciences), -pSTAT3-
APC and -VEGF165b-APC (Cell Signaling/Innova Biosciences), -VEGFR1-FITC or PE (clone 141522,
R&D), -VEGFR2-PE (clone 522302, R&D) , -STAT3-PeCy7 (polyclonal, Abcore, Ramona, CA).
Endothelial specific changes in phosphorylation of receptors were calculated as ratio of median

4
fluorescence intensity (MFI) of the phospho specific staining to MFI of total receptor after gating on
specific cell type.
Western blotting: At least 50ugms of tissue or cell lysates were resolved on SDS-PAGE, transferred
onto nitrocellulose membrane and western blotting as previously described with LICOR odyssey
imaging10, 11.
Immunoprecipitation: ~1µg of antibody was incubated in 50ugm of protein lysates for overnight at 4oC
in end-to-end mixer. Antibody-antigen complexes were isolated by adding Protein G sepharose beads
(Cat No: 17-0618-01, Amersham Biosciences, Uppsla, Sweden) for 2hr at room temperature. Antibody-
antigen complexes were separated by boiling in sample buffer (Cat No: 161-0791, Bio-Rad, Hercules,
CA) for 5-10min, resolved in SDS-PAGE and western blotted as previously described12, 13.
Immunostaining: Tissues were fixed in 4% paraformaldehyde overnight at 4oC followed by RT for
another 24hrs. Later tissues were processed and embedded in paraffin. Tissues embedded in paraffin
were later sectioned and 5µM thick paraffin sections were processed for immunostaining12-14. Briefly,
sections were processed through xylene and alcohol series followed by antigen retrieval in citrate buffer
(Cat no: H3300, Vector laboratories, Burlingame, CA) blocked in 5% goat serum for 30 mins followed by
incubation in a cocktail of pVEGFR1;CD31, pVEGFR1/pSTAT3 antibodies overnight at 4oC. Later
sections were washed (PBS) and incubated in cocktail of secondary antibodies conjugated to
AlexaFluor-555 (Cat No: A31572, TermoFisher Scientific, Grand Island, NY) and AlexaFluor-488 (Cat
No: A11006, TermoFisher Scientific, Grand Island, NY) for 2hrs at RT. Sections were washed (PBS)
and mounted using prolong gold anti fade mounting media (Cat No: P36935, ThermoFisher Scientific,
Grand Island, NY). Immunofluorescence was visualized and images obtained using Olympus BX51
fluorescent microscope.
Capillary Density: Sections were immunostained with CD31 antibody using di-amino benzidine and
photographed using Olympus BX51 fluorescent microscope. Vascular density was quantified as the
number of CD31+ cells per muscle fiber8, 9.

5
Terminal deoxy-Uridine Nick End Labeling (TUNEL) analysis: TUNEL assay was performed using
Click-iT® TUNEL Alexa Fluor® 488 Imaging Assay (Cat No: C10245) purchased from ThermoFisher
Scientific, Grand Island, NY according to manufacturer’s instructions. Four random images per section
were photographed on Olympus BX51 fluorescent microscope and quantified as an average of TUNEL+
cells per image between treatment groups.
RNA isolation: Cells and tissues were lysed in Trizol and RNA was extracted using Ambion PureLink
RNA mini kit (Cat No: 12183025) according to manufacturer’s instructions.
Cells and Cell culture: Human Umbilical Vein endothelial cells (HUVECs) were purchased and cultured
in All-in-one complete endothelial growth medium (Cat No: 211-500) from Cell Applications Inc. San
Diego, CA. HEK293 cells were cultured in Dulbecco’ modified eagles medium (DMEM) supplemented
with 10% fetal calf serum (FCS).
Hypoxia serum starvation (HSS): Cells were incubated in endothelial starvation medium purchased
from Cell applications Inc. (Cat No: 209-250) and subjected to hypoxia (2% O2) for 24-48hrs.
In vitro angiogenesis (capillary like tube (loop) formation) assay: Endothelial cells were treated with
VEGF165b (50ng/ml for 24hrs) or VEGF165b-Ab (10ugm/ml for 24hrs) in normal or HSS conditions and
equal number of cells (~15000) were plated on growth factor reduced matrigel (Cat No: 356231) to avoid
any interference from VEGF-A present in the matrigel. Capillary like tubes formed on the matrigel were
photographed at 1hr, 2hr and 3hr at the center concave and the number of capillary like tubes (loops)
were quantified by at least 2 people that were blinded to the treatments10, 11.
