
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript


Inte
rnat
iona
l Jou
rnal
of C
ase
Rep
orts
and
Imag
es
DisclaimerNeither International Journal of Case Reports and Images (IJCRI) nor its editors, publishers, owners or anyone else involved increating, producing or delivering International Journal of Case Reports and Images (IJCRI) or the materials contained therein,assumes any liability or responsibility for the accuracy, completeness, or usefulness of any information provided inInternational Journal of Case Reports and Images (IJCRI), nor shall they be liable for any direct, indirect, incidental, special,consequential or punitive damages arising out of the use of International Journal of Case Reports and Images (IJCRI) or itscontents. While the advice and information in this journal are believed to be true and accurate on the date of its publication,neither the editors, publisher, owners nor the authors can accept any legal responsibility for any errors or omissions that maybe made or for the results obtained from the use of such material. The editors, publisher or owners, make no warranty,express or implied, with respect to the material contained herein. (http://www.ijcasereportsandimages.com/disclaimer.php)
Contact Details:
Editorial Office
Email: [email protected]: +1-773-409-5040Website: www.ijcasereportsandimages.com
Guidelines for AuthorsFull instructions are available online at:www.ijcasereportsandimages.com/submit/instructions-for-authors
Manuscript submission:www.ijcasereportsandimages.com/submit
EDITORIAL BOARDInternational Journal of Case Reports and Images (IJCRI)
Dr. Achuta Kumar Guddati USA
Dr. Aditya Gupta USA
Dr. Antonio La Cava USA
Dr. Asher Bashiri Israel
Dr. Claudio Feliciani Italy
Dr. Emre Karasahin Turkey
Dr. Deepa Rastogi USA
Dr. Deepak Sharma USA
Dr. Gavin A. Falk USA
Dr. Gil Atzmon USA
Dr. Gokulakkrishna Subhas USA
Dr. Hajimi Orita Japan
Dr. Imtiaz Wani India
Dr. James Cheng-Yi Lin Taiwan
Dr. Jonathan D. Solomon USA
Dr. Makoto Adachi USA
Dr. Mohamed Radhi USA
Dr. Naila Khalil USA
Dr. Oner Dikensoy Turkey
Dr. Pengcheng Luo China
Dr. Radhika Muzumdar USA
Dr. Rajesh Pareta USA
Dr. Ricardo Correa USA
Dr. Sanju George UK
Dr. Saurabh Khakharia USA
Dr. Sergio Gabriel Susmallian Israel
Dr. Shashideep Singhal USA
Dr. Shilpa Jain USA
Dr. Siddharth Mathur USA
Dr. Sirinrath Sirivisoot USA
Dr. Stefan Hagmann USA
Dr. Tapas Saha USA
Dr. Teguh Haryo Sasongko Malaysia
Dr. Tun Hing Lui China
Dr. Yupeng Chen USA
Inte
rnat
iona
l Jou
rnal
of C
ase
Rep
orts
and
Imag
es
DisclaimerNeither International Journal of Case Reports and Images (IJCRI) nor its editors, publishers, owners or anyone else involved increating, producing or delivering International Journal of Case Reports and Images (IJCRI) or the materials contained therein,assumes any liability or responsibility for the accuracy, completeness, or usefulness of any information provided inInternational Journal of Case Reports and Images (IJCRI), nor shall they be liable for any direct, indirect, incidental, special,consequential or punitive damages arising out of the use of International Journal of Case Reports and Images (IJCRI) or itscontents. While the advice and information in this journal are believed to be true and accurate on the date of its publication,neither the editors, publisher, owners nor the authors can accept any legal responsibility for any errors or omissions that maybe made or for the results obtained from the use of such material. The editors, publisher or owners, make no warranty,express or implied, with respect to the material contained herein. (http://www.ijcasereportsandimages.com/disclaimer.php)
Contact Details:
Editorial Office
Email: [email protected]: +1-773-409-5040Website: www.ijcasereportsandimages.com
Guidelines for AuthorsFull instructions are available online at:www.ijcasereportsandimages.com/submit/instructions-for-authors
Manuscript submission:www.ijcasereportsandimages.com/submit
EDITORIAL BOARDInternational Journal of Case Reports and Images (IJCRI)
Dr. Achuta Kumar Guddati USA
Dr. Aditya Gupta USA
Dr. Antonio La Cava USA
Dr. Asher Bashiri Israel
Dr. Claudio Feliciani Italy
Dr. Emre Karasahin Turkey
Dr. Deepa Rastogi USA
Dr. Deepak Sharma USA
Dr. Gavin A. Falk USA
Dr. Gil Atzmon USA
Dr. Gokulakkrishna Subhas USA
Dr. Hajimi Orita Japan
Dr. Imtiaz Wani India
Dr. James Cheng-Yi Lin Taiwan
Dr. Jonathan D. Solomon USA
Dr. Makoto Adachi USA
Dr. Mohamed Radhi USA
Dr. Naila Khalil USA
Dr. Oner Dikensoy Turkey
Dr. Pengcheng Luo China
Dr. Radhika Muzumdar USA
Dr. Rajesh Pareta USA
Dr. Ricardo Correa USA
Dr. Sanju George UK
Dr. Saurabh Khakharia USA
Dr. Sergio Gabriel Susmallian Israel
Dr. Shashideep Singhal USA
Dr. Shilpa Jain USA
Dr. Siddharth Mathur USA
Dr. Sirinrath Sirivisoot USA
Dr. Stefan Hagmann USA
Dr. Tapas Saha USA
Dr. Teguh Haryo Sasongko Malaysia
Dr. Tun Hing Lui China
Dr. Yupeng Chen USA
Inte
rnat
iona
l Jou
rnal
of C
ase
Rep
orts
and
Imag
es
Dr. Achuta Kumar Guddati USADr. Aditya Gupta USADr. Adriana Handra-Luca FranceDr. Ali Soltani USADr. Antonio La Cava USADr. Asher Bashiri IsraelDr. Aziz Mustafa KosovoDr. Christopher CK Ho MalasiyaDr. Claudio Feliciani ItalyDr. Daniela Cabibi ItalyDr. Deepa Rastogi USADr. Deepak Sharma USADr. Emre Karasahin TurkeyDr. Federico Bizzarri ItalyDr. Gavin A. Falk USADr. Gerardo Gómez-Moreno SpainDr. Gil Atzmon USADr. Giovanni Leuzzi ItalyDr. Giovanni Tuccari ItalyDr. Gokulakkrishna Subhas USADr. Guo Wei USADr. Hajimi Orita JapanDr. Ho-Sheng Lin USADr. Imtiaz Wani IndiaDr. James Cheng-Yi Lin TaiwanDr. Jonathan D. Solomon USADr. Kyuzi Kamoi JapanDr. Luca Bertolaccini Italy
Dr. Makoto Adachi USADr. Mehmet Uludag TurkeyDr. Mohamed Radhi USADr. Mohannad Al-Qudah JordanDr. Morikuni Tobita USADr. Naila Khalil USADr. Natalya Semiletova USADr. Oner Dikensoy TurkeyDr. Ozlem Guneysel TurkeyDr. Paolo Cardelli ItalyDr. Paul Rea UK Dr. Pengcheng Luo ChinaDr. Piaray Lal Kariholu IndiaDr. Piraye Kervancioglu TurkeyDr. Radhika Muzumdar USADr. Rajesh Pareta USADr. Ranji Cui ChinaDr. Ricardo Correa USADr. Ricardo Macarenco BrazilDr. Sanju George UKDr. Saurabh Khakharia USADr. Sergio Gabriel Susmallian IsraelDr. Shashideep Singhal USADr. Shervin Assari USADr. Shilpa Jain USADr. Siddharth Mathur USADr. Sirinrath Sirivisoot USADr. Slobodan Marinkovic Slovenia

Inte
rnat
iona
l Jou
rnal
of C
ase
Rep
orts
and
Imag
es
DisclaimerNeither International Journal of Case Reports and Images (IJCRI) nor its editors, publishers, owners or anyone else involved increating, producing or delivering International Journal of Case Reports and Images (IJCRI) or the materials contained therein,assumes any liability or responsibility for the accuracy, completeness, or usefulness of any information provided inInternational Journal of Case Reports and Images (IJCRI), nor shall they be liable for any direct, indirect, incidental, special,consequential or punitive damages arising out of the use of International Journal of Case Reports and Images (IJCRI) or itscontents. While the advice and information in this journal are believed to be true and accurate on the date of its publication,neither the editors, publisher, owners nor the authors can accept any legal responsibility for any errors or omissions that maybe made or for the results obtained from the use of such material. The editors, publisher or owners, make no warranty,express or implied, with respect to the material contained herein. (http://www.ijcasereportsandimages.com/disclaimer.php)
Contact Details:
Editorial Office
Email: [email protected]: +1-773-409-5040Website: www.ijcasereportsandimages.com
Guidelines for AuthorsFull instructions are available online at:www.ijcasereportsandimages.com/submit/instructions-for-authors
Manuscript submission:www.ijcasereportsandimages.com/submit
EDITORIAL BOARDInternational Journal of Case Reports and Images (IJCRI)
Dr. Achuta Kumar Guddati USA
Dr. Aditya Gupta USA
Dr. Antonio La Cava USA
Dr. Asher Bashiri Israel
Dr. Claudio Feliciani Italy
Dr. Emre Karasahin Turkey
Dr. Deepa Rastogi USA
Dr. Deepak Sharma USA
Dr. Gavin A. Falk USA
Dr. Gil Atzmon USA
Dr. Gokulakkrishna Subhas USA
Dr. Hajimi Orita Japan
Dr. Imtiaz Wani India
Dr. James Cheng-Yi Lin Taiwan
Dr. Jonathan D. Solomon USA
Dr. Makoto Adachi USA
Dr. Mohamed Radhi USA
Dr. Naila Khalil USA
Dr. Oner Dikensoy Turkey
Dr. Pengcheng Luo China
Dr. Radhika Muzumdar USA
Dr. Rajesh Pareta USA
Dr. Ricardo Correa USA
Dr. Sanju George UK
Dr. Saurabh Khakharia USA
Dr. Sergio Gabriel Susmallian Israel
Dr. Shashideep Singhal USA
Dr. Shilpa Jain USA
Dr. Siddharth Mathur USA
Dr. Sirinrath Sirivisoot USA
Dr. Stefan Hagmann USA
Dr. Tapas Saha USA
Dr. Teguh Haryo Sasongko Malaysia
Dr. Tun Hing Lui China
Dr. Yupeng Chen USA
Inte
rnat
iona
l Jou
rnal
of C
ase
Rep
orts
and
Imag
es
DisclaimerNeither International Journal of Case Reports and Images (IJCRI) nor its editors, publishers, owners or anyone else involved increating, producing or delivering International Journal of Case Reports and Images (IJCRI) or the materials contained therein,assumes any liability or responsibility for the accuracy, completeness, or usefulness of any information provided inInternational Journal of Case Reports and Images (IJCRI), nor shall they be liable for any direct, indirect, incidental, special,consequential or punitive damages arising out of the use of International Journal of Case Reports and Images (IJCRI) or itscontents. While the advice and information in this journal are believed to be true and accurate on the date of its publication,neither the editors, publisher, owners nor the authors can accept any legal responsibility for any errors or omissions that maybe made or for the results obtained from the use of such material. The editors, publisher or owners, make no warranty,express or implied, with respect to the material contained herein. (http://www.ijcasereportsandimages.com/disclaimer.php)
Contact Details:
Editorial Office
Email: [email protected]: +1-773-409-5040Website: www.ijcasereportsandimages.com
Guidelines for AuthorsFull instructions are available online at:www.ijcasereportsandimages.com/submit/instructions-for-authors
Manuscript submission:www.ijcasereportsandimages.com/submit
EDITORIAL BOARDInternational Journal of Case Reports and Images (IJCRI)
Dr. Achuta Kumar Guddati USA
Dr. Aditya Gupta USA
Dr. Antonio La Cava USA
Dr. Asher Bashiri Israel
Dr. Claudio Feliciani Italy
Dr. Emre Karasahin Turkey
Dr. Deepa Rastogi USA
Dr. Deepak Sharma USA
Dr. Gavin A. Falk USA
Dr. Gil Atzmon USA
Dr. Gokulakkrishna Subhas USA
Dr. Hajimi Orita Japan
Dr. Imtiaz Wani India
Dr. James Cheng-Yi Lin Taiwan
Dr. Jonathan D. Solomon USA
Dr. Makoto Adachi USA
Dr. Mohamed Radhi USA
Dr. Naila Khalil USA
Dr. Oner Dikensoy Turkey
Dr. Pengcheng Luo China
Dr. Radhika Muzumdar USA
Dr. Rajesh Pareta USA
Dr. Ricardo Correa USA
Dr. Sanju George UK
Dr. Saurabh Khakharia USA
Dr. Sergio Gabriel Susmallian Israel
Dr. Shashideep Singhal USA
Dr. Shilpa Jain USA
Dr. Siddharth Mathur USA
Dr. Sirinrath Sirivisoot USA
Dr. Stefan Hagmann USA
Dr. Tapas Saha USA
Dr. Teguh Haryo Sasongko Malaysia
Dr. Tun Hing Lui China
Dr. Yupeng Chen USA
Inte
rnat
iona
l Jou
rnal
of C
ase
Rep
orts
and
Imag
es
Dr. Stefan Hagmann USADr. Stefano Romagnoli ItalyDr. Tapas Saha USADr. Teguh Haryo Sasongko MalaysiaDr. Tomoyuki Yano JapanDr. Tun Hing Lui ChinaDr. Yulin Li ChinaDr. Yupeng Chen USA

Cover Figure:
All Articles:
SUBMISSION INSTRUCTIONSAll manuscripts, including illustrations, should be submitted at: www.ijcasereportsandimages.com/submit or email to: submit@ijcasere-
portsandimages.com
Author Guidelines: www.ijcasereportsandimages.com/submit/instructions-for-authors.php
For any questions contact the Editorial Offi ce via e-mail at: [email protected] or Fax: 1-773-409-5040
Contents Vol. 4, No. 1 (January 2013)
International Journal of
Case Reports and Images
Cover Image
Case Series
Case Reports
Case in Images
Clinical Images
Figure 2: Magnetic resonance imaging of the brain with contrast (gadolinium) shows punctate pontine lesions slightly larger than on the prior study. Bilateral occipital abscesses have slightly decreased in size and edema as compared to the prior MRI.
1 Granular tumors of the central nervous system: A case series
Janese Trimaldi, Nicole D Riddle, Jeremy W Bowers, Harry R van Loveren, Kondi Wong
7 Magnetic resonance imaging confirmed clinical diagnosis of amyoplasia in two infants with arthrogryposis multiplex congenita
Ariam Diaz, Dominic Sia, Valerie May G Sia, Evelyn Erickson, Sergey Prokhorov, Menachem Gold
11 Multiple nocardial brain abscesses in an immunocompromised patient with myasthenia gravis
Ayesha Shaikh, Maria Lola Cevallos, Fang Lan, Jean Pratt Daniel
15 Sporadic pseudohypoaldosteronism: A challenging diagnosis
Suman Preet Kaur Bhullar, Raouf Seifeldin, Nikhil Hemady
19 Community acquired methicillin sensitive Staphylococcus aureus bacteremia, meningitis and brain abscess: A unique presentation
Carlos Gonzalez, Juan Roa, Nehad Shabarek
24 Reversible myeloneuropathy and pancytopenia related to copper deficiency from gastric bypass surgery: A case report
Laide Bello, Joseph Fiore
28 Spinal tuberculomas mimicking spinal dural arteriovenous fistula: A case report
Jyoti Sureka, Varsha Mary Khalkho, Binita Riya Chacko
32 Unusual case of ascites Guido Poggi, Benedetta Montagna, Pamela Di
Cesare, Erica Quaquarini, Emma Pozzi, Carlo Aprile
37 Feeding dystonia: A classical presentation of neuroacanthocytosis
Suman Kushwaha, Akhila Panda, Vachan Mehta, Seema Malik, Ishita Pant
41 A novel method of treating isolated unicondylar fracture of the head of the proximal phalanx: A case report
Aysha Rajeev, John Harrison
46 Pleuropulmonary blastoma in an adult: A case report
Nidhi Paliwal, Kumud Gupta, Shalini Mullick, Ravindra K Dewan, Sandeep Katiyar
51 Combined aortic and inferior vena cava injury
Basem Marcos, Yomayra Perez, Jennifer Matarlo, Jay A Yelon, Valerie Katz, Robert V Madlinger
55 Acute urinary retention in a female adolescent
Alberto Mendoza-Paredes, Antonio Pierre
58 Rib fractures: Accidental or non-accidental Muhammad Waseem, Evelyn Erickson
62 An unusual cause of hypertension Muhammad Waseem, Evelyn Erickson
66 A floppy infant Muhammad Waseem, Joel Gernsheimer,
Tae K Park, Fernando Jara, Evelyn Erickson
70 Intimal angiosarcoma of the thoracic aorta Michelle Forman, Michael E Mulligan
76 Neuroradiological imaging features of infratentorial cranial fossa tumors in a child
Muhammad Yunus Amran, Meryana Pauline, Andi Kurnia Bintang, Muhammad Akbar
80 Extensive maxillofacial and oral myiasis Felipe P Daltoé, André Ricardo Nosé,
Rodrigo C Mosca, Andrea Mantesso
83 Lesser-trélat sign in a patient with neoplasia of upper eyelid
Satyaki Ganguly, Kranti C Jaykar, Sambeet Kumar Mallik

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;(4):1 –6.www.ijcasereportsandimages.com
Granular tumors of the central nervous system: A caseseriesJanese Trimaldi, Nicole D Riddle, Jeremy W Bowers,Harry R van Loveren, Kondi Wong
ABSTRACTIntroduction: Granular cell tumors of thecentral nervous system are rare tumors. Todate, eight cases arising from cranial nerveshave been reported. Granular cell tumors havealso been found arising from theneurohypophysis and its stalk. Due to theirrarity and histological similarity to other centralnervous system (CNS) tumors with a granularappearance, they often pose a diagnosticconundrum. The differential diagnosis issurprisingly diverse and includes granular cellastrocytoma, infundibular granular cell tumor,spindle cell oncocytoma of theadenohypophysis, granular and oncocyticvariants of pituitary adenoma, meningioma,pituicytoma and intrasellar schwannoma.Distinguishing between the CNS tumors withgranular features is important because sometumors have an increased recurrence risk or apoor prognosis. Case Report: To highlight thehistological features of granular lesions of thecentral nervous system, including the
immunohistochemical profile and electronmicroscopic depiction, we review two cases eachwith a similar granular histology and a differentfinal diagnosis. Conclusion: Thorough onlineliterature search revealed several cases ofgranular cell lesions of the CNS, however,oftentimes the diagnosis is difficult to come byand the differential is long. Conclusion:Granular cell tumor and its variants, thoughuncommon, must be included in the differentialdiagnosis of CNS lesions.Keywords: Granular, Tumors, Central nervoussystem (CNS)
*********Trimaldi J, Riddle ND, Bowers JW, Loveren HRv, WongK. Granular tumors of the central nervous system: Acase series. International Journal of Case Reports andImages 2013;4(1):1–6.
*********doi:10.5348/ijcri201301247CS1
INTRODUCTIONGranular cell lesions of the central nervous system(CNS) are rather uncommon and may pose a diagnosticchallenge. Granular histology may be present as themain characteristic of a neoplasm or as a variation in anumber of benign and malignant lesions. Thedifferential diagnosis must always include a truegranular cell tumor when granular morphology isidentified, especially when it encompasses a majority ofthe lesion. The differential diagnosis of a CNS granularcell tumor is widely varied and includes infundibulargranular cell tumor, granular/oncocytic variants ofpituitary adenoma, meningioma, pituicytoma andintrasellar schwannoma, as well as spindle cell
CASE SERIES OPEN ACCESS
Janese Trimaldi1 , Nicole D Riddle2, Jeremy W Bowers3,Harry R van Loveren4, Kondi Wong1
Affi l iations: 1Department of Pathology and Cell Biology,University of South Florida College of Medicine, Tampa,FL, USA; 2Department of Pathology, University of TexasHealth Science Center, San Antonio, TX, USA;3Department of Pathology, Moffitt Cancer Center, Tampa,FL, USA; 4Department of Neurosurgery, University ofSouth Florida College of Medicine, Tampa, FL, USACorresponding Author: Nicole D Riddle, MD., Departmentof Pathology University of Texas Health Science Center,7703 Floyd Curl Dr, MC 7750 San Antonio, TX, USA; Ph:21 0-567-3748; Fax: 21 0-567-2478; Email :nriddlemd@gmail .com
Received: 1 4 March 201 2Accepted: 07 May 201 2Published: 01 January 201 3
Trimaldi et al. 1

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;(4):1 –6.www.ijcasereportsandimages.com Trimaldi et al. 2
oncocytoma of the adenohypophysis, which may beprone to recur and grow despite adjuvant therapy, andgranular cell astrocytoma, which has a poor prognosis.
CASE REPORTCase 1: A 37yearold male presented with a historyof depression, fatigue, and decreased libido for twoyears. Magnetic resonance imaging (MRI) wasperformed revealing a leftsided pituitary mass that wasenlarging the sella with suprasellar extension. Postcontrast images demonstrated an intensely enhancingmass in the right middle cranial fossa that washypointense on T2weighted images and isointense toadjacent gray matter on T1weighted images (Figure 1).It was centered in Meckel’s cave with involvement of thecisternal segment of the trigeminal nerve. There wasdistal extension of the mass along the inferior alveolarbranch of V3 into the mandibular foramen. The rightside of the pituitary gland appeared to be uninvolved.Surgical removal was performed through a transnasalendoscopic approach and yielded a 1.4x0.4x0.1 cmtumor. Frozen section analysis revealed a granularepithelial cell neoplasm. The differential diagnosis atthat time included: pituitary adenoma, xanthoma,infundibular granular cell tumor, langerhans cellhistiocytosis, hemangioblastoma and granular cellastrocytoma.Routine histological examination with hematoxylinand eosin (H&E) stained sections revealed diffuse,
densely granulated, eosinophilic cells (Figure 2). Specialstaining for PAS was also positive (Figure 3).Immunohistochemical studies showed the tumor cells tobe weakly positive for prolactin and stronglysynaptophysin positive. The tumor cells were nonreactive for CD68, inhibin, S100, chromogranin; as wellas pituitary markers such as ACTH, FSH, LH, GH, andTSH. Pancytokeratin and GFAP were also negative.Complete disruption of the native lobular reticulararchitecture was evident on reticulin stain, characteristicof a pituitary adenoma. A diagnosis of pituitaryadenoma with granular cell morphology, and evidenceof prolactin expression was given.Case 2: A 54yearold woman with a history of Bell’spalsy one year prior with recurrence nine months later,presented with numbness primarily in the third divisionof the right trigeminal nerve.MRI scan showed a tumor homogeneouslyenhancing in the gasserion ganglion with an extensioninto the foramen ovale (Figure 4). A right temporalcraniotomy and zygomatic osteotomy was performed,and the foramen ovale was opened for resection of a6.1x4.3x1.2 cm tanbrown mass with multinodularareas. Frozen section analysis suggested a possibleganglioneuroma. Following histological evaluation aFigure 1: Magnetic resonance imaging scan of pituitary masswith some suprasella extension. The mass can be seenoverlying the optic chiasm.
Figure 2: Monotonous cells with abundant granular cytoplasm(H&E, x200).
Figure 3: PAS with diastase showing abundant eosinophilicgranular cytoplasm (PAS with diastase, x200).

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;(4):1 –6.www.ijcasereportsandimages.com Trimaldi et al. 3
diagnosis of granular cell tumor of the right trigeminalganglion was rendered. Permanent section analysisrevealed peripheral nerve and entrapped ganglion cells(trigeminal ganglion) infiltrated by round, spindled andepithelioid cells with abundant, granular, PASpositivecytoplasm consistent with granular cell tumor (Figure 5).The tumor cells were strongly positive for CD 68 withabundant PAS positive cytoplasmic granules and strongreactivity to S100 (Figures 6 and 7). They were nonreactive for: synaptophysin, smooth muscle actin,muscle specific actin, melanA, myogenin, GFAP,smooth muscle myosin heavy, and desmin. Ki67demonstrated moderate proliferative activity as well asmoderate amounts of reticulin collagen and neuronspecific enolase (not shown). Electron microscopydemonstrated tumor cell cytoplasm filled and expandedwith dark, granular lysosomes, characteristic of granularcell tumor (Figure 8).
DISCUSSIONA number of granular or similarly oncocytic tumorshave remarkably similar histomorphology on initialstaining with hematoxylin and eosin. Differingintracranial locations may be helpful at times for
diagnosis, but at other times they may be misleading.Tumors containing granular cells can present in twoforms: (i) as a “pure” form composed entirely ofgranular cells (“granular cell tumor”), (ii) or as a focalchange that occurs in a neoplasm of a recognizable celltype [1]. In the latter form, one theory considersgranular cell change to be a degenerative phenomenon
Figure 4: Magnetic resonance imaging scan showing a mass inthe region of the gasserion ganglion.
Figure 5: Peripheral nerve and entrapped ganglion cellsinfiltrated by spindled and epithelioid cells with abundantgranular cytoplasm (H&E, 200x).
Figure 6: Lysomal granules strongly positive for CD68.
Figure 7: PAS with diastase showing abundant eosinophilicgranular cytoplasm (PAS with diastase, 200x).
Figure 8: (A) Tumor cells with cytoplasm filled and expandedwith dark, granular lysosomes, (B) Lysosomal vacuolescontaining membranous and electron dense materialconsistent with degenerating phagolysosomes.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;(4):1 –6.www.ijcasereportsandimages.com Trimaldi et al. 4
[2]. The hypothesis is that the granular cell phenotypeseems to be not specific of a certain tumor type, butrather a peculiar change characterized by an increase inintracellular lysosomes.Reported CNS tumors with granular cell or oncocyticchange include: astrocytoma, medulloblastoma,ganglioglioma, glioblastoma, meningioma,schwannoma, ependymoma, oligodendroglioma,neurofibroma, anterior and posterior pituitary [2–7].Spindle cell oncocytoma, granular cell astrocytoma, andpituicytoma are distinct entities and possibly theinfundibular granular cell tumor as well. Morphology onhematoxylin and eosin stain and special stains alongsidetumor immunophenotype are a valuable aide inpathologic evaluation and distinguishing the differenttumor types.There are distinguishing characteristics of differenttumor types. Granular cell astrocytomas are GFAPreactive in more than 95% cases [19] and have a poorprognosis. Spindle cell oncocytomas of theadenohypophysis have S100 reactivity, but are negativefor neuroglial markers and CD68 because they are filledwith mitochondria, not phagolysosomes. Spindle celloncocytomas are reactive for TTF1 (thyroidtranscription factor), but negative for thyroglobulin.Meningiomas are generally EMA (epithelial membraneantigen) reactive, at least focally, with a lobular reticulinpattern and meningeal pattern on ultrastructure.Granular/oncocytic pituitary adenomas lose the nativelobular reticulin architecture of the pituitary gland andare reactive for neuroendocrine markers. Intrasellarschwannoma may have granular cytoplasm, but can bedistinguished by the dense pericellular basal lamina onreticulin staining, diffuse S100 staining and nonreactivity for other neuroglial markers. Electronmicroscopy shows a profuse basal laminar or basementmembrane pattern with long spaced collagen or “Lusebodies” [20].Conventional granular cell tumors are similar anddiffer by primary location and possibly cell of origin.Eight cases of cranial nerve granular cell tumors havebeen reported in literature arising from the followingnerves: vagus, oculomotor, abducens, optic, facial; andan additional three arising from the trigeminal nerve[8–15]. Of the four arising from the trigeminal ganglion,two were reported as confined to Meckel’s cave, whileone reported the tumor originating from the brain stemand extending into Meckel’s cave [13–15]. All patientsexperienced symptoms of trigeminal neuralgia. Allmasses were composed of clusters or nests of cells, mostpolyhedral, with round nuclei, separated by connectivetissue ranging from dense strands of collagen to delicatereticulin fibers. Tumor in our case was nonreactive forsynaptophysin and GFAP, helping to rule out this entityin our differential diagnoses.Granular cell change occurs as a cell accumulateslysosomes and phagosomes. There are numerous typesof cytoplasmic accumulations that result in distinctivecell morphologies which correlate with the type ofmaterial that is being increased. Often a certain type ofaccumulated material favor a certain cell type. For
example, clear cell change is usually due to glycogenaccumulation, which is easily seen with a PAS stain withdiastase digestion. Epithelial cells are the primary typeof cell involved. Cytoplasmic vacuolization that leads toindentation of the nuclear membrane is due to lipidcontaining vacuoles, and is usually seen in sebaceousgland cells, adrenal cortex fasciculata cells, lipoblasts oradipose tissue neoplasms. In addition to phagosomesand lysosomes, other types of material such asneurosecretory granules or mast cell granules can leadto a granular appearance of the cytoplasm. In granularcell tumors, toluidine blue semithin sections of eponembedded tissue show cytoplasm filled with denselystaining, but variegated vacuolar material that prove tobe degenerated phagolysomes on electron microscopy[16]. Granular cell tumors may arise in theneurohypophysis or infundibulum of the pituitarygland. They have also been referred to as choristomas(microscopic nodules in the infundibulum) ormyoblastomas. They show a preference for the stalk, areusually suprasellar and composed of nests of large,eosinophilic cells with abundant granular cytoplasm dueto high lysosomal content. Most commonly, they are anincidental finding, usually at autopsy, composed ofsmall, round, eosinophilic, infundibular nodules. Theirorigin is presumed to be the pituicyte—a specializedtype of glial cell or modified astrocyte of theneurohypophysis [1]. Electron microscopy showspituicytes to possess variable amounts of electron denselysosomes, identical to those found in granular celltumors. Hence, a newly described tumor, thepituicytoma, may possibly be related. Isolated orclusters of granular cells are a normal part of thehistology of the neurohypophysis. It is rare for thesemicroscopic nodules to enlarge enough to becomesymptomatic. When they do, they are usuallysuprasellar, discrete, and show intense heterogenous orhomogenous enhancement with contrast. Commonsymptoms, including visual acuity deficits, or rarely,endocrine deficiency are usually the result of stalkcompression. They show a predilection for adultfemales. Grossly, the tumor is lobulated, soft butrubbery (firmer than an adenoma) and very vascular,with a grayyellow cut surface that usually lacksnecrosis, cystic degeneration or invasion of surroundingstructures. The tumor cells are polygonal, with smallnuclei containing evenly dispersed chromatin andinconspicuous nucleoli, low mitosis/proliferation, andcan grow in sheets, nodules, or in a spindled/fascicularpattern. The abundant cytoplasm contains granules thatare diastase resistant with PAS staining. “Atypical”tumors show increased mitosis (up to 5/10 HPF, andKi67 index of 7%), nuclear pleomorphism, prominentnucleoli and multinucleation. The significance of thisdifference is unknown. Giant cell tumors (GCTs) arepositive for: CD68, S100, alpha1antitrypsin, alpha1antichymotrypsin, and cathepsin B; and negative forneurofilament proteins, cytokeratins, chromogranin A,synaptophysin, desmin, smooth muscle actin andpituitary hormones. Though most are negative for glial