VEGFR1 Silencing, confirmation by qPCR and in vitro angiogenesis: HUVECs were transfected
with 150nM of VEGFR1 silencer select pre-designed siRNA (Cat no: 4392420, ThermoFisher) by
siPORT NeoFX (Cat No: AM4511, ThermoFisher) transfection reagent according to manufacturer’s
instructions for 48hrs. Transfection efficacy was determined by lysing the transfected HUVECs in trizol
and isolating RNA by Purelink RNA Mini Isolation Kit (Cat No: 12183025, ThermoFisher) and performing

6
qPCR by human VEGFR1-Taqman primer probes (Cat No: 4331182, ThermoFisher). Control cells
received same amount of scrambled SiRNA to account for off target effects. 48hrs post transfection
HUVECs were incubated in fresh growth medium with VEGF165b-Ab or isotype matched IgG (10µg/mL)
under normal conditions for an additional 24hrs or incubated in HSS medium with VEGF165b-Ab or IgG
(10µg/mL) and subjected to hypoxia serum starvation for 24hrs. Later, cells were trypsinized and equal
number of cells were plated on growth factor reduced matrigel to perform in vitro angiogenesis assay as
previously described.
VEGFR1 and VEGFR2 plasmid preparation: Full length Clone DNA of human VEGFR1 with C
terminal GFPSpark tag was purchased from Sinobiological Inc (Cat No: HG10136-ACG). mEmerald-
VEGFR2-N was a gift from Dr. Michael Davidson, National MagLab (Addgene plasmid # 54298).
Plasmid DNA was prepared using a Qiagen Endo-Free Plasmid Maxi-Prep kit according to the
manufacturer’s instructions.
VEGFR1 and VEGFR2 transfection: 70-80% confluent HEK293 cells were transfected with VEGFR1 or
VEGFR2 plasmids using lipofectamine-3000 (Thermofisher, Cat No: L3000015) according to
manufacturer’s instructions. 72hrs after transfection cells were examined for VEGFR1 or VEGFR2
expression either by immunoblotting or flow cytometry as described earlier.
VEGF165b plasmid preparation: HUVEC cDNA was PCR amplified using the following primers:
165b-Not-I-5' 5'-ATGCGGCCGCTATGAACTTTCTGCTGTCTTGGG-3'
165b-BamH-I-3’ 5'-TAGGATCCATCAGTCTTTCCTGGTGAGAGATCTGCAAGTACGTTCGTTTAAC-3'
PCR products were gel-purified using a Qiagen kit per manufacturer's instructions. The PCR Products
were blunt-end cloned using pCRII-Blunt-Topo (Invitrogen, Inc.). Resulting clones were mini-prepped
and sequenced to verify the lack of PCR-induced sequence errors. Clones with the correct sequence
were digested with Not-I and BamH-I, and gel-purified using a Qiagen kit. Plasmid
pAA.CMV.PI.EGFP.WPRE.bGH (Cat No: p0101, University of Pennsylvania Vector Core) was digested

7
with Not-I and BamH-I to remove the EGFP, and gel-purified with a Qiagen kit. Gel-purified inserts were
cloned into the digested, gel-purified vector preparation using T4 DNA ligase. Six colonies from each
reaction were sequenced to verify that they had received the correct insert, and that there were no
sequence errors.
VEGFR1 and VEGFR2 activation with VEGF165a and VEGF165b ligands in HUVECs. HUVECs were
incubated with 5, 25 or 50ng/mL of VEGF165a or VEGF165b ligands for 5, 15, and 30 min, in 1X PBS with
live/dead violet dye, fixed, permeabilized, stained for pVEGFR1Y1133 and pVEGFR2Y1175, and analyzed
by flow cytometry. Equal number of events (10, 000 cells) were acquired and MFI of pVEGFR1Y1333 and
pVEGFR2Y1175 were calculated at each dose and time point and then normalized to respective MFI at 0
min.