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;(4):1 –6.www.ijcasereportsandimages.com Trimaldi et al. 5
neural filament protein (GFAP), some variablereactivity has been reported. A presumed lineage frompituicytes may explain the conflicting results of IHCsince there are different types of pituicytes. Electronmicroscopy demonstrates phagolysosomes withunevenly distributed electron dense material andmembranous debris. Neurosecretory granules areabsent. Most are slow growing, noninvasive andclinically benign [17].Granular cell astrocytomas (GCA) are infiltrativeneoplasms and illdefined. They are considered anaggressive variant (WHO grade 3) despite the fact thatthey are cytologically bland with few mitosis. Themajority are found in cerebral hemispheres. Since theyare associated with high grade astrocytomas, theydisplay ring enhancement on imaging. Their cytoplasm,like other granular cell neoplasms, contains PASpositive, diastase resistant granules that often create aneccentric nucleus. Some tumor cells display a clearcentral cytoplasmic area and the granules accumulate atthe periphery beneath the cell membrane. Granular cellastrocytomas are positive for CD68, EMA, and S100 inthe majority of cases. GFAP is positive in over 95% oftumors (18, 19, 21). The malignant nuclei are larger andcourser than normal astrocytes, but are not ashyperchromatic or pleomorphic as in glioblastoma(WHO grade 4). The granular cell component can eitherbe mixed with an otherwise typical diffuse astrocytomaor be the exclusive cell type in a lesion. Diagnosis maybe particularly difficult in the diffuse exclusive casesand they are often mistaken for nonneoplasticprocesses such as infarcts or demyelinating diseases orGCTs of the infundibulum. Making this distinction,however, is paramount, as GCA’s have a poor prognosis,whereas, granular cell tumor of the neurohypophysis isa benign, slowly progressive tumor without an invasivegrowth pattern
CONCLUSIONGranular cell lesions of the central nervous systemare very rare. However, granular histology may be seenin a vast array of benign and malignant lesions.Histomorphology on hematoxylin and eosin stain andspecial stains with tumor immunophenotype can be ahelpful aid in distinguishing the various tumor types.The differential of a granular cell tumor must always beincluded when granular cell morphology is present.Also, a granular cell astrocytoma must be ruled out dueto prognostic implications.
*********Author ContributionsJanese Trimaldi – Conception and design, Acquisitionof data, Drafting the article, Final approval of theversion to be publishedNicole D Riddle – Conception and design, Acquisitionof data, Analysis and interpretation of data, Drafting thearticle, Final approval of the version to be published
Jeremy Bowers – Conception and design, Acquisition ofdata, Critical revision of the article, Final approval of theversion to be publishedHarry Van Loveren – Acquisition of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedKondi Wong – Conception and design, Analysis andinterpretation of data, Critical revision of the article,Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Janese Trimaldi et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distribution andreproduction in any means provided the original authorsand original publisher are properly credited. (Please seewww.ijcasereportsandimages.com/copyrightpolicy.phpfor more information.)
REFERENCES1. Cremonini A, Kuhn E, De Biase P, Franchi A. Welldifferentiated chondrosarcoma of the humerus withprominent granular cell component: a hithertounreported occurrence. Int J Surg Pathol2006;14(2):147–54.2. Geddes JF, Thom M, Robinson S, Revesz T. Granularcell change in astrocytic tumors. Am J Surg Pathol1996 January;20(1):55–63.3. Rickert CH, Kuchelmeiser K, Gullotta F.Morphological and immunohistochemicalcharacterization of granular cells in nonhypophyseal tumours of the central nervous system.Histopathology 1997;30(5):464–71.4. Yang KH, Lee MC, Lee YS, et al. Ependymoma withgranular cell changes: A case report. Presented at themeeting of American Association of Pathologists,Vancouver, BC, June 1996.5. Takei Y, Mirra SS, Miles ML. Eosinophilic granularcells in oligodendrogliomas. An ultrastructuralstudy. Cancer 1976;38(5):1968–76.6. Muller W, Dahmen HG. A combined neurofibromagranular cell tumor of the middle cranial fossa.Pathol Res Pract 1978;163(4):378–6.7. Tomita T, Gates E. Pituitary adenomas and granularcell tumors. Incidence, cell type, and location oftumor in 100 pituitary glands at autopsy. Am J ClinPathol 1999;111(6):817–25.8. Budzilovich GN. Granular cell “myoblastoma” ofvagus nerve. Acta Neuropathol. 1968;10(2):162–5.9. Inci S, Gulsen S, Soylemezoglu F, Kansu T, Ozgen T.Intracavernous granular cell tumor. Surg Neurol2004;61(4):384–90.10. Ogata S, Shimazaki H, Aida S, Miyazawa T, Tamai S.Giant intracranial granularcell tumor arising fromthe abducens. Pathology International2001;51(6):481–6.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;(4):1 –6.www.ijcasereportsandimages.com
11. Muller W, Dahmen HG. Granular cell tumor of theoptic nerve. Albrecht Von Graefes Arch Klin ExpOphthalmol 1978;207(3):181–8.12. May M, Beckford NS, Bedetti CD. Granular celltumor of facial nerve diagnosed at surgery foridiopathic facial nerve paralysis. Otolaryngol Headand Neck Surg 1985 February;93(1):122–6.13. Chimelli L, Symon L, Scaravilli F. Granular celltumor of the fifth cranial nerve: further evidence forschwann cell origin. J Neuropathol Exp Neurol1984;43(6):634–42.14. Rao TV, Puri R, Reddy GN. Intracranial trigeminalnerve granular cell myoblastoma. Case report. JNeurosurg 1983;59(4):706–9.15. Carvalho GA, Lindeke A, Tatagiba M, Ostertag H,Samii M. Cranial granularcell tumor of thetrigeminal nerve. Case report. J Neurosurg1994;81(5):795–8.16. Rosai J. The appearance, nature, and significance ofcytoplasmic accumulations, as exemplified by thegranular cell change. Int J Surg Pathol2006;14(2):109–11.
Trimaldi et al. 6
Access full text article onother devices Access PDF of article onother devices
17. AFIP Atlas of Tumor Pathology, Series 4. Tumors ofthe central nervous system. Burger PC, ScheithauerBW. The American Registry of Pathology 2007.18. Claassen U, Kuntz G, Schmitt HP. Malignantintracerebral granular cell tumor reacts positivelywith antialpha1antichymotrypsin and the MB2antibody: a clue to the histogenesis of the braingranular cell? Clin Neuropathol 1990;9(2):82–8.19. Brat DJ, Scheithauer BW, MedinaFlores R,Rosenblum MK, Burger PC. Infiltrativeastrocytomas with granular cell features (granularcell astrocytomas): a study of histopathologicfeatures, grading, and outcome. Am J Surg Pathol2002;26(6):750–7.20. Luse SA. Electron microscopic studies of braintumors. Neurology 1960;10:881–905.21. Geddes JF, Thom M, Robinson S, Revesz T. Granularcell change in astrocytic tumors. Am J Surg Pathol1996;20(1):55–63.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):7–1 0.www.ijcasereportsandimages.com
Magnetic resonance imaging confirmed clinical diagnosisof amyoplasia in two infants with arthrogryposis multiplexcongenitaAriam Diaz, Dominic Sia, Valerie May G Sia, Evelyn Erickson,Sergey Prokhorov, Menachem Gold
ABSTRACTIntroduction: We present two cases ofarthrogryposis multiplex congenita (AMC) withinvolvement of the lower extremities. In bothcases amyoplasia was confirmed by a magneticresonance imaging (MRI). The degree ofamyoplasia correlated with the severity ofarthrogryposis and determined the child’sprognosis. Case Series: Case 1 was a 16monthold male child with prenatally diagnosedKlinefelter syndrome was born at 36 weeksgestation. Brain MRI was reported as normal.Joint rigidity was detected in upper and lowerextremities. Amyoplasia was suspected at ninemonths of age since the lower limb muscleswere hardly palpable. Case 2 was a 5 1/2monthold female child and the first child of nonconsanguinous parents was noticed to have rigidright calcaneovalgus and left equinovarus feetdeformities as well as knee rigidity withlimitation of knee extension. Bilateral hipdisplacement was also diagnosed. Absence ofmuscles on thigh palpation prompted MRIstudy. Conclusion: Although amyoplasia is themost common type of arthrogryposis multiplexcongenita, muscle underdevelopment in these
patients remains puzzling for pediatricpractitioners. Amyoplasia congenita is usuallysymmetrical and involves either all extremitiesor selectively only the lower or upperextremities. Absence of muscle groups on MRIconfirms diagnosis of amyoplasia. Earlyrecognition of amyoplasia in children witharthrogryposis multiplex congenita can help intailoring their treatment and prognosis.Keywords: Amyoplasia, Arthrogryposismultiplex congenita (AMC)
*********Diaz A, Sia D, Sia VMG, Erickson E, Prokhorov S, GoldM. Magnetic resonance imaging confirmed clinicaldiagnosis of amyoplasia in two infants witharthrogryposis multiplex congenita. InternationalJournal of Case Reports and Images 2013;4(1):7–10.
*********doi:10.5348/ijcri201301248CS2
INTRODUCTIONArthrogriposis multiplex congenita (AMC) affectsabout 1/3000 birth in North America [1]. Amyoplasia(A=no, myo=muscle and plasia=growth) is the mostcommon type of arthrogryposis seen clinically [2]. Wepresent two cases of arthrogryposis multiplex congenitawith prominant involvement of the lower extremities. Inboth cases amyoplasia was confirmed by magneticresonance imaging (MRI) of the thighs. The degree ofamyoplasia correlated with the severity ofarthrogryposis and determined the child’s prognosis.MRI may be helpful in the differentiation betweenamyoplasia and other congenital myopathies andmuscular dystrophies [3].
CASE SERIES OPEN ACCESS
Ariam Diaz1 , Dominic Sia1 , Valerie May G Sia1 , EvelynErickson1 , Sergey Prokhorov1 , Menachem Gold2
Affi l iations: 1Department of Pediatrics, Lincoln Medical &Mental Health Center, Bronx, NY, United States;2Department of Radiology, Lincoln Medical & MentalHealth Center, Bronx, NY, United States.Corresponding Author: Sergey Prokhorov, MD Departmentof Pediatrics, Lincoln Medical & Mental Health Center, 234East 1 49th Street, Bronx, New York, USA-1 0451 ; Ph: 71 8-594-6501 ; Fax: 71 8-579-4700; Email :sproxy11 3@gmail .com
Received: 03 November 2011Accepted: 27 March 201 2Published: 01 January 201 3
Diaz et al. 7

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):7–1 0.www.ijcasereportsandimages.com Diaz et al. 8
CASE SERIESCase 1: The patient was a 16monthold male baby,prenatally diagnosed with Klinefelter syndrome born at36 weeks gestation from a nonconsanguineousmarriage. The delivery was by emergency cesareansection due to fetal heart rate deceleration. Apgar scorewas 1, 2 and 5 at 1, 5 and 10 minutes. During the firsttwo days of life the baby developed seizures. Brain MRIwas reported as normal. Joint contractures wererecognized on the fourth day of the baby’s life.Amyoplasia was suspected at nine months of age sincethe lower limb muscles were barely palpable. MRI of thethigh revealed paucity of muscles (Figures 1 and 2). Onexamination, the baby presented with microcephaly(head circumference 44 cm, <2%), bilateral epicanthusand clinodactily of the fifth fingers. The child was ableto hold sitting position, however, he was not able to situp by himself. There was stiffness in the wrists withlimitation of wrist extension. Hand grasp was weak. Thechild was unable to hold a spoon. Hip abduction waslimited to 300. Knee flexion was limited to 900. Therewas also equinovarus feet deformity. The child movedthe upper extremities well, but only slightly raised theextended lower extremities and minimally flexed themat the knees. He supported his weight on his legs whenin standing position with support. In the lower
extremities only hip adductors were slightly palpable.Gluteus muscle contraction was evident. Biceps,brachioradial and knee reflexes were normal. Plantarresponse was downgoing. There was no deficiencies insensation to the touch and pinprick.Case 2: A 51/2monthold female was the first childof nonconsanguinous parents was born after full termuneventful pregnancy by normal vaginal delivery. Apgarscore was 9 and 9 at 1 and 5 minutes. Birth weight was3.8 kg. At birth the baby was noticed to have rigid rightcalcaneovalgus and left equinovarus feet deformities, aswell as knee rigidity with limitation of knee extension.Bilateral hip displacement was also diagnosed. Currentneurological examination revealed peripheral right facialpalsy and torticollis due to shortened rightsternocleidomastoid muscle. The upper extremities werewithout any neurological deficiencies. The lowerextremities were in fixed frogleg position with rigid feetdeformity. Active movements manifested with veryslight hip adduction and minimal toe movements. Themuscles were not detectable on leg palpation. Knee andankle jerks were absent. Plantar response was mute.Sensation to pinprick and touch was preservedthroughout. The anus was closed and anal blink reflexwas brisk. The baby demonstrates good head control,raising the head and chest while being in prone position;grasping and transferring an object from one hand toanother with good visual attention to the grasped object.The baby had positive stranger anxiety. MRI of the hipsshowed absence of musculature with preserved fascialplanes, vessels, and adipose tissue (Figure 3).As part of management, both patients underwentsurgical correction of their tulip equinovarus. On followup at three years of age, their neurological evaluationshowed that they both were able to sit up, but not standup. Only the first patient was able to stand with support.
DISCUSSIONDespite the fact that amyoplasia is the most commontype of arthrogryposis multiplex congenital [2, 3],recognition of considerable muscle underdevelopmentin these patients remains surprising and puzzling forpediatric practitioners. Amyoplasia congenita is usually
Figure 1: Magnetic resonance imaging of the thigh revealedpaucity of muscles in patient from Case 1.
Figure 2: Another magnetic resonance imaging of the thighrevealed paucity of muscles in patient from Case 1.
Figure 3: Magnetic resonance imaging of the hips showedabsence of musculature in patient from Case 2.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):7–1 0.www.ijcasereportsandimages.com Diaz et al. 9
symmetrical and involves either all extremities orselectively only the lower or upper extremities [4]. Themuscle mass of the limbs with arthrogryposis isdiminished. In a study by Hall et al., histologicexamination of muscles showed the replacement ofmuscle with fibrofatty scar tissue [5]. Studies of spinalcord and muscles in patients with amyoplasia suggeststwo possible affected areas: either anterior horn cells ormuscles [6, 7]. Intrauterine vascular insult of spinalmotor neurons or limb muscles was hypothesized [8, 9].It is also known that arthrogryposis can be a geneticallyheterogeneous disorder as for instance, in distalarthrogryposis. Our first case can be considered as anarthrogryposis with all limb involvement even thoughthe wrist stiffness is the only finding in the upperextremities. The presence of a brisk knee jerk does notsupport the spinal origin of amyoplasia. Decrease offemoral muscle mass on thigh MRI is significant, butthe leg extensors remained strong enough to bear bodyweight. Klinefelter syndrome diagnosed in this childmost likely does not have any causal relation with theamyoplasia. Our second case presented witharthrogryposis only in the lower extremities. MRIshowed absence of femoral muscles bilaterally.Congenital facial palsy in this child is considered as apartial Moebius anomaly, which is a known comorbidity of amyoplasia congenita.Muscle weakness and multiple joint contractures inboth patients could have suggested congenital muscledystrophy (CMD) and congenital myopathy (CM).However, MRI revealed absence of whole musclegroups, with fibrofatty tissue instead and preservedfibrous planes. In patients with CMD and CM, MRIshows peculiar selectivity of muscle involvement, withdecreased muscle volume and abnormal MRI signalsfrom the muscles [3].
CONCLUSIONEarly recognition of amyoplasia with an assessmentof muscle underdevelopment via limb magneticresonance imaging in children with arthrogryposismultiplex congenita elucidates their clinicalpresentation and can help in tailoring their treatmentand in the prognostication of the degree of their futuredisability.
*********AcknowledgementsEvelyn Erickson, MD Technical contributorAuthor ContributionsAriam Diaz – Conception and design, Acquisition ofdata, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedDominic Sia – Conception and design, Acquisition ofdata, Analysis and interpretation of data, Drafting the
article, Critical revision of the article, Final approval ofthe version to be publishedValerie May G Sia – Conception and design, Acquisitionof data, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedEvelyn Erickson – Acquisition of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedSergey Prokhorov – Conception and design, Acquisitionof data, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedMenachem Gold – Conception and design, Acquisitionof data, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Ariam Diaz et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Hall JG. Arthrogryposis. In: Roger E Stevenson, HallJG, Goodman RM (Eds). Human Malformations andRelated Anomalies. Oxford monograph on medicalgenetics 1997.No.27; VolII:798–804.2. Hall JG. Genetic Aspects of Arthrogryposis. ClinicalOrthopedics and Related Research 1985Apr;(194):44–53.3. Mercuri E, Jungbluth H, Muntoni F. Muscle imagingin clinical practice: diagnostic value of musclemagnetic resonance imaging in inheritedneuromuscular disorders. Curr Opin Neurol2005;18(5):526–37.4. Hall JG. Arthrogryposis multiplex congenital:etiology, genetics, classification, diagnosticapproach, and general aspects. J Pediatr Orthop B1997;6(3):159–66.5. Sells JM, Jaffe KM, Hall JG. Amyoplasia, the mostcommon type of arthrogryposis: the pathophysiologyfor good outcome. Pediatr 1996:97(2):225–31.6. Brown LM, Robson MJ, Sharrard WJ. Thepathophysiology of arthrogryposis multiplexcongenita neurologica. J Bone Joint Surg Br 1980Aug;62(3):291–6.7. Drachan DB, Banket BQ. Arthrogryposis maltiplexcongenital. Arch Neurol 1991;5:77–933.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):7–1 0.www.ijcasereportsandimages.com Diaz et al. 1 0
8. Reid CO, Hall JG, Anderson C, et al. Association ofamyoplasia with gastroschisis, bowel atresia, anddefects of the muscular layer of the trunk. Am JMede Genet 1986;24(4):701–10.
9. Robertson WL, Glinski LP, Kirkpatrick SJ, PauliRM. Further evidence that arthrogryposis multiplexcongenital in the human is caused by an intrauterinevascular accident. Teratology 1992 Apr;45(4):345–1.
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 1 –1 4.www.ijcasereportsandimages.com
Multiple nocardial brain abscesses in animmunocompromised patient with myasthenia gravisAyesha Shaikh, Maria Lola Cevallos, Fang Lan, Jean Pratt Daniel
ABSTRACTIntroduction: Nocardia are gram positive,variably acidfast positive diphtheroidlike tobranched, filamentous, aerobic actinomycetes.Nocardiosis is an opportunistic infection thathas been noted in patients with malignancies,systemic lupus erythematosus, HIV infection,hematopoietic stem cell transplant recipientsand longterm steroid users. Case Report: A 32yearold female presented with history ofmyasthenia gravis on longterm glucocorticoidtherapy. During her last admission, woundculture of her left shoulder abscess showeddiptheroid organisms. Patient presented withsevere headache, nausea, vomiting and alteredmental status. She was initially diagnosed withmetastatic cerebral abscess and treated withempiric antimicrobial therapy. Imaging study ofthe brain showed bilateral occipital ringenhancing lesions. Biopsy results came back asculture positive for nocardia. Patient wassubsequently treated with intravenousantibiotics for a total of six months. Conclusion:Cases of nocardiosis may go undiagnosed, eitherbecause they respond to empiric antimicrobial
treatment or because Nocardia spp. may bedifficult to identify in cultures of clinicalspecimens. They may be mistaken fornonpathogenic microorganisms (diphtheroids)and discarded. High suspicion and early longterm institution of therapy are key to afavorable outcome of this disease which hashigh mortality rates.Keywords: Nocardiosis, Brain, Abscess, Steroiduse
*********Shaikh A, Cevallos ML, Lan F, Daniel JP. Multiplenocardial brain abscesses in an immunocompromisedpatient with myasthenia gravis. International Journal ofCase Reports and Images 2013;4(1):11–14.
*********doi:10.5348/ijcri201301249CR3
INTRODUCTIONNocardia can be found almost universally in soil andplants. Nocardia was first identified by Edmund Nocardin 1888 in bovine farcy. The first human disease wasdescribed by Eppinger in 1890. Nocardiosis is anopportunistic infection caused by gram positive, weaklyacidfast, filamentous, aerobic organisms [1, 2].Nocardia asteroides is the most common species tocause infection in humans [3–5]. There are at leastthirteen, Nocardia spp. but N. asteroides, N. farciniaand N. nova (N. asteroides complex) constitute about80–90% of the total cases [3, 4]. Though nocardiosis isa relatively rare bacterial infection, it is frequentlyassociated with immunosuppression. The majority ofinfections occur in patients with weakened cellmediated immunity. Infected population generallycomprises those who have received bone marrow or
CASE REPORT OPEN ACCESS
Ayesha Shaikh1 , Maria Lola Cevallos2, Fang Lan3, JeanPratt Daniel4
Affi l iations: 1PGY1 , Internal Medicine Lincoln Medical andMental Health Center, Bronx, NY, United States;2Department of Internal Medicine, Lincoln Medical & MentalHealth Center, Bronx, NY, United States; 3PGY2 , InternalMedicine, Lincoln Medical and Mental Health Center,Bronx, NY, United States; 4Department of InternalMedicine, Lincoln Medical & Mental Health Center, Bronx,NY, United States.Corresponding Author: Ayesha Shaikh, MD 234 East 1 49thStreet, Bronx, New York, USA-1 0451 ; Ph: 1 -71 8-579-6429; Fax: 1 -71 8-579-4836; Email : ashaikhdr@gmail .com
Received: 03 November 2011Accepted: 27 March 201 2Published: 01 January 201 3
Shaikh et al. 11

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 1 –1 4.www.ijcasereportsandimages.com Shaikh et al. 1 2
solid organ transplantation, patients onimmunosuppressive therapy, those with humanimmunodeficiency virus/acquired immunodeficiencysyndrome (HIV/AIDS), patients on longterm steroidtherapy and those with malignancies [3, 6].
CASE REPORTA 32yearold female was brought to the emergnencyroom (ER) with complaints of severe headache, nausea,vomiting and altered mental status. A diagnosis ofmyasthenia gravis had been made three years earlier,associated mainly with diplopia. Her medicationsincluded 60 mg of pyridostigmine and 60 mg ofprednisone daily. No history of seizures, fever orphotophobia was obtained. On neurological examination,she was confused and incoherent. There was generalizedweakness with no focal neurological deficits andpreserved deep tendon reflexes. The patient’s mentalstatus deteriorated to the point that she had to beintubated and placed on mechanical ventilation. Noncontrast computed tomography (CT) scan of brainshowed multiple ring lesions in both the hemispheres. Onreview of the chart, it was found that the patient had beenrecently discharged from surgical service after drainageof left shoulder abscess. Wound culture at that time hadgrown diptheroid organisms, and the patient wasdischarged home on oral augmentin after a brief courseof intravenous vancomycin in the hospital.In view of the immunocompromised status andrecent history of shoulder abscess, metastatic cerebralabscess was ascertained to be the most likely cause, andthe patient was started on empiric antimicrobial therapyincluding coverage for possible toxoplasmosis(ampicillin, vancomycin, ceftriaxone, metronidazole andtrimethoprimsulfamethoxazole (TMPSMX)).Magnetic resonance imaging (MRI) of the brain withcontrast was obtained for better evaluation of thecerebral lesions and showed bilateral occipital ringenhancing lesions (Figure 1, 2). Initial set of bloodcultures remained negative after 48 hr. Toxoplasmatiters were negative. A stereotactic biopsy of one of thecerebral lesion was done and frank pus was aspirated,however, gram stain failed to reveal any organisms.While awaiting biopsy culture results, empiric antibioticcoverage was continued with the exception ofampicillin.Serial interval neurological examinations continuedto be nonfocal and unchanged from presentation(Figure 3). Computed tomography scann of the chestand abdomen showed no abnormalities. The patient’smental status improved gradually and the patient wassuccessfully weaned off the ventilator on the seventhday of hospitalization. Aerobic and anaerobic bacterialcultures of the brain biopsy aspirate continued to showno growth. However, on the 19th day of hospitalization,fungal cultures were reported to be positive forNocardia. At this time, staphylococcal and anaerobiccoverage was discontinued and patient was continuedon IV ceftriaxone and IV TMPSMX.
Speciation and further identification confirmed theorganism to be Nocardia asteroides sensitive to theantibiotic regimen.A permanent intravenous access was established asprolonged antibiotic therapy was necessary and thepatient was discharged home with plan to continueintravenous antibiotics for total of six months accordingto current guidelines.The patient was subsequently followedup in the outpatient clinic and is doing well with some complaints ofoccasional sharp pain in left parietal area and no focalneurological deficits. The patient is presently in the fifthweek of antibiotic therapy.
DISCUSSIONNocardiae are gram positive, aerobic actinomycetesfound naturally in the soil, air and sewage. Nocardiaasteroides is the predominant species and the one mostcommonly associated with disseminated disease.Although there have been many reports of disseminatednocardiosis in immunocompromised patients, primarycerebral nocardiosis is a very rare presentation.
Figure 1: (A, B) Magnetic resonance imaging of the brain withcontrast (gadolinium) at presentation, showing bilateraloccipital ring enhancing lesions.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 1 –1 4.www.ijcasereportsandimages.com Shaikh et al. 1 3
Nocardial abscess associated mortality is reported tobe three times higher than in patients with otherbacterial brain abscesses. Management of nocardialbrain abscess remains a clinical challenge and isassociated with very high morbidity and mortality rates
(about 90%) [7–9]. A definitive diagnosis can only bemade with the isolation and identification of theorganism by invasive procedures and therefore, a highindex of suspicion is required as early institution oftherapy can be lifesaving. Cultures can take up to 13weeks to grow and speciation is difficult.All treatment modalities for nocardiosis generallyinvolve TMPSMX. However, there have been isolatedreports of benefit from amikacin and ceftriaxone.Aspiration has been recommended as the preferredmodality initially for nocardial brain abscess, withaggressive surgical management being reserved for thesmall proportion of patients who do not respond tominimally invasive surgery.Our patient was a housewife with no history ofrecent travel, trauma or engagement in water sports.Infection with Nocardia was therefore most likely viathe respiratory tract, which is the generally acceptedmode of inoculation. Her recent shoulder abscess was inall probability a Nocardial infection as Nocardia can bemisidentified for diptheroid organisms owing toanalogous gram staining and morphological features.This fact, coupled with the immunocompromised statusraised the suspicion for nocardial abscess initially.Nocardial cerebral abscess can give rise to focalneurological deficits depending on the intracraniallocation. In our patient, no motor or sensory focaldeficits were noted on serial neurological examinationsas all of the abscesses were localized to the occipital andposterior cortex. A good clinical response to thecombination therapy with intravenous ceftriaxone andTMPSMX was noted.
CONCLUSIONWe report a single case of multiple nocardial brainabscesses in an immunocompromised patient on longterm corticosteroid therapy for myasthenia gravis.Nocardiosis frequently goes undiagnosed initially as thepatient would have either responded to empiricantimicrobial treatment given for some other reason orbecause Nocardia can very easily be mistaken fornonpathogenic microorganisms (diphtheroids) anddiscarded on account of its morphological similarities.The diagnosis requires a high clinical suspicion withearly tissue and microbiological diagnosis. Prolongedantimicrobial therapy of 6–12 months and serialimaging is key in treatment and prevention of relapse.
*********AcknowledgementsWe would like to acknowledge and extend our heartfeltgratitude to the following persons who have made thecompletion of this case report possible: Rajan Khanna,MD; Cesar A Lopez, MDAuthor ContributionsAyesha Shaikh – Conception and design, Acquisition ofdata, Analysis and interpretation of data, Drafting the
Figure 2: (A, B) Magnetic resonance imaging of the brain withcontrast (gadolinium) shows punctate pontine lesions slightlylarger than on the prior study. Bilateral occipital abscesseshave slightly decreased in size and edema as compared to theprior MRI.
Figure 3: The magnetic resonance imaging of the patient postright occipital craniotomy with biopsy of one of the lesionsand aspiration of pus.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 1 –1 4.www.ijcasereportsandimages.com Shaikh et al. 1 4
article, Critical revision of the article, Final approval ofthe version to be publishedMaria Lola Cevallos – Conception and design,Acquisition of data, Analysis and interpretation of data,Drafting the article, Critical revision of the article, Finalapproval of the version to be publishedFang Lan – Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedJean Pratt Daniel – Conception and design, Analysisand interpretation of data, Critical revision of thearticle, Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Ayesha Shaikh et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Cortese I, Nath A. Case 11: a young woman with ringenhancing brain lesions. MedGenMed 2006 Jan5;8(1):3.
2. Mamelak AN, Obana WG, Flaherty JF, RosenblumML. Nocardial brain abscess: treatment strategiesand factors influencing outcome. Neurosurgery.1994;35(4):622–31.3. Sereti I, Holland SM. Disseminated nocardiosis in apatient with Xlinked chronic granulomatous diseaseand human immunodeficiency virus infection. ClinInfect Dis 2001;33(2):235–9.4. Barnaud G, Deschamps C, Manceron V, et al. BrainAbscess Caused by Nocardia cyriacigeorgica in aPatient with Human Immunodeficiency VirusInfection. J Clin Microbiol 2005;43(9):4895–7.5. Wakhlu A, Agarwal V, Dabadghao S, Prasad KN,Nityanand S. Nocardiosis in patients of chronicidiopathic thrombocytopenic purpura on steroids. JAssoc Phys India 2004;52:591–3.6. Talwar P, Chakrabarti A, Ayyagari A, et al. Brainabscess due to Nocardia. Mycopathologia1989;108(1):21–3.7. Skiest DJ. Focal Neurological Disease in Patientswith Acquired Immunodeficiency Syndrome. ClinicalInfectious Diseases 2002;34(1):103–15.8. Lee GY, Daniel RT, Brophy BP, Reilly PL. Surgicaltreatment of nocardial brain abscesses. Neurosurgery2002;51(3):668–71.9. Elmaci I, Senday D, Silav G, et al. Nocardial CerebralAbscess Associated with Mycetoma, Pneumonia, andMembranoproliferative Glomerulonephritis. J Clinmicrobiol 2007 June;45(6):2072–4.
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 5–1 8.www.ijcasereportsandimages.com
Sporadic pseudohypoaldosteronism: A challengingdiagnosisSuman Preet Kaur Bhullar, Raouf Seifeldin, Nikhil Hemady
ABSTRACTIntroduction: Pseudohypoaldosteronism (PHA)is a rare form of saltwasting syndrome, causedby peripheral resistance to aldosterone. PHA isof three types: PHA type 1, 2, 3.Pseudohypoaldosteronism type 1 (PHA1) isfurther differentiated into, (i) hereditary forms,autosomal recessive and dominant, which arecaused by epithelial sodium channel andmineralocorticoid receptor mutationsrespectively and (ii) secondary form which isassociated with urological problems. CaseReport: We present a case of a male infant whopresented with failure to thrive, vomiting, milddehydration and reflux. Evaluation revealedhyperkalemia with normal glucose and carbondioxide levels. A preliminary diagnosis of CAH(congenital adrenal hyperplasia) was made.Further workup showed high serum aldosteroneand renin levels with normal renal andadrenocortical functions. In line with theinvestigations the diagnosis ofpseudohypoaldosteronism was made. Thepatient was treated with sodiumsupplementation, which normalized his clinical
state and serum electrolytes. Followup revealedweight gain and improved status. Conclusion:Diagnosis of PHA1 is based on plasmaelectrolyte assessment, elevated renin activityand aldosterone levels with normal renalfunction. PHA1 results from a renal or systemicresistance to aldosterone. In our reportedpatient we suspected a renal form of PHA1,which is a milder form and responded well totreatment with salt supplements. Infants whopresent with electrolyte imbalance likehyperkalemia, hyponatremia and weight lossshould be evaluated for adrenocortical functionand need careful management. Though PHA is agroup of rare syndromes, a high degree ofsuspicion along with extensive laboratoryworkup should be pursued in cases withelectrolyte imbalances.Keywords: Pseudohypoaldosteronism,Congenital Adrenal Hyperplasia, Aldosterone
*********Bhullar SPK, Seifeldin R, Hemady N. Sporadicpseudohypoaldosteronism: A challenging diagnosis.International Journal of Case Reports and Images2013;4(1):15–18.
*********doi:10.5348/ijcri201301250CR4
INTRODUCTIONPseudohypoaldosteronism (PHA) is a rare form ofsaltwasting syndrome, caused by peripheral resistanceto aldosterone. PHA has three types: PHA type 1, 2, and 3.Pseudohypoaldosteronism type 1 (PHA1) is furtherdifferentiated into, (i) hereditary forms autosomalrecessive and dominant, which are caused by epithelial
CASE REPORT OPEN ACCESS
Suman Preet Kaur Bhullar1 , Raouf Seifeldin2, Nikhi lHemady3
Affi l iations: 1Resident, Department of Family Medicine,Doctors' Hospital of Michigan, Pontiac, MI , USA; 2AssociateProgram Director, Department of Family Medicine, Doctors'Hospital of Michigan, Pontiac, MI , USA; 3Program Director,Department of Family Medicine, Doctors' Hospital ofMichigan, Pontiac, MI , USA.Corresponding Author: Suman Preet Kaur Bhullar, MDResident, Department of Family Medicine, 461 W HuronStreet. Doctors' Hospital of Michigan. Pontiac, MI , USA; Ph:+1 -248-857-7200; Email : drsbhul lar@gmail .com
Received: 09 Apri l 201 2Accepted: 06 May 201 2Published: 01 January 201 3
Bhullar et al. 1 5

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 5–1 8.www.ijcasereportsandimages.com Bhullar et al. 1 6
sodium channel and mineralocorticoid receptormutations respectively and (ii) secondary form which isassociated with urological problems. PHA being a raredisorder, only about seventy cases have been reported inthe literature. We report a case of PHA which inspiteof being a rare disorder, can be potentially lifethreatening and also briefly discuss the types of PHA,diagnosis and treatment.
CASE REPORTA 5weekold Caucasian male infant was admitted tothe hospital for evaluation of failure to thrive. He wasborn at term via cesarean section and weighed 3.07 kgat birth. He was noted to have mild dehydration in spiteof feeding well along with on and off vomiting and somereflux. His weight was 2.96 kg and physical examinationwas normal at presentation to the hospital. Externalgenitalia were normal.Initial laboratory investigations showedhyperkalemia with potassium levels of 5.1 mmol/L, withsodium of 122 mmol/L. Repeat testing later on showedpotassium level of 8.1 mmol/L. He was resuscitated withD5 0.45 NS. Further biochemistry revealed low urineand serum osmolality, normal urine sodium and urinepotassium levels with trace of reducing substance in theurine. Upper gastrointestinal studies wereunremarkable. Results of urine analysis and renalfunctions were normal. Abdominal ultrasound wasnegative for renal and adrenal abnormalities. Apreliminary diagnosis of CAH (congenital adrenalhyperplasia) was made and patient was started onhydrocortisone 50 mg/m2, i.e 1.5 mg thrice a day.Further workup showed serum aldosterone level of2174 mg/dL (10–160 mg/dL), PRA (pre renal activity)of 103.9 ng/mL/hr (0.5–1.19 mg/mL), serum renin levelof 193998 ng/dL/hr (0.29–3.7 ng/dL/hr), carbondioxide of 19 mmol/L (normal levels in brackets).Normal levels were found for serum cortisol,androstenedione, 17OH progesterone, ACTH, FSH, LHand pregnenolone.In view of the laboratory results hydrocortisone wasdiscontinued and he was started on oral fludrocortisone0.1 mg twice daily along with NaCl supplements.Potassium lowering therapies were also used along withsodium supplementation. Patient responded to thetreatment and started gaining weight. He wasdischarged home with medications and weighed 3520gms. On followup he was found to be gainingappropriate weight and was kept on oral saltsupplements alone. Genetic analysis for PHA in thefamily was negative.
DISCUSSIONPseudohypoaldosteronism is a saltwastingsyndrome due to peripheral resistance to aldosterone.This may be either a primary (mutation of MR or ENaC)or a secondary (infection, uropathy, medication)
phenomenon. In all cases, sodium reabsorption andpotassium excretion are impaired in the principal cell ofthe collecting duct. The biological characteristics arehyponatraemia, hyperkalaemia and acidosis [1]. It isthus characterized by three essential features:hyperkalemia, metabolic acidosis and elevatedaldosterone concentration with normal glomerularfiltration rate (GFR). Volume depletion or hypervolemia,renal salt wasting or retention, hypotension orhypertension and elevated, normalhigh or low levels ofrenin and aldosterone may be observed in the variousconditions that result in differentiating this syndrome inthree types of PHA.Pseudohypoaldosteronism type 1: This was firstdescribed in 1958 by Cheek and Perry [2]. This raresyndrome starts during the neonatal period with a widespectrum [1]. PHA1 occurs in two genetic forms, (i) arenal form of autosomal dominant inheritance due to aheterozygous mutation of the mineralocorticoid receptor(MR) gene coding for the mineralocorticoid receptorand, (ii) a severe systemic form of autosomal recessiveinheritance due to a mutation of the epithelial sodiumchannel (EnaC) gene, which is a secondary form usuallyin association with urinary tract malformation and acutepyelonephritis. Autosomal dominant variant is RenalPHA1; while systemic one due to autosomal recessiveinheritance is also known as multiple target organdisorder (MTOD).Renal PHA1 or early childhood hyperkalemia isprobably due to a maturation disorder in the number orfunction of aldosterone receptors and also in sporadiccases. This form manifests with renal salt loss in infancyand failure to thrive and a gradual improvement withadvancing age.In systemic variant, other organs are involved, suchas the sweat glands, salivary glands and colon. Thefundamental abnormality in multiple target organ defect(MTOD) PHA1 is a lossoffunction mutation in thealpha or beta subunits of the epithelial sodium channel(ENaC) gene, resulting in defective sodium transport inmany organs containing the ENaC gene, (e.g., kidney,lung, colon, sweat and salivary glands). This amiloridesensitive member of the degenerin/epithelial sodiumchannel (Deg/ENaC) super family of ion channels iscomprised of three homologous units (alpha, beta andgamma) and is expressed in the apical membrane ofepithelial cells lining the airway, colon, and distalnephron. ENaC plays an essential role in transepithelialNa+ and fluid balance. PHA1 presents with potentiallifethreatening salt wasting and failure to thrive in earlyinfancy.Pseudohypoaldosteronism type 2: This is alsoknown as familial hyperkalemia and hypertension orGordon syndrome [3, 4]. The classification of thisheterogeneous syndrome as PHA is, however,controversial because plasma aldosteroneconcentrations are highly variable, usually almostnormal, and patients respond adequately tomineralocorticoid hormone [5]. The organ involvementand genetic abnormality in pseudohypoaldosteronismtype 2 (PHA2) is similar to PHA1. The hallmarks ofPHA2 are hypertension, hyperkalemia and correction of

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 5–1 8.www.ijcasereportsandimages.com Bhullar et al. 1 7
these abnormalities by low doses of thiazide diuretics[6, 7].Pseudohypoaldosteronism type 3: This istransient and secondary to various pathologies relatedto kidneys or other organs [7]. Rare cases of majorintestinal resection [8] or sweat gland dysfunctionassociated with excessive loss of sodium [9] have beendescribed as leading to PHA III. However, renal causesare encountered more frequently. Nephropathies suchas obstructive uropathy [10] or urinary tract infection[11] are reported as causes of transient aldosteroneresistance [7]. The main characteristic of this type ofPHA is a decreased GFR .In all cases of PHA, sodium reabsorption andpotassium excretion are impaired in the principle cellsof collecting ducts. The biological characteristics arehyponatremia, hyperkalemia and metabolic acidosis.After having excluded pseudohyperkalemia due tohemolysis, the diagnosis may be challenging. If serumchloride is normal while serum sodium has decreasedand GFR is not impaired, type4 renal tubular acidosiscan be ruled out. The normal hormone levels of ACTH,17OH progesterone and cortisol allows exclusion ofadrenal insufficiency. Finally, high aldosterone andplasma renin levels lead to the diagnosis of PHA [1].We diagnosed our patient as a case of PHA1 which ischaracterized by neonatal salt wasting, vomiting,dehydration and failure to thrive. We would like tosummarize our observations in the management of thispatient. In our case, diagnosis of PHA1 was based onplasma electrolyte assessment, high renin activity, highaldosterone levels, low level of carbon dioxide withnormal renal function resulting from a renal or systemicresistance to aldosterone. Normal levels of cortisol,17OH progestrone, ACTH and androstenedioneexcluded the diagnosis of congenital adrenalhyperplasia and other corticoid dysfunctions. By doingupper gastrointestinal studies, upper gastrointestinalabnormalities were excluded. Normal ultrasound ofkidneys and adrenals excluded anatomicalabnormalities. We suspected the sporadic form of PHA1in our patient and treated him with sodiumsupplementation which normalized his clinical stateand serum electrolytes. There was good response tosodium chloride supplementation and he was thrivingwell on follow up.
CONCLUSIONWhile evaluating any infant with suspected CAH,one should consider pseudohypoaldosteronism as oneof the differential and infants who present withelectrolyte imbalance like hyperkalemia, hyponatremiaand weight loss should be evaluated for adrenocorticalfunction and need careful management. Geneticanalysis should be done as the disease can have agenetic predisposition or may be sporadic. Though PHAis a group of rare syndromes, but a high degree ofsuspicion along with extensive laboratory workupshould be pursued in cases with electrolyte imbalances.
A multidisciplinary team approach including aneonatologist, an endocrinologist, genetic expert and adietician is essential for evaluation of longitudinalgrowth and neurological development in PHA patients.*********
Author ContributionsSuman Preet Kaur Bhullar – Conception and design,Acquisition of data, Analysis and interpretation of data,Drafting the article, Critical revision of the article, Finalapproval of the version to be publishedRauf Seifeldin – Conception and design, Acquisition ofdata, Analysis and interpretation of data, Drafting thearticle, Critical revision of the articleNikhil Hemady – Drafting the article, Critical revision ofthe article, Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Suman Preet Kaur Bhullar et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properlycredited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Belot A, Ranchin B, Fichtner C, et al.Pseudohypoaldosteronisms, report on a 10patientseries. Nephrol Dial Transplant 2008May;23(5):1636–41.2. Cheek DB, Perry JW. A salt wasting syndrome ininfancy. Arch Dis Child 1958;33(169):252–6.3. Gordon RD, Geddes RA, Pawsey CG, O'HalloranMW. Hypertension and severe hyperkalaemiaassociated with suppression of renin andaldosterone and completely reversed by dietarysodium restriction. Australas Ann Med1970;19(4):287–94.4. Gordon RD. Syndrome of hypertension andhyperkalemia with normal glomerular filtration rate.Hypertension 1986;8(2):93–102.5. Stokes JB. Disorders of the epithelial sodiumchannel: Insights into the regulation of extracellularvolume and blood pressure. Kidney Int1999;56(6):2318–33.6. DisseNicodeme S, Achard JM, Desitter I, et al. Anew locus on chromosome 12p13.3 forpseudohypoaldosteronism type II, an autosomaldominant form of hypertension. Am J Hum Genet2000;67(2):302–10.7. Olivier Bonny, Bernard C Rossier. Disturbances ofNa/K Balance: Pseudohypoaldosteronism Revisited.J Am Soc Nephrol 2002;13(9):2399–414.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 5–1 8.www.ijcasereportsandimages.com
8. Vantyghem MC, Hober C, Evrard A, et al. Transientpseudohypoaldosteronism following resection of theileum: normal level of lymphocytic aldosteronereceptors outside the acute phase. J EndocrinolInvest 1999;22(2):122–7.9. Anand SK, Froberg L, Northway JD, Weinberger M,Wright JC. Pseudohypoaldosteronism due to sweatgland dysfunction. Pediatr Res 1976;10(7):677–82.10. Bülchmann G, Schuster T, Heger A, Kuhnle U,Joppich I, Schmidt H. Transient
Bhullar et al. 1 8
Access full text article onother devices Access PDF of article onother devices
pseudohypoaldosteronism secondary to posteriorurethral valve–A case report and review of theliterature. Eur J Pediatr Surg 2001;11(4):277–9.11. PerezBrayfield MR, Gatti J, Smith E, Kirsch AJ.Pseudohypoaldosteronism associated withureterocele and upper pole moiety obstruction.Urology 2001;57(6):1178.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 9–23.www.ijcasereportsandimages.com
Community acquired methicillin sensitive Staphylococcusaureus bacteremia, meningitis and brain abscess: A uniquepresentationCarlos Gonzalez, Juan Roa, Nehad Shabarek
ABSTRACTIntroduction: Staphylococcus aureus causingisolate central nervous system (CNS) infection israre. It is mostly related to either neurosurgicalintervention or previous local or systemicinfection rather than spontaneous isolatedinfection in adults. Case Report: We report acase of a previously healthy 19yearold malewith no risk factors of Staphylococcus aureusinfection, who was found to have methicillinsensitive Staphylococcus aureus (MSSA)bacteremia, meningitis and cerebral abscess,without an apparent source of infection.Conclusion: Our case is a unique presentation ofa young male presenting with isolated CNSinfection by MSSA. This case highlights thechallenge of early diagnosis of brain abscess, anentity that presents very often with nonspecificsigns, the diagnosis of which can be easilymissed, with repercussions on longtermdisability and mortality.
Keywords: Staphylococcus aureus, Bacteremia,Meningitis, Brain abscess*********
Gonzalez C, Roa J, Shabarek N. Community acquiredmethicillin sensitive Staphylococcus aureus bacteremia,meningitis and brain abscess: A unique presentation.International Journal of Case Reports and Images2013;4(1):19–23.*********
doi:10.5348/ijcri201301251CR5
INTRODUCTIONStaphylococcus aureus is the most virulent of thestaphylococcal species with unique versatility to causevarious types of infections, from community acquiredmild skin infections to highly lethal infections such asnecrotizing pneumonia, endocarditis or central nervoussystem (CNS) infections. CNS infections caused byStaphylococcus aureus are in general uncommon.Various series report Staphylococcus aureus as thefourth or fifth most common etiology for neuroinfection; more commonly as cause of brain abscess andless commonly as an isolated cause of meningitis (lessthan 5%) [1–5, 6]. In either case, CNS infection causedby Staphylococcus aureus is related to recentneurosurgical intervention or associated with systemicor local infection classically described as a complicationof endocarditis or soft tissue infection [1–2, 6].Although there are a few case reports of CNS infectionin patients without an apparent source, most of themare related to community acquire methicillin resistantStaphylococcus aureus (CAMRSA) infections [7]. Todate, we have not found any reported case of confirmedmeningitis with brain abscess, caused by MSSAinfection in a previous healthy patient with no
CASE REPORT OPEN ACCESS
Carlos Gonzalez1 , Juan Roa2, Nehad Shabarek3
Affi l iations: 1PGY-1 Internal Medicine, Lincoln Medical andMental Health Center of Weil l Medical College at CornellUniversity, New York, USA; 2PGY-2 Internal Medicine,Lincoln Medical and Mental Health Center of Weil l MedicalCollege at Cornell University, New York, USA; 3Attendingphysian, associatted program director, Department ofInternal Medicine, Lincoln Medical and Mental HealthCenter of Weil l Medical College at Cornell University, NewYork, USA.Corresponding Author: Carlos Gonzalez, MD Departmentof Internal Medicine, Lincoln Medical and Mental HealthCenter, 234 E. 1 49th, St #8-32, Bronx, NY 1 0451 ; Ph:71 85794739; Fax: 71 85794836; Email :[email protected];carlos.gonzalez. [email protected]
Received: 08 Apri l 201 2Accepted: 28 July 201 2Published: 01 January 201 3
Gonzalez et al. 1 9

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 9–23.www.ijcasereportsandimages.com Gonzalez et al. 20
predisposing infection or risk factors. We report a caseof MSSA bacteremia with meningitis and brain abscessin a previous healthy young male without any riskfactors and no obvious source of infection, the detectionof which was delayed due to a normal brain noncontrast computed tomography (CT) scan findings.
CASE REPORTA 19yearold Hispanic male student, presented tothe emergency department (ED) complaining of dullfrontal and occipital headache associated with fever, fewepisodes of vomiting, poor oral intake and myalgia fortwo days. His past medical history was only remarkablefor a recent dental procedure. He denied any sickcontacts and drug abuse. Initial physical examinationwas remarkable for tachycardia of 123 bpm andtemperature of 38°C. No meningeal signs or focaldeficits were found on the initial presentation. Theremaining physical examination was unremarkable.Initial laboratory work up was positive for mildhyperglycemia with blood sugar 143 mg/dL, Na 132mEq/dL and neutrophilia 92% with WBC count11x103/mm3. Brain CT scan and lumbar puncture (LP)were performed to exclude meningitis or anyintracranial pathology. Both the investigations werenegative. The patient received symptomaticmanagement with IV fluids and non steroidal antiinflammatory drugs (NSAIDs) for fever, with notableimprovement. He was discharged home from the EDwith symptomatic management. One day after the initialpresentation, blood cultures were reported positive forMSSA. Multiple attempts to contact the patient and hisfamily were unfortunately unsuccessfully.Four days after the initial presentation, the patientwas brought to the ED again, after being foundunresponsive. Patient’s mother stated he was doing welluntil early morning when he complained of malaise,fever and headache. Physical examination was positivefor temperature of 40.5°C. Other vital signs werenormal. The patient was lethargic, found to have nuchalrigidity and was only responsive to painful stimuli.Glasgow coma scale (GCS) was 9/15. Mild patchyfolliculitis was noted in the suprapubic area withcrusted lesions. He was intubated and transfered to themedical intensive care unit (MICU) with a diagnosis ofpossible meningitis and MSSA bacteremia based on hisprevious blood culture. Repeat LP and blood culturewere performed. Lumbar puncture showedxanthochromia with RBC 11/mm3, WBC count 817/mm3
(neutrophils 90%, lymphocytes 2%, monocytes 8%),glucose 28 mg/dL and protein 126 mg/dL. Bloodcultures grew MSSA with the same pattern of sensitivityas the first culture. Noncontrast brain CT scan wasagain performed, with normal findings (Figure 1). Thepatient was initiated on bacterial meningitis treatmentwith ceftriaxone 2 g q12hr, nafcillin 2 g q4hr, anddexamethazone 4 mg q6hr. Ceftriaxone was switched togentamicin 60 mg qh8hr on day2.During the hospital course in MICU, transthoracicechocardiogram (TTE) was negative for endocarditis,
urine toxicology was negative and HIV status wasnegative. The patient was extubated four days afteradmission, at which time a left palpebral ptsosis wasnoted. His pupils were symmetric, 3 mm in diameter,and reactive. He remained lethargic and a contrastenhanced magnetic resonance imaging (MRI) wasordered due to his lack of neurological improvement.The MRI scan of brain showed a focal lesion centered atthe left anterior thalamus with contiguous extensioninto the subthalamic region and left cerebral peduncle ofthe left midbrain, with a small focus of contralateralextension into the right thalamus (left lesion measure:3.5 cm height, 2 cm anteroposterior and 1.8 transverse,right lesion 0.4 cm diameter) (Figure 2). These findingswere compatible with brain abscess.After eight days of medical treatment andobservation with the patient was sent for stereotacticbiopsy and aspiration of the abscess due to noneurological improvement. The procedure was donewithout complication and patient started showing signsof improvement. He became more alert and followedcommands. Pathology reported gliotic brain tissue withorganizing necrosis and hemorrhage without purulentinflammation. Fluid culture was negative.Ten days after treatment with nafcillin, the patientpresented with diffuse erythematous rash involving thepalm of the hands. Biopsy of the lesions was remarkablefor interface dermatitis, compatible with a drugeruption. Nafcillin was then switched to vancomycin1 g q12hr. Patient remained stable and was transferredto medical floor and subsequently to rehabilitation
Figure 1: Noncontrast computed tomography scan of brainshowing normal finding.
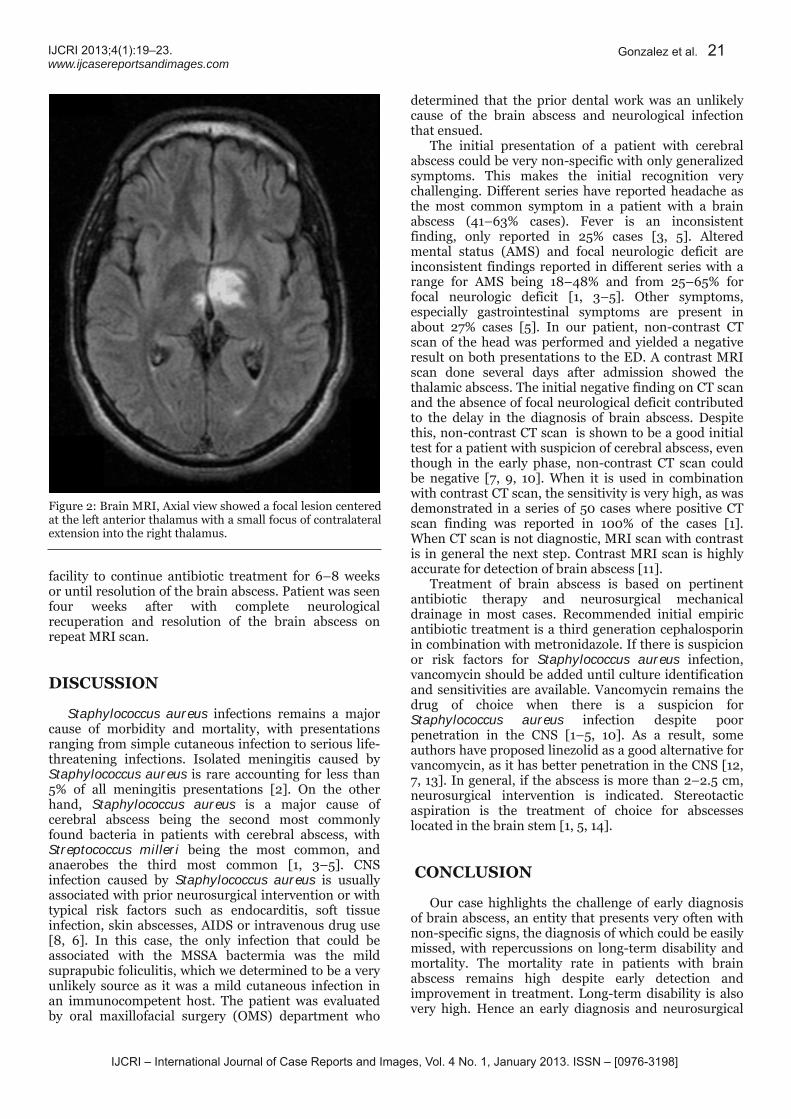
IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 9–23.www.ijcasereportsandimages.com Gonzalez et al. 21
facility to continue antibiotic treatment for 6–8 weeksor until resolution of the brain abscess. Patient was seenfour weeks after with complete neurologicalrecuperation and resolution of the brain abscess onrepeat MRI scan.
DISCUSSIONStaphylococcus aureus infections remains a majorcause of morbidity and mortality, with presentationsranging from simple cutaneous infection to serious lifethreatening infections. Isolated meningitis caused byStaphylococcus aureus is rare accounting for less than5% of all meningitis presentations [2]. On the otherhand, Staphylococcus aureus is a major cause ofcerebral abscess being the second most commonlyfound bacteria in patients with cerebral abscess, withStreptococcus milleri being the most common, andanaerobes the third most common [1, 3–5]. CNSinfection caused by Staphylococcus aureus is usuallyassociated with prior neurosurgical intervention or withtypical risk factors such as endocarditis, soft tissueinfection, skin abscesses, AIDS or intravenous drug use[8, 6]. In this case, the only infection that could beassociated with the MSSA bactermia was the mildsuprapubic foliculitis, which we determined to be a veryunlikely source as it was a mild cutaneous infection inan immunocompetent host. The patient was evaluatedby oral maxillofacial surgery (OMS) department who
determined that the prior dental work was an unlikelycause of the brain abscess and neurological infectionthat ensued.The initial presentation of a patient with cerebralabscess could be very nonspecific with only generalizedsymptoms. This makes the initial recognition verychallenging. Different series have reported headache asthe most common symptom in a patient with a brainabscess (41–63% cases). Fever is an inconsistentfinding, only reported in 25% cases [3, 5]. Alteredmental status (AMS) and focal neurologic deficit areinconsistent findings reported in different series with arange for AMS being 18–48% and from 25–65% forfocal neurologic deficit [1, 3–5]. Other symptoms,especially gastrointestinal symptoms are present inabout 27% cases [5]. In our patient, noncontrast CTscan of the head was performed and yielded a negativeresult on both presentations to the ED. A contrast MRIscan done several days after admission showed thethalamic abscess. The initial negative finding on CT scanand the absence of focal neurological deficit contributedto the delay in the diagnosis of brain abscess. Despitethis, noncontrast CT scan is shown to be a good initialtest for a patient with suspicion of cerebral abscess, eventhough in the early phase, noncontrast CT scan couldbe negative [7, 9, 10]. When it is used in combinationwith contrast CT scan, the sensitivity is very high, as wasdemonstrated in a series of 50 cases where positive CTscan finding was reported in 100% of the cases [1].When CT scan is not diagnostic, MRI scan with contrastis in general the next step. Contrast MRI scan is highlyaccurate for detection of brain abscess [11].Treatment of brain abscess is based on pertinentantibiotic therapy and neurosurgical mechanicaldrainage in most cases. Recommended initial empiricantibiotic treatment is a third generation cephalosporinin combination with metronidazole. If there is suspicionor risk factors for Staphylococcus aureus infection,vancomycin should be added until culture identificationand sensitivities are available. Vancomycin remains thedrug of choice when there is a suspicion forStaphylococcus aureus infection despite poorpenetration in the CNS [1–5, 10]. As a result, someauthors have proposed linezolid as a good alternative forvancomycin, as it has better penetration in the CNS [12,7, 13]. In general, if the abscess is more than 2–2.5 cm,neurosurgical intervention is indicated. Stereotacticaspiration is the treatment of choice for abscesseslocated in the brain stem [1, 5, 14].
CONCLUSIONOur case highlights the challenge of early diagnosisof brain abscess, an entity that presents very often withnonspecific signs, the diagnosis of which could be easilymissed, with repercussions on longterm disability andmortality. The mortality rate in patients with brainabscess remains high despite early detection andimprovement in treatment. Longterm disability is alsovery high. Hence an early diagnosis and neurosurgical
Figure 2: Brain MRI, Axial view showed a focal lesion centeredat the left anterior thalamus with a small focus of contralateralextension into the right thalamus.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 9–23.www.ijcasereportsandimages.com Gonzalez et al. 22
intervention could prove significant in the final outcomefor these patients.*********
Author ContributionsCarlos Gonzalez — Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedJuan Roa — Substantial contributions to conception anddesign, Acquisition of data, Analysis and interpretationof data, Drafting the article, Revising it critically forimportant intellectual content, Final approval of theversion to be publishedNehad Shabarek — Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Carlos Gonzalez et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distribution andreproduction in any means provided the original authorsand original publisher are properly credited. (Please seewww.ijcasereportsandimages.com/copyrightpolicy.phpfor more information.)
REFERENCES1. Lakshmi V, Rao RR, Dinakar I. Bacteriology of brainabscessobservations on 50 cases. J Med Microbiol1993 Mar;38(3):187–90.2. Roberts FJ, Smith JA, Wagner KR. Staphylococcusaureus meningitis: 26 years' experience at VancouverGeneral Hospital. Can Med Assoc J 1983 Jun15;128(12):1418–20.3. Sharma R, Mohandas K, Cooke RP. Intracranialabscesses: changes in epidemiology andmanagement over five decades in Merseyside.Infection 2009 Feb;37(1):39–43.4. Roche M, Humphreys H, Smyth E, et al. A twelveyear review of central nervous system bacterialabscesses; presentation and aetiology. Clin MicrobiolInfect 2003 Aug;9(8):803–9.5. Carpenter J, Stapleton S, Holliman R. Retrospectiveanalysis of 49 cases of brain abscess and review ofthe literature. Eur J Clin Microbiol Infect Dis 2007Jan;26(1):1–11.6. Aguilar J, UrdayCornejo V, Donabedian S, Perri M,Tibbetts R, Zervos M. Staphylococcus aureus
meningitis: case series and literature reviewe.Medicine (Baltimore) 2010 Mar;89(2):117–25.7. Naesens R, Ronsyn M, Druwé P, Denis O, Ieven M,Jeurissen A. Central nervous system invasion bycommunity acquired meticillinresistantStaphylococcus aureus. Journal of MedicalMicrobiology 2009;58(Pt 9):1247–51.8. Pintado V, Meseguer MA, Fortún J, et al. Clinicalstudy of 44 cases of Staphylococcus aureusmeningitis. Eur J Clin Microbiol Infect Dis 2002Dec;21(12):864–8.9. Lo BM, Erwin EA. Missed epidural brain abscessafter furunculosis. Am J Emerg Med. 2008May;26(4):522.e3–4.10. Muzumdar D, Jhawar S, Goel A. Brain abscess: Anoverview. Int J Surg 2011;9(2):136–44.11. Haimes AB, Zimmerman RD, Morgello S, et al. MRimaging of brain abscesses. AJR Am J Roentgenol1989;152(5):1073–85.12. Rupprecht TA, Pfister HW. Clinical experience withlinezolid for the treatment of central nervous systeminfection. Eur J nuerol 2005;12(7):536–42.13. Saito N, Aoki K, Sakurai T, et al. Linezolid treatmentfor intracranial abscesses caused by methicillinresistant Staphylococcus aureustwo case reports.Neurol Med Chir (Tokyo) 2010;50(6):515–7.14. Nishihara M, Sasayama T, Kudo H, Kohmura E.Morbidity of stereotactic biopsy for intracraniallesions. Kobe J Med Sci 2011 Jan 21;56(4):E148–53.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):1 9–23.www.ijcasereportsandimages.com Gonzalez et al. 23
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 3 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;3(1 ):24–27.www.ijcasereportsandimages.com
Reversible myeloneuropathy and pancytopenia related tocopper deficiency from gastric bypass surgery: A casereportLaide Bello, Joseph Fiore
ABSTRACTIntroduction: Weight loss surgery has becomean increasingly popular means of combating theobesity epidemic in modern society but like anyprocedure, it does not shy away from immediateand longterm complications. Copper deficiencyhas occasionally been reported to occur manyyears afterwards but with an increasedincidence of bariatric procedures and reducedawareness, the effects of this deficiency couldnow appear to favor an earlier onset. CaseReport: We report a case of a 56yearoldCaucasian female with a history of gastricbypass surgery five year ago; with an unsteadygait, weakness, decreased visual acuity, tinglingwith numbness in her hands and pancytopeniafor the last month. She was treated for copperdeficiency. Conclusion: Effects of copperdeficiency have been shown to cause a widearray of abnormalities related to inactivation ofenzymes such as cytochrome c oxidase,superoxide dismutase, dopamine betahydroxylase and metallothionein. This can leadto reduced nerve transmission within thecentral nervous system causing motor and
sensory polymyeloneuropathy and an overallreduction of energy required for blood cellformation. With early surveillance, suchanomalies can be detected and potentiallyreverse the effects of this micronutrientdeficiency.Keywords: Myeloneuropathy, Hemoglobinopathy,Micronutrient deficiency
*********Bello L, Fiore J. Reversible myeloneuropathy andpancytopenia related to copper deficiency from gastricbypass surgery: A case report. International Journal ofCase Reports and Images 2013;3(1):24–27.
*********doi:10.5348/ijcri201301252CR6
INTRODUCTIONMorbid obesity is one of the major risk factorsassociated with chronic diseases and conditions such asheart disease, cancer, stroke, diabetes and hypertension[1]. Given this impact on morbidity and mortality in the21st century, weight loss surgery options are bound tobe more prevalent than ever before and the RouxenYgastric bypass remains the most common type done inthe United States [2]. This procedure involves dividingthe stomach into a small upper pouch andanastomosing it to a distal segment of the jejunum thuscreating a gastrojejunostomy for drainage of gastricremnant contents, bile and pancreatic enzymes. Thisbypass potentially eliminates common micronutrientssuch as iron, vitamin B12, calcium and vitamin D frombeing absorbed through the latter stomach and initialpart of the small intestine [3–5]. There have been fewreports of other complications such as copper deficiencywhich caused detrimental longterm outcomes such as
CASE REPORT OPEN ACCESS
Laide Bello1 , Joseph Fiore2
Affi l iations: 1Department of Medicine, Steward CarneyHospital, TUFTS University School of Medicine, Boston,MA, 21 00 Dorchester Avenue, Boston, Massachusetts,USA; 2Division of Gastroenterology, Seton Medical OfficeBuilding, Steward Carney Hospital, TUFTS UniversitySchool of Medicine, Boston, MA 021 24, 211 0 DorchesterAvenue, Boston, Massachusetts, USA.Corresponding Author: Laide Bello, MD, MPH,Department of Medicine, Steward Carney Hospital, TUFTSUniversity School of Medicine, Boston, MA, 21 00Dorchester Avenue, Boston, Massachusetts, 021 24. ;Email : Laidebello@hotmail .com
Received: 21 May 201 2Accepted: 20 July 201 2Published: 01 January 201 3
Bello et al. 24

IJCRI – International Journal of Case Reports and Images, Vol. 3 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;3(1 ):24–27.www.ijcasereportsandimages.com Bello et al. 25
myeloneuropathies, anemia, leucopenia and sometimesthrombocytopenia, but with early surveillance theseconditions can be reversed [6, 7].
CASE REPORTA 56yearold Caucasian female presented to ouremergency room with complains of unsteady gait, visualdisturbance, dizziness, fatigue and recurrent tinglingwith numbness in her hands for the past month. Shelost 68.03 kg ever since her gastric bypass surgery fiveyears ago and more recently developed poor appetite,recurrent diarrhea and nausea despite being compliantwith once daily iron, thiamine and folic acidsupplements. Her gait had worsened to the point ofrequiring a cane for ambulation due to frequent falls. Arecent upper endoscopy showed a gastrojejunalanastomotic ulcer (H. Pylori negative) and colonoscopyrevealed mild diverticulosis with several previousexaminations in the past failing to show any clear cutetiology.The past medical history of the patient wassignificant for depression and chronic hepatitis C(Genotype 2). She was a retired home careadministrator, with no family history of gastrointestinalmalignancy and routinely took pantoprazole,citalopram, calcium and vitamin D supplements.On physical examination, patient appeared frail witha body mass index 17.5 kg/m2 (84% of ideal bodyweight), blood pressure 124/84 mmHg, heart rate90/min and respiratory rate 14/min. Neurologicalreview revealed reduced strength and sensation over herlower extremities, decreased ankle jerk, ataxic gait andmoderate loss of vibratory and joint position sense inthe toes. An ophthalmologic examination showedreduced visual acuity and protracted optic disc swelling.The rest of the physical examination was normal. Herlaboratory examination was unique for a white bloodcell count 2.2x103/mm3 (nadir 1.4x103/mm3),hemoglobin 8.9 g/dL, mean corpuscular volume (MCV)72 fl and platelet count 8.8x105/mm3. Her iron, percentsaturation and vitamin B12 levels were preserved in ahigh normal range at 76 μg/dL, 40% and 652 pg/mL,respectively. A hepatitis C viral load was undetectablewhile creatinine phosphokinase remained withinnormal limits. Serologies for lyme titer and syphiliswere undetectable. A brain and entire spine magneticresonance imaging was otherwise normal except for
minimal T2 hyperintensities within the periventricularwhite matter suggesting demyelination [8–10]. Hercerebrospinal fluid analysis was essentiallyunremarkable with no evidence of oligoclonal banding.An electroencephalogram recording revealed subtleslowing of the background suggestive of mildencephalopathy with no epileptiform activity and nerveconduction studies showed some motor and sensorypolyneuropathy affecting different parts of her upperand lower extremities. At that juncture given her historyof gastric bypass surgery, ongoing pancytopenia andcomplains of dizziness with unsteady gait, it was decidedto assess for copper deficiency. This was seen low at0.44 μg/mL (normal range 0.75–1.45 μg/mL) along withzinc at 0.37 μg/mL (normal range 0.66–1.1 μg/mL) buthad a normal ceruloplasmin level. The 24 hour urinecollection for copper was also low at 9 μg/L (normalrange 15–60 μg/L) and over the next three days shereceived a once daily intravenous infusion (containing 1mg of copper) in dextrose water along with a highpotency Women’s Ultra Mega vitamin supplement fourtimes a day. This contained 2 mg of copper, several fatand water soluble vitamins as well as trace elements likemanganese, chromium, selenium, magnesium and zinc.Over the next four to five days, her gait and visionimproved remarkably with increased acuity andresolution of the optic disc swelling on examination. Sheno longer required any assistance with ambulation aftera week, and was subsequently discharged on oral coppersupplements. Table 1 gives her followup laboratory dataafter the first and second months. On subsequent followups in the outpatient clinic, the patient had shown greatimprovement with her overall strength and ambulationbut still had some lingering but subtle tingling withnumbness in the hands.
DISCUSSIONCopper remains an essential nutrient serving as aceruloplasmin cofactor in the formation of transferring[11]. It thus facilitates iron uptake and ensures adequatered and white blood cell formation. Copper also plays acritical role in activating enzymes such as cytochrome coxidase, superoxide dismutase, dopamine betahydroxylase, metallothionein and its deficiency can leadto reduced nerve transmission within the centralnervous system and less adenosine triphosphateproduction for synthesis of hemoglobin [5]. The actual
Table 1: Laboratory data compared from admission and one month later after copper supplementation

IJCRI – International Journal of Case Reports and Images, Vol. 3 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;3(1 ):24–27.www.ijcasereportsandimages.com Bello et al. 26
mechanism on how neutropenia occurs is still unknown,but Lazarchick et al. suggested an inhibition ofdifferentiation and selfrenewal of CD34 positivehematopoietic progenitor cells as a likely cause [12].Most cases of copper deficiency myeloneuropathytypically occur after a few decades of gastric bypasssurgery but in our patient the symptoms were seen onlyafter a few years [8, 13]. Shorter gastrointestinal tractsmay cause reduced sites for reabsorption and a lack ofmicronutrient replacement can compound thisdeficiency. Zinc can interfere with copper metabolismsince they compete for absorption via the same site andO’Donnell et al. advised against simultaneoussupplementation in situations where both are found tobe deficient [14]. Previous studies have linkedhyperzincemia from toxic exposures as a potential causeof copper deficiency but this was not the case in ourpatient. With varying degrees of copper deficiency,patients may not necessarily have all the signs andsymptoms listed and in order to make the diagnosis aclinician would need to have a high index of suspicion,along with demonstrable low copper levels. Theoccurrence of longterm irreversible neurologicaldamage is not known and as such it is paramount toconsider early surveillance. Kumar et al. have alsostudied the value of urinary copper as a measure of itsdeficiency but concluded that a serum copper levelremains the best and most reliable assay [8]. An initialintravenous dose of 1 mg of copper is advised for thefirst three days after which patients can continue on oralsupplementation of 8 mg of copper gluconate daily [15].Our patient received these and blood levels for coppergradually normalized over the next two months alongwith other respective hematologic parameters. She didnot require any blood transfusions during her stay andthe abatement of her symptomatology was quiteimpressive over the immediate days to weeks ofcommencing therapy. In the absence of other causes forpancytopenia, blood levels usually improve or normalizeanywhere within three days to six months aftersupplementation [11, 15–17]. The actual thresholdbetween copper concentrations, tissue stores andneurological sequelae remains to be established andmore studies shall be required in the future to establishthis. The myeloneuropathy described here can alsomimic subacute combined degeneration typically seenwith vitamin B12 deficiency, as such this should also beassessed and treated promptly. Even though serial levelsof serum copper measured over time was seen to rise,potential confounding effects could exist with variousvitamins and trace elements contained in the brandedhigh potency Women's Ultra Mega vitamin supplement.
CONCLUSIONEarly surveillance for copper deficiency has itsbenefits and ought to be routinely evaluated after apatient undergoes gastric bypass surgery as this givesthe clinician an avenue to identify preventable andreversible causes of blood cell disorders, leukemic
transformation and polyneuropahthies that wouldotherwise have been termed idiopathic.*********
Author ContributionsLaide Bello – Substantial contributions to conceptionand design, Analysis and interpretation of data,Drafting the article, revising it critically for importantintellectual content, Final approval of the version to bepublishedJoseph Fiore – Substantial contributions to conceptionand design, Analysis and interpretation of data,Revising it critically for important intellectual content,Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Laide Bello et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Health statistics, United States 2009. Retrievedfromhttp://www.cdc.gov/nchs/data/hus/hus09.pdf#0472. John S, Hoegerl C. Nutritional Deficiencies aftergastric bypass surgery. Journal of AmericanOsteopathic Association 2009;109(11):601–4.3. AlvarezLeite JI. Nutrient deficiencies secondary tobariatric surgery. Current Opinion in ClinicalNutritional Metabolic Care 2004;7(5):569–75.4. Aills L, Blankenship J, Buffington C, Furtado M,Parrott J. ASMBS allied health nutritionalguidelines for the surgical weight loss patient.Surgical Obesity Related Diseases 2008:4(5Suppl):S73–108.5. Linder MC, HazeghAzam M. Copper biochemistryand molecular biology. American Journal of ClinicalNutrition 1996;63(5):797S–811S.6. Huff JD, Keung YK, Thakuri M, et al. Copperdeficiency causes reversible myelodysplasia.American Journal of Hematology2007;82(7):625–30.7. Kumar N, Elliott MA, Hoyer JD, Harper CM Jr,Ahlskog JE, Phyliky RL. "Myelodysplasia,"myeloneuropathy, and copper deficiency. MayoClinic Proceedings 2005;80(7):943–6.8. Kumar N, Gross JB Jr, Ahlskog JE. Copperdeficiency myelopathy produces a clinical picturelike subacute combined degeneration. Neurology2004;63(1):33–9.