VEGFR1 and VEGFR2 activation by VEGF165a and VEGF165b ligands in HEK293-VR1 and HEK293-
VR2 cells. HEK293-VEGFR1 or HEK293-VEGFR2 cells were treated with VEGF165a (50ng/ml),
VEGF165b (50ng/ml), VEGF165a (5ng/ml) + VEGF165b (5ng/ml), VEGF165a (25ng/ml) + VEGF165b (5ng/ml),
VEGF165a (50ng/ml) + VEGF165b (5ng/ml) for 5, 15 and 30 min in 1X PBS with live/dead violet dye,
fixed, permeabilized and stained for pVEGFR1Y1333 or pVEGFR1Y1175. Equal number of events (50, 000
cells) were acquired and MFI of pVEGFR1Y1333 and pVEGFR2Y1175 were calculated at each dose and
time point and then normalized to respective MFI at 0 min.
Statistics: GraphPad prism 6 was used to analyze the statistical significance of the data and generate
graphical representations. Statistical test used to determine statistical significance of a specific
experiment accompanied each figure legend. P<0.05 considered significant for all experiments. Data
presented as Mean±SEM for all experiments.
Reference List
(1) Duscha BD, Robbins JL, Jones WS, Kraus WE, Lye RJ, Sanders JM, Allen JD, Regensteiner JG, Hiatt WR, Annex BH. Angiogenesis in skeletal muscle precede improvements in peak

8
oxygen uptake in peripheral artery disease patients. Arterioscler Thromb Vasc Biol. 2011;31:2742-8.
(2) Jones WS, Duscha BD, Robbins JL, Duggan NN, Regensteiner JG, Kraus WE, Hiatt WR, Dokun AO, Annex BH. Alteration in angiogenic and anti-angiogenic forms of vascular endothelial growth factor-A in skeletal muscle of patients with intermittent claudication following exercise training. Vasc Med. 2012;17:94-100.
(3) Mitchell RG, Duscha BD, Robbins JL, Redfern SI, Chung J, Bensimhon DR, Kraus WE, Hiatt WR, Regensteiner JG, Annex BH. Increased levels of apoptosis in gastrocnemius skeletal muscle in patients with peripheral arterial disease. Vasc Med. 2007;12:285-90.
(4) Robbins JL, Jones WS, Duscha BD, Allen JD, Kraus WE, Regensteiner JG, Hiatt WR, Annex BH. Relationship between leg muscle capillary density and peak hyperemic blood flow with endurance capacity in peripheral artery disease. J Appl Physiol. 2011;111:81-6.
(5) Li Y, Hazarika S, Xie D, Pippen AM, Kontos CD, Annex BH. In mice with type 2 diabetes, a vascular endothelial growth factor (VEGF)-activating transcription factor modulates VEGF signaling and induces therapeutic angiogenesis after hindlimb ischemia. Diabetes. 2007;56:656-65.
(6) Akimoto T, Sorg BS, Yan Z. Real-time imaging of peroxisome proliferator-activated receptor-gamma coactivator-1alpha promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol. 2004;287:C790-C796.
(7) Dokun AO, Chen L, Okutsu M, Farber CR, Hazarika S, Jones WS, Craig D, Marchuk DA, Lye RJ, Shah SH, Annex BH. ADAM12: A Genetic Modifier of Pre-clinical Peripheral Arterial Disease. Am J Physiol Heart Circ Physiol. 2015;309:H790-803.
(8) Dokun AO, Keum S, Hazarika S, Li Y, Lamonte GM, Wheeler F, Marchuk DA, Annex BH. A quantitative trait locus (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation. 2008;117:1207-15.
(9) Hazarika S, Farber CR, Dokun AO, Pitsillides AN, Wang T, Lye RJ, Annex BH. MicroRNA-93 controls perfusion recovery after hindlimb ischemia by modulating expression of multiple genes in the cell cycle pathway. Circulation. 2013;127:1818-28.
(10) Chaitanya GV, Cromer WE, Parker CP, Couraud PO, Romero IA, Weksler B, Mathis JM, Minagar A, Alexander JS. A recombinant inhibitory isoform of vascular endothelial growth factor164/165 aggravates ischemic brain damage in a mouse model of focal cerebral ischemia. Am J Pathol. 2013;183:1010-24.
(11) Chaitanya GV, Minagar A, Alexander JS. Neuronal and astrocytic interactions modulate brain endothelial properties during metabolic stresses of in vitro cerebral ischemia. Cell Commun Signal. 2014;12:7.
(12) Chaitanya GV, Schwaninger M, Alexander JS, Babu PP. Granzyme-b is involved in mediating post-ischemic neuronal death during focal cerebral ischemia in rat model. Neuroscience. 2010;165:1203-16.

9
(13) Chaitanya GV, Eeka P, Munker R, Alexander JS, Babu PP. Role of cytotoxic protease granzyme-b in neuronal degeneration during human stroke. Brain Pathol 2011 January;21(1):16-30.
(14) Chaitanya GV, Babu PP. Activation of calpain, cathepsin-b and caspase-3 during transient focal cerebral ischemia in rat model. Neurochem Res 2008 November;33(11):2178-86.
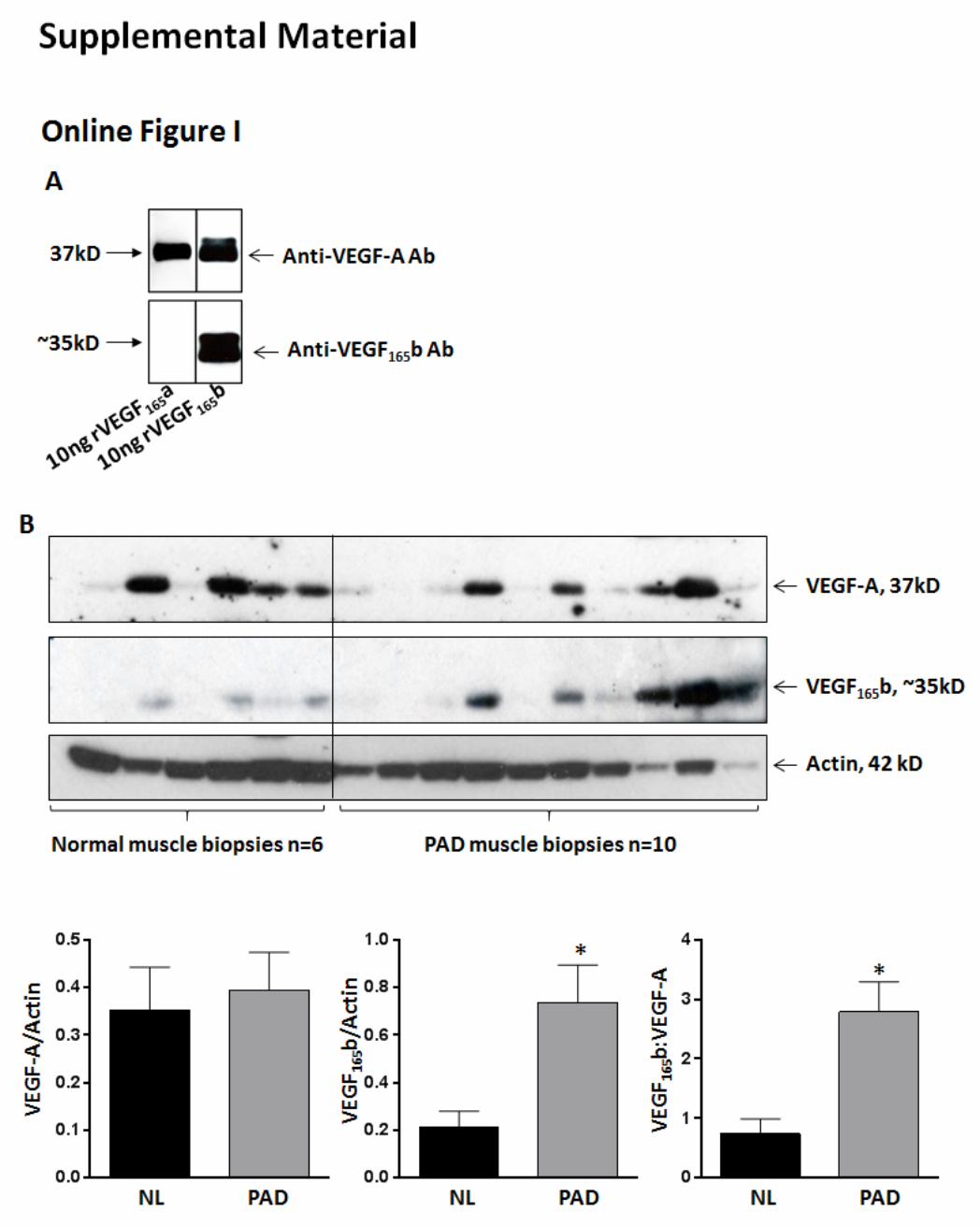

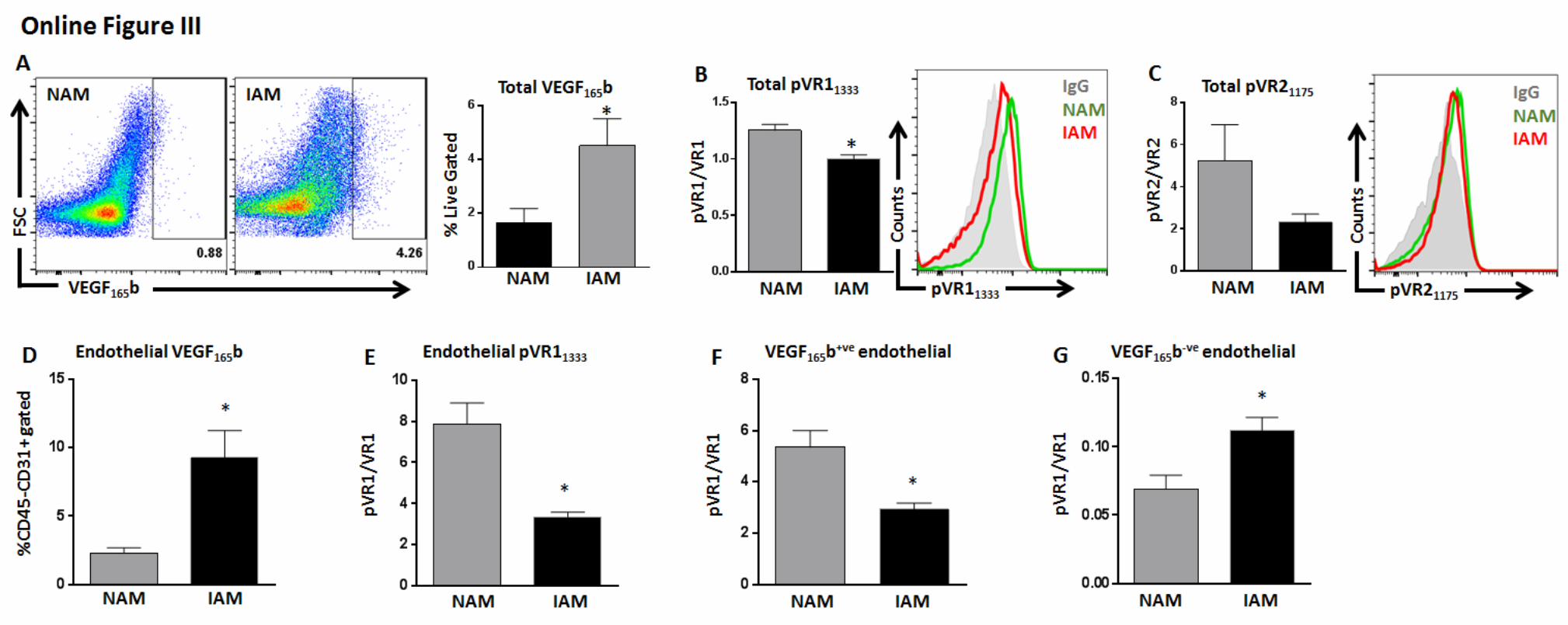

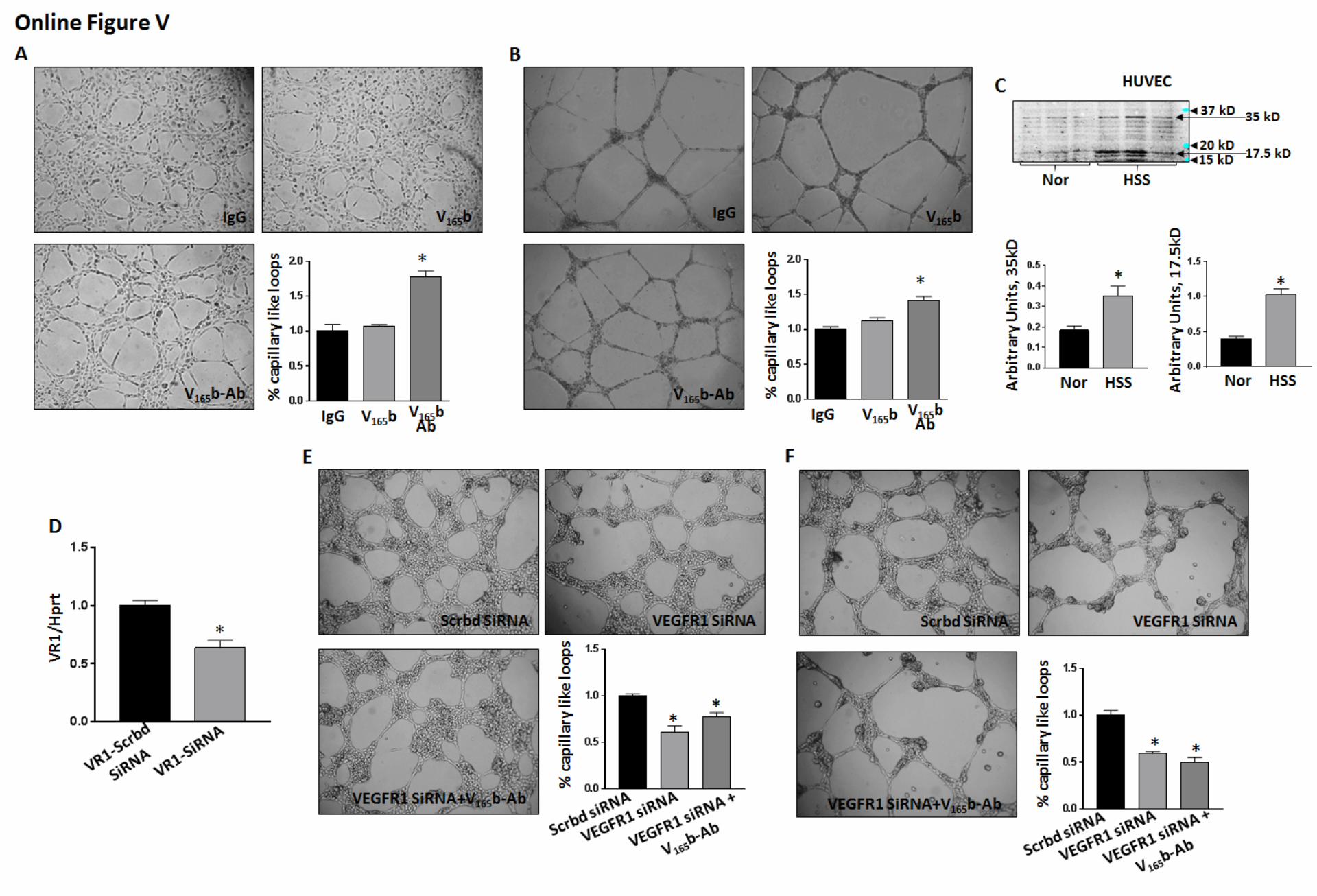








Online Figure I: A) Immunoblot analysis to confirm the specificity of isoform specific VEGF165b-Antibody. B)
Immunoblot analysis of VEGF165b-expression in human PAD. Normal (NL, n=6) and PAD (n=10) patients’
muscle-biopsies are immunoblotted for VEGF165b and total VEGF-A. Vertical line separates normal and PAD
samples. Unpaired T-test.
Online Figure II: Gating strategy for flow cytometry quantifying endothelial specific VEGFR1, VEGFR2 or
STAT3-activation. First, all doublets and dead cells were gated out. In live cell population endothelial cells
(CD31+ CD45-) were separated from leukocytes (CD45+CD31-) and activation of VEGFR1, VEGFR2 or STAT3
was checked in CD31+CD45- ECs by dividing median fluorescence intensity (MFI) of phosphorylated receptor
to MFI of total receptor.