IJCRI – International Journal of Case Reports and Images, Vol. 3 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;3(1 ):24–27.www.ijcasereportsandimages.com Bello et al. 27
9. Spinazzi M, De Lazzari F, Tavolato B, Angelini C,Manara R, Armani M. Myeloopticoneuropathy incopper deficiency occurring after partialgastrectomy. Do small bowel bacterial overgrowthsyndrome and occult zinc ingestion tip the balance?Journal of Neurology 2007;254(8):1012–7.10. Hedera P, Peltier A, Fink JK, Wilcock S, London Z,Brewer GJ. Myelopolyneuropathy and pancytopeniadue to copper deficiency and high zinc levels ofunknown origin II. The denture cream is a primarysource of excessive zinc. Neurotoxicology2009;30(6):996–9.11. http://www.drugs.com. Copper information fromDrugs.com; [Revised: 08/2011 Hospira, Inc.; Cited:2012 March 4]. Available from:http://www.drugs.com/pro/copper.html.12. Lazarchick J. Update on anemia and neutropenia incopper deficiency. Current Opinion on Hematology2012 Jan;19(1):58–60.13. Wu J, Ricker M, Muench J. Copper deficiency ascause of unexplained hematologic and neurologic
deficits in patient with prior gastrointestinal surgery.Journal of American Board of Family Medicine2006;19(2):191–4.14. O'Donnell KB, Simmons M. EarlyOnset CopperDeficiency Following RouxenY Gastric Bypass.Nutrition in Clinical Practice 2011;26(1):66–9.15. Shahidzadeh R, Sridhar S. Profound copperdeficiency in a patient with gastric bypass. AmericanJournal of Gastroenterology 2008;103(10):2660–2.16. Griffith DP, Liff DA, Ziegler TR, Esper GJ, WintonEF. Acquired copper deficiency: a potentially seriousand preventable complication following gastricbypass surgery. Obesity (Silver Spring)2009;17(4):827–31.17. Kelkar P, Chang S, Muley SA. Response to oralcopper supplementation in copper deficiencymyeloneuropathy. Journal of ClinicalNeuromuscular Disease 2008;10(1):1–3.
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):28–31 .www.ijcasereportsandimages.com
Spinal tuberculomas mimicking spinal dural arteriovenousfistula: A case reportJyoti Sureka, Varsha Mary Khalkho, Binita Riya Chacko
ABSTRACTIntroduction: Tuberculosis is a very commondisease in developing countries and has beenfound to affect almost all the parts of the body.We report the case of a patient who had spinalcord tuberculomas without evidence ofsymptoms of systemic tuberculosis. The lesionswere located at the surface of lower thoraciccord and mimicked a spinaldural arteriovenousfistula (SDAVF) on magnetic resonance imaging(MRI). Case Report: The patient was 45yearoldman who presented with a history of progressiveparaparesis with a clinical suspicion ofintramedullary tumor. First diagnosis was madeas SDAVF on MRI. Then he underwentdiagnostic and therapeutic digital substractionangiogram which was negative for the same.Again MRI was reviewed by a senior radiologistand a final diagnosis of spinal cord pial surfacetuberculomas was made, confirmed bycerebrospinal fluid analysis and treated byappropriate antitubercular therapy.Conclusion: Tuberculosis can mimic a numberof disease entitiles. It is important to be familiarwith various a typical radiological presentationsof tuberculosis.
Keywords: Tuberculoma, Spinal dural,Arteriovenous malformation*********
Sureka J, Khalkho VM, Chacko BR. Spinal tuberculomasmimicking spinal dural arteriovenous fistula: A casereport. International Journal of Case Reports andImages 2013;4(1):28–31.*********
doi:10.5348/ijcri201301253CR7
INTRODUCTIONTuberculosis (TB) is a very common disease indeveloping countries and has been found to affectalmost all the parts of the body. It can affect any organor organ system of the body. Tuberculosis primarilyaffects the chest and can involve multipleextrapulmonary sites like heart, bones, joints,gastrointestinal system, genitourinary system, centralnervous system, eyes, etc. Spinal TB accounts for morethan half of the musculoskeletal TB. The extraduralform of spinal TB is most common [1]. Uncommonly, itcan present as arachnoidal, intradural extramedullaryand intramedullary form. Spinal intraduralintramedullary tuberculoma is extremely rare entity cangive rise to a variety of clinical and radiologic featureswhich can mimic a number of other spinal cord lesionsparticularly intramedullary tumors [2, 3]. We reportthe case of a patient who had spinal cord tuberculomaswithout evidence of symptoms of systemic tuberculosis.The lesions were located at the surface of lower thoraciccord and mimicked a spinal dural arteriovenous fistula(SDAVF) on magnetic resonance imaging (MRI).
CASE REPORT OPEN ACCESS
Jyoti Sureka1 , Varsha Mary Khalkho2, Binita Riya Chacko3
Affi l iations: 1 Associate Professor, Department ofRadiology, Christian Medical College and Hospital,Vellore, Tamil Nadu, India; 2Tutor, Department ofRadiology, Christian Medical College and Hospital,Vellore, Tamil Nadu, India; 3Assistant Professor,Department of Radiology, Christian Medical College andHospital, Vellore, Tamil Nadu, IndiaCorresponding Author: Jyoti Sureka, Vellore, Tamil Nadu -632004, India: Ph: +91 9894584953; Fax: +91 04162232035; Email : drjyoticmch@rediffmail .com
Received: 28 May 201 2Accepted: 29 July 201 2Published: 01 January 201 3
Sureka et al. 28

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):28–31 .www.ijcasereportsandimages.com Sureka et al. 29
CASE REPORTA 45yearold male presented to the neurology outpatient department (OPD) with complaints of back painand progressive lower limb weakness since six months.There were no bowel or bladder difficulties. On physicalexamination, there was spine tenderness over the midand lower thoracic spine, muscle weakness andabnormal reflexes in lower limbs. Laboratoryinvestigations revealed positive results for humanimmunodeficiency virus (HIV) and hepatitis B virus(HBV). However, complete blood picture, ESR, Creactive protein and other routine blood examinationswere within normal limits. Chest Xray was also normal.Based on clinical findings possibility of intramedullarytumor was considered. Patient underwent gadoliniumenhanced MRI of spine. Sagittal and coronal T2weighted MRI of thoracolumbar spine showed multipletortuous flow voids along the surface of mid and lowerthoracic spinal cord. The lower thoracic cord was alsoslightly enlarged and showed increased intramedullarysignal intensity (Figure 1). Diagnosis of SDAVF withedema or ischemia of cord secondary to venoushypertension was made. For further management,patient underwent diagnostic and therapeutic selectivespinal DSA. The angiogram did not reveal any feedingvessels or nidus to suggest arteriovenous fistula (AVF)(Figure 2A–B). MRI images were further reviewed by asenior radiologist with a referring doctor. The coronalgadoliniumenhanced T1weighted images showedmultiple, small, almost similar sized ring and nodularenhancing lesions along the pial surface of the cord withcentral T2 hypointensity which appeared as a flow voidon T2weighted sagittal and coronal images (Figure 3).There was no focal enhancing lesion or abnormalenhancement in the region of high signal intensity ofthe lower thoracic spinal cord. Considering theimmunocompromised status, a final diagnosis of spinalcord pial surface tuberculomas with associated cordedema or myelitis was made. MRI of the brain did notreveal any lesion. Patient underwent CSF analysis whichgrew acidfast bacilli and isolated species wasMycobacterium tuberculosis and hence the diagnosiswas confirmed and first line antitubercular therapy(ATT) was started. The patient responded well to ATT.
DISCUSSIONCentral nervous system TB is commonly seen intropical countries [4]. In immunocompromisedindividuals, the presentation of tubercular lesions maybe atypical and can result in delayed diagnosis [5, 6].Imaging plays an important role in recognition of theseatypical cases that mimic other neurologic conditionsthus helps in early diagnosis and treatment, whichotherwise may result in irreversible neurological sequel[4–6]. A few case reports talk about the isolatedmeningeal or spinal tuberculoma mimicking spinaltumor. However, spinal TB mimicking a SDAVF not yetreported in literature. Extradural form is the most
common while arachnoiditis, intradural andintramedullary tuberculomas are uncommonpresentation of spinal TB [4, 7]. This atypical form of TBcan occur as a direct extension from the vertebrae, as adownward extension of intracranial tubercularmeningitis, and less commonly as tuberculous lesionsprimarily developing in spinal meninges [5, 8]. MRI isthe imaging modality of choice for these lesions. Spinalcord TB, generally, present as intramedullarytuberculomas with or without myelitis and syrinx.Clinically as well as radiologically, intramedullarytuberculomas may be difficult to differentiate fromspaceoccupying lesions such as primary and metastaticintramedullary spinal tumors and other chronicgranulomatous diseases (sarcoidosis, histiocytosis andbrucellosis) [9]. Commonly the intramedullarytuberculomas have specific MR features and can bediagnosed on imaging. It has a typical “target sign” onT2weighted imaging, demonstrating low signal center(caseous material) surrounded by high signal rim(peripheral infective granulation tissue), which helps todifferentiate tuberculoma from other intramedullarylesions [10, 11]. On intravenous contrast study, theselesions show rounded nodular and ring like peripheralenhancement with nonenhancing center of the lesion. Ifthe lesion has a typical appearance on MRI, and if thepatient has systemic tuberculosis, diagnosis oftuberculoma can be made easily [9–11]. If the patientdoes not have systemic tuberculosis and hasimmunocompromised status, MRI features can beatypical as seen in our case and diagnosis can bedifficult. In our case, all the lesions were of almostsimilar size and appeared as hypointense dots on thesurface of cord on T2weighted images and thus
Figure 1: A 45yearold male presented with progressiveworsening of lower limb weakness since several months. T2weighted (A) mid, (B) parasagittal, and (C) coronal MRI ofmid and lower thoracic spine demonstrate diffuse, lowerthoracic spinal cord, high signal intensity and mild swelling(stars) suggesting cord edema with multiple tortuoushypointense dots (open arrows in sagittal and black arrows incoronal images) giving appearance of flow voids along thesurface of mid and lower thoracic cord.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):28–31 .www.ijcasereportsandimages.com Sureka et al. 30
mimicked SDAVF. Further cord swelling, hyperintensityand involvement of lower thoracic cord again supportedthe diagnosis of SDAVF. During the initial MRIevaluation, gadolinium enhanced imaging wasoverlooked and thus the lesion was mistakenlydiagnosed as SDAVF and patient had to undergo DSA.Gadolinium enhanced images clearly demonstrated ringand nodular enhancement of all the T2 hypointenselesions thus helped in making the final diagnosis oftuberculomas.
CONCLUSIONTuberculosis has a variety of clinical and radiologicfeatures and can mimic a number of other diseaseentities. Therefore it is important to be familiar with thevarious atypical radiological presentations of TBparticularly in immunocompromised patients to ensureearly, accurate diagnosis and treatment and to avoidunnecessary invasive and costly investigation like DSA.Spinal cord tuberculomas can mimic SDAVF if thelesions are multiple, very small and similar in size andare located at the pial surface of the cord. The associatedcord swelling and hyperintensity can simulate venoushypertension associated with SDAVF. Gadoliniumenhanced imaging is extremely important. The coronalT1weighted post contrast imaging can show enhancingperiphery and nonenhancing center of the lesion whichis not seen in hypertrophied vessels of SDAVF.
*********Author ContributionsJyoti Sureka – Substantial contributions to conceptionand design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedVarsha Mary Khalkho – Analysis and interpretation ofdata, Revising it critically for important intellectualcontent, Final approval of the version to be publishedBinita Riya Chacko – Analysis and interpretation ofdata, Revising it critically for important intellectualcontent, Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Jyoti Sureka et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distribution andreproduction in any means provided the original authorsand original publisher are properly credited. (Please seewww.ijcasereportsandimages.com/copyrightpolicy.phpfor more information.)
Figure 2: A 45yearold male presented with progressiveworsening of lower limb weakness since several months.Spinal angiogram with selective runoff of (A) right thoracolumbar arteries (black arrows) from T4 to L4, and (B) leftthoracolumbar arteries (black arrows) T2 to L4, did notreveal any pial feeder vessel or other evidence of spinal cordarteriovenous malformation.
Figure 3: A 45yearold male presented with progressiveworsening of lower limb weakness since several months. T1weighted post gadolinium (A) sagittal, and (B) coronal MRI ofmid and lower thoracic spine demonstrated multiple smallnodular (white arrows) and ring (black arrows) enhancinglesions of almost same size along the surface of thoracic cord.No enhancement of cauda equina nerve roots or within thecord substance in the region of high signal intensity wasobserved.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):28–31 .www.ijcasereportsandimages.com
REFERENCES1. Harisinghani MG, McLoud TC, Shepard JA, Ko JP,Shroff MM, Mueller PR. Tuberculosis from head totoe. Radiographics 2000;20(2):449–70.2. Shah IU, Siddiqui UT, Imran M, Ashraf J, Mazhar S,Ghori SA. Intramedullary spinal tuberculoma. J CollPhysicians Surg Pak 2012;22(1):48–9.3. Mohit AA, Santiago P, Rostomily R. Intramedullarytuberculoma mimicking primary CNS lymphoma. JNeurol Neurosurg Psychiatry 2004;75(11):1636–8.4. Gupta RK, Kumar S. Central nervous systemtuberculosis. Neuroimaging Clin N Am2011;21(4):795–814.5. Sree Harsha CK, Shetty AP, Rajasekaran S.Intradural spinal tuberculosis in the absence ofvertebral or meningeal tuberculosis: a case report. JOrthop Surg (Hong Kong) 2006;14(1):71–5.6. Huebner RE, Castro KG. The changing face oftuberculosis. Annu Rev Med 1995;46:47–55.
Sureka et al. 31
7. Hristea A, Constantinescu RV, Exergian F, Arama V,Besleaga M, Tanasescu R. Paraplegia due to nonosseous spinal tuberculosis: report of three cases andreview of the literature. Int J Infect Dis2008;12(4):425–9.8. Poon TL, Ho WS, Pang KY, Wong CK. Tuberculousmeningitis with spinal tuberculous arachnoiditis.Hong Kong Med J 2003;9:59–61.9. Kadir KOTIL, Necmettin GUZEL. PrimaryIntramedullary Tuberculoma of The Spinal CordMimicking To Spinal Cord Tumor. Journal ofNeurological Sciences (Turkish) 2006;23(1):063–7.10. Lu M. Imaging diagnosis of spinal intramedullarytuberculoma: case reports and literature review. JSpinal Cord Med 2010;33(2):159–62.11. Jena A, Banerji AK, Tripathi RP, et al.Demonstration of intramedullary tuberculomas bymagnetic resonance imaging: a report of two cases.Br J Radiol 1991;64:555–7.
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):32–36.www.ijcasereportsandimages.com
Unusual case of ascitesGuido Poggi, Benedetta Montagna, Pamela Di Cesare,Erica Quaquarini, Emma Pozzi, Carlo Aprile
ABSTRACTIntroduction: Clostridium difficile infection isvery commonly related to antibiotic therapy.The spectrum of clinical manifestation of C.difficile infection may include, in an increasingorder of severity, absence of symptoms, colitiswithout formation of pseudomembranes andpseudomembranous colitis (PMC). PMC is asevere but rare complication of the infection. Itis related to the bacterial production ofenterotoxin A and B. Its clinical features includediarrhea, abdominal tenderness and fever. Inthe worst case, it may progress to toxicmegacolon and colonic perforation. Ascites is aninfrequent direct complication of most severecases of PMC. Case Report: We report a case ofascites arising two weeks after the resolution ofC. difficile infection, in whichhypoalbuminemia, caused by protein losingenteropathy, was the most likely pathogeneticmechanism. The patient recovered completelyafter human albumin intravenous support anddiuretics. Conclusion: Protein losing syndromerepresents a group of disorders in whichhypoproteinemia and edema occur in theabsence of either proteinuria or defects inprotein synthesis. It is a consequence of theintestinal epithelial cell damage and increased
mucosal permeability induced by C. difficileenterotoxin. The clinical manifestations areascites and low serum albumin levels. Thetreatment requires human albumin intravenousintegrations and diuretics. Ascites could be adirect complication of antibioticassociatedpseudomembranous colitis, but it could also bethe manifestation of a proteinlosingenteropathy, caused by inflammatory damageof gastrointestinal mucosa.Keywords: Clostridium difficile, Infection,Complications, Protein losing enteropathy,Ascites, MSSA infection
*********Poggi G, Montagna B, Di Cesare P, Quaquarini E, PozziE, Aprile C. Unusual case of ascites. InternationalJournal of Case Reports and Images 2013;4(1):32–36.
*********doi:10.5348/ijcri201301254CR8
INTRODUCTIONClostridium difficile is a Spore forming, grampositive bacillus. It is a wellrecognized cause ofantibiotic associated diarrhea. The spectrum of clinicalmanifestation of C. difficile infection may include, in anincreasing order of severity, absence of symptoms,antibiotic–associated colitis without the formation ofpseudomembranes and pseudomembranous colitis(PMC). PMC is a severe but rare complication of theinfection, related to the bacteria production ofenterotoxin A and B. Its clinical features includediarrhea, abdominal tenderness, fever, dehydration andleukocytosis. In the worst case, it may progress to toxicmegacolon and colonic perforation resulting from fullthickness colonic wall necrosis. The diagnosis of PMC
CASE REPORT OPEN ACCESS
Guido Poggi1 , Benedetta Montagna1 , Pamela Di Cesare1 ,Erica Quaquarini1 , Emma Pozzi1 , Carlo Apri le2
Affi l iations: 1Department of Oncology I I , IRCCS MaugeriFoundation, Pavia, I taly; 2Consultant, Department of NuclearMedicine, IRCCS S. Matteo Hospital, Pavia, I taly.Corresponding Author: Guido Poggi, MD, Department ofOncology I I , IRCCS Maugeri Foundation, 1 0, Maugeri Street,271 00 Pavia, I taly; Ph: ++39 0382 592675; Fax No: ++390382 592026; Email : guido.poggi@fsm. it
Received: 29 May 201 2Accepted: 20 August 201 2Published: 01 January 201 3
Poggi et al. 32

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):32–36.www.ijcasereportsandimages.com Poggi et al. 33
depends on the demonstration of C. difficile toxins inthe stool or on endoscopic evidence of adherent yellowplaques along colonic wall. Computed tomography (CT)scan is also useful for PMC diagnosis, showing colonicwall thickening, lowattenuation mural thickeningcorresponding to mucosal and submucosal edema andpericolonic stranding [1–3]. Early treatment with oralmetronidazole or oral vancomycin usually leads to aclinical improvement within 48–72 hours.Surgical intervention is mandatory in perforationand may be required in severe cases where medicaltreatment is not sufficient. Ascites is an infrequentdirect complication of most severe cases of PMC [4].We describe a case of ascites arising two weeks afterthe resolution of C. difficile infection, in whichhypoalbuminemia, secondary to protein losingenteropathy was the most likely pathogenic mechanism.
CASE REPORTA 54yearold man was admitted to our hospital withcomplaints of abdominal distension and bilateralperipheral edema. He had no previous history of suchillness. Ten weeks before his admission, he developed asoft tissue infection at the back of his left hand whichrapidly evolving to a necrotising fasciitis of the leftforearm and required surgical decompression followedby necrotic tissue debridements. Microbiological cultureof the tissue samples taken from the wound identified agrowth of a methicillin sensitive staphylococcus aureus(MSSA) producing pantonvalentine leukocidin toxin.Blood cultures were negative. The patient was treatedwith intravenous ciprofloxacin (200 mg/bid) andclindamycin (500 mg/bid) for ten days. After thetreatment, he was discharged home and continuedtherapy with oral ciprofloxacin (500 mg/bid) and oralrifampicin (600 mg/day). After one month oftreatment, he developed nonbloody, watery diarrheaconsisting of 812 stools daily, fever and abdominalpain. The patient was rehospitalized. Stool cultureswere negative for shigella, salmonella, campylobacterand yersinia bacteria while they were positive forC. difficile toxins. The EIAtest for C. difficile was alsonegative. The patient was treated with oral vancomycin(500 mg every 6 hours for 5 days), with progressiveclinical benefit and resolution of diarrhea, and he wasdischarged home.Due to the subsequent appearance of abdominalswelling and peripheral edema, occurring two weeksafter the resolution of abdominal symptoms, the patientwas readmitted to our hospital. On physicalexamination, the patient was alert and had littlediscomfort. Vital signs were normal and the patient hadno fever. Abdominal examination showed diffuseabdominal distension with active bowel sounds, a dullpercussion note and mild, diffuse tenderness withoutjaundice. The patient had pitting edema of lowerextremity. Lymphadenopathy was not present.Neurological and cardiac examinations were normal.Laboratory tests revealed normal hemoglobin values,
normal renal function tests, no electrolytesabnormalities, normal hepatic and coagulation function.Erythrocyte sedimentation rate and Creactive proteinwere negative. Total serum protein was 5.0 g/dL withmarked hypoalbuminemia (2.5 g/dL). Mild lymphopeniaand hypogammaglobulinemia were also detected.Hepatitis B surface antigen and hepatitis C antibodywere negative.Computed tomography scan showed thickenedcolonic wall and massive ascites without evidence ofhepatosplenomegaly, portal or hepatic vein thrombosisor peritoneal thickening (Figures 1, 2). Abdominalparacentesis was performed. Asatic fluid examinationshowed WBC count of 400 cells/mm3 with 65%polymorphonuclear leukocytes, total protein 3.0 g/dL,albumin 1.4 g/dL. Serumascites albumin gradient(SAAG) was 1.1 and ascitic fluid/serum LDH ratio was0.78. Cultures of ascitic fluid for bacteria andmycobacteria were negative and cytological examinationdid not find malignant cells.The C. difficile toxin was not detected in ascitic fluid.Urine collection over 24 hours failed to showproteinuria. A stool collection for fecal fat was normal.Serologic testing for celiac disease was negative. Fecal α1 antitrypsin (A1AT) clearance was found to be elevated(288 mg per 100 mL; normal value <30 mg/dL)suggesting that the severe hypoalbuminemia might becaused by protein losing enteropathy. Tc99m labeledhuman serum albumin scintigraphy demonstrated anabnormal excretion of Tc99m albumin into the largebowel confirming the diagnosis of protein loosingenteropathy (Figure 3).During the hospitalization, patient was treated withsalt restriction, human albumin intravenous infusionand diuretics. The patient gradually recovered and hewas discharged. Three weeks later, ascites hadcompletely disappeared as shown by ultrasoundexamination. At a followup visit one month later, hereported having no diarrhea and serum albumin levelswere normal. After three months, an abdominal CT scan
Figure 1: Abdominal computed tomography scan showingascites (marked by 'a' ).

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):32–36.www.ijcasereportsandimages.com Poggi et al. 34
showed complete resolution of increased thickness ofthe colonic wall and there was no evidence of ascites.
DISCUSSIONAscites is a clinical condition that may represent theinitial manifestation of a systemic disease or ofotherwise unsuspected abdominal disease. In mostcases, ascites appears as part of a wellrecognized
illness, for example cirrhosis, congestive heart failure,nephrosis or disseminated carcinomatosis. On occasion,ascites may develop as an isolated finding in the absenceof a clinically evident disease. In such cases, a carefulanalysis of ascitic fluid may suggest the etiology: theserum ascites–albumin gradient (SAAG) > or < 1 g/dLcan differentiate between a transudate or an exudatefluid. Serum ascitealbumin gradient >1 is usuallyrelated to uncomplicated chirrosis, alcoholic hepatitis,congestive heart failure and Budd–Chiari syndrome;while SAAG <1 is related to other causes, such asperitoneal carcinomatosis, tuberculous peritonitis,pancreatitis, serositis, pyogenic peritonitis, andnephritic syndrome [5].In our case, ascites was the late complication of a C.difficile associated diarrhea following a prolongedantibiotic therapy for a soft tissue infection. The patienthad no other systemic disease that could have justifiedthe occurrence of ascites, such as hepatic or cardiacfailure. SAAG was 1.1 g/dL with mild leukocytosis in thefluid and proteinuria was absent.A literature review revealed that ascites occurs morefrequently as an indirect complication of severe cases ofPMC. Systemic capillary leak is considered the mostlikely mechanism of ascites, occurring in the worst casesof PMC [6–8]. However, our patient did not developed asevere form of colitis and the onset of ascites occurredafter a few days from the complete resolution ofgastrointestinal symptoms. On investigating we foundthat, in this case, the prevalent mechanism of formationof ascites was a proteinlosing enteropathy.Proteinlosing enteropathy is not a specific diseasebut rather a group of gastrointestinal and nongastrointestinal disorders occurring withhypoproteinemia and edema in the absence of eitherproteinuria or defects in protein synthesis. Diseasescharacterized by excess protein loss into thegastrointestinal tract are caused by mucosal ulceration(e.g., ulcerative colitis, gastrointestinal carcinomas, andpeptic ulcer), by damage to mucosa without ulceration(e.g., celiac sprue and Ménétrier's disease) or bylymphatic dysfunction; either primary lymphatic diseaseor secondary to partial lymphatic obstruction (e.g.intestinal lymphangiectasia, mesenteric nodes orlymphoma, cardiac disease) [9].C. Difficile enterotoxins A and B induce intestinalepithelial cell damage, increase mucosal permeability,stimulate interleukin (IL)8 synthesis, and cause anacute inflammatory response characterized byneutrophil recruitment and tissue damage [10]. Thiscondition can lead to a protein losing syndrome withsevere hypoalbuminemia that can result in peripheraledema and ascites, as we described in this case report.The management of ascites involves, apart from thespecific antibiotic therapy for C. difficile infection,human albumin intravenous integrations and diuretics.
CONCLUSIONIn summary, ascites could be a direct complication ofantibioticassociated pseudomembranous colitis but it
Figure 2: Abdominal computed tomography image of colonicwall thickening (arrows).
Figure 3: 99mTclabeled human serum albumin scintigraphydemonstrated an abnormal excretion of 99mTc albumin intothe large bowel (after two and half hours) (thin arrows).