Online Figure III: A, B, C) Correlation of VEGF165b-expression with VEGFR1 and VEGFR2-activation in
adductor muscle (AM) from ischemic (IAM) and non-ischemic (NAM) Balb/c legs by flow cytometry, n=4. D, E)
Correlation between endothelial specific (CD31+CD45-) VEGF165b-expression and VEGFR1-activation in IAM
and NAM, n=4. F) Correlation between VEGF165b+ve ECs (CD45-CD31+ VEGF165b+ve) and VEGFR1 activation,
n=4. A-F) Unpaired T test
Online Figure IV: Flow cytometry of VEGFR1-activation in HUVECs treated with VEGF165b-Ab (10µg/ml) and
VEGF165b-Ab pre-adsorbed with VEGF165b ligand (1:1 w/w, overnight at 40C), n=3, One-Way ANOVA with
Dunnetts post-Test.
Online Figure V: A, B) In vitro capillary tube formation assay on growth factor reduced matrigel (GFRM) in
HUVECs treated with recombinant VEGF165b (50ng/ml), VEGF165b-Ab or IgG (10µg/ml) for 24hrs under normal
(A) or HSS (B), n=6, One-way ANOVA with Bonferroni select pair comparison. C) qPCR of VEGFR1-
expression normalized to Hprt in HUVECs treated with scrambled Si-RNA (scrbd siRNA) or VR1-SiRNA
(VEGFR1-SiRNA) for 48hrs. Unpaired T test. C) Immunoblot analysis of VEGF165b in normal or 48hrs HSS
HUVECs, n=4. Unpaired T test. D, E) In vitro capillary tube formation assay of HUVECs treated with
scrambled-SiRNA+IgG, VEGFR1-SiRNA+IgG or VEGFR1-SiRNA+VEGF165b-Ab (10µg/mL) on GFRM under

normal (D) or HSS (E) conditions, n=8, One-way ANOVA with Bonferroni select pair comparison.
Online Figure VI: Flow cytometry of VEGFR1 and VEGFR2 plasmid transfection efficiency in HEK293 cells.
Grey histogram represents untransfected HEK293 cells, bold black represents (A) HEK293-VR1 and (B)
HEK293-VR2 cells.
Online Figure VII: A) TUNEL assay to quantify apoptosis. B) Immunoblot analysis of P53. n=5 for IgG and n=7
for VEGF165b-Ab treatments. A, B) Unpaired T test. Full-length western blot images are presented in online
Figure XII.
Online Figure VIII: Immunoblot analysis of Jak1, Jak2 activation. n=5 for IgG and n=6 for VEGF165b-Ab
treatments. Unpaired T-Test. Full-length western blot images are presented in online Figure XII.
Online Figure IX: A) VEGFR1 immunoprecipitated complexes from VEGF165b-Ab and IgG treated HUVECs
under normal conditions. n=3. B) VEGFR1 immunoprecipitated complexes from IgG or VEGF165b-Ab treated
HUVECs under normal conditions. n=3. A,B) Unpaired T test. Full-length western blot images are presented in
online Figure XII.
Online Figure X: A) Representative immunoblot analysis of VEGFR1-expression in VEGFR1 plasmid
transfected HEK293 cells.
Online Figure XI: A) Flow cytometry analysis of STAT3-activation gated on total live cells from WT+IgG-HLI,
VEGFR1+/-+IgG-HLI and VEGFR1+/-+VEGF165b-Ab-HLI IAM day-3 post HLI. B) Flow cytometry analysis of
endothelial specific (CD31+CD45-) STAT3-activation from WT+IgG-HLI, VEGFR1+/-+IgG-HLI and VEGFR1+/-
+VEGF165b-Ab-HLI IAM day-3 post HLI. A, B) n=9 for WT+IgG, n=8 for VEGFR1+/-+IgG and n=6 for VEGFR1+/-
+VEGF165b-Ab treatments. One-Way ANOVA with Dunnetts post-test.
Online Figure XII: Full length western blot images of all the figures in the manuscript. Arrows point toward the
lanes that were spliced from the full length western blot and presented as representative lanes for the specific
treatment conditions.
Related Documents