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):32–36.www.ijcasereportsandimages.com Poggi et al. 35
could also be the manifestation of a protein losingenteropathy, caused by the inflammatory damage of thegastrointestinal mucosa.*********
Author ContributionsGuido Poggi – Substantial contributions to conceptionand design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedBenedetta Montagna – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedPamela Di Cesare – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedErica Quaquarini – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedEmma Pozzi – Substantial contributions to conceptionand design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedCarlo Aprile – Substantial contributions to conceptionand design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Finalapproval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Guido Poggi et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Kawamoto S, Horton KM, Fishman EK.Pseudomembranous colitis: spectrum of imagingfindings with clinical and pathologic correlation.RadioGraphic 1999;19(4):887–97.2. Boland GW, Lee MJ, Cats AM, Gaa JA, Saini S,Mueller PR. Antibiotic induced diarrhea: specificityof abdominal CT for the diagnosis of ClostridiumDifficile disease. Radiology 1994;191(1):103–6.3. Tsourous GI, Raftopoulos LG, Kafe EE, ManolerisEK, Makaritsis KP, Pinis SG. A case of
pseudomenbranous colitis presenting with massiveascites. Eur J Intern Med 2007 Jul;18(4):328–30.4. Ginès P, Cárdenas A, Arroyo V, Rodés J.Management of cirrhosis and ascites. N Engl J Med2004;350(16):1646–54.5. Rybolt AH, Bennett RG, Laughon BE, Thomas DR,Greenough WB 3rd, Bartlett JG. Proteinlosingenteropathy associated with Clostridium difficileinfection. Lancet 1989;1(8651):1353–5.6. Zuckerman E, Kanel G, Ha C, Kahn J, Gottesman BS,Korula J. Low albumin gradient ascites complicatingsevere pseudomembranous colitis. Gastroenterology1997;112(3):991–4.7. Spahr L, de Saussure P, Felley C, Pugin J, HadengueA. Pseudomembranous colitis: an unusual cause ofneutrocytic ascites. EJGH 1999;11(7):789–2.8. Parikh VA, Edlund JW. Ascites associated withantibioticassociated pseudomembranous colitis.South Med J 1997;90(4):460.9. Fauci AS, Braunwald E, Kasper DL, Hauser SL,Longo 192 D.L., Jameson J. et al. Harrison'sPrinciples of Internal Medicine, 17th Edition.McGraw Hill 2008.10. Farhadi A, Banan A, Fields J, Keshavarzian A.Intestinal barrier: an interface between health anddisease. J Gastroenterol Hepatol 2003;18(5):479–7.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):32–36.www.ijcasereportsandimages.com Poggi et al. 36
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):37–40.www.ijcasereportsandimages.com
Feeding dystonia: A classical presentation ofneuroacanthocytosisSuman Kushwaha, Akhila Panda, Vachan Mehta,Seema Malik, Ishita Pant
ABSTRACTIntroduction: Neuroacanthocytosis (NA) is aheterogeneous neurodegenerative geneticdisorder caused by disease specific geneticmutation. Being an extremely rare disorder,only a few thousand cases have been reportedtill date. This clinical entity was described byCitchley et al. and was initially namedLevine–Citchley syndrome. It is characterizedby movement disorder due to degeneration ofthe basal ganglia along with cognitive andbehavior changes. The classical clinicalpresentation includes the troublesomeabnormal involuntary movements in form ofchorea, dystonia and dyskinesia. Self mutilationof the lips and tongue is characteristic ofchoreoacanthocytosis. The NA syndromes havebeen broadly divided into two subtypes, (i) coreNA syndromes, and (ii) conditions withalterations in the lipoprotein metabolism. Thegenes of the different NA syndromes have beenidentified but the mechanism of these geneticmutations is not known. The management of NA
is primarily symptomatic and rehabilitative.Case Report: We are describing a case ofneuroacanthocytosis with typical phenotype ofchoreoacanthocytosis. The phenomenon offeeding dystonia is classically being discussed inthis report. Neuroimaging demonstrated theatrophy of the caudate nucleus resemblingHuntington’s chorea. Acanthocytic red bloodcells were seen in peripheral smear in ourpatients. Conclusion: The recognition ofneuroacanthocytosis is improved due to bettercharacterization of the clinical symptoms andinvestigations of this heterogeneous entity. Thepresented case describes the typical clinicalcharacteristics and investigations supportingthe diagnosis of this under diagnosed clinicalsyndrome.Keywords: Feeding dystonia, Chorea,Acanthocytes, Neuroacanthocytosis
*********Kushwaha S, Panda A, Mehta V, Malik S, Pant I.Feeding dystonia: A classical presentation ofneuroacanthocytosis. International Journal of CaseReports and Images 2013;4(1):37–40.
*********doi:10.5348/ijcri201301255CR9
INTRODUCTIONNeuroacanthocytosis (NA) is an uncommon,heterogeneous, genetic neurodegenerative disorder.This clinical entity was described by Citchley et al. andwas initially named Levine–Citchley syndrome. Theclinical manifestations include variety of movementdisorders and cognitive and behavioral changes closelyresembling Huntington’s disease. The orofaciolingual
CASE REPORT OPEN ACCESS
Suman Kushwaha1 , Akhila Panda2, Vachan Mehta2, SeemaMalik2, Ishita Pant3
Affi l iations: 1Associate Professor Neurology, DM Neurology,Department of Neurology, Institute of Human Behavior andAll ied Sciences, Delhi, India; 2Senior Resident Neurology,Institute of Human Behaviour and All ied Sciences, Delhi,India; 3Assistant Professor, Pathology, Institute of HumanBehavior and All ied Sciences, Delhi, IndiaCorresponding Author: Suman Kushwaha DM Neurology,Associate Professor Neurology, Room No. 11 2, AcademicBlock, Department of Neurology, Institute of HumanBehavior and All ied Sciences, Delhi - 95, India; Ph: 91 -986839681 4, Fax No: 91 -11 -22592337; Email :sumankushwaha@gmail .com
Received: 1 2 June 201 2Accepted: 1 7 September 201 2Published: 01 January 201 3
Kushwaha et al. 37

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):37–40.www.ijcasereportsandimages.com Kushwaha et al. 38
dyskinesia is prominent and characteristic ofneuroacanthocytosis. There is presence of thornydeformation in the lipid membrane of the circulating redblood cells called acanthocytosis. These acanthocytes aredistinct and unique for this syndrome. The recognitionof neuroacanthocytosis is improved due to bettercharacterization of the clinical symptoms andinvestigations of this heterogeneous entity. Thepresented case describes the typical clinicalcharacteristics and investigations supporting thediagnosis of this under diagnosed clinical syndrome.
CASE REPORTA 30yearold female presented with three yearshistory of progressive difficulty in eating. She attributedit to abnormal involuntary movement of tongue andsustained spasmodic movement of the neck. She useddifferent maneuvers during eating like pushing the boluswith the fingers inside the mouth and drinking waterand food in supine position. She sometimes developedchoking sensation and cough while eating food.Occasionally, she had nasal regurgitation while drinkingwater. She had abnormal, repetitive, nonpatterned,involuntary, arrhythmic, nonpurposeful movements ofthe whole body including head and neck. Thesemovements disappeared in sleep and were notsuppressed with voluntary action. Since last ninemonths she had developed abnormal behavior in theform of excessive and nonpurposeful talking, repeatingthe sentences and motor acts. The caregivers reportedthe habit of selfmutilation. She repeatedly chewed herlips which lead to severe injury of lower lip.There was no history of visual problems, limbweakness, sensory, cerebellar or autonomic disturbance.She had significant weight loss. Her personal historyrevealed poor maintenance of hygiene and history ofsecondary amenorrhea. Family history and past medicalhistory were noncontributory.General physical examination showed poor self careand cachexia. The vital parameters were normal. Asignificant lip biting mark was present in inner aspect oflower lip (Figure 1) suggestive of selfmutilation andrepeated friction of the lower lip due to orofacialdyskinesia. Other systemic examination was normal. Onneurological examination, she was conscious andoriented. Marked behavior changes in form of repetitionof words and perseveration of motor acts were present.Neuropsychological assessment shows high frontality onfrontal lobe assessment by frontal assessment battery(FAB) battery. Memory functions were normal. She hadfeatures of obsessive compulsive behavior and anxiety.Cranial nerve examination was normal. The limbs werehypotonic with power of 5/5 on Medical Research Council(MRC) scale in all the limbs. Generalized areflexia withflexor plantar response was present. All the primitivereflexes were absent. Sensory, cerebellar and autonomicsystem examination revealed no abnormality.Extra pyramidal system examination showedcombination of abnormal involuntary movements.
Chorea was prominent involving orolingual, neck, trunkand limbs. Tongue had classical jack in boxphenomenon. In addition to chorea, head and neckshows intermittent dystonic movements. Due tocombination of these abnormal movements of tongueand neck the patient had classical feeding difficultiesdescribed as feeding dystonia. She walked with bizarregait due to abnormal movements of the trunk. Theroutine investigations, complete blood count, kidneyand liver function tests, serum electrolytes, lipid profile,copper studies, HIV ELISA, blood VDRL were withinnormal limits.Creatinine phosphokinase was raised to 614 U/L.Peripheral blood acanthocytes were visible on screeningof three consecutive samples (Figure 2). Nerveconduction studies showed sensorimotor axonalneuropathy. Neuroimaging of brain using 3 Tesla MRIshowed diffuse cerebral atrophy with significant caudateatrophy (Figure 3).
Figure 1: Selfmutilation of lower lip. Severe injury due to lipbiting and dyskinesia causing repeated irritation by teeth.
Figure 2: Peripheral smear showing acanthocytes.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):37–40.www.ijcasereportsandimages.com Kushwaha et al. 39
DISCUSSIONNeuroacanthocytosis is a rare movement disordersyndrome. The prevalence is estimated to be less than1–5 million. Neuroacanthocytosis has varied clinicalpresentations ranging from involuntary hyperkineticmovements, neuromuscular involvement to cognitiveand behavioral changes. Onset of symptoms is usuallyin adulthood. The core NA syndrome includesautosomal recessive choreoacanthocytosis and XlinkedMcleod syndrome. The other subgroup includesneuroacanthocytosis with lipoprotein disorders e.g.abetalipoproteinemia (Bassen, Kornzweig disease). Thissymptom variation in presentation is responsible for theunder diagnosis of this clinical entity. Choreaacanthocytosis is caused by mutation of 73 exon gene onchromosome 9, VPS13A which codes for Chorein [1].Our patient had characteristic phenotype ofneuroacanthocytosis. She had presented in adulthoodwith typical orofacial and lingual dyskinesia troublingher in eating. Due to abnormal posturing of the neckand repeated tongue protrusion she had characteristicfeeding dystonia which has been described inneuroacanthocytosis [2]. Weight loss due to poornutritional status is a major concern in these patients.Lingual and orofacial dyskinesia causes irritation oflower lip in combination with lip biting resulting insevere lower lip injury. The patient had bizarremovements involving the whole body in form of violenttrunk spasms and head thrusting which were evident onwalking. This typical gait is described as "rubber man"gait [3]. As the disease progresses the hyperkineticmovement disorder evolves into the hypokinetic orbradykinetic state. These patients have major disabilitydue to combination of movement disorders likedystonia and chorea. Neuropsychiatric symptoms areprominent in neuroacanthocytosis and may appear
before the movement disorder.Seizure were absent in our patient while they areusually seen in approximately 40% patients [4]. Thepatients have elevated creatinine kinase levels andaxonal neuropathy. The neuromuscular involvement inchoreoacanthocytosis includes myopathy and axonalsensorymotor neuropathy [5]. McLeod syndromeshould be considered as a differential diagnosis ofchoreoacanthocytosis as neuromuscular involvement ismore common in this subtype of neuroacanthocytosis[6]. Hepatosplenomegaly and cardiac involvement isseen in McLeod syndrome. Panthothenatekinase–associated neurodegeneration (PKAN) anautosomal recessive disorder is another differentialdiagnosis. It is a rapidly progressive childhood disorder.Dystonia along with cognitive and behavior changes arethe prominent clinical manifestation. Huntington’sdisease is the closest differential diagnosis as thephenotype includes chorea, behavior and cognitivedeficits. The pattern of caudate atrophy onneuroimaging is similar in both these disorders. Thegenetic testing for Huntington’s disease is diagnostic.The classical phenotype of uncommon movementdisorder, neuroacanthocytosis presents withcombination of abnormal movements and feedingdystonia along with self mutilatilating behavior andcognitive deficits in our patient. The clinical diagnosis ofsporadic variant of choreoacanthocytosis is supportedby classical neuroimaging and presence ofacanthocytosis in the peripheral smear. Due to lack offacility for analysis of VPS13a or Chorein gene theconfirmation of diagnosis could not be reached in ourcase. The management remains mainly symptomatic.The incapacitating movements can be reduced withtypical antipsychotics and dopamine depletors liketetrabenezine. Choreoacanthocytosis progresses over15–30 years. Long term outcome of patient is poor.Neurorehabilitation and behavior therapy playsimportant role in functional improvement as well asmaintainance of better quality of life. Our patient ismaintaining well on treatment and regularly followedup in outpatient department.
CONCLUSIONOur case demonstrates the classical phenotype ofneuroacanthocytosis. The variation in symptomatologycausing the different phenotypes with significantoverlap makes the diagnosis difficult. The importantclinical differences exist among the phenotypes andshould be emphasized while making the clinicaldiagnosis. The better recognition and understanding ofthe phenomenology in neuroacanthocytosis willfacilitate us in diagnosis of this entity.
*********Author ContributionsSuman Kushwaha – Conception and design, Analysisand interpretation of data, Critical revision of the
Figure 3: Magnetic resonance imaging of brain showingcaudate atrophy resembling Huntington’s disease (3 tesla T2weighted coronal section).

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):37–40.www.ijcasereportsandimages.com Kushwaha et al. 40
article, Final approval of the version to be publishedAkhila Panda – Acquisition of data, Drafting the article,Final approval of the version to be publishedVachan Mehta – Acquisition of data, Drafting thearticle, Final approval of the version to be publishedSeema Malik – Acquisition of data, Drafting the article,Final approval of the version to be publishedIshita Pant – Analysis and interpretation of data,Drafting the article, Final approval of the version to bepublishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Suman Kushwaha et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properly
credited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Rampoldi L, DobsonStone C, Rubio JP, et al. Aconserved sorting associated protein is mutant inchorea acanthocytosis. Nat Genet2001;28(2):119–20.2. Walker RH, Jung HH, DobsonStone C, et al.Neurologic phenotype associated withacanthocytosis. Neurology 2007;68(2):92–8.3. Schnieder SA, Lang AE, Moro E, Bader B, Danek A,Bhatia KP. Characteristic head drops and axialextension in advanced choreaacanthocytosis.Movement disorder 2010;25(10):1487–91.4. Rampoldi L, Danek A, Monaco AP. Clinical featuresand molecular bases of neuroacanthocytosis. J MolMed (Berl) 2002;80(8):475–91.5. Hewer E, Danek A, Schoser BG, et al. McLeodmyopathy revisited: more neurogenic and lessbenign. Brain 2007;130(Pt 12):3285–96.6. Jung HH. McLeod syndrome: clinical andneuroradiological aspects. Mov Disorder2005;20:1677.
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):41 –45.www.ijcasereportsandimages.com
A novel method of treating isolated unicondylar fracture ofthe head of the proximal phalanx: A case reportAysha Rajeev, John Harrison
ABSTRACTIntroduction: The phalangeal fractures arecommon hand injuries. The unicondylarfractures of proximal phalanx are unique. Theyneed prompt and accurate treatment to have agood functional outcome. We present atechnique for managing a volar displaced (type4) unicondylar fracture of the proximal phalanx.This provides accurate reduction andstabilization until fracture union, with minimalsoft tissue tethering to allow early movement.Case Report: A 26yearsold male sustained aninjury to the right index finger while playingcricket. He attended fracture clinic complainingof pain and difficulty in moving the proximalinterphalangeal (PIP) joint of the index finger.The Xray showed a unicondylar fracture of theproximal phalanx in the coronal plane, with adisplaced volar fragment. The patient wastreated with open reduction and internalfixation with a single Kirschner wire (Kwire)passed from volar to dorsal aspect through thefragment. The patient commenced handphysiotherapy straight away and regained fullrange of movements. Conclusion: We report thetechnique of fixing the displaced unicondylarfractures of the proximal phalanx using singleKwire passed from the volar to dorsal aspect
after open reduction of the fragment through avolar approach. This technique will allow earlymobilization with very minimal soft tissuedissection thus preventing stiffness.Keywords: Unicondylar, Proximal phalanx,Type 4 fracture
*********Rajeev A, Harrison J. A novel method of treatingisolated unicondylar fracture of the head of the proximalphalanx: A case report. International Journal of CaseReports and Images 2013;4(1):41–45.
*********doi:10.5348/ijcri201301256CR10
INTRODUCTIONUnicondylar fractures of the phalangeal head arerelatively common injuries. They are more common inyoung, active patients and define a specific fracturepattern [1]. These are intraarticular fractures and fourtypes are described [2]. Class 1 and 2 occur between thecondyles (intercondylar) leading to angular deformity ofthe digit if displaced. Class 3 is a dorsal fragment in thecoronal plane. Class 4 fractures are less common and isa volar fragment. If displaced these fractures requireoperative management. Intercondylar fractures may betreated successfully with closed reduction andpercutaneous transverse fixation. Coronal planefractures may require open reduction and anymetalwork must not be left in the joint [3]. Accuratereduction and stabilization, allowing early motion arethe key aims for achieving good results with all types ofunicondylar fractures of the proximal phalanx. Whenstabilization is necessary, soft tissue trauma should beminimized to lessen the risk of scar formation andconsequent stiffness at the proximal interphalangeal
CASE REPORT OPEN ACCESS
Aysha Rajeev1 , John Harrison2
Affi l iations: 1FRCS, Associate Special ist,Trauma andOrthopaedics,Queen Elizabeth Hospital, Gateshead, UK;2FRCS, Consultant, Trauma and Orthopaedics,QueenElizabeth Hospital, Gateshead, UK.Corresponding Author: Aysha S Rajeev, 25,Fatfield Park,Washington, Tyne and Wear, NE38 8BW; Ph:0044741 4262665; Fax: 00441 91 4453874; Email :asrajeev1 8@gmail .com
Received: 23 June 201 2Accepted: 01 September 201 2Published: 01 January 201 3
Rajeev et al. 41

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):41 –45.www.ijcasereportsandimages.com Rajeev et al. 42
joint. Stiffness is the most frequent and seriousreported complication of unicondylar fractures [4].
CASE REPORTA 26yearsold male with right dominant hand wasseen in the fracture clinic following an injury to his rightindex finger. He worked as a computer salesrepresentative. He sustained a hyperextension injury tohis finger while playing cricket when the cricket ball hithis index finger tip. He complained about pain anddifficulty in moving the proximal interphalangeal (PIP)joint. He was otherwise fit and well.On examination, there was tenderness in the PIPjoint. With the finger extended there was no angulardeformity. Movement at the PIP joint was limited to0–40 degrees of flexion. Metacarpophalangeal (MCP)and distal interphalengeal (DIP) joint movements werefull. The Xray showed a unicondylar fracture of theproximal phalanx in the coronal plane (Figure 1), with adisplaced volar fragment. The patient was offeredsurgery after explaining the risk of stiffness andmalunion.Under general anaesthetic and tourniquet controlthe PIP joint was exposed through a Brunner's incision(Figure 2). The A3 pulley was released on the ulnar side.The condylar fragment was reduced and held with aKirschner wire (Kwire) passed from volar to dorsalthrough the fragment. The Kwire driver was thenmoved to the leading end of the wire where it protrudedthrough the skin and was then pulled through so thetrailing end of the Kwire was just under the articularsurface (Figure 3). The reduction was checked withimage intensifier, and full flexion of the PIP joint wasachieved.The patient was allowed to commence finger flexionfrom the first postoperative day. The patient wasfollowed up in the clinic after two weeks for woundinspection and check Xrays were satisfactory (Figure4). The patient was able to flex the PIP joint at this stageto 800. The Kwire was removed four weeks aftersurgery and the patient was referred for futher intensivephysiotherapy. He regained full range of movements ofthe PIP joint after eight weeks. The Xray showed thatthe fracture had healed in an anatomical position(Figure 5). He was discharged from the outpatientclinic.
DISCUSSIONVarious methods of open reduction and internalfixation of unicondylar fractures of the proximalphalanx have been described in literature. London et al.noted that some of these fractures were stable so thatthey can be treated with splints and early mobilization,whereas others needed open reduction and fixation [4].He also proposed a classification system for thesefractures. McCue et al. in a review of twenty casesreported that there was no predominance of either
radial or ulnar condyle involvement in these fractures[5]. They described that unicondylar fractures of PIPjoint were most commonly associated with sportsinjuries. Their treatment protocol was to fix all twentycases with two Kirschner wires which regained anaverage of 930 of PIP joint movements. The mechanismof distal unicondylar fractures of the proximal andmiddle phalanx were caused by a trochlear shear ratherthan compression as described by Soeur et al. [6].Ramos et al. developed a protocol for isolatedunicondylar fracture of the head of the proximalphalanx which included surgical fixation using lag screw
Figure 1: Xray showing class 4 unicondylar fracture with volardisplacement.
Figure 2: Intraoperative picture showing displaced volarfragment of proximal phalanx.

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):41 –45.www.ijcasereportsandimages.com Rajeev et al. 43
and immediate mobilization by use of a continouspassive motion and controlled active motion [7]. Theyused specially designed splints and Coban wrap tocontrol the position of the digit during the first sixmonths.Henry et al. described a wide array of treatmentoptions for these type of fractures of proximal phalanx,including Kirschner wires screws and plate fixation [8].He stated that early closed reduction is successful forunicondylar fractures of the head of the proximalphalanx.Blazar et al. in their study on fractures of proximalinterphalangeal joint stated that an understanding ofthe anatomy, the potential for joint instability and thetreatment options are essential to manage thesefractures [9]. They also described various treatmentoptions including extensionblock splinting,percutaneous pinning, traction, external fixation, openreduction and internal fixation and volarplatearthroplasty. A prompt recognition of the complexity ofthe injury and appropriate management are also
Figure 3: Intraoperative radiograph showing reduction offracture with the Kwire passed from dorsal aspect. Figure 4: Check radiograph showing accurate reduction offracture of head of the proximal phalanx.
essential for an optimal functional outcome.The objectives of techniques of internal fixation ofproximal phalangeal fractures are pain control and earlyfunctional restoration. When the fragment size permits,unstable and displaced proximal phalangeal jointfractures can be secured in an anatomic position byeither a direct method through the fragments or byindirect methods (buttress) like pinning or screwfixation techniques. Percutaneous or limited openreduction and internal fixation techniques are preferredin an effort to minimize additional soft tissue traumaand scarring [10].The classification of distal unicondylar fractures ofthe proximal phalanx was based on the mechanism ofinjury. There are four class of fractures [3]. The patientin our case had a class 4 fracture which involved a volarcoronal fragment. These injuries are caused by ashearing force across an extended proximal interphalangeal joint causing a transient volar subluxation ofthe PIP joint with the joint extended. Class 4 fractureshave the poorest final range of motion and this may be

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):41 –45.www.ijcasereportsandimages.com Rajeev et al. 44
Figure 5: Radiographs showing a well healed fracture of headof proximal phalanx (unicondylar).
due to imperfect reduction.
CONCLUSIONOur technique of fixing unicondylar fractures of theproximal phalanx used a single Kirschner wireintroduced from the volar aspect to engage the fracturefragment. We felt a screw placed from dorsal surfacemay not gain adequate fixation in the thin volarfragment and the prominent screw head may affect jointflexion, and if removal was necessary this would requiremore dissection than a subcutaneous Kirschner wire.
*********Author ContributionsAysha Rajeev – Conception and design, Acquisition ofdata, Analysis and interpretation of data, Drafting the
article, Critical revision of the article, Final approval ofthe version to be publishedJohn Harrison – Conception and design, Acquisition ofdata, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Aysha Rajeev et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distribution andreproduction in any means provided the original authorsand original publisher are properly credited. (Please seewww.ijcasereportsandimages.com/copyrightpolicy.phpfor more information.)
REFERENCES1. Glickel SZ, Barron OA. Proximal interphalangealjoint fracture dislocations. Hand Clin2000;16(3):333–44.2. Weiss AP, Hastings H 2nd. Distal UnicondylarFractures the Proximal Phalanx. Journal of HandSurgery Am 1993;18(4):594–9.3. Hastings H 2nd, Carroll C 4th. Treatment of closedarticular fractures of the metacarpophalangeal andproximal interphalangeal joints. Hand Clin1988;4(3):503–27.4. London PS. Sprains and fractures involving theinterphalangeal joints. Hand 1971;3(2):155–8.5. McCue FC, Honner R, Johnson MC, Gieck JH.Athletic injuries of the proximal interphalangealjoint requiring surgical treatment. J Bone Joint Surg1970;52(5):937–56.6. Soeur R. Fractures of the limbs: the relationshipbetween mechanism and treatment. Brussels: LaClinique Orthopedique 1981:542–3.7. Ramos LE, Becker GA, Grossman JA. A treatmentapproach for isolated unicondylar fracture of theproximal phalanx. Ann Chir Main Memb Super1997;16(4):305–9.8. Henry MH. Fractures of the proximal phalanx andmetacarpals in the hand: preferred methods ofstabilization. J Am Acad Orthop Surg2008;16(10):586–95.9. Blazar PE, Steiberg DR. Fractures of the proximalinterphanlangeal joint. J Am Acad Orthop Surg2000;8(6):383–90.10. Freeland AE, Benoist LA. Open reduction andinternal fixation method for fractures at theproximal onterphalangeal joint. Hand Clin1994;10(2):239–50.

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):41 –45.www.ijcasereportsandimages.com Rajeev et al. 45
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):46–50.www.ijcasereportsandimages.com
Pleuropulmonary blastoma in an adult: A case reportNidhi Paliwal, Kumud Gupta, Shalini Mullick,Ravindra K Dewan, Sandeep Katiyar
ABSTRACTIntroduction: Pleuropulmonary blastoma (PPB)is a dysontogenetic neoplasm of childhood thatappears as a pulmonary and/or pleuralbasedmass. PPB is of three types: cystic (type I),mixed (type II), or solid (type III). It is rarelyobserved in adults with only a few casesreported in literature. Case Report: We presenta case of a 30yearold male with complaints ofchest pain, dyspnea and low grade fever. Hiscomputed tompgraphy scan of chest showedthree multicystic lesions in right hemithorax.Cytomorphological examination of imprintsmears of biopsies taken from the massessuggested malignant lesion and on histologicalexamination, diagnosis of pleuropulmonaryblastoma type II was made. Conclusion:Pleuropulmonary blastoma is a rare andaggressive malignant tumor of childhood.Familial disposition and association of PPBtumor with other childhood malignancies hasbeen suggested in literature. Pathologically, thetumor tissue is composed of primitive blastemal
cells with focal areas of malignant mesenchyme.Therapy should include surgical tumorresection and subsequent chemotherapy andradiotherapy. PPB is very rare tumor in adultsand presentation as multiple lesions in onesided hemithorax has not been reported. PPBshould be considered in the clinical andradiographic differential diagnosis ofmulticystic lesions, even when the patient is ayoung adult.Keywords: Blastoma, Malignant,Pleuropulmonary
*********Paliwal N, Gupta K, Mullick S, Dewan RK, Katiyar S.Pleuropulmonary blastoma in an adult: A case report.International Journal of Case Reports and Images2013;4(1):46–50.
*********doi:10.5348/ijcri201301257CR11
INTRODUCTIONPleuropulmonary blastoma (PPB) is a uniquedysontogenetic neoplasm of childhood. Its primitive,sarcomatous features are analogous to those of otherdysontogenetic tumors such as Wilm's tumor,hepatoblastoma, neuroblastoma, and embryonalrhabdomyosarcoma [1]. PPB was classified into threegroups by Dehner et al. in 1995 as cystic (type I), mixed(type II), and solid (type III). Type I has a morefavorable prognosis than type II and III [2]. This rareand aggressive neoplasm arises from the lung, pleura, orboth and is a disease of the first decade of life [3]. PPB isobserved rarely in adults and clinical presentation as inour case has not been reported in literature. There is nooptimal defined treatment regimen for adult cases [4].
CASE REPORT OPEN ACCESS
Nidhi Paliwal1 , Kumud Gupta2, Shalini Mull ick3, Ravindra KDewan4, Sandeep Katiyar5
Affi l iations: 1Senior Resident, Pathology, LRS Institute of TB& Respiratory Diseases, New Delhi, India; 2Head, Pathology,LRS Institute of TB & Respiratory Diseases, New Delhi,India; 3DNB, Special ist, Pathology, LRS Institute of TB &Respiratory Diseases, New Delhi, India; 4Head, ThoracicSurgery, LRS Institute of TB & Respiratory Diseases, NewDelhi, India; 5Resident, Respiratory Medicine, LRS Instituteof TB & Respiratory Diseases, New Delhi, India.Corresponding Author: Nidhi Paliwal, Quarter no. 1 053,Sector-8, R K Puram, New Delhi, Delhi, India - 11 0022; Ph:91 -9999857039; Fax: 91 -26568227; Email :npal iwal1 983@gmail .com
Received: 07 July 201 2Accepted: 24 September 201 2Published: 01 January 201 3
Paliwal et al. 46

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):46–50.www.ijcasereportsandimages.com Paliwal et al. 47
We are reporting a case of PPB in a 30yearsold malepresenting with three multicystic lesions in righthemithorax and give a brief review of literature.
CASE REPORTA 30yearold male patient presented with chestpain, dyspnea, and low grade fever for last six months.Two years back he had taken antitubercular drugstherapy. He was a nonsmoker with no past history ofany malignancy. There was no family history of anymalignant lesion in his siblings or parents. Physicalexamination was unremarkable. His chest Xrayrevealed homogenous opacity in right mid and lowerzones (Figure 1). Computed tomography (CT) scan ofchest showed large, multiloculated collection in rightthoracic cage with a few foci of calcification in theseptations. Underlying lung was collapsed. Two smallloculated collections were also seen in right posteriorthoracic cavity (Figure 2). The possibilities of hydatidcyst or benign teratoma were considered based onclinical and radiological presentation.The patient underwent right thoracotomy.Intraoperatively, a large multicystic mass measuringmore than 10 cm in diameter, adherent to lower lobeand diaphragm was seen. Two masses measuring 5 cmand 4 cm in diameter were also present in middle andupper lobe of right lung, respectively. Biopsies frommasses were taken and sent to the pathologydepartment.Complete mobilization of largest mass was notpossible, so it could not be excised. As facility of frozensection was not available in our hospital, only imprintsmears from biopsies were made. All smears showedsimilar cytological picture and revealed small groupsand singly present round to oval to spindle cells withscanty to moderate cytoplasm. Some of the cells werebipolar (Figure 3A). A few cells showed high N:C ratio,moderate cytoplasm with indistinct outline, vesicularchromatin and prominent nucleoli (Figure 3B).Cytomorphological features were suggestive ofmalignancy, however, exact typing of the lesion was notpossible.Grossly, resected tissue pieces were 2.8x2.8x0.7 cmand 3.5x3.0x2.0 cm in dimensions, multicystic, greyishwhite to greyish red with multiple, noncommunicatingcysts, filled with clear fluid and thin intervening septaealong with few glistening, solid white nodules (Figure 4).The tissue was fixed in formalin and processedroutinely. Histopathological examination revealedcuboidal to low columnar epithelial lined clefts, cystsand fibroepithelial polyps (Figure 5A). Mesenchymalconnective tissue revealed disarray of spindle toelongated cells with mild to moderate atypia (Figure 5B).Cartilagenous islands were also present and some ofthem revealed hypercellularity of lacunae and focaloverlapping. Occasional focus of small darkly stainedangulated nuclei (blastemallike cells) was also seen(Figure 5C). On immunohistochemistry, the liningepithelial cells of cystic spaces were positive for
cytokeratin (DakoAE1/AE3). The stromal cells revealeddiffuse vimentin (DakoV9) positivity (Figure 5D) andfocal desmin (DakoD33) positivity. A diagnosis ofpleuropulmonary blastoma type II was made. One smallpiece of lung tissue with necrotic foci was also receivedwhich revealed a necrotizing granulomatous lesion. Thisfocus was negative for acidfast bacilli. Patient was
Figure 1: Xray of chest showing homogenous opacity in rightmid and lower zones, silhouetting right heart border andblunted right costophrenic angle (PA view).
Figure 2: Contrast enhanced computed tomography axialimage showing multiple, welldefined, heterogenouslyenhancing pleural based focal lesions in right hemithorax withenhancing internal septations within the lesions.
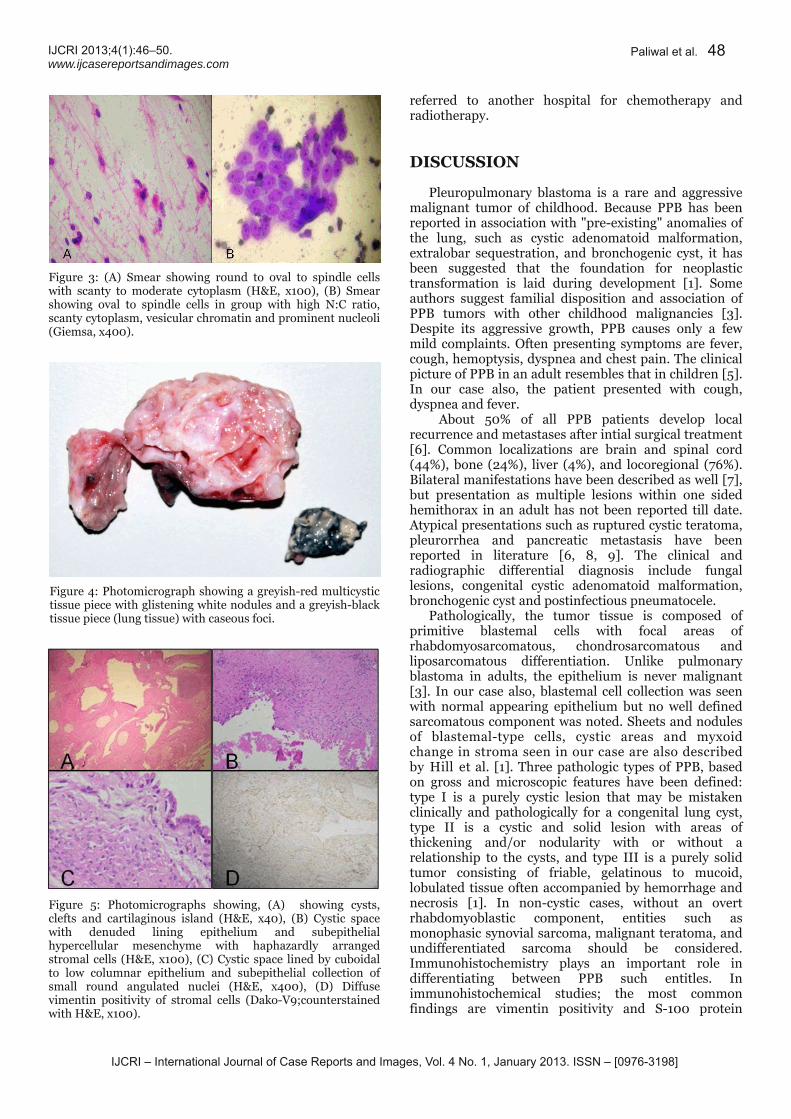
IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):46–50.www.ijcasereportsandimages.com Paliwal et al. 48
referred to another hospital for chemotherapy andradiotherapy.
DISCUSSIONPleuropulmonary blastoma is a rare and aggressivemalignant tumor of childhood. Because PPB has beenreported in association with "preexisting" anomalies ofthe lung, such as cystic adenomatoid malformation,extralobar sequestration, and bronchogenic cyst, it hasbeen suggested that the foundation for neoplastictransformation is laid during development [1]. Someauthors suggest familial disposition and association ofPPB tumors with other childhood malignancies [3].Despite its aggressive growth, PPB causes only a fewmild complaints. Often presenting symptoms are fever,cough, hemoptysis, dyspnea and chest pain. The clinicalpicture of PPB in an adult resembles that in children [5].In our case also, the patient presented with cough,dyspnea and fever.About 50% of all PPB patients develop localrecurrence and metastases after intial surgical treatment[6]. Common localizations are brain and spinal cord(44%), bone (24%), liver (4%), and locoregional (76%).Bilateral manifestations have been described as well [7],but presentation as multiple lesions within one sidedhemithorax in an adult has not been reported till date.Atypical presentations such as ruptured cystic teratoma,pleurorrhea and pancreatic metastasis have beenreported in literature [6, 8, 9]. The clinical andradiographic differential diagnosis include fungallesions, congenital cystic adenomatoid malformation,bronchogenic cyst and postinfectious pneumatocele.Pathologically, the tumor tissue is composed ofprimitive blastemal cells with focal areas ofrhabdomyosarcomatous, chondrosarcomatous andliposarcomatous differentiation. Unlike pulmonaryblastoma in adults, the epithelium is never malignant[3]. In our case also, blastemal cell collection was seenwith normal appearing epithelium but no well definedsarcomatous component was noted. Sheets and nodulesof blastemaltype cells, cystic areas and myxoidchange in stroma seen in our case are also describedby Hill et al. [1]. Three pathologic types of PPB, basedon gross and microscopic features have been defined:type I is a purely cystic lesion that may be mistakenclinically and pathologically for a congenital lung cyst,type II is a cystic and solid lesion with areas ofthickening and/or nodularity with or without arelationship to the cysts, and type III is a purely solidtumor consisting of friable, gelatinous to mucoid,lobulated tissue often accompanied by hemorrhage andnecrosis [1]. In noncystic cases, without an overtrhabdomyoblastic component, entities such asmonophasic synovial sarcoma, malignant teratoma, andundifferentiated sarcoma should be considered.Immunohistochemistry plays an important role indifferentiating between PPB such entitles. Inimmunohistochemical studies; the most commonfindings are vimentin positivity and S100 protein
Figure 3: (A) Smear showing round to oval to spindle cellswith scanty to moderate cytoplasm (H&E, x100), (B) Smearshowing oval to spindle cells in group with high N:C ratio,scanty cytoplasm, vesicular chromatin and prominent nucleoli(Giemsa, x400).
Figure 4: Photomicrograph showing a greyishred multicystictissue piece with glistening white nodules and a greyishblacktissue piece (lung tissue) with caseous foci.
Figure 5: Photomicrographs showing, (A) showing cysts,clefts and cartilaginous island (H&E, x40), (B) Cystic spacewith denuded lining epithelium and subepithelialhypercellular mesenchyme with haphazardly arrangedstromal cells (H&E, x100), (C) Cystic space lined by cuboidalto low columnar epithelium and subepithelial collection ofsmall round angulated nuclei (H&E, x400), (D) Diffusevimentin positivity of stromal cells (DakoV9;counterstainedwith H&E, x100).

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):46–50.www.ijcasereportsandimages.com Paliwal et al. 49
positivity in cartilagenous foci and desmin positivity inareas of rhabdomyoblastic differentiation. The onlytypical characteristic of PPB is vimentin positivity [4].In cytogenetic studies of childhood cases, chromosomalanomalies of trisomies 8 and 2 have been detected. Butas studied by Gonullu et al., in adult patients,interestingly, karyotypic abnormalities such astrisomies 8 and 2 were not demonstrated [4]. Germlinemutations in DICER1 has also been described in casesof familial PPB and ovarian sex cordstromal tumors [10].We were not provided with the opportunity to performcytogenetic studies in the current case.While tumor size >5 cm and mediastinal and/orpleural invasion are poor prognostic factors, the mostimportant prognostic factor is the total excision of themass with clear margins. Postoperative radiotherapy isrecommended in cases with incomplete resection [11].Cyclophosphamide, doxorubicin, ifosfamide, etoposide,vincristine are commonly used agents in the treatmentof PPB. The most common combination is vincristine,dactinomycin, cyclophosphamide (VAC) regimen [4].Therapy should include surgical tumor resection andsubsequent chemotherapy and radiotherapy. In areview of 50 cases of PPB in children by Priest et al.,overall survival for all patients was 63% after two yearsand 45% after five years. Median survival was calculatedand it was 15.5 months from diagnosis and 5.5 monthsfrom recurrence [6].
CONCLUSIONPleuropulmonary blastoma is very rare tumor inadults and its presentation as multiple lesions in onesided hemithorax has not been reported.Pleuropulmonary blastoma should be considered in theclinical and radiographic differential diagnosis whenhydatid cyst or benign teratoma is suspected in view ofits multicystic appearance, even when the patient is ayoung adult.
*********Author ContributionsNidhi Paliwal – Substantial contributions to conceptionand design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedKumud Gupta – Substantial contributions toconception and design, Acquisition of data, Analysisand interpretation of data, Drafting the article, Revisingit critically for important intellectual content, Finalapproval of the version to be publishedShalini Mullick – Substantial contributions toconception and design, Acquisition of data, Analysisand interpretation of data, Drafting the article, Revisingit critically for important intellectual content, Finalapproval of the version to be publishedRavindra K Dewan – Substantial contributions toconception and design, Acquisition of data, Analysis
and interpretation of data, Drafting the article, Revisingit critically for important intellectual content, Finalapproval of the version to be publishedSandeep Katiyar – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Nidhi Paliwal et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distribution andreproduction in any means provided the original authorsand original publisher are properly credited. (Please seewww.ijcasereportsandimages.com/copyrightpolicy.phpfor more information.)
REFERENCES1. Hill DA, Sadeghi S, Schultz MZ, Burr JS, Dehner LP.Pleuropulmonary blastoma in an adult: An initialcase report. Cancer 1999;85(11):2368–74.2. Dehner LP, Watterson J, Priest J. Pleuropulmonaryblastoma: a unique intrathroacicpulmonaryneoplasm of childhood. Perspect pediatr pathol1995;18:214–26.3. Manivel J, Priest J, Watterson J, et al.Pleuropulmonary blastoma. The socalledpulmonary blastoma of childhood. cancer1988;62(8):1516–26.4. Gonullu G, Evrense T, Kurt E, et al. Pleuropulmonaryblastoma in an adult patient: Report of a case.Turkish journal of cancer 2007;37(4):158–61.5. Walles T, Teebken OE, Bartels M, et al. Pancreaticmetastasis of a pleuropulmonary blastoma in anadult. Annals of oncology 2000;11(12):1609–11.6. Priest JR, McDermott MB, Bhatia S, Watterson J,Manivel JC, Dehner LP. Pleuropulmonary blastoma.A clinicopathologic study of 50 cases. Cancer1997;80(1):147–61.7. Lallier M, Bouchard S, Di Lorenzo M, et al.Pleuropulmonary blastoma: A rare pathology withan even rarer presentation. J Pediatr Surg1999;34(7):1057–9.8. Lee CH, Kim KI, Kim YD, et al. Pleuropulmonaryblastoma in a young adult presenting as a rupturedcystic teratoma in radiology. J Korean Med Sci 2003Aug;18(4):595–8.9. Liu AH, Zheng WY, WU L. [Pleuropulmonaryblastoma in an adult women with pleurorrhea as themajor clinical manifestation: report of a case]. NanFang Yi Ke Da Xue Xue Bao 2008Dec;28(12):2241–3.10. Schultz KAP, Pacheco MC, Yang J, et al. Ovarian sexcord –stromal tumors, pleuropulmonary blastomaand DICER1 mutations: A report from the

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):46–50.www.ijcasereportsandimages.com
International Pleuropulmonary blastoma Registry.Gynecol Oncol 2011 Aug;122(2):246–50.11. Parsons SK, Fishman SJ, Hoorntje LE, et al.Aggressive multimodal treatment of
Paliwal et al. 50
Access full text article onother devices Access PDF of article onother devices
Pleuropulmonary blastoma. Ann Thorac Surg2001;72(3):939–42.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):51 –54.www.ijcasereportsandimages.com
Combined aortic and inferior vena cava injuryBasem Marcos, Yomayra Perez, Jennifer Matarlo, Jay A Yelon,Valerie Katz, Robert V Madlinger
ABSTRACTIntroduction: Combined injuries to the aortaand inferior vena cava are among the mostsevere traumatic injuries where mortality ratescan approach 100%. Case Report: A 26yearoldmale presented with multiple small calibergunshot wounds to the right upper and lowerback, right posterior arm and right gluteal area.He was diagnosed with large retroperitonealhematoma on computed tomography (CT) scan.He underwent an exploratory laparotomy withexploration of a zone 1 retroperitonealhematoma. An injury to the inferior vena cava(IVC) at the level of the left renal vein wasidentified and repaired by lateral venorrhaphy.Massive transfusion protocol (MTP) wasactivated. The patient was returned back to theoperating room few hours later for recurrentbleeding. Aortic injury was identified onecentimeter distal to the renal arteries secondaryto ruptured pseudoaneurysm. The left renal vein
was ligated for exposure. The aortic injury wasrepaired. The patient was resuscitatedpostoperatively and was discharged homewithout major morbidity. Conclusion:Combined inferior vena cava and infrarenalaortic injury carries a mortality of around 73%.All zone 1 retroperitoneal hematomas should beexplored, with the exception of venoushematomas of the juxtahepatic vena cava.Mattox maneuver is the ideal exposure for asupramesocolic retroperitoneal hematoma andCattell–Brash maneuver is used forinframesocolic retroperitoneal hematomas.Zone 1 retroperitoneal hematoma resultingfrom combined injuries to the inferior venacava and aorta is highly lethal. Successfulmanagement requires early MTP, rapid andaggressive surgical management andapplication of elective vascular surgerytechniques.Keywords: Inferior vena cava injury, Aortainjury
*********Marcos B, Perez Y, Matarlo J, Yelon JA, Katz V,Madlinger RV. Combined aortic and inferior vena cavainjury. International Journal of Case Reports andImages 2013;4(1):51–54.
*********doi:10.5348/ijcri201301258CR12
INTRODUCTIONCombined injuries to the aorta and inferior venacava (IVC) are among the most severe traumatic injuriesand are associated with high mortality rates. Mortalityrates for such injuries can approach 100%. Adherence to
CASE REPORT OPEN ACCESS
Basem Marcos1 , Yomayra Perez2, Jennifer Matarlo3, Jay AYelon4, Valerie Katz5, Robert V Madlinger6
Affi l iations: 1Resident, Department of Surgery, LincolnMedical and Mental Health, Bronx, New York, USA;2Research Assistant, Department of Surgery, LincolnMedical and Mental Health, Bronx, New York, USA;3Trauma Coordinator, Department of Surgery, LincolnMedical and Mental Health, Bronx, New York, USA;4Chairman, Department of Surgery, Lincoln Medical andMental Health, Bronx, New York, USA; 5AssociateProgram Director, Department of Surgery, Lincoln Medicaland Mental Health, Bronx, New York, USA; 6Director ofTrauma Services, Department of Traumatology, LincolnMedical and Mental Health, Bronx, New York, USA.Corresponding Author: Basem Marcos, 234 East 1 49th,Bronx, New York, USA. 1 0451 ; Ph: 71 8-579-5900; Fax:71 8-579-4620; Email : [email protected]
Received: 26 October 2011Accepted: 24 Apri l 201 2Published: 01 January 201 3
Marcos et al. 51

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):51 –54.www.ijcasereportsandimages.com Marcos et al. 52
advanced trauma life support protocol, principles ofdamage control surgery, and implementation of anevidence based massive transfusion protocol (MTP) maylead to improved survival with a decrease in majormorbidity.
CASE REPORTA 26yearold male was brought to the emergencydepartment (ED) by emergency medical services afterhe was found with multiple small caliber gunshotwounds to the right upper and lower back, rightposterior arm and right gluteal area (Figure 1). Onarrival his vital signs were: heart rate 72/min,respiratory rate 22/min, blood pressure 100/48 mmHgand spO2 100%. He complained of abdominal pain. Onphysical examination the abdomen was soft, nondistended and moderately tender over the midabdomen without rebound, rigidity, or guarding. Bowelsounds were present. Focused abdominal sonographyfor trauma (FAST) was performed in the ED which didnot reveal any free fluid. Further diagnostic studiesincluded a computed tomography (CT) scan of theabdomen and pelvis was suspicious for injuries to theabdominal aorta and portal vein.The patient was immediately taken for operation.The MTP was activated in preparation for the possibilityof a major abdominal vascular injury. Massivetransfusion protocol in our institution is constituted ofpRBCs:FFPs in 1:1 ratio followed by giving single donorplatelets and 10 units of cryoprecipitate after 10 unitseach of pRBCs and FFPs. The patient underwent anexploratory laparotomy with exploration of a zone 1retroperitoneal hematoma (Figure 2). An injury to theIVC at the level of the left renal vein was identified andrepaired by lateral venorrhaphy. No bleeding was notedfrom the portal vein or the aorta. Hemorrhage wascontrolled, the abdomen was packed and left openbecause of midgut edema and the patient wastransferred to the surgical intensive care unit (SICU) forcontinued resuscitation. A few hours later, the patientdeveloped signs of hypovolemic shock and wasimmediately returned to the operating room. The MTPwas again activated. Supraceliac aortic control wasobtained and an aortic injury was identified one
centimeter distal to the renal arteries (this was thepseudoaneurysm that was present on the initial imagingand missed during the first laparotomy and progressedto subsequent rupture). The left renal vein was ligatedand divided to provide adequate exposure of the injury.The aortic injury was repaired by primary sutureclosure. The patient received a total of 53 units ofpacked red blood cells, 49 units of fresh frozen plasma, 7units of single donor platelets, and 40 units ofcryoprecipitate perioperatively. At the end of surgery,the patient’s lactate was 9.5 mmol/L and his base deficitwas 18. Resuscitation was continued and these findingsof hypoperfusion were corrected in less than 24 hours.The patient underwent a planned reexploration andabdominal washout with partial fascial closure onpostoperative day (POD)3. The abdomen was closed onPOD7. The patient developed acute cholecystitis andunderwent an open cholecystectomy on POD11. Atracheostomy was performed on POD14. The patientspent 23 days in the SICU, four days in the surgical wardand he was then transferred to an acute inpatientrehabilitation center. The patient was seen in thesurgery clinic following his discharge from inpatientrehabilitation. He was ambulating and tolerating a diet.His tracheostomy had been removed and the site of histracheostomy.
DISCUSSIONThe retroperitoneum is divided to zone 1 (around theabdominal aorta and IVC), zone 2 (around both thekidneys), and zone 3 (in the pelvis). Zone 1retroperitoneal hematoma is a sign of a majorabdominal vascular injury. Penetrating trauma accountsfor approximately 90% cases. The overall mortality rateis 57%. Mortality rates range from 30% for an infrarenalIVC injury, 50% for an infrarenal aortic injury, 60% foran injury to the suprarenal abdominal aorta and 100%for combined injuries to the suprarenal abdominal aortaand IVC. Our patient had an injury to the IVC as well asan infrarenal aortic injury, which carries approximately73% mortality rate. Factors associated with increasedFigure 1: Plain chest and abdominal Xray showing radioopaque bullet fragments.
Figure 2: Computed tomography scan of the abdomenshowing large zone 1 retroperitoneal hematoma.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):51 –54.www.ijcasereportsandimages.com Marcos et al. 53
mortality are presence of shock at the time ofadmission, bleeding without retroperitonealtamponade, acidosis, and suprarenal injury [1].This injury is more common in urban trauma centersthan it is in military conflicts. Debakey et al. reviewed2,471 patients with arterial injuries during World War II.Two percent patients had abdominal arterial injuries.Rich et al. reviewed 1,000 patients with arterial injuriessustained in the Vietnam War. Only 2.9% injuriesinvolved abdominal vessels. A thirtyyear review of5,760 cardiovascular injures seen at Ben Taub Hospital(Houston, TX, USA) found that 33.8% patients hadabdominal vascular injuries. This difference is thoughtto be due to the increased wounding power of militaryweapons, delayed transport and torso body armor [2].Patients can present clinically with one of twopictures: (i) contained hematoma with transienthypotension responding to crystalloid boluses, or (ii)free intraperitoneal rupture with marked hypotensionwhich does not respond to fluid boluses. Patients withcontained hematoma, like our patient, can remainhemodynamically normal until the hematoma is openedin the operating room. In one review, patients with acontained hematoma had a mean base deficit of 7.2,mean transfusion of 8.6 units of blood, and a survivalrate of 96%. Those with free intraperitoneal rupture hada mean base deficit of 14.7, mean transfusion of 15.1units and 43% survival [2]. In patients with penetratingabdominal trauma to the back and flank who arehemodinamically normal, CT scan of the abdomen canbe obtained for further evaluation of injuries prior tosurgery [3].In cases with penetrating abdominal trauma,standard trauma principles apply. These includeexploration via a midline incision, four quadrantpacking and evacuation of blood and blood clots.Proximal and distal vascular control is essential and canbe achieved by a variety of vascular techniques,including direct digital pressure, pressure withlaparotomy pads (packing) or pressure with spongesticks. Enteric injuries are addressed with clamps orstaples. An associated ureteral injury may be treatedinitially by ligating the ureter during damage controlsurgery.All zone 1 retroperitoneal hematomas should beexplored, with the exception of venous hematomas ofthe juxtahepatic vena cava [4]. A left medial visceralrotation (Mattox maneuver) is the ideal exposure for asupramesocolic retroperitoneal hematoma and the rightmedial visceral rotation or Cattell–Brash maneuver withkocherization of the duodenum is used forinframesocolic retroperitoneal hematomas [5].After hemorrhage control of the injured aorta or IVCis obtained, exploration of the hematoma is begun at thehighest point. Lateral repair (simple primary suturerepair) of the aorta and IVC should be attempted.Complex vascular repairs can be performed if there isloss of part of the arterial wall, but this is timeconsuming. The left renal vein can be ligated anddivided. This aids in exposure of injuries at this level. IfIVC repair is not possible, the IVC can be ligated. Lower
extremity fasciotomies may be needed along withaggressive resuscitation. The infrarenal aorta can beligated if it cannot be repaired with axillary bifemoralextraanatomic bypass performed later, if the patientsurvives [4].
CONCLUSIONZone 1 retroperitoneal hematoma resulting fromcombined injuries to the inferior vena cava and aorta ishighly lethal. Management should be individualizedbased on the patient’s presentation. Key maneuvers arehemorrhage control and exposure. Successfulmanagement of acute traumatic injury to both theabdominal aorta and inferior vena cava requires earlyactivation of massive transfusion protocol, rapid andaggressive surgical management, including damagecontrol techniques and application of techniques andprinciples developed for elective vascular surgery.
*********Author ContributionsBasem Marcos – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedYomayra Perez – Analysis and interpretation of data,Revising it critically for important intellectual content,Final approval of the version to be publishedJennifer Matarlo – Analysis and interpretation of data,Revising it critically for important intellectual content,Final approval of the version to be publishedJay A Yelon – Analysis and interpretation of data,Revising it critically for important intellectual content,Final approval of the version to be publishedValerie Katz – Analysis and interpretation of data,Revising it critically for important intellectual content,Final approval of the version to be publishedRobert V Madlinger – Analysis and interpretation ofdata, Revising it critically for important intellectualcontent, Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Basem Marcos et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):51 –54.www.ijcasereportsandimages.com
REFERENCES1. Coimbra R, Hoyt D, Winchell R, Simons R, FortlageD, Garcia J. The Ongoing Challenge ofRetroperitoneal Vascular Injuries. AmericanJournal of surgery 1996 Nov;172(5):541–4.2. Juan A Asencio, Donald D Trunkey. CurrentTherapy of Trauma & Surgical Critical Care2008:410.
Marcos et al. 54
Access full text article onother devices Access PDF of article onother devices
3. F. Charles Brunicardi, Dana K Andersen, Timothy RBilliar, David L Dunn, John G Hunter. SchwartzPrinciples of Surgery 2010(7):fig7–2.4. Hirshberg Asher, Kenneth Mattox. Top knife art &craft of trauma surgery 2005;9:131–46.5. Lenworth Jacobs, Stephen Luk. Advanced TraumaOperative Management. Surgical Strategies forPenetrating Trauma 2010: 257–72.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):55–57.www.ijcasereportsandimages.com
Acute urinary retention in a female adolescentAlberto MendozaParedes, Antonio Pierre
ABSTRACTIntroduction: Acute urinary retention requirestimely evaluation and management in order toprevent damage to the kidneys and urinarytract. Case Report: An 11yearold female childcame to the emergency department complainingof abdominal pain for three days and oliguriawith dysuria for the last 24 hours. Physicalexamination showed a palpable mass in lowerabdomen up to umbilical level and a bulgingmass in the introitus. A Foley catheter wasinserted, draining 500 mL of urine with relief ofthe abdominal pain. After emptying the bladder,a residual mass was palpated. Renal ultrasoundshowed no abnormalities and pelvic ultrasounddemonstrated a large homogeneous echogenicmass in the lower uterine region, diagnosed ashematocolpos. Further surgical hymenectomyresolved patient’s symptoms. Conclusion: Acuteurinary retention is relatively infrequent inchildren. Hematocolpos is a rare gynaecologicalabnormality that results from imperforatehymen. Retained blood in the vagina can causecompression of the urethra and consequenturinary retention. Hematometrocolpos isanother rare cause of acute urinary retention. Inthe evaluation of a premenarchal adolescent
with acute urinary retention and with tannerstage of development 3–4, a high index ofsuspicion should be placed towards finding ananomaly in the genital tract.Keywords: Urinary obstruction, Development,Tanner
*********MendozaParedes A, Pierre A. Acute urinary retentionin a female adolescent. International Journal of CaseReports and Images 2013;4(1):55–57.
*********doi:10.5348/ijcri201301259CR13
INTRODUCTIONAcute urinary retention (AUR) is a condition thatrequires timely evaluation and management in order toprevent damage to the kidneys and urinary tract.Although AUR in men due to benign prostatichyperplasia is well known and recognized, but in womenand especially in children, it is rare and has mostly beendescribed as case reports [1–3].
CASE REPORTAn 11yearold female child, came to the emergencydepartment (ED) complaining of abdominal pain forthree days localized to suprapubic and periumbilicalarea, concomitant dysuria, dribbling and oligouria forthe last 24 hours. The patient complained of tactile feverat home one day prior to ED visit, denied sexual activityand stated that she is premenarchal. On physicalexamination she was tanner stage 3 of development. Apalpable mass was localized in the lower abdomen up tothe umbilical level. In the costovertebral angle (CVA), a
CASE REPORT OPEN ACCESS
Alberto Mendoza-Paredes1 , Antonio Pierre2
Affi l iations: 1Resident – PGY3, Pediatrics, Lincoln Medicaland Mental Health Center, Bronx, NY, US; 2Attending -Pediatrics, Lincoln Medical and Mental Health Center,Bronx, NY, USCorresponding Author: Alberto Mendoza-Paredes, LincolnMedical and Mental Health Center, Pediatric Department,234E 1 49th St. Bronx, NY, US. 1 0451 ; Ph: 71 8-579-5800;Email : almendozapa@hotmail .com
Received: 28 October 2011Accepted: 04 March 201 2Published: 01 January 201 3
Mendoza-Paredes et al. 55

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):55–57.www.ijcasereportsandimages.com Mendoza-Paredes et al. 56
questionable tenderness was elicited. A bulging masswas observed in the introitus. A Foley catheter wasinserted which drained 500 mL of urine with relief ofabdominal pain. Beta human chorionic gonadotrophin(bHCG) was negative. Complete blood count and basicmetabolic profile were within normal limits. Urinalysisshowed no abnormalities and urine was sent for cultureand sensitivity testing. After emptying the bladder, aresidual mass was palpated two fingers above the pubicsymphysis. A renal ultrasound showed noabnormalities. An abdominal pelvic ultrasounddemonstrated a large homogeneous echogenic mass inthe lower uterine segment and the cervix (Figures 1Aand 1B), most consistent with hematometrocolpos. Thepatient was admitted to the pediatric ward and aspecialist in obstetrics and gynecology was consulted.The patient was taken to the operating room where acruciate incision was performed with evacuation of450 mL of old blood from vagina with subsequenthymenectomy. After the surgical procedure theabdominal pain resolved, the Foley catheter wasremoved and the patient was able to void freely. Urineculture followup was negative.
DISCUSSIONAcute urinary retention is relatively infrequent inchildren. There are a variety of causes that are poorlydefined in literature and they differ greatly from thoseseen most frequently in adults. Among the mainetiologies; neurological processes, severe voidingdysfunction, urinary tract infection, constipation,adverse drug effect, local inflammatory causes, locallyinvading neoplasms, benign obstructing lesions andidiopathic cases are included [4].Menarche is associated more with developmentalstages rather than chronological age. It usually occurs bytanner stage 4 of development, but it can be achieved intanner stage 3 [5], as in our patient.Imperforate hymen is reported at an approximaterate of 0.1% and occurs due to the incompletecanalization of the Mullerian and the urogenital system[6]. Collection of blood in the vagina (hematocolpos) is arare gynaecological abnormality that results fromimperforate hymen. However, vaginal atresia oriatrogenic injury can also result in hematocolpos [7].Retained blood in the vagina can cause compressionof the urethra and urinary retention [8–9]. It can also
Figure 1: (A) Transabdominal pelvic ultrasound (Transversal view), (B) Transabdominal pelvic ultrasound (Sagital view).
present as low back pain [10] or constipation [11]. Insuch cases thorough physical examination (sometimesdifficult in this particular age group due to personal andcultural reasons) will suggest the diagnosis and animaging test such as pelvic ultrasound or magneticresonance imaging will be confirmatory [12].
CONCLUSIONHematometrocolpos is a rare cause of acute urinaryretention. In the evaluation of a premenarchaladolescent with acute urinary retention and tanner
stage of development 3–4, a high index of suspicionshould be placed towards finding an anomaly in theurogenital tract.*********
Author ContributionsAlberto MendozaParedes – Conception and design,Acquisition of data, Analysis and interpretation of data,Drafting the article, Critical revision of the article, Finalapproval of the version to be publishedAntonio Pierre – Final approval of the version to bepublished

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):55–57.www.ijcasereportsandimages.com Mendoza-Paredes et al. 57
GuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Alberto MendozaParedes et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properlycredited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Yu TJ, Lin MC. Acute Urinary Retention in twoPatients with Imperforate Hymen. ScandinavianJournal of Urology and Nephrolog1993;27(4):543–4.2. Gyimadu A, Sayal B, Guven S, Gunalp GS.Hematocolpos causing severe urinary retention in anadolescent girl with imperforate hymen: anuncommon presentation. Arch Gynecol Obstet2009;280(3):461–3.3. Ercan CM, Karasahin KE, Alanbay I, Ulubay M,Baser I. Imperforate hymen causing hematocolposand acute urinary retention in an adolescent girl.Taiwan J Obstet Gynecol 2011;50(1):118–20.
Access full text article onother devices Access PDF of article onother devices
4. Gatti JM, PerezBrayfield M, Kirsch AJ, Smith EA,Massad HC, Broecker BH. Acute Urinary RetentionIn Children. The Journal of Urology2001;165(3):918–21.5. Marshall WA, Tanner JM. Variations in pattern ofpubertal changes in girls. Arch Dis Child1969;44(235):291–303.6. Stelling JR, Gray MR, Davis AJ, Cowan JM,Reindollar RH. Dominant transmission ofimperforate hymen. Fertility and Sterility2000;74(6):1241–4.7. Khan N, Qazi SA, Khan N. Congenitalhematometrocolpos in a circumcised girl: annormally superimposed by cultural mutilatingpractices. J Pak Med Assoc 1997;47(11):288–9.8. van Bommel PJ, Abdullo O. Imperforate hymen as acause of urnarv obstruction. Trop Doctor1996;26(3):133.9. Hall DJ. An unusual case of urinary retention due toimperforate hymen. J Accid Emerg Med1999;16(3):232–3.10. Buick RG, Chowdhary SK. Backache: a rare diagnosisand unusual complication. Pediatr Surg Int1999;15(8):586–7.11. Isenhour JL, Hanley ML, Marx JA.Hematocolpometra manifesting as constipation inthe young female. Acad Emerg Med1999;6(7):752–3.12. Adali E, Kurdoglu M, Yildizhan R, Kolusari A. Anoverlooked cause of acute urinary retention in anadolescent girl: a case report. Arch Gynecol Obstet2009;279(5):701–3.

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):58–61 .www.ijcasereportsandimages.com
Rib fractures: Accidental or nonaccidentalMuhammad Waseem, Evelyn Erickson
ABSTRACTIntroduction: We report an incidental discoveryof multiple rib fractures in a wheezing childwithout a history of an injury or the presence ofmetabolic bone disease. As a result, the childwas evaluated for the presence of nonaccidental trauma. Case Report: An 11monthold AfricanAmerican child was brought to theemergency department by his father with a 2day history of fever, cough and breathingdifficulty. After receiving nebulizer treatments,the child was still wheezing. A chest Xray wasobtained which showed bilateral fractures of theribs. No history of trauma was provided. Giventhe radiographic findings, Child ProtectiveServices was contacted and a report of childabuse was made. Conclusion: Child abuse is acomplex phenomenon. Any skeletal injury inyoung children can be due to abuse. Ribfractures are uncommon in the pliable chest of achild. When discovered, however, they raise thesuspicion of a nonaccidental trauma. They areoften uncovered during the assessment ofchildren who present to the emergencydepartment for unrelated reasons. Thephysician's ability to differentiate accidentalfrom nonaccidental trauma may depend on
gathered information. This report emphasizedthe importance to evaluate for nonaccidentaltrauma after the finding of bilateral ribfractures on a chest Xray. Nonaccidentaltrauma should be considered when there isevidence of injury without a history of trauma.Keywords: Nonaccidental trauma, Ribfractures, Child abuse
*********Waseem M, Erickson E. Rib fractures: Accidental ornonaccidental. International Journal of Case Reportsand Images 2013;4(1):58–61.
*********doi:10.5348/ijcri201301260CR14
INTRODUCTIONRib fractures are uncommon in infants and children.Nonaccidental trauma is a common cause of morbidityand mortality in young children, but the diagnosis is notalways apparent. Most abused children present withouta plausible explanation for their injuries. In the absenceof a documented history of significant injury or thepresence of metabolic bone disease, nonaccidentaltrauma is the most likely presumed diagnosis. Wereport an incidental discovery of multiple rib fracturesin a wheezing child.
CASE REPORTAn 11monthold AfricanAmerican, asthmatic childwas brought by his father to the emergency departmentduring the winter with a twoday history of fever, coughand breathing difficulty. On arrival, he was noted to bewheezing and was directly brought to the asthma room.
CASE REPORT OPEN ACCESS
Muhammad Waseem1 , Evelyn Erickson2
Affi l iations: 1Department of Emergency Medicine, LincolnMedical & Mental Health Center, Bronx, NY, United States;2PGY-1 Resident, Department of Pediatrics, LincolnMedical and Mental Health Center, Bronx, NY, UnitedStates.Corresponding Author: Muhammad Waseem, MD,Department of Emergency Medicine, Lincoln Medical &Mental Health Center, 234 East 1 49th Street, Bronx, NY,Unites States -1 0451 ; Ph: (71 8) 579-601 0; Fax: (71 8) 579-4822; E-mail ID: [email protected]
Received: 31 October 2011Accepted: 23 January 201 2Published: 01 January 201 3
Waseem et al. 58

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):58–61 .www.ijcasereportsandimages.com Waseem et al. 59
He had a temperature of 101.20F, heart rate of 112/min,respiratory rate of 30/min, and an oxygen saturation of95%. The rest of the physical examination was normal.The medical history revealed prior episodes ofwheezing in a developmentally appropriate child. Nohistory of trauma was given. The patient lived with hisfather and had not attended school or daycare. Therewere no other siblings. Physical examination revealedno bruising, swelling, abnormal marks or other signs oftrauma. His complete blood count and basic metabolicprofile were normal. Subsequent laboratoryinvestigations were as follows: serum phosphate level5.3 mg/dL (2.7–4.5 mg/dL), serum 25OH Vitamin D 31ng/ml (3–67 ng/mL) and serum alkaline phosphatase139 U/L (30–90 U/L). Normal range is given inparenthesis. A chest Xray was obtained due topersistent wheezing despite three nebulizer treatments.No infiltrate was noted but bilateral posterior fracturesof the 9th and 10th ribs were identified (Figures 1, 2). Asubsequent skeletal survey confirmed the presence ofbilateral healed fractures of the 9th and 10th ribs. Giventhe radiographic findings, Child Protective Services wasconsulted in the emergency department and a report ofchild abuse was made.
DISCUSSIONChild abuse is a complex phenomenon. Any skeletalinjury can be due to abuse. Rib fractures are uncommonin the pliable chest of a child. The presence of bilateralrib fractures in an infant should prompt a thoroughmedical and social evaluation for child abuse [1]. Whendiscovered, however, they raise the suspicion of nonaccidental trauma. These findings may be uncoveredduring the assessment of children who present to theemergency department for unrelated reasons. Nonaccidental trauma is a relatively common occurrenceand fractures are the second most common presentationof child abuse [2]. Bilateral rib fractures, particularly ininfants, should always raise the suspicion. Manychildren with nonaccidental trauma have healingfractures. Multiple rib fractures are considered a markerof serious injury in children.Often, a chest Xray is obtained in a wheezing childwith fever to rule out pneumonia. This child had anasthma exacerbation due to an upper respiratoryinfection and received care for his asthma. If the chestradiograph is not carefully reviewed, rib fractures maybe overlooked; this is especially true in a busyemergency department. The diagnosis of rib fractures isoften made by obtaining plain Xray of the chest, as inour patient. A dedicated rib series may better define thefracture including the age and location. As one mightexpect, location of rib fractures may provideinformation regarding the mechanism of injury. Theposterior fractures occur due to the mechanical stress atthe costovertebral junction as the child is grabbed andshaken. A detailed history of how the injury occurred is,therefore, essential. When nonaccidental trauma isbeing considered, it is imperative to evaluate the child
Figure 1: Chest Xray of an 11monthold infant showingbilateral rib fractures.
Figure 2: Xray showing bilateral posterior fractures of the 9thand 10th ribs in an 11monthold infant.

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):58–61 .www.ijcasereportsandimages.com Waseem et al. 60
for other fractures; a complete skeletal survey mayuncover additional injuries. It is uncommon to detectthese fractures in the acute phase as they are better seenwhen callus formation is advanced. A followup chestXray may, therefore, provide useful information inchildren with suspected nonaccidental injury and mayimprove detection [3, 4].The physician's ability to differentiate accidental
from nonaccidental trauma may depend on gatheredinformation. It is difficult to ascertain the cause of ribfractures when no plausible history to explain the injuryis offered. Often, the trauma is only recalled after thefracture is identified. Generally, most rib fractures innonaccidental trauma are the consequence of thoraciccompression [3]. Posterior rib fractures are consideredto have a strong association with nonaccidentaltrauma. Overall, a rib fracture had a positive predictivevalue of 95% for the diagnosis of nonaccidental trauma[3]. The compliance of the rib cage may allow significantinjury to occur with little apparent external signs oftrauma [3]. Because of the delay in clinical presentationin such cases, healing fractures with callus are moreprevalent than acute fractures [3]. Posterior ribfractures, in particular, have a wellknown associationwith nonaccidental injury [3].
Table 1 gives other causes of rib fractures in children[3, 5]. Rib fractures may be associated with birthtrauma. If birth related injuries are not identifiedinitially, they may later be attributed to nonaccidentaltrauma [3]. Rib fractures may occur withcardiopulmonary resuscitation but the possibility ofnonaccidental injury should be considered [3]. In thepresence of unexplained fractures, causes of bonefragility such as osteogenesis imperfecta and ricketsmust also be considered [3, 5–7]. The patients withrickets, laboratory evaluation usually reveals low to lownormal serum calcium, low serum phosphorus, and anelevated alkaline phosphatase level. The physical signsof rickets include growth retardation, metaphysealflaring, prominence of the costochondral junctions(rachitic rosary) and frontal bossing [8]. Our patient didnot have any physical findings consistent with thediagnosis of rickets. There was also no history of traumaor cardio pulmonary resuscitation.Table 1: Important Causes of Rib Fractures in Children
Trauma• Accidental (rare)• NonAccidentalMetabolic Bone Diseases• Osteogenesis Imperfecta• Rickets
CONCLUSIONThis case report provides an approach in theevaluation of child with evidence of injury but no history
of trauma. In the absence of a history of a significantaccidental trauma, evaluation of nonaccidental traumashould be performed. Determining whether a fracture isdue to accidental or nonaccidental trauma can bechallenging, but the future safety of the child, dependson a timely diagnosis and intervention.*********
Author ContributionsMuhammad Waseem – Conception and design,Acquisition of data, Analysis and interpretation of data,Drafting the article, Critical revision of the article, Finalapproval of the version to be publishedEvelyn Erickson – Acquisition of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Muhammad Waseem et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properlycredited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Melville JD, Lukefahr JL, Clarke EA. First RibFractures in Abused Infants: A Report of ThreeCases. Clin Pediatr (Phila): 2011 Dec 8.2. Bulloch B, Schubert CJ, Brophy PD, Johnson N,Reed MH, Shapiro RA. Cause and clinicalcharacteristics of rib fractures in infants. Pediatrics2000;105(4):E48.3. Maguire S, Mann M, John N, Ellaway B, Sibert JR,Kemp AM. Welsh Child Protection SystematicReview Group. Does cardiopulmonary resuscitationcause rib fractures in children? A systematic review.Child Abuse Negl 2006;30(7):739–51.4. Anilkumar A, Fender LJ, Broderick NJ, Somers JM,Halliday KE. The role of the followup chestradiograph in suspected nonaccidental injury.Pediatr Radiol 2006;36(3):216–8.5. Hoke RS, Chamberlain D. Skeletal chest injuriessecondary to cardiopulmonary resuscitation.Resuscitation 2004;63(3):327–8.6. Paterson CR. Osteogenesis imperfecta and otherbone disorders in the differential diagnosis ofunexplained fractures. J R Soc Med1990;83(2):72–4.7. Avarello JT, Cantor RM. Pediatric major trauma: anapproach to evaluation and management. EmergMed Clin North Am 2007;25(3):80336.

IJCRI – International Journal of Case Reports and Images, Vol. 4, No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):58–61 .www.ijcasereportsandimages.com Waseem et al. 61
8. Cadzow SP, Armstrong KL. Rib fractures in infants:red alert! The clinical features, investigations andchild protection outcomes. J Paediatr Child Health2000;36(4):322–6.
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):62–65.www.ijcasereportsandimages.com
An unusual cause of hypertensionMuhammad Waseem, Evelyn Erickson
ABSTRACTIntroduction: Hypertension is not common ininfants and young children. The etiology ofhypertension in this age group may be differentfrom older children and young adults.Hypertension may be associated withintussusception in young children. Case Report:A previously healthy 17monthold, illappearingand dehydrated girl was brought to theemergency department with fever and vomitingfor three days. The vomiting was nonbiliousand nonprojectile. Her temperature was 1030F,heart rate was 126/min, respiratory rate was32/min, oxygen saturation 98% and bloodpressure 122/77 mmHg. She had dry mucousmembranes. The abdomen was soft with mildtenderness in the left upper quadrant and nopalpable masses. She was diagnosed withileocolic intussusception. Conclusion:Intussusception should be considered adiagnostic possibility in infants who have ahistory of vomiting and in whom lethargy andhypertension are the presenting features. Thiscase report demonstrates the importance ofmeasuring blood pressure in illappearingchildren.Keywords: Hypertension, Intussusception,Diagnostic features
*********Waseem M, Erickson E. An unusual cause ofhypertension. International Journal of Case Reportsand Images 2013;4(1):62–65.
*********doi:10.5348/ijcri201301261CR15
INTRODUCTIONHypertension in children is defined as bloodpressure measurements above the 95th percentile forage, gender and height of the patient. Blood pressure isnot routinely measured in infants and young children inthe emergency department, though measurementshould be obtained in ill appearing young children.Unlike adults and adolescents, hypertension in an infantor young child is usually indicative of an underlyingcondition, therefore, a careful search should beconducted. Renal disorders and coarctation of the aortaare the two most common causes of hypertension inyoung children. The association of hypertension andintussusception has been described in the past, beingcharacterized as transient and usually resolving after thereduction of intussusception.
CASE REPORTA previously healthy 17monthold girl was broughtto the emergency department with fever and vomitingfor three days. The vomiting was nonbilious and nonprojectile. She had no diarrhea but parents reported hercrying more than usual for last two days. On the day ofpresentation, her parents also noted that “she was lessactive”. Her parents denied sick contacts and any travelhistory. Her immunization was current. There was nofamily history of hypertension.
CASE REPORT OPEN ACCESS
Muhammad Waseem1 , Evelyn Erickson2
Affi l iations: 1Associate Professor, Emergency Medicine,Lincoln Medical and Mental Health Center, Bronx, NY,USA; 2PGY-1 Resident, Pediatrics, Lincoln Medical andMental Health Center, Bronx, NY, USA.Corresponding Author: Muhammad Waseem, MD LincolnMedical and Mental Health Center, 234 E 1 49th Street,Bronx, NY, USA. 1 0451 ; Ph: (71 8) 579-601 0; Faxr: (71 8)579-4822; E-mail : [email protected]
Received: 31 October 2011Accepted: 28 March 201 2Published: 01 January 201 3
Waseem et al. 62

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):62–65.www.ijcasereportsandimages.com Waseem et al. 63
On arrival at the emergency department she wasdehydrated and illappearing. Her temperature was103°F, heart rate was 126/min, respiratory rate was32/min, oxygen saturation was 98% and blood pressurewas 122/77 mmHg. She had dry mucous membranes.There were no meningeal signs. Pupils were equal andreactive, and extraocular movements were intact. Herchest was clear. The abdomen was soft with mildtenderness in the left upper quadrant. There were nopalpable masses. Her initial stool was negative for occultblood. There was no peripheral edema. No skin lesionsor rashes were noted. There were no focal abnormalfindings on neurological examination.Laboratory studies showed a white blood cell countof 8.9x103/mm3 with a normal differential count and ahematocrit of 33.2%. Her urinalysis showed specificgravity of 1.043 and 3+ ketones. Serum biochemistriesrevealed sodium 141 mEq/L, potassium 4.6 mEq/L,chloride 107 mEq/L, CO2 19 mEq/L, urea 15 mg/dL,creatinine 0.6 mg/dL, glucose 74 mg/dL and calcium9.3 mg/dL. Due to persistent vomiting, a plain Xray ofabdomen was obtained, which showed nonspecificpattern and paucity of bowel gas (Figure 1).The patient was initially treated with intravenousfluids to correct dehydration. Her clinical conditionimproved after hydration but blood pressure remained120/76 mmHg. An abdominal ultrasound was obtainedwhich showed a normal urinary tract andintussusception in the mid transverse colon (Figure 2).Subsequently, a barium enema was performed whichsuccessfully reduced the ileocolic intussusception. Her
blood pressure returned to normal after reduction of theintussusception. She was discharged home in stablecondition two days after the procedure.
DISCUSSIONThe differential diagnosis of a child with fever,lethargy and vomiting is broad. In addition, thepresence of hypertension in a young child may makethis list more extensive. Since hypertension is not acommon problem in a pediatric emergency department,presence of hypertension in a young child may pose adiagnostic challenge.Unlike adults and adolescents, hypertension in aninfant or young child is usually indicative of anunderlying condition, therefore a careful search shouldbe conducted. Emergency physicians are often the firstto evaluate these children. It is important to recognizethe underlying causes of hypertension in this age group.Hypertension in children is defined as bloodpressure measurements above the 95th percentile forage, gender and height of the patient. Standardnomograms, based on the above factors, are necessaryfor the interpretations of blood pressure values.Inappropriate cuff size is the most common cause ofhypertension in children, thus selection of an arm cuffof the right size is necessary for accurate measurementof blood pressure [1]. An appropriate cuff size shouldhave an inflatable bladder width which is at least 40% ofthe arm circumference at a point midway between theolecranon and the acromion; and cuff bladder lengthshould also cover 80–100% of the circumference of thearm [2]. Recommended cuff sizes are as follows:neonates (2.5 cm), infants (5 cm), 1–8 years (9 cm) and9–14 years (12.5 cm).This case report raises the question whether bloodpressure is measured routinely in children less thanthree years of age. Generally, blood pressure is notroutinely measured in infants and young children in theemergency department [3, 4]. However, blood pressureFigure 1: Plain abdominal Xray showing nonspecific patternand paucity of bowel gas.
Figure 2: Abdominal ultrasound showing intussusception inthe mid transverse colon.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):62–65.www.ijcasereportsandimages.com Waseem et al. 64
measurement should be obtained in all ill appearingyoung children.Essential hypertension is very rare in children andshould be considered only after exclusion of othercauses. Renal disorders and coarctation of the aorta arethe two most common causes of hypertension in youngchildren. Blood pressure should be obtained in theupper and lower extremities to rule out coarctation ofthe aorta. In addition, a transient rise in blood pressuremay be seen in the presence of stress, crying or pain.Therefore, the diagnosis of hypertension in a childshould be made only after resolution of these causes.Hypertension may be associated withintussusception. The association of hypertension andintussusception has been described in the past, but onlya few case reports exist in literature [5, 6]. Hypertensionis transient and usually resolves after reduction ofintussusception. Therefore, intussusception should beconsidered a diagnostic possibility in infants who have ahistory of vomiting, and in whom lethargy andhypertension are the presenting features. This casereport highlights the importance of measuring bloodpressure in illappearing children.
CONCLUSIONThe association of hypertension and intussusceptionhas been described in the past. Intussusception shouldbe considered a diagnostic possibility in infants whohave a history of vomiting and in whom lethargy andhypertension are the presenting features. This casereport demonstrates the importance of measuring bloodpressure in illappearing children in the emergencydepartment.
*********Author ContributionsMuhammad Waseem – Conception and design,Acquisition of data, Analysis and interpretation of data,Drafting the article, Critical revision of the article, Finalapproval of the version to be publishedEvelyn Erickson – Acquisition of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Muhammad Waseem et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properly
credited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Mattoo TK. Arm Cuff in the Measurement of BloodPressure. Am J Hypertens 2002;15(2);67s–8s.2. National High Blood Pressure Education ProgramWorking Group on High Blood Pressure in Childrenand Adolescents. The Fourth Report on theDiagnosis, Evaluation, and Treatment of High BloodPressure in Children and Adolescents. Pediatrics2004;144(2 suppl 4th Report):555–76.3. Silverman MA, Walker AR, Nicolaous DD, Bono MJ.The Frequency of Blood Pressure measurements inChildren in Four EDs. Am J Emerg Med2000;18(7):784–8.4. Gilhotra Y, Willis F. Blood Pressure Measurementson Children in the Emergency Department. EmergMed Australas 2006;18(2):148–54.5. Barton LL, Chundu K. Intussusception associatedwith Transient Hypertention. Pediatr Emerg Care1988;4(4):249–50.6. Prichard JG, Pakula AS. Hypertension andintussuscpetion. Clin Pediatr 1987;26(4):196–8.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):62–65.www.ijcasereportsandimages.com Waseem et al. 65
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):66–69.www.ijcasereportsandimages.com
A floppy infantMuhammad Waseem, Joel Gernsheimer, Tae K Park,Fernando Jara, Evelyn Erickson
ABSTRACTIntroduction: Infant botulism is a relativelyuncommon but potentially life threateningcause of a septic appearing or lethargic infant.Case Report: A 6weekold male infantpresented to the emergency department with ahistory of poor feeding and fever for severaldays. His parents reported that he had been“more sleepy than usual” and had a weak cry.He had not passed any stool for five days. Hewas receiving a topical home herbal remedy forwhitish lesions in his mouth. The rest of hisreview of systems and past medical history wasnoncontributory. On arrival to emergencydepartment, he was ill appearing and lethargic.His vital signs were: temperature 1010F, heartrate 152/min, respiratory rate 44/min andoxygen saturation 99%. He had poor muscletone and generalized weakness. He wasdiagnosed with infant botulism. Conclusion: It isextremely important that the diagnosis of infantbotulism be suspected and appropriately treatedwhen any infant presents with progressiveweakness. Since infant botulism is a treatable
condition, prompt diagnosis is thereforeimportant in reducing morbidity and mortality.Keywords: Infant botulism, Clostridiumbotulinum, Weakness
*********Waseem M, Gernsheimer J, Park TK, Jara F, EricksonE. A floppy infant. International Journal of CaseReports and Images 2013;4(1):66–69.
*********doi:10.5348/ijcri201301262CR16
INTRODUCTIONInfant botulism is a relatively uncommon but,potentially life threatening cause of a very ill appearinginfant. We present the case of a six week old infant whopresented to the emergency department (ED) withprogressive neurological weakness that ultimatelyrequired ventilatory support. The differential diagnosisof weakness in the young infant is discussed and thepertinent literature is reviewed.
CASE REPORTA 6weekold male infant presented to theemergency department with a history of poor feedingand fever for several days. His parents reported that hehad been “more sleepy than usual” and had a weak cry.He had not passed any stool for five days. He wasreceiving a topical home herbal remedy for whitishlesions in his mouth. The rest of his review of systemsand past medical history was noncontributory.On arrival in the emergency department, he was illappearing and lethargic. His vital signs were:temperature 1010F, heart rate 152/min, respiratory rate
CASE REPORT OPEN ACCESS
Muhammad Waseem1 , Joel Gernsheimer2, Tae K Park1 ,Fernando Jara1 , Evelyn Erickson3
Affi l iations: 1Department of Emergency Medicine, LincolnMedical & Mental Health Center, Bronx, NY, United States,2Department of Emergency Medicine, Suny Downstate,Brooklyn, NY, United States; 3Department of Pediatrics,Lincoln Medical & Mental Health Center, Bronx, NY, UnitedStates.Corresponding Author: Muhammad Waseem, MDDepartment of Emergency Medicine, Lincoln Medical &Mental Health Center, 234 East 1 49th Street, Bronx, NY,United States - 1 0451 ; Ph: (71 8) 579-601 0; Fax: (71 8)579-4822; Email : waseemm2001@hotmail .com
Received: 03 November 2011Accepted: 28 March 201 2Published: 01 January 201 3
Waseem et al. 66

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):66–69.www.ijcasereportsandimages.com Waseem et al. 67
44/min and oxygen saturation 99%. His pupils wereboth equally dilated but reacted to light. His tympanicmembranes were normal. Examination of his pharynxwas normal. No whitish lesions were seen in his mouthand it is not known what the reported whitish lesionswere, although it was speculated that they could havebeen from candidiasis. His neck was supple. His chestwas clear with good bilateral breath sounds. Thecardiovascular examination was normal. His abdomenwas soft and nontender, but bowel sounds werediminished. The neurological evaluation revealed thathe was lethargic with a weak suck, cry and gag reflex. Hehad bilateral weakness of his facial muscles. He hadpoor muscle tone and weakness of his extremities. Thedeep tendon reflexes could not be elicited. His completeblood count, serum chemistries, urinalysis, chest Xrayand head CT scan were all normal. Lumbar puncturewas performed and analysis of his cerebrospinal fluidwas normal.While being monitored in the emergencydepartment, it was noted that he had frequent episodesof oxygen desaturation and apnea. He was intubatedand placed on a ventilator. The diagnosis of infantbotulism was suspected and subsequently wasconfirmed by stool studies. Clostridium botulinumspores were seen in his stool and botulinum toxin wasdetected in samples of his stool. He was givenintravenous botulism immune globulin. After receivingthe immunoglobulin, this infant improved graduallyover a period of three weeks. He was able to breathe onhis own, and was extubated two weeks after receivingthe immunoglobulin therapy. He was dischargedwithout any evidence of residual neurological deficitsthree weeks after his initial presentation. Unfortunately,this patient was lost to clinic followup, and we wereunable to contact the family to find out about thepatient’s current status.
DISCUSSIONThe diagnosis of infant botulism should be stronglysuspected in any infant with an acute onset of weaknessin sucking, swallowing or crying, ptosis, inactivity andconstipation. However, because infant botulism is an
uncommon disorder it is often missed, leading todisastrous consequences.Pathophysiology and Epidemiology: Botulismis a rare but potentially fatal paralytic disorder causedby a neurotoxin produced by Clostridium botulinum.This toxin, which is one of the most lethal poisons,causes an irreversible block of stimulation induced presynaptic cholinergic transmission. The toxin mainlyaffects the peripheral cholinergic nervous system.Because it does not affect adrenergic neuraltransmission and it does not readily cross the bloodbrain barrier. Botulism can be acquired in multipleways, such as ingestion of spores that colonize thegastrointestinal tract and produce the toxin, ingestion ofcontaminated food that already contains the toxin suchas sea food, sausages and canned foods, and infection ofa wound by Clostridium botulinum which thenproduces the toxin in the wound. An important exampleof this is the injection of contaminated “Black TarHeroin” [1, 2].Infant botulism is due to colonization of thegastrointestinal tract of the infant by Clostridiumbotulinum that then produces the neurotoxin whichspreads throughout the body via the circulation. Infantbotulism was first recognized in 1976, and since thenmany cases have been reported in the United Statesmaking it the most frequently recognized form ofbotulism [1–3].Infant botulism affects infants between one week andone year of age. Most cases occur within the first sixmonths of life with a peak incidence at 3–4 months ofage [3]. The majority of cases in the United Statesprobably are caused by ingested spores that are presentin dust that becomes contaminated by activities such asconstruction. The soil in some states such asPennsylvania, Utah and California are particularly richwith Clostridium botulinum. Although cases of infantbotulism from ingestion of Clostridium botulinumspores in raw honey or home canned foods have beenreported, this is less common than previously thought[4]. It was initially postulated that the home maderemedy that this baby was given may have containedraw honey or corn starch that was contaminated withClostridium botulinum spores, but the parents deniedthat these ingredients were in this home remedy and wewere unable to get a sample of it to test. It is much morelikely that the infection in this infant was fromClostridium botulinum spores in the soil that werereleased into the air from local construction, andentered the baby’s gastrointestinal tract. At the time ofthis case, there was a lot of construction being done inthe South Bronx, where this infant’s family lived.Ingestion of spores that are released into the air fromsoil due to construction is the most common method ofinfants developing botulism in the northeastern parts ofthe United States.The incubation period for Botulism is thought to beat least three days. There are eight Clostridiumbotulinum toxin types. The majority of the infantbotulism cases are caused by type A and B [4].Interestingly, breast fed infants appear to be protectedfrom botulism.
Table 1: Important Causes of Weakness in an infantSepsisMeningitis/encephalitisHypothyroidismPompe disease (Glycogen storage disease type II)Electrolyte disturbanceSpinal muscular atrophyNeonatal myasthenia gravisBotulismGuillainBarré syndromeCongenital myotonic dystrophy

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):66–69.www.ijcasereportsandimages.com Waseem et al. 68
Clinical Presentation: The clinical presentationof infant botulism includes progressive neuromuscularweakness which can be mild to severe. This may bemisinterpreted on examination as lethargy. Cranialnerves are affected first followed by the muscles of thetrunk, extremities and diaphragm [4]. This may causerespiratory failure. Lethargy and poor feeding are oftenthe initial presenting symptoms of infant botulism [1].Constipation and weak cry are other historical features.Occasionally, a history of ingestion of raw honey orcanned food may be present.This initial presentation is followed by progressivedescending weakness and hypotonia, so that the infantappears to be “floppy”. Bulbar involvement, often butnot always, presents with poor sucking and gag reflexes,dilated pupils with poor response to light andaccommodation, decreased eye movement, ptosis andfacial paralysis. Absent deep tendon reflexes, especiallywith type B, toxin often occurs [5]. Occasionally, younginfants may present with only a history of poor feedingfollowed by rapid collapse or deterioration [6].Differential Diagnosis: The differential diagnosisof the weak and floppy infant is extensive (Table 1) andincludes both neurologic and systemic diseases such assepsis, hypothyroidism, ingestions and metabolicdisorders. Because many disorders can mimic infantbotulism, which is a relatively rare condition, it is oftennot considered initially and is then missed [6].Sepsis is the most common diagnosis that mimicsinfant botulism. Most patients with infant botulism areusually afebrile. In addition, sepsis does not havecranial nerve and other neurologic findings that areoften present in patients with botulism. Electrolytedisturbances, including hypoglycemia may causelethargy and weakness.Several disorders that cause neuromuscularweakness deserve special mention. Tick paralysis iscaused by a neurotoxin secreted by a wood or dog tickthat prevents liberation of acetylcholine at theneuromuscular junction. The patient usually presentswith ataxia and then develops a rapidly progressiveascending flaccid paralysis that can cause respiratoryfailure and death. Bulbar findings, including dysphagia,dyarthria, facial paralysis and ocular muscle weaknesscan occur late in the course of this illness as comparedto botulism where bulbar findings occur early and thereis descending rather than ascending paralysis.Whenever a patient presents with rapidly progressiveparalysis, tick paralysis should be suspected and acareful search should be made for the presence of a tickwhich should then be removed if found, and thisremoval often results in rapid improvement.GuillainBarré syndrome is another differentialdiagnosis. It presents as a progressive symmetricalascending paralysis which starts in the lowerextremities. Its progression is comparatively slowerthan tick paralysis. Cranial nerves are rarely affected,although a MillerFisher variant causes facial paralysis.The pupils are not affected. Lumbar puncture showscells and high levels of protein in the CSF, whereas CSFanalysis is normal in botulism.
Poliomyelitis, which is now rare but may occur inunvaccinated children, presents with high fever,meningeal signs, asymmetrical weakness andlymphocytosis in the CSF.Myasthenia gravis is the most common disorder of theneuromuscular junction in children. It can occur astransient neonatal myasthenia gravis in infants who areborn to mothers who have myasthenia or as congenitalmyasthenia. Infants with myasthenia have generalizedweakness and hypotonia; however, deep tendon reflexesare present. Facial weakness and bulbar paralysis mayoften occur causing poor suck and swallowing and a weakcry. Ptosis may also be seen. Respiratory failurenecessitating ventilatory support may occur. Myastheniausually responds well to treatment with anticholinesteraseinhibitors, such as neostigmine or edrophonium.The diagnosis of infant botulism is clinical.Treatment should not be delayed pending laboratoryconfirmation. Although not pathognomonic and notalways present early in the disease, electromyographicfindings consistent with infant botulism stronglysupport this diagnosis in the presence of appropriateclinical setting. A clinical diagnosis is supported by theidentification of C. Botulinum spores in the stools andconfirmed by the identification of the toxin in the stool.Serum samples are often negative in patients with infantbotulism. Our patient had both spores and toxin in hisstool. Serum testing was not available at our institution.Toxin may be detected in contaminated food, if aspecific food is involved.Management: Supportive care, especiallyventilatory support as needed, with very closemonitoring is paramount in managment of botulisms.Botulism Immune Globulin IV (BIGIV) which is abotulinum antitoxin derived from humans is very safeand effective. It should be administered immediately inthe presence of a reasonably certain clinical suspicion.Botulism Immune Globulin Intravenous (Human)(BabyBIG, USA) was administered at a dose of 50 mg/kgto our infant, which at that time was the recommendeddose. It should be noted that since March of 2012, therecommended dose of BabyBIG is 75 mg/kg. BabyBIGinterrupts the progression of weakness by blocking theaccumulation of toxin in the nerve terminals. It reducescomplications, relapses, length of intubation andhospitalization. Antibiotics have not been shown toassist in the treatment of infant botulism. In fact, in thepast it was recommended that antibiotics should not beused in infant botulism because lysis of C. botulinum inthe gut would release more toxins into the gut and theninto the circulation. However, BIGIV therapy destroysall of the toxins in the gut and will block absorption oftoxin for at least six months. If needed antibiotics canthen be safely used to treat secondary infection.Antibiotics and debridement, as needed, are definitelyindicated to treat wound botulism. Penicillin ormetronidazole can be used, but aminoglycosides shouldnot be used as they worsen the effects of botulinumtoxin [6]. Given the age and septic appearingpresentation, many of these infants usually receivebroad spectrum antibiotics.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):66–69.www.ijcasereportsandimages.com
CONCLUSIONWe presented a case of an infant with botulism. It isextremely important that the diagnosis of infantbotulism be suspected and appropriately treated whenany infant presents with progressive weakness. Sinceinfant botulism is a treatable condition, promptdiagnosis is very important in reducing morbidity andmortality. Early recognition allows expeditious andappropriate treatment, which saves lives.
*********Author ContributionsMuhammad Waseem – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedJoel Gernsheimer – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedTae K Park – Substantial contributions to conceptionand design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedFernando Jara – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedEvelyn Erickson – Acquisition of data, Drafting thearticle, Final approval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.
Conflict of InterestAuthors declare no conflict of interest.Copyright© Muhammad Waseem et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properlycredited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Harrison MA, Garren DM, Huang YW, Gates KW.Risk of Clostridium botulinum type E toxinproduction in blue crab meat packaged in fourcommercialtype containers. J Food Prot1996;59(3):257–60.2. Hyytiä E, Hielm S, Korkeala H. Prevalence ofClostridium botulinum type E in Finnish fish andfishery products. Epidemiol Infect1998;120(3):245–50.3. Midura TF, Arnon SS. Infant botulism. Identificationof Clostridium botulinum and its toxins in faeces.Lancet 1976;2(7992):934–6.4. Pickett J, Berg B, Chaplin E, BrunstetterShafer MA.Syndrome of botulism in infancy: clinical andelectrophysiologic study. N Engl J Med1976;295(14):770–2.5. TsengOng L, Mitchell WG. Infant botulism: 20years' experience at a single institution. J ChildNeurol 2007;22(12):1333–7.6. Midura TF. Update: infant botulism. Clin MicrobiolRev 1996;9(2):119–25.
Waseem et al. 69
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):70–75.www.ijcasereportsandimages.com
Intimal angiosarcoma of the thoracic aortaMichelle Forman, Michael E Mulligan
ABSTRACTIntroduction: Sarcomas of the great vessels areuncommon, with aortic being the rarest. Only 30cases of true intimal aortic sarcomas (IAS) aredocumented. They tend to occur in theabdominal aorta, with less common occurrencesin the thoracic aorta. Their growth patterns,predispose them to a propensity for metastasesand cause embolic phenomenon. Case Report: A58yearold male presented with chest pain anddyspnea and was evaluated for pulmonaryembolus and coronary artery disease. Computedtomography angiography (CTA) demonstratedno pulmonary emboli; however, there wassevere atherosclerosis/thrombosis of the aorticarch. The process extended centrally, nearlyfilling the entire lumen. The surgery consultantadvised anticoagulation and strict bloodpressure control, recommending that thepatient come to the outpatient department forsurgery. Due to personal reasons, the patientfailed to return at the recommended time. Threemonths after initial presentation the patient wasadmitted for surgical replacement of the aorta.The surgeon reported the aorta as “chockfull offibrofatty material nearly obstructing itscourse”. The pathology report was aorticsarcoma of intimal origin. Conclusion: Aorticsarcomas are rare tumors, with the intimalsubtype in the thoracic aorta being even rarer.
Delay in diagnosis of these tumors often occurs,since the imaging features are nearly identicalto atherosclerotic disease. Since atheroscleroticdisease is clearly more frequent than intimalsarcoma of the aorta, it is not difficult tounderstand that this diagnosis is not usuallymade until after surgical resection or atautopsy. At presentation, nearly all of thepatients have metastatic disease.Keywords: Aorta, Sarcoma, Computedtomography
*********Forman M, Mulligan ME. Intimal angiosarcoma of thethoracic aorta. International Journal of Case Reportsand Images 2013;4(1):70–75.
*********doi:10.5348/ijcri201301263CII17
INTRODUCTIONPrimary sarcomas of the great vessels (aorta,pulmonary artery and vena cava) are extremelyuncommon tumors, with aortic sarcomas being therarest type; about 26% occurring in aorta compared toabout 37% each for pulmonary and vena caval sites.They are subclassified as either mural or intimaltumors, with intimal tumors characterized as poorlydifferentiated on histology [1, 2]. Latest reviews, usingthe strict histological definition of intimal sarcomareported that only about 21–30 cases of true intimalaortic sarcomas (IAS) have been documented, with themean age of presentation being 62.2 years [3, 4].Here, we present a case of undifferentiated intimalaortic sarcoma which is unusual both in location andpresentation. This patient’s tumor was located withinthe thoracic aortic arch, and he presented with chest
CASE IN IMAGES OPEN ACCESS
Michelle Forman1 , Michael E Mull igan1
Affi l iations: 1Department of Radiology, University of MarylandMedical Center 22 South Greene Street, Baltimore, MD21 201 .Corresponding Author: Michael E Mull igan, MD, Departmentof Radiology, University of Maryland Medical Center, 22South Greene Street, Baltimore, MD 21 201 ; Ph: 41 0-328-3477; Fax: 41 0-328-0641 ; Email : mmull [email protected]
Received: 1 1 June 201 2Accepted: 22 July 201 2Published: 01 January 201 3
Forman et al. 70

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):70–75.www.ijcasereportsandimages.com Forman et al. 71
pain, instead of the more typical secondary symptoms ofembolic phenomena.
CASE REPORTThe patient was a 58yearold AfricanAmericanmale with a past medical history significant for type twodiabetes mellitus. He presented with chest pain anddyspnea on June 4, 2010 and was subsequentlyevaluated for a pulmonary embolus as well as coronaryartery disease. Cardiac enzymes were negative forindication of ischemic changes. Computed tomographicangiography (CTA) of the chest demonstrated nopulmonary emboli; however, the imaging revealedsevere supposed atherosclerosis/thrombosis of theaortic arch extending to the descending thoracic aorta,stopping at the level of the diaphragm. The processextended centrally, nearly filling the entire aortic lumen.The adrenal glands were free of involvement(Figures 1–2).The vascular surgery consultant placed the patienton anticoagulation therapy (warfarin 5 mg) and strictblood pressure control (metoprolol 50 mg and lisinopril20 mg), recommending that he return as an outpatientfor surgery on the aortic arch and descending thoracicaorta. Due to personal and other preoperative medicalreasons (full mouth dental extraction), the patient’spresumed elective aortic surgery was delayed untilSeptember 2010.As a result of chest pain, he visited our emergencydepartment (ED) twice in the month of August 2010and each time was ruled out for cardiac ischemia. Bothtimes he was continued on his regimen of anticoagulation and blood pressure control. A computedtomography (CT) scan was performed on both ED visits,demonstrating a new right adrenal mass (2x2 cm) thatwas not present on initial CT scan, in addition to thepreviously identified severely diseased aorta (Figure 3).The second time he presented to the ED, he wasadmitted for one week.Three months after initial presentation, onSeptember 23, 2010 the patient was admitted forsurgical replacement of the descending thoracic aortawith a tube graft utilizing a right axillary arterycannulation site and left femoral artery and veincannulation site. The surgeon reported the thoracicaorta as “chockfull of fibrofatty material nearlyobstructing its course, blending to a more normalappearing aorta at the diaphragm and in the proximalaortic arch”.The pathology report described the specimen as"multiple fragments of opaque, yellowtan to pinkgrayfriable soft tissue in aggregate measuring 6.2x5.2x3.4 cm.Sectioning revealed markedly friable, partiallylaminated, yellowwhite to graybrown cut surfaces".The final pathology report unexpectedly revealedundifferentiated aortic pleomorphic sarcoma of intimalorigin (Figure 4).The patient was discharged from the hospital twoweeks later. However, he returned to the ED after one
week, complaining of neurologic symptoms (confusion,aphasia) and was admitted on October 13, 2010. CT scanand magnetic resonance imaging (MRI) of the braindemonstrated two intraaxial masses (4x3 cm in lefttemporal region and 2x2 cm in right occipital region).CT scan of the chest, abdomen and pelvis showedincreased size of the right adrenal mass (3x2 cm), a newleft adrenal mass (1x1 cm), indication of tumorthrombus in the IVC, and a peripherally enhancingparavertebral soft tissue mass involving the musculaturewhich, in hindsight, was present on the initial CT of thechest from June 2010 (Figures 5–8). A positronemission tomography (PET) performed a few days latershowed increased metabolic activity in the above lesionsas well as in the T3 and T4 vertebral bodies. Afterconsultations with medical oncology and radiationoncology specialists, a decision was made to first treatthe brain lesions with external beam radiation therapy(XRT). A course of systemic chemotherapy was plannedto follow. The brain lesions were treated with 6 mVphoton therapy for a total of 30 gray in 10 fractions. Twoweeks after completing the initial XRT, because ofworsening pain, the patient had his paraspinal lesiontreated with 6 mV photon therapy for a total of 25 grayin 5 fractions. Finally in mid December 2010, he wasable to begin three cycles of chemotherapy withgemcitabine and taxotere.With complaints of pain, nausea and vomiting fromthe metastatic disease and subsequent treatment, he wasadmitted multiple times in the following months.November 2010, January 2011, February 2011, andMarch 2011 admissions involved multiple followupimaging studies, all showing progression of the bilateraladrenal masses, invasion of the inferior vena cava (IVC),paravertebral soft tissue mass, and vertebral bodyinvolvement (Figures 9–10). New findings included rightlower lobe pulmonary emboli. A followup MRI of thespine in March 2011 showed epidural metastases at T3,where he also had bony disease. His MRI of the brain inMarch 2011 showed marked decrease in size of the brainmetastases and no new foci of metastasis.Early in presentation, the patient alreadydemonstrated metastatic disease. Metastatic disease wasconfirmed in a soft tissue lesion on the patient's back,again with pathology of metastatic undifferentiatedpleomorphic sarcoma/malignant fibrous histiocytoma.As of March 2011, the patient’s diagnosis was stage IVmetastatic, undifferentiated, pleomorphic sarcoma. Hereceived three cycles of the chemotherapy regimen ofgemcitabine and taxotere from December 2010 throughFebruary 2011, as well as supportive treatment for theneurological symptoms from the temporal and occipitallobe brain metastasis but died in July 2011 at hospice care.
DISCUSSIONAortic sarcomas are rare tumors, with the intimalsubtype occurring in the thoracic aorta, as the casereported here, being even more uncommon. Delay indiagnosis for these tumors often occurs as the imaging

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):70–75.www.ijcasereportsandimages.com Forman et al. 72
features appear nearly identical to atheroscleroticdisease. Seeing that atherosclerotic disease is clearly amore frequent diagnosis than intimal sarcoma of theaorta, it is not difficult to understand that this diagnosisis not usually made until after surgical resection or atautopsy.Sarcomas of the aorta tend to occur mostly in theabdominal aorta, with less common occurrences in thethoracic aorta. For example, in one case series, four outof 21 cases of sarcoma presented in the chest. Bydefinition, in contrast to the mural aortic sarcomas,intimal aortic sarcomas actually grow within thevascular lumen. Because of this growth pattern, theyhave a greater propensity for fragments to dislodge andbe carried in the blood stream and metastasize [5].Additionally, these tumors commonly present withsequelae from embolic phenomenon with symptomsranging from absent peripheral pulses to mesentericocclusion [1–4, 6, 7]. In one case series of 11 patients, all
the patients with aortic sarcoma, died within 16 monthsof diagnosis [1].Due to the rarity of this disease, there have been norandomized trials for the definitive treatment of IAS,with the therapies that are currently used being basedsolely on observational studies. Definitive treatmentmethods for IAS have not been delineated. Theliterature suggests that resection of the affected portion
Figures 1: (A, B) Computed tomography angiography imagingat time of initial diagnosis demonstrate marked luminalirregularity/filling defect of the aortic arch and descendingthoracic aorta (June 2010).
Figure 2: Small mass arising in the right posterior paraspinalmusculature at initial diagnosis (June 2010).
Figure 3: New right adrenal mass (August 2010).
Figure 4: Section of aortic wall showing intimal thickeningwith spindle cells that display nuclear pleomorphism andatypia (H&E, 10x).

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):70–75.www.ijcasereportsandimages.com Forman et al. 73
Figure 6: After surgical repair of the aorta, a moreconspicuous peripherally enhancing right posteriorparavertebral soft tissue mass is seen which wassubsequently removed (arrow) (October 2010).
Figure 5: Increased size of right adrenal mass, new small leftadrenal mass, and new tumor thrombus in the inferior venacava (October 2010).
Figures 7: (A, B) Nonenhanced computaed tomography scan ofthe brain demonstrating heterogeneous left temporal mass andright occipital mass with surrounding edema (October 2010).
Figure 8: Fluid attenuated inversion recovery (FLAIR) magneticresonance imaging demonstrating both left temporal and rightoccipital masses with surrounding edema (October 2010).
Figure 9: Increased size of right and left adrenal masses withexpansion and involvement of the inferior vena cava(November 2010).
of the aorta with placement of a graft and subsequentchemotherapy and radiation is the best approach [2].Endarterectomy is also a reported treatment choice [4].At presentation, nearly all of the cases of IAS haveevidence of metastatic disease.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):70–75.www.ijcasereportsandimages.com
Figure 10: Further involvement of the inferior vena cava andenlargement of the right and left adrenal masses (January2011).
CONCLUSIONOur case of intimal aortic sarcoma illustrated anumber of interesting facts. First, the tumor may occurin the thoracic aorta, more specifically at the aortic arch,which is an uncommon location. Second, the clinicalpresentation may not the classic one of symptoms fromembolic phenomenon; instead patient may present withsymptoms of chest pain likely from the primary tumor.Third, patients with intimal sarcoma of the aorta tend todie shortly after presentation. However, despite theevidence of metastatic disease early in presentation,patient may lived for many months after diagnosis andtreatment.
*********Author ContributionsMichelle Forman – Conception and design, Acquisitionof data, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedMichael Mulligan – Conception and design, Acquisitionof data, Analysis and interpretation of data, Drafting thearticle, Critical revision of the article, Final approval ofthe version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.
Conflict of InterestAuthors declare no conflict of interest.Copyright© Michelle Forman et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properlycredited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Burke AP, Virmani R. Sarcomas of the Great Vessels:A Clinicopathologic Study. Cancer 1993;71:1761–3.2. Shirani S, SoleymanzadehArdabili M, Arami M.Intimal Sarcoma of the Descending Aorta. ArcIranian Med 2007;10(2):253–4.3. Mohsen NA, Haber M, Urrutia VC, Nunes LW.Intimal sarcoma of the aorta. Am J Roentgenol2000;175:1289–90.4. Thalheimer A, Fein M, Geissinger E, Franke S.Intimal angiosarcoma of the aorta: Report of a caseand review of the literature. J Vascular Surg2004;40:548–3.5. Akiyama K, Nakata K, Negishi N, Henmi A. IntimalSarcoma of the Thoracic Aorta; Clinicalcourse andAutopsy Findings. Ann Thorac Cardiovasc Surg2005;11(2):135–8.6. Hashimoto M, Sashi R, Watarai J. Primary sarcomaof the aortic wall. Cardiovasc Intervent Radiol1997;20:322–3.7. Tucci M, Quatraro C, Calvani N, et al. PrimaryIntimal Sarcoma of the Thoracic Aorta. J Exp ClinCancer Res 2005;24(1):139–42.
Forman et al. 74

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):70–75.www.ijcasereportsandimages.com Forman et al. 75
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):76–79.www.ijcasereportsandimages.com
Neuroradiological imaging features of infratentorialcranial fossa tumors in a childMuhammad Yunus Amran, Meryana Pauline, Andi Kurnia Bintang,Muhammad Akbar
CASE REPORTAn 11yearold boy was admitted to our hospital withthe chief complaint of a generalized tonic seizure whichwas preceded by projectile vomiting, six hours prior tohis admission. There was no previous history of suchcomplaint. The patient’s right arm and right leg wererigid and extended while his left arm and left leg wererigid and flexed. Five years before admission, the patienthad complained of chronic headaches. One year beforeadmission, the patient had complained of blurred visionin his left eye followed by the same complaint in the
right eye. The blurred vision became progressivelychronic, accompanied by diplopia. Three months beforepresenting to us, he demonstrated truncal ataxia. Thepatient had no problems with either urination ordefecation. There was no history of fever, growthdisorders or developmental disorders. Neurologicalexamination showed neck stiffness. His pupils showedisochore mydriasis with decreased light reflexes andpapilledema; the patient’s visual acuity in both left andright eye was 1/300 with convergent strabismus in theleft eye, and left peripheral facial paresis. On motorexamination, we found dysmetria anddisdiadochokinesia in the extremities of the left side aswell as hypotonia on both the sides. Sensory andautonomic functions were within normal limits. Routineand blood chemistry analyses revealed the followingresults: Hemoglobin 12.5 g/dL, hematocrit 38.3%, WBCcounts 8x103/mm3, platelet count 3.13x105/mm3, bloodsugar (random) 156 mg/dL, SGOT 20 IU/L, SGPT 53IU/L, urea 15.53 mg/dL, creatinine 0.32 mg/dL, sodium135 mmol/dL, potassium 4.2 mmol/dL and chloride 101mmol/dL. Axial head computed tomography (CT) scansof both noncontrast and contrast (with administrationof intravenous gadolinium (GdDOTA)) wereperformed. The CT scan revealed a mass in theinfratentorial cranial fossa which suggested amedulloblastoma with a differential diagnosis of anependymoma (Figure 1A–B). After postcontrast themass density was enhanced. This result suggested anependymoma with noncommunicating hydrocephalus(Figure 1C–D). The patient was scheduled to undergoventriculoperitoneal (VP) shunting to relieve theincreased intracranial pressure due to mass effect fromthe tumor in the fourth ventricle. Subsequently, anmagnetic resonance imaging (MRI) scan was performedwith and without administration of intravenous GdDOTA. The MRI scan showed a round lesion with aclear border, irregular edges, hypo intensity in the axialand sagittal T1weighted images, and hypo intensity inthe T1WI noncontrast images, (Figure 2A–B) as well ashyper intensity in the T1WI post–contrast images(Figure 2C–D). Simultaneously, T2WI revealed hyperintensity, heterogeneous density and ring/rimenhancement, which is consistant with the description
CLINICAL IMAGES OPEN ACCESS
Muhammad Yunus Amran1 ,2, Meryana Pauline3, AndiKurnia Bintang4, Muhammad Akbar5
Affi l iations: 1MD,Ph.D, Candidate, Department ofBacteriology, Laboratory of Division Basic Medical Scienceand Molecular, Graduate School of Medical Sciences andSchool of Medicine, Kyushu University, Fukuoka, Japan;2MD, Clinical Assistant Professor and Lecturer,Hasanuddin University Hospital, Department of Neurology,Faculty of Medicine, Hasanuddin University, Makassar,South Sulawesi, Indonesia; 3MD, Clinical AssistantProfessor, Hasanuddin University Hospital, Department ofNeurology, Faculty of Medicine, Hasanuddin University,Makassar, South Sulawesi, Indonesia; 4MD, ClinicalAssociate Professor and Lecturer, Hasanuddin UniversityHospital, Department of Neurology, Faculty of Medicine,Hasanuddin University, Makassar, South Sulawesi,Indonesia; 5MD, Ph.D, Professor and Head of Department,Hasanuddin University Hospital, Department of Neurology,Faculty of Medicine, Hasanuddin University, Makassar,South Sulawesi, IndonesiaCorresponding Author: Muhammad Yunus Amran, MD,Department of Neurology, Hasanuddin University Hospital,Faculty of Medicine, Hasanuddin University, J l . PerintisKemerdekaan KM 11 (Pintu I I UNHAS), Makassar, 90245,South Sulawesi, Indonesia; Ph: +62-411 -585560; Fax No:+62-411 -582837; Office Email : [email protected];Email :[email protected]
Received: 07 June 201 2Accepted: 29 August 201 2Published: 01 January 201 3
Amran et al. 76

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):76–79.www.ijcasereportsandimages.com Amran et al. 77
of an ependymoma (Figure 3A–C) Fluidattenuatedinversion recovery (FLAIR) MRI further showed themass with hyper intensity and a heterogeneous signaldensity (Figure 4). After VPshunting, the headache wasrelieved and no more seizures occurred, after which atentative plan for tumour resection followed byradiotherapy was made. Unfortunately, the patient wasdischarged against medical advice from the hospitalbefore the surgical procedure could be carried out.
DISCUSSIONEpendymoma is a rare tumor derived from theneuroepithelia, constituting about 3–9% of all centralnervous system (CNS) tumors. In children,approximately twothirds of such tumors occur in theinfratentorial compartment while in adults thedistribution is normally supratentorial. Ependymomascan be located either intracranially or intraspinally.Intracranial ependymomas occur more commonly inchildren, while intraspinal ependymomas predominatein adults. Sixty percent of tumors are located in thefourth ventricle of the infratentorial cranial fossa, whichdevelop from the floor of the ventricle and may furtherextend into the foramen of Luschka and Magendie. Herewe reported a case of ependymoma in the infratentorial
Figure 1: Axial head computed tomography scan images (A,B) Noncontrast showing, a lesion with hyper density andcalcified spots, demarcated, with a density of 44,2 Hu in theinfra tentorial fossa pressing on the anterior aspect of fourthventricle causing dilatation of the lateral and third ventriclesand, (C, D) Postcontrast with intravenous gadolinium (GdDOTA) showing, a mass in the infra tentorial fossa, which issuspected to have arisen from the fourth ventricle. The mass islobulated and round shaped, demarcated with irregular edgesand enhanced density of 54.7 Hu postcontrast, causingdilatation of the lateral and third ventricle.
Figure 2: (A) Axial and (B) Sagittal head, Magnetic resonanceimaging (MRI) scan images (noncontrast) post VPshuntingshowing a round lesion with clear borders, irregular edges,size 4.87x5.43 cm in the area of the fourth ventricle and masswith hypo intensity on T1WI; (C) Axial and (D) Sagittal headMRI scan images (with contrast) post VPshunting showing around lesion with clear borders, irregular edges, size 4.87x5.43cm in the area of the fourth ventricle and mass with hyperintensity on T1WI.
Figure 3: (A, B) Axial head magnetic resonance imaging scanT2weighted image; post VPshunting, showing a mass withhyper intensity and heterogeneous density on T2WI signal,especially at the edge (ring/rim enhancement) which isconsistant with the description of an ependymoma, (C)Sagittal head MRI scann T2weighted image, post VPshunting, also showing a mass with hyper intensity andheterogeneous density on T2WI signal.
Figure 4: Sagittal head FLAIRMRI images post VPshunting,showing an intermediate flair signal relative to both gray andwhite matter.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):76–79.www.ijcasereportsandimages.com Amran et al. 78
cranial fossa with a differential diagnosis ofmedulloblastoma on head CT scan and MRI scan.In 1863, Virchow was the first to properly identify anependymoma derived from ependymal cells [1]. Thiswas followed by Bailey et al. in 1926, who described anependymoma and an ependymoblastoma, which havebeen considered the most primitive tumors of thenervous system. This tumor arises from the medullaryepithelial plate [2, 3]. Recently, researchers haveproposed that radial glial cells may be the candidatestem cells of this tumor [4, 5]. The annual incidence inthe countries of Central and South America as well asAsia is less than two per million in childhood [6]. InNorth America, Oceania and most of Europe, between2–4 cases per million was reported, while in Denmark,Sweden, Finland, former East Germany and Slovenia, atleast four cases per million are reported. Recently,McGuire et al. reported a comparison of the mean age oftumor incidence by tumor site in 237 children. Childrenwith 5±0.4 years of age had only 4.2–5.9% of tumorlocated infratentorially while those of 12.2±0.9 years ofage had 10.6–13.7% of tumor located in spinal column[7]. This indicated that the tumors would most likely belocated in the spinal column when diagnosed in childrenwith the average of age around 11 years; nevertheless,herein we have reported an 11yearold child with atumor located in the infratentorial space. According toWHO classification, ependymomas are classified intothree categories:(i) WHO grade I: subependymoma, myxopapillaryependymoma;(ii) WHO grade II: classic ependymoma (with thevariation of cellular, papillary, clear cell, and fibrillaryor tanycytic);iii) WHO grade III: anaplastic ependymoma [8]. Thesymptoms of ependymomas depend on the tumorlocation, rate of tumor growth, etc.In our patient the chief complaint was a generalizedtonic seizure, followed by several symptoms which areassociated with the location of the tumor in theinfratentorial cranial fossa, such as vomiting,headaches, double vision and unsteadiness.Fundoscopic examination and neuroradiology imagingindicated an increase in intracranial pressure.Ependymomas are the third most common pediatricbrain tumor (10.1%) after astrocytomas (47.3%) andmedulloblastoma (16.3%) [9]. In the above list, atypicalteratoidrhabdoid tumor (ATRT) should be consideredas well. It is very difficult to differentiate among thesetumors, especially between a medulloblastoma and anependymoma since both normally present as midlinetumors, whereas astrocytoma and ATRT are generallyeccentric. In order to distinguish between these tumors,additional examinations (CT scan and MRI) need to beperformed. Based on the results from a noncontrasthead CT scan, we made an initial diagnosis of amedulloblastoma with a differential diagnosis ofependymoma, although after an intravenous injectionwith contrast the results suggested that what the patienthad was in fact an ependymoma. Moreover, head MRIscans revealed the ring/rim enhancement that is very
typical for ependymomas. With regards to therapy, thesymptoms should be relieved by the use of anticonvulsants and antiedema agents. VPshunting mustbe done to release the increase in intracranial pressure,followed by tumour resection and radiotherapy. All ofthis is essential to improve the patient’s outcome [10].
CONCLUSIONDetailed history and physical examinations are veryimportant to diagnose an infratentorial cranial fossaoccupying lesion. Radiological imaging by computedtomography scan and magnetic resonance imaging scanis essential to support the diagnosis of ependymoma andto distinguish it from a medulloblastoma. Moreover,improved patient outcome requires total tumorresection whenever possible, followed by radiotherapy.
*********Amran MY, Pauline M, Bintang AK, Akbar M.Neuroradiological imaging features of infratentorialcranial fossa tumors in a child. International Journal ofCase Reports and Images 2013;4(1):76–79.
*********doi:10.5348/ijcri201301264CI18
*********Author ContributionsMuhammad Yunus Amran – Substantial contributionsto conception and design, Acquisition of data, Analysisand interpretation of data, Drafting the article, Revisingit critically for important intellectual content, Finalapproval of the version to be publishedMeryana Pauline – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedAndi Kurnia Bintang – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedMuhammad Akbar – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):76–79.www.ijcasereportsandimages.com
Copyright© Muhammad Yunus Amran et al. 2013; This article isdistributed under the terms of Creative CommonsAttribution 3.0 License which permits unrestricted use,distribution and reproduction in any means providedthe original authors and original publisher are properlycredited. (Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Virchow R. Die krankhaften Geschwülste.Hirschwald. 1863/1865. Berlin.2. Bailey P, Cushing H. A classification of the tumors ofthe glioma group on a histogenetic basis with acorrelated study of prognosis. JP Lippincott 1926.Philadelphia.3. Bailey P. A study of tumors arising from ependymalcells. Arch Neurol Psychiatry 1924;11:1–27.4. Taylor MD, Poppleton H, Fuller C, et al. Radial gliacells are candidate stem cells of ependymoma.Cancer Cell 2005;8(4):323–35.
Amran et al. 79
5. Poppleton H, Gilbertson RJ. Stem cells ofependymoma. Br J Cancer 2007 Jan 15;96(1):6–10.6. Parkin DM, Kramarova E, Draper JG, et al.International incidence of childhood cancer, Vol. II.IARC Scientific Pub 1998;(144):1–391.7. McGuire CS, Sainani KL, Fisher PG. Incidencepatterns for ependymoma: a surveillance,epidemiology, and end results study. J Neurosurg2009;110(4):725–9.8. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007WHO classification of tumours of the centralnervous system. Acta Neuropathol2007;114(2):97–109.9. Rickert CH, Paulus W. Epidemiology of centralnervous system tumors in childhood andadolescence based on the new WHO classification.Childs Nerv Syst 2001;17(9):503–11.10. Chan MD, McMullen KP. Multidisciplinarymanagement of intracranial ependymoma. CurrProbl Cancer 2012;36(1):6–19..
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):80–82.www.ijcasereportsandimages.com
Extensive maxillofacial and oral myiasisFelipe P Daltoé, André Ricardo Nosé, Rodrigo C Mosca,Andrea Mantesso
CASE REPORTA 28yearold homeless man was brought by a policeofficer to the emergency service of the Regional SulHospital (Sao Paulo, Brazil) for evaluation of anextensive destruction of the oral and maxillofacialtissues. The patient was a heavy smoker (three packs ofcigarettes per day) and according to his medical recordshe had a previous diagnosis of oral squamous cellcarcinoma, but decided not get it treated. Three yearslater, the surface of the swelling revealed an extensivenecrotic ulcer extending to the mouth, lips, nose andneck with live maggots visible and moving. Around 110larvae were surgically removed and the necrotic tissuewas debrided (Figure 1A–B). The patient was sent to theoncology service for a whole body evaluation, however,he passed away two weeks later due to systemiccomplications.
DISCUSSIONThe term myiasis is applied to the injurious actionthat a parasites of the order Diptera causes to the livingor dead tissue in which they grow in vertebrates organisms [1]. It is more common in animals and it hasbeen rarely reported in humans [2]. Moreover,considering that myiasis develops by direct infestationof tissues by larvae (maggots) laid by flies [1], the mouthis not a common place for its development comparedwith dermis or other tissues.Oral myiasis is usually associated with poor hygiene,wound healing, mouth breathing, mental impairment orsenility [3]. In our case, the patient was clearlypredisposed to the infestation considering the fact thathe was a homeless, had unhygienic living condition, andhad a previously untreated oral carcinoma.The treatment of oral myiasis in most cases includesonly surgical exploration to remove the larvae andnecrotic tissue [3]. Alternatively, use of medicines such asivermectin has also been proved efficacious, byenhancing parasitic death and their emergence to tissuesurface [4]. Initial infestation can easily mimic gingivalinflammations. Likewise, some cases of myiasis inassociation with oral tumors have also been reported [5].
CLINICAL IMAGES OPEN ACCESS
Felipe P Daltoé1 , André Ricardo Nosé2, Rodrigo C Mosca3,Andrea Mantesso1
Affi l iations: 1Department of Oral Pathology, School ofDentistry, University of São Paulo, Brazil ; 2Department ofBucomaxilofacial Surgery, School of Dentistry, PaulistaUniversity, Brazil ; 3Department of Biotechnology, Instituteof Energetic and Nuclear Research/National Committee ofNuclear Energy, University of São Paulo, Brazil .Corresponding Author: Dr. Andrea Mantesso; OralPathology Discipl ine – Dental School - University of SãoPaulo. , Av. Professor LineuPrestes, 2227 - CEP: 05508-900 - São Paulo/SP. Brazil ; Phe: + 55 11 3091 7902; Fax: +55 11 3091 781 4; Email : [email protected]
Received: 1 0 May 201 2Accepted: 04 June 201 2Published: 01 January 201 3
Daltoé et al. 80
Figure 1: (A, B) Clinical picture of the patient upon arrival atthe hospital. It was possible to observe an extensive necroticulcer extending to the mouth, lips, nose and neck with visiblelive maggots. Around one hundred ten larvae were surgicallyremoved. The patient was seriously compromised and requiredmechanical assistance to breath.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):80–82.www.ijcasereportsandimages.com Daltoé et al. 81
CONCLUSIONThe most effective action for prevention of humanmyiasis is by education and improvement of generalsanitary conditions. Unfortunately, in underdevelopedor developing countries like Brazil, some people still livein poor environment associated with compromisedhygiene and lack of information which can leads tohuman myiasis.
*********Daltoé FP, Nosé AR, Mosca RC, Mantesso A. Extensivemaxillofacial and oral myiasis. International Journal ofCase Reports and Images 2013;4(1):80–82.
*********doi:10.5348/ijcri201301265CI19
*********Author ContributionsFelipe P Daltoé – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedAndré Ricardo Nosé – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedRodrigo C Mosca – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedAndrea Mantesso – Substantial contributions toconception and design, Acquisition of data, Analysis andinterpretation of data, Drafting the article, Revising itcritically for important intellectual content, Finalapproval of the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Felipe P Daltoé et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Meinking TL, Burkhart CN, Burkhart CG. Changingparadigms in parasitic infections: commondermatological helminthic infections and cutaneousmyiasis. Clin Dermatol 2003;21(5):407–16.2. Kumar SL, Manuel S, John TV, Sivan MP. Extensivegingival myiasis Diagnosis, treatment, andprevention. J Oral Maxillofac Pathol 2011Sep;15(3):340–3.3. Vale DS, Cavalieri I, Araujo MM, et al. Myiasis inpalate by Cochliomyia hominivorax. J Craniofac Surg2011 Nov;22(6):e57–9.4. Gomez RS, Perdigão PF, Pimenta FJ, Rios Leite AC,Tanos de Lacerda JC, Custódio Neto AL. OralMyiasis by Screwworm Cochliomy Hominivorax. BrJ Oral Maxillofac Surg 2003;41(2):115–6.5. Carvalho RW, Santos TS, Antunes AA, LaureanoFilho JR, Anjos ED, Catunda RB. Oral andmaxillofacial myiasis associated with epidermoidcarcinoma: a case report. J Oral Sci 2008Mar;50(1):103–5.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):80–82.www.ijcasereportsandimages.com Daltoé et al. 82
Access full text article onother devices Access PDF of article onother devices

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):83–85.www.ijcasereportsandimages.com
Lessertrélat sign in a patient withneoplasia of upper eyelidSatyaki Ganguly, Kranti C Jaykar, Sambeet Kumar Mallik
CASE REPORTA 40yearold female presented to the dermatologyOPD with an ulcerating growth of the left upper eyelidfor the last three years. The growth was graduallyincreasing in size. On examination, there was anirregular erythematous swelling involving the whole ofleft upper eyelid with ulceration and areas of necrosis inthe lateral part. A provisional diagnosis of sebaceousgland carcinoma was made. A biopsy of the eyelidgrowth was advised to confirm the diagnosis. Along witheyelid lesion, numerous asymptomatic, darklypigmented papules were discovered over the face, trunkand extremities. These were more over the flexures likethe neck, axilla, submammary area and groin (Figures1, 2). On being questioned about the lesions, the patientsaid that these lesions have appeared rapidly over aperiod of last six months. Detailed haematological,biochemical investigations, chest Xray, uppergastrointestinal endoscopy, lower gastrointestinalendoscopy, mammography, abdominal ultrasound andbone marrow examination failed to reveal evidence ofany systemic malignancy. Based on the clinical findings,a diagnosis of Lesser–Trélat sign in association withskin malignancy was made. The patient was referred to
CLINICAL IMAGES OPEN ACCESS
Satyaki Ganguly1 , Kranti C Jaykar2, Sambeet KumarMall ik3
Affi l iations: 1Assistant Professor, Dept of Dermatology,Venereology and Leprosy Pondicherry Institute ofMedical sciences, Pondicherry; 2Assistant Professor, Deptof Dermatology, Venereology and Leprosy Katihar MedicalCollege, Katihar, Bihar; 3Junior resident, Dept ofDermatology, Venereology and Leprosy Katihar MedicalCollege, Katihar, Bihar.Corresponding Author: Dr. Satyaki Ganguly, Dept ofDermatology, Venereology and Leprosy PondicherryInstitute of Medical sciences, Pondicherry-60501 4; Email :[email protected]. in
Received: 1 9 November 2011Accepted: 1 3 Apri l 201 2Published: 01 January 201 3
Ganguly et al. 83
ophthalmology department for further management ofthe growth in the eyelid. Unfortunately, before a biopsycould be done the patient was lost to followup.
DISCUSSIONSeborrhoeic keratosis is a benign tumor, frequentlypigmented, more common in elderly and composed ofepidermal keratinocytes. The sudden appearance ofnumerous seborrhoeic keratoses in an adult may be acutaneous finding of internal malignancy. Internalmalignancy associated with the sudden development ofnumerous seborrhoeic keratoses in an eruptive fashion,with or without pruritus, is known as the sign ofLesser–Trélat [1]. Weakened subepithelial matrix—fromthe effects of neoplasm on the extracellular matrix of thehost—has been postulated as a possible cause of Lesser–Trélat sign. To be considered a case of Lesser–Trélat,the keratoses should begin at approximately the sametime as the development of cancer and run a parallelcourse in regard to growth and remission.
Figure 1: LesserTrélat sign: Upper eyelid tumour withnumerous seborrheic keratoses over face and neck.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):83–85.www.ijcasereportsandimages.com Ganguly et al. 84
Common malignancies associated with this sign areadenocarcinoma of stomach (most common), lung,colon, breast, prostate, lymphoma, leukemia, ovariancancer, nasopharyngeal carcinoma and transitional cellcarcinoma of the bladder [2]. It has been associated withskin malignancies like maligna melanoma [3],lymphocytoma cutis [4] Paget’s disease [5], and Sezarysyndrome [6]. A sudden eruption of many seborrhoeickeratoses may follow exfoliative erythroderma,erthrodermic psoriasis, erythrodermic drug eruption,lepromatous leprosy and HIV infection [7]. Sebaceousgland carcinomas are very rare tumors, usually arisesfrom the meibomian glands and majority of lesionsaffect the upper eye lid. The lesions are nodular andappear like a chalazion which lasts for more than sixmonths.
CONCLUSIONInternal malignancy associated with the suddendevelopment of numerous seborrhoeic keratoses in aneruptive fashion, with or without pruritus, is known asthe sign of LesserTrélat
*********
Figure 2: LesserTrélat sign: Upper eyelid tumor withnumerous seborrheic keratoses over axilla.
Ganguly S, Jaykar KC, Mallik SK. Lessertrélat sign in apatient with neoplasia of upper eye lid. InternationalJournal of Case Reports and Images; 4(1):83–85.*********
doi:10.5348/ijcri201301266CI20
*********Author ContributionsSatyaki Ganguly – Substantial contributions toconception and design, Acquisition of data, Drafting thearticle, Revising it critically for important intellectualcontent, Final approval of the version to be publishedKranti C Jaykar – Substantial contributions to analysisand interpretation of data, Drafting the article, Finalapproval of the version to be publishedSambeet Kumar Mallik – Substantial contributions toacquisition of data, Drafting the article, Final approvalof the version to be publishedGuarantorThe corresponding author is the guarantor ofsubmission.Conflict of InterestAuthors declare no conflict of interest.Copyright© Satyaki Ganguly et al. 2013; This article is distributedunder the terms of Creative Commons Attribution 3.0License which permits unrestricted use, distributionand reproduction in any means provided the originalauthors and original publisher are properly credited.(Please see www.ijcasereportsandimages.com/copyrightpolicy.php for more information.)
REFERENCES1. Sneddon IB, Roberts BM. An incomplete form ofacanthosis nigricans. J Br Soc Gastroent1962;3:269–72.2. Schwartz RA. Sign of LesserTrélat. J Am AcadDermatol 1996 Jul;35(1):88–95.3. Fanti PA, Metri M, Patrizi A. The sign of LeserTrélatassociated with malignant melanoma. Cutis 1989Jul;44(1):39–41.4. Halevy S, Sandbank M. Transformation oflymphocytoma cutis into a malignant lymphoma inassociation with the sign of LeserTrélat. Acta DermVenereol 1987;67(2):172–5.5. Shamsadini S, Wadji MB, Shamsadini A.Surrounding ipsilateral eruptive seborrheic keratosisas a warning sign of intraductal breast carcinomaand Paget's disease (Leser Trelat sign). DermatolOnline J 2006 Oct 31;12(6):27.6. Ikari Y, Ohkura M, Morita M, Seki K, Kubota Y,Mizoguchi M. LeserTrélat sign associated withSézary syndrome. J Dermatol 1995 Jan;22(1):62–7.

IJCRI – International Journal of Case Reports and Images, Vol. 4 No. 1 , January 201 3. ISSN – [0976-31 98]
IJCRI 201 3;4(1 ):83–85.www.ijcasereportsandimages.com
7. Flugman SL, McClain SA, Clark RA. Transienteruptive seborrheic keratoses associated witherythrodermic psoriasis and erythrodermic drugeruption: report of two cases. J Am Accad Dermatol2001 Dec;45(6 Suppl):S212–4.
Ganguly et al. 85
Access full text article onother devices Access PDF of article onother devices
Related Documents











